The Cellular Microenvironment and Metastases
Erinn B. Rankin, Janine Erler and Amato J. Giaccia
• Metastases are responsible for more than 90% of all cancer-related deaths.
• Gene mutations, the tumor microenvironment, and host cells drive the metastatic spread of tumor cells.
• Metastasis can be subdivided into four steps: invasion, intravasation, survival in circulation, and extravasation.
• Colonization of metastatic tumor cells requires the ability to proliferate in a foreign tissue and stimulate angiogenesis.
• The formation of a premetastatic niche is essential for the growth of extravasating metastatic tumor cells.
• Organ specificity of tumor metastases is determined both by blood flow and tissue-specific factors.
• Primary tumors possess stem cells that can recapitulate the tumor from a single cell, and a subset of these cancer stem cells may inherently possess altered gene expression changes with increased metastatic potential.
• Antimetastatic therapy will likely require the targeted inhibition of many pathways that control proliferation, invasion, and angiogenesis.
Mechanisms of Metastasis
Hematogenous Metastasis
Most tumors metastasize through the bloodstream. Metastatic dissemination through the vasculature involves a series of distinct cellular adaptations, including invasion and migration from the primary tumor; intravasation into the vasculature; arrest and extravasation from the vasculature into the distant organ; and proliferation and survival into the new tissue microenvironment (Fig. 3-1). The acquisition of these biological traits by tumor cells involves the coordination of both cellular intrinsic and extrinsic signals.
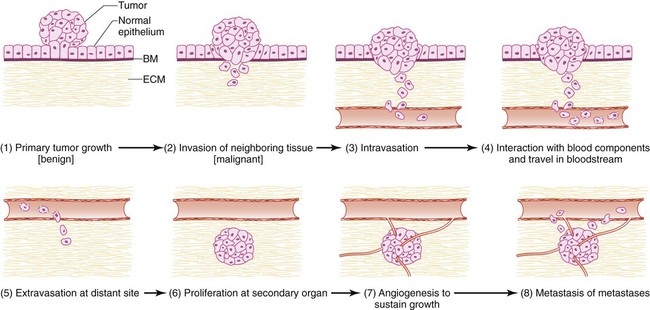
Invasion
Changes in Cell Adhesion
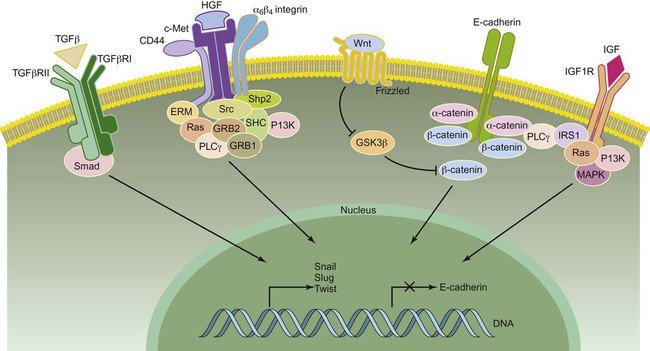
The first step of metastasis is invasion. Cells must undergo changes in their cell-cell and cell-matrix adhesion interactions to dissociate themselves from the tumor.1 Acquisition of an invasive phenotype requires changes in expression of genes that control cell-cell adhesion, as well as proteolytic degradation of the extracellular matrix (ECM).2 Cell-cell adhesions are mediated primarily by E-cadherin proteins expressed at junctions between cells.3 Cadherins bind cells through protein-protein interactions at their extracellular domains, whereas their intracellular domains signal to catenins and the actin cytoskeleton. Changes in E-cadherin expression allow cells to detach from their neighbors and begin their migratory route toward the circulatory or lymphatic system to seek out new terrain. Reduced expression of E-cadherin is often observed in aggressive cancers through epigenetic silencing, proteosomal degradation, proteolytic cleavage, or mutation. In fact, inactivating mutations of E-cadherin have been shown to predispose patients to gastric cancer, implicating E-cadherin as a tumor suppressor gene.1
Loss of E-cadherin is highly associated with epithelial to mesenchymal transition (EMT), a program that is essential for numerous developmental processes. 3 The acquisition of the invasive phenotype has many similarities to EMT, including loss of cell-cell adhesion mediated by E-cadherin repression and an increase in cell mobility. During EMT, a switch occurs from E-cadherin (an epithelial cell marker) to N-cadherin expression (a mesenchymal cell marker), which promotes cell-matrix adhesion instead of cell-cell adhesion.3
Several signal transduction pathways, such as the Ras-MAPK and WNT pathways, have been shown to regulate EMT (Fig. 3-2). In particular, the Ras-MAPK pathway activates two related transcription factors known as Snail and Slug.4,5 Both of these proteins act as transcriptional repressors of E-cadherin, and their expression induces EMT in cancer cells.6 Studies have indicated that Slug is an independent prognostic parameter for poor survival in patients with colorectal carcinoma. 7 Twist, another basic helix-loop-helix transcription factor that is necessary for proper embryonic development, has also been shown to induce EMT through the repression of E-cadherin. 8 Both Twist and Snail expression levels are elevated in patients with breast cancer and are associated with poor prognosis.8,9 Dysregulation of Wnt signaling is common in many types of human cancers and regulates EMT in part through Snail activation, an important early step in metastasis.10
Cell Motility
Cancer cells are able to take advantage of many mechanisms to migrate and invade, including both individual and collective cell-migration strategies.2 Most cancer cells of epithelial origin undergo EMT and acquire invasive migration capacity to enter the circulatory or lymphatic system. Invasive migration is a dynamic and complex process involving changes in cell-matrix adhesion and the cytoskeleton. Changes in cell-matrix adhesion are necessary for the leading edge of the cell to grab onto the matrix surrounding it and pull itself forward in a movement similar to an inchworm. This invasive migration can be viewed as cycles of adhesion and detachment, allowing the cell to bind, then detach after pulling forward. Cell-matrix adhesions are in large part regulated by integrin proteins. Integrins are heterodimers of one of 18 alpha and 8 beta transmembrane proteins that bind to specific components of the ECM. 11 They can transmit signals into or out of the cell and are important mediators of malignant transformation.
Integrins are stimulated when they come into contact with specific ECM substrates, or through growth factor–stimulated signaling where they interact with receptor tyrosine kinases.14–14 For example, hepatocyte growth factor (also known as scatter factor) influences invasion by signaling through its receptor c-met.15 Integrin stimulation also promotes formation of focal adhesion contacts, focal adhesion kinase (FAK) activation through phosphorylation, and formation of FAK-Src complexes.14 It is noteworthy that Src mutations that have been implicated in tumor cell motility are often observed in human cancers such as adenocarcinoma of the colon.16 Intracellular signaling mediated by FAK activates Rac, RhoC, cdc42, and other guanosine triphosphatases that mediate cellular changes required for invasion.12 These changes include actin-myosin contraction that propels the cell forward and recruitment of matrix metalloproteinases (MMPs) to focal adhesion sites where they degrade the ECM, allowing the cell to glide forward when focal adhesion complexes are disassembled after contraction.2 In addition, the remodeled matrix tracks left behind the cell have been shown to facilitate movement of subsequent cells, similar to the generation of ski tracks by the first cross-country skier that allow subsequent skiers to move more easily.17
Disruption of the Basement Membrane
The basement membrane provides a physical barrier between epithelial cells and the stroma. The basement membrane is composed of numerous glycoproteins and proteoglycans that provide ligands for integrins, permitting control of cell orientation and outside to inside signaling. Epithelial and stromal cells produce a mixture of these components that form a dense meshwork underlying the epithelial cells. Tumor cells overcome this barrier by altering the expression of their cell surface receptors such that they can now adhere to basement membrane components.2 For example, tumor cells will increase expression of integrins that can bind laminin and collagen IV.18 Increased CD44 expression permits cell binding to hyaluronan, a basement membrane proteoglycan, and is observed in several types of human cancer, including metastatic colon carcinomas.19 In addition, cancer cells modify the components of the basement membrane to ease penetration. For example, reduced laminin expression is observed in poorly differentiated human colon carcinomas.20
ECM protease activity is tightly regulated by proteins that inhibit their functions.21 Tumor cells proteolytically disrupt the basement membrane by altering the balance between ECM proteases and their inhibitory proteins. For example, elevated MMP expression and activity increases degradation of the basement membrane. MMP-1 (also known as collagenase or gelatinase) degrades collagen IV and is increased in highly metastatic cancer cells.22 MMP degradation of the ECM not only facilitates cell movement but also generates a large number of bioactive cleaved peptides and releases growth factors and chemokines trapped within the ECM mesh.21 For example, MMP activity releases active forms of proteoglycans including heparin, hyaluronate, and chondroitin sulfate.23,24
Intravasation
The entry of tumor cells into the circulation (intravasation) and the exit of tumor cells from the circulation (extravasation) to host tissue represent critical steps in the metastatic process. One clear difference between intravasation and extravasation has to do with the composition of the blood vessels. Tumor blood vessels are malformed and irregular, often possessing breaks in their thin lining that permit the easy access of tumor cells into the circulation. In contrast, the vasculature of normal tissue where tumor cells extravasate do not have these same features. This very observation suggests that the processes of intravasation and extravasation are distinct and probably require different gene functions. Our knowledge of the genetic determinants involved in intravasation is limited. Gradients of chemoattractant proteins such as chemokines have been proposed to guide cells toward the circulatory system.17 In addition, tumor cells move along collagen fibers produced by invading cells, a process facilitated by host macrophages.17
Survival in the Circulatory System
Before these tumor cells can extravasate into a target tissue, they must first survive the environment of the circulatory system. Tumor cells in the circulatory system are subjected to immune attack, circulatory forces, and anoikis (apoptosis induced by loss of adhesion).25 Circulating tumor cells bind to platelets to protect themselves from these dangers, thus increasing their chance of survival.28–28 Tumor cells also bind to coagulation factors including thrombin, fibrinogen, tissue factor, and fibrin, creating emboli.29 These tumor cell emboli are more resilient to both circulatory forces and immune attack and have been shown to have greater metastatic potential than single tumor cells.27 In the circulation, aggregates of tumor cells are termed homotypic clumps because they are homogeneous in their cellular composition, whereas those associated with platelets are termed heterotypic clumps and may possess greater metastatic potential for the reasons described previously.26,28
Arrest and Extravasation
Much of our knowledge of tumor cell extravasation is patterned after leukocyte transmigration through endothelium. It is well known that leukocytes arrest before transmigration. Similarly, tumor cell arrest can occur passively through mechanical lodging or can be allowed by cell-surface molecules.32–32 Endothelial cells are constantly shed from the blood vessel walls, creating temporary gaps to which tumor cells can more easily attach because basement membrane components are exposed.35–35 Vessel wall damage also attracts platelets and tumor cells associated with platelets, which is enhanced by fibrinogen expression on the endothelial cell surface.36,37 Fibrin blood clots at the sites of tumor cell arrest can further damage vessels, attracting more platelets and circulating tumor cells.38 Increased blood coagulation is often observed in patients with cancer as a result of elevated levels of thromboplastin, procoagulant A, and phosphatidylserine produced by tumor cells.39,40 The most severe manifestations of this hypercoagulation state were described by Trousseau many years ago. The induction of the enzymes involved in this state can also be enhanced by changes in the tumor microenvironment.41
Tumor cell arrest is allowed by endothelial cell P- and E-selectins that bind to the tumor cells and by tumor glycosylation patterns and cell-cell adhesion molecules such as integrins and CD44.42–47 Increased cell-surface expression of mucin carbohydrate is associated with increased metastatic potential in human colon carcinoma.48 Tumor clump formation additionally facilitates tumor cell arrest by increasing the number of adhesive interactions. ECM components such as fibronectin and laminin enhance tumor cell arrest, and administration of targeting peptides to fibronectin and laminin can reduce metastatic formation.49 Tumor cells may reside and grow within the intravascular space until the metastatic lesion physically breaks through the vessel.50 Tumor cells may also extravasate by inducing endothelial cell retraction, permitting cell attachment to the ECM.50 It is highly noteworthy that vascular endothelial growth factor (VEGF) increases vascular permeability and may permit extravasation through Src activation.51,52 Thus anti-VEGF therapy could potentially act to inhibit metastases by decreasing vascular permeability. In some cases, tumor cells direct their movement and invasion into new organ terrain by following migrating white blood cells and tissue motility factors.53
Proliferation
The final steps of metastasis involve the resumption of cell proliferation at the secondary site and induction of angiogenesis to supply oxygen and nutrients. The host tissue can influence tumor growth through autocrine, paracrine, and endocrine signals. However, it is the net balance of positive and negative signals that determines metastatic proliferation. This phenomenon can partially explain organ-specific metastasis, because only certain cells will be able to respond to tissue-specific proliferation-stimulating signals and leave their dormant state.54 For example, insulin-like growth factor–1 (IGF1), hepatocyte growth factor, and transforming growth factor–α (TGF-α) are highly expressed in the liver,57–57 and cancer cells from colon, breast, and bladder overexpress receptors for these ligands, such as epidermal growth factor receptor58–62 and c-met receptor,63 resulting in proliferation of metastatic cells in these tissues.
Angiogenesis
The formation of a new blood supply from preexisting vasculature is stimulated by an angiogenic “switch” that occurs when the ratio of inducers to inhibitors is increased. Inhibitors of angiogenesis include the ECM proteins thrombospondin and endostatin.66–66 Angiogenic inducers include VEGF, platelet-derived growth factor, basic fibroblast growth factor, TGF-β, and ephrin, along with their family members.67 Of these, VEGF is the best characterized and has successfully been targeted through the use of monoclonal antibodies and soluble receptors.68 VEGF increases angiogenesis by stimulating endothelial cells, mobilizing endothelial progenitor cells, stimulating outgrowth of pericytes that line the walls of mature blood vessels, and increasing vascular permeability, allowing macromolecules to traverse endothelium.69,70 Furthermore, VEGF is thought to be a key molecule for the homing of VEGF receptor (VEGFR)–positive bone marrow–derived progenitor cells involved in premetastatic niche formation (see the later discussion in this chapter)71 and for homing of VEGFR-positive tumor cells to metastatic sites.72 Recruitment of bone marrow–derived circulating endothelial cells additionally increases angiogenesis.73 Thus angiogenesis is important both for primary tumor and metastatic tumor growth, making it an attractive target.
Lymphatic Metastasis
The vascular and lymphatic systems have numerous connections, and metastasizing tumor cells can easily pass from one system to another. In tumors such as breast, melanoma, prostate, gastric, and colon cancer, metastasis to the nearby draining lymph node is often the first step in metastatic dissemination.74 Additionally, the presence of lymph node metastases correlates with decreased survival in these patient populations.75 Therefore examination of lymph nodes for metastases is an important parameter when determining staging, prognosis, and treatment options in patients with cancer.74
Tumor cells access lymphatic vessels via multiple mechanisms. The high internal pressure within tumors provides a net flow of interstitial fluid and dissociated tumor cells away from the tumor and into the lymphatics and the local draining lymph node.75 Additionally, a strong correlation exists between tumors that actively secrete prolymphangiogenic factors, including VEGF-C and VEGF-D, and the development of lymph node metastases.74 Lymphangiogenesis is a critical process driving lymph node metastasis. Overexpression of prolymphangiogenic factors by tumors is sufficient to promote lymph node and distant metastasis, whereas inhibition of lymphangiogenesis significantly inhibits lymph node metastasis.74 In addition to tumor cells, lymphatic endothelial cells (LECs) promote lymph metastases by actively recruiting tumor cells to the lymphatic vessels. LECs produce a variety of chemoattractant factors such as SDF1 and CCL21 that bind to cognate receptors CXCR4 and CCR7 expressed by tumor cells.74 Together these findings demonstrate that lymphatic metastasis is mediated at least in part by active processes that involve signaling between the tumor cells and lymphatic endothelial cells. Therefore therapeutic intervention of tumor cell–LEC signaling may offer therapeutic benefits in the treatment of metastatic cancer.
Transcoelomic Metastasis
In contrast to the majority of tumors that disseminate through the vascular or lymphatic systems, ovarian cancer predominately disseminates through the transcoelomic route. Ovarian cancer is a highly metastatic cancer, because approximately 70% of patients diagnosed with epithelial ovarian cancer present with stage III or IV disease in which the tumor has disseminated beyond the ovaries and pelvic organs to the peritoneum and mesothelial lining of abdominal organs.76 Transcoelomic metastases significantly contribute to morbidity in patients with ovarian cancer because these tumors are numerous and often obstruct vital organs in the abdomen, including the bowels.77 Additionally, transcoelomic metastases are associated with the formation of ascites that contribute to morbidity by increasing intraabdominal pressure, leading to impaired circulation and respiratory distress.77 Similar to hematogenous metastasis, transcoelomic metastasis occurs through a series of distinct steps that allow for successful colonization of tumor cells to mesothelial cells lining the abdominal organs.
Adhesion
The first step in transcoelomic metastasis is detachment from the primary tumor. Although this process in ovarian cancer is not completely understood, evidence suggests that EMT transition contributes to this process. Similar to the hematogenous tumors previously described, the EMT phenotype in ovarian cancer allows for the tumor cells to lose their cell-cell and cell-basement membrane adhesive properties. Loss of the cell adherin junction protein, E-cadherin, is thought to be a key event triggering peritoneal dissemination. E-cadherin loss is found in metastatic tumor cells and is associated with an invasive phenotype and poor patient survival in persons with ovarian cancer.78 In addition to active cell detachment, it has been proposed that ovarian tumor cells may also detach from the primary tumor through passive mechanisms in which tumor cells on the ovarian surface exfoliate off into the peritoneal fluid.77
Anoikis
Circulating tumors cells in the peritoneal fluid must resist anoikis for successful metastatic colonization. Several molecules have been shown to contribute to anoikis resistance in ovarian cancer. One of the first molecules to be linked to ovarian tumor cell resistance to anoikis is RAB25, a small guanosine triphosphatase that is increased in stages III and IV serous ovarian cancers.79 Increased RAB25 expression promotes anchorage-independent proliferation, prevents anoikis, and induces the aggressiveness of cancer cells in vivo. The immunomodulatory protein B7-H4 was also shown to protect ovarian cancer cells from anoikis. B7-H4 is an attractive therapeutic target for ovarian cancer therapy given the restricted expression of B7-H4 on normal tissue and its upregulation in the majority of ovarian tumors.80 Additional proteins including ADRB2 and ABHD4 have been shown to mediate resistance to anoikis in ovarian cancer cells and are additional therapeutic targets for the treatment of ovarian cancer.81,82
Immune Evasion
When circulating in the peritoneal fluid, ovarian tumor cells employ mechanisms to evade immunologic attack. Proinflammatory cytokines, complement proteins, and activated T cells are all found at significantly elevated levels in peritoneal fluid collected from patients with ovarian cancer.77 Perhaps the most striking evidence supporting a role for immunosurveillance in ovarian cancer is shown by clinical data demonstrating a 38% 5-year survival rate of patients who had CD3+ T cells contained within the ovarian tumor specimen compared with a 4.5% 5-year survival rate among patients without infiltrating T cells.83 Consistent with an immunologic response against tumors, patients with ovarian cancer produce antibodies against multiple tumor antigens including mutated p53, folate receptor α, IGF binding protein 2, HER2/neu, and topoisomerase II α.84 Patients with antibodies recognizing p53 have a significantly higher survival rate compared with those without p53 antibodies, further supporting the notion that ovarian tumors are under immunosurveillance.84
To overcome immunosurveillance, ovarian cancer cells have developed multiple mechanisms to evade and modulate the immune responses. Cancer cell spheroid formation is thought to protect tumor cells from immune attack by limiting the penetration of complement proteins and antibodies into the spheroids.77 Additionally, ovarian cancer cells actively suppress immune responses by expressing proteins that recruit regulatory T cells and myeloid-derived suppressor cells, and they produce proteins that inhibit complement activation.77
Peritoneal Implantation and Metastatic Growth
When establishing peritoneal implants, ovarian cancer cells attach to and invade into to the mesothelium covering the surface of all abdominal organs including the peritoneum, omentum, diaphragm, and bowel. The mesothelium is composed of a single layer of mesothelial cells that are attached to a basement membrane containing collagen types I and IV, fibronectin, and laminin.78 Integrins and the hyaluronic acid cell surface receptor CD44 are the key molecules mediating ovarian attachment to the mesothelial layer. Once attached, the MMP family mediates further adhesion and invasion into the basement membrane.78 Once at the distant site, the activation of VEGF and the induction of tumor angiogenesis plays a critical role in successful metastatic colonization. Together, these data demonstrate that many aspects of transcoelomic metastasis are mediated by active signaling processes between tumor cells and cells within the tumor microenvironment. Targeting these processes offers therapeutic strategies for the treatment of metastatic disease.85
Metastasis and Tumor Reseeding
Until recently, it was thought that tumor metastasis occurred in a unidirectional manner by which tumor cells leave the primary tumor site and colonize distant organs. Evidence now suggests that circulating tumor cells can also reseed the primary tumor. Kim and colleagues86 were among the first to demonstrate that circulating tumor cells return to the primary tumor site. Furthermore, they observed that the presence of circulating tumor cells in the primary tumor stimulated tumor growth, angiogenesis, and stromal cell recruitment.86 These data demonstrate that cells that have undergone metastatic selection respond to chemoattractant signals produced by the primary tumor and represent an aggressive subpopulation of tumor cells that promote tumor progression.
Tumor Microenvironment and Metastasis
Although the cellular intrinsic traits acquired by tumor cells as previously described are required for successful metastatic colonization, cellular and molecular factors within the tumor microenvironment also significantly contribute to metastatic progression. The tumor microenvironment contains a number of cell types that promote tumor progression, including cancer stem cells, endothelial cells and pericytes, bone marrow–derived and local stromal cells, immune cells, and fibroblasts (Fig. 3-3).87 Additionally, extracellular factors within the tumor microenvironment such as hypoxia promote tumorigenic phenotypes within these cell types.
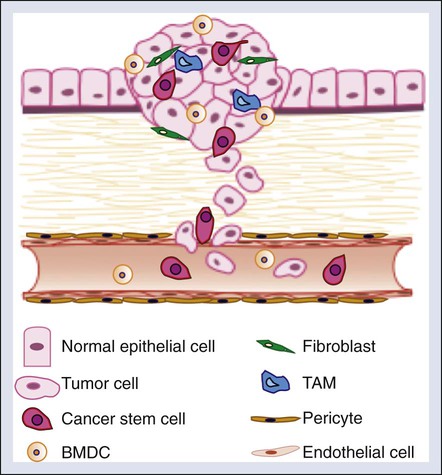
Cancer Stem Cells
The recent discovery of cancer stem cells has revolutionized our way of thinking about cancer. Our traditional view of tumors with a relatively homogenous population of tumorigenic cells has been significantly revised with the isolation and characterization of cancer stem cells. Stem cells are primal cells that retain the ability to renew themselves through cell division and can differentiate into a wide range of specialized cell types. Cells with stem cell properties have been identified in a variety of solid tumors including colon, breast, head and neck, and pancreatic tumors, glioblastomas, medulloblastomas, and melanoma.88 This often rare subpopulation of tumor cells exhibits enhanced tumor-initiating potential, and the ability to self-renew and differentiate into multiple cell types.
The origin of cancer stem cells remains unclear and may differ between tumor types. It has been hypothesized that cancer stem cells arise from either transformed resident stem and/or progenitors cells or may represent a dedifferentiated epithelial tumor cell. In intestinal cancer, mouse modeling studies have identified intestinal stem cells as the cells of origin. Cell type–specific deletion of APC in mice revealed that intestinal stem cell–specific deletion of APC leads to rapid cellular transformation with uncontrolled cellular growth and solid tumor formation. In contrast, deletion of APC in short-lived transit-amplifying cells was not sufficient to induce long-term tumor growth.89 Stem cell properties can also be acquired by tumor cells through EMT. Induction of EMT in immortalized human mammary epithelial cells was sufficient to induce the expression of stem cell markers, enhance self-renewal, and increase the number of tumor-initiating cells.90 Although the molecular mechanisms that drive the cancer stem cell phenotype in tumor cells remains largely unknown, a recent study shows that the transcription factors Slug and Sox9 drive EMT and the cancer cell phenotype in breast cancer cells.91 Future studies are eagerly awaited to further delineate the intracellular signaling pathways driving the cancer stem cell phenotype in vivo.
The role of cancer stem cells in multistage tumor progression, particularly with respect to metastasis, is poorly defined. Given that metastasis is an infrequent event that is achieved by only a small portion of cancer cells that reach distant sites, it has been hypothesized that cancer stem cells may be responsible for metastatic disease. Evidence to support this hypothesis has been generated in models of breast and pancreatic cancer. In the MMTV-PyMT model of breast cancer, Malanchi et al.92 observed that only the CD90+CD24+ population of cancer stem cells isolated from primary breast tumors contained cells with the ability to form metastases in the lung when introduced into the tail vein. Furthermore, in this study it was found that the extracellular matrix protein called periostin (POSTN), which is produced by fibroblasts and stromal tumor cells, is a critical factor in maintaining the cancer stem cell phenotype and metastatic potential in these cells.92 Similarly, in pancreatic cancer, lineage tracing studies showed that circulating pancreatic tumor cells exhibit EMT and cancer stem cell properties and initiate tumor formation. A critical role of inflammation in maintaining the ability of cancer stem cells to metastasize was observed in this study.93 Additionally, the extracellular protein tenascin C (TNC), which is expressed by stem cell niches and cancer cells, was also found to promote stem cell signaling and lung metastases in breast cancer cells.94 Collectively, these data strongly implicate the tumor microenvironment in promoting the cancer stem cell phenotype and the initiation of tumor metastasis.
Endothelial Cells and Pericytes
Tumor angiogenesis facilitates the hematogenous spread of metastatic tumors. Endothelial cells and pericytes are important structural and functional components of the tumor vasculature and thus both have been considered as potential targets in metastatic therapy.95 In addition to the important role that endothelial cells play in forming the framework of the vasculature, endothelial cells also play an active role in promoting metastasis by upregulating the expression of adhesion molecules, including E-selection, P-selectin, and vascular cell adhesion molecule 1, to induce tumor cell adhesion and migration.96
Similar to endothelial cells, pericytes are thought to play a protumorigenic role by supporting blood vessel maturation and function. Pericytes promote endothelial cell survival and stabilize the tumor vasculature.97 Therefore it has been proposed that targeting pericytes alone or in combination with endothelial cells may be an effective strategy in the treatment of cancer.98 However, clinical data indicate that low pericyte coverage is associated with metastasis and poor prognosis in a number of cancers.98 A recent study demonstrated that depletion of pericytes suppressed tumor growth and enhanced metastasis murine models of breast cancer.98 Enhanced metastasis was associated with increased levels of tumor hypoxia, EMT, and Met receptor activation.98 These findings indicate that pericyte coverage may be important to stabilize the tumor vasculature and prevent the hypoxic selection of aggressive tumor cells (see the discussion on hypoxia and metastasis later in this chapter). These findings raise concerns when considering antipericyte treatments in cancer therapy, especially in the setting of metastasis.
Immune Cells
A variety of inflammatory cells infiltrate tumors that exert both positive and negative effects on tumor progression and metastasis. As previously described, T cells, myeloid-derived suppressor cells, and dendritic cells contribute to immunosurveillance and immunosuppression. Recent studies have linked a unique subset of macrophages, termed “tumor-associated macrophages” (TAMs), to tumor progression and metastasis. Clinically, the presence of excessive macrophages is associated with poor prognosis in patients with breast, prostate, cervix, bladder, endometrial, and kidney cancer.99,100 TAMs express a variety of growth factors and cytokines that promote tumor angiogenesis, invasion, intravasation, and metastasis.96
Many questions remain regarding the factors that control the biological activities of TAMs within the tumor microenvironment. TAMs localize to regions of hypoxia, and thus it has been hypothesized that the hypoxic microenvironment may influence the unique characteristics and gene expression profiles of TAMs. Indeed, TAMs express high levels of the hypoxia-inducible transcription factor HIF2, which has been found to be an independent prognostic factor of outcome.101 When macrophages are exposed to hypoxia, they upregulate the expression of mitogenic and proangiogenic cytokines.102 Experimentally, deletion of myeloid HIF2 reduced TAM infiltration into tumors and impeded tumor proliferation and growth.102 In addition to regulating tumor angiogenesis and invasion, a subset of myeloid progenitor cells have been shown to promote immune evasion. Cells co-expressing the macrophage marker CD11b and Gr1 have been shown to inhibit cytotoxic T lymphocyte and natural killer activity.99 Thus targeting TAMs and CD11b cells may be a viable mechanism for antimetastatic therapies.
Fibroblasts
Accumulating evidence supports a critical role for cancer-associated fibroblasts (CAFs) in tumor progression and metastasis. Clinically it was observed that epigenetic changes and mutations commonly found in cancer cells, such as p53 and PTEN mutations, can also be found in cancer-associated stroma.103,104 These studies provided early evidence to suggest that alterations in the stroma may contribute to tumor progression. Gene expression profiling studies of CAFs in human specimens and murine tumor models revealed an “activated” proinflammatory gene signature.105 Among the cytokines produced by CAFs, the chemokine CXCL12 is of particular interest given its role in driving tumor cell migration and recruitment of endothelial progenitor cells expressing the CXCR4 receptor. Consistent with these data, an important role for CAFs in promoting tumor invasion has been observed in several murine models of cancer.105 In breast cancer, Hu and colleagues106 identified a role for CAFs in the transition of ductal in situ carcinoma to invasive carcinoma. Co-injection of fibroblasts with mammary ductal carcinoma in situ cells resulted in invasive carcinomas, whereas injection of ductal carcinoma in situ cells alone was only sufficient to induce mammary ductal carcinoma in situ.106 CAFs promote tumor cell invasion by establishing invasion-permissive tracks in the extracellular matrix through the secretion of factors that induce matrix remodeling.105,107 In addition to promoting tumor invasion, recent studies have found supportive roles for CAFs in the maintenance of cancer stem cell signaling and the establishment of the premetastatic niche.105 Further characterization of the distinct subsets of CAFs relevant to metastasis in various tumor types is needed. Additionally, further elucidation of the interplay between tumors and the stroma are warranted and may reveal novel strategies in the treatment of metastatic disease.
Hypoxia
Hypoxia is a potent microenvironmental factor driving metastatic tumor progression. Clinically, hypoxia is associated with metastasis and poor survival in a variety of patients with cancer.110–110 Hypoxia selects for cells with low apoptotic potential and increases genomic instability, allowing for rapid mutational adaptations.113–113 Additionally, hypoxia directly increases the expression of genes involved in glucose transport, angiogenesis, anaerobic metabolism, cell survival, invasion, and metastasis. All of these changes allow cells to adapt to oxygen-deprived conditions and permit cells to escape these conditions by establishing new blood supplies or by physically moving from an oxygen-poor environment to an oxygen-rich environment.
The primary molecular mediators of hypoxic signaling are the hypoxia-inducible transcription factors HIF1 and HIF2. In the presence of oxygen, the alpha subunits of HIF1 and HIF2 are rapidly degraded through the cooperative actions of prolyl hydroxylase enzymes (PHD1, 2, and 3) and the E3 ubiquitin ligase substrate recognition component VHL.114,115 Under hypoxia, HIF1 and HIF2 are stabilized and activate the expression of genes containing hypoxia response elements.108 More than 200 genes are activated in response to HIF1 and HIF2, which allows cells to survive and adapt to low oxygen tensions.
Consistent with the association between hypoxia and metastasis, HIF1 and HIF2 are highly expressed in primary tumors and metastases. Immunohistochemical analysis of human cancer specimens indicates that the majority of primary tumors and metastases have increased HIF1 and/or HIF2 protein compared with the normal adjacent tissue.110–110 Moreover, increased HIF expression is often associated with increased patient mortality.116 Experimentally, overexpression of HIF in tumor cells promotes metastasis,117 whereas inactivation of HIF significantly decreases the metastatic potential of metastatic tumor cells.120–120 These clinical and experimental findings indicate an important role for HIF in metastatic tumor progression.
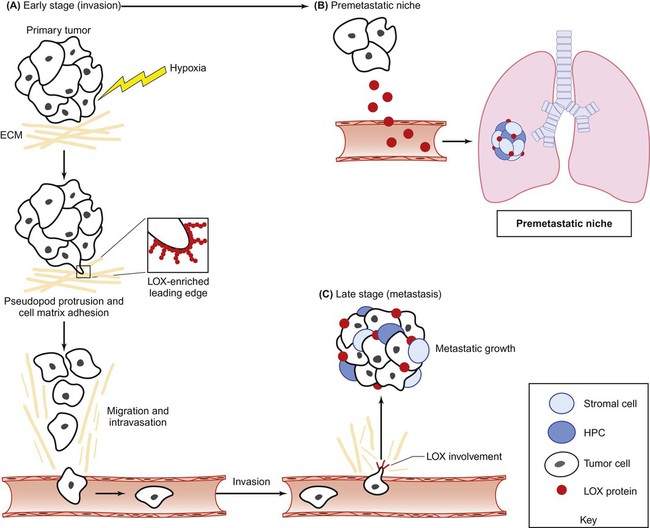
HIFs influences multiple steps within the metastatic cascade (Fig. 3-4). HIF1 and HIF2 activate the early stages of metastasis in the primary tumor by promoting EMT, invasion, and migration. HIFs promote invasion and migration through multiple mechanisms. First, HIF regulates the E- to N-cadherin switch during EMT phenotype through the activation of E-cadherin repressors, including Twist1, Zeb1/2, Snail, and TCF3.117,121,122 Second, HIF directly upregulates the expression of multiple factors driving cellular invasion and migration. Most notably, HIF drives the expression of the ECM protein lysyl oxidase (LOX).123 Increased LOX expression correlates with decreased distant metastasis-free survival and overall survival in patients with breast and head and neck cancer.123 In addition, LOX activation promotes the invasive and metastatic potential of breast cancer cells. Most importantly, Erler and colleagues123 demonstrated that genetic and pharmacologic inhibition of LOX in metastatic breast cancer is sufficient to prevent hypoxia-induced cell invasion and metastasis. These findings indicate that LOX is a promising therapeutic target for the treatment of metastatic disease.
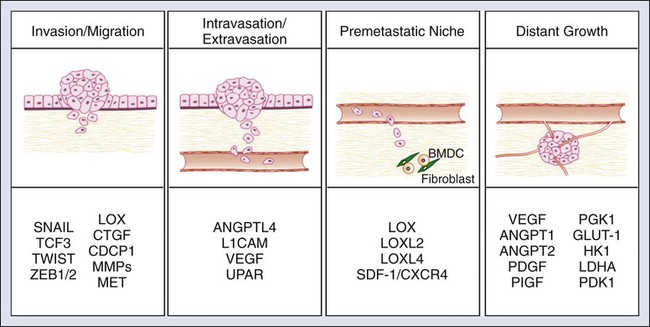
HIF also regulates tumor cell intravasation and extravasation from the vasculature. HIF activity in tumor cells results in the release of factors that modulate endothelial-endothelial cell and endothelial-tumor cell interactions. The upregulation of angiopoietin-like 4 (ANGPTL4) by HIF disrupts extracellular-extracellular interactions and allows for easy passage of tumor cells through blood vessels.119 Simultaneously, HIF strengthens tumor cell–endothelial cell interactions through the activation of L1 cell adhesion molecule.119 Another mechanism by which HIF promotes tumor cell intravasation and extravasation is through the activation of genes, which control vascular permeability. Hypoxic induction of VEGF, angiopoietin 2, MMPs, and urokinase type plasminogen activator receptor cooperatively act to destabilize the vascular wall and allow for tumor cell entry.124
Third, HIF activity in the primary tumor is involved in the formation of the metastatic niche. As previously mentioned, priming the premetastatic site for the recruitment and survival of metastatic tumor cells is a critical step in successful metastatic colonization. In breast cancer cells, HIF activity results in the upregulation and release of LOX and LOX-like proteins (LOXL2 and 4). These proteins recruit bone marrow–derived cells (BMDCs) into the lung and prime the lung for metastatic colonization.118,123,125 BMDCs produce chemokines that recruit tumor cells, as well as stimulate blood vessel invasion.71,126–128 Another mechanism by which HIF signaling controls the directional migration of metastatic tumor cells sites is through the upregulation of stromal-derived factor–1 (SDF1)/CXCR4 signaling.129,130 Stromal cells within target tissue sites produce SDF1, which recruits cancer cells expressing the receptor CXCR4.96 Although these studies indicate an important cellular intrinsic role for hypoxia and HIF signaling in the primary tumor, future studies are eagerly awaited to elucidate the role of HIF signaling in tumor support cells.
Finally, HIF promotes late stages of metastasis by metastatic growth at the distant site through the stimulation of angiogenesis. Similar to the primary tumor, angiogenesis is a critical step for metastatic tumor growth. VEGF-A is a proangiogenic factor produced by tumor cells that stimulates the recruitment and proliferation of endothelial cells and supports pericytes.96 VEGF-A is a well-established HIF target and is significantly induced by HIF signaling in both primary tumors and metastases.131 In summary, HIF affects multiple aspects of tumor metastasis, indicating that therapeutic targeting of HIF may be an effective strategy to selectively inhibit multiple aspects of metastasis.
Patterns of Metastasis
Seed and Soil Hypothesis
The patterns of colonization cannot solely be explained by the circulatory, lymphatic, and transcoelomic routes previously described. The propensity for certain tumors to seed in particular organs was first discussed as the “seed and soil” theory by Stephen Paget more than a century ago in 1889.132 For example, prostate cancer often metastasizes to the bones, colon cancer has a tendency to metastasize to the liver, and the primary site for ovarian metastasis is the omentum.133 Colonization is an extremely inefficient process that is heavily dependent on the interactions between “seeding” tumor cells and the “soil” microenvironment of the secondary site. Many factors, including formation of a premetastatic niche and interactions between circulating tumor cells and the distant microenvironment, determine patterns of colonization.
Premetastatic Niche
During the past decade, studies have convincingly shown that formation of a premetastatic niche is essential for the growth of extravasating metastatic tumor cells.71 Soluble factors and bone marrow derived cells are recruited to the distant site prior to the arrival of tumor cells to establish a permissive environment for malignant colonization. The mechanisms by which the premetastatic niche is formed remain largely unknown. However, recent studies have shown that factors secreted by the primary tumor are directly involved in establishing a permissive ECM environment and in recruiting bone marrow derived cells to the distant site.
BMDCs expressing VEGFR1, c-kit, CD133, and CD134 have been detected at distant sites and increase angiogenesis at the premetastatic sites. Targeted inhibition of VEGFR1 prevented niche formation and subsequent metastatic progression. This tissue preconditioning may thus represent a key step that could be targeted therapeutically, although studies with anti-VEGF therapy fail to show significant benefit in preventing metastatic growth for long periods. The role of hematopoietic progenitor cells and other BMDCs in tumor progression is reviewed by Kaplan and colleagues134 and shown in Figure 3-5.
An additional function of the premetastatic niche is to guide metastases to specific organs. Kaplan and coworkers71 demonstrated that injection of secreted factors collected from cancer cells that metastasize to multiple organs could permit cancer cells that only metastasize to the lung when grown as subcutaneous tumors in mice to display widespread metastasis through governing BMDC accumulation. Elevated fibronectin expression by fibroblasts and fibroblast-like cells resident at premetastatic sites seems to be a critical factor in the development of the premetastatic niche. The key tumor-secreted factors that determine metastatic sites and mediate premetastatic niche formation have yet to be identified, although a role for TNF-α), TGF-β, and VEGF-α pathways has been demonstrated.135
MMPs may also play an important role in this process. For example, VEGFR1 signaling has been shown to be required for premetastatic induction of MMP9 expression in endothelial cells and macrophages of the lungs by distant primary tumors.136 This process is thought to make the lung microenvironment more compliant for invasion of metastasizing cells. This concept is supported by the finding that pericyte recruitment and angiogenesis are not observed in tumor-bearing mice with MMP9 knockout bone marrow cells.137 Furthermore, stromal-derived MMP2 and MMP9 have also been shown to contribute to the establishment and growth of metastases.138 Although evidence indicates that MMPs play multiple roles in metastases, clinical trials with MMP inhibitors have failed to show significant efficacy. In large part, this outcome has been due to unexpected normal tissue toxicities and conflicting roles in metastases.
Organ Specificity
The organ distribution of metastases from a primary tumor is not random. Minn and colleagues139 used bioluminescence imaging to reveal patterns of metastasis formation by human breast cancer cells in mice. They also showed that individual cells from the pleural effusion of a patient with breast cancer showed distinct patterns of organ-specific metastasis.140 Single-cell progenies derived from this population demonstrated different abilities to metastasize to the bone, lung, or adrenal medulla. These studies indicate that particular requirements exist for circulating tumor cells to colonize specific organs. The factors contributing to tissue-specific colonization include cellular intrinsic factors within the disseminated tumor cells and specific factors within the tissue microenvironment. Some of the key cellular intrinsic molecules determining organ-specific metastasis have been identified and are briefly discussed in the following section. Additionally, we are beginning to unravel the mechanisms by which infiltrating tumor cells “educate” the normal tissue stroma at distant sites to support metastatic growth. Elucidating the complex signaling networks that exist between tumor cells and their tissue microenvironment offer new opportunities for targeted therapy against cancer.
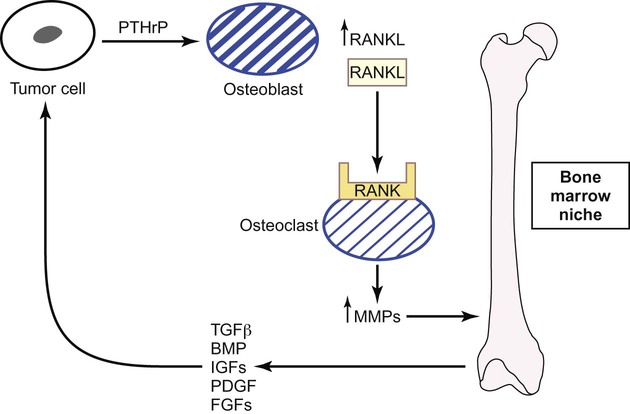
Metastases to the Bone
Two types of bone metastases occur: osteoblastic and osteolytic.141 Osteoblastic metastases are observed in patients with advanced prostate cancer. Both the differentiation of osteoblastic precursors and the activity of osteoblast cells are stimulated by tumor and microenvironmental signals such as bone morphogenetic protein, fibroblast growth factor receptors, and IGF1R.142 Runx2, a key transcription factor that regulates the differentiation of osteoblasts and osteoblastic precursor cells, represents a potential new target for inhibiting osteoblastic metastases by preventing osteoblastic precursor differentiation.143 In contrast, osteolytic metastases are observed in patients with breast cancer or multiple myeloma, and in these patients, interactions between tumor cells and the bone microenvironment result in bone resorption and metastatic growth due to the unique interplay between osteoblasts and osteoclasts (Fig. 3-5).141,144 Parathyroid hormone–related protein secreted by the tumor cells stimulates osteoblasts to produce receptor activator of nuclear factor (NF)–κB (RANK) ligand (RANKL). Consequently, bone-resorbing osteoclast cells are activated by RANKL when it binds to the RANK receptor. Activated osteoclasts upregulate MMPs, which degrade the bone matrix–releasing growth factors such as TGF-β, IGFs, platelet-derived growth factor, fibroblast growth factor, and bone morphogenetic protein.141,145,146 These factors stimulate tumor cells to release parathyroid hormone–related protein, thus restarting this pathway of bone resorption. Gene profiling has identified other important mediators of osteoclastic bone metastases including CXCR4, MMP1, CTGF, and osteopontin.147 Tumor cells also induce osteoclast formation by overexpressing interleukins (ILs) such as IL-8 and IL-11 and by downregulating macrophage colony-stimulating factor.148,149 All of these latter factors represent new targets for metastases, although the importance of each factor in osteoclastic bone metastases requires further clarification.
Metastases to the Brain
Brain metastases are most commonly observed in patients with breast cancer, lung cancer, and melanoma. Vascular access to the brain is strictly regulated by the blood-brain barrier, an endothelial layer surrounding the brain connected by tight junctions and further lined by a basement membrane, pericytes, and astrocytes.150 Macromolecules are not usually able to traverse the blood-brain barrier, and it remains unclear how tumor cells are able to penetrate this barrier. However, once the tumor cells are within the brain parenchyma, glial cells permit establishment and growth of metastases by secreting chemokines, cytokines, and growth factors.151 Other neurotransmitter hormones in the brain such as norepinephrine have also been reported to increase tumor cell motility and metastatic spread.152
Little is known about the key factors that determine colonization of the brain, mostly because there is a lack of good in vivo models of brain metastasis. Overexpression of Stat3 increases melanoma metastasis to the brain and increases invasion of the melanoma cells and angiogenesis, although the pathways modulated by Stat3 signaling require elucidation.153 The dependence of brain metastases on VEGF has been demonstrated experimentally in animals through inhibition studies where VEGF neutralization reduces brain metastases.154,155
In general, patients with brain metastases have an extremely poor prognosis. It is of concern that there has been an increase in the incidence of brain metastases in patients whose systemic disease is well controlled.158–158 For example, patients with breast tumors that overexpress HER2 and who are treated with HER2-targeting trastuzumab (see the later discussion in this chapter) have an incidence of brain metastases twice that of patients with breast cancer who are treated with other agents.157 This result is thought to occur because the brain offers a sanctuary when systemic disease is being controlled.67 The development of drugs that can cross the blood-brain barrier and target brain metastases are of paramount importance in the development of new targeted therapies to tackle this problem. Currently, the best treatment for oligometastases to the brain is radiosurgery.
Metastases to the Lung
Pulmonary metastases are frequently observed in patients with sarcoma, breast, melanoma, gastrointestinal, and kidney cancers. Because cardiac output from the pulmonary artery circulates through the lungs, a high incidence of pulmonary metastases in patients with cancer can be expected on the basis of blood flow alone. Metastases therefore often initiate in pulmonary arterioles and later traverse the basement membrane into the lung parenchyma. TGF-β and NF-κB facilitate this process in breast cancer, as does osteopontin in hepatocellular cancer and ezrin in osteosarcoma and breast cancer.159–164 In vivo studies have identified a gene expression signature for lung metastasis including several membrane-localized and secreted proteins that has been validated in patients with breast cancer.139 Interestingly, this group of genes was able to induce lung metastasis when expressed together but not individually, suggesting essential cooperation between proteins. Increased expression of antiapoptotic proteins such as Bcl2 and Bcl-xl is also observed in lung metastases, facilitating survival and providing resistance to therapy.165–169
Adding to the complexity of signaling interactions needed for successful metastatic colonization in the lung, recent studies have revealed that interactions between breast cancer stem cells and the lung stroma are necessary for metastatic colonization. The ECM components TNC and POSTN have recently been shown to be essential factors required for tumor-initiating cells to form metastases in the lung.92,94,170 Malanchi and colleagues92 showed that tumor-initiating cells induce POSTN expression in lung myofibroblasts to initiate colonization and maintain their stem cell phenotype. Similarly, Oskarsson and colleagues demonstrated a role for the extracellular protein TNC in supporting the breast cancer cell stem cell phenotype and metastases in the lung.94 Interestingly, previous studies have shown that TNC and POSTN form complexes together with collagen type I and fibronectin in the ECM. It was shown that POSTN promotes the incorporation of TNC into the ECM to strengthen the ECM architecture, suggesting that these factors may act through similar pathways in promoting cancer stem cell metastasis.171 In support of this notion, maintenance of the cancer stem cell phenotype by TNC and POSTN was mediated at least in part through the induction of WNT signaling. These studies suggest that targeting signaling pathways mediated through the ECM may be an effective strategy against cancer stem cell–driven metastasis.
Metastases to the Liver
Liver metastases are observed in patients with breast, lung, and pancreatic cancers. However, liver metastases are most commonly found in patients with metastatic colorectal cancer, because the liver is the first capillary bed encountered by the metastasizing cells. The circulatory system of the liver, in particular the liver sinusoids, does not have a barrier limiting macromolecule flux, and it is well perfused and highly permeable, permitting metastasizing cancer cells to establish themselves and grow. Thus tumor cell invasion and survival are probably the key determinants in metastatic colonization of the liver.172 Two types of liver metastases occur: a nonangiogenic “replacement” of liver cells with tumor cells that preserves the stroma, and a “pushing” type of metastasis whereby the liver stroma is not preserved and has higher levels of endothelial cell proliferation.175–175 In light of the “pushing” type of metastases that stimulate angiogenesis, targeting the VEGF pathway experimentally in vivo has been shown to prevent liver metastases and is effective when combined with cytotoxic agents in patients with metastatic colon cancer.178–178 Other molecules thought to be important in colonization to the liver and that could be targeted therapeutically are COX2, integrins, and Src.179–182
Clinical Relevance and Applications
Many of the initial targeted therapies developed for tumor metastasis inhibit factors driving tumor cell invasion and migration. Preclinical studies have demonstrated that agents that inhibit MMPs, the receptor tyrosine kinase AXL, miR-10b, and fascin are effective in blocking the initiation and/or progression of metastatic tumors in mice (for a recent review, see references 183 and 184).183,184 Of all of these agents, MMP inhibitors are the most advanced and have been tested in clinical trials. Disappointingly, these inhibitors failed to increase survival in patients with advanced cancer and were associated with adverse effects.185 Important lessons regarding clinical trial design were learned from these trials. Because MMP inhibitors and other inhibitors of invasion are likely to function at the early stages of metastasis, it is important to thoughtfully identify patient populations that may benefit most from these types of therapies. Additionally, it is likely that the combination of targeted agents controlling distinct aspects of metastasis may prove to be the most effective when targeting metastatic cancer.
An emerging strategy in the treatment of metastasis involves the therapeutic targeting of cellular and molecular constituents within the tumor microenvironment. As previously mentioned, hypoxia and HIF signaling are critical drivers of both the tumorigenic and metastatic phenotypes. HIF and hypoxia-induced proteins represent therapeutic targets that have the potential to be tumor specific because these proteins are highly elevated in both the primary tumor and metastases in comparison with normal tissue.186 Additionally, targeting HIF is an attractive strategy for the treatment of metastatic disease because HIF controls multiple aspects of metastasis, including EMT, invasion, migration, metastatic niche formation, and metastatic tumor growth. In this regard, a number of small molecule HIF inhibitors, including digoxin and acriflavine, have been identified and have shown success in preclinical studies by preventing lung metastasis in mice bearing primary breast tumors.187 Although the efficacy of these inhibitors in treating established metastatic disease remains unknown, these compounds do inhibit metastatic xenograft growth after tumor implantation.119 In addition to targeting HIF directly, a number of HIF-induced proteins have been targeted for metastatic therapy. One very promising target is LOX, a hypoxia-induced secreted protein involved in multiple stages of metastasis (Fig. 3-6).123 LOX contributes to tumor cell invasion by cross-linking collagens in the ECM, which stimulates integrin-mediated cell-matrix adhesion and activation of FAK, and additionally provides a route (“highway”) by which tumor cells may travel. Furthermore, LOX is involved in the formation and maintenance of the metastatic niche, allowing metastatic dissemination and growth. Targeting secreted LOX through antibody or small-molecule inhibition significantly reduced formation and growth of metastases to the lung, liver, bone, and brain in several preclinical studies. Another promising hypoxia-induced target is connective tissue growth factor (CTGF).188 CTGF is an ECM protein that exerts a number of prometastatic activities, including migration, invasion, angiogenesis, and anoikis.189 Both genetic and therapeutic inhibition of CTGF has shown efficacy in primary and metastatic tumor growth inhibition of preclinical models of pancreatic cancer.190,191 The results of clinical trials targeting LOX and CTGF are eagerly awaited.
Among the therapies targeting the cellular components of the tumor microenvironment, the ones that target endothelial cells are the most advanced and have shown success within the clinic.96 Targeting VEGF signaling on vascular and lymphatic endothelial cells has shown clinical benefits in patients with metastatic cancer.192 A variety of VEGF inhibitors have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of metastasis. The humanized anti-VEGF-A monoclonal antibody, bevacizumab, has been approved as a first-line therapy in combination with 5-fluorouracil for metastatic colon cancer.176 Additionally, in patients with metastatic breast cancer and non–small-cell lung cancer, bevacizumab increased survival in combination with chemotherapy.193 In addition to biological therapies, small molecule VEGFR inhibitors have also been developed for the treatment of metastasis. Both sorafenib and sunitinib are approved by the FDA for the treatment of metastatic renal cancer. Phase 3 clinical trials demonstrated that sorafenib monotherapy resulted in a significant increase in progression-free survival in this patient population. The results of future clinical trials targeting additional key cellular components of the tumor microenvironment are eagerly awaited.