[level-membership-for-surgery-category]
Chapter 8A Pancreatic cancer and premalignant tumors
Molecular aspects
Pancreatic Cancer Overview
Pancreatic ductal adenocarcinoma (PDA), commonly known as pancreatic cancer, is the fourth leading cause of cancer death in the United States, preceded only by lung, colon, breast (in women), and prostate (in men) cancers (American Cancer Society, 2009). The diagnosis of PDA is devastating because it has one of the highest mortality rates of any cancer (American Cancer Society, 2009). To underscore this point, in 2009 approximately 42,470 Americans were diagnosed with PDA, and about 35,240 will die from it (American Cancer Society, 2009). A diagnosis of PDA is met with the daunting statistic that the patient has an overall 5-year survival rate of less than 5%.
The past 2 decades have seen an exponential increase in our understanding of the molecular basis and etiology behind PDA (Hruban et al, 2001; Jones et al, 2008, 2009). Still, the clinical management of this disease—including primary prevention, early detection, and better targeted treatment options—has not changed significantly over the past decade. Currently, the only cure for this disease is surgical resection. Unfortunately, only 20% of the patient population presents with resectable disease (Winter et al, 2002).
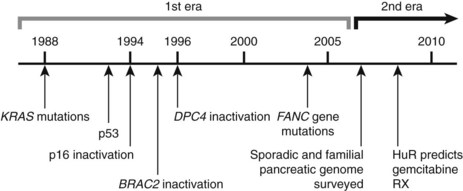
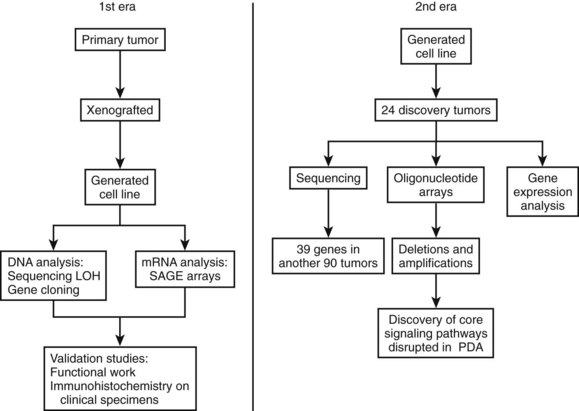
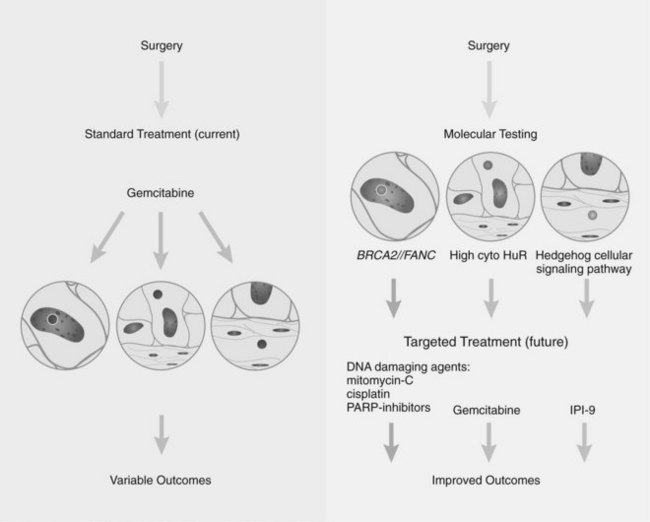
Research studying PDA can be simplistically divided into two eras (Fig. 8A.1). The first is highlighted by the discovery and development of technologies such as gene cloning and isolation of pure tumor tissue (i.e., xenografting primary tumors postresection; Fig. 8A.2, left). This era included landmark studies defining the genomic region of BRCA2 (Goggins et al, 1996), discovering SMAD4 (Hahn et al, 1996) as a tumor suppressor in PDA, activation of KRAS (Almoguera et al, 1988) as an early and frequent event in pancreatic tumorigenesis, and silencing of the CDKN2A (Caldas et al, 1994a) gene. As the result of advances in a number of molecular techniques, we are now upon the early stages of a new second era in genetic pancreatic cancer research, which combines high-throughput sequencing, gene expression and comparative genomic hybridization (CGH) arrays with statistical computational analysis, and large clinical databases (Fig. 8A.2, right) (Jones et al, 2008, 2009). This second era has already produced landmark publications that include a precise survey of multiple pancreatic cancer genomes both in sporadic and familial forms of the disease. This chapter aims to 1) provide a summary of the past two decades of pancreatic cancer research; 2) show that even though every tumor has a number of common molecular events, it is the differences between tumors that have clinical implications (Fig. 8A.3); and, finally, 3) set the stage for the future, which will include a discussion of successful early detection and treatment strategies for this deadly disease (Fig. 8A.3).
Progression Model of Pancreatic Ductal Adenocarcinoma
In the first era of pancreatic cancer research (see Figs. 8A.1 and 8A.2), the fields of molecular biology and pathology combined to establish a paradigm that PDA culminates from a multistep progression model (Hruban et al, 2001). This slow, sequential process may be the reason PDA is primarily a disease of people in their sixth and seventh decades of life. Definable pathologic markers on this stepwise progression—which follows a similar model first developed in colon carcinogenesis—are lesions referred to as pancreatic intraepithelial neoplasia (PanIN) (Hruban et al, 2001; Figs. 8A.4A and B). These lesions are believed to be precursor lesions to pancreatic cancer. PanIN lesions are thought to develop years before the emergence of PDA and are pathologically graded from low-grade PanIN lesions, PanIN-1 (Fig. 8A.4A), to high-grade lesions, PanIN-3 (see Fig. 8A.4B). Although low-grade PanIN lesions are common incidental findings, high-grade PanIN lesions are more common in pancreata with PDA.
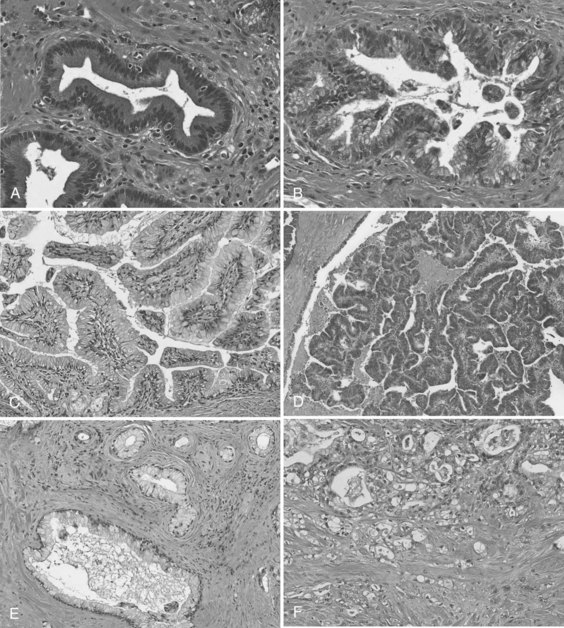
Key evidence that PanIN lesions are precursors to PDA is that similar hallmark molecular defects are found in PanIN lesions adjacent to invasive cancers (Biankin et al, 2003; Luttges et al, 2001a; Maitra et al, 2002; van Heek et al, 2002; Wilentz et al, 2000a). However, although KRAS mutations are frequently found in early PanIN lesions (Shi et al, 2009), genes involved in DNA repair mechanisms, such as TP53 and BRCA2, are altered late in this progression model. Because these data are based on limited sample numbers and data points, it remains somewhat counterintuitive that an oncogenic stimulus or stimuli (e.g., KRAS activation) precedes DNA damage (mutations). Recently, it was shown that higher density PanIN lesions were found in familial PDA patients compared with patients with sporadic forms of PDA, again arguing in favor of the hypothesis that PanIN lesions are true precursors to PDA (Shi et al, 2009).15
Intraductal Papillary Mucinous Neoplasm
Intraductal papillary mucinous neoplasms (IPMNs) are a well-accepted clinical and pathologic entity (Fig. 8A.4C and D; see Chapter 57) (Hruban et al, 2004). IPMNs typically produce radiographically identifiable pancreatic ductal dilation, which may predominantly involve the main pancreatic ducts (main-duct type IPMN), the secondary ducts (branch-duct type IPMN), or both types of ducts (mixed type). The distinction between the branch-duct type and main-duct type is important, because the former arise in younger patients, are more likely to involve the head and uncinate process of the pancreas, and are associated with lower-grade dysplasia and fewer invasive carcinomas. Approximately 30% to 40% of resected IPMNs harbor an invasive adenocarcinoma, and adenocarcinoma is most strongly associated with main-duct IPMNs. Approximately half of invasive carcinomas arising within IPMNs are so-called colloid (mucinous) carcinomas, and most of the remainder are tubular adenocarcinomas, the latter being histologically indistinguishable from invasive ductal adenocarcinomas that arise in the setting of PanINs (Adsay et al, 2002). Colloid carcinomas associated with IPMNs have a relatively good prognosis compared with other pancreatic carcinomas of the ductal type, which have a 5-year survival of 60% (Maire et al, 2002).
PanINs and IPMNs show some overlapping features, such that both are inherently intraductal lesions composed predominantly of columnar, mucin-producing cells that may grow in a flat configuration or may produce papillae; these lesions show a range of cytologic and architectural atypia and can give rise to invasive adenocarcinomas of the pancreas (Fig. 8A.4E and F). An important feature that distinguishes the two lesions is that PanINs are microscopic lesions, and IPMNs are macroscopic. Nevertheless, recognition of an IPMN and its distinction from a PanIN lesion is important for two reasons: 1) IPMN-associated colloid carcinomas have a significantly better prognosis than either PanIN- or IPMN-associated tubular adenocarcinomas (Adsay et al, 2001), and 2) IPMNs have a propensity to be multifocal lesions, therefore patients who undergo partial pancreatectomy and are left with a remnant pancreas need to be followed for life, even when the lesion originally resected was a noninvasive IPMN (Sohn et al, 2004).20
Genetic analyses of IPMNs have disclosed abnormalities in many of the same genes altered in conventional ductal adenocarcinoma, including mutations in the KRAS2, TP53, and CDKN2A genes, although the frequency and stage of neoplastic progression at which these alterations occur in IPMNs differ from PanINs (Hruban et al, 2004; Sessa et al, 1994). For example, the Peutz–Jeghers gene STK11 is inactivated in one third of IPMNs, and some patients with Peutz–Jeghers syndrome develop IPMNs (Sahin et al, 2003); this is in contrast to conventional ductal adenocarcinomas, in which loss of STK11 is a rare event. In contrast to ductal adenocarcinomas and PanIN-3 lesions, abnormalities in the SMAD4 gene seem to be rare in IPMNs (Iacobuzio-Donahue et al, 2000).
Another distinction between PanINs and IPMNs relates to the expression of the caudal differentiation factor CDX2, a marker of intestinal differentiation. Most IPMNs express CDX2, in particular IPMNs associated with an invasive colloid carcinoma, whereas this is uncommon both in PanINs and in the subset of IPMNs that give rise to invasive cancers resembling ductal adenocarcinomas (Adsay et al, 2004). CDX2 expression in IPMNs usually is associated with expression of MUC2, an intestinal epithelial apomucin, whereas absence of CDX2 usually is associated with expression of MUC1, a biliary apomucin, and concomitant lack of expression of MUC2. These findings have suggested that there may be two divergent pathways of carcinogenesis within the pancreatic ducts (Adsay et al, 2002). The first is a so-called intestinal pathway that gives rise to CDX2– and MUC2-expressing IPMNs that progress to the better-prognosis colloid carcinomas. The second is a pancreatobiliary pathway that gives rise to CDX2-negative, MUC2-negative, MUC1-expressing PanINs and a subset of IPMNs, both of which can progress to the poorer-prognosis conventional ductal adenocarcinomas. More recently, it has been observed that IPMNs with moderate dysplasia had reduced or absent pp32 nuclear expression (Brody et al, 2007). This may be functionally significant, because ANP32A acts as a tumor suppressor in pancreatic and other cancers, and the absence of this protein may facilitate the tumorigenesis process.
Genetics of Pancreatic Ductal Adenocarcinoma
Genomic (DNA) Alterations in Pancreatic Cancer
The multitude of genetic abnormalities in pancreatic cancer have many characteristics similar to other solid tumors, thus they include point mutations in critical genes, chromosomal (copy number) aberrations, mitochondrial DNA mutations, telomeric abnormalities, and epigenetic silencing by methylation of defined promoter DNA sequences. Recently, it was estimated that an individual pancreatic tumor contains, on average, 63 genetic alterations, primarily point mutations. It is also believed that only a small subset of these mutations is required for tumorigenesis (Jones et al, 2008).
The field of analyzing the genetics of pancreatic cancer can be broken down chronologically (see Fig. 8A.1). First, landmark studies starting in the late 1980s and spanning nearly two decades are highlighted by the discovery of KRAS activation, SMAD4 and BRCA2 mutations, and CDKN2A silencing (see Fig. 8A.1 and Fig. 8A.2, left). Some of these discoveries not only opened up lines of investigation in the field of pancreatic cancer but also opened up avenues with other tumor types and specific classification of PDA subtypes (Iacobuzio-Donahue et al, 2009). Then, in 2008 and 2009, Jones and colleagues, with the help of advanced DNA sequencing technology, were able to perform high-throughput genetic analysis on various pancreatic cancer genomes (see Figs. 8A.1 and 8A.2, right).
Copy-Number Aberrations
Although now considered primitive, valuable cytogenetic analysis performed over a decade ago found that chromosomal aberrations occur in virtually every pancreatic cancer. Cytogenetic analyses of pancreatic cancers have shown multiple, nonrandom numerical and structural changes (Griffin et al, 1995; Sirivatanauksorn et al, 2001). The most common numerical abnormalities include losses of chromosomes 6, 12, 13, and 18 and gains of chromosomes 7 and 20. Structural abnormalities (intrachromosomal break points) frequently involve 1p and 1q, 3p, 4q, 6q, 7q, 17p, 11p, 11q, 15q, 16q, and 19q (Rigaud et al, 2000). The technical limitations of conventional cytogenetics has presented challenges for identifying genes that are affected by chromosomal breaks. Allelotyping identifies areas of gross chromosomal loss by using polymorphic microsatellite markers to determine regions of genomic loss compared with matched normal tissues, also known as loss of heterozygosity (LOH) analysis. Allelotyping operates on the basic principle of the two-hit hypothesis, which postulates that tumor suppressor genes require biallelic inactivation. This most commonly happens by intragenic mutation in one allele, followed by loss of genetic material in the other allele. Identifying regions of single or biallelic loss or mutation holds the potential to understand the role of neighboring novel and well-known tumor suppressor genes. A landmark allelotype analysis of pancreatic cancers was performed using approximately 80 pancreatic cancer xenografts and 386 microsatellite markers (Iacobuzio-Donahue et al, 2004). This work discovered allelic losses in chromosome regions in proximity to tumor suppressor genes CDKN2A, TP53, and SMAD4. Allelotype analysis of PanIN lesions also has been performed using microdissected samples, and as expected, LOH is seen in many of the same chromosomal regions as invasive cancer, including 9p, 17p, and 18q (Luttges et al, 2001b; Yamano et al, 2000). Although the changes are conserved in most synchronous precursor lesions (i.e., the same allele is lost in PanIN and associated cancers), there is occasional clonal divergence between high-grade PanIN lesions harboring genetically distinct changes from the synchronous invasive cancer (Yamano et al, 2000). These findings may have important clinical implications in regard to tumor heterogeneity and clonal cancer cell drug resistance.
CGH identifies genomic amplifications and deletions and differentially labels normal and tumor genomic sequences with different dyes. The relative ratio of the two dyes indicates regions of cancer-associated losses or gains, with a ratio of 1 : 1 consistent with no change in copy number compared with normal DNA. Conventional CGH is performed on metaphase spreads and suffers from low resolution and inability to precisely map the various regions of amplifications and deletions (Mahlamaki et al, 1997). The resolution of array CGH is significantly better than the conventional technique, ranging from 500 kb to 30 kb, permitting the precise mapping of deletion and amplicon boundaries and genes targeted therein. Array technology also provides the ability to more efficiently use probes to study amplification of a larger number of genes. Array CGH analysis of pancreatic cancers has identified numerous recurrent copy-number aberrations, including amplifications of c-MYC (8q), EGFR (7p), KRAS (12p), AKT2 (19q), and NCOA3 (20q) and deletions of SMAD4 (18q), CDKN2A (9p), FHIT (3p), and MAP2K4 (17p) (Aguirre et al, 2004; Calhoun et al, 2006; Holzmann et al, 2004).
Utilizing high-density single-nucleotide polymorphism arrays, Calhoun and colleagues surveyed all the commercially available pancreatic cancer cell lines. In brief, this study provided high resolution and detailed break-point mapping of these cell lines and found two subclasses of cancer cell lines, original chromosomal instability (CIN) and holey CIN genotypes (Calhoun et al, 2006). Perhaps global classification of tumor cells with high-density arrays will become part of a prognostic or predictive molecular signature panel in the future.
Specific Gene Mutations
Much like other solid tumors, genes altered in pancreatic cancer include three functional classes: oncogenes, tumor suppressor genes, and caretaker genes. A family of caretaker genes recognized as being disrupted in pancreatic cancer are genes of the Fanconi anemia complementation group, which are involved in homologous recombination-based DNA damage repair (D’Andrea et al, 2003). Patients with Fanconi anemia present with a wide variety of clinical issues, including aplastic anemia and a high risk of developing cancer. BRCA2 is a member of this DNA repair pathway and is mutated in a subset of familial pancreatic cancers (Murphy et al, 2002). This has led to the search for mutations in other Fanconi anemia genes in pancreatic cancer. Somatic mutations of two genes in the core complex, FANCC and FANCG, were discovered but are rare in sporadic pancreatic cancers (van der Heijden et al, 2003). Through other modern techniques, Jones and colleagues (2009) discovered FANCN (PALB2) as another mutated gene found in familial pancreatic cancers.
Mutations in this core complex and in this DNA repair mechanism have major therapeutic implications (Fig. 8A.5, Table 8A.1; see the Familial Pancreatic cancer section below) (van der Heijden et al, 2004). Unlike most in vivo experiments, xenografted mice with isogenic cell lines (FANCC deficient and proficient) experienced regression of tumor after a single dose of the available mitomycin C intrastrand cross-linking drug (van der Heijden et al, 2005). Although other mutations in the Fanconi complementation group have not yet been described—other than FANN, FANCC, and FANCG—and the frequency of these mutations in PDA appear to be low, it is likely that defects in other FANC genes yet to be thoroughly investigated (i.e., FANCA) are the direct cause of some familial and sporadic PDAs.
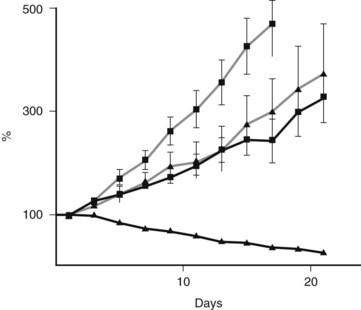
FIGURE 8A.5 Preclinical model shows an example of a successful targeted treatment strategy against a Fanconi-deficient tumor. A single-dose treatment with mitomycin C (5 mg/kg) of pancreatic cancer cell lines xenografted into nude mice. Note the hypersensitivity and tumor regression in the FANCC-deficient PL11 cells (squares) compared with the retrovirally corrected FANC-proficient PL11 cells (triangles). Solid lines indicate treated mice; gray lines indicate no treatment controls. Similar sensitivity was seen in the BRCA2-deficient CAPAN1 xenografted cells (van der Heijden et al, 2005).

Oncogenes
Perhaps the best evidence that KRAS activation is an early and important event in tumorigenesis comes from decades of research on pancreatic cancer. The KRAS2 oncogene on chromosome 12p is the most commonly altered oncogene, with up to 90% of pancreatic cancers containing mutations on codons 12, 13, and 61 (Caldas et al, 1994b). Activating mutations impairs the intrinsic GTPase activity of the KRAS2 gene product, resulting in a protein that is constitutively active in intracellular signal transduction. Inactivation of KRAS2 happens early in the pathway to oncogenesis, with approximately 30% of PanIN-1 lesions harboring KRAS2 mutations (Hruban et al, 2000; Moskaluk et al, 1997). The first mouse model of pancreatic cancer was generated by constitutive overexpression of mutant KRAS2 in the murine pancreatic ductal epithelium, underscoring its importance in pancreatic oncogenesis (Hingorani et al, 2003). This model was further developed into a powerful and useful preclinical model for PDA progression (Hingorani et al, 2005). Several good sources on mouse modeling and pancreatic cancer have been published (Frese et al, 2007; Karreth et al, 2009; Olive et al, 2006; Tuveson et al, 2005).
Rarely, pancreatic cancers with wild-type KRAS2 genes harbor point mutations of BRAF, another gene in the RAS/RAF/MAP kinase signaling pathway, thereby explaining why mutations of these genes occur in mutually exclusive patterns in pancreatic cancer (Calhoun et al, 2003). This highlights the importance of identifying different molecular targets that lead to similar pathways in pancreatic cancer development and of finding a drug that can target one pathway, not one gene. Studies have shown that targeting KRAS may have potential in modulating angiogenesis in tumorigenesis. Matsuo and colleagues showed that oncogenic overexpression of KRAS increases production of angiogenesis, promoting CXC chemokines and VEGF from human pancreatic duct epithelial cells. This upregulation acts through the mitogen-activated protein kinase pathway and c-JUN signaling (Matsuo et al, 2009). KRAS mutation has been shown to be associated with increased VEGFA expression and poorer prognosis in pancreatic carcinoma (Ikeda et al, 2001). Yet to date, targeting KRAS activation in PDA patients has shown no success. Perhaps KRAS activation is a critical early event in pancreatic tumorigenesis, but once cells become malignant, there is no need for constitutive KRAS activation or for oncogenic addiction for that matter.
Other oncogenes implicated in pancreatic cancers include MYC and EGFR, which can be amplified in various subsets of cancers. Overexpression of MYC transcripts occurs in approximately 50% to 60% of pancreatic cancers (Aguirre et al, 2004; Han et al, 2002). Therapeutic targeting using monoclonal antibodies or small-molecule tyrosine kinase inhibitors makes use of the constitutive activation of the EGFR signaling pathway in pancreatic cancers via amplification of the EGFR gene (Holzmann et al, 2004; Li et al, 2004). Lack of a rigorous and reliable immunohistochemical assay to detect overexpression of EGFR has proven to be difficult and limiting.
Tumor Suppressor Genes
CDKN2A, on chromosome 9p, is the most commonly inactivated gene in pancreatic cancers, occurring in 90% of patients (Caldas et al, 1994a; Schutte et al, 1997). CDKN2A belongs to the cyclin-dependent kinase inhibitor family and inhibits cell-cycle progression through the G1-S checkpoint mediated by cyclin-dependent kinases such as CDK4 and CDK6. Homozygous deletions (40%), intragenic mutation with loss of the second allele (40%), and epigenetic silencing by promoter methylation (10% to 15%) all contribute to gene inactivation. Immunolabeling for nuclear p16 protein expression is a reliable surrogate for CDKN2A genetic status; however, such immunolabeling does not identify specific genetic mechanisms of inactivation (Geradts et al, 2000). Loss of CDKN2A function occurs throughout the process of oncogenesis, with lesions appearing in different PanINs: 30% of PanIN-1A and PanIN-1B, 55% of PanIN-2, and 71% of PanIN-3 lesions show loss of nuclear p16 protein expression (Wilentz et al, 1998). The CDKN2A homozygous deletions encompass the MTAP gene in approximately 30% of pancreatic cancers, which offers potential therapeutic benefit, because targeted therapies have been developed that specifically inhibit the growth of MTAP-deficient cells (Chen et al, 1996).
Approximately 50% to 75% of pancreatic cancers contain an inactivation of the TP53 gene on chromosome 17p. Such inactivation almost always occurs via intragenic mutation combined with loss of the second allele. The TP53 (p53) protein leads to cell-cycle arrest and activates apoptosis in the presence of DNA damage. It is believed that loss of TP53 function allows cells to survive and divide, despite the presence of damaged DNA, leading to accumulation of additional genetic abnormalities and eventually to neoplasia. Nuclear overexpression of the TP53 protein does not correlate well with mutation status. By immunohistochemistry, TP53 accumulation is seen only in the advanced PanIN-3 lesions, consistent with TP53 being a “late” genetic event in pancreatic cancer progression (Maitra et al, 2003). PDA cells with a mutant TP53 have been shown to have a greater propensity for metastases (Morton et al, 2010). Of note, in an experimental model, the BRCA2 gene could not be artificially disrupted in cancer cells with intact wild-type TP53 status (Hucl et al, 2008; Callmeier et al, 2007). This interplay between TP53 and a genome maintenance pathway may have therapeutic value (see discussion on synthetic lethality later in this chapter).
Inactivation of the SMAD4 gene on chromosome 18q21 occurs in approximately 55% of pancreatic cancers by homozygous deletions (30%) or by intragenic mutations and loss of the second allele (25%). Loss of SMAD4 function interferes with intracellular signaling cascades downstream of the transforming growth factor (TGF) family of cell surface receptors, leading to decreased growth inhibition and uncontrolled proliferation. SMAD4 is only rarely mutated in other types of cancer (Schutte et al, 1996). Similar to TP53, loss of SMAD4 function is a late genetic event in pancreatic carcinoma progression, with loss of SMAD4 seen only in a few PanIN-3 lesions (Maitra et al, 2003). Examination of resected tumor specimens found that SMAD4 inactivation portends a poorer prognosis and greater potential to metastasize (Iacobuzio-Donahue et al, 2009; Blackford et al, 2009). In brief, upon autospy evaluation, PDA patients died, with either locally destructive pancreatic cancer (30%) or widespread metastatic disease (70%). At autopsy, intact DPC4 staining (wild-type DPC4 status) of the tumor tissue significantly correlated with a locally advanced tumor and no detectable widespread metastatic disease phenotype (22%), although the majority of patients who had widespread metastases, numbering in the hundreds and thousands, had loss of DPC4 (78%) (Iacobuzio-Donahue et al, 2009). Identification of downstream targets might allow restoration of DPC4-dependent signaling in pancreatic cancer, yielding an improved prognosis (Cao et al, 2008).
Several tumor suppressor genes are inactivated in smaller numbers (5% to 10%) of pancreatic cancers, including STK11 (chromosome 19p) (Sahin et al, 2003), TGFBR1 (chromosome 9q), TGFBR2 (chromosome 3p), RB1 (chromosome 13q) (Jaeger et al, 1997), and MAP2K4 (chromosome 17p) (Su et al, 1998). MAP2K4s function has been explored in a number of models, yet the main reason for its loss in pancreatic cancer is still unknown (Cunningham et al, 2006).
Other Caretaker Genes
In addition to classic oncogenes or tumor suppressor genes, caretaker genes (beyond Fanconi anemia–related genes) have been shown to play a role in oncogenesis. In theory, caretaker genes do not influence cell growth and proliferation directly but rather prevent the accumulation of DNA damage and cumulative mutations within key exonic sequences that make up the human genome. Loss of function of the DNA damage repair genes (MLH1, hMSH2) occurs in a small subset of pancreatic cancers in the familial setting but can also be seen in occasional, sporadic, nonfamilial cases (Ghimenti et al, 1999). Histologically, these microsatellite instability (MSI) cancers comprise poorly differentiated cancers with a syncytial growth pattern, expanding tumor margins, extensive necrosis, and intratumoral lymphocytic infiltrates. This uncommon variant has been termed medullary cancer to distinguish it from the more common pancreatic ductal adenocarcinoma (Wilentz et al, 2000b). Although findings in colon cancer have attempted to correlate MSI status with response to 5-fluorouracil, other studies have questioned these claims (Brody et al, 2009a).
Telomere Length Abnormalities
Telomeres are hexameric repeats of the sequence TTAGGG at the ends of chromosome arms that confer stability to chromosomes during cell division and prevent the ends from becoming “promiscuous” (Gisselsson, 2003). In other words, intact telomere structure guards against chromosomal fusion and thus may prevent chromosomal instability (Greenberg et al, 2005; Raynaud et al, 2008). In fact, telomeric dysfunction has been hypothesized to be one of the more important gateways of chromosomal instability, a signature of most solid cancers characterized by aneuploidy and extensive chromosomal rearrangements. The development of direct visualization of in situ telomere length was a breakthrough for understanding telomere length abnormalities and cancer development (Meeker et al, 2004). A study by van Heek and colleagues showed that telomere length abnormalities are one of the earliest demonstrable genetic aberrations in pancreatic cancer, with greater than 90% of the lowest grade PanIN lesions showing marked shortening of telomeres, as compared with normal duct epithelium (van Heek et al, 2002). It has been hypothesized that intact telomeres may serve as “caretakers” in the pancreatic ducts, and that the loss of telomeres in PanIN lesions sets the stage for progressive accumulation of chromosomal abnormalities, eventually culminating in neoplasia. Of note, Dr. Carol Greider, the lead investigator on a number of landmark studies involving telomerase and telomere function, recently (2009) won the Nobel prize based on her work with telomeres. This underscores the importance of this molecule’s involvement in both cancer and the aging process (Harley et al, 1994).
Alternative Genetic Silencing: Epigenetic Abnormalities
Although the classic two-hit hypothesis postulated that tumor suppressor gene silencing occurs by a combination of intragenic mutations and allelic loss, it has become apparent since the 1990s that epigenetic mechanisms of silencing are probably as important in terms of frequency and prevalence in many cancers (Baylin et al, 2000). Epigenetic silencing occurs predominantly through hypermethylation of so-called CpG islands in the promoter region of tumor suppressor genes, leading to transcriptional abrogation. Gene silencing by promoter methylation occurs physiologically at many sites in the human genome, where it is called imprinting. The most basic example is the one that occurs on an X chromosome in females, known as X inactivation, in which one of the two X chromosomes in females becomes silenced. In cancers, preferential hypermethylation of the promoter occurs in the neoplastic cells with consequent downregulation of gene expression, but this does not occur in the corresponding normal counterpart. Epigenetic silencing is seen frequently in pancreatic cancers and tends to involve genes that function in tumor suppression or in critical homeostatic pathways (e.g., CDKN2A, E-cadherin, retinoic acid β, osteonectin, SOCS1) or in both (Sato et al, 2003a; Ueki et al, 2000).
Aberrant methylation of genes is also found in precursor lesions of pancreatic cancers and tends to occur in intermediate- or late-stage lesions (PanIN-2 and PanIN-3) (Fukushima et al, 2002). The presence of promoter hypermethylation in noninvasive precursor lesions implies that the detection of aberrantly methylated DNA sequences in clinical samples might be useful as a strategy for early detection of pancreatic cancer. Methylated proenkephalin sequences can be detected in approximately 60% of pancreatic juice samples obtained from patients with pancreatic cancer but not in patients with nonneoplastic periampullary diseases, including chronic pancreatitis (Fukushima et al, 2003). The development of tumor-specific early detection panels using aberrantly methylated DNA sequences is currently an area of active investigation for many human cancers, including pancreatic cancer.
Although there has been extensive work on the role of promoter hypermethylation in the pathogenesis of cancers, more recent data suggest that promoter hypomethylation in candidate genes also may be important in cancer development and progression. Genes showing preferential hypomethylation in pancreatic cancers—SERPINB5, S100P, MSLN, PSCA, and CLDN4—are usually overexpressed in the cancers compared with normal pancreas, suggesting that epigenetic mechanisms can affect gene expression in either direction (Sato et al, 2003b).
Aberrant epigenetic silencing by promoter methylation also has been reported in IPMNs, including methylation of the SOCS1 and CDKN2A genes (Sato et al, 2002). Global analyses of gene expression in IPMNs have revealed the overexpression of LCN2, LGALS3, CTSE, CLDN4, and three members of the trefoil factor family, TFF1, TFF2, and TFF3 (Sato et al, 2004a; Terris et al, 2002). These global analyses also have shown that CLDN4, CXCR4, S100A4, and MSLN all are expressed at significantly higher levels in invasive IPMNs than in noninvasive IPMNs, suggesting that these proteins may contribute to the process of invasion (Sato et al, 2004b).
Second ERA of Molecular Biology
Core Signaling Pathways Disrupted in Pancreatic Cancer
The modern era of molecular biology has progressed to allow high-throughput surveying of the pancreatic cancer genome (Figs. 8A.1 and 8A.2, right), bringing some clarity to our understanding of the interactions of molecular pathways in tumorigenesis. This high-throughput analysis revealed that pancreatic cancers contain an average of 63 genes that are genetically altered (Jones et al, 2008). In this work, Jones and colleagues used a combination of modern molecular techniques to report that 67% to 100% of all pancreatic cancer genomes surveyed had a genetic abnormality in 12 core signaling pathways and processes. These pathways include apoptosis, DNA damage control, regulation of G1–S phase transition, Hedgehog signaling, hemophilic cell adhesion, integrin signaling, c-JUN N-terminal kinase signaling, KRAS signaling, regulation of invasion, small GTPase-dependent signaling (other than KRAS), TGFB signaling, and WNT/NOTCH signaling.
Unlike certain subtypes of leukemia that are driven by single “targetable” oncogenes, we have learned that pancreatic cancers result from genetic alterations of large numbers of genes that function through a distinct number of pathways. The study by Jones and colleagues suggests that it may be beneficial to target the physiologic effects of disrupted pathways rather than individual target genes.4 In fact, targeting multiple pathways or multiple points in the pathway may be in line with the early preclinical and clinical success stories of the concept of “synthetic lethality” (Ashworth, 2008). This promising direction in therapeutic development is extracted from fruit fly (Drosophila melanogaster) and yeast genetic studies, in which deletion of multiple genes in the same pathway was found to be deleterious to the organism, although disruption in just one gene in the pathway is not. Thus, targeting an additional gene in an already disrupted pathway in a tumor may be deleterious for tumor cells and may thus be a promising therapeutic strategy.
Familial Pancreatic Cancer
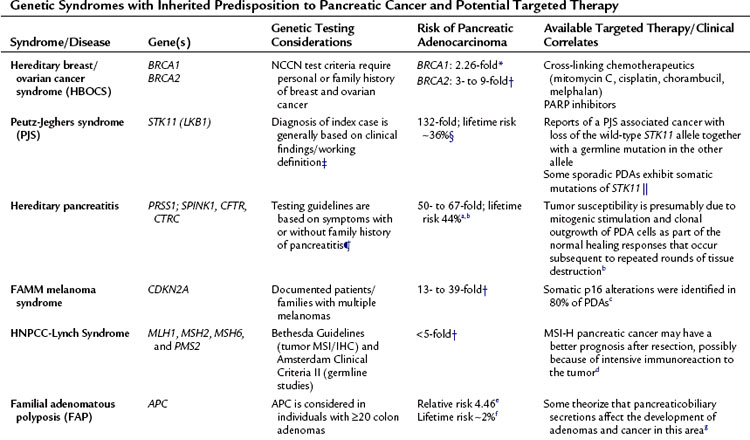
Approximately 10% of pancreatic cancers show familial aggregation (Petersen et al, 2003). The presence of two first-degree family members with pancreatic cancer confers a 6- to 18-fold increased risk of the disease in other first-degree relatives. This risk is increased 32- to 57-fold in families with three or more first-degree relatives with pancreatic cancer (Klein et al, 2004; Tersmette et al, 2001). Only a minority of these familial cancers is caused by a recognized cancer syndrome associated with germline mutations in known genes. Included in Table 8A.1 are possible “targeted therapies” for the genes and disorders germane to pancreatic cancer. Thus, this table underscores the significance for understanding the inherited lesion that may have contributed to the pancreatic cancers found in these families.
For example, as mentioned earlier in this chapter, Fanconi anemia is characterized as a rare, autosomally recessive cancer syndrome that results initially from a mutation in one of the multiple FANC/BRCA complementation groups in the FANC/BRCA pathway (Mathew, 2006). One gene in this pathway, BRCA2, is associated with a greatly increased risk of cancer when deleted via a biallelic mutation. The BRCA genes play a critical role in DNA repair via RAD51 repair pathways (Gudmundsdottir et al, 2006). Loss of functional BRCA1 and BRCA2 conveys chromosomal instability to cells by impairing the critical function of DNA double-stranded break repair (Ashworth, 2008). It has been well established that DNA damaging agents such as mitomycin c or cisplatin effectively kill cells with loss of BRCA2 or related genes (see Fig. 8A.5) (Hussain et al, 2004).
Currently, poly (ADP-ribose) polymerase (PARP) inhibitors have similar promising results as intrastrand cross-linking agents in early phase trials in other cancer types (ovarian, breast) (Farmer et al 2005). Certainly, pancreatic cancer is a logical tumor system in which to test novel PARP inhibitors in combination with other DNA-damaging agents. Targeting the FANC-BRCA pathway with PARP inhibitors similarly has been shown to lead to synthetic lethality, creating a convenient and fortuitous therapeutic window (Ashworth, 2008). The makeup of this therapeutic window relies on the fact that normal cells will have an intact DNA repair mechanism and thus will be capable of managing and repairing the damage put forth by a DNA-damaging agent. In contrast, the tumor will be unable to repair such damage due to loss of a key aspect of this repair mechanism (i.e., BRCA2; see Fig. 8A.5).
Identification of mutations in the FANC-BRCA pathway in familial cancers has the potential to shed light on the treatment of sporadic cancers as well. It has been suggested that perhaps up to 25% of sporadic breast and ovarian cancers manifest a BRCA-like phenotype. (Turner et al, 2004). The data have been derived from BRCA1, FANCC, FANCG, and FANCF methylation studies (Turner et al, 2004). Further studies are warranted, but future banking of sporadic pancreatic cancers to study all of the tumor characteristics—posttranscriptional modification, polymorphisms, CGH analysis—along with thorough analysis of family history may reveal a “BRCAness” among certain sporadic pancreatic tumors, aiding in the personalization of therapy (Martin et al, 2010). Focused DNA repair microarray analysis may also shed light on other cancer susceptibility genes.
Of note, a recent report demonstrated that PALB2, formerly known as FANCN, is a gene inherited in a mutant form that produces a stop codon in a small percentage of familial PDAs. This gene was discovered to be mutated in three of 96 familial pancreatic cancers, each producing a different stop codon (Jones et al, 2009). Truncating mutations in PALB2 were not found in any of the 1084 patients of similar ethnicity used as a control cohort in a similar study, thus ruling out a polymorphic sequence variant. This information suggests that next to BRCA2, PALB2 is the second most commonly mutated gene in hereditary pancreatic cancer (Jones et al, 2009). Thus most familial pancreatic cancers have no known genetic bases at this time, although many believe an autosomal dominant inheritance of a rare mutant allele is the most likely cause of these cancers. Of apparently “sporadic” (nonfamilial) pancreatic cancer patients, 7% harbor germline mutations in the BRCA2 gene, and this low-penetrance pattern is peculiar to cancers arising in the Ashkenazi Jewish population (Goggins et al, 1996). Perhaps no single gene is responsible for the other familial forms of PDA, carcinogenesis of which may be the best example that core pathways collaborate with the environment (Yeo et al, 2009); thus no single gene from one pathway will prove to be disrupted in the complex process of tumorigenesis in this familial form of pancreatic cancer.
Finding Familial Patients
Canto and colleagues published a list of arguments for and against screening patients with a family history of pancreatic cancer. Arguments for screening include the fact that once the disease is symptomatic, it is often not curable, and that high-risk patients can be identified and are often very compliant with follow-up. Arguments against screening include lack of a standard protocol on screening patients with two or fewer first-degree relatives with PDA and technical limitations in screening (Steinberg et al, 2009).
Transcriptomic (RNA) Abnormalities in Pancreatic Cancer
Although the presence of clonal genomic abnormalities in some ways defines the pancreatic cancer phenotype, recent years also have seen an explosion of new data characterizing the transcriptomic (RNA) abnormalities in human cancers. This cataloging has been greatly facilitated by the development of global expression analysis platforms, such as oligonucleotide microarrays, cDNA microarrays, and serial analysis of gene expression (SAGE). Several studies have analyzed pancreatic cancers and compared their gene expression profile with normal pancreas tissue to identify differentially overexpressed and underexpressed genes (Argani et al, 2001a; Crnogorac-Jurcevic et al, 2001; Geng et al, 1998; Iacobuzio-Donahue et al, 2002, 2003a, 2003b; Ryu et al, 2001, 2002). A comprehensive analysis of pancreatic cancer using high-density oligonucleotide microarrays identified 217 genes as being overexpressed threefold or higher in cancers versus normal tissue (Iacobuzio-Donahue et al, 2003a). Of these genes, 40 were determined to be overexpressed by more than one global expression profiling platform, and six genes—keratin 19, retinoic acid–induced 3, secretory leukocyte protease inhibitor, stratifin, tetraspan 1, and transglutaminase 2—were found to be overexpressed in pancreatic cancer by all three platforms: oligonucleotide and cDNA microarrays and SAGE. The future role of one or all of these six genes in early detection or therapy remains to be elucidated.
The identification of differentially expressed genes not only serves the further understanding of the basic biology of pancreatic cancers, it also provides a fertile ground to identify markers for early diagnosis, imaging, and novel therapeutic strategies. Mesothelin (MSLN) was identified by SAGE as a gene overexpressed in pancreatic cancers, and it was confirmed by immunohistochemistry to be restricted to the neoplastic epithelium (Argani et al, 2001b). These data were corroborated by a number of laboratories that included surveying of tumor tissues from diverse and distinct tumor banks (Showalter et al, 2008). This identification led to the development of a pancreatic cancer vaccine targeted to the mesothelin antigen and the development of antimesothelin antibody-conjugated immunotoxins, both of which are undergoing clinical trials in pancreatic cancer (Thomas et al, 2004). Moreover, a therapeutic strategy that delivers a suicide gene, DT-A, to pancreatic cancer cells under the regulation of the mesothelin promoter has showed early success (Showalter et al, 2008). Hucl and colleagues were able to define the exact sequence motif within the promoter region that was responsible for driving cancer-specific transcription, termed CanScript. This region was critical for binding of the TEF-1 transcription factor (Hucl et al, 2007). Interestingly, in the comprehensive SAGE analysis performed in the Jones and colleagues publication (see Fig. 8A.2) (Jones et al, 2009), mesothelin was labeled as not overexpressed, because it did not exceed the arbitrary tenfold-over-normal threshold. Future bench-work studies on these proteins should elucidate the functional significance of these overexpressed genes. Moreover, better tools in the laboratory will show that haploinsufficiency and/or modestly reduced and overexpressed molecules may have important effects on tumor phenotype and invasiveness.
More recently, the misexpression of two “embryonic” signaling pathways, Notch and Hedgehog, was described in most pancreatic cancers (Berman et al, 2003; Miyamoto et al, 2003; Thayer et al, 2003). These pathways are highly expressed in multiple tissues during in utero development, hence the term “embryonic”; but these are for the most part turned off in adult somatic cells, including those of the normal pancreas. Overexpression of genes in both the NOTCH and HEDGEHOG pathways have been described in invasive cancers and PanIN lesions but not in normal pancreatic duct epithelium (Miyamoto et al, 2003; Thayer et al, 2003). The neoplasia-specific activation of these pathways makes them an attractive target for therapy using specific small-molecule inhibitors. In fact, the HEDGEHOG small-molecule inhibitor cyclopamine has been shown to cause regression of pancreatic cancer growth in vitro and in vivo (Berman et al, 2003).
Posttranscriptional Regulation
In recent years, strong evidence has shown that posttranscriptional regulation of genes can directly affect both the tumorigenesis process (Lopez de Silanes et al, 2003, 2005) and cancer cell susceptibility to chemotherapy (Brody et al, 2009a; Constantino et al, 2009; Gorospe, 2003). Posttranscriptional gene regulation can have the same effect on gene expression as a genetic mutation or methylation of a promoter. One potent mechanism of posttranscriptional regulation involves RNA-binding proteins. One such RNA-binding protein that has been shown to be important in a number of tumor systems is Hu antigen R (HuR), a ubiquitously expressed member of the HU family that mediates cellular response to stress and DNA damage by posttranscriptional regulation (Hinman et al, 2008). Elevated HuR cytoplasmic expression is detected in tumors with poor pathologic features and poor predicted outcomes (Lopez de Silanes et al, 2005). It is has been shown that during times of certain cellular stressors—brought on by agents such as ultraviolet C (UVC) irradiation, heat shock, hypoxia, tamoxifen (Hostetter et al, 2008), and actinomycin D—HuR can bind to certain apoptotic or survival mRNA transcripts by binding to AU-rich elements in the 3′ untranslated region (UTR) of these mRNAs. In regard to tumorigenesis, HuR has been shown to bind to and stabilize proteins such as p21, p53, and cyclin A (Gorospe, 2003). For instance, Gorospe and colleagues showed that HuR can enhance translation of proteins such as p53 under stress from UVC irradiation (Gorospe, 2003). Thus HuR’s role in cellular stress and damage gives it a likely pivotal role in both the tumorigenesis process and in the acute cellular response to chemotherapy in pancreatic cancer cells.
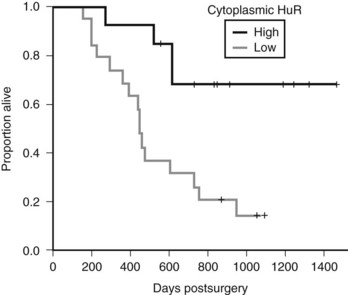
As an example, Costantino and colleagues showed that pancreatic cancer cell lines that overexpressed HuR were dramatically sensitive to gemcitabine, the standard chemotherapeutic treatment for pancreatic cancer, when compared with controls (Constantino et al, 2009). Treatment of cells with gemcitabine caused an increase in cytoplasmic HuR levels, and HuR associates with deoxycytidine kinase (dCK) mRNA and in turn regulates dCK protein levels (Constantino et al, 2009). This is important, because DCK encodes the protein that metabolizes gemcitabine. Clinically, cytoplasmic HuR levels in pancreatic cancer patients predicted response to gemcitabine therapy. In brief, patients who had low cytoplasmic HuR levels had a sevenfold increase in mortality compared with patients who had elevated cytoplasmic HuR levels (Fig. 8A.6) (Constantino et al, 2009). Prospective studies and attempts to manipulate HuR expression levels in tumor cells to optimize gemcitabine therapy are currently being investigated.
MicroRNAs
MicroRNAs (miRNAs) are defined as short, noncoding regions of RNA sequences (22 nucleotides) that can potently regulate gene expression patterns; miRNAs have been shown to regulate a number of disease- and developmental-related genes, and they are tissue specific in expression (Rosenfeld et al, 2008). These miRNA-specific attributes make them putative, powerful, and unique candidate biomarkers (Mardin et al, 2009). Discovering the presence of miRNAs in pancreatic cancer may be extremely valuable, although understanding the significance of these miRNAs may be more difficult and tedious, as miRNAs have been shown to distinguish between various disease states and tissues including pancreatitis, PDA, IPMN, and normal specimens (Mardin et al, 2009). Further, it has been shown that a miRNA molecular signature can stratify long- and short-term survivors (Bloomstron et al, 2007).
Future Perspectives on the Transcriptome
Genetic alterations have been proven over countless investigations to be critical for the development of PDA. Now that we have the technology to identify subtle mRNA expression changes, and also the ability to comprehend the complexity of numerous transcriptional regulatory mechanisms, we can begin to utilize this information for our understanding of PDA biology and therapeutic resistance mechanisms. Understanding the special plasticity of the pancreatic cancer transcriptome—not only in response to drugs (Brody et al, 2009b) but also in response to cancer-specific environmental concerns, such as hypoxia and radiation—will most likely be critical for our successful management of pancreatic cancer patients.
Stromal Content in Pancreatic Cancer
A number of studies have been conducted using cell co-culturing (Sato et al, 2004b) as well as genetic evaluation (Walter et al, 2008) of the stromal component of pancreatic cancer tissue. For example, Olive and colleagues studied the differences in gemcitabine delivery in mice that had tumors implanted in their tissue compared with mice that were genetically engineered (KPC mice) to conditionally express endogenous mutant KRAS and TP53 alleles similar to pancreatic cancer cells (Olive et al, 2009). Mice with transplanted tumors showed greater tissue accumulation of the active metabolites of gemcitabine than the KPC mice. Visual microscopic studies revealed that the KPC mice had a much greater stromal content and reduced blood vessel density than the tumor-implanted mice, therefore increasing the space between neoplastic cells and blood flow. The decreased stromal content in the tumor-implanted mice apparently led to greater therapeutic efficiency of gemcitabine delivery. To pursue this issue, the investigators treated mice with IPI-926, an inhibitor of SMO that is normally upregulated in the HEDGEHOG pathway in stromal cells. Mice treated with IPI-926 exhibited depletion of desmoplastic stroma and increased tumor vascularity. In turn, the concentration of gemcitabine in KPC tumors was elevated by 60% after 10 days of pretreatment with the combined IPI-926/gemcitabine treatment. KPC mice treated with the combination therapy showed decreased tumor size and increased survival benefit when compared with mice treated with IPI-926 or gemcitabine alone (Olive et al, 2009). This study is a nice example of exploring and exploiting a potential target in stroma cells, and its results sheds light on the importance of identifying pathways (i.e., stromal development) that lead to tumorigenesis or impact drug delivery. Treating the stromal component of the tumor with IPI-926 and taking advantage of the tumor cell’s HuR status is an example of how we may be able to powerfully target both the stromal and epithelial components of pancreatic cancer at the same time.
Final Thoughts and Perspectives
We are currently at an interesting and critical time in studying the molecular aspects of pancreatic cancer. The research community has incredible resources at its disposal, ranging from patient databases to complex sequencing equipment. This coming of age of pancreatic cancer research must include surgeons, pathologists, molecular biologists, and medical oncologists collaborating toward ultimately better and more personalized patient care. It is also time for the National Institutes of Health and other funding agencies to provide increased support and to step up their efforts at this exciting time in pancreatic cancer research. Hopefully, those in the field will take the time to connect the dots with all the rigorous and voluminous research that has been performed over the last few decades (see Fig. 8A.1), allowing future landmark progress. Current research will need to provide better early detection markers, so that physicians can have more opportunities to prevent cancer from forming, instead of attempting to cure it before it is too late. Our hope is that the treatment of pancreatic cancer will follow a paradigm similar to that of infectious disease specialists treating bacterial infections. Specific bacterial infections are easily cleared with specific antibiotics. Following this logical course, and based on the past decades of research, an individual pancreatic cancer patient of the future will likely benefit from personalized treatment. In the coming decade, we must translate our molecular research efforts into a better understanding of familial risk, earlier detection, and logical but innovative therapies in order to generate better outcomes for our patients.
Adsay NV, et al. Colloid (mucinous noncystic) carcinoma of the pancreas. Am J Surg Pathol. 2001;25(1):26-42.
Adsay NV, et al. The dichotomy in the preinvasive neoplasia to invasive carcinoma sequence in the pancreas: differential expression of MUC1 and MUC2 supports the existence of two separate pathways of carcinogenesis. Mod Pathol. 2002;15(10):1087-1095.
Adsay NV, et al. Pathologically and biologically distinct types of epithelium in intraductal papillary mucinous neoplasms: delineation of an “intestinal” pathway of carcinogenesis in the pancreas. Am J Surg Pathol. 2004;28(7):839-848.
Aguirre AJ, et al. High-resolution characterization of the pancreatic adenocarcinoma genome. Proc Natl Acad Sci U S A. 2004;101(24):9067-9072.
Almoguera C, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53(4):549-554.
American Cancer Society. Cancer Facts and Figures 2009. Accessed online at http://www.cancer.org/downloads/STT/500809web.pdf, 2009.
Argani P, et al. Discovery of new markers of cancer through serial analysis of gene expression: prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. 2001;61(11):4320-4324.
Argani P, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res. 2001;7(12):3862-3868.
Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26(22):3785-3790.
Baylin SB, et al. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16(4):168-174.
Berman DM, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425(6960):846-851.
Biankin AV, et al. Molecular pathogenesis of precursor lesions of pancreatic ductal adenocarcinoma. Pathology. 2003;35(1):14-24.
Blackford A, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 2009;15(14):4674-4679.
Bloomston M, et al. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA. 2007;297(17):1901-1908.
Brody JR, et al. Reduction of pp32 expression in poorly differentiated pancreatic ductal adenocarcinomas and intraductal papillary mucinous neoplasms with moderate dysplasia. Mod Pathol. 2007;20(12):1238-1244.
Brody JR, et al. Limits to thymidylate synthase and TP53 genes as predictive determinants for fluoropyrimidine sensitivity and further evidence for RNA-based toxicity as a major influence. Cancer Res. 2009;69(3):984-991.
Brody JR, et al. The “RNA-binding ome”: future implications for chemotherapeutic efficacy. Future Oncol. 2009;5(9):1317-1319.
Cao D, et al. Differential expression of multiple genes in association with MADH4/DPC4/SMAD4 inactivation in pancreatic cancer. Int J Clin Exp Pathol. 2008;1(6):510-517.
Caldas C, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8(1):27-32.
Caldas C, et al. Detection of K-ras mutations in the stool of patients with pancreatic adenocarcinoma and pancreatic ductal hyperplasia. Cancer Res. 1994;54(13):3568-3573.
Calhoun ES, et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am J Pathol. 2003;163(4):1255-1260.
Calhoun ES, et al. Identifying allelic loss and homozygous deletions in pancreatic cancer without matched normals using high-density single-nucleotide polymorphism arrays. Cancer Res. 2006;66(16):7920-7928.
Chen ZH, et al. Gene deletion chemoselectivity: codeletion of the genes for p16(INK4), methylthioadenosine phosphorylase, and the alpha- and beta-interferons in human pancreatic cell carcinoma lines and its implications for chemotherapy. Cancer Res. 1996;56(5):1083-1090.
Costantino CL, et al. The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer Res. 2009;69(11):4567-4572.
Crnogorac-Jurcevic T, et al. Gene expression profiles of pancreatic cancer and stromal desmoplasia. Oncogene. 2001;20(50):7437-7446.
Cunningham SC, et al. Targeted deletion of MKK4 in cancer cells: a detrimental phenotype manifests as decreased experimental metastasis and suggests a counterweight to the evolution of tumor-suppressor loss. Cancer Res. 2006;66(11):5560-5564.
D’Andrea AD, et al. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer. 2003;3(1):23-34.
Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917-921.
Frese KK, et al. Maximizing mouse cancer models. Nat Rev Cancer. 2007;7(9):645-658.
Fukushima N, et al. Aberrant methylation of preproenkephalin and p16 genes in pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am J Pathol. 2002;160(5):1573-1581.
Fukushima N, et al. Diagnosing pancreatic cancer using methylation specific PCR analysis of pancreatic juice. Cancer Biol Ther. 2003;2(1):78-83.
Gallmeier E, et al. Gene-specific selection against experimental fanconi anemia gene inactivation in human cancer. Cancer Biol Ther. 2007;6(5):654-660.
Geng M, et al. Isolation of differentially expressed genes by combining representational difference analysis (RDA) and cDNA library arrays. Biotechniques. 1998;25(3):434-438.
Geradts J, et al. Immunohistochemical p16INK4a analysis of archival tumors with deletion, hypermethylation, or mutation of the CDKN2/MTS1 gene: a comparison of four commercial antibodies. Appl Immunohistochem Mol Morphol. 2000;8(1):71-79.
Ghimenti C, et al. Microsatellite instability and mismatch repair gene inactivation in sporadic pancreatic and colon tumours. Br J Cancer. 1999;80(1-2):11-16.
Gisselsson D. Chromosome instability in cancer: how, when, and why? Adv Cancer Res. 2003;87:1-29.
Goggins M, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56(23):5360-5364.
Gorospe M. HuR in the mammalian genotoxic response: post-transcriptional multitasking. Cell Cycle. 2003;2(5):412-414.
Greenberg RA, et al. Telomere structural dynamics in genome integrity control and carcinogenesis. Adv Exp Med Biol. 2005;570:311-341.
Griffin CA, et al. Consistent chromosome abnormalities in adenocarcinoma of the pancreas. Cancer Res. 1995;55(11):2394-2399.
Gudmundsdottir K, et al. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25(43):5864-5874.
Hahn SA, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271(5247):350-353.
Han H, et al. Identification of differentially expressed genes in pancreatic cancer cells using cDNA microarray. Cancer Res. 2002;62(10):2890-2896.
Harley CB, et al. Telomerase, cell immortality, and cancer. Cold Spring Harb Symp Quant Biol. 1994;59:307-315.
Hingorani SR, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437-450.
Hingorani SR, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469-483.
Hinman MN, et al. Diverse molecular functions of Hu proteins. Cell Mol Life Sci. 2008;65(20):3168-3181.
Holzmann K, et al. Genomic DNA-chip hybridization reveals a higher incidence of genomic amplifications in pancreatic cancer than conventional comparative genomic hybridization and leads to the identification of novel candidate genes. Cancer Res. 2004;64(13):4428-4433.
Hostetter C, et al. Cytoplasmic accumulation of the RNA binding protein HuR is central to tamoxifen resistance in estrogen receptor–positive breast cancer cells. Cancer Biol Ther. 2008;7(9):1496-1506.
Hruban RH, et al. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6(8):2969-2972.
Hruban RH, et al. Molecular pathology of pancreatic cancer. Cancer J. 2001;7(4):251-258.
Hruban RH, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28(8):977-987.
Hucl T, et al. High cancer-specific expression of mesothelin (MSLN) is attributable to an upstream enhancer containing a transcription enhancer factor dependent MCAT motif. Cancer Res. 2007;67(19):9055-9065.
Hucl T, et al. A syngeneic variance library for functional annotation of human variation: application to BRCA2. Cancer Res. 2008;68(13):5023-5030.
Hussain S, et al. Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum Mol Genet. 2004;13(12):1241-1248.
Iacobuzio-Donahue CA, et al. Dpc-4 protein is expressed in virtually all human intraductal papillary mucinous neoplasms of the pancreas: comparison with conventional ductal adenocarcinomas. Am J Pathol. 2000;157(3):755-761.
Iacobuzio-Donahue CA, et al. Exploring the host desmoplastic response to pancreatic carcinoma: gene expression of stromal and neoplastic cells at the site of primary invasion. Am J Pathol. 2002;160(1):91-99.
Iacobuzio-Donahue CA, et al. Highly expressed genes in pancreatic ductal adenocarcinomas: a comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res. 2003;63(24):8614-8622.
Iacobuzio-Donahue CA, et al. Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays. Am J Pathol. 2003;162(4):1151-1162.
Iacobuzio-Donahue CA, et al. Large-scale allelotype of pancreaticobiliary carcinoma provides quantitative estimates of genome-wide allelic loss. Cancer Res. 2004;64(3):871-875.
Iacobuzio-Donahue CA, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27(11):1806-1813.
Ikeda N, et al. The association of K-ras gene mutation and vascular endothelial growth factor gene expression in pancreatic carcinoma. Cancer. 2001;92(3):488-499.
Jaeger D, et al. The role of synaptic and voltage-gated currents in the control of Purkinje cell spiking: a modeling study. J Neurosci. 1997;17(1):91-106.
Jones S, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;33:1801-1806.
Jones S, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324(5924):217.
Karreth FA, et al. Modelling oncogenic Ras/Raf signalling in the mouse. Curr Opin Genet Dev. 2009;19(1):4-11.
Klein AP, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004;64(7):2634-2638.
Li J, et al. Gefitinib (“Iressa,” ZD1839), a selective epidermal growth factor receptor tyrosine kinase inhibitor, inhibits pancreatic cancer cell growth, invasion, and colony formation. Int J Oncol. 2004;25(1):203-210.
Lopez de Silanes I, et al. Role of the RNA-binding protein HuR in colon carcinogenesis. Oncogene. 2003;22(46):7146-7154.
Lopez de Silanes I, et al. HuR: post-transcriptional paths to malignancy. RNA Biol. 2005;2(1):11-13.
Luttges J, et al. The immunohistochemical mucin expression pattern distinguishes different types of intraductal papillary mucinous neoplasms of the pancreas and determines their relationship to mucinous noncystic carcinoma and ductal adenocarcinoma. Am J Surg Pathol. 2001;25(7):942-948.
Luttges J, et al. Allelic loss is often the first hit in the biallelic inactivation of the p53 and DPC4 genes during pancreatic carcinogenesis. Am J Pathol. 2001;158(5):1677-1683.
Mahlamaki EH, et al. Comparative genomic hybridization reveals frequent gains of 20q, 8q, 11q, 12p, and 17q, and losses of 18q, 9p, and 15q in pancreatic cancer. Genes Chromosomes Cancer. 1997;20(4):383-391.
Maire F, et al. Prognosis of malignant intraductal papillary mucinous tumours of the pancreas after surgical resection. Comparison with pancreatic ductal adenocarcinoma. Gut. 2002;51(5):717-722.
Maitra A, et al. Cyclooxygenase 2 expression in pancreatic adenocarcinoma and pancreatic intraepithelial neoplasia: an immunohistochemical analysis with automated cellular imaging. Am J Clin Pathol. 2002;118(2):194-201.
Maitra A, et al. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol. 2003;16(9):902-912.
Mardin WA, et al. MicroRNAs: novel diagnostic and therapeutic tools for pancreatic ductal adenocarcinoma? Ann Surg Oncol. 2009;16(11):3183-3189.
Martin SA, et al. Genomic instability and the selection of treatments for cancer. J Pathol. 2010;220(2):281-289.
Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene. 2006;25(43):5875-5884.
Matsuo Y, et al. K-Ras promotes angiogenesis mediated by immortalized human pancreatic epithelial cells through mitogen-activated protein kinase signaling pathways. Mol Cancer Res. 2009;7(6):799-808.
Meeker AK, et al. Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clin Cancer Res. 2004;10(10):3317-3326.
Miyamoto Y, et al. Notch mediates TGF alpha–induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3(6):565-576.
Morton JP, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107(1):246-251.
Moskaluk CA, et al. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997;57(11):2140-2143.
Murphy KM, et al. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17%. Cancer Res. 2002;62(13):3789-3793.
Olive KP, et al. The use of targeted mouse models for preclinical testing of novel cancer therapeutics. Clin Cancer Res. 2006;12(18):5277-5287.
Olive KP, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457-1461.
Petersen GM, et al. Familial pancreatic cancer: where are we in 2003? J Natl Cancer Inst. 2003;95(3):180-181.
Raynaud CM, et al. Telomere length, telomeric proteins and genomic instability during the multistep carcinogenic process. Crit Rev Oncol Hematol. 2008;66(2):99-117.
Rigaud G, et al. Allelotype of pancreatic acinar cell carcinoma. Int J Cancer. 2000;88(5):772-777.
Rosenfeld N, et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 2008;26(4):462-469.
Ryu B, et al. Invasion-specific genes in malignancy: serial analysis of gene expression comparisons of primary and passaged cancers. Cancer Res. 2001;61(5):1833-1838.
Ryu B, et al. Relationships and differentially expressed genes among pancreatic cancers examined by large-scale serial analysis of gene expression. Cancer Res. 2002;62(3):819-826.
Sahin F, et al. Loss of Stk11/Lkb1 expression in pancreatic and biliary neoplasms. Mod Pathol. 2003;16(7):686-691.
Sato N, et al. Aberrant methylation of CpG islands in intraductal papillary mucinous neoplasms of the pancreas. Gastroenterology. 2002;123(1):365-372.
Sato N, et al. Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high-throughput microarrays. Cancer Res. 2003;63(13):3735-3742.
Sato N, et al. Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma. Cancer Res. 2003;63(14):4158-4166.
Sato N, et al. Gene expression profiling identifies genes associated with invasive intraductal papillary mucinous neoplasms of the pancreas. Am J Pathol. 2004;164(3):903-914.
Sato N, et al. Gene expression profiling of tumor-stromal interactions between pancreatic cancer cells and stromal fibroblasts. Cancer Res. 2004;64(19):6950-6956.
Schutte M, et al. DPC4 gene in various tumor types. Cancer Res. 1996;56(11):2527-2530.
Schutte M, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57(15):3126-3130.
Sessa F, et al. Intraductal papillary-mucinous tumours represent a distinct group of pancreatic neoplasms: an investigation of tumour cell differentiation and K-ras, p53 and c-erbB-2 abnormalities in 26 patients. Virchows Arch. 1994;425(4):357-367.
Shi C, et al. KRAS2 mutations in human pancreatic acinar-ductal metaplastic lesions are limited to those with PanIN: implications for the human pancreatic cancer cell of origin. Mol Cancer Res. 2009;7(2):230-236.
Showalter SL, et al. Nanoparticulate delivery of diphtheria toxin DNA effectively kills mesothelin-expressing pancreatic cancer cells. Cancer Biol Ther. 2008;7(10):1584-1590.
Sirivatanauksorn V, et al. Non-random chromosomal rearrangements in pancreatic cancer cell lines identified by spectral karyotyping. Int J Cancer. 2001;91(3):350-358.
Sohn TA, et al. Intraductal papillary mucinous neoplasms of the pancreas: an updated experience. Ann Surg. 2004;239(6):788-797.
Steinberg WM, et al. Should patients with a strong family history of pancreatic cancer be screened on a periodic basis for cancer of the pancreas? Pancreas. 2009;38(5):e137-e150.
Su GH, et al. Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor suppressor gene. Cancer Res. 1998;58(11):2339-2342.
Terris B, et al. Characterization of gene expression profiles in intraductal papillary-mucinous tumors of the pancreas. Am J Pathol. 2002;160(5):1745-1754.
Tersmette AC, et al. Increased risk of incident pancreatic cancer among first-degree relatives of patients with familial pancreatic cancer. Clin Cancer Res. 2001;7(3):738-744.
Thayer SP, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425(6960):851-856.
Thomas AM, et al. Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med. 2004;200(3):297-306.
Turner N, et al. Hallmarks of “BRCAness” in sporadic cancers. Nat Rev Cancer. 2004;4(10):814-819.
Tuveson DA, et al. Ductal pancreatic cancer in humans and mice. Cold Spring Harb Symp Quant Biol. 2005;70:65-72.
Ueki T, et al. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000;60(7):1835-1839.
van der Heijden MS, et al. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003;63(10):2585-2588.
van der Heijden MS, et al. Functional defects in the fanconi anemia pathway in pancreatic cancer cells. Am J Pathol. 2004;165(2):651-657.
van der Heijden MS, et al. In vivo therapeutic responses contingent on Fanconi anemia/BRCA2 status of the tumor. Clin Cancer Res. 2005;11(20):7508-7515.
van Heek NT, et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol. 2002;161(5):1541-1547.
Walter K, et al. Pancreatic cancer associated fibroblasts display normal allelotypes. Cancer Biol Ther. 2008;7(6):882-888.
Wilentz RE, et al. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res. 1998;58(20):4740-4744.
Wilentz RE, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60(7):2002-2006.
Wilentz RE, et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: a newly described and characterized entity. Am J Pathol. 2000;156(5):1641-1651.
Winter JM, et al. 1423 pancreaticoduodenectomies for pancreatic cancer: a single-institution experience. J Gastrointest Surg. 2002;10(9):1199-1210.
Yamano M, et al. Genetic progression and divergence in pancreatic carcinoma. Am J Pathol. 2000;156(6):2123-2133.
Yeo TP, et al. Assessment of “gene-environment” interaction in cases of familial and sporadic pancreatic cancer. J Gastrointest Surg. 2009;13(8):1487-1494.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 8A Pancreatic cancer and premalignant tumors
Molecular aspects
Pancreatic Cancer Overview
Pancreatic ductal adenocarcinoma (PDA), commonly known as pancreatic cancer, is the fourth leading cause of cancer death in the United States, preceded only by lung, colon, breast (in women), and prostate (in men) cancers (American Cancer Society, 2009). The diagnosis of PDA is devastating because it has one of the highest mortality rates of any cancer (American Cancer Society, 2009). To underscore this point, in 2009 approximately 42,470 Americans were diagnosed with PDA, and about 35,240 will die from it (American Cancer Society, 2009). A diagnosis of PDA is met with the daunting statistic that the patient has an overall 5-year survival rate of less than 5%.
The past 2 decades have seen an exponential increase in our understanding of the molecular basis and etiology behind PDA (Hruban et al, 2001; Jones et al, 2008, 2009). Still, the clinical management of this disease—including primary prevention, early detection, and better targeted treatment options—has not changed significantly over the past decade. Currently, the only cure for this disease is surgical resection. Unfortunately, only 20% of the patient population presents with resectable disease (Winter et al, 2002).
Research studying PDA can be simplistically divided into two eras (Fig. 8A.1). The first is highlighted by the discovery and development of technologies such as gene cloning and isolation of pure tumor tissue (i.e., xenografting primary tumors postresection; Fig. 8A.2, left). This era included landmark studies defining the genomic region of BRCA2 (Goggins et al, 1996), discovering SMAD4 (Hahn et al, 1996) as a tumor suppressor in PDA, activation of KRAS (Almoguera et al, 1988) as an early and frequent event in pancreatic tumorigenesis, and silencing of the CDKN2A (Caldas et al, 1994a) gene. As the result of advances in a number of molecular techniques, we are now upon the early stages of a new second era in genetic pancreatic cancer research, which combines high-throughput sequencing, gene expression and comparative genomic hybridization (CGH) arrays with statistical computational analysis, and large clinical databases (Fig. 8A.2, right) (Jones et al, 2008, 2009). This second era has already produced landmark publications that include a precise survey of multiple pancreatic cancer genomes both in sporadic and familial forms of the disease. This chapter aims to 1) provide a summary of the past two decades of pancreatic cancer research; 2) show that even though every tumor has a number of common molecular events, it is the differences between tumors that have clinical implications (Fig. 8A.3); and, finally, 3) set the stage for the future, which will include a discussion of successful early detection and treatment strategies for this deadly disease (Fig. 8A.3).
Progression Model of Pancreatic Ductal Adenocarcinoma
In the first era of pancreatic cancer research (see Figs. 8A.1 and 8A.2), the fields of molecular biology and pathology combined to establish a paradigm that PDA culminates from a multistep progression model (Hruban et al, 2001). This slow, sequential process may be the reason PDA is primarily a disease of people in their sixth and seventh decades of life. Definable pathologic markers on this stepwise progression—which follows a similar model first developed in colon carcinogenesis—are lesions referred to as pancreatic intraepithelial neoplasia (PanIN) (Hruban et al, 2001; Figs. 8A.4A and B). These lesions are believed to be precursor lesions to pancreatic cancer. PanIN lesions are thought to develop years before the emergence of PDA and are pathologically graded from low-grade PanIN lesions, PanIN-1 (Fig. 8A.4A), to high-grade lesions, PanIN-3 (see Fig. 8A.4B). Although low-grade PanIN lesions are common incidental findings, high-grade PanIN lesions are more common in pancreata with PDA.
Key evidence that PanIN lesions are precursors to PDA is that similar hallmark molecular defects are found in PanIN lesions adjacent to invasive cancers (Biankin et al, 2003; Luttges et al, 2001a; Maitra et al, 2002; van Heek et al, 2002; Wilentz et al, 2000a). However, although KRAS mutations are frequently found in early PanIN lesions (Shi et al, 2009), genes involved in DNA repair mechanisms, such as TP53 and BRCA2, are altered late in this progression model. Because these data are based on limited sample numbers and data points, it remains somewhat counterintuitive that an oncogenic stimulus or stimuli (e.g., KRAS activation) precedes DNA damage (mutations). Recently, it was shown that higher density PanIN lesions were found in familial PDA patients compared with patients with sporadic forms of PDA, again arguing in favor of the hypothesis that PanIN lesions are true precursors to PDA (Shi et al, 2009).15
Intraductal Papillary Mucinous Neoplasm
Intraductal papillary mucinous neoplasms (IPMNs) are a well-accepted clinical and pathologic entity (Fig. 8A.4C and D; see Chapter 57) (Hruban et al, 2004). IPMNs typically produce radiographically identifiable pancreatic ductal dilation, which may predominantly involve the main pancreatic ducts (main-duct type IPMN), the secondary ducts (branch-duct type IPMN), or both types of ducts (mixed type). The distinction between the branch-duct type and main-duct type is important, because the former arise in younger patients, are more likely to involve the head and uncinate process of the pancreas, and are associated with lower-grade dysplasia and fewer invasive carcinomas. Approximately 30% to 40% of resected IPMNs harbor an invasive adenocarcinoma, and adenocarcinoma is most strongly associated with main-duct IPMNs. Approximately half of invasive carcinomas arising within IPMNs are so-called colloid (mucinous) carcinomas, and most of the remainder are tubular adenocarcinomas, the latter being histologically indistinguishable from invasive ductal adenocarcinomas that arise in the setting of PanINs (Adsay et al, 2002). Colloid carcinomas associated with IPMNs have a relatively good prognosis compared with other pancreatic carcinomas of the ductal type, which have a 5-year survival of 60% (Maire et al, 2002).
PanINs and IPMNs show some overlapping features, such that both are inherently intraductal lesions composed predominantly of columnar, mucin-producing cells that may grow in a flat configuration or may produce papillae; these lesions show a range of cytologic and architectural atypia and can give rise to invasive adenocarcinomas of the pancreas (Fig. 8A.4E and F). An important feature that distinguishes the two lesions is that PanINs are microscopic lesions, and IPMNs are macroscopic. Nevertheless, recognition of an IPMN and its distinction from a PanIN lesion is important for two reasons: 1) IPMN-associated colloid carcinomas have a significantly better prognosis than either PanIN- or IPMN-associated tubular adenocarcinomas (Adsay et al, 2001), and 2) IPMNs have a propensity to be multifocal lesions, therefore patients who undergo partial pancreatectomy and are left with a remnant pancreas need to be followed for life, even when the lesion originally resected was a noninvasive IPMN (Sohn et al, 2004).20
Genetic analyses of IPMNs have disclosed abnormalities in many of the same genes altered in conventional ductal adenocarcinoma, including mutations in the KRAS2, TP53, and CDKN2A genes, although the frequency and stage of neoplastic progression at which these alterations occur in IPMNs differ from PanINs (Hruban et al, 2004; Sessa et al, 1994). For example, the Peutz–Jeghers gene STK11 is inactivated in one third of IPMNs, and some patients with Peutz–Jeghers syndrome develop IPMNs (Sahin et al, 2003); this is in contrast to conventional ductal adenocarcinomas, in which loss of STK11 is a rare event. In contrast to ductal adenocarcinomas and PanIN-3 lesions, abnormalities in the SMAD4 gene seem to be rare in IPMNs (Iacobuzio-Donahue et al, 2000).
Another distinction between PanINs and IPMNs relates to the expression of the caudal differentiation factor CDX2, a marker of intestinal differentiation. Most IPMNs express CDX2, in particular IPMNs associated with an invasive colloid carcinoma, whereas this is uncommon both in PanINs and in the subset of IPMNs that give rise to invasive cancers resembling ductal adenocarcinomas (Adsay et al, 2004). CDX2 expression in IPMNs usually is associated with expression of MUC2, an intestinal epithelial apomucin, whereas absence of CDX2 usually is associated with expression of MUC1, a biliary apomucin, and concomitant lack of expression of MUC2. These findings have suggested that there may be two divergent pathways of carcinogenesis within the pancreatic ducts (Adsay et al, 2002). The first is a so-called intestinal pathway that gives rise to CDX2– and MUC2-expressing IPMNs that progress to the better-prognosis colloid carcinomas. The second is a pancreatobiliary pathway that gives rise to CDX2-negative, MUC2-negative, MUC1-expressing PanINs and a subset of IPMNs, both of which can progress to the poorer-prognosis conventional ductal adenocarcinomas. More recently, it has been observed that IPMNs with moderate dysplasia had reduced or absent pp32 nuclear expression (Brody et al, 2007). This may be functionally significant, because ANP32A acts as a tumor suppressor in pancreatic and other cancers, and the absence of this protein may facilitate the tumorigenesis process.
Genetics of Pancreatic Ductal Adenocarcinoma
Genomic (DNA) Alterations in Pancreatic Cancer
The multitude of genetic abnormalities in pancreatic cancer have many characteristics similar to other solid tumors, thus they include point mutations in critical genes, chromosomal (copy number) aberrations, mitochondrial DNA mutations, telomeric abnormalities, and epigenetic silencing by methylation of defined promoter DNA sequences. Recently, it was estimated that an individual pancreatic tumor contains, on average, 63 genetic alterations, primarily point mutations. It is also believed that only a small subset of these mutations is required for tumorigenesis (Jones et al, 2008).
The field of analyzing the genetics of pancreatic cancer can be broken down chronologically (see Fig. 8A.1). First, landmark studies starting in the late 1980s and spanning nearly two decades are highlighted by the discovery of KRAS activation, SMAD4 and BRCA2 mutations, and CDKN2A silencing (see Fig. 8A.1 and Fig. 8A.2, left). Some of these discoveries not only opened up lines of investigation in the field of pancreatic cancer but also opened up avenues with other tumor types and specific classification of PDA subtypes (Iacobuzio-Donahue et al, 2009). Then, in 2008 and 2009, Jones and colleagues, with the help of advanced DNA sequencing technology, were able to perform high-throughput genetic analysis on various pancreatic cancer genomes (see Figs. 8A.1 and 8A.2, right).
Copy-Number Aberrations
Although now considered primitive, valuable cytogenetic analysis performed over a decade ago found that chromosomal aberrations occur in virtually every pancreatic cancer. Cytogenetic analyses of pancreatic cancers have shown multiple, nonrandom numerical and structural changes (Griffin et al, 1995; Sirivatanauksorn et al, 2001). The most common numerical abnormalities include losses of chromosomes 6, 12, 13, and 18 and gains of chromosomes 7 and 20. Structural abnormalities (intrachromosomal break points) frequently involve 1p and 1q, 3p, 4q, 6q, 7q, 17p, 11p, 11q, 15q, 16q, and 19q (Rigaud et al, 2000). The technical limitations of conventional cytogenetics has presented challenges for identifying genes that are affected by chromosomal breaks. Allelotyping identifies areas of gross chromosomal loss by using polymorphic microsatellite markers to determine regions of genomic loss compared with matched normal tissues, also known as loss of heterozygosity (LOH) analysis. Allelotyping operates on the basic principle of the two-hit hypothesis, which postulates that tumor suppressor genes require biallelic inactivation. This most commonly happens by intragenic mutation in one allele, followed by loss of genetic material in the other allele. Identifying regions of single or biallelic loss or mutation holds the potential to understand the role of neighboring novel and well-known tumor suppressor genes. A landmark allelotype analysis of pancreatic cancers was performed using approximately 80 pancreatic cancer xenografts and 386 microsatellite markers (Iacobuzio-Donahue et al, 2004). This work discovered allelic losses in chromosome regions in proximity to tumor suppressor genes CDKN2A, TP53, and SMAD4. Allelotype analysis of PanIN lesions also has been performed using microdissected samples, and as expected, LOH is seen in many of the same chromosomal regions as invasive cancer, including 9p, 17p, and 18q (Luttges et al, 2001b; Yamano et al, 2000). Although the changes are conserved in most synchronous precursor lesions (i.e., the same allele is lost in PanIN and associated cancers), there is occasional clonal divergence between high-grade PanIN lesions harboring genetically distinct changes from the synchronous invasive cancer (Yamano et al, 2000). These findings may have important clinical implications in regard to tumor heterogeneity and clonal cancer cell drug resistance.
CGH identifies genomic amplifications and deletions and differentially labels normal and tumor genomic sequences with different dyes. The relative ratio of the two dyes indicates regions of cancer-associated losses or gains, with a ratio of 1 : 1 consistent with no change in copy number compared with normal DNA. Conventional CGH is performed on metaphase spreads and suffers from low resolution and inability to precisely map the various regions of amplifications and deletions (Mahlamaki et al, 1997). The resolution of array CGH is significantly better than the conventional technique, ranging from 500 kb to 30 kb, permitting the precise mapping of deletion and amplicon boundaries and genes targeted therein. Array technology also provides the ability to more efficiently use probes to study amplification of a larger number of genes. Array CGH analysis of pancreatic cancers has identified numerous recurrent copy-number aberrations, including amplifications of c-MYC (8q), EGFR (7p), KRAS (12p), AKT2 (19q), and NCOA3 (20q) and deletions of SMAD4 (18q), CDKN2A (9p), FHIT (3p), and MAP2K4 (17p) (Aguirre et al, 2004; Calhoun et al, 2006; Holzmann et al, 2004).
Utilizing high-density single-nucleotide polymorphism arrays, Calhoun and colleagues surveyed all the commercially available pancreatic cancer cell lines. In brief, this study provided high resolution and detailed break-point mapping of these cell lines and found two subclasses of cancer cell lines, original chromosomal instability (CIN) and holey CIN genotypes (Calhoun et al, 2006). Perhaps global classification of tumor cells with high-density arrays will become part of a prognostic or predictive molecular signature panel in the future.
Specific Gene Mutations
Much like other solid tumors, genes altered in pancreatic cancer include three functional classes: oncogenes, tumor suppressor genes, and caretaker genes. A family of caretaker genes recognized as being disrupted in pancreatic cancer are genes of the Fanconi anemia complementation group, which are involved in homologous recombination-based DNA damage repair (D’Andrea et al, 2003). Patients with Fanconi anemia present with a wide variety of clinical issues, including aplastic anemia and a high risk of developing cancer. BRCA2 is a member of this DNA repair pathway and is mutated in a subset of familial pancreatic cancers (Murphy et al, 2002). This has led to the search for mutations in other Fanconi anemia genes in pancreatic cancer. Somatic mutations of two genes in the core complex, FANCC and FANCG, were discovered but are rare in sporadic pancreatic cancers (van der Heijden et al, 2003). Through other modern techniques, Jones and colleagues (2009) discovered FANCN (PALB2) as another mutated gene found in familial pancreatic cancers.
Mutations in this core complex and in this DNA repair mechanism have major therapeutic implications (Fig. 8A.5, Table 8A.1; see the Familial Pancreatic cancer section below) (van der Heijden et al, 2004). Unlike most in vivo experiments, xenografted mice with isogenic cell lines (FANCC deficient and proficient) experienced regression of tumor after a single dose of the available mitomycin C intrastrand cross-linking drug (van der Heijden et al, 2005). Although other mutations in the Fanconi complementation group have not yet been described—other than FANN, FANCC, and FANCG—and the frequency of these mutations in PDA appear to be low, it is likely that defects in other FANC genes yet to be thoroughly investigated (i.e., FANCA) are the direct cause of some familial and sporadic PDAs.
FIGURE 8A.5 Preclinical model shows an example of a successful targeted treatment strategy against a Fanconi-deficient tumor. A single-dose treatment with mitomycin C (5 mg/kg) of pancreatic cancer cell lines xenografted into nude mice. Note the hypersensitivity and tumor regression in the FANCC-deficient PL11 cells (squares) compared with the retrovirally corrected FANC-proficient PL11 cells (triangles). Solid lines indicate treated mice; gray lines indicate no treatment controls. Similar sensitivity was seen in the BRCA2-deficient CAPAN1 xenografted cells (van der Heijden et al, 2005).
Oncogenes
Perhaps the best evidence that KRAS activation is an early and important event in tumorigenesis comes from decades of research on pancreatic cancer. The KRAS2 oncogene on chromosome 12p is the most commonly altered oncogene, with up to 90% of pancreatic cancers containing mutations on codons 12, 13, and 61 (Caldas et al, 1994b). Activating mutations impairs the intrinsic GTPase activity of the KRAS2 gene product, resulting in a protein that is constitutively active in intracellular signal transduction. Inactivation of KRAS2 happens early in the pathway to oncogenesis, with approximately 30% of PanIN-1 lesions harboring KRAS2 mutations (Hruban et al, 2000; Moskaluk et al, 1997). The first mouse model of pancreatic cancer was generated by constitutive overexpression of mutant KRAS2 in the murine pancreatic ductal epithelium, underscoring its importance in pancreatic oncogenesis (Hingorani et al, 2003). This model was further developed into a powerful and useful preclinical model for PDA progression (Hingorani et al, 2005). Several good sources on mouse modeling and pancreatic cancer have been published (Frese et al, 2007; Karreth et al, 2009; Olive et al, 2006; Tuveson et al, 2005).
Rarely, pancreatic cancers with wild-type KRAS2 genes harbor point mutations of BRAF, another gene in the RAS/RAF/MAP kinase signaling pathway, thereby explaining why mutations of these genes occur in mutually exclusive patterns in pancreatic cancer (Calhoun et al, 2003). This highlights the importance of identifying different molecular targets that lead to similar pathways in pancreatic cancer development and of finding a drug that can target one pathway, not one gene. Studies have shown that targeting KRAS may have potential in modulating angiogenesis in tumorigenesis. Matsuo and colleagues showed that oncogenic overexpression of KRAS increases production of angiogenesis, promoting CXC chemokines and VEGF from human pancreatic duct epithelial cells. This upregulation acts through the mitogen-activated protein kinase pathway and c-JUN signaling (Matsuo et al, 2009). KRAS mutation has been shown to be associated with increased VEGFA expression and poorer prognosis in pancreatic carcinoma (Ikeda et al, 2001). Yet to date, targeting KRAS activation in PDA patients has shown no success. Perhaps KRAS activation is a critical early event in pancreatic tumorigenesis, but once cells become malignant, there is no need for constitutive KRAS activation or for oncogenic addiction for that matter.
Other oncogenes implicated in pancreatic cancers include MYC and EGFR, which can be amplified in various subsets of cancers. Overexpression of MYC transcripts occurs in approximately 50% to 60% of pancreatic cancers (Aguirre et al, 2004; Han et al, 2002). Therapeutic targeting using monoclonal antibodies or small-molecule tyrosine kinase inhibitors makes use of the constitutive activation of the EGFR signaling pathway in pancreatic cancers via amplification of the EGFR gene (Holzmann et al, 2004; Li et al, 2004). Lack of a rigorous and reliable immunohistochemical assay to detect overexpression of EGFR has proven to be difficult and limiting.
Tumor Suppressor Genes
CDKN2A, on chromosome 9p, is the most commonly inactivated gene in pancreatic cancers, occurring in 90% of patients (Caldas et al, 1994a; Schutte et al, 1997). CDKN2A belongs to the cyclin-dependent kinase inhibitor family and inhibits cell-cycle progression through the G1-S checkpoint mediated by cyclin-dependent kinases such as CDK4 and CDK6. Homozygous deletions (40%), intragenic mutation with loss of the second allele (40%), and epigenetic silencing by promoter methylation (10% to 15%) all contribute to gene inactivation. Immunolabeling for nuclear p16 protein expression is a reliable surrogate for CDKN2A genetic status; however, such immunolabeling does not identify specific genetic mechanisms of inactivation (Geradts et al, 2000). Loss of CDKN2A function occurs throughout the process of oncogenesis, with lesions appearing in different PanINs: 30% of PanIN-1A and PanIN-1B, 55% of PanIN-2, and 71% of PanIN-3 lesions show loss of nuclear p16 protein expression (Wilentz et al, 1998). The CDKN2A homozygous deletions encompass the MTAP gene in approximately 30% of pancreatic cancers, which offers potential therapeutic benefit, because targeted therapies have been developed that specifically inhibit the growth of MTAP-deficient cells (Chen et al, 1996).
[/not-level-membership-for-surgery-category]
