[level-membership-for-hematology-oncology-and-palliative-medicine-category]92
Sarcomas of Bone
Megan E. Anderson, R. Lor Randall, Dempsey S. Springfield and Mark C. Gebhardt
• More than 2800 new cases of bone sarcoma are diagnosed annually in the United States.
• No specific etiologic agents are identified in the majority of cases.
• Secondary neoplasms are related to known oncogenic factors. (e.g., ionizing radiation, alkylating chemotherapy agents, combinations of both).
• Hereditary cancer syndromes (tumor suppressor genes) are responsible for some cases.
Diagnosis and Radiographic Staging
• Plain radiographs are recommended.
• Magnetic resonance imaging (MRI) scan of primary tumor is the best radiographic study to obtain.
• Chest computed tomography (CT) is indicated to evaluate for pulmonary metastases.
• Whole-body technetium-99m (99mTc) bone scan is indicated to evaluate for metastases to other bones.
• Positron emission tomography (PET) scanning is controversial and has yet to be generally accepted.
• Needle or open biopsy is necessary for a tissue-specific diagnosis and to determine histologic grade.
• In the pathology review, immunohistochemistry and cytogenetics are important.
• Electron microscopic tissue occasionally is required.
• Metastasis at presentation is a worse prognostic finding.
• Histologic grade is the next most significant prognostic indicator.
• Size is less significant, but lesions larger than 10 cm in diameter have a poor prognosis.
• Tumor response to neoadjuvant chemotherapy is important in osteosarcoma and Ewing sarcoma.
• Surgical margins of resection (minimum of a “wide” margin).
• The American Joint Committee on Cancer now monitors grade (high and low), and size (8 cm); designates “skip” lesions (T3); and separates metastasis to bone from other sites (Mla, Mlb).
• The Musculoskeletal Tumor Society monitors location. (intracompartmental and extracompartmental), grade (high and low), and metastasis (“skip” lesions, nodal, bone, and lung are all lumped together).
• A wide surgical margin is recommended.
• Limb-sparing procedures are appropriate for 70% to 90% of patients.
• Adjuvant irradiation is not routinely used for bone sarcomas.
• Local recurrence rates for limb-sparing procedures approach 5% or less.
• Reconstruction methods can be tailored to patients’ needs.
• New and improved biocompatible implant materials and improved designs are available.
• The search continues for new drugs, drug schedules, potentiating agents, and improved dose intensity.
• Identification of risk factors (e.g., cytogenetic, molecular genetic, and signal transduction abnormalities) will improve to identify new methods of potential treatment.
Introduction
It is estimated that 2890 new malignant tumors of bone (excluding multiple myeloma) were diagnosed in 2012 in the United States (http://seer.cancer.gov). The femur is the most common site, but primary sarcoma can occur in any bone. Osteosarcoma, Ewing sarcoma, and chondrosarcoma account for approximately 90% of all primary sarcomas of bone. The management of osteosarcoma and Ewing sarcoma includes chemotherapy and surgery, whereas chondrosarcoma is treated by surgery alone.1 The management of these patients, from initial evaluation and biopsy through surgical therapy and long-term follow-up, is labor intensive and technically demanding. Patients with a bone sarcoma should be treated in a center that has expertise in the management of these tumors.
Surgical Staging System
Currently, the staging system adopted by the Musculoskeletal Tumor Society (MSTS) in 1980 and modified in 1986 is accepted by most musculoskeletal oncologists.4–4 Malignant tumors are divided into only two histologic grades: low-grade malignant (G1) and high-grade malignant (G2). Low-grade malignant lesions (G1), comprising Broder I and II lesions, have a low probability of metastasis (25%). The majority of these tumors can be managed by relatively conservative surgical procedures and do not require chemotherapy. High-grade lesions (G2), Broder III and IV tumors, have a significantly higher incidence of metastases and require more radical surgical procedures and possibly neoadjuvant and/or adjuvant chemotherapy. Table 92-1 is a representative grouping of both low- and high-grade malignant tumors of bone and soft-tissue origin.
Table 92-1
| Low (G1) | High (G2) |
| Parosteal osteosarcoma | Classic intramedullary osteosarcoma |
| Periosteal osteosarcoma (typically intermediate grade) | High-grade surface osteosarcoma |
| Low-grade central osteosarcoma | Paget sarcoma of bone |
| Radiation sarcoma | |
| Secondary chondrosarcoma | Primary chondrosarcoma |
| Clear cell chondrosarcoma | Dedifferentiated chondrosarcoma |
| Mesenchymal chondrosarcoma | |
| Fibrosarcoma | Fibrosarcoma |
| Atypical malignant fibrous histiocytoma of bone | Malignant fibrous histiocytoma (MFH) of bone |
| Adamantinoma | |
| Undifferentiated primary sarcoma of bone | |
| Giant cell sarcoma of bone | |
| Hemangioendothelioma | Angiosarcoma |
| Chordoma | Neurofibrosarcoma/malignant peripheral nerve sheath tumor |
| Ewing sarcoma family of tumors |
Radical margins of a bone sarcoma are achieved when the entire bone is removed. This process usually requires an amputation. A radical margin is rarely necessary.5
The American Joint Committee on Cancer (7th edition) has adapted the TNM (tumor–nodes–metastases) staging system to bone. The topography (T) of the primary tumor now includes size based on relevant published reviews, in which the greatest dimension (8 cm) has replaced the compartment concept. Also, T3 has now been assigned to patients with “skip” metastases (Table 92-2).
Table 92-2
Definition of TNM Primary Tumor (T)
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor ≤8 cm in greatest dimension |
| T2 | Tumor >8 cm in greatest dimension |
| T3 | Discontinuous tumors in the primary bone site |
Adapted from Edge SB, Byrd DR, Compton CC, et al., editors. AJCC cancer staging manual. 7th ed. New York: Springer Verlag; 2010.
The problem of defining histopathological grade (G) has been addressed and now essentially consists of low- and high-grade lesions (Table 92-3). G1 and G2 have been combined into low-grade and G3 and G4 into high-grade histopathology. Currently, all Ewing tumors are classified as G4 or high-grade. This grouping is now identical to the G1 and G2 of the MSTS staging system. Table 92-4 shows the stage groupings. Here, the committee has appropriately addressed the difference in prognosis of patients who have sustained metastases to lung (Mla) and to other sites, including bone (Mlb).
Table 92-3
| GX | Grade cannot be assessed |
| G1 | Well differentiated—low grade |
| G2 | Moderately differentiated—low grade |
| G3 | Poorly differentiated—high grade |
| G4* | Undifferentiated—high grade |
*Ewing sarcoma is classified as G4.
Adapted from Edge SB, Byrd DR, Compton CC, et al., editors. AJCC cancer staging manual. 7th ed. New York: Springer Verlag; 2010.
Table 92-4
| Stage 1A | T1 | N0 | M0 | G1, 2 low grade, GX |
| Stage 1B | T2 | N0 | M0 | G1, 2 low grade, GX |
| T3 | N0 | M0 | G1, 2 low grade, GX | |
| Stage IIA | T1 | N0 | M0 | G3, 4 high grade |
| Stage IIB | T2 | N0 | M0 | G3, 4 high grade |
| Stage III | T3 | N0 | M0 | G3, 4 high grade |
| Stage IVA | Any T | N0 | M1a | Any G |
| Stage IVB | Any T | N1 | Any M | Any G |
| Any T | Any N | Mlb | Any G |
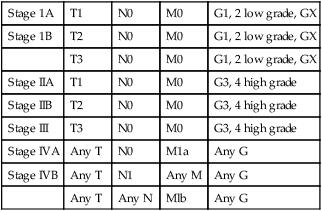
Adapted from Edge SB, Byrd DR, Compton CC, et al., editors. AJCC cancer staging manual. 7th ed. New York: Springer Verlag; 2010.
Radiographic Staging
Conventional bone radiography remains the single most useful initial study for bone tumor evaluation. The study should include anteroposterior and lateral projections of the lesion and ideally the whole bone in which the lesion is present. Malignant neoplasms usually result in ill-defined or “poorly marginated” radiographic margins with little or no reactive bone, loss of medullary trabeculation, and endosteal cortical erosion at the tumor–host bone interface, suggesting an active and destructive process. The pathological process biologically overwhelms the normal time-dependent reactive processes of bone formation. Therefore, the radiographic presence or absence of a reactive rim of bone is often useful in predicting the biological aggressiveness of the pathological process (Table 92-5). Neoplastic bone formation is often seen in osteosarcoma, and calcification is often seen in chondrosarcoma.
Table 92-5
Radiographic Techniques Available

Computed tomography (CT) is superior to magnetic resonance imaging (MRI) only for evaluating a small lesion in the cortex, subtle bone formation, or calcification; otherwise, MRI is the study of choice. CT remains the standard for evaluation of the chest for occult metastases. The cross-sectional display usually provides sufficient resolution (<0.5 cm) to demonstrate subpleural metastases long before they become evident on plain chest films. Before definitive therapy or local management of a potentially malignant lesion, a staging CT evaluation of the chest and mediastinum should be performed.6
Currently, MRI of the primary tumor appears somewhat predictive of tumor response to neoadjuvant therapy. Changes in the T2-weighted image signal intensity correlate with an obvious reduction in tumor volume (especially in Ewing sarcoma) and appear predictive of tumor necrosis.7 The addition of contrast enhancement does not appear to provide more viable tumor/necrotic tumor contrast than do T2-weighted images; however, the absence of contrast enhancement appears to be an indicator of tumor necrosis.7–10
Positron emission tomography (PET) has been used to predict response to chemotherapy for Ewing sarcoma and osteosarcoma. PET scan might be valuable in the initial screening, but this has yet to be determined.11,12
Staging Biopsy
The staging biopsy might well be the most important and difficult procedure that is performed in the patient’s management (Box 92-1). The placement, length, and orientation of the biopsy scar and the anatomic compartments that are contaminated during the biopsy procedure dictate which tissues and how many surgical compartments will require removal for local tumor management and limb-sparing surgery. A thorough knowledge of the soft-tissue anatomic planes and muscle compartments is mandatory before proceeding with bone biopsy. Consideration should be given to the location and type of biopsy to be used, whether fine-needle aspiration biopsy, core-needle biopsy, or open biopsy. In addition, it is critical that adequate diagnostic tissue be obtained so that an accurate diagnosis can be made. Usually, a core-needle biopsy is sufficient.
Osteosarcoma
High-grade osteosarcoma used to be a fatal neoplasm leading to metastases and death of 90% of patients despite aggressive local control, including radical amputations and/or radiotherapy. Previous versions of this chapter outlined the evolution of the use of adjuvant chemotherapy and the dramatic improvement in survival and event-free survival (EFS) that followed. Initially, the benefits of adjuvant chemotherapy were questioned, leading to the need for a randomized study comparing surgical management alone with surgery followed by multiagent chemotherapy. The active drugs were shown to be doxorubicin, high-dose methotrexate, and cisplatin. A randomized study, including both the randomly assigned patients and those who chose whether or not to have adjuvant chemotherapy, clearly demonstrated the benefits of adjuvant chemotherapy.13,14
Surgical advances paralleled the advances in medical management of osteosarcoma patients. Originally, disarticulation or resection of the entire involved bone was recommended for surgical management of osteosarcoma.15 This recommendation was partly a result of the intramedullary origin of the tumor with proximal intramedullary growth and the reported 25% incidence of intramedullary “skip” metastases.16 Later studies reviewing the local recurrence rates for patients whose primary management was transmedullary amputation alone revealed local recurrences in approximately 5% to 10%, suggesting that the incidence of “skip” or intraosseous metastasis was probably lower than originally believed. The general standard of surgical management of patients with extremity osteosarcoma in 1980 included transmedullary amputation approximately 5 to 7 cm proximal to the intramedullary extent of the tumor.17
One event that led to the popularization of limb salvage was the use of preoperative chemotherapy. Initially implemented during the time it would take to manufacture a custom prosthesis, it became apparent that the tumor showed clinical and radiographic “response” to the preoperative (neoadjuvant) chemotherapy. This seemed to make the subsequent surgery easier, if not safer, and gave the surgeon 10 to 12 weeks to work with the patient to decide on the best surgical option. There was initial concern that the delay in the resection might worsen the prognosis, but this issue was addressed in a randomized trial that showed no apparent advantage to having the surgery initially compared to the neoadjuvant mode of administration.18 In addition, it was learned that the histologic response to the preoperative chemotherapy was a predictor of outcome and, second only to the presence of metastases at diagnosis, was the best predictor of survival.18,19
Epidemiology
Conventional, or classic, osteosarcoma makes up the majority of all osteosarcomas. It occurs primarily in the metaphyses of adolescents with open physes or in young adults. Most patients with classic osteosarcoma are younger than age 30 years, and many have no apparent predisposing factors.20 The lesion most often arises in the larger, more active metaphyses (e.g., distal femur, proximal tibia, proximal humerus), but also can arise in the flat bones of the pelvis, skull, scapula, and ribs, and in the spine. Overall, the majority of the lesions develop in the extremities and pelvis.21
An epidemiological study conducted in Sweden between 1971 and 1984 investigated possible changes in the typical features of 227 conventional osteosarcomas. The mean annual incidence was 2.1 per million. The male-to-female ratio of 1.6 : 1 remained unchanged over the study period, as did the location and distribution of the tumors. In the United States, the incidence of osteosarcoma is considered to be higher in males than in females, occurring at a rate of 5.4 per million persons per year in males versus 4.0 per million in females, with a higher incidence in blacks (6.8 per million persons per year) and Hispanics (6.5 per million), than in whites (4.6 per million).22
The only clear change over the study period was an increase in the age of patients beyond the classic peak age range of 10 to 29 years.23 In a recent study in the United States, the incidence rates and 95% confidence intervals of osteosarcoma for all races and both sexes are 4.0 (3.5 to 4.6) for the range 0 to 14 years and 5.0 (4.6 to 5.6) for the range 0 to 19 years per year per 1 million persons. Among childhood cancers, osteosarcoma occurs eighth in general incidence. Osteosarcoma has a bimodal age distribution, having the first peak during adolescence and the second peak in older adulthood. The first peak is in the 10- to 14-year-old age group, coinciding with the pubertal growth spurt. This suggests a close relationship between the adolescent growth spurt and osteosarcoma.24
Osteosarcoma develops in 10% of patients after the age of 60 years. This group composes the second peak of the bimodal age distribution curve. In these older patients, the anatomic region of presentation differs substantially from the sites of classic osteosarcoma. Whereas lesions develop in the region of the knee (the largest and most active physes) in more than 50% of patients with classic osteosarcoma, osteosarcoma at that site develops in only 15% of the older patients. Moreover, osteosarcomas in the older population characteristically present in regions that have had previous radiotherapy, underlying Paget disease of bone, fibrous dysplasia, or some other pathological abnormality. In many ways, the older group can be thought of as having “secondary” osteosarcoma.25
An estimated 2000 malignant bone tumors are diagnosed in the United States each year. Approximately 750 of these patients have classic or conventional osteosarcomas. Males are affected slightly more often than females. Classic osteosarcoma develops in females slightly earlier than in males, and there appears to be no race predilection.26 Although the common histologic presentation of malignant cells producing osteoid would suggest a homogenous group of tumors, the morphologic appearance can vary considerably, ranging from classic osteoblastic osteosarcoma (45% of cases) through fibroblastic (9%), chondroblastic (27%), anaplastic (17%), telangiectatic, low-grade central, and other osteosarcomas (2%).27
Etiological and Biological Considerations
Osteosarcoma is considered a sporadic complex genotype sarcoma, as distinguished from the balanced translocation-associated sarcomas (e.g., Ewing sarcoma). Many cell-cycle regulatory factors have been implicated, including p53, Rb, and others. Certain populations may, in fact, be at risk for developing osteosarcoma.24,28 Herein we provide a brief overview of the major regulatory pathways of the cell cycle with implications for sarcomagenesis in osteosarcoma.
The p53-ARF-MDM2 Pathway
In conjunction with the CDK4/6 inhibitor p16INK4A, mutations of the TP53 gene are the most recurrent genetic alterations associated with cancer.29 The TP53 gene encodes the well-known tumor-suppressor p53, which plays an important role in various regulatory processes involved with cell-cycle progression. Most importantly, p53 acts as an important negative regulator, facilitating growth arrest, senescence, and/or apoptosis when cells are exposed to genotoxic, cytotoxic, and/or physiological stresses.30,31 p53 is a transcription factor possessing two transcriptional activation domains and a DNA-binding domain that recognizes specific sequences. Following exposure to cellular stress, p53 activates expression of CDKN1A, which encodes the cyclin-dependent inhibitor p21CIP1. It is through the expression of p21 that p53 negatively regulates the cell cycle.32–35
The expression level and activity of p53 is regulated by two different proteins: MDM2 and p14ARF. MDM2 is an E3 ubiquitin ligase, which facilitates the degradation of p53 in a ubiquitin- and proteasome-dependent manner.36 Interestingly, MDM2 is a transcriptional target of p53, whose expression is increased in concert with increased levels of p53.37 In addition to promoting degradation, MDM2 can also inhibit p53 function through a direct protein–protein interaction, suppressing its transcriptional activity, as well as translation of the messenger RNA transcript itself. To combat these negative regulatory effects, p14ARF is able to positively regulate p53, in part through the negative regulation of MDM2. p14ARF can actually be found at the same gene locus as CDKN2A, which encodes the CKI p16.37,38 The two gene sequences do overlap but possess alternate reading frames and are independently regulated. ARF binds MDM2, preventing it from interacting with p53.7 Consequently, p53 becomes stabilized and its overall activity increases within the cell.39
The CIP/KIP Family of Cyclin-Dependent Kinase Inhibitors
The CIP/KIP family of proteins is the second group of cyclin-dependent kinase inhibitors (CKIs). There are three different members: p21CIP1, p27KIP1, and p57KIP2. Similar to the INK4 family of CKIs, CIP/KIP proteins inhibit the kinase activity of cyclin-dependent kinases (CDKs) by preventing their association with cyclin subunits as well as adenosine triphosphate molecules, both of which are required for the phosphorylation of target substrates.32,40 Unlike the INK4 family, however, CIP/KIP proteins are able to functionally inhibit multiple CDKs. For instance, both p27 and p57 can inhibit the kinase activity of CDK4/6 and CDK2, whereas p21 acts to control the function of both CDK2 as well as CDK1.33,41 Of the three family members, p21 is the most diverse, performing a variety of functions in addition to controlling CDK activity. For example, it was previously mentioned that p21 is a transcriptional target of p53.32–35 Following exposure to various stresses, p53 activates the expression of p21, thus inducing a DNA damage response.33 In addition, p21 has been shown to localize to the cytoplasm where it acts to inhibit the induction of apoptosis by interacting with proteins involved in mediating this process.
Other Important Players
The last two cell-cycle regulators that are discussed are C-MYC and Ki-67. Both of these proteins play crucial roles during cell proliferation but do so in very different ways. C-MYC influences several processes involved in cell-cycle regulation via its function as a transcription factor. For example, C-MYC, when bound to its partner MAX, has been shown to induce the expression of cyclins D1 and D2 as well as CDK4, subsequently promoting G1 phase progression.40,42 The MYC-MAX heterodimer can also support continued cell-cycle progression through the repression of multiple CKIs, including p15, p18, p21, and p27.43 Furthermore, C-MYC can increase the expression of E2F2 and cyclin A2, both of which affect the S-phase of cell cycles and contribute to overall proliferation.45–45 Similar to C-MYC, Ki-67 has been shown to be vital for cell proliferation.46 Ki-67 is a cell proliferation-associated nuclear antigen that is thought to contribute to cell-cycle progression via its involvement in ribosomal RNA and ribosome synthesis.11,46,47 Interestingly, Ki-67 is expressed in all of the phases of cell cycle (excluding G0), but whether or not it participates in other such related processes is currently unknown.
A small subset of osteosarcomas is hereditary.48 Osteosarcoma in siblings occurs in fewer than 1 in 1000 to 1 in 3000 osteosarcoma patients.49,50 Observation of two or more affected siblings in a family indicates an underlying genetic predisposition.50–57 When siblings in multiple generations are affected, an autosomal dominant disorder is most likely responsible. One example would be the hereditary form of retinoblastoma. Individuals with hereditary retinoblastoma (germline retinoblastoma gene mutation) have a 2000-fold risk for osteosarcoma in the second decade of life when compared with the general population.58–63
The gene for retinoblastoma (RB) has been localized to the long arm of chromosome 13 (13q14). The RB gene is recognized as the prototype of a tumor suppressor gene and has been implicated in the pathogenesis of a number of human neoplasms.64,65 A tumor suppressor gene normally functions by restraining cell (tumor) growth, so loss of function or inactivation of a tumor suppressor gene results in tumor growth. Loss of 13q14 (the RB gene) is thought to be responsible for the development of RB.64–68 A two-hit kinetic model for this class of genes was proposed by Knudson.69 For hereditary RB, the primary mutation in one RB locus occurs in germinal cells; for sporadically occurring RB, the primary mutation exists in somatic cells. The second step, responsible for malignant transformation, is the loss of function of the remaining normal homolog in somatic cells by some chromosomal rearrangement or mutation identified as loss of heterozygosity for markers in or around the RB gene.58,60–72 Molecular analyses of both sporadic osteosarcomas and osteosarcomas from patients with RB have revealed homozygous loss of RB gene function in a high percentage of cases.58,59,65–77 Assessment of loss of heterozygosity at the RB gene in a study by Feugeas and colleagues78 revealed that RB gene locus loss of heterozygosity could be an early predictive feature for osteosarcomas with a potentially unfavorable outcome. Osteosarcoma develops in 12% of patients with bilateral RB, yet as many as 70% of osteosarcomas have a dysfunctional RB gene product.79,80–85 Thus other oncogenes are likely implicated in the oncogenesis of osteosarcoma.
Several investigators have demonstrated that the mutational profiles of the RB gene in osteosarcoma are basically the same as those for RB and that mutation of the RB gene plays an essential role in the development of osteosarcoma.65,73 Besides loss of gene function at the locus on chromosome 13, however, loss of heterozygosity for other chromosomal loci, such as 3q, 17p, and 18q, has been implicated.73,86–91
Pathology and Pathways of Spread
Osteosarcoma is a high-grade sarcoma comprising malignant osteoblasts that vary in size and shape and have bizarre mitoses. Proposed histologic grading systems for osteosarcoma appear to be of little value.92 Attempting to grade an osteosarcoma presents many difficulties that limit the usefulness of any grading system. For example, many tumors are heterogeneous, and tissues sampled from separate areas of the same tumor may give different impressions. The number of mitoses, the degree of cellularity, and cellular anaplasia or pleomorphism can differ from site to site within the same tumor. Tumors of identical histologic appearance often differ in their clinical behavior. All “classic” or conventional osteosarcomas are considered high-grade. Osteosarcoma is a vascular tumor, and the tumor osteoblasts produce tumor osteoid or woven bone (Fig. 92-1, A and B). These tumors are poorly differentiated and may take on a fibroblastic or chondroblastic appearance on light microscopy (Fig. 92-1, C and D), but if there are areas of bone formation, they are considered osteosarcomas. In fact, a high-grade chondroblastic sarcoma in a child or adolescent on biopsy is considered to be an osteosarcoma (and treated as such) until proved otherwise from examination of the entire specimen (chondrosarcomas are extremely unusual in children). The tumor usually originates in the metaphysis of the bone and percolates between the preexisting trabeculae of bone, incompletely destroying the existing bone, presumably because of its rapid growth (seen well in Fig. 92-1A). The tumor eventually follows the vascular Haversian and Volkmann canals in the cortex, partially resorbing the normal cortex and replacing it with tumor bone as it spreads to the adjacent soft tissue. The periosteum is lifted and tries to respond, but the response is incomplete, leading to the appearance of the Codman triangle on a radiograph. It may also cause perpendicular striations of bone, the so-called starburst appearance of osteosarcoma. Proximally, the tumor ends fairly sharply in the medullary cavity, but “skip metastases” may be detectable in the marrow surrounding the tumor in a small proportion of patients. The physis or growth plate is a relative barrier to tumor spread, but because the open physis has vascular channels, it is well documented that the tumor will cross the growth plate and enter the epiphysis. The articular cartilage is a more definitive barrier, and osteosarcomas seldom cross the articular cartilage unless there has been a fracture. It may spread into the joint at the periphery of the cartilage or enter the joint along ligaments such as the cruciate ligaments of the knee, but this is a relatively rare event (Fig. 92-2).
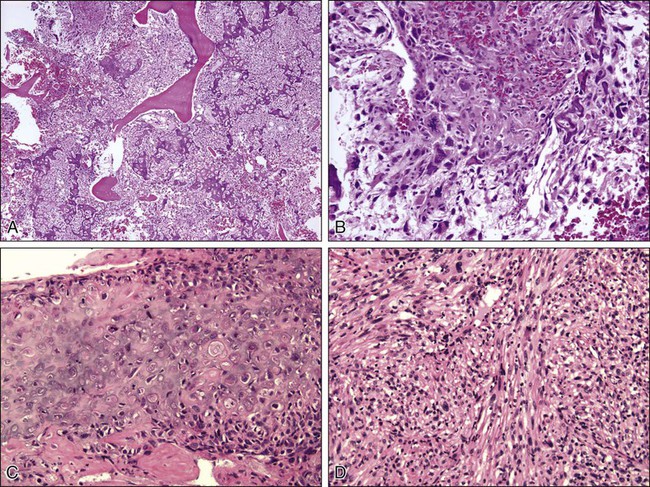
Telangiectatic osteosarcoma is a predominantly radiolucent, destructive osteosarcoma variant. Histologically, it is composed of single or multiple dilated spaces containing blood or degenerated tumor cells and lined by anaplastic, mitotically active sarcoma cells.79 Telangiectatic osteosarcoma must be differentiated from aneurysmal bone cyst, to which it can be similar in appearance both radiographically and histologically.
A review of 124 patients with telangiectatic osteosarcoma spanning the years 1921 to 1979 suggested no differences in survival compared with patients with conventional osteosarcoma. Further analysis demonstrated that the favorable outcome in 17 of the patients with telangiectatic osteosarcoma was related to their being treated with multiagent chemotherapy. Twenty-five patients had received this therapy, and 17 were free of disease at 5.5 years, demonstrating the response to chemotherapy in this highly vascular tumor.93A more recent evaluation of 323 patients with telangiectatic osteosarcoma revealed that although these tumors may be at higher risk for pathological fracture, with multimodal therapy, outcomes were similar to other osteosarcomas even with the negative prognostic factor of the fracture itself.93a
Clinical Manifestations, Patient Evaluation, and Staging
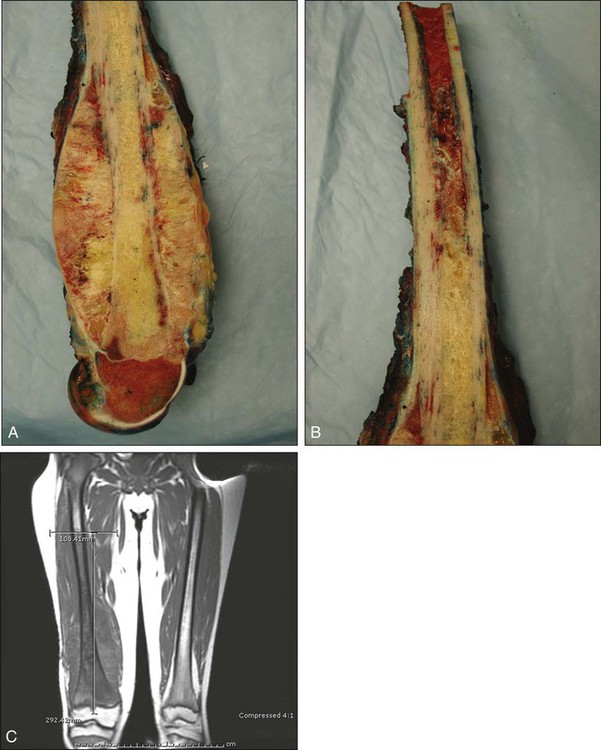
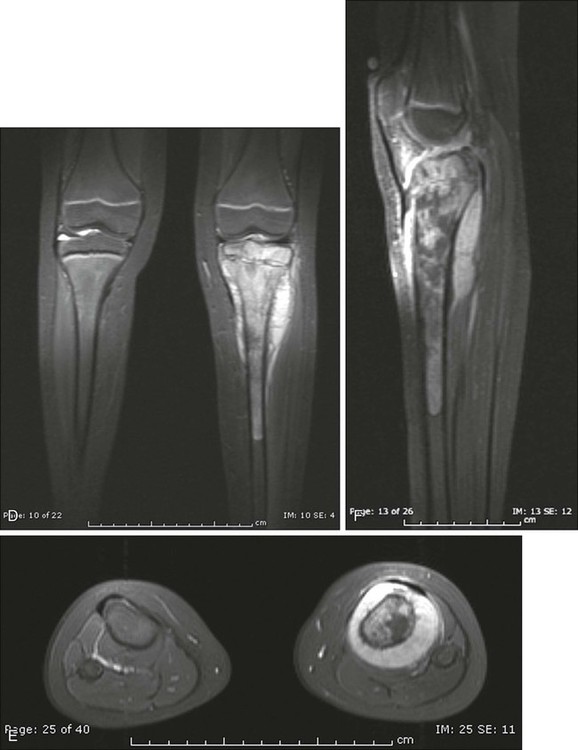
The plain radiograph is the best diagnostic tool. Osteosarcomas may either completely destroy the bone (radiolucent lesion) or replace the bone with a blastic response (radiodense), but they most often do both. The radiograph shows areas of destruction of the host bone and blotchy densities of new (tumor) bone production. The lesion is most frequently in the metaphysis of a long bone in the adolescent or child and in the flat bones in the adult, but any bone can be involved at any age (Fig. 92-3, A and B). The tumor is usually large, destroys the cortex, and is associated with a soft-tissue mass. The mineralization of the matrix is often apparent, but because these tumors can contain areas of chondroblastic and fibroblastic as well as osteoblastic differentiation, the pattern on mineralization might not be that of bone.
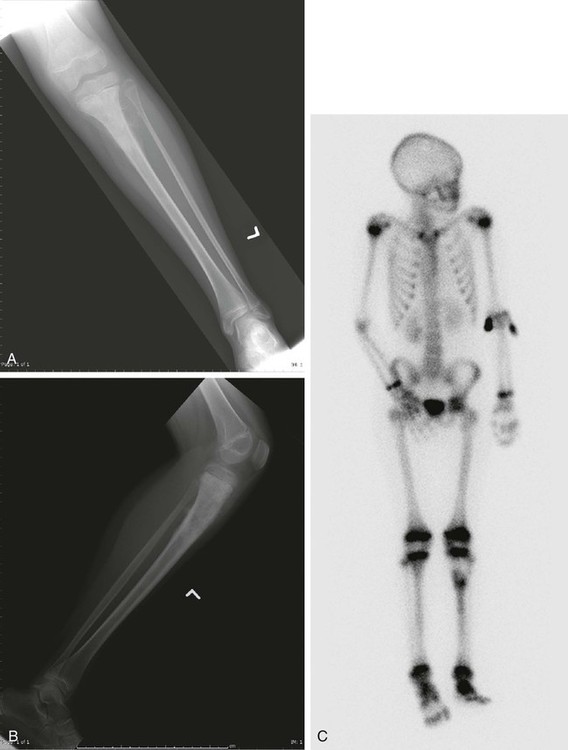
There are no blood laboratory studies that aid in the diagnosis of osteosarcoma, but it has been shown in several large series of patients that an elevated level of alkaline phosphatase and/or lactic dehydrogenase is associated with a worse prognosis.96–96
MRI is the single most useful study in evaluating the intraosseous and extraosseous extent of the primary tumor and in detecting intramedullary or transarticular “skip” metastases. It has been well demonstrated that MRI better identifies the edema (high water content) in and around the reactive zone of the pseudocapsule illuminating the potential surgical margin. MRI has also been shown to be superior to CT in displaying the medullary canal extent of the tumor, suspected “skip” lesions, soft-tissue extension, and overall anatomic location of an extremity tumor (see Fig. 92-3, D to F).97 MRI, with or without magnetic resonance angiography, is the single most valuable tool for planning limb-sparing surgical procedures.100–100 The use of MRI has significantly improved radiologic staging and presurgical planning.101,102 Serial MRI studies are less reliable, however, in evaluating tumor response to primary chemotherapy, and are more predictive of a poor rather than a good response. By demonstrating an increase in size of the tumor, more bone destruction, and soft-tissue invasion on serial studies, MRI is more accurate as a measure of a poor chemotherapy response.7,103,104
Metastases to bone and/or lung are usually assessed by a whole-body 99mTc bone scan and a complete CT of the chest and mediastinum. The 99mTc bone scan has been considered superior to other imaging studies for surveying the skeleton for metastatic or multiple lesions and for later detecting the development of skeletal metastases (see Fig. 92-3C).105
PET scans have been used in an attempt to stage and separate high-grade from low-grade tumors.106 Brenner and associates107 summarized the current usefulness of 18F-fluorodeoxyglucose (18F-FDG) PET in patients with osteosarcoma. High-resolution CT has been shown to be superior to 18F-FDG PET for detecting pulmonary metastases and is not recommended to detect bone metastases except when a suspected “skip” lesion has been identified on MRI. PET does not make it possible to differentiate between high- and low-grade osteosarcoma. It could be useful, however, in determining the appropriate area to biopsy to identify viable representative tumor tissue and in distinguishing benign aggressive lesions from other lesions where local recurrence is likely.
18F-FDG PET may be most useful to determine the response to neoadjuvant chemotherapy and to demonstrate a region of viable tumor.108 The timing between the initiation of preoperative chemotherapy and the point at which 18F-FDG PET becomes predictive remains uncertain. The response to soft-tissue postchemotherapy inflammation and healing surrounding the tumor must be characterized before this technique becomes predictive of preoperative response to chemotherapy. PET also has potential usefulness in patient follow-up, differentiating postoperative tissue changes and possible tumor recurrence especially in patients with metallic reconstructions. Orthopedic implants often hamper assessment by CT or MRI, and sequential 18F-FDG PET scans could differentiate between recurrent tumors and the normal healing process. Also, because 18F-FDG PET provides scanning of the entire patient, it may be useful when combined with CT of the chest to detect first evidence of pulmonary metastasis.109A recent study showed that PET correlates only moderately to histologic response, but may be predictive of progression-free survival.11 Once the staging workup is complete, a biopsy is performed. The procedure can be either an open biopsy or a needle biopsy (core-needle or fine-needle aspirate) depending on the experience of the surgeon, the interventional radiologist, and the pathologist. If an open biopsy is performed, it should be done by the surgeon who will be responsible for local control, with a view to placement in a site that can be resected with the definitive specimen. If a core-needle biopsy is performed by an interventional radiologist (which is becoming more frequent), the radiologist and the surgeon should agree on the placement of the needle track. Communication between the surgeon and interventional radiologist is essential, as is communication with the entire multidisciplinary team to determine whether tissue should be sent for cytogenetics or other special biological studies. Table 92-6 provides a diagnostic workup algorithm.
Table 92-6
Diagnostic Workup Algorithm for Bone Sarcomas

Once the biopsy results are available, the sarcoma can be staged as discussed in the introduction.
Primary Therapy
Adjuvant Chemotherapy
The use of adjuvant chemotherapy for osteosarcoma was introduced in the 1970s, and after initial concern relative to its efficacy, a randomized trial confirmed its benefit. It is now established as an essential part of the treatment of osteosarcoma. Initially used as an adjuvant following amputation or resection of a tumor, chemotherapy is now used in the neoadjuvant setting, although no clear survival benefit to that approach has been documented.18,110 The use of neoadjuvant chemotherapy does allow the assessment of histologic response to the chemotherapy, which has prognostic implications, allows time for planning of limb salvage operations, and makes those resections easier and probably safer.
A number of single- and multiinstitutional studies have reported the results of treatment protocols, including multiagent neoadjuvant chemotherapy and limb-sparing surgery.113–113 The specific drugs and their methods of administration are important. Delépine and colleagues114 published a meta-analysis of the relationship of total dose and dose intensity of methotrexate from nine single-institution and nine multiinstitution randomized trials. They concluded that both methotrexate dose and dose intensity had major prognostic value. Intravenous administration appears to be as effective as the intraarterial route of administration.110,115 A meta-analysis of 16 regimens published by a group from the National Cancer Institute found that dose intensity was the most important determinant of a favorable outcome, defined as a good histopathological response to neoadjuvant chemotherapy.116 Other studies likewise support the importance of doxorubicin treatment in patients with osteosarcoma.117,118
The use of preoperative chemotherapy was extended to selected patients in an attempt to contain growth of the primary tumor while awaiting construction of a custom prosthesis (usually 12 to 16 weeks).111,113,119 The sequence of several courses of primary chemotherapy and subsequent surgery afforded the opportunity to examine and histopathologically grade the tumor tissue response to multiple chemotherapy agents. Patients with greater than 90% tumor necrosis (good) were shown to have a better disease-free survival than those with a poor (<90%) response to chemotherapy.19,120,121 Goorin and associates18 completed a study of 106 patients admitted to the Pediatric Oncology Group Study 8651 who were randomly assigned to immediate surgery or to preoperative chemotherapy with high-dose methotrexate, doxorubicin, and cisplatin for 2 cycles (10 weeks). Six patients were excluded from analysis. Of the remaining 100 patients, 45 were randomly assigned to immediate chemotherapy, and 55 were randomly assigned to immediate surgery. At 5 years follow-up, 67 patients remained disease-free. At 5 years, the projected EFS rate was 65% for immediate surgery and 61% for presurgical chemotherapy. There was no apparent advantage to the group that received preoperative chemotherapy. This was a difficult study to complete because at the time, there was a surgical bias toward neoadjuvant chemotherapy for limb salvage patients, making accrual slow. The number of limb-preserving procedures was nearly equal in both groups (55% with immediate surgery and 50% with presurgical chemotherapy). Although the percentage of patients in the two groups was similar, the overall percentage of patients receiving limb-sparing procedures was much lower than the usual 60% to 90% limb-preserving procedures reported. Only one local recurrence was reported among all 100 patients.
Surgical resection of the primary tumor is usually planned for 10 to 12 weeks following induction chemotherapy. Limb preservation is elected if the tumor responds favorably; that is, there is evidence of a clinically favorable response, including diminished local pain; radiographic evidence of mineralization of the previously unmineralized soft-tissue portion of the tumor on radiographs; and MRI evidence of retention of a normal fatty tissue plane between vascular and neural structures and the tumor. However, if the lesion enlarges and the vessels become involved secondarily (poor response), ablative surgery is recommended. Once the operative wound heals (usually within 2 to 3 weeks), adjuvant chemotherapy is initiated and is maintained for approximately 40 weeks, depending on the particular study.122–127 Wilkins and colleagues128 described their results at two institutions with a dose-intensified neoadjuvant protocol using intravenous doxorubicin and intraarterial cisplatin administered until a maximum angiographic response was observed (usually four courses). Despite showing results similar to others previously reported, it requires extraordinary resources, making it unlikely that a similar study could be carried out in an extensive cooperative manner.129,130
Long-term outcomes were studied by Bacci and coauthors,131 who reported the results of treatment of 164 patients with nonmetastatic extremity osteosarcoma who were followed up for a minimum of 10 years. Preoperative chemotherapy consisted of high-dose methotrexate, cisplatin, and Adriamycin. Postoperatively, good responders (≥90% tumor necrosis) received the same three drugs, whereas poor responders (<90% tumor necrosis) received ifosfamide and etoposide in addition to the three-drug chemotherapy regimen. Follow-up showed that 101 patients (61%) remained continuously free of disease, 61 had relapsed, and two had died of Adriamycin-related cardiotoxicity. There were no differences in outcome between good and poor responders.
In 1997, Bramwell132 conducted a review to provide answers to important questions about the role of chemotherapy in the management of patients with nonmetastatic osteosarcoma of the extremities. Many studies were analyzed, including a study by Link and coworkers14 that clearly demonstrated the role of adjuvant chemotherapy in the treatment of patients with conventional osteosarcoma. The role of adjuvant chemotherapy was also confirmed by another study that provided additional objective evidence for the efficacy of multiagent chemotherapy in preventing and/or delaying relapse.133 The five studies that were reviewed by Bramwell14,134–137 approached the results of Rosen and associates121; three were multicenter studies and two were reports from a single institution. Bramwell concluded that although the Rosen T10 regimen is complex and toxic, it can be given in a multicenter setting without apparent major compromise in efficacy.
Bramwell concluded that reports of outcomes with multiagent chemotherapy were similar to regimens containing the most active drugs (doxorubicin and cisplatin) that were used in two consecutive European Osteosarcoma Intergroup protocols when compared with results from multicenter studies using the T10 regimen.137,138
It is apparent that histopathological response to neoadjuvant chemotherapy correlates with improved survival. The reports by Picci and colleagues139 (Bologna, 355 patients), Kempf-Bielack and coworkers140 (Cooperative Osteosarcoma Study Group, 504 patients), and Delépine and associates141 (Paris, 112 patients) all demonstrated that a good response to neoadjuvant chemotherapy was an independent prognostic factor. Meyers and colleagues142 reported the relationship between duration of preoperative chemotherapy and histopathological response. In univariate analysis, the duration of preoperative chemotherapy did not correlate with relapse-free survival. With longer preoperative treatment, a greater proportion of patients had a favorable histopathological response to therapy, but the correlation of the response with outcome decreased. Bramwell132 postulated that with prolonged preoperative chemotherapy, a good response to chemotherapy might lose its prognostic significance. The CCG-782 study published by Provisor and coworkers143 involving 268 patients with nonmetastatic osteosarcoma of the extremity used the resected tumor histologic response to neoadjuvant chemotherapy to determine postoperative chemotherapy. In 206 patients, the tumor was morphometrically assessed for residual viable tumor; 28% displayed a good (<5% viable tumor) histologic response, whereas the remaining patients were judged to have a poor histologic response (>5% residual viable tumor). The patients who had a good response had an 8-year postoperative EFS rate of 81% and a survival rate of 81%. Patients with a poor histologic response had an 8-year postoperative EFS rate of 46% and an overall survival rate of 52%. They concluded that event-free and overall survival appeared to be related directly to histologic response to neoadjuvant chemotherapy. If that is true, then increasing the percent necrosis by intensifying chemotherapy should improve outcome even further. A recent study compared standard chemotherapy with an intensified arm to assess this contention. Conventional treatment consisted of six 3-week cycles of cisplatin (100 mg/m2 by 24-hour infusion) and doxorubicin (25 mg/m2 per day by 4-hour infusion for 3 days). Intensified therapy was treatment with identical total doses of cisplatin and doxorubicin, planned as six 2-week cycles supported by granulocyte colony-stimulating factor. In this study, 497 eligible patients were evaluated, and good histologic response (>90% tumor necrosis) was observed in 36% of standard arm and 50% of the intensified arm. However, there was no evidence of a difference in overall survival between the two treatment regimens. The study found that intensification of chemotherapy could increase the percentage of necrosis but not progression-free survival or overall survival.144 Another study looked at increasing the dose of chemotherapy with the same drugs in 196 osteosarcoma patients. The authors failed to find a difference in percentage of necrosis, 5-year EFS, or overall survival, suggesting that response to chemotherapy is related to the specific drugs used but that increasing the intensity is of little benefit.145 The tumors either respond or do not, suggesting biological differences in the tumors.
Currently, there are no strong data showing that changing drugs in the poor histologic responders improves the survival of those patients. A retrospective study reported by Benjamin and associates146 compared outcomes from three consecutive cohorts of patients receiving intraarterial cisplatin and intravenous doxorubicin between 1980 and 1992. In cohort 1 (37 patients), the postoperative chemotherapy was the same. In cohort 2 (59 patients), the postoperative chemotherapy for poor responders consisted of high-dose methotrexate, bleomycin, cyclophosphamide, and dactinomycin alternating between doxorubicin and dacarbazine. In cohort 3 (28 patients between 1988 and 1992), poor responders were managed with three alternating regimens of high-dose methotrexate, ifosfamide, and alternating doxorubicin and dacarbazine. The significant 5-year relapse-free survival rate for poor responders for the three cohorts was 13%, 34%, and 67%. Bramwell believes that although the results appear significant, they could also be explained on the basis of small sample size, increasing dose intensity, total dose, and increased duration of preoperative chemotherapy.147
A recent meta-analysis of North American and European studies of nonmetastatic osteosarcoma studies showed that three drug regimens had better event-free and overall survival rates compared with two drug regimens (EFS 48% and 58%, and overall survival 62% and 70%, respectively). Adding a fourth drug (e.g., ifosfamide) did not improve outcome further, but dose intensification improved histologic response to chemotherapy. There was no benefit to intraarterial chemotherapy.110
A randomized multiinstitutional study of patients from North America and Europe is attempting to address this question further by randomly assigning poor responders to continued preoperative therapy or an intensified arm in which ifosfamide and etoposide are added to the experimental arm.148 The good responders will be treated with continued preoperative therapy with or without the addition of interferon-α. This is the first study to randomize the treatment of patients following determination of histologic response. Results are pending.
The issue of whether other agents incorporated into intensive multiagent regimens will improve survival further is less clear. Preliminary trials incorporating ifosfamide into multiagent chemotherapy appeared promising,149–153 but a recent study of the Children’s Oncology Group patients who were randomly assigned to multiagent regimens that either contained or did not contain ifosfamide shed some doubt on this.154 In this study, 677 nonmetastatic osteosarcoma patients were treated with one of four prospectively randomized treatments. All patients received identical cumulative doses of cisplatin, doxorubicin, and high-dose methotrexate and underwent definitive surgical resection of the primary tumor. They were randomly assigned to receive or not to receive ifosfamide and/or muramyl tripeptide (MTP), a pulmonary macrophage stimulant. The addition of ifosfamide in this dose schedule to standard chemotherapy did not enhance EFS. The addition of MTP to the ifosfamide arm appeared to add further benefit, but the study was not designed to test this, and this question remains uncertain but intriguing. Whether MTP should be included into standard treatment protocols remains a topic of debate at this time.155 There is a suggestion in a small cohort of patients with metastatic osteosarcoma, that addition of liposomal muramyl tripeptide phosphatidylethanolamine (L-MTP-PE) may enhance event-free and overall survival, but this study was not of sufficient size to be definitive.156
To test whether intensified ifosfamide therapy might be of benefit, a study of 182 patients treated with of 2 cycles of high-dose ifosfamide (15 g/m2), methotrexate (12 g/m2), cisplatin (120 mg/m2), and doxorubicin (75 mg/m2) was conducted. Postoperatively, patients received 2 cycles of doxorubicin (90 mg/m2) and 3 cycles each of high-dose ifosfamide, methotrexate, and cisplatin (120 to 150 mg/m2). Granulocyte colony-stimulating factor support was mandatory after the high-dose ifosfamide+cisplatin+doxorubicin combination. No disease progression was recorded during primary chemotherapy. With a median follow-up of 55 months, the 5-year probability of EFS was 64%, and the overall survival rate was 77%. The addition of high-dose ifosfamide to methotrexate, cisplatin, and doxorubicin in the neoadjuvant setting was found to be feasible, but associated with major renal and hematologic toxicities. Survival rates were similar to those obtained with four-drug regimens by using standard dose ifosfamide.157 Growth factors do appear to offer a benefit in increasing dose intensity. In a pilot study, the European Osteosarcoma Intergroup demonstrated the use of granulocyte colony-stimulating factor–supported increased dose intensity, making chemotherapy every 2 weeks feasible.158 Thrombocytopenia remained dose limiting, and whether attainable dose intensification improves survival remains uncertain.
The histologic subtypes of osteosarcoma also appear to influence response rates. Bacci and colleagues159 at the Rizzoli Institute in Bologna correlated the histopathological response to preoperative chemotherapy in 1058 patients with conventional osteosarcoma of the extremity. They classified the tumors as osteoblastic (70%), chondroblastic (13%), fibroblastic (9%), and telangiectatic (6%). At diagnosis, 911 patients had localized disease, and 147 had resectable pulmonary metastases. The response to preoperative chemotherapy was good (90% or more tumor necrosis) in 59% of patients and poor (<90% tumor necrosis) in 41%. Notably, the rate of good responders was significantly higher (P = 0.0001) in patients with fibroblastic (83%) and telangiectatic tumors (80%) than in those with osteoblastic (62%) and chondroblastic (60%) tumors. In all subtypes (excepting the chondroblastic), the 5-year overall survival rate was significantly higher (P = 0.0001) in good responders (68%) than in poor responders (52%).
Essential to the improvement in survival in osteosarcoma is the combination of effective surgical resection along with the use of chemotherapy. Jaffe and colleagues160 in the Department of Pediatrics at the University of Texas M.D. Anderson Cancer Center attempted the cure of 31 patients with nonmetastatic osteosarcoma. Their protocol for selection included initial treatment with chemotherapy comprising high-dose methotrexate and leucovorin rescue (MTX-LF) in three patients and intraarterial cisplatin in 28 patients. After response at 3 months, entry into the study was permitted, and chemotherapy treatment was maintained for a total of 18 to 21 months with a combination of MTX-LF, intraarterial cisplatin, and doxorubicin. Only 3 of 31 patients (10%) were cured exclusively with chemotherapy. Four additional patients requested surgical extirpation of the tumor after the cessation of chemotherapy. Histopathological examination revealed no evidence of viable tumor. Adding these patients to the three mentioned previously yielded a total of seven patients (23%) who had a cure from chemotherapy alone. As the expected cure rate with conventional strategies is 50% to 65%, the authors concluded that their results do not justify the option of current forms of chemotherapy as exclusive treatments for osteosarcoma.
In summary, although great improvements have been made since the 1970s in the outcome of patients with nonmetastatic osteosarcoma, it seems that a plateau has been reached with little new improvement in outcome in recent years. It is likely that future progress will result from novel treatments capitalizing on knowledge of the molecular biology of osteosarcoma and the development of new, targeted agents to specific molecular targets. Many have been proposed, but none are in routine clinical use nor have they been successfully studied in clinical trials. This remains an area of intense interest and research.161,162
Morphometric analysis of pathological specimens was instituted after it was recognized that chemotherapy-induced necrosis correlated with clinical outcome. Picci and coauthors163 described their methodology in 50 patients. Necrosis was divided into three categories: good (100% to 80% necrosis), fair (80% to 50% necrosis), and poor (<50% necrosis). Other authors have used different classifications. Depending on the system that is used, tumor necrosis ranging from 60% to 95% is common. The information gained has prognostic significance. Winkler and associates119 were early to report that patients with unfavorable pathological responses to preoperative chemotherapy experienced a poorer (49%) disease-free survival than did patients with a favorable pathological response (87%; P = 0.005). Glasser and coworkers164 later reviewed 279 consecutive patients with stage II osteosarcoma of the appendicular skeleton who were treated between 1976 and 1986. Continuous disease-free survival for the overall group was 70% at 5 years and 69% at 10 years. The only independent predictor of a favorable outcome was found to be the histopathologic response to chemotherapy as defined by pathological review of the surgical specimen.
A literature review by Davis and colleagues165 attempted to identify prognostic factors that could influence survival in patients with nonmetastatic high-grade osteosarcoma of the extremities. Eight previously reported large series of patients included sufficient data to evaluate the numerous identified variables. Only two variables proved significant to univariate analysis: tumor size and chemotherapy-induced tumor necrosis after primary chemotherapy. Only tumor necrosis remained significant after multivariate analysis, however. Other large series have demonstrated similar prognostic responses to neoadjuvant chemotherapy.111,120,123–127,142,143,164
Table 92-7 summarizes recent data regarding treatments for nonmetastatic osteosarcoma reported by 10 internationally recognized institutions. The table suggests that results have improved owing to management by multiagent neoadjuvant chemotherapy combined with adequate local surgery performed by experienced musculoskeletal surgeons. Several conclusions can be drawn from these studies:
Table 92-7
Summary of Recent Studies of Patients Treated for Nonmetastatic Osteosarcoma
| Institution | No. of Nonmetastatic Extremity Total* | Resection* | Rotationplasty* | Amputation* | Local Control* | Disease-Free Survival |
| M.D. Anderson | 60 | 31 | — | 28 | 4 (7%) | 74% (TIOS-III) |
| Dana Farber | 74 | 36 | — | 38 | 1 (1%) | 87% |
| Memorial Sloan-Kettering | 271 | 159 | 9 | 103 | 18 (7%) | 77% |
| Vienna University Clinic | 73 | 41 | 22 | 10 | 2 (3%) | 76.7% |
| Birmingham Service | 99 | 74 | — | 15 | 4 (4%) | 48% |
| French Study | 100 | 79 | 3 | 18 | 1 (1%) | 82% |
| Brazil Group | 92 | 34 | — | 58 | 6 (7%) | 41.1% |
| Mie Japan | 52 | 25 | — | 27 | 2 (4%) | 58.9% |
| COSS 86 | 159 | 65 | 39 | 44 | 3 (2%) | 84% |
| Rizzoli Institute | 125 | 106 | 9 | 10 | 1 (0.08%) | 87% |
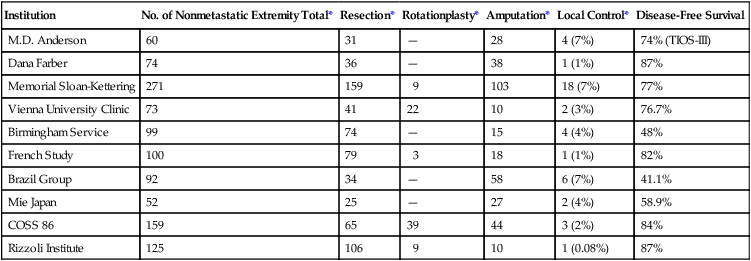
• Patients who request limb-sparing operations do not appear to be at greater risk for development of local or distant relapse than patients who have transmedullary amputations.
• The administration of sequential multiagent adjuvant chemotherapy has significantly improved the disease-free survival for patients without metastasis.
• The risk of local recurrence appears to be no greater in patients who complete limb-preserving procedures than for those who have an amputation.123,125,142
We do seem to have reached a plateau, however, and few improvements in survival have been made recently. This situation has led to the refinement of prognostic factors and the search for potential biological factors that might provide targets for better treatment. A recent study looked at 1054 patients who had been enrolled in cooperative trials in North America and showed that patients older than 18 years of age had a statistically worse survival outcome that could not be explained by tumor size, histologic response to chemotherapy, or metastatic disease at presentation.28
Relationship of Surgical Margins, Neoadjuvant Chemotherapy, and Local Recurrence
The role of neoadjuvant chemotherapy in facilitating limb preservation appears to be well established.139,166 The relationship between margins, chemotherapy, and local recurrence is not as straightforward. Although in general local recurrence is higher in patients with inadequate margins, some patients who are so treated do not have recurrence, whereas some patients with wide or radical margins do recur or persist locally. The effect of neoadjuvant chemotherapy in this regard is suspected to be beneficial, but only retrospective data are available. Four of the institutions reporting data shown in Table 92-7 also provided data on the pathological margins achieved at surgery and correlated the surgical margins with locally recurrent disease. Of the 271 margins reported by Memorial Sloan-Kettering, 266 were adequate (261 wide and 5 radical), and five were inadequate (three marginal and two intralesional). Of the 18 locally persistent tumors, however, all developed in patients who were thought to have wide margins. There were no locally recurrent tumors from the five known inadequate surgical margins.125 The University of Vienna group reported 61 margins as wide, with 12 inadequate (nine marginal and three intralesional) margins. Only two local recurrences developed, one in a patient having an intralesional amputation and the other with a marginal resection and reconstruction.
A similar review from Birmingham, England, revealed 23 marginal and 15 intralesional margins in 99 patients, with a 4.5% recurrence rate.125 A French study reported 100 patients with nonmetastatic osteosarcoma managed by neoadjuvant chemotherapy and surgery with “numerous” marginal margins but no intralesional margins. They reported one local recurrence.126 A Rizzoli Institute study, reporting 125 patients retrospectively, identified 15 patients with inadequate margins. Only one patient had a local recurrence.123
Investigators from the Rizzoli Institute in Bologna have shown by multivariate analysis that the incidence of local recurrence in 355 patients was related closely to surgical margins (P < 0.0001) and response to preoperative chemotherapy (P < 0.0001). There were 28 patients who experienced local recurrence (7%), three of whom survived (11%). Six of 10 patients who did not receive preoperative chemotherapy had local recurrence. In the other limb salvage procedures, 110 patients had wide margins, 12 were marginal, seven had intralesional margins, and seven had wide contaminated margins. Although 27 patients had inadequate margins, only three developed local recurrence, all within the first 2 years after diagnosis.127
To better illustrate the relationship between surgical margins and local recurrence, Picci and colleagues139 reported on a single-institution study retrospectively reviewing 355 patients with nonmetastatic high-grade osteosarcoma of the pelvis and extremities. The average length of follow-up was 65 months for surviving patients.
Pathological review demonstrated less-than-wide margins in 65 of the 355 patients. The most common anatomic site for inadequate margins was the popliteal region near the vascular and neural structures, where 20 of 140 patients were found to have inadequate margins (either marginal or intralesional). Only 7 of the 20 patients had local recurrence, however. Locally persistent disease developed in only 3 of 15 patients with inadequate margins associated with lesions around major joints locally persistent disease. The intramedullary canal was the site of inadequate margins in 20 of 237 patients, and local recurrence developed in 6 patients. It is of interest that 7 of the 11 patients with intralesional surgical margins and 16 of the 21 with contaminated margins did not have local recurrence. These findings could not be explained on the basis of poor survival or early death. The observation is believed to be related to the effectiveness of preoperative chemotherapy at producing tumor necrosis and the development of a “mature” capsule surrounding the tumor where satellite tumor nodule formation had been suppressed.167
An analysis of a more recent group, 164 patients with nonmetastatic osteosarcoma of the extremities, also came from the Rizzoli Institute. Limb-sparing procedures were performed in 136 patients (83%), 18 patients (11%) had amputation, and 10 (5%) had rotationplasty. The surgical margins were reviewed. In amputations, 18 margins were wide or greater; in rotationplasty, nine were wide and one was intralesional. In the limb salvage procedures, 110 patients had wide margins, 12 were marginal, seven had intralesional margins, and seven had wide contaminated margins. Although 27 patients had inadequate margins, only three experienced local recurrence, all within the first 2 years after diagnosis.127
Surgical Treatment
To identify the potential risks of awaiting limb-sparing surgery versus immediate amputation, the results of data on 279 patients treated at Memorial Sloan-Kettering between 1975 and 1984 were reviewed retrospectively. Sixty-three patients who completed primary surgery and adjuvant chemotherapy were compared with patients who had primary chemotherapy followed by surgery and adjuvant chemotherapy. Univariate analysis showed no difference in outcome between patients who had limb-sparing procedures and those who had amputations (P = 0.34).142
A multiinstitutional retrospective study evaluating patients with nonmetastatic osteosarcoma of the distal femur reported treatment results for 227 patients who were managed by limb-sparing procedures, above-knee amputation, and hip disarticulation between July 1975 and June 1980. The Kaplan-Meier estimates for the three surgical groups revealed no significant difference in continuous disease-free and ultimate survival rates for each group (Mantel-Cox test, P = 0.8) after a median follow-up of 5.5 years. The continuous disease-free survival rate for the entire group was 42%, with an overall survival rate of 55% at 5 years. The researchers concluded that limb-sparing procedures for osteosarcoma of the distal end of the femur did not compromise either disease-free survival or overall survival. Outcome results showed that one-third of patients with limb-sparing procedures required at least one additional surgical procedure, and one-fourth eventually required amputation.168
A follow-up of the aforementioned study 8 years later reviewed the original 227 patients. In this study, 213 patients had been classified as having stage IIB osteosarcomas. Seventy-three patients had limb-preserving procedures, 115 had above-knee amputations, and 39 had hip disarticulations. Eighty-four percent of patients were followed up for a minimum of 10 years. The Kaplan-Meier estimate of the disease-free survival rate for all patients at 10 years was 41%. Fourteen of the original 17 patients experiencing a local recurrence in the first study did so within the first 2 years after the index procedure, and only one of the original 17 patients survived. There were nine local recurrences after above-knee amputations and eight after limb-sparing procedures. No patient who had a hip disarticulation (radical margin) experienced a local recurrence. Although the function of patients with limb-sparing procedures was superior to that of both the amputation and disarticulation groups, no differences could be identified regarding patient acceptance or psychosocial outcome (quality of life) among the three operative groups.169
Limb-preserving procedures are currently performed in approximately 60% to 90% of osteosarcoma patients with nonmetastatic extremity tumors, in contrast to the previously reported high amputation rates. Advances in chemotherapy, imaging technology, implant design and materials, and subspecialization in orthopedic oncology have reversed the trends of previous decades. Limb preservation, however, has not altered disease-free survival rates when compared with ablative procedures.171–171 Local recurrence rates after neoadjuvant chemotherapy and resection or amputation appear to be similar; these are low-frequency but still serious events (0.8% to 7%). Although limb-sparing surgery in appropriately selected patients appears to be safe from an oncologic viewpoint, it is not clear that functional outcome and quality of life (QOL) are superior in limb salvage patients compared with those who have undergone amputation. The long-term outcomes and reoperation rate (including eventual amputation) of the various limb salvage constructs are unknown. Most patients prefer to keep their limbs, however, and limb-sparing procedures are now routinely offered.
Problems associated with limb sparing include an increased early complication rate of 25% to 35%.123,127,172–174 Many reconstruction alternatives are available, and the method that is chosen depends on such variables as patient age and employability, tumor location and size, and the potential of the elected procedure to provide curative margins. Of paramount importance are recognition of the patient’s desires and discussion of realistic expectations. In addition, the surgeon’s experience and expertise play a role in the choice of procedure.
Lindner and associates,175 from the University of Muenster, reported their results of a study with 133 patients who had high-grade osteosarcoma of the extremities treated with intravenous neoadjuvant chemotherapy and surgery between 1978 and 1994. Seventy-nine patients had limb-preserving procedures, including 32 with endoprosthesis, 39 with allograft replacement, six with autograft reconstruction, and two with shortening procedures. Twenty-one patients had rotationplasty, and 33 patients elected amputation. With the MSTS (1993) functional evaluation scale, major complications were experienced after all procedures; 20 of 32 patients with endoprosthetic procedures experienced a major complication, and six of the 20 required removal of the prosthesis. Twenty of the 39 patients with allografts experienced a major complication, and six of the 20 patients required removal as well. Ten of the 21 patients with rotationplasty also experienced major complications, but none required revision to amputation. Eight of the 33 patients who were treated by transmedullary amputation experienced a major complication, and three of the eight required a more proximal reamputation. Lindner and associates concluded that the extent of preoperative primary tumor necrosis, surgical margins, and tumor volume were the most important oncologic prognostic factors and that functional outcome after rotationplasty was superior to that of amputation and other limb-preserving techniques.
Rotationplasty
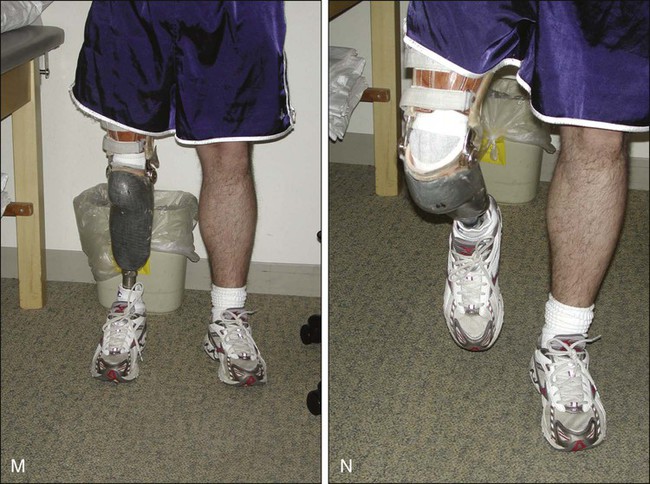
Rotationplasty for tumors around the knee and hip is a very functional alternative to amputation (Fig. 92-4, A to E), but carries a cosmetic disadvantage. For skeletally immature patients, patients with a pathological fracture, or patients with large tumors, especially of the distal femur, a rotationplasty should be considered. A similar procedure of hip rotationplasty has been described and may be used at times for proximal femoral tumors.176
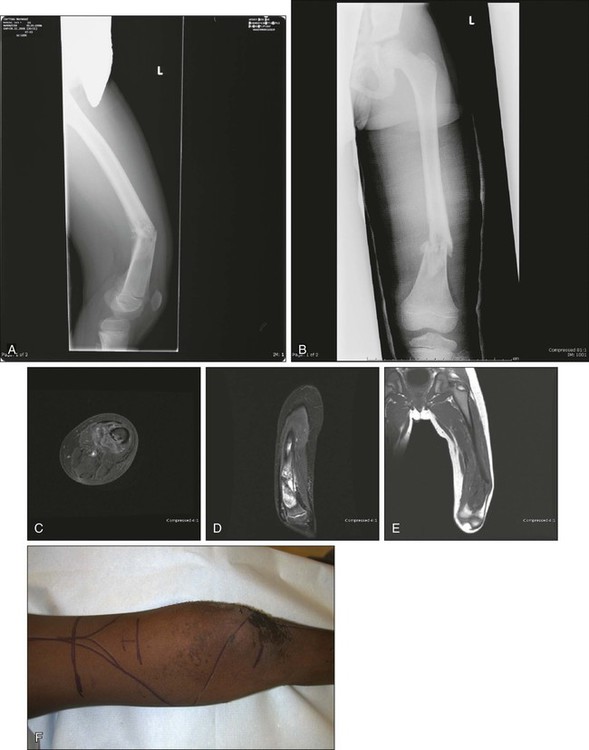
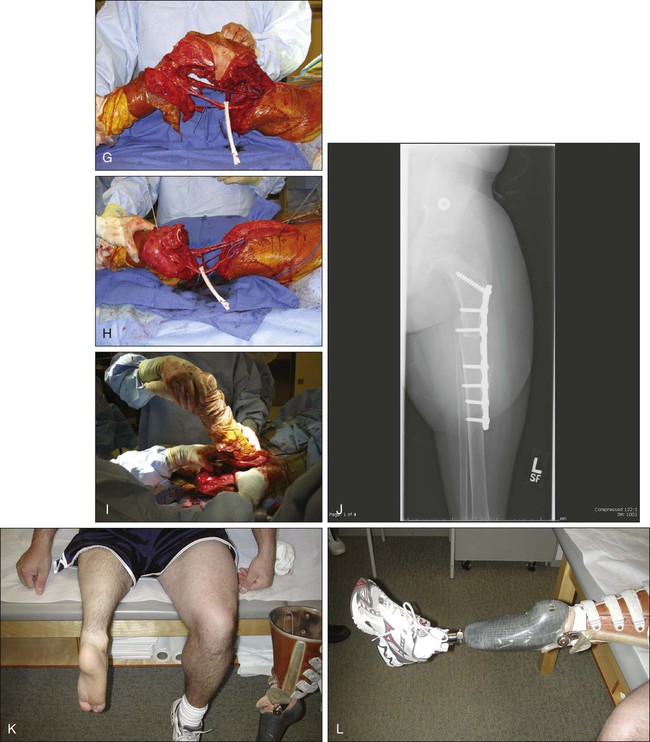
Tibial rotationplasty was first described by Borggreve in 1930 for management of a limb that was markedly shortened by tuberculous involvement of the knee.177 In 1950, Van Nes178 and others extended the use of the procedure to treat congenital femoral focal limb deficiencies.181–181 The first tibial rotationplasty used for reconstruction after a radical resection of the distal femur for osteosarcoma was performed by Dr. Martin Salzer and coworkers in Vienna in 1974 and reported in 1981.182
The largest collective experience, reported in 1991, involved 70 patients with sarcoma of bone who were managed with rotationplasty.183 Forty-seven patients had stage IIB osteosarcoma; other conditions included MFH of bone, parosteal osteosarcoma, chondrosarcoma, Ewing sarcoma, giant cell tumor of bone, periosteal osteosarcoma, and undifferentiated sarcoma of bone. Sixty-two of the lesions originated in the distal femur, six involved the diaphysis, and two originated in the tibia. At a mean follow-up of 4.3 years, Kaplan-Meier analysis revealed a 70% probability of survival and a 58% probability of disease-free survival.184
The surgical oncology principles of tibial rotationplasty yield surgical margins that are comparable to those of a transmedullary amputation.183,184 In patient selection, the only absolute requirement is the absence of tumor involvement of the sciatic nerve and a previously untraumatized, essentially normally functioning foot and ankle (see Fig. 92-4, F to N). The procedure is applicable for lesions that involve the distal half of the femur and the proximal third of the tibia that spare both the peroneal and tibial divisions of the nerve. Large, locally invasive lesions of the distal femur, even with knee joint involvement, are often amenable to tibial rotationplasty.
The procedure is ideally used in young, skeletally immature patients in whom the anticipated adult limb length inequality (associated with the loss of the distal femoral and proximal tibial epiphyses) would be substantial if other reconstructions were used. The desired final effect is to have the axis of rotation of the rotated ankle joint slightly proximal to the axis of rotation of the normal knee at skeletal maturity. A recent study from Italy showed that the QOL in survivors as assessed by the Short Form-36 was very similar, and sometimes better, than the general Italian population, although the study stressed the importance of support from family and other.185
Resection and Distraction Osteogenesis
Borrowing from techniques of the pediatric orthopedic surgeon, distraction osteogenesis has been used by some for limb salvage in children. This technique involves slowly transporting a segment of vascularized bone with an external fixation devise to fill the gap that tumor resection creates. It requires the ability to preserve the articular cartilage, and the potential risks include close margins and infection from the percutaneous pins in patients receiving chemotherapy. The results of treating patients who have a local resection for tumor of bone and treatment by using bone transport (10 patients), shortening-distraction (three patients), and distraction osteogenesis (six patients) have been reported,186 but distraction osteogenesis has not had widespread acceptance in this country.
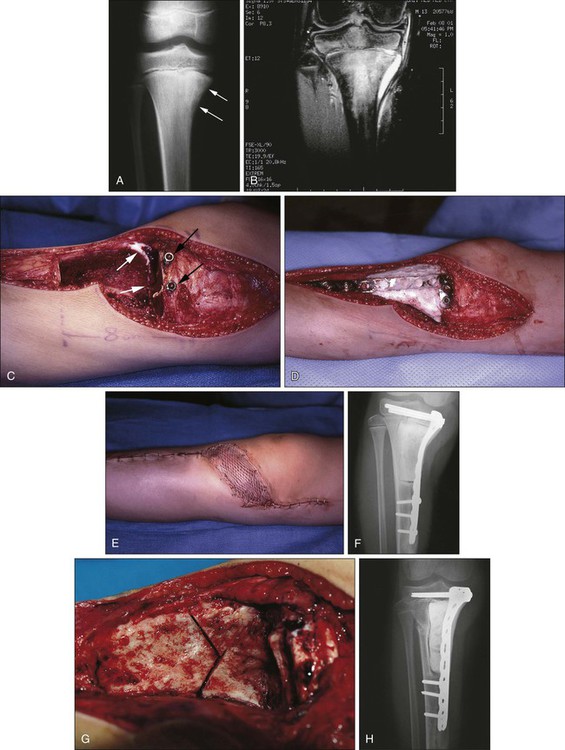
Canadell and colleagues187 described an innovative physeal-sparing procedure in skeletally immature patients in whom the primary tumor was limited to the metaphysis. During the neoadjuvant chemotherapy phase, distraction forces were applied to the epiphysis, “pulling” the tumor away from the epiphysis and providing new, widened uninvolved metaphyseal bone for a margin of resection without sacrificing the adjacent joint. The procedure was used in 20 patients with a mean follow-up of 54 months without evidence of local recurrence (Fig. 92-5).
Expandable Prostheses
Limb length is an issue to be considered in a skeletally immature patient with a tumor of the lower extremity. In general, patients younger than 8 years of age are probably best treated by amputation or, when possible, rotationplasty. Those between the ages of 8 and 10 years (in girls) and 12 years (in boys) have significant growth remaining such that resection of a physis about the joint will lead to significant limb length discrepancy. One alternative for managing this issue in children is to use standard allografts or endoprostheses and rely on techniques of epiphysiodesis or subsequent limb lengthening to equalize the extremities. Another alternative is to use expandable prostheses to deal with limb length. There are several on the market, some of which require operations to expand the prostheses and others that can be lengthened by using electromagnetic devices in a radiology suite (Fig. 92-6).190–190 The early reports of these prostheses are encouraging, especially to parents, but the long-term outcomes are unknown. They are very expensive and complex prostheses, and few data are available with respect to two key issues: whether a very young child can reach skeletal maturity with equal limb lengths and good function, and whether the results of the required revision to an adult prosthesis at skeletal maturity will be successful. Early reports show that lengthenings can be achieved and that the complication rates are similar to those of adult endoprostheses. A higher rate of flexion contractures had been noted, because young children are not as cooperative with physical therapy, and loosening and failure of the implant also occur in these complex prostheses.190–190 The optimal method of fixation of the stems to the host bone remains controversial. It is also unclear whether sufficient bone stock will remain when the patient needs to be converted to an adult prosthesis, because stress shielding of the stem in these immature bones. A recent study shows that children treated with an expandable prosthesis have high emotional acceptance by assessment with the Pediatric Outcomes Data Collection Instrument, although only 12 of 26 potential families completed the assessment.191
Amputation
Ablation or primary amputation may be considered for patients with pathological fracture and for patients who are younger than 9 years of age in whom lower-extremity limb length inequality would be considerable.192,193 Also, stage IIB lesions that arise below the knee and below the elbow may be best treated by primary amputation. It should be recognized, however, that selected patients with favorable lesions that arise below the knee and elbow might also be candidates for limb-sparing procedures. Also, some pathological fractures have been shown to heal during intensive neoadjuvant chemotherapy.194 We have become so accustomed to salvaging limbs that amputations are probably not being used enough. It is wise to remember that with modern prosthetics for the lower extremity, function and QOL can be quite good, and perhaps equivalent to or better than that of limb `salvage for tumors of the distal femur 195 and below. Improvements such as bony ingrowth mechanisms for attachment of the prosthesis directly to bone are being evaluated clinically. Amputation should not be viewed as a failure or sign of defeat until we develop better methods for limb reconstruction.
Surgical Options for Limb Salvage Reconstruction
Endoprostheses
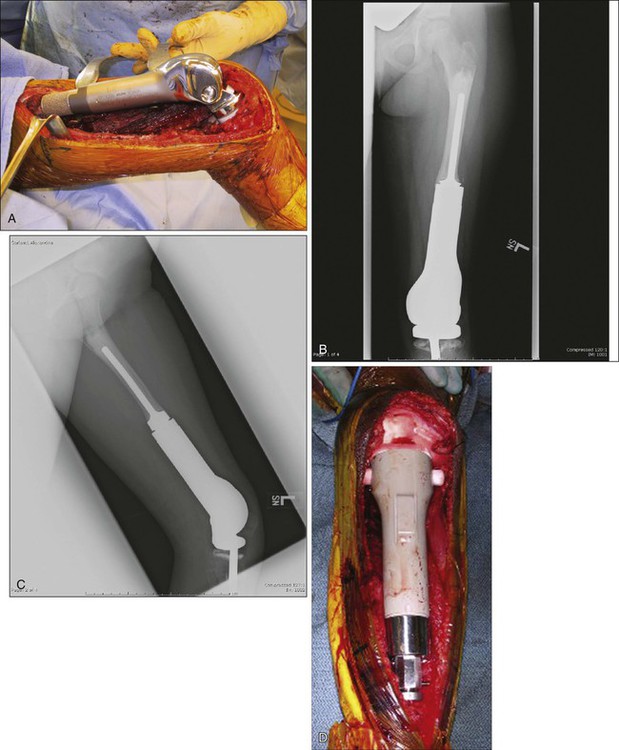
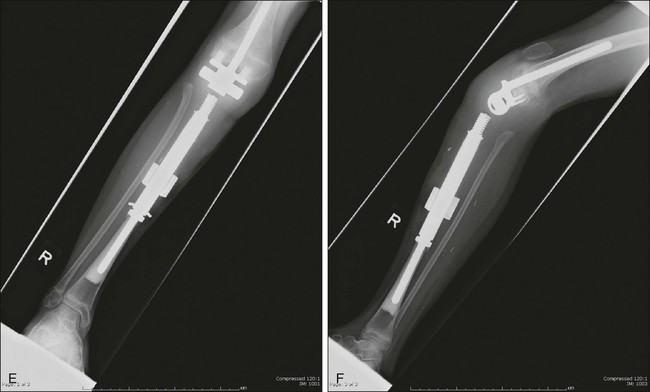
Early experience with metallic reconstructions for limb salvage of tumors required custom-made implants, which took time to manufacture and had design features that limited longevity. Now there are modular prostheses for most anatomic sites of the upper and lower extremities that allow the surgeon to reconstruct individual defects at the time of the resection without custom implants (Fig. 92-7).196–202,202a Design improvements in rotating knee hinges and cementless stem options will likely lead to increased longevity of these implants. The advantages of endoprostheses include their relative ease of use, a lower reported infection rate, and a more rapid return to function because there is no waiting for union as in bone allografts. The main drawbacks include the concern for longevity of the implants and their fixation to the host bone. The complications and functional outcomes of these implants have been included in several articles.174,202,202a,203–205 Recently, a multicenter study classified a large group of endoprosthetic complications into five categories: soft-tissue complications, aseptic loosening, structural failures, infection, and tumor failure, with infection being the most common complication.206
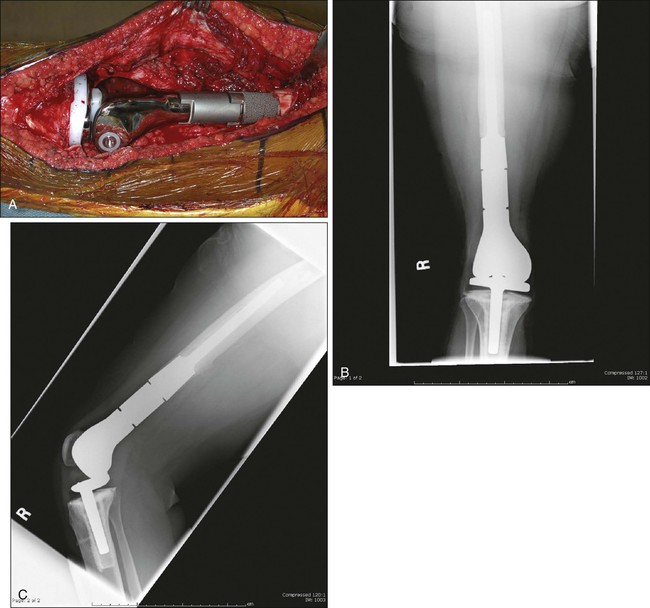
A recent stem design uses constant compression forces and a cementless fixation system that is purported to induce bone hypertrophy and lessen stress shielding, but results are preliminary.207 In young children, it is unreasonable to expect a complex implant to last the patient’s lifetime if the patient survives the disease, and multiple revisions are to be expected. Advances in fixation of implants to the host and design improvements in the joints will likely improve the longevity and usefulness of these implants.
Bone Allografts
Bone allografts are another alternative to limb salvage for sarcomas. They offer the potential advantage of incorporation by the host and greater longevity but are technically more demanding than prostheses. Availability of a bone bank with a variety of shapes and sizes of allografts is a prerequisite, although several large commercial bone banks that are accredited by the American Association of Tissue Banks are now available. Younger patients are more suitable because the recovery period is longer because of the need to protect the osteosynthesis site until it heals. Allografts can be used as osteoarticular grafts (Fig. 92-8), replacing one side of a joint, or can be combined with standard prosthesis to create allograft-prosthetic composites (Fig. 92-9). Intercalary defects are particularly suited to allografts, because the joint on either end of the diaphyseal defect is intact (Fig. 92-10). An arthrodesis of a shoulder, knee, or hip can also be created by using allografts. The main drawbacks are the potential for disease transmission and the relatively high reported complication rates compared with endoprostheses.
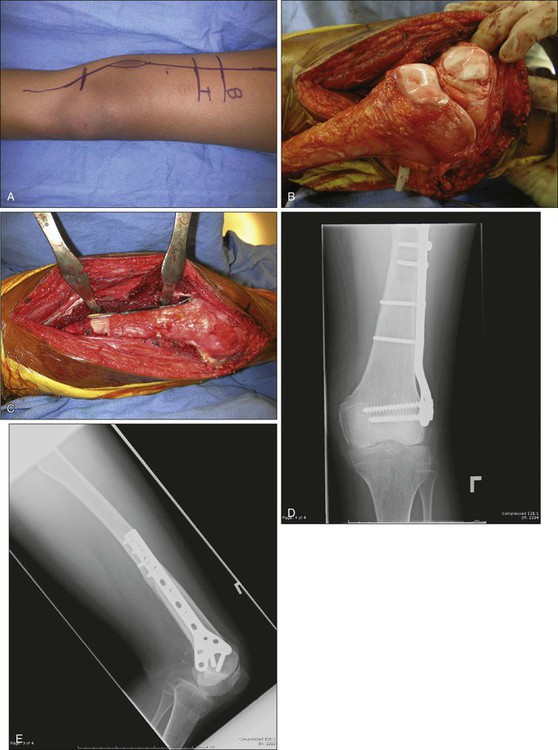
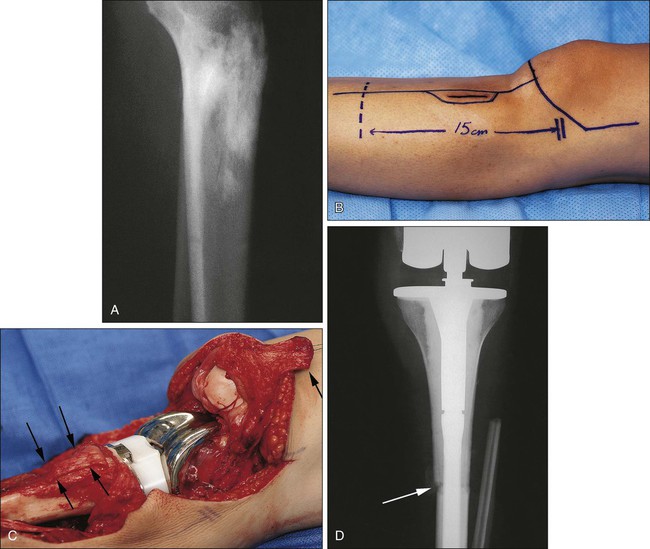
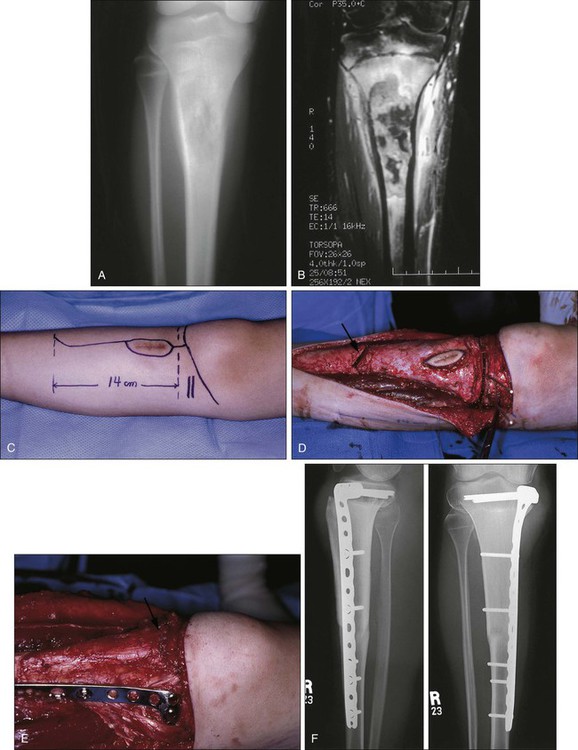
The experience with large-bone allografts for reconstruction in high-grade osteosarcoma at the Rizzoli Institute in Bologna was recently reviewed by Donati and colleagues.208 Between 1986 and 1994, 112 large-bone allograft reconstructions were performed. Forty-one were used for arthrodesis of the knee, and three were used in the ankle. Thirty-nine were used as intercalary grafts (see Fig. 92-10), and 22 were used as osteoarticular allografts (three humeral, six in the distal femur, and 13 in the proximal tibia). Seven composite constructs were reported: two were in the proximal and distal humerus, I in the proximal femur, and four in the proximal tibia. Complications included delayed union (greater than 1 year without radiographic union) in 49% of patients and fracture in 30%. There were no deep infections. With the modification of the Mankin scale by Gebhardt and coworkers,209 good to excellent functional results were reported in 74% of the 92 patients who were available for review. Examples of these reconstructions at various sites are briefly presented and recent articles that address these reconstructions can be found in the following references.210–214
Proximal Humerus
Proximal humeral osteosarcomas can frequently be resected while preserving all or part of the scapula. The motion, strength, and functional outcomes of these reconstructions vary depending on whether or not the rotator cuff muscles and deltoid can be preserved. A patient in whom the glenoid, rotator cuff, and deltoid can be preserved should expect reasonable function, whereas a patient who requires resection of the deltoid, rotator cuff, and portions of the scapula will have less good shoulder function.212 If the neurovascular supply to the hand can be safely preserved, however, the function is superior to amputation. By using current prosthetic designs augmented with muscle rotation flaps and synthetic grafting materials (Dacron, Gore-Tex, and others), a stable shoulder can often be achieved, and preservation of an innervated hand is paramount.
Wittig and coauthors215 described the single-surgeon experience of 23 patients with osteosarcoma of the proximal humerus that included one stage IIA lesion, 18 stage IIB lesions, and four stage III lesions. Twenty-two of the 23 patients were treated surgically with an extraarticular resection that included the deltoid muscle and the rotator cuff, whereas one patient was treated with a transarticular resection that spared the shoulder abductors. The patients ranged in age from 10 to 77 years. Before 1988, 10 patients received custom proximal humeral replacements, and after 1988, 13 modular segmental prosthetic replacements permitting intraoperative sizing were used. Prosthetic survival for the 15 survivors was 100% at a median follow-up of 120 months (range: 24 to 234 months). No prosthesis required revision. There was one instance of aseptic loosening. Complications included transient nerve palsies. All nerve palsies resolved within 6 to 12 months after surgery. Functional outcome as recorded by the MSTS Extremity Functional Scores ranged from 24 to 27 (80% to 90%). All shoulders were stable, and all patients could perform the activities of daily living with the involved extremity.
Osteoarticular allografts (Fig. 92-11) or allograft prosthetic composites can also be used at this site.212,216–220 They offer the advantage of having sites for attachments of muscle tendons (rotator cuff and deltoid), and because this is a non–weight-bearing bone, the likelihood of fracture is less than that in the lower extremity. Allograft arthrodeses can also be used.
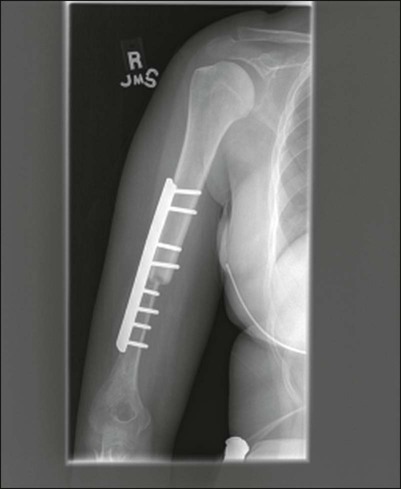
The experience of the Mayo Clinic was reviewed by O’Connor, Sim, and Chao,218 who described the treatment of 53 patients with malignant bone tumors of the shoulder girdle with an average follow-up of 5.3 years (median: 4.6 years). A variety of surgical procedures and reconstruction methods were used to achieve wide margins in 40 of 53 patients, with 13 patients having marginal resection margins. Four patients (two after wide resection, two after marginal resection) experienced locally recurrent disease. The functional results are the subject of another report.221
Rödl and colleagues,222 in 2002, retrospectively compared osteoarticular allografts (11), clavicula pro humeri (15), and a tumor prosthesis (19) for reconstruction following resection of the proximal humerus. The glenoid was resected in 25 patients, and the axillary nerve was resected in 42. The functional results of all three types of reconstruction were similar, but the complication rate was lowest in the prosthetic group, leading the researchers to conclude that the prostheses were the most reliable reconstruction.
Pelvis
The pelvis is the most challenging site for resection, although in many cases, the tumor can be resected without a hind-quarter amputation (hemipelvectomy). It should also be remembered that the prognosis of pelvic osteosarcoma remains poor despite modern multimodality treatment regimens, including neoadjuvant chemotherapy.223 The difficulty comes in reconstruction when the acetabulum is resected. Removing the iliac wing or pubic ramus and ischium often requires no reconstruction and results in relatively good function if the sciatic nerve and acetabulum can be preserved. If the acetabulum is resected, the reconstruction options include leaving the hip flail, attempting an arthrodesis, or replacing the defect with an allograft or custom metallic prosthesis.
Schwameis and associates224 described the results of the Austrian experience of reconstruction of the pelvis in children and adolescents. Thirty patients, ages 19 years or younger, with malignant tumors of the pelvic bones were treated by using an endoprosthesis in 10 patients, and in 20 patients, reconstruction was completed by using autologous grafts (seven patients), allograft/prosthetic composites (two patients), methyl methacrylate reconstruction (one patient), iliosacral arthrodesis (one patient), modified Girdlestone procedure (three patients), or resection/arthroplasty (six patients). After a mean follow-up of 52 months (range: 2 to 241 months), 15 patients were continuously free of disease, two were alive with disease, and 13 had died of their disease. MSTS ratings revealed an 81% functional status after autograft, 73% functional status after allograft, and 60% functional status after endoprosthetic reconstruction. Reoperations were significant in that the patients who received allograft reconstruction required an average of 3.5 reoperations, whereas those who received endoprosthetic reconstruction required an average of 2.5 reoperations. Only 0.8 reoperations per patient were necessary after the other described methods of reconstruction.
Satcher and coworkers225 described the results of 15 patients who underwent reconstruction for pelvic deficits associated with tumor resection in and about the acetabulum in a slightly older population. The review was limited to periacetabular resections and included patients with primary malignancies and metastatic disease. The authors described a reconstruction technique using numerous Steinman pins placed within the residual and remaining bone to reconstruct the acetabular structures with methyl methacrylate and (occasionally) autoclaved autograft bone in combination with total hip arthroplasty. After tumor resection, the pelvis was reconstructed by using autoclaved autograft or the cement and Steinman technique. Functional results were graded overall excellent or good for 87% of the patients. Their recovery time in general approximated that of patients with routine total hip arthroplasty.
Pelvic allografts have had some success but are demanding procedures and, if the soft tissues are not ideal, subject to a high infection rate. One study of 13 patients looked at the results of reconstruction by massive allografts (some were combined with metal hip prostheses) after resection of a malignant tumor (primary in 10 patients and metastatic in three patients). Postoperative complications were one infection and two dislocations. At 3 years, one cup loosening and one acetabular fatigue fracture required surgery. The functional result was excellent in two patients whose gluteal muscles could be spared, good (allowing a normal family life) in six patients, fair in two patients, and poor in two patients. Seven patients had a MSTS rating greater than 60% of normal (the mean rating in 12 patients was 56.4%). The authors concluded that reconstruction of the hemipelvis with massive allografts and arthroplasty is a rewarding but demanding procedure that should be reserved for physically active patients who are in good general health and are expected to have a response to anticancer therapy.226
Despite the complexity of the pelvic resections or amputations, in patients without evidence of metastases, an attempt at resection seems reasonable. A study of 67 patients with pelvic osteosarcoma (15 of whom had metastases at diagnosis) who were registered in the Cooperative Osteosarcoma Study Group were analyzed. All patients received chemotherapy according to that group’s protocols. Thirty-eight patients underwent limb-sparing surgery, 12 patients underwent hemipelvectomy, and 17 patients did not undergo definitive surgery. Local failure occurred in 47 of all 67 patients (70%) and in 31 of 50 patients (62%) who underwent definitive surgery. Five-year overall survival and progression-free survival rates were 27% and 19%, respectively. Primary metastasis, large tumor, no or intralesional surgery, no radiotherapy, and existence of primary metastasis were independent poor prognostic factors. The researchers concluded that an operative approach with wide or marginal margins improves local control but that if the surgical margin is intralesional or excision is impossible, additional radiotherapy has a positive influence on prognosis.227
Recently, Sherman and associates228 retrospectively reviewed 60 patients treated with pelvic limb salvage before 1989. They updated a previous study of 38 survivors by medical records and received questionnaires from 15 of those patients. Follow-up was a median of 30 years; (range: 23 to 38 years). Overall survival rate of the 60 patients was related to the stage of the disease, with low-grade neoplasms surviving longer than high grade. Patient-reported function declined over time to a MSTS score of 23% in the 15 patients who completed a questionnaire. Two patients had further reconstructive procedures. The authors concluded that pelvic limb salvage is a reasonable treatment if satisfactory margins can be achieved.
Proximal Femur
Two types of proximal femoral reconstruction are currently used:
1. Megaprosthesis, a large metallic prosthesis, often modular in design, that is used to replace the entire proximal femur and occasionally the acetabulum. Reattachment of ligamentous structures to the prosthesis is facilitated by holes or rings designed into the prosthesis, to which the ligaments can be sutured securely. There is very little evidence to suggest secure long-term reattachment other than to the scar enveloping the prosthesis.
2. Allograft/prosthetic composite, consisting of a long-stemmed proximal femoral prosthesis placed within a proximal femoral allograft, securely cemented into the allograft and remaining shaft of the host bone (Fig. 92-12). Advantages over megaprosthesis include the ability to repair host ligaments to the retained ligaments on the allograft. There is histologic evidence that the host and allograft ligamentous structures do unite by scar formation, with less-than-normal tensile strength.
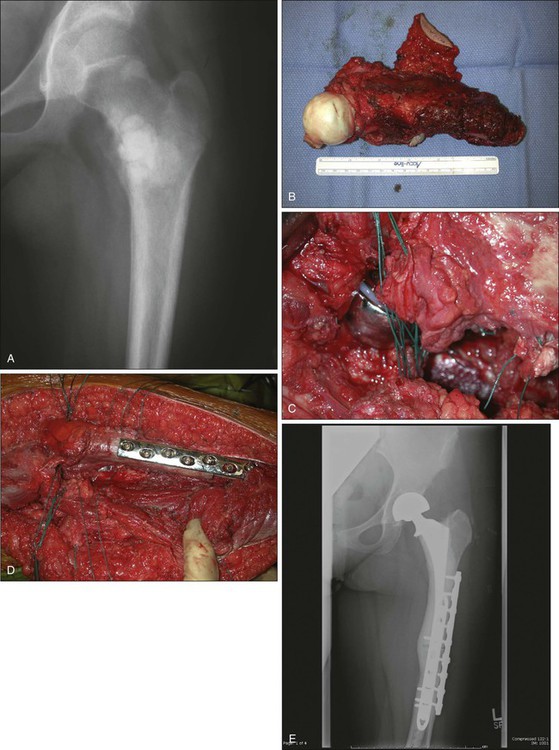
Survival of prostheses that replace the proximal femur is generally reported at a rate between 88% and 100% at 5 years.231–231 A direct comparison of megaprostheses and allograft/prosthetic composites used to reconstruct the proximal femur has demonstrated a survival advantage for composites.232 Stability of the hip remains a concern, with dislocation rates reported at between 2% and 14%, whereas aseptic loosening remains low for proximal femoral reconstruction.205,229–231,233–236
Distal Femoral Prosthetic Replacement
Two prosthetic designs are available:
1. The simple hinged-knee prosthetic does not allow for rotation, but only for flexion and extension.
2. The kinematic rotation hinge-knee prosthetic allows for rotation, flexion, and extension through an offset hinge that provides stability in full extension.
The most important cause of failure for distal femoral prosthetic replacement remains aseptic loosening, which is found in 0% to 11% of patients.231,237,238 In 218 reconstructions, the overall survival rate for patients with the simple hinge design was 80% at 5 years, 65% at 10 years, and 53% at 20 years.237 The rotating kinematic hinge design has a better record, with 5- and 10-year survival rates of 90% and 80%, respectively.230 It is suggested that the rotation of the normal knee simulated by the improved design diminishes impact torsional loading of the cement/bone interface, which often is responsible for loosening.
A novel compliant, osteointegration fixation device (Biomet, Warsaw, ID) is now available, designed to address the aseptic loosening issue with massive endoprostheses.239–244 Advocates argue that the device enables bone regeneration as well as integration. Revision is also advantaged compared with conventional stemmed technique (cemented and press fit) because of the short fixation arm. Early results are encouraging, but further follow-up is necessary.
At present, several modular tumor prosthetic systems are available for reconstruction of tumor resection about the knee (see Fig. 92-7). Results with one modular tumor system used in large numbers by two separate groups have recently been published. Mittermayer and associates245 reported on the University of Vienna’s treatment of 100 patients with primary lower-extremity uncemented reconstruction by using the Kotz Modular Femur Tibia Reconstruction System between 1982 and 1989. Forty-one patients were followed up for a mean of 138 months (51 patients had died, and eight were lost to follow-up). Nine patients required revision for aseptic loosening, and two required two such revisions. Four patients experienced deep infections. Fourteen patients required reoperation for early failure of the polyethylene bushings.
A large multicenter Canadian study reported by Malo and colleagues246 addressed the question of stem fixation and fixed versus rotation hinge design of the knee in the Kotz design prosthesis. Thirty-one patients with Kotz fixed hinge and uncemented stem constructs were compared with 25 patients with the knee modular replacement system cemented stem and rotating hinge design by using the MSTS (1987 and 1993) functional assessment, the Toronto Extremity Salvage Score, and the Short Form-36 Physical Component Score for a follow-up period of at least 1 year. Results in the two groups of implants were comparable, except that the uncemented group had an average of 2 cm greater length of resection. The outcomes of the MSTS (1993; P = 0.006) and the Toronto Extremity Salvage Score (P = 0.03) assessment suggest that cemented stem fixation and a less constrained knee design were preferable, even though the study was designed to assess functional issues from the patient’s perspective. The size of the stem seems to matter, and one recent report suggested that the largest size stem possible was preferable.204
No direct comparison between endoprostheses and allografts for distal femoral reconstruction is available, and allografts remain a reasonable alternative, especially in young patients. A study of 118 knee osteoarticular allografts in 114 patients was reported with long-term follow-up.247 Ten patients were lost to follow-up, and 18 patients without allograft failures died of complications related to the tumor. Twenty-six allografts failed because of infection (13 allografts), local recurrence (eight allografts), massive resorption (three allografts), and fractures (two allografts). Sixty-four allografts were still in place at a mean of 98 months after implantation. The Kaplan-Meier 5-year survival rate for the knee osteoarticular allografts was 73%, and the limb preservation rate was 93%. Five patients required joint resurfacing to preserve the original allograft. Most of the allograft failures occurred during the first 4 years, and the allograft survival rate for this series remained unchanged after 5 years. Others217 have reported more dismal results, attesting to the demanding technical aspects of this surgery and the need for experience with the surgical techniques and tissue banking.248,249
Proximal Tibial Prosthetic Replacement
1. Difficulty in obtaining good soft-tissue coverage
2. Poor reconstructive options for extensor mechanism reconstitution
Allograft/prosthetic composites appear to have improved survivorship and function compared with megaprostheses and remain a favorite method of reconstruction because the composites provide the ability to reconstruct the host patellar tendon with the remaining allograft patellar tendon. The routine use of the rotation of a medial gastrocnemius flap either with or without skin has improved coverage of the prosthesis and local nutrition. However, there remains an inordinately high risk of infection, up to 31% (see Fig. 92-9).229 Although the debate continues about fixation of the implant to the host bone, one recent study showed good longevity and survival of the implant by using cemented stems and modular prostheses. Fifteen-year survival rate of 88% in 29 modular prostheses was reported.203
Validation of a Functional Evaluation System
Lee and coworkers250 evaluated the MSTS functional evaluation system using the Nottingham Health Profile, the Short Form-36, and the EuroQOL protocol to measure the QOL of patients with malignant musculoskeletal tumors. Forty-nine patients were assessed, and osteosarcoma about the knee was the most common condition. Prosthetic reconstructions had been completed in 55.1% of patients.
All of the items of the lower extremity were related to the same function: walking ability. The MSTS system had a strong correlation with other QOL measures in the construct validity and reliability, but the sociologic domains of the system were so comprehensive that it could not represent the QOL properly. The patients’ preoperative and postoperative status could not be compared by using this system. The overall validity and reliability of the MSTS system appeared to be appropriate; however, the system could require the additional development of domains for evaluating the QOL in patients with musculoskeletal tumors. Other systems, such as the Short Form-36 and the Toronto Extremity Salvage Score, have also been used and validated for sarcoma patients. These issues have been more recently examined in the review by Barr and Wunder.195
Not many studies have compared amputees to limb-sparing procedures for sarcoma. Renard and colleagues,251 from the Netherlands, reported retrospective results in 77 patients treated between 1975 and 1995 for sarcomas in the lower extremity. They compared functional results using the MSTS (1993) criteria and the QOL for groups of 52 patients who had limb-sparing surgery and 25 who had ablative treatment. Treatment of tumors involving the pelvis to distal femur and proximal tibia were included. When all anatomic groups of limb-preserving procedures were combined and these patients were compared with patients who were managed by amputation, the functional score of the limb-sparing group after a mean duration of 97 months follow-up was significantly better (P = 0.0001) than that of patients who had an ablative procedure.
A recent study compared QOL and subjective well-being between patients who underwent either amputation or limb salvage procedures for lower-extremity sarcomas. Sixty-six patients were evaluated at least 1 year after surgery and systemic therapy for lower-extremity sarcomas. Self-report questionnaires such as the Quality of Life Questionnaire (QLQ-C30), the Life Satisfaction Questionnaire (FLZ), and the Enneking Score (MSTS) were used. There were no differences in QOL and subjective well-being between the two groups. Small differences in scores were found in social functioning, but not in scores of physical, cognitive, and emotional functioning. Global subjective well-being in both groups was also similar (219.5 vs. 223.7). High satisfaction was found in marital life and partnership, as well as in self-assessment. The least satisfaction was found in finances. Results in functional outcome after limb salvage procedures were better than for amputation (77% vs. 65%), although more complications were reported after limb salvage procedures. They demonstrated that the type of surgery had no influence on QOL and subjective well-being after treatment of lower-extremity sarcomas.252 A study of 408 patients with sarcomas, who were treated with either limb salvage or amputation, had postoperative follow-up of 2 years or greater when they completed a QOL self-report questionnaire. Limb salvage offered a functional advantage particularly at proximal tumor locations.253
Refaat and coworkers254 used a computerized questionnaire that was automatically adjusted for gender, age, diagnosis, site, and treatment that was sent to 2200 patients with high-grade sarcomas who had been treated during the past 25 years. They compared the response from 66 patients who had an amputation and 342 patients with a limb-sparing procedure for a lower-extremity neoplasm. The outcomes for the two groups were similar. Patients who had limb-sparing procedures did not have a significant improvement in the ability to ambulate, climb stairs, drive a car, or be employed. More of the patients with amputations required walking aids; however, they participated in sports activities at almost the same rate as the patients who had limb-sparing procedures. Patients who had amputations had no more anxiety, drug dependence, depression, sleep problems, or limitation of sexual performance than patients who retained their limbs. The patients with amputations had more children and fewer menstrual problems; however, they were far less satisfied with their status at early ages than at later periods in their lives.
Oxygen consumption studies of gait suggest that patients with rotationplasty compare favorably with patients who have had a below-knee amputation and function better than most with an above-knee amputation.255–258
Harris and coauthors193 studied the function of 22 patients who had surgical management for malignant neoplasms around the knee. Seven had an above-knee amputation, nine had resection/arthrodesis, and six had replacement arthroplasty. All patients were available for physical examination and measurement of consumption of oxygen, and all answered questionnaires on function and psychological effects.
Physical Capacity
Arthroplasty
Patients who had undergone arthroplasty walked better in crowds and on snow, sat easily and could rise from a chair with ease, and could drive automobiles normally. Half of the patients required a hand rail while descending steps one at a time. More could flex the limb sufficiently to squat on the opposite (normal) knee. They often refrained from kneeling because of pressure on the prosthesis. These patients were the most protective of all, limiting their recreational activity, and were the most sedentary of any of the groups.193
Rotationplasty
Most of the rotationplasty patients were very active, with general disregard for the well-being of the limb or prosthesis. All of them shared problems associated with patients with a below-knee amputation (e.g., difficulty walking on steep slopes or uneven ground, inability to squat with ease). Oxygen consumption studies of gait suggest that patients with rotationplasty compare favorably with patients who have had a below-knee amputation, functioning better than most of those with an above-knee amputation and as well as those who have had endoprosthetic replacement.256–259
Activities and Quality of Life After Rotationplasty
At least two studies have examined general activities and QOL for people who have undergone rotationplasty. Hillman and associates259 compared functional results in 34 patients treated with endoprostheses and 33 patients treated with rotationplasty. The rotationplasty patients were younger than 24 years and had a minimum follow-up of 2 years. Results were evaluated by using the 1993 version of the MSTS functional scoring system and the core QOL questionnaire of the European Organization for Research and Treatment of Cancer. Patients who were treated by rotationplasty had better scores related to role functioning that included hobbies and work, had better ability to function without external aids, and were less inhibited when performing daily and sporting tasks. Most were less concerned than their counterparts about restrictions of activities involving sporting activities and work. Patients who were treated by endoprosthetic replacement had slightly more complaints of discomfort, and more (6 of 34 patients) used external aids than did those with rotationplasty (1 of 33 patients). Neither group was unhappy; however, some patients with rotationplasty appeared more self-conscious.
Veenstra and colleagues,260 from the Netherlands, assessed the QOL and psychosocial functioning of 33 of 34 patients with a minimum age of 16 years who had large malignancies of the distal femur treated by rotationplasty between 1981 and 1994. The mean follow-up period was 6.3 years (range: 1 to 11 years). The general QOL was assessed with the European Organization for Research and Treatment of Cancer’s QLQ-C30 cancer-specific QOL questionnaire. When compared with healthy counterparts, patients with rotationplasty had significantly poorer performance functionally, but were superior psychosocially. Nine of 33 patients reported a negative effect on social contact, while 17 of 30 reported that the operation exerted no influences on social contacts. Seventeen did not feel limited in initiating intimate relationships, while 13 felt slightly to moderately limited, and one felt very limited. There was no apparent difference between males and females regarding the effect of surgery. Twenty-one of 33 patients reported that they were sexually active during the months before surgery. Eleven patients reported no sexuality problems, whereas 10 reported a small-to-moderate degree of inhibition. Physical function and prosthetic use assessment revealed that 21 of 33 patients did not use extended aids, and 8 of 33 used a single crutch periodically. No patient was confined to a wheelchair. Twelve patients used a normal bicycle regularly, and 15 required a shortened crank. Thirty-two of 33 patients reported wearing their prostheses from early morning to late at night, and all but two reported no difficulty sitting. The researchers concluded that rotationplasty was an excellent operative procedure for managing large tumors of the distal femur. Although the patients’ level of physical functioning was below that of their normal peers, patients who were treated with rotationplasty experienced an overall QOL similar to that of their normal peers and functioned comparably in a psychosocial context.
Locally Advanced Disease (Local Recurrence)
Two studies indicate that local recurrence within 18 to 24 months of surgery has a substantial negative influence on long-term survival. Ferrari and colleagues,261 from three Italian institutions, reviewed the data on patients who were treated for nonmetastatic osteosarcoma of the extremities between October 1986 and June 1995, finding 162 patients with recurrence or relapse. The main prognostic factors for postrelapse survival were the relapse-free interval, site of metastasis, and number of pulmonary nodules. Complete surgical resection of the recurrence was found to be pivotal in the strategy of treatment. When the risk factors were combined, patients with a relapse-free interval longer than 24 months and with only one or two pulmonary nodules had a postrelapse survival rate of 72%. Patients with a short relapse-free interval and three or more lung pulmonary nodules had a poor postrelapse survival rate of 5%. Patients with unresectable recurrence did receive benefit from second-line chemotherapy, but the authors’ data did not support the generalized use of chemotherapy after complete surgical resection of the first recurrence.
Weeden and colleagues262 reported the effect of local recurrence on survival of 559 patients entered into two randomized controlled trials of the European Osteosarcoma Intergroup using preoperative chemotherapy, but there was no survival benefit between chemotherapy arms. A landmark analysis allowed the assessment of 440 patients at 18 months, and 22 patients (5%) had experienced a local recurrence. Patients with a local recurrence had a fourfold greater risk of death during the 18 months after surgery. Similar analyses were conducted with landmark time points at 1 and 2 years. With a multivariate proportional hazards model, 368 patients were assessed to investigate the relative prognostic importance of histologic response and local recurrence. It appears clear that histologic response to chemotherapy and local recurrence had a significant effect on survival. A study from St. Jude’s Research Hospital analyzed prognostic factors for survival in 26 patients with a local recurrence of osteosarcoma who were treated between 1970 and 2000. The initial surgical procedure was amputation in 20 patients (76.9%) and limb salvage in six patients (23.1%). Eleven patients (42.3%) developed an isolated local recurrence, and 15 patients (57.7%) developed local and distant recurrence. The 5-year estimate of post recurrence survival (PRS) for the 26 patients was 19.2%. The factors that affected survival after recurrence were as follows:
• Recurrence 2 or more years from the time of diagnosis (5-year PRS of 50.0% ± 20.4% compared with earlier recurrence (10.0% ± 5.5%) (P = 0.037).
• Patients with negative margins after initial surgery were found to have improved survival (5-year PRS of 33.3% ± 13.6%) compared with patients with positive margins (7.1% ± 4.9%) (P = 0.015).
• Patients who underwent complete surgical resection at the time of recurrence were found to have a better PRS (5-year PRS of 41.7% ± 14.2%) compared with patients who did not undergo surgery (0% ± 0%) (P < 0.001).
This demonstrated that the prognosis for patients after local recurrence of osteosarcoma is poor, but attempts at salvage should be made. Complete surgical resection at the time of recurrence is essential for survival.263
A study from the European Osteosarcoma Intergroup of 202 patients assessed the primary surgical treatment alternatives. Three centers differed in the rates of limb salvage, and all local recurrences arose in limbs with attempted limb salvage. Local recurrence was closely related to the adequacy of the margins of excision and to the chemotherapeutic response. Interestingly, patients who had undergone limb-salvage surgery and who experienced local recurrence still had a better survival rate than did patients who had primary amputation (37% vs. 31% survival at 5 years), suggesting that they had a less-aggressive tumor. Of patients who relapsed, 31% of those with local recurrence alone were cured by further treatment, compared with only 10% of those with metastases.264
Another retrospective analysis of the management and outcome of 44 patients who experienced local recurrence after neoadjuvant chemotherapy of osteosarcoma of the extremities was performed in a single institution between 1983 and 1999. In 24 patients (54.5%), local recurrence was the first sign of recurrence; in eight patients (18.2%), local recurrence followed systemic recurrence, and in 12 patients (27.3%), the two events were concurrent. The only prognostic factor that was identified for disease-free survival of patients following local recurrence was the presence of systemic recurrence at the time of diagnosis of the local recurrence or before. The 5-year post-recurrence EFS rate was 29.1% for patients without metastases at the time of local recurrence versus 0% for those with metastases (P = 0.02). The analysis confirmed that patients with osteosarcoma of the extremities who experience local recurrence are at a significantly high risk for metastatic disease and dying of the tumor despite the use of effective neoadjuvant chemotherapy.265 Another study looked at 407 patients and found 23 patients with resectable local recurrence. Median time to local recurrence was 13 months. All patients were treated with chemotherapy following recurrence. The 5-year and 10-year survival rates in the recurrent cases were 29% and 10%, respectively. Increased risk of local recurrence (P < 0.0001) was strongly correlated with positive margins of resection. The strongest correlates with poor survival were local recurrence within the first year after primary resection (P = 0.001) and, as in the Bacci study, metastasis at the time of first local recurrence (P = 0.04). Failure to achieve clinical remission after disease recurrence (P = 0.04) was also a poor prognostic factor.266
To determine whether inappropriate surgical procedures based on an initial misdiagnosis affected recurrence and survival rates, one group retrospectively reviewed the surgical treatment and results of 117 patients with high-grade osteosarcomas.267 Nine patients had intralesional curettage performed at other institutions based on an erroneous diagnosis of a benign lesion. Two of the nine patients had amputations, and seven patients had limb salvage procedures. Of the 108 patients who were not misdiagnosed, six patients had amputations and 102 patients had limb salvage procedures. All patients received neoadjuvant therapy. Fifteen of the 117 patients had local recurrences. Patients who had inappropriate surgical procedures at diagnosis had an increased risk of local recurrence and lower 10-year survival rates.267
A recent study assessed the prognostic factors for survival in 449 patients with extremity osteosarcoma without metastatic disease at initial diagnosis and treatment (38 with local recurrence, 411 without local recurrence). They compared the survival difference between patients with local recurrence (n = 38) and without local recurrence (control, n = 76) matched for age, location, initial tumor volume, and tumor volume change after chemotherapy, and assessed prognostic factors in this subgroup. In a cohort study, multivariate analysis revealed initial tumor volume, tumor enlargement, inadequate margin, and local recurrence predicted poor survival. In the case-control study, the 10-year metastasis-free survival rates of two groups were 13.1% ± 10.7% and 19.3% ± 9%, respectively. Tumor enlargement and initial tumor volume showed multivariate significance. They concluded that local recurrence has a small impact on survival in patients with high-risk osteosarcoma.268
Management of Patients with High-Grade Osteosarcoma and Pathological Fractures
The incidence of the development of pathological fracture of the extremity in patients with high-grade osteosarcoma varies from 10% to 13%.269,270 The occurrence of a pathological fracture is associated with a poorer prognosis and, in the past, was felt to demand an immediate amputation.165 More recently, limb-sparing procedures have been used if there is a good response to preoperative chemotherapy.
Before the advent of neoadjuvant chemotherapy, the majority of patients who experienced pathological fractures were treated by amputation. Jaffe and colleagues,269 however, observed that some pathological fractures would heal while under the influence of systemic chemotherapy. They observed “reduction of the associated soft-tissue mass, repair of periosteum, and deposition of new mineral in the tumor and about the fracture.” These observations made the closed management of some selected pathological fractures feasible during treatment with neoadjuvant chemotherapy and in preparation for a limb-preserving procedure.
• The patient must have the ability to tolerate conventional intensive chemotherapy.
• The fracture should be treatable by closed means—either slings (usually upper-extremity fractures) or casts, external fixators that are well removed from the tumor margins, fracture braces, or traction for lower-extremity fracture management. In some femoral lesions, open reduction and provisional fixation might be appropriate to stabilize the bone during neoadjuvant chemotherapy if it is determined that the procedure won’t contaminate the tumor bed more extensively than the fracture itself. This is a complex decision requiring an experienced surgeon and an informed patient.
• The fracture should be monitored by physical examination (e.g., decreased fracture motion and pain) and the demonstration of a radiographically favorable response (e.g., increased maturation of the tumor, increased mineral deposition, subperiosteal new bone). If no evidence of improvement or favorable tumor response can be demonstrated after chemotherapy, then consideration must be given to immediate limb-preserving surgery or amputation.
Current small retrospective reviews suggest that patients with pathological fracture who can tolerate both the proposed method of fracture treatment and intensive multiagent chemotherapy do not appear to have a greater risk of an unfavorable outcome compared with fracture patients who are treated with immediate amputation, unless the fracture demonstrates poor healing and unfavorable clinical response to treatment.272–272 The proximal femur may be a particularly difficult site for a pathological fracture, as shown by the dismal 5-year survival rate of 14% in a recent study.273
Treatment of Metastatic Disease
Metastatic disease is principally the result of hematogenous spread to the lungs and bone, followed by secondary involvement of the kidney, liver, and brain. Regional lymph nodes are involved in fewer than 10% of cases. Metastatic disease at diagnosis does not preclude long-term disease-free survival when the disease is sensitive to chemotherapy and all sites of disease can be surgically resected.276–276
Aggressive surgical treatment of pulmonary metastases (often requiring more than one procedure) can yield disease-free survival rates from 17% to as high as 40%.279–279 Generally, favorable prognostic indicators include metastasis later than 1 year from original diagnosis, unilateral disease, presence of fewer than five nodules, and completeness of resection. Early pulmonary metastases, unresectable bilateral disease, and hilar, nodal, or pleural-based lesions have a poor prognosis.
Bacci and coauthors,280 from the Rizzoli Institute, reported their results of treatment of 44 patients with osteosarcoma of the extremity with detectable pulmonary metastases between January 1993 and June 1995. Twenty-three patients were evaluable, having completed primary chemotherapy consisting of methotrexate, cisplatin, doxorubicin, and ifosfamide as defined by their protocol. After primary chemotherapy, lung metastases disappeared in three of the 23 evaluable patients, whereas in four patients, the pulmonary disease was assessed as unresectable. All seven patients received surgical therapy for the primary tumor only. In the remaining 16 patients, simultaneous resection of the primary tumor and of the pulmonary metastases was performed. Remission was complete in 15 of the 16 patients and incomplete in one patient. Ten patients (55.5%) remained continuously free of disease at a mean follow-up of 30 months.
Kaste and coworkers281 reported the experience of St. Jude Children’s Research Hospital in evaluating 215 patients with extremity osteosarcoma, of whom 32 (15%) had evidence of metastatic disease. Thirty-one patients demonstrated evidence of pulmonary metastases, and only one demonstrated metastases to bone. The histologic subtype that was most commonly associated with metastases was the osteoblastic subtype (N = 17). Both the number of nodules and the number of lobes that were involved were found to be significant predictors of survival (P = 0.0009 and P = 0.04, respectively); multiple nodes were bilateral in 61% of patients. In the nonmetastatic cohort, the 5-year survival rate plus or minus standard error was 69% ± 4%, and the 5-year EFS rate plus or minus standard error was 52% ± 4%. This is in contrast to the results of the metastatic group (N = 32), in which the 5-year survival plus or minus standard error was 29% ± 8%, and the 5-year event-free rate was 14% ± 7%. Notably, the risk of death increased 1.4-fold for each additional lobe that was involved with pulmonary metastases (95% confidence interval: 1.018). The authors found a 2-year EFS rate similar to those previously reported, in spite of the inclusion of cases accrued over a 20-year span. Bacci and colleagues280 have reported an estimated 2-year survival rate of 45% for patients with resected pulmonary metastases. The 2-year survival rate estimated for their cohort was 52%. Meyer and colleagues282 reported an estimated 5-year survival rate of 11% compared with the reported cohort estimate of 14% of patients presenting with metastases. They concluded that not only the number of pulmonary metastases but also their distribution among pulmonary lobes affected survival.
The Cooperative German-Austrian-Swiss Osteosarcoma Study Group reported their findings with patients with proven high-grade osteosarcoma of the extremities or trunk who were registered between 1979 and 1998.283 Of the total 1765 patients, 202 (11.4%) had clinically detected metastatic osteosarcoma. In univariate analysis, survival of the patients with metastases was correlated significantly with patient age, site of primary tumor, number and location of metastases, number of involved organ systems, histologic response of the primary tumor to preoperative chemotherapy, and completeness and time point of surgical resection of all tumor sites. After Cox multivariate regression analysis, however, only multiple metastases at diagnosis and macroscopically incomplete surgical resection remained significantly associated with poor outcomes. At a median follow-up of 1.9 years, 60 patients were alive, and 37 of the original 202 patients were in complete and continuous surgical remission. The overall actuarial survival rate at 5 years was 29%, and at 10 years it was 24%. The researchers concluded that the number of metastases and the completeness of surgical resection of all clinically detected tumor sites are of independent prognostic value in patients with proven primary metastatic osteosarcoma. A more recent study of 21 patients with metastatic osteosarcoma showed a cumulative 5-year survival rate after complete resection of 34.2%. Complete resection was found to be a significant prognostic factor for survival following metastasectomy (P = 0.04). The authors concluded that every attempt should be made to completely resect all clinically detected metastases even if it meant repeat thoracotomy in recurrent disease.284
Attempts have been made to substitute newer drugs in an attempt to improve survival of patients with metastatic osteosarcoma. To date, many of these have been disappointing. Substitution of cisplatin with carboplatin does not appear to be effective.285,286 The use of topotecan is similarly disappointing.287 One recent ray of hope was the use of L-MTP-PE in newly diagnosed osteosarcoma patients treated on the Children’s Oncology Group Intergroup-0133 (INT-0133). They compared three-drug chemotherapy with cisplatin, doxorubicin, and high-dose methotrexate (Regimen A) with the same three drugs with the addition of ifosfamide (Regimen B). The 5-year EFS for patients who received L-MTP-PE (n = 46) was 42% versus 26% for those who did not (n = 45, P = 0.23), and the 5-year overall survival for patients who received MTP-PE versus no MTP-PE was 53% and 40%.156
Controversies, Problems, Challenges, and Late Effects of Therapy
In addition to the psychosocial outcomes of therapy discussed previously, late effects of chemotherapy, including cardiomyopathy from doxorubicin,288 ototoxicity from cisplatin,289–293 and thyroid disorders,294 remain problematic in survivors. The addition of dexrazoxane has been shown to reduce the toxic effects of doxorubicin on the myocardium.295 Infertility is another concern for patients who receive chemotherapy especially with alkylating agents. A study recently showed that although patients who undergo chemotherapy should be informed of the possibility of infertility, many survivors of pediatric sarcoma treatment can be expected to have successful childbirths.296 Teenage males might want to consider sperm banking.
Metachronous Osteosarcoma
Aung and colleagues297 described a single-institution assessment of patients who experienced metachronous skeletal osteosarcoma between 1973 and May 2000. A retrospective review of records from 426 patients with nonmetastatic high-grade and primary osteosarcoma showed 23 patients, with a median age of 18.7 years, who experienced metachronous osteosarcoma. Initial therapy included combination chemotherapy and surgery. Treatment of the metachronous relapse consisted of chemotherapy or radiation alone or surgery with or without additional individualized chemotherapy.
Rodriguez and associates298 described the treatment of five patients in whom primary osteosarcoma had been treated effectively with chemotherapy and resection of the primary tumor and in whom another osteosarcoma developed at another site but without pulmonary metastases. The interval between management of the primary tumor and the subsequent development of another, apparently unrelated metachronous osteosarcoma ranged from 99 to 150 months. All patients remained otherwise free of disease 24 to 96 months after treatment of the metachronous lesion, except for one patient who experienced acute myelogenous leukemia, most likely as a late effect from chemotherapy. Fitzgerald, Dohlin, and Sim299 reported that among 800 patients, 12 experienced metachronous osteosarcoma, and none experienced pulmonary metastases, a quite different response from that of patients who were reported with primary metastatic osteosarcoma. Others have observed that aggressive treatment of metachronous osteosarcoma is indicated and suggested that the etiology in some might be related to hereditary RB or Li-Fraumeni syndrome.300
Second Malignant Neoplasms
Aung and associates301 retrospectively reviewed the experience of the Memorial Sloan-Kettering Cancer Center with long-term survivors of osteosarcoma and the subsequent development of secondary malignancies. The patients were treated between February 1973 and March 2000, and all had received chemotherapy and/or surgery. Chemotherapy consisted of a combination of agents that included high-dose methotrexate, doxorubicin, bleomycin, cyclophosphamide, dactinomycin, vincristine, cisplatin, and ifosfamide. Of 509 patients, 14 were identified with secondary malignant neoplasms. The time interval from diagnosis of the primary osteosarcoma to development of the secondary malignant neoplasm ranged from 1.3 years to 13.1 years (median: 5.5 years). The second neoplasms occurred in the central nervous system in four patients. There were two cases of acute myeloid leukemia and one case each of myelodysplastic syndrome, non-Hodgkin lymphoma, high-grade pleomorphic sarcoma, leiomyosarcoma, fibrosarcoma, carcinoma of the breast, and mucoepidermoid carcinoma. The standard incidence was 4.6% when patients with a history of RB or Rothmund-Thompson syndrome were excluded.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]92
Sarcomas of Bone
Megan E. Anderson, R. Lor Randall, Dempsey S. Springfield and Mark C. Gebhardt
• More than 2800 new cases of bone sarcoma are diagnosed annually in the United States.
• No specific etiologic agents are identified in the majority of cases.
• Secondary neoplasms are related to known oncogenic factors. (e.g., ionizing radiation, alkylating chemotherapy agents, combinations of both).
• Hereditary cancer syndromes (tumor suppressor genes) are responsible for some cases.
Diagnosis and Radiographic Staging
• Plain radiographs are recommended.
• Magnetic resonance imaging (MRI) scan of primary tumor is the best radiographic study to obtain.
• Chest computed tomography (CT) is indicated to evaluate for pulmonary metastases.
• Whole-body technetium-99m (99mTc) bone scan is indicated to evaluate for metastases to other bones.
• Positron emission tomography (PET) scanning is controversial and has yet to be generally accepted.
• Needle or open biopsy is necessary for a tissue-specific diagnosis and to determine histologic grade.
• In the pathology review, immunohistochemistry and cytogenetics are important.
• Electron microscopic tissue occasionally is required.
• Metastasis at presentation is a worse prognostic finding.
• Histologic grade is the next most significant prognostic indicator.
• Size is less significant, but lesions larger than 10 cm in diameter have a poor prognosis.
• Tumor response to neoadjuvant chemotherapy is important in osteosarcoma and Ewing sarcoma.
• Surgical margins of resection (minimum of a “wide” margin).
• The American Joint Committee on Cancer now monitors grade (high and low), and size (8 cm); designates “skip” lesions (T3); and separates metastasis to bone from other sites (Mla, Mlb).
• The Musculoskeletal Tumor Society monitors location. (intracompartmental and extracompartmental), grade (high and low), and metastasis (“skip” lesions, nodal, bone, and lung are all lumped together).
• A wide surgical margin is recommended.
• Limb-sparing procedures are appropriate for 70% to 90% of patients.
• Adjuvant irradiation is not routinely used for bone sarcomas.
• Local recurrence rates for limb-sparing procedures approach 5% or less.
• Reconstruction methods can be tailored to patients’ needs.
• New and improved biocompatible implant materials and improved designs are available.
• The search continues for new drugs, drug schedules, potentiating agents, and improved dose intensity.
• Identification of risk factors (e.g., cytogenetic, molecular genetic, and signal transduction abnormalities) will improve to identify new methods of potential treatment.
Introduction
It is estimated that 2890 new malignant tumors of bone (excluding multiple myeloma) were diagnosed in 2012 in the United States (http://seer.cancer.gov). The femur is the most common site, but primary sarcoma can occur in any bone. Osteosarcoma, Ewing sarcoma, and chondrosarcoma account for approximately 90% of all primary sarcomas of bone. The management of osteosarcoma and Ewing sarcoma includes chemotherapy and surgery, whereas chondrosarcoma is treated by surgery alone.1 The management of these patients, from initial evaluation and biopsy through surgical therapy and long-term follow-up, is labor intensive and technically demanding. Patients with a bone sarcoma should be treated in a center that has expertise in the management of these tumors.
Surgical Staging System
Currently, the staging system adopted by the Musculoskeletal Tumor Society (MSTS) in 1980 and modified in 1986 is accepted by most musculoskeletal oncologists.4–4 Malignant tumors are divided into only two histologic grades: low-grade malignant (G1) and high-grade malignant (G2). Low-grade malignant lesions (G1), comprising Broder I and II lesions, have a low probability of metastasis (25%). The majority of these tumors can be managed by relatively conservative surgical procedures and do not require chemotherapy. High-grade lesions (G2), Broder III and IV tumors, have a significantly higher incidence of metastases and require more radical surgical procedures and possibly neoadjuvant and/or adjuvant chemotherapy. Table 92-1 is a representative grouping of both low- and high-grade malignant tumors of bone and soft-tissue origin.
Table 92-1
| Low (G1) | High (G2) |
| Parosteal osteosarcoma | Classic intramedullary osteosarcoma |
| Periosteal osteosarcoma (typically intermediate grade) | High-grade surface osteosarcoma |
| Low-grade central osteosarcoma | Paget sarcoma of bone |
| Radiation sarcoma | |
| Secondary chondrosarcoma | Primary chondrosarcoma |
| Clear cell chondrosarcoma | Dedifferentiated chondrosarcoma |
| Mesenchymal chondrosarcoma | |
| Fibrosarcoma | Fibrosarcoma |
| Atypical malignant fibrous histiocytoma of bone | Malignant fibrous histiocytoma (MFH) of bone |
| Adamantinoma | |
| Undifferentiated primary sarcoma of bone | |
| Giant cell sarcoma of bone | |
| Hemangioendothelioma | Angiosarcoma |
| Chordoma | Neurofibrosarcoma/malignant peripheral nerve sheath tumor |
| Ewing sarcoma family of tumors |
Radical margins of a bone sarcoma are achieved when the entire bone is removed. This process usually requires an amputation. A radical margin is rarely necessary.5
The American Joint Committee on Cancer (7th edition) has adapted the TNM (tumor–nodes–metastases) staging system to bone. The topography (T) of the primary tumor now includes size based on relevant published reviews, in which the greatest dimension (8 cm) has replaced the compartment concept. Also, T3 has now been assigned to patients with “skip” metastases (Table 92-2).
Table 92-2
Definition of TNM Primary Tumor (T)
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor ≤8 cm in greatest dimension |
| T2 | Tumor >8 cm in greatest dimension |
| T3 | Discontinuous tumors in the primary bone site |
Adapted from Edge SB, Byrd DR, Compton CC, et al., editors. AJCC cancer staging manual. 7th ed. New York: Springer Verlag; 2010.
The problem of defining histopathological grade (G) has been addressed and now essentially consists of low- and high-grade lesions (Table 92-3). G1 and G2 have been combined into low-grade and G3 and G4 into high-grade histopathology. Currently, all Ewing tumors are classified as G4 or high-grade. This grouping is now identical to the G1 and G2 of the MSTS staging system. Table 92-4 shows the stage groupings. Here, the committee has appropriately addressed the difference in prognosis of patients who have sustained metastases to lung (Mla) and to other sites, including bone (Mlb).
Table 92-3
| GX | Grade cannot be assessed |
| G1 | Well differentiated—low grade |
| G2 | Moderately differentiated—low grade |
| G3 | Poorly differentiated—high grade |
| G4* | Undifferentiated—high grade |
*Ewing sarcoma is classified as G4.
Adapted from Edge SB, Byrd DR, Compton CC, et al., editors. AJCC cancer staging manual. 7th ed. New York: Springer Verlag; 2010.
Table 92-4
| Stage 1A | T1 | N0 | M0 | G1, 2 low grade, GX |
| Stage 1B | T2 | N0 | M0 | G1, 2 low grade, GX |
| T3 | N0 | M0 | G1, 2 low grade, GX | |
| Stage IIA | T1 | N0 | M0 | G3, 4 high grade |
| Stage IIB | T2 | N0 | M0 | G3, 4 high grade |
| Stage III | T3 | N0 | M0 | G3, 4 high grade |
| Stage IVA | Any T | N0 | M1a | Any G |
| Stage IVB | Any T | N1 | Any M | Any G |
| Any T | Any N | Mlb | Any G |
Adapted from Edge SB, Byrd DR, Compton CC, et al., editors. AJCC cancer staging manual. 7th ed. New York: Springer Verlag; 2010.
Radiographic Staging
Conventional bone radiography remains the single most useful initial study for bone tumor evaluation. The study should include anteroposterior and lateral projections of the lesion and ideally the whole bone in which the lesion is present. Malignant neoplasms usually result in ill-defined or “poorly marginated” radiographic margins with little or no reactive bone, loss of medullary trabeculation, and endosteal cortical erosion at the tumor–host bone interface, suggesting an active and destructive process. The pathological process biologically overwhelms the normal time-dependent reactive processes of bone formation. Therefore, the radiographic presence or absence of a reactive rim of bone is often useful in predicting the biological aggressiveness of the pathological process (Table 92-5). Neoplastic bone formation is often seen in osteosarcoma, and calcification is often seen in chondrosarcoma.
Table 92-5
Radiographic Techniques Available
Computed tomography (CT) is superior to magnetic resonance imaging (MRI) only for evaluating a small lesion in the cortex, subtle bone formation, or calcification; otherwise, MRI is the study of choice. CT remains the standard for evaluation of the chest for occult metastases. The cross-sectional display usually provides sufficient resolution (<0.5 cm) to demonstrate subpleural metastases long before they become evident on plain chest films. Before definitive therapy or local management of a potentially malignant lesion, a staging CT evaluation of the chest and mediastinum should be performed.6
Currently, MRI of the primary tumor appears somewhat predictive of tumor response to neoadjuvant therapy. Changes in the T2-weighted image signal intensity correlate with an obvious reduction in tumor volume (especially in Ewing sarcoma) and appear predictive of tumor necrosis.7 The addition of contrast enhancement does not appear to provide more viable tumor/necrotic tumor contrast than do T2-weighted images; however, the absence of contrast enhancement appears to be an indicator of tumor necrosis.7–10
Positron emission tomography (PET) has been used to predict response to chemotherapy for Ewing sarcoma and osteosarcoma. PET scan might be valuable in the initial screening, but this has yet to be determined.11,12
Staging Biopsy
The staging biopsy might well be the most important and difficult procedure that is performed in the patient’s management (Box 92-1). The placement, length, and orientation of the biopsy scar and the anatomic compartments that are contaminated during the biopsy procedure dictate which tissues and how many surgical compartments will require removal for local tumor management and limb-sparing surgery. A thorough knowledge of the soft-tissue anatomic planes and muscle compartments is mandatory before proceeding with bone biopsy. Consideration should be given to the location and type of biopsy to be used, whether fine-needle aspiration biopsy, core-needle biopsy, or open biopsy. In addition, it is critical that adequate diagnostic tissue be obtained so that an accurate diagnosis can be made. Usually, a core-needle biopsy is sufficient.
Osteosarcoma
High-grade osteosarcoma used to be a fatal neoplasm leading to metastases and death of 90% of patients despite aggressive local control, including radical amputations and/or radiotherapy. Previous versions of this chapter outlined the evolution of the use of adjuvant chemotherapy and the dramatic improvement in survival and event-free survival (EFS) that followed. Initially, the benefits of adjuvant chemotherapy were questioned, leading to the need for a randomized study comparing surgical management alone with surgery followed by multiagent chemotherapy. The active drugs were shown to be doxorubicin, high-dose methotrexate, and cisplatin. A randomized study, including both the randomly assigned patients and those who chose whether or not to have adjuvant chemotherapy, clearly demonstrated the benefits of adjuvant chemotherapy.13,14
Surgical advances paralleled the advances in medical management of osteosarcoma patients. Originally, disarticulation or resection of the entire involved bone was recommended for surgical management of osteosarcoma.15 This recommendation was partly a result of the intramedullary origin of the tumor with proximal intramedullary growth and the reported 25% incidence of intramedullary “skip” metastases.16 Later studies reviewing the local recurrence rates for patients whose primary management was transmedullary amputation alone revealed local recurrences in approximately 5% to 10%, suggesting that the incidence of “skip” or intraosseous metastasis was probably lower than originally believed. The general standard of surgical management of patients with extremity osteosarcoma in 1980 included transmedullary amputation approximately 5 to 7 cm proximal to the intramedullary extent of the tumor.17
One event that led to the popularization of limb salvage was the use of preoperative chemotherapy. Initially implemented during the time it would take to manufacture a custom prosthesis, it became apparent that the tumor showed clinical and radiographic “response” to the preoperative (neoadjuvant) chemotherapy. This seemed to make the subsequent surgery easier, if not safer, and gave the surgeon 10 to 12 weeks to work with the patient to decide on the best surgical option. There was initial concern that the delay in the resection might worsen the prognosis, but this issue was addressed in a randomized trial that showed no apparent advantage to having the surgery initially compared to the neoadjuvant mode of administration.18 In addition, it was learned that the histologic response to the preoperative chemotherapy was a predictor of outcome and, second only to the presence of metastases at diagnosis, was the best predictor of survival.18,19
Epidemiology
Conventional, or classic, osteosarcoma makes up the majority of all osteosarcomas. It occurs primarily in the metaphyses of adolescents with open physes or in young adults. Most patients with classic osteosarcoma are younger than age 30 years, and many have no apparent predisposing factors.20 The lesion most often arises in the larger, more active metaphyses (e.g., distal femur, proximal tibia, proximal humerus), but also can arise in the flat bones of the pelvis, skull, scapula, and ribs, and in the spine. Overall, the majority of the lesions develop in the extremities and pelvis.21
An epidemiological study conducted in Sweden between 1971 and 1984 investigated possible changes in the typical features of 227 conventional osteosarcomas. The mean annual incidence was 2.1 per million. The male-to-female ratio of 1.6 : 1 remained unchanged over the study period, as did the location and distribution of the tumors. In the United States, the incidence of osteosarcoma is considered to be higher in males than in females, occurring at a rate of 5.4 per million persons per year in males versus 4.0 per million in females, with a higher incidence in blacks (6.8 per million persons per year) and Hispanics (6.5 per million), than in whites (4.6 per million).22
The only clear change over the study period was an increase in the age of patients beyond the classic peak age range of 10 to 29 years.23 In a recent study in the United States, the incidence rates and 95% confidence intervals of osteosarcoma for all races and both sexes are 4.0 (3.5 to 4.6) for the range 0 to 14 years and 5.0 (4.6 to 5.6) for the range 0 to 19 years per year per 1 million persons. Among childhood cancers, osteosarcoma occurs eighth in general incidence. Osteosarcoma has a bimodal age distribution, having the first peak during adolescence and the second peak in older adulthood. The first peak is in the 10- to 14-year-old age group, coinciding with the pubertal growth spurt. This suggests a close relationship between the adolescent growth spurt and osteosarcoma.24
Osteosarcoma develops in 10% of patients after the age of 60 years. This group composes the second peak of the bimodal age distribution curve. In these older patients, the anatomic region of presentation differs substantially from the sites of classic osteosarcoma. Whereas lesions develop in the region of the knee (the largest and most active physes) in more than 50% of patients with classic osteosarcoma, osteosarcoma at that site develops in only 15% of the older patients. Moreover, osteosarcomas in the older population characteristically present in regions that have had previous radiotherapy, underlying Paget disease of bone, fibrous dysplasia, or some other pathological abnormality. In many ways, the older group can be thought of as having “secondary” osteosarcoma.25
An estimated 2000 malignant bone tumors are diagnosed in the United States each year. Approximately 750 of these patients have classic or conventional osteosarcomas. Males are affected slightly more often than females. Classic osteosarcoma develops in females slightly earlier than in males, and there appears to be no race predilection.26 Although the common histologic presentation of malignant cells producing osteoid would suggest a homogenous group of tumors, the morphologic appearance can vary considerably, ranging from classic osteoblastic osteosarcoma (45% of cases) through fibroblastic (9%), chondroblastic (27%), anaplastic (17%), telangiectatic, low-grade central, and other osteosarcomas (2%).27
Etiological and Biological Considerations
Osteosarcoma is considered a sporadic complex genotype sarcoma, as distinguished from the balanced translocation-associated sarcomas (e.g., Ewing sarcoma). Many cell-cycle regulatory factors have been implicated, including p53, Rb, and others. Certain populations may, in fact, be at risk for developing osteosarcoma.24,28 Herein we provide a brief overview of the major regulatory pathways of the cell cycle with implications for sarcomagenesis in osteosarcoma.
The p53-ARF-MDM2 Pathway
In conjunction with the CDK4/6 inhibitor p16INK4A, mutations of the TP53 gene are the most recurrent genetic alterations associated with cancer.29 The TP53 gene encodes the well-known tumor-suppressor p53, which plays an important role in various regulatory processes involved with cell-cycle progression. Most importantly, p53 acts as an important negative regulator, facilitating growth arrest, senescence, and/or apoptosis when cells are exposed to genotoxic, cytotoxic, and/or physiological stresses.30,31 p53 is a transcription factor possessing two transcriptional activation domains and a DNA-binding domain that recognizes specific sequences. Following exposure to cellular stress, p53 activates expression of CDKN1A, which encodes the cyclin-dependent inhibitor p21CIP1. It is through the expression of p21 that p53 negatively regulates the cell cycle.32–35
The expression level and activity of p53 is regulated by two different proteins: MDM2 and p14ARF. MDM2 is an E3 ubiquitin ligase, which facilitates the degradation of p53 in a ubiquitin- and proteasome-dependent manner.36 Interestingly, MDM2 is a transcriptional target of p53, whose expression is increased in concert with increased levels of p53.37 In addition to promoting degradation, MDM2 can also inhibit p53 function through a direct protein–protein interaction, suppressing its transcriptional activity, as well as translation of the messenger RNA transcript itself. To combat these negative regulatory effects, p14ARF is able to positively regulate p53, in part through the negative regulation of MDM2. p14ARF can actually be found at the same gene locus as CDKN2A, which encodes the CKI p16.37,38 The two gene sequences do overlap but possess alternate reading frames and are independently regulated. ARF binds MDM2, preventing it from interacting with p53.7 Consequently, p53 becomes stabilized and its overall activity increases within the cell.39
The CIP/KIP Family of Cyclin-Dependent Kinase Inhibitors
The CIP/KIP family of proteins is the second group of cyclin-dependent kinase inhibitors (CKIs). There are three different members: p21CIP1, p27KIP1, and p57KIP2. Similar to the INK4 family of CKIs, CIP/KIP proteins inhibit the kinase activity of cyclin-dependent kinases (CDKs) by preventing their association with cyclin subunits as well as adenosine triphosphate molecules, both of which are required for the phosphorylation of target substrates.32,40 Unlike the INK4 family, however, CIP/KIP proteins are able to functionally inhibit multiple CDKs. For instance, both p27 and p57 can inhibit the kinase activity of CDK4/6 and CDK2, whereas p21 acts to control the function of both CDK2 as well as CDK1.33,41 Of the three family members, p21 is the most diverse, performing a variety of functions in addition to controlling CDK activity. For example, it was previously mentioned that p21 is a transcriptional target of p53.32–35 Following exposure to various stresses, p53 activates the expression of p21, thus inducing a DNA damage response.33 In addition, p21 has been shown to localize to the cytoplasm where it acts to inhibit the induction of apoptosis by interacting with proteins involved in mediating this process.
Other Important Players
The last two cell-cycle regulators that are discussed are C-MYC and Ki-67. Both of these proteins play crucial roles during cell proliferation but do so in very different ways. C-MYC influences several processes involved in cell-cycle regulation via its function as a transcription factor. For example, C-MYC, when bound to its partner MAX, has been shown to induce the expression of cyclins D1 and D2 as well as CDK4, subsequently promoting G1 phase progression.40,42 The MYC-MAX heterodimer can also support continued cell-cycle progression through the repression of multiple CKIs, including p15, p18, p21, and p27.43 Furthermore, C-MYC can increase the expression of E2F2 and cyclin A2, both of which affect the S-phase of cell cycles and contribute to overall proliferation.45–45 Similar to C-MYC, Ki-67 has been shown to be vital for cell proliferation.46 Ki-67 is a cell proliferation-associated nuclear antigen that is thought to contribute to cell-cycle progression via its involvement in ribosomal RNA and ribosome synthesis.11,46,47 Interestingly, Ki-67 is expressed in all of the phases of cell cycle (excluding G0), but whether or not it participates in other such related processes is currently unknown.
A small subset of osteosarcomas is hereditary.48 Osteosarcoma in siblings occurs in fewer than 1 in 1000 to 1 in 3000 osteosarcoma patients.49,50 Observation of two or more affected siblings in a family indicates an underlying genetic predisposition.50–57 When siblings in multiple generations are affected, an autosomal dominant disorder is most likely responsible. One example would be the hereditary form of retinoblastoma. Individuals with hereditary retinoblastoma (germline retinoblastoma gene mutation) have a 2000-fold risk for osteosarcoma in the second decade of life when compared with the general population.58–63
The gene for retinoblastoma (RB) has been localized to the long arm of chromosome 13 (13q14). The RB gene is recognized as the prototype of a tumor suppressor gene and has been implicated in the pathogenesis of a number of human neoplasms.64,65 A tumor suppressor gene normally functions by restraining cell (tumor) growth, so loss of function or inactivation of a tumor suppressor gene results in tumor growth. Loss of 13q14 (the RB gene) is thought to be responsible for the development of RB.64–68 A two-hit kinetic model for this class of genes was proposed by Knudson.69 For hereditary RB, the primary mutation in one RB locus occurs in germinal cells; for sporadically occurring RB, the primary mutation exists in somatic cells. The second step, responsible for malignant transformation, is the loss of function of the remaining normal homolog in somatic cells by some chromosomal rearrangement or mutation identified as loss of heterozygosity for markers in or around the RB gene.58,60–72 Molecular analyses of both sporadic osteosarcomas and osteosarcomas from patients with RB have revealed homozygous loss of RB gene function in a high percentage of cases.58,59,65–77 Assessment of loss of heterozygosity at the RB gene in a study by Feugeas and colleagues78 revealed that RB gene locus loss of heterozygosity could be an early predictive feature for osteosarcomas with a potentially unfavorable outcome. Osteosarcoma develops in 12% of patients with bilateral RB, yet as many as 70% of osteosarcomas have a dysfunctional RB gene product.79,80–85 Thus other oncogenes are likely implicated in the oncogenesis of osteosarcoma.
Several investigators have demonstrated that the mutational profiles of the RB gene in osteosarcoma are basically the same as those for RB and that mutation of the RB gene plays an essential role in the development of osteosarcoma.65,73 Besides loss of gene function at the locus on chromosome 13, however, loss of heterozygosity for other chromosomal loci, such as 3q, 17p, and 18q, has been implicated.73,86–91
Pathology and Pathways of Spread
Osteosarcoma is a high-grade sarcoma comprising malignant osteoblasts that vary in size and shape and have bizarre mitoses. Proposed histologic grading systems for osteosarcoma appear to be of little value.92 Attempting to grade an osteosarcoma presents many difficulties that limit the usefulness of any grading system. For example, many tumors are heterogeneous, and tissues sampled from separate areas of the same tumor may give different impressions. The number of mitoses, the degree of cellularity, and cellular anaplasia or pleomorphism can differ from site to site within the same tumor. Tumors of identical histologic appearance often differ in their clinical behavior. All “classic” or conventional osteosarcomas are considered high-grade. Osteosarcoma is a vascular tumor, and the tumor osteoblasts produce tumor osteoid or woven bone (Fig. 92-1, A and B). These tumors are poorly differentiated and may take on a fibroblastic or chondroblastic appearance on light microscopy (Fig. 92-1, C and D), but if there are areas of bone formation, they are considered osteosarcomas. In fact, a high-grade chondroblastic sarcoma in a child or adolescent on biopsy is considered to be an osteosarcoma (and treated as such) until proved otherwise from examination of the entire specimen (chondrosarcomas are extremely unusual in children). The tumor usually originates in the metaphysis of the bone and percolates between the preexisting trabeculae of bone, incompletely destroying the existing bone, presumably because of its rapid growth (seen well in Fig. 92-1A). The tumor eventually follows the vascular Haversian and Volkmann canals in the cortex, partially resorbing the normal cortex and replacing it with tumor bone as it spreads to the adjacent soft tissue. The periosteum is lifted and tries to respond, but the response is incomplete, leading to the appearance of the Codman triangle on a radiograph. It may also cause perpendicular striations of bone, the so-called starburst appearance of osteosarcoma. Proximally, the tumor ends fairly sharply in the medullary cavity, but “skip metastases” may be detectable in the marrow surrounding the tumor in a small proportion of patients. The physis or growth plate is a relative barrier to tumor spread, but because the open physis has vascular channels, it is well documented that the tumor will cross the growth plate and enter the epiphysis. The articular cartilage is a more definitive barrier, and osteosarcomas seldom cross the articular cartilage unless there has been a fracture. It may spread into the joint at the periphery of the cartilage or enter the joint along ligaments such as the cruciate ligaments of the knee, but this is a relatively rare event (Fig. 92-2).
Telangiectatic osteosarcoma is a predominantly radiolucent, destructive osteosarcoma variant. Histologically, it is composed of single or multiple dilated spaces containing blood or degenerated tumor cells and lined by anaplastic, mitotically active sarcoma cells.79 Telangiectatic osteosarcoma must be differentiated from aneurysmal bone cyst, to which it can be similar in appearance both radiographically and histologically.
A review of 124 patients with telangiectatic osteosarcoma spanning the years 1921 to 1979 suggested no differences in survival compared with patients with conventional osteosarcoma. Further analysis demonstrated that the favorable outcome in 17 of the patients with telangiectatic osteosarcoma was related to their being treated with multiagent chemotherapy. Twenty-five patients had received this therapy, and 17 were free of disease at 5.5 years, demonstrating the response to chemotherapy in this highly vascular tumor.93A more recent evaluation of 323 patients with telangiectatic osteosarcoma revealed that although these tumors may be at higher risk for pathological fracture, with multimodal therapy, outcomes were similar to other osteosarcomas even with the negative prognostic factor of the fracture itself.93a
Clinical Manifestations, Patient Evaluation, and Staging
The plain radiograph is the best diagnostic tool. Osteosarcomas may either completely destroy the bone (radiolucent lesion) or replace the bone with a blastic response (radiodense), but they most often do both. The radiograph shows areas of destruction of the host bone and blotchy densities of new (tumor) bone production. The lesion is most frequently in the metaphysis of a long bone in the adolescent or child and in the flat bones in the adult, but any bone can be involved at any age (Fig. 92-3, A and B). The tumor is usually large, destroys the cortex, and is associated with a soft-tissue mass. The mineralization of the matrix is often apparent, but because these tumors can contain areas of chondroblastic and fibroblastic as well as osteoblastic differentiation, the pattern on mineralization might not be that of bone.
There are no blood laboratory studies that aid in the diagnosis of osteosarcoma, but it has been shown in several large series of patients that an elevated level of alkaline phosphatase and/or lactic dehydrogenase is associated with a worse prognosis.96–96
MRI is the single most useful study in evaluating the intraosseous and extraosseous extent of the primary tumor and in detecting intramedullary or transarticular “skip” metastases. It has been well demonstrated that MRI better identifies the edema (high water content) in and around the reactive zone of the pseudocapsule illuminating the potential surgical margin. MRI has also been shown to be superior to CT in displaying the medullary canal extent of the tumor, suspected “skip” lesions, soft-tissue extension, and overall anatomic location of an extremity tumor (see Fig. 92-3, D to F).97 MRI, with or without magnetic resonance angiography, is the single most valuable tool for planning limb-sparing surgical procedures.100–100 The use of MRI has significantly improved radiologic staging and presurgical planning.101,102 Serial MRI studies are less reliable, however, in evaluating tumor response to primary chemotherapy, and are more predictive of a poor rather than a good response. By demonstrating an increase in size of the tumor, more bone destruction, and soft-tissue invasion on serial studies, MRI is more accurate as a measure of a poor chemotherapy response.7,103,104
Metastases to bone and/or lung are usually assessed by a whole-body 99mTc bone scan and a complete CT of the chest and mediastinum. The 99mTc bone scan has been considered superior to other imaging studies for surveying the skeleton for metastatic or multiple lesions and for later detecting the development of skeletal metastases (see Fig. 92-3C).105
PET scans have been used in an attempt to stage and separate high-grade from low-grade tumors.106 Brenner and associates107 summarized the current usefulness of 18F-fluorodeoxyglucose (18F-FDG) PET in patients with osteosarcoma. High-resolution CT has been shown to be superior to 18F-FDG PET for detecting pulmonary metastases and is not recommended to detect bone metastases except when a suspected “skip” lesion has been identified on MRI. PET does not make it possible to differentiate between high- and low-grade osteosarcoma. It could be useful, however, in determining the appropriate area to biopsy to identify viable representative tumor tissue and in distinguishing benign aggressive lesions from other lesions where local recurrence is likely.
18F-FDG PET may be most useful to determine the response to neoadjuvant chemotherapy and to demonstrate a region of viable tumor.108 The timing between the initiation of preoperative chemotherapy and the point at which 18F-FDG PET becomes predictive remains uncertain. The response to soft-tissue postchemotherapy inflammation and healing surrounding the tumor must be characterized before this technique becomes predictive of preoperative response to chemotherapy. PET also has potential usefulness in patient follow-up, differentiating postoperative tissue changes and possible tumor recurrence especially in patients with metallic reconstructions. Orthopedic implants often hamper assessment by CT or MRI, and sequential 18F-FDG PET scans could differentiate between recurrent tumors and the normal healing process. Also, because 18F-FDG PET provides scanning of the entire patient, it may be useful when combined with CT of the chest to detect first evidence of pulmonary metastasis.109A recent study showed that PET correlates only moderately to histologic response, but may be predictive of progression-free survival.11 Once the staging workup is complete, a biopsy is performed. The procedure can be either an open biopsy or a needle biopsy (core-needle or fine-needle aspirate) depending on the experience of the surgeon, the interventional radiologist, and the pathologist. If an open biopsy is performed, it should be done by the surgeon who will be responsible for local control, with a view to placement in a site that can be resected with the definitive specimen. If a core-needle biopsy is performed by an interventional radiologist (which is becoming more frequent), the radiologist and the surgeon should agree on the placement of the needle track. Communication between the surgeon and interventional radiologist is essential, as is communication with the entire multidisciplinary team to determine whether tissue should be sent for cytogenetics or other special biological studies. Table 92-6 provides a diagnostic workup algorithm.
Table 92-6
Diagnostic Workup Algorithm for Bone Sarcomas
Once the biopsy results are available, the sarcoma can be staged as discussed in the introduction.
Primary Therapy
Adjuvant Chemotherapy
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
