Pediatric Solid Tumors
Jeffrey S. Dome, Carlos Rodriguez-Galindo, Sheri L. Spunt and Victor M. Santana
• The incidence of osteosarcoma is four new cases per 1 million population per year among children younger than 15 years of age.
• Osteosarcoma is the most common bone tumor in children and adolescents.
• Other lytic bone lesions, including eosinophilic granuloma and giant cell tumor, must be excluded.
• At histopathological examination, tumors must be differentiated from fibrosarcoma and chondrosarcoma.
• Staging evaluation includes a complete history and physical examination, complete blood cell count (CBC), serum chemistry analysis (including determination of alkaline phosphatase level), and imaging studies of the primary tumor and chest.
• The primary tumor is managed surgically with a limb-sparing operation or amputation.
• Adjuvant chemotherapy incorporating high-dose methotrexate is used preoperatively for presumed micrometastatic disease; response will guide further therapy.
• Methotrexate responders have a more than 80% chance of cure.
• Primary pulmonary metastatic disease is managed surgically, although adjuvant chemotherapy may reduce the extent of resection.
• Pulmonary metastatic disease is managed surgically.
• Postthoracotomy adjuvant chemotherapy is of unproven benefit.
• Local recurrence is managed surgically.
• Further adjuvant chemotherapy with non–cross-resistant agents is of uncertain benefit.
• The incidence of Ewing sarcoma family tumors is 2.8 new cases per 1 million population per year among children younger than 15 years of age.
• Ewing sarcoma is the second most common bone tumor among children and adolescents.
• Osteomyelitis must be ruled out, especially when the patient has a fever.
• Lesions to be excluded include benign tumors of bone that manifest as lytic lesions (e.g., eosinophilic granuloma, giant cell tumor), malignant tumors (e.g., osteosarcoma, primary lymphoma of bone), and metastatic lesions from a nonosseous tumor (e.g., neuroblastoma).
• Staging evaluation includes a complete history and physical examination, CBC, serum chemistry analysis (including determination of lactate dehydrogenase level), bone marrow biopsy, and imaging studies of the primary tumor, bones, and chest.
• The primary tumor should always be treated with multimodality therapy consisting of chemotherapy, radiation therapy, surgery, or a combination of these treatments.
• Specific local treatment depends on the primary site. Surgical extirpation may be considered with tumors in expendable bones (proximal part of the fibula, rib, clavicle, iliac wing).
• Unresectable tumors generally necessitate a combined approach of chemotherapy, radiation therapy, and surgery.
• Localized disease is curable with combined therapy in more than 70% of cases. Metastatic disease is curable in 30% to 40% of cases.
• Effective second-line therapy has not been established.
• Local recurrence may be amenable to surgical extirpation.
• Among white children younger than 15 years of age, 10.5 new cases occur per 1 million population per year. Among black children, the incidence is 8.8 new cases per 1 million population per year.
• Neuroblastoma is the most common extracranial solid tumor in children.
• Disseminated bone disease can resemble systemic infection, inflammatory disease, osteomyelitis, or rheumatoid arthritis.
• Paraneoplastic syndromes associated with neuroblastoma (vasoactive intestinal peptide syndrome, opsoclonus-myoclonus-ataxia syndrome) must be differentiated from primary inflammatory bowel disease and neurologic disease.
• Neuroblastoma must be differentiated from other small, round, blue cell neoplasms of childhood (e.g., Ewing sarcoma, primitive neuroectodermal tumor, non-Hodgkin lymphoma, undifferentiated soft-tissue sarcoma).
• In as many as 10% of tumors, catecholamines are not produced. In 1%, the absence of an obvious primary lesion confounds the diagnosis.
• Staging evaluation includes a complete history and physical examination, CBC, serum chemistry analysis (including determination of lactate dehydrogenase level), quantitative urine catecholamines, bone marrow examination, radionuclide scintigraphy, and imaging studies of the primary tumor and chest.
• Specific treatment depends on stage of disease, age of the patient, and biological features of the tumor.
• With few exceptions (completely resected primary tumor, localized tumor with/without complete excision and favorable tumor biological features, infants with stable stage 4S disease), multiple-agent chemotherapy is the backbone of multimodality treatment.
• Survival depends on stage and biological features of the tumor (e.g., histopathological subtype, MYCN gene amplification).
• The overall survival rate for asymptomatic patients with favorable biology stage 1, 2a, or 2b tumors is greater than 90%.
• Overall survival among patients with advanced-stage 3 or 4 with MYCN gene amplification disease is poor. Less than one-third of children with high risk disease are long-term survivors.
• No second-line therapy has proved to increase survival.
• The incidence of Wilms tumor is eight cases per 1 million population per year among children younger than 15 years of age.
• Mean age at diagnosis is 44 months for unilateral tumors and 31 months for bilateral tumors.
• Familial cases account for 1.5% of cases of Wilms tumor.
• Associated with Wilms tumor is a syndrome comprising aniridia, genitourinary anomalies, and mental retardation (WAGR syndrome); Denys-Drash syndrome; and Beckwith-Wiedemann syndrome.
• No firmly established environmental factors have been identified.
• The main histologic subtypes are favorable and anaplastic.
• Major implicated genes and loci include WT1 (11p13), IGF2 (11p15), FAM123B/WTX (Xq11), and CTNNB1/β-catenin (3p22).
• Mutations of the p53 gene (TP53) are associated with anaplastic tumors.
• Loss of heterozygosity (LOH) at chromosomes 1p and 16q and gain of chromosome 1q are associated with adverse prognosis.
• An asymptomatic abdominal mass is found in most patients.
• Other characteristic features include abdominal pain, hematuria, hypertension, and congenital anomalies (genitourinary malformations, aniridia, and hemihypertrophy).
• Neuroblastoma is the main consideration in the differential diagnosis.
• Other tumors to be excluded are renal neoplasms (clear cell sarcoma, rhabdoid tumor, congenital mesoblastic nephroma, renal cell carcinoma).
• Benign renal processes (nephrogenic rests, multicystic or polycystic kidneys, hydronephrosis, renal carbuncles, hemorrhage) also should be ruled out.
• Evaluation begins with complete history and physical examination, with careful attention to blood pressure and assessment for associated congenital anomalies.
• Other components of the staging evaluation include CBC, serum chemistry analysis, urinalysis, abdominal ultrasonography, abdominal/pelvic computed tomography (CT) or magnetic resonance imaging (MRI), and chest CT.
• Surgery: Surgical resection of the primary tumor usually precedes chemotherapy in the approach taken by the Children’s Oncology Group (COG). Presurgical chemotherapy is used in the approach taken by the International Society of Pediatric Oncology (SIOP).
• Chemotherapy: Agents used depend on disease stage and favorable versus anaplastic histology: stages I and II, favorable histology—vincristine, actinomycin D; stages III and IV, favorable histology, and stage I, anaplastic histology—vincristine, doxorubicin, actinomycin D; stages II through IV, anaplastic histology—vincristine, cyclophosphamide, doxorubicin, carboplatin, etoposide.
• Radiation therapy: Radiation therapy is included in management for stages III and IV, favorable histology, and stages II through IV, anaplastic histology.
• Recurrent disease is effectively managed with radiation therapy and chemotherapy with agents not used for initial treatment.
• Patients who initially received aggressive treatment may respond to cyclophosphamide or ifosfamide-, carboplatin-, and etoposide-based regimens.
• Renal failure is seen in less than 1% of patients with unilateral tumors and 12% in patients with bilateral tumors.
• Congestive heart failure may occur in patients who receive doxorubicin (occurs in 4.4% of patients).
• Pregnancy-related complications in adulthood have occurred in girls who receive flank irradiation.
• A second malignant neoplasm also may develop (6.7% cumulative incidence of solid malignancies at age 40 years).
• With favorable histology, outcomes include 85% 4-year relapse-free survival rate and 90% 4-year overall survival rate.
• With anaplastic histology, outcomes include 50% 4-year relapse-free survival rate and 50% 4-year overall survival rate.
• The incidence of renal cell carcinoma (RCC) is four cases per 10 million population per year among children younger than 20 years of age.
• Mean age at diagnosis in pediatric series is 9 years.
• Other renal tumors of childhood, primarily Wilms tumor, should be ruled out.
• Staging evaluation includes a complete history and physical examination, CBC, serum chemistry analysis, and imaging studies of the abdomen, pelvis, and chest.
• Nephrectomy of the involved kidney constitutes definitive therapy for primary tumors.
• For metastatic disease, enrollment in clinical studies of investigational agents is recommended. Tyrosine kinase inhibitors have shown benefit for translocation renal cell carcinoma.
• Among children younger than 20 years, the incidence of rhabdomyosarcoma is 4.7 new cases per 1 million population per year.
• Rhabdomyosarcoma is the most common soft-tissue sarcoma among children and adolescents.
• Other benign and malignant soft-tissue tumors must be excluded.
• At pathological examination, rhabdomyosarcoma must be differentiated from the other small, round, blue cell tumors of childhood (e.g., Ewing sarcoma, neuroblastoma, non-Hodgkin lymphoma).
• Staging evaluation includes a thorough history and physical examination; CBC; serum chemistry analysis; imaging studies of the primary tumor, regional lymph nodes, lungs, and bones; and bone marrow examination.
• Primary therapy is chemotherapy with surgery, radiation therapy, or both. Specific treatment depends on age of the patient, tumor primary site and histologic subtype, and extent of disease.
• Mutilating surgery usually can be avoided because the tumor is sensitive to both chemotherapy and radiation therapy.
• The overall survival rate exceeds 70%, but survival will depend on extent of disease. More than 90% of patients with localized, resectable, favorable histology tumors survive, but less than 20% of patients with metastatic disease survive.
• Cure rarely is possible except for patients with botryoid tumors and those with embryonal tumors whose tumor arose in a favorable site and was completely resected at initial diagnosis.
Nonrhabdomyosarcoma Soft-Tissue Sarcoma
• Among persons younger than 20 years, 7.7 new cases occur per 1 million population per year.
• Peaks in incidence occur among infants and among children older than 10 years of age.
• Other tumors to be ruled out include benign soft-tissue tumors, rhabdomyosarcoma, and extraosseous Ewing sarcoma.
• Staging evaluation includes a thorough history and physical examination and imaging studies of the primary tumor and lungs.
• Imaging of regional lymph nodes, liver, bones, and brain is indicated in some clinical settings.
• Surgical excision with or without radiation therapy is indicated for patients with resectable tumors.
• Chemotherapy may provide some benefit for patients with high-grade tumors larger than 5 cm in diameter and for those with unresectable or metastatic tumors.
• Survival depends on the size and grade of the tumor and extent of disease. Patients with localized tumors 5 cm or less in diameter or localized low-grade tumors larger than 5 cm in diameter that can be resected have a survival rate exceeding 85%. Approximately 50% of patients with high-grade tumors larger than 5 cm or with unresectable disease survive. The survival rate is approximately 10% among patients with metastatic disease.
• Cure generally is possible only for patients with local recurrence amenable to surgical extirpation and for those with distant recurrence of low-grade tumor that proves to be surgically resectable.
• Among children younger than 5 years of age,11 new cases occur per 1 million population per year.
• The two clinical forms of retinoblastoma are as follows: (1) Hereditary, bilateral or multifocal (40% of cases)—This form, characterized by germline mutations of RB1, may be inherited from an affected survivor or a silent carrier parent or may be the result of a new germline mutation. (2) Nonhereditary, unilateral or unifocal (60% of cases)—15% of unilateral cases represent germline mutations.
Clinical Manifestations and Differential Diagnosis
• Clinical manifestations: Leukocoria is seen in more than 50% of cases, strabismus in 20% to 25%.
• Differential diagnosis: Coats disease, retinopathy of prematurity, persistent hyperplastic primary vitreous, Toxocara uveitis, and toxoplasmosis need to be ruled out.
• Approach to staging depends on tumor size and on the presence or absence of intraocular and extraocular extension.
• Indirect ophthalmoscopic examination of both eyes with the patient under general anesthesia is essential.
• Imaging studies helpful in staging include ultrasonography, orbital and cerebral CT, and MRI.
• Bone marrow biopsy and cerebrospinal fluid examination are reserved for patients with extraocular disease, optic nerve involvement, or choroid invasion.
• Treatment must be individualized and depends on laterality, potential for vision, and tumor extent.
• Enucleation is reserved for cases in which no potential for useful vision remains. Cryotherapy and photocoagulation are useful for management of small primary or recurrent tumors.
• Radiation therapy is the treatment modality of choice for controlling local disease and preserving vision with larger tumors.
• Use of chemotherapy is restricted to patients with advanced intraocular disease or with extraocular disease.
• Among children younger than 15 years of age, 2.2 new cases occur per 1 million population per year.
• Mean age at diagnosis is 18 months.
• The tumor is associated with familial adenomatous polyposis and Beckwith-Wiedemann syndrome.
• An association with very low birth weight has been found.
• The major histologic subtypes are fetal, embryonal, macrotrabecular, and small cell undifferentiated.
• The tumor is associated with mutations of the APC and β-catenin genes and loss of heterozygosity at 11p15, the locus of the IGF2 gene.
• An asymptomatic abdominal mass is present in most patients.
• Other features may include anorexia, weight loss, vomiting, and precocious puberty (seen in 2% of cases).
• Other malignant tumors to be excluded include hepatocellular carcinoma, embryonal sarcoma, rhabdomyosarcoma, angiosarcoma, and teratoma.
• Benign tumors to be ruled out include hemangioma, hemangioendothelioma, hamartoma, and adenoma.
• Staging evaluation includes a complete history and physical examination, with careful assessment for any congenital abnormalities or signs of precocious puberty.
• Laboratory tests typically include CBC, serum chemical analysis, and α-fetoprotein and α-human chorionic gonadotropin assays.
• Diagnostic imaging may include CT or MRI of the abdomen and chest CT.
• Cure is possible only when complete surgical excision is performed. If complete excision is not feasible, liver transplantation should be considered.
• Adjuvant chemotherapy (cisplatin-based, in conjunction with doxorubicin or 5-fluorouracil) is useful preoperatively for achieving resectability and postoperatively for preventing distant metastasis.
• Radiation therapy has a limited and ill-defined role.
• Recurrent disease confers a poor prognosis, but repeated resection of local and metastatic recurrences has lengthened the survival period.
• Hearing loss and nephrotoxicity (related to cisplatin), cardiac toxicity (related to doxorubicin), and development of second malignant neoplasms may occur.
• With localized disease, outcomes include a 60% to 70% 5-year relapse-free survival rate and a 70% to 80% 5-year overall survival rate.
• With metastatic disease, outcomes include a 25% to 30% 5-year relapse-free survival rate and a 50% to 60% 5-year overall survival rate.
• The incidence of adrenocortical carcinoma is two to three new cases per 10 million population per year in the United States.
• A 10- to 15-fold higher incidence has been observed in southern Brazil.
• Associated with germline TP53 mutations and Li-Fraumeni syndrome.
• Other conditions associated with pathological androgen or cortisol production, such as Cushing syndrome or ovarian or testicular tumors, should be ruled out.
• Evaluation for staging includes imaging studies of the abdomen, pelvis, and chest; skeletal scintigraphy; and measurement of blood and urine concentrations of adrenocortical hormones.
• Complete surgical removal of the tumor is indicated.
• Mitotane-based therapy or chemotherapy with cisplatin also is given.
• Recurrent disease can be managed with further surgery, or experimental agents can be tried.
Osteosarcoma
Epidemiology
Osteosarcoma, a malignant neoplasm derived from primitive mesenchymal cells and characterized by the presence of osteoid-producing spindle cell stroma, is the most common malignant bone tumor in the pediatric age group.1 Osteosarcoma ranks tenth among all newly reported pediatric cancers in the United States, accounting for 2.6% of all neoplasms in children. The estimated annual incidence is 3.9 cases per 1 million population among white children and 4.5 per 1 million population among African American children.2 Most osteosarcomas occur during the first 2 decades of life, a period characterized by rapid skeletal growth. Boys are affected more commonly than girls. Several observations support the association between skeletal growth velocity and osteosarcoma. First, patients with osteosarcoma tend to be taller than their counterparts without this disease. Second, osteosarcoma develops at an earlier age in female patients than in male patients, perhaps because of differences in the timing of onset of puberty and the growth spurt.3
Tumor Biology
Unlike osteosarcoma in adults, in whom more than 25% of tumors are associated with preexisting pathological osseous conditions such as Paget disease or fibrous dysplasia, most pediatric osteosarcomas arise spontaneously in areas of bone without any abnormality.1 Irradiation is the best-characterized etiologic factor contributing to the development of secondary osteosarcoma. In a study involving 91 patients with second malignant bone sarcomas, osteosarcoma accounted for 72 cases, 52 (72%) of these tumors arising within previously irradiated fields.4 The median time for development of the secondary tumor was 9.6 years after irradiation. Osteosarcoma as a second malignancy is often associated with retinoblastoma; osteosarcoma is the most common malignancy in survivors of retinoblastoma, both in the irradiated and the nonirradiated areas, and it accounts for 25% to 40% of all second neoplasms in this population.5,6 One half to two-thirds of osteosarcomas occur in the irradiated fields of the skull and face, one-third of tumors develop in the extremities, and less than 10% occur in the trunk. Osteosarcoma is also a very frequent malignancy in individuals with germline TP53 mutations (Li-Fraumeni syndrome)7 and in patients with REC helicase-associated disorders (Rothmund-Thomson, Werner, and Bloom syndromes.8
Consistent with the association of osteosarcoma with retinoblastoma survivors and Li-Fraumeni syndromes, alterations in components of the cell-cycle control system appear to characterize the ontogeny of osteosarcoma. Studies of the retinoblastoma gene (RB1) have shown that alterations affect the RB1 gene in as many as 80% of cases and that other events, such as CDK4 alterations, also may result in RB1 inactivation.9,10 Genetically engineered mice based on osteoblast-restricted deletion of p53 and pRb (retinoblastoma gene product) develop short-latency high-grade osteosarcoma that reproduces many of the defining features of human osteosarcoma, including cytogenetic complexity and comparable gene expression signatures, histology, and metastatic behavior.11 Other genetic contributions to the pathogenesis of osteosarcoma are less known; multiple cytogenetic abnormalities have been described, suggesting the presence of additional gene pathways involved in the multistep process of tumor development and progression.12
The peak incidence of osteosarcoma coincides with the adolescent growth spurt. This finding has led to the hypothesis that the altered hormonal milieu typical of adolescence may play a role in development of osteosarcoma. It is therefore possible that the insulin-like growth factor–1 (IGF1)—insulin-like growth factor–1 receptor (IGF1R) axis may be involved in the unregulated proliferation of osteoblasts that occurs in osteosarcoma. IGF1 functions as a mitogen in human and mouse osteosarcoma cells, and osteosarcoma cell lines depend on IGF1 for in vitro growth.13 Although the levels of IGF1 and its insulin-like growth factor binding protein–3 (IGFBP3) are not elevated in patients with osteosarcoma, other components of the IGF1 signaling pathway may be involved in the development and progression of osteosarcoma.14
Pathology
Osteosarcoma is characterized by the presence of spindle cell stroma that produces osteoid. Conventional osteosarcoma can be subdivided histologically into three major groups depending on the predominant cell type. Approximately 50% of tumors are categorized as osteoblastic, because the predominant extracellular element is osteoid, whereas 25% are chondroblastic, with a prominent cartilaginous component. Approximately 25% have a herringbone pattern similar to that observed in fibrosarcoma, and are therefore called fibroblastic. No significant differences in overall outcome are apparent among these three histologic subtypes.1 In one report, however, patients with fibroblastic histology showed better histologic response and better overall outcome than were observed for patients with osteoblastic or chondroblastic histology.15
Clinical Manifestations
Pain is the most common symptom in children and adolescents with osteosarcoma.1 Onset of pain often is insidious, and the pain usually involves the area affected by tumor. Severe pain of sudden onset commonly is associated with pathological fracture. Swelling around the affected bone is the second most common clinical finding. The tumor may be easily palpable when located in areas such as the anterior surface of the femur but may manifest only as leg edema when occurring in difficult-to-appreciate areas such as the popliteal fossa. A painful limp that increases with weight bearing is the third most common symptom. Systemic signs and symptoms such as fever and weight loss are uncommon.
Osteosarcoma most commonly involves the long bones, most tumors occurring around the knee. The most frequent sites of involvement are the distal part of the femur, the proximal portion of the tibia, and the proximal part of the humerus. The axial skeleton, including the pelvis, is rarely affected in children (fewer than 10% of cases) but more frequently is involved in patients older than 60 years.1,16 Overt macroscopic metastatic disease occurs in 20% of cases and carries a grave prognosis.17
Laboratory and Radiologic Evaluation
Laboratory evaluation often is unrevealing. Elevations of serum lactate dehydrogenase (LDH) and alkaline phosphatase levels are the most common laboratory abnormalities. The latter appears to correlate with osteoblastic activity and thus has proved useful in monitoring response to therapy.18
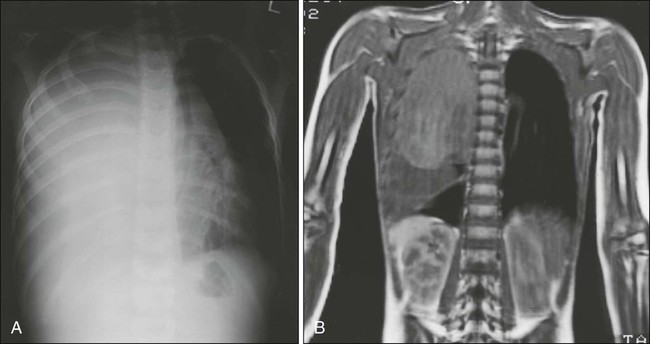
Radiologic evaluation of a patient with osteosarcoma must include assessment of the primary site as well as a search for distant metastatic lesions.19 Plain radiography is the most effective method of detection of bone tumors. The main limitation of plain radiography is accurate delineation of local tumor extent. Characteristic radiologic findings in osteosarcoma commonly include a metaphyseal permeative lesion with periosteal new bone formation and destruction of preexisting cortical bone. A soft-tissue mass is present in more than 90% of cases. Other radiologic signs commonly associated with osteosarcoma include cumulus cloud-like density and the presence of Codman’s triangle (Fig. 95-1A). A baseline chest radiograph should be obtained to search for distant metastatic lesions. Angiography usually is reserved for patients who receive intraarterial chemotherapy or for those who need optimal vessel visualization before limb salvage.
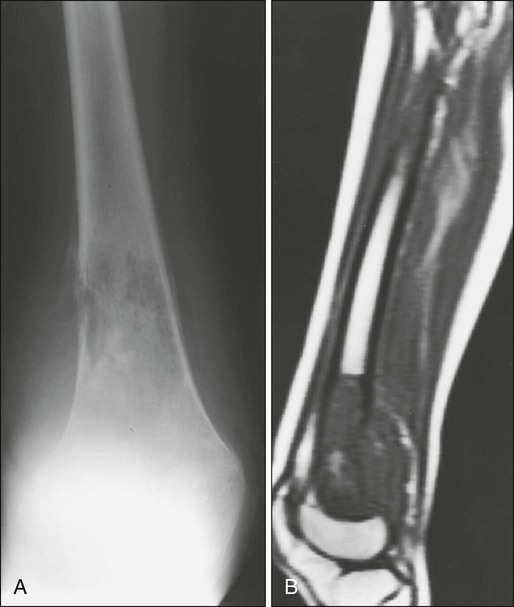
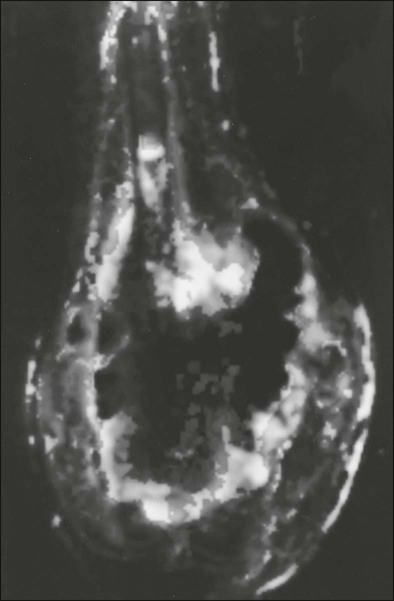
Computed tomography (CT) of the primary tumor is accurate in assessment of degree of tumor calcification and ossification, which are important in assessment of response to therapy. Chest CT always should be performed at the time of diagnosis for documentation of metastatic disease. Findings by magnetic resonance imaging (MRI) offer the best estimate of intramedullary tumor extension, joint and vascular involvement, detection of “skip” metastatic lesions, and delineation of the soft-tissue component (see Fig. 95-1B). The blood supply and vascularity of the tumor can be better appreciated with administration of gadopentetate dimeglumine, or gadolinium-diethylenetriamine pentaacetic acid (Gd-DTPA), a paramagnetic contrast material. MRI is helpful in assessing response to chemotherapy, as evidenced by changes in signal intensity on T2-weighted images and alterations in the enhancement of tumor tissue after administration of Gd-DTPA (Fig. 95-2). Dynamic contrast-enhanced MRI (DEMRI) is a valuable method for assessing microcirculation in osteosarcoma.20 DEMRI can be used to evaluate changes in regional contrast access during chemotherapy. The findings appear to correlate accurately with histologic response and outcome, allowing early identification of patients at risk for recurrence. Radionuclide bone scans with technetium-99m (99mTc)-labeled bone-seeking phosphate compounds are particularly useful in detection of metastatic bone lesions.19 Fluorodeoxyglucose positron emission tomography (FDG-PET) is also a useful tool in diagnosis and response assessment.21
Osteosarcoma Subtypes
Telangiectatic Osteosarcoma
Telangiectatic osteosarcoma is characterized microscopically by blood-filled spaces divided by septa containing neoplastic sarcomatous cells. Both radiologically and histologically, it is difficult to differentiate from aneurysmal bone cyst. Telangiectatic osteosarcoma accounts for less than 4% of cases of osteosarcoma. Age and anatomic distributions are similar to those in conventional osteosarcoma. On imaging studies, telangiectatic osteosarcoma manifests as a purely lytic lesion with a permeative destructive growth pattern; it usually disrupts the cortex, but with minimal or no periosteal new bone formation, which often is multilayered, in an onionskin pattern. Overall, the radiographic appearance is not that of a typical osteosarcoma, which usually has a mixture of blastic and lytic areas. Indeed, a purely lytic radiographic appearance is a diagnostic requirement for its diagnosis. This pattern can simulate aneurysmal bone cyst.22,23 Histologically, telangiectatic osteosarcoma is very hemorrhagic, similar to the gross appearance of aneurysmal bone cysts. Microscopically, this tumor consists of cystlike spaces divided by septa, which are composed of highly atypical sarcomatous tissue. Unlike aneurysmal bone cysts, these cystic spaces have no endothelial lining, and the tumor cells are in direct contact with areas of hemorrhage.22 In the past, it was thought that telangiectatic osteosarcoma carried a worse prognosis than for conventional osteosarcoma. With appropriate multimodality therapy, however, the outcome is similar to or better than that for conventional osteosarcoma.15
Low-Grade Intramedullary Osteosarcoma
Low-grade intramedullary osteosarcoma is a very rare variant of osteosarcoma, accounting for less than 1% of cases. Most cases are diagnosed during the third decade of life. Its anatomic distribution is similar to that of conventional osteosarcoma, with predilection for the distal femur and proximal tibia. In contrast with conventional osteosarcoma, symptoms typically develop over many months or even years before the patient comes to medical attention. Imaging studies usually show a variable pattern of lytic foci and dense areas, poorly demarcated. Periosteal new bone formation is minimal. Histologically, it shows a predominantly differentiated fibroblastic and osseous component, similar to what is seen in parosteal osteosarcoma (see “Surface Osteosarcomas” below). The differential diagnosis must distinguish this lesion from fibrous dysplasia. Treatment includes a complete resection of the lesion. Incomplete resection invariably results in local recurrence, and dedifferentiation increases with each recurrence.24
Surface Osteosarcomas
Parosteal osteosarcoma is a low-grade tumor that grows predominantly on the surface of long bones, in an exophytic pattern. Because it is derived from the outer layer of the periosteum, it grows without causing elevation of the periosteum or evidence of periosteal new bone formation. Parosteal osteosarcomas account for 3% of all osteosarcomas. They tend to occur in skeletally mature patients, with diagnosis during the third and fourth decades of life. More than 80% of these tumors are located in the distal portion of the femoral shaft, in its posterior aspect, within the superior popliteal area. Because of their slow growth, parosteal osteosarcomas usually manifest as a painless mass. Imaging shows a tumor growing on the surface of the bone, with a broad base, in a mushroom-like fashion. The mass typically is densely mineralized and has lobulated outlines. Microscopically, these tumors are characterized by the presence of a spindle cell fibroblastic component, with variable osteoid production, low mitotic rate, and no atypical features.25,26 The treatment is surgical, and a complete resection is mandatory. Incomplete resections invariably lead to local recurrences, and the risk of high-grade transformation increases with local recurrence.25
Prognostic Factors
The most important adverse prognostic factor in patients with osteosarcoma is the presence of metastatic disease.17 In addition, primary tumor location is associated with outcome. Children with primary tumors of the tibia and distal femur appear to have a more favorable prognosis than those with axial primary tumors. This finding highlights the importance of complete surgical resection in the management of this malignant disease.17 For patients with localized disease, factors associated with poor prognosis include measures of tumor burden, such as tumor size, and levels of alkaline phosphatase and LDH,17,18,27 as well as more biological measures, such as poor histologic response to preoperative chemotherapy,28 hyperdiploidy,29 and increased expression of P-glycoprotein30 or Ki-67.31 The percentage of tumor necrosis after preoperative chemotherapy is the most consistent and important factor associated with outcome in children and adolescents with localized osteosarcoma. A favorable response (more than 90% tumor necrosis) correlates with excellent overall survival. Patients who have less than 90% tumor necrosis are considered poor responders, for whom the prognosis usually is poor.17,28,32 Because of this strong correlation between degree of histologic response to preoperative chemotherapy and outcome, noninvasive methods such as DEMRI, used for monitoring tumor response, can be used to evaluate changes in regional contrast access during chemotherapy.20 Finally, a proportion of patients with extremity osteosarcoma are seen with a pathological fracture, or a pathological fracture develops after institution of therapy. This has been considered a poor prognostic factor and an indication for immediate amputation. It is possible, however, that with the use of preoperative chemotherapy and judicious use of limb-sparing techniques, a selected group of patients with pathological fracture may still do well without amputation.
Treatment
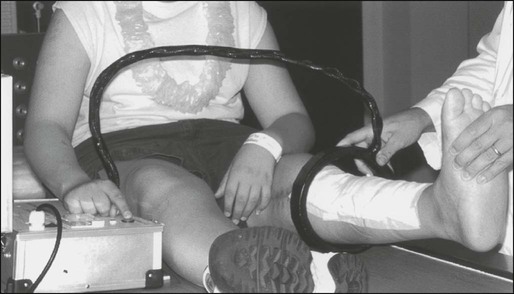
Optimal management of osteosarcoma consists of multiple-agent chemotherapy and local control measures, including amputation or limb-sparing surgical procedures. Box 95-1 outlines the standard approach to management of osteosarcoma used at most institutions. Before the development of limb-sparing procedures, amputation was the standard surgical method used for curative treatment of osteosarcoma. Amputation now generally is reserved for primary tumors deemed unresectable. Limb function after below-the-knee amputation usually is excellent. Over the past several years, the role of limb-sparing procedures has increased dramatically. As a result of refinements in neoadjuvant chemotherapy, bioengineering, and imaging techniques, it is estimated that as many as 80% of patients with osteosarcoma will eventually be candidates for limb-sparing procedures.33 The criteria for limb-sparing procedures include (a) absence of major neurovascular involvement by tumor, (b) feasibility of wide surgical excision to include a normal muscle cuff in all directions and en bloc removal of all biopsy sites, (c) resection of the adjacent joint and capsule, (d) adequate motor reconstruction with regional muscle transfer, and (e) adequate soft-tissue coverage.34 In the past, immature skeletal age and primary tumor of the humerus were relative contraindications; however, new expandable prosthetic devices may help overcome this problem.33,35 More recent improvements have been concentrated on achieving noninvasive extension of prostheses. One such method is the Phenix technology (Repiphysis). The basic principle involves storage of energy in a spring maintained in compressed form by a locking system. Prosthetic lengthening is performed through exposure to an external electromagnetic field that pilots the locking system and allows controlled release of the spring energy (Fig. 95-3). Prosthetic expansion of several millimeters can be achieved with each procedure, and the total duration of the procedure is less than 30 seconds (Fig. 95-4).36 Thus limb-sparing procedures are considered feasible in the care of most children and adolescents with osteosarcoma. When these procedures are appropriately performed, the risk of local recurrence is low (less than 5%).37 Long-term functional outcome, however, must be carefully compared with that obtained with amputation alone. Complications of limb-sparing surgery include infection, nonunion, fracture, and unstable joints.

Before the introduction of adjuvant chemotherapy, fatal metastatic disease developed in more than 80% of patients with osteosarcoma.38 Trials of single-agent chemotherapy began in the 1960s and early 1970s and established, in a nonrandomized manner, a role for the use of chemotherapy in the management of osteosarcoma. Responses with single-agent high-dose methotrexate or doxorubicin occurred in 20% to 40% of patients with metastatic disease.38,39 Since then, different combinations of platinum compounds, doxorubicin, and high-dose methotrexate have formed the basis of standard chemotherapy regimens that lead to cure in 50% to 75% of patients with nonmetastatic disease (Table 95-1).
Table 95-1
Summary of Most Relevant Chemotherapy Protocols for the Management of Osteosarcoma
| Treatment | |||||
| Protocol/Author | N | Preoperative | Postoperative* | Outcome | Comment(s) |
| T-10/Meyers et al.28 | 31 | MTX-BCD | GR: + DOX PR: + CDDP/DOX | 5-Year EFS: 73% | PR: No advantage of intensified postoperative treatment |
| T-12/Meyers et al.27 | 36 | MTX-BCD-DOX-CDDP | Same | 5-Year EFS: 78% | No advantage of intensified neoadjuvant treatment |
| MIOS/Link et al.44 | 77 | None | MTX-BCD-DOX-CDDP | 2-Year DFS: 66% | Randomized study demonstrating need for chemotherapy |
| 36 | Surgery alone | 2-Year DFS: 17% | |||
| CCG-782/Provisor et al.43 | 268 | MTX-BCD | GR: + DOX PR: + DOX-CDDP | 8-Year EFS: 53% GR: 81% PR: 46% | PR: No advantage of intensified postoperative treatment |
| COSS-82 arm A/Winkler et al.45 | 59 | MTX-BCD | GR: Same PR: + CDDP/DOX | 4-Year EFS: 49% | |
| COSS-82 arm B/Winkler et al.45 | 66 | MTX-DOX-CDDP | GR: Same PR: + IFO | 4-Year EFS: 68% | PR: IFO not advantageous |
| COSS-86 low risk/Fuchs et al.47 | 41 | MTX-DOX-CDDP | Same | 10-Year EFS: 66% | No difference for IA vs. IV administration of CDDP |
| COSS-86 high risk/Fuchs et al.47 | 128 | MTX-DOX-CDDP-IFO | Same | 10-Year EFS: 67% | Use of IFO for high-risk patients |
| EOI-1/Bramwell et al.46 | 142 | CDDP-DOX × 3 | CDDP-DOX × 3 | 5-Year DFS: 57% | Importance of a two-drug short regimen with CDDP-DOX |
| 140 | CDDP-DOX-MTX × 2 | CDDP-DOX-MTX × 2 | 5-Year DFS: 41% | ||
| EOI-2/Souhami et al.41 | 192 | MTX-DOX | + BCD-CDDP | 5-Year PFS: 44% | Two-drug short regimen may be better than longer; more complex protocols |
| 199 | CDDP-DOX | Same | 5-Year PFS: 44% | ||
| IOR-1/Ferrari et al.42 | 127 | MTX-CDDP | GR: + DOX-BCD PR: MTX-DOX-BCD | 12-Year DFS: 46% | HD MTX better than MD MTX |
| IOR-2/Bacci et al.70 | 164 | MTX-CDDP-DOX | GR: Same | 5-Year DFS: 63% | Importance of dose intensity |
| PR: + IFO/ETO | GR: 67% | PR: IFO/ETO good salvage | |||
| PR: 56% | |||||
| IOR-3/Bacci et al.50 | 139 | MTX-CDDP-DOX | GR: Same PR: + IFO | 3-Year DFS: 60% | PR: No benefit of IFO |
| IOR-4/Bacci et al.70 | 133 | MTX-CDDP-DOX-IFO | Same | 5-Year EFS: 56% | No benefit of intensified therapy with IFO |
| SJ-OS91/Meyer et al.51 | 47 | CBP-IFO | Same + DOX-MTX | 3-Year EFS: 72% | CBP is a good alternative to CDDP for nonmetastatic disease |
| INT 0133/Meyers et al.53 | 577 | Randomized to: | 5-Year. EFS: | Possible synergistic effect between IFO and MTP-PE | |
| A: MTX-CDDP-DOX | 64% | ||||
| A+: MTX-CDDP-DOX + L-MTP-PE | 63% | ||||
| B: MTX-CDDP-DOX + IFO | 56% | ||||
| B+: MTX-CDDP-DOX + IFO + L-MTP-PE | 72% | ||||
| SSG VIII/Smeland et al.71 | 113 | MTX-CDDP-DOX | GR: Same | 5-Year EFS: 68% | |
| PR: + IFO-ETO | 5-Year EFS: 53% | PR: Lack of benefit of modifying postoperative therapy | |||
| SJ-OS99/Daw et al.52 | 72 | CBP-IFO-DOX | Same | 5-Year EFS 66.7% | Treatment without HD-MTX and CDDP |
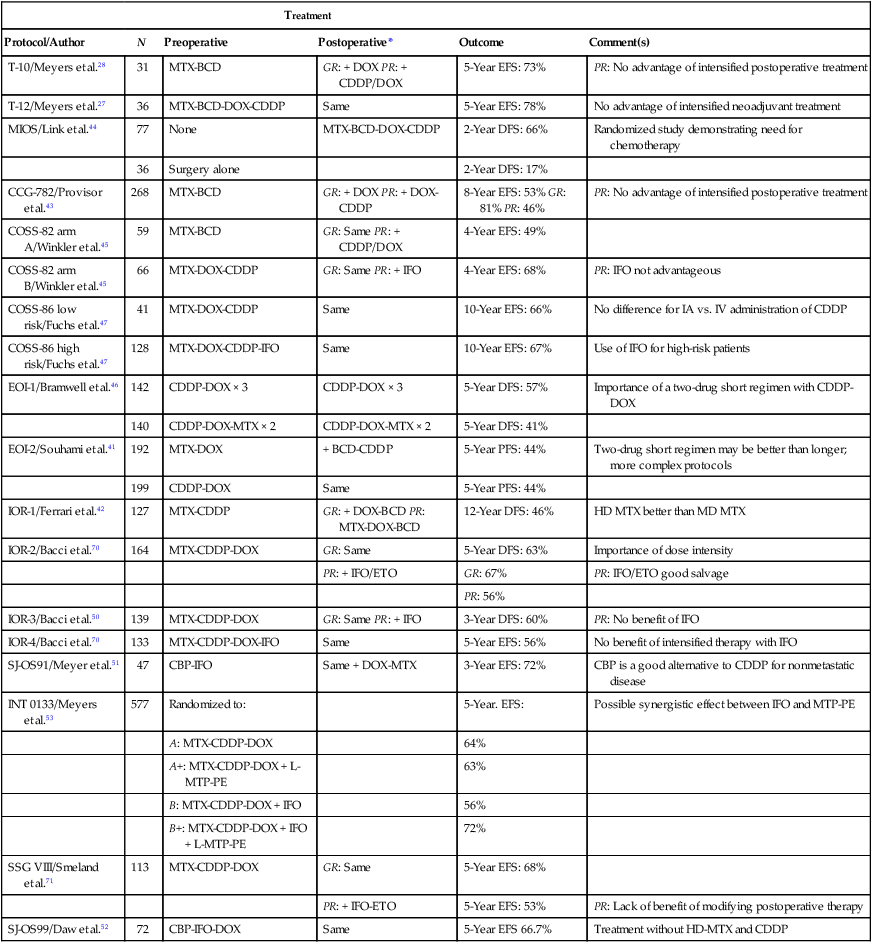
*Postoperative regimens are either the same as or in addition to preoperative regimens, or as specified.
For more than 2 decades, therapy for nonmetastatic osteosarcoma has followed the basic guidelines of the T-10 protocol,40 and most of the current treatment strategies have evolved from lessons learned with it. The T-10 protocol and its variants consist of a multiple-agent regimen of high-dose methotrexate, doxorubicin, cisplatin, and a combination of bleomycin, cyclophosphamide, and dactinomycin. When the T-10 protocol guidelines are used, the 5-year actuarial event-free survival (EFS) rate may approach 70%.27,28 These results are not always reproducible, however, and multiinstitutional U.S. and European studies conducted according to similar guidelines have shown lower EFS rates, usually 50% to 60%.41–45 The lack of reproducibility of the results of the original study may be attributed in part to the complexity of the treatment. In a study performed by the European Osteosarcoma Intergroup, patients were randomly assigned to receive the T-10 protocol or a much simpler treatment of 6 courses of a combination of cisplatin and doxorubicin.41 Only half of the patients in the T-10 protocol-like group completed all scheduled therapy, whereas 94% of the patients in the short-treatment group received the 6 courses of chemotherapy. The results for both treatment groups were identical, suggesting that a simple, two-drug regimen of cisplatin and doxorubicin can cure more than half of patients with nonmetastatic osteosarcoma.46
A major contribution of the T-10 protocol and its predecessor T-7 is that the histologic response to neoadjuvant chemotherapy has been identified as the most important prognostic factor in the care of patients with nonmetastatic disease.28 Intensification of postoperative28,42,43 or preoperative27 chemotherapy with cisplatin, doxorubicin, and ifosfamide, however, has not improved outcome. To increase the proportion of good histologic responders, some researchers have investigated intraarterial administration of cisplatin. In the context of an aggressive, multiple-agent treatment, however, intraarterial infusion of cisplatin does not offer any significant advantages.47
In recent years, ifosfamide has been incorporated into therapeutic regimens for osteosarcoma. After early reports showed that ifosfamide as a single agent achieved response rates of 10% to 60% in patients with advanced or treatment-refractory osteosarcoma,48,49 some investigators began to use the agent in salvage therapy regimens for poor responders.45,50 More recently, ifosfamide has been incorporated into frontline therapy in many regimens.50–52 However, whether incorporating the agent into standard treatment will improve the cure rate is unknown. The Children’s Oncology Group (COG) investigated the role of ifosfamide in a randomized study.53,54 Because giving an alkylating agent with etoposide has been shown to produce a synergistic antitumor effect,55 several investigators have administered ifosfamide and etoposide together on a fractionated dosing schedule over 3 to 5 days. In patients with refractory osteosarcoma, some of whom had previously received ifosfamide, the combination of ifosfamide and etoposide achieved response rates of 15% to 48%.56,57 Thus this combination is increasingly used to manage osteosarcoma. Investigators in the Pediatric Oncology Group (POG) have reported response rates of 59% in patients with untreated metastatic osteosarcoma receiving this combination.58 The combination of ifosfamide and etoposide appears to be more effective than ifosfamide alone in patients with untreated metastatic osteosarcoma. These results support performance of additional studies of this combination in treatment for patients with nonmetastatic disease. The use of the combination of ifosfamide and etoposide is increasingly being used for the salvage of patients with poor histologic responses; however, evidence is lacking for improved outcome in this group of patients. The randomized European-American Osteosarcoma (EUROS) trial will attempt to answer this question.
In the cooperative POG-Children’s Cancer Group (CCG) Intergroup INT0133 trial, patients with localized osteosarcoma received treatment with a standard methotrexate-cisplatin-doxorubicin regimen, and underwent a double randomization. The first randomization was to receive ifosfamide; the second randomization was to receive MTP (muramyl tripeptide), a derivative of the Bacillus Calmette-Guérin (BCG) cell wall that is known to stimulate the immune system, with the objective of decreasing pulmonary failures. Therefore four regimens were compared. The addition of ifosfamide or MTP alone to the standard regimen did not seem to provide any benefit. However, the addition of both ifosfamide and MTP-PE (muramyl tripeptide phosphatidylethanolamine) resulted in a significantly better EFS when compared with the other three regimens.53 When the long-term outcome of same cohort of patients was analyzed, those patients who received MTP had improved overall survival (although not improved EFS) compared to those that did not.54 The significance of these findings is unclear.
Methotrexate is the classical antifolate, and blocks the action of dihydrofolate reductase. Methotrexate was among the first drugs reported to have an antitumor effect in osteosarcoma and it has been a major component of the osteosarcoma treatment ever since.38 Furthermore, there is evidence that suggests that the pharmacokinetics of this agent may influence outcome, although this influence is minor in the context of more intensive multiagent protocols. However, the role of methotrexate appears to be limited to the administration of high doses (usually 12 g/m2), because the administration of lower doses appears to have a lesser therapeutic effect. The administration of high-dose methotrexate requires close monitoring of serum levels, which cannot be performed in many institutions, particularly those of developing countries, and extensive supportive measures, including hyperhydration, urine alkalinization, and leucovorin rescue, which must be adjusted to the methotrexate serum levels. The development of acute renal failure, mediated by the perception of methotrexate and its metabolites in the renal tubules, is a potentially life-threatening complication. Even among patients receiving all available monitoring and supportive care, the rate of severe nephrotoxicity is 2%, and the mortality rate is 4.4%. Dialysis methods have limited effectiveness in removing methotrexate, compared with the rapid reductions in plasma concentrations that can be achieved with carboxypeptidase G2. Studies have shown the possibility of obtaining comparable outcomes without the use of methotrexate, provided intensification of platinum, ifosfamide, and doxorubicin is performed.52
Cisplatin is one of the most active agents against osteosarcoma. The toxicity of this agent is substantial and includes hearing loss and renal impairment that can be permanent for some patients. Substitution of carboplatin for cisplatin was investigated at St. Jude Children’s Research Hospital. In the context of a multiple-agent chemotherapeutic approach with high-dose methotrexate, ifosfamide, and doxorubicin, use of carboplatin resulted in a 3-year EFS rate of 72%, an outcome comparable with that obtained with cisplatin-based therapy but with less long-term toxicity.51 Incorporation of carboplatin in future osteosarcoma trials needs further evaluation, however, because in some reports, carboplatin as a single agent has been found to have poor antitumor effect in the treatment of metastatic disease.59
Approximately 20% of patients with osteosarcoma have clinically detectable metastatic disease at diagnosis, and their outcome usually is very poor.17 Optimal treatment for these patients entails a very aggressive multimodality approach that combines intensive preoperative and postoperative chemotherapy with resection of both the primary tumor and metastatic lesions. When these guidelines are followed, contemporary protocols that incorporate ifosfamide or the combination of ifosfamide and etoposide, along with high-dose methotrexate, doxorubicin, and cisplatin, result in 2- to 5-year progression-free survival rates of 25% to 45%.58,60 For patients with recurrent osteosarcoma, a very aggressive surgical approach is recommended; the 5-year postrelapse survival rate for patients in whom a complete resection of all macroscopic disease can be achieved is close to 40%.61
In a small number of patients (3% to 5%), metachronous osteosarcoma can develop after primary treatment. This condition has been associated with the presence of germline TP53 (Li-Fraumeni) or RB1 gene mutations. These second primaries usually develop within 3 years from initial diagnosis, and with systemic chemotherapy and surgery, 30% to 40% of these patients can be cured.62
Lung metastasis develops in most patients in whom therapy fails. The ability to control pulmonary micrometastatic disease after completion of therapy would certainly result in a significant improvement in outcome. In an animal model, administration of liposome-encapsulated muramyl tripeptide phosphatidylethanolamine (L-MTP-PE) resulted in activation of pulmonary macrophages and eradication of pulmonary micrometastatic lesions.63 However, as indicated above, the clinical impact of the addition of this agent in the frontline of osteosarcoma therapy is not well defined. Other strategies under evaluation include use of new agents such as sequential administration of gemcitabine and docetaxel, administration of aerosolized granulocyte-macrophage colony-stimulating factor or 9-nitrocamptothecin, and use of various gene-modified interleukins.64,65
Some researchers have reported overexpression of HER2/erbB-2 (measured by immunohistochemistry studies) in approximately 40% of osteosarcoma tumor samples and correlation of this overexpression with more aggressive behavior and adverse outcome.66 If this correlation holds true, the use of anti-HER2 monoclonal antibodies might be an attractive therapeutic strategy for this group of high-risk patients. Nevertheless, studies with either immunofluorescence67 or fluorescence in situ hybridization68 have not shown amplification of the HER2/neu gene in patients with osteosarcoma.
Finally, administration of high doses of the bone-seeking radio-pharmaceutical samarium-153 ethylenediamine tetramethylene phosphonate (153Sm-EDTMP) may provide good pain palliation with minimal nonhematologic toxicity for patients with local recurrences or bone metastasis of osteosarcoma, and its role may be expanding.69
Ewing Sarcoma Family Tumors
Epidemiology
The term Ewing sarcoma family tumors (ESFTs) defines a group of small, round cell neoplasms of neuroectodermal origin that manifest as a continuum of neurogenic differentiation. On this continuum, Ewing sarcoma of bone represents the least differentiated (most primitive) form of neuroectodermal tumors, and peripheral neuroepithelioma represents the most differentiated form. Ewing sarcoma is the second most common malignant bone tumor in children and adolescents. The estimated incidence among white children younger than 15 years is 2.8 cases per 1 million population.1 The tumor is rare in the nonwhite population, and boys are predominantly affected in most series; most cases are diagnosed during the second decade of life. The location of the tumor varies. Although most Ewing sarcomas arise in bone, a significant proportion arise in soft tissue. The most common locations for this tumor are the chest wall, pelvis, and extremities, but any bone can be involved.2–4
Tumor Biology
The histogenesis of Ewing sarcoma has been a source of controversy since the first description of the tumor in 1921. Various hypotheses have been proposed in an attempt to identify the possible cell of origin in Ewing sarcoma. Among these, cells of endothelial, pericytic, myeloid, mesenchymal, and neuroectodermal origin have been suggested.5 The existence of either a mesenchymal stem cell or an early primitive neuroectodermal cell that has retained its ability for multilineage differentiation is the currently accepted hypothesis. It is now well accepted that ESFTs constitute a single group of neurally derived neoplasms that share unique immunocytochemical, cytogenetic, and molecular markers.5,6
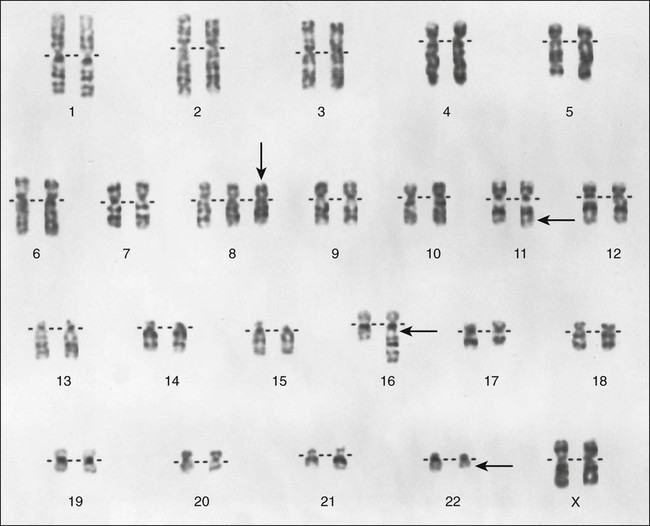
Nearly all ESFTs have a reciprocal translocation that involves the EWS gene in chromosome 22q12.7 The t(11;22) is the most commonly observed translocation (85% to 95% of cases) and juxtaposes the DNA-binding domain of the human homologue of the murine Fli1 gene in chromosome 11 with the 5′ end of EWS in chromosome 22 (Fig. 95-5). The most common fusions occur between exon 7 of EWS and exon 6 of FLI1 (type 1; 55% to 60% of cases) and between exon 7 of EWS and exon 5 of FLI1 (type 2; 25% of cases). The t(11;22) appears to play a pivotal role in development of ESFTs because the EWS-FLI1 fusion transcript can transform NIH 3T3 cells.8 As many as 10% of ESFTs contain an alternative translocation between chromosomes 21 and 22. This translocation fuses the ERG gene on chromosome 21 and the EWS gene on chromosome 22, producing an ERG-EWS fusion transcript.7 Finally, rare cases of ESFT contain a t(7;22)(p22;q12) that fuses the ETV1 and EWS genes.9 The mechanism by which these fusion transcripts become tumorigenic is poorly understood, although it has been postulated that this chimeric transcript can transcriptionally deregulate members of the manic fringe family of genes, which are instrumental in somatic development.10 The IGF1/IGF1R pathway is actively involved in the cell transformation and inhibition of apoptosis induced by EWS-FLI1. 11–13
Pathology
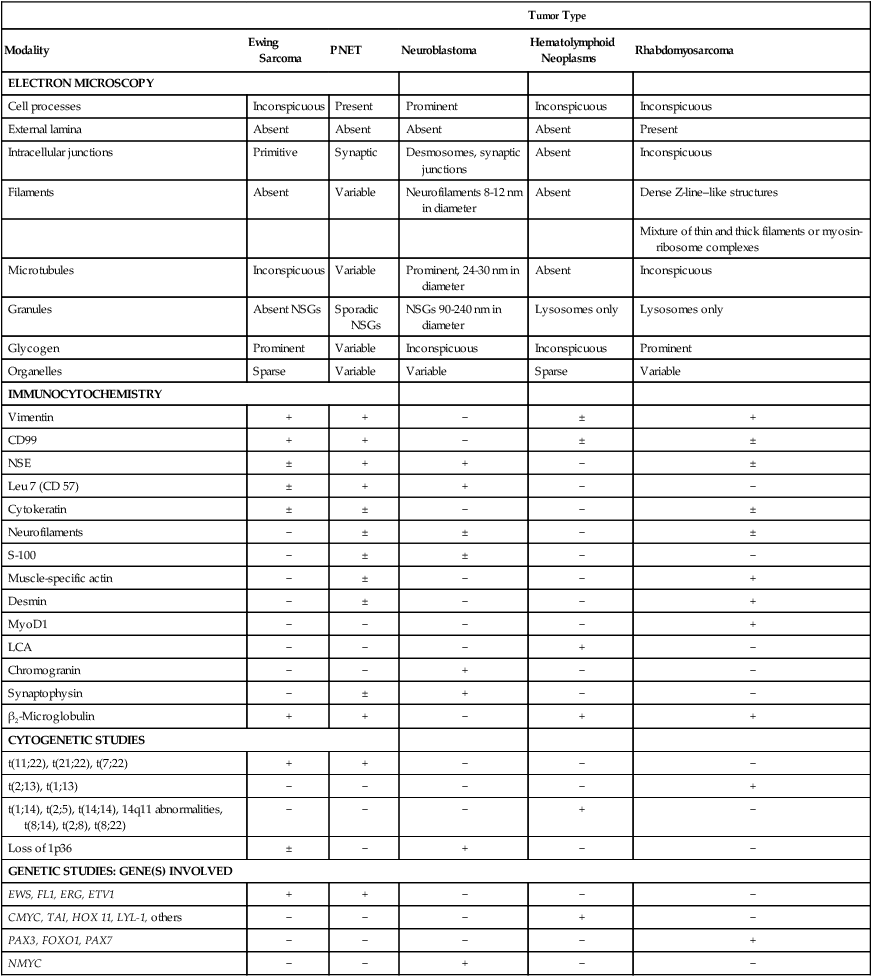
Microscopic examination shows that Ewing sarcoma is the prototypical small, round, blue cell tumor of childhood. Table 95-2 shows the ultrastructural and immunocytochemical characteristics of this and other small round cell tumors of childhood. Ewing sarcoma is characterized by the presence of a dimorphic pattern of densely packed cells with variable amounts of large clear cytoplasm. Individual cells have an ellipsoid nucleus without distinct cytoplasmic outlines. The cells are primitive, show a paucity of organelles, and often contain large amounts of intracellular glycogen. Various microscopic patterns, including the diffuse, lobular, organoid, and filigree patterns, have been described.14 Immunocytochemical analysis shows vimentin and CD99 reactivity. The latter monoclonal antibody specifically recognizes the cell surface antigen p30/p32MIC-2, which normally is expressed as a component of the T-cell receptor complex.14,15 When overt neural differentiation is present, the term peripheral neuroectodermal tumor or peripheral neuroepithelioma is used (Table 95-2). In such cases, the cells are round with more abundant cytoplasm, but do not show mature neural elements, such as nerve bundles and mats of neuropile.14 The term extraosseous Ewing sarcoma has been reserved for neoplasms that cannot be differentiated from Ewing sarcoma at light microscopic examination but arise exclusively in soft tissue. Patients with this histologic variety accounted for 5% of all subjects in the Intergroup Rhabdomyosarcoma Study,3 although extraosseous tumors are now managed in the same manner as for classic Ewing sarcoma of bone. Despite these histologic distinctions, Ewing sarcoma of bone, extraosseous Ewing sarcoma, primitive neuroectodermal tumors, and peripheral neuroepithelioma are unified by the presence of the same oncogenic events and should therefore be considered the same neoplasm and managed in a similar manner. The advent of molecular diagnostic techniques has allowed development of sensitive tests, such as reverse transcriptase-polymerase chain reaction (RT-PCR) analysis, which is accurate in detection of EWS-FLI1 and EWS-ERG fusion transcripts at levels well below those commonly found with routine cytogenetic studies.16 RT-PCR assay is a useful adjunct in the diagnosis of Ewing sarcoma, particularly in differentiation from other soft-tissue small, round cell tumors, such as rhabdomyosarcoma.
Table 95-2
Differential Diagnosis for Small, Blue, Round Cell Tumors with Electron Microscopy, Immunocytochemistry, and Molecular Cytogenetics
| Tumor Type | |||||
| Modality | Ewing Sarcoma | PNET | Neuroblastoma | Hematolymphoid Neoplasms | Rhabdomyosarcoma |
| ELECTRON MICROSCOPY | |||||
| Cell processes | Inconspicuous | Present | Prominent | Inconspicuous | Inconspicuous |
| External lamina | Absent | Absent | Absent | Absent | Present |
| Intracellular junctions | Primitive | Synaptic | Desmosomes, synaptic junctions | Absent | Inconspicuous |
| Filaments | Absent | Variable | Neurofilaments 8-12 nm in diameter | Absent | Dense Z-line–like structures |
| Mixture of thin and thick filaments or myosin-ribosome complexes | |||||
| Microtubules | Inconspicuous | Variable | Prominent, 24-30 nm in diameter | Absent | Inconspicuous |
| Granules | Absent NSGs | Sporadic NSGs | NSGs 90-240 nm in diameter | Lysosomes only | Lysosomes only |
| Glycogen | Prominent | Variable | Inconspicuous | Inconspicuous | Prominent |
| Organelles | Sparse | Variable | Variable | Sparse | Variable |
| IMMUNOCYTOCHEMISTRY | |||||
| Vimentin | + | + | − | ± | + |
| CD99 | + | + | − | ± | ± |
| NSE | ± | + | + | − | ± |
| Leu 7 (CD 57) | ± | + | + | − | − |
| Cytokeratin | ± | ± | − | − | ± |
| Neurofilaments | − | ± | ± | − | ± |
| S-100 | − | ± | ± | − | − |
| Muscle-specific actin | − | ± | − | − | + |
| Desmin | − | ± | − | − | + |
| MyoD1 | − | − | − | − | + |
| LCA | − | − | − | + | − |
| Chromogranin | − | − | + | − | − |
| Synaptophysin | − | ± | + | − | − |
| β2-Microglobulin | + | + | − | + | + |
| CYTOGENETIC STUDIES | |||||
| t(11;22), t(21;22), t(7;22) | + | + | − | − | − |
| t(2;13), t(1;13) | − | − | − | − | + |
| t(1;14), t(2;5), t(14;14), 14q11 abnormalities, t(8;14), t(2;8), t(8;22) | − | − | − | + | − |
| Loss of 1p36 | ± | − | + | − | − |
| GENETIC STUDIES: GENE(S) INVOLVED | |||||
| EWS, FL1, ERG, ETV1 | + | + | − | − | − |
| CMYC, TAI, HOX 11, LYL-1, others | − | − | − | + | − |
| PAX3, FOXO1, PAX7 | − | − | − | − | + |
| NMYC | − | − | + | − | − |
Clinical Manifestations
Ewing sarcoma commonly manifests during the second decade of life (median age: 13 years) with localized pain and a visible palpable mass.17 Fewer than 3% of cases occur in children younger than age 3 years.18 Boys are more commonly affected than girls. Pathological fractures may be present in as many as 15% of children and adolescents before diagnosis.19 Back pain, extremity weakness, or altered sensation should raise suspicion for the presence of primary or metastatic disease. Systemic manifestations such as fever are more frequent than in osteosarcoma. Almost half of patients have signs and symptoms referable to the primary tumor for more than 3 months before the diagnosis is made. Ewing sarcoma has a tendency to involve the shaft of long tubular bones, pelvis, and ribs, but almost every bone can be affected. More than 50% of the tumors arise from axial bones, the pelvis being the most commonly involved (25%); one-third of the tumors originate in the lower extremities, and less than 10% in the upper extremities. Chest wall Ewing sarcoma is known as Askin tumor (Fig. 95-6). Approximately 20% to 25% of patients with this tumor have metastatic disease when they are seen for evaluation. The most common sites of metastatic disease are in the lungs, followed by the bones and bone marrow. Metastatic disease appears to be associated with older age and with large tumor or pelvic primaries.
Laboratory and Radiologic Evaluation
Patients with suspected Ewing sarcoma should be thoroughly evaluated to define the extent of local disease and the presence of metastatic lesions. Initial laboratory studies include complete blood cell count and erythrocyte sedimentation rate; measurement of serum electrolytes; LDH assay; renal and liver function tests; determination of alkaline phosphatase, calcium, phosphorus, and magnesium levels; and coagulation profile. In Ewing sarcoma, elevation of erythrocyte sedimentation rate and of serum LDH is not uncommon. Bone marrow aspiration and biopsy should be performed, and evaluation with molecular techniques such as RT-PCR is recommended. Important imaging studies are chest radiography, plain radiography of primary and metastatic sites, bone scintigraphy, CT of the chest, and MRI of the primary site with T1– and T2-weighted sequences, as well as DEMRI.20,21
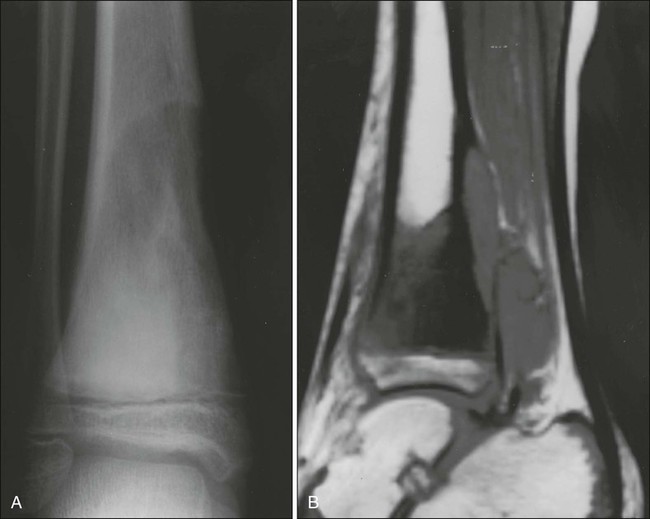
In Ewing sarcoma, plain radiographs typically show a diaphyseal destructive lesion with a laminated periosteal reaction and large soft-tissue mass (Fig. 95-7A). St. Jude Children’s Research Hospital favors MRI over CT for defining the intramedullary component of the primary tumor and the extent of soft-tissue mass (Fig. 95-7B). In contrast with the situation in osteosarcoma, DEMRI findings are not a reliable prognostic indicator.22 Newer techniques such as positron emission tomography (PET) appear to be useful in noninvasive evaluation of response to chemotherapy.23
Prognostic Factors
A variety of initial clinical features have been correlated with outcome of therapy for Ewing sarcoma. Several groups have analyzed factors that are prognostically important in Ewing sarcoma.2,4,24 The results confirmed the classic clinical features associated with poor prognosis, such as metastatic disease, older age, large tumor size, and trunk and pelvic primary sites. However, with refinement in the multidisciplinary approach to this disease that entails newer and more intensive chemotherapeutic regimens and superior local control measures, some of these classic prognostic factors are being redefined.
Although large tumor size classically has been associated with worse prognosis, this feature may be less important with more aggressive treatment. For example, in the first Cooperative Ewing Sarcoma Study (CESS-81), tumors larger than 100 cm3 were associated with worse outcome.25 With treatment improvements, the tumor size associated with worse prognosis has increased to 200 cm.26
Tumor location also seems to be losing its prognostic significance with newer treatments, although pelvic and axial tumor locations classically were associated with worse outcome in the early studies,27–29 differences in outcome were minimal in subsequent studies.30–32 The first POG–CCG Ewing sarcoma trial (POG-8850/CCG-7881), an investigation of the effect of addition of ifosfamide and etoposide to the standard VACD regimen (vincristine, actinomycin D, cyclophosphamide, and doxorubicin), showed that addition of the drug pair abrogated the negative prognostic implications of large tumor size (greater than 8 cm in diameter) and pelvic location. Of note, the benefit of addition of ifosfamide and etoposide was not seen in patients older than 18 years.33
A newer prognostic factor is degree of histologic response to chemotherapy.34 European studies consistently have shown the prognostic value of histologic response, the significance of which stands across protocols and appears to be independent of the drugs used. Patients with good histologic responses had a significantly better outcome than those with poor responses in the consecutive REN-1, -2, and -3 trials in Italy34 and in the CESS-81 and CESS-86 trials in Germany.35 Because ESFTs manifest a continuum of neuroectodermal differentiation, the histologic diversity may reflect different biological behaviors. Evidence is lacking, however, for correlation of degree of neuroectodermal differentiation with prognosis.36 The most important prognostic factor remains the presence of metastatic disease at diagnosis.24 Advances in management of ESFTs have resulted in only very modest improvement in outcome among patients with metastatic lesions. Even among patients with metastatic disease, heterogeneity is common. With an appropriately intensive treatment that includes bilateral lung irradiation, the European Intergroup Cooperative Ewing Sarcoma Studies (EICESSs) have shown that patients with isolated metastatic lesions in the lung may have a better prognosis, albeit still inferior to that for patients with localized disease. Patients with extrapulmonary metastasis have a worse prognosis.37,38
With the use of molecular techniques in the staging of ESFTs, it is evident that a significant proportion of patients with localized disease (20% to 30%) have micrometastatic disease in the bone marrow detected by polymerase chain reaction (PCR) assay.39–41 In patients with lung metastasis and those with bone metastasis, this proportion is 40% and 90%, respectively.40 Although clinical stage remains the most powerful prognostic indicator in patients with ESFT, detection of minimal disease in the bone marrow by RT-PCR has been associated with metastatic disease and appears to connote a more adverse clinical outcome in a subset of patients with apparently localized disease.42 The utility of detecting circulating neoplastic cells by RT-PCR in patients with ESFT is less clear.
Treatment
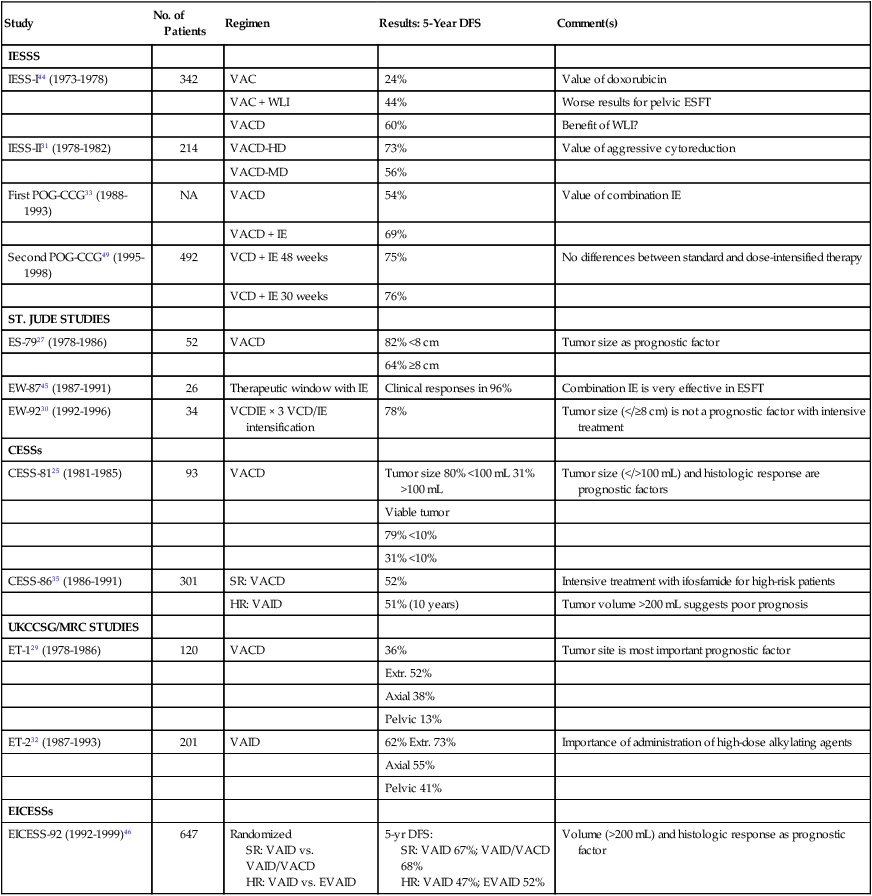
Before the introduction of systemic chemotherapy, less than 20% of children with Ewing sarcoma treated with either surgery or radiation therapy alone were expected to be long-term survivors.43 In the past 3 decades, major advances have been made in the management of Ewing sarcoma. These advances derive largely from cooperative trials (Table 95-3). The first studies conducted by the Intergroup Ewing Sarcoma Study (IESS) showed the importance of adjuvant chemotherapy that included a combination of alkylating agents and anthracyclines (IESS-I and IESS-II).31,44 Thereafter, conventional management of Ewing sarcoma included local control measures in combination with administration of VACD.27,29,30 More recently, incorporation of ifosfamide (VAID [vincristine, actinomycin D, ifosfamide, doxorubicin]) has resulted in modest benefit for patients with high-risk features.32,35 Combined administration of ifosfamide and etoposide (IE) has been shown to be very active in patients with Ewing sarcoma who have not received previous treatment.45 Two randomized studies were conducted to investigate the effect of adding etoposide to VACD or VAID regimens.33,46 In EICESS-92, patients with localized high-risk disease (tumor diameter greater than 200 cm3) receiving VAID did not seem to benefit from the addition of etoposide.46 The first POG–CCG Ewing sarcoma trial (POG-8850/CCG-7881) was an investigation of incorporation of the IE combination in frontline management of ESFTs. Patients were randomly assigned to receive VACD with or without IE; patients receiving IE plus VACD appeared to have a more favorable outcome.33
Table 95-3
Management of Nonmetastatic Ewing Sarcoma Family of Tumors
| Study | No. of Patients | Regimen | Results: 5-Year DFS | Comment(s) |
| IESSS | ||||
| IESS-I44 (1973-1978) | 342 | VAC | 24% | Value of doxorubicin |
| VAC + WLI | 44% | Worse results for pelvic ESFT | ||
| VACD | 60% | Benefit of WLI? | ||
| IESS-II31 (1978-1982) | 214 | VACD-HD | 73% | Value of aggressive cytoreduction |
| VACD-MD | 56% | |||
| First POG-CCG33 (1988-1993) | NA | VACD | 54% | Value of combination IE |
| VACD + IE | 69% | |||
| Second POG-CCG49 (1995-1998) | 492 | VCD + IE 48 weeks | 75% | No differences between standard and dose-intensified therapy |
| VCD + IE 30 weeks | 76% | |||
| ST. JUDE STUDIES | ||||
| ES-7927 (1978-1986) | 52 | VACD | 82% <8 cm | Tumor size as prognostic factor |
| 64% ≥8 cm | ||||
| EW-8745 (1987-1991) | 26 | Therapeutic window with IE | Clinical responses in 96% | Combination IE is very effective in ESFT |
| EW-9230 (1992-1996) | 34 | VCDIE × 3 VCD/IE intensification | 78% | Tumor size (</≥8 cm) is not a prognostic factor with intensive treatment |
| CESSs | ||||
| CESS-8125 (1981-1985) | 93 | VACD | Tumor size 80% <100 mL 31% >100 mL | Tumor size (</>100 mL) and histologic response are prognostic factors |
| Viable tumor | ||||
| 79% <10% | ||||
| 31% <10% | ||||
| CESS-8635 (1986-1991) | 301 | SR: VACD | 52% | Intensive treatment with ifosfamide for high-risk patients |
| HR: VAID | 51% (10 years) | Tumor volume >200 mL suggests poor prognosis | ||
| UKCCSG/MRC STUDIES | ||||
| ET-129 (1978-1986) | 120 | VACD | 36% | Tumor site is most important prognostic factor |
| Extr. 52% | ||||
| Axial 38% | ||||
| Pelvic 13% | ||||
| ET-232 (1987-1993) | 201 | VAID | 62% Extr. 73% | Importance of administration of high-dose alkylating agents |
| Axial 55% | ||||
| Pelvic 41% | ||||
| EICESSs | ||||
| EICESS-92 (1992-1999)46 | 647 | Randomized SR: VAID vs. VAID/VACD HR: VAID vs. EVAID |
5-yr DFS: SR: VAID 67%; VAID/VACD 68% HR: VAID 47%; EVAID 52% |
Volume (>200 mL) and histologic response as prognostic factor |
Incorporation of granulocyte colony-stimulating factor into treatment regimens for many types of cancer has allowed modest dose intensification of multiple-agent chemotherapy by either increasing the total dose per cycle30,47 or shortening the time between treatments.48 For ESFTs, this strategy is based on use of high cumulative doses of alkylating agents and topoisomerase-II inhibitors. When these general treatment guidelines are followed, the disease-free survival rate for patients with localized, low-risk disease approaches 70% to 75%, and the overall survival rate for this group of patients may be greater than 80%.24
The importance of dose intensification in the management of ESFT was evaluated in the second POG–CCG Ewing sarcoma trial (POG-9354/CCG-7942). In that trial, patients were randomly assigned to receive alternating courses of vincristine, doxorubicin, and cyclophosphamide with IE over either 48 or 30 weeks. The early results of that randomized trial demonstrated no difference in outcome between the standard and the dose-intensified treatment groups.49 An alternative to increasing dose intensity is decreasing the intervals between cycles while maintaining the same dose per cycle with the use of granulocyte colony-stimulating factor.48 In the United States, this is the approach taken by the COG in the recently completed AEWS-0031 study, in which patients were randomly assigned to receive alternating cycles of VCD (vincristine, cyclophosphamide, doxorubicin) and IE every 3 weeks or 2 weeks, which results in 33% dose intensification.
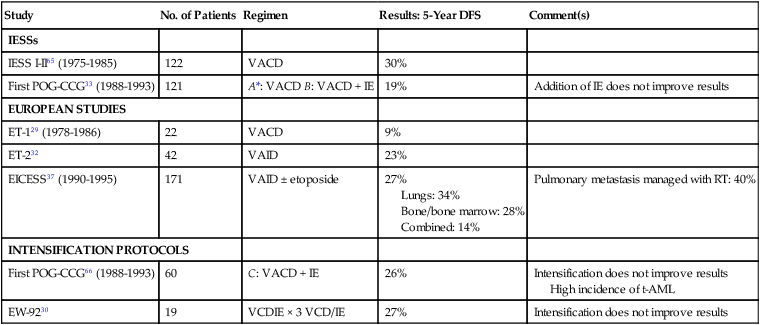
Despite improvement, the prognosis for patients with metastatic disease continues to be very poor, and only 20% to 25% survive24 (Table 95-4). Several institutions have used treatment intensification, by which very high doses of different agents are administered in a short time. In the case of ESFTs, this is a very attractive alternative, because ESFTs are highly sensitive to alkylating agents, which have a steep dose-response curve. Although most patients with ESFTs treated in this manner may exhibit good clinical and histologic responses, the final results are not better than those obtained with conventional therapy.
Table 95-4
Management of Metastatic Ewing Sarcoma Family of Tumors Without Hematopoietic Stem Cell Transplantation
| Study | No. of Patients | Regimen | Results: 5-Year DFS | Comment(s) |
| IESSs | ||||
| IESS I-II65 (1975-1985) | 122 | VACD | 30% | |
| First POG-CCG33 (1988-1993) | 121 | A*: VACD B: VACD + IE | 19% | Addition of IE does not improve results |
| EUROPEAN STUDIES | ||||
| ET-129 (1978-1986) | 22 | VACD | 9% | |
| ET-232 | 42 | VAID | 23% | |
| EICESS37 (1990-1995) | 171 | VAID ± etoposide | 27% Lungs: 34% Bone/bone marrow: 28% Combined: 14% |
Pulmonary metastasis managed with RT: 40% |
| INTENSIFICATION PROTOCOLS | ||||
| First POG-CCG66 (1988-1993) | 60 | C: VACD + IE | 26% | Intensification does not improve results High incidence of t-AML |
| EW-9230 | 19 | VCDIE × 3 VCD/IE | 27% | Intensification does not improve results |
Use of protocols that include intensification of alkylators and topoisomerase-II inhibitors has resulted in a significant increase in the incidence of treatment-related leukemia—specifically, acute myelogenous leukemia (AML)—and myelodysplastic syndrome (t-AML/MDS) (Box 95-2). This therapeutic strategy appears to be strongly leukemogenic, and patients are at increased risk for both alkylator-related and etoposide-related t-AML/MDS.50–52 This increased risk of t-AML/MDS appears to be related to both the increase in the total cumulative doses and dose intensification. The role of hematopoietic growth factors in this complication is unknown.
New drugs and new drug combinations continue to be investigated in the care of patients with Ewing sarcoma. Although phases I and II studies of topotecan and irinotecan as single agents have shown little or no activity in patients with refractory disease, recent studies suggest that their combination with alkylating agents may be more promising. Phase II studies of the combination of topotecan (0.75 mg/m2 per day for 5 days) and cyclophosphamide (250 mg/m2 per day for 5 days) resulted in responses in 36% of patients with recurrent disease53 and in 56% of patients with untreated metastatic disease.54 The COG is currently investigating the incorporation of this combination in the frontline treatment of ESFTs, given in alternating cycles with VCD and IE. Also recently, the combination of irinotecan on a protracted schedule (10 to 20 mg/m2 per day for 5 days in 2 consecutive weeks) with temozolomide (100 mg/m2 per day for 5 days) has shown excellent response rates.55
Advances in management of ESFTs have resulted in only modest improvement in outcome among patients with metastatic disease.24 With appropriately intensive treatment that includes bilateral lung irradiation, however, the prognosis for patients with isolated lung metastasis may be better than for patients with extrapulmonary metastasis.37,38 For patients with bone and bone marrow metastasis, therefore, more intense therapies may have a role. ESFTs are very sensitive to alkylators, a group of agents with a very steep dose-response curve, which provides the basis for the use of consolidation with myeloablative therapy and autologous hematopoietic stem cell transplantation (HSCT). The results of treatment with megatherapy and HSCT for patients with high-risk ESFTs must be analyzed with caution because of the lack of randomized studies and the heterogeneity of patients and treatments. Results of most retrospective European and American studies do not seem to support the use of this approach.56–59 In more recent results reported by the European Bone Marrow Transplant Registry, however, conditioning regimens that incorporate high doses of alkylating agents, generally busulfan and melphalan, confer an apparent survival advantage.60 Total-body irradiation does not seem to provide any additional benefit and only adds toxicity.59 The European Cooperative Group is conducting a randomized study to evaluate the role of HSCT in the care of patients with metastatic Ewing sarcoma.
Improvement in outcome among patients with ESFT also must be attributed to improvement in local control, which is largely related to advances in radiation therapy and better surgical approaches. Advances in systemic therapy by means of incorporation of new drugs and treatment intensification also appear to contribute to better local control.61 With current multimodality intensive protocols, rates of local recurrence have decreased significantly, and little difference in efficacy has been observed between surgery and radiation therapy for local control.24,61–64 Surgery continues to offer slightly better results, but this observation is biased by the fact that small lesions are more likely to be managed surgically. For unresectable tumors or in case of gross residual disease, recommended doses are 55 to 60 Gy. Doses of 40 to 45 Gy are used for microscopic disease. The risk of secondary sarcoma after radiation therapy is not negligible.52 This risk is dose-dependent, but use of lower doses of radiation therapy has been associated with higher local recurrence rates.61
Neuroblastoma
Epidemiology
Neuroblastoma is the most common extracranial solid tumor of childhood, accounting for 8% to 10% of all pediatric cancers.1 The incidence of neuroblastoma varies in different regions of the world, with a greater prevalence in developed countries. In the United States, the annual incidence is approximately 8.4 new cases per 1 million children per year with 650 newly diagnosed cases. The median age at diagnosis is approximately 18 months. Most (85%) of the cases are diagnosed by the age of 5 years; it is extremely rare in children older than 10 years. Familial cases occur and a small number are inherited in an autosomal dominant manner (5%).2 Recent studies have linked germline mutations in the anaplastic lymphoma kinase (ALK) gene or in the PHOX2B gene to familial neuroblastoma.3,4 Neuroblastoma also has been found in patients with neurofibromatosis, Hirschsprung’s disease, Beckwith-Wiedemann syndrome, fetal hydantoin syndrome and central hypoventilation syndrome.3,5–6 Microscopic neuroblast nodules are found in the adrenal glands of 2% to 3% of infants who die of nonmalignant causes before 3 months of age.7 It is uncertain whether these nodules represent spontaneous regression of congenital neuroblastoma or maturation of the tumor into asymptomatic benign tumors. Prenatal ultrasonography can identify in utero cases as early as 31 weeks’ gestation.
Mass screening programs for detection of neuroblastoma in early infancy with assays for urinary catecholamine metabolites have been conducted in Japan, Germany, and the United States.8–10 In general, patients with neuroblastoma detected in these programs have early stage disease and enjoy otherwise excellent outcome. Mass screening, however, has not resulted in a reduction in deaths from neuroblastoma. In addition, early detection has not changed outcome among patients with tumors that have biological features associated with a poor prognosis.
Tumor Biology
Neuroblastoma originates in neural crest cells of the sympathetic nervous system and, not unexpectedly, secretes a variety of neurogenically derived substances, including catecholamines, neuron-specific enolase (NSE), and ferritin.11–14 Urinary excretion of abnormally high levels of catecholamine metabolites occurs in 75% to 90% of patients and is related to the degree of tumor differentiation. The metabolites most often measured in evaluation of suspected neuroblastoma are urinary vanillylmandelic acid and homovanillic acid.15 Levels of these catecholamines are sensitive indicators of disease status. At diagnosis, other biological markers, such as NSE, serum ferritin, and ganglioside GD2, are present in high concentration in the serum of most patients with neuroblastoma. None of these biological markers is specific, but their presence at diagnosis may be associated with the extent of disease.
Cytogenetic studies of neuroblastoma have demonstrated chromosomal abnormalities in most cases. The most common cytogenetic abnormalities in neuroblastoma are double-minute chromatin bodies (DMs), homogeneously staining regions (HSRs), and nonrandom deletion or loss of heterozygosity of the short arm of chromosome 1p36.16,17 DMs and HSRs both represent cytogenetic manifestations of amplified sequences of the MYCN cellular oncogene.18 Amplified MYCN (more than 10 copies per cell) is a feature in approximately 25% to 30% of neuroblastomas. It is associated with the presence of advanced disease at diagnosis and poor outcome, even in patients who have early stage disease or who come to medical attention in infancy.19,20 The DNA content of the tumor (ploidy) has been shown to correlate with therapeutic outcome and survival among infants with this tumor. Infants with hyperdiploid tumors (DNA index greater than 1.0) have a more favorable outcome than those with diploid tumors (DNA index 1.0).21–23 Results suggest that the presence of either MYCN amplification or diploid cellular DNA tumor content identifies whether an infant has a poor prognosis with current therapeutic regimens, independent of stage. For children older than 24 months with disseminated disease, however, the DNA content of the tumor does not appear to have prognostic importance.23 Recent studies suggest that additional cytogenetic imprints, such as loss of heterozygosity at 1p36 and 11q23, confer an aggressive phenotype.24 Activating mutations in the tyrosine kinase domain of the anaplastic lymphoma kinase (i) oncogene have recently been noted to account for most cases of hereditary neuroblastoma, and may be somatically acquired in 5% to 10% of neuroblastoma.4
Pathology
Neuroblastoma is one of the small, round, blue cell tumors of childhood. It originates from neural crest cells from within the sympathetic nervous system. In 1999, the International Neuroblastoma Pathology Classification (INPC) was established to standardize the terminology and criteria for the prognostic evaluation of the morphologic features of neuroblastic tumors in an age-linked framework.25 INPC categorizes neuroblastic tumors into favorable and unfavorable histology based on the degree of differentiation, Schwannian stroma content, mitosis-karyorrhexis index, and age at diagnosis. The INPC distinguishes four histologic categories: neuroblastoma (Schwannian stroma-poor); ganglioneuroblastoma intermixed (Schwannian stroma-rich); ganglioneuroma (Schwannian stroma-dominant); and ganglioneuroblastoma nodular (composite Schwannian stroma-rich/stroma-dominant and stroma-poor) (Fig. 95-8).
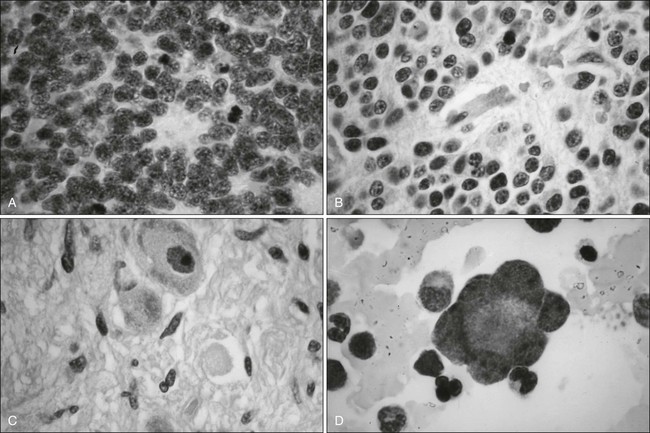
These subgroups appear to recapitulate stages in the normal differentiation of neural crest stem cells. For example, neuroblastoma is the most primitive entity and is characterized by diffuse growth of undifferentiated neuroblastic cell nests irregularly separated by thin fibrovascular septa (see Fig. 95-8A).25 By contrast, benign ganglioneuroma consists of mature ganglion cells embedded in bulky stroma composed of Schwann cell sheets enveloping neuritic processes and perineural and endoneural elements (see Fig. 95-8C). Between these two extremes is the transitional form known as ganglioneuroblastoma (see Fig. 95-8B). These transitional forms have been subclassified into intermixed and nodular diffuse ganglioneuroblastoma. Composite (nodular) tumor is a ganglioneuroma that contains one or more discrete nodules of pure neuroblastoma. Intermixed (diffuse) tumor contains a mixture of primitive and differentiating neuroblasts with bizarre, immature, and mature ganglion cells. Patients frequently have tumors with mixtures of these cell types, and evidence supports the clinical observation that neuroblastoma can sometimes mature into benign ganglioneuroma. Spontaneous regression and therapy-induced maturation can occur.26
Clinical Manifestations
The clinical manifestations of neuroblastoma are varied. Nonspecific constitutional signs and symptoms, such as fever, general malaise, and pain, are frequent initial features. The most common sites of primary tumors are the abdomen (adrenal gland or paraspinal ganglia) and the thorax (usually the posterior mediastinum). In infants, the distribution is slightly different in that a higher proportion of primary tumors occur in the thoracic cavity than is the case in older children. Common manifestations include a hard, painless mass in the neck, a localized intrathoracic mass found incidentally on a chest radiograph, and a palpable abdominal mass, according to the location of the tumor. A palpable abdominal mass can result from an enlarging primary adrenal or retroperitoneal tumor or from hepatomegaly secondary to tumor metastasis. Children may appear chronically ill and irritable and have periorbital ecchymosis, scalp nodules, and bone pain from widespread metastasis to the bone marrow or bone (Fig. 95-9; see also Fig. 95-8D). Lower-limb paresis secondary to epidural extension of a primary paraspinal tumor growing through the intervertebral foramen can cause signs of spinal cord compression. Intermittent abdominal pain, malaise, failure to gain weight, and recurrent, unexplained fever may be present for prolonged periods before neuroblastoma is diagnosed.
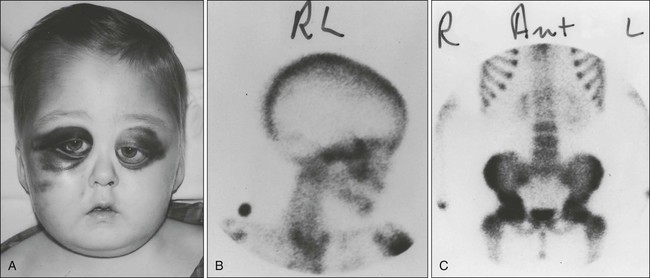
Seventy-five percent of patients with neuroblastoma have metastatic disease at the time of diagnosis. The most common sites of metastasis are lymph nodes (local or distant), bone marrow, liver, skin, orbit, and bone (facial bones, skull, appendicular skeleton). Nearly half of patients have widespread skeletal metastasis at diagnosis. The bones of the skull and orbit frequently are affected, so proptosis, ecchymosis, and masses beneath the scalp are frequent findings (see Fig. 95-9). Lung metastatic lesions are extremely uncommon when the patient is first examined, but can be seen after autologous HSCT.27 Horner syndrome (ipsilateral miosis, ptosis, and anhidrosis) may be present in patients with lesions originating in the cervical or upper thoracic sympathetic ganglia. Less frequent is the syndrome of opsomyoclonus and ataxia, manifested as acute cerebellar encephalopathy, truncal ataxia, and rapid and random conjugate eye movements (so-called dancing eyes, dancing feet). The pathophysiological mechanism of this syndrome is unknown, but metabolic and immunologic causes have been invoked. A syndrome of chronic watery diarrhea also may occur in patients with neuroblastoma. Increased serum levels of vasoactive intestinal peptide, which lead to increased intestinal motility and secretions, have been found in some cases.
Laboratory and Radiologic Evaluation
Laboratory studies include complete blood cell count, renal and liver function studies, coagulation screen, urinalysis, and urine catecholamine assay. Serum often is obtained for NSE, ferritin, and ganglioside GD2 determinations because the results may be helpful in prognosis.28–30
Staging of neuroblastoma requires radiologic studies and marrow aspiration and biopsy to determine the extent of local disease and distant spread. The minimal evaluation for metastatic disease includes radionuclide skeletal scintigraphy and bone marrow aspiration. CT generally has replaced intravenous pyelography, arteriography, and inferior vena cavography (Fig. 95-10). Ultrasonographic studies of the abdomen and pelvis may be useful in evaluation of mass lesions and the degree of compression of vital structures causing secondary complications. MRI may further improve the accuracy of determination of the anatomic extent of disease, especially in evaluation of tumors impinging on the spinal cord and assessment for the presence of hepatic metastasis. CT- or MRI-based myelography is necessary in evaluation of all patients with neurologic evidence of cord involvement. Scans performed with radionuclides such as 131I-m-iodoben-zylguanidine (MIBG) may be more accurate and sensitive for detection of nonosseous as well as osseous disease.31 Newer modalities such as FDG-PET combined with CT scanning may provide additional value in the management and diagnosis of MIBG-negative neuroblastoma, and in visualization of hepatic metastases and soft-tissue lesions not readily visualized with MIBG scan.32 When tumor material is obtained, it should be submitted for determination of DNA content (tumor cell ploidy), detection of MYCN genomic amplification, cytogenetic analysis, and preserved for additional genomics evaluation.
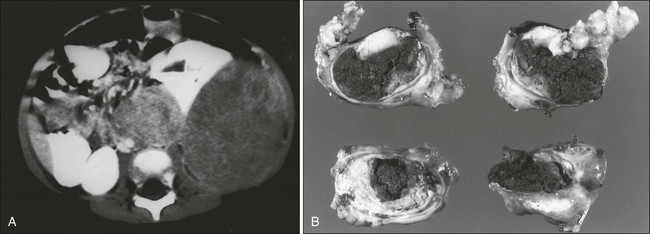
Several staging systems have been used to classify disease extent. The Evans and POG staging systems historically were the most widely used in the United States before 1999.33,34 To facilitate comparison of clinical studies, the International Neuroblastoma Staging System (INSS), which is based on clinical and surgical evaluations, was developed by consensus of major pediatric oncology groups in the United States, Europe, and Japan.33,35 The INSS differentiates between INSS stages 1, 2A, 2B, 3, 4, and 4S, based on surgical excision, lymph node involvement, and metastatic sites (Table 95-5). Because the INSS is a surgically based staging system, the stage for patients with locoregional disease can vary based on degree of surgical resection. Noteworthy, patients with localized disease who will not undergo surgery cannot be adequately staged. For these reasons, the International Neuroblastoma Risk Group (INRG) Task Force developed a new pretreatment staging system in 2005 based on clinical criteria and specific image-defined risk factors.36–37 Required imaging modalities include either CT or MRI, as well as MIBG scintigraphy. The International Neuroblastoma Risk Group Staging System (INRGSS) differentiates between L1, L2, M, and MS stage. The INRGSS differs from the INSS in that the INRGSS is based on preoperative imaging characteristics and not on surgical resection. The age for the INRGSS MS stage is set at 547 days (18 months) compared with 12 months in the INSS 4S stage. Table 95-5 compares INSS and INRGSS. The INRGSS not only allows preoperative staging, but also permits reassessment during treatment.
Table 95-5
Comparison Between INSS and INRGSS
| INSS | INRGSS |
| Stage 1: Localized tumor with complete gross excision; ± microscopic residual disease; representative ipsilateral lymph node negative for tumor microscopically | Stage L1: Localized tumor not involving vital structures as defined by IDRFs and confined to one body compartment |
| Stage 2A: Localized tumor with incomplete gross excision; representative ipsilateral lymph node negative for tumor microscopically | Stage L2: Locoregional tumor with presence of one or more IDRFs |
| Stage 2B: Localized tumor with or without complete gross excision; ipsilateral lymph node positive for tumor microscopically; enlarged contralateral lymph nodes should be negative microscopically | Equals stage L2 |
| Stage 3: Unresectable unilateral tumor infiltrating across the midline; ± regional lymph node involvement; or localized unilateral tumor with contralateral regional lymph node involvement or midline tumor with bilateral extension by infiltration (unresectable) or by lymph node involvement | Equals stage L2 |
| Stage 4: Any primary tumor with dissemination to distant lymph nodes, bone, bone marrow, liver, skin, or other organs | Stage M: Distant metastatic disease (except stage MS). Distant lymph node involvement is metastatic disease. Ascites and pleural effusion, even if malignant cells are present, do not constitute metastatic disease unless they are remote from the primary tumor |
| Stage 4S: Localized primary tumor in infants younger than age 1 year (localized as in stage 1, 2A, or 2B) with dissemination limited to skin, liver, or bone marrow (<10% malignant cells) | Stage MS: Metastatic disease in children younger than 547 days (18 months) of age with metastases confined to skin, liver, and/or bone marrow (<10% malignant cells); MIBG scan must be negative in bone and bone marrow. Primary tumor can be L1 or L2 with no limitations in terms of crossing or infiltration of the midline |
Prognostic Factors
Although many factors have been investigated and purported to have prognostic significance in neuroblastoma, the most significant in prediction of cure are patient age and stage at diagnosis. Children with low-stage disease have a good prognosis, regardless of age. In general, extent of disease at diagnosis is inversely related to cure. In infants, the DNA content of tumor cells has been reported to be predictive of response to chemotherapy and ultimate outcome. Infants with hyperdiploid tumors fare better than do those with diploid tumors.22 In children with metastatic disease, the most significant clinical prognostic variable is age at diagnosis. Children younger than 12 months of age at diagnosis have a significantly greater chance of cure.38 In older children with disseminated disease, the prognosis is dismal, although improvements in response and survival have been reported with modern treatment modalities
A variety of biological markers of neuroblastoma have been used to identify whether a patient is at prognostically low, intermediate, or high risk. These markers include tumor markers such as urinary catecholamine excretion and serum levels of ferritin, NSE, LDH, and ganglioside GD2. Histologic subclassification based on degree of differentiation or presence or absence of stroma, mitoses, or karyorrhexis also has been used as a prognostic indicator (Shimada histopathological classification). Identification of various biological and genetic features, such as the DNA index determined by flow cytometry, MYCN copy number, deletion of chromosomal regions on 1p together with clinical features has defined subsets of patients with different outcomes after therapy.23,36–40 For example, MYCN amplification can be used as a marker of poor outcome independent of age. Ongoing clinical studies continue to redefine the most informative combination of biological markers, age, and stage to develop appropriate risk-directed therapies (Table 95-6).
Table 95-6
The International Neuroblastoma Risk Group Classification Scheme
| Stage | Age, Months | Histologic Category/Grade of | MYCN | 11q Aberration | Ploidy | Pretreatment Risk Group* |
| L1/L2 | — | GN maturing, GNB intermixed | — | — | — | A: Very low |
| L1 | — | Any, except GN maturing or GNB intermixed | NA | — | — | B: Very low |
| Amp | — | — | K: High | |||
| L2 | <18 | Any, except GN maturing or GNB intermixed | NA | No | — | D: Low |
| Yes | — | G: Intermediate | ||||
| ≥18 | GNB nodular, differentiating NB, differentiating | NA | No | — | E: Low | |
| GNB nodular, poorly differentiated, or undifferentiated; NB, poorly undifferentiated, or undifferentiated | NA | Yes | — | H: Intermediate | ||
| Amp | — | — | N: High | |||
| M | <18 | — | NA | — | Hyperdiploid† | F: Low |
| <12 | — | NA | — | Diploid‡ | I: Intermediate | |
| 12 to <18 | — | NA | — | Diploid‡ | J: Intermediate | |
| <18 | — | Amp | — | — | O: High | |
| ≥18 | — | — | — | — | P: High | |
| MS | <18 | NA | No | — | C: Very low | |
| Yes | — | Q: High | ||||
| Amp | R: High |
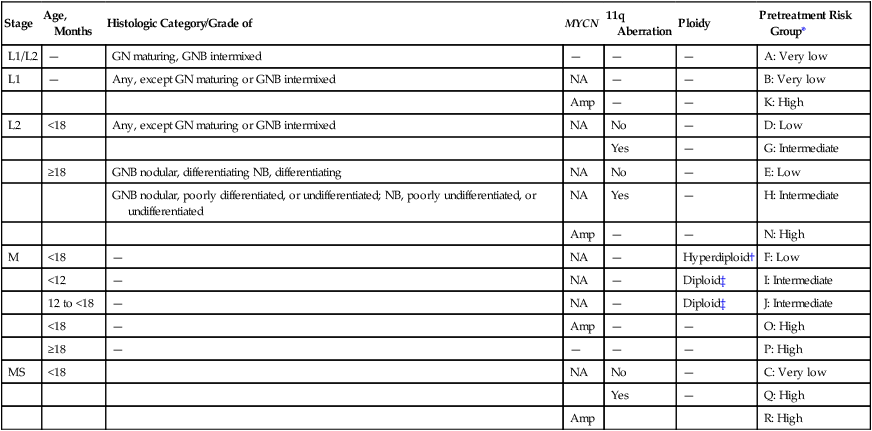
Amp, Amplified; GN, ganglioneuroma; GNB, ganglioneuroblastoma; NA, not amplified; NB, neuroblastoma.
*Five-year event-free survival by International Neuroblastoma Risk Group: very low risk, >85%; low risk, >75% to ≤85%; intermediate risk, ≥50% to ≤75%; high risk, <50%.
†DNA index >1.0 and includes near-triploid and near-tetraploid tumors.
Adapted from Cohn SL, Pearson AD, London WB, et al; INRG Task Force. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 2009;27:289-297.
Treatment
A complete pathological evaluation in conjunction with assessment of clinical features is important, because management of neuroblastoma is based on extent of disease. Accurate staging of the tumor at diagnosis is essential. Staging includes diagnostic imaging, pathological assessment of lymph node involvement in locoregional disease, liver biopsy in the case of primary abdominal disease in infants, and determination of the presence of hematogenous dissemination (usually to bone or to bone marrow).41 Patients with tumors localized to one side of the midline or that cross the midline without encasement of major blood vessels are candidates for primary surgical resection.42,43 With local disease, complete gross surgical resection gives an excellent chance of cure without additional therapy.42 If the patient has localized unresectable disease, surgical procedures that carry a high potential for morbidity should be avoided because of the excellent outcome with chemotherapy in this setting.44 If the tumor cannot be excised because it encases major blood vessels, surgery is performed for diagnostic purposes or for palliation if the tumor is compressing a vital organ. With dumbbell tumors with spinal cord compression, for example, rapid response to chemotherapy generally obviates laminectomy.45 In patients who have disseminated disease when they come to medical attention, initial aggressive surgical approaches have been shown to be of no significant benefit. The potential benefit of earlier resection of bulky primary tumors after some period of initial reductive chemotherapy in disseminated disease is currently under investigation.
Chemotherapy is the primary modality of treatment for most children with neuroblastoma, because the disease frequently is widespread at diagnosis.46,47 Box 95-3 outlines the approach to management of neuroblastoma used at St. Jude Children’s Research Hospital. A variety of single agents (cyclophosphamide, doxorubicin, cisplatin, epipodophyllotoxin, vincristine, topotecan) produce responses in patients with neuroblastoma, but significant and durable responses have been achieved only with combination chemotherapy.48–50 In general, pairs of these drugs, such as cyclophosphamide plus doxorubicin or cisplatin plus etoposide, delivered in a cytokinetically rational manner, are more effective in producing responses than single agents. Although improvement in survival of infants with advanced-stage disease and children with locally unresectable disease has resulted from implementation of various chemotherapeutic regimens, the outlook for older children with advanced disease remains a challenge. Less than 50% of these patients are alive at 5 years from diagnosis. Among older children, however, the complete response rate and disease-free interval have increased with more intensive combination chemotherapy, encouraging further development and refinement of this therapy.50 Recent studies report a subset of 12- to 18-month-old children with favorable tumor biological features who have a much greater chance of survival with intensive therapy.36 Furthermore, treatment for children with high-risk neuroblastoma in first response after induction chemotherapy, myeloablative therapy and stem cell rescue followed with immunotherapy (ch14.18 antibody combined with granulocyte-macrophage colony-stimulating factor and interleukin-2) and isotretinoin has resulted in a notable increase in EFS at 2 years (66%).51 Some current therapeutic regimens for neuroblastoma are so intensive that myelosuppression can be the dose-limiting toxicity. This problem can be overcome with HSCT. High-dose chemotherapy with bone marrow transplantation may be useful in the care of patients with a poor long-term prognosis who have achieved complete remission or at least substantial partial remission with chemotherapy.50,52 Results with various transplantation protocols, both autologous and allogeneic, suggest a modest survival advantage for these aggressive regimens.52
Additional investigational strategies presently under study include use of MIBG, which targets the human norepinephrine transporter (expressed in the majority of neuroblastoma tumors) and small-molecule inhibitors that target anaplastic lymphoma kinase. In addition, parallel clinical studies of newer biological therapies, such as those with next-generation GD2-targeted immunotherapy, in a setting of minimal residual disease or molecularly targeted therapy are expected to form the basis for future therapeutic gains.53
Wilms Tumor
Epidemiology
The annual incidence of Wilms tumor is seven cases per 1 million children younger than age 15 years, representing approximately 5% of cases of childhood cancer.1 In the United States, approximately 500 new cases are diagnosed each year, making Wilms tumor the fifth most common pediatric cancer by specific histologic type. The incidence of Wilms tumor varies with race and ethnic group. The range is 2.5 cases per 1 million Chinese children to 10.9 cases per 1 million African American children.2 Girls have a slightly increased risk for Wilms tumor, with a male-to-female ratio of 0.92 : 1. The mean age at diagnosis is 44 months for unilateral disease and 31 months for bilateral disease. According to the Knudson two-hit model of tumorigenesis, the earlier age at onset of bilateral Wilms tumor represents a genetic predisposition to the disease. Wilms tumor in the adult population is rare, although cases have been reported.3,4 Although results of older studies indicate that the prognosis for adult Wilms tumor is unfavorable, newer reports demonstrate cure of both localized and advanced disease with treatment regimens similar to those used for children.5,6
Familial Wilms tumor is uncommon, occurring in only 1.5% of affected patients.7 Most cases of familial Wilms tumor occur in distant relatives, rather than in parents or siblings. Sixteen percent of cases of familial Wilms tumor are bilateral, compared with 7% of sporadic cases. Unlike retinoblastoma, familial Wilms tumor is bilateral in only a small number of cases. Conversely, only a small proportion (3%) of cases of bilateral Wilms tumor is familial. The mean ages at diagnosis of familial unilateral and bilateral disease are 35 months and 16 months, respectively.
Tumor Biology
Although Wilms tumor was one of the original paradigms of the Knudson two-hit model of cancer formation,8 it has become apparent that several genetic events participate in tumorigenesis for this neoplasm. WT1 was the first Wilms tumor gene identified and is the most completely characterized Wilms tumor gene to date. The discovery of WT1 began with the observation that patients with a combination of aniridia, genitourinary anomalies, and mental retardation are at high (greater than 30%) risk for development of Wilms tumor and WAGR (Wilms tumor, aniridia, genitourinary anomalies, and mental retardation) syndrome. Cytogenetic analysis in persons with WAGR syndrome revealed large deletions at chromosomal band 11p13, which later was found to encompass a contiguous set of genes, including PAX6, the gene responsible for aniridia,9 and WT1.10–12 Patients with sporadic aniridia (PAX6 defect with normal WT1) are not at increased risk for Wilms tumor.13 WT1 encodes a transcription factor that is critical to normal kidney and gonadal development, but the precise role of which in tumorigenesis is undefined. Although germinal deletions or mutations in WT1 have been documented in almost all patients with WAGR syndrome and the related Denys-Drash and Frasier syndromes, only a small number of patients with seemingly sporadic Wilms tumor carry WT1 mutations in the germline (5%) or in tumor tissue (6% to 18%).14–17 Although WT1 is a bona fide tumor suppressor gene, its role in sporadic Wilms tumor development is limited.
A second Wilms tumor-associated condition is Beckwith-Wiedemann syndrome, which is an overgrowth disorder that manifests as high birth weight, macroglossia, organomegaly, hemihypertrophy, neonatal hypoglycemia, abdominal wall defects, and ear pits and creases. Patients with this syndrome have a 5% to 10% risk for development of Wilms tumor but also are predisposed to the development of other malignant tumors, such as hepatoblastoma, adrenocortical carcinoma, neuroblastoma, and rhabdomyosarcoma.18 Beckwith-Wiedemann syndrome maps to chromosomal band 11p15, sometimes called WT2, because loss of heterozygosity at this locus has been detected in Wilms tumor.19,20 Molecular characterization of the Beckwith-Wiedemann syndrome locus has revealed that the locus contains two domains of genomically imprinted genes, which are genes that are normally expressed solely from either the maternal or paternal allele. The expression of the imprinted genes is determined by the methylation patterns at two imprinting centers. The first imprinting center, called DMR1 (differentially methylated region 1), regulates insulin-like growth factor–2 (IGF2) and H19 expression. The second center, DMR2, regulates CDKN1C, KCNQ10T1, and KCNQ1 expression. The phenotype of Beckwith-Wiedemann syndrome may result from one of several alterations within DMR1 or DMR2, but it seems that alterations of IGF2 expression are most strongly associated with Wilms tumor.21–28 The role of IGF2 in the pathogenesis of Wilms tumor was recent confirmed by the observation that genetically engineered mice with IGF2 overexpression and WT1 ablation develop Wilms tumor, though mice with either genetic lesion in isolation did not develop tumors.29 Other genes have been implicated in the molecular pathogenesis of Wilms tumor. In two studies, investigators found that approximately 15% of Wilms tumors have activating mutations of CTNNB1, which encodes β-catenin, a central effector of the Wnt signaling pathway.30,31 Of interest, the CTNNB1 mutations were strongly associated with WT1 mutations, indicating that these two genes may collaborate in the genesis of Wilms tumor. A Wilms tumor gene on the X-chromosome, FAM123B (more commonly called WTX), was recently found to be mutated in 20% to 30% of sporadic Wilms tumors.32,33 In girls, the mutations are found exclusively on the active X-chromosome, providing a variation on the two-hit hypothesis (only one hit needed). Like β-catenin, WTX participates in the Wnt signaling pathway.34
Genetic linkage analysis has identified two familial Wilms tumor loci, called FWT1 and FWT2, on chromosomes 17 and 19, respectively.35,36 Identifying the pertinent genes at these loci is an area of active investigation. It was recently recognized that some cases of familial Wilms tumor are associated with biallelic BRCA2 mutations. Whereas monoallelic BRCA2 mutations are associated with breast and ovarian cancers, individuals with biallelic mutations have the D1 type of Fanconi anemia and are predisposed to the development of Wilms tumor and brain tumors.37 Mutations in PALB2, which encodes a binding partner of BRCA2, have also been described in families with Fanconi anemia and Wilms tumor predisposition.38 Other syndromes (and associated genes) that predispose to Wilms tumor include Li-Fraumeni syndrome (TP53), Simpson-Golabi-Behmel syndrome (GPC3), Perlman syndrome, mosaic variegated aneuploidy (BUB1B), hereditary hyperparathyroid jaw tumor syndrome (HRPT2), Bloom syndrome (BLM), Mulibrey nanism (TRIM37), trisomy 18, and trisomy 13.39
Certain genetic mutations are associated with unfavorable prognosis in Wilms tumor. Loss of heterozygosity at 1p and 16q has been found in 10% to 20% of Wilms tumors. Loss at both loci is associated with adverse prognosis and is used for treatment stratification in the current COG studies.40 Gain of chromosome 1q has recently emerged as an indicator of adverse outcome but this marker requires validation before it is incorporated into clinical risk stratification schema.41,42 Mutations of TP53 are observed in most cases of anaplastic histologic features of Wilms tumor, implicating a role for this gene in progression from favorable to anaplastic histologic type.43–45
Pathology
Classic Wilms tumor consists of blastemal, stromal, and epithelial elements, although tumors do not necessarily contain all three (Fig. 95-11). Because Wilms tumor can be recognized with standard hematoxylin-and-eosin staining, the role of ultrastructural or immunohistochemical studies is limited. Other childhood renal neoplasms that must be considered in the differential diagnosis for Wilms tumor are clear cell sarcoma of the kidney, rhabdoid tumor of the kidney, congenital mesoblastic nephroma, renal cell carcinoma, and soft-tissue sarcoma of the kidney.
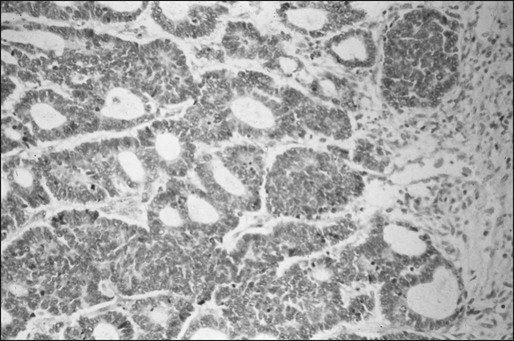
An important advance in the care of patients with Wilms tumor has been appreciation of the prognostic importance of histologic subtype. In 1978, Beckwith and Palmer published the results of a detailed histopathological review of the cases of patients entered in the first National Wilms Tumor Study (NWTS).46 Approximately 6% of the Wilms tumor specimens contained anaplasia, a term used to describe nuclear enlargement and atypia with irregular mitotic figures (Fig. 95-12). The presence of anaplasia was prognostically significant: 11 (44%) of 25 patients with anaplasia died of tumor, whereas only 26 (7.1%) of 364 patients without anaplasia died of tumor. Results of subsequent studies by NWTS investigators, studies by the International Society of Pediatric Oncology (SIOP), and a United Kingdom Wilms tumor study confirmed the adverse prognostic significance of anaplastic histologic features.47,48
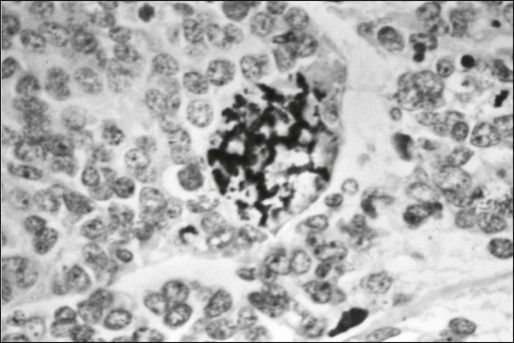
Patients treated on SIOP studies receive 4 to 6 weeks of chemotherapy before undergoing nephrectomy. The microscopic appearance of the tumor after chemotherapy has prognostic significance. Approximately 5% to 10% of Wilms tumors are completely necrotic after chemotherapy, a finding associated with a 98% 5-year relapse-free survival rate.49 By contrast, Wilms tumors with a predominance of blastemal cells after chemotherapy, defined as viable cells in more than one third of the tumor mass and blastemal cells in at least two thirds of the viable component, have relapse rates nearly as high as those for anaplastic Wilms tumor.50 Based on these observations, the SIOP histologic risk classification schema divides renal tumors into three risk categories based on the viability and cell type after chemotherapy: low risk, intermediate risk, and high risk.
Nephrogenic rests are foci of embryonal kidney cells that persist abnormally into postnatal life. They are present in approximately 1% of newborn kidneys and usually regress or differentiate by early childhood.51 Because nephrogenic rests are present in the kidneys of approximately 40% of patients with Wilms tumor, it is presumed that the rests represent Wilms tumor precursors. Current models of tumorigenesis for this neoplasm propose that a mutation in a tumor suppressor gene, such as WT1, predisposes to nephrogenic rests, which may sustain additional mutations and transform into a Wilms tumor.52
Clinical Manifestations and Patterns of Spread
Wilms tumor typically manifests as an asymptomatic abdominal mass discovered by a parent or a health care practitioner. The mass is smooth and firm, is fixed in position, and often extends across the midline. Abdominal pain, fever, anemia, hematuria, and hypertension are other common signs and symptoms observed in 20% to 30% of children with Wilms tumor.53 Constitutional signs and symptoms (e.g., weight loss, cachexia, bone pain) are unusual manifestations of Wilms tumor.
Laboratory and Radiologic Evaluation
The goal of imaging before Wilms tumor therapy is to define the extent of disease, assess the contralateral kidney, and determine whether tumor thrombus is present. Ultrasonography with Doppler technique is the recommended first-line study for Wilms tumor because it allows panoramic examination of the abdomen, including the patency of the inferior vena cava, in a safe and painless manner (Fig. 95-13). CT can depict pelvic and abdominal structures as well as lymph nodes (Fig. 95-14). CT is especially useful in detection of bilateral tumors. Intravenous pyelography and MRI typically are not necessary in the evaluation of Wilms tumor, although MRI may facilitate differentiation between nephrogenic rests and Wilms tumor.54 Because Wilms tumor metastasizes to the lungs, preoperative chest radiography is imperative. Plain radiographs of the chest are the traditional means of evaluation. Although chest CT is more sensitive than plain radiography in the detection of pulmonary metastasis, it can be associated with false-positive findings and a high interreader variability among radiologists.55–57 However, a recent study from the COG demonstrated that patients with small pulmonary nodules detected by chest CT and not by chest x-ray had a lower relapse rate when treated with doxorubicin, although the administration of lung radiation did not have an impact. This finding suggests that CT scans do provide additional prognostic information beyond that of plain x-rays.58,59 On the current COG AREN0533 study, CT scan is mandated and all patients with lung nodules, regardless of size, are treated with doxorubicin.
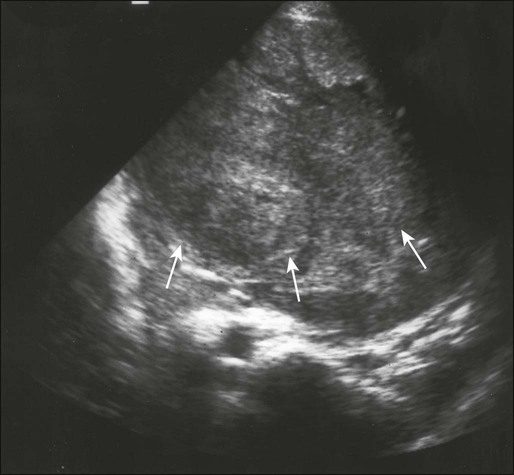
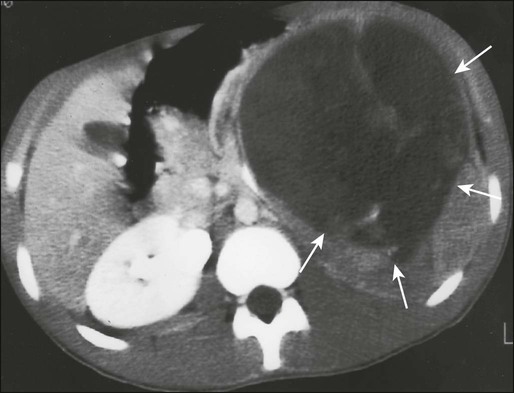
Treatment
Wilms tumor surgery should be performed by an experienced pediatric surgeon through a transverse abdominal incision. The peritoneal surface, liver, and lymph nodes are inspected for tumor involvement. Biopsy is performed on suspicious lesions. A lymph node sample should be obtained whether or not the node appears involved. Although the NWTS group previously recommended inspection of the contralateral kidney, less than 0.3% of patients enrolled in NWTS-4 had lesions detected during surgery that were not detected on preoperative imaging studies.60 With the availability of modern imaging techniques, routine exploration of the contralateral kidney is no longer recommended. The tumor is removed en bloc with the kidney, hilar structures, and a generous segment of ureter. The adrenal gland is included in the resection if the tumor is adherent to the gland or if the tumor originates in the upper pole of the kidney. Caution should be exercised to avoid capsular rupture and tumor spillage, which could adversely influence staging and alter therapy. If a tumor is deemed inoperable because of size or invasion of vital structures, biopsy is performed and adjuvant therapy is administered before definitive surgery. The COG staging system for Wilms tumor is based on surgical and histopathological findings (Table 95-7).61
Table 95-7
Children’s Oncology Group Clinicopathological Staging for Wilms Tumor
| Stage | Description |
| I | Tumor limited to kidney and completely excised. No penetration of the renal capsule or involvement of renal sinus vessels. |
| II | Tumor extends beyond the kidney but is completely excised with negative margins and lymph nodes. At least one of the following has occurred: (a) penetration of the renal capsule, (b) invasion of the renal sinus vessels. |
| III | Gross or microscopic residual tumor remains postoperatively, including inoperable tumor, positive surgical margins, tumor spillage occurring preoperatively or intraoperatively, regional lymph node metastasis, and transected tumor thrombus. |
| IV | Hematogenous metastasis (lung, liver, bone, brain) or lymph node metastasis outside the abdominal or pelvic cavities. |
| V | Bilateral renal tumors at diagnosis. |
Consecutive trials of the NWTS beginning in the late 1960s provided critical insights into the role of adjuvant therapy for Wilms tumor.62 The most recently completed study, NWTS-5, sought to capitalize on previous successes. The aim was to curtail therapy in low-risk groups, improve cure rates for high-risk groups, and identify novel prognostic markers. Table 95-8 shows the treatment algorithms used in NWTS-5.61 One of the primary objectives was to evaluate whether adjuvant chemotherapy provides benefit to children younger than 24 months who have small stage I tumors with favorable histologic features, a group with an outstanding prognosis. In 75 patients who met the eligibility criteria, the tumor was treated with surgical resection and close observation only. Eleven (14.7%) of these 75 patients experienced relapse or were found to have metachronous disease in the contralateral kidney, prompting early closure of this study arm.63 Additional follow-up indicated that salvage therapy was very successful in this group and nearly all patients survived.64,65 The current COG AREN0532 study is reassessing whether the outstanding survival rate can be replicated in these very low-risk patients. A recent gene expression array analysis demonstrated that small stage I favorable histology Wilms tumors may be divided into subsets with distinct gene expression profiles.66 These profiles may enable clinicians to predict which patients are likely to experience recurrence. Loss of heterozygosity (LOH) at 11p15 and WT1 mutation correlated with relapse.67 The AREN0532 study will enable validation of these findings and could pave the way for molecular definition of very low-risk rather than relying on clinical features with arbitrary cutoffs (e.g., age <24 months, tumor weight <550 g). A second primary objective of NWTS-5 was to test the hypothesis that tumor LOH at chromosomal loci 1p and 16q is associated with unfavorable prognosis. More than 2000 patients on NWTS-5 had informative LOH results. LOH at 1p and 16q was prognostically significant for Wilms tumor of favorable histology, but not for Wilms tumor of anaplastic histology. The greatest effect on prognosis was seen in tumors that had LOH at both loci (Table 95-9).68 The current COG AREN0532 and AREN0533 studies are evaluating the benefit of augmenting therapy for patients whose tumors harbor LOH at both 1p and 16q. The biological mechanism for the association between LOH and unfavorable outcome is under investigation.
Table 95-8
Treatment Algorithms for Nonanaplastic Wilms Tumor
| NWTS-5 | SIOP 93-01 | ||||
| Stage | Chemotherapy | Radiation Therapy | Chemotherapy | Radiation Therapy | |
| Preoperative | Postoperative | ||||
| I | VA × 18 weeks | none | VA × 4 weeks | VA × 4 weeks* | None |
| II | VA × 18 weeks | none | VA × 4 weeks | VDA × 27 weeks | Node-negative: none Node-positive: 15 Gy |
| III | VDA × 24 weeks | 10.8 Gy (flank/abdomen) | VA × 4 weeks | VDA × 27 weeks | 15 Gy (flank/abdomen) |
| IV | VDA × 24 weeks | 12 Gy lung (if lung metastasis) 10.8 Gy flank (if local stage III) |
VDA × 6 weeks | CR after 9 weeks: VDA × 27 weeks No CR after 9 wks: ICED × 34 weeks |
None if lung lesions disappear by week 9, otherwise 12 Gy |
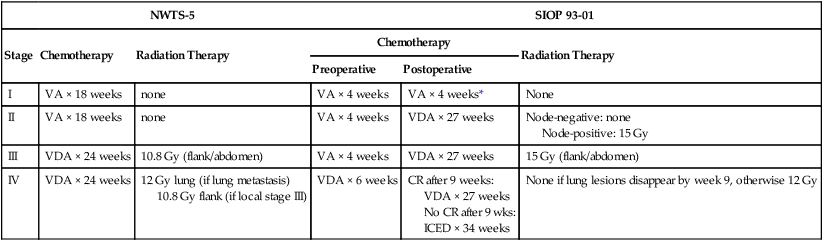
*Patients were randomly assigned to receive postoperative VA for 4 or 18 weeks. Treatment with VA was no less effective for 4 weeks than for 18 weeks.
Table 95-9
Results of National Wilms Tumor Study 5
| Stage | 4-Year Relapse-Free Survival Rate (%) | 4-Year Overall Survival Rate (%) |
| FAVORABLE HISTOLOGIC FEATURES WITHOUT LOH 1P | ||
| I (age <24 months/tumor weight <550 g) | 95.6 | 100 |
| I (age ≥24 months/tumor weight ≥550 g) | 94.2 | 98.4 |
| II | 86.2 | 97.7 |
| III | 86.5 | 94.4 |
| IV | 76.4 | 86.1 |
| V | 64.8 | 87.1 |
| FAVORABLE HISTOLOGIC FEATURES—STAGES I/II | ||
| No LOH | 91.2 | 98.4 |
| LOH 1p only | 80.4 | 91.2 |
| LOH 16q only | 82.5 | 98.1 |
| LOH 1p and 16q | 74.9 | 90.5 |
| FAVORABLE HISTOLOGIC FEATURES—STAGES III/IV | ||
| No LOH | 83.0 | 91.9 |
| LOH 1p only | 89.0 | 97.6 |
| LOH 16q only | 85.3 | 92.0 |
| LOH 1p and 16q | 65.9 | 77.5 |
| DIFFUSE ANAPLASTIC HISTOLOGIC FEATURES | ||
| I | 68.4 | 78.9 |
| II | 82.6 | 81.5 |
| III | 64.7 | 66.7 |
| IV | 33.3 | 33.3 |
| V | 25.1 | 41.6 |
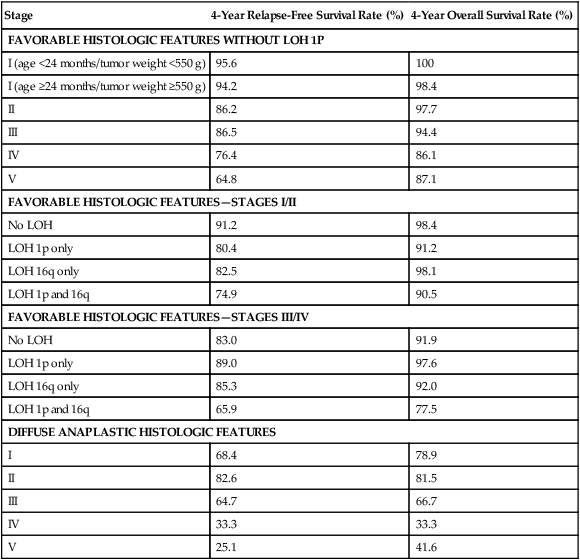
A third objective of NWTS-5 was to improve outcomes of patients with stages II to IV Wilms tumor of diffuse anaplastic histology. Based on the finding that patients with diffuse anaplastic Wilms tumor benefited from cyclophosphamide, a new treatment regimen consisting of chemotherapy with vincristine, doxorubicin, cyclophosphamide, and etoposide, and radiation therapy was developed. The outcomes for patients who received this regimen were improved compared with historic controls but still were clearly inferior to outcomes in patients with tumors of favorable histology (see Table 95-9).69 Patients with stage I anaplastic Wilms tumor received a two-drug regimen of vincristine plus dactinomycin, with the idea that completely resected tumors do not require intensive treatment. However, these patients did not fare as well as expected (see Table 95-9). The current COG AREN0321 study is augmenting therapy for patients with stages II to IV diffuse anaplastic Wilms tumor by adding carboplatin to the treatment regimen. Patients with stage I anaplastic Wilms tumor receive doxorubicin and flank irradiation in addition to vincristine+dactinomycin. Box 95-4 outlines the approach to management of Wilms tumor used at St. Jude Children’s Research Hospital.
In parallel to the NWTS, the SIOP Renal Tumor Committee conducted a series of trials, beginning in 1971, that optimized an approach by using prenephrectomy chemotherapy.62 The SIOP-9 study demonstrated that histology after treatment is highly predictive of outcome.50 Both anaplastic histology and blastemal predominance after chemotherapy were shown to have a strong association with adverse outcome. Conversely, complete necrosis portended outstanding outcome.49 The most recently completed SIOP 93-01 study demonstrated that reduction of postoperative chemotherapy for stage I Wilms tumor to 4 doses of vincristine and 1 dose of dactinomycin was as effective as the standard 18-week regimen.70 Table 95-8 summarizes the treatment regimens used on the SIOP 93-01 study.
Bilateral Wilms Tumor
Bilateral Wilms tumor occurs in approximately 5% to 10% of patients with Wilms tumor. These children pose a therapeutic challenge because of difficulty in obtaining local control while sparing renal parenchyma. The approach to patients with bilateral Wilms tumor is to administer preoperative chemotherapy to elicit tumor shrinkage and then to perform partial nephrectomy, whenever possible, or complete nephrectomy. Patients with tumors that cannot be resected with clear margins receive localized radiation therapy. On NWTS-4, the 8-year EFS and overall survival (OS) rates for patients with bilateral favorable histology Wilms tumor were 74% and 89%, respectively. The 8-year EFS and OS for bilateral anaplastic Wilms tumor were 40% and 45%, respectively.71 Compared with patients with unilateral Wilms tumor, patients with bilateral tumors have an increased rate of renal failure, with a long-term risk estimated to be 12% in the absence of congenital genitourinary anomalies.72 The most common cause of renal failure in this patient group is tumor progression or recurrence necessitating nephrectomy, not therapy-related effects.
Recurrent Wilms Tumor
Despite excellent outcome among most patients with Wilms tumor, approximately 10% to 15% of patients with disease with favorable histologic features and 50% of patients with anaplastic disease experience primary progression or tumor recurrence. The most common sites of recurrence are the lungs, liver, opposite kidney, and intraabdominal sites, including the original tumor bed. Wilms tumor occasionally recurs in the brain, bone, and distant lymph nodes. Most relapses are diagnosed within the first 2 years after the original diagnosis. Factors associated with favorable prognosis after recurrence include favorable histologic features, initial treatment with only vincristine and actinomycin D, recurrent disease involving the lungs only, recurrent disease arising in the abdomen of a patient who did not receive abdominal irradiation, and relapse more than 12 months after the original diagnosis.73,74 With aggressive therapy, approximately 60% to 80% of patients with tumors with favorable prognostic features can be cured. Salvage regimens are not as successful for patients with tumors with at least one unfavorable prognostic feature. Salvage regimens include ifosfamide, cyclophosphamide, carboplatin, etoposide, and topotecan.74–77 The overall 4-year survival rate after relapse for patients whose initial treatment regimen consisted of vincristine and actinomycin D was 81% after treatment with a regimen containing vincristine, doxorubicin, cyclophosphamide, and etoposide, and radiation therapy.78 For patients whose initial treatment regimen included agents in addition to vincristine and actinomycin D, the 4-year OS rate was 48% after treatment with cyclophosphamide, carboplatin, etoposide, and irradiation.79 Several groups of investigators have used high-dose chemotherapy followed by autologous stem cell rescue in patients with recurrent Wilms tumor.80–82 Results are promising, but it is unclear whether high-dose therapy is superior to conventional-dose chemotherapy with modern agents.
Late Effects of Therapy
The late effects of Wilms tumor treatment have received considerable attention because Wilms tumor usually is curable, and the number of long-term survivors is growing. Late complications can result from chemotherapy, radiation therapy, or the primary nephrectomy itself. Although most Wilms tumor survivors have only one kidney, less than 1% of patients with unilateral Wilms tumor treated in NWTS-1 through NWTS-4 were found to have renal failure.83 The median interval from diagnosis to onset of renal failure was 21 months. Renal failure is most prevalent in patients with bilateral Wilms tumor. Another recognized long-term effect of Wilms tumor therapy is congestive heart failure, which was found to have a cumulative frequency of 4.4% 20 years after diagnosis of Wilms tumor in patients whose initial treatment regimen included doxorubicin.84 The frequency of congestive heart failure was higher among patients who received doxorubicin as part of a salvage chemotherapy regimen. Risk factors for congestive heart failure included increasing cumulative doxorubicin dose, female gender, and radiation to the lung and left hemiabdomen (but not the right hemiabdomen). An analysis of pregnancy outcome among Wilms tumor survivors revealed that women who received flank radiation therapy were at increased risk for hypertension complicating pregnancy, fetal malposition, premature labor and having low-birth-weight infants.85 The cumulative incidence of solid tumors at age 40 years for subjects who survived free of second malignant neoplasms to age 15 years was 6.7%. Leukemia risk was also increased compared with the general population, but was much less common than the solid tumor risk. The cases of leukemia occurred much earlier than those of solid malignancies.86