Chapter 6 Liver fibrogenesis
Mechanisms and clinical relevance
Liver Fibrogenesis
Liver fibrosis represents a scarring response to either acute or chronic cellular injury. Following acute liver injury, parenchymal cells regenerate to successfully preserve hepatocellular mass and function. This acute process is associated with an inflammatory and fibrogenic response but limited deposition of extracellular matrix (ECM). In contrast, prolonged liver injury leads to sustained production of growth factors, proteolytic enzymes, angiogenic factors, and fibrogenic cytokines. These events culminate in the accumulation of ECM, forming septa that coalesce into broad bands of scar tissue that encircle nodules of hepatocytes and lead to altered microvascular structure (Friedman, 2004). This late stage of fibrosis, termed cirrhosis, ultimately impairs liver function and leads to portal hypertension and its complications (see Chapter 70A).
Progression of fibrosis to cirrhosis typically evolves over decades before clinical events ensue, but disease may progress more rapidly following repeated episodes of severe acute alcoholic hepatitis; subfulminant hepatitis, especially as a result of drug toxicity; and fibrosing cholestasis in patients with hepatitis C virus (HCV) reinfection after liver transplantation (Berenguer et al, 2003). In addition, rapidly progressive acute HCV with fibrosis in men coinfected with human immunodeficiency virus (HIV) has been reported recently (Fierer et al, 2008).
Genetic and environmental factors also influence the course of the disease. For example, polymorphisms in a number of candidate genes involving the inflammatory (TLR4; Guo et al, 2009) or immune response (specific human leukocyte antigen-II [HLA-II] alleles; Powell et al, 2000) may influence the progression of liver fibrosis in humans. Validated groups of single nucleotide polymorphisms (SNPs) can be used to calculate a disease-specific risk assessment for fibrosis progression (HCV: DDXminor allele, DDX-5POLG2 haplotype [Huang et al, 2006]; alcohol-related or chronic cholestatic disorders: TNF-α, interleukin [IL]-1B [Jarvelainen et al, 2001; Donaldson et al, 2001]). These findings have led to the development of a seven-gene signature with good prognostic value in assessing the risk for cirrhosis development in HCV patients (Huang et al, 2007).
The main etiologies of liver fibrosis in Western countries are chronic HCV and HBV infection, alcohol abuse, and nonalcoholic steatohepatitis (NASH; see Chapters 64 and 65). As a generalized tissue response to chronic injury, fibrosis also occurs in many other organs—including the heart, lung, and kidneys—and typically represents the result of ongoing inflammation. Remarkably, up to 45% of all deaths are related to some kind of fibrosis, which emphasizes the importance of this response and explains the growing interest in this field of research (Fig. 6.1).
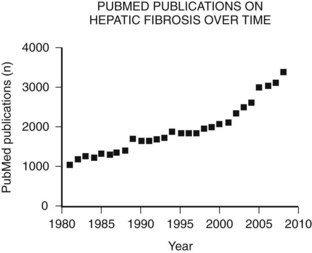
For decades fibrosis was seen as an irreversible disease that progresses to cirrhosis, with a greater risk for developing hepatocellular carcinoma (see Chapters 8C and 80) and liver failure. This meant the only potential treatment for liver fibrosis once it had progressed to cirrhosis was transplantation. Research over the past 30 years has yielded increasing insight into the cellular and molecular mechanisms of this disease, uncovering an orchestrated pathophysiology and identifying the hepatic stellate cell (HSC) as the central cell type in fibrogenesis (Friedman, 1985). Most importantly, this revealed the potential reversibility of this disease and the discovery of potential therapeutic targets.
Molecular and Cellular Mechanisms of Fibrosis
The anatomical arrangement of parenchymal and nonparenchymal cells of the liver contributes to its unique role as an immunity-modulating organ (see Chapter 9) and helps explain how the liver responds to an insult. The liver is composed primarily of epithelial cells, hepatocytes and cholangiocytes, and resident nonparenchymal cells that include hepatic macrophages (Kupffer cells), sinusoidal endothelium, and HSCs. In addition to Kupffer cells, a growing list of specialized immune cells have been characterized, including dendritic cells (DCs), natural killer (NK) cells, and natural killer T (NKT) cells, which indicate that the liver represents a key organ in the regulation of innate immunity.
Common Triggers of Hepatic Fibrogenesis
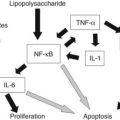
Ongoing insult to the liver will lead to an increased inflammatory state with activation of hepatic stellate cells, which ultimately tilts the profibrotic–antifibrotic balance toward fibrosis. Ethanol, viral infection, reactive oxygen species (ROS), and bile acids are among the most common stress signals for the liver (Fig. 6.2). An in vitro study further suggests that free fatty acids promote fibrogenesis by indirect activation of HSCs (Wobser, 2009). Less common causes of hepatocellular injury are autoimmune hepatitis, Wilson disease, or hemochromatosis.
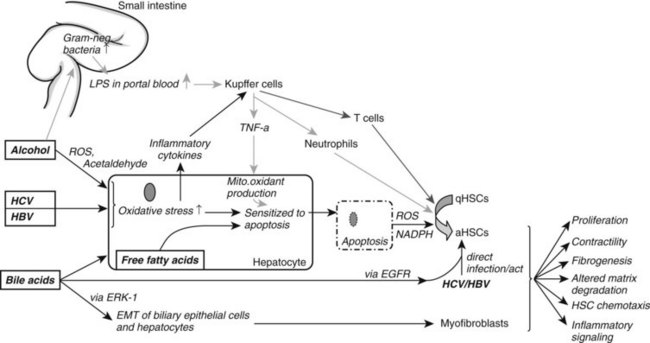
Alcohol decreases gut motility, increases epithelial permeability, and promotes overgrowth of gram-negative bacteria. Consequently, lipopolysaccharide concentration is elevated in portal blood, which activates Kupffer cells through the Toll-like receptor (TLR) 4 complex to generate ROS via reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Wheeler, 2001). Oxidants then upregulate NF-κB in Kupffer cells, which leads to increased tumor necrosis factor (TNF)-α production. TNF-α in turn induces neutrophil infiltration and stimulates mitochondrial oxidant production in hepatocytes, which are then sensitized to undergo apoptosis. Furthermore, acetaldehyde, the main degradation product of alcohol, and ROS both activate HSCs and stimulate inflammatory signals (Maher et al, 1994).
In HCV infection, the virus escapes the HLA-II immune response and infects hepatocytes. This causes oxidative stress, again leading to recruitment of inflammatory cells and HSC activation. HSCs can also be directly activated by either HCV, through membrane receptors (Mazzocca et al, 2005, Schulze-Krebs et al, 2005), or by HBV (Martin-Vilchez et al, 2008).
Bile acids are hepatotoxic agents that typically target hepatocytes, but they may also injure biliary epithelium (Higuchi & Gores, 2003). Damaged hepatocytes turn apoptotic or necrotic, thereby releasing ROS (Nieto et al, 2002) and NADPH oxidase, which both activate HSCs (Canbay et al, 2004a). Injured hepatocytes also release inflammatory cytokines and soluble factors that activate Kupffer cells and stimulate the recruitment of activated T cells. This inflammatory milieu further stimulates the activation of resident HSCs. Bile acids also directly stimulate proliferation of HSCs by activating the epidermal growth-factor receptor (Svegliati-Baroni et al, 2005). In contrast to hepatocytes, HSCs are protected from bile acid–induced apoptosis by excluding bile acids (Svegliati-Baroni et al, 2005).
Nonalcoholic fatty liver disease (NAFLD) is increasingly prevalent as a result of increased obesity in the United States and Western Europe. NAFLD can progress to NASH with consequent fibrosis and cirrhosis (Carter-Kent et al, 2009). The pathogenesis is not fully understood; however, a two-hit model has been proposed with hyperglycemia and insulin resistance representing the first hit, leading to elevated serum levels of free fatty acids. Histologically this correlates to hepatic steatosis. Additional oxidative stress or proinflammatory cytokines are thought to promote hepatocyte apoptosis and recruitment of inflammatory cells, thereby enhancing fibrogenesis.
Hepatic Stellate Cell Activation and Hepatic Myofibroblasts (MFBs)
The HSC has emerged as a central regulator of the liver’s fibrotic and repair responses (Friedman, 2000). In normal liver the HSC is a quiescent cell type that contains cytoplasmic retinoid droplets, representing the major storage site for vitamin A in the body, and expresses the markers desmin and glial fibrillary acidic protein (GFAP; Friedman, 2008).
During liver injury, HSCs undergo activation in response to a range of inflammatory and injury signals (Casini, 1997) produced by damaged hepatocytes and biliary cells; changes in the composition of the ECM; proangiogenic growth factors, like VEGF and angiopoietin; and fibrogenic cytokines that include TGF-β1, angiotensin II, and leptin.
Activation of HSCs is accompanied by loss of retinoid droplets and accumulation of α-smooth muscle actin, a myogenic filament that confers increased cellular contractility. Activated HSCs are characteristically desmin- and α-SMA–positive cells. Highly activated subsets of HSCs are known as hepatic myofibroblasts (MFBs; Friedman, 2008), a cell type that is also characteristic of wound healing in a range of tissues: skin, kidney, lung, bone marrow, and pancreas, among others.
HSC activation can be divided conceptually into two phases: initiation begins with early changes in gene expression and phenotype that result from paracrine stimulation, primarily due to changes in surrounding ECM and exposure to lipid peroxides and products of damaged hepatocytes (Fig. 6.3); perpetuation results from the effects of these stimuli on maintaining the activated phenotype and generating fibrosis. Within the nucleus, a growing number of transcription factors regulate HSC behavior, including peroxisomal proliferator–activated receptors (PPARs), retinoid receptors, NF-κB, JUND, Kruppel-like factor 6 (KLF6), and FOXF1 (Mann & Mann, 2009). A range of general and cell type–specific membrane receptors and signaling pathways also control HSC biology, including receptor tyrosine kinases, chemokine receptors, and integrins (Dodig et al, 2007).
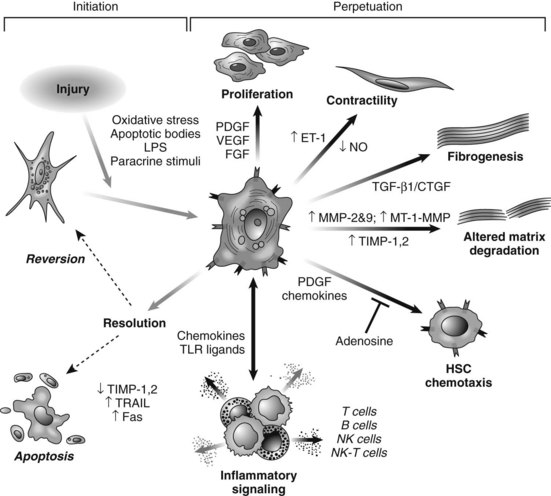
Portal fibroblasts (Beaussier et al, 2007; Ramadori & Saile, 2004) and bone marrow–derived MFBs (Russo et al, 2006) have also been identified as collagen-producing cells in the injured liver. Another emerging source of fibrogenic cells is epithelial-mesenchymal transition (EMT), in which adult hepatocytes of biliary epithelium transdifferentiate into fibrogenic cells (Rygiel et al, 2008). EMT has been extensively characterized in kidney and lung fibrosis, in subsequent animal models and human samples of liver fibrosis, and in the context of hepatocarcinogenesis. Interestingly, key signals regulating EMT also drive activation of HSCs, including TGF-β, RAS, extracellular signal-regulated kinase 1 (ERK1; Zhong et al, 2009), SMAD7, and SHH (Dooley et al, 2008; Lahsnig et al, 2009; Syn et al, 2009).
The relative importance of each cell type in liver fibrogenesis may depend on the origin of the liver injury. In cholestatic liver diseases and ischemia, portal fibroblasts may be especially important (Clouzeau-Girard et al, 2006), whereas hepatic MFBs may be of predominant importance in alcoholic liver disease. In chronic liver injury, progressive recruitment of bone marrow–derived cells may occur over time. However, bone marrow–derived cells may have both fibrogenic and antifibrotic effects (Karlmark et al, 2009). To date, the quantitative contribution to fibrogenesis of nonstellate cell–derived fibroblasts remains unclear.
Functions of Hepatic MFBs
Hepatic MFBs have functions that are distinct from their quiescent cells of origin. They are profibrogenic and promitotic, play a chemotactic and vasoregulatory role, and control the degradation of the ECM (see Fig. 6.3). They also have important immune and phagocytic functions (Friedman, 2008). The regulation of ECM accumulation and degradation by HSCs is reviewed in the next section.
Fibrogenesis
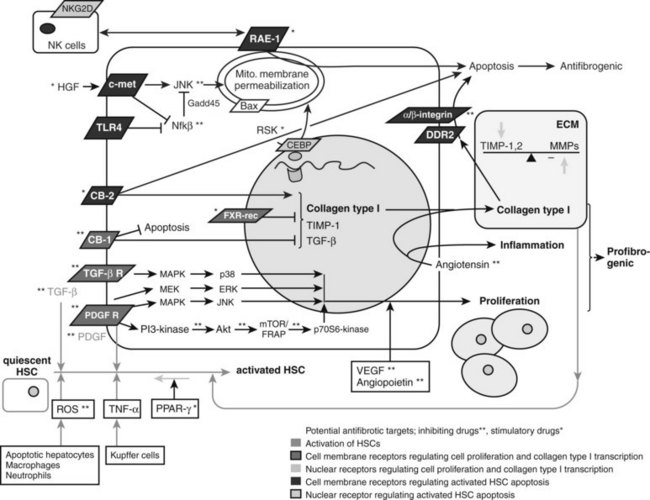
The major profibrogenic signal in the liver is the cytokine TGF-β1 (Bachem et al, 1989a). TGF-β1 is secreted mainly by MFBs (Bissell et al, 1995) but also by platelets (Bachem et al, 1989b) and Kupffer cells (Bilzer et al, 2006). It acts by activating the TGF-βII receptor, which recruits the TGF-βI receptor. SMAD2 and SMAD3 then associate with the TGF-βI receptor (Dooley et al, 2001), are phosphorylated, and recruit SMAD4. This triheteromeric complex then translocates to the nucleus, where it activates profibrogenic transcription factors. TGF-β also activates the MAPK p38 pathway (Cao et al, 2002), which leads to additional, SMAD-independent collagen type 1 synthesis and, in contrast to the SMAD-dependent collagen type 1 synthesis, also leads to a posttranscriptionally regulated stabilization of the collagen type 1 mRNA (Tsukada et al, 2005; Fig. 6.4).
Proliferation
The predominant stimulus to MFB proliferation is the mitogen platelet-derived growth factor (PDGF; Borkham-Kamphorst et al, 2004), in addition to other mitogens that include EGF, VEGF, and FGF. All pathways downstream of PDGF receptor activation promote proliferation. First, JNK is stimulated through MAPK (Schnabl et al, 2001); second, PDGF receptor stimulates the Ras/Raf complex, followed by MEK and ERK engagement (Schnabl et al, 2001); and third, activation of the PI3K pathway leads to Akt activation and phosphorylation of the 70s6 kinase (see Fig. 6.4; Reif, 2003).
Immunoregulation
The liver is a microenvironment of diminished immunogenicity, which is necessary to cope with the high exposure of antigens from the portal vein (Crispe, 2003). This feature also accounts for the tolerance of liver transplantation across ABO barriers and may contribute to the chronic nature of hepatitis B and C, where the virus persists despite the development of an immune response. Upon entry of antigen into the sinusoid, classical antigen-presenting cells (Kupffer cells, dendritic cells) are first encountered. Subsequently, HSCs in the space of Disse may contact antigens. Indeed, HSCs display a wide range of immunoregulatory functions and are an essential part of the local immunogenicity (Gao et al, 2008; Mehal, 2007; see Chapter 9).
Hepatic MFBs produce a range of proinflammatory (e.g., TNF-α; Tiggelman et al, 1995) and antiinflammatory IL-10 cytokines (Tiegs & Lohse, 2009; Safadi et al, 2004) and they recruit lymphocytes through secretion of chemokines (MCP-1, IL-8, CCL21, RANTES; Marra et al, 1998, 1993; Maher et al, 1998), thus amplifying the inflammatory response. However, upon activation, they exert a profound immunosuppressive activity by inducing T-cell apoptosis (Yu et al, 2004). In the setting of liver transplantation, MFB-induced T-cell apoptosis via programmed death ligand-1 (Yu et al, 2004) may enable local immunotolerance of the liver. In liver fibrosis, MFBs may further regulate the contribution of lymphocytes to the course of hepatic fibrosis by ingesting disease-associated lymphocytes (Muhanna et al, 2008).
The interaction between HSCs and immune cells is bidirectional. T cells activate HSCs by IFN-γ, which upregulates both stimulatory (CD80, CD86, CD54) and inhibitory (B7-H1) surface molecules and enhances both inflammatory and suppressive cytokines. However, the inhibitory molecules are thought to override the stimulatory counterparts, resulting in immunosuppression. Lymphocytes can also mediate hepatic fibrosis by activating HSCs. CD8-positive T lymphocytes are more fibrogenic toward stellate cells than CD4-positive T lymphocytes (Safadi et al, 2004). This may explain in part why patients coinfected with HIV and HCV have accelerated fibrosis, as their CD4–CD8 cell ratios are reduced. Of the CD4-positive T lymphocytes, previously called T helper cells, the humoral-mediated immunity by T helper-2 (Th2) cells is profibrogenic in liver injury, whereas the cell-mediated immunity by T helper-1 (Th1) cells via IFN-γ, TNF-α, and IL-2 is antifibrogenic (Shi, 1997).
HSCs can also function as antigen-presenting cells (Winau et al, 2007). They can interact with bacterial lipopolysaccharides directly via TLR4, which amplifies their activation. TLR4 signaling leads to downregulation of a TGF-β pseudoreceptor, BAMBI, which thereby amplifies fibrogenic activity of MFBs (Seki & Brenner, 2008). This might contribute to an accelerated progression of liver cirrhosis, as microbial translocation from the gut to the liver is increased, which is the case in alcoholic liver disease.
Vasoregulation
MFBs play an important role in the regulation of sinusoidal blood flow and may contribute to portal hypertension that is characteristic of advanced liver disease. The release of endothelin-1 (ET-1) can stimulate their contraction through the ETA receptor (Shi-Wen et al, 2004), thereby promoting tissue contraction, increasing portal resistance, and generating portal hypertension. On the other hand, MFBs and endothelial cells also secrete nitric oxide (NO), which is the physiologic antagonist of ET-1.
Structural Features of Hepatic Fibrogenesis
In hepatic fibrosis the total amount of collagen is increased up to sixfold, whereas the parenchymal mass (e.g., hepatocytes) is progressively diminished. The composition of the ECM changes with progression of disease. Collagen type IV in the space of Disse is replaced by interstitial collagen types I and III. Additionally, the discontinuous basal membrane beneath the sinusoidal endothelial cells is replaced by a continuous basement membrane, and sinusoidal fenestrations are reduced. This decreased porosity, also known as capillarization, combined with perisinusoidal fibrosis, scar contraction, and formation of intrahepatic shunts contributes to increased hepatic venous pressure and portal hypertension. Fibrillar collagens produced by MFBs interact with MFBs via discoidin domain receptors and integrins (Olaso et al, 2001), thereby inhibiting apoptosis and increasing MFB proliferation.
With the maturation of the fibrotic scar, not only is the amount of collagen increased, but the scar also becomes increasingly insoluble through chemical cross-linking by transglutaminase, and assembly of collagen fibrils through increased generation of collagen monomers by metalloproteinases from the “ADAM metallopeptidase with thrombospondin type 1 motif”; in addition, metalloproteinase with thrombospondin-type repeats metalloproteinase with thrombospondin type I motif (ADAMTS2; Kesteloot et al, 2007) family. This makes the fibrous septa progressively resistant to proteolysis by matrix metalloproteinases (MMPs). The long-standing clinical dogma—the slower the pace of injury, the less reversible the scar—is supported by animal studies in which even advanced fibrosis of short duration is reversible. Thus, the reversibility of a scar may be limited primarily by the extent of collagen cross linking. Clinically, increased septal thickness and smaller nodule size, both of which reflect more advanced stages of fibrosis, are significant predictors of worse clinical outcomes (Nagula et al, 2006).
Regulation of Collagen Deposition and Degradation
The deposition and degradation of collagen is tightly regulated. MMPs are the key enzymes that degrade fibrillar collagens (types I and III) and noncollagenous ECM substrates. The tissue inhibitor metalloproteinases (TIMPs) are their major antagonists by inactivating proteases and by inhibiting MFB apoptosis (Murphy et al, 2002).
Both decreased levels of interstitial collagenases and increased levels of MMP inhibitors in liver injury create an imbalance that favors reduced degradation of fibrillar collagens in hepatic fibrosis. The interstitial collagenases MMP-1, MMP-8, and MMP-13 in humans and MMP-13 in rodents (Emonard & Grimaud, 1990) unwind the triple-helical collagen type I, which is the principal collagen in the fibrotic liver (Williams & Olsen, 2009), so that each α-chain is presented to the active site of the enzyme (Chung et al, 2004), which cleaves the collagen. Other MMPs (e.g., MMP-2) cannot unwind the triple-helical collagen and thus cannot degrade intact collagen type I alone. In early liver injury, MMP-2 degrades the low-density basement membrane present in the subendothelial space (Zhou et al, 2004a). Its replacement with fibril-forming matrix impairs hepatocyte differentiation and function. During progressive fibrosis, expression of MMP-1 (humans) or MMP-13 (rodents) is decreased, and MMP-2 expression increases (Preaux et al, 1999; Milani et al, 1994). In parallel, the expression of TIMP-1 and TIMP-2, which inhibit the collagen-degrading matrix metalloproteinases, are increased (Kossakowska et al, 1998; Murawaki et al, 1993).
Hepatic macrophages also regulate matrix remodeling (Mitchell et al, 2009), and they are an important source of proteases, including MMP-13 (Hironaka et al, 2000; Fallowfield et al, 2007) in rodents. Macrophages might even be more critical to ECM degradation than HSCs, whose degradation potential is impaired during activation by elevation of the inhibitor TIMP-1. Kupffer cells can further stimulate TIMP-1 expression by hepatic MFBs (Wang et al, 2009), leading to inhibition of MMP activity and also protection of MFBs from apoptosis. In mouse models, macrophages augment fibrogenesis during progression of liver fibrosis, whereas during resolution they hasten matrix degradation through increased production of MMP-13 (Fallowfield et al, 2007).
Although both macrophages and MFBs secrete MMP-1 in humans and MMP-13 in rodents, it is not clear which is the major interstitial collagenase in fibrosis regression, because MMP-1 is only expressed at low levels in liver. Moreover, the cellular origin of those MMPs deemed essential to fibrosis regression remains controversial. Dendritic cells and T lymphocytes are also potential sources, as both can secrete MMP-9 (Chabot et al, 2006; Owen et al, 2003).
Diagnosis and Clinical Monitoring of Hepatic Fibrosis
Many patients with chronic liver disease may initially present with late-stage fibrosis, as earlier stages are often asymptomatic. Thus clinicians must have a high index of suspicion for occult fibrosis, especially in patients with unexplained elevations of liver enzymes, splenic enlargement, stigmata of liver disease, or laboratory or imaging findings suggestive of portal hypertension. When chronic liver disease is suspected, liver biopsy remains the gold standard for diagnosing and staging liver fibrosis, but it is an invasive procedure with risk of adverse events (Bravo et al, 2001; Poynard et al, 2000) and, equally important, a high likelihood of sampling variability (Bedossa et al, 2003) and interpathologist and intrapathologist variability (Bedossa & Poynard, 1996). At least one third of biopsies may deviate by one fibrosis stage between the right and left hepatic lobes in HCV (Regev et al, 2002). Shorter biopsies are associated with an increase in reported diagnoses of mild and moderate fibrosis at the cost of more severe fibrosis, representing an understaging of fibrosis (Colloredo et al, 2003); the smaller the tissue sample, the milder the apparent disease.
Several commonly used histologic staging systems for fibrosis exist. The Histology Activity Index (HAI) score reported by Knodell includes three stages (Knodell et al, 1981), and the Ishak score differentiates six stages, including two stages of cirrhosis, incomplete and complete (Ishak et al, 1995). The METAVIR score, developed by the French METAVIR Cooperative Study Group, is a simple, widely applied five-stage scoring system (Bedossa & Poynard, 1996) that is most commonly used worldwide. It incorporates the fibrosis scores F0 through F4 (F0, no fibrosis; F1, fibrosis without septa; F2, few septa; F3, numerous septa without cirrhosis; F4, cirrhosis) and activity scores A0 through A3 that assess the amount of necroinflammation (A0, no necroinflammatory activity; A1, mild; A2, moderate; A3, severe). All these systems were developed primarily for scoring fibrosis associated with viral hepatitis, with the main emphasis on the degree of necroinflammation. The fewer fibrosis stages within a scoring system, the higher the reproducibility among observers.
Scoring systems have also been specifically developed for different etiologies of liver fibrosis: Ludwig and colleagues proposed a four-stage system to describe fibrosis stages for both primary biliary cirrhosis (Ludwig et al, 1978) and sclerosing cholangitis (Ludwig et al, 1981), and Kleiner and colleagues (Kleiner, 2005) quantified fibrosis in NAFLD with a seven-stage system. The Kleiner stages are stage 0, no fibrosis; stage 1, perisinusoidal or periportal fibrosis (1a: mild, zone 3; 1b: moderate, zone 3; 1c: portal/periportal); stage 2, periportal and perisinusoidal fibrosis; stage 3, bridging fibrosis; and stage 4, cirrhosis.
There is a great need for reliable, quantitative, noninvasive diagnostics of fibrosis, and recent studies indicate steady progress. Cross-sectional imaging studies, such as those provided by computed tomographic (CT) and magnetic resonance imaging (MRI) scans, can reliably demonstrate features of advanced liver disease, including nodularity and signs of portal hypertension (splenomegaly, enlarged caudate lobe, esophageal varices). Diffusion-weighted MRI measures the apparent diffusion coefficient of water, a parameter that depends upon tissue structure, and it has compared favorably to other noninvasive measures for determining advanced fibrosis (Lewin et al, 2007).
Biochemical Tests
The most studied combination serum tests are the AST platelet ratio index (APRI; Wai et al, 2003), the FIB-4 index (Sterling et al, 2006), the Forns test (Forns et al, 2002), and the proprietary FibroTest (Imbert-Bismut et al, 2001). Newer, proprietary biomarker scores include the HepaScore (Adams et al, 2005) and the FibroMeter (Cales et al, 2005). The factors included in the various tests were chosen upon multivariate analysis and are included in Table 6.1. All these biochemical tests are sufficient to excellent in ruling out significant fibrosis (F3 to F4) when the proper cutoff value is chosen, but they are less useful in distinguishing mild from moderate fibrosis. The sensitivities of these tests vary based on the etiology of the liver disease. A further difficulty with these tests involves the absence of an ideal “gold standard” in view of the liver biopsy’s significant sampling variability and interlaboratory differences (Gressner et al, 2009). A key advantage of the biochemical tests, on the other hand, is that the patients most at risk for false-positive results are well defined, so that the results from these individuals can be evaluated more critically or complemented with further diagnostics.
Table 6.1 Common Noninvasive Tests for Detecting and Staging Liver Fibrosis
| Test | Content |
|---|---|
| FIB-4 (Sterling, 2006) | Age, AST, platelets |
| APRI (Wai, 2003) | AST, platelets |
| Forns (Forns, 2002) | Age, GGT, cholesterol, platelets |
| FibroTest (Imbert-Bismut, 2001) | GGT, haptoglobin, α2-macroglobulin, bilirubin, apolipoprotein A1, adjusted by age and gender |
| FibroTest-ActiTest (Imbert-Bismut, 2001) | ALT, GGT, haptoglobin, α2-macroglobulin, bilirubin, apolipoprotein A1, adjusted by age and gender |
| Hepascore (Adams, 2005) | Total bilirubin, GGT, α2-macroglobulin, hyaluronic acid, adjusted by age and gender |
| Fibrometer (Cales, 2005) | P1, α2-macroglobulin, hyaluronic acid, age |
| SHASTA (Kelleher, 2005) | Hyaluronic acid, AST, albumin |
| ELF (Guha, 2008) | TIMP-1, hyaluronic acid, amino-terminal peptide of pro-collagen III (P3NP) |
| Fibroscan (Sandrin, 2003) | Liver elasticity measurement (in kilo-Pascals) by pulse-echo ultrasound (50 MHz) |
AST, Aspartate aminotransferase; GGT, gamma-glutamyl transferase; ALT, alanine aminotransferase; TIMP, tissue inhibitor of metaloproteinases.
Of the biochemical tests, the FibroTest has been shown to be equal to or better than the others (Thabut et al, 2003; Le Calvez et al, 2004). In particular, it is more sensitive for discriminating between F1 and F2 stages and is more linearly correlated to fibrosis stages when compared with the other markers. Further improvement in prognostic value can be achieved by combining the FibroTest with the ActiTest and/or the FibroScan. The FibroTest combines the parameters gamma-glutamyl transferase (GGT), haptoglobin, α2-macroglobulin, bilirubin, and apolipoprotein A1, corrected for age and gender; it has a high predictive value for significant fibrosis in patients with chronic hepatitis C and B and nonfatty liver disease (Imbert-Bismut et al, 2001; Poynard et al, 2007) and in patients with chronic alcoholic liver disease (Naveau et al, 2005; Thabut et al, 2006). In contrast to the other tests, the FibroTest has also been evaluated in a longitudinal study in which patients with HCV were followed up by a liver biopsy after α-interferon treatment. Promisingly, FibroTest results correlated well with the progression or regression of fibrosis after antiviral therapy, and it compared favorably with liver biopsy (Poynard et al, 2002a).
The diagnostic value of the FibroTest has been compared to those of the HepaScore and FibroMeter in patients with chronic HCV (Halfon et al, 2007; Leroy et al, 2007), and the diagnostic and prognostic value was evaluated in patients with alcoholic liver disease (Naveau et al, 2009). All three tests have similar diagnostic accuracy in discriminating between advanced and nonadvanced fibrosis and between cirrhotic and noncirrhotic stages, however, the FibroTest was significantly better than the HepaScore or FibroMeter at predicting 10-year mortality in patients with alcoholic liver disease, and it was similar or even slightly better than the liver biopsy.
Serum Assays of Extracellular Matrix Molecules
Serum assays have been developed that measure circulating molecules involved either in the deposition of ECM or its degradation as well as cytokines involved in fibrogenesis. The best validated of these tests is the Enhanced Liver Fibrosis (ELF) panel (Guha et al, 2008). This test incorporates markers of ECM turnover: TIMP-1, hyaluronic acid, and aminoterminal peptide of procollagen III (P3NP). The ELF test was better at identifying minimal, moderate, or severe fibrosis than a clinical-biochemical panel that considered age, body mass index, presence of diabetes or impaired fasting glucose, aspartate aminotransferase/alanine aminotransferase (AST/ALT) ratio, platelets, and albumin. The combination of both clinical and biochemical scores showed further improved area under the receiver operating characteristic curve (AUROC) values in the subset of patients with an altered clinical-biochemical panel in reference to liver biopsies that were staged according to the Kleiner score (Angulo et al, 2007). If the ELF test was used to delineate any fibrosis, using thresholds with a sensitivity and specificity of 90%, the authors concluded that liver biopsy could have been avoided in 48% of patients.
Cytokines and Chemokines Associated with Hepatic Fibrosis
Of the cytokines and chemokines associated with hepatic fibrosis, TGF-β1 is the dominant stimulus for the production of ECM by HSCs. Hepatic mRNA levels of TGF-β1 are increased in chronic liver disease in association with increases in mRNA levels of type I collagen (Annoni et al, 1992). In HCV-related chronic liver disease, serum TGF-β levels correlate with the Knodell fibrosis score but not with clinical, biochemical, or virological parameters (Nelson et al, 1997). Interestingly, TGF-β levels also decreased in a group of HCV patients with histologic decreases in necroinflammation in the absence of changes in fibrosis following interferon treatment (Roulot et al, 1995). Thus TGF-β serum levels not only correlate with fibrosis scores but may also indicate necroinflammation (see Chapter 10). PDGF is upregulated following liver injury (Ikura et al, 1997), and at least one study suggested that the amount of PDGF correlates with the severity of fibrosis (Shiraishi et al, 1994).
Proteomics and Glycomics
Proteomics and glycomics are promising new technologies suitable as noninvasive diagnostic methods. Patterns of proteins or glycoprotein can be assessed by mass spectroscopy of serum samples (Petricoin et al, 2004). Profiles of serum protein N-glycans were found to have AUROC values similar to the FibroTest for the diagnosis of compensated cirrhosis. The combination of serum protein N-glycans with the FibroTest (FT) was able to increase the sensitivity and specificity of glycoprotein assessment alone (Callewaert et al, 2004).
FibroScan
An alternative approach to fibrosis assessment is the measurement of liver stiffness using a device that performs transient elastography. Currently, FibroScan (FS) is the only device to utilize this approach. It measures liver elasticity by using a pulse-echo ultrasound. A vibration of low frequency induces an elastic shear wave that propagates through the liver. The stiffer the liver tissue, the faster the shear wave propagates. Results are expressed in kilopascals (kPas; (Sandrin et al, 2003; Kettaneh et al, 2007). With FS, the virtual cylinder of tissue that is assessed is at least 200 times larger than a biopsy sample and therefore is far more representative of the hepatic parenchyma; thus the FS may provide a more accurate and reproducible picture of cirrhosis than liver biopsy. There is also potential value of FS in early stages of disease, when fibrosis may be unevenly distributed and thus underestimated by liver biopsy. Furthermore, FS has very low interobserver variability, although its accuracy is limited in patients with obesity, ascites, or acute hepatitis. FS has been evaluated extensively (Ziol et al, 2005; Cross et al, 2009; Sanchez-Conde et al, 2009; Boursier et al, 2009), is licensed in several European countries, and is currently being evaluated by the Food and Drug Administration (FDA) in the United States. When FS was combined with the FT in HCV patients, both tests agreed in 70% to 80% of subjects, with increasing concordance in higher stages of liver fibrosis. Compared with the liver biopsy, results were confirmed in 84% to 94% of cases with a tendency of FS/FT to underestimate fibrosis (Castera et al, 2005).
Although the combination of FT, FS, the ActiTest, and the ELF panel in NAFLD patients seems to be a good noninvasive alternative to liver biopsy, the value of these tests in individual patient management over time needs to be established. These tests may even be superior to liver biopsy at correctly staging and grading fibrosis (Poynard et al, 2004), but proving this remains a challenge with liver biopsy being the gold standard. A way to surmount this problem may be to assess the prognostic value of a test with the hard clinical end point of liver-related mortality compared with liver biopsy.
Therapeutic Strategies
Reversibility of Fibrosis: The Point of No Return
The concept of fibrosis as an irreversible and constantly progressing state is no longer accurate. Liver fibrosis of various etiologies is usually reversible by removing the causative agent (Friedman & Bansal, 2006). For example, a decrease in the viral load of HBV patients; clearance of HCV with pegylated interferon and ribavirin (Poynard et al, 2002b), but not maintenance interferon monotherapy (Di Bisceglie et al, 2008); cessation of ethanol intake; weight loss or bariatric surgery in patients with NASH; and decrease in iron or copper or immunosuppressive therapy in autoimmune diseases have been shown experimentally and clinically to limit fibrosis progression and to even regress cirrhosis in some cases. In fact, removing the causative agent is still the most effective therapy.
Although fibrosis, inflammation, and bile duct proliferation decrease when the damaging stimulus is withdrawn, regenerative nodules may become autonomous and grow progressively (Quinn & Higginson, 1965). This raises a key question, whether a “point of no return” exists, wherein even complete clearance of the underlying disease will no longer yield an improvement in cirrhosis. Increased cross linking of the collagen fibrils over time makes the fibrous septa progressively resistant to proteolysis by metalloproteinases, and hypoxia stimulates secretion of proangiogenic factors such as VEGF and angiopoietin-1 by HSCs, which induce proliferation and motility (Nakamura et al, 2007). These findings suggest there may be a positive feedback loop between angiogenesis and fibrogenesis, which persists even when the primary etiology is cleared, leading to sustained abnormalities of the intrahepatic vasculature.
Prevention of Hepatocyte Apoptosis in Liver Injury
Caspase Inhibitors
Apoptosis is a functional antagonist to mitosis, and together these two processes regulate homeostatic cell turnover. The two main pathways of apoptosis are the extrinsic pathway, which is death ligand–death receptor mediated and dependent on caspase 8, and the intrinsic pathway, which is regulated by Bcl2-induced mitochondrial dysfunction and downstream activation of caspase 9 and its effector caspases 3, 6, and 7. Blocking caspases downstream of the mitochondria, however, only delays apoptosis; it does not inhibit it (Chen et al, 1998; Strobel et al, 1996). Thus pancaspase inhibitors are being used to assess the therapeutic effect of apoptosis inhibition. In preclinical studies, the pancaspase inhibitor ZVAD-fmk was able to reduce mortality in rats with acute hepatic failure after major hepatic resection (Yoshida et al, 2007). Other caspase inhibitors have reduced I/R injury in rodents (Natori et al, 1999, 2003; Hoglen et al, 2007), decreased fibrogenesis in mice after bile duct ligation (Canbay et al, 2004), and decreased fibrogenesis in a diet deficient in methionine and choline, induced in a NASH mouse model in which hepatic steatosis and fibrogenesis improved without improvement of liver injury (Witek et al, 2009).
Only one clinical trial has been done in which a pancaspase inhibitor was administered in a dose-ranged manner to patients with liver disease of different etiologies. However, this agent, IDN-6556, also inhibits caspase 1, which has an inflammatory effect; thus the drug should be antiinflammatory. In the two weeks of treatment, in the subset of patients with HCV as their underlying disease, AST and ALT levels were significantly reduced without affecting the HCV mRNA levels. No adverse events occured (Pockros et al, 2007). Caspase inhibitors thus far are considered safe, as animals with genetic deletions of death receptors do not show increased spontaneous tumors (Adachi et al, 1995; Pfeffer et al, 1993; Yue et al, 2005). Nonetheless, the potential emergence of tumors following long-term administration remains a lingering concern.
Inhibition of HSC Activation or Inactivation of MFBs
Oxidative stress in the form of ROS released by injured hepatocytes through the action of NADPH represent a major fibrogenic stimulus. Thus antioxidants including vitamin E, silymarin, CYP2E1 inhibitors (Nieto et al, 2002), phosphocholine, or S-adenosyl-L-methionine may benefit fibrosis, particularly in patients with alcohol-induced liver disease and NASH, where oxidative stress plays an especially important role. Efforts to establish the activity of antioxidants are confounded by the uneven quality of commercially available products, because these compounds are typically sold over the counter, and their potency is not monitored.
Immune manipulation also holds promise but is not yet ready for clinical testing. Th1 cells that mediate cellular immunity through IFN-γ, TNF-α, and IL-2 are antifibrogenic, whereas Th2 cells that promote humoral immunity are more profibrogenic via IL-4, IL-5, IL-6, and IL-13 (Shi et al, 1997; Lee et al, 2001). Thus a shift toward the Th1 cells, away from the Th2 cells, might be beneficial for fibrosis development.
Given their widespread use in diabetes, PPARγ agonists are now being tested in clinical trials both in NASH and HCV (Belfort et al, 2006). PPARγ nuclear receptors are expressed in HSCs, and PPARγ overexpression can regress MFBs to their quiescent state experimentally (Hazra et al, 2004). However, the total amount of HSCs detected by αSMA- or desmin-positive staining is decreased in animals with fibrosis regression (Kim et al, 2005), which suggests that MFB apoptosis, and not reversion into the quiescent form, is the predominant mechanism in vivo that leads to fibrosis regression. Synthetic PPARγ ligands downregulate HSC activation (Zhao et al, 2006).
Wnt signaling has been implicated in pulmonary and renal fibrosis and has also been reported to promote hepatic fibrosis by enhancing HSC activation and survival. This suggests that Wnt antagonism may be a useful target in liver fibrosis (Cheng et al, 2008).
Induction of MFB Apoptosis
Because the natural resolution of fibrosis leads to apoptosis and clearance of MFBs, approaches that exploit these native pathways of resolution merit attention. More specifically, because TIMP is antiapoptotic and blocks matrix proteases, reduced expression or neutralization favors clearance of MFBs through increased apoptosis and enhanced breakdown of scar (Iredale et al, 1998). The translation of this approach to humans will be challenging, however, as human cells appear to have more enhanced antiapoptotic activity through increased expression of the antiapoptotic protein Bcl-2 (Novo et al, 2006).
NF-κB is a nuclear transcription factor that inhibits apoptosis of MFBs, thus any compound that inhibits NF-κB merits evaluation (see Fig. 6.4). For example, bortezomib, a proteosomal inhibitor, prolongs the half-life of IκB-α, which is the naturally occurring cytosolic inhibitor of NF-κB (Oakley et al, 2003), preventing it from translocating to the nucleus and acting as a transcription factor. Related compounds, including gliotoxin or sulfalazine, exert similar effects by regulating transcription of NF-κB. For example, treatment of rodents with the NF-κB–inhibitor gliotoxin that specifically targets MFBs, with the help of targeting technologies (C1-3) selectively induced MFB apoptosis and accelerated regression of fibrosis in experimental liver injury (Douglass et al, 2008). Further, an NF-κB decoy increased TNF-α–induced MFB apoptosis and decreased CCl4-induced fibrosis in comparison to controls (Son et al, 2007).
Affecting the phosphorylation of CCAAT/enhancer-binding protein β (C/EBP-β) may lead to caspase activation and enhanced HSC apoptosis (see Fig. 6.4). C/EBP-β is a transcription factor predominantly expressed in adipose, hepatic, and immune tissue. When C/EBP-β is phosphorylated by ribosomal S-6 kinase (RSK), this leads to activation of caspase 8, which leads to MFB apoptosis (Buck & Chojkier, 2007). Thus, activating RSK or caspase 8 directly will lead to MFB apoptosis.
There are also receptor-ligand–mediated pathways of MFB apoptosis that are potential drug targets. For example, the cannabinoid-1 (CB1) receptor leads to increased collagen deposition and protects MFBs from apoptosis, whereas the cannabinoid-2 (CB2) receptor is proapoptotic via induction of intracellular oxidative stress (see Fig. 6.4). Correspondingly, CB1-knockout mice, CB1 antagonist–treated mice (Teixeira-Clerc et al, 2006), and CB2 receptor–stimulated mice (Munoz-Luque et al, 2008) display less fibrosis and more MFB apoptosis than controls after CCl4 or TAA treatment or bile duct ligation, and CB2-knockout mice showed increased CCl4-induced fibrosis (Julien et al, 2005). Whereas trials of a systemic CB1-receptor antagonist for obesity and NASH were discontinued because of central nervous system (CNS) effects, a new generation of peripheral CB1 antagonists that do not enter the CNS are under development and could play a major role in an antifibrotic strategy.
Natural killer (NK) cells directly interact with the RAE-1 ligand expressed by early-activated MFBs and can lead to apoptosis (Radaeva et al, 2006). This effect is lost, however, in mature MFBs, as these cells lose RAE-1 expression (Radaeva et al, 2007), thereby limiting the potential of therapeutic NK cell–mediated MFB death in advanced fibrosis.
The adipokines adiponectin and leptin are natural counterregulators. Leptin is produced by MFBs, which contributes to their activation (Marra, 2002; Ikejima et al, 2002), and it has a profibrogenic effect on MFBs in hepatic injury (Saxena et al, 2002). In contrast, adiponectin promotes MFB apoptosis (Ding et al, 2005) and inhibits liver fibrogenesis in vitro and in vivo (Kamada et al, 2003). Adiponectin may become a useful antifibrotic agent, particularly in NASH.
Blocking MFB–ECM Interactions
MFBs and ECM interact in a positive feedback mechanism that could be amenable to therapeutic antagonism. MFBs interact with ECM via α- and β-integrins (Zhou et al, 2004b), thereby decreasing apoptosis and increasing proliferation of MFBs. This leads to increased collagen type I deposition, which further promotes survival of fibrogenic MFBs (Issa et al, 2003); thus blocking the MFB–ECM interaction could lead to increased MFB apoptosis (see Fig. 6.4). This has been confirmed by α3β2-integrin disruption with echistatin, neutralizing antibodies, or small interfering RNA (Zhou et al, 2004b). Similar studies exploring antagonism of integrin α5β6 are ongoing.
Antagonizing Compounds That Mediate Inflammation
As inflammation precedes and stimulates liver fibrosis, the use of antiinflammatory drugs has been proposed. A number of agents have antiinflammatory activity. For example, corticosteroids have been used for decades to treat autoimmune hepatitis, and pentoxifylline may exert its antifibrotic activity by downregulating TGF-β1 and connective tissue growth factor signaling (Raetsch et al, 2002). However, pentoxifylline can upregulate TIMP-1, thereby reducing its antifibrotic effect. It also inhibits NF-κB in Kupffer cells, thereby reducing TNF-α production, the impact of which is uncertain.
The renin–angiotensin system may also amplify inflammation, and its role in hepatic fibrosis research has grown. Angiotensin II is a vasoconstrictive peptide expressed by activated HSC in chronically injured livers (Paizis et al, 2002; Bataller et al, 2003a). It induces hepatic inflammation and stimulates fibrogenic actions of HSCs including cell proliferation and migration, secretion of proinflammatory cytokines, and collagen synthesis (Bataller et al, 2000, 2003b, 2003c; see Fig. 6.4). Inhibitors of this system have been in clinical use for antihypertensive therapy for a substantial time, which makes their use in humans attractive. Preliminary studies in patients with chronic hepatitis C and nonalcoholic steatohepatitis suggest a positive effect on fibrosis progression by administering blocking agents (Yokohama et al, 2004), and several human trials are currently underway (Oakley et al, 2009, Colmenero et al, 2009).
Ursodeoxycholic acid (UDCA) has a beneficial effect on fibrosis in primary biliary cirrhosis. Similarly, a nitric oxide–releasing derivative of UDCA reduces inflammation, fibrosis, and portal pressure in an animal model (Fiorucci et al, 2003). Interestingly, UDCA also activates the pregnane X receptor (PXR), which has antifibrotic properties (Beuers et al, 2009). More recently, ligands of the farnesoid X receptor (FXR), another nuclear receptor, have been developed that are also antifibrotic in animal models (Zhang et al, 2009).
Selectively Antagonizing Pathways of HSC Activation
Fibrogenic, proliferative, proangiogenic, vasoconstrictive, and proinflammatory mediators work synergistically toward hepatic fibrogenesis in the setting of chronic liver injury. Thus, efforts are underway to antagonize the specific mediators driving these pathways. Multiple approaches have been directed toward blocking the profibrogenic TGF-β signaling pathway. The effects of soluble TGF-β receptor type II (George et al, 1999), TGF-β–blocking antibodies, TGF-β–antisense oligonucleotides, or agents that interfere with TGF-β downstream signal transduction have been assessed experimentally (see Fig. 6.4). Systemically blocking the TGF pathway has theoretical limitations; however, because apart from stimulating wound healing and fibrosis, TGF-β is also a central inhibitor of uncontrolled inflammation, and it is essential in inducing epithelial differentiation and in triggering apoptosis. This raises safety concerns for the general and long-term use of TGF-β inhibition, especially in patients with chronic hepatic inflammation. The concern regarding use of TGF-β–blocking agents has further increased as a result of the first clinical study using CAT-192, a recombinant human antibody that neutralizes TGF-β in patients with cutaneous systemic sclerosis, in which a significant increase in morbidity and mortality was shown, with no evidence of a therapeutic effect (Denton et al, 2007). Nonetheless, there are currently two monoclonal antibodies against TGF-β, lerdelimumab and meselimumab, in clinical Phase I through phase III trials for pulmonary fibrosis, systemic sclerosis, and postoperative scarring in glaucoma patients (Yingling et al, 2004).
To antagonize PDGF, administration of a PDGF kinase inhibitor with an HSC-selective carrier (mannose-6-phosphate–modified human serum albumin) significantly reduced bile duct ligation–induced fibrosis (Gonzalo et al, 2007). Imatinib mesylate, a clinically used PDGF receptor tyrosine kinase inhibitor, also attenuates proliferation, migration, and expression of αSMA and α2-(I)-procollagen mRNA in MFBs in a dose-dependent manner (Yoshiji et al, 2005). Furthermore, rapamycin, an immunosuppressive drug used after liver transplantation, has the added benefit of inhibiting HSC proliferation. It acts by inhibiting the p70s kinase, which is a downstream target of PDGF and leads to HSC proliferation (Gabele, 2005; see Fig. 6.4).
Proangiogenic growth factors, such as VEGF-A and angiopoietin 1, induce HSC proliferation and motility. The efficacy of the small-molecule VEGF receptor antagonist sunitinib in hepatocellular carcinoma (HCC) has been complemented by evidence of antifibrotic activity in an animal model (Tugues et al, 2007; see Fig. 6.4).
Apart from blocking HSC-stimulating factors, activating HSC-inhibitory factors is yet another possibility. Hepatocyte growth factor (HGF) inhibits HSC activation, decreases TGF-β, and increases HSC apoptosis (Kim et al, 2005; see Fig. 6.4). However, potential procarcinogenic effects are likely to limit its therapeutic use. Clinical trials with HGF deletion variants and mimetics are underway.
Bone marrow–derived mesenchymal stem cells have an antifibrotic effect, on the one hand increasing HSC apoptosis through secretion of HGF and on the other decreasing proliferation of HSCs through release of IL-10 and TNF-α upon stimulation by IL-6 from hepatic MFBs (Parekkadan et al, 2007). This makes stem cell therapy an interesting direction for fibrosis therapies. However, the bone marrow–derived mesenchymal stem cells also contribute to scar-forming MFBs in various organs, including the liver (Russo et al, 2006).
In rodents the blockade of the ETA receptor, which leads to vasoconstriction or scar contraction upon binding of ET-1, and the administration of vasodilators (prostaglandin E2 and nitric oxide donors) have antifibrotic qualities (Cho et al, 2000). This effect has yet to be confirmed in human studies.
Enhancing ECM Degradation
A number of rational approaches are under development to increase ECM degradation rather than solely block its production. As mentioned earlier, the two main families regulating ECM turnover are the matrix metalloproteinases (MMPs) that degrade the collagenous and noncollagenous ECM substrates and the TIMPs that inhibit MMP activities and also have an antiapoptotic effect on MFBs (Murphy et al, 2002; see Fig. 6.4). Blocking TIMP-1 with a monoclonal antibody reverses CCl4-induced fibrosis (Parsons et al, 2004).
Other experimental ways to shift the balance toward collagen degradation are the administration of uroplasminogen activator (uPA); it promotes formation of plasminogen out of plasmin, which upregulates MMP-9 synthesis (Hsiao et al, 2008; Hu et al, 2009; see Fig. 6.4). The administration via viral vector of recombinant proteolytically active MMP has shown antifibrotic potential in rodents (Iimuro et al, 2003; Siller-Lopez et al, 2004). However, there is a theoretical concern that MMP administration will lead to increased tumor formation. An advance using proteolytically inactive MMP-9 to solely neutralize TIMP-1 showed promising results without having this potential side effect (Roderfeld et al, 2006, 2007).
Adachi M, et al. Targeted mutation in the Fas gene causes hyperplasia in peripheral lymphoid organs and liver. Nat Genet. 1995;11(3):294-300.
Adams LA, et al. Hepascore: an accurate validated predictor of liver fibrosis in chronic hepatitis C infection. Clin Chem. 2005;51(10):1867-1873.
Angulo P, et al. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology. 2007;45(4):846-854.
Annoni G, Weiner FR, Zern MA. Increased transforming growth factor-beta 1 gene expression in human liver disease. J Hepatol. 1992;14(2-3):259-264.
Bachem MG, et al. Transforming growth factors (TGF alpha and TGF beta 1) stimulate chondroitin sulfate and hyaluronate synthesis in cultured rat liver fat-storing cells. FEBS Lett. 1989;257(1):134-137.
Bachem MG, Melchior R, Gressner AM. The role of thrombocytes in liver fibrogenesis: effects of platelet lysate and thrombocyte-derived growth factors on the mitogenic activity and glycosaminoglycan synthesis of cultured rat liver fat-storing cells. J Clin Chem Clin Biochem. 1989;27(9):555-565.
Bataller R, et al. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology. 2000;118(6):1149-1156.
Bataller R, et al. Activated human hepatic stellate cells express the renin–angiotensin system and synthesize angiotensin II. Gastroenterology. 2003;125(1):117-125.
Bataller R, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112(9):1383-1394.
Bataller R, et al. Prolonged infusion of angiotensin II into normal rats induces stellate cell activation and proinflammatory events in liver. Am J Physiol Gastrointest Liver Physiol. 2003;285(3):G642-G651.
Beaussier M, et al. Prominent contribution of portal mesenchymal cells to liver fibrosis in ischemic and obstructive cholestatic injuries. Lab Invest. 2007;87(3):292-303.
Bedossa P, Dargere D, Paradis V. Sampling variability of liver fibrosis in chronic hepatitis C. Hepatology. 2003;38(6):1449-1457.
Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology. 1996;24(2):289-293.
Belfort R, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355(22):2297-2307.
Berenguer M, et al. Severe recurrent hepatitis C after liver retransplantation for hepatitis C virus–related graft cirrhosis. Liver Transpl. 2003;9(3):228-235.
Beuers U, et al. Medical treatment of primary sclerosing cholangitis: a role for novel bile acids and other (post-)transcriptional modulators? Clin Rev Allergy Immunol. 2009;36(1):52-61.
Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006;26(10):1175-1186.
Bissell DM, et al. Cell-specific expression of transforming growth factor-beta in rat liver: evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest. 1995;96(1):447-455.
Borkham-Kamphorst E, et al. Antisense strategy against PDGF B-chain proves effective in preventing experimental liver fibrogenesis. Biochem Biophys Res Commun. 2004;321(2):413-423.
Boursier J, et al. The combination of a blood test and Fibroscan improves the non-invasive diagnosis of liver fibrosis. Liver Int. 2009;29(10):1507-1515.
Bravo AA, Sheth SG, Chopra S. Liver biopsy. N Engl J Med. 2001;344(7):495-500.
Buck M, Chojkier M. C/EBPbeta phosphorylation rescues macrophage dysfunction and apoptosis induced by anthrax lethal toxin. Am J Physiol Cell Physiol. 2007;293(6):C1788-C1796.
Cales P, et al. A novel panel of blood markers to assess the degree of liver fibrosis. Hepatology. 2005;42(6):1373-1381.
Callewaert N, et al. Noninvasive diagnosis of liver cirrhosis using DNA sequencer–based total serum protein glycomics. Nat Med. 2004;10(4):429-434.
Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39(2):273-278.
Canbay A, et al. The caspase inhibitor IDN-6556 attenuates hepatic injury and fibrosis in the bile duct–ligated mouse. J Pharmacol Exp Ther. 2004;308(3):1191-1196.
Cao Q, Mak KM, Lieber CS. DLPC decreases TGF-beta1–induced collagen mRNA by inhibiting p38 MAPK in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2002;283(5):G1051-G1061.
Carter-Kent C, et al. Nonalcoholic steatohepatitis in children: a multicenter clinicopathological study. Hepatology. 2009;50(4):1113-1120.
Casini A, et al. Neutrophil-derived superoxide anion induces lipid peroxidation and stimulates collagen synthesis in human hepatic stellate cells: role of nitric oxide. Hepatology. 1997;25(2):361-367.
Castera L, et al. Prospective comparison of transient elastography, Fibrotest, APRI, and liver biopsy for the assessment of fibrosis in chronic hepatitis C. Gastroenterology. 2005;128(2):343-350.
Chabot V, et al. CCL5-enhanced human immature dendritic cell migration through the basement membrane in vitro depends on matrix metalloproteinase-9. J Leukoc Biol. 2006;79(4):767-778.
Chen J, et al. Induction of caspase-3–like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18(13):4914-4928.
Cheng JH, et al. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G39-49.
Cho JJ, et al. An oral endothelin-A receptor antagonist blocks collagen synthesis and deposition in advanced rat liver fibrosis. Gastroenterology. 2000;118(6):1169-1178.
Chung L, et al. Collagenase unwinds triple-helical collagen prior to peptide bond hydrolysis. EMBO J. 2004;23(15):3020-3030.
Clouzeau-Girard H, et al. Effects of bile acids on biliary epithelial cell proliferation and portal fibroblast activation using rat liver slices. Lab Invest. 2006;86(3):275-285.
Colloredo G, et al. Impact of liver biopsy size on histological evaluation of chronic viral hepatitis: the smaller the sample, the milder the disease. J Hepatol. 2003;39(2):239-244.
Colmenero J, et al. Effects of losartan on hepatic expression of non-phagocytic NADPH oxidase and fibrogenic genes in patients with chronic hepatitis C. Am J Physiol Gastrointest Liver Physiol. 2009;297(4):G726-G734.
Crispe IN. Hepatic T cells and liver tolerance. Nat Rev Immunol. 2003;3(1):51-62.
Cross TJ, et al. Prospective comparison of Fibroscan, King’s score and liver biopsy for the assessment of cirrhosis in chronic hepatitis C infection. J Viral Hepat. 2009;17(8):546-554.
Denton CP, et al. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007;56(1):323-333.
Di Bisceglie AM, et al. Prolonged therapy of advanced chronic hepatitis C with low-dose peginterferon. N Engl J Med. 2008;359(23):2429-2441.
Ding X, et al. The roles of leptin and adiponectin: a novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am J Pathol. 2005;166(6):1655-1669.
Dodig M, et al. Differences in regulation of type I collagen synthesis in primary and passaged hepatic stellate cell cultures: the role of alpha5beta1-integrin. Am J Physiol Gastrointest Liver Physiol. 2007;293(1):G154-G164.
Donaldson P, et al. HLA and interleukin 1 gene polymorphisms in primary biliary cirrhosis: associations with disease progression and disease susceptibility. Gut. 2001;48(3):397-402.
Dooley S, et al. Expression of Smads during in vitro transdifferentiation of hepatic stellate cells to myofibroblasts. Biochem Biophys Res Commun. 2001;283(3):554-562.
Dooley S, et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta–mediated fibrogenesis and protects against liver damage. Gastroenterology. 2008;135(2):642-659.
Douglass A, et al. Antibody-targeted myofibroblast apoptosis reduces fibrosis during sustained liver injury. J Hepatol. 2008;49(1):88-98.
Emonard H, Grimaud JA. Matrix metalloproteinases: a review. Cell Mol Biol. 1990;36(2):131-153.
Fallowfield JA, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178(8):5288-5295.
Fierer DS, et al. Liver fibrosis during an outbreak of acute hepatitis C virus infection in HIV-infected men: a prospective cohort study. J Infect Dis. 2008;198(5):683-686.
Fiorucci S, Antonelli E, Morelli A. Nitric oxide and portal hypertension: a nitric oxide–releasing derivative of ursodeoxycholic acid that selectively releases nitric oxide in the liver. Dig Liver Dis. 2003;35(Suppl 2):S61-S69.
Forns X, et al. Identification of chronic hepatitis C patients without hepatic fibrosis by a simple predictive model. Hepatology. 2002;36(4 Pt 1):986-992.
Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275(4):2247-2250.
Friedman SL. Mechanisms of disease: mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol. 2004;1(2):98-105.
Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88(1):125-172.
Friedman SL, et al. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci U S A. 1985;82(24):8681-8685.
Friedman SL, Bansal MB. Reversal of hepatic fibrosis: fact or fantasy? Hepatology. 2006;43(2 Suppl 1):S82-S88.
Gabele E, et al. The role of p70S6K in hepatic stellate cell collagen gene expression and cell proliferation. J Biol Chem. 2005;280(14):13374-13382.
Gao B, Jeong WI, Tian Z. Liver: an organ with predominant innate immunity. Hepatology. 2008;47(2):729-736.
George J, et al. In vivo inhibition of rat stellate cell activation by soluble transforming growth factor beta type II receptor: a potential new therapy for hepatic fibrosis. Proc Natl Acad Sci U S A. 1999;96(22):12719-12724.
Gonzalo T, et al. Local inhibition of liver fibrosis by specific delivery of a platelet-derived growth factor kinase inhibitor to hepatic stellate cells. J Pharmacol Exp Ther. 2007;321(3):856-865.
Gressner OA, et al. Impact of quality control accepted inter-laboratory variations on calculated Fibrotest/Actitest scores for the non-invasive biochemical assessment of liver fibrosis. Clin Chim Acta. 2009;409(1-2):90-95.
Guha IN, et al. Noninvasive markers of fibrosis in nonalcoholic fatty liver disease: validating the European Liver Fibrosis Panel and exploring simple markers. Hepatology. 2008;47(2):455-460.
Guo J, et al. Functional linkage of cirrhosis-predictive single nucleotide polymorphisms of Toll-like receptor 4 to hepatic stellate cell responses. Hepatology. 2009;49(3):960-968.
Halfon P, et al. Comparison of test performance profile for blood tests of liver fibrosis in chronic hepatitis C. J Hepatol. 2007;46(3):395-402.
Hazra S, et al. PPAR gamma and hepatic stellate cells. Comp Hepatol. 2004;3(Suppl 1):S7.
Higuchi H, Gores GJ. Mechanisms of liver injury: an overview. Curr Mol Med. 2003;3(6):483-490.
Hironaka K, et al. Enhanced interstitial collagenase (matrix metalloproteinase-13) production of Kupffer cell by gadolinium chloride prevents pig serum–induced rat liver fibrosis. Biochem Biophys Res Commun. 2000;267(1):290-295.
Hoglen NC, et al. A caspase inhibitor, IDN-6556, ameliorates early hepatic injury in an ex vivo rat model of warm and cold ischemia. Liver Transpl. 2007;13(3):361-366.
Hsiao Y, et al. Disruption of tissue-type plasminogen activator gene in mice aggravated liver fibrosis. J Gastroenterol Hepatol. 2008;23(7 Pt 2):e258-e264.
Hu PF, et al. Adenovirus-mediated transfer of siRNA against PAI-1 mRNA ameliorates hepatic fibrosis in rats. J Hepatol. 2009;51(1):102-113.
Huang H, et al. Identification of two gene variants associated with risk of advanced fibrosis in patients with chronic hepatitis C. Gastroenterology. 2006;130(6):1679-1687.
Huang H, et al. A 7-gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology. 2007;46(2):297-306.
Iimuro Y, et al. Delivery of matrix metalloproteinase-1 attenuates established liver fibrosis in the rat. Gastroenterology. 2003;124(2):445-458.
Ikejima K, et al. Leptin receptor–mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology. 2002;122(5):1399-1410.
Ikura Y, et al. Expression of platelet-derived growth factor and its receptor in livers of patients with chronic liver disease. J Gastroenterol. 1997;32(4):496-501.
Imbert-Bismut F, et al. Biochemical markers of liver fibrosis in patients with hepatitis C virus infection: a prospective study. Lancet. 2001;357(9262):1069-1075.
Iredale JP, et al. Mechanisms of spontaneous resolution of rat liver fibrosis: hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102(3):538-549.
Ishak K, et al. Histological grading and staging of chronic hepatitis. J Hepatol. 1995;22(6):696-699.
Issa R, et al. Mutation in collagen-1 that confers resistance to the action of collagenase results in failure of recovery from CCl4-induced liver fibrosis, persistence of activated hepatic stellate cells, and diminished hepatocyte regeneration. Faseb J. 2003;17(1):47-49.
Jarvelainen HA, et al. Promoter polymorphism of the CD14 endotoxin receptor gene as a risk factor for alcoholic liver disease. Hepatology. 2001;33(5):1148-1153.
Julien B, et al. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology. 2005;128(3):742-755.
Kamada Y, et al. Enhanced carbon tetrachloride–induced liver fibrosis in mice lacking adiponectin. Gastroenterology. 2003;125(6):1796-1807.
Karlmark KR, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50(1):261-274.
Kelleher TB, Mehta SH, Bhaskar R, et al. Prediction of hepatic fibrosis in HIV/HCV co-infected patients using serum fibrosis markers: the SHASTA index. J Hepatol. 2005;43:78-84.
Kesteloot F, et al. ADAM metallopeptidase with thrombospondin type 1 motif 2 inactivation reduces the extent and stability of carbon tetrachloride–induced hepatic fibrosis in mice. Hepatology. 2007;46(5):1620-1631.
Kettaneh A, et al. Features associated with success rate and performance of FibroScan measurements for the diagnosis of cirrhosis in HCV patients: a prospective study of 935 patients. J Hepatol. 2007;46(4):628-634.
Kim WH, et al. Growth inhibition and apoptosis in liver myofibroblasts promoted by hepatocyte growth factor leads to resolution from liver cirrhosis. Am J Pathol. 2005;166(4):1017-1028.
Kleiner DE, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313-1321.
Knodell RG, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1(5):431-435.
Kossakowska AE, et al. Altered balance between matrix metalloproteinases and their inhibitors in experimental biliary fibrosis. Am J Pathol. 1998;153(6):1895-1902.
Lahsnig C, et al. ILEI requires oncogenic Ras for the epithelial to mesenchymal transition of hepatocytes and liver carcinoma progression. Oncogene. 2009;28(5):638-650.
Le Calvez S, et al. The predictive value of Fibrotest vs. APRI for the diagnosis of fibrosis in chronic hepatitis C. Hepatology. 2004;39(3):862-863. author reply 863
Lee CG, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med. 2001;194(6):809-821.
Leroy V, et al. Prospective comparison of six non-invasive scores for the diagnosis of liver fibrosis in chronic hepatitis C. J Hepatol. 2007;46(5):775-782.
Lewin M, et al. Diffusion-weighted magnetic resonance imaging for the assessment of fibrosis in chronic hepatitis C. Hepatology. 2007;46(3):658-665.
Ludwig J, Dickson ER, McDonald GS. Staging of chronic nonsuppurative destructive cholangitis (syndrome of primary biliary cirrhosis). Virchows Arch A Pathol Anat Histol. 1978;379(2):103-112.
Ludwig J, et al. Morphologic features of chronic hepatitis associated with primary sclerosing cholangitis and chronic ulcerative colitis. Hepatology. 1981;1(6):632-640.
Maher JJ, Lozier JS, Scott MK. Rat hepatic stellate cells produce cytokine-induced neutrophil chemoattractant in culture and in vivo. Am J Physiol. 1998;275(4 Pt 1):G847-G853.
Maher JJ, Zia S, Tzagarakis C. Acetaldehyde-induced stimulation of collagen synthesis and gene expression is dependent on conditions of cell culture: studies with rat lipocytes and fibroblasts. Alcohol Clin Exp Res. 1994;18(2):403-409.
Mann J, Mann DA. Transcriptional regulation of hepatic stellate cells. Adv Drug Deliv Rev. 2009;61(7-8):497-512.
Marra F, et al. Cultured human liver fat-storing cells produce monocyte chemotactic protein-1: regulation by proinflammatory cytokines. J Clin Invest. 1993;92(4):1674-1680.
Marra F, et al. Increased expression of monocyte chemotactic protein-1 during active hepatic fibrogenesis: correlation with monocyte infiltration. Am J Pathol. 1998;152(2):423-430.
Marra F. Leptin and liver fibrosis: a matter of fat. Gastroenterology. 2002;122(5):1529-1532.
Martin-Vilchez S, et al. The hepatitis B virus X protein induces paracrine activation of human hepatic stellate cells. Hepatology. 2008;47(6):1872-1883.
Mazzocca A, et al. Binding of hepatitis C virus envelope protein E2 to CD81 up-regulates matrix metalloproteinase-2 in human hepatic stellate cells. J Biol Chem. 2005;280(12):11329-11339.
Mehal WZ, Friedman SL, Gershwin ME, Vierling JM, Manns MP, editors. Liver Immunology, vol 2. Tatowa, NJ: Humana Press. 2007:99-109.
Milani S, et al. Differential expression of matrix-metalloproteinase-1 and -2 genes in normal and fibrotic human liver. Am J Pathol. 1994;144(3):528-537.
Mitchell C, et al. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol. 2009;174(5):1766-1775.
Muhanna N, et al. Activation of hepatic stellate cells after phagocytosis of lymphocytes: a novel pathway of fibrogenesis. Hepatology. 2008;48(3):963-977.
Munoz-Luque J, et al. Regression of fibrosis after chronic stimulation of cannabinoid CB2 receptor in cirrhotic rats. J Pharmacol Exp Ther. 2008;324(2):475-483.
Murawaki Y, et al. Serum tissue inhibitor of metalloproteinases in patients with chronic liver disease and with hepatocellular carcinoma. Clin Chim Acta. 1993;218(1):47-58.
Murphy FR, et al. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: implications for reversibility of liver fibrosis. J Biol Chem. 2002;277(13):11069-11076.
Nagula S, et al. Histological-hemodynamic correlation in cirrhosis: a histological classification of the severity of cirrhosis. J Hepatol. 2006;44(1):111-117.
Nakamura T, et al. Significance and therapeutic potential of endothelial progenitor cell transplantation in a cirrhotic liver rat model. Gastroenterology. 2007;133(1):91-107. e101
Natori S, et al. Apoptosis of sinusoidal endothelial cells occurs during liver preservation injury by a caspase-dependent mechanism. Transplantation. 1999;68(1):89-96.
Natori S, et al. The caspase inhibitor IDN-6556 prevents caspase activation and apoptosis in sinusoidal endothelial cells during liver preservation injury. Liver Transpl. 2003;9(3):278-284.
Naveau S, et al. Biomarkers for the prediction of liver fibrosis in patients with chronic alcoholic liver disease. Clin Gastroenterol Hepatol. 2005;3(2):167-174.
Naveau S, et al. Diagnostic and prognostic values of noninvasive biomarkers of fibrosis in patients with alcoholic liver disease. Hepatology. 2009;49(1):97-105.
Nelson DR, et al. Transforming growth factor-beta 1 in chronic hepatitis C. J Viral Hepat. 1997;4(1):29-35.
Nieto N, Friedman SL, Cederbaum AI. Stimulation and proliferation of primary rat hepatic stellate cells by cytochrome P450 2E1–derived reactive oxygen species. Hepatology. 2002;35(1):62-73.
Novo E, et al. Overexpression of Bcl-2 by activated human hepatic stellate cells: resistance to apoptosis as a mechanism of progressive hepatic fibrogenesis in humans. Gut. 2006;55(8):1174-1182.
Oakley F, et al. Basal expression of IkappaBalpha is controlled by the mammalian transcriptional repressor RBP-J (CBF1) and its activator Notch1. J Biol Chem. 2003;278(27):24359-24370.
Oakley F, et al. Angiotensin II activates I kappaB kinase phosphorylation of RelA at Ser 536 to promote myofibroblast survival and liver fibrosis. Gastroenterology. 2009;136(7):2334-2344. e2331
Olaso E, et al. DDR2 receptor promotes MMP-2–mediated proliferation and invasion by hepatic stellate cells. J Clin Invest. 2001;108(9):1369-1378.
Owen JL, et al. Up-regulation of matrix metalloproteinase-9 in T lymphocytes of mammary tumor bearers: role of vascular endothelial growth factor. J Immunol. 2003;171(8):4340-4351.
Paizis G, et al. Up-regulation of components of the renin–angiotensin system in the bile duct–ligated rat liver. Gastroenterology. 2002;123(5):1667-1676.
Parekkadan B, et al. Immunomodulation of activated hepatic stellate cells by mesenchymal stem cells. Biochem Biophys Res Commun. 2007;363(2):247-252.
Parsons CJ, et al. Antifibrotic effects of a tissue inhibitor of metalloproteinase-1 antibody on established liver fibrosis in rats. Hepatology. 2004;40(5):1106-1115.
Petricoin E, et al. Clinical proteomics: revolutionizing disease detection and patient tailoring therapy. J Proteome Res. 2004;3(2):209-217.
Pfeffer K, et al. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73(3):457-467.
Pockros PJ, et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology. 2007;46(2):324-329.
Powell EE, et al. Host genetic factors influence disease progression in chronic hepatitis C. Hepatology. 2000;31(4):828-833.
Poynard T, Ratziu V, Bedossa P. Appropriateness of liver biopsy. Can J Gastroenterol. 2000;14(6):543-548.
Poynard T, et al. Biochemical markers of liver fibrosis in patients infected by hepatitis C virus: longitudinal validation in a randomized trial. J Viral Hepat. 2002;9(2):128-133.
Poynard T, et al. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology. 2002;122(5):1303-1313.
Poynard T, et al. Prospective analysis of discordant results between biochemical markers and biopsy in patients with chronic hepatitis C. Clin Chem. 2004;50(8):1344-1355.
Poynard T, et al. Meta-analyses of FibroTest diagnostic value in chronic liver disease. BMC Gastroenterol. 2007;7:40.
Preaux AM, et al. Matrix metalloproteinase-2 activation in human hepatic fibrosis regulation by cell-matrix interactions. Hepatology. 1999;30(4):944-950.
Quinn PS, Higginson J. Reversible and irreversible changes in experimental cirrhosis. Am J Pathol. 1965;47:353-369.
Radaeva S, et al. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor–related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130(2):435-452.
Radaeva S, et al. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am J Physiol Gastrointest Liver Physiol. 2007;293(4):G809-G816.
Raetsch C, et al. Pentoxifylline downregulates profibrogenic cytokines and procollagen I expression in rat secondary biliary fibrosis. Gut. 2002;50(2):241-247.
Ramadori G, Saile B. Portal tract fibrogenesis in the liver. Lab Invest. 2004;84(2):153-159.
Regev A, et al. Sampling error and intraobserver variation in liver biopsy in patients with chronic HCV infection. Am J Gastroenterol. 2002;97(10):2614-2618.
Reif S, et al. The role of focal adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in hepatic stellate cell proliferation and type I collagen expression. J Biol Chem. 2003;278(10):8083-8090.
Roderfeld M, et al. Inhibition of hepatic fibrogenesis by matrix metalloproteinase-9 mutants in mice. FASEB J. 2006;20(3):444-454.
Roderfeld M, Hemmann S, Roeb E. Mechanisms of fibrinolysis in chronic liver injury (with special emphasis on MMPs and TIMPs). Z Gastroenterol. 2007;45(1):25-33.
Roulot D, et al. Quantitative analysis of transforming growth factor beta 1 messenger RNA in the liver of patients with chronic hepatitis C: absence of correlation between high levels and severity of disease. Hepatology. 1995;21(2):298-304.
Russo FP, et al. The bone marrow functionally contributes to liver fibrosis. Gastroenterology. 2006;130(6):1807-1821.
Rygiel KA, et al. Epithelial-mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Lab Invest. 2008;88(2):112-123.
Safadi R, et al. Immune stimulation of hepatic fibrogenesis by CD8 cells and attenuation by transgenic interleukin-10 from hepatocytes. Gastroenterology. 2004;127(3):870-882.
Sanchez-Conde M, et al. Comparison of transient elastography and liver biopsy for the assessment of liver fibrosis in HIV/hepatitis C virus-coinfected patients and correlation with noninvasive serum markers. J Viral Hepat. 2010;17(4):280-286.
Sandrin L, et al. Transient elastography: a new noninvasive method for assessment of hepatic fibrosis. Ultrasound Med Biol. 2003;29(12):1705-1713.
Saxena NK, et al. Leptin in hepatic fibrosis: evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology. 2002;35(4):762-771.
Schnabl B, et al. TAK1/JNK and p38 have opposite effects on rat hepatic stellate cells. Hepatology. 2001;34(5):953-963.
Schulze-Krebs A, et al. Hepatitis C virus–replicating hepatocytes induce fibrogenic activation of hepatic stellate cells. Gastroenterology. 2005;129(1):246-258.
Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48(1):322-335.
Shiraishi T, et al. Increased release of platelet-derived growth factor from platelets in chronic liver disease. Eur J Clin Chem Clin Biochem. 1994;32(1):5-9.
Shi Z, Wakil AE, Rockey DC. Strain-specific differences in mouse hepatic wound healing are mediated by divergent T helper cytokine responses. Proc Natl Acad Sci U S A. 1997;94(20):10663-10668.
Shi-Wen X, et al. Endothelin-1 promotes myofibroblast induction through the ETA receptor via a rac/phosphoinositide 3-kinase/Akt-dependent pathway and is essential for the enhanced contractile phenotype of fibrotic fibroblasts. Mol Biol Cell. 2004;15(6):2707-2719.
Siller-Lopez F, et al. Treatment with human metalloproteinase-8 gene delivery ameliorates experimental rat liver cirrhosis. Gastroenterology. 2004;126(4):1122-1133. discussion 1949
Son G, et al. Selective inactivation of NF-kappaB in the liver using NF-kappaB decoy suppresses CCl4-induced liver injury and fibrosis. Am J Physiol Gastrointest Liver Physiol. 2007;293(3):G631-G639.
Sterling RK, et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology. 2006;43(6):1317-1325.
Strobel T, et al. BAX enhances paclitaxel-induced apoptosis through a p53-independent pathway. Proc Natl Acad Sci U S A. 1996;93(24):14094-14099.
Svegliati-Baroni G, et al. Bile acids induce hepatic stellate cell proliferation via activation of the epidermal growth factor receptor. Gastroenterology. 2005;128(4):1042-1055.
Syn WK, et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology. 2009;137(4):1478-1488. e1478
Teixeira-Clerc F, et al. CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat Med. 2006;12(6):671-676.
Thabut D, et al. Noninvasive prediction of fibrosis in patients with chronic hepatitis C. Hepatology. 2003;37(5):1220-1221. author reply 1221
Thabut D, et al. The diagnostic value of biomarkers (AshTest) for the prediction of alcoholic steato-hepatitis in patients with chronic alcoholic liver disease. J Hepatol. 2006;44(6):1175-1185.
Tiegs G, Lohse AW. Immune tolerance: what is unique about the liver. J Autoimmun. 2010;34(1):1-6.
Tiggelman AM, et al. Interleukin-6 production by human liver (myo)fibroblasts in culture: evidence for a regulatory role of LPS, IL-1 beta and TNF alpha. J Hepatol. 1995;23(3):295-306.
Tsukada S, et al. SMAD and p38 MAPK signaling pathways independently regulate alpha1(I) collagen gene expression in unstimulated and transforming growth factor-beta–stimulated hepatic stellate cells. J Biol Chem. 2005;280(11):10055-10064.
Tugues S, et al. Antiangiogenic treatment with sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology. 2007;46(6):1919-1926.
Wai CT, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology. 2003;38(2):518-526.
Wang J, et al. Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology. 2009;137(2):713-723.
Wheeler MD, et al. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic Biol Med. 2001;31(12):1544-1549.
Williams KE, Olsen DR. Matrix metalloproteinase-1 cleavage site recognition and binding in full-length human type III collagen. Matrix Biol. 2009;28(6):373-379.
Winau F, et al. Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity. 2007;26(1):117-129.
Witek RP, et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology. 2009;50(5):1421-1430.
Wobser H, et al. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell Res. 2009;19(8):996-1005.
Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004;3(12):1011-1022.
Yokohama S, et al. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology. 2004;40(5):1222-1225.
Yoshida N, et al. Improvement of the survival rate after rat massive hepatectomy due to the reduction of apoptosis by caspase inhibitor. J Gastroenterol Hepatol. 2007;22(11):2015-2021.
Yoshiji H, et al. Imatinib mesylate (STI-571) attenuates liver fibrosis development in rats. Am J Physiol Gastrointest Liver Physiol. 2005;288(5):G907-G913.
Yu MC, et al. Inhibition of T cell responses by hepatic stellate cells via B7-H1–mediated T cell apoptosis in mice. Hepatology. 2004;40(6):1312-1321.
Yue HH, Diehl GE, Winoto A. Loss of TRAIL-R does not affect thymic or intestinal tumor development in p53 and adenomatous polyposis coli mutant mice. Cell Death Differ. 2005;12(1):94-97.
Zhang S, et al. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol. 2009;51(2):380-388.
Zhao C, et al. PPARgamma agonists prevent TGFbeta1/Smad3-signaling in human hepatic stellate cells. Biochem Biophys Res Commun. 2006;350(2):385-391.
Zhong W, et al. Inhibition of extracellular signal-regulated kinase 1 by adenovirus-mediated small interfering RNA attenuates hepatic fibrosis in rats. Hepatology. 2009;50(5):1524-1536.
Zhou X, et al. Expression of matrix metalloproteinase-2 and -14 persists during early resolution of experimental liver fibrosis and might contribute to fibrolysis. Liver Int. 2004;24(5):492-501.
Zhou X, et al. Engagement of alphavbeta3 integrin regulates proliferation and apoptosis of hepatic stellate cells. J Biol Chem. 2004;279(23):23996-24006.
Ziol M, et al. Noninvasive assessment of liver fibrosis by measurement of stiffness in patients with chronic hepatitis C. Hepatology. 2005;41(1):48-54.