Cancer Pharmacology
• Cancer pharmacology encompasses the spectrum of therapeutic issues from everyday clinical treatment to the earliest stages of new drug discovery.
• The number of drugs approved for treating patients with cancer is rapidly rising.
• The era of oral drugs for cancer treatment is well underway.
• Drug actions, both desired anticancer effects and unfortunate adverse effects, are informed by the discipline of pharmacodynamics, which includes efforts to customize treatment for the specific tumor pathways in patients with cancer.
• Drug delivery issues, including selection and adjustment of doses, routes, and schedules of administration, are addressed by the discipline of pharmacokinetics.
• Navigating the explosion of information about cancer biology and the specific properties of drugs for cancer treatment will continue to be a challenge for oncologists and related health professionals.
Introduction
One of the most obvious metrics for the importance of cancer pharmacology is the rapid and continuing expansion in the number of drugs available to treat cancer. More than 100 agents are marketed for oncology, including 11 that were approved by the Food and Drug Administration (FDA) in 2012 (Box 29-1). As a point of reference, at the first meeting of the American Society for Clinical Oncology in 1964, only 12 drugs were marketed for oncology.
Although this chapter focuses on small, synthetic molecules, one of the most exciting developments in cancer pharmacology is the successful transition of antibodies from the research stage to full integration with small molecules in the everyday treatment of patients. An increasing role is also developing for linkage of toxins that are guided to tumors by antibodies (see Chapter 32, “Therapeutic Antibodies and Immunologic Conjugates”). Although very different in terms of their size and manufacturing techniques, the principles of development for these new therapeutic classes have the same goals as for small molecules—for example, modulation of specific molecular targets and linkage to a diagnostic test for that target. Trastuzumab embodies all of these elements, including its extension into the realm of antibody-drug conjugates with the 2013 FDA approval of ado-trastuzumab emtansine.
Fundamental Science
Principles of Cancer Drug Action
Genetics is a fundamental part of the study of cancer research (see Chapter 1, “Molecular Tools in Cancer Research”). The range of applications for genetics in cancer include hereditary predisposition and interactions of heredity with nonhereditary factors (see Chapter 13, “Genetic and Epigenetic Changes in Cancer”).
As described in the preceding chapter (“Therapeutic Targeting of Cancer Cells”) and elsewhere (e.g., Chapter 8, “Vascular and Interstitial Biology of Tumors”), our understanding of the physiology and molecular biology of tumors has provided a wealth of potential targets for anticancer therapy. Although the relative intensity has magnified, the concept of molecular targeting and linkage to diagnostics for selection and monitoring of individual patients has a long history. The selection of hormonal therapies for patients whose tumors express the estrogen receptor was among the first successful uses of molecular targeting. Among several recent successful examples in the current era of targeted cancer pharmacology, two noteworthy cases are the approval and everyday use of imatinib only in patients with chronic myelogenous leukemia (CML) that is Philadelphia chromosome–positive and trastuzumab only in patients with HER2-positive tumors.
In addition to the obvious attraction of matching drugs to the molecular characteristics of the tumor, the therapeutic index can also be improved by examining host tissues. In his last major research publication,1 Dr. Abeloff was a leader in a large multicenter study of the PG for cyclophosphamide. The goal was to determine if the germline DNA of patients altered the toxicity profile of cyclophosphamide. A subgroup of women was found who had variant GSTP1 alleles that were associated with less susceptibility to adverse hematologic toxicity in regimens containing cyclophosphamide.
Another large cooperative group clinical study examined germline determinants of paclitaxel neurotoxicity.2 Wheeler et al.2 used genome-wide analyses coupled with well-characterized data from a cell bank to support a polygenic approach to understanding the variation in host tissue sensitivity to the sensory peripheral neuropathy associated with paclitaxel.
Combinations of Drugs
As described in the various chapters relating to specific malignancies in Part III of this book, it is rare for a drug to be used as a single agent. The development of combinations would be an extensive topic in itself. Box 29-2 provides a set of points to consider for combinations. Within a combination chemotherapy regimen, the drugs are intended to interact at the PD level. Originally, a major strategy was to combine cancer drugs with nonoverlapping toxicities. Increasingly, more detailed knowledge regarding pathways for cancer drugs permits the design and evaluation of combination strategies that target parallel and/or sequential pathways.
Most combinations are based on a preexisting scientific rationale or by addition of a new agent to an established regimen. However, it would be presumptuous to assume that all possibilities are already understood. One tool to expand the scope of hypothesis generation is the systematic study of all cancer drugs in combination with each other, or in combination with approved agents outside oncology, or testing every investigational agent versus all approved cancer drugs.3 Although this screening approach begins as an empiric exercise, the successful combinations become challenges for explaining the underlying mechanisms of action. The focus on drugs that are already approved provides a potentially fast route to clinical implementation.
Molecular Imaging of Cancer Drug Action
With the widening availability of fluorine-18 fluorodeoxyglucose (18F-FDG), a consensus was building in the late 1990s that positron emission tomography (PET) would emerge as a complementary tool to anatomic imaging modalities.4 Since then, FDG has been highly successful as a general probe in many tumor types, providing additional information to help separate malignant from benign lesions and to monitor response to therapy.
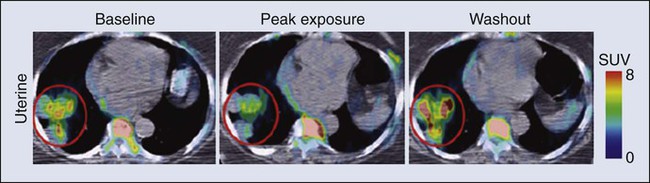
FDG is now an approved drug for imaging. Its success has spurred interest in the development of other probes for PET imaging that are currently in the investigational stage of clinical evaluation, including 18F-fluorothymidine (FLT). Figure 29-1 shows the results for FLT in a patient with uterine cancer.5 At baseline, the FLT image shows major uptake by the tumor, indicative of active proliferation. During treatment with sunitinib, uptake of FLT in the tumor decreased, which was interpreted as reduced proliferation. The image following the withdrawal of treatment exhibited a flare, that is, intensity that rebounded above the baseline value. Anatomic imaging by computed tomography (CT) readily identified the tumor and could monitor regression if that occurred, but FLT provided the ability to follow molecular pathways in real time.
Principles of Cancer Drug Delivery
Clearance is at the top of the list for drug delivery parameters in Box 29-3 because it is the factor with the greatest impact on dose-adjustment in cancer drug delivery. As stated in Box 29-4, clearance is the summation of all mechanisms for a drug to be removed from the systemic circulation, sometimes called total body clearance. Several specific applications of clearance for dosage adjustment will be covered in the next few sections.
Dosage Adjustment Based on Clearance
Compared with other medical areas in which fixed doses are prevalent, dosing in cancer pharmacology has long been titrated on the basis of each patient’s body weight or body surface area. These measurements are intended to serve as first approximations for clearance in the absence of data, but their incremental value compared with a standard dose is unfortunately small, and most current trials have adopted fixed doses, that is, “flat dosing.”6
The role of clearance in dosage adjustment is summarized in Box 29-4. The premise is that the standard dose was selected on the basis of the needs of patients with average drug clearance. If a patient has lower clearance than the average, then using the standard dose will lead to higher than average systemic exposure, with the probability of higher than average toxicity. Using a dose reduction to match the lower clearance will normalize the exposure of this patient relative to the average in the population. Similarly, a patient with higher than average clearance will have lower systemic drug exposure than will patients with the average exposure for the population. Although this lower systemic drug exposure will probably reduce the frequency or severity of toxicity, it has the potentially lethal consequence of reducing the response of the tumor.
This overall strategy for dosage adjustment has been elegantly demonstrated for the first scenario in Box 29-3, namely, patients with impaired renal function. The seminal clinical study of carboplatin by Egorin et al.7 in patients with variable renal function remains the prototype for dosage adjustment based on impaired organ function. For the other two areas in Box 29-3 (drug-drug interactions and pharmacogenetic factors), many practical cases of dosage adjustment exist. Specific examples will be discussed in the following sections.
Drug-Drug Interactions
Drug-drug metabolic interactions are possible for parenteral routes of drug delivery, but they are far more common for the oral route. Because metabolism is the primary determinant of clearance for most drugs, it is the dominant controller for changes in plasma concentrations. Following the paradigm in Box 29-4, inhibition of drug metabolism via a drug-drug interaction will produce lower clearance and thus higher systemic exposure. A reduction in dose may be necessary to avoid increased toxicity. Conversely, drugs such as phenytoin can induce the expression of certain metabolizing enzymes, which would decrease systemic concentrations for the drugs cleared via those enzymes, because the rate of their metabolism would be faster. Decreased exposure increases the probability of inadequate delivery of the cancer drug to the tumor. In the section on Oral Cancer Drugs, Table 29-1 provides some examples of the enzymatic pathways that metabolize cancer drugs.
Table 29-1
Interaction of Drugs with Enzymes and Transporters for Recently Approved Oral Cancer Drugs
| Drug | Is This Drug Affected by Inducers/Inhibitors? | Does This Drug Cause Change for Other Drugs?* |
| Imatinib | 3A | 3A, 2D6 |
| Lapatinib | 3A | 3A, 2D6, ABCB1 |
| Pazopanib | 3A | 3A, 2D6, 2C8 |
| Ruxolitinib | 3A | Not reported |
| Vemurafenib | No data | 3A, 1A2, 2D6 |
| Crizotinib | 3A | 3A |
| Abiraterone | No data | 2D6 |
| Bosutinib | 3A | No data |
| Regorafenib | 3A | No data |
| Enzalutamide | 2C8, 3A | 3A, 2C9, 2C19 |
*The drug metabolism pathways are: 1A2 = CYP1A2; 3A = CYP3A; 2D6 = CYP2D6; and various members of the CYP2C family: 2C8, 2C9, 2C19. For transport, the ABCB1 (MDR1) pathway is cited.
Data retrieved from Food and Drug Administration approved labeling, found at http://www.accessdata.fda.gov/scripts/cder/drugsatfda/.
Drug-Drug Transporter Interactions
In cancer pharmacology, the primary focus for transport of drugs has been the flux across the cell membranes of tumors. A major mechanism of drug resistance has been described for tumors with high expression of efflux pumps that reduce the availability of cancer drugs to their targets. Investigations into the influx of cancer drugs into tumors have also been conducted. White et al.8 demonstrated that patients with CML had more long-term benefit from imatinib if expression of its influx pump, OCT-1, was higher.
In addition to the interactions of cancer drugs with transporters in the tumor, interactions occur among cancer drugs and cellular transport systems in host tissues that control uptake and elimination of drugs from the GI tract, the liver, and the kidneys. The expression of transporter function can be modulated by other drugs in a manner similar to drug-drug metabolic interactions. Indeed, some of the same inducers of metabolism (phenytoin or rifampin) are also major inducers of transporters. In contrast, ketoconazole, itraconazole, and certain psychopharmacologic agents are inhibitors. This focus on drug transport is important regardless of the route of drug delivery, but similar to metabolic interactions, additional factors exist that are related to control of absorption of cancer drugs from the GI tract. Transporters are also the underpinning of the blood-brain barrier and control the entry of cancer drugs into the brain to treat metastases or primary brain tumors.9
Interactions Via Self-Medication
The source of drugs for patients with cancer is not restricted to those prescribed by oncologists, or even medicines prescribed by other medical practitioners. Self-medication with over-the-counter drugs and alternative therapies is prevalent in patients with cancer and includes substances that interact with cancer drugs (see Chapter 33, “Complementary and Alternative Medicine”).
Oral Cancer Drugs
Parenteral administration will continue to be important, both for the many legacy drugs that remain part of regiments with established therapeutic value and for occasional cases in which an agent cannot be successfully delivered via the oral route. However, based on the up-front goal to achieve oral delivery for most new cancer drugs under development, especially for chronic daily dosing, it is easy to project that the ratio of oral to IV cancer drugs will continue to rise. As shown in Box 29-1, the FDA approved 11 cancer drugs in 2012. Overall, 7 of these 11 drugs are administered orally, including 7 of the 9 small synthetic compounds.
As indicated in Box 29-5, adherence to the prescribed regimen is one of the first questions to explore. Although patients with cancer are highly motivated to carefully follow their dosing instructions, it is naïve to expect complete compliance and especially to ignore the problems of persistence over a long period. Marin et al.10 concluded that poor adherence may be the predominant reason for failure to achieve adequate molecular responses with imatinib. Partridge et al.11 examined the complex scenario of elderly patients with cancer, a population that has a high frequency of polypharmacy and perhaps a fading memory. For adjuvant therapy of early stage breast cancer with tamoxifen, 25% of patients took fewer than 80% of the expected doses, which is problematic. This study included no patients younger than 65 years, but the authors reported that adherence was not dependent on age for persons ranging from 65 to 89 years.
Systemic drug exposure ratio, at equal doses:
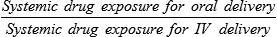
Although ease of oral cancer drug administration is generally a major advantage, it can become a liability when co-morbid conditions make swallowing drugs a major problem (see Chapter 43, “Oral Complications.” Cancer chemotherapy can produce emesis (see Chapter 42, “Nausea and Vomiting”), which thwarts the reliability of retaining swallowed doses. In addition, the treatment of these complications of cancer therapy can include the use of additional drugs to complicate the drug-drug interaction scenarios.
For the representative set of 10 cancer drugs described in Table 29-2, the advice for four drugs (ruxolitinib, vemurafenib, crizotinib, and enzalutamide) is that the timing of food intake is not important. The other six drugs are split evenly between recommendations to take while fasting versus taking with a meal. For two of these six drugs, the magnitude of the food effect is provided.
Table 29-2
Impact of Food on Drug Absorption for Recently Approved Oral Oncology Drugs*
| Drug | Whether Drugs Should Be Taken with Food | Size of Effect |
| Imatinib | Yes | ?? |
| Lapatinib | No | Fourfold |
| Pazopanib | No | ?? |
| Ruxolitinib | ± | NS |
| Vemurafenib | ± | NS |
| Crizotinib | ± | NS |
| Abiraterone | No | Tenfold |
| Bosutinib | Yes | ?? |
| Regorafenib | Yes | ?? |
| Enzalutamide | ± | NS |
*If no preference is provided for taking with food or fasting, ± is indicated, and the effect size is not significant (NS). If no advice is provided, ?? is indicated.
Data retrieved from Food and Drug Administration approved labeling, found at http://www.accessdata.fda.gov/scripts/cder/drugsatfda/.
If these dosing recommendations are unintentionally or intentionally reversed, potentially serious issues of reduced efficacy or increased toxicity occur, along with potential economic issues. The fourfold increase in absorption of lapatinib with food has been the source of substantial public discussion,12 which has been extended to cover other oral cancer drugs.13 For abiraterone, the tenfold difference in absorption is sufficiently large to capture everyone’s attention. Perhaps abiraterone has a wider therapeutic index than most cancer drugs, but a tenfold difference in exposure seems unlikely to be without consequences.
In addition to the general effects of food on solubility of drugs and passage into the body, a few food substances have highly specific effects on cancer drug metabolism and cancer drug transporters. The most notable example is the interaction of grapefruit juice with metabolism via the CYP3A family and the ABC family of transporters.14 The specificity of effects with grapefruit and drugs closely resembles the drug-drug interactions described later.
For the same set of 10 representative oral cancer drugs described in Table 29-2 for food effects, Table 29-1 provides information from the FDA Web site for drug interactions with enzymes and transporters. The entries in Table 29-1 indicate that all of these drugs have at least some information about enzymatic pathways. The largest category of potential interactions is with the CYP3A family of enzymes, which are present in both intestines and liver and have potential interactions with 9 of the 10 drugs. Scattered information is presented for other members of the CYP superfamily: 1A2, 2C8, 2C9, 2C19, and 2D6. Interest is expanding rapidly in the role of transporters15 interacting with cancer drugs, but only one drug in this set, lapatinib, has information about the transporter pathways.
Clinical Relevance and Applications
Regional Cancer Drug Delivery
Intraarterial catheterization has been technically feasible for delivery of cancer drugs to a variety of places in the body. The use of an implanted pump for the intraarterial delivery of fluorodeoxyuridine is approved for the treatment of liver metastases from colorectal cancer and is the most common application of this approach.16
Intraperitoneal delivery of drugs for treatment of localized ovarian cancer and other intraperitoneal malignancies has undergone many evaluations. Three randomized phase 3 studies have shown a survival benefit for intraperitoneal delivery compared with the IV route.17 Despite this success, the lack of familiarity with the catheters, uniform distribution within the peritoneal cavity, and other factors have slowed the penetration of this modality into routine practice.
Shortages of Cancer Drugs
Speculation that many shortages could be bypassed by substituting drugs that are “almost as good” was dramatically dashed by the study of Metzger et al.18 Because of a lack of supply of mechlorethamine, cyclophosphamide was substituted in the multidrug regimen for children with Hodgkin lymphoma. In only 2 years, more than a doubling occurred in the number of patients who no longer had event-free survival.
Development and Discovery of Cancer Drugs
Within the context of recent trends for new cancer drugs (Box 29-6), the processes for development and discovery need to be clearly understood. However, the actual research that comprises these phases needs to adapt to the continuous stream of new findings in cancer biology. Further, the optimal approach to information gathering during human evaluation of cancer drugs is continually reevaluated, and the relevance of preclinical models to the clinical realities is continually improved.
Clinical Phases of Drug Development
As indicated in Box 29-7, the clinical stages of drug development have been divided into phases that are related to the primary goals. These stages have historical and regulatory significance, but the boundaries are always being pushed, and it is not possible to assign every compound into the same bins. For patients with cancer, this view of clinical development is continually challenged by the compelling need to provide therapeutic options when no treatments have been established. The boundaries that are calibrated for less serious diseases may be a bit more flexible in oncology, just as the definition of maximum tolerated dose is a relative term that depends on the medical scenario. Phase 1, phase 2, and phase 3 are long-established terms. Phase 4 was created by a formal change in FDA regulations. Phase 0 is a less-formal term used by some investigators to describe early explorations to seek information before making a decision to launch full-scale development.
Phase 2 clinical trials are the setting for determining if an agent or combination of agents has sufficient activity in well-defined groups of patients who have no established treatments.19 In the majority of phase 2 studies, there is only a single treatment group, but randomized designs of two or more groups have been increasingly used. The goal is to determine if the activity of the agent or combination is sufficient to move into much larger phase 3 trials with comparison to standard treatments. Unfortunately, areas within oncology remain for which no standard treatment has been established. In such cases, phase 2 studies can be sufficient for regulatory approval.
Phase 0 clinical studies are an approach to first-in-human studies that extends the preclinical search for specific types of information into the clinic. Frequently, there are concentrations that are considered essential to activity.20,21 For oral therapy, concerns exist about the consistency of absorption.
What the Future Holds
With the large and increasing number of drugs available for use (see the Appendix)22, it is not feasible to remember all the details about each one. The widespread availability and use of online resources, including FDA databases, supplements traditional textbooks. Assembling the key pieces of data is only the first step. Professional interpretation and judgment remain as central features.
| Drug Name | Drug Class and/or Mechanism | Pharmacokinetics/Metabolism | Toxicity | Indications | Dosing |
| Abiraterone acetate (Zytiga) | Inhibits androgen biosynthesis | Do not take with food; inhibits CYP2D6 | Joint swelling or discomfort; edema; monitor liver enzymes | Metastatic prostate cancer; castration resistant with prior use of docetaxel | 1000 mg once a day orally with 5 mg prednisone BID |
| Ziv-aflibercept (Zaltrap) | Fusion protein binding VEGF and related ligands | No studies for renal, hepatic, or drug-drug interactions | Hemorrhage, GI perforation, wound healing problems, fistula, hypertension, arterial thrombosis, proteinuria | Metastatic colorectal cancer resistant or progressed while taking oxaliplatin | 4 mg/kg via 1-hr IV (not bolus) every 2 wk |
| Altretamine (Hexalen), hexamethylmelamine, HMM | Alkylating agent | Well absorbed by mouth, metabolized in the liver; half-life 4-13 hr; metabolism may be slowed by cimetidine or enhanced by phenobarbital | Myelosuppression is dose limiting; leukopenia, thrombocytopenia, nausea, and vomiting are common; neurologic toxicity, including confusion, lethargy, weakness, and sensory changes, is common | Refractory ovarian carcinoma | 4-12 mg/kg/day in divided doses for 3-6 wk or 150 mg/m2/day for 14 days each cycle; higher doses have been used |
| Amifostine (Ethyol), WR- 2721, ethiofos | Cytoprotectant; free-radical scavenger | After IV infusion, the drug is metabolized to a thiol metabolite, which is responsible for its beneficial activity | Transient hypotension is dose limiting; nausea, vomiting, and somnolence are common; sneezing, hypocalcemia, and flushing can be seen | Pretreatment with cisplatin; useful as a bone marrow, kidney, and nerve cytoprotectant; useful with other alkylators as well; also FDA approved as a radiation protectant to reduce xerostomia | 740 mg/m2 IV infusion over 15 min given 15-30 min before the cytotoxic agent or radiation; lower doses and subcutaneous administration have also been used |
| Anastrazole (Arimidex) | Nonsteroidal aromatase inhibitor; blocks estrogen production selectively | Well absorbed from GI tract, maximum plasma levels within 2 hr; half-life is 50 hr; extensively metabolized in liver; despite hepatic and renal clearance being important, no adjustments needed for abnormal function of these organs because of the wide therapeutic index of this drug | Very well tolerated; asthenia, headache, and hot flashes occur in fewer than 15% of women; diarrhea, abdominal pain, anorexia, nausea, and vomiting occur in 10% or fewer; thrombophlebitis has been reported | Indications: As adjuvant therapy of breast cancer and for treatment of postmenopausal women with breast carcinoma who have progressed while being treated with tamoxifen | 1 mg PO every day; higher doses are no more effective |
| Arsenic trioxide (Trisenox) | Novel arsenical differentiating agent | Half-life of this compound is unknown; it is methylated in the liver and eliminated in the urine | The “differentiation syndrome” is dose limiting and includes leukocytosis, fever, dyspnea, chest pain, tachycardia, hypoxia, and sometimes death; corticosteroids seem to benefit this syndrome; QT prolongation is common; common adverse effects include rash, pruritus, headache, arthralgias, anxiety, bleeding, nausea, and vomiting; liver and renal toxicity are uncommon | Relapsed acute promyelocytic leukemia | 0.15 mg/kg/day in 100-250 mL of D5W until remission, not to exceed 60 doses, then up to 25 doses over 5 wk for consolidation starting 3 to 6 wk after achievement of remission |
| l-Asparaginase (Elspar), colaspase | Naturally occurring enzyme derived from Escherichia coli or Erwinia carotovora that cleaves asparagine, an essential amino acid required by rapidly proliferating cells | After IV or IM injection, the drug is metabolized intravascularly by proteolysis; elimination half-life of 8-30 hr | Hypersensitivity can be life- threatening, requiring anaphylaxis precautions and a 2-unit test dose; coagulopathy is common and requires monitoring; nausea, vomiting, abdominal cramps, anorexia, elevated liver function tests, and transient renal insufficiency are common; lethargy, somnolence, fatigue, depression, and confusion are seen, as are pancreatitis and fever | ALL; also used in AML, late-stage CML, CLL, and non-Hodgkin lymphomas | After a 2-unit intradermal test dose, an IM dose of 6000-10,000 IU/m2 every 3 days for 9 doses, or 1000 IU/kg/day IV over 30 min for 10 days, has been used |
| PEG-Asparaginase (Oncaspar)—pegaspargase | Naturally occurring enzyme, covalently linked to polyethylene glycol to reduce immunogenicity, slow metabolism, and prolong half-life; the enzyme cleaves asparagine, an essential amino acid required by rapidly proliferating cells | When given by IM injection, it has an elimination half-life of approximately 5 days; clearance is not dependent on renal or hepatic function | Although less immunogenic that the non-PEGylated form, hypersensitivity and anaphylaxis can still occur; toxicities similar to those of the non-PEGylated forms are seen, including elevated liver enzymes, coagulopathy, hypercholesterolemia, pancreatitis, hyperglycemia, fever, chills, anorexia, lethargy, confusion, headache, seizures, and azotemia | ALL, and like asparaginase, it is also used for other leukemias and non-Hodgkin lymphomas | 2,500 IU/m2 IM every 14 days with other chemotherapy agents for induction or maintenance |
| Asparaginase Erwinia chrysanthemi (Erwinaze) | Substitute for the naturally occurring enzyme | Not characterized | Severe hypersensitivity reactions, including anaphylaxis | ALL | 25,000 IU IM 3×/wk for 6 doses per cycle |
| Axitinib (Inlyta) | Avoid strong CYP3A inhibitors or reduce dose | Decrease dose 50% for CYP3A inhibitors, renal, hepatic impairment | Diarrhea, hypertension, fatigue, nausea | Advanced renal cell carcinoma after one treatment failure | 5 mg BID oral |
| Azacytidine (Vidaza) | Antimetabolite; induces hypomethylation of DNA, either inducing apoptosis or restoring normal function; at higher doses, acts as a cytidine analog | Not orally bioavailable; metabolized by the liver and excreted in urine; elimination half-life of 4 hr | Myelosuppression is dose limiting; leukopenia, thrombocytopenia, and transient elevation of liver function tests are common; nausea and vomiting and abdominal pain are common | Myelodysplastic syndromes | 75-100 mg/m2 for 7 days; repeated every 4 wk for 4 to 6 cycles |
| Bacillus Calmette- Guérin (TICE BCG, TheraCys), BCG | Immunostimulant/vaccine; induces a cellular immune response at the site of instillation | BCG is a live, attenuated bacteria culture; it does not enter the body in viable form; it has no detectable pharmacokinetic fate; in rare cases, a clinical infection can result from treatment, indicating invasion of the body at site of administration into systemic circulation | Urinary symptoms predominate, including dysuria, hematuria, hesitancy, urgency, frequency, and secondary infection; other toxicities include fever, chills, malaise, myalgias/arthralgias, anorexia, nausea, vomiting, and anemia; clinical mycobacterial infection is rare and generally seen only in immunocompromised patients | Intravesical instillation for noninvasive bladder cancer after removal of papillary tumors; also used for some experimental vaccine programs as an adjuvant to the vaccine | 81 mg per treatment, in 53 mL total volume, instructions as previously given; given once weekly for 6 doses and then at 3, 6, 12, 18, and 24 mo after the induction |
| Bendamustine (Treanda) | Alkylating agent | Reduce dose for hematologic toxicity | Nausea, pyrexia, vomiting, hematologic abnormalities | CLL and B-cell non-Hodgkin lymphoma | CLL: 100 mg/m2 over 60 min, day 1, 2 of 28-day cycle; B-cell: 120 mg/m2 over 30 min on day 1, 2 of 21-day cycle |
| Bevacizumab (Avastin) | Recombinant humanized monoclonal antibody that binds to all forms of VEGF, preventing binding to its receptors | Administered by IV infusion; half-life is 20 days; the fate of parent drug and metabolites is unknown | Asthenia, pain, nausea/vomiting, diarrhea, anorexia, stomatitis, dermatitis, hypertension, proteinuria; infusion-related reactions rare; hemoptysis, hemorrhage, delayed wound healing, GI perforations; increased risk of thromboembolic events can be severe or fatal | Metastatic colorectal cancer and NSCLC; renal cell carcinoma | 5-15 mg/m2/day IV every 3 wk |
| Bexarotene (Targretin) | Synthetic retinoid, differentiating agent | Good oral bioavailability increased by a high-fat meal; metabolized in the liver to oxidative metabolites by cytochrome P450 3A4, glucuronidated, eliminated in bile | Hyperlipidemia is dose limiting and should be monitored during therapy and treated as appropriate; pruritus, leukopenia, diarrhea, fatigue, headache, and liver function test elevation can also be dose limiting; rash, edema, fever, chills, and nausea are uncommon; excessive bleeding and back or abdominal pain are rare | Cutaneous T-cell lymphoma (mycosis fungoides) refractory to at least one prior therapy | 300 mg/m2/day orally, dose adjusted for toxicity |
| Bicalutamide (Casodex) | Nonsteroidal antiandrogen | Well absorbed orally; highly protein bound; converted to inactive metabolites in liver via oxidation and glucuronidation; half-life of several days | Constitutional symptoms predominate, including hot flashes, decreased libido, depression, weight gain, edema, gynecomastia, early disease-site pain (flare reaction), and constipation; nausea, vomiting, anorexia, diarrhea, and dizziness are uncommon; dyspnea, anemia, fever, and rashes are rare | Stage D2 prostate cancer, in combination with an LHRH agonist agent | 50 mg PO daily, in combination with an LHRH agonist agent |
| Bleomycin (Blenoxane), Bleo | Antitumor antibiotic; causes DNA strand breaks directly in normal and neoplastic cells | After an IV infusion, it has an elimination half-life of 3-5 hr; bleomycin is incompletely metabolized by intracellular aminopeptidases; it is excreted in the kidney as unchanged drug and metabolites | Pulmonary toxicity, including reversible and irreversible fibrosis, is dose limiting; other common toxicities include fever, chills, rash, exfoliation, and anorexia; nausea, vomiting, myelosuppression, anaphylaxis, and mucositis are rare | Germ cell tumors, Hodgkin disease, and squamous cell cancers; used off-label for melanoma, ovarian cancer, and Kaposi sarcoma; also used as a sclerosing agent for malignant pleural or pericardial effusions | Observe 1-6 hr; after 2-unit IV test dose over 15 min, the full dose can be given; usual dose is 10-20 units/m2 IV, IM, or SC 1-2×/ wk, or 15-20 units/m2/day as a continuous infusion over 3-7 days; As a sclerosing agent, 60 units generally used |
| Bosutinib | Kinase inhibitor | Take with food; reduce dose to 200 mg if hepatic impaired; substrate for CYP3A | GI toxicity should be monitored; myelosuppressive | Philadelphia chromosome–positive CML, resistant or intolerant to prior therapy | 500 mg/day oral |
| Brentuximab vedotin (Adcetris) | CD30-directed antibody conjugate | Monitor patients taking CYP3A inducers or inhibitors | Progressive multifocal leukoencephalopathy; peripheral neuropathy; neutropenia | Hodgkin lymphoma and anaplastic large cell lymphoma | 1.8 mg/kg IV infusion over 30 min every 3 wk |
| Buserelin (Suprefact), HOE766 | LHRH agonist; shuts off luteinizing LH and FSH secretion, producing chemical castration | Intravascular and extravascular proteolysis | Flare reactions, which can be prevented; castration symptoms such as hot flashes and decreased libido common; other nonspecific symptoms include headache, nausea, vomiting, diarrhea, constipation, and weakness | Prostatic cancer | 500 µg SC TID for the first wk, then 200 µg/day, or intranasally 800 µg TID followed by 400 µg TID |
| Busulfan (Myleran-oral), (Busulfex-iv), BSF | Alkylating agent | Excellent oral bioavailability, with peak levels in serum occurring at about 1 hr; elimination half-life of 2.5 hr; metabolized partially in liver; parent drug and metabolites excreted in the urine; also available IV | Myelosuppression, partly chronic and cumulative, is dose limiting; other common toxicities include nausea, vomiting, anorexia, mucositis, hyperpigmentation, and elevated liver function tests (or venoocclusive disease of the liver at transplant doses); neurologic toxicity, including blurred vision, dizziness, and confusion, and interstitial lung disease are less common | Oral: palliative treatment of chronic myelogenous (myeloid, myelocytic, granulocytic) leukemia; IV: in combination with cyclophosphamide as a conditioning regimen prior to allogeneic hematopoietic progenitor cell transplantation for CML | 4-8 mg per day oral initially, decreasing based on blood counts, with 1-3 mg daily used for maintenance; IV conditioning dose 0.8 mg/kg every 6 hr for 4 days |
| Capecitabine (Xeloda) | Oral antimetabolite prodrug | Readily absorbed by the GI tract, metabolized in vivo to fluorouracil in the liver by carboxylesterase and cytidine deaminase, and then in turn in the peripheral tissues and tumor tissue by thymidine phosphorylase | Myelosuppression and palmar-plantar erythrodysesthesia are dose limiting; diarrhea, fatigue, stomatitis, and hyperbilirubinemia are uncommon; nausea, vomiting, and rash are rare | Metastatic breast cancer and metastatic colorectal cancer; used also in head and neck squamous cell cancer | 1250 mg/m2 every 12 hr for 14 days every 21 days; dose reductions are often required; treatment delays are sometimes required; other doses and schedules have been used |
| Cabazitaxel (Jevtana) | Microtubule inhibitor | Caution for patients taking strong CYP3A inducers or inhibitors | Neutropenia and hypersensitivity | Metastatic hormone refractory prostate cancer when docetaxel fails | 25 mg/m2 every 3 wk 1-hr IV infusion with oral prednisone 10 mg daily |
| Cabozantinib | Inhibits many receptor tyrosine kinases | 55-hour half-life; take without food | Perforations, fistulas, and hemorrhage | Metastatic medullary thyroid cancer | 140 mg daily |
| Carboplatin (Paraplatin), Carbo, CBDCA | Atypical alkylator; produces intrastrand and interstrand cross-links in DNA via association bonds with the platinum molecule, leading to DNA strand breakage during replication | Rapidly cleared from the bloodstream after IV infusion, with a terminal half-life of 2.5 hr; it is cleared largely as unchanged drug by the kidneys | Hemorrhage; thrombocytopenia | Ovarian cancer and used extensively in testicular cancer, squamous cell cancers of the head and neck and cervix, and lung cancer | Dosing can be performed on a per-meter-squared basis or through several formulas that take into account renal function and desired level of thrombocytopenia (such as Calvert’s formula); typical doses with normal renal function are in the 300- to 500- mg/m2 range as IV infusion |
| Carfilzomib | Proteosome inhibitor | Drug-drug interactions unlikely; no adjustment for renal impairment; no experience with hepatic impairment | Fatigue, anemia, nausea, thrombocytopenia, dyspnea, diarrhea, and pyrexia; monitor for cardiac, pulmonary, tumor lysis, infusion reactions, thrombocytopenia | Multiple myeloma | 20 mg/m2/day IV over 2-10 min, days 1, 2, 8, 9, 15, 16; rest period (days 17-28); 27 mg/m2/day other cycles if tolerated |
| Carmustine (BiCNU), BCNU, bis-chloronitrosourea | Alkylator agent in the nitrosourea class; cell cycle independent mechanism | After an IV infusion, the drug is rapidly taken up by tissues, including the CNS; extensively metabolized in the liver; the serum half-life is only 15-20 min | Myelosuppression is slow in onset and cumulative and dose limiting; nausea and vomiting are common, can be severe; hyperpigmentation and renal toxicity can be seen; interstitial lung disease, including fibrosis, is rare but can occur with any dose; transplant doses can cause severe liver toxicity and more frequent lung toxicity | Melanoma, stomach cancer, colon cancer, and liver cancer; indications: FDA approved for brain tumors, multiple myeloma, Hodgkin disease, lymphoma; also used for breast cancer | Single-agent dose is 150-200 mg/m2 every 6 wk; for transplant, the dose is as high as 600 mg/m2, along with other drugs |
| Carmustine impregnated wafer (Gliadel), polifeprosan 20 with carmustine implant | Novel delivery mechanism for classical nitrosourea alkylating agent | More than 70% of the copolymer degrades by 3 wk; minimal systemic exposure to carmustine | None | Patients with newly diagnosed high-grade malignant glioma as an adjunct to surgery and radiation; also, patients with recurrent glioblastoma multiforme as an adjunct to surgery | Up to 8 wafers are placed in the resection cavity at the time of craniotomy and operative resection |
| Cetuximab (Erbitux) | Recombinant humanized monoclonal antibody, targeting EGFR; competitively inhibits growth factor binding; inhibits autophosphorylation and cell signaling | Metabolism poorly understood; half-life 5-7 days with minimal clearance by kidneys or liver | Infusion reaction, characterized by rapid-onset dyspnea, fever, chills, urticaria, flushing, angioedema, and hypotension, is seen in 40%-50% of patients; acneiform rash is common; constitutional symptoms; hypomagnesemia; interstitial lung disease is rare | Metastatic colorectal cancer, head and neck cancer in combination with radiation | 400 mg/m2 IV, followed by 250 mg/m2 weekly |
| Chlorambucil (Leukeran) | Alkylating agent; cell cycle–independent | Excellent oral bioavailability; maximum plasma level at 1 hr; half-life of 1-2 hr; extensively metabolized in liver to active and inactive metabolites | Myelosuppression is dose limiting and universal, and it can be cumulative; nausea, vomiting, and diarrhea are mild and uncommon; sterility and alopecia occur in a minority of patients; pulmonary fibrosis and neurologic adverse effects are quite rare | CLL and low-grade lymphomas; also used for Waldenstrom macroglobulinemia, multiple myeloma, hairy cell leukemia, and rarely in some solid tumors | 16 mg/m2/day for 5 days every 4 wk, or 0.4 mg/kg every 2-4 wk, or 0.1-0.2 mg/kg/day for 3-6 wk |
| Cisplatin (Platinol)—cDDP, DDP, cisplatinum, cis-diamminedichloro-platinum (II) | Atypical alkylator; produces intrastrand and interstrand cross-links in DNA via association bonds with the platinum molecule, leading to DNA strand breakage during replication | After IV infusion, rapid distribution to tissues takes place, and the drug is over 90% protein bound | Nephrotoxicity is dose limiting for an individual dose, and neurotoxicity, especially painful peripheral neuropathy, is dose limiting for cumulative doses; myelosuppression is mild; nausea and vomiting are common but manageable, and anorexia and diarrhea are common; cumulative ototoxicity is also common; chronic renal magnesium and potassium wasting is common and sometimes not reversible; elevated liver transaminases can be seen, whereas alopecia and cardiac conduction abnormalities are rare; adequate renal perfusion and urine output are critical for minimizing renal toxicity; therefore prehydration and adequate posttreatment hydration are used, usually with normal saline solution with or without mannitol, potassium, and magnesium | Used for almost every class of solid tumor and lymphoma; FDA approved for testicular and ovarian cancer and transitional cell carcinoma | Cisplatin can be given all in one IV infusion or daily as an IV infusion for several days, which are somewhat better tolerated; total dose per cycle ranges from 80-160 mg/m2; continuous infusion can also be used; dose should be reduced for creatinine clearance below 60 mL/min along with the cisplatin; cisplatin 100-200 mg/m2 is also used intraperitoneally for ovarian cancer |
| Crizotinib (Xalkori) | ALK kinase inhibitor | Avoid strong CYP3A inhibitors, inducers, or substrates | Hepatotoxicity; pneumonitis; QT interval prolonged | ALK-positive NSCLC | 500 mg once daily; reduce or interrupt based on toxicity |
| Dasatinib (Sprycel) | Kinase inhibitor | Use with caution in patients with hepatic impairment; CYP3A4 inhibitors: may increase dasatinib drug levels; avoid; otherwise, monitor closely and consider reducing dose; CYP3A4 inducers: may decrease dasatinib drug levels; avoid, or consider increasing dose; antacids: may decrease dasatinib drug levels; avoid simultaneous administration; if needed, administer the antacid at least 2 hr prior to or 2 hr after the dose; H2 antagonists/proton pump inhibitors: may decrease dasatinib drug levels; consider antacids in place of H2 antagonists or proton pump inhibitors | Myelosuppression, bleeding events, fluid retention, diarrhea, headache, musculoskeletal pain, and rash | (Ph+) chronic myeloid leukemia (CML) in chronic phase, or chronic, accelerated, or myeloid or lymphoid blast phase Ph+ CML with resistance or intolerance to prior therapy including imatinib, or Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) with resistance or intolerance to prior therapy | The recommended starting dosage of SPRYCEL for chronic phase SML is 100 mg administered orally once daily. The recommended starting dosage of SPRYCEL for accelerated phase CML, myeloid or lymphoid blast phase CML, or Ph+ ALL is 140 mg administered orally once daily |
| Daunorubicin | Anthracycline antitumor antibiotic; pleiotropic effects including free radical formation, topoisomerase II inhibition; altered mitochondrial function | After IV bolus, widely distributed and metabolized in liver to active and inactive metabolites; half-life of the parent drug is 18 hr, and 25 hr for active metabolite daunorubicinol | Daunorubicin is a vesicant; precautions are necessary; myelosuppression is dose limiting; alopecia, nausea, vomiting, and stomatitis are common; diarrhea, rash, elevated liver function tests, and transient arrhythmias are uncommon; dose-related cardiomyopathy is uncommon below cumulative doses of 400-500 mg/m2 | AML and ALL | Single IV injection daily for 1-5 days; total dose per course up to 150 mg/m2; a typical dose would be 45 mg/m2/day for 3 days |
| Daunorubicin, liposomal (DaunoXome) | Novel liposomal preparation of the anthracycline that modifies the pharmacokinetics and toxicities of the drug | Liposomes are rapidly cleared from plasma into peripheral tissues; low levels of metabolites are detected in plasma, likely because of slow distribution of parent drug from peripheral tissues to liver for metabolism; metabolic fates are the same as for conventional drugs | Milder adverse effect and toxicity profile than conventional daunorubicin; myelosuppression is mild but still dose limiting; acute syndrome of back pain, chest tightness, and flushing can occur uncommonly during administration, which can usually be treated symptomatically; other cardiac adverse effects are rare; skin rashes are rare; nausea, vomiting, and alopecia are rare | AIDS-associated Kaposi sarcoma; some experience in other solid tumors | Dosing: 100 mg/m2 IV infusion over 60 min every 2 wk, with reduction and delay for significant myelosuppression |
| Decitabine (Dacogen) | Antimetabolite; inhibits DNA methyltransferase, causing hypomethylation of DNA; this may induce apoptosis or restore normal function to genes that control cellular differentiation and proliferation | Deaminated by cytidine deaminase, found in liver, granulocytes, gut, and blood; elimination half-life is 30 min | Myelosuppression; nausea, vomiting, abdominal pain; constitutional symptoms; elevated liver functions, blood sugar, low serum magnesium, low serum potassium; respiratory toxicity | Myelodysplastic syndromes | 15 mg/m2 by continuous infusion over 3 hr, every 8 hr for 3 days, repeated every 6 wk for a minimum of 4 cycles |
| Degarelix (Firmagon) | GnRH receptor antagonist | No adjustment for mild to moderate renal or hepatic impairment; drug interactions unlikely | Injection site reactions; hot flashes, increased weight, increases in serum levels of transaminases and GGT; long-term QT risk for antiandrogens | Prostate cancer | SC only; 240 mg given as two injections of 120 mg each; maintenance doses of 80 mg as single injection every 28 days |
| Denileukin diftitox (Ontak) | Recombinant DNA peptide fusion product combining interleukin-2 and a diphtheria toxin, allowing relative specificity of diphtheria toxin toward interleukin-2 receptor expressing cells | After IV administration, the plasma half-life of denileukin diftitox is about 80 min; radiolabeling studies show that the drug accumulates in the vasculature, liver, and kidneys, but its specific metabolic fates are unknown; antibodies against the drug have been shown to slow its clearance | Broad range of toxicities similar to other peptide biological response modifiers, with hypotension and other manifestations of vascular leak syndrome being dose limiting; fever, chills, edema, rash, fatigue, headache, nausea, vomiting, anorexia, and diarrhea are common; dyspnea, cough, arthralgias, myalgias, and pharyngitis are uncommon; infections associated with drug administration are common; arrhythmias and significant neurologic, hepatic, or renal complications are rare | Recurrent cutaneous T-cell lymphoma (mycosis fungoides) | 9 or 18 µg/kg/day for 5 days as an IV infusion over at least 15 min, repeated every 21 days |
| Denosumab (Xgeva) | Monoclonal antibody; RANK ligand inhibitor | No drug interaction studies; more hypocalcemia for creatinine clearance <30 mL/min | Use calcium and vitamin D to treat or prevent hypocalcemia; fatigue/asthenia, hypophosphatemia, and nausea; osteonecrosis of jaw | Bone metastases from solid tumors; not in multiple myeloma | 120 mg every 4 weeks as SC injection in the upper arm, upper thigh, or abdomen |
| Dexamethasone (Decadron), Dex, DXM | Corticosteroid that has pleiotropic properties in various body tissues; directly toxic to benign and malignant lymphocytes; potent antiinflammatory action | Well absorbed by the GI tract; metabolized in the liver; elimination half-life is 3-4 hr; elimination of metabolites is primarily renal, with some biliary component | Toxicities are shared with other corticosteroids and include leukocytosis, hyperglycemia, mood changes, euphoria, insomnia, increased appetite, weight gain, dyspepsia, exacerbation of peptic ulcer disease, cataracts, adrenal suppression, edema, and osteoporosis | Used for treatment of multiple myeloma, CLL and ALL, non-Hodgkin lymphoma, immune thrombocytopenic purpura, and hemolytic anemia; also used to alleviate symptoms from brain or spinal cord metastases and other metastatic sites where edema and inflammation exist; used as an adjunctive antiemetic medication | Oral and parenteral dosing are equivalent; dosage for acute indications or active treatment involves total daily doses of 16-40 mg, sometimes with an initial “bolus” dose of up to 100 mg; tapering treatments will decrease down to 1-2 mg/day; as an antiemetic, 10-20 mg is the standard dose |
| Dexrazoxane (Zinecard)—ADR- 529, ICRF-187 | Iron-chelating agent that serves as a free-radical scavenger/cytoprotectant; widespread and rapid | Metabolism is mostly hepatic; half-life is 3-4 hr; parent drug and metabolites are excreted by the kidneys; hepatic impairment not studied; renal impairment suggests 50% dose reduction | Dexrazoxane appears to worsen slightly the leukopenia induced by doxorubicin; mild nausea and vomiting are common; fever, stomatitis, fatigue, anorexia, and hypotension are uncommon; seizure, respiratory arrest, deep venous thrombosis, and significant liver toxicity are rare | Prevent doxorubicin-induced cardiomyopathy | Administered just prior to a dose of doxorubicin as a 15- to 30-min infusion at a dose of 500-1000 mg/m2 |
| Docetaxel (Taxotere), RP- 56976 | Docetaxel is a semisynthetic taxane, a class of compounds that inhibit the mitotic spindle apparatus by stabilizing tubulin polymers, leading to death of mitotic cells | After a 1-hr infusion, docetaxel is widely distributed, with elimination half-life of 1 hr and a terminal half-life of 18 hr ; extent and by- products of metabolism are not well known; the main excretion route is biliary | Myelosuppression is universal and dose limiting; alopecia is also universal; edema and fluid accumulation, including pleural effusions and ascites, are common and can be dose limiting; fluid accumulation is partially preventable with corticosteroid treatment before and after each cycle of docetaxel; mild sensory or sensorimotor neuropathy is common; mucositis and diarrhea are common and usually mild; hypersensitivity reactions are uncommon and can largely be prevented through premedication with corticosteroids and antihistamines; rash and elevated liver function tests are uncommon | Metastatic breast cancer and first- and second-line NSCLC | Standard dose is 100 mg/m2 IV over 1 hr every 3 wk; higher doses and other schedules have been used |
| Doxorubicin (Adriamycin, Rubex), Adria, hydroxydaunorubicin | Anthracycline antitumor antibiotic; pleiotropic effects including free radical formation, topoisomerase II inhibition; altered mitochondrial function | After an IV dose, it is widely distributed in tissues and is 70% protein bound; it is metabolized in the liver to active and inactive forms; it has an elimination half-life of 18 hr or more; most of the drug and metabolites are excreted through the biliary route | Doxorubicin is a potent vesicant, and extravasation precautions are a must; myelosuppression is universal and usually dose limiting with each individual cycle; cardiotoxicity is common and can be dose limiting, although usually subclinical; chronic, cumulative cardiomyopathy is expected when the total dose exceeds 450 mg/m2; this toxicity can be lessened by the addition of dexrazoxane or by longer infusions; acute cardiac effects, including arrhythmias, are less often seen and are unpredictable; nausea and vomiting are common but manageable; diarrhea and stomatitis are common but usually mild; alopecia, rash, and hyperpigmentation are common | Approved for a variety of cancers and used for many more; most commonly used for breast carcinoma, adult sarcomas, pediatric solid tumors, Hodgkin disease, non-Hodgkin lymphomas, and ovarian cancer | Standard doses range from 60-90 mg/m2 IV as a bolus or continuous infusion over 48-72 hr every 3-4 wk; weekly and biweekly schedules are also used, with lower doses; doses are usually reduced for elevated bilirubin levels |
| Doxorubicin, liposomal (Doxil) | Novel liposomal preparation of the anthracycline doxorubicin | Doxorubicin metabolized in the liver; significant plasma levels of the principal metabolite, doxorubicinol, not observed with the liposomal preparation, likely because of slow distribution of free doxorubicin to liver; half-life of liposomes in plasma is 55 hr | Myelosuppression is mild but dose limiting; palmar plantar erythrodysesthesia is common and can occasionally be severe and dose limiting; stomatitis and nausea are common but usually mild; alopecia is uncommon; acute infusion reactions including chest pain, back pain, dyspnea, and wheezing can occur uncommonly | Recurrent metastatic ovarian cancer and AIDS-related Kaposi sarcoma; also used commonly in metastatic breast cancer and multiple myeloma | 50 mg/m2 IV infusion over 1 hr for ovarian cancer, 20 mg/m2 IV infusion over 30 min for Kaposi sarcoma |
| Enzalutamide (XTANDI) | Androgen receptor signaling inhibitor | Take with or without food; 2C8 substrate; no other pathways need adjustments | Fatigue, musculoskeletal pain, dizziness, weakness | Metastatic castrate-resistant prostate carcinoma | 160 mg/day oral |
| Epirubicin (Ellence) | Anthracycline; pleiotropic effects including free radical formation, topoisomerase II inhibition; altered mitochondrial function | Metabolized primarily by liver; parent drug and metabolites glucuronidated and excreted in bile much more than via renal clearance; reduce doses for mild to moderate hepatic dysfunction; severe hepatic dysfunction is contraindicated; half-life is 30-35 hr | Myelosuppression is universal and dose limiting; alopecia is expected; this drug is a vesicant, and precaution must be taken to avoid extravasation into soft tissue around veins; nausea and vomiting are common but usually manageable; stomatitis is common; fatigue is common; detectable cardiac dysfunction at somewhat higher cumulative doses than doxorubicin; secondary leukemia is rare | Adjuvant therapy after optimal surgical treatment of localized breast cancer with involved axillary lymph nodes | 100-120 mg/m2 by IV infusion every 3-4 wk; usually combined with cyclophosphamide and 5-FU |
| Eribulin mesylate (Halaven) | Microtubule inhibitor | Reduce dose in patients with hepatic impairment and moderate renal impairment | Neutropenia, peripheral neuropathy, monitor for QT prolongations, nausea, fatigue | Persons with metastatic breast cancer who have previously received at least two chemotherapeutic regimens for the treatment of metastatic disease; prior therapy should have included an anthracycline and a taxane in either the adjuvant or metastatic setting | 1.4 mg/m2 intravenously over 2-5 min on days 1 and 8 of a 21-day cycle |
| Erlotinib (Tarceva) | Targeted agent | Inhibits the tyrosine kinase domain of the EGFR, leading to inhibition of EGFR autophosphorylation and signaling | Acneiform rash; diarrhea; interstitial lung disease | Second- or third-line therapy of NSCLC; pancreatic cancer, in combination with gemcitabine | Lung cancer: 150 mg/day; pancreatic cancer: 100 mg/day |
| Erythropoietin (Epogen, Procrit), EPO, epoetin alpha | Hematopoietic growth factor; stimulates erythrocytic precursors | Detectable in plasma for 24 hr after dosing; distributed to volume approximating total blood volume; degraded by proteolysis within blood compartment; half-life is 4-27 hr; onset of therapeutic effect takes at least 7 days; excretion of intact peptide is negligible | Hypertension is common but usually mild and not dose limiting; injection site pain is common but mild; flu-like syndrome and diaphoresis are uncommon; nausea and vomiting are rare; seizures have been reported in patients undergoing dialysis who receive the drug; iron deficiency anemia can occur after prolonged therapy, and concomitant iron dosing may increase effectiveness of erythropoietin; hematocrit values should be monitored closely during therapy to prevent polycythemia and hyperviscosity | The oncology indication is chemotherapy-induced anemia that is symptomatic; also used for anemia of chronic renal failure and HIV-associated anemia | Starting doses of 150 units/kg SC 3×/wk were recommended, with increases up to 300 units/kg if there is suboptimal effect after 6-8 wk, although 40,000 units weekly with increases to 60,000 units is the most commonly used program |
| Estramustine (Emcyt) | Conjugate of estrogen and an alkylating moiety; appears to work through estrogen-binding proteins to kill malignant cells through a nonalkylator mechanism, perhaps by inhibition of microtubules | Well absorbed by mouth, subject to hepatic metabolism, with a terminal half-life of about 20 hr; excretion route is not clearly delineated | Nausea and vomiting are common and dose limiting but diminish over time; headache, edema, decreased libido, and impotence are common; gynecomastia and breast tenderness can be seen; rash, alopecia, myelosuppression, hepatic toxicity, and thromboembolic events are rare | Prostate cancer; not used commonly for any other types of cancer | The usual dose for prostate cancer is 15 mg/kg/day, which is typically given as 420 mg PO TID for most men |
| Etoposide (Vespid), VP-16, epipodophyllotoxin; also available as etoposide phosphate (Etopophos) | Plant alkaloid; topoisomerase II inhibitor; partially cell- cycle dependent | Etoposide phosphate is rapidly converted to etoposide after IV infusion; etoposide itself is extensively protein bound, is metabolized in liver, and has half-life of about 10 hr; 50% of oral etoposide is absorbed via GI tract, requiring oral doses to be twice as high as parenteral doses; excreted both unchanged in urine and as metabolites in bile | Myelosuppression, primarily leukopenia, is universal and dose limiting; nausea and vomiting are common with PO administration but rare when the drug is given by IV; stomatitis and diarrhea are rare with normal doses but common with high doses; alopecia is mild or absent; hepatic toxicity and neurologic effects (peripheral neuropathy and CNS changes) are rare; hypotension can occur with rapid administration of etoposide but does not occur commonly when etoposide phosphate is infused over 5 min; secondary AML has been reported after etoposide | Germ cell tumors and SCLC; also used for lymphomas, AML, brain tumors, non-SCLC, and as high-dose therapy in the transplant setting for breast cancer, ovarian cancer, and lymphomas | Can be given either over several days or at lower doses over many days; typical doses are 50-120 mg/m2/day for 3-5 days IV; oral doses are generally twice IV doses; a typical protracted oral course would be 50 mg/m2/day for 21 days, every 28 days; transplant doses up to 1200 mg/m2 over 1-3 days have been used |
| Everolimus (Afinitor) | m-TOR inhibitor | Hepatic impairment: For advanced HR+ BC, advanced PNET, advanced RCC, or renal angiomyolipoma with TSC patients with hepatic impairment, reduce the starting dose. Strong CYP 3A4 inhibitors: Avoid concomitant use. Moderate CYP 3A4 and/or PgP inhibitors: If combination is required, use caution and reduce dose. Strong CYP 3A4 inducers: Avoid concomitant use. If combination cannot be avoided, increase dose. | Noninfectious pneumonitis: Monitor for clinical symptoms or radiological changes; fatal cases have occurred. Infections: Increased risk of infections, some fatal. Monitor for signs and symptoms, and treat promptly. Oral ulceration: Mouth ulcers, stomatitis, and oral mucositis are common. Cases of renal failure (including acute renal failure), some with a fatal outcome, have been observed. Laboratory test alterations: Elevations of serum creatinine, blood glucose, and lipids may occur. Decreases in hemoglobin, neutrophils, and platelets may also occur. Monitor renal function, blood glucose, lipids and hematologic parameters prior to treatment and periodically thereafter. Avoid live vaccines and close contact with those who have received live vaccines. Advanced HR+ BC, advanced PNET, advanced RCC: Most common adverse reactions include stomatitis, infections, rash, fatigue, diarrhea, edema, abdominal pain, nausea, fever, asthenia, cough, headache, and decreased appetite. Renal angiomyolipoma with TSC: Most common adverse reaction is stomatitis. SEGA with TSC: Most common adverse reaction (incidence ≥ 30%) are stomatitis and respiratory tract infection. | Postmenopausal women with advanced hormone receptor-positive, HER2 negative breast cancer (advanced HR+ BC) in combination with exemestane after failure of treatment with letrozole or anastrozole. Adults with progressive neuroendocrine tumors of creatic origin (PNET) that are unresectable, locally advanced, or metastatic. Adults with advanced renal cell carcinoma (RCC) after failure of treatment with sunitinib or sorafenib. Adults with renal angiomyolipoma and tuberous sclerosis complex (TSC) not requiring immediate surgery. Pediatric and adult patients with tuberous sclerosis complex (TSC) who have subependymal giant cell astrocytoma (SEGA) that requires therapeutic intervention but cannot be curatively resected. | Advanced HR+ BC, advanced PNET, advanced RCC, or renal angiomyolipoma with TSC: 10 mg once daily with or without food. SEGA with TSC: 4.5 mg/m2 once daily; adjust dose to attain trough concentrations of 5-15 ng/mL. Assess trough concentrations approximately 2 weeks after initiation of treatment, a change in dose, a change in co-administration of CYP 3A4 and/or PgP inducers or inhibitors, a change in hepatic function. |
| Exemestane (Aromasin) | Hormonal agent, steroidal aromatase inhibitor | 40% of oral exemestane is absorbed from GI tract, and increased by a fatty meal; it is highly protein bound in plasma; exemestane is metabolized in liver by cytochrome P450 3A4 and aldoketoreductases | Although generally well tolerated, exemestane is expected to cause or exacerbate hot flashes or intermittent flushing in some women; fatigue and mild nausea are common; vomiting, headache, and dyspnea are uncommon; it is teratogenic and should not be used in premenopausal women | Estrogen-responsive metastatic breast cancer in postmenopausal women whose disease has progressed while undergoing prior hormonal therapy | 25 mg orally once daily after a meal |
| Filgrastim (Neupogen), G- CSF | Hematopoietic growth factor, relatively specific for the granulocyte lineage | After a bolus SC injection, peak plasma levels of filgrastim occur in 2-6 hr, whereas the elimination half-life is generally 7 hr or less; metabolism is via proteolysis in the blood compartment; the intact molecule is largely absent from bile or urine | Mild bone pain is common; low-grade fever, myalgias, arthralgias, and transient hypotension are uncommon, as are hyperuricemia and elevations of lactate dehydrogenase and alkaline phosphatase; leukocytosis leading to hypoxia or capillary leak syndrome has been reported; anaphylaxis or allergic reaction are rare | Approved for minimization of granulocytopenia after myelosuppressive chemotherapy; also used to speed recovery of granulocytes in the setting of neutropenic fever after chemotherapy, for myelodysplastic syndromes, for congenital agranulocytosis, for cyclic neutropenia, and for mobilization of peripheral blood stem cells from patients or donors for transplant | Starting dose 5 µg/kg/day until neutrophil recovery (discontinue drug after an absolute neutrophil count of 10,000 or greater achieved), although generally either the whole 300 µg or 480 µg vial is used; for posttransplant or high-dose chemotherapy applications, 10 µg/kg/day is typical dose; no known maximum dose |
| Floxuridine (FUDR), FdUR, fluorodeoxyuridine | Pyrimidine nucleotide analog, antimetabolite; cell cycle dependent | After infusion into the hepatic artery, the drug is phosphorylated to the active monophosphate form and incorporated into cells; further hepatic metabolism to inactive forms is rapid; the elimination half-life is 30 min; metabolites are cleared by the kidneys | When given as a bolus, myelosuppression is dose limiting, whereas diarrhea and stomatitis are the dose-limiting toxicities of the more common protracted infusions; other GI toxicities, all rare, include nausea, vomiting, anorexia, gastritis, cramping, enteritis, and duodenal ulcers; liver toxicity, usually a cholestatic picture, is dose limiting with intrahepatic arterial infusions; serious neurologic adverse effects, including ataxia and visual changes, are rare, as is fever | FDA approved for regional (intraarterial) treatment of GI adenocarcinomas metastatic to the liver; sometimes used intravenously for the same tumors | Protracted intraarterial infusions are generally given at 0.1-0.6 mg/kg/day until grade III toxicity, sometimes according to a circadian schedule; IV doses range up to 60 mg/kg/wk by various infusion schedules |
| Fludarabine (Fludara),FAMP | Purine nucleotide analog antimetabolite; only partially cell cycle– dependent | Fludarabine is available only by the parenteral route; after IV administration, the drug is metabolized to 2-fluoroara-A and widely distributed in tissues; it has an elimination half-life of 9-10 hr; the drug and metabolite are excreted primarily by the kidneys | Neurotoxicity, including cortical blindness, confusion, somnolence, coma, and demyelinating lesions, is dose limiting, but the lower doses that are conventionally used rarely produce these adverse effects; at these doses, mild myelosuppression is the most common toxicity, cumulative lymphopenia being the most clinically important; nausea, vomiting, and other GI toxicities are rare; alopecia and rash are also rare | Approved for the treatment of CLL; also used for low-grade lymphomas and for AML | The standard regimen is 25 mg/m2/day for 5 days by short IV infusion; prolonged infusions have also been used |
| 5-Fluorouracil (Adrucil, Efudex), 5-FU | Pyrimidine antimetabolite; inhibitor of thymidylate synthase; partially cell cycle dependent | 80% of IV drug is metabolized to inactive dihydro-5-FU by dihydropyrimidine dehydrogenase in the liver; the rest of the drug is activated to fluorodeoxyuridine monophosphate in target cells; elimination half-life is 20 min; excretion is via the kidneys | GI toxicities, primarily mucositis for bolus injections and diarrhea for prolonged infusions, are dose limiting; rare patients with dihydropyrimidine dehydrogenase deficiency have excessive GI toxicity; myelosuppression is generally less with continuous infusion schedules; nausea and vomiting are uncommon and mild; dermatitis and other cutaneous toxicities, including hand-foot syndrome, are common; cerebellar ataxia and myocardial ischemia are rare | Approved for colon, rectum, gastric, pancreas, and breast carcinomas and used for a wide range of other neoplasms in combination regimens; used for intrahepatic arterial infusion for liver metastases from GI tumors; also used topically for various cutaneous neoplasms and disorders | IV dosing schemes include weekly bolus, 5 days of bolus every 28 days, 4-to 5-day continuous infusions; doses range from 300-3000 mg/m2/day depending on the dosing scheme and schedule |
| Fluoxymesterone (Halotestin, Oro- Testryl) | Synthetic steroidal androgen; antagonizes estrogenic effects in estrogen-dependent target cells | The drug is available by the oral route, is metabolized in the liver, and has an elimination half-life of about 10 hr; route of excretion is unknown | Androgenic effects predominate; hirsutism, amenorrhea, hoarseness, acne, and increased libido occur in women; men may have gynecomastia; mild edema is common; liver abnormalities, including transaminitis, fatty change, cholestatic jaundice, and rarely carcinoma, are not uncommon; polycythemia may occur | Hormone-sensitive breast cancer and for hypogonadism in males | The total daily dose for breast cancer is usually between 10-40 mg, divided into 2 or 3 doses per day |
| Flutamide (Eulexin) | Nonsteroidal antiandrogen | Good oral bioavailability, with peak plasma levels after an oral dose at 1 to 2 hr; metabolized to active and inactive forms in the liver; half-life is 8-10 hr | Generally well tolerated; gynecomastia, galactorrhea, and impotence are common; nausea, vomiting, diarrhea, mild myelosuppression, myalgias, and elevated liver function tests are rare | Prostate carcinoma | 250 mg PO 3× daily; often given in conjunction with an LHRH agonist such as leuprolide to create complete androgen blockade |
| Fulvestrant (Faslodex) estrogen receptor antagonist | Binds to the ER, leading to degradation and loss of ER from the cell | Peak plasma levels reached in 7 days, half-life is 40 days; metabolized by the liver microsomal P4503A4 systems | Constitutional symptoms, including hot flashes; peripheral edema; nausea, vomiting | ER+ metastatic breast cancer in postmenopausal women | 250 mg IM monthly |
| Gallium Nitrate (Ganite) | Heavy metal that antagonizes iron metabolism in tumor cells preferentially; causes hypocalcemia by a similar mechanism | This drug is not metabolized and has an elimination half-life of about 5 hr; cleared unchanged in the urine | Renal toxicity, including glomerular and tubular defects, is dose limiting but partly preventable with adequate hydration during therapy; hypocalcemia is expected and common and can be dose limiting; nausea, vomiting, diarrhea, and anorexia are not uncommon; mild myelosuppression, rashes, hearing loss or tinnitus, visual disturbances, and transient neurologic symptoms are rare | Malignancy-related hypercalcemia; also used for advanced bladder carcinoma | The standard dose and schedule is 300 mg/m2/day for 7 days by continuous IV infusion in a volume of 1000 mL of normal saline solution |
| Gemcitabine (Gemzar) | Antimetabolite; gemcitabine is a nucleoside analog that exhibits cell cycle– dependent and S-phase– specific cytotoxicity, likely because of inhibition of DNA synthesis | After IV infusion, the drug is rapidly distributed and has a half-life of less than 2 hr; it is metabolized throughout the body to inactive forms; parent drug and metabolite are excreted principally by the kidneys | Myelosuppression, including anemia, is mild but dose limiting; nausea and vomiting are mild but common; diarrhea and edema are sometimes seen; elevated transaminases are common, as is fever during drug administration; hematuria and proteinuria are uncommon; acute dyspnea and rash are uncommon; paresthesias and CNS depression are rare | Advanced pancreatic adenocarcinoma, NSCLC, and metastatic breast cancer; also extensively used in bladder cancer | 1000 mg/m2 in pancreatic cancer as an IV bolus weekly for up to 7 wk, followed by a week of rest before another cycle is begun; similar doses in combination with or without platinating agents are used for other indications |
| Gemtuzumab ozogamicin (Mylotarg) | Novel toxin-conjugated monoclonal antibody directed at myeloid lineage cells | After IV dosing, the total calicheamicin (ozogamicin released from the antibody by hydrolysis) has a half-life of 45 hours for the first dose and 60 hours after the second dose; metabolism of the toxin is hepatic; the elimination routes of the toxin and the antibody are unknown | This peptide antibody linked to a toxin, given in a group of patients with poor prognosis who are often medically fragile, can have marked acute toxicities; these include somewhat common typical antibody infusion adverse effects, including fever, chills, hypotension, dyspnea, and wheezing, and other uncommon toxicities including tachycardia, renal insufficiency, hepatic compromise (including hepatic venoocclusive disease), dizziness, headache, and rash; leukopenia is expected and can be prolonged, causing a high risk of bacterial, fungal, and sometimes viral infections; thrombocytopenia and anemia are common also; nausea and vomiting and diarrhea are common; serious coagulopathy or hemorrhage are rare | Relapsed AML | 9 mg/m2 as a 2-hr IV infusion given once up front and then again in 14 days |
| Goserelin Acetate (Zoladex) | LHRH that inhibits pituitary-gonadal axis function; this drug causes steroid hormone withdrawal from dependent tissues, including prostate cancer and breast cancer cells | After injection SC into adipose tissue, the depot of drug is slowly released over 28 days, peaking at 12 to 15 days; the elimination half-life is 4 hr, and the drug is not appreciably metabolized; excretion is almost entirely by the urinary route | Toxicity is mild; endocrine adverse effects are most prominent and include hot flashes, diminished libido, impotence, gynecomastia, amenorrhea, and breakthrough vaginal bleeding; other toxicities include flares of pain early during treatment in sites of disease, local tenderness at injection sites, headache, nausea, depression, and elevated cholesterol levels | FDA approved for advanced prostate cancer; used also in persons with metastatic breast cancer | 3.6 mg SC usually in the abdomen, every 28 days |
| Hydroxyurea (Hydrea)—hydroxycarbamide | Antimetabolite; inhibitor of ribonucleotide reductase, which converts nucleotides to the deoxyribose forms for DNA synthesis; cell cycle– dependent | The drug is well absorbed; drug levels peak in the blood 2 hr after the dose is administered; half-life is 2-5 hr; metabolism to inactive forms in the liver; renal excretion is the route of elimination | Myelosuppression is common and dose limiting; other toxicities include rash, headache, fever, and hyperuricemia; nausea and vomiting are uncommon; liver toxicity and serious neurologic toxicity are rare | FDA approved for CML; commonly used for other myeloproliferative disorders; also used occasionally for metastatic melanoma, refractory ovarian carcinoma, and squamous cell carcinoma of the cervix and the head and neck | For CML, the dose is 1000-3000 mg/day; in solid tumors, the dose is either 80 mg/kg every third day or 1.25 g/m2 every 8 hr for 5 doses once a week |
| Ibritumomab tiuxetan—Yttrium-90 (Zevalin) | Monoclonal antibody directed to the B-cell surface antigen CD20 linked to beta-emitting radionuclide yttrium-90 | Optimal ibritumomab/Y-90 binding and clinical effect require pretreatment with unconjugated ibritumomab; the physical half-life of Y-90 is 64 hr, but the biological half-life of the agent in the body in terms of radioactivity detected is 30 hr; metabolic and excretory fates of the radionuclide and antibody are not known | Serious infusion reactions, prolonged and severe cytopenias, and severe cutaneous and mucocutaneous reactions; antibody toxicities can include fever, chills, dyspnea, wheezing, urticaria, and rash; radionuclide or total agent adverse effects include lymphopenia, and myelosuppression of other cell lines, which can be prolonged; infection risk is increased accordingly; nausea, vomiting, and diarrhea are uncommon, as are arthralgias, myalgias, or neurologic adverse effects; previously required imaging study, no longer used | Relapsed and/or transformed follicular B-cell lymphomas | Administer rituximab 250 mg/m2 IV; on day 7, 8, or 9: administer rituximab, 250 mg/m2 IV infusion; if platelets >150,000/mm3, within 4 hr after rituximab infusion, administer 0.4 mCi/kg (14.8 MBq per kg) Y-90 Zevalin IV; if platelets >100,000 but <149,000/mm3 in relapsed or refractory patients: within 4 hr after rituximab infusion, administer 0.3 mCi/kg (11.MBq per kg) Y-90 Zevalin IV |
| Idarubicin (Idamycin), 4-demethoxy-daunorubicin | Anthracycline; ; pleiotropic effects including free radical formation, topoisomerase II inhibition; altered mitochondrial function | After an IV dose, the drug is metabolized in the liver to active and inactive forms; the elimination half-life of the parent compound is 13-26 hr | Myelosuppression is common and generally dose limiting for each dose; the cumulative dose-limiting toxicity is cardiomyopathy, but idarubicin is less cardiotoxic than daunorubicin or doxorubicin; nausea and vomiting are common but usually mild; diarrhea and stomatitis are sometimes seen; idarubicin is a weak vesicant or irritant | Approved for the treatment of AML | The standard dose as part of a “7 plus 3” regimen (with cytarabine) is 12 mg/m2/day for 3 days for induction or reinduction/intensification; other doses have been used |
| Ifosfamide (Ifex) | Classic alkylating agent; not cell cycle–dependent. | After an intravenous dose, ifosfamide is activated by hepatic microsomal enzymes; it is then converted to inactive metabolites in the liver; the active form of the drug is the same as that for cyclophosphamide; the elimination half-life of the drug is 7-15 hr; the metabolites and some unchanged drug are excreted in the urine | Myelosuppression, hemorrhagic cystitis, and CNS toxicity are all fairly common and can be dose limiting; hemorrhagic cystitis can largely be prevented by co-administration of the uroprotective agent mesna, and nausea and vomiting are minimized with modern antiemetic regimens; the CNS toxicity, including lethargy, stupor, coma, myoclonus, and seizures, is usually mild and completely reversible; it is worse with impaired renal function; renal dysfunction, usually reversible, is also seen with ifosfamide; hepatic toxicity, diarrhea, and rash are rare | Approved for the treatment of recurrent germ cell tumors; used for many other tumor types, including adult sarcomas, lymphoma, Hodgkin disease, breast cancer, and ovarian cancer | IV over 3-5 days with total dose of 8-12 g/m2/cycle, repeated every 3-4 wk; can be given as a short infusion each day or as a continuous infusion; mesna is given IV concurrently, also by short infusion or continuous infusion; hydration of greater than 3 L/day total, with saline or alkali solutions, is also recommended |
| Interferon-alpha (IntronA, Roferon) | Biological response modifier, antiviral, immunostimulant | After parenteral administration, peak levels of IFN-α in the blood occur in 30 min–8 hr depending on the route; the elimination half-life is 2-9 hr; IFN-α is catabolized throughout the body through proteolysis but primarily in the renal tubules; excretion of intact drug is minimal and not significantly affected by organ function | Constitutional symptoms are predominant adverse effects and are dose limiting in both the short and long term in lower dose schedules; acute adverse effects include fever, chills, nasal congestion, diarrhea, and malaise; chronic adverse effects include fatigue, anorexia, weight loss, and depression; neutropenia and thrombocytopenia, both of which are transient, are dose limiting at higher doses; anemia may also occur, albeit with more chronic administration; cardiac toxicity, including congestive heart failure and arrhythmias, is rare and almost always reversible; serious CNS toxicity, including delirium and psychosis, or peripheral neuropathies are also rare and reversible; hypocalcemia and hyperglycemia can also occur | Nonmalignant conditions and malignancies including melanoma, CML, hairy cell leukemia, Kaposi sarcoma, and cutaneous T-cell lymphoma; also used in multiple myeloma and low-grade lymphomas | Dose depends on both the diagnosis and the brand or type of IFN- α; the doses for malignant conditions range from 2 million up to 30 million units/m2 by the SC, IM, or IV route from 3×/wk to every day; adjustments are made on the basis of patient tolerance and laboratory parameters |
| Interleukin-2 (Proleukin)—aldesleukin, IL-2 | IL-2 is a glycoprotein cytokine, previously known as T-cell growth factor, that stimulates antigen-specific and nonspecific T-cell and other lymphocyte subsets and also triggers an inflammatory cytokine cascade; its antineoplastic effects are dependent on an intact immune system | Elimination half-life of 30-60 min; it is catabolized by proteolysis throughout the body; negligible amounts of intact drug are found in urine or bile | IL-2 has a wide range of moderate to severe toxicities that are both dose and schedule dependent; toxicities tend to follow immediately after a bolus dose but gradually accumulate during a continuous infusion; toxicities are higher for a given dose with continuous infusion compared with bolus dosing; capillary leak syndrome, which is dose limiting for most IL-2 administration schedules, results in hypotension, edema, pulmonary congestion, renal insufficiency, arrhythmias, diarrhea, and possibly some of the CNS and hepatic toxicity seen with IL-2; transient myelosuppression or more prolonged anemia occurs commonly, as does transient hyperbilirubinemia, elevation of transaminases, and electrolyte imbalances; other constitutional symptoms that occur with IL-2 include fever, chills, malaise, arthralgia/myalgias, erythroderma, nasal congestion/rhinorrhea, and nausea/vomiting; other serious and less common toxicities include lethargy or delirium, angina pectoris, congestive heart failure, frank respiratory failure, and infections, particularly gram-positive bacteremia | High-dose bolus treatment of metastatic renal cell cancer and metastatic melanoma; also used at lower doses for metastatic melanoma and for maintenance treatment of acute myeloid leukemia | The FDA-approved dose for renal cell carcinoma and melanoma is a 600,000-720,000 IU/kg IV bolus every 8 hr for a maximum of 14 doses on days 1 to 5 and 11 to 15 every 6 wk; lower doses are more commonly used, especially continuous infusions of 3-18 million IU/m2/day for 96 hr; SC administration at similar daily doses has also been attempted with reasonable patient tolerance |
| Peginterferon alfa-2b (Sylatron) | Pleiotropic cytokine; the mechanism by which it exerts its effects in patients with melanoma is unknown | Increase frequency of monitoring for fatigue, increased ALT, increased AST, pyrexia, headache, anorexia, myalgia, nausea, chills, and injection site reaction | Depression and other neuropsychiatric disorders; SYLATRON toxicity in patients with moderate and severe renal impairment | Melanoma | 6 µg/kg/wk subcutaneously for 8 doses followed by 3 µg/kg/wk SC for up to 5 yr |
| Ipilimumab (Yervoy) | Human CTLA-4–blocking antibody | No formal pharmacokinetic drug interaction studies have been conducted; no clinically important differences in clearance of ipilimumab were found between patients with renal impairment and patients with normal renal function; no clinically important differences in clearance of ipilimumab were found between patients with mild hepatic impairment and normal hepatic function | Immune-mediated adverse reactions; fatigue, diarrhea, pruritus, rash, and colitis | Unresectable or metastatic melanoma | 3 mg/kg IV over 90 min every 3 wk for a total of 4 doses |
| Irinotecan (Camptosar)—CP T-11 | Semisynthetic camptothecin, which functions as a topoisomerase I inhibitor; -partly cell cycle– dependent | Converted partially from active lactone form to inactive carboxylate form; active metabolite of irinotecan, SN-38, also exists in lactone and inactive carboxylate form in equilibrium in plasma; SN- 38 is inactivated by glucuronidation in the liver; SN-38 is responsible for majority of antitumor activity attributed to parent drug; half-life of irinotecan is 8 hr, whereas half-life of SN-38 is 12 hr | Myelosuppression, primarily neutropenia, is common and dose limiting; diarrhea is also common and can be dose limiting; diarrhea can occur as part of a cholinergic syndrome, along with cramping, nausea, and vomiting, during or immediately after drug administration or for several days after drug administration; anticholinergic and antidiarrheal agents will curtail the immediate diarrhea and other GI symptoms partially but are less effective in treating the delayed diarrhea; flushing, rash, and alopecia are common; significant hepatic, renal, neurologic, or pulmonary toxicities are rare | Irinotecan is FDA approved for refractory or recurrent metastatic colon cancer, and it has now been used in other malignancies, including lung cancer, ovarian cancer, and lymphoma | The recommended dosage for recurrent colon cancer is 125 mg/m2 as a 90-minute IV infusion every wk for 4 wk, with this cycle repeated every 6 wk; other doses and schedules have been used |
| Isotretinoin (Accutane), 13-cis- retinoic acid, 13- CRA | Isotretinoin is a retinoid derivative of vitamin A that binds to specific nuclear receptors and leads to changes in gene expression; this results in apoptosis or differentiation of many malignant or premalignant cell lines | Oral bioavailability is about 25%, and the drug is highly protein bound in plasma; it is metabolized in the liver and has an elimination half-life of 10-20 hr; parent compound and metabolite are excreted in both the urine and feces | Isotretinoin is teratogenic and should not be given to women of childbearing age without adequate contraception; mucocutaneous adverse effects are common and dose limiting and include xerostomia, stomatitis, conjunctivitis, dry skin, pruritus, cheilitis, rash, patchy alopecia, fragility of nails and skin, photosensitivity, and epistaxis; other less common adverse effects include elevations in transaminases and bilirubin or frank hepatitis, hyperlipidemia, nausea, vomiting, anorexia, diarrhea, headache, fatigue, depression, and myalgias/arthralgias; anemia and pseudotumor cerebri are rare | Approved for acne vulgaris; has shown some effectiveness in chemoprevention of aerodigestive malignancies; ongoing studies are testing its chemopreventive potential in other malignancies | Daily oral doses of 0.5-4 mg/kg/day have been used in the chemoprevention trials for durations of 2-6 mo |
| Ixabepilone (Ixempra) | Microtubule inhibitor | In combination with capecitabine, must not be given to patients with AST or ALT >2.5 × ULN or bilirubin >1 × ULN; dose reduction is recommended when administering IXEMPRA as monotherapy to patients with hepatic impairment; no formal renal impairment study; inhibitors of CYP3A4 may increase plasma concentrations of ixabepilone; dose of IXEMPRA must be reduced with strong CYP3A4 inhibitors; inducers of CYP3A4 may decrease plasma concentrations of ixabepilone; alternative therapeutic agents with low enzyme induction potential should be considered | Peripheral neuropathy: monitor for symptoms of neuropathy, primarily sensory; neuropathy is cumulative, generally reversible, and should be managed by dose adjustment and delays; myelosuppression: primarily neutropenia; monitor with peripheral blood cell counts and adjust dose as appropriate; hypersensitivity reaction: must premedicate all patients with an H1 antagonist and an H2 antagonist before treatment; fatigue/asthenia, myalgia/arthralgia, alopecia, nausea, vomiting, stomatitis/mucositis, diarrhea, and musculoskeletal pain | Combination with capecitabine is indicated for the treatment of metastatic or locally advanced breast cancer in patients after failure of an anthracycline and a taxane; as monotherapy is indicated for the treatment of metastatic or locally advanced breast cancer in patients after failure of an anthracycline, a taxane, and capecitabine | 40 mg/m2 infused intravenously over 3 hr every 3 wk |
| Ketoconazole (Nizoral) | Oral antifungal agent that also acts as an androgen antagonist at high doses | Ketoconazole has good oral bioavailability; its level peaks in the plasma in about 2 hr; it has a terminal half-life of about 8 hr; it is metabolized in the liver to several inactive metabolites; the metabolites are excreted in the bile | This drug is not strictly an antineoplastic drug and is generally very well tolerated; nausea, vomiting, headache, dizziness, fever, chills, impotence, gynecomastia, leukopenia, hemolytic anemia, urticaria, and anaphylaxis are all rare; hepatic toxicity is also rare; it has been fatal in unusual cases; administration with terfenadine and astemizole has resulted in prolonged QT interval, arrhythmias, and deaths in rare cases; other potential drug interactions are possible | FDA approved for fungal infections, primarily yeast infections; used in doses of up to 1200 mg/day for androgen dependent or independent prostate cancer | As per preceding instructions, used alone or in combination with chemotherapy, including doxorubicin |
| Lapatinib (Tykerb) | EGRF and HER2 tyrosine kinase inhibitor | Extensive metabolism spread over several enzymes | Hepatotoxicity; decreased left ventricular ejection fraction; diarrhea; some QT interval prolongation | HER2+ breast cancer, with capecitabine; treatment with anthracycline, taxane, trastuzumab failed | 1250 mg once daily, without food |
| Lenalidomide (Revlimid) | Antiangiogenesis agent, immunomodulator | Mechanism of action not fully characterized; an immunomodulatory agent that inhibits angiogenesis in some cells; inhibits bone marrow secretion of IL-6, VEGF, and TNF-α | Myelosuppression; diarrhea, rash, fatigue; increased risk of DVT and PE | Transfusion-dependent anemia due to low- or intermediate-risk myelodysplasia with 5q deletion; multiple myeloma | For MDS: 10 mg daily; for myeloma: 25 mg daily for 21 of 28 days |
| Letrozole (Femara) | Nonsteroidal aromatase inhibitor | Letrozole has nearly 100% bioavailability, is metabolized in the liver, glucuronidated, and excreted by the kidneys; half-life is 2 days; with daily administration, steady- state plasma levels are reached in 2-6 wk | This drug is generally well tolerated; muscle aches and nausea are uncommon; hot flashes and fatigue are uncommon; weight change, urticaria, and dyspepsia are rare | Metastatic estrogen responsive breast cancer in postmenopausal patients | 2.5 mg orally once daily |
| Leucovorin Calcium (Wellcovorin) citrovorum factor, folinic acid, FA, LV | Tetrahydrofolate derivative and enzyme cofactor for thymidylate synthase and other purine and pyrimidine synthesis steps; bypasses the dihydrofolate reductase step, which is inhibited by methotrexate and therefore can be used to “rescue” normal cells from the toxicity of methotrexate after high doses are administered; in addition, leucovorin potentiates the toxicity of fluoropyrimidines such as fluorouracil by strengthening the association of the drug | Leucovorin has excellent bioavailability by the oral or parenteral route; it is oxidized in cofactor reactions throughout the body, and is also partly metabolized; it has an elimination half-life of 2-4 hr; excreted in the urine | Leucovorin is generally very well tolerated; it occasionally causes stomach upset or nausea, rash, diarrhea, and headache; allergic reactions have been reported | Used for rescue of high-dose methotrexate therapy for a variety of neoplasms and as a potentiator of fluoropyrimidines therapy in GI malignancies, particularly colorectal cancer | For rescue from methotrexate, the usual dose is 10-25 mg/m2 orally or IV every 6 hr starting up to 24 hr after the methotrexate, until methotrexate levels are less than 1 × 10−8 molar; when used to potentiate 5-FU, doses ranging from 20 to 500 mg/m2, usually given IV, have been used, depending on the 5-FU dose |
| Leuprolide acetate (Lupron)—leuprorelin acetate | Gonadotropin-releasing hormone agonist, which serves to paradoxically shut down the pituitary release of gonadotropins with chronic exposure; this results in a dramatic decrease in gonadal estrogens and androgens and growth inhibition of hormone-dependent neoplasms | After an SC injection, about 90% of the drug is eventually absorbed; the depot form of the drug is absorbed slowly over days, whereas the injectable solution is absorbed over several hours; the elimination half- life of the drug once in the serum is 3 hr; metabolism and excretion are not well delineated but are clinically unimportant | Usually well tolerated, but adverse effects can affect many systems, including endocrine (hot flashes, impotence, gynecomastia, breast tenderness, diminished libido, amenorrhea, atrophic vaginitis, increased cholesterol); GI (nausea, constipation, anorexia, diarrhea); hepatic (elevation of transaminases); dermatologic (rash, changes in body hair composition, pruritus); and neuropsychiatric (insomnia, depression, emotional lability, lethargy, memory loss); significant cardiac toxicity is rare | Approved for the treatment of hormone-dependent advanced prostate cancer; also used for breast cancer and endometriosis | The usual dose for prostate cancer is 7.5 mg of the depot form by SC injection once every month or 1 mg of the injectable solution SC daily |
| Levoleucovorin (Fusilev) | Single-isomeric form of the tetrahydrofolate derivative and enzyme cofactor for thymidylate synthase and other purine and pyrimidine synthesis steps | Pharmacokinetics of levoleucovorin after 15 mg IV dose was studied in healthy male volunteers; after rapid intravenous administration, the mean time to peak was 0.9 hr; half-life for total-THF and (6S)-5- methyl-5,6,7,8 tetrahydrofolate was 5.1 and 6.8 hr, respectively | Allergic reactions were reported in patients; vomiting (38%), stomatitis (38%), and nausea (19%) were reported in patients after high-dose methotrexate therapy; the most common adverse reactions (>50%) in patients with advanced colorectal cancer in combination with 5-FU were diarrhea, nausea, and stomatitis | Rescue after high-dose methotrexate in osteosarcoma and to reduce toxicity of methotrexate | Dosed at 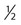 the usual dose of racemic d,l-leucovorin the usual dose of racemic d,l-leucovorin |
| Lomustine (CeeNU), CCNU | Nitrosourea alkylating agent; cell cycle– independent | Well absorbed after oral dose; widely distributed in body, including cerebrospinal fluid; metabolized extensively in liver to active metabolites; half-life is 72 hr | Myelosuppression is dose limiting and tends to be cumulative; nausea and vomiting are common but usually mild to moderate; anorexia is also common but short lived; pulmonary fibrosis can occur with long-term administration; other toxicities, including CNS effects, hepatic or renal dysfunction, and secondary leukemia, are rare | Approved for primary brain tumors and Hodgkin disease; also used in melanoma, multiple myeloma, other lymphomas, and breast cancer | The recommended dose for brain tumors is 100-130 mg/m2 orally every 6 wk; other doses and schedules have been used |
| Mechlorethamine (Mustargen), nitrogen mustard, HN2 | Classic alkylating agent; cell cycle– independent | Mechlorethamine is not orally bioavailable; after an IV dose, the drug is rapidly deactivated in the blood by reaction with biomolecules; it has an elimination half-life of 15 min and has no significant organ metabolism; virtually no excretion of the drug is detected in urine or stool | This drug is a powerful vesicant, so optimal extravasation precautions are a must, as well as rapid infusion; tissue necrosis will occur if the drug extravasates, although sodium thiosulfate is a somewhat effective antidote; vein discoloration and scarring are common; nausea and vomiting are common, potentially severe, and often dose limiting; myelosuppression is expected and also often dose limiting; alopecia and infertility are often seen; less common toxicities include anorexia, diarrhea, jaundice, tinnitus, and skin rash (common with topical treatment); secondary leukemia and permanent hearing loss are rare | Approved for a variety of hematologic malignancies and solid tumors but generally used less in the last decade; still used for Hodgkin disease and topically for cutaneous T-cell lymphoma | The standard dose for Hodgkin disease as part of the MOPP regimen is 6 mg/m2 IV over 1-5 min on day 1 and day 8 of a 28-day cycle |
| Medroxyprogesterone acetate (Provera, Depo- Provera) | Steroidal progestational agent | Good oral bioavailability; it is metabolized in the liver to inactive metabolites and has an elimination half-life of up to 60 hr; parent drug and metabolites are excreted in the urine and bile | Toxicities are mostly constitutional and not dose limiting; they include menstrual changes, amenorrhea, gynecomastia, hot flashes, edema, weight gain, fatigue, acne, hirsutism, anxiety, depression, sleep disturbance, and headache; nausea, significant skin reactions or allergy, jaundice, and thrombophlebitis are uncommon | Approved for treatment of advanced endometrial or renal cell carcinoma; also used occasionally for breast or prostate cancer | Loading doses of up to 1000 mg IM weekly and 400 mg IM every month have been used, while oral doses range from 100-300 mg/day; much lower doses are used for gynecologic indications |
| Megestrol Acetate (Megace), megestrol | Steroidal progestational agent | Well absorbed by mouth, metabolized in liver to inactive compounds; half-life is 15-20 hr | Toxicities are similar to those of other progestins as noted previously; they include menstrual changes, hot flashes, edema, weight gain, fatigue, acne, hirsutism, anxiety, depression, sleep disturbance, and headache; urinary frequency can also occur; nausea, vomiting, diarrhea, skin rash or allergy, jaundice, and thrombophlebitis are uncommon | Approved for treatment of breast and endometrial carcinoma; also used for renal cell carcinoma and for appetite stimulation in HIV disease and patients with cancer | The standard dose for cancer treatment is 160 mg/day in divided doses or a single dose; the dose for appetite stimulation may be as high as 800 mg/day, which is where the concentrated oral solution is useful |
| Melphalan (Alkeran), l-PAM, l-phenylalanine mustard, l-sarcolysin | Classical alkylating agent; cell cycle–independent | Melphalan has unpredictable GI absorption, is highly protein bound, and is rapidly autometabolized by hydrolysis in the plasma; it has an elimination half-life of about 2 hr; 10%-15% is excreted as unchanged drug in the urine | Myelosuppression is expected and is dose limiting; recovery may be prolonged, and effects can be cumulative; large doses may cause significant nausea and vomiting; diarrhea and stomatitis are uncommon; vein reactions including scarring may occur, but this agent is not known as a vesicant; other skin reactions are uncommon, as are pulmonary fibrosis, vasculitis, infertility, alopecia, and secondary leukemia | Used primarily for multiple myeloma, but also FDA approved for ovarian carcinoma; may also be useful in high-dose chemotherapy/transplant settings and in regional perfusion of extremities for melanoma and sarcoma | Doses for myeloma are typically in the range of 0.1 mg/kg/day for 2-3 wk or up to 6 mg/m2/day for 5 days every 6 wk; transplant doses (IV or PO) range up to 140 mg/m2 total; doses for perfusion are either 0.45-0.9 mg/kg or dosed for a certain concentration in the perfusate |
| Mercaptopurine (Purinethol), 6-MP, 6-mercaptopurine | Purine analog antimetabolite; predominantly S-phase specific | 6-MP has good oral bioavailability, undergoes extensive first-pass metabolism in the liver, and has an elimination half-life of about 7 hr; intact drug and metabolites are excreted by the kidneys | Myelosuppression is common and dose limiting; nausea and vomiting occur occasionally but are usually mild; diarrhea, anorexia, and stomatitis are less common; headache and rash are uncommon; fulminant hepatic toxicity is very rare, but lesser degrees of cholestasis and hepatitis are sometimes seen | Mercaptopurine is approved for treatment of acute lymphoblastic leukemia; it is occasionally used for other hematologic malignancies | The usual dose is 70 -100 mg/m2/day for a defined period of days during induction or maintenance |
| Mesna (Mesnex), mercaptoethane sulfonate sodium, Uromitexan | Thiol uroprotectant; binds to and inactivates acrolein, the highly reactive metabolite of cyclophosphamide and ifosfamide, helping to prevent hemorrhagic cystitis | Mesna has an oral bioavailability of about 50% and is usually given by IV; after an IV dose, mesna is converted in the plasma to dimesna, is filtered by the kidneys, and is converted back into mesna in the urine; it has an elimination half-life of 1 hr | Mesna is usually very well tolerated; it has been described to occasionally cause nausea, vomiting, diarrhea, rash, fatigue, headache, hypotension, or arthralgias | Approved for use as a uroprotectant when administering ifosfamide; LSO effective for high-dose cyclophosphamide | Daily dose of mesna is 60% of daily milligram amount of ifosfamide, via IV bolus before, 4 hr after, and 8 hr after the chemotherapy or as a continuous infusion with a loading dose before chemotherapy; mesna may be continued for up to 24 hr after the chemotherapy has been completed |
| Methotrexate (Mexate, Folex, others)—MTX, amethopterin | Antifolate antimetabolite; interferes with nucleotide synthesis by inhibiting dihydrofolate reductase; cell cycle–dependent | Good oral bioavailability at low doses; after oral or IV dosing, it is distributed throughout the body water compartment; it will accumulate in “third-space” fluid compartments and exhibit prolonged toxicity and therefore should be used with caution, if at all, in patients with significant pleural or peritoneal fluid; minimally metabolized in the liver; half-life is 3 hr; even low concentrations of drug after most of the drug is eliminated can contribute to significant toxicity; therefore dosing based on renal function is critical; excretion of this drug is mostly renal | Myelosuppression is expected and is usually dose limiting; stomatitis and diarrhea are common; nausea and vomiting are uncommon; renal toxicity is uncommon and usually reversible but can be severe; many types of skin reactions can occur but are uncommon; pulmonary fibrosis and hepatic fibrosis are rare; encephalopathy is rare with moderate- to low-dose therapy but is more common with high doses, intrathecal administration, or concomitant CNS radiation; it can be severe and permanent | Approved for a wide spectrum of malignant and nonmalignant diseases; most often used for acute leukemias, lymphomas, breast cancer, bladder cancer, squamous cell cancers, and sarcomas | For malignant conditions, doses up to 100 mg/m2 are considered low dose, 100-1000 mg/m2 are a moderate dose, and >1000 mg/m2 is high dose; moderate and high doses require leucovorin rescue; doses can be given weekly or at longer intervals; IV infusions can be 30 minutes or longer, including 24-hour continuous infusions; methotrexate is also commonly given intrathecally, usually as a 12-mg dose in 10 mL of preservative-free saline solution |
| Mitomycin C (Mutamycin) | Antitumor antibiotic; inhibits DNA and RNA synthesis | After an IV dose, mitomycin C is rapidly metabolized to inactive forms in the liver, spleen, and kidneys, with an elimination half-life of about 1 hr; parent drug and inactive metabolites are excreted in the urine | Mitomycin C is a vesicant; extravasation precautions are a must; myelosuppression is expected and is dose limiting, with a white blood cell nadir at 4 wk and full recovery at 6-7 wk; mild nausea, vomiting, anorexia, and fatigue are common; uncommon toxicities include diarrhea, stomatitis, rash, fever, and renal insufficiency; rare toxicities include venoocclusive disease of the liver, hemolytic-uremic syndrome, and interstitial pneumonitis | Approved for adenocarcinomas of the stomach and pancreas; also used commonly in breast cancer and lung cancer | The usual dose is 10-20 mg/m2 IV over 2-5 min every 6-8 wk |
| Mitotane (Lysodren), o,p′-DDD | Adrenal cortical cytotoxin | Moderate oral bioavailability, with peak plasma level 4 hr after oral dose; significant therapeutic effect is not seen until up to 4 wk of continuous usage; metabolized in the liver and has variable elimination half-life (due to storage of the drug in adipose tissue) of up to 160 hr; mitotane is eliminated in the urine and bile | Adrenal insufficiency is expected and must be abrogated with concomitant oral glucocorticoid usage (and sometimes mineralocorticoids as well); anorexia, nausea, vomiting, sedation, and lethargy are common; hypercholesterolemia and elevation of liver function tests are also common; rash is seen frequently but is usually mild; myelosuppression, diarrhea, fever, wheezing, changes in blood pressure, and flushing are uncommon; permanent CNS changes, retinopathy, nephrotoxicity, and hemorrhagic cystitis are rare | Adrenal cortical carcinoma | The initial dose is usually 1 g/day in 4 divided doses; this is increased up to 10 g/day as tolerated |
| Mitoxantrone (Novantrone), DHAD, dihydroxyanthrace nedione | Topoisomerase II inhibitor | After an IV dose, it exhibits a large volume of distribution, undergoes metabolism in the liver, and has an elimination half-life of 24-37 hr; mitoxantrone is eliminated through the bile | Mitoxantrone is not a tissue vesicant; myelosuppression, mostly limited to leukopenia, is dose limiting; nausea, vomiting are common but mild, stomatitis is common, and diarrhea and anorexia are less common; elevated liver function tests are common, but significant hepatic toxicity is rare; cardiotoxicity is uncommon and dose dependent; pulmonary or neurologic toxicity is rare | Approved for AML and prostate carcinoma; also used for breast cancer, lymphoma, and hepatocellular carcinoma | For AML, the typical dose is 10-12 mg/m2/day for 3 days by 30-min infusion, along with AraC; for solid tumors, 12 mg/m2 is given over 30 min every 3-4 wk |
| Nelarabine (Arranon) | Antimetabolite | A prodrug of 9-beta-D-arabino- furanosylguanine (ara-G); after demethylation by adenosine deaminase to ara-G, it is converted to its active triphosphate form ara-GTP; incorporation of ara-GTP into DNA inhibits DNA synthesis and function | CNS toxicity is dose limiting, with somnolence, headache, dizziness, peripheral neuropathy, seizures, and coma, which may be severe and permanent; myelosuppression; GI toxicities; respiratory toxicity; fatigue, fever, asthenia, blurred vision, and edema | Refractory or resistant T-cell acute lymphoblastic leukemia/lymphoma as third-line therapy | 1500 mg/m2 IV over 2 hr on days 1, 3, and 5 every 21 days |
| Nilotinib (Tasigna) | Kinase inhibitor | Dose adjustment may be required for hematologic and nonhematologic toxicities and drug interactions; lower starting dose is recommended in patients with impairment (at baseline); do not consume food for at least 2 hr before the dose is taken and for at least 1 hr after | Prolongs QT interval; rash, pruritus, headache, nausea, fatigue, myalgia, nasopharyngitis, constipation, diarrhea, abdominal pain, vomiting, arthralgia, pyrexia, upper respiratory tract infection, back pain, cough, and asthenia; hematologic adverse drug reactions include myelosuppression: thrombocytopenia, neutropenia, and anemia | Newly diagnosed adult patients with Philadelphia chromosome–positive chronic myeloid leukemia (Ph+ CML) in chronic phase; also, the treatment of CP and AP Ph+ CML in adult patients resistant to or intolerant to prior therapy that included imatinib | Newly diagnosed Ph+ CML-CP: 300 mg orally twice daily; resistant or intolerant Ph+ CML-CP and CML-AP: 400 mg orally twice daily; administer Tasigna approximately 12 hr apart and must not take with food |
| Nilutamide (Nilandron) | Orally administered nonsteroidal antiandrogen | Rapid and complete GI absorption; elimination half-life is about 45 hr; the drug is metabolized in the liver and eliminated in the urine | Hot flashes, body hair loss, fatigue, loss of libido, and weight gain are common but usually mild; loss of visual adaptation to the darkness is common but transient, and nausea and fever and dyspepsia are uncommon; interstitial pneumonitis is rare | Metastatic prostate cancer | The usual dose is 300 mg once daily for 30 days followed by 150 mg daily |
| Octreotide, Octreotide long-acting (Sandostatin, Sandostatin LA), l- cysteinamide | Synthetic peptide analog of somatostatin; inhibits other GI peptide actions, such as serotonin, insulin, glucagon, and gastrin | Not orally bioavailable but rapidly absorbed after SC administration; metabolized by hydrolysis throughout the body; no active metabolites; half-life of elimination is about 1.5 hr; intact drug is cleared via the kidneys | GI adverse effects are dose limiting and include abdominal pain, vomiting, loose stool, occasional fat malabsorption, bloating, and cholelithiasis; elevations of liver function tests can also occur, but frank hepatitis is rare; skin reactions, such as pain at the injection site or flushing, rash, or skin thinning, are sometimes seen; constitutional symptoms, including rhinorrhea, xerostomia, sweating, throat discomfort, and vertigo, can be bothersome; hyperglycemia or hypoglycemia can occur; cardiac side effects, including angina, congestive heart failure, and hypotension or hypertension, are uncommon; anxiety, depression, fatigue, and anorexia are uncommon, and seizures are rare | Approved for carcinoid tumors causing carcinoid syndrome and for vasoactive peptide-secreting tumors; also used for refractory diarrhea, either cancer related or treatment related, in patients with cancer | Doses from 50 µg twice a day to 1000 µg 4× a day injected SC have been used; continuous IV infusions or administration of drug in total parenteral nutrition solutions has also been used |
| Ofatumumab (Arzerra) | CD20-directed cytolytic monoclonal antibody | Creatinine clearance at baseline did not have a clinically important effect on ofatumumab pharmacokinetics in patients with calculated creatinine clearance values ranging from 33-287 mL/min; age did not significantly influence ofatumumab pharmacokinetics in patients ranging from 21-86 years of age; no pharmacokinetic data are available in pediatric patients | Infusion reactions, cytopenias, progressive multifocal leukoencephalopathy; neutropenia, pneumonia, pyrexia, cough, diarrhea, anemia, fatigue, dyspnea, rash, nausea, bronchitis, and upper respiratory tract infections | CLL refractory to fludarabine and alemtuzumab | 300 mg initial dose, followed 1 wk later by 2000 mg weekly for 7 doses, followed 4 wk later by 2000 mg every 4 wk for 4 doses; premedicate with oral acetaminophen, oral or intravenous antihistamine, and IV corticosteroid |
| Omacetaxine mepesuccinate (Synribo) | The mechanism of action of omacetaxine mepesuccinate has not been fully elucidated but includes inhibition of protein synthesis and is independent of direct Bcr-Abl binding | Omacetaxine mepesuccinate is absorbed following subcutaneous administration, and maximum concentrations are achieved after approximately 30 min; primarily hydrolyzed to 4′-DMHHT via plasma esterases with little hepatic microsomal oxidative and/or esterase-mediated metabolism in vitro | Myelosuppression: severe and fatal thrombocytopenia, neutropenia and anemia; monitor hematologic parameters frequently; bleeding: severe thrombocytopenia and increased risk of hemorrhage; fatal cerebral hemorrhage and severe, nonfatal GI hemorrhage; hyperglycemia: glucose intolerance and hyperglycemia including hyperosmolar nonketotic hyperglycemia; diarrhea, nausea, fatigue, asthenia, injection site reaction, pyrexia, infection, and lymphopenia | Chronic or accelerated phase CML with resistance and/or intolerance to two or more tyrosine kinase inhibitors | Induction dose: 1.25 mg/m2 administered SC twice daily for 14 consecutive days of a 28-day cycle; maintenance dose: 1.25 mg/m2 administered SC twice daily for 7 consecutive days of a 28-day cycle; dose modifications are needed for toxicity |
| Oprelvekin (Neumega)—inter leukin-11, IL-11 | Recombinant polypeptide cytokine molecule; multiple cellular actions, including stimulation of megakaryocyte proliferation and platelet production from megakaryocytes | With SC administration, it is absorbed into the circulation with a peak plasma concentration of 3 hr and has an elimination half- life of about 7 hr; this polypeptide agent is metabolized throughout the body by proteolysis; excretion of drug is not substantial because of degradation | Headache, fever, malaise, dyspnea, rash, conjunctival irritation, fluid retention, and edema are common during administration but are not usually severe; oral thrush, dizziness, diarrhea, pleural effusions, and transient anemia are uncommon; paresthesias, ocular hemorrhage, atrial arrhythmias, and exfoliative dermatitis are rare | Severe chemotherapy-related thrombocytopenia | 50 µg/kg/day SC injection until the postnadir platelet count is greater than 50,000/mm3, starting 1 day after the completion of chemotherapy |
| Oxaliplatin (Eloxatin) | Platinating agent; DNA disrupted via intrastrand and interstrand cross-links with two strong platinum association bonds in the molecule, which induces apoptosis beyond a certain level of DNA damage in malignant cells | After IV administration, rapid distribution into tissues occurs, as well as rapid spontaneous conversion via hydrolysis into active drug and metabolites; the terminal half-life is long (>300 hr) but represents minimal plasma levels of the hydrolyzed drug; elimination of platinum metabolites is via the kidneys | Neurotoxicity, in the form of a transient neuropathy with each dose and a persistent, cumulative typical sensory polyneuropathy, is very common and dose limiting; myelosuppression is expected but mild and only sometimes dose limiting; fatigue and nausea are common but mild; diarrhea, stomatitis, edema, cough, hypersensitivity reactions, and extravasation injury are rare | Approved for metastatic colorectal cancer in combination with 5-FU/leucovorin | 85 mg/m2 IV every 2 wk as a 2-hr infusion in 250-500 mL of D5W |
| Paclitaxel (Taxol, Onxol) | Naturally occurring taxane molecule; inhibits depolymerization of tubulin in the spindle apparatus, thereby inducing apoptosis in dividing cells | After IV administration, the drug exhibits a large volume of distribution and undergoes metabolism in the liver; the elimination half-life is 15-50 hr; excretion of drug and metabolites is predominantly via the bile | Paclitaxel is an irritant or mild vesicant when extravasated into subcutaneous tissue; myelosuppression, predominantly neutropenia, is expected and is dose limiting; shorter infusions of the same dose produce less neutropenia; mucositis is also very common, particularly with longer infusions; peripheral neuropathy is common, usually mild, and increases with cumulative dose; acute neuromyopathy is also common and occurs for several days after each dose; this syndrome may require opiate analgesics to control pain; cardiovascular adverse effects, including hypertension, hypotension, premature contractions, and bradyarrhythmias, are common but rarely require intervention; hypersensitivity reactions to paclitaxel, including urticaria, wheezing, chest pain, dyspnea, and hypotension, are common but are reduced in frequency and severity by premedication with corticosteroids and histamine-1 and histamine-2 antihistamines (the recommended regimen is dexamethasone, 20 mg PO 12 and 6 hr prior to paclitaxel and diphenhydramine, 50 mg, and cimetidine | Approved for salvage therapy in ovarian cancer and for breast cancer in both the metastatic and adjuvant settings; used also in persons with lung cancer, head and neck cancer, and bladder cancer | 135-250 mg/m2 IV over 3 hr or 24 hr every 3 wk; weekly schedules and longer infusions have also been used |
| Paclitaxel, Protein bound (Abraxane) | Taxane; an albumin-bound form of paclitaxel with a mean particle size of approximately 130 nanometers | No change in patients with mild to moderate hepatic dysfunction | Neutropenia, thrombocytopenia, anemia, hypersensitivity reactions, hypotension, cardiovascular events, dyspnea, cough, sensory neuropathy, arthralgias/myalgias, nausea/vomiting, asthenia | Metastatic breast cancer after failure of combination chemotherapy or relapse with 6 mo of adjuvant chemotherapy; also approved for NSCLC | 260 mg/m2 IV over 30 min every 3 wk |
| Pamidronate (Aredia), APD, aminohydroxypropylidene diphosphonate | Organic bisphosphonate; inhibitor of bone resorption by osteoclasts | After IV administration, the drug concentrates in the bone, spleen, and liver; its metabolism is not well characterized; it has a terminal half-life of about 27 hr; 50% of the parent drug is eliminated in the urine | Pamidronate is usually quite well tolerated; hypotension, syncope, tachycardia, and even atrial fibrillation have been reported uncommonly during the infusion; hypocalcemia, hypophosphatemia, hypokalemia, and hypomagnesemia occur commonly but only rarely require intervention; nausea, vomiting, and somnolence are rare | Approved for malignancy-induced hypercalcemia; may lead to pain relief and even tumor shrinkage of bone metastases in multiple myeloma, breast cancer, and prostate cancer | 60-90 mg/m2 IV over 24 hr, although the clinical experience with infusions of 1-3 hr is extensive; treatment may be repeated every 1-3 wk; peak effect occurs 3-7 days after a dose |
| Panitumumab (Vectibix) | Recombinant, fully humanized IgG2 kappa monoclonal antibody that binds specifically to EGFR, competitively inhibiting ligand binding to the receptor | Steady-state levels are reached by the third infusion. Elimination half-life is about 7.5 days | Rash, which may be severe; diarrhea; infusion reactions; hypomagnesemia; pulmonary fibrosis | Previously treated EGFR expressing metastatic colorectal cancer | 6 mg/m2 IV every 2 wk |
| Pazopanib (Votrient) | Inhibits large number of tyrosine kinases | Take without food; reduce dose for mild to moderate hepatic impairment; avoid severe hepatic impairment; interacts with many metabolizing enzymes | Diarrhea, hypertension, nausea, anorexia, vomiting, hair color depigmentation | Renal cell carcinoma; also approved for soft tissue sarcoma | 800 mg once daily |
| Pegfilgrastim (Neulasta) | Long-acting (PEGylated) recombinant DNA granulocytic growth factor polypeptide | PEGylation of filgrastim increases its half-life by decreasing renal clearance; after SC administration, half-life is between 15 and 80 hr, with biological activity lasting much longer | Toxicity and adverse effect profile is essentially no different than those of filgrastim; bone pain is common and usually mild and treatable but can be dose limiting; nausea, fatigue, weakness, and diarrhea are rare; very rare instances of adult respiratory distress syndrome, sickle cell crisis, splenic rupture, and severe allergic reaction have been seen with the parent drug filgrastim | Prevention of severe granulocytopenia from cytotoxic chemotherapy for nonmyeloid malignancies | 6 mg SC after each chemotherapy cycle (generally used for a 3- or 4-week cycle duration) |
| Pemetrexed (Alimta) | Antimetabolite; inhibits folate-dependent enzymes thymidylate synthetase, dihydrofolate reductase, and glycinamide ribonucleotide formyltransferase, which are involved in the de novo synthesis of thymidine and purine nucleotides, leading to inhibition of DNA and RNA synthesis and function; metabolized intracellularly to its highly active polyglutamated form | Excreted in urine with 90% of drug unchanged | Myelosuppression; nausea, vomiting, diarrhea; rash; dyspnea and fatigue | Mesothelioma and NSCLC | 500 mg/m2 IV every 3 wk; note: all patients must be given folic acid and vitamin B12 supplementation to decrease toxicity, starting 1 wk prior to therapy |
| Pentostatin (Nipent) | Antimetabolite, purine antagonist; inhibits enzyme adenosine deaminase, found in high concentration in lymphocytes, leading to accumulation of deoxyadenosine and dATP, which are cytotoxic to lymphocytes; elevated dATP levels inhibit ribonucleotide reductase, which inhibits DNA synthesis and function | Undergoes little metabolism, and >90% of the drug is excreted in the urine with an elimination half-life of about 5 hr | Myelosuppression; increased risk of opportunistic infections; hypersensitivity reactions; CNS toxicity; ophthalmologic and otologic complications | Hairy cell leukemia, CLL, and cutaneous T-cell leukemia | 4 mg/m2 IV every 2 wk |
| Plerixafor (Mozobil) | Hematopoietic stem cell mobilizer | Renal impairment: if creatinine clearance is <50 mL/min, decrease dose by one third to 0.16 mg/kg | May mobilize leukemic cells and should not be used in leukemia patients; hematologic effects: increased circulating leukocytes and decreased platelet counts have been observed; monitor blood cell counts and platelet counts during use; potential for tumor cell mobilization: tumor cells may be released from marrow during HSC mobilization; effect of reinfusion of tumor cells is unknown; potential for splenic rupture: evaluate patients who report left upper abdominal and/or scapular or shoulder pain | Combination with G-CSF to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation in patients with non-Hodgkin lymphoma and multiple myeloma | Initiate Mozobil treatment after the patient has received G-CSF once daily for 4 days; repeat Mozobil dose up to 4 consecutive days; select dose based on 0.24 mg/kg actual body weight; administer SC approximately 11 hr prior to initiation of apheresis |
| Pertuzumab (Perjeta) | HER2/neu receptor antagonist antibody; HER2 testing: perform using FDA-approved tests by laboratories with demonstrated proficiency | Of 402 patients who received PERJETA in the randomized trial, 60 patients (15%) were 65 years of age and 5 patients (1%) were 75 years of age; no overall differences in efficacy and safety of PERJETA were observed between these patients and younger patients; based on a population pharmacokinetic analysis, no significant difference was observed in the pharmacokinetics of pertuzumab between patients <65 yr (n = 306) and patients ≥ 65 years (n = 175) | Left ventricular dysfunction: monitor LVEF and withhold dosing as appropriate; reactions, hypersensitivity reactions/anaphylaxis: monitor for signs and symptoms; if a significant infusion- associated reaction occurs, slow or interrupt the infusion and administer appropriate medical therapies; in combination with trastuzumab and docetaxel: diarrhea, alopecia, neutropenia, nausea, fatigue, rash, and peripheral neuropathy | First-line HER2+ metastatic breast cancer | Initial dose is 840 mg administered as a 60-min IV infusion, followed every 3 wk thereafter by 420 mg administered as a 30- to 60-min IV infusion |
| Ponatinib (ICLUSIG) | Kinase inhibitor | For strong CYP3A inhibitors: reduce dose if co-administration cannot be avoided; renal or hepatic impairment not studied | Arterial thrombosis and hepatotoxicity have boxed warnings; nonhematologic adverse reactions (≥ 20%) were hypertension, rash, abdominal pain, fatigue, headache, dry skin, constipation, arthralgia, nausea, and pyrexia; hematologic adverse reactions included thrombocytopenia, anemia, neutropenia, lymphopenia, and leukopenia | CML and Philadelphia chromosome–positive acute ALL | 45 mg taken orally once daily with or without food |
| Pralatrexate (Folotyn) | Folate analog metabolic inhibitor | Co-administration with probenecid or other drugs that may affect relevant transporter systems (e.g., NSAIDs) requires close monitoring for signs of systemic toxicity; not studied in patients with renal impairment | Thrombocytopenia, neutropenia, and anemia: monitor blood counts and omit and/or reduce dose for hematologic toxicities; mucositis: monitor at least weekly; if ≥ grade 2 mucositis is observed, omit and/or reduce dose; dermatologic reactions: reactions, including fatal reactions, have occurred and may be progressive and increase in severity with further treatment; monitor closely, and omit and/or reduce dose or discontinue drug; tumor lysis syndrome: anticipate, monitor, and treat promptly; hepatic toxicity: monitor for toxicity; for liver function test abnormalities grade 3 or greater, omit until recovery then reduce dose or discontinue therapy as required; risk of increased toxicity with renal impairment: patients with moderate to severe renal function impairment may be at greater risk for increased exposure and toxicity; monitor patients for renal function and systemic toxicity and adjust dosing accordingly; avoid use in patients with end stage renal disease including those undergoing dialysis unless the potential benefit justifies the potential risk; most common adverse | Relapsed, refractory peripheral T-cell lymphoma | 30 mg/m2 administered as an IV push over 3-5 min once weekly for 6 wk in 7-wk cycles |
| Prednisone (Deltasone, others) | Corticosteroid | Good oral bioavailability; extensively metabolized in liver, primarily to active form of drug, prednisolone; half-life of 4 hr; liver disease may decrease conversion to the active form, requiring use of prednisolone instead of prednisone; routes of excretion are not well delineated | Toxicity is mostly in the form of constitutional symptoms, including mood changes (depressive, anxious, or euphoric), insomnia, indigestion, enhanced appetite, weight gain, acne, and cushingoid features; other adverse effects may be more serious but are less common; hyperglycemia and increased stomach acid predisposing to ulceration occur acutely, and osteopenia, cataracts, skin atrophy, and adrenal insufficiency occur with prolonged use | Approved for a wide variety of malignant and nonmalignant conditions; used in oncology for lymphoid malignancies, for palliative care, and for management of adverse effects/toxicities | Lympholytic doses are generally in the range of 50-100 mg/m2/day for 5-14 days; higher or lower doses are also used, depending upon the indication |
| Procarbazine (Matulane), N-methylhydrazine | Alkylating agent; cell cycle–independent | Well absorbed by the oral route, reaching peak plasma levels in 1 hr, with good distribution to the cerebrospinal fluid; procarbazine is metabolized by the liver and has an elimination half-life of about 1 hr; largely excreted in the urine | Myelosuppression is expected and dose limiting, but anemia is uncommon; nausea and vomiting are common and can be dose limiting as well; rash, hives, and photosensitivity sometimes occur; other adverse effects are uncommon and include anorexia, diarrhea, stomatitis, hypotension, tachycardia, syncope, flu-like syndrome, interstitial pneumonitis, CNS excitation including seizures, and secondary malignancies | Approved for Hodgkin disease and may also be useful in non-Hodgkin lymphoma, multiple myeloma, brain tumors, melanoma, and lung cancer | In Hodgkin disease regimens such as MOPP, the dose is 100 mg/m2/day for 14 days during each cycle |
| Rasburicase (Elitek) | Recombinant urate oxidase | No drug interaction studies have been conducted in humans; of the total No. of adults treated with Elitek (n = 434) in clinical studies, 30% were 65 yr and older and 8% were 75 yr and older; no overall differences in pharmacokinetics, safety, and effectiveness were observed between the elderly and younger patients | Anaphylaxis, hemolysis, methemoglobinemia are boxed warnings | Management of plasma uric acid levels in pediatric and adult patients with leukemia, lymphoma, and solid tumor malignancies | 0.2 mg/kg as an IV infusion over 30 min daily for up to 5 days; G6PD deficiency is a contraindication |
| Regorafenib (Stivarga) | Inhibits many membrane and intracellular enzymes | Take with food; decreased exposure with strong inducers of CYP3A | Hepatotoxicity, fatigue, decreased appetite, hand-foot skin reaction | Previously treated metastatic colorectal cancer | 160 mg once daily for first 21 days of 28-day cycle |
| Rituximab (Rituxan) | Monoclonal antibody directed against the B-cell surface antigen CD20 | When given IV, it is taken up by B lymphocytes and then degraded throughout the body by proteolysis, with a wide ranging serum half-life of 11-105 hr (mean 60 hr) with the first dose; there is no appreciable excretion of this polypeptide | Fever, chills, and malaise are common during administration, even with premedication with acetaminophen and diphenhydramine; other infusion- related symptoms include nausea, vomiting, flushing, urticaria, angioedema, hypotension, dyspnea, bronchospasm, fatigue, headache, rhinitis, and pain at disease sites; these symptoms are generally self-limited, improve with slowing of the infusion, and resolve after infusion; short-lived myelosuppression, abdominal pain, and myalgia are uncommon; arrhythmias and angina pectoris are rare | Relapsed or refractory low-grade or follicular, CD20-positive, B-cell lymphomas | The recommended dose is 375 mg/m2 by IV infusion (starting at 50 mg/hr and increasing to 400 mg/hr maximum) weekly for 4 wk; higher doses, more doses, and longer courses are being used in other lymphoid malignancies |
| Romidepsin (Istodax) | (HDAC) inhibitor | Carefully monitor PT and INR in patients concurrently taking coumadin derivatives; strong CYP3A4 inhibitors may increase pralatrexate concentrations and should be avoided; potent CYP3A4 inducers may decrease concentrations of pralatrexate and should be avoided | The most common adverse reactions were neutropenia, lymphopenia, thrombocytopenia, infections, nausea, fatigue, vomiting, anorexia, anemia, and ECG T-wave changes | Treatment of peripheral or CTCL in patients who have received at least one prior systemic therapy | 14 mg/m2 administered IV over a 4-hr period on days 1, 8, and 15 of a 28-day cycle; repeat cycles every 28 days provided that the patient continues to benefit from and tolerates the drug |
| Ruxolitinib (Jakafi) | JAK kinase signaling inhibitor | With or without food; reduce dose 50% with strong CYP3A inhibitors, moderate renal or hepatic impairment | Thrombocytopenia, anemia, bruising, dizziness, headache | Intermediate or high-risk myelofibrosis | 20 mg BID oral |
| Sargramostim (Leukine, Leukomax), GM-CSF | Cytokine; exhibits pleiotropic stimulatory effects on bone marrow progenitor cells | Similar bioavailability when given IV or SC; degraded throughout the body, predominantly in the liver and kidneys, with an elimination half-life of 2 hr; no appreciable excretion of this peptide occurs | Constitutional symptoms, which tend to decrease over time, predominate at standard doses; higher doses may cause capillary leak syndrome; adverse effects include flushing, hypotension (or hypertension), dyspnea, fever, nausea, vomiting, fatigue, myalgias, bone pain, headache, and skin rash; thrombocytopenia may also occur; fluid retention and edema rarely occur at standard doses; progression of myelodysplastic syndrome has been documented in patients on GM-CSF | FDA approved for the treatment of myelosuppression after ABMT; may be useful to minimize myelosuppression after standard-dose chemotherapy or to shorten the course of neutropenic fever | 250 µg/m2/day for 21 days or 5 µg/ kg/day for 10-14 days |
| Sipuleucel-T (Provenge) | Autologous vaccine | No studies performed | Chills, fatigue, fever, back pain, nausea, joint ache, and headache | Metastatic castrate resistant (hormone refractory) prostate cancer | 3 IV doses of 50 million cells each, 2 wk apart |
| Sorafenib (Nexavar) | Oral multikinase inhibitor that interacts with multiple intracellular (CRAD, BRAF) and cell surface kinases (KIT, FLT-3, VEGFR-2, VEGFR-2, and PDGFR; may inhibit angiogenesis | Metabolized in the liver; drug and metabolites are excreted primarily in the feces | Skin toxicity with rash and hand-foot syndrome; nausea, vomiting, anorexia, diarrhea; asthenia, pain, arthralgias; hypertension; bleeding events, especially in anticoagulated patients; cardiac ischemia; myelosuppression | Advanced renal cell carcinoma | 400 mg twice daily |
| Streptozocin (Zanosar) | Alkylating agent; cell cycle– independent | Metabolized primarily in the liver and has an elimination half-life of <1 hr; parent drug and metabolites are excreted in the urine | GI adverse effects (nausea, vomiting, and cramping) or nephrotoxicity (glomerular and tubular damage) are common and potentially dose limiting; myelosuppression is less often dose limiting; elevated liver function tests can occur occasionally but are rarely clinically significant; fever, delirium, and depression occur rarely; streptozocin is an irritant if extravasated into perivenous soft tissue | Approved for metastatic islet cell carcinoma and may also be useful for advanced carcinoid tumor, pancreatic carcinoma, and Hodgkin disease | The usual dose is 500-1000 mg/m2/day by IV bolus for 5 days every 4 wk |
| Sunitinib maleate (Sutent) | Inhibits PDGFR-A and PDGFR-B, VEGFR1–3, stem cell factor receptor, FLT-3, CSF-1R, and the neurotrophic factor receptor; this inhibition inhibits tumor growth and metastases | Metabolized in the liver with elimination primarily in the feces; half-life is 40-60 hr for the drug and 80-110 hr for its primary metabolite | Cardiotoxicity—usually reversible; bleeding event, epistaxis most common; hypertension; myelosuppression; nausea, vomiting; rash; liver function test alterations | Advanced renal cell carcinoma; second-line therapy for GIST cell tumors; also approved for pancreatic neuroendocrine tumors | 50 mg daily for 4 wk, with a 2-wk rest |
| Tamoxifen (Nolvadex) | Nonsteroidal antiestrogen; cytostatic effects on estrogen- dependent and nondependent malignant cells | Tamoxifen has good oral bioavailability, is metabolized in the liver, and has an elimination half-life of about 7 days; neither tamoxifen nor its major metabolite is found in the bile or urine | Tamoxifen is usually very well tolerated; constitutional symptoms are most prevalent and usually dose limiting; hot flashes, sweating, mood changes, weight gain or loss, and stomach upset are most common; nausea, vomiting, diarrhea, and constipation are less common; menstrual changes, including significant vaginal bleeding, are uncommon; venous thromboembolism, myelosuppression, and retinopathy are rare | Approved for the treatment of breast cancer, generally in postmenopausal patients or those with ER+ tumors; the same dose has been approved for chemoprevention of breast cancer in high risk individuals; higher doses are used for melanoma and pancreatic cancer | The standard dose for breast cancer is 10 mg PO BID (or 20 mg once a day) |
| Temozolomide (Temodar) | Atypical alkylator (semiselective DNA methylator) drug sharing the same active metabolite as dacarbazine, MTIC but, unlike dacarbazine, is spontaneously converted to MTIC and also penetrates the blood-brain barrier effectively | Temozolomide had good bioavailability, enhanced by an empty stomach; after absorption into the bloodstream, it is spontaneously converted to the active moiety MTIC; peak plasma concentrations occur in about 1 hr; the elimination half-life is about 1.8 hr; parent drug, MTIC, and other metabolites are eliminated in the urine | Myelosuppression is expected and dose-limiting; it may be cumulative; nausea is common but generally mild and treatable; headache and fatigue are common; rash or other cutaneous reactions are uncommon; infections are common in this population, and some are probably caused by immunosuppression that is known to occur with temozolomide | Approved for treatment of recurrent high-grade astrocytomas; used commonly for other gliomas and also for metastatic melanoma | 200 mg/m2 PO on an empty stomach daily for 5 days on a 28-day cycle; other doses and schedules have been used with similar clinical results |
| Temsirolimus (Torisel) | Inhibitor of mTOR | Antihistamine pretreatment is recommended; dose reduction is required in patients with mild hepatic impairment; strong inducers of CYP3A4/5 and inhibitors of CYP3A4 may affect concentrations of the primary metabolite, sirolimus; if alternatives cannot be used, dose modifications are recommended | Hypersensitivity/infusion reactions (including some life- threatening and rare fatal reactions) can occur early in the first infusion; patients should be monitored throughout the infusion; monitor for symptoms or radiographic changes of ILD; if ILD is suspected, discontinue drug, and consider use of corticosteroids and/or antibiotics; bowel perforation may occur; evaluate fever, abdominal pain, bloody stools, and/or acute abdomen promptly; elderly patients may be more likely to experience certain adverse reactions, including diarrhea, edema and pneumonia | Advanced renal cell carcinoma | The recommended dose of TORISEL is 25 mg infused over a 30- to 60-min period once a week; treat until disease progression or unacceptable toxicity; contraindicated in patients with bilirubin >1.5 × ULN |
| Teniposide (Vumon), VM-26, PTG | Inhibitor of topoisomerase II; similar in action to etoposide | Teniposide is only available by the IV route; it is extensively protein bound in the plasma and undergoes near-complete metabolism in the liver; it has an elimination half-life of 5 hr; metabolites are excreted in the bile and urine | Myelosuppression, predominantly leukopenia, is universal and dose limiting; otherwise usually well tolerated; nausea, vomiting, diarrhea, stomatitis, and anorexia are uncommon; alopecia is generally mild; elevated liver function tests can occur but are not usually clinically significant; allergic reactions, hypotension, fatigue, seizures, somnolence, fever, renal insufficiency, and secondary leukemia are all rare | Approved for childhood ALL; not used commonly for other malignancies, but does have activity against SCLC | 100 mg/m2 once or twice weekly or 20-60 mg/m2/day for 5 days as a slow IV infusion (at least 30 min) |
| Thalidomide (Thalomid) | Novel antiangiogenic and immunomodulating agent | Although it has acceptable bioavailability, thalidomide is slowly and incompletely absorbed from the GI tract; metabolism appears to be via spontaneous hydrolysis, with an elimination half-life of about 6 hr; exact quantification of elimination routes are unknown | Historical teratogenicity has led to required strict evaluation and monitoring for those taking thalidomide, called the S.T.E.P.S. Program; strict procedures to prevent conception include barrier contraception for men, because the drug is present in semen of men who are taking it; fatigue and peripheral neuropathy are the main toxicities and are dose limiting; myelosuppression is uncommon and usually mild; rash and headache uncommon | Approved for cutaneous leprosy and multiple myeloma; used in oncology for renal cell carcinoma, metastatic melanoma, and malignant gliomas | Oncology doses range from 50 mg/day up to 1200 mg/day |
| Thioguanine (Tabloid), 6-TG, aminopurine-6-thiol hemihydrate | Purine analog antimetabolite; cell cycle–dependent | Thioguanine has modest but slow oral route absorption; it is almost completely metabolized in the liver and has an elimination half-life of up to 11 hr; metabolites are excreted in the urine | Thioguanine is usually well tolerated; leukopenia and thrombocytopenia are common and dose limiting; nausea and vomiting, stomatitis, diarrhea, rash, elevated liver function tests, hyperuricemia, and renal insufficiency are uncommon | Approved for AML in all phases of treatment; may be useful in other leukemias | The usual dose for leukemias is 2-3 mg/kg/day as part of an ongoing multidrug regimen |
| Thiotepa (TESPA), triethylenethiophosphoramide, TSPA | Classical alkylating agent; cell cycle–independent | Extensive metabolism occurs in the liver, and the drug has an elimination half-life of 2-3 hr; metabolites are excreted in the urine | Myelosuppression, predominantly leukopenia, is expected and dose limiting and may be cumulative; nausea, vomiting, anorexia, stomatitis, and diarrhea are uncommon; infertility, fever, and angioedema or urticaria are uncommon; second malignancies such as acute leukemia are rare; with high-dose therapy and bone marrow rescue, stomatitis and cognitive impairment can be severe; intravesical administration leads to predominant urinary symptoms, including pain, hematuria, hemorrhagic cystitis, and rare ureteral obstruction | Approved for the treatment of breast and ovarian carcinoma, as well as Hodgkin disease and non-Hodgkin lymphoma; used for intravesical therapy of superficial bladder cancer and may also be used for intracavitary and intrathecal administration; used in the transplant setting for ovarian and breast carcinoma | The usual dose is 12-16 mg/m2 IV over 10 min every 1-4 wk; in the transplant setting, doses up to 900 mg/m2 have been used; the bladder instillation dose is 30-60 mg once weekly for 4 wk; the intrathecal dose is 1-10 mg/m2 1-2×/wk |
| Topotecan (Hycamtin), hycamptamine | Semisynthetic camptothecin molecule; an inhibitor of topoisomerase I, which is required by cells for both transcription and replication | After IV administration, the drug is not extensively metabolized, and it has an elimination half-life of about 3 hr; a significant portion of the drug is excreted unchanged in the urine | Myelosuppression, especially leukopenia, is expected and dose limiting; thrombocytopenia and anemia are common but mild; nausea, vomiting, and diarrhea are common but usually not severe; headache, fever, fatigue, anorexia, malaise, and elevated liver function tests are also common; hypertension, tachycardia, urticaria, renal insufficiency, hematuria, neuropathy, and mucositis are uncommon | Approved for the treatment of refractory, relapsed ovarian carcinoma and for relapsed small cell lung cancer; also used in myeloid leukemias | The standard dose for ovarian cancer is 1.5 mg/m2/day for 5 days as a 30-min infusion |
| Toremifene (Fareston) | Nonsteroidal antiestrogen; cytostatic effects on estrogen-dependent and nondependent malignant cells | Good oral bioavailability and extensively bound to plasma proteins; metabolized in liver to active metabolites; half-life of 5 days; parent drug and metabolites are excreted in bile | Toremifene is usually very well tolerated; hot flashes, nausea, sweating, dizziness, and fatigue are the most common adverse effects; vomiting, diarrhea, anorexia, vaginal discharge, vaginal bleeding, and headache are less common; venous thrombosis and pulmonary embolism are rare | Postmenopausal or ER+ metastatic breast cancer | 60 mg PO every day |
| Trastuzumab (Herceptin) | Genetically engineered humanized mouse monoclonal antibody directed against HER2/neu growth factor receptor that is overexpressed in many invasive breast carcinomas; mechanism of action for clinical activity in breast cancer is unknown but may be complement mediated cell lysis, antibody- dependent cellular cytotoxicity, or induction of apoptosis | Binding studies show strong binding to cells overexpressing HER2/neu molecules; very little else is known regarding the distribution and metabolic fates of this molecule; half-life should be very short with minimal distribution outside the vascular compartment and minimal clearance by kidneys or liver (similar to other monoclonal agents) | Common toxicities include acute fever, chills, nausea, vomiting, and headache; trastuzumab seems to worsen leukopenia, anemia, and diarrhea when given with chemotherapy compared with chemotherapy alone; also, trastuzumab may have uncommon acute cardiotoxicity, which may add to the more common anthracycline- induced cardiotoxicity; therefore, the use of trastuzumab with doxorubicin is not indicated by the FDA | Approved for HER2/neu overexpressing metastatic or locally advanced breast cancer; has shown clinical benefit as a single agent and in conjunction with paclitaxel based chemotherapy; also approved for gastric cancer | Loading dose of 250 mg or 4 mg/kg by IV infusion followed by weekly IV infusions of 100 mg or 2 mg/kg for up to 10 wk (or longer) |
| Tretinoin (Vesanoid), ATRA, all-trans- retinoic acid | A naturally occurring retinoid; induces differentiation and apoptosis of malignant promyelocytes in acute promyelocytic leukemia | This drug has good oral bioavailability and a very short elimination half-life of about 40 min; it induces its own metabolism in the liver, leading to decreased levels and clinical effect with continued administration; no appreciable excretion of the parent compound is evident | Tretinoin is teratogenic, so women of childbearing age who take this drug must take optimal contraceptive measures; leukostasis and hemorrhage due to leukocytosis are dose limiting but uncommonly life threatening if the drug is stopped; “retinoic acid syndrome,” although not common, can be dose limiting and consists of fever, chest pain, dyspnea, hypoxia, pulmonary infiltrates, and pleural/pericardial effusions; it can be lethal but improves with cessation of the drug and is treatable with corticosteroids; dry skin, exfoliation, xerostomia, and cheilitis are common; elevations in liver function tests and hyperlipidemias are also common; headache is often seen, but pseudotumor cerebri or other neurologic occurrences are uncommon | Approved induction therapy for acute promyelocytic leukemia; also of benefit in the maintenance phase of this disorder and may have clinical activity in other hematologic malignancies | For induction, the dose is 45 mg/m2/day PO for 30-90 days, depending on the clinical response |
| Vandetanib (Caprelsa) | kinase inhibitor | The concomitant use of known strong CYP 3A4 inducers may reduce drug levels of CAPRELSA and should be avoided (7.1). No clinically significant drug interaction was shown with CAPRELSA and the potent CYP 3A4 inhibitor, itraconazole (7.2). The administration of CAPRELSA with agents that may prolong the QT interval should be avoided. In total, 18% of medullary thyroid cancer patients treated with CAPRELSA were age 65 years or older, and 3% were 75 years and older. No overall differences in safety and efficacy were observed between elderly and younger patients. No adjustment in starting dose is required for patients over 65 years of age. There are limited data for patients over the age of 75 years. Limited data for renal or hepatic impairment point towards dose reductions | QT prolongation: sudden death has been reported. Monitor electrocardiograms and levels of serum potassium, calcium, magnesium, and TSH at baseline, 2-4 weeks and 8-12 weeks after starting treatment, and every 3 months thereafter and following dose adjustments. Dose reduce as appropriate. Stevens-Johnson syndrome resulting in death has been observed. Severe skin reactions may prompt permanent discontinuation of drug. Interstitial lung disease resulting in death has been reported. Interrupt dosing and investigate unexplained dyspnea, cough, and fever. Appropriate measures should be taken for ILD. Ischemic cerebrovascular events, hemorrhage, heart failure, diarrhea, hypothyroidism, hypertension, and reversible posterior leukoencephalopathy syndrome, have been observed. Other common adverse events: diarrhea, rash, acne, nausea, hypertension, headache, fatigue, upper respiratory tract infections, decreased appetite, and abdominal pain. The most common laboratory abnormalities were decreased calcium, increased ALT, and decreased glucose. | Thyroid cancer unresectable locally advanced or metastatic | 300 mg once daily |
| Vemurafenib (Zelboraf) | Inhibits V600E serine-threonine and other kinases | Take with or without food; inhibits CYP1A2, 2D6; enhances CYP3A | Produces cutaneous squamous cell carcinoma, hypersensitive and dermatologic reagins, QP prolonged, liver lab abnormalities | Metastatic BRAF+ melanoma | 960 mg BID oral |
| Vinblastine (Velban, Velsar, others), VLB, vincaleukoblastine | Vinca alkaloid; inhibitor of tubulin polymerization and thereby mitosis; G2- phase specific | After an IV dose, the drug undergoes deacetylation in the liver to an active metabolite, followed by further metabolism; the elimination half-life is about 20 hr; excretion is predominantly via the bile | Vinblastine is a soft tissue vesicant, requiring extravasation precautions during administration; myelosuppression, especially leukopenia, is expected and dose limiting; anemia and thrombocytopenia are less common; peripheral and autonomic neuropathy are less common than that observed with vincristine; nausea and vomiting are uncommon, but constipation is more often seen; acute reactions during administration, including dyspnea, wheezing, chest pain, tumor pain, and fever, are uncommon; syndrome of inappropriate antidiuretic hormone secretion occurs rarely, as does angina pectoris | Approved for multiple hematologic and solid neoplasms; most often used for Hodgkin disease, non-Hodgkin lymphoma, germ cell tumors, and breast cancer | Typical doses are between 6 and 10 mg/m2 by IV push every 2-4 wk, combined with other drugs; can also be given as a continuous infusion over 96 hr at a dose of 1.7-2.0 mg/m2/day |
| Vincristine (Oncovin, Vincasar), leurocristine, VCR | Vinca alkaloid; inhibitor of tubulin polymerization and thereby mitosis; G2- phase specific | Metabolized by the liver; the elimination half-life is variable but usually greater than 10 hr; parent drug and metabolites are primarily excreted in the bile | Vincristine is a vesicant and should be administered with extravasation precautions; neurotoxicity is dose limiting in the form of peripheral neuropathy, which is related to total cumulative dose; autonomic neuropathy is less common, and CNS toxicity is rare; myelosuppression is mild; nausea and vomiting are rare, but constipation is fairly common; acute cardiopulmonary or pain symptoms occurring during administration are uncommon; transient elevation of liver function tests is sometimes seen | Approved for Hodgkin disease and other lymphomas, acute leukemias, rhabdomyosarcoma, neuroblastoma, and Wilms tumor; used for many other neoplasms as well | The usual dose is 0.5-1.4 mg/m2 IV push every 1-4 wk; a continuous infusion of 0.5 mg/m2/day over 96 hr has also been used |
| Vinorelbine (Navelbine), 5′-noranhydrovinblastine, NVB | Semisynthetic vinca alkaloid; inhibitor of tubulin polymerization and thereby mitosis; G2- phase specific | Metabolized by the liver and has an elimination half-life of about 24 hr; excretion is predominantly in the bile | Vinorelbine is a mild vesicant, requiring extravasation precautions; myelosuppression, mostly leukopenia, is expected and dose limiting; significant nausea and vomiting are uncommon; neurotoxicity in the form of neuropathy is less common and milder than that seen with vincristine; tumor pain during administration has been reported; acute reaction such as dyspnea, chest pain, and wheezing have occurred during administration and may be prevented by premedication with corticosteroids | Approved for the treatment of relapsed metastatic breast cancer and for NSCLC as a single agent or combined with a platinating agent | The recommended dose is 30 mg/m2 IV over 20 min every week, with dose adjustments based on leukocyte counts |
| Vismodegib (Erivedge) | Hedgehog pathway inhibitor | Safety and effectiveness have not been established for hepatic or renal impairment; absorption is saturable as evidenced by the lack of dose proportional increase in exposure after a single dose of 270 mg or 540 mg vismodegib; may be taken without regard to meals because the systemic exposure of vismodegib at steady state is not affected by food | The most common adverse reactions (incidence of ≥10%) are muscle spasms, alopecia, dysgeusia, weight loss, fatigue, nausea, diarrhea, decreased appetite, constipation, arthralgias, vomiting, and ageusia. Advise patients not to donate blood or blood products while receiving vismodegib and for at least 7 months after the last dose | Metastatic basal cell carcinoma, or locally advanced basal cell carcinoma that has recurred following surgery or patients who are not candidates for surgery, and who are not candidates for radiation | 150 mg orally once daily |
| Zoledronic Acid (Zometa) | Bisphosphonate inhibitor of bone metastases | Zoledronic acid is poorly absorbed by the GI tract and is therefore given as an IV infusion; it is not metabolized and is excreted by the kidneys; it has a plasma terminal elimination half-life of about 150 hr | Zoledronic acid is generally well tolerated; the most common infusional adverse effect is fever, which is usually mild and treatable; nausea and constipation are also common; dyspnea, fatigue, diffuse pain, rash, and headache are uncommon; renal insufficiency is uncommon and generally reversible after discontinuation of the drug, but it is more likely with higher doses than with the approved and recommended 4-mg dose | Approved for treatment of hypercalcemia of malignancy and for prevention of pathologic fractures in multiple myeloma and solid tumors with known bone metastases | 4 mg IV injection over 15 min once monthly |
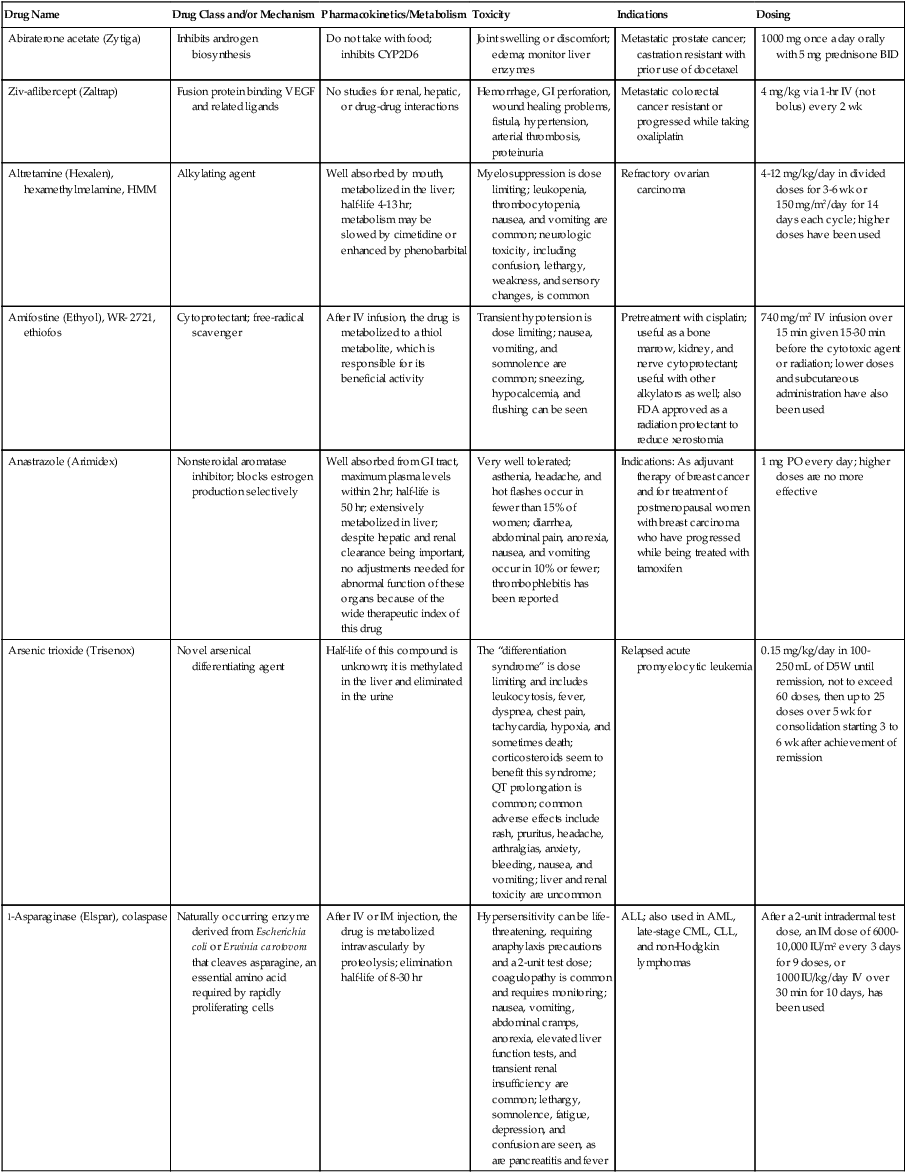
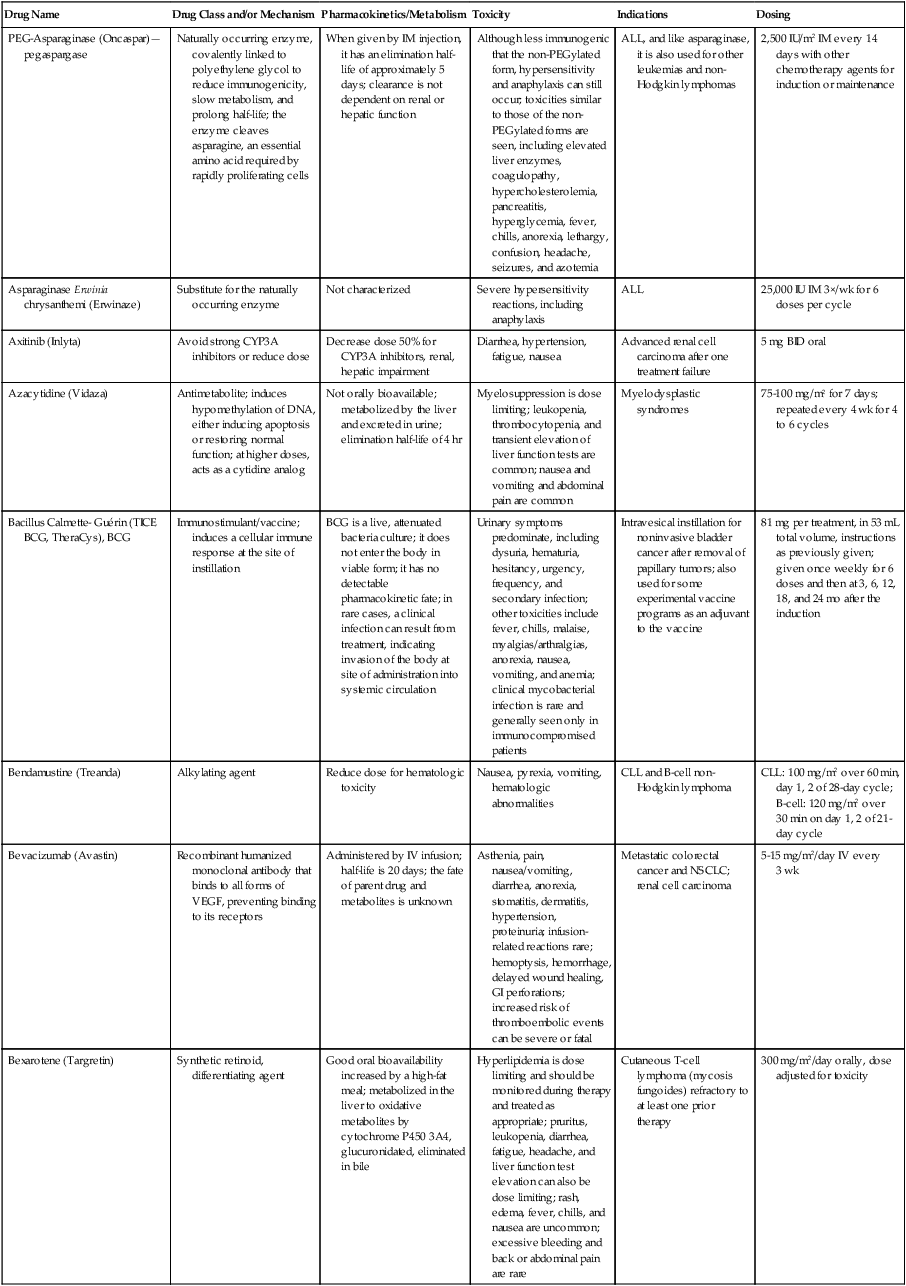
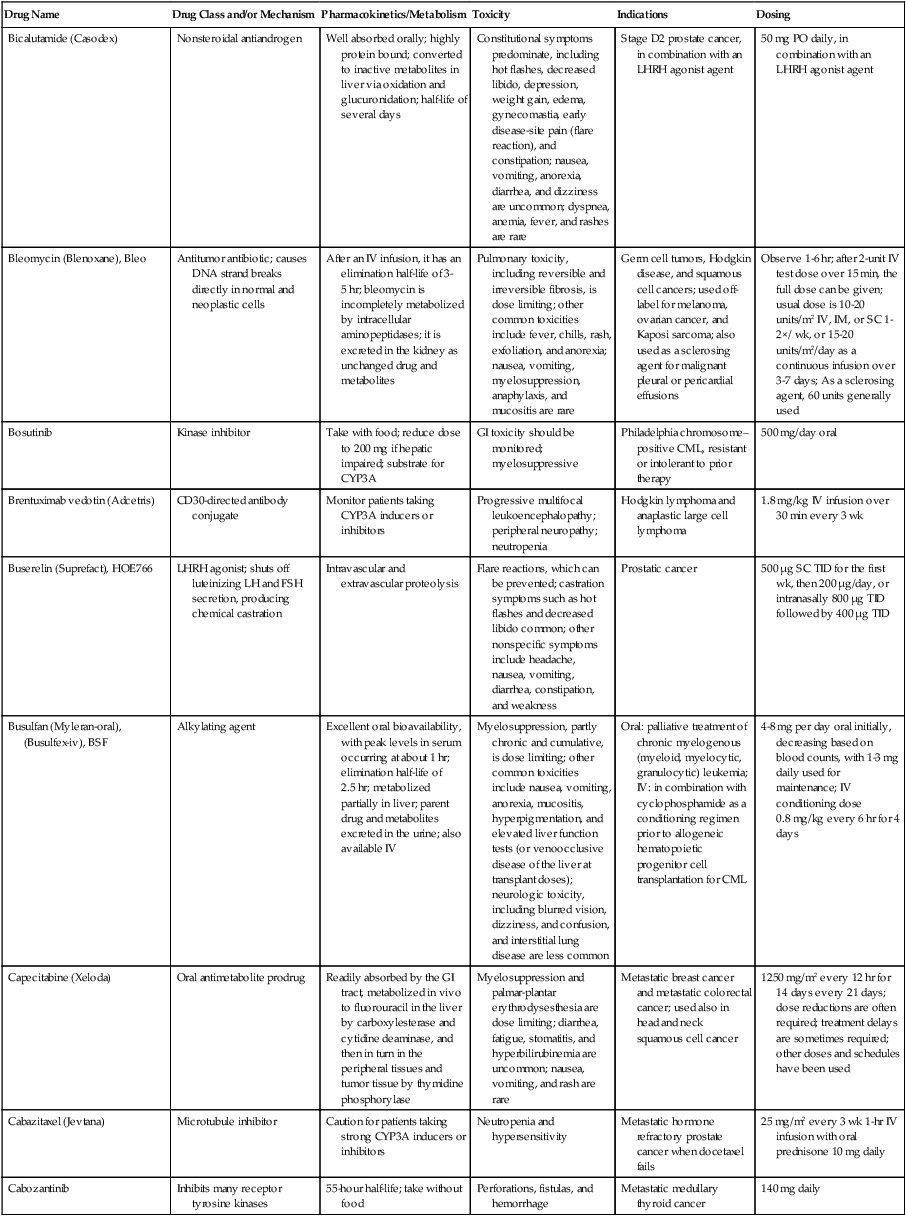
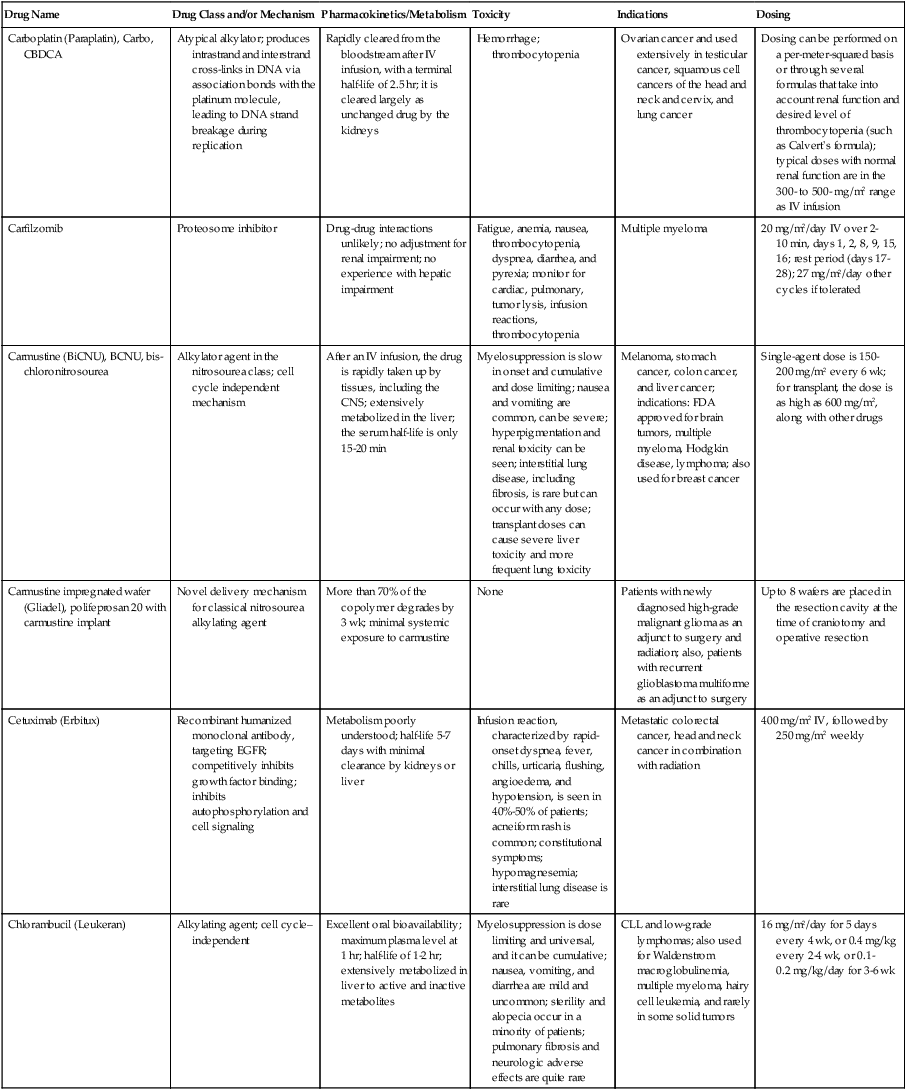
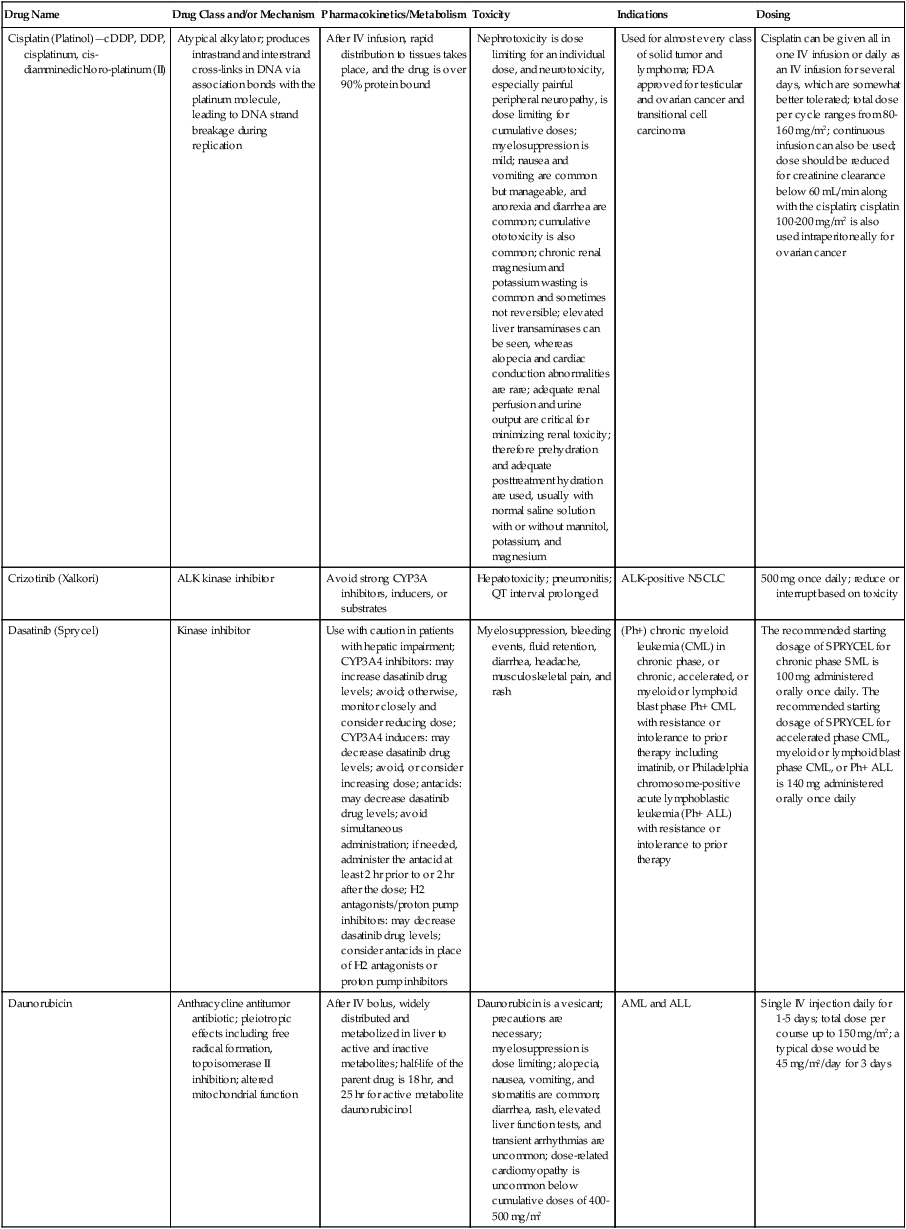
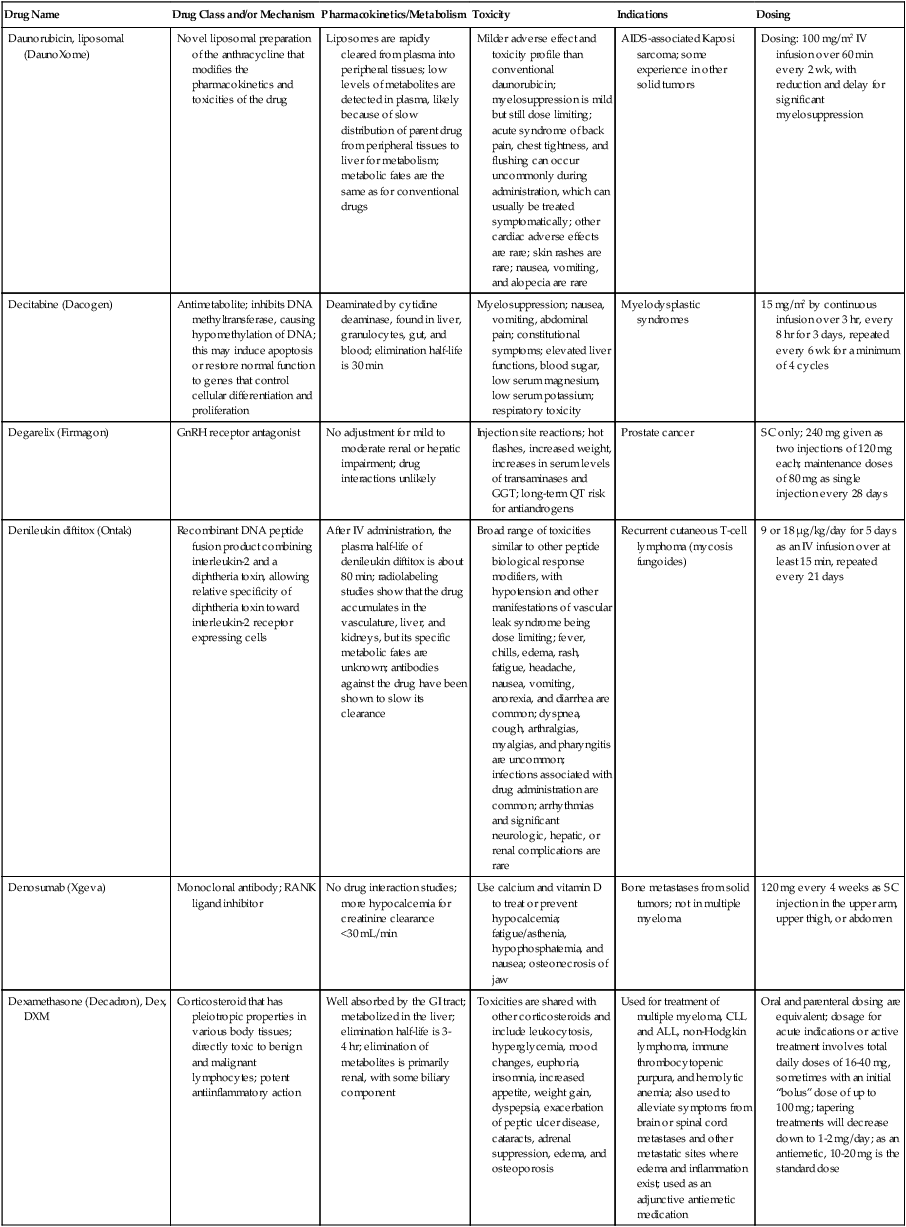
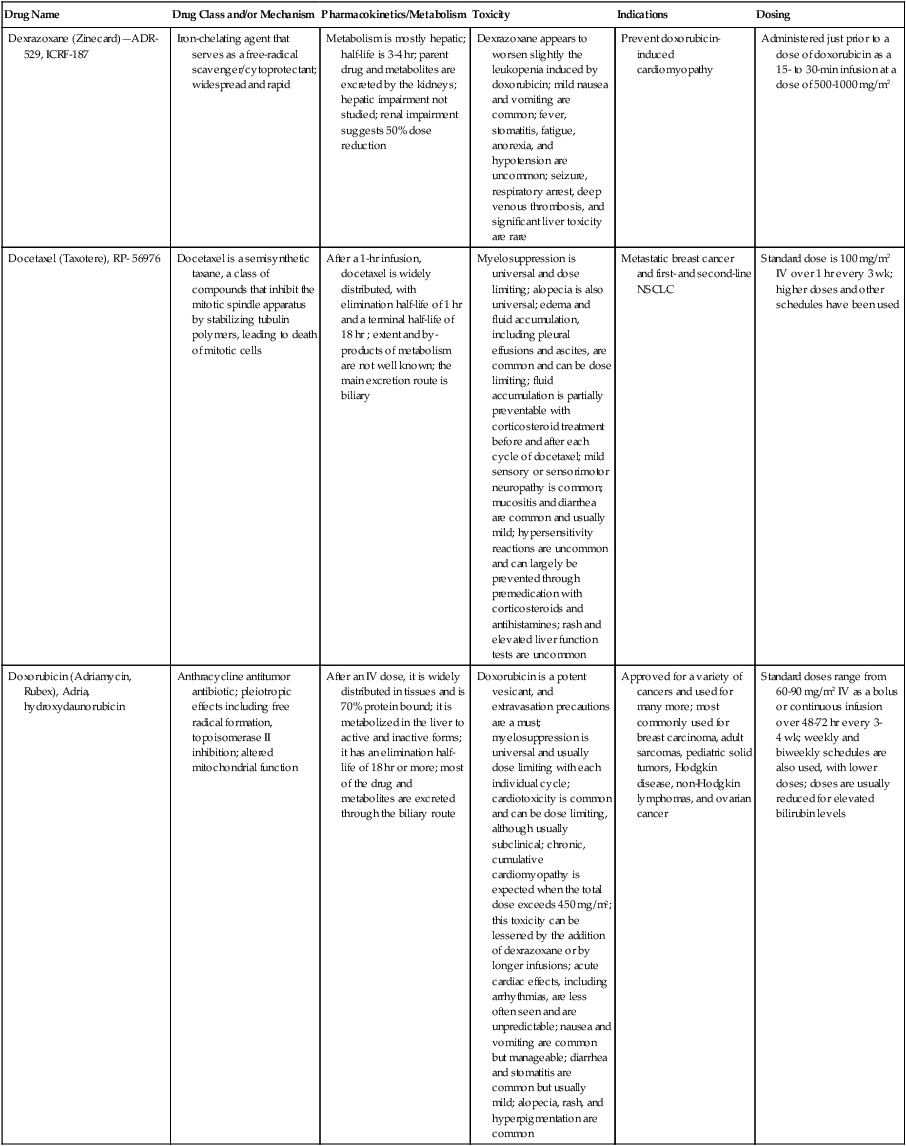
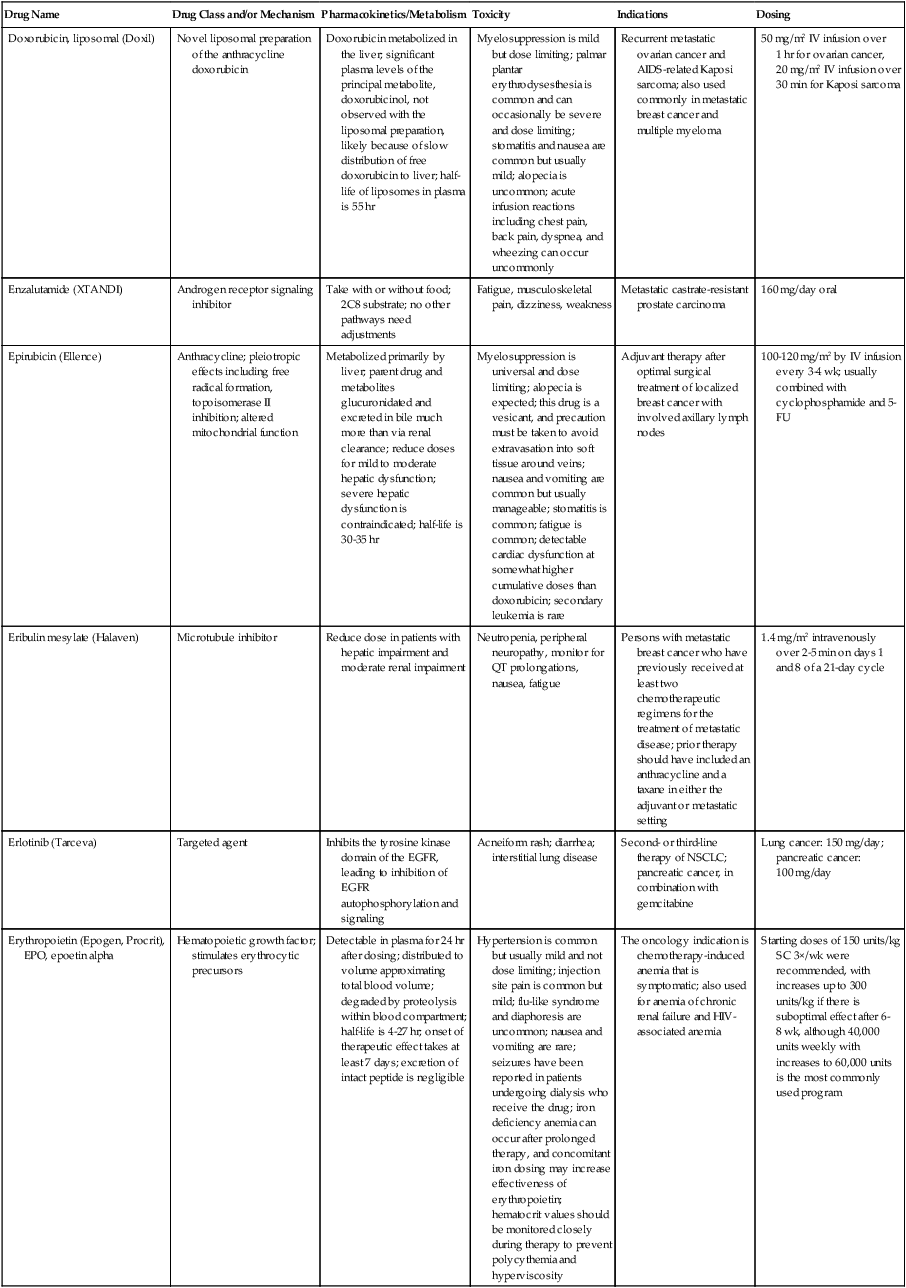
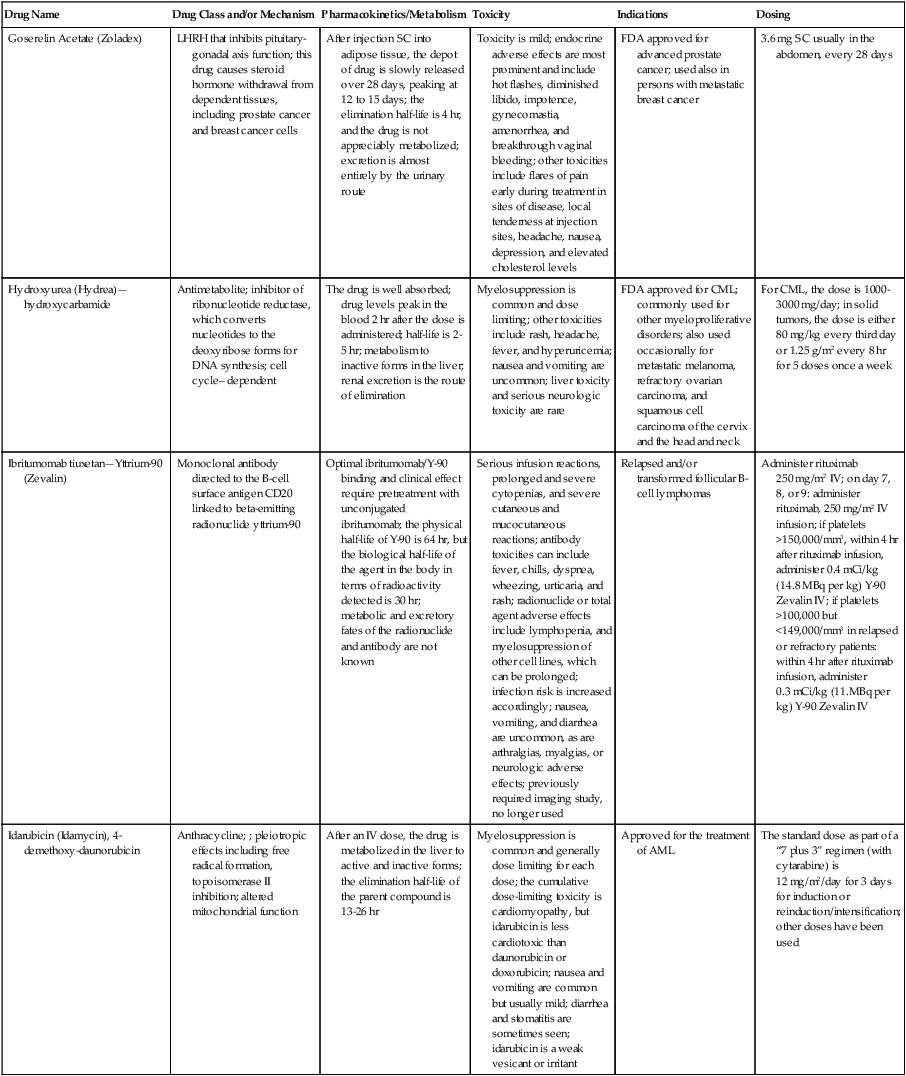
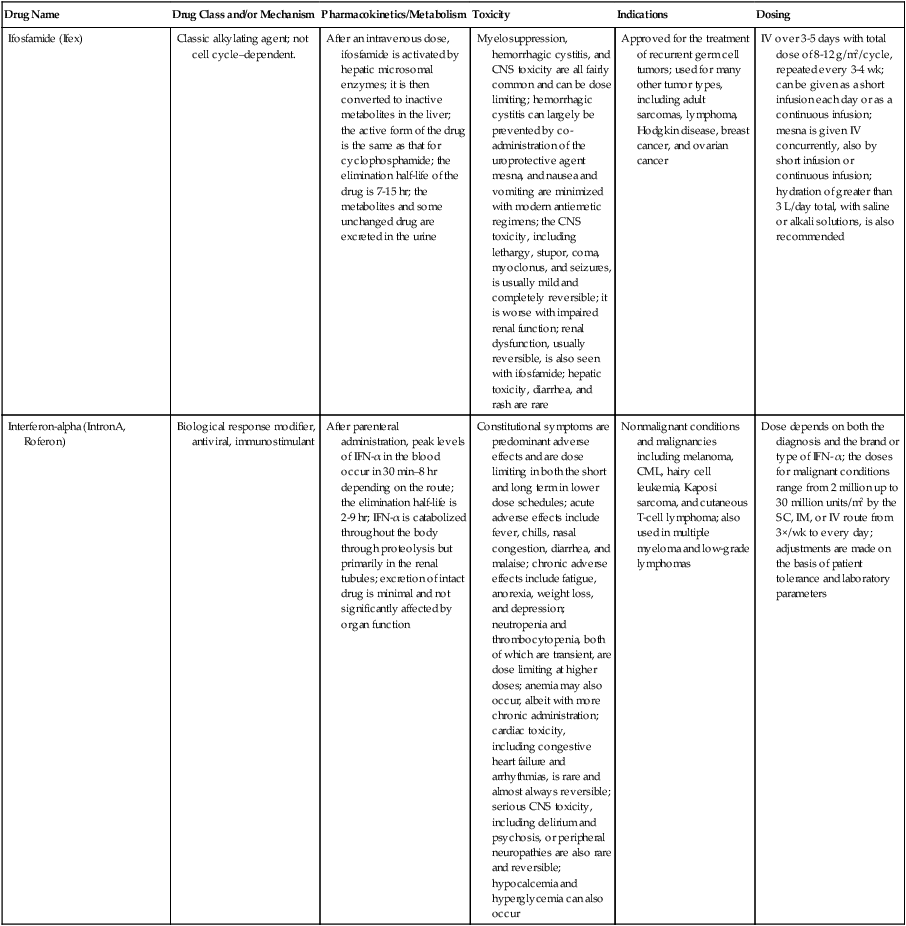
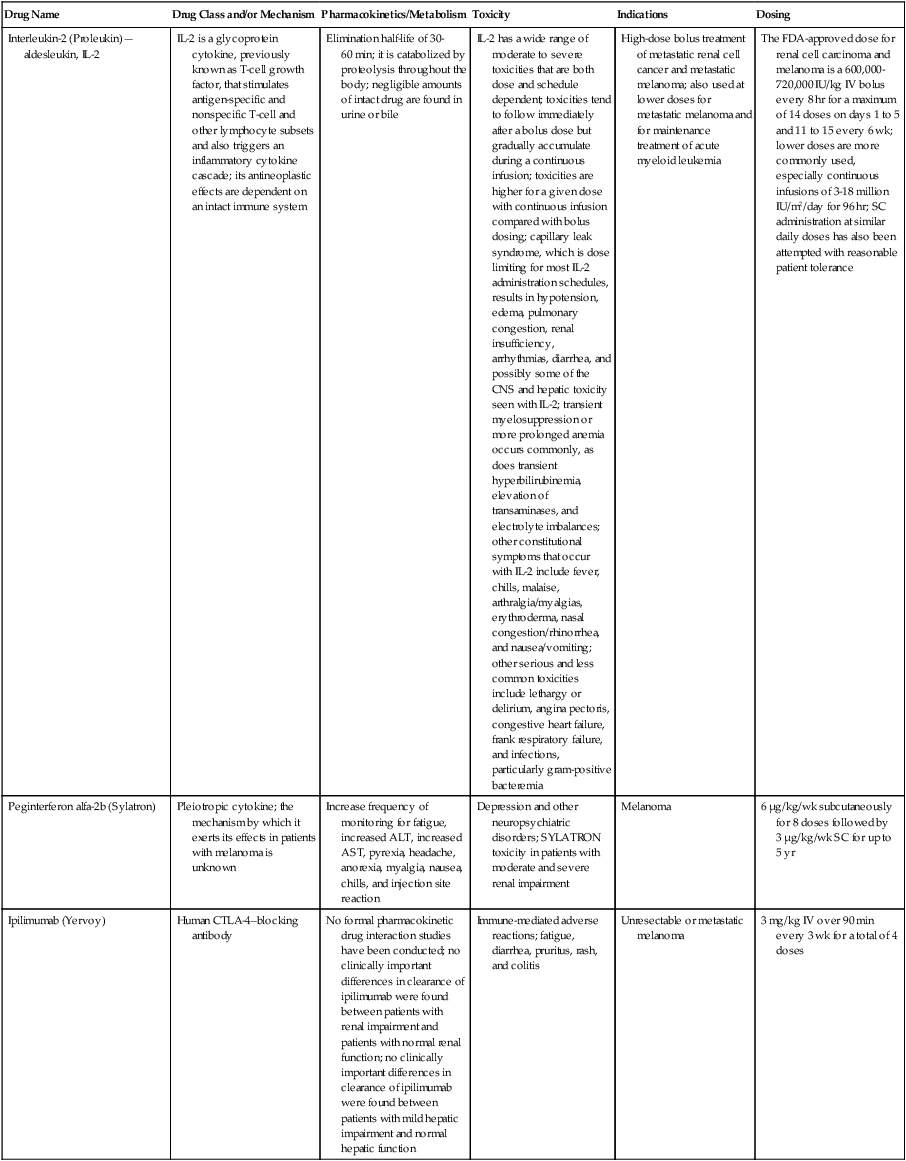
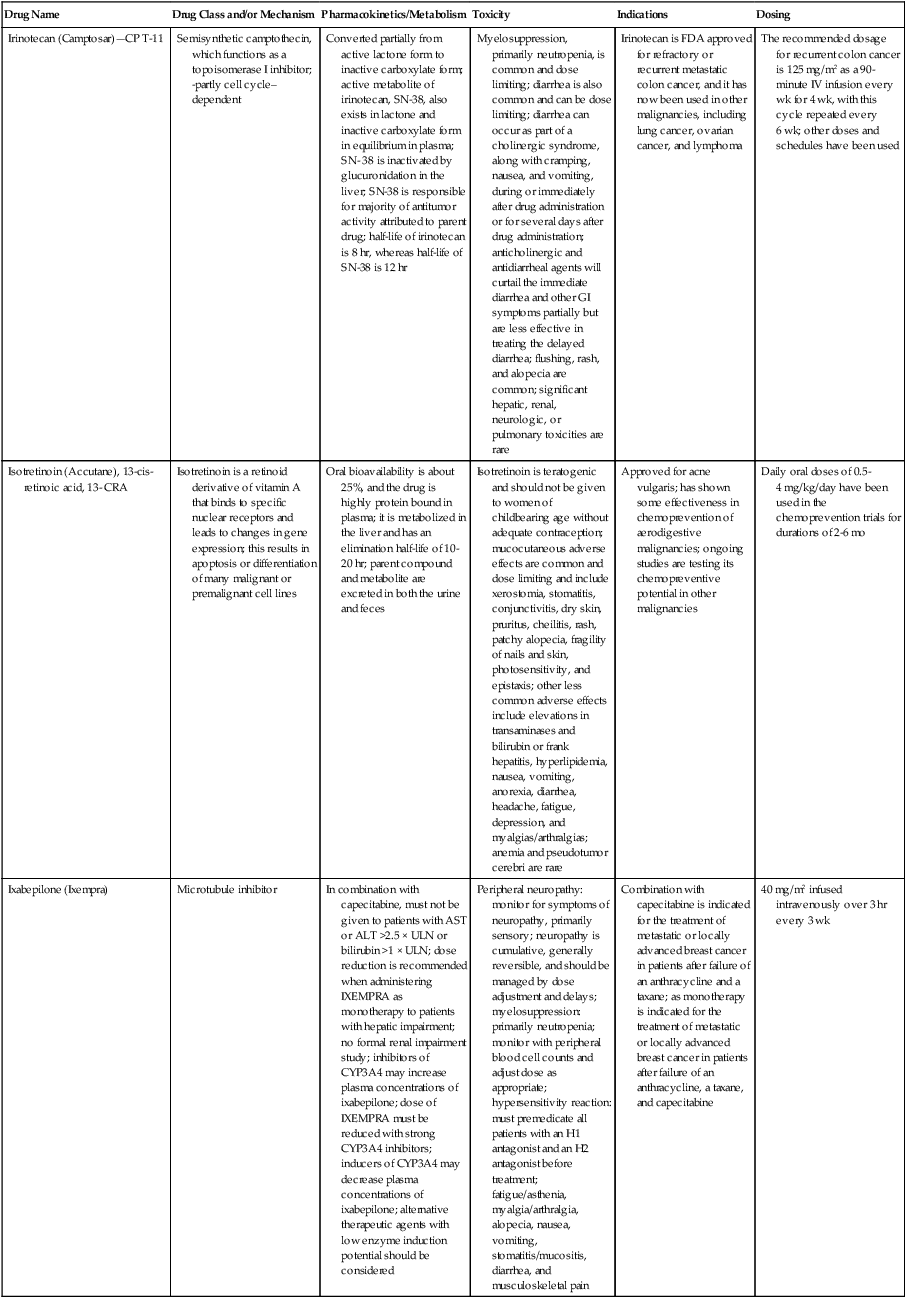
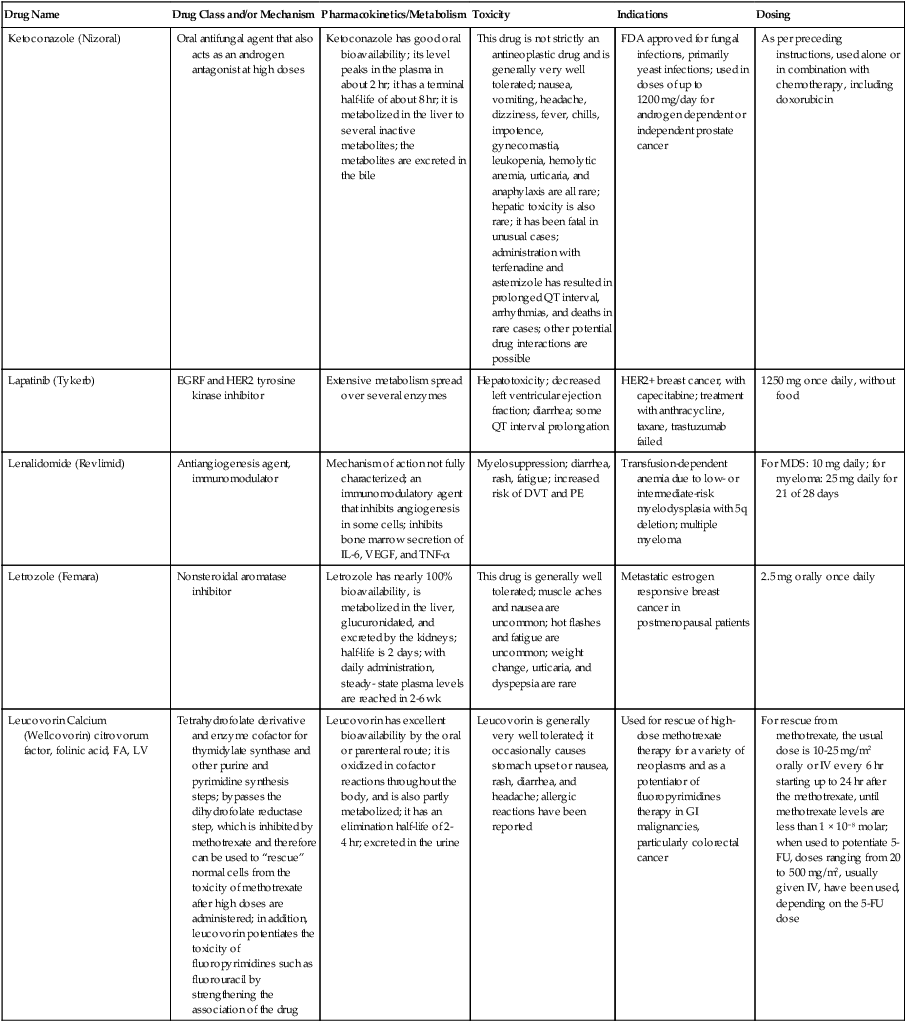
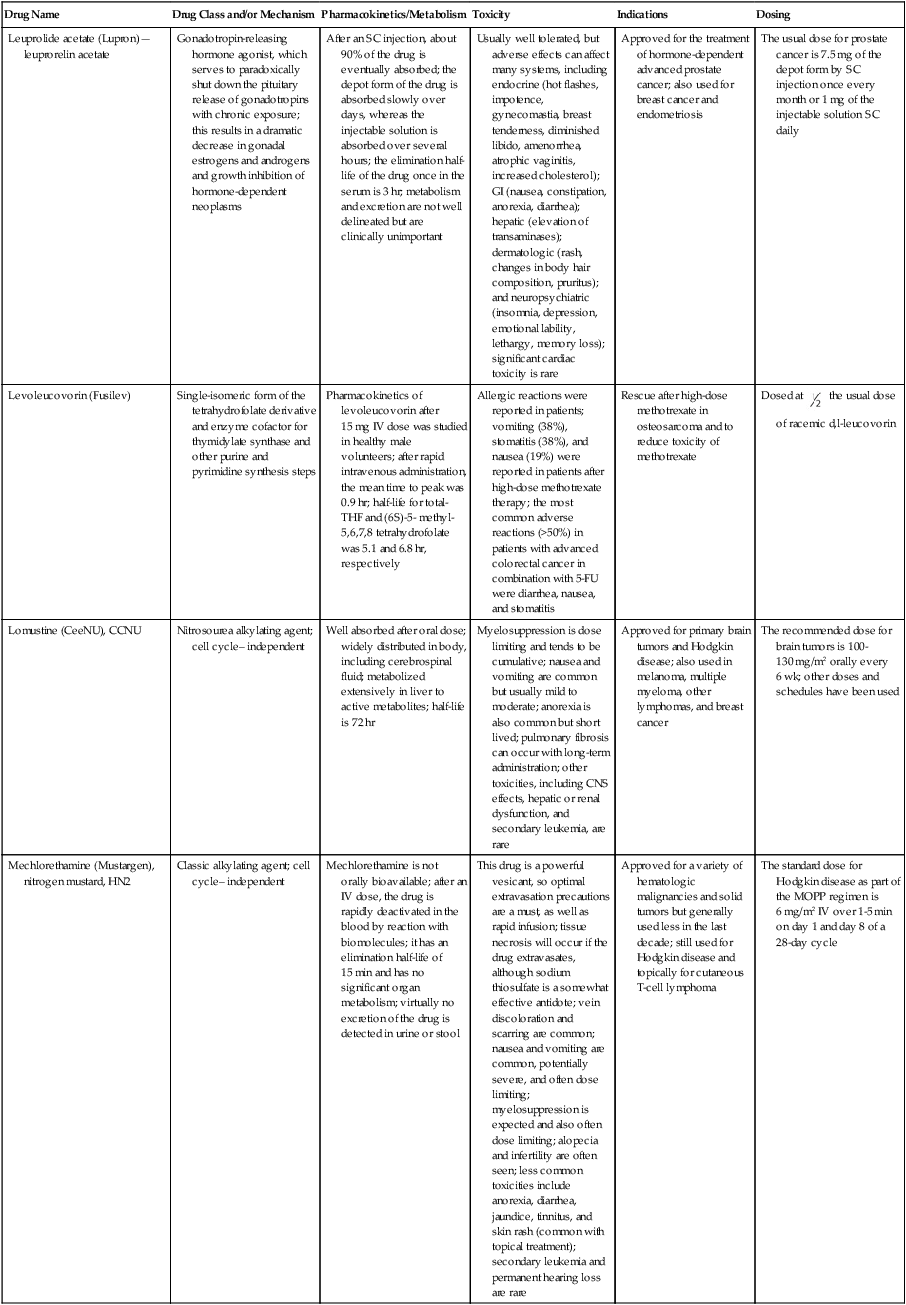
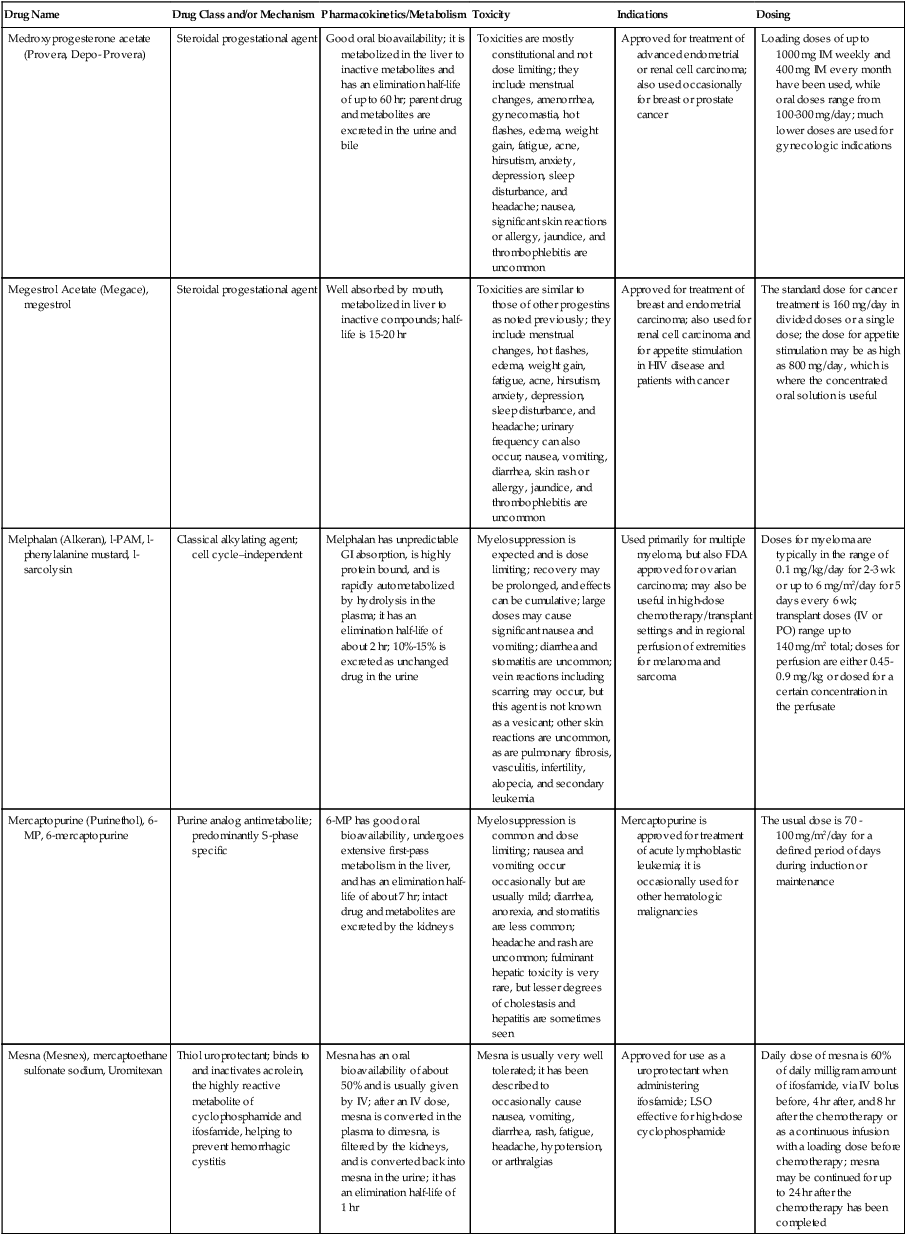
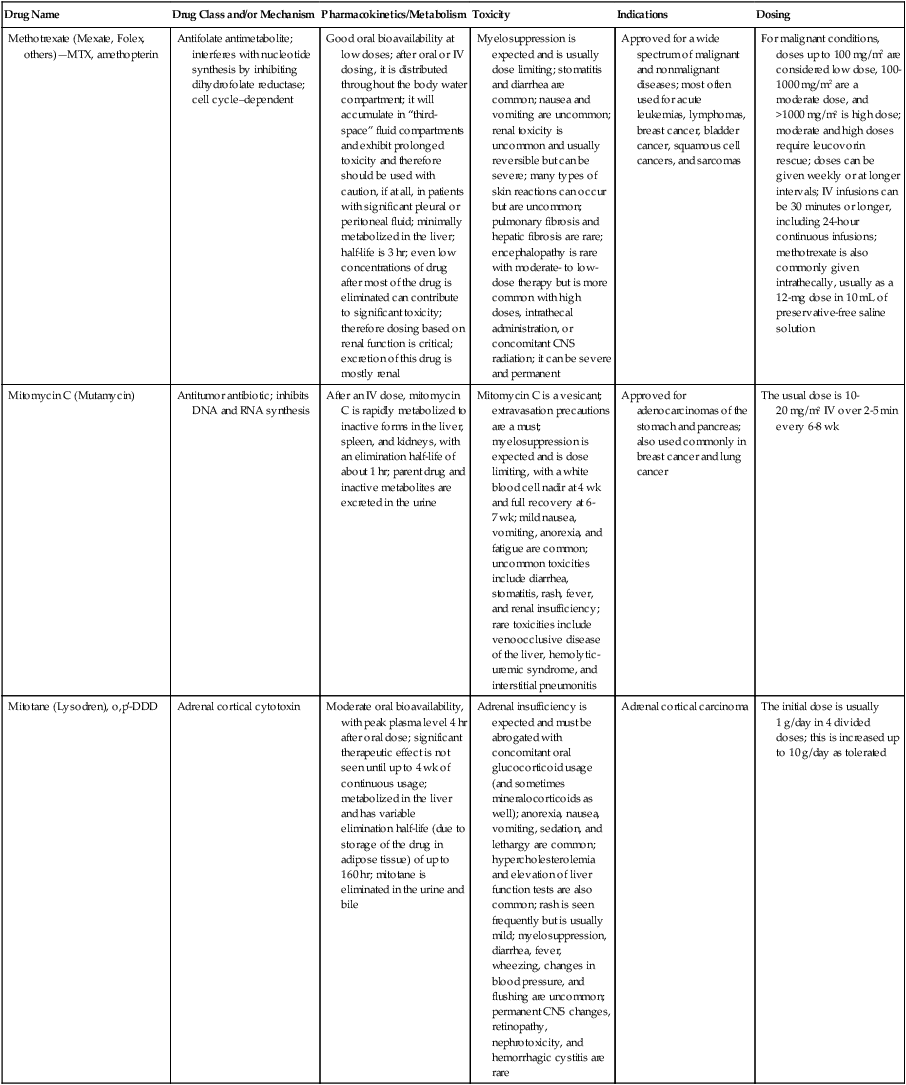
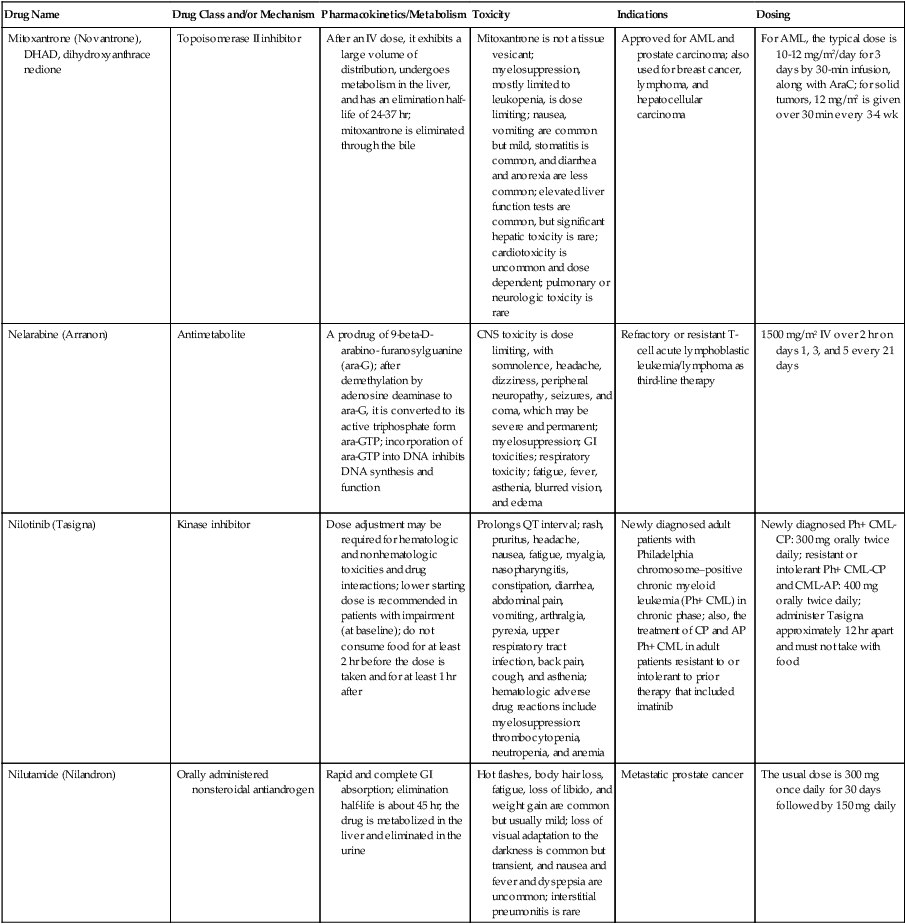
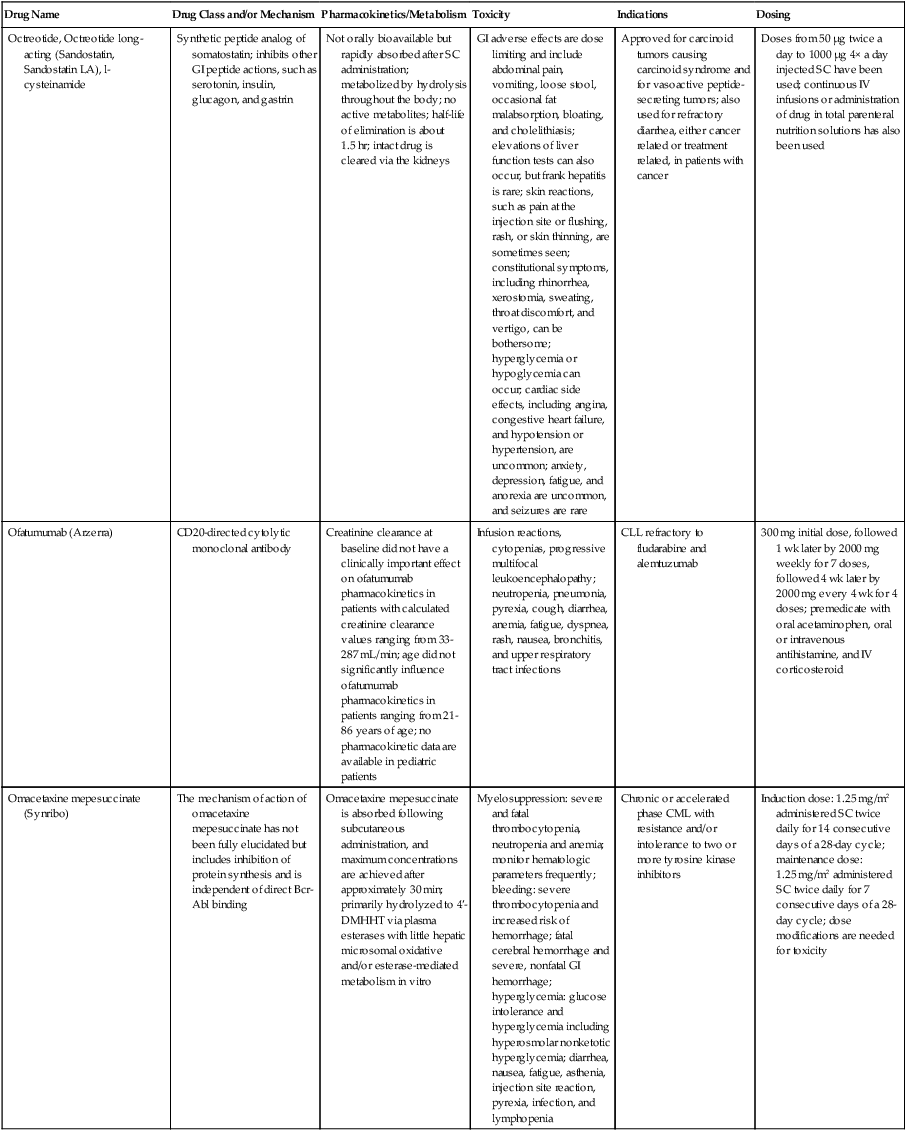
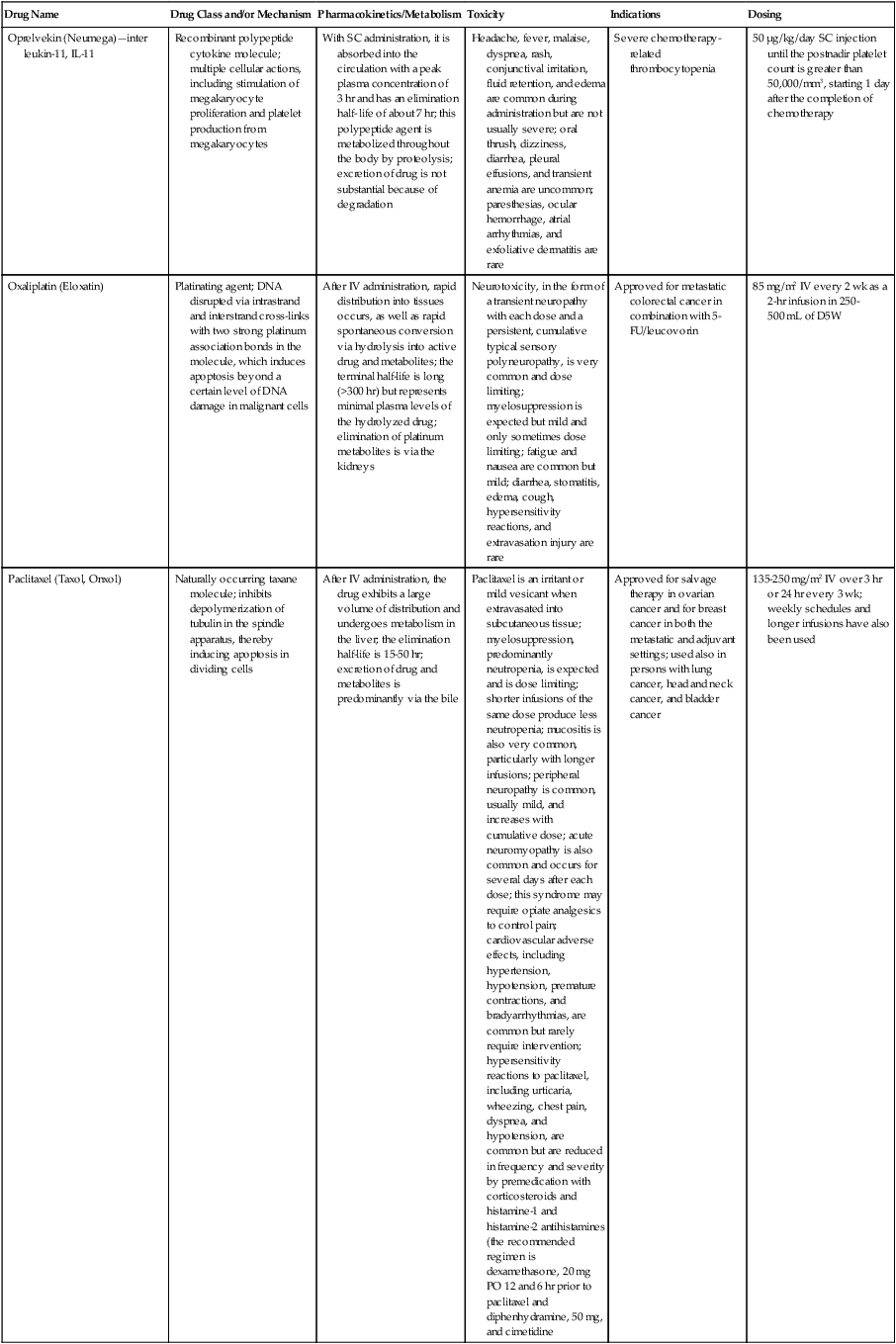
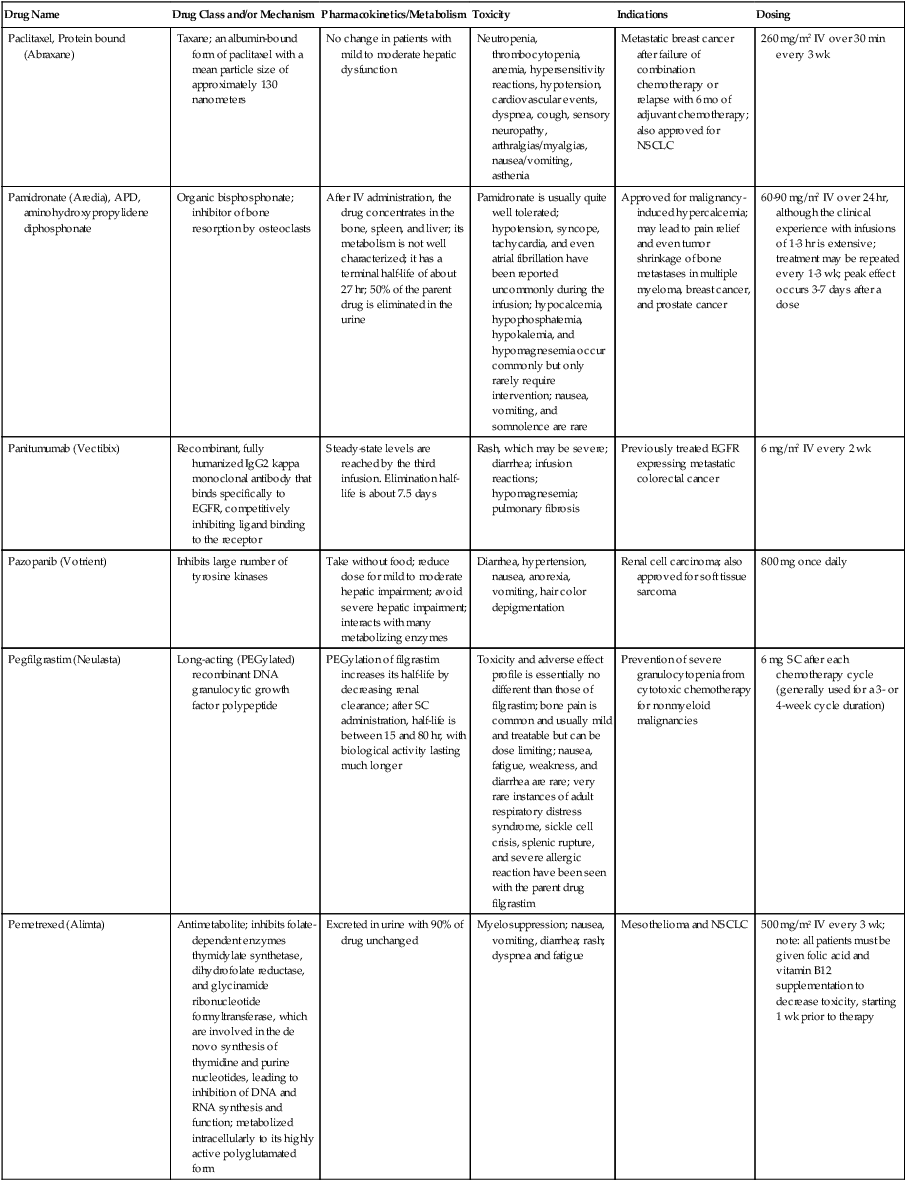
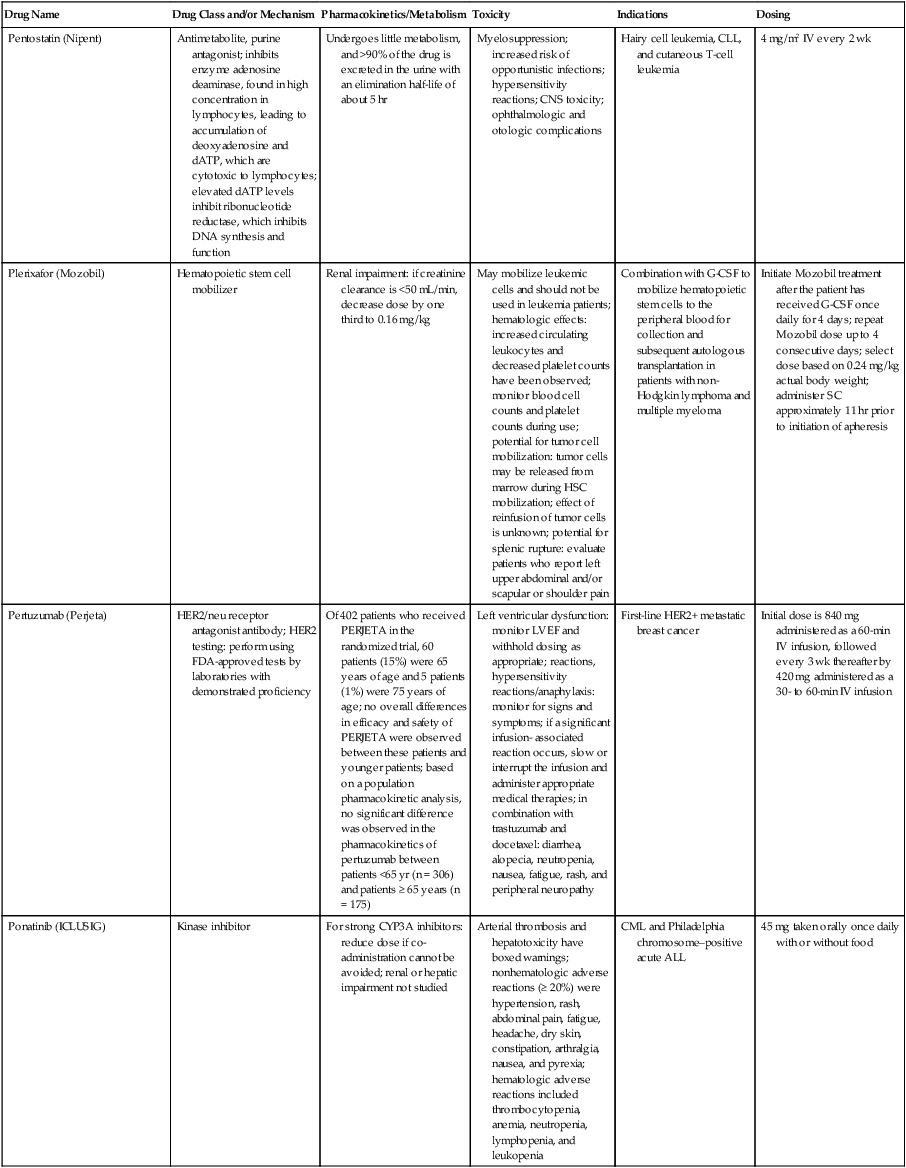
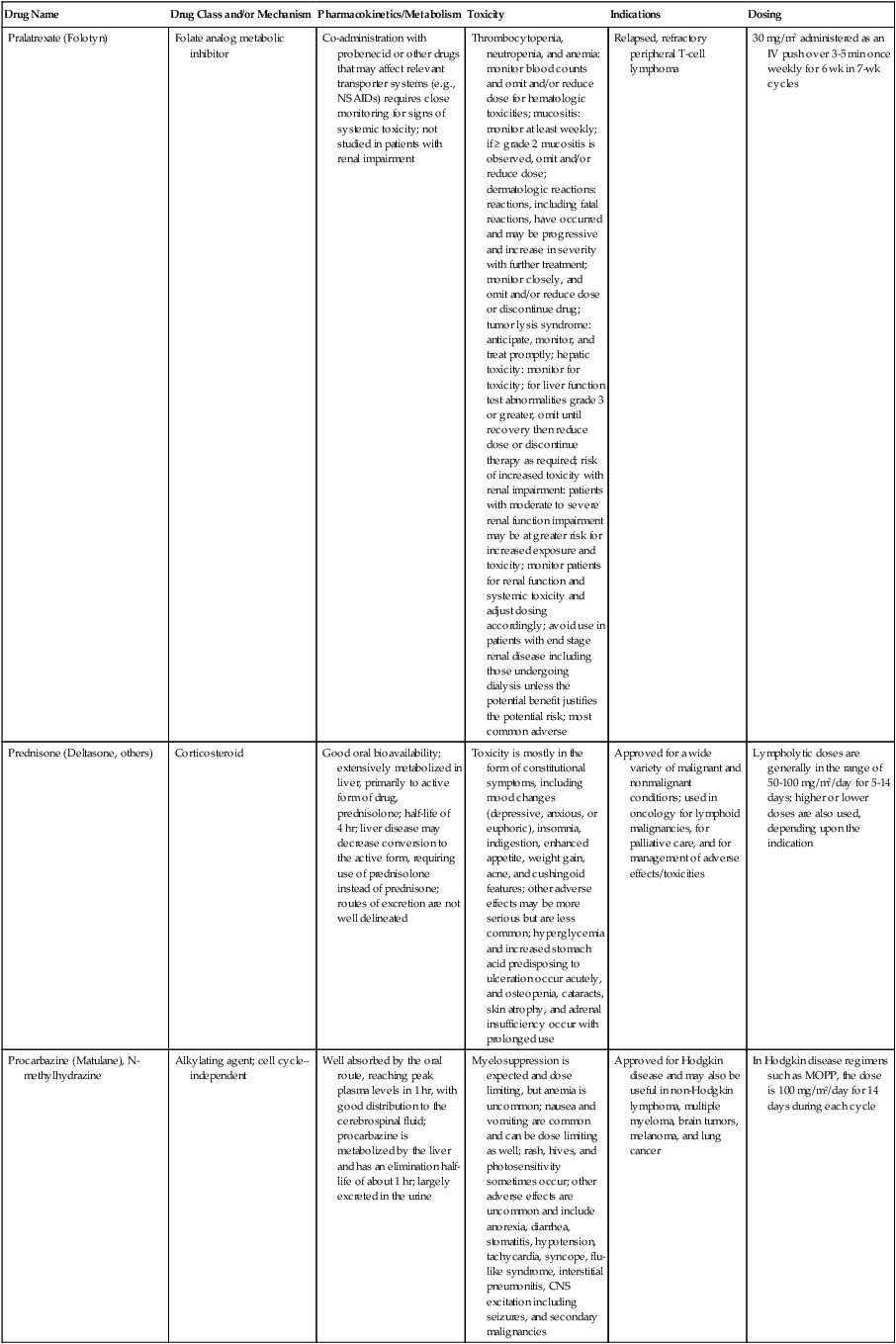
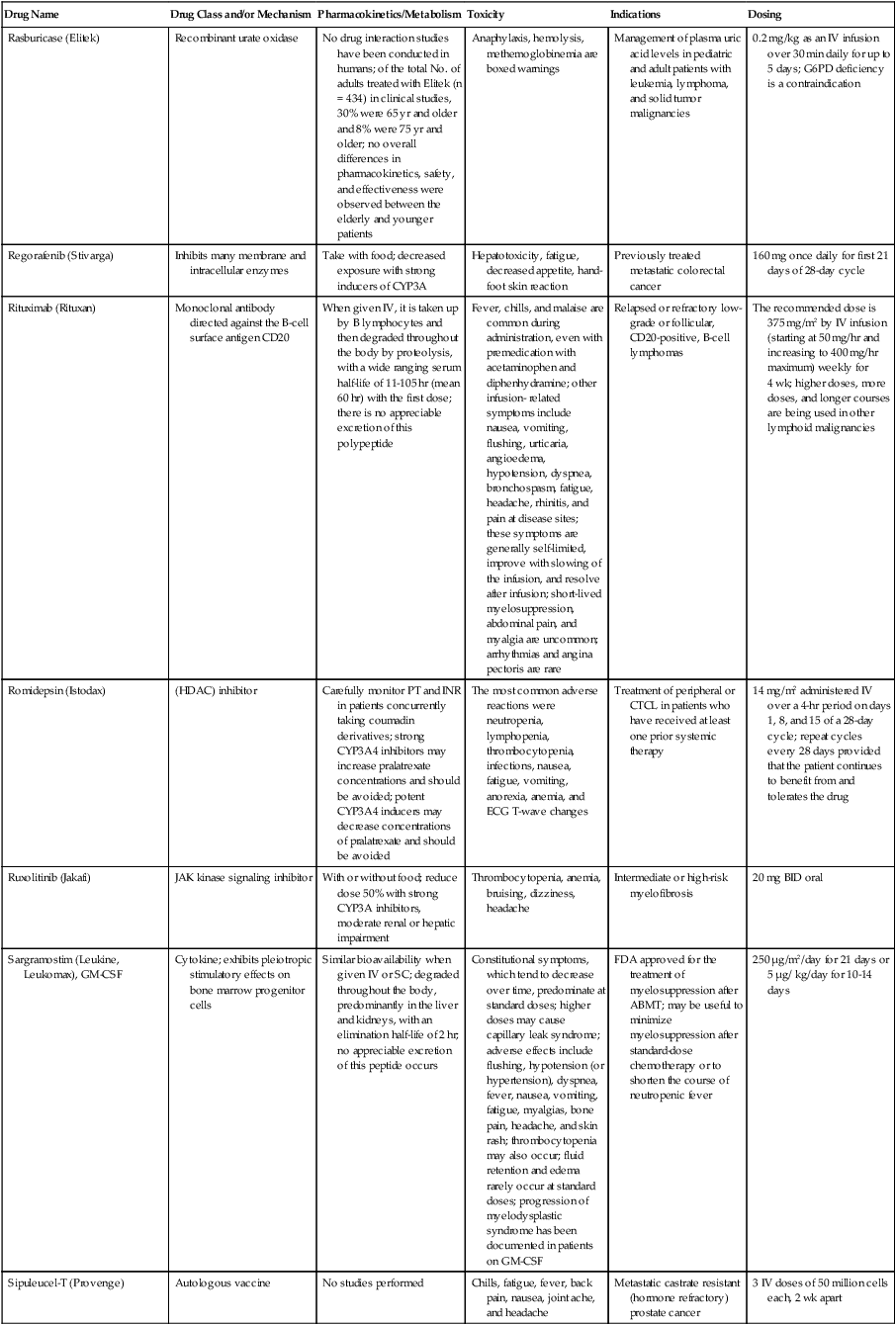
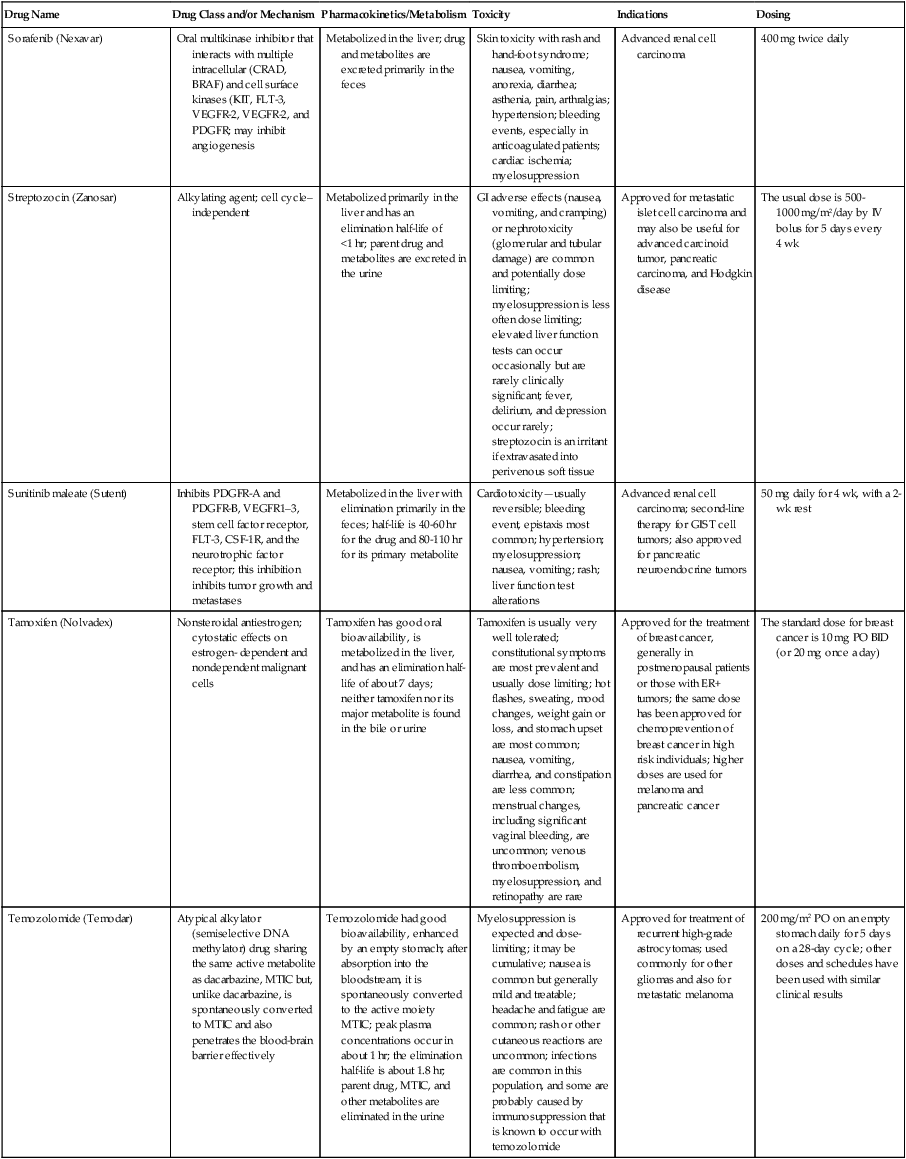
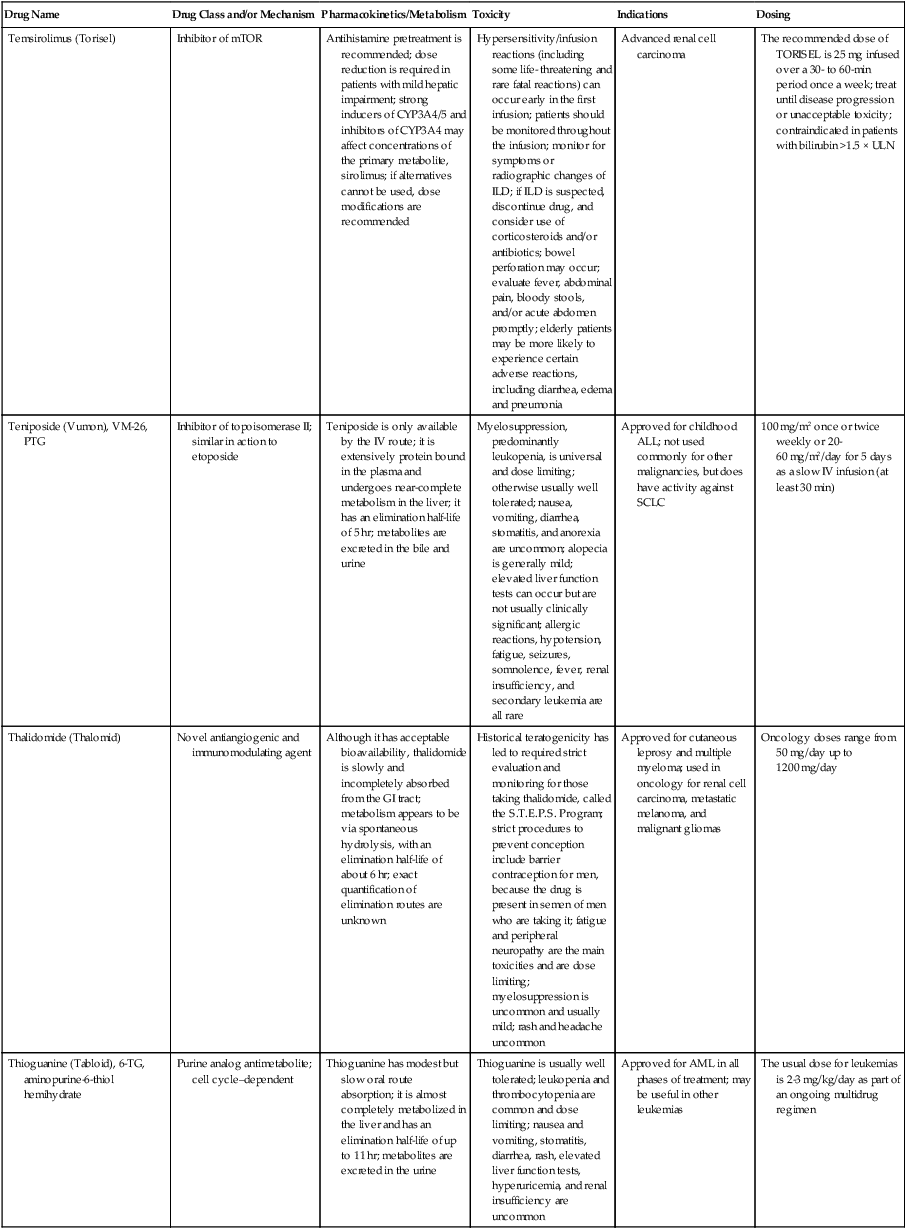
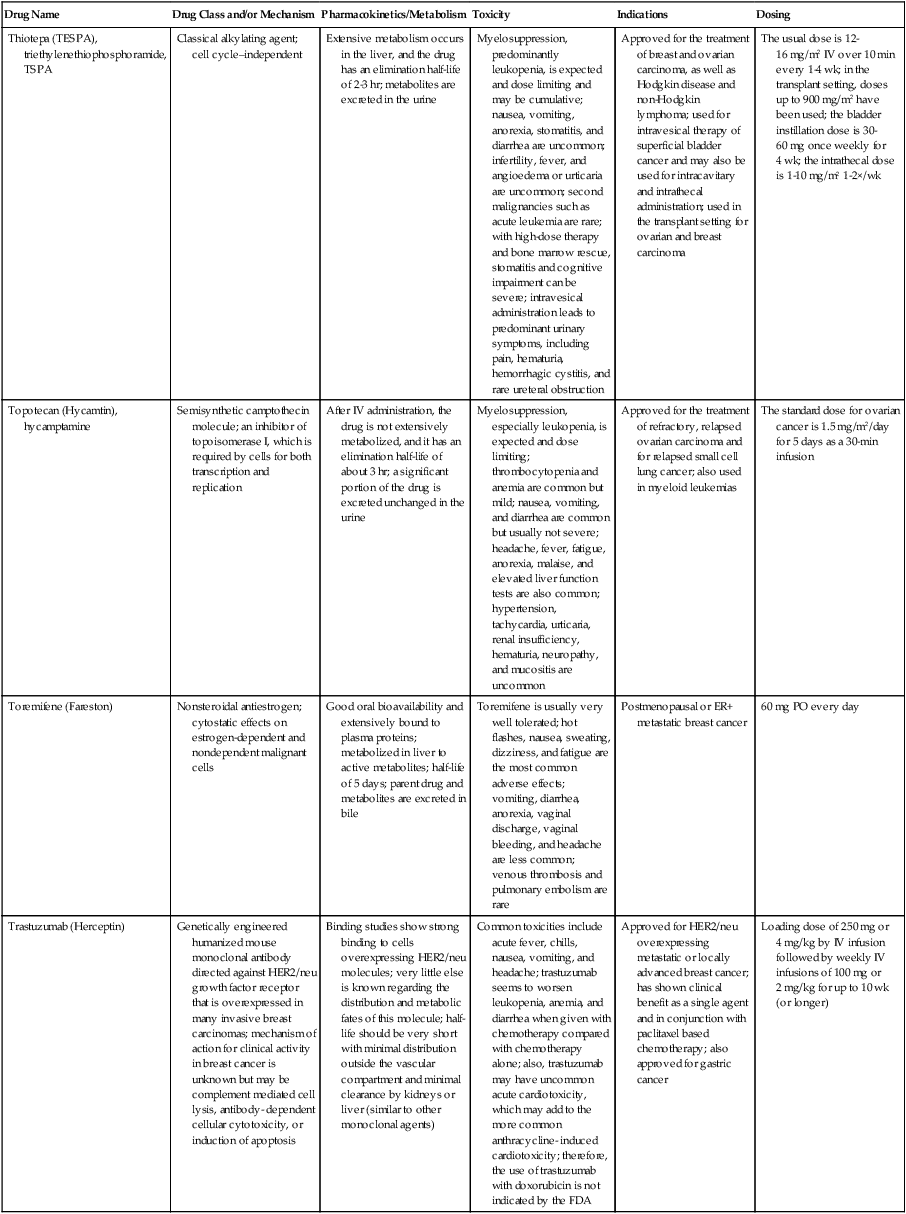
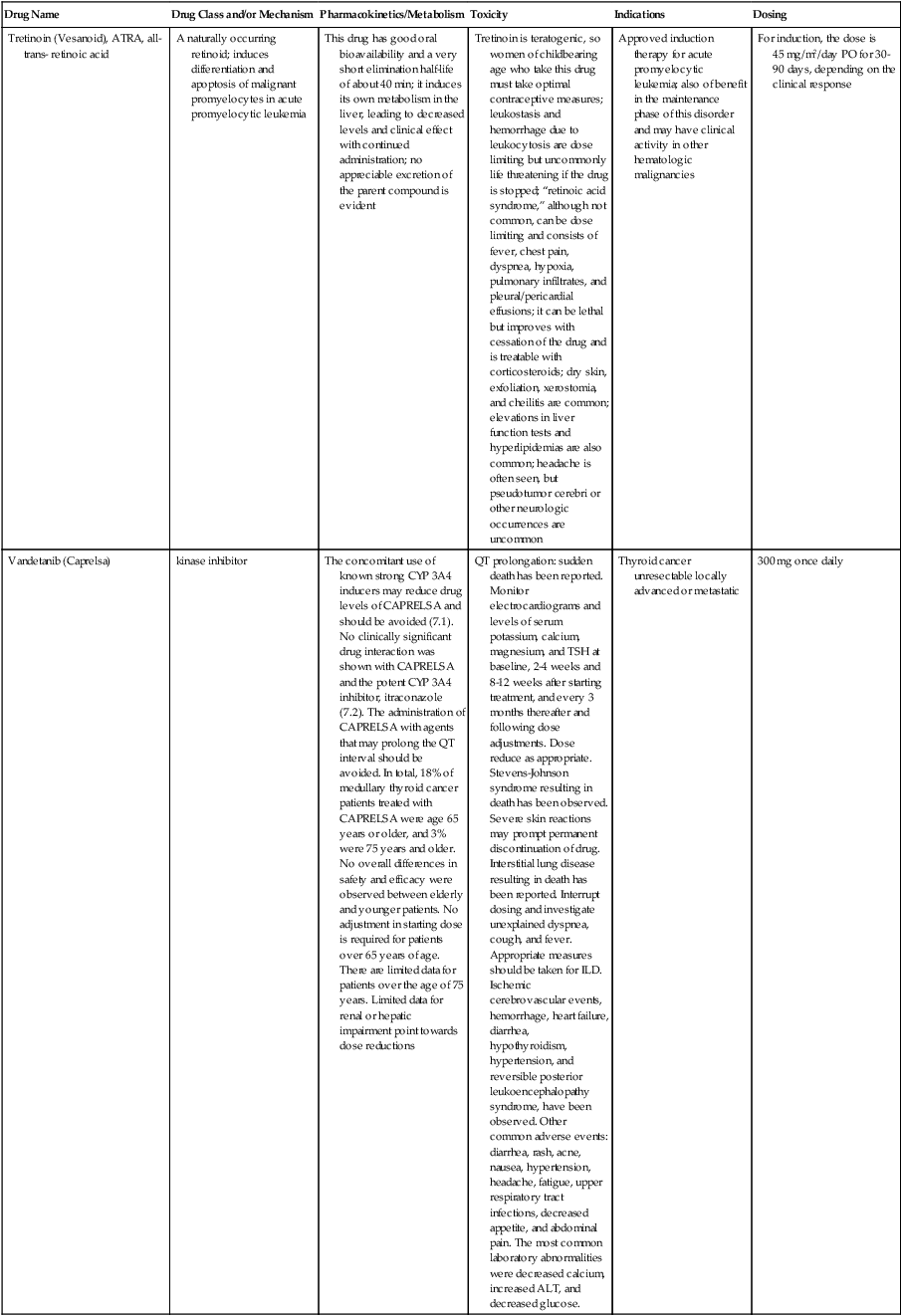
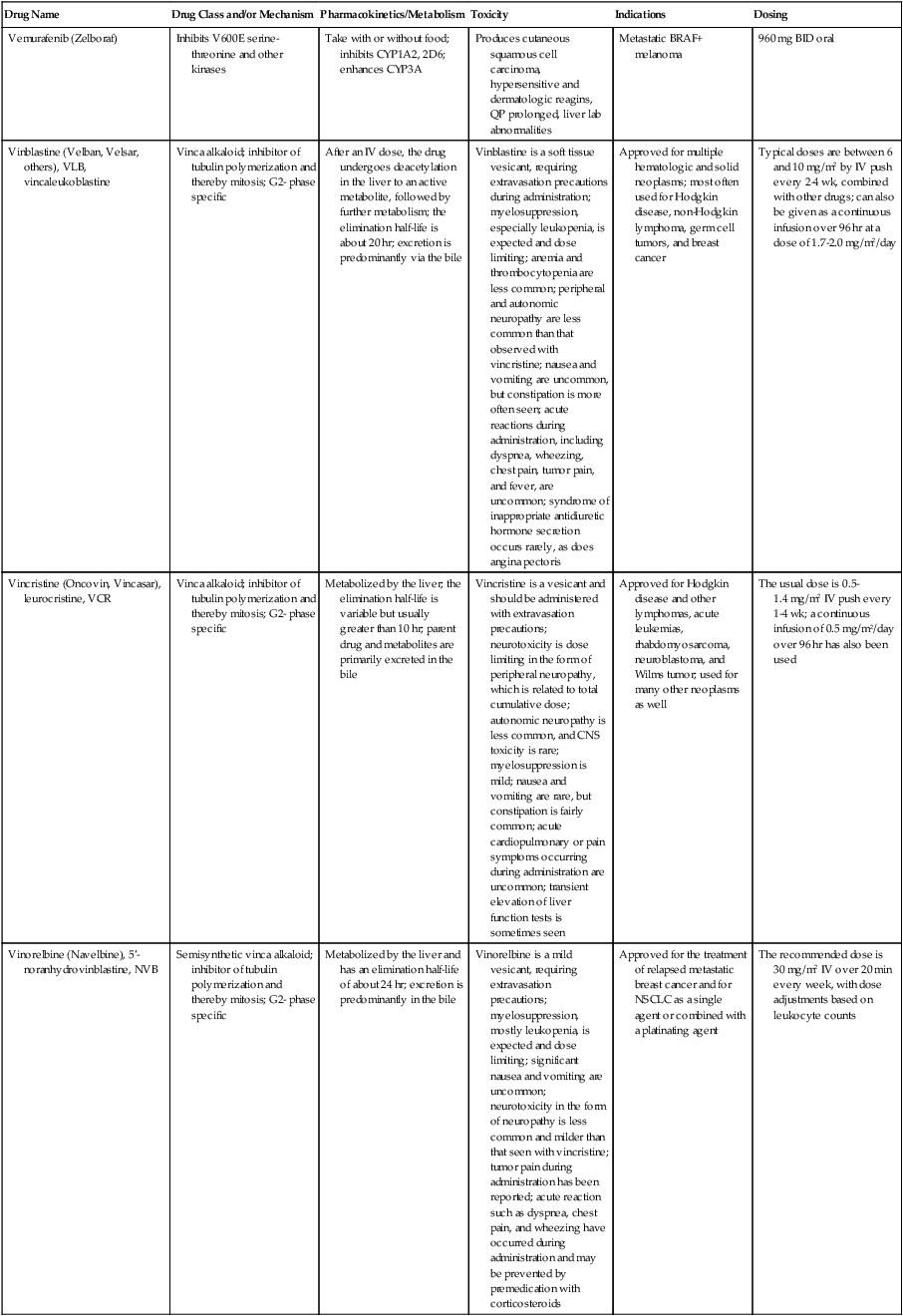
*As indicated throughout this chapter, the pace has accelerated for approvals of new drugs and new indications. Updated information should be checked on the FDA Web site (www.fda.gov/drugs) or other authoritative sources.
Adapted and updated from Freter C, Perry W. Systemic therapy. In: Abeloff MD, Armitage JO, Niederhuber JE, Kastan MB, McKenna WG, editors. Abeloff’s clinical oncology. 4th ed. Philadelphia: Churchill Livingston; 2008.