Pathophysiology of Cancer Cell Death
Lorenzo Galluzzi, Oliver Kepp and Guido Kroemer
• Regulated cell death can occur via extrinsic apoptosis, intrinsic apoptosis, or regulated necrosis. Autophagy operates as a bona fide cell death mechanism in a few (mostly developmental) settings.
• Oncogenesis results from multiple molecular alterations, one of which frequently impairs the ability of cancer cells to die in response to exogenous or endogenous signals.
• Several oncoproteins and oncosuppressor proteins regulate the molecular machinery for apoptotic or necrotic cell death either directly or in an indirect fashion.
• Targeting deregulated cell death signaling pathways in cancer is a clinical reality and underlies promising approaches for the development of novel anticancer regimens.
Introduction
Perhaps biased by their focus on the cell’s vital functions, biologists have disregarded the existence of programmed cell death (PCD; defined later) for a long time. Sporadic observations of PCD have been made throughout the nineteenth century by scientists such as Carl Vogt, August Weismann, Ludwig Stieda, Elie Metschnikoff, Walther Flemming, Sigmund Mayer, and John Beard.1 Nonetheless, the concept of PCD has been theorized as late as in the 1960s, thanks to the work of Sir Richard Lockshin.2 In 1972, John Kerr, Alastair Currie, and Andre Wyllie introduced the Greek term “apoptosis” (apo: from/off/without; ptosis: falling) to describe one type of cell death that manifests with peculiar morphologic traits.3 At that time and for the subsequent 30 years, apoptosis was thought to be the only form of PCD—an oversimplistic notion that recently has been invalidated.4, 5 In the mid-2000s, it became clear that other subroutines of cell death, notably necrosis, can occur in a regulated fashion and account for some instances of PCD.6
Regulated cell death constitutes a conserved mechanism whose utility trespasses evolutionistic barriers. For instance, in unicellular organisms that grow in colonies (such as yeast), the controlled demise of old and damaged cells increases the probability that fit individuals will survive adverse environmental conditions and hence will perpetuate their genes.7 Conversely, in metazoan (including humans), PCD is critical for embryonic and postembryonic development, as well as for the maintenance of adult tissue homeostasis.8 In line with this notion, defects in the molecular mechanisms that mediate cell death contribute to the development of a wide array of human diseases. On one hand, the excessive demise of postmitotic cells decisively contributes to the pathogenesis of diseases encompassing ischemia (of the heart and brain) and neurodegeneration. On the other hand, insufficient rates of cell death have been associated with autoimmune disorders and cancer.9
The first hint that the molecular machinery for cell death is involved in oncogenesis came in the mid 1980s, when it was found that a translocation between chromosomes 14 and 18 (t14;18) that is common in persons with lymphoma leads to the overexpression of the protein BCL-2.10 In subsequent work it was clarified that BCL-2 promotes lymphomagenesis by inhibiting the programmed demise of excessive B cells rather than by stimulating their proliferation.11 This work was the first demonstration that disabled cell death constitutes one of the hallmarks of cancer, irrespective of the histologic origin of malignant cells.12 During the subsequent two decades, it rapidly became clear that defects in the signaling pathways that lead to cell death not only contribute to oncogenesis and tumor progression but also determine, at least in some instances, the resistance of neoplastic cells to chemotherapy and radiotherapy.13
Fundamental Science—Mechanisms of Cell Death
In cell death research, the word “programmed” is used to highlight the implication of one particular instance of cell death in developmental programs (and hence its physiological relevance), whereas “regulated” is used to stress the notion that one particular instance of cell death can be inhibited by targeted pharmacologic or genetic interventions (and hence is mediated by precise molecular mechanisms).4 Thus all instances of PCD are by definition regulated, but not vice versa. Additional recommendations on how to define specific cell death subroutines based on morphologic5 or biochemical4 parameters have been formulated recently and will be followed throughout this chapter.
According to currently accepted models, two main types of cell death occur: apoptosis and necrosis. Additional cell death subroutines with very specific biochemical traits have been described, but they often constitute particular cases of apoptosis and necrosis and have a limited pathophysiologic relevance.4 Macroautophagy (hereafter referred to as autophagy) also has been indicated as a potential mechanism of cell death, a notion that remains highly debated.14
Apoptosis
For a long time, instances of cell death have been catalogued as apoptotic on the basis of purely morphologic manifestations, including cytoplasmic and nuclear shrinkage (pyknosis), nuclear breakdown (karyorrhexis), and plasma membrane blebbing.5 This morphologic definition, reflecting the original observations by Kerr, Wyllie, and Currie,3 is progressively being abandoned in favor of a biochemical one.4 Thus apoptosis can occur as a result of the activation of either of two distinct but not mutually exclusive signaling pathways. On one hand, extrinsic apoptosis depends on a peculiar class of cysteine proteases called caspases and is always initiated by an extracellular stimulus acting on plasma membrane receptors. On the other hand, intrinsic apoptosis invariably follows the permeabilization of mitochondrial membranes but does not always require caspases.4 The enzymatic activity of executioner caspases, encompassing caspase-3, -6 and -7, is responsible for multiple (but not all) of the classic morphologic and biochemical manifestations of apoptosis, including karyorrhexis and the internucleosomal degradation of DNA.15
Extrinsic Apoptosis
Extrinsic apoptosis is frequently elicited by the ligand-induced activation of plasma membrane proteins of the death receptor family, such as CD95/FAS and the tumor necrosis factor α receptor 1.16 Alternatively, apoptotic signals can be transduced by the so-called dependence receptors (such as patched 1) when the extracellular concentration of trophic factors, such as netrin-1, falls below a critical threshold level. This means that—at odds with death receptors—dependence receptors deliver proapoptotic signals in the absence, rather than in the presence, of their ligands.17 Death receptors undergo spontaneous trimerization because of the so-called preligand assembly domain.18 Ligand binding stabilizes this configuration and hence allows for the recruitment of several proteins at the cytoplasmic tails of death receptors, including (but in some instances not limited to) pro-caspase-8, receptor-interacting protein kinase 1 (RIPK1, also known as RIP1), FAS-associated protein with a death domain, and cellular inhibitor of apoptosis proteins (cIAP1 and cIAP2, which are E3 ubiquitin ligases that also inhibit apoptosis as a result of their ability to interfere with caspase activation).19 This multiprotein platform is known as “death-inducing signaling complex” and stimulates the conversion of pro-caspase-8 into caspase-8, which proteolytically activates caspase-3 and hence initiates the degradative cascade that mediates the execution of extrinsic apoptosis.20 Of note, the components of the signaling pathways that emanate from specific death receptors exhibit some degree of variation, yet all converge (in apoptosis-permissive conditions, as described later) to the activation of caspase-8. The molecular mechanisms that link dependence receptors to caspase-3 activation are not precisely understood, yet they appear to involve caspase-9 and the adaptor proteins DRAL and TUCAN (Fig. 5-1).21
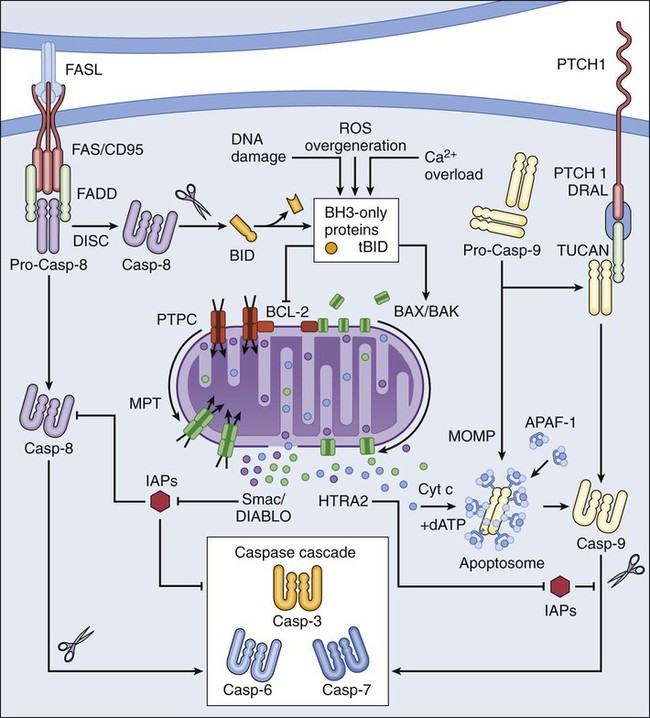
Intrinsic Apoptosis
Intrinsic apoptosis (also known as mitochondrial apoptosis) can be activated by a plethora of stimuli, including intracellular damage (to virtually any of the subcellular compartments) and by the so-called oncogenic stress (described later). Cells are equipped with a heterogeneous set of intracellular sensors that respond to perturbations first by activating signaling pathways for the reestablishment of homeostasis and the repair of damage and then, if damage is irreparable, by igniting intrinsic apoptosis.22 The central step in this cascade is regulated by the integrity of mitochondrial structure and function. Indeed, if proapoptotic signals are predominant, mitochondrial membranes get permeabilized, resulting in the abrupt cessation of adenosine triphosphate (ATP) synthesis and other metabolic functions, as well as in the spillage of several proteins that normally are confined in the mitochondrial intermembrane space.22 These proteins include cytochrome c (a semisoluble component of the mitochondrial respiratory chain), apoptosis-inducing factor (which normally contributes to the stability and function of respiratory complex I), endonuclease G, Smac/DIABLO, and HTRA2.23–27 Once in the cytosol, cytochrome c—together with dATP and the adaptor protein APAF1—drives the assembly of the apoptosome, a molecular platform for the activation of caspase-9.27 Smac/DIABLO and HTRA2 also stimulate caspase activation, though indirectly, by sequestrating and/or degrading cytosolic caspase inhibitors of the IAP family.23,24 Conversely, both apoptosis-inducing factor and endonuclease G translocate to the nucleus and mediate large-scale DNA degradation independently of caspases.25
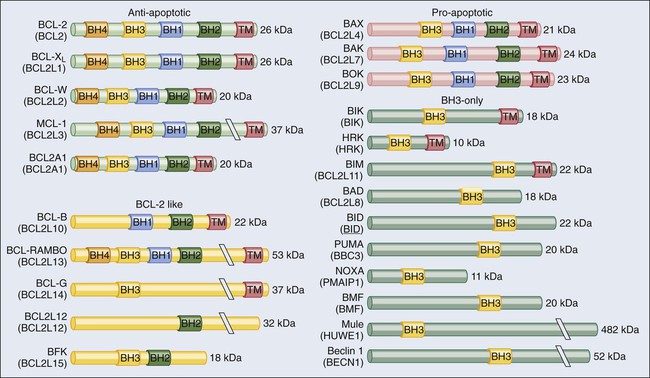
According to current models, mitochondrial outer membrane permeabilization (MOMP) can proceed via two non–mutually exclusive mechanisms. First, MOMP can be initiated at the mitochondrial outer membrane (OM) by the pore-forming activity of proapoptotic multidomain proteins of the BCL-2 family.28 These proteins, such as BAX and BAK, contain several BCL-2 homology (BH) domains and a transmembrane domain that allow for their constitutive or inducible insertion into the OM. MOMP execution by BAK and BAX is regulated by other members of the same protein family. In particular, antiapoptotic proteins such as BCL-2, BCL-XL, and MCL-1 inhibit MOMP by binding to BAK and BAX and hence by maintaining them in an inactive conformation.29 Conversely, the so-called BH3-only proteins (small members of the BCL-2 family that often contain only the BH3 domain) can promote the pore-forming activity of BAK and BAX by two different mechanisms. Thus BH3-only proteins can either stimulate the conformational activation of BAX and BAK in a direct fashion or competitively displace BAX, BAK, or other BH3-only proteins from inhibitory interactions with BCL-2, BCL-XL, and MCL-1. BH3-only proteins (e.g., BID, BAD, and PUMA) are regulated at the transcriptional level (for instance, the gene coding for PUMA can be transactivated by the stress-responsive transcription factor p53), as well as by rapid posttranslational modifications (e.g., phosphorylation and proteolytic processing), de facto constituting sensors of intracellular stress that directly impinge on the regulation of intrinsic apoptosis.30
Second, MOMP can be initiated by an abrupt increase in the permeability to ions and small solutes of the mitochondrial inner membrane (IM), a process known as mitochondrial permeability transition (MPT). Although the actual physiological relevance of MPT and the underlying molecular mechanisms remain a matter of debate, MPT has been ascribed to the activity of a multiprotein protein complex that is assembled at the juxtaposition sites between the OM and the IM, the so-called “permeability transition pore complex” (PTPC).31 The main components of the PTPC, including the voltage-dependent anion channel of the OM, the adenine nucleotide translocase of the IM, and cyclophilin D (a protein of the mitochondrial matrix), normally mediate physiological functions. For instance, adenine nucleotide translocase catalyzes the exchange of adenosine diphosphate and ATP between the cytosol and the mitochondrial matrix. However, in response to specific lethal triggers, including cytosolic Ca2+ overload and the overgeneration of reactive oxygen species, the PTPC has been proposed to assume a high-conductance conformation, leading to the osmotic swelling of the mitochondrial matrix and consequent MOMP.22 Thus far, mouse knockout experiments failed to attribute a critical role in the regulation of intrinsic apoptosis to specific components of the PTPC, perhaps because of the existence of multiple and (at least partially) redundant isoforms of these proteins.34–34 One notable exception is represented by cyclophilin D, the absence of which has been shown to limit pathological cell death in multiple circumstances, in vitro and in vivo.35,36
Of note, the signaling pathways leading to extrinsic and intrinsic apoptosis exhibit some degree of cross talk. Thus, whereas in some cells including lymphocytes (type I cells), the activation of death receptors leads to pro-caspase-3 processing and apoptosis without any mitochondrial involvement,37 in other cells such as hepatocytes (type II cells), caspase-8 not only activates caspase-3 but also mediates the proteolytic cleavage of BID, generating a MOMP-inducing fragment.38 Thus in type II cells, MOMP functions as an amplifier of apoptotic signaling by eliciting the caspase-9-mediated activation of caspase-3. This cross talk is pathophysiologically relevant in vivo, as demonstrated by the fact that the hepatocytes of Bid-/- mice are partially protected from FAS-induced apoptosis (Fig. 5-1).39
Necrosis
Classically, necrosis was defined as an instance of cell death lacking the peculiar morphologic manifestations of apoptosis and the accumulation of cytoplasmic vacuoles that characterize autophagic cells. Somehow, this definition was in line with the belief that necrosis would always proceed in an unregulated fashion and would only terminate accidental instances of cell death.5 In the late 1990s, the group of investigators led by Peter Vandenabeele demonstrated that the engagement of FAS does not always lead to cell death via extrinsic apoptosis.40,41 This observation instilled in some researchers the suspicion that, similar to apoptosis, necrosis also might be orchestrated by a refined molecular machinery and ignited an intense wave of research that has not yet come to an end.6
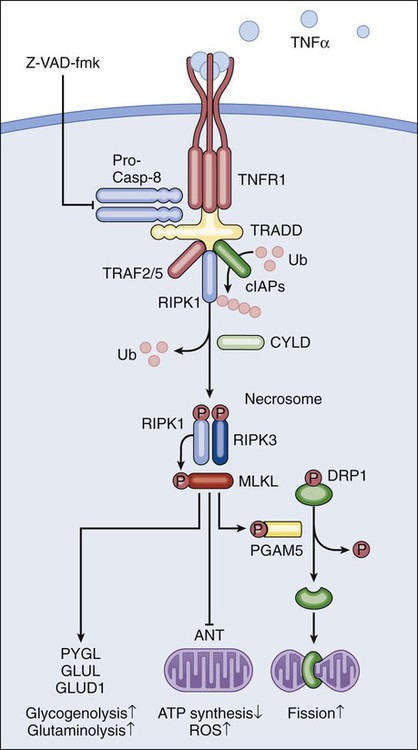
The best-characterized pathway of regulated necrosis, which is also known as necroptosis, is elicited by the ligation of death receptors in conditions in which caspase-8 is inhibited (either by pharmacologic or by genetic interventions). In this context, death-inducing signaling complex–bound RIPK1 does not get degraded by caspase-8 as it occurs during extrinsic apoptosis; rather, it recruits and functionally interacts with its homolog RIPK3, generating the so-called necrosome.44–44 Recently it has been shown that the mixed-lineage kinase like protein and the mitochondrial phosphatase PGAM5 may contribute to the execution of regulated necrosis downstream of RIPK1 and RIPK3.45,46 These results confirmed previous suspicions regarding the involvement of mitochondria in regulated necrosis, driven by the prominent role exerted by reactive oxygen species and by metabolic circuitries that (at least in part) localize to mitochondria, such as glutaminolysis.44,47 Stimuli other than death receptor ligands (e.g., alkylating DNA damage) reportedly trigger regulated necrosis.48 However, it remains unclear whether these instances of regulated necrosis also require RIPK1 and RIPK3 and proceed via PGAM5-dependent mitochondrial fragmentation (Fig. 5-2).46 Regulated necrosis occurs during mammalian development, in particular at the bone growth plate (i.e., the zone of the bone that controls its length), as well as during adult tissue homeostasis, for instance, in the lower regions of intestinal crypts.49,50 Moreover, RIPK1/RIPK3-dependent necroptosis has been involved in the pathophysiology of several diseases, including viral infection, neurodegeneration, ischemia, and others.51
Autophagy
Autophagy entails the engulfment of intracellular structures (including organelles, protein aggregates, and portions of cytoplasm) by double-membraned vacuoles called autophagosomes. Autophagosomes normally fuse with lysosomes, leading to the degradation of their content by lysosomal hydrolases. Baseline levels of autophagy contribute to the maintenance of intracellular homeostasis by ensuring the removal of old and damaged (and hence potentially dangerous) organelles, notably mitochondria.52,53 In addition, autophagy is upregulated in response to a wide array of stressful conditions, including nutrient deprivation, hypoxia, and the presence of xenobiotics, such as anticancer agents.54
For a while it was thought that the continuative activation of autophagy eventually would lead to the exhaustion of cellular resources and cell death. Such an “autophagic cell death” was defined morphologically by an extensive vacuolization of the cytoplasm, representative of an elevated number of autophagosomes and autolysosomes (the organelles that are generated by the autophagosomal-lysosomal fusion).5,14 However, the association between the accumulation of autophagosomes and cell death has rarely if ever been proved to be causal, in particular in settings of stress-induced cancer cell death. Indeed, the inhibition of autophagy by pharmacologic or genetic means often accelerates (rather than inhibits) cell death, suggesting that autophagy constitutes a stress response mechanism that attempts (but fails) to avoid the cellular demise.55 These results cast doubts on the appropriateness of the term “autophagic cell death,” which inadvertently suggests a cause-effect relationship between these two phenomena.14 Thus far autophagy has been demonstrated to mediate cell death in several developmental scenarios, notably during the metamorphosis of insects.56,57 Moreover, at least in some instances, autophagy appears to contribute to the execution of human cancer cells succumbing to specific experimental cell death inducers in vitro.58 These observations de facto justify the use of the expression “autophagic cell death” under selected circumstances.4 Defects of the autophagic machinery have been associated with a plethora of human pathophysiologic conditions, including accelerated aging, neurodegeneration, and cancer.59,60 However, this association appears to be more strictly related to the role that autophagy exerts in the regulation of intracellular homeostasis rather than as a bona fide cell death mechanism52 and hence will not be treated here in further detail.
Fundamental Science—Cell Death and Cancer
According to classic models, single molecular alterations are per se unable to fully transform normal cells into highly aggressive cancer cells. Rather, oncogenesis seems to proceed along with a progressive increase in genetic instability and with the accumulation of several molecular defects. Often, if not always, one of these alterations consists of the interruption of the signaling cascades that ensure the homeostatic death of continuously proliferating cells.61 As they evolve, premalignant and malignant cells are indeed subjected to elevated levels of stress, in part as a result of the overactivation of cell-intrinsic oncogenic signaling pathways (so-called oncogenic stress) and in part because of microenvironmental conditions, which often are characterized by hypoxia and nutrient shortage (especially in rapidly proliferating neoplasms).62 Thus in virtually all scenarios, carcinogenesis requires the (at least partial) suppression of cell death signaling pathways. This suppression can result from loss-of-function mutations in proteins that transduce lethal signals or execute cell death (as with many oncosuppressor proteins) or from gain-of-function alterations in molecules that normally deliver prosurvival signals (as with several oncoproteins). Defects in the molecular pathways that regulate cell death also are instrumental to tumors when it comes to resistance to chemotherapy and radiotherapy.13
Oncogenes and Cell Death Regulation
Oncogenes (i.e., genes that stimulate malignant transformation) were originally identified in tumorigenic viruses and then were shown to exist as inactive variants (or proto-oncogenes) in the human genome.63 Proto-oncogenes including MYC and NRAS are involved in the regulation of mitogenic signals and hence play a critical role in the control of tissue homeostasis. Proto-oncogenes are not intrinsically tumorigenic and must acquire gain-of-function alterations to become so. This process can originate from alterations as gross as chromosomal translocations that bring proto-oncogene coding sequences in the proximity of strong transcriptional regulators (as in the case of MYC, which is often rearranged in persons with lymphoma) or as specific as point mutations that render proto-oncogene products constitutively active (as in the case of NRAS, which is affected by hyperactivating mutations in 20% to 25% of all cancers).64,65 By transducing strong and persistent mitogenic signals, constitutively active MYC and NRAS—as well as other oncoproteins such as the epidermal-growth factor receptor (which is often overactivated in lung and colon cancer) and the Abelson tyrosine kinase (ABL, which is fused to BCR upon the t(9;22) translocation in chronic myelogenous leukemia)—facilitate the escape of malignant precursors from the growth control that usually guarantees tissue homeostasis. As previously mentioned, however, one single alteration of this kind is insufficient to convert normal cells into their malignant counterparts. The overactivation of mitogenic pathways indeed causes consistent functional alterations and drives cells into a state of constant stress, featuring significant metabolic rewiring and increased generation of reactive oxygen species. Because oncogenic stress normally would lead to cell death, only premalignant cells in which the cell death signaling pathways are blocked can progress toward tumorigenesis.61
Premalignant cells resist oncogenic stress by activating one (or more) among several pathways that (1) directly counteract the execution of cell death (because of the deregulation of additional proto-oncogenes) or (2) facilitate the management of intracellular stress (as a result of the elicitation of per se nononcogenic stress response pathways). This response often leads to two distinct phenomena of dependence, which are called “oncogene addiction” and “nononcogene addiction.”62 In the former scenario, not only the tumorigenic phenotype but also the survival of cancer cells becomes dependent on the overactivation of one or more proto-oncogenes. This dependency has provided a rationale for the use of multiple oncogene-targeted agents in anticancer therapy, some of which (such as the BCR-ABL–targeting compound imatinib) have been associated with impressive rates of therapeutic responses.61 In the latter scenario, cancer cells become dependent on the activation of intrinsically nononcogenic mechanisms of intracellular stress management, such as those centered around molecular chaperones of the heat-shock protein family.62 Major examples of prosurvival systems that often get overactivated throughout (and contribute to) oncogenesis are provided by antiapoptotic BCL-2 family members, the nuclear factor–κB (NF-κB) pathway, and the serine/threonine protein kinase AKT.
Antiapoptotic BCL-2 proteins have been shown to regulate cell death by multiple mechanisms, including the sequestration of their proapoptotic counterparts into inactive complexes, as well as Ca2+-related and metabolic effects (Fig. 5-3).29,66,67 Following the discovery that the overexpression of BCL-2 as a result of the t(14;18) translocation underlies multiple instances of lymphomagenesis,10 preclinical and clinical evidence has accumulated to demonstrate that not only BCL-2 but also its antiapoptotic homologues BCL-XL and MCL-1 exert bona fide oncogenic functions.68 The overexpression of BCL-2 shortens the latency period required for transgenic mice that overexpress MYC in B cells to develop lymphomas.11 Along similar lines, both BCL-XL and MCL-1 have been shown to cooperate with MYC in murine models of hematopoietic malignant transformation.69,70 However, consistent with the notion of multistep oncogenesis previously described, mice engineered for the overexpression of BCL-2, BCL-XL, or MCL-1 in hematopoietic cells exhibit perturbed myelopoiesis or lymphopoiesis but are only slightly more prone to the development of age-related lymphomas than are their normal counterparts.69,71,72 Of note, the protein expression levels of BCL-2, BCL-XL, and MCL-1 are elevated in a wide range of human tumors.73
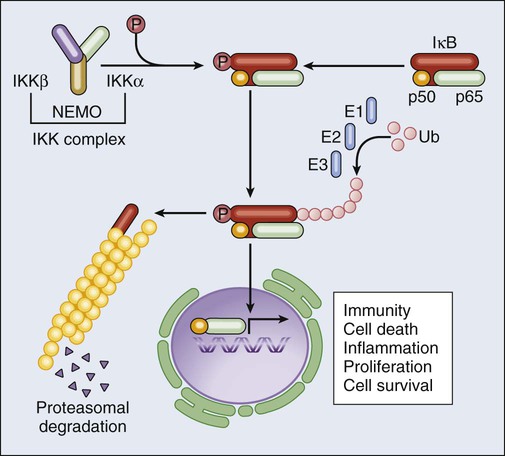
NF-κB is a transcription factor that participates in the cellular response to a wide array of conditions, including (but not limited to) cytokine stimulation, infection, and oxidative stress. Under normal circumstances, NF-κB subunits such as p65RELA and p50NFKB1 are held in check in the cytoplasm by inhibitory interactions with IκBα. In the presence of NF-κB–inducing conditions, however, IκBα is phosphorylated by the so-called IκB kinase (IKK, a multiprotein complex including two catalytic subunits, i.e., IKKα and IKKβ, and one regulatory component, i.e., IKKγ/NEMO), resulting in its proteasomal degradation and in the nuclear translocation of NF-κB.74 Nuclear NF-κB homodimers and heterodimers control the expression of more than 150 target genes, including genes that code for mitogens, other transcription factors (including MYC), inhibitors of extrinsic apoptosis (e.g., the so-called FLIPs), IAPs, BCL-2 and BCL-XL (Fig. 5-4).75 Thus, the activation of NF-κB orchestrates the response of cells to stress while delivering a strong prosurvival (and in some cases mitogenic) signal. Multiple lines of evidence demonstrate that the NF-κB system exerts genuine oncogenic functions. First, REL genes are homologues of the avian reticuloendotheliosis virus v-rel oncogene, which is strictly required for virus-induced malignant transformation.76,77 Second, the human leukemia/lymphoma virus type I can transform target cells via a mechanism that depends, at least in part, on the activation of cellular IKK by the viral protein Tax.78 Finally, several components of the NF-κB pathway get overexpressed or overactivated during malignant transformation. For instance, REL genes are amplified or affected by gain-of-function mutations in a constituent proportion of B-cell lymphomas,79 and the IkB-related protein BCL-3 (which stimulates the nuclear translocation and activity of NF-κB dimers)80 is involved in a chromosomal rearrangement, t(14;19), that is found in some hematologic malignancies.81 Of note, the NF-κB system appears to play a critical role in the handling of oncogenic stress, as demonstrated by the fact that multiple oncoproteins, including RAS and BCR-ABL, stimulate NF-κB nuclear translocation and transcriptional activity.82,83
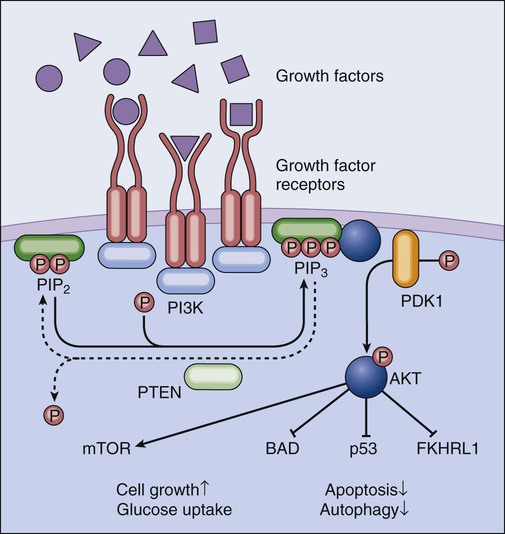
One of the hallmarks of cancer cells as originally formulated by Hanahan and Weinberg in 2000 consists of their ability to circumvent the absence of extracellular growth factors and de facto emancipate from homeostatic tissue regulation.84 Several growth factors, including interleukin-2, the platelet-derived growth factor, and the insulin-like growth factor, promote proliferation and survival via transmembrane receptors that activate the phosphoinositide-3-kinase (PI3K, also known as phosphatidylinositol-3-kinase). Active PI3K generates plasma membrane–bound phosphatidylinositol-3,4,5-trisphophate, in turn stimulating the activation of the serine/threonine kinase AKT (also known as protein kinase B). By phosphorylating multiple substrates, AKT transduces prosurvival signals via distinct mechanisms. For example, AKT inhibits the BH3-only protein BAD by facilitating its sequestration by 14-3-3 proteins, stimulates the degradation of the oncosuppressor protein p53 (discussed later), promotes glucose uptake and hence favors the maintenance of bioenergetic homeostasis, and blocks the nuclear translocation of proapoptotic Forkhead transcription factors such as FKHRL1.85 Moreover, AKT indirectly activates the mammalian target of rapamycin (mTOR), thus stimulating cell growth and proliferation while inhibiting autophagy (Fig. 5-5).86 Several preclinical and clinical studies have demonstrated the importance of AKT in tumorigenesis. In transgenic mice expressing constitutively active AKT under the control of a T cell–restricted promoter, lymphoma develops early in life.87 Amplification of AKT genes or gain-of-function AKT mutations have been detected in several neoplasms, including (but not limited to) breast and gastric cancers. In addition, loss of function of the oncosuppressor PTEN (coding for the phosphatase that directly antagonizes PI3K and hence inhibits AKT, discussed later) is common across a wide array of tumors.88 Of note, the inhibition of mTOR limits tumor formation in Pten+/- mice,89 demonstrating that AKT-driven oncogenesis relies, at least in part, on mTOR overactivation. This observation is particularly interesting in view of the fact the mTOR inhibition stimulates autophagy, which is considered a legitimate oncosuppressor mechanism during early tumorigenesis.90
Oncosuppressors and Cell Death Regulation
The existence of tumor suppressor genes (also known as oncosuppressor genes), that is, genes whose products actively counteract tumorigenesis, was first suspected in the 1960s, when Harris and colleagues observed that highly malignant Ehrlich ascites cells lose the ability to form tumors upon fusion with virtually nontumorigenic cells.91 However, the molecular characterization of the first tumor suppressor gene lagged until the 1980s, when the complementary DNA encoding the protein product of the RB1 gene (a negative regulator of cell cycle progression involved in the development of pediatric retinoblastoma) was isolated and sequenced.92 Since then, several other oncosuppressor proteins have been identified that encompass negative regulators of mitogenic signals, such as PTEN, pro-apoptotic BCL-2 family members like BAX, and proteins that couple the management of cellular stress to cell death induction, such as ATM and p53. Oncosuppressor genes are inactivated during oncogenesis not only upon loss-of-function mutations and deletions but also after epigenetic gene silencing. Thus promoter hypermethylation at CpG islands or histone deacetylation can turn off oncosuppressor gene expression as a result of local chromatin remodeling and impaired access for essential transcription factors.93 These instances are exemplified by the loss of caspase-8 expression in pediatric neuroblastomas or by the loss of p14ARF (an upstream activator of p53; discussed later) expression in multiple tumor types.94,95 Irrespective of the underlying mechanisms, in most cases, both alleles of one particular tumor suppressor gene must be inactivated for the establishment of tumor-permissive conditions. In this context, germline mutations in oncosuppressor genes are associated with familial tumor syndromes (as one copy of the gene is lost in the whole organism), whereas somatic mutations are detected frequently in sporadic tumors (in this case, both alleles have been inactivated during tumorigenesis).96
As previously mentioned, the protein product of PTEN (phosphatase and tensin homolog) functions as a phosphatase, catalyzing the conversion of phosphatidylinositol-3,4,5-trisphosphate into phosphatidylinositol-4,5-diphosphate, hence essentially antagonizing the enzymatic activity of PI3K and negatively regulating AKT (Fig. 5-5).97 PTEN was discovered in the search for an oncosuppressor gene on chromosome 10 that often is lost in glioblastoma, breast, endometrial, and prostate cancer.98 Subsequent preclinical and clinical observations supported the notion that PTEN loss of function sustains tumorigenesis in many different organs.99,100 Recently the oncosuppressive functions of PTEN have been shown to be particularly sensitive to gene dosage, even beyond the classic concept of haploinsufficiency.101 Of note, the frequency of PTEN inactivation in human tumors is exceeded only by that of p53 (discussed later).88
Pro-apoptotic BCL-2 family members, in particular BAX and BAK, exert crucial oncosuppressive functions as they mediate the execution of cell death in response to adverse conditions, including oncogenic stress (Fig. 5-3). Results from multiple murine models support the notion that BAX and BAK operate in vivo to counteract oncogenesis independent of p53. For instance, murine cells expressing adenoviral E1A (a proliferative factor) and dominant negative p53 are unable to form tumors in mice unless Bax and Bak are lost.102 In addition, loss-of-function mutations in BAX and BAK have been detected in many hematopoietic and solid malignancies.73,103 By acting as stress sensors that activate intrinsic apoptosis, BH3-only proteins also could exert, at least theoretically, prominent oncosuppressive functions. That said, evidence implicating them as legitimate tumor suppressors is scant, and only a few articles report their loss in patients with cancer.104 The reasons underlying this apparent discrepancy have not yet been clarified but perhaps are linked to the fact that whereas BAX and BAK operate at the convergence of multiple lethal signaling pathways, single BH3-only proteins are involved in highly specific signaling cascades and perhaps are relatively redundant among each other.
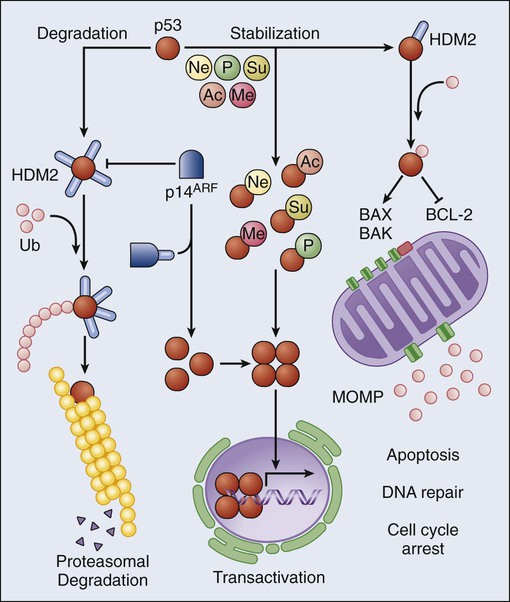
p53 originally was identified in 1979 by two methodologically distinct approaches, that is, as a cellular protein coimmunoprecipitating with the large-T antigen in SV40-transformed cells105,106 and as the target of antibodies isolated from the serum of immunodeficient mice bearing human tumors.107,108 Since then, p53 has been subjected to an intense wave of investigation, leading to the discovery that p53 regulates an ever-growing list of cellular processes.109 The first (and perhaps best) characterized function of p53 is that of a stress-responsive transcription factor. In physiologic conditions, the intracellular levels of p53 are regulated by HDM2, an E3 ubiquitin ligase that polyubiquitinates p53 and hence targets it to proteasomal degradation.110 In response to a variety of adverse conditions, including DNA damage and oncogenic stress, p53 escapes HDM2-mediated degradation and accumulates in the cytoplasm. This response can be due to posttranslational modifications of p53 that reduce its affinity for HDM2 or to the expression of HDM2 inhibitors such as p14ARF.111 Irrespective of the underlying molecular mechanisms, stabilized p53 assembles into transcriptionally proficient tetramers that can regulate the expression of distinct sets of genes, leading to highly diverse functional outcomes.112 Among the most common p53-regulated responses are reversible cell cycle arrest, senescence, and cell death. The latter is accomplished via the upregulation of multiple genes involved in the execution of apoptosis, including APAF1, BAX, FAS, and PUMA,112–115 as well as via transcription-independent mechanisms whereby cytoplasmic p53 stimulates MOMP by interacting with both proapoptotic and antiapoptotic members of the BCL-2 family (Fig. 5-6).116–119 The inactivation of p53, be it mutational, due to the overexpression/overactivation of p53 negative regulators such as HDM2, or resulting from the inactivation of upstream p53-activating factors such as p14ARF, is the most common molecular alteration in human cancer, affecting more than 50% of all neoplasms. In addition, the p53 status has been shown to influence prognosis and/or therapeutic outcome in multiple oncologic settings, including (but not limited to) breast, lung, and colorectal cancer.120 These clinical data reflect a huge number of preclinical observations, demonstrating that p53 exerts legitimate oncosuppression in vivo.121 Of note, recent research has focused on physiological aspects of the p53 biology, unveiling a critical role for baseline p53 levels in the maintenance of bioenergetic, redox, and genomic homeostasis.122–125 Thus p53 appears to exert oncosuppressive functions both as it regulates intracellular homeostasis, thereby preventing malignant transformation, and as it orchestrates the elimination of potentially tumorigenic cells.
Clinical Relevance and Applications
As previously discussed, genetic, epigenetic, and functional alterations of the mechanisms that underlie regulated cell death (1) are highly prevalent in human cancer, (2) are often required for oncogenesis and/or tumor progression, and (3) underlie many instances of cancer chemoresistance and radioresistance. The clinical relevance of these alterations is therefore dual. On one hand, the characterization of the genotype and functional phenotype of specific neoplasms can provide prognostic and/or predictive information. This information enables the stratification of patients into precise risk groups and, at least in some instances, enables the implementation of personalized therapeutic regimens.126 On the other hand, many anticancer agents that specifically target alterations in cell death-regulating signaling pathways have been developed recently, and some of them have successfully entered clinical routines. At odds with conventional chemotherapeutics, which frequently kill tumor cells because of their elevated proliferative potential, targeted anticancer agents act on cancer cell–specific defects and therefore are generally associated with a low incidence and severity of adverse effects.127 These agents include compounds that interrupt oncogene or nononcogene addiction, as well as strategies that attempt to reconstitute the lost function of tumor suppressors. In most cases, these approaches lead to the apoptotic or necrotic demise of cancer cells, although in some cases the therapeutic benefit results from the activation of cellular senescence (an irreversible arrest in cell cycle progression).128 Cell death mechanisms also have attracted much attention for the development of strategies for chemosensitization and radiosensitization. In this case, restoring the proficiency of cancer cells to undergo stress-induced cell death is not therapeutic per se but restores the tumor sensitivity to conventional chemotherapeutics, which often is lost during tumor progression.129 Of note, the success of targeted anticancer agents appears to depend, at least in part, on the activation of immune effector mechanisms,130 as demonstrated in multiple settings, in vitro and in vivo, including models of pure oncogene addiction.131
The list of successful approaches that target deregulated cell death signaling in cancer cells is continuously growing and encompasses, among others, the proteasomal inhibitor bortezomib, which blocks NF-κB activation and is approved by the Food and Drug Administration (FDA) for the treatment of multiple myeloma and mantle cell lymphoma132; oblimersen, an antisense oligonucleotide that targets BCL-2 and recently has provided promising results in advanced clinical trials133,134; so-called BH3 mimetics, that is, small molecules that inhibit multiple antiapoptotic members of the BCL-2 protein family, such as ABT-737135; rapamycin, an mTOR inhibitor that is approved by the FDA for transplant rejection and is now being evaluated in combination therapies against multiple neoplasms136; and p53-reconstituting strategies, aimed at either reintroducing wild-type p53 in p53-deficient cancer cells (by gene therapy) or blocking HDM2 in p53-proficient cells (by pharmacologic inhibitors).137,138 Of note, a recombinant adenovirus engineered to express wild-type p53 (known as Gendicine) is the first gene therapy product approved for clinical use in humans.139 In addition to targeted approaches, such as those previously listed, promising results have been obtained with epigenetic regulators such as suberoylanilide hydroxamic acid (SAHA, also known as Vorinostat), a histone deacetylase inhibitor that is currently approved by the FDA for the treatment of cutaneous T-cell lymphoma,140 and azacitidine or decitabine, methyltransferase inhibitors that currently are used for the therapy of myelodysplastic syndromes.141
What the Future Holds
Finely manipulating cell death mechanisms in cancer, a strategy that 20 years ago appeared to be a distant and hardly reachable goal, is now a reality with crucial clinical implications. Future investigations will need to provide further insights into the molecular cascades that underlie the execution of cell death in both physiological (homeostatic cell death) and pathological (in response to oncogenic stress) scenarios and unravel the mechanisms through which these cascades are disabled during oncogenesis and tumor progression. This research will be instrumental not only for the discovery of novel drug targets and hence for the development of new anticancer agents but also for the precise identification of patients who are likely to respond to particular therapeutic regimens. Furthermore, strategies for the therapeutic induction of alternative cell death modes, including regulated necrosis, or oncosuppressive signaling pathways that operate in multiple ways, such as mitotic catastrophe,122 should be elaborated. These approaches will be particularly relevant for the treatment of chemoresistant and radioresistant cancers, which very often exhibit alterations in the molecular machinery that initiates or executes apoptotic cell death. Finally, it will be indispensable to understand how distinct cell death subroutines cross talk (i.e., the extent to which they can substitute for each other in scenarios in which one is specifically disabled by tumorigenic alterations) and to what extent the immune system contributes to the therapeutic efficacy of currently used anticancer regimens. In this context, the induction of immunogenic cell death, which converts dying tumor cells into a vaccine that is capable of eliciting a tumor-specific immune response,130 may constitute a particularly interesting therapeutic goal.