Myeloproliferative Neoplasms
• Each of the three classic BCR-ABL1− myeloproliferative neoplasms (MPNs)—that is, polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF)—are estimated to occur at the rate of 0.5 to 2.5 per 100,000 people per year.
• All three classic BCR-ABL1− MPNs may be considered as diagnoses of exclusion, because a specific diagnostic marker is currently lacking.
• The presence of a JAK2 mutation distinguishes PV from all other causes of polycythemia, and ET and PMF from reactive thrombocytosis or myelofibrosis, respectively.
• Evaluation of suspected PV should start with peripheral blood screening for JAK2V617F and serum erythropoietin (Epo) measurement.
• More than 90% of PV patients are expected to display the JAK2 mutation, whereas a low serum Epo level should capture most of the JAK2V617F− cases, which might display other JAK2 mutations such as an exon 12 mutation.
• Bone marrow examination remains essential in the diagnosis of ET and PMF.
• Both PV and ET display a near-normal life expectancy in the first decade of the disease. The major problem during this period is thrombosis that may occur in as many as 30% of the patients.
• A history of thrombosis or age older than 60 years is associated with a high-risk of thrombosis.
• Platelet count by itself has not been significantly associated with thrombosis in either PV or ET.
• The presence of advanced age, anemia, red cell transfusion-dependency, thrombocytopenia, leukocytosis, severe constitutional symptoms, circulating blasts or unfavorable karyotype are all adverse risk factors in PMF.
• Phlebotomy, to a hematocrit target of below 45%, remains the mainstay of therapy in PV.
• Low-risk patients (age younger than 60 years and no history of thrombosis) with either PV or ET have not been shown to benefit from cytoreductive therapy.
• Treatment with hydroxyurea has been shown to reduce thrombosis risk in high-risk patients with both ET and PV.
• Microvascular symptoms, including headache and erythromelalgia (painful and burning sensation of the feet or hands associated with erythema and warmth), are easily treated with low-dose aspirin, which is indicated in the absence of extreme thrombocytosis.
• Drug therapy in PMF is currently palliative, and effective agents include corticosteroids, erythropoiesis-stimulating agents (ESAs), androgen preparations, danazol, thalidomide, lenalidomide, hydroxyurea and ruxolitinib.
• Splenectomy continues to have a palliative role in PMF.
• Involved-field radiation therapy in PMF is most effective in the setting of nonhepatosplenic extramedullary hematopoiesis.
• Hematopoietic stem cell transplantation has a therapeutic role in high-risk PMF but is associated with substantial regiment-related toxicity including death and chronic morbidity.
Introduction
The World Health Organization (WHO) classification system for hematopoietic tumors recognizes five categories of myeloid malignancies including acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPNs), MDS/MPN overlap, and PDGFR/FGFR1-rearranged myeloid/lymphoid neoplasm with eosinophilia.1 “BCR-ABL1-negative MPN” is an operational subcategory of MPN that includes polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF).2 In addition, the WHO MPN category includes chronic myeloid leukemia (CML), chronic neutrophilic leukemia, chronic eosinophilic leukemia-not otherwise specified, mastocytosis, and MPN unclassifiable (MPN-U). The latter refers to a clinically and histologically MPN-like phenotype that does not fulfill the standard diagnostic criteria for the other seven MPN variants.
The WHO MDS/MPN category includes chronic myelomonocytic leukemia (CMML), juvenile myelomonocytic leukemia, atypical CML-BCR-ABL1-negative, and “MDS/MPN, unclassifiable (MDS/MPN-U).”2 Atypical CML is characterized by the absence of BCR-ABL1 and the presence of left-shifted granulocytosis with granulocytic dysplasia. MDS/MPN-U refers to a clinical phenotype that displays histologic characteristics of both MDS and MPN without fulfilling the diagnostic criteria for CMML, juvenile myelomonocytic leukemia, or atypical CML-BCR-ABL1-negative.The WHO provisional entity of “refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T)” is included in the category of MDS/MPN-U.
Table 100-1 represents an alternative semimolecular classification scheme for chronic myeloid neoplasms that uses the term “classic MPNs” to refer to the original four “MPD” variants including CML, PV, ET, and PMF.3 In this operational classification system, all remaining MPNs are assigned the term “nonclassic MPNs.”4 This chapter focuses on the classic BCR-ABL1− MPNs: PV, ET, and PMF.
Table 100-1
Modern Classification of Myeloproliferative Neoplasms
| Main Categories | Subcategories | Molecular Signatures |
| Classic myeloproliferative disorders | Chronic myeloid leukemia | 100% BCR-ABL+ |
| Polycythemia vera | ~95% JAK2V617F+ | |
| ~4% JAK2 exon 12 mutations+ | ||
| Essential thrombocythemia | ~50% JAK2V617F+ | |
| ~1% MPLW515L/K+ | ||
| Primary myelofibrosis | ~50% JAK2V617F+ | |
| ~5% MPLW515L/K+ | ||
| Nonclassic myeloproliferative neoplasms | ||
| Chronic neutrophilic leukemia | ~20% JAK2V617F+ | |
| Chronic eosinophilic leukemia, not otherwise specified Mastocytosis Myeloproliferative neoplasms-unclassifiable |
Polycythemia Vera
Vaquez and Osler are credited for the initial description of PV, as a primary erythrocythemic process, in 1892 and 1903, respectively.5,6 Reported incidence figures in PV range from approximately 0.5 to 2.6 per 100,000 population.7,8 A higher disease incidence has been suggested in persons of Jewish ancestry,9 as well as among parent-offspring pairs.10 Median age at diagnosis of PV is approximately 60 years with a slight (1.2 : 1) male preponderance.11 Approximately 7% of patients are diagnosed before age 40 years,11 and there are rare cases of children affected with PV.12
Pathogenesis
PV is a clonal stem cell disease with trilineage myeloid involvement.13 In addition, some studies suggest clonal heterogeneity, including clonal involvement of B lymphocytes,14 as well as polyclonal granulopoiesis in certain cases.15 In vitro, erythroid colony formation in patients with PV does not require the addition of exogenous erythropoietin (Epo).16 This phenomenon is called endogenous erythroid colony growth and does not occur in either normal controls or in patients with nonclonal polycythemia. In addition, erythroid progenitor cells in PV display growth factor hypersensitivity to Epo, insulin-like growth factor (IGF)–1, and other cytokines.17,18 The consistently observed IGF1 hypersensitivity of erythroid cells in PV has been attributed to alterations in IGF1 binding proteins.19 However, several studies show that growth factor–independent or growth factor–hypersensitive colony formation is specific to neither PV nor erythroid progenitor cells.18
The Epo receptor gene as well as its protein are intact in PV.20 On the other hand, several postreceptor molecular abnormalities have been reported and include increased baseline phosphorylation of the IGF1 receptor,21 decreased activity of SH-PTP1 (a tyrosine phosphatase),22 increased activity of membrane-associated SH-PTP,23 constitutive activation of STAT3,24 upregulation of negative control elements of the cell cycle (p16/p14),25 and abundance, in erythroid precursors, of antiapoptotic proteins (Bcl-xL).26 However, these observations have not been always reproducible, and none of them have been shown to be specific to PV, as opposed to other MPNs.
In 2005, a novel Janus kinase 2 (JAK2) mutation (JAK2V617F) was described in association with PV, ET, and PMF.27 JAK2V617F occurs in approximately 95% of PV patients but also in approximately 50% of those with ET or PMF.28–31 In addition, the mutation is found in a small proportion of patients with other myeloid neoplasms including nonclassic MPNs and MDS.32,33 Other JAK2 mutations (i.e., JAK2 exon 12 mutations) were subsequently described in the majority of patients with JAK2V617F− PV or “idiopathic erythrocytosis,”34 thus raising the possibility that a JAK2 mutation is essential for the PV phenotype. On the other hand, neither JAK2V617F nor JAK2 exon 12 mutations have so far been reported in lymphoid disorders,35–38 solid tumor,41–41 or secondary myeloproliferation including congenital or acquired polycythemia.44–44
JAK2V617F is a G-to-T somatic mutation, at nucleotide 1849, in exon 14, resulting in the substitution of valine by phenylalanine at codon 617.28–31 JAK2 exon 12 mutations (F537-K539delinsL, H538QK539L, K539L, N542–E543del) include both in-frame deletions and tandem point mutations.34 Both exon 14 and exon 12 JAK2 mutations induce cytokine-independent/hypersensitive proliferation in Epo receptor–expressing cell lines and a PV-like phenotype in mice.34 Whereas homozygosity for JAK2V617F, which results from mitotic recombination, can be demonstrated in the majority of patients with PV,45 exon 12 JAK2 mutations are often heterozygous.34 The precise pathogenetic role of JAK2 mutations in PV and related MPNs continues to be investigated.
Diagnosis
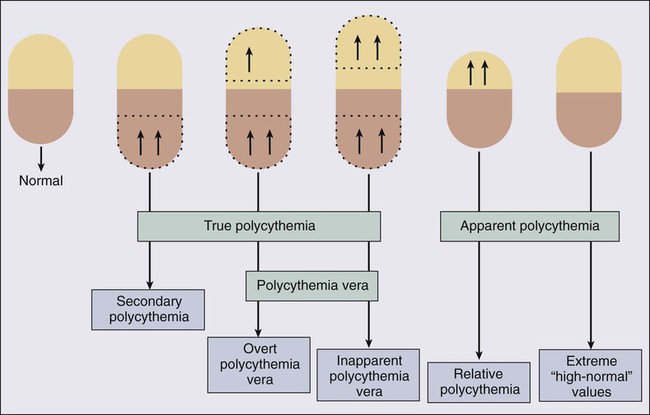
In clinical practice, the term polycythemia is used to indicate the possible occurrence of an increased erythrocyte volume or red blood cell mass (RCM). Such a perception might be either real (true polycythemia) or spurious (apparent polycythemia; Fig. 100-1).18 True polycythemia may represent either PV or a nonclonal increase in RCM that is often, but not always, mediated by Epo (secondary polycythemia; Fig. 100-2). Apparent polycythemia may result from either a reduction in plasma volume (relative polycythemia) or an inaccurate perception of an elevated RCM that results from not appreciating high normal values of hemoglobin/hematocrit.46 Inapparent polycythemia is the converse of apparent polycythemia and indicates a true increase in RCM that is masked by a normal hemoglobin/hematocrit value secondary to a concomitant increase in plasma volume (see Fig. 100-1).47
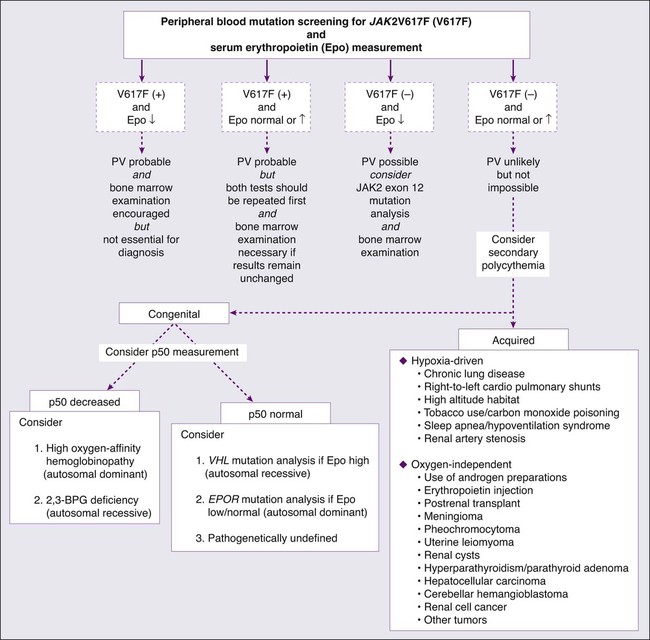
In 1975, the PV study group published a set of “diagnostic criteria” that were primarily used to ensure the exclusion, to treatment protocols, of patients with secondary or apparent polycythemia.48 These criteria required the demonstration of increased RCM by blood volume measurement using labeled erythrocytes as well as the demonstration of normal hemoglobin oxygen saturation. In 2001, the WHO published a refined diagnostic criteria for PV and related MPNs that recognized the value of both bone marrow histology and “MPN-specific” biological parameters.49 The recent discovery of the almost invariable association between PV and a JAK2 mutation has led to revised WHO diagnostic criteria for PV (Table 100-2)50 and a refined diagnostic algorithm (see Fig. 100-2),51 both of which incorporate JAK2 mutation screening.
Table 100-2
Revised World Health Organization Criteria for Polycythemia Vera
Diagnosis requires the presence of both major criteria and one minor criterion or the presence of the first major criterion together with two minor criteria.
MAJOR CRITERIA
Hemoglobin >18.5 g/dL in men, 16.5 g/dL in women, or other evidence of increased red cell volume*
Presence of JAK2V617F or other functionally similar mutation such as JAK2 exon 12 mutation
MINOR CRITERIA
Bone marrow biopsy showing hypercellularity for age with trilineage growth (panmyelosis) with prominent erythroid, granulocytic, and megakaryocytic proliferation
Serum erythropoietin level below the reference range for normal
Endogenous erythroid colony formation in vitro
*Hemoglobin or hematocrit >99th percentile of method-specific reference range for age, sex, and altitude of residence, or hemoglobin >17 g/dL in men, 15 g/dL in women if associated with a documented and sustained increase of at least 2 g/dL from an individual’s baseline value that cannot be attributed to correction of iron deficiency, or elevated red cell mass >25% above mean normal predicted value.
Data from Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocytopenia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood 2007;110:1092–1097.
Because more than 95% of patients with PV carry the JAK2V617F mutation, which is absent in both secondary and apparent polycythemia, it is most effective to initiate the workup of a patient with suspected PV with peripheral blood mutation screening for JAK2V617F (see Fig. 100-2). Furthermore, to minimize the consequences of false-positive or false-negative molecular test results, as well as capture the few cases of PV that are JAK2V617F−, concomitant measurement of serum Epo level is recommended.52 If the results of both tests are suggestive of PV (i.e., mutation-positive and low serum Epo), then the diagnosis is likely and bone marrow examination is encouraged but not essential for making the diagnosis. If the JAK2V617F and serum Epo test results are both not consistent with the diagnosis of PV (i.e., mutation-negative and either normal or increased Epo), then further investigation for PV is not advised unless dictated otherwise by the clinical scenario.
If there is discrepancy between the molecular test result and serum Epo level, one should first repeat both tests and then proceed with bone marrow examination if the results are unchanged (see Fig. 100-2). In a patient with acquired erythrocytosis and a subnormal serum Epo level, the possibility of exon 12 JAK2 mutations should be entertained. In this regard it should be noted that some cases with JAK2 exon 12 mutation–associated PV might not display overt PV-characteristic features, even at the level of bone marrow histology.34
When congenital polycythemia is suspected, initial laboratory testing should include measurement of the oxygen tension at which hemoglobin is 50% saturated (i.e., p50).51 Left-shifted oxygen dissociation curve, suggested by decreased p50, suggests the presence of either high oxygen-affinity hemoglobinopathy (autosomal dominant)53 or 2,3-bisphosphoglycerate (2,3-BPG) deficiency, usually a consequence of BPG mutase mutation (autosomal recessive).54 If the p50 is normal, then the possibility of germline mutations of molecules that either enhance Epo effect (e.g., EPOR mutations)55 or govern intracellular oxygen sensing (e.g., mutations involving the von Hippel-Lindau [VHL] tumor suppressor or hypoxia-inducible factor 1a [HIF-1a] prolyl hydroxylase genes)56,57 should be considered (see Fig. 100-2). VHL mutations are usually associated with increased serum Epo level and constitute the most frequent mutations in congenital polycythemia (e.g., Chuvash polycythemia).58 In contrast, serum Epo level is often subnormal in patients with EPOR mutations.59
Bone marrow histology, to the experienced hematopathologist, is often revealing of characteristic changes of a MPN that include hypercellularity, increased number of megakaryocytes including cluster formation, the presence of giant megakaryocytes and pleomorphism in megakaryocyte morphology, mild reticulin fibrosis, and decreased bone marrow iron stores.60 In contrast, cytogenetic studies in PV disclose abnormalities (trisomies of chromosomes 9 and 8 and deletions of the long arms of chromosomes 13 and 20) in only 13% to 18% of patients at diagnosis, and hence have limited diagnostic value.61
Treatment
The natural history of PV is characterized by a lifelong propensity for thrombohemorrhagic complications, late-onset disease transformation into post-PV myelofibrosis and/or AML, and a shortened life expectancy.61 Specific treatment has been shown to positively influence the risk of both macrovascular and microvascular complications but not that of clonal evolution to post-PV myelofibrosis or AML.
Phlebotomy is the cornerstone of therapy in PV and is the only treatment modality that has improved survival in affected patients (Box 100-1). It is reported that median survival may be as low as 2 years in the absence of such treatment.63 Based on limited retrospective studies in PV that showed a progressive increase in the incidence of vascular occlusive episodes above a hematocrit level of 44%,64 as well as other studies that showed suboptimal cerebral blood flow in ranges of hematocrit values between 46% and 52%,65 the therapeutic target hematocrit level has, for a long time, been set at or below 45%. This particular practice is now further supported by the results of a recent randomized study where 365 adults with PV were treated with a target hematocrit, less than 45% or 45% to 50%.66 After a median follow-up of 31 months, the primary end point of thrombotic events or deaths from cardiovascular causes was recorded in 5 of 182 patients in the low-hematocrit group (2.7%) and 18 of 183 patients in the high-hematocrit group (9.8%) (P = 0.007). Furthermore, because of the physiological difference in hematocrit values between the two genders as well as among different races, it is reasonable, though not evidence-based, to target an even lower hematocrit level (i.e., 42%) in women and African-Americans.67
Role of Drug Therapy in Polycythemia Vera
Observations from Randomized Studies
In the first controlled study in PV, the PV study group randomized 431 patients to treatment with either phlebotomy alone or phlebotomy supplanted by either oral chlorambucil or intravenous radioactive phosphorus (32P). The results favored treatment with phlebotomy alone with a median survival of 12.6 years as compared with 10.9 and 9.1 years for treatment with 32P and chlorambucil, respectively (P = 0.008). The difference in survival was attributed to an increased incidence of AML in patients treated with chlorambucil or 32P compared with those treated with phlebotomy alone (13.2% vs. 9.6% vs. 1.5% over a period of 13 to 19 years).68 Furthermore, 3.5% of the patients treated with chlorambucil developed large cell lymphoma, and the incidence of gastrointestinal and skin cancer was increased in those patients treated with either chlorambucil or 32P.
In another controlled study, the European Organization for Research on Treatment of Cancer randomized 293 patients to treatment with either 32P or oral busulfan. The results favored busulfan in terms of both first-remission duration (median: 4 years vs. 2 years) and overall survival (10-year survival rates of 70% vs. 55%; P = 0.02). At a median follow-up period of 8 years, there was not a significant difference in the risk of leukemic transformation (2% vs. 1.4%), nonhematologic malignancy (2.8% vs. 5%), vascular complications (27% vs. 37%), or transformation into post-PV myelofibrosis (4.8% vs. 4.1%) between the busulfan and 32P arms, respectively.69
Other randomized studies in PV have compared hydroxyurea against pipobroman (a significant difference favoring pipobroman in the incidence of transformation into post-PV myelofibrosis but no difference in survival, incidence of thrombosis, or the rate of leukemic conversion),70 and 32P alone against 32P plus hydroxyurea (no difference in survival, incidence of thrombosis, or risk of transformation into post-PV myelofibrosis, but 32P alone was associated with significantly fewer incidences of both acute leukemia and other cancers).71 More importantly, in the final analyses from the aforementioned French study that compared hydroxyurea to pipobroman in 285 patients younger than age 65 years,72 after a median follow-up of 16.3 years, median survival was 20.3 years for the hydroxyurea arm, and 15.4 years for the pipobroman arm (P = 0.008). At 10, 15, and 20 years, cumulative incidence of AML/MDS was 6.6%, 16.5%, and 24% in the hydroxyurea arm, and 13%, 34%, and 52% in the pipobroman arm (P = 0.004). Cumulative myelofibrosis incidence at 10, 15, and 20 years according to main treatment received was 15%, 24%, and 32% with hydroxyurea versus 5%, 10%, and 21% with pipobroman (P = 0.02). These results suggest a leukemogenic potential for pipobroman.
Randomized studies involving aspirin therapy include one study where 32P plus phlebotomy was compared to phlebotomy plus high-dose aspirin (900 mg/day) in combination with dipyridamole (225 mg/day); the addition of antiplatelet agents provided no benefit in terms of thrombosis prevention but increased the risk of gastrointestinal bleeding.73 However, a more recent randomized study of PV (112 patients) using lower doses of aspirin (40 mg/day) did not show an increased bleeding diathesis.74 Furthermore, the results of the PV study group aspirin study may have been influenced by the fact that 27% of the patients randomized to the phlebotomy-aspirin-dipyridamole arm had a prior history of thrombosis compared with 13% in the other arm. This contention was confirmed by the most recent European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study that showed safety as well as antithrombotic activity of low-dose aspirin in a randomized study.75
Observations from Nonrandomized Studies
In a nonrandomized study by the PV study group, treatment with hydroxyurea was associated with a lower incidence of early thrombosis compared with a historical cohort treated with phlebotomy alone (6.6% vs. 14% at 2 years). Similarly, the incidence of AML in patients treated with hydroxyurea, compared with a historical control treated with either chlorambucil or 32P, was significantly lower (5.9% vs. 10.6% vs. 8.3%, respectively, in the first 11 years of treatment).76 Other studies have confirmed the low incidence of AML in PV patients treated with hydroxyurea (1% to 5.6%).79–79
Many studies have reported on the use of pipobroman as a single agent in PV.80,81 In one of these studies involving 163 patients, the drug was effective in more than 90% of the patients and median survival exceeded 17 years.80 In the first 10 years, the incidences of thrombotic events, acute leukemia, post-PV myelofibrosis, and other malignancies were 16%, 5%, 4%, and 8%, respectively. Favorable outcome has also been reported in single-arm studies using oral busulfan.82,83 In 65 busulfan-treated patients with PV followed between 1962 and 1983, overall median survival was 11.1 years and 19 years in patients whose disease was diagnosed before age 60 years.82 Only two patients (3.5%) treated with busulfan alone developed acute leukemia.
Most recently, alfa-interferon (IFN-α) was shown to control erythrocytosis in approximately 76% of the patients with PV receiving subcutaneous drug in doses ranging from 4.5 to 27 million units per week (usual dose is 3 million units subcutaneously 3 times a week).84,85 A similar degree of benefit is appreciated in terms of reduction in spleen size or relief from intractable pruritus. Furthermore, a variable degree of JAK2V617F allele burden reduction was recently associated with IFN-α therapy, but the relevance in overall outcome of the disease is not known.86 A randomized study (which is currently ongoing) is needed to determine if IFN-α is superior to hydroxyurea as a cytoreductive agent in PV, especially in view of its higher cost and toxicity profile. Finally, anagrelide, an oral imidazoquinazoline derivative that inhibits platelet aggregation at higher than therapeutic drug concentrations but displays a species-specific platelet-lowering effect in humans at therapeutic concentrations, controls thrombocytosis in PV.87 However, the drug is currently not recommended in the treatment of PV, because it was associated with increased incidence of arterial thrombosis and post-ET myelofibrosis, when compared with hydroxyurea, in patients with ET.88
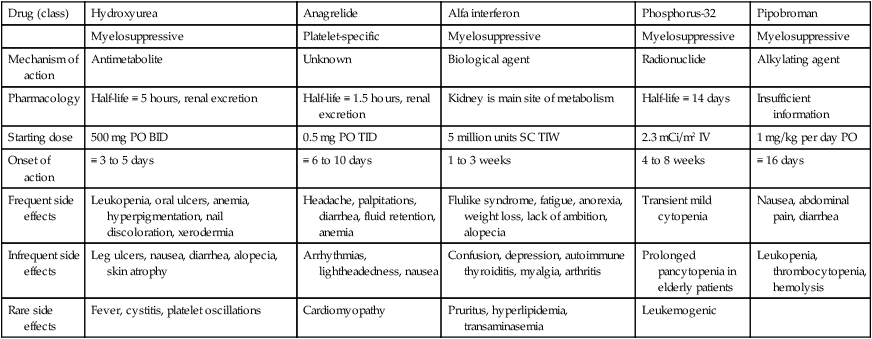
In summary, the results from single-arm studies support those of the randomized studies and show that busulfan is a valuable treatment agent in PV and might be considered as an alternative to hydroxyurea. Pipobroman is currently not available in the United States and a recent French study suggested a leukemogenic potential for the particular drug.72 It is currently not clear what additional value is gained by substituting the aforementioned traditional cytoreductive agents by the newer drugs (i.e., IFN-α and anagrelide), especially in view of their unfavorable toxicity and cost profile (Table 100-3), but randomized studies are currently ongoing, in this regard.89
Table 100-3
Clinical Properties of Popular Cytoreductive Agents Used in Polycythemia Vera or Essential Thrombocythemia
| Drug (class) | Hydroxyurea | Anagrelide | Alfa interferon | Phosphorus-32 | Pipobroman |
| Myelosuppressive | Platelet-specific | Myelosuppressive | Myelosuppressive | Myelosuppressive | |
| Mechanism of action | Antimetabolite | Unknown | Biological agent | Radionuclide | Alkylating agent |
| Pharmacology | Half-life ≡ 5 hours, renal excretion | Half-life ≡ 1.5 hours, renal excretion | Kidney is main site of metabolism | Half-life ≡ 14 days | Insufficient information |
| Starting dose | 500 mg PO BID | 0.5 mg PO TID | 5 million units SC TIW | 2.3 mCi/m2 IV | 1 mg/kg per day PO |
| Onset of action | ≡ 3 to 5 days | ≡ 6 to 10 days | 1 to 3 weeks | 4 to 8 weeks | ≡ 16 days |
| Frequent side effects | Leukopenia, oral ulcers, anemia, hyperpigmentation, nail discoloration, xerodermia | Headache, palpitations, diarrhea, fluid retention, anemia | Flulike syndrome, fatigue, anorexia, weight loss, lack of ambition, alopecia | Transient mild cytopenia | Nausea, abdominal pain, diarrhea |
| Infrequent side effects | Leg ulcers, nausea, diarrhea, alopecia, skin atrophy | Arrhythmias, lightheadedness, nausea | Confusion, depression, autoimmune thyroiditis, myalgia, arthritis | Prolonged pancytopenia in elderly patients | Leukopenia, thrombocytopenia, hemolysis |
| Rare side effects | Fever, cystitis, platelet oscillations | Cardiomyopathy | Pruritus, hyperlipidemia, transaminasemia | Leukemogenic |
Thrombohemorrhagic Risk Factors in Polycythemia Vera
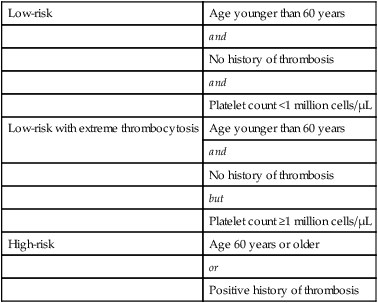
The series of studies performed by the PV study group revealed the following: (a) a significantly higher incidence of thrombotic events, in the first 3 years, in patients treated with phlebotomy alone; (b) a lack of correlation between thrombosis and either platelet count or hematocrit value; and (c) a significant association between thrombosis risk and either age older than 70 years or a prior history of thrombosis. In addition, in patients treated with phlebotomy alone, thrombosis risk was associated with an increased frequency of phlebotomy (maintenance phlebotomy of more than once every 3 months). Other studies have confirmed the detrimental effect of advanced age (>60 years) and thrombosis history in PV.18 As such, patients who are 60 years old or older or have a history of thrombosis are considered at high-risk (Table 100-4). In contrast, the degree of thrombocytosis has never been correlated with thrombosis risk. However, some patients with extreme thrombocytosis (platelet count at or above 1 million platelets/µL), carry an acquired bleeding diathesis that results from an abnormal adsorption and catabolism of large-molecular-weight von Willebrand factor.90 The prognostic influence of extreme thrombocytosis, in the absence of acquired von Willebrand disease, is unknown. Equally uncertain is the prognostic relevance of cardiovascular risk factors.
Table 100-4
Risk Stratification in Polycythemia Vera and Essential Thrombocythemia
| Low-risk | Age younger than 60 years |
| and | |
| No history of thrombosis | |
| and | |
| Platelet count <1 million cells/µL | |
| Low-risk with extreme thrombocytosis | Age younger than 60 years |
| and | |
| No history of thrombosis | |
| but | |
| Platelet count ≥1 million cells/µL | |
| High-risk | Age 60 years or older |
| or | |
| Positive history of thrombosis |
Current Treatment Recommendations
The mainstay of therapy in PV remains phlebotomy for all patients to keep hematocrit at or below 45% in male whites and the appropriate corresponding value for females and other races.66 Additional drug therapy depends on an individual patient’s risk for thrombohemorrhagic complications (see Table 100-4). In general, there is good evidence to advocate the use of cytoreductive agents in high-risk patients (Table 100-5). In this regard, and based on the results of the aforementioned studies, my current choice of chemotherapy is hydroxyurea (starting dose of 500 mg twice daily) or busulfan (starting dose of 4 mg/day) in case of nontolerance to hydroxyurea. Side effects of hydroxyurea that might necessitate the use of an alternative agent include neutropenia and mucocutaneous changes such as ulcers in the mouth and lower extremities. In using busulfan, one should recognize the potential, but infrequent, toxicity to the lungs (pulmonary fibrosis)91 and the bone marrow (aplasia).92 Intermittent treatment with drug holidays and withholding treatment for impending cytopenia are recommended.
Table 100-5
Treatment Algorithm in Polycythemia Vera
| Risk Category | Age <60 Years | Age ≥60 Years | Women of Childbearing Age |
| Low-risk | Phlebotomy + low-dose aspirin | Not applicable | Phlebotomy + low-dose aspirin |
| Low-risk with extreme thrombocytosis | Phlebotomy + low-dose aspirin* | Not applicable | Phlebotomy + low-dose aspirin* |
| High-risk | Phlebotomy + hydroxyurea + | Phlebotomy + hydroxyurea + | Phlebotomy + IFN-α*† |
| low-dose aspirin | low-dose aspirin* | + low-dose aspirin |
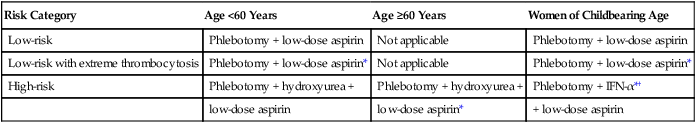
*Screening for acquired von Willebrand disease is encouraged before the use of aspirin in patients with extreme thrombocytosis (platelet count ≥1 million cells/µL). Use of aspirin is discouraged if ristocetin cofactor activity is <50%.
In the younger but high-risk patient group, some investigators are concerned about drug leukemogenicity associated with long-term treatment with either hydroxyurea or busulfan. However, there is currently not hard evidence to support this particular concern.79 Regardless, IFN-α (starting dose of 3 million units subcutaneously 3 times a week) or pegylated IFN is a reasonable alternative in this instance.84 IFN-α is also the treatment of choice in women of childbearing age (see Table 100-5) because of the theoretical risk of teratogenicity associated with the use of other cytoreductive agents.85
It is currently unclear whether any specific drug therapy influences clonal evolution in PV. Under current treatment strategies, the incidences of transformation into post-PV myelofibrosis or AML, in the first decade of disease, are estimated at 10% and 5%, respectively.68 The risk beyond the first decade increases progressively.93
Treatment of Non–Life-Threatening Complications in Polycythemia Vera
Generalized pruritus that is often exacerbated by hot bath is a characteristic feature of PV and occurs in 48% of patients either at diagnosis or at a later stage of the disease.94 Etiology of PV-associated pruritus remains to be determined and treatment responses to antihistamines have been both unpredictable and variable.94 Interestingly, a recent study demonstrated a greater than 80% response rate in PV-associated pruritus treated with paroxetine, which is a selective serotonin reuptake inhibitor.95 Other treatment modalities that have been used in PV-associated pruritus include IFN-α, psoralen photochemotherapy, cholestyramine and ruxolitinib.5,18
Essential Thrombocythemia
Among the classic BCR-ABL1− MPNs, ET is the most recently described.96 Reported incidence figures range from 0.2 to 2.5 per 100,000 population.9,97–99 With a median age at diagnosis of 60 years, approximately 20% of the patients with ET are diagnosed before age 40 years and in the young age group of patients, the incidence is higher in women than in men.100,101 There are well-documented cases of ET in children, although some of the reported cases may have represented familial thrombocytosis.102,103
Pathogenesis
Trilineage clonal myeloproliferation has been demonstrated in the majority of patients with ET using X chromosome–linked DNA or gene product analysis.104,105 However, X-linked clonal assays have revealed both polyclonal hematopoiesis in a substantial minority of patients with ET106 and “monoclonal” hematopoiesis in normal elderly controls.107 Furthermore, in some cases, the clonal process in ET was shown to include lymphocytes14 or be restricted to megakaryocytes.105 The clonal basis of ET was recently underlined by JAK2V617F mutation analysis that revealed the presence of the mutation even in those patients who feature “polyclonal” hematopoiesis by X-linked clonality studies.106,108 However, the primary clonogenic event in ET remains undefined despite the recent descriptions of two gain-of-function mutations that occur in 50% (JAK2V617F)109 and 1% (MPLW515L/K)110 of patients with ET, respectively.
Other biological features in ET include in vitro growth factor independence/hypersensitivity of both erythroid and megakaryocyte progenitor cells,111,112 low serum Epo level,113 altered megakaryocyte/platelet Mpl expression,114,115 increased neutrophil PRV-1 expression,116,117 and decreased platelet serotonin content.118 Laboratory studies in ET have demonstrated myeloid growth factor hypersensitivity to interleukin-3119 as well as thrombopoietin (Tpo).112 Growth factor independence of myeloid progenitor cells in ET and related myeloproliferative disorders has now been attributed to mutations involving molecules of the JAK-STAT pathway including the aforementioned JAK2V617F and MPLW515L/K. In patients with ET,120 PV,121 and myelofibrosis,122 serum Tpo levels are usually normal or elevated despite an increased megakaryocyte mass. This has been attributed to the markedly decreased megakaryocyte/platelet expression of myeloproliferative leukemia (MPL), although the extent of this particular phenotypic abnormality is significantly less in ET as compared with PV or PMF.114,115,123,124
In addition to clonal myeloproliferation, ET is also characterized by microvascular symptoms (e.g., headaches and erythromelalgia) as well as an increased risk of both thrombosis and bleeding.125 Pathogenesis in the former might involve abnormal thromboxane A2 generation and small vessel–based platelet-endothelial interactions that are inhibited by aspirin therapy.126,127 On the other hand, there is increasing information that implicates granulocytes but not platelets as the thrombogenic culprit in ET.88,128–130 Consistent with these observations, a recent study has identified leukocytosis as an independent risk factor for thrombosis in ET.131 Bleeding diathesis in ET is currently believed to involve an acquired von Willebrand syndrome that becomes apparent in the presence of extreme thrombocytosis.90 The mechanism of acquired von Willebrand syndrome in ET is currently believed to involve a platelet count-dependent increased proteolysis of high-molecular-weight von Willebrand protein.90 Other qualitative platelet defects in ET are believed to play a minor role in disease-associated hemorrhage and include defects in epinephrine-, collagen-, and adenosine diphosphate–induced platelet aggregation, decreased adenosine triphosphate secretion, and acquired storage pool deficiency that results from abnormal in vivo platelet activation.125
Diagnosis
Although thrombocytosis is the hallmark of ET, more than 85% of cases with thrombocytosis seen in routine clinical practice are reactive (secondary thrombocytosis) and associated with other comorbid conditions (Table 100-6).132 The degree of thrombocytosis is a poor discriminator of ET from secondary thrombocytosis. However, the clinical scenario is often helpful in distinguishing ET from secondary thrombocytosis. In routine clinical practice, it is important to exclude the contribution of either iron-deficiency anemia or a hyposplenic state as possible causes of an otherwise unexplained thrombocytosis (Box 100-2). This is accomplished by the measurement of serum ferritin concentration and examination of a peripheral blood smear looking for Howell-Jolly bodies, respectively. The possibility of secondary thrombocytosis associated with an occult inflammatory or malignant process is addressed by the measurement of C-reactive protein or other acute-phase reactants.133 In other words, an uncomplicated ET should be accompanied by a normal serum ferritin, mostly unremarkable peripheral smear, and a normal serum C-reactive protein. In general, plasma Tpo levels are not helpful in distinguishing secondary thrombocytosis from ET.120
Table 100-6
SECONDARY CAUSES
ACUTE
Postsurgery
Bleeding
Hemolysis
Infections
Tissue damage
Chemotherapy rebound effect
Coronary artery bypass surgery
PRIMARY CAUSES
Essential thrombocythemia
Polycythemia vera
Primary myelofibrosis
Chronic myeloid leukemia
Myelodysplastic syndrome
CHRONIC
Iron-deficiency anemia
Surgical or functional asplenia
Metastatic cancer or lymphoma
Chronic inflammatory process
Renal failure, nephrotic syndrome
The typical bone marrow finding in ET consists of a mild to moderate increase in cellularity and the presence of megakaryocyte clusters that are often absent in secondary thrombocytosis (Fig. 100-3). It should be remembered that CML and the cellular phase of PMF could both mimic ET in presentation.134 With respect to CML, a peripheral blood or bone marrow fluorescence in situ hybridization study may be needed, in addition to cytogenetic analysis, so as to exclude the possibility of karyotypically occult CML.135 Similarly, bone marrow histology should be carefully scrutinized for the presence of intense marrow cellularity with florid atypical megakaryocytic hyperplasia suggesting cellular phase PMF. Cytogenetic abnormalities are rare in ET (<5%) and diagnostically not helpful.136 Taking all the preceding information into consideration, bone marrow examination remains central to the diagnosis of ET, and its importance is underlined in the newly revised WHO criteria for the diagnosis of ET (Table 100-7).
Table 100-7
Revised World Health Organization Criteria for Essential Thrombocythemia
DIAGNOSIS REQUIRES MEETING ALL FOUR CRITERIA
Sustained platelet count ≥450 ×109 platelets/L*
Bone marrow biopsy specimen showing proliferation mainly of the megakaryocytic lineage with increased numbers of enlarged, mature megakaryocytes. No significant increase or left-shift of neutrophil granulopoiesis or erythropoiesis is seen.
Not meeting WHO criteria for polycythemia vera,† primary myelofibrosis,‡ chronic myelogenous leukemia,§ myelodysplastic syndrome, or other myeloid neoplasm
or other myeloid neoplasm
Demonstration of JAK2V617F or other clonal marker, or, in the absence of a clonal marker, no evidence for reactive thrombocytosis¶
†Requires the failure of iron-replacement therapy to increase hemoglobin level to the polycythemia vera range in the presence of decreased serum ferritin. Exclusion of polycythemia vera is based on hemoglobin and hematocrit levels and red cell mass measurement is not required.
‡Requires the absence of relevant reticulin fibrosis, collagen fibrosis, peripheral blood leukoerythroblastosis, or markedly hypercellular marrow for age accompanied by megakaryocyte morphology that is typical for primary myelofibrosis—small to large with an aberrant nuclear-to-cytoplasmic ratio and hyperchromatic, bulbous, or irregularly folded nuclei and dense clustering.
§Requires the absence of BCR-ABL.
 Requires absence of dyserythropoiesis and dysgranulopoiesis.
Requires absence of dyserythropoiesis and dysgranulopoiesis.
¶Causes of reactive thrombocytosis include iron deficiency, splenectomy, surgery, infection, inflammation, connective tissue disease, metastatic cancer, and lymphoproliferative disorders. However, the presence of a condition associated with reactive thrombocytosis does not exclude the possibility of essential thrombocythemia if the first three criteria are met.
Data from Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocytopenia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood 2007;110:1092–1097.
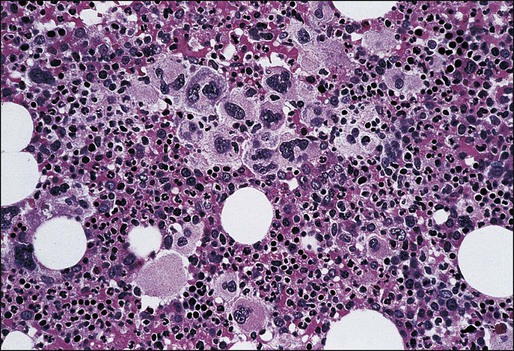
Treatment
The majority of patients with ET either are asymptomatic or suffer from non–life-threatening microvascular disturbances (headache, visual symptoms, lightheadedness, atypical chest pain, acral dysesthesia, erythromelalgia) that are effectively treated with low-dose aspirin.137
Approximately 20% of patients experience, usually nonfatal, thrombohemorrhagic complications, and less than 5% progress into AML or post-ET myelofibrosis during the first decade of the disease.131,138–141 Therefore it is reasonable to expect long survival in the majority of patients with ET.
In an international collaborative study of 1104 patients with clinically suspected ET,142 diagnosis was confirmed as true WHO-defined ET in 891 patients (81%) and was revised to early/prefibrotic PMF in 180 (16%). In early/prefibrotic PMF compared with ET, the 10-year survival rates (76% and 89%, respectively) and 15-year survival rates (59% and 80%, respectively), leukemic transformation rates at 10 years (5.8% and 0.7%, respectively) and 15 years (11.7% and 2.1%, respectively), and rates of progression to overt myelofibrosis at 10 years (12.3% and 0.8%, respectively) and 15 years (16.9% and 9.3%) were significantly worse. Multivariable analysis confirmed these findings and also identified age older than 60 years leukocyte count greater than 11 × 109/L, anemia, and thrombosis history as additional risk factors for survival. Furthermore, survival in ET was similar to the sex- and age-standardized European population. In analysis of the 891 patients with ET, after a median follow-up of 6.2 years, 109 (12%) patients experienced arterial (n = 79) or venous (n = 37) thrombosis. In multivariable analysis, predictors of arterial thrombosis included age more than 60 years, thrombosis history, cardiovascular risk factors including tobacco use, hypertension, or diabetes mellitus, leukocytosis (>11 × 109/L), and presence of JAK2V617F.143 In contrast, only male gender predicted venous thrombosis. Interestingly, a platelet count greater than 1000 × 109/L was associated with a lower risk of arterial thrombosis.
The relatively low incidence figures of thrombosis and hemorrhage as well as the occurrence of both short-term and long-term drug side effects are the basis for carefully selecting the patients with ET who require specific treatment. Risk stratification in ET is similar to that of PV (see Table 100-4). Table 100-8 outlines risk-adjusted treatment guideline in ET that is further elaborated in the following sections.
Table 100-8
Treatment Algorithm in Essential Thrombocythemia
| Risk Category | Age <60 Years | Age ≥60 Years | Women of Childbearing Age |
| Low-risk | Low-dose aspirin* | Not applicable | Low-dose aspirin* |
| Low-risk with extreme thrombocytosis | Low-dose aspirin† | Not applicable | Low-dose aspirin† |
| High-risk | Hydroxyurea + low-dose aspirin | Hydroxyurea + low-dose aspirin | IFN-α‡ + low-dose aspirin |
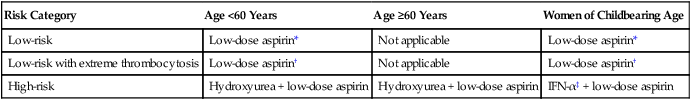
†One must rule out the presence of clinically relevant (ristocetin cofactor activity >50%) acquired von Willebrand disease before using aspirin.
Antiplatelet Therapy in Essential Thrombocythemia
Unlike the case with higher doses (≥500 mg/day), low-dose aspirin (81 to 325 mg/day) may not increase the bleeding diathesis of patients with ET and is currently recommended as a supplement to cytoreductive therapy in high-risk patients as well as an optional consideration in most patients with ET (see Table 100-8).144 However, it is important to exclude the possibility of clinically significant (ristocetin cofactor activity of <30%) acquired von Willebrand disease before using aspirin in those patients with platelet counts above 1 million cells/µL.
Cytoreductive Therapy in Essential Thrombocythemia
The use of cytoreductive therapy to reduce the risk of thrombosis in ET is appropriate and evidence-based as long as it is applied to those patients with an increased risk for thrombosis (see Table 100-4).130 In this instance it may be necessary to reduce the platelet count to 400,000 cells/µL or lower.145 A less-aggressive platelet control, with a cytoreductive drug, may also be indicated in patients who manifest either aspirin-resistant microvascular disturbances or symptomatic acquired von Willebrand disease.90,146 A recent report suggested that twice-a-day aspirin may work better than a once-daily dose in such apparently aspirin-resistant cases.147 In all other instances the decision to use a platelet-lowering agent in ET is not supported by hard data. Specifically, neither the degree of thrombocytosis nor the presence of cardiovascular risk factors has consistently been correlated with an increased thrombotic risk in ET.137 The frequently cited association of extreme thrombocytosis and increased gastrointestinal bleeding is based on anecdotal observation and may in some instances be attributed to occult acquired von Willebrand disease. A retrospective study of young, low-risk ET patients with extreme thrombocytosis revealed no difference in the risk of either thrombosis or bleeding between patients receiving or not receiving cytoreductive therapy.148
Table 100-3 summarizes the practical pharmacologic and clinical information regarding currently used platelet count–lowering agents in ET. It is important to note that only hydroxyurea, as a treatment agent, has been shown in a prospective study to be associated with a reduced risk of thrombosis in ET.130 Hydroxyurea was also found to be superior to anagrelide in a head-to-head comparative study of high-risk patients with ET88; patients treated with anagrelide displayed statistically higher incidence of arterial thrombosis and transformation into post-ET myelofibrosis. Finally, the concern regarding hydroxyurea leukemogenicity in ET is currently unsubstantiated,141 and the indiscriminate use of new drugs that are not tested in a controlled setting is unwarranted.
Pregnancy and Essential Thrombocythemia
First-trimester spontaneous abortion rate in ET (37%) is significantly higher than the 15% rate expected in the control population and does not appear to be influenced by specific treatment.149 Late obstetric complications, as well as maternal thrombohemorrhagic events, are relatively infrequent. Neither the platelet count nor treatment with aspirin seems to affect either maternal morbidity or pregnancy outcome. In fact, several studies have shown a spontaneous lowering of platelet counts during pregnancy in ET. Therefore cytoreductive treatment is currently not recommended for low-risk women with ET that are either pregnant or wish to be pregnant. In contrast, high-risk women require cytoreductive therapy to minimize the risk of recurrent thrombosis and anecdotal evidence of safety has encouraged a preference for the use of IFN-α in case of pregnancy in such patients.85
Primary Myelofibrosis
PMF was first described in 1879.150 The clinical course of PMF is characterized by progressive anemia, marked hepatosplenomegaly, cachexia, development of nonhepatosplenic extramedullary hematopoiesis, and evolution into acute leukemia.151 Similar to the situation in both PV and ET, the disease-initiating genetic lesion in PMF has not been identified, and current diagnosis is based on characteristic, but not specific, clinical and laboratory features that include bone marrow fibrosis (Table 100-9).151
Table 100-9
Revised World Health Organization Criteria for Primary Myelofibrosis
| Diagnosis requires meeting all three major criteria and two minor criteria. |
| MAJOR CRITERIA |
| Presence of megakaryocyte proliferation and atypia*, usually accompanied by either reticulin and/or collagen fibrosis, or, in the absence of significant reticulin fibrosis, the megakaryocyte changes must be accompanied by an increased bone marrow cellularity characterized by granulocytic proliferation and often decreased erythropoiesis (i.e., prefibrotic cellular-phase disease). |
| Not meeting WHO criteria for polycythemia vera†, chronic myelogenous leukemia‡, myelodysplastic syndrome§, or other myeloid neoplasm. |
Demonstration of JAK2V617F or other clonal marker (e.g., MPLW515L/K), or, in the absence of a clonal marker, no evidence of bone marrow fibrosis caused by underlying inflammatory or other neoplastic diseases . . |
| MINOR CRITERIA |

*Small to large megakaryocytes with an aberrant nuclear-to-cytoplasmic ratio and hyperchromatic, bulbous, or irregularly folded nuclei and dense clustering.
†Requires the failure of iron-replacement therapy to increase hemoglobin level to the polycythemia vera range in the presence of decreased serum ferritin. Exclusion of polycythemia vera is based on hemoglobin and hematocrit levels. Red cell mass measurement is not required.
‡Requires the absence of BCR-ABL.
§Requires absence of dyserythropoiesis and dysgranulopoiesis.

¶Degree of abnormality could be borderline or marked.
Data from Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocytopenia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood 2007;110:1092–1097.
Among the BCR-ABL1− classic MPNs, PMF is the least frequent, with an incidence figure that ranges from 0.3 to 1.5 per 100,000 population.98,99 The median age at diagnosis is 60 years, and approximately 10% of the patients are diagnosed at age 45 years or below.152 Children are not spared of PMF, but their clinical course may be less aggressive.153
Pathogenesis
In addition to clonal myeloproliferation, the bone marrow in PMF might display excess collagen fibrosis, new bone formation (osteosclerosis), and angiogenesis. These changes have been associated with alterations in both cellular and extracellular levels of various fibrogenic and angiogenic cytokines including transforming growth factor-β (TGF-β), basic fibroblast growth factor, and platelet-derived growth factor.154 The demonstration of polyclonal fibroblast proliferation in PMF is the basis for the current assumption that the bone marrow stromal aberration in PMF is reactive155 and mediated by the aforementioned cytokines derived from the resident clonal megakaryocytes and monocytes (Fig. 100-4).156
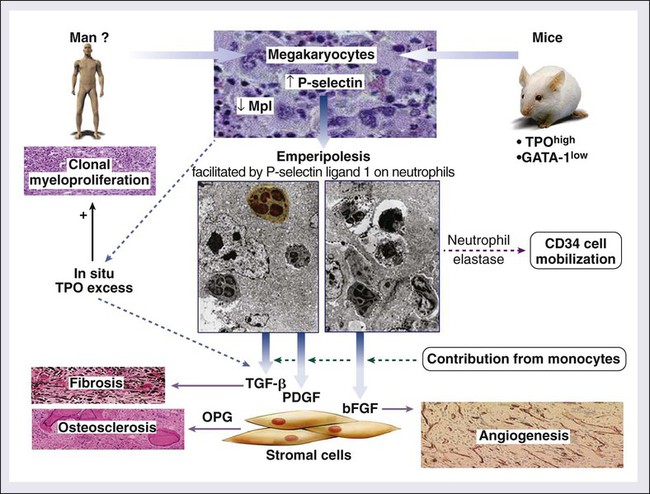
Current evidence strongly supports the stem cell origin of the clonal myeloproliferation in PMF.157,158 However, the primary clonogenic mutation(s) has not been identified, although much attention has been given to recently described novel gain-of-function mutations involving the JAK2 tyrosine kinase (JAK2V617F) and Tpo receptor (MPLW515L/K).31,159 JAK2V617F represents a G-to-T somatic mutation of JAK2, at nucleotide 1849, in exon 14, resulting in the substitution of valine to phenylalanine at codon 617.31 MPLW515L mutation represents a G-to-T transition at nucleotide 1544 resulting in a tryptophan-to-leucine substitution at codon 515 of the transmembrane region of the MPL receptor.159
JAK2V617F occurs not only in PMF (~50% mutational frequency) but also in ET with a similar mutational frequency and in PV where it is present in more than 90% of affected patients.28–31 In contrast, MPLW515L/K seems to be specific to PMF or ET, although mutational frequency is substantially lower (~5% and 1%, respectively).110 As mentioned before, JAK2V617F induces constitutive JAK-STAT activation, cytokine hypersensitivity, and/or independence in cell lines, and a PV-like disease in mice.31,160,161 JAK-STAT is similarly hyperactivated by MPLW515L/K, but this mutation induces a PMF-like disease in mice.159 In any case, about half of the patients with PMF do not display either mutation, and the precise pathogenetic role of these mutations, when they are present, remains to be clarified. Furthermore, since the description of JAK2V617F in 2005, many other mutations involving LNK, CBL, TET2, ASXL1, IDH, IKZF1, EZH2, DNMT3A, TP53, SF3B1, or SRSF2 have been described in PMF and other MPNs. However, none of them appears to garner the disease specificity or pathogenetic relevance otherwise displayed by BCR-ABL1. Nevertheless, the phenotype of clonal erythrocytosis might require the presence of a mutation in JAK2 (JAK2V617F or JAK2 exon 12 mutation)162 or its negative regulators such as LNK.163 Similarly, there appears to be a close association between SF3B1 mutations and presence of bone marrow ring sideroblasts in MPN.
An animal model of PMF has been established in mice that are either chronically overexposed to Tpo164 or carriers of a mutant GATA-1 gene that results in reduced expression of a transcription factor that plays a role in erythroid and megakaryocyte differentiation.165 These mice display characteristic features of PMF, including megakaryocytic hyperplasia, bone marrow fibrosis, osteosclerosis, and extramedullary hematopoiesis. The increased megakaryocyte accumulation in these experimental animals has been attributed to either a direct effect of Tpo or the lack of a negative feedback from mature elements as a result of impaired megakaryocyte differentiation, respectively. In both instances it is possible that the abnormal accumulation of megakaryocytes and their sequestered cytokines might be central to the pathogenesis of the associated stromal reaction. In this regard, the development of Tpo-induced bone marrow fibrosis in mice has been temporally associated with elevated TGF-β levels,166 whereas it was abrogated in TGF-β knockout experiments.167 Similar experiments may decipher the individual roles of other cytokines, including basic fibroblast growth factor and platelet-derived growth factor, that have been implicated in PMF.168 It is currently not clear whether the aforementioned animal models of PMF involve pathogenetic mechanisms that are operating in the human form of the disease.
Diagnosis
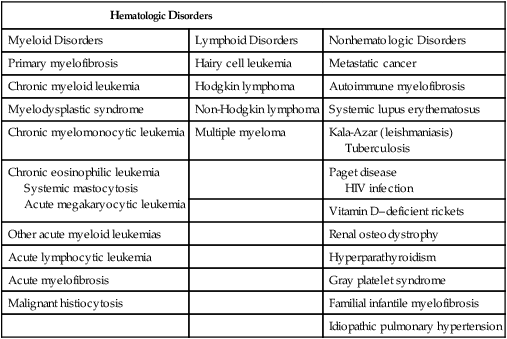
The typical presentation of PMF includes anemia (from ineffective erythropoiesis), marked splenomegaly (from extramedullary hematopoiesis), and a myelophthisic peripheral blood smear. Myelophthisis (the presence of nucleated red blood cells, granulocyte precursors, and teardrop-shaped erythrocytes) suggests a bone marrow infiltrative process, and the differential diagnosis includes bone marrow fibrosis, metastatic cancer, granulomatous infection, and lymphoma (Box 100-3). In PMF, peripheral blood myelophthisis is associated with bone marrow megakaryocytic hyperplasia, collagen fibrosis, osteosclerosis, and intramedullary sinusoidal hematopoiesis (Fig. 100-5). It should be noted, however, that bone marrow fibrosis may accompany not only PMF but also other hematologic and nonhematologic disorders, including systemic mast cell disease, eosinophilic disorders, and hairy cell leukemia (Table 100-10). Therefore before embarking on treatment recommendations, one has to consult with an experienced hematopathologist to confirm a specific diagnosis. Extra caution should be taken not to misdiagnose CML or hairy cell leukemia as PMF. These issues are addressed and JAK2V617F mutation screening incorporated in the newly revised WHO diagnostic criteria for PMF (see Table 100-9).50
Table 100-10
Causes of Bone Marrow Fibrosis
| Hematologic Disorders | ||
| Myeloid Disorders | Lymphoid Disorders | Nonhematologic Disorders |
| Primary myelofibrosis | Hairy cell leukemia | Metastatic cancer |
| Chronic myeloid leukemia | Hodgkin lymphoma | Autoimmune myelofibrosis |
| Myelodysplastic syndrome | Non-Hodgkin lymphoma | Systemic lupus erythematosus |
| Chronic myelomonocytic leukemia | Multiple myeloma | Kala-Azar (leishmaniasis) Tuberculosis |
| Chronic eosinophilic leukemia Systemic mastocytosis Acute megakaryocytic leukemia |
Paget disease HIV infection |
|
| Vitamin D–deficient rickets | ||
| Other acute myeloid leukemias | Renal osteodystrophy | |
| Acute lymphocytic leukemia | Hyperparathyroidism | |
| Acute myelofibrosis | Gray platelet syndrome | |
| Malignant histiocytosis | Familial infantile myelofibrosis | |
| Idiopathic pulmonary hypertension | ||
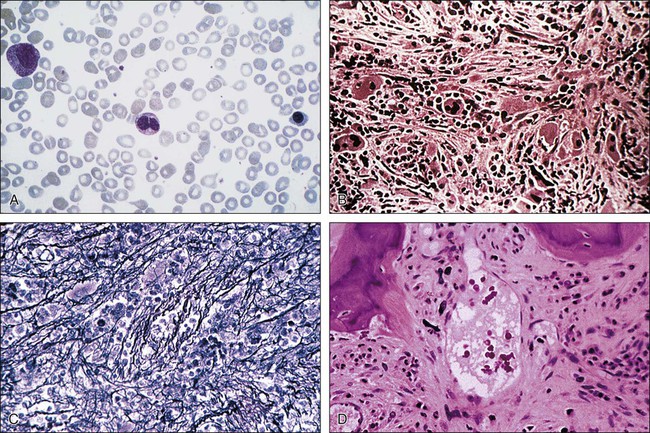
PMF should be distinguished from other closely related myeloid neoplasms, including CML, PV, ET, MDS, CMML, and “acute myelofibrosis.” The presence of dwarf megakaryocytes raises the possibility of CML and should be pursued with BCR-ABL1 cytogenetic or molecular testing. Prefibrotic PMF can mimic ET in its presentation and careful morphologic examination is necessary for distinguishing the two; megakaryocytes in ET are large and mature-appearing whereas those in prefibrotic PMF display abnormal maturation with hyperchromatic and irregularly folded nuclei169; the distinction between ET and prefibrotic PMF is prognostically relevant.170 MDS should be suspected in the presence of dyserythropoiesis or dysgranulopoiesis.171 CMML is a possibility in the presence of peripheral blood monocyte count of greater than 1 × 109/L. Patients with acute myelofibrosis (either acute panmyelosis with myelofibrosis or acute megakaryoblastic leukemia) usually present with severe constitutional symptoms, pancytopenia, mild or no splenomegaly, and increased circulating blasts.172
Treatment
Prognostic Factors
Among the BCR-ABL1− classic MPNs, PMF has the worst prognosis, with an approximate median survival of 5 years.173 However, several studies have identified both clinical and laboratory parameters that are used to identify good-risk as well as high-risk patient categories.151 The most recently revised prognostic scoring system in PMF is called “DIPSS-Plus”174 (Dynamic International Prognostic Scoring System–Plus) and can be applied at any point during the disease course and uses eight independent predictors of inferior survival: age older than 65 years, hemoglobin <10 g/dL, leukocytes >25 × 109/L, circulating blasts ≥1%, constitutional symptoms, red cell transfusion dependency, platelet count <100 × 109/L and unfavorable karyotype (i.e., complex karyotype or sole or two abnormalities that include +8, −7/7q−, i(17q), inv(3), −5/5q−, 12p- or 11q23 rearrangement). The presence of 0, 1, “2 or 3,” and ≥4 adverse factors defines low, intermediate-1, intermediate-2, and high-risk disease with median survivals of approximately 15.4, 6.5, 2.9, and 1.3 years, respectively. Furthermore, a greater than 80% 2-year mortality rate was predicted by monosomal karyotype (two or more autosomal monosomies or a single autosomal monosomy associated with at least one structural abnormality), inv(3)/i(17q) abnormalities, or any two of circulating blasts greater than 9%, leukocytes 40 × 109/L or greater, or other unfavorable karyotype.175 Patients with the latter characteristics are operationally assigned a “very high risk” category and might be better served by immediate consideration for allogeneic stem cell transplantation. More recent data suggest inferior survival in PMF associated with nullizygosity for JAK2 46/1 haplotype,176 low JAK2V617F allele burden,177,178 or presence of IDH,179 EZH2,180 SRSF2,181 or ASXL1182 mutations. Survival in PMF was also affected by increased serum IL-8 and IL-2R levels as well as serum-free light chain levels, both independent of DIPSS-Plus.66,183,184
Risk factors for leukemia-free survival include 3% or greater circulating blasts, platelet count less than 100 × 109/L, unfavorable karyotype, and presence of certain mutations, including IDH and SRSF2.179,181,185,186
Hematopoietic Stem Cell Transplantation
At present, only allogeneic hematopoietic stem cell transplantation offers a potentially curative treatment modality in myelofibrosis.187 PMF patients with high or intermediate-2 risk disease should be considered for investigational drug therapy or allogeneic stem cell transplantation. In considering allogeneic stem cell transplantation as a treatment modality, one should be acutely aware of the risks involved. In one of the largest studies of allogeneic stem cell transplantation in PMF,188 5-year disease-free survival and treatment-related mortality were 33% and 35% for match-related and 27% and 50% for unrelated transplantations, respectively. Of note, outcome did not appear to be favorably affected by reduced-intensity conditioning. In another reduced-intensity conditioning transplant study, 5-year disease-free survival was estimated at 51%; chronic graft-versus-host disease occurred in 49% of the patients and relapse (29%) was predicted by high-risk disease and prior splenectomy.189 In the earlier study,188 the respective chronic graft-versus-host disease and relapse rates for match-related transplantations were 40% and 32%, and history of splenectomy did not affect outcome. More recent outcome reports on allogeneic stem cell transplantation in myelofibrosis (MF) were more encouraging: 100-day mortality 13%, a relapse rate of 11%, and a 7-year survival of 61%.190
Conventional Drug Treatment of Anemia
MF-associated anemia is usually treated with androgens (e.g., testosterone enanthate 400 to 600 mg intramuscular weekly, oral fluoxymesterone 10 mg 3 times daily),191 prednisone (0.5 mg/kg per day), danazol (600 mg/day),192 thalidomide (50 mg/day) ± prednisone193,194,195 or lenalidomide (10 mg/day) ± prednisone196,197 (10 mg/day). I do not use erythropoiesis-stimulating agents because they are ineffective in transfusion-dependent patients and could exacerbate splenomegaly.198 Response rates to each of the aforementioned drugs are in the vicinity of 15% to 25%, and response durations average about 1 to 2 years. Lenalidomide works best in the presence of del(5q31).199 Drug side effects include hepatotoxicity and virilizing effects for androgens, peripheral neuropathy for thalidomide, and myelosuppression for lenalidomide.
Management of Splenomegaly and Other Extramedullary Hematopoiesis
Hydroxyurea is the drug of choice for controlling splenomegaly, leukocytosis, or thrombocytosis.200 Other drugs that have been used in a similar setting include busulfan,201 melphalan,202 and 2-chlorodeoxyadenosine.203 In contrast, IFN-α has limited therapeutic value in myelofibrosis.204 Most recently, ruxolitinib (a JAK1 and JAK2 inhibitor) has been shown to be effective in hydroxyurea-resistant splenomegaly and PMF-associated constitutional symptoms (see Investigational Treatment” below for further details). Many patients with myelofibrosis experience recurrent splenic infarcts that may be associated with debilitating left upper quadrant pain that may be preceded by referred left shoulder discomfort. A computed tomography scan may but does not always show a hypodense region without contrast enhancement. Symptoms from splenic infarcts are usually managed by opiate analgesics, and drug-refractory cases may require splenectomy.
Drug-refractory anemia and symptomatic splenomegaly (portal hypertension, severe hypercatabolic symptoms) may necessitate splenectomy that often alleviates splenomegaly associated symptoms and may also benefit approximately 25% of patients with transfusion-dependent anemia.205 The surgical procedure is associated with an approximately 9% mortality rate, and as many as 25% of patients may experience accelerated hepatomegaly and extreme thrombocytosis/leucocytosis after splenectomy. In the presence of severe portal hypertension, which is primarily a result of intrahepatic obstruction rather than increased portal flow from marked splenomegaly, portal-systemic shunt surgery may be performed concomitantly with splenectomy, and has been shown to be a useful therapeutic option.206
In patients with mechanical splenic discomfort, splenic irradiation (100 to 500 cGy in 5 to 10 fractions) may be considered as an alternative treatment to splenectomy.85 However, the benefit of splenic irradiation is transient (median response duration is 6 months), and the procedure is associated with more than 10% mortality rate resulting from severe and prolonged cytopenias that occur in as many as 25% of treated patients. The outcome from hepatic irradiation is even worse, and the procedure is generally not recommended.207
Radiation therapy is most useful in the treatment of nonhepatosplenic extramedullary hematopoiesis.208 Symptomatic pulmonary hypertension that is not secondary to a thromboembolic process has been associated with myelofibrosis and is believed to arise from diffuse pulmonary extramedullary hematopoiesis. Diagnosis is confirmed by technetium-99m sulfur colloid scintigraphy, which shows diffuse pulmonary uptake, and treatment with single-fraction (100 cGy) whole-lung irradiation has been shown to be effective.209,210 Low-dose irradiation is also effective for the treatment of paraspinal/epidural extramedullary hematopoiesis (1000 cGy in 5 to 10 fractions), as well as extramedullary hematopoiesis resulting in pleural and peritoneal effusions (100 to 500 cGy in 5 to 10 fractions).208,211
Investigational Treatment
The demonstration of intense bone marrow angiogenesis in myelofibrosis prompted a series of studies using thalidomide as a therapeutic agent. When used as a single agent and at doses that average about 200 mg/day, thalidomide results in clinically relevant improvement of anemia (20%), splenomegaly (23%), and thrombocytopenia (71%).212 However, approximately 20% of patients experience substantial thrombocytosis or leukocytosis in addition to the usual side effects of thalidomide. The use of low-dose thalidomide (50 mg/day) in combination with a tapering dose of prednisone (0.5 mg/kg per day) is associated with a higher rate of response in both anemia (62%) and clinically relevant thrombocytopenia (75%), as well as a better toxicity profile.213 Interestingly, some of the patients responding to thalidomide-based therapy displayed unmaintained remissions.213 However, treatment response to thalidomide is not associated with reduced myelofibrosis or bone marrow angiogenesis, and the mechanism of action may involve other effects of thalidomide, including immune modulation and tumor necrosis factor antagonism. The latter possibility is supported by another study that demonstrated a favorable response in constitutional symptoms with the use of a soluble tumor necrosis factor receptor, etanercept.214
Lenalidomide and pomalidomide are structurally related to thalidomide but belong to a new line of immunomodulatory drugs with a more potent antiangiogenic and antiinflammatory activity. Among myeloid neoplasms, the most impressive treatment results with lenalidomide have been reported in del(5q)-associated MDS—45% complete cytogenetic response and an even higher rate of hematologic response.215 In primary or post-PV/ET myelofibrosis, lenalidomide treatment was associated in approximately 20% to 30% response rate in both anemia and splenomegaly.216 Lenalidomide response rates were higher and quality of responses most impressive in myelofibrosis patients with the del(5q) abnormality.217
Clinical trials using pomalidomide in myelofibrosis have recently been reported. In a phase II randomized study, 25% of patients with anemia responded to the drug used alone (2 mg/day) or in combination with prednisone (0.5 or 2 mg/day).218 In a subsequent phase II study of single agent pomalidomide (0.5 mg/day),219 anemia response was documented only in the presence of JAK2V617F (24% vs. 0%) or absence of marked splenomegaly (38% vs. 11%). Platelet response was seen in 58% of patients but the drug had limited activity in reducing spleen size. Drug-associated neuropathy or myelosuppression was infrequent but possible.
JAK2 inhibitor ATP mimetics that are currently in clinical trials include ruxolitinib (INCB018424), SAR302503 (TG101348), CYT387, lestaurtinib (CEP-701), SB1518, AZD1480, BMS911543, LY2784544, and XL019 (www.clinicalTrials.gov). Among these, the results of two promising drugs have now been published in full (ruxolitinib and SAR302503). Ruxolitinib is a JAK1/JAK2 inhibitor. Two randomized studies comparing ruxolitinib with either placebo or best supportive care have now been published.220,221 In the COMFORT-1 trial that compared the drug with placebo (n = 309),220 the spleen response rate was approximately 42% for ruxolitinib versus less than 1% for placebo. In addition, approximately 46% of patients experienced substantial improvement in their constitutional symptoms. However, the benefit of the drug was antagonized by ruxolitinib-associated anemia (31% vs. 13.9%) and thrombocytopenia (34.2% vs. 9.3%). In the COMFORT-2 trial that compared the drug with “best available therapy” (n = 219),221 the spleen response was 28.5% with ruxolitinib versus 0% otherwise, but the drug was detrimental in terms of thrombocytopenia (44.5% vs. 9.6%), anemia (40.4% vs. 12.3%), and diarrhea (24.0% vs. 11.0%). The long-term outcome of ruxolitinib therapy in MF was recently reported and disclosed a very high treatment discontinuation rate (92% after a median time of 9.2 months) and the occurrence of severe withdrawal symptoms during ruxolitinib treatment discontinuation (“ruxolitinib withdrawal syndrome”) characterized by acute relapse of disease symptoms, accelerated splenomegaly, worsening of cytopenias, and occasional hemodynamic decompensation, including a septic shock-like syndrome.222
SAR302503, a selective JAK2 inhibitor, was evaluated in 59 patients with PMF or post-PV/ET MF, in a phases I/II study.223 The dose limiting toxicity was a reversible and asymptomatic increase in serum amylase/lipase and the maximum tolerated dose was 680 mg/day. Grade 3 or 4 adverse events were all reversible and dose-dependent, and included nausea (3%), vomiting (3%), diarrhea (10%), asymptomatic mild increases in serum lipase (27%), transaminases (27%) or creatinine (24%), thrombocytopenia (24%), and anemia (35%). By 6 or 12 months of treatment, 39% and 47% of patients, respectively, experienced a 50% or greater decrease in palpable spleen size. In addition, the majority of patients with early satiety, fatigue, night sweats, cough, or pruritus reported a durable resolution of their symptoms. Almost all patients with thrombocytosis and the majority with leukocytosis had normalization of their counts. Among 23 patients with a baseline JAK2V617F allele burden of greater than 20%, nine (39%) had a 50% or greater decrease in allele burden. Effect on bone marrow pathology was limited. In general, response was not affected by the presence of JAK2V617F. The results of a SAR302503 phase III study are currently being awaited.
Conclusion
Table 100-11 provides a risk-based management strategy in PMF. At present it is reasonable to consider all high-risk patients (using the DIPSS-Plus prognostic system)174 for allogeneic stem cell transplantation if they are transplant-eligible. On the other hand, it is acceptable to monitor low-risk or asymptomatic intermediate-1 risk patients without specific therapeutic intervention. Management in intermediate-2 or symptomatic intermediate-1 risk patients should be individualized and is often dictated by age and performance status. Such patients are also ideal candidates for investigational drug therapy including treatment with JAK inhibitors. At present, I recommend clinical trial participation with newer JAK inhibitors or other novel drugs, rather than treatment with ruxolitinib (an FDA-approved JAK1 and JAK2 inhibitor), as the drug does little to modify the natural history of the disease and can possibly make matters worse by inducing anemia or thrombocytopenia.
Table 100-11
Risk Stratification and Risk-Adapted Therapy in Primary Myelofibrosis
| DIPSS-Plus224 Risk Groups of PMF | Median Survival | Management of PMF |
| Low-risk (No risk factors*) | ~15.4 years | Observation or Conventional drugs† |
| Intermediate-1 risk (1 risk factor*) | ~6.5 years | Observation or Conventional drugs† or Experimental drugs |
| Intermediate-2 risk (2 or 3 risk factors*) | ~2.9 years | Allogeneic stem cell transplantation or Experimental drugs |
| High-risk (≥4 risk factors*) | ~1.3 years | Allogeneic stem cell transplantation or Experimental drugs |
DIPSS, Dynamic International Prognostic Scoring System.
*DIPSS-Plus14 uses eight risk factors for inferior survival: age older than 65 years, hemoglobin <10 g/dL, leukocyte count >25 × 109/L, circulating blasts ≥1%, presence of constitutional symptoms, presence of unfavorable karyotype, platelet count <100 × 109/L and presence of red cell transfusion need. Please note that a transfusion-dependent patient automatically has two risk factors because of transfusion need (1 risk point) and hemoglobin <10 g/dL (1 risk point).
†Androgen preparations or thalidomide with prednisone for anemia; hydroxyurea for sym.