Chapter 98C Liver transplantation in children and segmental transplantation
Historical Overview
Thomas Starzl attempted the first human liver transplantation in 1963 in a 3-year-old child with biliary atresia. Unfortunately, the child died in the operating room from uncontrollable hemorrhage (Starzl, 1992), but subsequently, in 1967, Starzl successfully performed liver transplantation in eight children. All survived the operation, and half survived more than 1 year (Starzl et al, 1968).
In 2007, there were 5887 adult liver transplantations and 602 pediatric transplantations (Organ Procurement and Transplantation Network/Scientific Registry of Transplant Recipients [OPTN/SRTR], 2008). Currently, approximately 15,500 adults and 540 children are awaiting liver transplantation (United Network for Organ Sharing [UNOS], 2009). Potential recipients who are infants, toddlers, or young children have narrow constraints with regard to graft size. Historically, the lack of size-matched organ availability, coupled with the disproportionate number of adults on the waiting list, has significantly disadvantaged children awaiting transplantation. Compared with adults who have liver disease, children have much less time to await transplantation; an infant with decompensated liver disease from cirrhosis typically cannot survive on a waiting list for several years (McDiarmid, 2002). Furthermore, waiting-list mortality for children younger than 1 year continues to far exceed that of other children and adults (OPTN/SRTR, 2008), in part because of the lack of size-matched organs.
Progressive improvements in surgical technique have evolved such that now the left lateral section and the right trisegment graft each can be used, meaning that a single liver potentially can be divided and transplanted into two recipients. With this innovative technique, the split-liver procedure, important progress in the effort to expand donors available to children has been accomplished without disadvantaging adult recipients. Except in unusual circumstances, the use of a reduced-size liver allograft in which the remnant is discarded is theoretically rarely justified. In 2007, partial or split-liver transplantations accounted for approximately 4% of all deceased-donor liver transplantations (OPTN/SRTR, 2008). The use of reduced-size liver grafts declined from 23% to 16% of pediatric liver transplantations over the last decade, but during this same time, split-liver allografts increased from 9% to 16% (Anderson et al, 2008).
Favorable outcomes transplanting reduced-size and split livers led to the development of techniques for using liver segments from living donors for pediatric grafts. With increasing experience, donations from living donor livers are no longer confined to left lateral sections or left hepatic lobes but have expanded to include full right hepatic lobe grafts, which contributes to the adult organ pool as well (Hong et al, 2008). Today, living-related and unrelated living-donor liver transplantation (LDLT) accounts for approximately 10% of all pediatric liver transplantations and 4% of adult liver transplants (OPTN/SRTR, 2008), and it has been one of the most exciting and challenging advances in liver transplantation.
Indications
Box 98C.1 lists common indications for pediatric liver transplantation. Biliary atresia is a progressive, inflammatory, fibrosing cholangiopathy of unclear pathogenesis (see Chapter 40). It occurs in only 1 in 8000 to 18,000 live births; and although it is rare, it remains the most common cause of infant death owing to hepatic disease and accounts for approximately 50% of all pediatric liver transplantations (Bassett & Murray, 2008; Bezerra, 2005; Perlmutter & Shepherd, 2002). Most children with this disease lack an extrahepatic biliary tree, resulting in impaired bile flow, conjugated hyperbilirubinemia, acholic stools, and hepatomegaly. The hepatic parenchyma becomes congested, and progressive damage leads to secondary biliary cirrhosis. Although portoenterostomy, also known as the Kasai procedure (Kasai & Suzuki, 1959), may yield some clinical improvement and is the first-line treatment (Ohi, 2000; Ryckman et al, 1998), approximately 70% to 80% of children with biliary atresia eventually require liver transplantation (Karrer et al, 1996; Lowell et al, 1996).
Box 98C.1 Indications for Pediatric Liver Transplantation
Other causes of cholestasis in children include idiopathic neonatal hepatitis; infection by both viral and bacterial pathogens, including toxoplasmosis and syphilis; progressive familial intrahepatic cholestasis (Byler disease); metabolic and genetic diseases; familial arteriohepatic dysplasia (Alagille syndrome); choledochal cyst; and ischemia-reperfusion injury. Cholestasis may progress to cirrhosis and has also been associated with parenteral nutrition in children with short bowel syndrome (Baker et al, 1998).
Pediatric liver transplantation is also indicated in cases of noncholestatic liver failure secondary to infectious, metabolic, genetic, and neoplastic etiologies. Although less frequent in children than in adults, postnecrotic liver cirrhosis is the indication for transplantation in approximately 10% of pediatric patients and is most commonly the result of viral hepatitis or idiopathic cryptogenic cirrhosis (Malatack et al, 1983).
Metabolic liver diseases are genetic disorders that lead to the production of aberrant transport proteins or enzymes and altered metabolic pathways. These inborn errors of metabolism, such as α1-antitrypsin (AAT) deficiency or Wilson disease, may cause direct injury to the liver and result in liver failure, with or without injury to other organs; it may also lead to abnormal liver metabolism, including urea cycle disorders or oxalosis, that can lead to injury of other organs. Altogether, such diseases account for approximately 10% of all pediatric liver transplantations (Hansen & Horslen, 2008).
AAT deficiency is a genetic disorder that leads to defective production of the serine protease α1-antitrypsin (see Chapter 70A), an enzyme produced in the liver to protect the lungs from neutrophil elastase. The abnormal protein accumulates in the liver, apparently causing liver damage, although the mechanism of injury remains unclear. Synthetic AAT has some utility in the treatment of AAT deficiency–related lung disease, but it has no role in the treatment of the associated liver disease (Köhnlein & Welte, 2008; Silverman & Sandhaus, 2009).
Wilson disease is an inborn error of metabolism characterized by defective copper excretion and the accumulation of toxic amounts of copper in the liver, basal ganglia, kidney, and cornea, which may lead to the characteristic Kayser-Fleischer rings (see Chapter 70A). The defect is located on chromosome 13, is inherited in an autosomal recessive fashion, and affects 1 in 50,000 births. Initial symptoms are nonspecific and include lethargy, anorexia, vague abdominal pain, and weight loss. Some patients present with asymptomatic hepatomegaly, and others present with fulminant hepatic failure (FHF) (Riordan & Williams, 2001).
CN syndrome is caused by mutations of the gene that codes uridine diphosphate glucuronosyltransferase-1, resulting in unconjugated hyperbilirubinemia. Untreated neonates develop a condition of severe neuronal damage called kernicterus. Homozygous familial hypercholesterolemia (HFH) is a rare disease caused by mutations in the gene that codes the low-density lipoprotein (LDL) receptor. The receptor may be defective or completely absent, and affected individuals have dramatically elevated plasma cholesterol levels, leading to accelerated atherosclerosis, childhood coronary artery disease, and premature death from myocardial infarction (Hansen & Horslen, 2008). Liver transplantation corrects these conditions by providing the required cellular machinery to synthesize the correct gene product or metabolite (Florman & Shneider, 2001; Meyburg & Hoffmann, 2005). Occasionally, auxiliary hepatic transplantation has been performed in these circumstances (Bismuth et al, 1996; Sze et al, 2009; Van Hoek et al, 1999). In AAT deficiency, the potential for development of hepatocellular carcinoma (HCC) precludes auxiliary liver transplantation and mandates total hepatectomy.
Cystic fibrosis (CF) is an autosomal recessive multiorgan disorder that is the most common lethal inherited disease affecting the white population. As management of these patients continues to improve, CF liver disease (CFLD) is increasingly recognized as a significant cause of morbidity and mortality. Approximately 5% to 10% of CF patients will progress to cirrhosis, with the most frequently recognized complications attributable to portal hypertension with an associated nutritional and pulmonary decline. A significant number of patients with CFLD may be helped with an isolated liver transplantation or a concomitant liver and lung transplantation; however, the debate is ongoing in the literature regarding the timing of liver transplantation in these patients (Colombo et al, 2006; Fridell et al, 2003; Nash et al, 2008).
Although more common in adult patients, chronic Budd-Chiari syndrome with severe hepatic congestion and focal areas of liver fibrosis or cirrhosis may be an indication for liver transplantation in older children (see Chapter 77). Pretransplantation evaluation should be performed to recognize predisposing factors or underlying diseases such as myeloproliferative disorders, primary hepatic protein deficiencies—of proteins C and S, antithrombin III, and activated protein C resistance—or secondary protein deficiencies, such as increased intestinal protein loss in inflammatory bowel disease (Slakey et al, 2001).
FHF (see Chapters 73 and 97C) is associated with a mortality rate greater than 70%. It is essential to transfer children with FHF to a transplant center immediately for management and urgent evaluation and listing for liver transplantation. Because cerebral edema develops rapidly, careful monitoring and intensive supportive care are required. Mild elevation of intracranial pressure (ICP) may be managed acutely by hyperventilation and elevation of the patient’s head, although maintenance likely requires strict sodium and osmolar and intravascular volume control. Placement of an ICP monitor should be considered, but regardless, attention to maintenance of cerebral perfusion pressure (CPP) is paramount. Transplantation in patients with signs of cerebral edema is urgent, as edema may lead to irreversible brain damage or death (Tanaka et al, 1994). Poor prognostic factors for liver transplantation in children with FHF include age younger than 10 years, liver disease other than viral hepatitis, grade 2 or 3 hepatic encephalopathy, coagulopathy (prothrombin time >30 seconds), and increasing jaundice (bilirubin >9 mg/dL) (Uemoto et al, 2000).
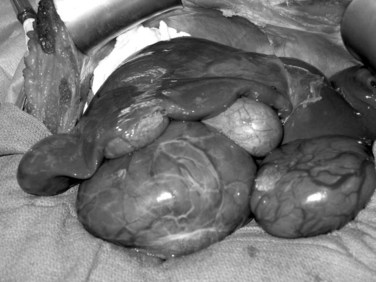
Liver tumors account for less than 3% of all indications for pediatric liver transplantation (Austin et al, 2006). Hepatoblastoma, HCC, and fibrolamellar HCC represent the most frequent tumors (see Chapter 82). In contrast to adults, the predisposing factors for malignant liver tumors in children are not viral hepatitis or alcoholism but are more often metabolic disorders, such as AAT deficiency, tyrosinemia, or glycogen storage disease. In patients without cirrhosis, hepatic resection is the treatment of choice. Liver transplantation should be considered in the case of unresectable tumors confined to the liver (Chen et al, 2006; Otte et al, 2004). In patients with advanced cirrhosis and HCC meeting Milan criteria, transplantation is indicated (Austin et al, 2006; Beaunoyer et al, 2007; Perilongo et al, 2004). Rarely, giant arteriovenous malformations and benign liver tumors—when they replace the whole liver, or when they have the potential for malignancy (e.g., hepatic adenomatosis or multiple adenomas in glycogen storage disease [Fig. 98C.1])—may be indications for total hepatectomy and liver transplant (Malatack et al, 1987; Wellen et al, 2009; see Chapter 79A).
Evaluation of the Potential Recipient
A multidisciplinary evaluation of a child with decompensated liver disease should be completed with the involvement of surgeons, hepatologists, nurses, anesthesiologists, psychologists, and social workers. Early referral to a transplant center allows maximum time to develop a management strategy and to optimize pretransplantation clinical status. The timing of liver transplantation is crucial, because late referral of patients with significant hepatic complications and malnutrition results in a poorer outcome. The expected waiting time on the transplant list may vary based on the geographic region, and this should be taken into account. In infants younger than 2 years who weigh less than 10 kg, it is often difficult to maintain metabolic and nutritional support. Ideally, children should be considered for liver transplantation before the complications of malnutrition, such as weight loss and growth failure, occur (Kimura et al, 2004; McDiarmid et al, 2004). With chronic liver disease, prolonged clotting times, intractable ascites, recurrent variceal bleeding secondary to portal hypertension, and recurrent cholangitis with severe cholestasis are indications for liver transplantation (Hendrickson et al, 2004).
The prognosis after liver transplantation for chronic liver disease is influenced by preoperative comorbidities. The advent of the Pediatric End-Stage Liver Disease (PELD) and Model for End-Stage Liver Disease (MELD) scores (Malinchoc et al, 2000; Wiesner et al, 2001) has made it possible to better predict mortality and progression of liver disease while awaiting transplantation (Desai et al, 2004; Barshes, et al, 2006). The pertinent factors in the PELD score are the international normalized ratio (INR), total bilirubin, serum albumin, age younger than 1 year, and weight or length less than 2 standard deviations (SDs) from the mean for age and gender. The PELD score is used for patients younger than 12 years, and the scores range from a negative value (−10) to 50. Adolescents aged 13 to 18 years are allocated organs based on the MELD score, similar to adults. Box 98C.2 presents the formulas for calculating the MELD and PELD scores.
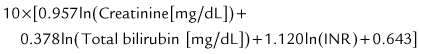
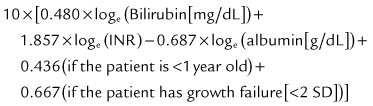
Comparing the periods before and after the introduction of the MELD and PELD scores, a similar percentage of children are being transplanted, but mortality rates have decreased (Freeman et al, 2004; Yao et al, 2004). Small size and young age are no longer contraindications, and many transplantations for biliary atresia are now done in infants who weigh less than 10 kg (Colombani et al, 1996; Sokal et al, 1990). Whereas posttransplantation mortality has improved, waiting-list mortality for infants younger than 1 year remains high, unfortunately (OPTN/SRTR, 2008). Consideration should be given to referring these young and small infants to pediatric specialty centers that have expertise with these challenging cases. Contraindications to pediatric hepatic transplantation are listed in Box 98C.3.
Recipient Hepatectomy
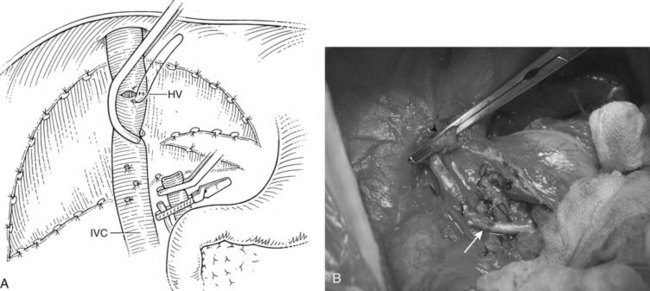
Before beginning the transplantation, a Broviac central venous catheter is placed for intraoperative and postoperative fluid resuscitation, monitoring, and postoperative laboratory draws, the same as an arterial catheter. Pressure points are well padded, and convective heating blankets are used. The abdomen is entered through a curved transverse subcostal incision. Meticulous hemostasis must be maintained throughout the procedure, because ongoing bleeding may lead to dilutional coagulopathy and fibrinolysis. Abdominal wall venous collaterals must be carefully controlled as they are encountered, and adhesions to the liver are carefully dissected to allow access to the portal region (Fig. 98C.2).
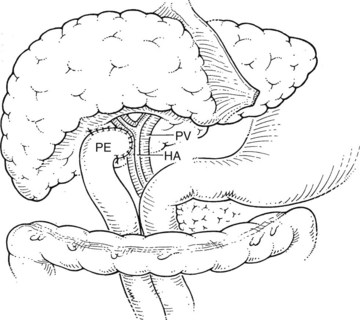
For orthotopic whole-liver transplantation, the small retrohepatic caval segment can be removed with the native liver, or the vena cava can be preserved and the donor liver anastomosed in a piggyback fashion to the confluence of the recipient’s right, middle, and left hepatic veins. With a segmental graft, the recipient inferior vena cava must be preserved. A vascular clamp is placed across the hepatic veins so that they can be joined to form a common opening to sew to the donor suprahepatic cava or, in case of segmental grafts, the left hepatic vein (Fig. 98C.3). Complete hemostasis of the retroperitoneum should be accomplished before removing the donor liver from cold storage.
Graft Procurement and Engraftment
The techniques used for recovering whole, deceased-donor livers for transplantation from children are similar to the techniques used in adult-adult transplantation. Special attention should be paid to donor serum sodium levels and to dynamic liver function tests. High sodium levels (>170 mmol/L) may be associated with severe reperfusion injury and poor graft function (Totsuka et al, 1999).
The arterial anastomosis is performed with sutures of fine (7-0 or 8-0) monofilament material using microsurgical loupes. A microscope may be useful in anastomosing arteries with a diameter smaller than 1 to 2 mm. The anastomosis is performed most commonly in an end-to-end fashion directly to the native hepatic artery. If the native artery is not of adequate size, or if there is an intimal dissection, an extension allograft using saphenous vein or donor iliac artery from the infrarenal or supraceliac aorta may be used (Yamaoka, 1996).
Reduced-Size Segmental Liver Transplantation and Split-Liver Transplantation
Reduced-Size Segmental Liver Transplantation
The use of reduced-size grafts for pediatric recipients is one of the most important advances in liver transplantation. The disproportionate shortage of grafts for very young children, compared with older children, motivated surgeons to develop innovative operative techniques to overcome the restrictions imposed by donor and recipient size mismatch. With these techniques, livers can be reduced to a functional unit of appropriate size for the recipient. The optimal ratio between the required volume of liver and the actual “tailored” hepatic mass is between 1 : 1 and 1 : 2. In this way, livers from donors 10 or 20 times the size of the recipient can be used (Broelsch et al, 1988).
The liver comprises eight anatomically defined segments, each with its own arterial and portal blood inflow and separate biliary drainage (Strasberg, 1997; see Chapter 1B). Similarly, segmental hepatic venous outflow is independent, but major liver veins run along sectional borderlines, receiving blood from several adjacent segments (Couinaud, 1954). For purposes of transplantation, the liver can be divided into several functional grafts. The most commonly used reduced-size graft is the left lateral section graft, composed of segments II and III (Fig. 98C.4; Botero & Strasberg, 1998). To preserve a single donor portal vein, the liver is transected just to the right of the falciform ligament, and branches of the ascending (umbilical) portion of the left portal vein to segment IV are carefully suture ligated. The left hepatic vein is used for hepatic venous drainage. It is anastomosed directly to the recipient’s vena cava, which must be preserved during hepatectomy. The full left graft (segments I, II, III, and IV or segments II, III, and IV) is used in small adults or older children who require a graft slightly larger than the left lateral section.
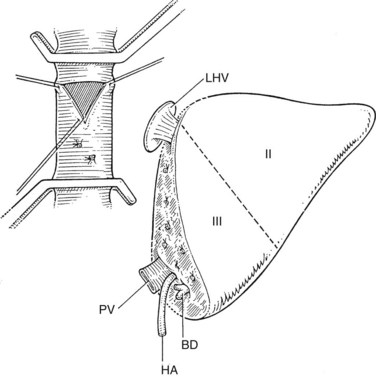
Bismuth and Houssin (1984) first reported success with transplantation of a left hepatic allograft; in the same year, Broelsch and colleagues (1984) reported the successful grafting of the left lateral section. Since then, most pediatric transplant centers have adopted segmental transplant techniques using reduced-size livers. The results after these segmental grafts are comparable to the results achieved with whole-organ orthotopic liver transplants (Hong et al, 2008). Although reduced-size grafts can overcome the donor-recipient size discrepancy by distributing cadaveric livers to the advantage of children, and to small adults, they do not increase the absolute number of grafts. The advent of the split-liver technique not only allowed the donor liver to be tailored to fit a small child, but it also allowed for the remaining donor graft to be used, which increased the actual organ pool; however, routine use of reduced-size grafts is discouraged except in unique situations.
Split-Liver Transplantation
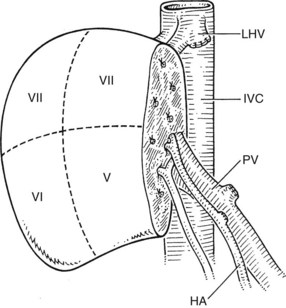
In the split-liver procedure, a single cadaveric liver is divided into two grafts to be transplanted into two patients (Cintorino et al, 2006; Strasberg et al, 1999; Zamir et al, 2002). The first successful split-liver transplantation was performed by Pichlmayr and colleagues in 1988, utilizing a right extended graft (segments IV, V, VI, VII, and VIII) and a left lateral graft (segments II and III). Leaving segment IV with the right graft rarely leads to an impaired arterial and portal venous perfusion, despite the fact that the vascular supply of this segment belongs to the left liver. Very rarely, resection of segment IV may be required because of poor perfusion. The liver also may be dissected along the principal plane, dividing it into right and left livers. The vena cava and the full length of main portal vein and celiac artery typically are included with the right graft (segments V through VIII), which is then anastomosed to the recipient in a manner similar to a whole-liver graft (Fig. 98C.5). By contrast, the left lobe has a left hepatic vein, left hepatic artery, and left portal vein, which have been divided at the branching points from the vena cava, common hepatic artery, and portal vein.
Broelsch and coworkers (1990) reported the first successful split-liver transplantation series. Since then, other groups have developed and refined the technique (Azoulay et al, 2001a, 2001b; Busuttil & Goss, 1999; Rela et al, 1998); however, the initial enthusiasm for this procedure faded, because the preliminary results were inferior, mainly owing to technical and organ-preservation problems. In addition, progress in split-liver transplantation was hampered by tremendous practical and logistical problems, because this procedure requires two full teams of surgeons, anesthesiologists, and operating room staff to be available almost simultaneously. The initial reported patient survival at 1 year was 67%, and graft survival was reduced to 55% (Broelsch et al, 1990). The most frequent complications during the initial experience with this procedure were bleeding, primary poor function, and biliary fistulae and strictures. In adult recipients of right-lobe grafts, liver insufficiency became a major problem because of long ischemic times and borderline size matching.
With surgical refinements of liver splitting and transplantation techniques and better patient selection, results improved (Goss et al, 1997; Malago et al, 1997; Yersiz et al, 2006). The introduction of the in situ split-liver technique, developed by Rogiers in 1994, significantly reduced ischemic time of the liver (Rogiers et al, 1996). Using this technique, the liver is divided in situ in the heart-beating deceased donor before cold perfusion starts (Yersiz et al, 2003). While arterial and portal blood supply and venous drainage are preserved, the liver is divided by careful transection of the hepatic parenchyma, most commonly by separating the future left lateral section allograft from the future right trisegment graft. The splitting procedure requires meticulous hemostasis, which also affects the recipient operation, because little or no bleeding occurs at the cut surface of the graft after reperfusion.
Although the split-liver technique remains a procedure confined to large transplant centers that have experience in segmental grafting, the possibility of transferring a partial graft to another center has now become reality. Today, split-liver transplantation has gained wide acceptance, with results comparable to those of whole-organ transplantation (Deshpande et al, 2002; Diamond et al, 2007; Hong et al, 2009); now a standard procedure, it represents an important technical advance in expanding the total number of transplantable organs. Together with living-related liver transplantation, the split-liver technique has had a substantial impact on the inequities of the pediatric donor organ shortage.
Living Donor Transplantation
The success of many centers performing reduced-size and split-liver grafting provided a strong surgical basis for using segments from living donors (Strong et al, 1990; see Chapter 98B), particularly in the treatment of small children with ESLD (Broelsch et al, 1994; Edmond et al, 1993; Otte, 1995; Otte et al, 1998; Raia et al, 1989). Donors are most commonly the parents or other close relatives of the recipient, but nonrelated living donor liver transplantation (LDLT) procedures also are performed (OPTN/SRTR, 2008).
For ethical reasons, adequate and thoughtful selection of donors is a major prerequisite of LDLT, and most centers have adopted stringent donor evaluation protocols (Chen et al, 2003; Surman & Hurtl, 2003). The segmental liver donation must be deemed by the transplant team to be a purely voluntary, altruistic act with no motive of secondary gain, and a complete psychologic and social evaluation of the donor is performed. Potential donors and their families are extensively counseled, and they are evaluated by an independent physician, who acts as the donor advocate and explains the risks of the procedure in detail (Trotter, 2000). Any objections regarding the psychosocial background of the potential donor result in withdrawal from consideration for organ donation (Singer et al, 1989; Surman, 2002; Ghobrial et al, 2008).
Of paramount concern, however, is the medical risk to healthy donors; everything possible must be done to minimize risks of donor morbidity and mortality. As much as possible, the donor must be fully apprised of all the risks, possible complications, and potential sequelae; with the donor so informed, they must accept these possibilities before undertaking the procedure (see Donor Risk in Living Donor Liver Transplantation).
The success of LDLT depends partly on adequate graft volume (Kiuchi et al, 1999). The required graft volume should be determined individually and can be assessed with the use of computed tomography (CT) or magnetic resonance imaging (MRI) (Kamel et al, 2000, 2001). The left lateral section provides the most straightforward dissection, but full left- and right-lobe grafts have been used successfully. The size of the living donor’s left lateral section is assessed by volumetric CT or MRI (Urata et al, 1995), the hepatic vasculature is assessed by CT angiography or MRI angiography, and the biliary anatomy is assessed by MR cholangiography or endoscopic retrograde cholangiopancreatography (ERCP). Rarely, percutaneous liver biopsy is done, but only if imaging suggests hepatic steatosis in the donor. The type and size of graft should be decided on an individual basis. In general, a full left-lobe liver graft (segments II through IV) can overcome a disparity between recipient and donor size of about 1 : 4. With a left lateral segment graft, a disparity of 1 : 10 or more can be managed. In infants weighing less than 10 kg, left lateral segment grafts are used almost exclusively.
Procedure
The donor liver is approached in the usual manner, through a standard bilateral subcostal incision or a right subcostal incision with upper midline extension. Recently, laparoscopic-assisted or hybrid laparoscopic and open techniques have been described (Baker et al, 2009; see Chapter 98B). Regardless of method, the surgical procedure begins by identifying the structures in the hilum. It is imperative to dissect close to the falciform ligament to avoid injuries to the hilar structures running to the right lobe. The left hepatic artery is isolated and followed to the liver parenchyma, and the left portal vein is dissected free and encircled with a vessel loop. The dissection continues by dividing portal and arterial branches to the caudate, which increases the length of the left hepatic artery and left portal vein. The portal branches to segment IV are ligated, and the left hepatic vein is identified above the liver and encircled with a vessel loop as it enters the vena cava. The hepatic parenchyma is transected just to the right of the falciform ligament, with suture ligation of all vessels and ducts, until the left lateral section is completely separated from the rest of the liver; the segment is still perfused with arterial and portal blood and has drainage via the left hepatic vein.
The parenchymal transection is performed without any vascular control of the graft or the remaining liver to maintain maximal viability. Hepatic transection continues until the hilar plate is reached. At this point, the section II and III bile duct is divided sharply, and its distal end is oversewn with fine, absorbable monofilament suture. With the plane of transection 0.5 to 1.0 cm to the right of the umbilical fissure, approximately 90% of left lateral section grafts will be obtained with a single section II and III bile duct (Renz et al, 2000).
As discussed previously, during the recipient hepatectomy, the vena cava is left in situ, and a temporary portacaval shunt may be used. The recipient hepatic vein–vena cava junction is modified to accommodate the anastomosis with the donor hepatic vein. A generous triangular venous anastomosis to ensure adequate drainage of the graft is recommended; the donor hepatic venous cuff should be kept short to provide an anastomosis that is not susceptible to kinking. Portal vein reconstruction is done in an end-to-end fashion. In children with biliary atresia, the recipient’s portal vein is often small and may be phlebosclerotic. In these cases, an anastomosis to the confluence of the superior mesenteric and splenic veins may be preferred (Saad et al, 1998). Hepatic artery reconstruction usually can be achieved by an end-to-end anastomosis to the recipient’s proper hepatic artery via a microsurgical technique using surgical loupes or a microscope (Inomata et al, 2000). In some cases, a venous interposition graft from the recipient’s infrarenal aorta, using donor saphenous vein, is needed to achieve a tension-free arterial anastomosis.
Care must be taken to identify correctly the bile ducts from segments II and III (Fig. 98C.6); such ducts are not obtained with a common trunk up to 10% of the time (Renz et al, 2000; Salvalaggio et al, 2005). The biliary drainage is fashioned using a Roux-en-Y loop of the jejunum with or without a stent. After completion of all anastomoses, the liver rotates slightly to the right and occupies a more central position than it does in the donor (Fig. 98C.7). This slight shift must be taken into account when performing the vascular and biliary anastomoses. The graft should be resuspended to the anterior abdominal wall by the falciform ligament to prevent further right shift and vascular torsion.
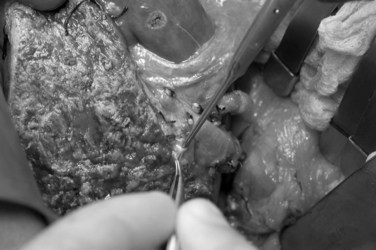
FIGURE 98C.6 Common trunk formed by segments II and III bile ducts from a left lateral section graft.
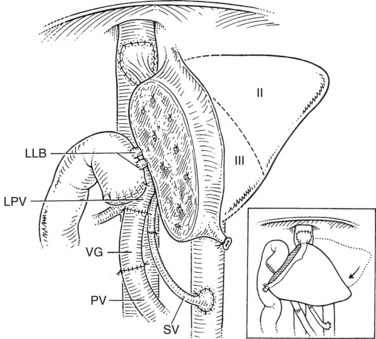
Donation and transplantation of a living donor full left liver lobe (segments II, III, and IV with or without segment I) is similar to that of a left lateral section graft, with a few technical variations. After cholecystectomy, the resection follows the principal plane. The venous outflow of the graft can be provided by the left hepatic vein or preferentially by use of a common patch of the left and middle hepatic vein (Makuuchi & Sugawara, 2003). Devascularization of the main left hepatic bile duct is less of a concern when the caudate is preserved with the allograft.
Although the primary goal of LDLT is the relief of organ shortage, numerous additional benefits are associated with this procedure (Sindhi et al, 1999). The most important advantage for the recipient of a living-donor transplant is a much shorter waiting period leading up to transplantation (Tanaka et al, 1994). Timing of the procedure can be planned, and the operation can be performed under elective conditions, which results in decreased operative risk. Careful donor evaluation and a short cold ischemic time offer uniformly good grafts and can minimize the risk for initial nonfunction (Farmer et al, 2001).
Adults
After the encouraging results of living donor left lateral section transplantation in children, this innovative technique was extended to try to solve another problem: the shortage of donor organs for adults (Renz et al, 2003; Strasberg et al, 1999). Especially in countries where cadaveric donor organ transplantation has not been established, either by law or for religious or cultural reasons, living-donor organ donation offers the only chance of liver transplantation for patients with ESLD. In 1991, the first LDLT to an adult was performed in Kyoto by Tanaka, using a left liver graft (Hashikura et al, 1994). This technique has been expanded to the use of full right hepatic lobe grafts, which are now the preferred grafts in adult-adult living donor transplants (Kawasaki et al, 1998; Seek et al, 2002; Brown, 2008).
The right lobe graft (segments V through VIII) comprises approximately 50% to 60% of total liver volume and presents the advantage of fitting exactly in the right hepatic fossa (Fig. 98C.8). Problems with this graft include a large cut parenchymal surface and an irregular biliary, arterial, portal, and hepatic venous anatomy (Trotter et al, 2002). Variations of portal vein anatomy, such as a trifurcation, and variations in arterial anatomy, such as an accessory right hepatic artery, are relative contraindications to living donation of segments V through VIII (Erbay et al, 2003; Imamura et al, 2000). Most often, the right hepatic artery has a large diameter, unless there is an additional right artery arising from the superior mesenteric artery. The hepatic artery and portal vein usually can be anastomosed directly to the recipient’s vessels. In most cases, venous drainage of the right graft is achieved exclusively via the right hepatic vein, and a careful anastomosis of this vessel with the vena cava or the orifice of the recipient’s right hepatic vein must be performed to prevent compromised venous outflow and congestion of the graft. If necessary, the orifice should be enlarged longitudinally to provide an exact anastomosis.
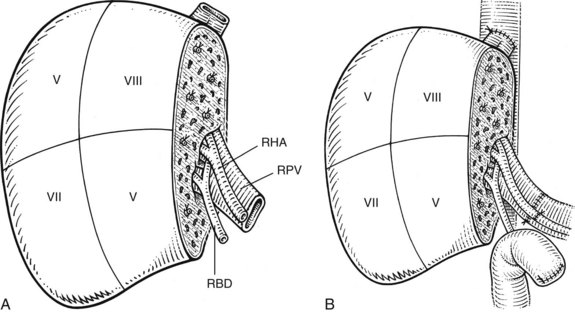
In the case of a second right hepatic vein, such as an inferior right hepatic vein, two independent anastomoses are recommended for uncompromised venous drainage (Ghobrial et al, 2001). The inclusion of the donor middle hepatic vein with the graft is controversial (Fan et al, 2003). Biliary reconstruction can be challenging, because bile ducts from segments V through VIII may not form a common trunk, and two or more ducts may be present (Fan et al, 2002). Reconstruction with a Roux-en-Y hepaticojejunostomy is standard. A direct biliary anastomosis to the recipient’s bile duct using a T-tube may be considered when a large, single right hepatic bile duct is present.
One dilemma of LDLT in adults centers on the maximum extent of liver resection that can be safely tolerated by the donor and the minimum amount of hepatic mass necessary for the recipient to survive (Fan et al, 2000). The definition of small-for-size grafts is vague, because the minimal amount of liver compatible with life is unknown, and variables such as ischemia-reperfusion injury, steatosis, or rejection may reduce further the initially transplanted liver mass (Hill et al, 2009). Many reports have indicated that grafts corresponding to no more than 40% of the recipient’s standard volume may not be expected to function well (Pomfret, 2003). At present, a graft/body weight ratio of 0.8% or a graft volume greater than 50% of the standard liver volume are suggested values for successful grafting (Kiuchi et al, 1999; Sugawara et al, 2001). The use of an extended right liver with the middle vein retained with the graft lowers the risk of liver insufficiency in the recipient but is associated with greater donor morbidity and possible mortality. Because this procedure requires a major hepatectomy with the donation of about two thirds of total liver volume, transient impairment of donor liver function is common (Lo et al, 1999; Pomfret et al, 2003).
Prolonged cholestasis in the absence of rejection or mechanical bile duct obstruction is a hallmark of small-for-size grafting. In such patients, the perioperative management is focused on avoiding any further damage to the graft. Unless retransplantation is considered, optimal supportive care is currently the only therapy for small-for-size liver transplantation. Temporary liver support systems, such as artificial liver devices or extracorporeal perfusion, are under investigation, but their clinical use is not yet available in the United States (Demetriou et al, 2004; see Chapters 73 and 97C). In the future, it may be desirable to combine temporary liver support systems with small-for-size organs. This could be one option to improve organ shortage in adults by an extended use of LDLT.
Although there are several advantages for the recipient of an LDLT, an adult-adult LDLT subjects the donor, a healthy individual, to all of the risks of a major abdominal operation. Risk minimization begins with careful selection of appropriate candidates during the donor evaluation; however, despite careful donor and patient selection, LDLT still has significant risks for the donor, as well as potential added complications to the recipient, compared with a full-sized cadaveric donor graft (Man et al, 2003; Sugawara et al, 2003).
Donor and Recipient Risk in Living Donor Liver Transplantation
Early enthusiasm for adult-adult LDLT has been tempered by growing concern for donor morbidity and mortality. Following one high-profile donor death reported in 2002 (Trotter et al, 2006), the annual number of LDLTs in the United States declined steadily, from a high of 522 in 2001 to only 266 in 2007 (OPTN/SRTR, 2008). Quantifying the risk associated with LDLT has been difficult, because no formal international registry exists to prospectively collect data on donors worldwide. UNOS has only recently mandated reporting of outcomes for all LDLT donors who are taken to the operating room for a donor hepatectomy, but this data set is not yet mature enough to be informative (Trotter et al, 2006). Most published reports of complications associated with adult-adult LDLT have come from single-center experience, and reports vary widely, with complication rates as low as 9% and as high as 67% (Ghobrial et al, 2008; Marsh et al, 2009). Estimates of donor mortality rates range from 0.1% for left lateral section grafts to 0.5% for right lobe grafts (Ringe & Strong, 2008).
In an effort to provide an unbiased assessment of the risks and severity of complications for both donors and recipients of LDLT, the nine-center, NIH-funded Adult-to-Adult Living Donor Liver Transplantation (A2ALL) cohort study was initiated in 2002 (Ghobrial et al, 2008). The first phase of the A2ALL study utilized a retrospective, observational design and included 405 potential donors accepted for LDLT from 1998 through 2003. Among this cohort, 393 underwent donation, 12 procedures were aborted (3%), and 148 donors (38%) experienced a total of 220 complications. The most common postoperative complication was bacterial infection (12.5%), followed by biliary leak (9.2%), incisional hernia (6%), pleural effusion requiring drainage (5%), neuropraxia (4%), repeat laparotomy (3%), wound infection (3%), and intraabdominal abscess (2%). One patient died from gastric gas gangrene, sepsis, and multiorgan failure on the third postoperative day; another died from a drug overdose, and a third died from suicide, more than a year after operation; and a fourth donor died in a nonsuicide pedestrian-train accident 2 years after operation (Ghobrial et al, 2008; Trotter et al, 2006).
In addition to examining donor morbidity, the A2ALL cohort study examined complications to recipients of adult-adult LDLT relative to recipients of deceased-donor liver transplantation (DDLT). Overall, the complication rate after LDLT was not significantly higher than after DDLT (82.8% vs. 78.2%; P = .17), but the severity of complications was greater. Among a cohort of 384 LDLTs and 216 DDLTs, the complications that occurred more frequently in recipients of LDLT included bile leak (31.8% vs. 10.2%), unplanned laparotomy (26.2% vs. 17.1), hepatic artery thrombosis (6.5% vs. 2.3%), and portal vein thrombosis (2.9% vs. 0%). Complications leading to retransplantation or death with or without retransplantation also occurred more often after LDLT than after DDLT (15.9% vs. 9.3%); however, in a separate analysis that factored in center experience, the authors found a significant decline in LDLT complications after a center had performed at least 20 procedures (Freise et al, 2008).
Postoperative Management and Complications after Pediatric Liver Transplantation (See Chapter 100)
Primary nonfunction of the graft occurs in 2% of patients and requires immediate retransplantation (Doyle et al, 1996). With regard to pediatric recipients, retransplantation with a reduced size graft from a cadaveric donor or a living donor may offer emergency options (Seiders et al, 2002). The most frequent complication after liver transplantation is hemorrhage, which may occur during the transplant procedure or in the immediate perioperative period. If a child becomes unstable or hypotensive in the postoperative course, it is prudent to reexplore immediately. The use of the protease inhibitor aprotinin (Trasylol) and an antifibrinolytic agent such as aminocaproic acid (Amicar) may help reduce diffuse bleeding, but the debate continues concerning potential increased risk of arterial thrombosis after their use (Himmelreich et al, 1992; Kang et al, 1987; Xia & Steadman, 2005).
Hepatic artery thrombosis (HAT) is the most common vascular complication in pediatric OLT and is a major cause of early graft failure and retransplantation. Previously published rates of HAT were over 25% (Dalgic et al, 2003; Heffron et al, 2003), but more recent large series report rates ranging from 5% to 19% (D’Alessandro et al, 2007; Diamond et al, 2007; Duffy et al, 2009; Farmer et al, 2007; Salvalaggio et al, 2005; Yilmaz et al, 2007). Decreases in the incidence of HAT are attributed to improved surgical techniques. Careful anticoagulation and rheologic management and avoidance of rejection also may diminish the risk of HAT. Doppler ultrasound, used routinely or with a low threshold based on clinical indications, is essential for early diagnosis and possible thrombectomy and revascularization. Portal vein thrombosis occurs less frequently than HAT but is estimated to occur in 3% to 8% of all pediatric OLTs, and incidence increases with segmental grafts (Chardot et al, 1997; Diamond et al, 2007; Millis et al, 1996).
Similar to HAT, graft rejection or preservative injury with swelling of the liver increases the risks for portal vein thrombosis. Large-for-size transplantation is another risk factor for thrombosis (Kiuchi et al, 1999), if elevated intraabdominal pressure is not lowered using a patch abdominoplasty. Early portal vein thrombosis rarely may be treated successfully by emergency thrombectomy and revascularization. Late portal vein thrombosis with complications of portal hypertension may be an indication for retransplantation (Dalgic et al, 2003; Vilca-Melendez & Heaton, 2004).
Biliary strictures are the most common complication of segmental OLT (10% to 35%) (Bhatnagar et al, 1995; Reichert et al, 1998). In partial grafts, the biliary complication rate can be as high as 40% (Anderson et al, 2010; Diamond et al, 2007; Salvalaggio et al, 2005). It is important to recognize biliary complications early and to treat them aggressively, because biliary complications account for approximately 8% of late graft loss in pediatric OLTs. With widespread use of percutaneous radiologic intervention, such as transhepatic balloon dilation and stenting, the need for surgical reintervention and reconstruction has decreased (Heffron et al, 2003; Ko et al, 2008; Moreira et al, 2010).
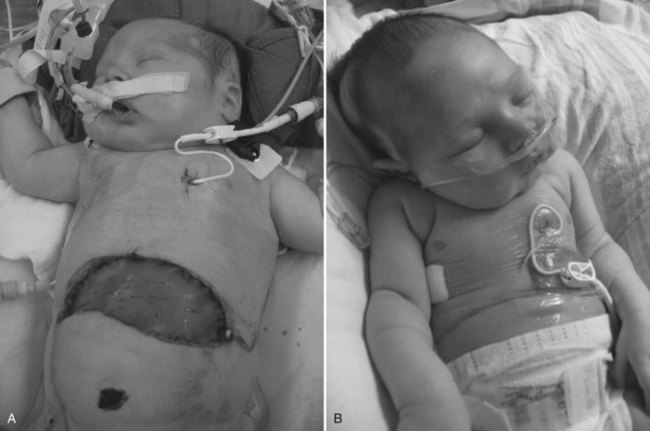
Many children who undergo liver transplantation have had a prior portoenterostomy and are at risk for iatrogenic bowel injury as a result of the adhesiolysis during native hepatectomy. A high index of suspicion and early repeat laparotomy and primary closure of any perforation are crucial to prevent systemic complications of sepsis. Many children have compromised pulmonary function before the transplant procedure because of significant arteriovenous pulmonary shunting, pulmonary infections, or pleural effusions. Postoperatively, respiratory problems may be worsened as a result of diaphragmatic splinting or temporary right hemidiaphragm paralysis, pulmonary edema secondary to perioperative fluid shifting, or atelectasis. In children with high intraabdominal pressure secondary to a relatively large liver, a tension-free temporary closure of the abdomen with a prosthetic graft is advisable (Fig. 98C.9). This abdominal wall graft may be reduced in size over several days, similar to when an abdominal wall silo is used in newborns with gastroschisis (Cacciarelli et al, 1997; Noble-Jamieson & Barnes, 1999).
Peritransplant impairment of renal function is common but is usually reversible. Reduction or temporary cessation of nephrotoxic immunosuppressive medications (e.g., cyclosporine or tacrolimus) may help to improve renal function. Less than 5% of children need temporary dialysis after transplantation, unless this was already a pretransplantation requirement (Bartosh et al, 1997; Berg et al, 2001).
Septic complications and infections are lifelong risks in an immunocompromised patient. In the initial postoperative course, bacterial infections with Enterococcus faecalis, Pseudomonas species, and Staphylococcus aureus are most common, often related to central venous lines or bacterial translocation from the gut (Deen & Blumberg, 1993; Fishman, 1999). Viral infections, which usually occur in children immunologically naive to cytomegalovirus (CMV) or Epstein-Barr virus (EBV) before transplantation, typically manifest about 6 weeks after transplantation and may be effectively prevented or treated by ganciclovir. Fungal infections may occur frequently in patients seen with acute hepatic failure, and the use of prophylactic oral antifungal medication is recommended (Gladdy et al, 1999; Kahn et al, 1988).
Immunosuppression
Much of the progress in pediatric liver transplantation is attributable to improvements in immunosuppression (Evrard et al, 2004; see Chapter 97B), and treatment regimens are constantly in development, even as new drugs become available. Children generally have a higher incidence of rejection and require higher levels of immunosuppressive agents compared with adults. Baseline immunosuppression consists of a corticosteroid and a calcineurin inhibitor (cyclosporine or tacrolimus; van Buren et al, 1998). Tacrolimus has the advantage of requiring lower steroid dosage than cyclosporine for the same level of immunosuppression and less risk of hirsutism and gingival hyperplasia. Mycophenolate commonly is used to minimize the nephrotoxic effects of cyclosporine and tacrolimus. The initial 12-hour trough levels should be about 250 to 300 ng/mL for cyclosporine and about 10 ng/mL for tacrolimus.
In the early postoperative period, levels of immunosuppressive drug should be maintained at a higher level to avoid rejection periods; elevated drug levels can result in severe graft edema with subsequent arterial or portal vein thrombosis, so drug levels are tapered over time (Mazariegos et al, 1997), and most children can be weaned off the steroid component within 1 year. Recent studies have shown excellent graft survival in pediatric OLT patients maintained on tacrolimus (Prograf) monotherapy. Additional benefits of reduced immunosuppression include decreased infection rates and improvements in renal function and growth (Turmelle et al, 2009) with reports of complete withdrawal of immunosuppressants in children several years after transplantation (Diem et al, 2003; Reding et al, 2003; Riordan & Williams, 1999).
Results of Pediatric Liver Transplantation
The application of split-liver grafts and LDLT has led to significant relief of the organ shortage in pediatric liver transplantation, particularly among small infants with ESLD (Hong et al, 2009; Sindhi et al, 1999). In the decade from January 1998 through December 2007 in the United States, 5762 pediatric liver transplantations were performed. LDLT accounted for approximately 14% of these transplantations (OPTN/SRTR, 2008). The overall survival rates after liver transplantation in children are good, and most centers report 1-year rates greater than 80% (OPTN/SRTR, 2008). In the United States, 5- and 10-year actuarial survival of children who have undergone liver transplantation for biliary atresia is 87.2% and 85.8%, respectively. For elective transplantation in children with stable metabolic disease, the 1-year survival is 95% (Abt et al, 2004; SPLIT Research Group, 2001; OPTN/SRTR, 2010).
Extremely young age, low body weight, and anatomic abnormalities all increase the complexity and complication rate of pediatric OLT (Anderson et al, 2008). A series from Hamburg of 12 infants weighing less than 5 kg had a survival of 75% at 1 year (Lang et al, 2000; Rogiers et al, 1997). This series showed the feasibility of liver transplantation in small infants. Children with FHF who undergo emergency hepatic transplantation have 1-year survival rates of approximately 60% (Lee et al, 2001; Uemoto et al, 2000). Likewise, infants with vascular and other anomalies associated with biliary atresia have increased complication rates and decreased survival (Anderson et al, 2008).
There is no doubt that outcome in liver transplantation is related directly to the severity of illness and the physical condition of the child (Farmer et al, 2007; Kimura et al, 2004). Early referral to a pediatric transplant center, along with early liver grafting before the clinical condition deteriorates significantly, remains the most decisive factor for success.
Abt PL, et al. Survival among pediatric liver transplant recipients: impact of segmental grafts. Liver Transpl. 2004;10:1287-1293.
Anderson CD, et al. The effect of recipient-specific surgical issues on outcome of liver transplantation in biliary atresia. Am J Transplant. 2008;8:1197-1204.
Anderson CD, et al. Biliary strictures in pediatric liver transplant recipients: early diagnosis and treatment results in excellent graft outcomes. Pediatr Transplant. 2010;14:358-363.
Austin M, et al. Liver transplantation for childhood hepatic malignancy: a review of the United Network for Organ Sharing (UNOS) database. J Ped Surg. 2006;41:182-186.
Azoulay D, et al. Split-liver transplantation for two adult recipients: feasibility and long-term outcomes. Ann Surg. 2001;233:565-574.
Azoulay D, et al. Ex situ splitting of the liver: the versatile Paul Brousse technique. Arch Surg. 2001;136:956-961.
Baker A, et al. Who needs a liver transplant? New disease specific indications. Arch Dis Child. 1998;79:460-464.
Baker T, et al. Laparoscopy-assisted and open living donor right hepatectomy: a comparative study of outcomes. Surgery. 2009;146(4):817-823.
Barshes N, et al. The Pediatric End-stage Liver Disease (PELD) model as a predictor of survival benefit and postransplant survival in pediatric liver transplant recipients. Liver Transpl. 2006;12:475-480.
Bartosh SM, et al. Renal outcomes in pediatric liver transplantation. Clin Transpl. 1997;11:354-360.
Bassett M, Murray K. Biliary atresia: recent progress. J Clin Gastroenterol. 2008;42(6):720-729.
Beaunoyer M, et al. Outcomes of transplantation in children with primary hepatic malignancy. Pediatr Transplant. 2007;11:655-660.
Berg UB, et al. Renal function before and long after liver transplantation in children. Transplantation. 2001;72:631-637.
Bezerra J. Potential etiologies of biliary atresia. Pediatr Transplant. 2005;9:646-651.
Bhatnagar V, et al. The incidence and management of biliary complications following liver transplantation in children. Transpl Int. 1995;8:388-391.
Bismuth H, Houssin D. Reduced-sized orthotopic liver graft in hepatic transplantation in children. Surgery. 1984;95:367-370.
Bismuth H, et al. Auxiliary partial orthotopic liver transplantation for fulminant hepatitis: the Paul Brousse experience. Ann Surg. 1996;224:712-726.
Botero AC, Strasberg SM. Division of the left hemiliver in segments, sectors, or sections. Liver Transpl. 1998;4:226-231.
Broelsch CE, et al. Orthotopic transplantation of hepatic segments in infants with biliary atresia. In: Koslowski, L, editor. Chirurgisches Forum 84f. Experim U. Klinische Forschung. Berlin-Heidelberg: Springer; 1984:105-109.
Broelsch CE, et al. Liver transplantation with reduced size donor organs. Transplantation. 1988;45:519-524.
Broelsch CE, et al. Application of reduced-size liver transplants as split grafts, auxillary orthotopic grafts, and living-related segmental transplants. Ann Surg. 1990;212:368-375.
Broelsch CE, et al. Living donor for liver transplantation. Hepatology. 1994;20:49S-55S.
Brown RSJr. Live donors in liver transplantation. Gastroenterology. 2008;134(6):1802-1813.
Busuttil RW, Goss JA. Split liver transplantation. Ann Surg. 1999;229:313-321.
Cacciarelli TV, et al. Factors affecting survival after orthotopic liver transplantation in infants. Transplantation. 1997;64:242-248.
Chardot C, et al. Portal vein complications after liver transplantation for biliary atresia. Liver Transpl. 1997;3:351-358.
Chen L, et al. Liver transplantation and chemotherapy in children with unresectable primary hepatic malignancies: development of a management algorithm. J Pediatr Gastroenterol Nutr. 2006;43(4):487-493.
Chen YS, et al. Evaluation of living liver donors. Transplantation. 2003;75(3 Suppl):S16-S19.
Cintorino D, et al. In situ split liver transplantatin for adult and pediatric recipients: an answer to organ shortage. Transplant Proc. 2006;38(4):1096-1098.
Colombani P, et al. Liver transplantation in infants younger than 1 year of age. Ann Surg. 1996;223(6):658-662.
Colombo C, et al. Liver disease in cystic fibrosis. J Pediatr Gastroenterol Nutr. 2006;43(Suppl 1):S49-S55.
Couinaud C. Lobes et segments hépatiques: notes sur l’architecture anatomique et chirurgicale du foie. Presse Med. 1954;62:709.
D’Alessandro AM, et al. Liver transplantation in pediatric patients: twenty years of experience at the University of Wisconsin. Pediatr Transplant. 2007;11(6):661-670.
Dalgic A, et al. Clinical approach to graft hepatic artery thrombosis following living related liver transplantation. Pediatr Transplant. 2003;7:149-152.
Deen JL, Blumberg DA. Infectious disease considerations in pediatric organ transplantation. Semin Pediatr Surg. 1993;2:218-234.
Demetriou AA, et al. Prospective, randomized, multicenter, controlled trial of a bioartificial liver in treating acute liver failure. Ann Surg. 2004;239:660-670.
Desai NM, et al. Predicting outcome after liver transplantation: utility of the model for end-stage liver disease and a newly derived discrimination function. Transplantation. 2004;77:99-106.
Deshpande RR, et al. Results of split liver transplantation in children. Ann Surg. 2002;236:248-253.
Diamond IR, et al. Impact of graft type on outcome in pediatric liver transplantation: a report from Studies of Pediatric Liver Transplantation (SPLIT). Ann Surg. 2007;246(2):301-310.
Diem HV, et al. Steroid withdrawal after pediatric liver transplantation: a long-term follow-up study in 109 recipients. Transplantation. 2003;75:1664-1670.
Doyle HR, et al. Hepatic retransplantation: an analysis of risk factors associated with outcome. Transplantation. 1996;61:1499-1505.
Duffy J, et al. Vascular complications of orthotopic liver transplantation: experience in more than 4,200 patients. J Am Coll Surg. 2009;208:896-905.
Edmond JC, et al. Improved results of living-related liver transplantation with routine application in a pediatric program. Transplantation. 1993;55:835-840.
Erbay N, et al. Living donor liver transplantation in adults: vascular variants important in surgical planning for donors and recipients. AJR Am J Roentgenol. 2003;181:109-114.
Evrard V, et al. Impact of surgical and immunological parameters in pediatric liver transplantation: a multivariate analysis in 500 consecutive recipients of primary grafts. Ann Surg. 2004;239:272-280.
Fan ST, et al. Safety of donors in live donor liver transplantation using right lobe grafts. Arch Surg. 2000;135:336.
Fan ST, et al. Biliary reconstruction and complications of right lobe live donor liver transplantation. Ann Surg. 2002;236:676-683.
Fan ST, et al. Safety and necessity of including the middle hepatic vein in the right lobe graft in adult-to-adult live donor liver transplantation. Ann Surg. 2003;238:137-148.
Farmer DG, et al. Early graft function after pediatric liver transplantation: comparison between in situ split liver grafts and living-related liver grafts. Transplantation. 2001;72:1795-1802.
Farmer DG, et al. Predictors of outcomes after pediatric liver transplantation: an analysis of more than 800 cases performed at a single institution. J Am Coll Surg. 2007;204:904-916.
Fishman JA. Infection in the organ transplant patient. Graft Organ Cell Transpl. 1999;2(Suppl):96-100.
Florman S, Shneider B. Living-related liver transplantation in inherited metabolic liver disease: feasibility and cautions. J Pediatr Gastroenterol Nutr. 2001;33:520-521.
Freeman RBJr, et al. Improving liver allocation: MELD and PELD. Am J Transplant. 2004;4(Suppl 9):114-131.
Freise CE, et al. Recipient morbidity after living and deceased donor liver transplantation: findings from the A2ALL retrospective cohort study. Am J Transplant. 2008;8:2569-2579.
Fridell JA, et al. Liver transplantation in children with cystic fibrosis: a long-term longitudinal review of a single center’s experience. J Pediatr Surg. 2003;38:1152-1156.
Ghobrial RM, et al. Technical challenges of hepatic venous outflow reconstruction in right lobe adult living donor liver transplantation. Liver Transpl. 2001;7:551-555.
Ghobrial RM, et al. Donor morbidity after living donation for liver transplantation. Gastroenterology. 2008;135:468-476.
Gladdy RA, et al. Candida infection in pediatric liver recipients. Liver Transpl Surg. 1999;5:16-24.
Goss JA, et al. In situ splitting of the cadaveric liver for transplantation. Transplantation. 1997;64:871-877.
Hansen K, Horslen S. Metabolic liver disease in children. Liver Transpl. 2008;14:713-733.
Hashikura Y, et al. Successful living-related partial liver transplantation to an adult patient. Lancet. 1994;343:1233.
Heffron TG, et al. Hepatic artery thrombosis in pediatric liver transplantation. Transplant Proc. 2003;35:1447-1448.
Hendrickson RJ, et al. Pediatric liver transplantation. Curr Opin Pediatr. 2004;16:309-313.
Hill M, et al. Graft weight/recipient weight ratio: how well does it predict outcome after partial liver transplants? Liver Transpl. 2009;15:1056-1062.
Himmelreich G, et al. Different aprotinin applications influencing changes in orthotopic liver transplantation. Transplantation. 1992;53:132-136.
Hong J, et al. Long-term outcomes for whole and segmental liver grafts in adult and pediatric liver transplant recipients: a 10-year comparative analysis of 2,988 cases. J Am Coll Surg. 2008;208:682-691.
Hong JC, et al. Long-term outcomes for whole and segmental liver grafts in adult and pediatric liver transplant recipients: a 10-year comparative analysis of 2,988 cases. J Am Coll Surg. 2009;208(5):682-689.
Imamura H, et al. Anatomical keys and pitfalls in living donor liver transplantation. J Hepatobiliary Pancreat Surg. 2000;7:380-394.
Inomata Y, et al. Right lobe graft in living donor liver transplantation. Transplantation. 2000;69:258-264.
Kahn D, et al. An analysis of the causes of death after pediatric liver transplantation. Transplant Proc. 1988;20:613-615.
Kamel IR, et al. Living adult right lobe liver transplantation: imaging before surgery with multidetector multiphase CT. AJR Am J Roentgenol. 2000;175:1141-1143.
Kamel IR, et al. Impact of multidetector CT on donor selection and surgical planning before living adult right lobe liver transplantation. AJR Am J Roentgenol. 2001;176:193-200.
Kang Y, et al. Epsilon-aminocaproic acid for treatment of fibrinolysis during liver transplantation. Anesthesiology. 1987;66:766-773.
Karrer FM, et al. Long-term results with the Kasai operation for biliary atresia. Arch Surg. 1996;131:493-496.
Kasai M, Suzuki S. A new operation for noncorrectable biliary atresia: hepatic portoenterostomy. Shujutsu. 1959;13:733-739.
Kawasaki S, et al. Living-related liver transplantation in adults. Ann Surg. 1998;227:269-274.
Kimura T, et al. Optimal timing for living-related liver transplantation in children. Clin Transplant. 2004;18:497-501.
Kiuchi T, et al. Impact of graft size mismatching on graft prognosis in liver transplantation from living donors. Transplantation. 1999;67:321-327.
Ko G, et al. Percutaneous transhepatic treatment of hepaticojejunal anastomotic biliary strictures after living donor liver transplantation. Liver Transpl. 2008;14(9):1323-1332.
Köhnlein T, Welte T. Alpha-1 antitrypsin deficiency: pathogenesis, clinical presentation, diagnosis, and treatment. Am J Med. 2008;121:3-9.
Lang H, et al. Liver transplantation in children and segmental transplantation. In: Blumgart, LH, Fong, Y. Surgery of the Liver and Biliary Tract. Philadelphia: Saunders; 2000:2107-2119.
Lee WS, et al. Fulminant hepatic failure in children in the United Kingdom: etiology and outcome. Hepatology. 2001;3(Suppl 1):A291.
Lo CM, et al. Applicability of living donor liver transplantation to high-urgency patients. Transplantation. 1999;67:73.
Lowell JA, et al. Long-term results with the Kasai operation for biliary atresia. Arch Surg. 1996;131:1235.
Makuuchi M, Sugawara Y. Living-donor liver transplantation using the left liver, with special reference to vein reconstruction. Transplantation. 2003;75(Suppl):S23-S24.
Malago M, et al. Liver splitting and living donor techniques. Br Med Bull. 1997;52:860-867.
Malatack JJ, et al. Liver transplantation for type I glycogen storage disease. Lancet. 1983;1:1073-1075.
Malatack JJ, et al. Choosing a pediatric recipient for orthotopic liver transplantation. J Pediatr. 1987;111:479-489.
Malinchoc M, et al. A model to predict poor survival in patients undergoing transjugular intrahepatic portosystemic shunts. Hepatology. 2000;31:865-871.
Man K, et al. Graft injury in relation to graft size in right lobe live donor liver transplantation: a study of hepatic sinusoidal injury in correlation with portal hemodynamics and intragraft gene expression. Ann Surg. 2003;237:256-264.
Marsh JW, et al. Complications of right lobe living donor liver transplantation. J Hepatology. 2009;51:715-724.
Mazariegos GV, et al. Weaning of immunosuppression in liver transplant recipients. Transplantation. 1997;63:243-249.
McDiarmid SV. New liver allocation policies and their potential effect on pediatric patients awaiting liver transplantation. Pediatr Transplant. 2002;6:180-186.
McDiarmid SV, et al. Selection of pediatric candidates under the PELD system. Liver Transpl. 2004;10:S23-S30.
Meyburg J, Hoffmann G. Liver transplantation for inborn errors of metabolism. Transplantation. 2005;80:S135-S137.
Millis JM, et al. Portal vein thrombosis and stenosis in pediatric liver transplantation. Transplantation. 1996;62:748-754.
Moreira A, et al. Long-term results of percutaneous bilioenteric anastomotic stricture treatment in liver transplanted children. Cardiovasc Intervent Radiol. 2010;33:90-96.
Nash KL, et al. A single centre experience of liver disease in adults with cystic fibrosis, 1995-2006. J Cyst Fibros. 2008;7:252-257.
Noble-Jamieson G, Barnes N. Diagnosis and management of late complications after liver transplantation. Arch Dis Child. 1999;81:446-451.
Ohi R. Biliary atresia: a surgical perspective. Clin Liver Dis. 2000;4:779-804.
www.ustransplant.org, 2008. Annual Report 1998-2007, HHS/HRSA/HSB/DOT. Available at
Otte JB. Is it right to develop living related liver transplantation? Do reduced and split livers not suffice to cover the needs? Transplant Int. 1995;8:6973.
Otte JB, et al. Pediatric liver transplantation: from the full-size liver graft to reduced, split, and living related liver transplantation. Pediatr Surg Int. 1998;13:308-318.
Otte JB, et al. Liver transplantation for hepatoblastoma: results from the International Society of Pediatric Oncology (SIOP) study SIOPEL-1 and review of the world experience. Pediatr Blood Cancer. 2004;42:74-83.
Perilongo G, et al. Risk-adapted treatment for childhood hepatoblastoma: final report of the second study of the International Society of Pediatric Oncology-SIOPEL 2. Eur J Cancer. 2004;40:411-421.
Perlmutter D, Shepherd R. Extrahepatic biliary atresia: a disease or a phenotype? Hepatology. 2002;36(6):1297-1304.
Pichlmayr R, et al. Transplantation einer Spenderlebr auf zwei Empfanger (splitting-transplantation): eine neue Methode in der Weiterent-wicklung der Lebersegmenttransplantation. Langenbecks Arch Chir. 1988;76:172-230.
Pomfret EA. Early and late complications in the right-lobe adult living donor. Liver Transpl. 2003;9(10 Suppl 2):S45-S49.
Pomfret EA, et al. Liver regeneration and surgical outcome in donors of right-lobe liver grafts. Transplantation. 2003;76:5-10.
Raia SJ, et al. Liver transplantation from live donors. Lancet. 1989;2:497.
Reding R, et al. Steroid-free liver transplantation in children. Lancet. 2003;362:2068-2070.
Reichert P, et al. Biliary complications of reduced-organ transplantation. Liver Transpl. 1998;4:343-349.
Rela M, et al. Split liver transplantation: King’s College Hospital experience. Ann Surg. 1998;227:282-288.
Renz JF, et al. Biliary anatomy as applied to pediatric living donor and split-liver transplantation. Liver Transpl. 2000;6:367-369.
Renz JF, et al. Changing faces of liver transplantation: partial-liver grafts for adults. J Hepatobiliary Pancreat Surg. 2003;10:31-44.
Ringe B, Strong RW. The dilemma of living liver donor death: to report or not to report? Transplantation. 2008;85(6):790-793.
Riordan SM, Williams R. Tolerance after liver transplantation: does it exist and can immunosuppression be withdrawn? J Hepatol. 1999;31:1106-1119.
Riordan SM, Williams R. The Wilson’s disease gene and phenotypic diversity. J Hepatol. 2001;34:165-171.
Rogiers X, et al. In situ splitting of cadaveric livers: the ultimate expansion of a limited donor pool. Ann Surg. 1996;224:331-339.
Rogiers X, et al. The Hamburg liver transplant program. Clin Transpl. 1997:183-190.
Ryckman FC, et al. Biliary atresia: surgical management and treatment options as they relate to outcome. Liver Transpl Surg. 1998;4(Suppl 1):S24-S33.
Saad S, et al. Portal vein reconstruction in pediatric liver transplantation from living donors. Ann Surg. 1998;227:275-281.
Salvalaggio P, et al. Presence of multiple bile ducts in the liver graft increases the incidence of biliary complications in pediatric liver transplantation. Liver Transpl. 2005;11(2):161-166.
Seek AL, et al. Transplantation of the right hepatic lobe. N Engl J Med. 2002;347:615-618.
Seiders E, et al. Graft loss after pediatric liver transplantation. Ann Surg. 2002;235:125-132.
Silverman E, Sandhaus R. Alpha-1 antitrypsin deficiency. N Engl J Med. 2009;360(26):2749-2757.
Sindhi R, et al. Impact of segmental grafts on pediatric liver transplantation: a review of the United Network for Organ Sharing Scientific Registry Data (1990-1996). J Pediatr Surg. 1999;34:107-111.
Singer PA, et al. Ethics of liver transplantation with living donors. N Engl J Med. 1989;321:620-622.
Slakey D, et al. Budd-Chiari syndrome: current management options. Ann Surg. 2001;233:522-527.
Sokal E, et al. Liver transplantation in children less than 1 year of age. J Pediatr. 1990;117(2 Pt 1):205-210.
SPLIT Research Group. Studies of pediatric liver transplantation (SPLIT): year 2000 outcomes. Transplantation. 2001;72:463-476.
Starzl TE. Memoirs of a Transplant Surgeon: The Puzzle People. Pittsburgh: University of Pittsburgh Press; 1992.
Starzl TE, et al. Orthotopic homotransplantation of the human liver. Ann Surg. 1968;168:392-415.
Strasberg SM. Terminology of liver anatomy and liver resections: coming to grips with hepatic Babel. J Am Coll Surg. 1997;184:413-434.
Strasberg SM, et al. Reducing the shortage of donor livers: what would it take to reliably split livers for transplantation into two adult recipients? Liver Transpl Surg. 1999;5:437-450.
Strong RW, et al. Successful liver transplantation from a living donor to her son. N Engl J Med. 1990;322:1505-1507.
Sugawara Y, et al. Small-for-size grafts in living-related liver transplantation. J Am Coll Surg. 2001;192:510-513.
Sugawara Y, et al. MELD score for selection of patients to receive a left liver graft. Transplantation. 2003;75:573-574.
Surman OS. The ethics of partial-liver donation. N Engl J Med. 2002;346:1038.
Surman OS, Hertl M. Liver donation: donor safety comes first. Lancet. 2003;362:674-675.
Sze Y, et al. Pediatric liver transplantation for metabolic liver disease: experience at Kings College Hospital. Transplantation. 2009;87(1):87-93.
Tanaka K, et al. Living-related liver transplantation for fulminant hepatic failure in children. Transpl Int. 1994;7(Suppl 1):S108.
Totsuka E, et al. Influence of high donor serum sodium levels on early postoperative graft funcion in human liver transplantation: effect of correction of donor hypernatremia. Liver Transpl. 1999;5:421-428.
Trotter JF. Selection of donors and recipients for living donor liver transplantation. Liver Transplant. 6(Suppl 2), 2000.
Trotter JF, et al. Adult-to-adult transplantation of the right hepatic lobe from a living donor. N Engl J Med. 2002;346:1074.
Trotter JF, et al. Documented deaths of hepatic lobe donors for living donor liver transplantation. Liver Transpl. 2006;12:1485-1488.
Turmelle YP, et al. Towards minimizing immunosuppression in pediatric liver transplant recipients. Pediatr Transplant. 2009;13(5):553-559.
Uemoto S, et al. Living donor liver transplantation for fulminant hepatic failure. Transplantation. 2000;70:152-157.
Urata K, et al. Calculation of child and adult standard liver volume for liver transplantation. Hepatology. 1995;21(5):1317-1321.
U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients. OPTN/SRTR Annual Report: Transplant Data 2000-2009. Available at http://www.srtr.org/annual_reports/2010/912b_agecat_li.htm, 2010.
van Buren D, et al. Renal function in primary liver transplant recipients receiving neoral (cyclosporine) versus Prograf (tacrolimus). Transplant Proc. 1998;30:1401-1402.
Van Hoek B, et al. Auxilliary versus orthotopic liver transplantation for acute liver failure. EURALT Study Group. European Auxilliary Liver Transplant Registry. J Hepatol. 1999;30:699-705.
Vilca-Melendez H, Heaton ND. Pediatric liver transplantation: the surgical view. Postgrad Med J. 2004;80:571-576.
Wellen J, et al. The role of liver transplantation for hepatic adenomatosis in the pediatric population: case report and review of the literature. Pediatr Transplant. 2009;14(3):E16-E19.
Wiesner RH, et al. MELD and PELD: application of survival models to liver allocation. Liver Transpl. 2001;7:567-580.
Xia VW, Steadman RH. Antifibrinolytics in orthotopic liver transplantation: current status and controversies. Liver Transpl. 2005;11:10-18.
Yamaoka Y. Experiences of 120 microsurgical reconstructions of hepatic artery in living related liver transplantation. Surgery. 1996;119:20-26.
Yao FY, et al. Liver transplantation for hepatocellular carcinoma: lessons from the first year under the Model of End-stage Liver Disease (MELD) organ allocation policy. Liver Transpl. 2004;10:621-630.
Yersiz H, et al. The conventional technique in in-situ split-liver transplantation. J Hepatobiliary Pancreat Surg. 2003;10:11-15.
Yersiz H, et al. Split liver transplantation. Transplant Proc. 2006;38(2):602-603.
Yilmaz A, et al. Vascular complications in living-related and deceased donation pediatric liver transplantation: single center’s experience from Turkey. Pediatr Transplant. 2007;11(2):124-126.
Zamir G, et al. Toward further expansion of the organ pool for adult liver recipients: splitting the cadaveric liver into right and left lobes. Transplantation. 2002;74:1757-1761.