Chapter 61 Endocrine tumors of the pancreas
Clinical picture, diagnosis, and therapy
Overview
Pancreatic endocrine tumors (PETs) are rare neoplasms that comprise 2% to 4% of all clinically detected pancreatic tumors (see Chapter 56). The origin of these tumors is not completely understood. Based on the most recent evidence, it has been suggested that these tumors arise from an endocrine cell–derived gastrointestinal epithelium. The pathogenesis of these tumors is not completely understood. Earlier theorists and modern geneticists have suggested that PETs arise from omnipotent endocrine stem cells present within the gastrointestinal epithelium. PETs are frequently divided into two groups based on the presence or absence of hormone function. Nonfunctioning, or non–hormone producing, tumors account for up to 30% or more of all neuroendocrine tumors; they express neurosecretory granules but are not associated with a clinically evident endocrinopathy, and most are discovered incidentally through the use of imaging studies done for other reasons or because of nonspecific abdominal symptoms. Functional tumors produce hormones that result in well-described endocrinopathies directly attributable to the hormones they secrete. The majority of PETs are sporadic, but they can be associated with hereditary syndromes, such as multiple endocrine neoplasia type 1 (MEN-1), von Hippel–Lindau (VHL) syndrome, tuberous sclerosis complex (TSC), and neurofibromatosis 1 (NF-1). The genetic predisposition for pancreatic endocrine tumors is strongest with MEN-1, which is characterized by the development of multigland parathyroid hyperplasia, pancreatic islet cell tumors, and pituitary adenomas. Almost half of all patients with MEN-1 develop symptomatic pancreatic endocrine tumors, and 100% of patients are shown to harbor small, nonfunctioning pancreatic endocrine neoplasms when the gland is examined histologically (Muscarella & Ellison, 2004).
Clinical management of PETs involves a multidisciplinary approach. Biochemical diagnosis and radiologic localization are the cornerstone of preoperative planning. Surgical resection is the only treatment that is curative, especially in an early stage of disease, but as many as 75% of patients present with advanced disease. Nonoperative therapies include chemotherapy, radiofrequency ablation (RFA), radionuclide therapy, biotherapy, and chemoembolization (see Chapters 81B and 83). These therapeutic modalities are useful for patients with unresectable or metastatic disease in whom symptomatic control of the hormonal effects and arrest of tumor progression can provide palliation and in some instances prolong survival.
Classification and Prognosis
The World Health Organization (WHO) developed a classification of pancreatic tumors that predicts malignant behavior based on histologic criteria (Heitz et al, 2004). This classification system assesses tumor localization, extension, proliferative capacity, and the presence of vascular or perineural invasion (Table 61.1). Some authors have challenged the clinical relevance of the WHO classification system (Schindl et al, 2000). Other studies have shown to have strong prognostic value and the ability to predict response to adjuvant treatments (Artale et al, 2005; Bajetta et al, 2005). The tumor-node-metastasis (TNM) staging system for pancreatic exocrine adenocarcinoma has been applied to the staging of PETs and was recently revised to include grades relative to mitotic counts or Ki-67 index (Table 61.2). This was recently validated in a retrospective study looking at 202 foregut neuroendocrine tumors, of which 131 were PETs. This system was found to be equivalent but not superior to the WHO classification system (Pape et al, 2008).
Table 61.1 WHO Classification of Pancreatic Endocrine Tumors
| Well-Differentiated Endocrine Tumor | |
| Type 1: Benign Behavior | Type 2: Uncertain Behavior |
| Confined to the pancreas<2 cm in diameter<2 mitoses per high-powered field<2% Ki-67–positive cellsNo vascular or perineural invasion | Confined to the pancreas and one of the following: |
| >2 cm in diameter | |
| >2 mitoses per high-powered field | |
| >2% Ki-67–positive cells | |
| Vascular or perineural invasion | |
| Well-Differentiated Endocrine Carcinoma | |
Table 61.2 TNM Classification for Pancreatic Neuroendocrine Tumors
| T: Primary Tumor | |
| T0 | No evidence of cancer |
| Tis | Carcinoma in situ |
| T2 | Tumor limited to the pancreas, size >2 cm |
| T3 | Tumor extends beyond the pancreas but does not involve the celiac axis or superior mesenteric artery |
| T4 | Tumor involves celiac axis or superior mesenteric artery (unresectable primary tumor) |
| N: Regional Lymph Nodes | |
| N0 | No regional lymph nodes involved |
| N1 | Regional lymph nodes involved |
| M: Distant Metastases | |
| M0 | No distant metastases |
| M1 | Distant metastases |
| Stages | |
| 0 | TisN0M0 |
| IA | T1N0M0 |
| IB | T2N0M0 |
| IIA | T3N0M0 |
| IIB | T1N1M0, T2N1M0, T3N1M0 |
| III | T4, any N, M0 |
| IV | Any T, any N, M1 |
From AJCC Cancer Staging Manual, 7th ed. Chicago, 2010, American Joint Committee on Cancer.
The prognostic value of newer systems has yet to be validated in a prospective way. In a large retrospective study of PET patients from the National Cancer Database, variables predictive of outcomes included age, grade, type of resection, distant metastases, and tumor functionality. Tumor size, nodal status, and margins were not associated with survival (Bilimoria et al, 2008). The limitations of retrospective studies, usually with a small cohort size, and heterogeneity among these tumors likely explain why a standard classification system for PETs still does not exist.
Insulinoma
Diagnosis
Insulin is synthesized in the β-cells as a larger molecule called proinsulin. C-peptide is cleaved from proinsulin to make the active form of insulin. Elevated levels of insulin with undetectable proinsulin and C-peptide indicate factitious hypoglycemia from the exogenous administration of insulin. Factitious hypoglycemia can also be caused by the ingestion of sulfonylureas, which cause elevated levels of all the β-cell polypeptides. Factitious hypoglycemia often occurs in health care workers who have access to insulin or oral hypoglycemics. So, during a monitored 72-hour fast, both C-peptide and sulfonylureas should also be measured in the patient’s plasma and urine. Monitored testing can detect up to 99% of insulinomas (Table 61.3; Service et al, 2000). Provocative testing with tolbutamide, glucagon, or intravenous calcium is rarely required for the diagnosis of insulinoma; furthermore, their use can cause harmful hypoglycemia with permanent neurologic damage. Autoimmune syndromes are rare causes of hypoglycemia that result from elevated levels of insulin in the presence of antiinsulin antibodies or antiinsulin receptor antibodies. Screening for these rare conditions can be done with serum measurements of the respective antibodies. Treatment is nonsurgical and sometimes warrants plasmapheresis.
Localization
Imaging for preoperative localization of insulinomas should take place only after the biochemical diagnosis has been confirmed. Most patients with insulinomas have a solitary benign adenoma (Friesen, 1982). Patients with familial syndromes, however, are more likely to have multifocal disease. Adenomas occur with equal frequency in the head, body, and tail of the pancreas. The majority of lesions are less than 1.5 cm in size and can be below the limit of detection for many conventional imaging modalities. Localization of insulinomas preoperatively remains a challenge for present-day clinicians. Traditional imaging techniques include ultrasound (US), computed tomography (CT) (see Chapter 16), and magnetic resonance imaging (MRI) (see Chapter 17). Transabdominal US, although widely available and used routinely, has low sensitivity in detecting insulinomas because of limitations from body habitus, surrounding intraabdominal organs, and the retroperitoneal depth of the pancreatic head.
Endoscopic ultrasound (EUS) (see Chapter 14) has substantially improved preoperative imaging of pancreatic neoplasms, but it is invasive. The sensitivity of EUS for localizing insulinomas has been reported to be 65% to 94%, with a mean sensitivity of 84% to 89% (Sotoudehmanesh et al, 2007). EUS visualization of the pancreatic head and uncinate process is superior to that of conventional US and provides the option of image-guided biopsy. The sensitivity of EUS is variable and depends on the location of the tumor. Sotoudehmanesh and colleagues (2007) reported sensitivity of EUS for detection of lesions in the pancreatic head, body, and tail as 92.6%, 78.9%, and 40.0%, respectively. This is understandable because of a blind spot in the distal body and tail where the stomach does not directly overly the pancreas.
Because of its wide availability, CT is usually the first localizing study performed; it can detect up to two thirds of lesions. When available, multidetector-row CT has been shown to be more sensitive for detecting small insulinomas (Liu et al, 2009). Most insulinomas are vascular and can be visualized on arterial phase imaging. One series comparing the use of CT, EUS, and the two in combination found the sensitivity of CT with EUS was superior to either modality done separately (Gouya et al, 2003). MRI alone or in combination with other imaging modalities has demonstrated higher sensitivity and rates of detection than were originally reported. More recent use of rapid tri-phase breath-holding imagery allows better fat suppression and reduction in motion artifact in both the arterial and venous phases (Catalano et al, 1999; Thoeni et al, 2000). At present, MRI is considered a second-line modality in the evaluation of insulinomas because of its greater expense and more limited availability.
Because only 30% of insulinomas possess somatostatin type II receptors, the value of somatostatin receptor scintigraphy (SRS) is limited in this tumor type (Kisker et al, 1997). Intraarterial calcium stimulation with hepatic venous sampling is one of the most sensitive localization modalities but also one of the most invasive (Guettier et al, 2009). It is a modification of the selective arterial secretin injection test developed by Imamura and colleagues (1993) and used to localize gastrinomas. This technique involves selective infusion of calcium into branches of the celiac axis and superior mesenteric artery with sampling of the hepatic venous effluent for insulin. It is an invasive, expensive, time-consuming, and technically difficult procedure that should be reserved for patients with persistent or recurrent disease, or when other tests are unsuccessful.
Despite these advances, preoperative localization may still not be successful in up to a third of patients. Current research suggests that future improvements will continue. A recent study showed that glucagon-like peptide-1 (GLP-1) receptor was expressed in more than 90% of insulinomas and at twice the density of somatostatin receptors (Reubi et al, 2003). Exendin-3, the analogue of GLP-1, is known to enhance insulin secretion in β-cells and has been introduced in the treatment of type 2 diabetes (Gallwitz et al, 2006). In a pilot trial of six patients, scintigraphy done with GLP-1 radioligands was able to identify all the insulinomas prior to operative exploration (Christi et al, 2009).
The extent of imaging necessary to ensure operative cure has not been clearly defined. Some institutions have suggested that preoperative localization is not necessary beyond the evaluation for metastatic disease (Hashimoto et al, 1999). This philosophy is based on the observation that the combination of surgical exploration and intraoperative US can identify more than 90% of insulinomas.
Therapy
Although the definitive treatment for patients with insulinomas is resection of the tumor, presurgical therapy to alleviate the symptoms and neurologic affects of hypoglycemia should be instituted. The medical management of insulinomas consists of dietary measures to minimize the occurrence of dangerous hypoglycemia. This involves taking small, frequent meals and closely monitoring blood glucose levels throughout the day. A number of insulin antisecretagogues can be used, such as diazoxide, verapamil, octreotide, or dilantin. Diazoxide, the most commonly prescribed drug, works by directly inhibiting the release of insulin from β-cells by stimulating their α-adrenergic receptors. Diazoxide also promotes glycogenolysis by inhibiting cyclic adenosine monophosphate phosphodiesterase. It can offer symptom control in up to 50% to 60% of patients (Boukhman et al, 1998). Octreotide, a somatostatin analogue, has had variable results and alleviates symptoms in approximately 40% of patients (Arnold et al, 2002). Its use is limited by side effects such as bloating, malabsorption, cholelithiasis, and eventual tachyphylaxis. Dilantin and verapamil have been used alone or in combination with other drugs, but their duration of action and side effects limit their efficacy for long-term medical therapy.
Most sporadic adenomas are amenable to enucleation regardless of their location. Surgical exploration starts by gaining access to the pancreas in the lesser sac. Lesions located in the head of the pancreas should be explored via a wide kocherization of the duodenum. In experienced hands, intraoperative US in combination with palpation detects up 98% of insulinomas (Norton et al, 1988). Care must be taken not to injure the pancreatic duct when performing an enucleation or segmental resection. Intraoperative US is useful to identify the location of the pancreatic duct for lesions in the head that are being enucleated. The insulinoma can be approached anteriorly or posteriorly depending on findings from palpation and intraoperative US. After an insulinoma is fully enucleated, it is essential to inspect the area for ductal leaks. Should a leak be identified, a suture repair should be attempted if feasible, and a suction drain should be left in place; postoperative management of the drain is identical to that for any pancreatic leak. The most common complication following resection for an insulinoma is a pancreatic leak. Rates of pancreatic fistula have been reported between 18% and 38% (Espana-Gomez et al, 2009; Nikfarjam et al, 2008). Fistulas are seen more commonly among patients treated with enucleation procedures than those undergoing formal resection.
Most insulinomas are solitary, but in patients with MEN-1, multifocal lesions are typical. In MEN-1 patients—in whom multiple, subcentimeter tumors often coalesce—segmental resection rather than enucleation is preferred (see Chapter 62A). The goal of surgery in MEN-1 patients is to remove only the tumors, preserving as much normal pancreas as possible. After preoperative imaging, if the tumor cannot be identified by operative exploration with complete mobilization of the pancreas—including kocherization to allow bimanual palpation of the entire gland—or intraoperative US, blind resection is not recommended. When the lesion is not found, a pancreatic biopsy specimen should be obtained to rule out adult nesidioblastosis, which generally can be treated successfully by subtotal pancreatectomy. The biopsy specimen is obtained by removing a small portion of the most distal tail.
Numerous studies have shown that laparoscopy is a safe and feasible approach for the treatment of pancreatic insulinomas (see Chapter 62B). Solitary lesions localized preoperatively are ideally suited for this approach. In the absence of bimanual palpation, laparoscopic US assists in tumor localization. Whereas preoperative localization has an accuracy rate of 45% for laparoscopic resection alone, when combined with laparoscopic US, the accuracy rises to 96% (Luo et al, 2009). Conversion rates to an open procedure range between 7% and 44% (Ayav et al, 2005; Jaroszewski et al, 2004; Luo et al, 2009), and the most common reason for conversion is failure to localize the tumor. Limited data suggest that fistula rates are lower among patients treated by laparoscopic compared to open approaches (Cunha et al, 2007).
The rarity of malignant insulinoma limits definitive statements about therapeutic strategies and outcome. In patients with metastatic disease, surgical resection of the tumor and metastatic lesions should be considered for palliation of symptoms of hypoglycemia. Because of the often indolent nature of malignant insulinomas, extended survival is possible. Multimodal therapy that includes surgical debulking, chemoembolization, radiofrequency thermoablation, and liver transplantation has been shown to prolong survival (Begu-Le Corroller et al, 2008). Median disease-free survival has been reported to be approximately 4 years in patients who underwent curative resection for malignant disease (Danforth et al, 1984; Hirshberg et al, 2005). Chemotherapy of malignant insulinomas consists of streptozotocin in combination with other agents, including 5-fluorouracil or doxorubicin. Reports regarding the success of these agents in clinical trials are understandably scarce given the rarity of this condition.
Gastrinoma (Zollinger-Ellison Syndrome)
Gastrinomas are the second most common functioning islet cell tumor of the pancreas. The overall incidence of gastrinomas is 0.5 to 3 per million population per year (Jensen et al, 2006). The peak age of onset is 50 years, and there is a slight male predominance. Despite their slow-growing nature, more than 60% of gastrinomas are malignant. Because of their indolent growth pattern, even in the presence of metastases, the 10-year survival approaches 90%. Approximately two thirds of gastrinomas are sporadic, with the remainder associated with familial MEN-1. Gastrinomas in MEN-1 patients tend to be multifocal, and as many as 50% of patients have lymph node, liver, or distant metastases at the time of presentation (Andersen, 1989).
Clinical Picture
Gastrinomas are so named because they produce excess levels of the gastrointestinal hormone gastrin. This results in acid hypersecretion and the subsequent development of intractable gastrointestinal ulcers. Zollinger-Ellison syndrome (ZES) is the name given to this condition, and it includes the presence of a gastrin-producing tumor in the presence of acid hypersecretion. With the advent of effective antisecretory medications, patients may or may not have intractable ulcer disease at the time of diagnosis. ZES patients come to medical attention with symptoms of abdominal pain, heartburn, nausea, and often weight loss. Diarrhea is a common problem in ZES, occuring in close to 80% of patients (Jensen et al, 2006), and 70% of patients being seen for evaluation of ZES have a confirmed history of peptic ulcer disease. Patients with ZES are also likely to exhibit sequelae of gastric acid hypersecretion, such as esophageal stricture or prominent gastric folds, documented by endoscopic gastroduodenoscopy (EGD). A suspicion of ZES is not appropriate for all patients who present with peptic ulcer disease (PUD). Approximately 0.2% of patients with PUD will have ZES, whereas 2% of patients with recurrent PUD will have ZES (Modlin et al, 1982). Other than recurrent ulcers, a higher suspicion of ZES should be held in patients who present with 1) PUD with diarrhea; 2) ulcers in unusual locations, including the distal duodenum and jejunum; 3) ulcers that are refractory to medical management or those associated with complications, such as bleeding and perforation; 4) disease at a young age; and 5) hyperparathyroidism, pituitary disorders, or a familial history of endocrinopathies. Given the nonspecific symptoms and relative rarity of this disorder, the mean time from the onset of symptoms to diagnosis is 5.9 years (Roy et al, 2000; Berna et al, 2006a). A high index of suspicion is necessary to diagnose these patients.
Diagnosis
The diagnosis of ZES is established by measuring a fasting serum gastrin (FSG) level and documenting the presence of acid hypersecretion (Table 61.4). Prior to testing, patients should be off all antisecretory medications at least 3 to 7 days. Proton pump inhibitors should be stopped for at least 1 week, and H2 receptor antagonists should be stopped for 2 days prior to testing. A fasting serum gastrin (FSG) level above 100 pg/mL or 10 times greater than the upper limit of normal (ULN) is highly suggestive of ZES. However, only one third of ZES patients have an FSG level 10 times higher than the ULN, and a number of conditions that are not associated with ZES can cause an elevated FSG level (Table 61.5). To help differentiate these conditions, the presence of gastric acid hypersecretion should be documented. Excess gastric acid production is demonstrated by having a gastric pH of less than 2.1 or a basal acid output level greater than 15 mEq/h.
| Fasting gastrin level | >100 pg/mL or >10 times higher than upper limit of normal |
| Basic acid output level | >15 mEq/h |
| Secretin stimulation testing | Increase of >200 pg/mL |
| Calcium infusion provocative testing | Rise >395 pg/mL |
Patients should be off antisecretory agents a minimum of 3 to 7 days.
Table 61.5 Differential Diagnosis of Hypergastrinemia
| High Acid Output | Normal Acid Production |
|---|---|
| Zollinger-Ellison syndrome | Atrophic gastritis |
| G-cell hyperplasia | Proton pump inhibitors |
| Retained gastric antrum | Postvagotomy syndrome |
| Gastric outlet obstruction | Renal failure |
The magnitude of the increased FSG has been shown to correlate with certain tumor features (Berna et al, 2006a). Patients with pancreatic primary tumors are more likely than those with duodenal primary tumors to have an FSG level higher than 10 times the ULN (50% of pancreatic vs. 25% of duodenal primaries). FSG level also correlates with tumor size and extension. Patients with larger tumors (>3 cm) are more likely to have an FSG level greater than 10 times the ULN when compared with smaller tumors (40% vs. 23%, respectively). Fifty percent of patients with liver metastases will have an FSG level more than 10 times the ULN, compared with only a third of those who do not.
Two thirds of ZES patients will have equivocal FSG testing, and provocative testing should be performed. Three types of provocative tests are available for the diagnosis of ZES: 1) the secretin stimulation test, 2) the calcium infusion test, and 3) the standard meal test. The secretin stimulation test is performed by intravenously administering a 2 IU/kg bolus of secretin to the patient and serially measuring serum gastrin levels at specific time points. A serum gastrin rise of 200 pg/mL (range, >100 to 335 pg/mL) is consistent with ZES (Frucht et al, 1989b; McGuigan et al, 1980). The secretin stimulation test is highly sensitive and useful in patients who cannot tolerate being off their antisecretory medications. The calcium infusion provocative test is performed by administering an intravenous infusion of calcium gluconate 5 mg/kg/h over a 3-hour period (Deveney et al, 1977). Serial time points of gastrin and calcium are measured. A rise of more than 1.5 mEq/L of calcium is used to validate the test. A rise in serum gastrin above 395 pg/mL is considered diagnostic (range, 326 to 450 pg/mL). This test should be avoided in patients with renal disease, hypercalcemia, and cardiac disease. The standard meal test is performed by serving the patient a meal composed of 30 g protein, 20 g fat, and 35 g carbohydrate (Frucht et al, 1989a). Serum gastrin levels are again measured serially and are considered diagnostic of ZES if levels rise to either more than 50% of the zero time point level or to a maximal gastrin level greater than 500 pg/mL. This test is not performed in patients who have had a prior gastrectomy or those who cannot tolerate oral feedings.
Berna and colleagues (2006b) compared these three provocative tests for ZES and showed that the secretin test was more sensitive than the calcium infusion test (94% vs. 62%). However, 38% to 50% of patients with normal secretin test values went on to have positive calcium infusion tests. Fewer than 25% of ZES patients had an appropriate rise in their postprandial serum gastrin levels with provocative meal testing. The authors concluded that secretin stimulation should be the first-line provocative test, followed by calcium testing if the secretin test results are normal in the presence of a strong suspicion for ZES. Meal provocative testing should not be used given the high percentage of equivocal studies. Once a diagnosis of ZES is established, all patients should be screened for MEN-1 by measuring serum calcium, parathyroid hormone, and prolactin levels.
Localization
In contrast to insulinomas, SRS has proven effective in detecting gastrinomas. A radiolabeled form of the somatostatin analogue octreotide can localize tumors that express high-affinity somatostatin receptors. SRS can also identify metastatic lesions elsewhere in the body. Current improvements in CT imaging have raised reported localization sensitivities from 30% to 55% through the increased use of multiphase detector CT scanning and the use of thin-slice imaging (Klose et al, 2007; Noone et al, 2005). CT imaging provides anatomic information, and it is useful for identifying metastatic disease. With MRI, gastrinomas have high signal sensitivity on T2-weighted fat-suppressed sequences. MRI can localize lesions smaller than 1 cm, but its overall sensitivity for gastrinomas is only 20% to 30% (Klose et al, 2007). Arterial angiography is now rarely used and has highly variable sensitivities because of its user-dependent variability.
As with insulinomas, EUS is extremely useful in detecting lesions within the pancreatic head but is less sensitive at identifying duodenal lesions (75% vs. 50%; Ruzzniewski et al, 1995). One promising technology being investigated for use with positron emission tomography is (68)Ga-DOTATOC, a somatostatin tracer (Hofmann et al, 2001). Preliminary studies have shown that 68 Ga-DOTATOC positron emission tomography has a higher sensitivity for detecting pancreatic endocrine tumors compared with both CT and SRS (Gabriel et al, 2007). Gastrinoma patients should first be evaluated by SRS and cross-sectional imaging (CT or MRI). If these tests fail to localize the lesions, EUS or angiography with secretin stimulation can be considered. When preoperative localization studies fail to detect the gastrinoma, surgical exploration with intraoperative palpation, US, and endoscopy with transillumination of the duodenum should be attempted, especially with sporadic gastrinomas.
Therapy
Surgical resection is the recommended treatment for patients with sporadic, resectable disease (see Chapter 62A). As with all PETs, the only chance for cure is complete surgical resection. This is achieved in only a minority of patients (Norton et al, 1999). The majority of gastrinomas are located within the “gastrinoma triangle,” as was described by Stabile and colleagues (Stabile et al, 1984; Howard et al, 1990). The gastrinoma triangle is defined by a line that joins the confluence of the cystic and common bile duct superiorly, the junction of the second and third portion of the duodenum inferiorly, and the junction of the neck and body of the pancreas medially (Fig. 61.1). More than 80% of gastrinomas are located within this anatomic triangle (Abood et al, 2009).
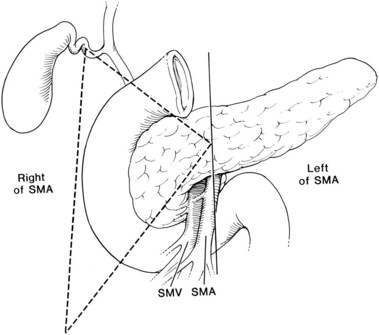
FIGURE 61.1 Gastrinoma triangle. SMA, superior mesenteric artery; SMV, superior mesenteric vein.
(From Howard TJ, et al, 1990: Anatomic distribution of pancreatic endocrine tumors. Am J Surg 159:258-264.)
The primary goal of treatment in ZES is to control acid production, remove the primary tumor, and prevent malignant progression. Historically, ZES patients were offered total gastrectomy to control acid production, but this has become unnecessary since the development of effective pharmacologic therapies for gastric hypersecretion. Resection of the primary is undertaken by performing a complete exploration of the gastrinoma triangle, which begins with an extended Kocher maneuver to mobilize the duodenum, gaining full exposure of the pancreas in the lesser sac. Bimanual palpation with the use of intraoperative US assists in identifying small and multifocal lesions located within the pancreas. Lesions within the pancreatic head should be treated with enucleation, and those in the tail or body should be treated with spleen-preserving distal pancreatectomy or enucleation. A duodenotomy should be performed routinely in all patients undergoing operative exploration for ZES (Morrow et al, 2009). Routine duodenotomy in the exploration for ZES doubles the cure rate from 30% to 60% (Morrow et al, 2009; Norton et al, 2004, 2006). Duodenal tumors less than 5 mm can be enucleated, but larger tumors should be resected by full-thickness excision of the duodenal wall. Laparoscopy has been attempted in patients with sporadic gastrinomas. Because more than 50% of these lesions occur in the duodenum or tend to be larger than other PETs, its use is limited to patients with lesions localized to the distal pancreas or those amenable to enucleation (Fernandez Cruz et al, 2008; Pierce et al, 2007).
The operative role and appropriate procedure in MEN-1 patients with ZES is controversial. This is because more than 50% of these patients are initially seen with evidence of metastases; thus MEN-1 patients are rarely cured by surgery. The goal of surgery in MEN-1 patients is not cure but prevention of metastatic disease. Surgical resection of the primary tumor may prevent the development of liver metastases and prolong disease free-survival among patients with MEN-1 ZES (Fraker et al, 1994; Motellaro et al, 2009). The larger the tumor, the more likely the patient is to develop liver metastases. Among patients with tumors less than 1 cm, only 4% are likely to have liver metastases compared with more than 60% of patients with tumors greater than 3 cm (Bartsch et al, 2007; Weber et al, 1995a).
The main predictor of survival in MEN-1 patients is the extent of liver metastasis (Weber et al, 1995b). ZES patients with MEN-1 who have tumors smaller than 2 cm may be observed. Tumors larger than 2 cm should have resection of their primary tumors with full duodenal and pancreatic exploration for multifocal disease. The use of pancreatoduodenectomy remains controversial in the management of MEN-1 ZES. This approach is recommended for young patients or those who have large isolated pancreatic head tumors (Fendrich et al, 2007; Morrow et al, 2009).
Glucagonoma
The first patient with a glucagonoma was described in 1942 by Becker and colleagues, who reported a 45-year-old woman with a diffuse, erythematous, vesicular, necrotic dermatosis in addition to diabetes mellitus, stomatitis, anemia, weight loss, and thromboembolism. Her autopsy revealed a pancreatic islet cell tumor. More than 20 years later, with the benefit of radioimmunoassay techniques, McGavran and colleagues (1966) showed elevated glucagon levels in the blood and pancreatic islet α-cell tumor tissue in a similar patient with “bullous and eczematoid dermatitis,” diabetes, and anemia. Glucagonomas are rare, frequently large (>4 cm) at diagnosis, and almost always found within the pancreas. Patients typically are seen in their 40s, with an equal distribution between genders (Wermers et al, 1966). Most of the tumors (60% to 70%) are malignant, and association with MEN-1 is rare.
Clinical Picture
The clinical manifestations of a glucagonoma encompass what is sometimes referred to as the “four D syndrome,” which includes diabetes mellitus type 2, dermatitis (necrolytic migratory erythema), deep vein thrombosis (30% of patients), and depression. Other symptoms can include chronic anemia, stomatitis, weight loss, and generalized weakness. Less common features include mental changes, diarrhea, arterial thrombosis, and neurologic abnormalities (Prinz et al, 1981). Necrolytic migratory erythema (NME) is present in 70% of patients (Fig. 61.2). This rash is characteristic of a glucagonoma but not definitively diagnostic. The distribution of NME is highly variable across regions of the body. It typically occurs where there is frequent friction or inadvertent trauma, such as the feet, lower legs, buttocks, or perineum, but lesions can be present anywhere. The rash is commonly confused with psoriasis, pemphigus, or eczema. To differentiate NME from these other rashes, a biopsy can be performed. Histopathology shows necrolysis, pallor of the epidermis, and mild lymphocytic and histiocytic infiltration. The biopsy should be taken from an early lesion and preferably from its edge. The reason for this is that older lesions with blisters in various stages of healing reveal nonspecific findings of dyskeratosis and acanthosis (Kovacs et al, 2006). These features make it more difficult to distinguish NME from other causes of subacute dermatitis. After resection, patients can have complete regression of NME.
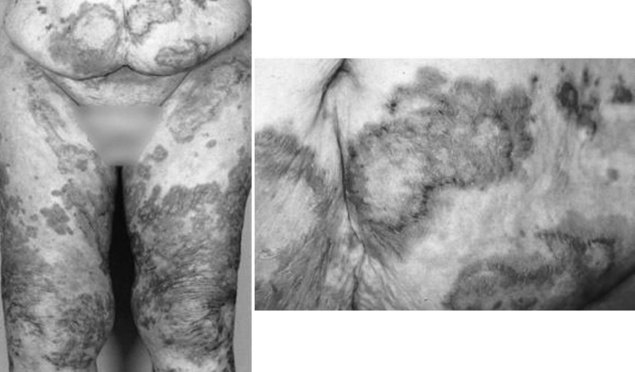
Diagnosis and Localization
Laboratory findings of a glucagonoma include elevated plasma glucose level, normochromic normocytic anemia, and a generalized decrease in plasma amino acids. The diminished levels of amino acids stem from the stimulation of gluconeogenesis and amino acid oxidation by glucagon. Fasting serum glucagon levels greater than 1000 pg/mL (normal is 0 to 150 pg/mL) are virtually diagnostic. Other conditions that can cause elevated glucagon levels (usually <500 pg/mL) include fasting, sepsis, pancreatitis, abdominal surgery, Cushing syndrome, and renal or hepatic failure. Because the diagnosis is often delayed, patients with glucagonomas typically are seen with large tumors (>5 cm). CT scan with intravenous contrast administration is often sufficient for localization and has been reported to detect 86% of tumors (Wermers et al, 1966). Abdominal CT is also useful for identifying metastatic disease, which can be present in up to 50% of patients at the time of presentation (Stacpoole, 1981). EUS is usually unnecessary for localization, but it can be useful for US-guided needle biopsy. SRS has been used more for long-term follow-up of these patients and can demonstrate metastatic disease (Lipp et al, 2000). Other imaging modalities, such as angiography and portal venous sampling, are rarely needed for localization purposes.
Therapy
Glucagonomas cause a prolonged and profound catabolic state. Preoperative nutritional status should be assessed, and the patient’s metabolic deficits should be corrected before operation. Somatostatin analogues should be considered for the preoperative preparation because they can markedly diminish circulating levels of glucagon and consequently lessen or reverse its catabolic effects. The majority of tumors are located in the body or tail of the pancreas and are potentially amenable to laparoscopic spleen-preserving distal pancreatectomy, if diagnosed early. Unfortunately, most are very large at diagnosis. Glucagonomas are malignant in 60% to 70% of patients, and the likelihood of cure is small. An R0 resection is achievable in only 30% of patients (Abood et al, 2009). Nevertheless, resection of the primary and even metastatic lesions can offer substantial palliation if the tumor tissue can be removed safely. Like most islet cell carcinomas, glucagonomas frequently have an indolent course. Thus, reoperative surgery for recurrent disease is a reasonable option if the resection can be performed safely. Control of the hormonal effects through debulking procedures can help manage symptoms and prolong survival (Altimari et al, 1986). When widely metastatic disease is present and surgical debulking is not possible, medical therapy can alleviate hormonal symptoms (Nightingale et al, 1999). Somatostatin analogues such as octreotide can reduce serum glucagon levels and improve NME, diabetes, diarrhea, and neurologic symptoms. Dacarbazine and streptozotocin have been used to treat metastatic glucagonomas with some success. As with other pancreatic endocrine cell tumors, metastatic disease to the liver has been treated by resection, hepatic artery embolization, and liver transplantation (Dousset et al, 1995).
Vasoactive Intestinal Peptide Tumor
Vasoactive intestinal peptide (VIP) tumors are rare, with an annual incidence in the United States of 1 in 100,000,000 people. The majority of VIP tumors are benign and typically consist of a solitary lesion with a mean size of 3 to 5 cm. The mean age at presentation is 48 years, with a slight male predominance (Peng et al, 2004). In 1958, Verner and Morrison first described this syndrome of watery diarrhea (WD), hypokalemia (H), and achlorhydria (A) associated with a non–β-cell tumor of the pancreas (Verner & Morrison, 1958). As a result, this complex is often referred to as WDHA syndrome or Verner-Morrison syndrome, and 98% of patients are first seen with diarrhea. Hypokalemia is present in 67% to 100% of patients. The prevalence of flushing and weight loss is much more variable. The secretory diarrhea is high in volume (6 to 8 L/day) but painless and odorless; its color resembles tea, and it characteristically persists despite fasting. In VIP tumors, the diarrhea does not decrease with nasogastric fluid aspiration, whereas in ZES diarrhea will cease (Mansour & Chen, 2004). Over time, with large-volume gastrointestinal fluid losses, patients become hypokalemic and acidotic. The fluid and electrolyte deficits in VIP tumor patients can be profound and must be corrected before proceeding to surgical or other treatment.
The differential diagnosis for diarrhea is very long, so the diagnosis of a VIP tumor is typically made several months to years after the onset of symptoms. The average duration of symptoms before diagnosis is 3 years (Thomason et al, 2000). To confirm the diagnosis, a fasting serum VIP level should be greater than 200 pg/mL; average levels are close to 1000 pg/mL in patients with VIP tumors (O’Dorisio et al, 1989). The majority of pancreatic VIP tumors are located within the tail of the pancreas (72%) and thus are readily identified by conventional imaging. Radiologic examination begins with a CT scan of the abdomen and pelvis. Because of their relatively large size, sensitivity for localizing these lesions approaches 100%. Other radiographic adjuncts include MRI and SRS. Unlike insulinomas, more than 85% of VIP tumors are somatostatin receptor positive. SRS can be useful in detecting primary lesions outside the pancreas, where one third of VIP tumors are located. Compared with pancreatic tumors, extrapancreatic lesions are typically smaller, less likely to be malignant (33% vs. 64%), and less likely to present with metastases (29% vs. 56%) (Ghaferi et al, 2008).
Somatostatinoma
Somatostatinomas are rare, representing only 1% of all pancreatic neuroendocrine tumors (O’Toole et al, 2006). In 1977, Ganda and colleagues and Larsson and colleagues independently reported the first patients with a somatostatinoma. Somatostatinoma syndrome was described by Krejs and coworkers in 1979 and includes hyperglycemia, cholelithiasis, steatorrhea, and diarrhea. The mean age of patients at diagnosis is 50 years, with an equal distribution between men and women. In most patients (>90%), the lesions are solitary and generally average between 5 and 6 cm. Extrapancreatic somatostatinomas occur in 44% of patients, most commonly within the duodenum, but they have also been reported to arise from the biliary tract or other parts of the small bowel. The inhibition of insulin secretion leads to mild hyperglycemia, and decreased gallbladder contractility from the inhibition of cholecystokinin results in the formation of gallstones. The inhibition of pancreatic enzyme and bicarbonate secretion and intestinal absorption causes diarrhea and steatorrhea. Patients also may have gastric hypochlorhydria from the inhibition of gastrin release (Jensen & Norton, 1995). These clinical manifestations are rarely present with somatostatinomas arising in the duodenum. Instead, patients with duodenal somatostatinomas are initially seen with obstructive symptoms that include abdominal pain and jaundice. Duodenal somatostatinomas often are identified as an incidental finding (Mao et al, 1995). They are malignant in 60% to 70% of patients, with nearly two thirds of patients having evidence of metastases at diagnosis (Nesi et al, 2007). Lesions greater than 2 cm are more likely to be associated with metastatic disease (Tanaka et al, 2000).
Most tumors located in the head of the pancreas are large. Localization of pancreatic somatostatinomas often can be accomplished by CT or US, whereas EUS, MRI, and SRS play a role in localization of smaller or metastatic tumors. Because many duodenal somatostatinomas are small (<1 cm), EUS can be especially useful for their localization. Surgical resection offers the only chance for cure, but many somatostatinomas have metastasized by the time they are discovered. Debulking can provide symptomatic relief, and cholecystectomy should be performed at the time of operation because of the high incidence of cholelithiasis. In unresectable disease, octreotide and interferon-alfa may improve symptoms. In patients without metastatic disease, the mean 5-year survival is 100%. Those patients with metastatic disease who undergo radical resection or debulking procedures have a mean 5-year survival of 60% (Abood et al, 2009).
Nonfunctional Pancreatic Endocrine Tumors
Nonfunctional pancreatic neuroendocrine tumors (NF-PETs) histologically express endocrine differentiation but lack a clinical syndrome of hormone overproduction (Prinz et al, 1983). They represent more than 75% of all pancreatic endocrine tumors (Halfdanarson et al, 2008). The mean age at presentation is 45 years, and an equal distribution is found between men and women. Traditionally, NF-PETs are described as large tumors, and more than 60% to 80% are metastatic at the time of diagnosis. With the widespread use of high-resolution abdominal imaging, however, these lesions are being increasingly identified as small, asymptomatic tumors. Approximately 60% are malignant at the time of diagnosis (Ehehalt et al, 2009), and 8% of NF-PETs occur in association with MEN-1; of patients with MEN-1, approximately 55% of tumors will be NF-PETs (Thomas-Marques et al, 2006).
Diagnosis and Localization
Although these tumors are hormonally “silent,” they often release inert precursor hormones into the systemic circulation. Blood testing for such inert tumor markers include pancreatic polypeptide, neurotensin, protein S, neuron-specific enolase, and chromogranin A. Measurement of these levels prior to resection may help establish a baseline for tumor burden and provide a possible marker for follow-up for tumor recurrence. Localization of the primary tumor and establishment of the extent of metastatic disease is best achieved with a triple-phase—arterial, portal, and venous phase—CT scan (see Chapter 16). Because these tumors are typically large, EUS is only necessary for biopsy or to identify subcentimeter disease. SRS is recommended to determine receptor expression status for the postsurgical treatment with somatostatin analogues. Because a small percentage of these lesions can be associated with MEN-1, patients who have a medical or family history suggestive of MEN-1 should undergo testing for this familial syndrome’s associated endocrinopathies.
Therapy
In patients with sporadic NF-PETs, resection should be geared toward cure when possible. When the lesion is limited to the pancreas, a standard oncologic resection should be performed (see Chapter 62A). In the presence of limited, synchronous hepatic metastases, concomitant liver resection should be considered if the patient is a good surgical candidate. In patients with extensive liver disease, resection of the primary tumor may be considered in conjunction with partial hepatectomy combined with locoregional ablative techniques, such as transarterial chemoembolization (TACE) and radiofrequency ablation (RFA).
In MEN-1 patients, the indications and approach to surgery remain controversial. MEN-1 patients have a field defect that makes the entire pancreas at risk of neoplastic disease, such that resection of a primary lesion does not necessarily prevent recurrence in the remnant gland. The goal of surgery for MEN-1 NF-PETs should be to resect all gross disease while also trying to preserve pancreatic function. Surveillance EUS performed in these patients often reveals small PETs of uncertain importance. The threshold for operation varies across institutions. Bartsch and colleagues (2005) advocate that patients undergo early resection at a size threshold of 10 mm, based on the observation that lesions greater than 10 mm carry a 20% risk of having locoregional metastases. Some groups advocate surgical exploration based on biochemical evidence alone, even if imaging studies fail to localize lesions (Skogseid et al, 1991). The recommended procedure includes enucleation of lesions in the pancreatic head followed by prophylactic spleen-preserving distal pancreatectomy.
Metastatic Disease
Unlike exocrine pancreatic neoplasms, patients with endocrine tumors may benefit from reoperation for resectable recurrences or metastatic disease. This is likely because of the relatively indolent nature of these tumors. Observational studies have shown that many patients live for several years with untreated metastatic disease (Weber et al, 1995a). The debilitating aspects of these tumors are related to the excess hormone secretion in patients with functional PETs or to obstructive symptoms caused by large NF-PETs. The predominant site of metastatic spread is the liver. Resection, chemotherapy, and other treatment modalities may not cure the patient, but they do offer substantial palliation and may extend survival. Compared with medical management, patients treated surgically or with ablative therapy have improved overall survival (see Chapter 81B; Chamberlain et al, 2000).
Resection
With advanced metastatic disease that includes hepatic metastases, resection remains the gold standard of treatment in these patients when feasible. The biochemical response rate is 96%, and the 5-year survival is reported as 80% (Steinmuller et al, 2008). Patients with advanced disease at initial presentation should undergo radical resection with an attempt at cure if it can be done safely. In study by Norton and colleagues (2003), in which the mean tumor size was 8 cm and 40% of patients had liver metastases, a curative resection was achieved in 75% of patients. Procedures included pancreatoduodenectomy, liver resection, nodal dissection, and total pancreatectomy. No perioperative deaths were reported, and the mean follow-up period was 19 months. During that time the actuarial overall 5-year survival rate was 80%, which supports the conclusion that patients with advanced disease at presentation may benefit from a complete resection or a debulking procedure. Musunuru and colleagues (2006) retrospectively reviewed 48 patients with neuroendocrine liver metastases only and evaluated their 3-year survival based on medical management, ablation, and surgical resection. The surgical group had a 3-year survival of 83% compared with 31% in the patients treated with medical therapy or embolization. No difference was reported in the palliation of symptoms among the three treatment groups.
Because of the limited patient population, none of these studies is likely to be validated by a prospective randomized controlled trial. Case-based studies and retrospective reviews are the basis for the current recommendations. Such studies recommend that curative resection in conjunction with an anatomic liver resection should be attempted, when it appears that at least 90% of the tumor burden can be removed based on preoperative imaging (Morrow et al, 2009).
Liver transplantation is an effective therapy for patients with hepatocellular cancer. In PET patients with isolated liver metastases, organ transplantation remains controversial. Lehnert (1998) reported 103 patients with metastatic neuroendocrine tumors who received liver transplants. The overall survival at 1 and 5 years was 61% and 30%, respectively, for the 48 patients with pancreatic islet cell tumors. Recurrence-free survival for all 103 patients was 24% at 5 years. Pretransplant treatment with a somatostatin analogue was found to be a favorable prognostic factor (Lehnert, 1998). The morbidity associated with these procedures can be high as a result of opportunistic infections, and a 10% operative mortality rate has been reported (Marin et al, 2007). There is a high risk of recurrence in the transplanted liver, and liver transplantation is generally reserved for young patients with unresectable isolated liver metastases as the single site of disease and for those who have poor symptom control on medical therapy.
Ablative Therapies for Neuroendocrine Hepatic Metastases
Although surgery remains the most effective therapy for PETs, many patients will be seen later with recurrent disease. The median time to recurrence is 21 months, with 5-year recurrence rates of 84% (Atwell et al, 2005; Sarmiento et al, 2003). At the time of recurrence, less than 10% of patients are candidates for reoperative surgery (Schurr et al, 2007). For this subset of unresectable patients, ablative cytoreductive measures have been attempted. Ablative therapies that include RFA, cryoablation, TACE, and radioembolization have been effective in reducing tumor burden in marginally resectable patients (see Chapters 83, 84A, and Chapter 85A, Chapter 85B, Chapter 85C, Chapter 85D ; Maithel et al, 2009).
Ablative techniques are most efficacious when used for tumors smaller than 4 cm and for deep lesions, for which resection would sacrifice a great deal of normal liver parenchyma; ablative techniques are also beneficial as an adjunct in patients with bilobar disease who undergo anatomic resection of the lobe with the greatest tumor burden. The largest series on RFA therapy showed 95% symptom relief and a 5-year survival rate of 48% (Maithel et al, 2009). This was comparable to resections in which Sarmiento and colleagues (2003) showed a 5-year survival of 61% after aggressive operative removal. Hepatic artery embolization with or without direct infusion of chemotherapeutic agents has been used as an alternative therapy for patients who are not candidates for hepatic resection. Tumors of the liver receive most of their blood supply from the hepatic artery, whereas normal hepatic parenchyma can be sustained mainly from the portal venous circulation. Chemotherapeutic agents used include doxorubicin, cisplatin, mitomycin C, streptozotocin, and 5-fluorouracil (Mansour & Chen, 2004). In retrospective reviews of patients treated with TACE for unresectable liver metastases, response rates range from 66% to 100%, and 5-year survival rates of up to 83% are reported (Roche et al, 2003; Kim et al, 1999). A comparison between simple arterial embolization versus chemoinfused embolization showed no difference in symptom improvement or median overall survival between the two groups (Pitt et al, 2008).
Somatostatin Analogues and Systemic Chemotherapy
Approximately 80% of PETs express somatostatin receptors, and treatment with somatostatin analogues can alleviate symptoms associated with excessive hormone activity in patients with functional PETs. Subtypes of PETs most amenable to treatment with octreotide are glucagonomas, VIP tumors, and, to a lesser extent, gastrinomas and insulinomas. Positive SRS imaging may help predict which patients will respond to somatostatin analogue therapy (Oberg et al, 2004). The most commonly prescribed regimen starts with subcutaneous administration of 50 µg dosed three times per day. Lanreotide, a long-acting somatostatin analogue administered every 10 to 14 days, has been shown to have efficacy similar to that of octreotide (Ricci et al, 2000; Scherubl et al, 1994). Depot formulations of somatostatin analogues are also available that can be administered no more frequently than 4 to 6 weeks apart. These drugs are more difficult to titrate given their long-acting effects. It is not uncommon for patients treated with depot formulations to have “breakthrough” symptoms such that immediate, shorter acting medications must be given until a steady state is achieved.
Somatostatin analogues such as octreotide are generally well tolerated, safe, and have minimal side effects, although an increased incidence of cholelithiasis (up to 50%) is reported with continuous use of these drugs. However, the incidence of acute cholecystitis requring emergent cholecystecomy is rare (0% to 12%). Although it remains controversial, mounting evidence suggests that somatostatin analogues have cytoreductive properties in addition to their ability to control hormone symptoms (Clements et al, 1985; Imtiaz et al, 2000). An interim report at 6 months from an ongoing multiinstitutional, randomized, placebo-controlled trial has shown that 67% of patients receiving therapy had radiographic evidence of stable disease compared with 37% in the placebo group (Rinke et al, 2009). Of the patients included in this trial, 66% had undergone resection of the primary tumor, 74% were octreoscan positive, and 62% had some evidence of liver involvement. When unresectable disease is found at laparotomy and it is likely that a somatostatin analogue will be used later, cholecystectomy should be considered.
Several trials have studied the role of interferon (IFN) in the treatment of pancreatic endocrine neoplasms. In one series, 57 patients treated with monotherapy IFN had a radiologic response of only 12% and biochemical control in 47%. IFN-alfa stimulates T-cell function and can control hormone secretion by the tumor. Oberg (2000) pooled 13 studies that examined a total of 383 patients with malignant neuroendocrine tumors; the biochemical and tumor response rates were 44% and 11%, respectively, with a disease stabilization rate of 35%. A randomized, prospective study of 80 patients with metastatic neuroendocrine gastroenteropancreatic tumors showed comparable antiproliferative effects among patients treated with somatostatin analogues, IFN-alfa, or a combination of the two (Faiss et al, 2003).
The role for systemic chemotherapy in the treatment of PETs is still undefined. Several different chemotherapeutic agents have been studied, both as single agents and in combination regimens. The most common single agents include streptozotocin, doxorubicin, capecitabine, fluorouracil, and dacarbazine. In the early trials, overall response rates for these single-agent regimens ranged from 20% to 50% (Broder et al, 1973; Moertel et al, 1982; Ramanathan et al, 2001). Combination regimens have shown even more promising outcomes, with overall response rates as high as 81% (Strosberg et al, 2011a). A newer agent, temozolomide, has been shown to have promise in Phase II trials. The combination of this drug with capecitabine in chemo-naive patients illustrated an objective radiographic response rate of 70%, with a median progression-free survival of 18 months (Strosberg et al, 2011b). The limitation of these drugs, either in combination or as single agents, is their associated toxicity. In several of these trials, patients were unable to tolerate the regimens because of complications such as lymphopenia or development of opportunistic infections.
Ki-67 is an immunohistochemical marker of high tumor proliferation. The Ki-67 index has been used to determine tumor grade and prognosis. In fact, a high Ki-67 index has been suggested as a measure to predict which patients would do better with antiproliferative or chemotherapy, even as a first-line treatment (Vilar et al, 2007).
New Drugs and Targeted Therapies
External-beam radiation has been used for the treatment of bulky hepatic metastases. The normal liver has a low tolerance for the levels of radiation needed to kill tumor cells. To minimize this, patients have been treated with targeted radiotherapy through the use of radiolabeled somatostatin analogues. These agents are conjugated to an injectable microsphere that allows high-dose intraarterial administration of the radiolabeled drug in a fashion similar to that used for TACE/transarterial embolization. Results of these studies are highly variable, but partial response rates ranging from 32% to 87% have been reported (Kennedy et al, 2008; King et al, 2008; McStay et al, 2005; Safford et al, 2004). Complete responses were seen in 5% to 18% of patients, and symptomatic improvement occurred in 49% to 61% of patients. The most common symptoms associated with toxicity were fatigue and nausea (Kennedy et al, 2008).
Novel molecular targets are also being studied for their therapeutic potential. PETs are highly vascular tumors that may be inhibited by the use of angiogenesis inhibitors such as bevacizumab, an antibody to vascular endothelial growth factor. In a Phase II trial of 44 patients with advanced carcinoid tumors, the reported progression-free surival rate over 18 weeks was 95% with bevacizumab versus 67% in those treated with IFN (Yao et al, 2008). In a Phase II study, rapamycin, a serine threonine kinase known as mTOR (mammalian target of rapamycin), was recently reported to achieve stable disease in 80% of patients treated during a 9-month period. The median progression-free survival was 17 months. As more is understood about the tumor biology of pancreatic endocrine neoplasms, it is likely more multimodal therapies will be developed.
Abood G, et al. The surgical and systematic management of neuroendocrine tumors of the pancreas. Surg Clin N Am. 2009;9:249-266.
Altimari AF, et al. Use of somatostatin analogue (SMS 201e995) in the glucagonoma syndrome. Surgery. 1986;100:989-996.
Andersen DK. Current diagnosis and management of Zollinger–Ellison syndrome. Ann Surg. 1989;210:685-703.
Arnold R, et al. Somatostatin analogues in the treatment of endocrine tumours of the gastrointestinal tract. Expert Opin Pharmacother. 2002;3:643-656.
Artale S, et al. Treatment of metastatic neuroendocrine carcinomas based on WHO classification. Anticancer Res. 2005;25:4463-4469.
Atwell TD, et al. Treatment of neuroendocrine cancer metastatic to the liver: the role of ablative tachniques. Cardiovasc Invtervent Radiol. 2005;28:409-421.
Ayav A, et al. Laparoscopic approach for solitary insulinoma: a multicentre study. Lagenbecks Arch Surg. 2005;390:134-140.
Bajetta E, et al. Is the new WHO classification of neuroendocrine tumours useful for selection an appropriate treatment? Ann Oncol. 2005;16:1374-1380.
Bartsch D, et al. Surgical aspects of gastrinoma in multiple endocrine neoplasia type 1. Wien Klin Wochenschr. 2007;119:602-608.
Bartsch DK, et al. Outcome of duodenopancreatic resctions in patients with multiple endocrine neoplasia type 1. Ann Surg. 2005;242:757-764.
Becker SW, et al. Cutaneous manifestations of internal malignant tumors. Arch Dermatol Syphilol. 1942;45:1068-1080.
Begu-Le Corroller A, et al. Aggressive multimodal therapy of sporadic malignant insulinoma can improve survival: a retrospective 35-year study of 12 patients. Diabetes Metabol. 2008;34:343-348.
Berna M, et al. Serum gastrin in Zollinger–Ellison syndrome: I. Prospective study of fasting serum gastrin in 309 patients from the National Institutes of Health and comparison with 2229 cases from the literature. Medicine. 2006;85:295-330.
Berna M, et al. Serum gastrin in Zollinger–Ellison syndrome: II. Prospective study of gastrin provocative testing in 293 patients from the National Institutes of Health and comparison with 537 cases from the literature: evaluation of diagnostic criteria, proposal of new criteria, and correlation with clinical and tumoral features. Medicine. 2006;85:331-364.
Bilimoria KY, et al. Prognostic score predicting survival after resection of pancreatic neuroendocrine tumors. Ann Surg. 2008;247:490-500.
Boukhman M, et al. Insulinoma-experience from 1950 to 1995. West J Med. 1998;169:98-104.
Broder LE, et al. Pancreatic islet cell carcinoma: results of therapy with streptozotocin in 52 patients. Ann Intern Med. 1973;79:108-118.
Catalano C, et al. Localization of pancreatic insulinoma with MR imaging at 0.5 T. Acta Radiol. 1999;40:644-648.
Chamberlain RS, et al. Hepatic neuroendocrine metastases: does intervention alter outcomes? J Am Coll Surg. 2000;190:432.
Christi E, et al. Glucagon-like peptide-1 receptor imaging for localization of insulinomas. J Clin Endocrinol Metab. 2009;94:4398-4405.
Clements D, et al. Regression of metastatic VIPoma with somatostatin analogue SMS 201-995. Lancet. 1985;1:874-875.
Cunha A, et al. Laparoscopic versus open approach for solitary insulinoma. Surg Endosc. 2007;21:103-108.
Danforth DN, et al. Metastatic insulin-secreting carcinoma of the pancreas: clinical course and the role of surgery. Surgery. 1984;96:1027-1036.
Deveney CW, et al. Use of calcium and secretin in the diagnosis of gastrinoma (Zollinger–Ellison syndrome). Ann Intern Med. 1977;87:680-686.
Dousset B, et al. Metastatic endocrine tumors: is there a place for liver transplantation? Liver Transpl Surg. 1995;1:111-117.
Ehehalt F, et al. Neuroendocrine tumors of the pancreas. Oncologist. 2009;14:456-467.
Espana-Gomez M, et al. Pancreatic insulinoma: a surgical experience. World J Surg. 2009;33:1966-1970.
Faiss S, et al. Prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors: the International Lanreotide and Interferon Alfa Study Group. J Clin Oncol. 2003;21:2689-2696.
Fendrich V, et al. Management of sporadic and multiple endocrine neoplasia type 1 gastrinomas. Br J Surg. 2007;94:1334-1341.
Fernandez Cruz L, et al. Is laparoscopic resection adequate in patients with neuroendocrine pancreatic tumors? World J Surg. 2008;32:904-917.
Fraker D, et al. Surgery in Zollinger–Ellison syndrome alters the natural history of gastrinoma. Ann Surg. 1994;220:320-328.
Friesen SR. Tumors of the endocrine pancreas. N Engl J Med. 1982;306:580-590.
Frucht H, et al. Prospective study of the standard meal provocative test in Zollinger–Ellison syndrome. Am J Med. 1989;87:528-536.
Frucht H, et al. Secretin and calcium provocative tests in the Zollinger–Ellison syndrome: a prospective study. Ann Intern Med. 1989;111:713-722.
Gabriel M, et al. 68Ga-DOTA-Tyr3-octreotide PET in neuroendocrine tumors: comparison with somatostatin receptor scintigraphy and CT. J Nuc Med. 2007;48:508-518.
Gallwitz B, et al. Exenatide in type 2 diabetes: treatment effects in clinical studies and animal study data. Int J Clin Pract. 2006;60:1654-1661.
Ganda OP, et al. Somatostatinoma: a somatostatin-containing tumor of the pancreas. N Engl J Med. 1977;296:963-967.
Ghaferi AA, et al. Pancreatic VIPomas: subject review and one institutional epxerience. J Gastrointest Surg. 2008;12:382-393.
Gouya H, et al. CT, endoscopic sonography, and combined protocol for preoperative evaluation of pancreatic insulinomas. Am J Roentgenol. 2003;181:987-992.
Guettier JM, et al. Localization of insulinomas to regions of the pancreas by intraarterial calcium stimulation: the NIH experience. J Clin Endocrinol Metab. 2009;94:1074-1080.
Halfdanarson TR, et al. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis, and recent trend toward improved survival. Ann Oncol. 2008;19:1727-1733.
Hashimoto LA, et al. Preoperative localization of insulinomas is not necessary. J Am Coll Surg. 1999;189:368-373.
Heitz PU, et al. Pancreatic endocrine tumors: introduction. In: DeLellis, DA, Lloyd, RV, Heitz, PU. Pathology and Genetics of Tumours of Endocrine Organs: WHO Classification of Tumours. Lyon, France: IARC Press; 2004:177-182.
Hirshberg B, et al. Malignant insulinoma: spectrum of unusual clinical features. Cancer. 2005;104:264-272.
Hofmann M, et al. Biokinetics and imaging with the somatostatin receptor PET radioligand 68 Ga-DOTATOC: preliminary data. Eur J Nucl Med. 2001;8:1751-1757.
Howard TJ, et al. Anatomic distribution of pancreatic endocrine tumors. Am J Surg. 1990;59:258.
Imamura M, et al. Use of selective arterial secretin injection test to guide surgery in patients with Zollinger–Ellison syndrome. World J Surg. 1993;17:433-438.
Imtiaz KE, et al. Complete histological regression of metastatic carcinoid tumour after treatment with octreotide. Clin Endocrinol. 2000;53:755-758.
Jaroszewski D, et al. Laparoscopic localization and resection of insulinomas. Arch Surg. 2004;139:270-274.
Jensen RT, Norton JA. Endocrine neoplasms of the pancreas. In: Yamada, T, editor. Textbook of Gastroenterology. Philadelphia: Lippincott; 1995:2131.
Jensen R, et al. Gastrinoma (duodenal and pancreatic). Neuroendocrinology. 2006;84:173-182.
Kennedy AS, et al. Radioembolization for unresectable neuroendocrine hepatic metastases using resin 90Y-microspheres: early results in 148 patients. Am J Clin Oncol. 2008;31(3):271-279.
Kim YH, et al. Selective hepatic arterial chemoembolization for liver metastases in patients with carcinoid tumors or islet cell carcinoma. Cancer Invest. 1999;17:474-478.
King J, et al. Radioembolization with selective internal radiation microspheres for neuroendocrine liver metastases. Cancer. 2008;113:921-929.
Kisker O, et al. The value of somatostatin receptor scintigraphy in newly diagnosed endocrine gastroenteropancreatic tumors. J Am Coll Surg. 1997;184:487-492.
Klose K, et al. Localization and staging of gastrin producing tumors using cross-sectional imaging modalities. Wien Klin Wochenschr. 2007;19:588-592.
Kovacs R, et al. Necrolytic migratory erythema. J Cutan Pathol. 2006;33:242-245.
Krejs G, et al. Somatostatinoma syndrome: biochemical, morphologic, and clinical features. N Engl J Med. 1979;301:285-292.
Larsson LI, et al. Pancreatic somatostatinoma. Clinical features and physiological implications. Lancet. 1977;1:666-668.
Lehnert T. Liver transplantation for metastatic neuroendocrine carcinoma: an analysis of 103 patients. Transplantation. 1998;66:1307-1312.
Lipp R, et al. Scintigraphic long-term follow-up of a patient with metastatic glucagonoma. Am J Gastroenterol. 2000;95:1818-1820.
Liu Y, et al. The value of multidetector-row CT in the preoperative detection of pancreatic insulinomas. Radiol Med. 2009;114:1232-1238.
Luo Y, et al. Laparoscopic surgery for pancreatic insulinoma: a single-institution experience of 29 cases. J Gastrointest Surg. 2009;13:945-950.
Maithel SK, et al. Hepatic ablation of neuroendocrine tumor metastases. J Surg Oncol. 2009;100:635-638.
Mansour JC, Chen H. Pancreatic endocrine tumors. J Surg Res. 2004;120:139-161.
Mao C, et al. Von Recklinghausen’s disease associated with duodenal somatostatinoma: contrast of duodenal versus pancreatic somatostatinomas. J Surg Oncol. 1995;59:67-73.
Marin C, et al. Role of liver transplantation in the management of unresectable neuroendocrine liver metastases. Transplant Proc. 2007;39:2302-2303.
McAuley G, et al. Multimodality preoperative imaging of pancreatic insulinomas. Clin Radiol. 2005;60:1039-1050.
McGuigan J, et al. Secretin injection test in gastrinoma. Gastroenterology. 1980;79:1324-1331.
McStay MK, et al. Large-volume liver metastases from neuroendocrine tumors: hepatic intraarterial 90Y-DOTA lanreotide as effective palliative therapy. Radiology. 2005;237:718-726.
Modlin I, et al. The early diagnosis of gastrinoma. Ann Surg. 1982;196:512-517.
Moertel CG, et al. Phase II trial of doxorubicin therapy for advanced islet cell carcinoma. Cancer Treat Rep. 1982;66:1567-1569.
Morrow E, et al. Surgical management of Zollinger–Ellison Syndrome: state of the art. Surg Clin North Am. 2009;89:1091-1103.
Motellaro V, et al. Long-term results of selective surgical approach to management of Zollinger–Ellison syndrome in patients with MEN-1. Am Surg. 2009;75:730-733.
Muscarella P, Ellison EC. Pancreatic islet cell tumors excluding gastrinoma. In: Cameron, JL, editor. Current Surgical Therapy. 8th ed. Philadelphia: Mosby; 2004:520-524.
Musunuru S, et al. Metastatic neuroendocrine hepatic tumors: resection improves survival. Arch Surg. 2006;141:1000-1004.
Nesi G, et al. Somatostatinoma: clinico-patholgocial features of three cases and literature reviewed. J Gastroenterol Hepatol. 2007;23:521-526.
Nightingale KJ, et al. Glucagonoma syndrome: survival 24 years following diagnosis. Digest Surg. 1999;16:68-71.
Nikfarjam M, et al. Improved contemporary surgical management of insulinomas: a 25-year experience at the Massachusetts General Hospital. Ann Surg. 2008;4:165-172.
Noone T, et al. Imaging and localization of islet-cell tumours of the pancreas on CT and MRI. Best Pract Res Clin Endocrinol Metab. 2005;9:195-211.
Norton JA, et al. Intraoperative ultrasonographic localization of islet cell tumors: a prospective comparison to palpation. Ann Surg. 1988;207:160-168.
Norton J, et al. Surgery to cure Zollinger–Ellison syndrome. N Engl J Med. 1999;341:635-644.
Norton JA, et al. Morbidity and mortality of aggressive resection in patients with advanced neuroendocrine tumors. Arch Surg. 2003;138:859-866.
Norton J, et al. Does the use of routine duodenotomy (DUODX) affect the rate of cure, development of liver metastases, or survival in patients with Zollinger–Ellison syndrome? Ann Surg. 2004;239:617-625.
Norton J, et al. Surgery increases survival in patients with gastrinoma? Ann Surg. 2006;44:590-598.
Oberg K. Interferon in the management of neuroendocrine GEP-tumors: a review. Digestion. 2000;62:92-97.
Oberg K, et al. Consensus report on the use of somatostatin analogs for the management of neuroendocrine tumors of the gastroenteropancreatic system. Ann Oncol. 2004;15:966-973.
O’Dorisio TM, et al. Medical therapy of VIPomas. Endocrinol Metab Clin N Am. 1989;18:545.
O’Toole D, et al. Rare functioning pancreatic endocrine tumors. Neuroendocrinology. 2006;84:189-195.
Pape UF, et al. Prognostic relevance of a novel TNM classification system for upper gastroenteropancreatic neuroendocrine tumors. Cancer. 2008;113:256-265.
Peng SY, et al. Diagnosis and treatment of VIPoma in China: diagnosis and treatment of VIPoma. Pancreas. 2004;28:93-97.
Pierce R, et al. Outcomes analysis of laparoscopic resection of pancreatic neoplasms. Surg Endos. 2007;21:579-586.
Pitt SC, et al. Hepatic neuroendocrine metastases: chemo- or bland embolization? J Gastrointest Surg. 2008;12:1951-1960.
Prinz RA, et al. Clinical aspects of glucagon-producing islet cell tumors. Am J Gastroenterol. 1981;76:125-131.
Prinz RA, et al. “Non-functioning” islet cell carcinoma of the pancreas. Am Surg. 1983;49:345-349.
Ramanathan RK, et al. Phase II trial of dacarbazine (DTIC) in advanced pancreatic islet cell carcinoma: study of the Eastern Cooperative Oncology Group-E6282. Ann Oncol. 2001;12:1139-1143.
Reubi JC, et al. Concomitant expression of several peptide receptors in neuroendocrine tumors: molecular basis for in vivo multireceptor tumor targeting. Eur J Nucl Med. 2003;30:781-793.
Ricci S, et al. Octreotide acetate long-acting release in patients with metastatic neuroendocrine tumors pretreated with lanreotide. Ann Oncol. 2000;11:1127-1130.
Rinke A, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID study group. J Clin Oncol. 2009;27:4656-4663.
Roche A, et al. Trans-catheter arterial chemoembolization as first-line treatment for hepatic metastases from endocrine tumors. Eur Radiol. 2003;13:136-140.
Roy P, et al. Zollinger–Ellison syndrome: clinical presentation in 261 patients. Medicine. 2000;79:379-411.
Ruzzniewski P, et al. Localization of gastrinomas by endoscopic ultrasonography in patients with Zollinger–Ellison syndrome. Surgery. 1995;17:629-635.
Safford SD, et al. Iodine-131 metaiodobenzylguanidine treatment for metastatic carcinoid: results in 98 patients. Cancer. 2004;101:1987-1993.
Sarmiento JM, et al. Surgical treatment of neuroendocrine metastases to the liver: a plea for resection to increase survival. J Am Coll Surg. 2003;197:29-37.
Scherubl H, et al. Treatment of carcinoid syndrome with a depot formulation of the somatostain analogue lanreotide. Eur J Cancer. 1994;20:1590-1591.
Schindl M, et al. Is the new classification of neuroendocrine pancreatic tumors of clinical help? World J Surg. 2000;24:1312-1318.
Schurr PG, et al. Aggressive surgery improves long-term survival in neuroendocrine pancreatic tumors: an institutional experience. Ann Surg. 2007;245:273-281.
Service FJ, et al. The prolonged fast. J Clin Endocrinol Metab. 2000;85:3973-3974.
Skogseid B, et al. Multiple endocrine neoplasia type 1: a 10-year prospective screening study in four kindreds. J Clin Endocrinol Metab. 1991;73:281-287.
Sotoudehmanesh R, et al. Endoscopic ultrasonography in the localization of insulinoma. Endocrine. 2007;31:238-241.
Stabile BE, et al. The gastrinoma triangle: operative implications. Am J Surg. 1984;47:25-31.
Stacpoole PW. The glucagonoma syndrome: clinical features, diagnosis and treatment. Endocr Rev. 1981;2:347-361.
Steinmuller T, et al. Consensus guidelines for the management of patients with liver metastases from digestive (neuro) endocrine tumors: foregut, midgut, hindgut, and unknown primary. Neuroendocrinology. 2008;87:47-62.
Strosberg JR, et al. A review of systemic and liver-directed therapies for metastatic neuroendocrine tumors of the gastroenteropancreatic tract. Cancer Control. 2011;18:127-137.
Strosberg JR, et al. First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer. 2011;117:268-275.
Tanaka S, et al. Duodenal somatostatinoma: a case report and review of 31 cases with special reference to the relationship between tumor size and metastasis. Pathol Int. 2000;50:146-152.
Thoeni RF, et al. Detection of small functional islet cell tumours in pancreas: selection of MR imaging sequences for optimal sensitivity. Radiology. 2000;214:483-490.
Thomas-Marques L, et al. Prospective endoscopic ultrasonographic evaluation of the frequency of nonfunctioning pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. Am J Gastroentrol. 2006;101:266-273.
Thomason JW, et al. Somatostatin receptor scintigraphy: the definitive technique for characterizing vasoactive intestinal peptide-secreting tumors. Clin Nucl Med. 2000;9:661-664.
Verner JVJr, Morrison AB. Islet cell tumor and a syndrome of refractory watery diarrhea and hypokalemia. Am J Med. 1958;25:374-380.
Vilar E, et al. Chemotherapy and role of the proliferation marker Ki-67 in digestive neuroendocrine tumors. Endocrine-Related Cancer. 2007;14:221-232.
Weber HC, et al. Determinant of metastatic rate and survival in patients with Zollinger–Ellison syndrome: a prospective long-term study. Gastroenterology. 1995;108:1637-1649.
Weber HC, et al. Diagnosis and management of Zollinger–Ellison syndrome. Semin Gastrointest Dis. 1995;6:79-89.
Wermers RA, et al. The glucagonoma syndrome: clinical and pathologic features in 21 patients. Medicine. 1966;75:53-63.
Yao JC, et al. Targeting vascular endothelial growth factor in advanced carcinoids tumor: a random assignment phase II study of depot octreotide with bevacizumab and pegylated interferon alpha-2b. J Clin Oncol. 2008;26:1316-1323.