Chapter 3 Pancreatic physiology and functional assessment
The Pancreas
The pancreas is a complex organ. In addition to nerves, blood vessels, and connective tissue, it is composed of endocrine and exocrine elements. The endocrine pancreas consists of 105 to 106 islets of Langerhans, each of which contains approximately a thousand endocrine cells. Together the islets constitute approximately 2% of the normal human pancreas. The exocrine pancreas includes the acinar cells, which account for 75% to 90% of the pancreatic mass, and the duct cells, which account for approximately 5%. Most of the currently available evidence indicates that all endocrine and exocrine pancreatic cells are derived from a common stem or progenitor cell (Fishman & Melton, 2002; Zaret & Grompe, 2008). Commitment to become either a duct cell or an acinar cell is believed to occur relatively early during embryogenesis. Subsequently, the endocrine cells undergo terminal differentiation into islet and acinar cells, but some also still may be capable of differentiating into duct cells (Gu et al, 2003). Conversely, some duct cells may be capable of differentiating into endocrine cells.
Endocrine Pancreas
Structure and Innervation
The islets of Langerhans are composed of four different cell types evenly distributed throughout the pancreas. Approximately 15% of islet cells are α cells, which secrete glucagon; 70% to 80% are β cells, which secrete insulin; 5% to 10% are δ cells, which secrete somatostatin; and 15% to 25% are pancreatic polypeptide (PP) cells, which secrete pancreatic polypeptide (Kulkarni, 2004). The PP cell type is only sparsely present in the dorsal pancreas, neck, body, and tail; most PP cells are found in the portion of the pancreas derived from the ventral pancreatic bud (the head and uncinate process). Rodent islets consist of a core composed of β cells surrounded by the other cell types, but in humans, the various cell types are intermixed within each islet. These islets contain a rich capillary network, and they receive substantial blood flow.
The islets of Langerhans are innervated by parasympathetic and sympathetic nerves. Vagal stimulation increases insulin release, and vagal stimulation may play a key role in mediating the cephalic phase of insulin secretion; that is, insulin release in response to the sweet taste of glucose. Although a cephalic phase of insulin secretion has been shown to occur in rats and dogs, it is not certain that a cephalic phase occurs in humans. Stimulation and inhibition of insulin release have been associated with sympathetic nervous stimulation in experimental animals, but the role of sympathetic nerves in regulating insulin release in humans has not been established (Nauck, 1998).
Synthesis and Storage of Insulin
Insulin is assembled within the endoplasmic reticulum as preproinsulin (Madsbad et al, 1992; Malaisse, 1992; Steiner & James, 1992). With cleavage of a signal sequence, the intermediate product proinsulin is formed, packaged within Golgi-derived vesicles that evolve into immature storage granules and eventually become mature secretory granules. During this process of granule maturation, proteolytic processing of insulin continues, liberating mature insulin from its C-peptide. Mature insulin, its C-peptide fragment, and other processing intermediates are stored together within the secretory granules, until they are all discharged at the cell surface. Normally, that secretion occurs primarily in response to external stimuli (regulated secretion). In insulinomas, insulin and its processing intermediates often are released continuously, however, without the need for external stimuli (constitutive secretion; see Chapter 61).
Stimulus–Secretion Coupling for Insulin Secretion
Glucose, certain amino acids (e.g., arginine), acetylcholine, and incretins such as gastric inhibitory peptide (GIP) and glucagon-like peptide (GLP-1) are the most important regulators of insulin release by β cells (Nauck, 1998). To varying degrees, the intracellular mechanisms responsible for insulin release in response to each of these types of regulators are different (Fig. 3.1). The “first phase” of insulin release seems to involve mechanisms that differ from the mechanisms in the later phase of sustained insulin secretion. This review focuses on events believed to underlie that first phase of insulin secretion (Rutter, 2001).
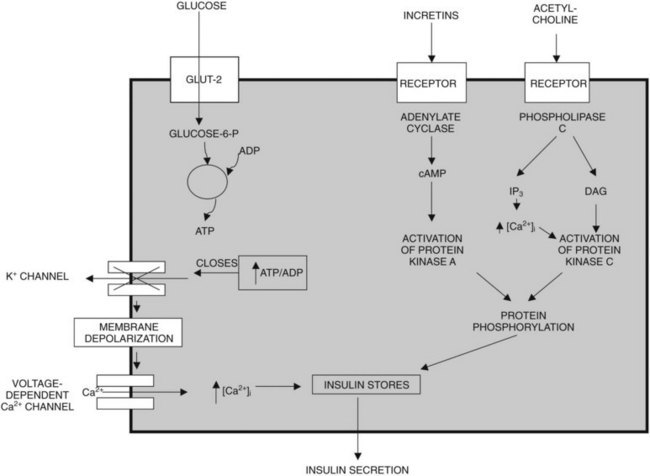
Glucose, the most important regulator of insulin release, enters β cells via a highly efficient glucose transporter, GLUT-2, which for the most part equalizes intracellular and extracellular glucose concentrations (Unger, 1991). Subsequent to its uptake by the cell, glucose is phosphorylated by glucokinase (Meglasson & Matschinsky, 1986), which, because of its high michaelis-menten constant (Km), acts as a glucose sensor. Glucokinase catalyzes the formation of glucose-6-phosphate, and as a result, glucose-6-phosphate accumulates within the cell at concentrations closely related to the intracellular concentration of glucose. Glucose-6-phosphate is metabolized, leading to formation of adenosine triphosphate (ATP); this results in an increased ATP–ADP (adenosine diphosphate) ratio, which causes closure of an ATP-dependent potassium channel in the plasma membrane of β cells (Henquin, 1988). Closure of this channel results in membrane depolarization, which leads to activation of voltage-dependent L-type calcium channels and calcium influx into the cell (Misler et al, 1992). The resulting increase in cytoplasmic calcium levels triggers exocytosis of the secretory granules that contain insulin (Eliasson et al, 2008). In contrast to the initial phase of insulin secretion, it is believed that the later phase of sustained secretion does not involve closure of ATP-dependent potassium channels (Rutter, 2001).
Amino acids such as arginine also upregulate insulin secretion by being metabolized and causing ATP levels to increase, increasing the ATP–ADP ratio and triggering calcium influx. The incretins, including GIP and GLP-1, are believed to upregulate insulin secretion by activating adenylate cyclase, increasing cyclic adenosine monophosphate (cAMP) levels, activating protein kinase A, and causing phosphorylation and activation of exocytosis-related proteins; cAMP also may promote insulin secretion by mechanisms that involve activation of L-type calcium channels and accelerated calcium influx. Insulin secretion also can be stimulated by acetylcholine released from cholinergic nerves. Acetylcholine binds to receptors on the cell surface, causing activation of phospholipase C and generation of the secondary messengers, inositol triphosphate (IP3), and diacylglycerol (DAG). IP3 triggers release of calcium (Ca2+) from intracellular storage pools, which causes release of insulin. DAG binds to and activates protein kinase C, which, like protein kinase A, promotes insulin secretion by phosphorylating and activating exocytosis-related proteins (Persaud et al, 1989; Prentki & Matschinsky, 1987).
Incretins and the Regulation of Insulin Secretion
Insulin secretion in response to orally administered glucose and other nutrients is 25% to 50% greater than that after intravenous administration (Creutzfeldt, 1979; Tillil et al, 1988). This so-called incretin effect is mediated by hormones released from the gut, primarily GIP and GLP-1 (Fiesler et al, 1995; Nauck et al, 1993). GIP is produced by K cells in the duodenum and upper jejunum, and it is released into the circulation after ingestion of nutrients. GLP-1 is synthesized in enteroglucagon or L cells, which also contain peptide YY (PYY) and enteroglucagon. These L cells are primarily located in the distal small bowel, colon, and rectum. Of the two hormones GIP and GLP-1, GIP is probably the major physiologic incretin. Glucose and amino acids are considered to be “primary” or “direct” stimulants of insulin secretion, but insulin secretion is also modulated by the incretins. These incretins cause little or no insulin release in the absence of glucose or amino acids, but they can alter insulin release dramatically in response to either glucose or amino acids.
Glucagon and Other Islet Hormones
In addition to insulin, the islets of Langerhans secrete somatostatin, pancreatic polypeptide, glucagon, and possibly ghrelin. Clinically, pancreatic diseases generally are not associated with disorders of somatostatin, because somatostatin is synthesized and released by many other organs besides the pancreas. Pancreatic polypeptide, released from the ventral region of the pancreas in response to food, does not seem to have any specific physiologic function, and as a result, abnormal pancreatic polypeptide secretion is not associated with clinically recognizable abnormalities. Glucagon, released from islet α cells, is a functionally important hormone, and disorders of glucagon secretion can result in clinically recognizable functional changes (Lefebvre, 1992).
Glucagon is a 29–amino acid hormone that functions to regulate hepatic glucose output. Acting via adenylate cyclase and cAMP, glucagon stimulation of hepatocytes accelerates glycogenolysis and gluconeogenesis, causing blood glucose levels to increase. Hypoglycemia promotes glucagon release, and hyperglycemia inhibits glucogon release (Lefebvre, 1992; Unger, 1985). In some species, glucagon has other effects, including accelerating lipolysis, promoting gastric emptying, and stimulating insulin secretion. In type 1 diabetes, there is a low insulin–glucagon ratio, and this worsens hyperglycemia by promoting hepatic glucose output. In type 2 diabetes, glucagon secretion seems to be increased, and it is not suppressed by hyperglycemia. As a result, plasma glucagon levels are normal or elevated. With total pancreatectomy, patients become profoundly glucagon deficient and consequently prone to hypoglycemia. This leads to so-called brittle diabetes, which is characterized by swings in blood glucose levels, from marked hyperglycemia to marked hypoglycemia.
Endocrine Pancreas in Diabetes
Type 1 diabetes is a chronic autoimmune disease that leads to the destruction of β cells (Powers & Eisenbarth, 1995). Consequently, disordered insulin secretion in type 1 diabetes is primarily related to progressive loss of β cells, and clinical diabetes occurs when 80% to 90% of β cells are destroyed. In type 1 diabetes, serum insulin and C-peptide levels are usually extremely low (Polonsky et al, 1986). In contrast, the islets of Langerhans usually appear histologically normal in patients with type 2 diabetes, or the cells may be normal, but the islet may contain deposits of islet amyloid polypeptide. Despite this appearance, however, β-cell mass may be reduced by 50% in patients with type 2 diabetes, especially in patients who require insulin therapy. For the most part, patients with type 2 diabetes secrete increased amounts of insulin, proinsulin, and insulin conversion intermediates such as C-peptide, and elevated circulating levels of these factors can be detected (Porte, 1991).
Endocrine Pancreas in Chronic Pancreatitis
In chronic pancreatitis, there is progressive scarring of the pancreas with replacement of the gland by fibrous tissue. The islets become embedded in these areas of fibrosis, and eventually islets and islet cell mass are lost. This loss leads eventually to the loss of β cells and α cells (Diem, 2002). Functional testing may reveal a reduced response to oral and intravenous glucose and to glucagon and arginine. Approximately 10% of patients with exocrine pancreatic insufficiency secondary to chronic pancreatitis have clinically significant diabetes, whereas 30% have impaired glucose tolerance (Larsen et al, 1987; Sjoberg & Kidd, 1989).
Exocrine Pancreas
On a tissue-weight basis, the exocrine pancreas synthesizes and secretes more protein per day than any tissue in the body other than the lactating breast, and approximately 90% of that newly synthesized protein consists of digestive enzymes. On a daily basis, the normal human pancreas secretes 6 to 20 g of digestive enzyme protein in 2.5 L of bicarbonate-rich fluid (Case, 1998). Derangements of exocrine pancreatic function commonly are observed in patients with pancreatic disease, and pancreatic functional assessment mostly involves tests of exocrine pancreatic function. The structure of the exocrine pancreas, its neurohormonal control, and the cell physiology of exocrine pancreatic secretion are reviewed in this section, and when possible, this review emphasizes information known to be valid for humans. Most of the literature concerning these issues is based, however, on work performed using various nonprimate species—rats, mice, guinea pigs, cats, rabbits, dogs, and pigeons—or work performed in vitro using isolated cells. To a great extent, this literature has indicated that there may be important differences in pancreatic exocrine function and physiology among the various species as well as important differences between the in vivo and in vitro setting. Although many of the overall concepts covered in this section are no doubt relevant to the human in vivo setting, there may be important exceptions to this generalization (Case, 1998).
Structure
The exocrine pancreas is composed of two functional elements: a ductal tree and a mass of pancreatic acini (Case, 1998; Konturek et al, 2003). Each acinus consists of a grapelike cluster of acinar cells that surround a central or acinar lumen, which is continuous with the ductal space. Acinar cells constitute 75% to 90% of the gland. They are highly polarized cells that possess abundant, rough endoplasmic reticulum, located almost entirely within the basolateral region of the cell. The nucleus also is located in the basolateral region, and a prominent Golgi complex is located near the center of the cell. Secretory vesicles consisting of zymogen granules and their immature condensing vacuole precursors fill the apical portion of acinar cells.
The pancreas is innervated by extrinsic and intrinsic parasympathetic and sympathetic nerves. These autonomic nerves have been shown to use a vast array of neurotransmitters in addition to acetylcholine and catecholamines, including nitric oxide, vasoactive intestinal peptide (VIP), gastrin-releasing peptide, neuropeptide Y, galanin, substance P, calcitonin gene-related peptide, gastrin/cholecystokinin (CCK), and enkephalins (Case, 1998; Case & Argent 1993). From a purely functional standpoint, the most important of the neurotransmitters seems to be acetylcholine, which in addition to functioning as a neurotransmitter also acts as a paracrine hormone released near acinar cells to regulate their function. Evidence indicates that many of the intrapancreatic cholinergic vagal nerves possess CCK receptors, and that at least in humans, the hormonal effects of CCK on pancreatic acinar cells may be mediated by vagal afferents in response to CCK stimulation (Owyang & Logsdon, 2004). Whether or not CCK can directly stimulate acinar cell secretion of enzymes in humans is a controversial issue (Murphy et al, 2008; Owyang & Logsdon, 2004).
Neurohormonal Regulation of Exocrine Pancreatic Function
The exocrine pancreatic response to a meal traditionally is considered to involve three phases (Konturek et al, 2003): 1) the cephalic phase, triggered by the sight or smell of food, accounts for 10% to 20% of the pancreatic secretion during a meal; 2) the gastric phase, triggered by food entering the stomach and by gastric distention, accounts for 15% to 20% of meal-stimulated pancreatic secretion; and 3) the intestinal phase, triggered by food and acid entering the duodenum and proximal jejunum, accounts for 60% to 70% of meal-stimulated pancreatic secretion (Table 3.1). In humans, the cephalic and gastric phases are thought to be vagally mediated and to primarily involve stimulation of digestive enzyme secretion rather than bicarbonate secretion (Anagnostides et al, 1984; Case, 1998; Konturek et al, 2003). The intestinal phase is neurally and hormonally regulated, however, and it involves secretion of a pancreatic juice that is rich in bicarbonate and digestive enzymes.

The intestinal phase of meal-stimulated pancreatic secretion has been the most fully characterized phase (Fig. 3.2). The entry of gastric chyme into the duodenum and proximal jejunum triggers vagovagal (centrally acting) reflexes and enteropancreatic neural reflexes that together primarily stimulate digestive enzyme secretion from pancreatic acinar cells. Acidification of the duodenum (pH <4.5) causes specialized cells within the mucosa to release secretin into the circulation, and secretin potently stimulates duct cell secretion of fluid and bicarbonate (Case & Argent, 1993). Release of secretin and stimulation of fluid/bicarbonate secretion also are elicited by various products of digestion, especially fatty acids and bile salts (Rhodes et al, 1988; Riepl et al, 1990). Fatty acids and polypeptides released by the partial digestion of food also stimulate release of CCK from other specialized cells within the duodenal and jejunal mucosa, and elevated CCK levels cause acinar cells to secrete digestive enzymes and other proteins into the pancreatic juice (Dale et al, 1989).
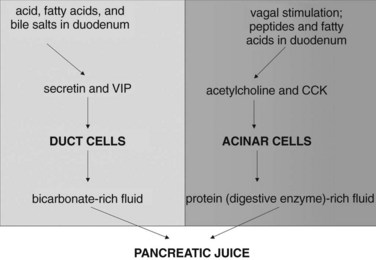
Pancreatic duct cells express secretin receptors, and it is generally believed that secretin stimulation of fluid/bicarbonate secretion by duct cells reflects the direct effect of secretin—and possibly other hormones, such as VIP—on those duct cells. Acinar cells express receptors for acetylcholine and several other candidate secretagogues, and it is generally believed that neurally stimulated acinar cell secretion of digestive enzymes reflects the direct effects of those neurally released factors on acinar cells, acting via their specific receptors. However, the mechanisms responsible for CCK-stimulated acinar cell secretion seem to be more complex and may be species specific. The acinar cells of many species (e.g., mice, rats) express CCK receptors, and in those species, CCK may stimulate secretion by directly interacting with those acinar cell receptors (Owyang & Logsdon, 2004; Soudah et al, 1992). For many years, it was widely believed that human acinar cells do not express CCK receptors (Ji et al, 2001). This has been recently challenged by Murphy and colleagues (2008), who suggested that human acinar cells may express functional CCK receptors coupled to digestive enzyme secretion. On the other hand, it has long been known that human CCK-stimulated digestive enzyme secretion can be almost entirely prevented by cholinergic blockade (Li & Owyang, 1993; Li et al, 1997), which would suggest that CCK-stimulated secretion is mediated via cholinergic mechanisms and that CCK receptors on acinar cells, even if they do exist, are unlikely to play a major role in mediating meal-stimulated secretion in humans.
Diversion of pancreatic juice from the duodenum causes circulating levels of secretin and CCK to increase. This increase stimulates pancreatic secretion of fluid, bicarbonate, and digestive enzymes. The mechanisms behind this phenomenon have been the subject of considerable interest, and at present, diversion-induced upregulated secretion is believed to reflect the presence of trypsin-sensitive “releasing factors” for secretin and CCK within the duodenal lumen. These factors are believed to be released into the duodenum from specific cells of the mucosa (Lu et al, 1989; Miyasaka et al, 1989) or secreted into pancreatic juice (Iwai et al, 1987) and degraded by luminally active trypsin when food-stimulated secretion is adequate; as a result, the stimulus for further secretion is dampened. When food-stimulated secretion is inadequate, however, all of the secreted trypsin is bound to proteins within the duodenal lumen; as a result, trypsin-induced degradation of the releasing factors does not occur, and CCK or secretin release into the circulation is increased.
The physiologic importance of this “feedback” mechanism for regulating pancreatic secretion in humans is unclear, but it has been invoked to explain the observation that, at least in some series, patients with painful, chronic pancreatitis seem to obtain pain relief by ingesting exogenous pancreatic enzyme preparations that contain trypsin. It is argued that patients with chronic pancreatitis secrete inadequate amounts of digestive enzymes (trypsin) into the duodenum, and as a result, levels of intraluminal CCK-releasing factor may be chronically elevated, leading to continuous CCK-induced stimulation of the pancreas and possibly to chronic pain. In this setting, provision of exogenous enzyme preparations that include trypsin could terminate that signal for pancreatic secretion by promoting intraluminal degradation of the CCK-releasing factors, and this could ameliorate the pain of chronic pancreatitis (Toskes, 2002). Most patients with painful chronic pancreatitis and steatorrhea continue to have pain, however, despite ingesting exogenous pancreatic enzymes (Mossner et al, 1992); this has led many investigators to question the role of the feedback mechanism in the regulation of human pancreatic function and the role of CCK in the genesis of chronic pancreatitis pain.
Electrolyte Secretion
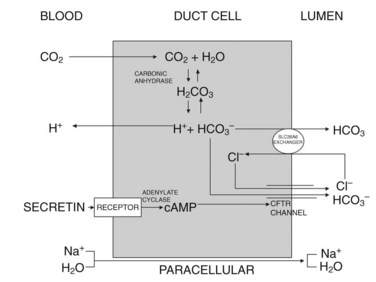
Currently, the most widely accepted model for duct cell secretion in humans is the one shown in Figure 3.3 (Case & Argent 1993; Ishiguro et al, 2009). Initially, carbon dioxide diffuses into the cell from the circulation, and it is hydrated by carbonic anhydrase to form H2CO3 (carbonic acid), which dissociates into H+ and HCO3−. The H+ back-diffuses across the basolateral cell membrane and is cleared into the circulation. The HCO3− is carried across the luminal cell surface by a Cl−/HCO3− exchanger, SLC26A6, the activity of which depends on the availability of Cl− in the duct lumen; this luminal Cl− is maintained by the cAMP-stimulated opening of the cystic fibrosis transmembrane regulator (CFTR), which is a Cl− channel. In this system, cAMP is generated by secretin-induced activation of adenylate cyclase at the basal surface of the cell. In many species, including mice, the maximal concentration of HCO3− in pancreatic juice is only 80 mmol/L, which can be achieved by secretion of HCO3− via the HCO3−/Cl− exchanger SLC26A6. In humans and guinea pigs, however, maximal HCO3− concentrations in pancreatic juice can reach 140 mmol/L; to achieve these high concentrations, another mechanism is required—secretion via CFTR in response to activation by cAMP (Ishiguro et al, 2009). In an overall sense, the secretion of HCO3− at the luminal surface generates a net negative transepithelial current and an ionic gradient, and these changes draw Na+ and water into the lumen via paracellular pathways.
Stimulus-Secretion Coupling in Duct Cells
Duct cells express seven-transmembrane G protein–coupled receptors for secretin, acetylcholine, and other possible regulators of fluid and electrolyte secretion. Occupancy of secretin receptors leads to G protein changes that trigger activation of adenylate cyclase, generation of cAMP, and activation of protein kinase A. As noted earlier, this leads to the activation of CFTR and to the opening of the Cl− channel, which may mediate both Cl− secretion and HCO3− secretion at the luminal cell surface. Occupancy of acetylcholine receptors leads to changes in other G proteins that trigger activation of phospholipase C, generation of IP3 and DAG, elevation of intracellular free calcium levels, and activation of protein kinase C, but it is not clear how this ultimately promotes fluid and electrolyte secretion into the ductal space (Case, 1998).
Acinar Cell Biology
Amino acids enter the acinar cell, against a concentration gradient, through at least four membrane transporters, and they bind to vacant transfer RNA in the cytoplasm. They are used to assemble newly synthesized proteins within the rough endoplasmic reticulum by a complex, extensively studied process that involves docking to the endoplasmic reticulum surface and elongation of the nascent polypeptide chain within the cisternae of the rough endoplasmic reticulum. On reaching a termination sequence, translation ceases and cleavage occurs, leaving the newly synthesized protein free to assume its tertiary and quaternary structure within the cisternae of the endoplasmic reticulum. After their synthesis, many of the newly synthesized proteins within acinar cells, particularly the proteins destined for secretion, are transported to the Golgi stacks in small vesicles; within those stacks, they are posttranslationally modified, usually by glycosylation or phosphorylation or both, and sorted into three groups: secretory, lysosomal, or structural. Secretory proteins, including digestive enzymes and their inactive zymogens, are packaged in immature secretory vesicles (condensing vacuoles), which migrate toward the cell surface as they mature into more dense zymogen granules. At the cell surface, and in response to secretory stimulation, the limiting membranes of the zymogen granules fuse with the luminal plasmalemma, secretory or fusion pores develop, and the granule contents are discharged into the acinar lumen (Palade, 1975; Rothman, 1994).
Digestive Enzyme Activation
The acinar cells of the pancreas synthesize and secrete numerous digestive enzymes. Some (amylase, lipase, RNase, and DNase) are synthesized as active enzymes, whereas others (trypsinogen, chymotrypsinogen, procarboxypeptidase, and proelastase) are synthesized as inactive proenzymes or zymogens. Zymogen activation usually involves proteolytic cleavage of the zymogen, exposing the activated enzyme after an activating peptide has been released. For the most part, the digestive enzyme zymogens are not activated until they reach the duodenum, where the brush-border enzyme enteropeptidase activates trypsinogen, and trypsin activates the other zymogens. Considerable evidence suggests that the synthesis of specific digestive enzymes may be controlled to some extent by diet—in other words, that ingestion of a high-fat diet promotes synthesis of lipolytic enzymes, ingestion of a high-protein diet favors synthesis of proteolytic enzymes, and ingestion of a starch-rich diet increases the synthesis of amylolytic enzymes—but the mechanisms underlying this control are unclear (Case, 1998).
Stimulus–Secretion Coupling in Acinar Cells
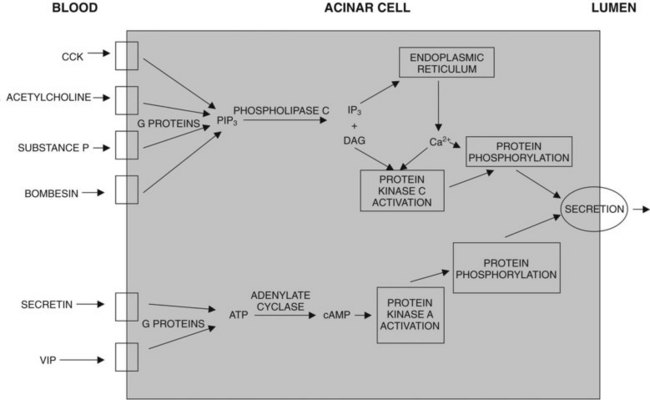
Many neurohormones can stimulate acinar cell digestive enzyme secretion; but to a great extent, the mechanisms involved differ, depending on the hormone involved (Fig. 3.4; Williams, 2001, 2002). Secretin and VIP bind to their specific G protein–coupled receptors and cause activation of adenylate cyclase, generation of cAMP, and activation of protein kinase A. Acetylcholine, bombesin/gastrin-releasing peptide, CCK, PAR-2–activating proteases, and substance P bind to their specific G protein–coupled receptors. CCK appears to bind to high-affinity CCK-A receptors, which in humans may be located on intrapancreatic cholinergic nerves; in other species these are on the surface of acinar cells. The receptors for acetylcholine and substance P are on acinar cells, and they are type 4 muscarinic receptors and NK-1 receptors, respectively. Activation of those receptors causes activation of phospholipase C, leading to the generation of DAG and IP3; release of calcium from intracellular stores, leading to a rise in cytoplasmic calcium levels; and activation of protein kinase C. The ultimate process of secretion is believed to be regulated by specific proteins that are phosphorylated (activated) by the activated protein kinases A and C (Williams, 2001, 2002; Yule & Williams, 1994).
Assessment of Pancreatic Function
Endocrine Function
In the past, numerous attempts were made to use changes in circulating glucagon or pancreatic polypeptide levels or both in response to stimulation as a diagnostic test for patients with suspected mild to moderate chronic pancreatitis; because of very poor sensitivity, those tests were abandoned and are of historical interest only (Toskes, 1986). At present, functional assessment of the endocrine pancreas in patients suspected of having chronic pancreatitis involves evaluation of insulin secretion and glucose metabolism. Endocrine pancreatic insufficiency in chronic pancreatitis may eventually result in clinical diabetes mellitus, because with time, functional islet cell mass and insulin secretion are lost; approximately 90% of β-cell mass must be lost before clinical diabetes develops. Twenty-five years after the onset of chronic pancreatitis, the incidence of clinical diabetes mellitus has been reported to exceed 80%, and more than 50% of the patients have insulin-dependent diabetes mellitus (Malka et al, 2000). With careful testing, many patients with lesser degrees of endocrine loss can be identified at an earlier time after the onset of chronic pancreatitis (Diem, 2002). In addition to an abnormal glucose tolerance test, these patients have depressed C-peptide secretion; consequently, circulating and urinary C-peptide levels may be low. In most cases, insulin-dependent diabetes develops, but in some patients, non–insulin-dependent diabetes may be seen; in these patients, C-peptide secretion may be normal. Diabetic ketoacidosis is unusual in patients with diabetes caused by chronic pancreatitis, but glucagon secretion is also often depressed, and a “brittle” form of diabetes may evolve (Angelopoulos et al, 2005). Pancreatic endocrine and exocrine insufficiencies in chronic pancreatitis evolve with different time dependencies, and in most cases, clinically apparent exocrine insufficiency precedes endocrine insufficiency by many years (Layer et al, 1994). Exocrine insufficiency is more closely correlated with morphologic changes (fibrosis, calcifications) than is endocrine insufficiency.
Assessment of Exocrine Function
Tests of exocrine pancreatic function can be divided into two major groups (Table 3.2): indirect tests monitor the intestinal effects of secreted pancreatic digestive enzymes; direct tests monitor the actual secretion of pancreatic exocrine products (enzymes, fluid, and bicarbonate). The indirect tests are the least invasive and most widely available of the tests, but they also are the least sensitive, and such tests are most likely to be normal in patients with mild degrees of pancreatic functional loss.
| Indirect Tests | Direct Tests |
|---|---|
| Fecal fat staining | Noninvasive tests |
| 72-hour fecal fat excretion | Serum trypsinogen |
| Bentiromide test | Fecal chymotrypsin |
| Pancreolauryl test | Fecal elastase |
| MRCP function tests | |
| Invasive tests | |
| Secretin test | |
| Secretin-CCK test | |
| Intraductal secretin test | |
| Lundh test |
MRCP, magnetic resonance cholangiopancreatography; CCK, cholecystokinin.
Indirect Pancreatic Function Tests
Conceptually, fecal fat analysis is the simplest of the indirect pancreatic function tests. It is based on the fact that pancreatic lipase is the enzyme responsible for most fat digestion, and diminished lipase secretion results in fat malabsorption. Fecal fat analysis can be accomplished by staining stool samples for fat with Sudan stain or by quantifying fecal fat when the patient is on a controlled-fat diet (Chowdhury & Forsmark, 2003; Lieb & Draganov, 2008). In the latter case, the patient is placed on a diet consisting of 100 g of fat per day for 5 days. Stool is collected on days 3 to 5, and fat content is measured. Fecal fat output of greater than 7 g/day is considered to be abnormal and diagnostic of steatorrhea; but fecal fat measurement is notoriously insensitive for the diagnosis of chronic pancreatitis, and it is most commonly abnormal only in patients with overtly symptomatic steatorrhea.
The bentiromide and the pancreolauryl tests are noninvasive, indirect pancreatic function tests. The former involves ingestion of the chymotrypsin substrate bentiromide, which is hydrolyzed by chymotrypsin to yield paraaminobenzoic acid, which is absorbed in the small intestine, conjugated in the liver, and excreted in the urine. The test is completed by collecting urine for 6 hours and quantifying urinary paraaminobenzoic acid recovery, which is considered to be abnormal if less than 50% (Niederau & Grendell, 1985). The pancreolauryl test involves ingestion of fluorescein dilaurate, which is hydrolyzed by pancreatic esterases to yield lauric acid and free fluorescein. The fluorescein is absorbed, conjugated in the liver, and excreted in the urine. Similar to the bentiromide test, the pancreolauryl test is completed by collecting urine, in this case for 10 hours, and measuring fluorescein excretion; in this test, excretion is compared with the patient’s excretion of orally ingested free fluorescein several days later. The bentiromide and pancreolauryl tests may be falsely abnormal in the face of liver or kidney disease and when certain drugs are present. The pancreolauryl test is not available in the United States, but the bentiromide test is commercially available and widely used, primarily because it is easy to perform; however, low sensitivities of 37% to 46% have been reported in patients with mild degrees of chronic pancreatitis (Naruse et al, 1980; Toskes, 1983; Ventrucci et al, 1983), so this test is of only limited value.
Other indirect tests of pancreatic function include the triglyceride breath tests (Loser et al, 1998) and the dual-label Schilling test (Brugge et al, 1980; Chen et al, 1989). Both tests have been the subject of numerous clinical studies, but for the most part, their sensitivity and reproducibility have been poor. At present, they are not used clinically for the diagnosis of chronic pancreatitis.
Direct Pancreatic Function Tests
Direct pancreatic function tests can be subdivided further into noninvasive and invasive tests. The noninvasive tests involve measuring fecal or serum levels of pancreas-derived digestive enzymes (serum trypsinogen, fecal chymotrypsin, and fecal elastase). Recently, direct function tests combining MRCP with secretagogue stimulation have been proposed (Schneider et al, 2006; Czako, 2007). These MRCP functional tests aim to either quantify juice flow into the duodenum or to provide contrast enhancement of the pancreatic parenchyma after hormonal stimulation; to date, however, the overall sensitivity and specificity of these MRCP-based tests remain to be determined, and their overall value as diagnostic tests for early chronic pancreatitis is unproven. The invasive tests involve placing a collecting device into the duodenum or pancreatic duct, stimulating pancreatic exocrine secretion, and measuring the output or concentration of exocrine pancreatic products.
Circulating levels of trypsinogen are easily measured and frequently low in patients with severe pancreatic insufficiency (Jacobson et al, 1984). Although measurement of serum trypsinogen may be helpful in evaluating the severity of chronic pancreatitis, the test has low sensitivity for the diagnosis of mild pancreatitis. Fecal levels of chymotrypsin and elastase also can be measured and used to assess exocrine pancreatic function (Dominguez-Munoz et al, 1995; Dominici & Franzini, 2002; Goldberg, 2000; Katschinski et al, 1997; Loser et al, 1996; Luth et al, 2001). Because trypsin is degraded as it passes through the intestinal tract, measurement of fecal trypsin levels is not a useful pancreatic function test. Fecal levels of chymotrypsin and elastase can be measured easily using commercially available assays; the levels of these enzymes are reduced in patients with advanced chronic pancreatitis; however, the sensitivity of fecal chymotrypsin and elastase measurement in diagnosing mild or moderate pancreatic insufficiency is only 40% to 60%.
The invasive, direct pancreatic function tests are the most sensitive of the tests used to identify patients with mild to moderate chronic pancreatitis. In these tests, pancreatic secretions are continuously aspirated from either the duodenum or the pancreatic duct after administration of a pancreatic stimulant; this stimulant varies among the different tests. In some, secretin is administered to stimulate pancreatic secretion, and bicarbonate in duodenal juice is measured. In others, a combination of secretin and CCK or one of its analogs is used, and bicarbonate and protein (or pancreatic enzymes) in duodenal juice are measured (Chowdhury & Forsmark, 2003). The Lundh test involves stimulating pancreatic secretion by administration of a standardized meal and measuring pancreatic enzymes in the aspirated duodenal juice (Lundh, 1962). Secretin usually is used as the stimulant when pancreatic secretions are collected directly from the cannulated pancreatic duct; these so-called intraductal pancreatic function tests usually involve measurement of bicarbonate (Denyer & Cotton, 1979; Ochi et al, 1997).
Hundreds of reports have described these various invasive, direct pancreatic function tests and their seemingly endless minor variations (Chowdhury & Forsmark, 2003). Most tests that involve aspiration of duodenal juice require the use of multilumen tubes that permit the investigator to aspirate separately and discard gastric secretions to prevent the secretions from diluting or neutralizing the secreted pancreatic juice. Some tests involve measurement of either bicarbonate or enzyme output (amount/time), and in those tests, markers such as polyethylene glycol or distally occluding balloons or both may be needed to ensure high recovery rates. The markers add an additional level of complexity, and for this reason, those tests of output are less commonly employed than tests that simply measure the peak concentration of the secreted element (bicarbonate).
Perhaps the most frequently employed and most reproducible of the invasive, direct pancreatic function tests is the secretin test performed using a double-lumen (Dreiling) tube. Under fluoroscopic control, the tube is passed into the third or fourth portion of the duodenum, while the “discard port” is positioned in the stomach. Passage can be time consuming and difficult for the investigator and the patient, and for this reason, I prefer to pass the tube over a wire that is itself passed through an endoscope. In my experience using this approach, the total time needed to pass and position the tube properly rarely exceeds 10 minutes (Waxman et al, 1996). When the tube is properly positioned, secretin is intravenously administered, and duodenal juice is collected over four consecutive 15-minute periods. Peak bicarbonate concentration is measured in each of the four samples, and a positive test diagnostic of pancreatic insufficiency is defined as one in which the peak bicarbonate concentration does not exceed 60 mEq/L. Borderline values are defined as ranging from 60 to 80 mEq/L; a normal response is defined as one in which at least one of the values exceeds 80 mEq/L. The sensitivity of this secretin test in diagnosing chronic pancreatitis has been reported to be 60% to 100%, but to a great extent, the sensitivity depends on the gold standard being used to confirm the diagnosis of chronic pancreatitis—endoscopic retrograde cholangiopancreatography (ERCP)—and on the case mix of pancreatitis severity.
General Approach to Exocrine Pancreatic Function Testing
Tests of pancreatic function have little or no role in the diagnosis and management of patients with advanced chronic pancreatitis and well-established morphologic changes that can be shown by imaging studies, although in this setting, exocrine function testing may play a role in gauging the adequacy of enzyme therapy. Function tests may be useful in a patient with symptoms suggestive of chronic pancreatitis (i.e., “pancreatic” pain), whose imaging studies show no changes and who have no obvious symptoms of pancreatic maldigestion (clinical steatorrhea). In that setting, an initial noninvasive test might be preferred, and because of its simplicity, measurement of fecal elastase-1 or the bentiromide test may be the most appropriate of those tests. Measurement of fecal fat is aesthetically unpleasant to patients and technicians, and it requires administration of a diet with measured fat content. Because of the poor sensitivity, however, chronic pancreatitis cannot be excluded in patients with normal fecal elastase-1 or bentiromide tests, and in this setting, performance of an invasive pancreatic function test would be appropriate. For this purpose, the secretin test is generally considered to be the gold standard pancreatic function test in the United States. In Europe, many clinicians prefer the secretin-cerulein test or the intraductal secretin test (Boeck et al, 2001). Performance of magnetic resonance cholangiopancreatography (MRCP) or ERCP should be considered in patients with convincing symptoms and a normal or borderline invasive pancreatic function test.
Anagnostides A, et al. Sham feeding and pancreatic secretion: evidence for direct vagal stimulation of enzyme output. Gastroenterology. 1984;87:109-114.
Angelopoulos N, et al. Endocrine pancreatic insufficiency in chronic pancreatitis. Pancreatology. 2005;5:122-131.
Boeck WG, et al. Pancreatic function tests: when to choose, what to use. Curr Gastroenterol Rep. 2001;3:95-100.
Brugge WR, et al. Development of a dual-label Schilling test for pancreatic exocrine function based on the differential absorption of cobalamin bound to intrinsic factor and R protein. Gastroenterology. 1980;78:937-949.
Case RM. Pancreatic exocrine secretion: mechanisms and control. In: Beger, HG, et al. The Pancreas. Berlin: Blackwell; 1998:63-100.
Case RM, Argent BE. Pancreatic duct cell secretion: control and mechanisms of transport. In: Go, VLW, et al. The Pancreas: Biology, Pathobiology, and Disease. 2nd ed. New York: Raven Press; 1993:301-350.
Chen WL, et al. Clinical usefulness of dual-label Schilling test for pancreatic exocrine function. Gastroenterology. 1989;96:1337-1345.
Chowdhury RS, Forsmark CE. Review article: pancreatic function testing. Aliment Pharmacol Ther. 2003;17:733-750.
Creutzfeldt W. The incretin concept today. Diabetologia. 1979;16:75-85.
Dale WE, et al. Role of cholecystokinin in intestinal phase and meal-induced pancreatic secretion. Am J Physiol. 1989;257:G782-G790.
Czako L. Diagnosis of early-stage chronic pancreatitis by secretin-enhanced magnetic resonance cholangiopancreatography. J Gastroenterol. 2007;42(Suppl 17):113-117.
Denyer E, Cotton PB. Pure pancreatic juice studies in normal subjects and patients with chronic pancreatitis. Gut. 1979;20:89-97.
Diem P. Pathogenesis and treatment of diabetes secondary to chronic pancreatitis. In: Buchler, MW, et al. Chronic Pancreatitis: Novel Concepts in Biology and Therapy. Heidelberg: Blackwell; 2002:353-355.
Dominguez-Munoz JE, et al. Fecal elastase test: evaluation of a new noninvasive pancreatic function test. Am J Gastroenterol. 1995;90:1834-1837.
Dominici R, Franzini C. Fecal elastase-1 as a test for pancreatic function: a review. Clin Chem Lab Med. 2002;40:325-332.
Eliasson L, et al. Novel aspects of the molecular mechanisms controlling insulin secretion. J Physiol. 2008;586:3313-3324.
Fiesler P, et al. Physiological augmentation of amino acid–induced insulin secretion by GIP and GLP-1 but not by CCK-8. Am J Physiol. 1995;268:E949-E955.
Fishman MP, Melton DA. Pancreatic lineage analysis using a retroviral vector in embryonic mice demonstrates a common progenitor for endocrine and exocrine cells. Int J Dev Biol. 2002;46:201-207.
Goldberg DM. Proteases in the evaluation of pancreatic function and pancreatic disease. Clin Chim Acta. 2000;291:201-221.
Gu G, et al. Direct lineage tracing reveals the ontogeny of pancreatic cell fates during mouse embryogenesis. Mech Dev. 2003;120:35-43.
Henquin JC. ATP-sensitive K channels may control glucose-induced electrical activity in pancreatic β cells. Biochem Biophys Res Commun. 1988;156:769-775.
Ishiguro H, et al. CFTR functions as a bicarbonate channel in pancreatic duct cells. J Gen Physiol. 2009;133:315-326.
Iwai K, et al. Purification and sequencing of a trypsin-sensitive cholecystokinin-releasing peptide from rat pancreatic juice. J Biol Chem. 1987;262:8956-8959.
Jacobson DG, et al. Trypsin-like immunoreactivity as a test for pancreatic insufficiency. N Engl J Med. 1984;310:1307-1308.
Ji B, et al. Human pancreatic acinar cells lack functional responses to cholecystokinin and gastrin. Gastroenterology. 2001;121:1380-1390.
Katschinski M, et al. Duodenal secretion and fecal excretion of pancreatic elastase-1 in healthy humans and patients with chronic pancreatitis. Pancreas. 1997;15:191-200.
Konturek SJ, et al. Neuroendocrinology of the pancreas: role of brain–gut axis in pancreatic secretion. Eur J Pharmacol. 2003;481:1-14.
Kulkarni RN. The islet β cell. Int J Biochem Cell Biol. 2004;36:365-371.
Larsen S, et al. Metabolic control and β-cell function in patients with insulin-dependent diabetes mellitus secondary to chronic pancreatitis. Metabolism. 1987;36:964-967.
Layer P, et al. The different courses of early and late-onset idiopathic and alcoholic chronic pancreatitis. Gastroenterology. 1994;107:1481-1487.
Lefebvre PJ. Biosynthesis, secretion and action of glucagon. In: Alberti, KGMM, et al. International Textbook of Diabetes Mellitus. Chichester: Wiley; 1992:333-339.
Li Y, Owyang C. Vagal afferent pathway mediates physiological action of cholecystokinin on pancreatic enzyme secretion. J Clin Invest. 1993;982:418-424.
Li Y, et al. High-affinity CCK-A receptors on the vagus nerve mediate CCK-stimulated pancreatic secretion in rats. Am J Physiol. 1997;273:G679-G685.
Lieb JGII, Draganov PV. Pancreatic function testing: here to stay for the 21st century. World J Gastroenterol. 2008;14:3149-3158.
Loser C, et al. Fecal elastase-1: a novel highly sensitive and specific tubeless pancreatic function test. Gut. 1996;39:580-586.
Loser CHR, et al. Comparative clinical evaluation of the 13C-mixed triglyceride breath test as an indirect pancreatic function test. Scand J Gastroenterol. 1998;33:327-334.
Lu L, et al. A cholecystokinin releasing peptide mediates feedback regulation of pancreatic secretion. Am J Physiol. 1989;256:G430-G435.
Lundh G. Pancreatic exocrine function in neoplastic and inflammatory disease: a simple and reliable new test. Gastroenterology. 1962;42:275-280.
Luth S, et al. Fecal elastase-1 determination: “gold standard” of indirect pancreatic function tests? Scand J Gastroenterol. 2001;10:1092-1099.
Madsbad S, et al. C-peptide and proinsulin. In: Alberti, KGMM, et al. International Textbook of Diabetes Mellitus. Chichester: Wiley; 1992:303-332.
Malaisse WJ. Insulin biosynthesis and secretion in vitro. In: Alberti, KGMM, et al. International Textbook of Diabetes Mellitus. Chichester: Wiley; 1992:261-283.
Malka D, et al. Risk factors for diabetes mellitus in chronic pancreatitis. Gastroenterology. 2000;119:1324-1332.
Meglasson MD, Matschinsky FM. Pancreatic islet glucose metabolism and regulation of insulin secretion. Diabetes Metab Rev. 1986;2:163-214.
Misler S, et al. Electrophysiology of stimulus-secretion coupling in human B cells. Diabetes. 1992;41:1221-1228.
Miyasaka K, et al. Feedback regulation by trypsin: evidence for intraluminal CCK-releasing peptide. Am J Physiol. 1989;257:G175-G181.
Mossner J, et al. Treatment of pain with pancreatic extracts in chronic pancreatitis: results of a prospective placebo-controlled multi-center trial. Digestion. 1992;53:54-66.
Murphy JA, et al. Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology. 2008;135:632-641.
Naruse S, et al. Diagnosis of pancreatic disease by a synthetic peptide. In: Masuda, M, editor. Pancreatic Function Diagnosis. New York: Igaku-Shoin; 1980:88-92.
Nauck MA, et al. Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon-like peptide-1-(7-36) amide infused at near physiological insulinotropic hormone and glucose concentrations. J Clin Endocrinol Metab. 1993;76:912-917.
Nauck MA. Physiology and pathophysiology of endocrine pancreatic secretion. In: Beger, HG, et al. The Pancreas. Berlin: Blackwell; 1998:101-137.
Niederau C, Grendell JH. Diagnosis of chronic pancreatitis. Gastroenterology. 1985;88:1973-1995.
Ochi K, et al. Intraductal secretin test is as useful as duodenal secretin test in assessing exocrine pancreatic function. Dig Dis Sci. 1997;42:492-496.
Owyang C, Logsdon CD. New insights into neurohormonal regulation of pancreatic secretion. Gastroenterology. 2004;127:957-969.
Palade G. Intracellular aspects of the process of protein synthesis. Science. 1975;189:347-358.
Persaud SJ, et al. The role of protein kinase C in cholinergic stimulation of insulin secretion from rat islets of Langerhans. Biochem J. 1989;264:753-758.
Polonsky KS, et al. Use of biosynthetic human C-peptide in the measurement of insulin secretion rates in normal volunteers and type 1 diabetic patients. J Clin Invest. 1986;77:98-105.
Porte DJr. The β cell in type 2 diabetes mellitus. Diabetes. 1991;40:166-180.
Powers AC, Eisenbarth GS. Autoimmunity to islet cells in diabetes mellitus. Ann Rev Med. 1995;36:533-544.
Prentki M, Matschinsky FM. Ca2+, cAMP, and phospholipid-derived messengers in coupling mechanisms of insulin secretion. Physiol Rev. 1987;67:1185-1248.
Rhodes RA, et al. Acid-independent release of secretin and cholecystokinin by intraduodenal infusion of fat in humans. Pancreas. 1988;3:391-398.
Riepl RL, et al. Effect of intraduodenal bile and Na-taurocholate on exocrine pancreatic secretion and on plasma levels of secretin, pancreatic polypeptide, and gastrin in man. Scand J Gastroenterol. 1990;25:45-53.
Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55-63.
Rutter GA. Nutrient-secretion coupling in the pancreatic islet β cell: recent advances. Mol Asp Med. 2001;22:247-284.
Sjoberg RJ, Kidd GS. Pancreatic diabetes mellitus. Diabetes Care. 1989;12:715-724.
Schneider AR, et al. Does secretin-stimulated MRCP predict exocrine pancreatic insufficiency? A comparison with noninvasive exocrine pancreatic function tests. J Clin Gastroenterol. 2006;40:851-855.
Soudah HC, et al. Cholecystokinin at physiological levels evokes pancreatic enzyme secretion via a cholinergic pathway. Am J Physiol. 1992;263:G102-G107.
Steiner DF, James DE. Cellular and molecular biology of the beta cell. Diabetologia. 1992;36(Suppl 2):41-48.
Tillil H, et al. Dose-dependent effects of oral and intravenous glucose on insulin secretion and clearance in normal humans. Am J Physiol. 1988;254:E349-E357.
Toskes PP. Bentiromide as a test of exocrine pancreatic function in adult patients with pancreatic exocrine insufficiency: determination of appropriate dose and urinary collection interval. Gastroenterology. 1983;85:365-369.
Toskes PP. Endocrine parameters in the diagnosis of chronic pancreatitis. In: Malfertheiner, P, Ditschuneit, H. Diagnostic Procedures in Pancreatic Disease. Berlin: Springer–Verlag; 1986:259-261.
Toskes PP. Treatment of pain in chronic pancreatitis: inhibition of enzyme secretion. In: Buchler, MW, et al. Chronic Pancreatitis: Novel Concepts in Biology and Therapy. Heidelberg: Blackwell; 2002:389-394.
Unger H. Diabetic hyperglycemia: link to impaired glucose transport in pancreatic β cells. Science. 1991;251:1200-1205.
Unger RH. Glucagon physiology and patholophysiology in the light of new advances. Diabetologia. 1985;28:574-578.
Ventrucci M, et al. Pancreolauryl test for pancreatic exocrine insufficiency. Am J Gastroenterol. 1983;73:86-89.
Waxman I, et al. Endoscopically assisted direct pancreatic function testing: a simplified technique. Gastrointest Endosc. 1996;44:630.
Williams JA. Intracellular signaling mechanisms activated by cholecystokinin regulating synthesis and secretion of digestive enzymes in pancreatic acinar cells. Annu Rev Physiol. 2001;63:77-97.
Williams JA. Receptor biology and intracellular regulatory mechanisms in pancreatic acinar secretion. Curr Opin Gastroenterol. 2002;18:529-535.
Yule DL, Williams JA. Stimulus-secretion coupling in the pancreatic acinus. In: Johnson, LR, editor. Physiology of the Gastrointestinal Tract. 3rd ed. New York: Raven Press; 1994:1447-1472.
Zaret KS, Grompe M. Generation and regeneration of cells of the liver and pancreas. Science. 2008;322:1490-1494.