[level-membership-for-surgery-category]
Chapter 71 Portal hypertension in children
Special Features of Portal Hypertension in Children
Portal hypertension secondary to chronic liver disease and prehepatic or posthepatic vascular events is a major cause of morbidity and death in both adults and children (see also Chapter 70A). However, children with these conditions provide a different set of challenges in the understanding and management of portal hypertension because of a predominance of congenital etiologies combined with growth and developmental considerations.
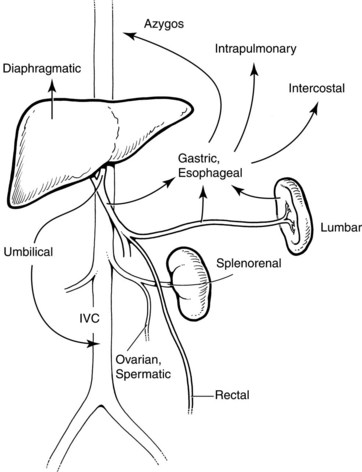
In general, portal hypertension is the result of a combination of increased portal blood flow and increased portal resistance; it occurs when portal blood pressure rises above approximately 10 mm Hg. Signs and symptoms are primarily the result of decompression of this elevated pressure through portosystemic collaterals, but special developmental vascular alterations apply in children (Fig. 71.1). The major problems seen in children are bleeding varices, ascites, and malnutrition. Encephalopathy, portopulmonary hypertension, and hepatorenal syndrome, although important when they do occur, are seen less frequently in children. Splenomegaly and hypersplenism rarely require specific intervention.
Causes of Portal Hypertension in Children
Portal hypertension may arise as a result of an extrahepatic (portal vein), posthepatic (hepatic vein), or intrahepatic block; the block may be presinusoidal, sinusoidal, or postsinusoidal. Causes in children are shown in Box 71.1.
Extrahepatic Causes
Although possibly declining in frequency, extrahepatic portal venous obstruction, caused by a congenital thrombotic or atretic process, is an important cause of noncirrhotic portal hypertension, especially in developing countries but certainly not confined there (Sarin & Agarwal, 2002). Septic or traumatic umbilical vein injury from omphalitis and/or catherization accounts for some cases, but most cases are idiopathic or represent a congenital malformation of the portal venous system. Portal vein stenosis or thrombosis may also occur in the context of portal vein anastomoses in liver transplantation. In the congenital form, the portal vein is transformed into a cavernoma, resulting in portal hypertension and esophagogastic varices. Importantly, from a treatment standpoint, splenic vein or more extensive portal system involvement results in somewhat different hemodynamics, with an extensive collateral circulation involving a preponderance of gastric varices (Shah et al, 2003) and sometimes ectopic varices involving paracholecystic, paracholedochal, and pancreatoduodenal veins. Esophageal bleeding, or sometimes gastric variceal bleeding, is thus the most important consequence, although the presence of naturally occurring shunts may reduce the risk over time, usually by the second decade of life. Besides variceal bleeding, which is the commonest presentation, hypersplenism is common, and anemia, easy bruising from thrombocytopenia, and abdominal pain may be presenting features. Some patients may develop symptomatic portal biliary obstruction. Growth retardation from malabsorption as a result of failure of the enteropancreatic and enterohepatic circulation is not uncommon. Overt encephalopathy as a result of shunting appears uncommon, but subclinical signs may occur, including disturbed neurocognitive function, particularly attention and short-term memory problems (Mack et al, 2006). Although the liver may appear normal, reversible decompensation may be seen after an acute variceal hemorrhage, and functional compromise may develop over the long term.
Intrahepatic Causes
Portal hypertension may result from a range of presinusoidal, sinusoidal, and postsinusoidal causes of increased portal bed resistance within the liver. Presinusoidal conditions, such as congenital hepatic fibrosis, do not result in impaired liver function. Congenital hepatic fibrosis is a developmental disorder that belongs to the family of hepatic ductal plate malformations and is characterized histologically by a variable degree of periportal fibrosis and irregularly shaped proliferating bile ducts (Summerfield et al, 1986). Liver biopsy is highly specific for the diagnosis. In most patients, the first manifestations of the disease are signs or symptoms related to portal hypertension, especially splenomegaly and varices, often with spontaneous gastrointestinal (GI) bleeding that occurs from early childhood and sometimes even into adulthood.
Increased sinusoidal resistance and portal hypertension occur almost invariably in cirrhosis in children. In children, common causes include forms of infant cholestasis, such as biliary atresia, as well as a range of metabolic disorders, infections, toxins, and vascular and nutritional diseases. Cirrhosis is a chronic, diffuse disease characterized by irreversible widespread hepatic fibrosis with regenerative nodule formation (see Chapter 6, Chapter 70A, Chapter 70B ). The prominent fibrous tissue contains vascular anastomoses, which cause hemodynamic alterations and portosystemic shunting. This diffuse pathology superimposed on the primary liver disease often obscures the nature of the original insult. The major pathophysiologic consequences are the result of impaired hepatic function and portal hypertension. Progression to cirrhosis and its complications in pediatric liver diseases is highly variable and represents an important consideration in management, particularly when considering surgical options that may compromise later outcomes. In some conditions, such as neonatal extrahepatic biliary atresia, the development of cirrhosis can be extraordinarily rapid, occurring by 12 to 16 weeks of age with liver failure as early as 24 weeks of age. Early diagnosis and surgical treatment by hepatoportoenterostomy improves outcome, but in most cases, liver transplant becomes the only available treatment option. Other disorders, such as cystic fibrosis–associated focal biliary cirrhosis, can be compatible with normal liver function for many years, presenting with signs of portal hypertension in the second decade of life.
Postsinusoidal intrahepatic conditions, such as venoocclusive disease, are rare in children and usually occur only in the context of chemotherapy for childhood cancers (Gharib et al, 2006) or occasionally related to toxin ingestion, such as from eating poisonous mushrooms. However, posthepatic portal hypertension is a significant condition in children (see Box 71.1). Obstruction to hepatic venous outflow, such as from hepatic vein anastomotic stenosis, can occur after liver transplantation, with Budd-Chiari syndrome, or as a result of cardiac lesions that cause an increase in right atrial pressure and/or chronic systemic venous hypertension. Acute Budd-Chiari syndrome is rare in children; although it may occur with some thrombophilic disorders, it is usually idiopathic and is not associated with the causes seen in adults, such as myeloproliferative disorders. A cavopulmonary or atriopulmonary shunt, known as a Fontan procedure, allows life-saving systemic-pulmonary blood flow in single-ventricle syndromes in neonates but results in chronic systemic venous hypertension (pressures may be >20 mm Hg) and eventually to portal hypertension (Narkewicz et al, 2003).
Effects of Portal Hypertension in Children
The major pathologic effect from portal hypertension is the development of collaterals that carry blood from the portal venous system to the systemic circulation in the upper part of the stomach, esophagus, rectum, and falciform ligament. These collaterals may drain into the IVC via the umbilical vein remnant, or they may drain into the left renal vein (see Fig. 71.1). Absence or disconnection of the IVC or interruption to the azygos system, such as occurs in some cases of bilary atresia, may cause special concern. Similarly, in extrahepatic portal venous malformations, the splenic vein can be small and possibly thrombosed.
Although changes in vascular resistance to flow of blood between the splanchnic bed and the right atrium appear to be the initial events in the development of portal hypertension, a number of other hemodynamic changes contribute to and amplify the increased portal blood pressure. There is a hyperdynamic circulatory state with increased cardiac and decreased splanchnic arteriolar tone, both of which increase portal inflow. Changes in intravascular volume also play an important role, as do alterations in adrenergic tone in the splanchnic system. These observations have led to new experimental and clinical studies that suggest possible pharmacologic treatments for portal hypertension (Boyer, 2001). However, because the major clinical effect is that of bleeding from esophageal varices, direct treatment of variceal hemorrhage, or shunt surgery in selected cases, remains the primary approach, except in the presence of liver decompensation, in which case the treatment of choice is liver transplantation (see Chapters 97A and 98A).
Clinical Features
The main clinical features are splenomegaly, nutritional growth failure, and ascites, along with esophageal, gastric, and rectal varices (Box 71.2).
Signs of hepatic encephalopathy are subtle and uncommon in children with portal hypertension. Malnutrition with reduced lean tissue and fat stores as well as poor linear growth is a well-recognized and important feature resulting from malabsorption and impaired protein synthesis (Chin et al, 1992). Spontaneous bruising caused by impaired hepatic production of clotting factors and thrombocytopenia as a result of hypersplenism are signs of advanced disease. Cirrhosis with decompensation may also be associated with changes in the systemic and pulmonary circulations and with arteriolar vasodilation, increased blood volume, a hyperdynamic circulatory state, and cyanosis as a result of intrapulmonary shunting. Renal failure is a late but serious event.
Diagnosis
Angiography, either direct venography or computed tomography (CT) angiography, can provide important information about the site of the blockage, the size and patency of major veins in the portal system, and relationship to the coronary esophageal or other varices (see Chapters 16 and 19). Suspected obstruction to hepatic venous outflow requires venography and/or cardiac catheterization, which are the diagnostic procedures of choice in such cases. Pressure gradient measurements may be useful across venous blocks and to determine the magnitude of the portal pressure.
Management (See Chapter 74, Chapter 75a, Chapter 75b, Chapter 75c, Chapter 76a, Chapter 76b, Chapter 76c, Chapter 76d, Chapter 76e )
Emergency Therapy
Bleeding esophageal varices in children require emergency treatment (Fig. 71.2); no matter how small the hemorrhage, admission to the nearest hospital with blood transfusion facilities is advised. As soon as the blood transfusion available and the patient has a secure intravenous infusion line and is hemodynamically stable, referral to a tertiary center with experience in the management of variceal hemorrhage in children is recommended. Initial melena or other signs of a sentinel hemorrhage may precede sudden hematemesis and shock, which require rapid blood transfusion to prevent death. Significant bleeding with hypotension impairs hepatic perfusion, often causes deterioration of liver function, and precipitates ascites and encephalopathy. Initial fluid management in the form of crystalloids followed by red blood cell transfusion is important, and any coagulopathy should be corrected with vitamin K and fresh frozen plasma. Pharmacologic therapy with a short-acting splanchnic vasoconstrictor may be useful (Boyer, 2001). Octreotide is the drug of choice in this circumstance (maximum dose 1 µg/kg/h IV or 2 to 4 µg/kg/8 h subcutaneously for 24 h, or until bleeding has ceased) because of fewer side effects (Moitinho, 2001). Alternatives are vasopressin (0.3 U/kg as a bolus over 20 minutes followed by continuous infusion of the same amount on an hourly basis) or its inactive precursor, glypressin (0.01 mg/kg bolus q4-6h or 0.05 mg/kg infusion over 6 h for 24 to 48 h). Common side effects include skin pallor, abdominal colic, and chest pain. Studies in adults have indicated that an adjunctive vasodilator, such as nitroglycerine in the form of a 10-mg patch, may reduce these hemodynamic complications. Nasogastric intubation is an essential part of management, allowing the documentation of ongoing bleeding and the removal of blood that could precipitate encephalopathy.
When the patient is hemodynamically stable, endoscopy is indicated. A significant percentage of patients with known varices have bleeding from sources other than the varices, including duodenal or gastric ulceration. In addition, meta-analysis of multiple controlled adult studies clearly shows that endoscopic treatment plus pharmacologic treatment is currently the best measure to achieve initial control of significant acute variceal bleeding (Banares et al, 2002).
Endoscopic Treatment of Acute Variceal Bleeding
Episodes of minor variceal hemorrhage may spontaneously terminate, but endoscopic treatment by sclerotherapy or band ligation is often necessary (Fig. 71.3). Both techniques are well described in children (Goenka et al, 1993; Goncalves et al, 2000; Zargar et al, 2002). In adults, comparative trials of sclerotherapy and ligation indicate equal efficacy in controlling bleeding, reducing rebleeding, and ablating varices, but fewer adverse effects are reported with banding. The two procedures can be complementary, and access to both procedures may be warranted. Ligation may be technically difficult in an esophagus awash with blood or for small varices, particularly in small infants, where entrapment of part of the esophageal wall with perforation or bleeding can occur. In these circumstances, sclerotherapy is more appropriate.
Emergency Surgical Approaches and Emergency Portosystemic Shunts (See Chapters 75A to 76E)
Emergency creation of portosystemic shunts or other surgical therapy is usually a last resort for persistent, acute, exsanguinating variceal hemorrhage. Patients with shunts who come to the emergency department often have gastric variceal bleeding. Considerations in these circumstances include potential death, and the range of techniques available include transjugular intrahepatic portosystemic shunting (TIPS), surgical shunting, esophageal transection, and esophagogastric devasculation with splenectomy. TIPS is an attractive approach because it does not require major surgery. It has been used effectively in critically ill adults to control bleeding before liver transplantation, and it has also been used in some children with bleeding stomal varices (Lagier et al, 1994). The procedure appears to decrease portal pressure acutely, although 60% occlude within 3 to 12 months; therefore it is best viewed as a bridging procedure only. Pediatric application is limited by size constraints, but in experienced hands and in selected children older than 2 to 5 years, the procedure has application and is preferable to major shunt surgery for hepatic causes of portal hypertension.
Prophylaxis Against the First Gastrointestinal Bleed
In recognized cases of portal hypertension with varices, controversy exists regarding whether any therapy reduces the risk or prevents the occurrence of GI bleeding. However, in all cases it is reasonable to be prepared for the possibility by ensuring that the child’s caregivers understand the importance of seeking early medical advice by attending the nearest hospital for blood cross-matching and appropriate referral to a tertiary unit. β-Blockers, such as propranolol and the more selective atenolol, reduce hepatic arterial and portal vein blood flow and have been studied with respect to reduction in portal pressures to less than 12 mm Hg, thereby reducing the risk of an initial hemorrhage (Shasidhar et al, 1999). Under certain circumstances, prophylactic sclerotherapy may be indicated, but the potential in children for bleeding of known varices that have never bled is conjectural. Goncalves and colleagues (2000) compared sclerotherapy to no treatment in a controlled clinical trial in children and found a reduced risk of bleeding but an increase in portal gastropathy.
Prevention of Recurrent Gastrointestinal Bleeding
Intrahepatic Causes of Portal Hypertension
Direct Obliteration of Varices
Where bleeding has occurred from intrahepatic causes of portal vein obstruction, direct obliteration of the varices is the initial treatment of choice, although consideration of the underlying liver disease is the major determinant of long-term management. Randomized controlled trials in adults have shown a reduction in the frequency of bleeding and improved survival. Although no randomized controlled trials have been performed in children, several large studies of sclerotherapy or banding in children with portal hypertension indicate that both procedures are safe, and both reduce the chance of rebleeding (Fox et al, 1995; Goenka et al, 1993; McKiernan et al, 2002). Neither technique reduces portal pressure, but both obliterate the dangerous varices. The procedures described above may cause some interference with the vascular hemodynamics; hypersplenism and portal gastropathy temporarily gets worse, but ultimately, with time, spontaneous portosystemic shunts will arise to reduce portal pressures.
In multiple controlled trials in adults, several vasoactive drugs have also now been documented to reduce the risk of rebleeding by reducing portal and systemic pressures (Boyer, 2001). A combination of a nonselective β-blockers and certain nitrates, such as isosorbide 5-mononitrate, are the drugs of choice, aiming at a 25% reduction in resting heart rate. Major adverse effects of β-blockers include reactive airway disease and heart block, in which cases they are contraindicated.
Surgery
Where there is active liver disease and a significant risk of death from bleeding, surgical management of portal hypertension is a major consideration (Box 71-3). Such patients should ideally be evaluated and treated in a transplant center, where the range of surgical options can be assessed. Surgical portosystemic shunts may reduce the risk of GI bleeding, but they cause a decrease in portal blood flow and hepatic perfusion, and they carry the risks of hepatic decompensation and encephalopathy, which either enhances the difficulty of liver transplantation or precludes it entirely. Moreover, randomized controlled trials in adult patients have not shown any significant improvement in survival with portosystemic shunts in patients with intrahepatic causes of portal hypertension. If a shunt operation is contemplated in a critically ill patient, the use of TIPS as a bridging procedure is probably the best choice. Ultimately, the surgical procedure of choice for uncontrolled portal hypertension as a result of intrahepatic disease is liver transplantation. In nonshunt, nontransplant candidates, a Sugiura procedure (esophageal disconnection and devascularization) may be life saving, with the added advantage of a low-risk of encephalopathy.
Special consideration should be given to shunt surgery in some cases of liver disease associated with cystic fibrosis, in which there is a slow evolution of hepatic dysfunction (Debray et al, 1999), and in congenital hepatic fibrosis, a presinusoidal cause of portal hypertension not associated with the occurrence of liver synthetic dysfunction. In these cases, the choice of the type of shunt is determined by the vascular anatomy, the size of the veins, the risk of thrombosis and failure, and the risk of encephalopathy. Selective shunts are preferred, such as distal splenorenal or distal splenoadrenal shunts (see Chapter 76A, Chapter 76B, Chapter 76C, Chapter 76D, Chapter 76E ; Valayer et al, 1985; Kato et al, 2000); the latter often allows better size matching of the splenic and enlarged adrenal vein and avoids renal vein clamping.
Extrahepatic Portal Venous Obstruction
Portal hypertension as a result of extrahepatic portal venous obstruction in children is associated with a better long-term prognosis and quality of life than portal hypertension associated with intrahepatic diseases, and management considerations are quite different. Acute variceal bleeding is usually well tolerated because of normal liver function, but recurrent variceal bleeds can be associated with significant morbidity (Zargar et al, 2004). Although long-term outcomes of individual center approaches have been documented (Bambini et al, 2000; Orloff et al, 2002; Zargar et al, 2004), no controlled trials have compared long-term outcomes from the two main approaches to therapy: direct variceal ablation and shunt surgery. This is compounded by the fact that some patients never bleed at all, and anecdotal and published data suggest that some patients develop spontaneous decompression of their varices over time (Goenka et al, 1993; Lykaverios et al, 2000).
Direct Variceal Ablation
Endoscopic therapy has gained increasing acceptance in the treatment of bleeding esophageal varices in children with extrahepatic portal hypertension. Zargar and colleagues (2004) reported a 15-year follow-up of 69 children. Almost 90% had no rebleeding after eradication, which was achieved in 90% of patients. Maksoud and colleagues (1991) reported that 7% to 8% of patients did not respond to sclerotherapy and subsequently underwent shunt surgery. Stringer and Howard (1994) reported that only 4 (11%) of 36 patients required shunt surgery after variceal sclerotherapy over an 8-year follow-up. All these studies had little or no morbidity or deaths, but several repeat sessions of sclerotherapy were necessary during follow-up for complete variceal eradication. The main reason for long-term failure was the occurrence of gastric varices, but clearly this does not occur in all cases, and some studies suggest that the occurrence of spontaneous shunts after successful sclerotherapy negates the need for shunt surgery altogether (Goenka et al, 1993).
Surgery
Surgery may be required for patients with extrahepatic portal hypertension with preserved liver function for recurrent bleeding or, rarely, for massive hypersplenism. Surgical options in the management of extrahepatic portal venous obstruction include portosystemic shunts, Rex shunts (mesenteric left portal bypass), devascularization, and transection (see Box 71.3).
Although earlier outcomes of portosystemic shunts were disappointing, some recent reports of have shown recurrence of bleeding in only 2.5% to 10%, low mortality rates, a continued shunt thrombosis rate of 7% to 13%, and an apparently low risk of encephalopathy (Botha et al, 2004; Kato et al, 2000; Orloff et al, 2002). Nonselective mesocaval shunts and the more selective distal splenorenal shunt have been the procedures of choice in the past, although a central splenorenal shunt with splenectomy is sometimes advocated if massive hypersplenism and pain from splenic infarcts are present. However, these shunts may not be technically possible because of thrombotic involvement of splenic or mesenteric veins, and in some cases, particularly where varices are derived from the cavernoma, certain shunts may not alter variceal pressures to the coronary esophageal veins.
The Rex shunt, usually an internal jugular vein graft and mesenteric to left portal vein bypass, has the major theoretical advantage of restoring portal flow to the liver and reducing risk of encephalopathy, and it obviates the technical problems associated with splenic vein thrombosis (Bambini et al, 2000; de Ville de Goyet, 1999). First introduced for portal vein thrombosis after liver transplantation, this procedure may become the surgical procedure of choice for all causes of extrahepatic portal vein obstruction. Thrombosis and stenosis rates appear to be similar to those of portosystemic shunts. Patency of the intrahepatic portal system can be difficult to prove before the procedure. The use of a preprocedural portal venogram to ensure patency antegrade to the liver is important when performing any type of shunt surgery for extrahepatic venous obstruction, particularly where a cavernoma is present; selective division of the portal system (e.g., coronary branch), where there are residual collaterals, must also be considered.
Bambini DA, et al. Experience with the Rex shunt (mesenterico-left portal bypass) in children with extrahepatic portal hypertension. J Pediatr Surg. 2000;35(1):13-18.
Banares R, et al. Endoscopic treatment versus endoscopic plus pharmacologic treatment for acute variceal bleeding: a meta-analysis. Hepatology. 2002;35(3):609-615.
Botha JF, et al. Portosystemic shunts in children: a 15-year experience. J Am Coll Surg. 2004;199(2):179-185.
Boyer TD. Pharmacologic treatment of portal hypertension: past, present, and future. Hepatology. 2001;34(4 Pt 1):834-839.
Chin SE, et al. The nature of malnutrition in children with end-stage liver disease awaiting orthotopic liver transplantation. Am J Clin Nutr. 1992;56(1):164-168.
Debray D, et al. Outcome of cystic fibrosis-associated liver cirrhosis: management of portal hypertension. J Hepatol. 1999;31(1):77-83.
de Ville de Goyet J, et al. Treatment of extrahepatic portal hypertension in children by mesenteric-to-left portal vein bypass: a new physiological procedure. Eur J Surg. 1999;165(8):777-781.
Fox VL, et al. Endoscopic ligation of esophageal varices in children. J Pediatr Gastroenterol Nutr. 1995;20(2):202-208.
Gharib MI, et al. Venous occlusive disease in children. Thromb Res. 2006;118(1):27-38.
Goenka AS, et al. Therapeutic upper gastrointestinal endoscopy in children: an audit of 443 procedures and literature review. J Gastroenterol Hepatol. 1993;8(1):44-51.
Goncalves ME, Cardoso SR, Maksoud JG. Prophylactic sclerotherapy in children with esophageal varices: long-term results of a controlled prospective randomized trial. J Pediatr Surg. 2000;35(3):401-405.
Kato T, et al. Portosystemic shunting in children during the era of endoscopic therapy improved postoperative growth parameters. J Pediatr Gastroenterol Nutr. 2000;30(4):419-425.
Lagier E, et al. Treatment of bleeding stomal varices using transjugular intrahepatic portosystemic shunt. J Pediatr Gastroenterol Nutr. 1994;18:501-503.
Lebrec D. Review: pharmacotherapeutic agents in the treatment of portal hypertension. J Gastroenterol Hepatol. 1997;12(2):159-166.
Lykaverios P, et al. Risk of gastrointestinal bleeding during adolescence and early adulthood in children with portal vein obstruction. J Pediatr. 2000;136(6):805-808.
Mack CL, et al. Surgically restoring portal blood flow to the liver in children with primary extrahepatic portal vein thrombosis improves fluid neurocognitive ability. Pediatrics. 2006;117(3):405-412.
Maksoud JG, et al. The endoscopic and surgical management of portal hypertension in children: analysis of 123 cases. J Pediatr Surg. 1991;26(2):178-181.
McKiernan PJ, Beath SV, Davison SM. A prospective study of endoscopic esophageal variceal ligation using a multiband ligator. J Pediatr Gastroenterol Nutr. 2002;34(2):207-211.
Moitinho E, et al. Multicenter randomized controlled trial comparing different schedules of somatostatin in the treatment of acute variceal bleeding. J Hepatol. 2001;35(6):712-718.
Narkewicz MR, et al. Hepatic dysfunction following the Fontan procedure. J Pediatr Gastroenterol Nutr. 2003;36(3):352-357.
Orloff MJ, et al. Bleeding esophagogastric varices from extrahepatic portal hypertension: 40 years’ experience with portal-systemic shunt. J Am Coll Surg. 2002;194(6):717-728.
Sarin SK, Agarwal SR. Extrahepatic portal vein obstruction [review]. Semin Liver Dis. 2002;22(1):43-58.
Shah SR, Deshmukh HL, Mathur SK. Extensive portal and splenic vein thrombosis: differences in hemodynamics and management. Hepatogastroenterology. 2003;50(52):1085-1089.
Shasidhar H, Langhans N, Grand RJ. Propranolol in prevention of portal hypertensive hemorrhage in children: a pilot study. J Pediatr Gastroenterol Nutr. 1999;29:12-17.
Stringer MD, Howard ER. Long-term outcome after injection sclerotherapy for oesophageal varices in children with extrahepatic portal hypertension. Gut. 1994;35(2):257-259.
Summerfield JA, et al. Hepatobiliary fibropolycystic diseases: a clinical and histological review of 51 patients. J Hepatol. 1986;2(2):141-156.
Valayer J, et al. Shunt surgery for treatment of portal hypertension in children. World J Surg. 1985;9(2):258-268.
Zargar SA, et al. Endoscopic ligation compared with sclerotherapy for bleeding esophageal varices in children with extrahepatic portal venous obstruction. Hepatology. 2002;36(3):666-672.
Zargar SA, et al. Fifteen-year follow up of endoscopic injection sclerotherapy in children with extrahepatic portal venous obstruction. J Gastroenterol Hepatol. 2004;19(2):139-145.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 71 Portal hypertension in children
Special Features of Portal Hypertension in Children
Portal hypertension secondary to chronic liver disease and prehepatic or posthepatic vascular events is a major cause of morbidity and death in both adults and children (see also Chapter 70A). However, children with these conditions provide a different set of challenges in the understanding and management of portal hypertension because of a predominance of congenital etiologies combined with growth and developmental considerations.
In general, portal hypertension is the result of a combination of increased portal blood flow and increased portal resistance; it occurs when portal blood pressure rises above approximately 10 mm Hg. Signs and symptoms are primarily the result of decompression of this elevated pressure through portosystemic collaterals, but special developmental vascular alterations apply in children (Fig. 71.1). The major problems seen in children are bleeding varices, ascites, and malnutrition. Encephalopathy, portopulmonary hypertension, and hepatorenal syndrome, although important when they do occur, are seen less frequently in children. Splenomegaly and hypersplenism rarely require specific intervention.
Causes of Portal Hypertension in Children
Portal hypertension may arise as a result of an extrahepatic (portal vein), posthepatic (hepatic vein), or intrahepatic block; the block may be presinusoidal, sinusoidal, or postsinusoidal. Causes in children are shown in Box 71.1.
Extrahepatic Causes
Although possibly declining in frequency, extrahepatic portal venous obstruction, caused by a congenital thrombotic or atretic process, is an important cause of noncirrhotic portal hypertension, especially in developing countries but certainly not confined there (Sarin & Agarwal, 2002). Septic or traumatic umbilical vein injury from omphalitis and/or catherization accounts for some cases, but most cases are idiopathic or represent a congenital malformation of the portal venous system. Portal vein stenosis or thrombosis may also occur in the context of portal vein anastomoses in liver transplantation. In the congenital form, the portal vein is transformed into a cavernoma, resulting in portal hypertension and esophagogastic varices. Importantly, from a treatment standpoint, splenic vein or more extensive portal system involvement results in somewhat different hemodynamics, with an extensive collateral circulation involving a preponderance of gastric varices (Shah et al, 2003) and sometimes ectopic varices involving paracholecystic, paracholedochal, and pancreatoduodenal veins. Esophageal bleeding, or sometimes gastric variceal bleeding, is thus the most important consequence, although the presence of naturally occurring shunts may reduce the risk over time, usually by the second decade of life. Besides variceal bleeding, which is the commonest presentation, hypersplenism is common, and anemia, easy bruising from thrombocytopenia, and abdominal pain may be presenting features. Some patients may develop symptomatic portal biliary obstruction. Growth retardation from malabsorption as a result of failure of the enteropancreatic and enterohepatic circulation is not uncommon. Overt encephalopathy as a result of shunting appears uncommon, but subclinical signs may occur, including disturbed neurocognitive function, particularly attention and short-term memory problems (Mack et al, 2006). Although the liver may appear normal, reversible decompensation may be seen after an acute variceal hemorrhage, and functional compromise may develop over the long term.
Intrahepatic Causes
Portal hypertension may result from a range of presinusoidal, sinusoidal, and postsinusoidal causes of increased portal bed resistance within the liver. Presinusoidal conditions, such as congenital hepatic fibrosis, do not result in impaired liver function. Congenital hepatic fibrosis is a developmental disorder that belongs to the family of hepatic ductal plate malformations and is characterized histologically by a variable degree of periportal fibrosis and irregularly shaped proliferating bile ducts (Summerfield et al, 1986). Liver biopsy is highly specific for the diagnosis. In most patients, the first manifestations of the disease are signs or symptoms related to portal hypertension, especially splenomegaly and varices, often with spontaneous gastrointestinal (GI) bleeding that occurs from early childhood and sometimes even into adulthood.
Increased sinusoidal resistance and portal hypertension occur almost invariably in cirrhosis in children. In children, common causes include forms of infant cholestasis, such as biliary atresia, as well as a range of metabolic disorders, infections, toxins, and vascular and nutritional diseases. Cirrhosis is a chronic, diffuse disease characterized by irreversible widespread hepatic fibrosis with regenerative nodule formation (see Chapter 6, Chapter 70A, Chapter 70B ). The prominent fibrous tissue contains vascular anastomoses, which cause hemodynamic alterations and portosystemic shunting. This diffuse pathology superimposed on the primary liver disease often obscures the nature of the original insult. The major pathophysiologic consequences are the result of impaired hepatic function and portal hypertension. Progression to cirrhosis and its complications in pediatric liver diseases is highly variable and represents an important consideration in management, particularly when considering surgical options that may compromise later outcomes. In some conditions, such as neonatal extrahepatic biliary atresia, the development of cirrhosis can be extraordinarily rapid, occurring by 12 to 16 weeks of age with liver failure as early as 24 weeks of age. Early diagnosis and surgical treatment by hepatoportoenterostomy improves outcome, but in most cases, liver transplant becomes the only available treatment option. Other disorders, such as cystic fibrosis–associated focal biliary cirrhosis, can be compatible with normal liver function for many years, presenting with signs of portal hypertension in the second decade of life.
Postsinusoidal intrahepatic conditions, such as venoocclusive disease, are rare in children and usually occur only in the context of chemotherapy for childhood cancers (Gharib et al, 2006) or occasionally related to toxin ingestion, such as from eating poisonous mushrooms. However, posthepatic portal hypertension is a significant condition in children (see Box 71.1). Obstruction to hepatic venous outflow, such as from hepatic vein anastomotic stenosis, can occur after liver transplantation, with Budd-Chiari syndrome, or as a result of cardiac lesions that cause an increase in right atrial pressure and/or chronic systemic venous hypertension. Acute Budd-Chiari syndrome is rare in children; although it may occur with some thrombophilic disorders, it is usually idiopathic and is not associated with the causes seen in adults, such as myeloproliferative disorders. A cavopulmonary or atriopulmonary shunt, known as a Fontan procedure, allows life-saving systemic-pulmonary blood flow in single-ventricle syndromes in neonates but results in chronic systemic venous hypertension (pressures may be >20 mm Hg) and eventually to portal hypertension (Narkewicz et al, 2003).
Effects of Portal Hypertension in Children
The major pathologic effect from portal hypertension is the development of collaterals that carry blood from the portal venous system to the systemic circulation in the upper part of the stomach, esophagus, rectum, and falciform ligament. These collaterals may drain into the IVC via the umbilical vein remnant, or they may drain into the left renal vein (see Fig. 71.1). Absence or disconnection of the IVC or interruption to the azygos system, such as occurs in some cases of bilary atresia, may cause special concern. Similarly, in extrahepatic portal venous malformations, the splenic vein can be small and possibly thrombosed.
Although changes in vascular resistance to flow of blood between the splanchnic bed and the right atrium appear to be the initial events in the development of portal hypertension, a number of other hemodynamic changes contribute to and amplify the increased portal blood pressure. There is a hyperdynamic circulatory state with increased cardiac and decreased splanchnic arteriolar tone, both of which increase portal inflow. Changes in intravascular volume also play an important role, as do alterations in adrenergic tone in the splanchnic system. These observations have led to new experimental and clinical studies that suggest possible pharmacologic treatments for portal hypertension (Boyer, 2001). However, because the major clinical effect is that of bleeding from esophageal varices, direct treatment of variceal hemorrhage, or shunt surgery in selected cases, remains the primary approach, except in the presence of liver decompensation, in which case the treatment of choice is liver transplantation (see Chapters 97A and 98A).
Clinical Features
The main clinical features are splenomegaly, nutritional growth failure, and ascites, along with esophageal, gastric, and rectal varices (Box 71.2).
[/not-level-membership-for-surgery-category]
