Viruses and Human Cancer
Paul F. Lambert and Bill Sugden
• More than 15% of human cancers are known today to be caused by viruses.
• A hallmark of virally induced cancers is that they are associated with persistent viral infections.
• Although some viruses encode oncogenes that directly contribute to the cancers they cause, other viruses are thought to result in cancer indirectly by causing chronic destruction of the target organ from which the cancer arises.
• The etiologic role of viruses in cancer is established through a combination of epidemiological and molecular evidence.
• In many cases, the cancers caused by the virus represent dead-end streets for the virus; that is, the virus is no longer able to replicate in the cancer.
• Virally induced cancers can be prevented through the use of effective prophylactic vaccines.
• Viral genes expressed in associated cancers represent targets for therapeutic vaccines and viral-specific anticancer drugs.
Introduction
Viruses cause cancers in people. More than 15% of all human cancers are thought to have a viral etiology,1 and this fraction is likely to grow as we investigate additional cancers for a potential viral cause and identify new human viruses. Identifying a human cancer as having a viral etiology has substantive consequences both for its treatment and its prevention. The known virally caused human cancers often express virally encoded products in the tumor cells. These viral products are potential targets for antiviral, tumor-specific therapies. Viral infections can be prevented by vaccines; therefore it may be possible to eliminate the human cancers that require viral contributions for their development.
The search for human tumor viruses has been propelled by a long appreciation that viruses can cause cancers in birds and rodents. Viruses were isolated as filterable extracts from avian tumors in the first decade of the last century and were shown to induce tumors in susceptible animals.2,3 Parallel findings were made in mice in the 1940s.4 These animal tumor viruses were in the retrovirus family and led researchers to look for retroviruses as human tumor viruses in the 1960s and 1970s. However, most of the human tumor viruses subsequently identified are in different virus families and do not conform to some of the expectations derived from the study of highly oncogenic animal retroviruses. For example, highly oncogenic tumor viruses induce tumors in animals rapidly. The inoculation of Rous sarcoma virus into the wing web of newborn chicks can induce fatal sarcomas within 2 weeks in 100% of susceptible animals.5 In addition, highly oncogenic tumor viruses are oncogenic because they have acquired and express potent derivatives of cellular proto-oncogenes. In fact, many of the known human proto-oncogenes were first identified as homologues of the oncogenes transduced by the highly oncogenic animal retroviruses.6 Known human tumor viruses, however, usually do not induce cancers rapidly; often 15 to 50 years will elapse between the primary infection and tumor development. Nor do human tumor viruses express cellularly derived oncogenes; rather, some of them have evolved to inhibit cellular tumor suppressor genes. These differences between highly oncogenic animal viruses and human tumor viruses probably have contributed to the delay in our recognition that viruses do cause cancers in people.
A second obstacle in our recognition that viruses can be tumorigenic in their human host is that we lack convincing animal models in which to test these viruses directly. For all practical purposes, all known human tumor viruses infect only people. In addition, we now know that human viruses found not to be tumorigenic in people are tumorigenic when experimentally introduced into test animals. For example, human adenovirus 12, which causes only respiratory infections in people, is highly oncogenic when inoculated into newborn hamsters.7 The lack of an experimentally tractable animal host for human tumor viruses has required multiple lines of evidence to affirm that a given virus can contribute to a given human cancer. In particular, epidemiological findings have been combined with genetic and molecular analyses in cell culture to identify human tumor viruses. Experiments with mice transgenic for viral genes also have supported these identifications.
Epstein-Barr Virus
Epstein-Barr Virus (EBV) was identified through the insight and advocacy of Denis Burkitt. As a young surgeon, having identified Burkitt lymphoma as a new disease entity, he analyzed the geographic and climatic distribution of this childhood lymphoma, determined that it overlapped with that of malaria, and postulated that it had an infectious etiology.8 At his urging and with his proffered biopsy samples, Tony Epstein and colleagues identified EBV in Burkitt lymphoma–derived cells.9 To do so, they developed the expertise to propagate these cells in culture.10 Cell lines derived from EBV-positive Burkitt lymphomas proved to be powerful tools in associating EBV with different human diseases. Different EBV-positive cell lines express different viral antigens and thereby have served as test samples for patients’ expression of antibodies to EBV-encoded antigens.
The analyses of these antibodies by serology led the Henles in Philadelphia to propose EBV as the cause of infectious mononucleosis.11,12 A colleague in their laboratory who had lacked antibodies to EBV-encoded antigens developed those antibodies upon presenting with infectious mononucleosis. The etiologic role for EBV in this “self-limiting lymphoproliferation” was subsequently established by careful, prospective epidemiological studies in which serology was used to demonstrate that only immunologically naive people were at risk of the development of infectious mononucleosis; upon its development, they would first express antibodies to EBV-encoded antigens of the immunoglobulin (Ig) M class, and only later to those of the IgG class.13 Thus about 85% of infectious mononucleosis cases were shown to arise from a primary infection with EBV. Serologic studies also allowed the Henles to propose that nasopharyngeal carcinoma (NPC) might be caused by EBV because patients with NPC were characterized by having atypically high titers to EBV-associated antigens.14 However, the data that linked EBV causally to Burkitt lymphoma and NPC by the early 1970s were only “guilt by association.” Although EBV caused most infectious mononucleosis upon primary infection, serology also had demonstrated that children in the parts of Africa in which Burkitt lymphoma is endemic and adults in the parts of China in which NPC is prevalent all had been infected with EBV, that is, were “EBV seropositive,” long before these cancers developed.
The serologic analyses of EBV in the 1960s and 1970s illustrate a conundrum for viruses and human cancers: “How can many people be infected with a given virus, and yet how can that virus contribute to tumor development in only a few infected subjects after long periods?” This apparent paradox applies, in fact, to most cancers associated with human tumor viruses and explains a major reluctance to consider viruses as etiologic agents for human cancers. The World Health Organization (WHO), without resolving this conundrum, sponsored a prospective epidemiological survey in Uganda to assay 42,000 youngsters serologically for evidence for or against the contribution of EBV causally to Burkitt lymphoma. The region studied had a high incidence of this cancer. Samples of blood were collected from children, their serum was stored, and for children later identified as having Burkitt lymphoma, blood samples were collected again and their titers to EBV antigens were determined. In this prospective survey, Burkitt lymphoma developed in 14 youngsters during the 5 years they were monitored, and those in whom the lymphoma developed had, prior to tumor development, on average a 3.4-fold higher titer of antibodies to one class of EBV antigens than did the children in whom the lymphoma did not develop.15 That is, for children in the portions of the world in which EBV-associated Burkitt lymphoma is endemic, a high titer of antibodies to a given set of EBV-encoded antigens represents a thirtyfold risk factor for the development of Burkitt lymphoma.15
Do these findings prove that EBV causes Burkitt lymphoma? No; proof in such cases for which direct experiments are not feasible ultimately comes from the accretion of supporting findings in the absence of confounding data. However, the WHO study did make it unlikely that EBV is a passenger virus that merely replicates well in tumor cells, because the antibody titers were elevated 7 to 54 months before tumor detection.15 Similar prospective surveys were carried out in China, which identified antibodies of the IgA class to the same set of EBV antigens as a risk factor for the development of NPC.16
The association of EBV with Burkitt lymphoma and NPC and the demonstration that EBV causes the bulk of infectious mononucleosis have led researchers to consider other diseases with which EBV might be associated. During the past 20 years, EBV also has been linked to posttransplant lymphoproliferation disease (PTLD)17; oral hairy leukoplakia18; approximately one third to one half of cases of Hodgkin disease19,20; one third of AIDS-related diffuse large B-cell lymphomas (DLBCL)21; and one tenth of gastric carcinomas.22 These linkages have been made not only through serology but also by molecular genetic analyses that render the linkages more robust. The latter analyses have been made possible by the elucidation of the molecular virology of EBV in cell culture.23
EBV is a herpesvirus; it has a double-stranded DNA of 165,000 to 170,000 base pairs24 and encodes more than 100 genes,25 with 25 of these being pre-microRNAs (miRNAs), which encode up to 50 miRNAs26–29 (Fig. 11-1). As with other herpesviruses, EBV has two distinct phases to its life cycle. It can infect cells, express a small subset of its genes (see Fig. 11-1), and cohabit with the cell without killing it (its “latent” phase). EBV also can emerge from its latency, express all or most of its genes, amplify its DNA, assemble progeny virions, and kill its host cell by lysis (its “lytic” phase). Unlike neurotropic herpesviruses such as herpes simplex virus type 1 and Varicella zoster virus, EBV in its latent phase need not be maintained in a nonproliferating host cell. Rather, it has the capacity to both initiate and maintain proliferation in at least the B lymphocytes it infects in cell culture and at early stages of primary infection in vivo.30,31 It is the ability of EBV to affect proliferation and survival of its infected host cell that likely renders it oncogenic.
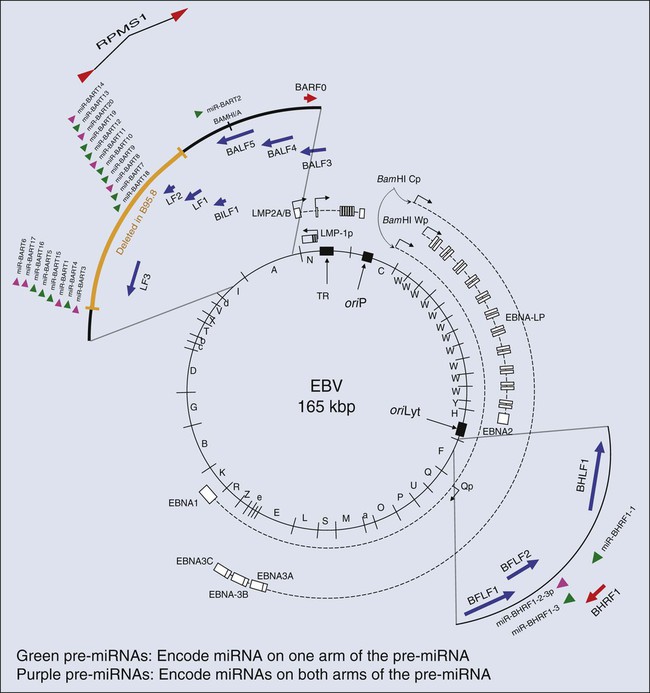
EBV clearly can induce and maintain proliferation of infected B cells. In genetic experiments in which two viral genes, EBNA2 and LMP1 (see Fig. 11-1), are expressed conditionally within the context of the virus, it has been demonstrated that each gene product when assayed alone needs to function for infected B cells to continue to proliferate.32,33 These observations are particularly telling because they help to explain the multistep evolution of Burkitt lymphoma. Many other genetic analyses have shown that four additional viral genes—EBNA1, EBNA3a, EBNA3b, and EBNA3c (see Fig. 11-1)—affect some facet of cell proliferation or survival.34 EBNA1 is essential for the viral genome to be maintained as a plasmid in cells.35 Its inhibition leads to the loss of the viral DNA from tumor cells and to their death.36 EBNA3a acts at the stage of initiation of proliferation.37 EBNA3b is a tumor suppressor.38 EBNA3a and EBNA3c inhibit p16INK4 and p14ARF.39 A seventh viral gene, LMP2a, can substitute for the signaling provided by a functional B-cell receptor (BCR) to support the survival of murine and human B cells that lack functional BCRs.40,41,42 This activity of LMP2a likely underlies one contribution of EBV to the tumor cells in Hodgkin disease that fail to express functional BCRs.43 Two additional genes of EBV are expressed as nontranslated, small RNAs, termed “EBERs.” They contribute to the efficiency with which EBV induces and maintains proliferation of B lymphocytes infected in culture44; they also induce type I interferon45 and mediate other activities that potentially contribute to tumor phenotypes.46 The EBERs usually are highly expressed in EBV-positive tumor cells and thus provide a convenient, sensitive assay for identifying these tumor cells.47,48
All of the transforming genes of EBV that are expressed as proteins are recognized by the host’s T-cell response.49 EBNA1, which can be the only viral protein detected in some EBV-positive tumors,50 is recognized in an atypical response involving CD4+ T cells and its endogenous major histocompatibility complex class II processing.51,52 The host’s cytotoxic response is sufficiently robust that patients recovering from infectious mononucleosis lack B cells that detectably express RNAs that encode these transforming genes. The surviving EBV-infected B cells are in distinct, differentiated states in which they no longer proliferate and express only LMP2a (see Fig. 11-1) detectably, a viral gene product not directly required for cellular proliferation.53,54
A failure of this robust immune response to EBV’s transforming proteins contributes to PTLD. The infected proliferating B cells in these immunosuppressed patients express all of EBV’s transforming proteins, as well as the EBERs.55 Two types of successful treatments for PTLD demonstrate the critical role of the patient’s immune response in failing to limit this “iatrogenic” tumor. First, if immunosuppression can be reduced for the patient such that the transplant is still tolerated, PTLD may regress. Second, several investigative groups have amplified the donor’s T cells that are cytotoxic for EBV’s transforming proteins before bone marrow transplantation. Treatment of patients with PTLD who have these syngenic, specific T-killer cells has cured the disease.56 These encouraging findings underscore the important role of the immune response in limiting survival of EBV-infected cells.
Two startling features of tumor cells freshly isolated from Burkitt lymphoma biopsies help to explain the evolution of this tumor when considered in the context of EBV’s transforming genes and the immune response to them. These tumor cells express only EBNA1 among the required viral transforming proteins, along with the EBERs and miRNAs, yet they proliferate.50 They also display a chromosomal translocation between one of three human immunoglobulin loci and the c-myc proto-oncogene.57,58 These translocations are likely fostered by expression of the activation-induced cytidine deaminase gene, which is essential for immunoglobulin class switching.59 The juxtaposition of an immunoglobulin locus to c-myc drives expression of the proto-oncogene in these B cells as it does in murine plasmacytomas that display similar translocations.60 These observations can be arranged to provide a satisfying though necessarily speculative model for the genesis of Burkitt lymphoma. First, EBV infects young children living in regions of central Africa in which malaria is endemic.61 The malaria is a T-cell immunosuppressive that decreases the children’s ability to limit proliferation of infected cells62 and increases their viral loads.63 Although the youngsters with severe infections have increased antibody titers to viral antigens, they have more proliferating B cells and are at increased risk for the chromosomal translocations, fostered by the recombination mechanism, that occur in B cells and use signals at immunoglobulin loci.64 A rare immunoglobulin/c-myc translocation provides the cell some undefined selective advantages. In addition, multiple viral genes including LMP1, EBNA2, EBNA3a, and EBNA3c are shut off and the cell proliferates, acquires additional mutations (often mutations inactivating p53), and evolves rapidly into a Burkitt lymphoma.65
Two recent findings affect this speculative model for Burkitt lymphoma and have implications for the other EBV-associated cancers as well. Studies of the replication of EBV’s plasmid genome have revealed unexpectedly that only 84% of the viral DNAs on average are synthesized each cell cycle.66 This defect in DNA synthesis occurs in all cell types tested and indicates that EBV DNA must be lost from proliferating cells.66 Proliferating populations of cells can only remain EBV positive if the daughter cells that retain EBV genomes are provided with selective advantages, such that they outgrow their sisters that lose EBV. All EBV-associated tumors maintain EBV DNA as plasmids in most of the tumor cells; therefore EBV must provide all of its associated tumors one or more selective advantages. This profound insight means that EBV provides lymphomas such as AIDS-related diffuse large B-cell lymphoma with selective advantages, even though the EBV-negative and EBV-positive forms of the disease appear similar,21 or that EBV provides selective advantages to NPC tumors in vivo even though these tumors generally lose EBV upon explanting into cell culture. EBV, when found as a plasmid in tumor cells, cannot merely be a passenger.
This insight has been tested with two forms of Burkitt lymphomas and PTLD. Tumor cells were engineered to force the loss of their EBV plasmids. All of these tumors died by apoptosis upon the loss of EBV.36 Viral miRNAs rescued the Burkitt lymphomas that expressed the fewest viral genes.66a These observations underscore the role of EBV in maintaining its associated tumors and the importance of the miRNAs it encodes in oncogenesis.
Treatments for Burkitt lymphoma have evolved to favor high-dose, short-term chemotherapies that can yield greater than 90% 5-year survival for children with localized disease.67 This high rate has not been achieved, however, in some centers in Africa, where the rate of event-free survival over 12 months has been found to be only 33% for patients at all stages of the disease.68
All EBV-associated cancers contain EBV DNA and express EBNA1, EBERs, and all or a subset of EBV’s miRNAs (see Fig. 11-1).55 We know less about the genesis of these other tumors, but findings with NPC provide evidence for an unexpected contribution of EBV to its etiology. Chan and colleagues have shown that EBV infects cells that already can be distinguished as being “preneoplastic” in the evolution of NPC.69 This finding might lead one to think that EBV is merely a passenger in this tumor. However, the fact that 100% of NPC tumors are infected with EBV and that most tumor cells retain viral plasmid genomes means that EBV must provide these tumor cells with selective advantages so that the cells with EBV outgrow those that inevitably lose the plasmid. It is not known what this selective advantage is, but it is reasonable to hypothesize that EBV could provide these cells with proliferative or survival signals, as it does with Burkitt lymphoma cells. Viral gene expression in NPC biopsies correlates with decreased expression of host cell genes involved in antigen display, which likely facilitates the growth of these cells in vivo as well.70 Some of EBV’s miRNAs are particularly well expressed in biopsies of NPCs relative to their levels in Burkitt lymphomas and cells infected in vitro.71,72 The viral miRNAs are thus likely candidates for providing NPC tumor cells with selective advantages that act in vivo.
Hepatitis B Virus
HBV was identified by virtue of its being recognized as an antigen in the sera of donors detected by antibodies in sera of other infected donors.73 Thoughtful analyses by Blumberg correlated the presence of the antigen with hepatitis, a correlation strengthened by the seroconversion of a laboratory member who contracted hepatitis.73 By the late 1960s, blood donated to blood banks was screened for the antigen, positive samples were removed, and only negative samples were used for transfusions. This early insightful intervention led to a significant reduction in transfusion-associated hepatitis.74 Blumberg also demonstrated a striking association between HBV, antibodies to its antigens, and hepatocellular carcinoma (HCC).73 These early findings have been built upon to demonstrate that HBV does cause HCC, which is either the fifth or sixth most common cancer in people today. Of the approximately 500,000 new cases of HCC in the world each year, HBV is estimated to cause between 50% and 70% of them.75,76 Most of the rest of these cases are attributable to hepatitis C virus (HCV), a member of the flavivirus family.
Two types of data have established HBV’s causal role in HCC. A prospective survey of 22,707 male civil servants in Taiwan was initiated at the end of 1975.77 Of these subjects, 3454 were found to be positive for HBV’s surface antigen (HBsAg), indicating that they were chronically infected with HBV. The entire group of 22,707 members was observed on average for 8.9 years. By the end of 1986, HCC had developed in 152 of the 3454 HBsAg-positive men, whereas it had developed in only 9 of the 19,253 HBsAg-negative men. The relative risk of the HBsAg-positive cohort for the development of HCC was therefore 100 times greater than that for the HBsAg-negative group.78 This prospective epidemiological study provides robust data that the presence of HBV is strongly associated with the development of HCC.
The second type of data demonstrating that HBV can cause HCC has been derived by removing HBV from a population and determining whether the incidence of HCC declines. Taiwan began vaccinating children in 1984, first with a plasma-derived antigen and eventually with a recombinant antigen. Between 1984 and 1994, the incidence of infection as monitored by the presence of HBsAg had dropped from 9.8% to 1.3% among children 12 years or younger.79 Similar findings have been made in the Gambia, where children vaccinated during their first year develop into only 10% as many chronically infected 9-year-olds as do unvaccinated children.80 HCC is a cancer that peaks between 50 and 60 years of age and rarely occurs in children from 6 to 14 years of age. The incidence of HCC in this latter population in Taiwan dropped from 0.64 per 100,000 per year when averaged from 1981 to 1990 to 0.36 per 100,000 per year when averaged between 1990 and 1994, a finding that is statistically significant (P < .01).81 This decline presumably reflects the eightfold reduction of chronic HBV infection in children at risk for the development of HCC. Removal of a virus from a population with a subsequent decrease in an associated cancer in that population provides compelling evidence that the virus contributes causally to the cancer. We can expect that a decline of HCC in the vaccinated adult population will be more striking in decades to come.
Although it is clear that HBV causes HCC in people, it is not clear how it does so. Researchers now invoke two distinct contributions, direct or indirect, to explain HBV’s oncogenesis. HBV encodes one gene, pX, (Fig. 11-2), which can affect viral and cellular transcription and has been proposed to contribute directly to oncogenesis. HCC in general evolves in patients with marked liver cirrhosis. HBV can contribute to that cirrhosis by providing targets for T-cell killing and thereby could contribute indirectly to oncogenesis.
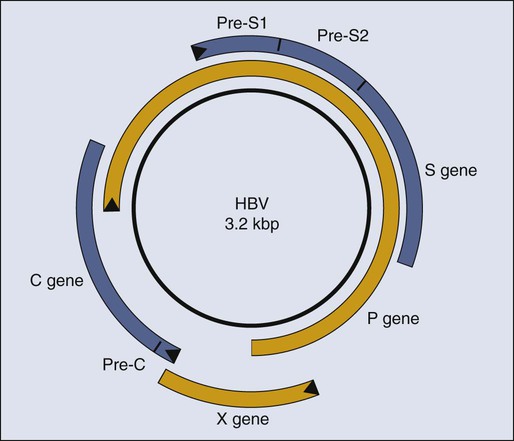
Molecular virology studies of HBV have illuminated the viral life cycle but have yet to identify its mode of oncogenesis.82 HBV is a small, enveloped virus with a double-stranded DNA genome, one strand of which is incomplete. The complete viral duplex DNA is 3.2 kilobase pair (kbp) in length (see Fig. 11-2), serves as a template for transcription by RNA polymerase II, and is replicated via reverse transcription of a greater than full-length RNA transcript of approximately 3.4 kb. All members of the hepadnavirus family preferentially infect hepatocytes. This tropism is apparently mediated by a cellular receptor expressed in hepatocytes and by viral transcription that is controlled in part by cellular transcription factors principally expressed in hepatocytes. Unlike most DNA viruses, HBV undergoes its complete life cycle to yield progeny virions, which exit from hepatocytes via secretory pathways without killing the host cell. This anomalous behavior of hepadnaviruses means that, in the absence of an exogenous function of the host, an infected hepatocyte could survive, carry out its normal functions, and release large amounts of infectious HBV for long periods. Accordingly, some chronically infected people have large amounts of HBV in their sera.
Mammalian hepadnaviruses encode pX, which is not found in the avian species; only the mammalian members are known to cause HCC in their hosts. This correlation has focused interest on pX as being likely to contribute to the oncogenesis of mammalian hepadnaviruses. It is difficult to gauge pX’s potential role in the oncogenesis of HBV; the literature includes much information about it, yet no ready synthesis of this information explains such a role. Most HCC tumor biopsies retain viral sequences encoding pX,83 but few express the protein detectably.84 It has been proposed that pX associates with p53 and inhibits its activation of apoptosis85; however, pX is not detected in HCC biopsies,84 and between 30% and 90% of such biopsies have mutations in p53.83,84 The viral protein pX in studies in cell culture can bind one subunit of RNA polymerase II, as well as transcription factor IIB.86,87 It also associates with Smad4, an integral member of the TGFβ signaling pathway, to foster this pathway’s signaling.88 How these different transcriptional activities of pX might contribute to the evolution of HCC is unclear. Recently, a study reported that pX can induce markers for “stemness” in hepatocytes by activating β-catenin and epigenetic upregulation of miR-181,89 leading to the hypothesis that pX may contribute to the induction of cancer stem cells in the context of HCC.
HBV is often integrated in HCC tumors,90 and it has been proposed that integration of the viral DNA could affect transcription of nearby cellular genes. This suggestion has been strengthened by the recognition that the woodchuck member of the hepadnavirus family contributes to HCC via insertional mutagenesis.91 However, HBV has not been found to integrate at sites that can be interpreted to affect its oncogenesis. In addition, HBV DNAs cloned from HCC biopsies have been tested and found not to score as enhancer sequences in hepatoma cells in culture.92
The hypothesis that HBV contributes to the development of HCC indirectly by inducing rounds of cirrhosis and subsequent liver regeneration is appealing. HBV infection can be acute or chronic; it appears that acute infection correlates with a robust cytotoxic T-cell response to all viral antigens, whereas chronic infection correlates with a weak T-cell response.93 These cytotoxic responses do lead to the death of hepatocytes; however, experiments in mice transgenic for HBV genes and in infected chimpanzees indicate that a potent noncytotoxic mechanism also exists for limiting viral expression in infected hepatocytes.94,95 In these experiments, immune cells release γ-interferon, which by some means inhibits viral gene expression and promotes loss of viral DNA from infected cells.94,95 The administration of interleukin (IL)-18 limits viral replication efficiently in a transgenic mouse model by inducing production of both type 1 and type 2 interferons.96 Chronic but not acute infection correlates with the eventual development of HCC. How these two modes of eliminating infected cells would be balanced to yield long-term or chronic infection, the resulting cirrhosis, and the concomitant hepatocellular regeneration required for the accumulation of mutations predisposing to HCC is not known.
A role for cytotoxic T cells in the evolution of HCC has been modeled in mice transgenic for HBV surface proteins (see Fig. 11-2). These mice are tolerant to these viral antigens, but when the mice are reconstituted with syngeneic, nontransgenic bone marrow and subsequently challenged with syngeneic, immune, nontransgenic splenocytes, cirrhosis develops, and they maintain cytotoxic T cells specific for HBsAg.97 These animals have long-term liver damage, and by 18 to 20 months of age, HCC develops.
Human Papillomaviruses
Cervical cancer is caused by human papillomaviruses (HPVs). Approximately 200 genotypes of HPVs have been identified to date. Most HPV genotypes infect squamous epithelia lining the skin; a subset are mucosotropic and infect stratified, squamous epithelia lining the anogenital tract and oral cavity. A subset of these mucosotropic HPVs, the so-called “high-risk” HPVs, are associated with more than 99% of human cervical cancers, other anogenital cancers, and a subset of squamous carcinomas of the head and neck, particularly those of the oropharynx. HPV-16 and HPV-18, the high-risk HPVs most common in cancers, are present in more than 85% of human cervical carcinomas. The mucosotropic HPVs are transmitted sexually and represent the most frequently detected sexually transmitted disease. The association of specific mucosotropic HPVs with human cancers was first recognized in the 1980s when zur Hausen and associates at the German Cancer Research Institute in Heidelberg detected the presence of then-novel HPV genotypes in human cervical cancers and in cell lines derived from such cancers.98,99 In these cell lines, which include HeLa cells, the HPV DNA often is integrated into the host genome, and only a subset of viral genes, E6 and E7, is expressed.100,101 This discovery led to the hypothesis that HPV E6 and E7 genes contribute to cervical cancer, a premise now well supported by experimental research.
An association of papillomaviruses with cancer was first demonstrated in the early 1930s with the recognition that a subcellular, transmittable (i.e., infectious) agent causes squamous carcinomas in cottontail rabbits.102 The agent was later identified to be a virus, cottontail rabbit papillomavirus (CRPV), that induces warts in the rabbits. In a subset of these infected rabbits, cancers develop at the original site of the CRPV infection. Other animal papillomaviruses induce frank cancer. Bovine papillomaviruses (BPVs), which represent a class of papillomaviruses that induce fibropapillomas and are characterized by hyperplasia of both the dermal fibroblasts and epidermal epithelial cells, can induce epithelial tumors of the alimentary canal in cows.103 Such tumors are thought to arise when animals ingest bracken fern, which contains a potent chemical carcinogen, quercetin.106–106 Thus papillomaviruses and chemical carcinogens act together to induce tumors in cattle.
The study of papillomaviruses in the laboratory began in earnest in the late 1970s when Doug Lowy, Peter Howley, and their colleagues at the National Institutes of Health in Bethesda discovered that BPV-1 infects and transforms a mouse fibroblast cell line, C127, in cell culture.109–109 The parental C127 cells, although immortalized, are contact inhibited. Infection by BPV-1 or transfection of C127 cells with a bacterial recombinant plasmid containing the entire BPV-1 genome yields foci of cells that are no longer contact inhibited. Transformed C127 cells harbor the viral genome as a nuclear plasmid and express early viral genes. The viral gene products E5 (E referring to early and 5 referring to the fifth largest translational open reading frame), E6, and E7110–115 contribute to this transformation (Fig. 11-3). Thus by the time HPVs were recognized as potential etiologic agents in human cervical cancers in the mid-1980s, a wealth of information pointing to the transforming potential of animal papillomaviruses in tissue culture had been established.
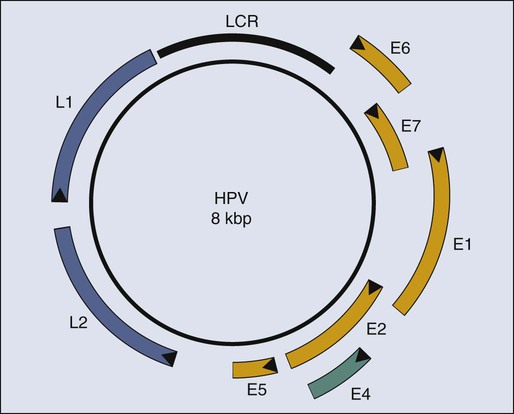
In 1985, both the zur Hausen and Howley laboratories reported that HPV DNAs (see Fig. 11-3) were integrated in chromosomal DNAs in cell lines derived from cervical cancers.100,101 This finding initially led to speculation that HPVs contribute to cervical cancer by integrating in or nearby to disrupt the function of cellular genes that protect against cancer (i.e., tumor suppressor genes) or activate the cellular genes that can promote cancers (i.e., proto-oncogenes), akin to the mechanism of oncogenesis by certain oncogenic avian and rodent retroviruses. However, a role of HPV as an insertional mutagen in cancer is not consistent with its different sites of integration in different cancers, which rarely are found to be near known or suspected tumor suppressor genes or proto-oncogenes. Rather, it is likely that the integration of the HPV genome leads to the selective, and in some cases, upregulated expression of two viral genes, E6 and E7 (see Fig. 11-3), which encode gene products that directly contribute to cancer. The mechanism for this upregulation remains poorly understood but may reflect (1) derepression of E6 and E7 expression from the viral promoter resulting from the disruption of a viral transcription factor, E2, that can repress their transcription116; (2) an increase in the stability of the E6 and E7 mRNAs resulting from the disruption upon integration of an mRNA instability element present in the 3′ end of the E6 and E7 mRNAs117; or (3) increased transcriptional initiation from the viral promoter directing expression of E6 and E7 after integration of the viral DNA. The recognition of the increased expression of E6 and E7 in cervical carcinomas, coupled with the knowledge that E6 and E7 contribute to the transforming potential of BPV-1 in mouse fibroblasts, provided the impetus to examine the tumorigenic activities of these two viral genes.
Evidence for a critical role of increased expression of HPV E6 and E7 in the genesis of cervical cancers comes from multiple studies: (1) cervical epithelial cells harboring integrated HPV-16 DNA have a selective growth advantage over cells harboring normal extrachromosomal viral genomes, and this growth advantage correlates with the increased expression of E6 and E7118; (2) E6 and E7 bind and inactivate the tumor suppressor gene products p53 and retinoblastoma protein (pRB), respectively119,120; (3) p53 and pRB are wild type in cell lines derived from HPV-positive cervical cancers, whereas they are mutated in HPV-negative cervical cancer–derived cell lines121; and (4) the expression of the E6 and E7 viral genes is required for survival of cervical cancer–derived cell lines,122–127 and in the case of E7, maintenance of a tumor phenotype in the context of mouse models for HPV-driven cervical carcinogenesis is required.128,129 Together, these observations strongly support the hypothesis that E6 and E7 contribute causally to human cervical cancers at least by blocking the functions of cellular tumor suppressors.
The E6 and E7 genes from the high-risk HPVs are transforming in tissue culture. They act independently or synergistically to immortalize multiple cell types, including human foreskin keratinocytes, cervical epithelial cells, or mammary epithelial cells.130–135 In addition, E7 cooperates with an activated ras to transform baby rat kidney or human cervical epithelial cells.138–138 The oncogenic properties of high-risk HPV E6 and E7 in vivo have been validated through the characterization of HPV transgenic mice in which expression of E6 and E7, as well as a third viral oncogene, E5, predisposes animals to epithelial cancers of the skin, cervix, anus, and head/neck region.139–149
E6 and E7 are best known for their ability to associate with the cellular tumor suppressors p53 and pRB, respectively.119,120 The discovery of these interactions represents a major advance in our understanding of the mechanisms of oncogenesis: the inhibition of tumor suppressors predisposes cells to evolve into tumors. E6 induces degradation of p53 via recruitment of a ubiquitin ligase, E6AP.150,151 E6 inhibits p53 protein’s transcriptional regulatory activities in tissue culture cells.152,153 Association of E7 with pRB also promotes the degradation of pRB154,155 and disrupts the capacity of pRB to bind and functionally inactivate the cellular E2F transcription factors.138,156 Although these abilities of E6 and E7 to inactivate p53 and pRB, respectively, likely play an important role in their oncogenic potentials, it is important to recognize that E6 and E7 both can bind additional cellular factors, and these interactions also may contribute to HPV-associated carcinogenesis. Studies in HPV-16 transgenic mice strongly support this premise.157–160 Which of these many interactions contribute to their oncogenic potential largely remains to be determined.
E6 proteins from multiple papillomaviruses bind to cellular proteins other than p53 and E6AP. These other interacting partners include p300161,162; paxillin163,164; E6 target protein 1 (E6TP1)165; interferon regulatory factor 3 (IRF3)166; E6 binding protein 1 (E6BP1)167; Bak168; protein kinase PKN169; myc170; the mammalian homologue of Drosophila disk-large tumor suppressor gene product (DLG)171,172; Scribble173; MAGI1174; and MUPP1.175 In addition, E6 can induce expression of telomerase activity at least in part through its interaction with multiple isoforms of NFX1,176–179 a property that correlates with the immortalizing potential of E6. In many cases, including but not limited to p53, the proteins E6 interacts with are targeted for proteasomal degradation largely through E6‘s recruitment of a ubiquitin ligase, E6AP. The importance of E6AP in E6-mediated cervical carcinogenesis has been clearly demonstrated.180
In addition to binding and degrading pRB, E7 can bind to other cellular proteins, p107 and p130, that are related to pRB protein181 and interact with different members of the E2F family of transcription factors.182,183 The inactivation of the pocket proteins drives carcinogenesis in the head/neck region184 but is insufficient to do so in the cervix.184a E7 also is argued to associate with cyclins181,185–187 and to inactivate the cyclin-associated kinase inhibitors p21 and p27.190–190 A role for E7’s inactivation of p21 in cervical carcinogenesis has been demonstrated.159 Thus E7 can associate with and/or alter the activities of multiple cellular factors that themselves interact normally and thereby contribute to the normal regulation of the cell cycle. Still other interactions have been identified between E7 and cellular factors, including the S4 subunit of the 26 S proteasome191; Mi2beta, a component of the NuRD histone deacetylase complex192; the fork-head domain transcription factor MPP2193; the transcription factor AP1194; insulin-like growth factor-binding protein 3195; TATA box-binding protein (TBP)196,197; TBP-associated factor-110198; and a novel human DnaJ protein, hTid1.199 Recently, E7 has been shown to alter proteins that modify chromatin,192,200–202 raising the possibility that E7 causes global changes in host gene expression. It remains unclear whether any of these targets of E7 contribute to E7’s oncogenic potential.
Appreciation of the role of HPV is growing, not only in anogenital cancers such as cervical cancer, but also in head and neck cancers of the oral cavity and in skin cancers.203–206 The multiple interactions of E6 and E7 with cellular proteins that have regulatory functions is consistent with the contribution of E6 and E7 to cancers through multiple mechanisms. Studies in tissue culture strongly support the hypothesis that continued expression of E6 and E7 is required for the continued growth of cervical cancer cells.122–126 Studies in mouse models also support this premise.128,129 These studies point to the potential value of developing drugs that interfere with the expression or function of these viral oncogenes as means for treating HPV-associated cancers.
Cervical cancers take decades to arise in most patients after initial infection with high-risk HPVs. During this time, the HPV must persist in the patient. Strategies that can interfere with viral persistence may prove effective in preventing the development of cancer. E6 and E7 are important to the replicative phase of the HPV life cycle and therefore for viral persistence, indicating that they are also appropriate targets for antiviral and antitumor drug development. Recent studies have used organotypic tissue culturing to recapitulate the life cycle of HPVs in fully differentiating, stratified squamous epithelial cells. These studies have implicated E7 in reprogramming cells within the terminally differentiating compartment of the epithelia to support the amplification of the viral DNA genome, likely through its inactivation of pRB.207 The inactivation of p53 by E6 may be necessary for viral replication by inhibiting cellular stress responses elicited by E7’s inactivation of pRB.208 At least two more HPV proteins, E1 and E2 (see Fig. 11-3), contribute to the replication of the viral genome.209 E1 and E2 bind to an origin-of-DNA replication on the viral genome.210 E1 is a DNA helicase that unwinds the viral double-stranded DNA genome at its origin and, together with E2, recruits cellular DNA replication proteins that then synthesize the viral DNA.212–212 Thus E1 and E2 both represent potentially useful targets for intervening in the viral life cycle.
Human T-Cell Leukemia Virus-I
Adult T-cell leukemia/lymphoma (ATLL), a tumor of CD4+ T-cells, is caused by human T-cell leukemia virus I (HTLV-I), the only retrovirus now accepted as being oncogenic in people. HTLV-I is found worldwide, with approximately 10 to 20 million people estimated to be infected today. This number is likely an overestimate because of a failure to distinguish serologically between HTLV-I and HTLV-II.213 HTLV-I is particularly prevalent in restricted sites, including southern Japan, the Caribbean, and West Central Africa. ATLL is prevalent in areas in which HTLV-I is common, and it is estimated that ATLL will develop in as many as 5% of HTLV-I–positive carriers in their lifetime.214 HTLV-I also causes a progressive, paralytic myelopathy termed HTLV-I–associated myelopathy or tropical, spastic paraparesis. This neurological disorder appears to arise preferentially in patients whose human leukocyte antigen (HLA) haplotypes fail to limit viral load.215
The data that link HTLV-I causally to ATLL are varied. ATLL can occur in familial clusters. It is characterized by an average age at onset of 56 years and proves rapidly fatal, with 50% of patients dying within 6 months of diagnosis.214 Patients generally have antibodies to HTLV-I–encoded proteins,216 and in 88 of 88 primary biopsies of ATLL examined, all had single copies of integrated HTLV-I proviruses.217 The presence of the provirus of HTLV-I in all of the tumors makes it likely that infection with the virus is an early, contributing event in the evolution of the tumor. The presence of ATLL in familial clusters is consistent with the routes of transmission of HTLV-I. HTLV-I is passed from male to female via semen and from mother to child by breast milk. The latter route has been demonstrated prospectively. Encouraging carrier mothers to refrain from breastfeeding has decreased transmission of HTLV-I to their children by 80%.218 It appears highly likely that this form of public health intervention will lead to a corresponding decrease in ATLL in Japan in the future. Such a decrease would constitute formal proof of the oncogenic role of HTLV-I in ATLL.
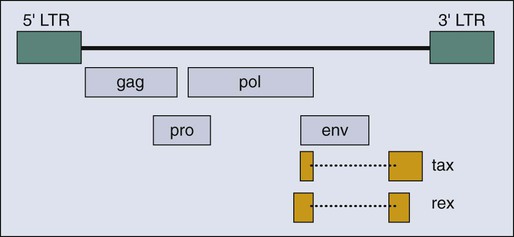
HTLV-I clearly differs from the highly oncogenic animal retroviruses that encode oncogenes derived from cellular proto-oncogenes. HTLV-I is a complex retrovirus that encodes multiple open reading frames in addition to the gag, pol, and env genes common to simple retroviruses (Fig. 11-4). However, none of these additional viral genes is obviously related to known proto-oncogenes, and all are thought to affect the viral life cycle either directly or indirectly by affecting the host cell.219,220 It is accepted that one viral protein, Tax (Fig. 11-4), which regulates both viral and cellular gene expression, is a major contribution of HTLV-I to leukemogenesis.221 It is also evident that viral infection precedes onset of ATLL by 50 years or more, that many cells are infected, and that this clonal tumor develops in only a minority of infected people. These combined observations indicate that multiple rare events in an HTLV-I–infected CD4+ T-cell must occur for that cell to evolve into ATLL.
Multiple approaches demonstrate that Tax can transform cells in culture and be oncogenic in animal models. The introduction of a vector expressing Tax into established, adherent rodent cells can transform them to grow in an anchorage-independent fashion.220 Strains of rodent cells can be transformed with Tax in combination with the ras oncogene to yield cells tumorigenic in nude mice.222 Tax also has been recombined into herpesvirus samiri and introduced into resting human T cells. These infected cells can proliferate and yield infected, immortalized progeny, whereas the Tax-negative parental virus cannot do so.223 In accordance with these data, variants of HTLV-I from which the Tax gene has been deleted no longer can immortalize human T cells in culture.224 These results in culture are paralleled and bolstered by others in transgenic animal models. Expression of Tax from the HTLV-I long terminal repeat (the viral promoter) in transgenic mice leads to mesenchymal tumors.225 When its expression is directed to lymphoid cells, leukemias develop in the transgenic animals.226 However, the exact means by which Tax transforms cells in culture or is oncogenic in animal models is not obvious.
Tax can be considered a paradigm for viral proteins that affect host cells in that it has multiple, distinct functions not found in any one cellular protein. Apparently HTLV-I during its evolution has assimilated multiple cellular activities in this one gene product. Tax can be viewed as having at least three kinds of activities: it activates transcription via nuclear factor (NF)-κB and CBP, the cyclic adenosine monophosphate response element-binding (CREB) protein; it inhibits transcription, perhaps through binding histone deacetylase 1 (HDAC1), and it inhibits several tumor suppressor gene products.220,227–231 Tax potently activates transcription from HTLV-I’s own long terminal repeat (LTR)232 and activates the promoters for IL-2, IL-2 receptor α chain, and c-fos.235–235 This transcriptional activation obviously could contribute to proliferation of an infected T cell. It also can activate the promoter for bcl-x and thereby help to inhibit apoptosis.236 Tax can positively regulate transcription by binding CREB and CBP, as well as some members of the NF-κB family.237,238 Tax not only binds members of the NF-κB family directly, but it also can activate NF-κB’s homing to the nucleus by binding to IkappaBα and promoting its degradation.239,240 Some of Tax’s protein : protein associations have been functionally validated through chromatin immunoprecipitations that have documented the binding of Tax, CREB, and CBP to HTLV-I’s LTR in intact, HTLV-I–transformed T cells.241
Tax also can inhibit transcription. It has been shown to inhibit both expression of the β-DNA polymerase gene228 and some promoters regulated by CBP/p300.242 It has been proposed that the latter inhibition occurs by Tax’s binding to CBP and effectively sequestering CBP such that it is unavailable to bind other DNA-binding transcriptional factors. It also has been shown that Tax binds HDAC1 as measured by co-immunoprecipitations.225 Were Tax to tether HDAC1 to a promoter, the resulting localized histone deacetylation presumably would lead to its decreased support of transcription.243
A third activity of Tax is its inhibition of some cellular tumor suppressors. Tax has been shown to bind to and inhibit the function of p16INK4A.231 p16INK4A binds cyclin-dependent kinase 4 (CDK4), a kinase that, when activated, can phosphorylate pRb, yielding release of E2F transcription factors and promotion of the G1 to S transition of the cell cycle. Tax, upon binding p16INK4A, can increase the kinase activity of CDK4 kinase.231 p16INK4A often is found to be mutationally inactivated in tumors. Consistent with Tax’s functionally inactivating p16INK4A, p16INK4A was shown to be wild type in sequence in two HTLV-I infected T-cell lines in which Tax is expressed but deleted in four uninfected T-cell lines.231 Tax also appears to bind cyclin D3 and thereby may foster, by a second means, the activity of CDK4 and CDK6.229 Tax’s binding to p16INK4A and cyclin D3 would inhibit control of the cell cycle and promote cell proliferation. Tax also can inhibit the transcriptional activity of p53 by an NF-κB–dependent mechanism that culminates in the phosphorylation of p53 as certain residues.230 Such an inhibition of p53 is likely to limit its induction of apoptosis and increase the rate of survival of HTLV-I infected, proliferating T cells.
The multiple activities of Tax likely contribute to HTLV-I’s associated leukemogenesis but must be insufficient for the development of ATLL. Multiple T cells are initially infected by HTLV-I, but over the course of the 50 to 60 years of its development, only one infected cell and its progeny give rise to the tumor. The additional genetic and epigenetic events necessary for this evolution are not known. It also is clear that the expression of viral genes in infected cells is surprisingly low; measurements of RNAs encoding Tax indicate that between 0.1% and 10% of freshly harvested, infected T cells express any Tax message in vivo.244 The fact that Tax expression is absent in approximately 60% of ATLL indicates that a role of this viral gene in cancer development must be transient in nature,245 and this finding has led to additional studies, described later, that have identified another HTLV-I gene that is potentially as important, if not more important, in ATLL.
One of the hallmarks of ATLL is that, whereas the 5′ untranslated region (UTR) of HTLV-I that drives expression of Tax is often disrupted in ATLL, the 3′ UTR is not disrupted.245 Analysis of HTLV-I transcripts led to the realization that the viral genome gives rise to antisense transcripts,246 one arising from the 3′ UTR that is spliced, and the other arising from within the Tax gene that is unspliced.247 Both encode for highly related proteins referred to as HTLV-I bZIP factor (HBZ). Importantly, HBZ is found expressed in all ATLLs.248 The two forms of HBZ, which differ by only nine amino acids at their N terminus, contain three domains: an N-terminal activation domain, a central domain, and basic ZIP domain in the C terminus. The version of HBZ arising from the spliced mRNA, sHBZ, is more abundant than that arises from the unspliced mRNA, usHBZ. HBZ originally was discovered to inhibit expression of the sense mRNAs, including those that encode Tax249 through is interaction with CREB proteins250 and p300/CBP.251 Of relevance to ATLL, knockdown of expression of HBZ in ATLL-derived cell lines led to suppression of proliferation.248 Overexpression of the spliced HBZ could induce the proliferation of ATLL cells, whereas overexpression of usHBZ could not induce such proliferation.247 In the context of transgenic mice, expression of the HBZ gene from the CD4 promoter/enhancer led to increases in the frequency of CD4+ T cells, systemic inflammation, and the spontaneous development of T-cell lymphomas after a long latent period in more than a third of the mice,252 all hallmarks of human patients infected with HTLV-I. These recent studies highlight the potential importance of antisense transcripts arising from human retroviruses, contributing to their pathogenicity.
Human Hepatitis C Virus
Human HCV is accepted as an etiologic agent for HCC along with HBV. Approximately half of the cases in the United States253 and 25% globally254 are ascribed to HCV. Infection with HCV constitutes a twentyfold risk for men in Taiwan of developing HCC.255 HCV represents the most common chronic viral infection among bloodborne pathogens in the United States, where the rate of infection during the period from 1988 to 1994 was estimated to be 1.8%.256 The WHO estimates the worldwide infection rate at 3%, yielding more than 170 million infected persons.257 The rates of infection vary widely, with rates as low as 0.6% in Germany to as high as 22% in Egypt.256 Infection is thought to arise primarily from the use of shared needles among intravenous drug users, blood transfusions, and contaminated syringes in medical settings in developing countries.256 The establishment of sensitive tests for identifying contaminated blood and blood products fortunately has now greatly reduced the rate of infection in the general population in developed countries. As with people chronically infected with HBV, HCC in HCV-positive individuals correlates with chronic hepatitis and cirrhosis. Di Bisceglie258 has estimated that cirrhosis develops in 20% of people chronically infected with HCV per decade, and by two decades, HCC develops in 2% to 7% of chronically infected people. The hyperproliferative state induced by chronic hepatitis and cirrhosis is argued to lead to an accumulation of genetic changes and contribute to the onset of liver cancer. What specifically HCV contributes to this scheme is not known.
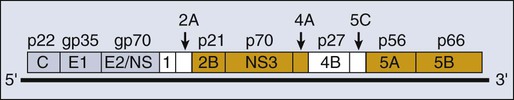
HCV is a flavivirus, indicating that it is unique among the human tumor viruses for having RNA as its only genetic material (Fig. 11-5). It was identified in 1989 during a search for the causal agent of non-A, non-B hepatitis.259 This flavivirus contains a 9.6-kb, positive-stranded RNA as its genome, which encodes a single translation product of approximately 3000 amino acids (see Fig. 11-5). This polyprotein is cleaved by both cellularly and virally encoded proteases to yield at least 10 proteins.260 Inhibitors of these viral proteases are being developed, which do reduce the levels of HCV RNA in the plasma of treated patients for short times.261,262 The study of HCV has been daunting for at least two reasons. Its sequence varies in infected people such that there are six recognized genotypes with multiple subtypes among patients and a spectrum of quasispecies within any one infected individual.263 The different genotypes vary substantially in the success with which they can be treated.264 Members of these quasispecies within one patient vary by 1% to 2% in their sequence.265 In addition, until quite recently, no cell culture for HCV has been available. Researchers have made heroic efforts to overcome this latter hurdle and have met with partial success.
In 1999, Lohmann and colleagues266 described the replication of a subgenomic derivative of HCV in a cell line derived from a human hepatocellular carcinoma. These subgenomic replicons were difficult to establish, but once established, they exhibited bona fide characteristics of flaviviral nucleic acid replication. One to five thousand molecules of plus-strand RNA were present in each cell; minus-strand RNA was present at 10% to 20% of the level of the plus-strand RNA; and this viral RNA replication was insensitive to treatment of cells with actinomycin D.266 This mode of nucleic acid replication in cells would allow the elucidation of the cis and trans elements it requires, but with an unexpected twist. The efficient replication of the subgenomic derivatives of HCV requires mutations in at least two viral genes that enhance replication in cells but abrogate infectivity of intact HCV RNA.267 Chimpanzees are the only nonhuman host for HCV. The parental strain of HCV used to derive the subgenomic replicons is infectious in this primate host, whereas a derivative of it, which contains the mutations required for the efficient replication of the subgenomic replicons, is not infectious in this primate host.267 This finding must limit the conclusions to be drawn from the analyses of replication of these mutated subgenomic replicons in cells. This finding also indicates that the generation of the many sequence variants that constitute the quasispecies within any one infected patient may contribute to the successful replication of HCV.
Earlier attempts at developing cell cultures that support the replication of HCV have been substantively advanced by the identification of an isolate of HCV genotype 2a, JFH1, which replicates in a human hepatocellular carcinoma–derived cell line after transfection of its RNA.268,269 A chimeric HCV genome consisting of a portion of this isolate and a portion of a different isolate of genotype 2a strain has been found to be infectious in cell culture, and the virus derived in cell culture has been found to be infectious in chimpanzees and in mice reconstituted with human hepatocytes.270 These findings will now pave the way to dissect HCV’s replicative functions molecularly. They also should facilitate the identification and initial testing of drugs targeted to inhibit HCV’s replication, such as the inhibitors of its encoded proteases.
A second important insight has been confirmed from the analyses of HCV propagated in cell culture and animal models. Virus isolated from infected animals is more infectious on a “per particle basis” than virus isolated from cell culture; this increased specific infectivity correlates with a lower buoyant density of the virus isolated from animals.270 Although the cellular receptors used by HCV to bind to and enter cells have not yet been fully elucidated, it is clear that HCV particles on binding serum molecules such as high-density lipoprotein become more infectious.271 This binding would lower their buoyant density and mediate binding to a cellular receptor such as the human scavenger receptor class B type I.271,272 Virus grown in cell culture would not be exposed to these serum molecules and thus would be less infectious.
One promising path to study HCV infections in cell culture is to generate hepatocytes by inducing their differentiation from either human embryonic stem cells (hESCs) or induced pluripotent stem cells (iPSCs). Wu and colleagues273 have generated hepatocytes through differentiation of both types of stem cells in culture and infected these hepatocyte-like cells with derivatives of HCV isolated from cell culture and with serum isolates. Both forms of the virus yielded successful infections, each of which secreted core antigens.273 The success of infecting these hepatocyte-like cells with serum isolates of genotypes 1A and 1B is particularly desirable because other culture systems do not support infection by serum isolates in general and those of genotypes 1A and 1B in particular. The general application of hESCs and iPSCs after their differentiation to hepatocyte-like cells for study of infection by HCV will depend both on the efficiency of this differentiation and the practicality of carrying out this differentiation with large numbers of precursors.
Evidence for a direct role of HCV in HCC has come from studies of the HCV Core protein. The HCV Core protein can cooperate with an activated form of the ras oncogene to transform primary rodent cells in tissue culture.274 HCC develops in mice transgenic for the HCV Core protein.275 Several potential mechanisms have been invoked to explain the transforming potential of HCV Core protein. It has been found to enhance cell proliferation through the stimulation of the mitogen-activated protein kinase.276,277 In addition, HCV Core protein can inactivate a transcription factor, lZIP, and this inactivation correlates with transformation in rodent cells.278 More recently, the Core protein has been found to activate STAT3, and this activation may contribute to its transforming potential.279 Whether the HCV Core protein contributes directly to human HCC is uncertain. Neither replication of chimeric HCV in cell culture nor infection of chimeric mice populated with human hepatocytes will allow ready testing of the possible role of the Core protein in HCV’s oncogenesis.
HCV infection alone is not sufficient to give rise to HCC, indicating that other events must occur that contribute to this cancer. Exome sequence analyses have begun to identify changes in cellular genes that contribute to HCC. One such study280 identified mutations in a number of known cancer-related genes such as p53 and CTNNB1, which previously had been identified as being mutated in HCC, as well as mutations in ARID2, DMXL1, and NLRP1. The mutations in ARID2 were all found to disrupt the coding for its gene product, which is consistent with ARID2 being a tumor suppressor in HCC. ARID2 is a subunit of the polybromo- and BRG1-associated factor chromatin remodeling complex, which facilitates ligand-dependent transcriptional activation by nuclear receptors. Interestingly, mutations in ARID2 selectively arise in HCV-associated HCC and are infrequently observed in HBV-associated HCC.280 The significance of this finding remains unclear.
Treatments for HCV have been unsatisfactory, but new ones are on the horizon. The combination of interferon α-2b, which is bound to polyethyleneglycol to increase its half-life (pegylated interferon α), with ribavirin, a nucleotide analogue, has been tested in 1500 adults infected with one of three genotypes of HCV.281 Sustained virologic responses, defined as “undetectable HCV RNA in the serum at a 24-week follow-up,” were achieved in 42% of patients infected with genotype I and 80% of patients infected with genotypes 2 and 3 who received the highest dose of the drugs.281 The mechanism by which these two drugs work in concert is not known, but they remain the treatment that currently is most often used.282 In the United States, approximately 10,000 deaths are attributed to HCV each year, and today infection with HCV is the leading indication for liver transplantation.283
Currently 40% to 50% of 5000 people in the United States receiving liver transplants each year are recipients because of HCV infections.284 More than twice this number are waiting for donors, indicating that transplantation is not a practical treatment for most people infected with HCV. It is also clear that new livers in HCV-positive recipients become infected and limit the success of the transplant.284 Several new drugs have been tested in phase II and III trials and appear promising even by people infected with genotype 1 HCV. These new drugs, Boceprevir and Telaprevir, inhibit the NS3/4A protease of HCV, and in combination with pegylated interferon α and ribavirin, have yielded sustained virologic responses of up to 75%.285,286 It is likely that these new treatments also will be used to limit or abrogate HCV in patients with HCC before they are recipients of liver transplants. Such treatment should increase the long-term success of this transplant therapy.
Kaposi Sarcoma Herpesvirus
The identification of Kaposi sarcoma herpesvirus (KSHV) represents the culmination of much scientific detective work. Kaposi sarcoma (KS) was known before the AIDS epidemic,287,288 but it increased markedly among HIV-positive people. Researchers therefore looked for molecular evidence of an infectious agent present in KS lesions. In 1994, Chang and colleagues289 used a newly developed enrichment procedure based on polymerase chain reaction to identify DNA sequences present in KS but absent in normal cells. They identified a new herpesvirus, KSHV (also termed human herpesvirus 8, or HHV-8), which is related to EBV and herpesvirus saimiri.289 Retrospective studies indicate that KSHV was prevalent in different parts of the world prior to the spread of HIV and that in Africa, for example, the prevalence of KSHV has not changed. What has changed there is the incidence of KS, so that in regions where it was formerly infrequent, the incidence of KS rose threefold between 1988 and 1996.290 Similarly, the frequency of infection with KSHV of certain cohorts in the United States has not altered with the advent of HIV,291 but the incidence of KS has changed.292 KSHV also has been detected in one class of lymphomas termed body-cavity–based lymphomas or primary effusion lymphomas (PEL)293 and in an atypical lymphoproliferative disorder termed “multicentric Castleman disease.”294
KSHV can be transmitted sexually, as evidenced by its prevalence in a population being related to the number of sex partners of its members.295 Its most frequent route of transmission is via saliva.296 KSHV also is transmitted among intravenous drug users, with 30% of this group being infected in parts of China.297 Its prevalence worldwide is clearly nonuniform, with a low prevalence in the United States and Northern Europe, intermediate levels around the Mediterranean, and high levels in the Middle East and in sub-Saharan Africa.297 Although KS was a rather rare tumor prior to the AIDS epidemic, it has evolved to become common in areas of the world in which people are co-infected with KSHV and HIV. The immunosuppressive role of HIV makes it likely that immunosuppression, along with KSHV, contributes to the development of KS.298 Treating HIV-positive people with highly active antiretroviral therapy (HAART) not only decreases a person’s load of HIV but also decreases the incidence of KS and, importantly, leads to a regression of the tumor in patients with KS.299 These observations indicate that immunosuppression not only likely contributes to the development but also the maintenance of KS, along with infection with KSHV. HAART also has led to a decrease in the incidence of overall AIDS-related non-Hodgkin lymphomas, as well as an increased tolerance for their treatment with chemotherapy.299,300
KSHV is now accepted as contributing causally to KS, PEL, and multicentric Castleman disease. For example, a prospective study in HIV-infected adults has identified a significant elevation in titers of antibodies against KSHV in those in whom KS develops.301 KS and PEL also display intriguing features likely to reflect their viral etiology. Single-cell assays of early KS lesions have detected KSHV in a minority of cells that surround their vascular spaces, whereas in the more advanced, nodular lesions, more than 90% of the spindle cells characteristic of these lesions are KSHV-antigen positive.302 The viral antigen-positive cells also stain with antibodies against the VEGF-receptor 3, indicating that they are lymphatic or proliferating endothelial cells.302 That only a subset of the cells characteristic of early KS lesions appear to be infected with KSHV likely indicates that infected cells affect the development of their neighbors. This possibility is supported by the multiple cellular homologues of cytokines and receptors encoded by KSHV that may allow infected cells to interact with adjacent, uninfected cells.303,304 PEL cells are B cells in origin, and in six of seven cases examined, they have nongermline immunoglobulin mRNAs, indicating that they are likely derived from B cells that have encountered antigen.305 However, they often fail to express B-cell activation antigens.306 PEL cells usually but not always are co-infected with EBV.293,306 The role of EBV in the etiology of this lymphoma is not yet determined, but for cases in which EBV is retained as a plasmid in the PELs, it must be providing the tumor cells with one or more selective advantages.
KSHV encodes many genes with clear homologies to cellular genes, some of which are candidates for contributing to viral oncogenesis (Fig. 11-6). Three categories of these cellular homologues are particularly likely to be important for tumor development: cyclins; inhibitors of apoptosis; and cytokines and receptors. KSHV encodes its own cyclin. Cellular cyclins bind specific, dependent kinases (CdKs) whose activities promote progression through the cell cycle and are controlled by cellular inhibitors, including p16INK4A, p21CIP1, and p27Kip1. The KSHV-encoded cyclin can promote cellular proliferation in part by overcoming these cellular inhibitors in its complex with CdK6.307
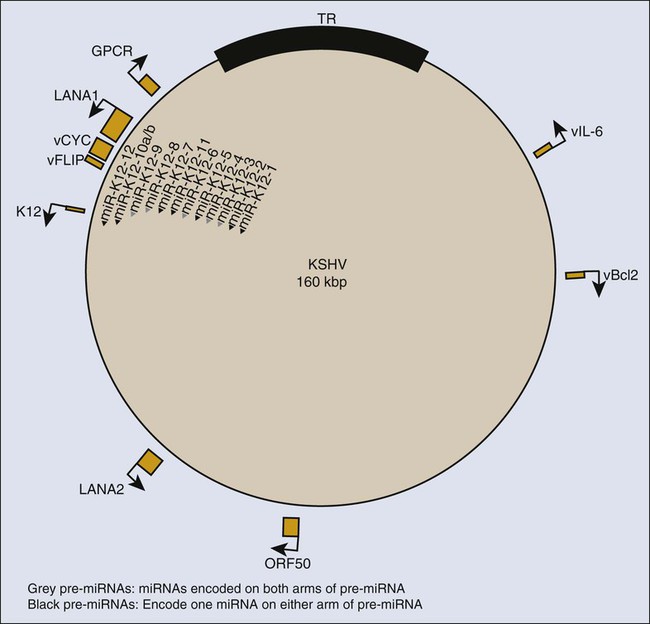
KSHV also encodes its own inhibitors of apoptosis, viral Bcl-2 and viral FLIP (an inhibitor of cellular FLICE, a protein that mediates Fas ligand-induced cell death) (see Fig. 11-6). The viral Bcl-2 shares its limited sequence homology with cellular Bcl-2 in the regions critical for inhibiting apoptosis and fails to dimerize with cellular Bcl-2 and Bcl-XL, thus avoiding regulation by potential cell-binding partners.308 It can inhibit the apoptosis induced by efficient expression of KSHV’s cyclin.309 Viral FLIP can inhibit apoptosis mediated by the Fas pathway and induced by cytotoxic T lymphocytes.310 Viral FLIP also activates NF-κB signaling by binding to its inhibitor, IKKγ,311,312 which fosters expression of cytokines in infected cells.307
Finally, KSHV encodes homologues of cytokines, cytokine receptors, and an intriguing protein that stabilizes cellular cytokine mRNAs (see Fig. 11-6). Its viral IL-6 may promote proliferation of PEL cells, although they appear to be dependent on cellular IL-6 and not viral IL-6 for their continued growth.313 This dependence may be facilitated by KSHV’s Kaposin gene, which is synthesized by an atypical translational initiation to yield a protein formed from 23 residue repeats. Kaposin activates a protein kinase to stabilize mRNAs that encode cellular cytokines, including IL-6.314 KSHV also encodes a G protein–coupled receptor, viral GPCR, which is likely to be pivotal for the development of KS. The expression of viral GPCR in endothelial cells in mice leads to KS-like tumors, whereas the individual expression of KSHV cyclin, KSHV Bcl-2, or KSHV FLIP does not lead to such tumors.315 What is particularly exciting is that cells inoculated into mice that express viral GPCR promote tumor formation by co-inoculated cells that individually express viral cyclin or viral FLIP, or both.315 These findings support a model in which viral GPCR contributes to the development of KS by affecting neighboring cells not infected by KSHV. The model is also supported by the findings that viral GPCR induces expression of the cellular vascular endothelial growth factor (VEGF)-receptor-2 in endothelial cells that proliferate in the presence of VEGF and that viral GPCR induces expression of VEGF.316 Clearly, these observations define an auto-stimulatory loop in which KSHV GPCR alone can maintain proliferation of endothelial cells. They are consistent with findings in which mouse bone marrow cells transfected with recombinant KSHV grow as “KS-like” tumors in nude mice.317 The inhibition of expression of viral GPCR in these KSHV-positive cells inhibits tumor formation when the engineered cells are transplanted into recipients.317 These data indicate that although the viral GPCR may be expressed only in a minority of infected endothelial cells, it makes an essential contribution to oncogenicity by KSHV.
KSHV limits its host’s immune response. The virus suppresses Toll-like receptor 4 (TLR4), which decreases the infected cell’s innate immune response.318 It inhibits expression of HLA-1 molecules, which would help to avoid recognition of infected cells by cytotoxic T cells319 but would increase the likelihood of their being recognized by NK cells. KSHV infection also limits expression of receptors on NK cells needed to recognize their infected targets,320 showing that it uses multiple avenues to evade the immune response. This immune evasion also likely contributes to its oncogenicity.
Recent studies of cell lines derived from PEL and human endothelial cells are providing the foundation for understanding infections by KSHV in vivo. PEL cells maintain KSHV DNA extrachromosomally and consistently express the viral protein LANA-1 (see Fig. 11-6), which is required for viral plasmid replication.321,322 In general, these cells support the latent phase of KSHV’s life cycle and also express the viral cyclin and the viral inhibitor of cellular FLICE, FLIP, but few other viral proteins. Some PEL cell lines support an inefficient spontaneous conversion to the lytic phase of the viral cycle, which can be further induced by treatment of cells with tetradecanoyl phorbol acetate or introduction of a plasmid including ORF50 (see Fig. 11-6), a viral inducer of the lytic cycle.323 The released virus is infectious in human dermal microvascular endothelial cells (DMVECs).324,325 These DMVEC cultures can be passaged to yield populations in which all cells are infected, express LANA-1, assume a spindle-cell morphology, and can proliferate indefinitely.324 The spindle shape is characteristic of cells in KS lesions in vivo. Staining of these infected DMVEC-derived spindle cells indicates that 5% to 10% of them spontaneously support early stages of KSHV’s lytic cycle and 1% to 2% express genes diagnostic of the late stages of the viral life cycle.324 If endothelial cells infected in vivo by KSHV display a similar distribution of cells supporting the latent, early, and late lytic phases of the viral life cycle, then the enigma of the oncogenic contribution of viral genes expressed only during the lytic cycle may be resolved. A sustained, spontaneous conversion of 5% to 10% of KSHV-infected cells to support the viral lytic cycle might allow enough infected cells to express enough KSHV GPCR to promote the bystander-dependent tumor evolution proposed by Bais and colleagues.316
Merkel Cell Polyomavirus
The association of viruses with AIDs-related malignancies drove Chang and colleagues to search for a virus associated with a second AIDs-related malignancy, Merkel cell carcinoma (MCC). This rare but highly aggressive skin cancer arises in elderly and in immunosuppressed and immunodeficient patients, often at sun-exposed regions of the body.326 In the general population, more than a third of patients with MCC will die of their cancer327; in immunocompromised patients, the percentage rises to greater than 50%. MCC is distinguished by a unique pattern of expression of neuroendocrine and epithelial-specific markers expressed in Merkel cells, which are specialized mechanoreceptors cells within epithelia that transduce mechanical stimuli from the skin (reviewed in references 328–331). Feng and colleagues332 used a state of the art “digital transcriptome subtraction” (DTS) approach to identify nonhuman sequences present in MCCs. DTS involves deep sequencing of complementary DNAs followed by in silico subtraction of host transcripts to divulge nonhuman sequences present in a sample.332 Using DTS, Feng and colleagues332 identified nonhuman sequences in four MCC samples that had similarity but were not identical to the large tumor antigen genes of previously identified human polyomaviruses, which led them to a full-length clone of a novel polyomavirus that they called Merkel cell polyomavirus (MCPyV). MCPyV is present in approximately 80% of cases of MCC.335–335 Like other human polyomaviruses, MCPyV has a circular double-stranded DNA genome containing a well-conserved replication origin and transcriptional control region and translational open reading frames encoding for small and large tumor antigens on one strand, and capsid proteins VP1, VP2, and VP3 on the other strand. Serology studies indicate that most of the human population is exposed to MCPyV,336–340 likely in early childhood.336,337 The virus has been detected in a number of different cell types, including non-MCC cells of the skin.341–344 The identification of this ubiquitous novel human polyomavirus in the skin led other groups to screen for other human polyomaviruses in the skin using a technique for cloning small circular DNAs called “rolling circle amplification.” This screening led to the identification of many additional novel human polyomaviruses, most of which have not been linked to any specific disease.
In their analyses of MCC, Shuda and colleagues335 made another important observation, akin to that made by zur Hausen in the context of HPV’s role in cervical cancer; the MCPyV genome is found integrated into the host genome in the cancers. Importantly, these integrated copies of MCPyV carried mutations in the large tumor antigen gene that resulted in expression of truncated large tumor antigen gene products (these mutations did not alter the small tumor antigen gene, which is also expressed in MCC). The truncated large tumor antigen proteins expressed in MCC retain the domain within the N-terminal half of the full-length protein that mediates interaction with the cellular tumor suppressor, pRb, but they are missing part or all of the highly conserved adenosine triphosphatase/DNA helicase domain required for viral DNA replication. This finding led to the hypothesis that in MCC, there is selection for replication-defective MCPyV genomes that retain an ability to inactivate pRb. Consistent with this hypothesis, knockdown of expression of the MCPyV tumor antigens (both large and small tumor antigens) in MCC-derived cell lines led to cell growth arrest and death.345 Follow-up studies by Shuda and colleagues,346 however, led to the further discovery that specifically knocking down expression of MCPyV small tumor antigen, although not affecting expression of MCPyV large tumor antigen, led to growth arrest, although not death, of MCPyV-positive MCC cell lines, leading to the revised hypothesis that both small and large tumor antigens contribute to MCC. MCPyV small tumor antigen was found to transform cells at least in part by acting downstream of the mammalian target of rapamycin signaling pathway to preserve eukaryotic translation initiation factor 4E–binding protein 1 hyperphosphorylation, resulting in dysregulated cap-dependent translation.346 That continued expression of MCPyV large tumor antigen is specifically necessary for the survival of MCPyV-positive MCC cells and appears to correlate with large tumor antigen, causing an increase in levels of expression of survivin.347 A drug that inhibits survivin was shown to induce programmed cell death of MCPyV-positive MCC cells in tissue culture and prolong the survival of immunodeficient mice in which these tumor-forming MCC cells have been engrafted,347 providing a potential new therapeutic approach for treating patients with this deadly cancer.
Treatment and Prevention of Viral Tumors
Tumors associated causally with viruses are treated variously; surgical resections, where appropriate, are used in combination with chemotherapy and/or radiation therapy. These treatments are disappointing in that overall survival rates are often low and thus far have not been able to capitalize on any specific antiviral therapies. Current survival rates after different therapies obviously vary with the malignancy, its stage, and the setting in which it is treated. For example, youngsters with Burkitt lymphoma have a 4-year overall survival rate of 65%.348 Approximately 45% of patients with NPC remain disease-free after treatment for 10 years, but this value is highly dependent on the stage at which the NPC was diagnosed.172 In the United States, 26% of HBV-positive patients with HCC that has been surgically resected patients have a 5-year, local disease-free survival rate compared with 38% for the analogous HCV-positive group.349 A retrospective study of German patients with a mix of HBV- and HCV-associated HCC found a median survival rate of 7 years.350 The 5-year median survival rate for patients with cervical carcinoma ranges from 65%351 to 75% to 90%352 in different studies. The median survival rate for patients who have been diagnosed with ATLL is only 10 months.353 The overall survival rates of patients with KS who are HIV positive have changed with the introduction of HAARTs. Before this therapy was available, median overall survival was 13 months; with HAART, it surpasses 28 months.354 The median survival for patients with PEL has been described as “dismal,” but case reports indicate that some combination therapies are helpful.355 These actuarial findings indicate that the development of antiviral therapies directed at the viral gene products that maintain tumor phenotypes is a highly desirable goal. It also obviously is desirable to develop vaccines to prevent infections by tumor viruses. The development of effective vaccines that can prevent an initial infection or induce the elimination of viral persistence is being pursued for all human tumor viruses. Success has been achieved for two of them.
HBV Vaccine
Although four serotypes of HBV exist, highly effective vaccines for HBV have been developed that consist only of its surface antigen, HBsAg. Today this subunit vaccine is usually synthesized with a recombinant DNA expressed in yeast. Vaccines for HBV began to be used in the early 1980s, and since 2001, 129 countries throughout the world have routinely vaccinated infants and/or adolescents against HBV.356 These vaccinations are effective. The fraction of children infected with HBV has been reported to have declined between 7.5-fold and tenfold in Taiwan and the Gambia, respectively.79,80 This decline has been paralleled by a detectable drop in the frequency of HCC in children in Taiwan, an age group for which this viral tumor is rare.81
The current HBV vaccines apparently are free of unwanted adverse effects but can select for variants of HBV resistant to the neutralizing antibodies they elicit.356 One domain of the HBsAg in particular elicits neutralizing antibodies efficiently, and mutations in it can allow for viral escape. The frequency of these escape mutants in populations of vaccinated children has risen significantly.357 One means to overcome the selection for such escape mutants would be to generate vaccines for HBV that include more than the HBsAg as an immunogen; such vaccines are now being developed.
Human Papillomavirus Vaccine
The second family of human tumor viruses for which an effective prophylactic vaccine can now be envisioned are the human papillomaviruses associated with cervical cancer, other anogenital cancers, and certain head and neck cancers, in particular HPV-16 and HPV-18. These two viruses account for approximately 85% of cervical cancers worldwide, and in a recent study, 16% of new infections.358 Two highly effective vaccines that prevent infections by HPV-16 and HPV-18 have been approved for use in the United States and other countries. These vaccines are based on the production of virus-like particles (VLPs) composed of the major capsid protein of HPV-16 and HPV-18, L1, which self-assembles into icosahedrons (see Fig. 11-3). These L1-based VLPs induce neutralizing antibodies in vaccinated individuals. Preclinical studies with CRPV and canine oral papillomavirus demonstrated the effectiveness of VLP-based vaccines in protecting animals from CRPV and canine oral papillomavirus infection.359,360 Phases I and II clinical trials likewise demonstrated the effectiveness of HPV-16 VLP-based vaccines in inducing neutralizing antibodies specific for HVP-16 and in preventing HPV-16 infections.361,362 One issue of relevance to the effectiveness of these vaccines is the specificity of immune responses invoked by VLP-based vaccines for specific HPV genotypes. Approximately a dozen HPV genotypes are associated with anogenital cancers. VLP-based vaccines induce protective immunity that is highly specific for the genotype from which the VLP is generated. This specificity has led to the development of polyvalent vaccines composed of a mixture of VLPs generated with L1 proteins from multiple mucosotropic HPV genotypes. One of the available vaccines, Gardasil, is a multivalent vaccine composed of HPV-16, HPV-18, HPV-6, and HPV-11 VLPs. HPV-6 and HPV-11 are low-risk mucosotropic HPVs that induce genital warts but do not contribute to cervical cancer. The other available vaccine, Cervarix is composed of just HPV-16 and HPV-18 VLPs. It remains to be seen whether, once a population is protected from HPV-16– and HPV-18–induced cancers, other HPV genotypes arise to induce a greater incidence of cervical cancers than they currently cause. For this reason, the development of vaccines that provide protection against a wider variety of high-risk HPVs is now being considered. One approach is to add L1 VLPs specific for other genotypes to the current vaccines. Alternatively, incorporation of the minor capsid protein L2 may be of benefit, because neutralizing antibodies induced against L2 tend to be more generally effective at neutralizing multiple genotypes.363 In this regard, a stand-alone L2 vaccine might be effective. Concern exists about whether the HPVs, particularly HPV-16 and HPV-18, will evolve to become resistant to VLP-based vaccines. Assuming the high effectiveness of the vaccines currently under development and their worldwide distribution, and given the long latency of cervical cancer, the WHO estimates that prophylactic HPV vaccines will lead to a reduction of deaths due to cervical cancer no earlier than 2040. Given that HPVs also cause other anogenital cancers, as well as head and neck cancers that afflict both men and women, the available vaccines are now being considered for use both in vaccinating young men and women in the United States and elsewhere in the world.