Nonmelanoma Skin Cancers
Basal Cell and Squamous Cell Carcinomas
Gary S. Wood, Yaohui Gloria Xu, Juliet L. Aylward, Vladimir Spiegelman, Erin Vanness, Joyce M.C. Teng and Stephen N. Snow
• More than 2 million new cases of nonmelanoma skin cancer (NMSC) occur annually in the United States, including 80% basal cell carcinomas (BCCs), 20% squamous cell carcinomas (SCCs), and a few rarer types.
• Incidence is increasing 2% to 3% per year.
• Fifteen percent to 43% of solid organ transplant recipients will develop NMSC within 10 years.
• Ultraviolet radiation from sun exposure is a major risk factor, causes mutations in key genes and explains the predilection of NMSC for sun-exposed skin.
• Hedgehog signaling pathway mutations are involved in BCC pathogenesis.
• p53 Mutations are involved in both SCC and BCC pathogenesis, as well as in the development of actinic keratoses, which are the precursors of SCCs.
• There are several histopathologic subtypes of each NMSC.
• The more infiltrative or poorly differentiated variants are more clinically aggressive (e.g., morpheaform BCC and spindle cell SCC).
• TNM staging classifications exist for most types of NMSC and depend on clinical characteristics, pathological features and radiologic evaluation of the primary tumor, adjacent structures, lymph nodes, and viscera.
• BCCs that are large, deep, or infiltrative may be locally aggressive and recurrent, but metastasize only rarely (<0.05%).
• SCCs have a greater metastatic rate, especially those that are large, deep, have perineural invasion, or are located on the dorsal hands, lips, ears, penis, or sites of chronic infection, ulceration, or radiation.
• Primary treatment for both BCCs and SCCs is surgical. Mohs surgery is preferred for ill-defined or aggressive lesions because it allows microscopic control of tumor margins.
• The 5-year recurrence rate for BCC is 1% for Mohs surgery compared to 5% for other types of surgical excision.
• Alternative primary therapies include various forms of physical destruction and radiation therapy.
• Interferons and inducers of interferons (e.g., imiquimod) are useful in selected cases. Retinoids, hedgehog pathway agents, and difluoromethylornithine are other promising chemopreventive and adjunctive modalities.
Locally Advanced and Metastatic Disease
• Local recurrence is a problem for large, deep, or histologically infiltrative variants. SCCs with these features may also metastasize. The 5-year survival for patients with metastatic SCC is <50%.
• The rarer forms of nonmelanoma skin cancer have a significantly more aggressive clinical course as compared with BCC and SCC. These include sebaceous carcinoma, Merkel cell carcinoma, dermatofibrosarcoma protuberans (DFSP), and cutaneous angiosarcoma.
• Combinations of surgery, radiation therapy, and chemotherapy can be used for metastatic disease.
• Palliation is directed mainly at managing complications such as local recurrence, destruction of adjacent structures, scarring, and loss of function.
Genetics of Nonmelanoma Skin Cancer
As in other malignancies, tumor suppressor genes and protooncogenes are two basic classes of genes that undergo mutations leading to skin cancer. The mutations in these genes lead to pathophysiologic changes, such as sustaining proliferative signaling, evading growth suppressors, resisting cell death, replicative immortality, inducing angiogenesis, and activating invasion and metastasis, described as hallmarks of cancer.1 Examples of tumor suppressor genes (whose loss of function contributes to tumor development) include the Patched (PTCH1 and PTCH2) genes involved in development of BCC, p53 involved in development of SCC, and the xeroderma pigmentosum genes involved in development of BCC, SCC, and melanoma.2–5
Protooncogenes are another class of genes whose mutations contribute to skin cancer formation. They become oncogenes after acquiring “gain-of-function” genetic alteration. These are generally growth-signaling molecules that once mutated can perpetually cause normal cells to become malignant cells by altering cellular growth. Some examples are the ras oncogene implicated in the development of SCC, and Smoothened (SMO) mutated in a subset of BCC.7–7
Fundamental Science and Clinical Relevance
Hedgehog Signaling Pathway
The role of the hedgehog (Hh) signaling pathway has been well documented in vertebrate embryonic development, stem cell maintenance, and tumorigenesis (reviewed in references 8 and 9). Activation of the Hh signaling is a driving force in the development of both hereditary and sporadic cases of BCC. These tumors are the most common of all skin cancers, with an estimated 1 million new cases per year in the United States alone.10 Each of the three Hh genes—Sonic hedgehog (SHH), Desert hedgehog (DHH), Indian hedgehog (IHH)—encodes a secreted signaling peptide that binds to a membrane bound 12-span transmembrane receptor called Patched (PTCH1 or PTCH2). Binding of Hh ligand to the PTCH receptor on the target cell alleviates PTCH-mediated suppression of the 7-span transmembrane protein called Smoothened (SMO). SMO activation initiates an intracellular signal transduction cascade that ultimately activates the expression of Hh target genes. Transcriptional activation of Hh target genes in mammals occurs through the actions of three related proteins: GLI1, GLI2, and GLI3.11,12 GLI3 acts primarily as a transcriptional repressor,13 whereas GLI2 is reported to be the primary activator of Hh signaling, and GLI1 is a secondary target, downstream of GLI2, that also acts as a transcriptional activator.14
Aberrant regulation of the Hh pathway contributes to the development of many human cancers. Activating mutations of SMO or suppressing mutations of PTCH have been shown to constitutively activate the Hh signaling pathway (reviewed in reference 15). Involvement in human BCC was found through analysis of nevoid BCC kindreds (Gorlin syndrome, basal cell nevus syndrome).16 Persons with this autosomal dominant disease have odontogenic cysts, skeletal defects, palmar pits, various associated visceral tumors (medulloblastoma, meningioma, fibrosarcoma, cardiac fibroma, and ovarian fibroma), and multiple BCCs by a median age of 20 years. Early analyses mapped the defect to a tumor suppressor gene on chromosome 9q22–q31.17 This site proved to be the location of the PTCH1 gene. It was later elucidated that patients with nevoid BCC syndrome inherit one defective chromosome 9q region with loss of heterozygosity in the PTCH1 locus. Many BCCs from these patients show inactivation of the remaining PTCH1 gene through acquired somatic mutations, consistent with the view that the BCC phenotype develops once both alleles are nonfunctional. Thus, PTCH1 acts as a classic tumor suppressor gene in the skin. Mutations in PTCH1 also are common in many sporadic BCCs, as are mutations in other Hh pathway genes, including SMO, PTCH2, and SHH (reviewed in reference 18).
p53 Mutations
The protein p53 is encoded by the TP53 gene on chromosome 17p and is an important regulator of cell cycle, DNA repair, and apoptosis.19 Inactivation of the p53 gene seems to play a principal role in the development of both premalignant actinic keratosis and SCC.20 SCC is the second most common skin cancer, accounting for approximately 200,000 cases per year and 2000 deaths per year.10 p53 mutations associated with SCC are usually UV induced, and many are pyrimidine alterations with CC to TT (cysteine to threonine) changes.21,22 Both the loss of the tumor suppressor ability of p53 and the ability of UV irradiation to affect induction of apoptosis by p53 are linked to SCC development.20
Mutations of TP53 have also been implicated in development of sporadic BCC.23 It seems that UV-induced alterations similar to those in SCC are involved in BCC induction. Many of the mutations are CC to TT or C to T alterations, consistent with UV damage.
ras Mutations
Mutations in the family of ras protooncogenes have been implicated in development of sporadic BCC, premalignant actinic keratosis, and sporadic SCC. They are among the most common mutations in human malignant disease.24 ras Proteins are small G-proteins responsible for transducing intracellular signaling. Activation of ras occurs only when guanosine triphosphate (GTP) is bound. The signal is attenuated by hydrolysis of GTP to guanosine diphosphate (GDP). Mutations in ras alter the rate of this hydrolysis, resulting in activated protein that constitutively activates tumor promoting Ras-Raf-MAP kinase and PI3K-AKT signaling pathways.
Mutations of Other Genes Predisposing to Squamous Cell Carcinoma
XP is a collection of autosomal recessive disorders characterized by severe photosensitivity with the onset of cutaneous malignant lesions at a very early age. BCC, actinic keratosis, SCC, and melanoma develop during the first decade of life in persons without adequate photoprotection. Mutations in eight genes have been implicated in different XP phenotypes, which vary in severity of cutaneous neoplasia and frequency of neurologic delay.25 All XP-associated genes encode proteins that are part of a DNA repair process known as nucleotide excision repair, which responds to UV-induced DNA damage.4 These proteins recognize the damaged DNA, unwind the coiled DNA structure, and repair the damaged strand. Germline mutations in these genes result in defects in the repair process and their genomic caretaker role; however, actual tumor production is still caused by mutagenic inactivation of tumor suppressor genes such as TP53 and activation of oncogenes such as ras.26,27
Muir-Torre syndrome (MTS) is an autosomal dominant syndrome characterized by various sebaceous gland tumors and internal malignant lesions. The sebaceous gland tumors range from benign sebaceous adenoma to malignant sebaceous gland carcinoma, predominantly on the face. Keratoacanthoma is another cutaneous neoplasm of variable malignant potential that has been reported in as many as 20% of MTS patients.28 In initial studies of MTS families, investigators identified germline mutations in the human MSH2 gene.29,30 Subsequently, mutations in human MLH1 have also been identified in patients with MTS.31 It seems that, like other caretaker genes, human MSH2 and MLH1 encode a type of DNA-mismatch repair enzyme involved in repairing errors in DNA replication that occur naturally at a low rate. In cells that have this defect, the result is varying lengths of repetitive DNA sequences known as microsatellite instability.31 This microsatellite instability can result in functional gene mutations and has been observed in keratoacanthoma and sebaceous tumors from MTS patients.32
Dyskeratosis congenita is a progeroid disease associated with abnormalities in telomere function. The mutations in TERT, TERC, DKC1, TINF2, NHP2, TCAB1, and NOP10 may lead to shortening of telomeres resulting in premature aging, chromosomal rearrangements, and altered cell proliferation (reviewed in reference 33). The cutaneous SCC in dyskeratosis congenita patients were reported often when the patients were in their teens or twenties, with about half of the patients having SCC by the age of 50 years.34 This syndrome has a varied pattern of inheritance depending on the gene affected: DKC1—X-linked recessive; TERC and TINF2—autosomal dominant; NHP2, TCAB1 and NOP10—autosomal recessive; and TERT can be autosomal dominant or autosomal recessive.
Fanconi anemia is associated with mutations in one of 15 genes involved in DNA cross-link repairs. This syndrome is characterized by early onset of a variety of hematologic and solid malignancies, including SCC.35,36 These patients have increased susceptibility to UV radiation (and other DNA–cross-linking agents), as the cells have altered ability to excise pyrimidine dimers.37 In the vast majority of cases of Fanconi anemia, there is an autosomal-recessive pattern of inheritance (with the exception of rare cases associated with mutations of FANCB where there is an X-linked recessive pattern).
SCC is also a common feature in syndromes that are caused by structural abnormalities in skin, such as dystrophic epidermolysis bullosa with mutations in the gene encoding type VII collagen (COL7A1) that normally connect the basement membrane to the dermis,38,39 or junctional epidermolysis bullosa with mutations in genes encoding basic subunits of laminin 332 (LAMA3, LAMB3, and LAMC2), or COL7A1.40,41
Because melanin protects keratinocytes from UV radiation, the number of inherited disorders that affect melanin synthesis, melanosomal and lysosomal storage, and pigment granule transport are associated with predisposition to SCC. These syndromes are characterized by an autosomal recessive pattern of inheritance and include oculocutaneous albinism that is usually associated with mutations in TYR, OCA2, or TYRP1 that affect melanin synthesis42,43; Hermansky-Pudlak syndrome characterized by abnormal melanosome and lysosome transport (mutations in HSP1, HSP3, HSP4, HSP5, HSP7, HSP8, and HSP9 genes)44; Chediak-Higashi syndrome, characterized by abnormal lysosome transport (mutations in LYST); Griscelli syndrome, characterized by abnormal pigment granule transport (mutations in MYO5A or RAB27A).45
In addition to mutations in the ras family described above, alterations in other genes involved in signal transduction were shown to play causative role in the development of SCC. Loss of function mutations in transforming growth factor-β receptor 1 (TGFBR1) are associated with multiple self-healing squamous epitheliomata (Ferguson-Smith syndrome),46 and mutations in EVER1 and EVER2 are associated with increased susceptibility to HPV types 5 and 8 in epidermodysplasia verruciformis.47
Basal Cell Carcinoma
Epidemiology
BCC accounts for approximately 80% of all skin cancers. The American Cancer Society reports more than 2 million cases of BCC and SCC yearly in the United States. Worldwide, the incidence for BCC varies widely, with the highest rates in Australia (>1000/100,000 person-years) and the lowest rates in parts of Africa (<1/100,000 person-years).48 In Australia, the annual incidence of BCC is slightly over 2% in males and 1% to 1.5% in females, while that of SCC is approximately 1% in males, and 0.5% to 0.8% in females (calculated from “Table 70-1. Age-Standardized Rates of NMSC in Whites per 100,000 Population from Australia, United States, and Europe [Selected Studies after 1990]” in reference 49).49 The incidence of BCC has increased over the past decades in a manner similar to the increase in melanoma. It is estimated that the incidence of NMSC is increasing 2% to 3% yearly.50 One study, conducted in New Hampshire, showed an annual increase in the incidence of SCC of approximately 10% and of BCC of approximately 5%.51
Etiology and Biological Characteristics
Exposure to ultraviolet radiation is the major risk factor for the development of BCC, especially severe sunburn during childhood and adolescence.52 Interestingly, the risk of BCC is significantly increased by recreational, intermittent intense exposure to the sun, whereas SCC appears to be strongly related to cumulative sun exposure.54–54 Other associated risk factors are Fitzpatrick skin types 1 and 2, red hair, freckling in childhood, family history of skin cancer, male sex, and Celtic ancestry.55 BCC is more common in lighter skinned African Americans than in darker persons.56 Immunodeficiency secondary to acquired immunodeficiency syndrome or organ transplantation also is associated with increased risk of BCC.57,58 Other uncommon risk factors include exposure to arsenic,59 oral methoxsalen (psoralen) and UVA radiation,60 and ionizing radiation,61 especially among patients who have received radiation for acne.
Keratin pattern and immunohistochemical results suggest the origin of BCC is the outer root sheath of the hair follicle below the isthmus.62 Recent work using genetic mouse models of BCC has shown that stem and progenitor cells are the most probable sources of BCC initiation.63 The locally invasive characteristic of BCC could be related to the presence of an abnormal hemidesmosome-anchoring fibril complex.64 Genetic abnormalities and mutations are thought to play a major role in development of BCC, especially in inherited syndromes of BCC. Patients with XP cannot repair the UV-induced DNA mutations; therefore, they are at increased risk of cutaneous carcinogenesis. Mutations in the Hh signaling pathway genes and TP53 are important in the pathogenesis of sporadic BCC and those arising in patients with the basal cell nevus syndrome (BCNS) and XP.20,65–68
Prevention and Early Detection
Patients need to be educated on self-skin examination and about protection against sun exposure. Photoprotection is essential to prevent NMSCs,69 and should begin at a young age to reduce the cumulative damage induced by the sun. Recently, chemopreventive agents for BCC have been explored, including retinoids,70 nonsteroidal antiinflammatory drugs,71,72 and difluoromethylornithine.73 Targeted skin-cancer screening of high risk populations is effective for early detection of BCC.74
Pathology and Pathways of Spread
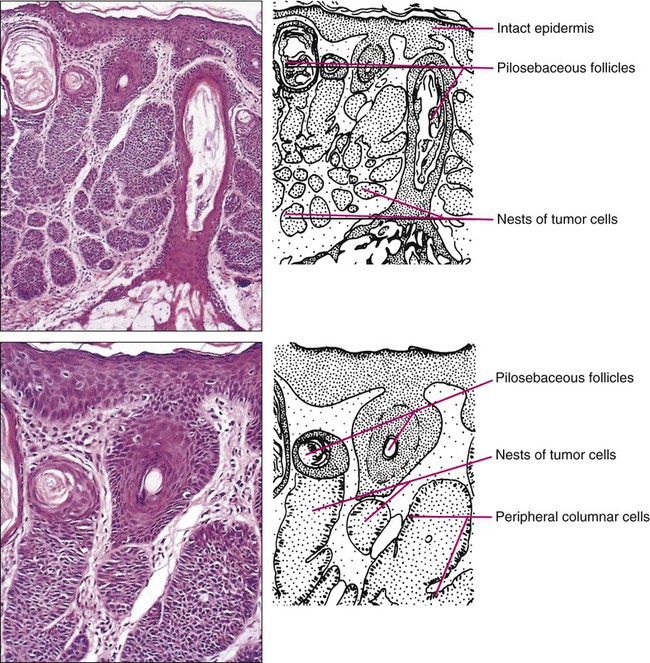
In common nodular BCC, nodular masses of basaloid cells extend from the epidermis or outer root sheath into the dermis with surrounding connective tissue stroma (Fig. 70-1). A palisade arrangement of cells is present in the periphery. Sometimes as a result of tumor necrosis and disintegration, cystic spaces form. The surrounding stroma may retract from the tumor mass forming the typical lacunae, a sign that aids in diagnosis. In the pigmented type of BCC, large amounts of melanin are produced in the melanocytes that colonize the tumor. The many melanophages in the surrounding stroma also contribute to pigmentation. The superficial type of BCC shows multifocal small nests of basaloid tumor cells budding off the epidermis and adnexa. Morpheaform BCC is different in that strands of tumor cells are embedded in a dense fibrous tissue stroma. These strands often extend into the deeper dermis. Micronodular BCCs contain small nodules of tumor cells that invade surrounding stroma. The morpheaform and micronodular types are generally the most locally invasive variants of BCC. It is common for BCCs to show mixed histologic patterns of these various types. Even the less-invasive variants may invade deeply when located in regions of embryonic fusion planes, such as around the nose and ears.
Clinical Manifestations/Patient Evaluation/Staging
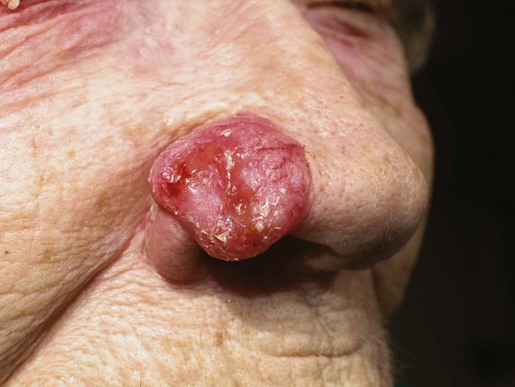
The most common location for BCC is the head and neck, especially the nose. BCC occurs on hair-bearing skin; mucosal surfaces are not involved. The major types of BCC include nodular, pigmented, superficial, and morpheaform. The typical nodular BCC is a dome-shaped, pearly papule with a telangiectatic surface and translucent rolled borders (Fig. 70-2). The surface might become ulcerated (Fig. 70-3). In darker-skinned individuals, especially African Americans, the more darkly pigmented BCCs may be misdiagnosed as seborrheic keratosis or nodular melanoma. Superficial BCC is a well-demarcated erythematous scaly plaque with elevated borders that often occurs on the trunk or extremities. It might be confused with Bowen disease or nummular eczema. The most difficult BCC to diagnose and manage is the morpheaform or sclerosing variant. This ill-defined, white, indurated plaque can be mistaken for a scar or localized patch of scleroderma, and therefore is ignored by patient and physician, with resulting wide subclinical extension.
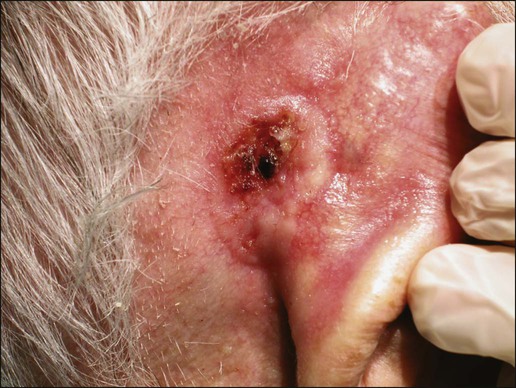
Patients with lesions clinically suspicious for BCC should undergo skin biopsy for histologic evaluation. Pertinent history is taken, such as the rate of growth, prior therapy, local neurologic symptoms, and evidence of immunosuppression, and a thorough skin examination is recommended. No formal staging system has been developed specifically for BCC, as metastasis of BCC is rare, with rates ranging from 0.003% to 0.55%.75 The American Joint Committee on Cancer (AJCC) staging system for non-eyelid skin cancer (Table 70-1) can be used for BCC as well as SCC. Regardless, it is important to stratify BCC into high-risk or low-risk lesions to guide treatment. Criteria for high-risk BCC include size greater than 2 cm, location in the midface (H zone) or ear, morpheaform or other aggressive histologic pattern, long duration, incomplete excision, perineural or perivascular involvement, and recurrent lesion.76
Table 70-1
TNM Staging System for Cutaneous Squamous Cell Carcinoma
| T: PRIMARY TUMOR | |||
| TX | Primary tumor cannot be assessed | ||
| T0 | No evidence of primary tumor | ||
| Tis | Carcinoma in situ | ||
| T1 | Tumor ≤2 cm in greatest dimension with <2 high-risk features* | ||
| T2 | Tumor >2 cm in greatest dimension with or without one additional high-risk feature,* or any size with ≥2 high-risk features* | ||
| T3 | Tumor with invasion of maxilla, mandible, orbit, or temporal bone | ||
| T4 | Tumor with invasion of skeleton (axial or appendicular) or perineural invasion of skull base | ||
| *High-risk features include depth (>2-mm thickness; Clark level ≥ IV); perineural invasion; location (primary site ear; primary site nonglabrous lip); and differentiation (poorly differentiated or undifferentiated) | |||
| N: REGIONAL LYMPH NODES | |||
| NX | Regional lymph nodes cannot be assessed | ||
| N0 | No regional lymph node metastasis | ||
| N1 | Metastasis in single ipsilateral lymph node, ≤3 cm in greatest dimension | ||
| N2 | Metastasis in single ipsilateral lymph node, >3 cm but not >6 cm in greatest dimension; or in multiple ipsilateral lymph nodes, none >6 cm in greatest dimension; or in bilateral or contralateral lymph nodes, none >6 cm in greatest dimension | ||
| N2a | Metastasis in single ipsilateral lymph node, >3 cm but no >6 cm in greatest dimension | ||
| N2b | Metastasis in multiple ipsilateral lymph nodes, none >6 cm in greatest dimension | ||
| N2c | Metastasis in bilateral or contralateral lymph nodes, none >6 cm in greatest dimension | ||
| N3 | Metastasis in lymph node, >6 cm in greatest dimension | ||
| M: METASTASIS | |||
| MX | Distant metastasis cannot be assessed | ||
| M0 | No distant metastasis | ||
| M1 | Present distant metastasis | ||
| Staging for Squamous Cell Carcinoma | |||
| Stage 0 | In situ | N0 | M0 |
| Stage I | T1 | N0 | M0 |
| Stage II | T2 | N0 | M0 |
| Stage IIIA | T3 | N0 or N1 | M0 |
| Stage IIIB | T1 or T2 | N1 | M0 |
| Stage IVA | T1, T2, or T3 | N2 | M0 |
| Stage IVB | Any T | N3 | M0 |
| Stage IVC | T4 | Any N | M0 |
| Stage IVD | Any T | Any N | M1 |
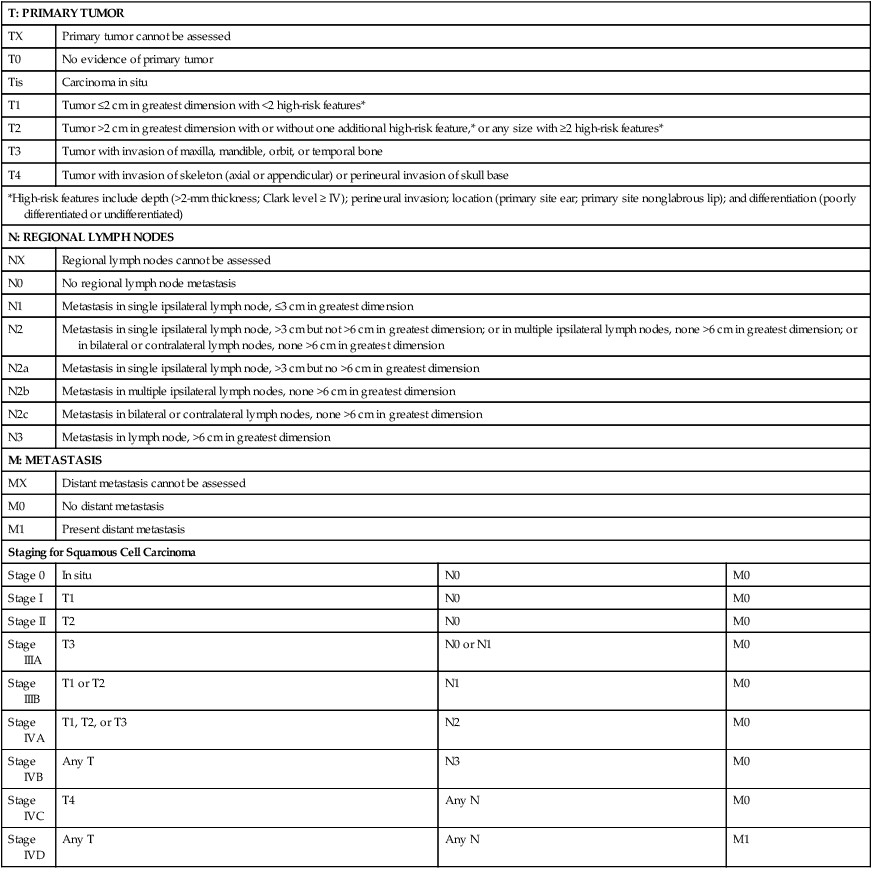
From American Joint Committee on Cancer Staging Manual, Seventh Edition, 2010; and Farasat S, Yu SS, Neel VA, et al. A new American Joint Committee on Cancer staging system for cutaneous squamous cell carcinoma: creation and rationale for inclusion of tumor (T) characteristics. J Am Acad Dermatol 2011;64:1051-1059.
Two-thirds of BCC recurrences occur during the first 3 years after treatment.77 The risk of development of another BCC is approximately 45% within 5 years.78 The risk of development of another NMSC is associated with the number of previous NMSCs. In an Australian study of patients with three to nine previous NMSCs, the risk of development of a new cancer was 93%.79 Patients treated for BCC need to be examined at least once a year for the first few years, preferably for 5 years after the last cancer was diagnosed. For patients with a history of multiple skin cancers, more frequent followup examinations are recommended.
Primary Therapy
BCC is rarely metastatic but can be locally invasive (Fig. 70-4). Consequently, eradicating the primary tumor is the goal of therapy. Several treatment options are available, both surgical and nonsurgical. Selection depends on the tumor type, patient profile, size and location of tumor, recurrence, physician’s experience, and patient preference. Surgical modalities include Mohs micrographic surgery, surgical excision, cryosurgery, and electrodessication and curettage (EDC). Nonsurgical options include radiation therapy and photodynamic therapy.82–82 Other treatment modalities, such as immunotherapy with intralesional interferon83 and topical 5% imiquimod (Aldara),84 chemotherapy with 5-fluorouracil,85 and retinoids,86 have been reported with variable success (Fig. 70-5).
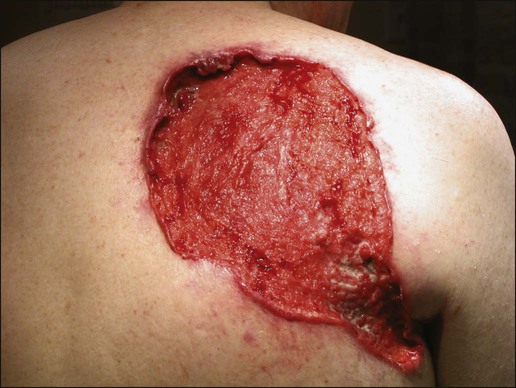
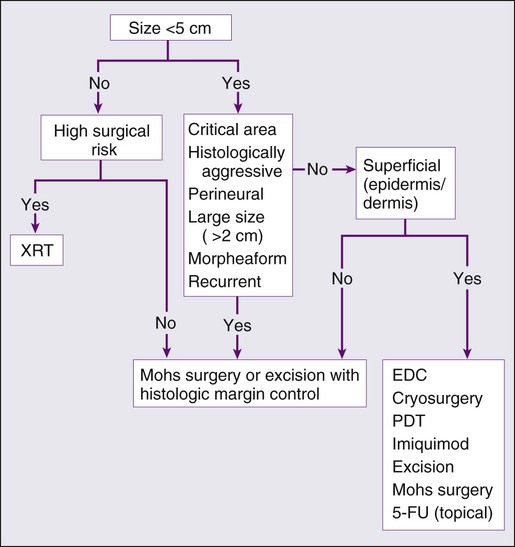
A systematic review of studies in which investigators reported recurrence rates of BCC after different treatment modalities showed that the mean 5-year recurrence rates after Mohs surgery and surgical excision were approximately 1% and 5.3%, respectively.87 High-risk BCC with increased risk for recurrence should be completely resected, preferably by Mohs technique or excision with margin control. Mohs surgery should be used in areas where preserving maximum tissue is important, such as eyelids, nose, and lips. It is also indicated for recurrent BCC and tumors with ill-defined clinical margins. The American Academy of Dermatology (AAD) has developed its first appropriate use criteria (AUC) on Mohs micrographic surgery in collaboration with the American College of Mohs Surgery (ACMS), American Society for Mohs Surgery (ASMS), and American Society for Dermatologic Surgery (ASDS). The Mohs AUC was recently published in journal of the American Academy of Dermatology88 and Dermatologic Surgery.89 If simple excision is used, the margin for excision, according to the newest National Comprehensive Cancer Network (NCCN) guidelines, should be at least 4 mm for low-risk tumors, and 10 mm for high-risk tumors. If there are contraindications to surgery, or a low-risk tumor that is small and located on less-critical sites, such as the trunk, cryotherapy or EDC can be used with good outcome. Because of the less-favorable long-term cosmetic results and the possibility of secondary radiation-induced skin cancer, radiation therapy is best avoided in the care of relatively young patients.
Treatment of Locally Advanced and Metastatic Disease
Very large or poorly controlled BCC may necessitate a coordinated approach of standard surgical excision, Mohs surgery, radiation therapy, and immunotherapy or chemotherapy.90 Targeted therapy suppressing the Hh signaling pathway at the level of SMO, or at the final effectors, GLI transcription factors, has been proven effective. GDC-0449 (Vismodegib), a synthetic oral inhibitor of SMO, is the first FDA-approved drug in this category. GDC-0449 is indicated for adults with metastatic BCC, or with locally advanced BCC that has recurred following surgery or who are not candidates for surgery, and who are not candidates for radiation.63,91
Controversies/Problems/Challenges/Future Possibilities and Clinical Trials
Other potential new BCC therapies include vitamin D3, which works through blocking Hh signaling, topical treatment with the RARB/RARG-selective retinoid, tazarotene,63 and molecules targeting epidermal growth factor receptor (EGFR) pathway.92 It remains to be determined whether the strategy of inhibiting Hh signaling will truly result in the eradication of BCC and why certain tumors are refractory to such treatment.63
Squamous Cell Carcinoma and Bowen Disease
Etiology and Biological Characteristics
There are many risk factors for SCC, the most important being solar radiation. The incidence of SCC is increasing especially on the head and neck area as a result of exposure to sunlight.94 SCC is thought to be correlated with recent (in the 10 years preceding diagnosis) chronic sunlight exposure95 and cumulative sun exposure.96 Phototherapy with PUVA (psoralen + ultraviolet A) increases the risk of SCC. Other risk factors for SCC include fair skin, red hair, albinism, and Celtic origin. Nonsolar risk factors include exposure to chemicals (insecticides and herbicides),97 arsenic, organic hydrocarbons, chronic thermal injury and scars, ionizing radiation, and chronic immunosuppression.98 The incidence of SCC is increased by 65 times in organ transplant patients, as compared to the tenfold increase of BCC.99 This increase of risk is correlated with the type of organ transplantation and elapsed time after transplantation. Heart and lung transplant recipients have a higher risk of NMSC than kidney transplant recipients, possibly because of more intense immunosuppression.99,100 SCCs in organ transplant patients are usually more aggressive, with an increased risk of metastases of approximately 8%, as compared to the risk of 0.5% to 5% in the general population.101 Tobacco is a risk factor for oral SCC. Viruses, especially human papillomavirus (HPV), have been linked to epithelial malignancies, including SCC. This is especially true among patients with epidermodysplasia verruciformis, who have an underlying immunodeficiency and can develop SCCs within HPV-infected warts.98
Carcinogens, such as ultraviolet radiation (UVR), certain chemicals, and viruses play a role in the pathogenesis of SCC by damaging keratinocyte DNA and other cellular contents. It is known that UVR, especially UVB, causes mutations in DNA of keratinocytes.102 Early repair of these mutations is important for the prevention of SCC. Patients with NMSC are thought to have decreased ability for DNA repair compared with controls, thus making them more susceptible to the effects of UVR.103 One example is the effect of UVR on DNA alterations in patients with XP who are genetically unable to repair these defects. UVR also causes cutaneous immunosuppression, weakening the host’s immune response against tumor cells and promoting tumor growth.104 Studies of SCCs have shown that mutations in the TP53 tumor suppressor gene are an early event.3,105 Most of the mutations occur at dipyrimidine sequences, in the form of UV-induced cyclobutane pyrimidine dimers.106 The development of SCC does not occur by a simple single step, but rather through a multistage process. Conversion of susceptible keratinocytes to premalignant cells and then progression to carcinoma occurs as a result of successive genetic hits.107 In the initiation stage of carcinogenesis, there is clonal expansion of premalignant cells. The next stage is increased proliferation of premalignant cells and subsequent chromosomal aberrations. The final stage is conversion to SCC. DNA aneuploidy with a single peak was detected by flow cytometry in lesions of Bowen disease, suggesting a monoclonal proliferation of abnormal keratinocytes and a clonal basis for skin cancer.108 Aberrant expression of P-cadherin, and changes in expressions of cytokeratins and transforming growth factors, all play a role in tumor progression.109
Prevention and Early Detection
Prevention of SCC is similar to that of BCC, that is, an emphasis on photoprotection is essential. Interestingly, one study showed that regular application of sunscreen has prolonged preventive effects on SCC but with no statistically significant benefit in reducing BCC.110 Treatment of precursor actinic keratoses with field therapy such as photodynamic therapy, topical 5- fluorouracil and imiquimod cream may reduce the number of lesions transformed into SCC. Promising chemopreventive agents for lesions in the actinic keratosis–SCC spectrum include retinoids, cyclooxygenase (COX)-2 inhibitors, difluoromethylornithine, and epigallocatechin gallate (EGCG), a green tea catechin.111 In organ transplant recipients, systemic retinoids and reduction of immunosuppression are used as preventative means in reducing the incidence of NMSCs.100 One study showed that high-dose topical tretinoin, however, is ineffective at reducing risk of NMSCs in high-risk patients.70
Pathology and Pathways of Spread
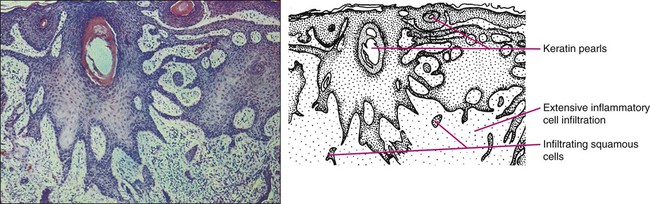
A deep shave or punch biopsy that includes the base of the tumor is needed to distinguish SCC in situ from invasive SCC. Bowen disease is SCC in situ. The stratum corneum is thickened, and epidermis is hyperplastic with disordered maturation of keratinocytes. Mitotic figures, multinucleated keratinocytes, and dyskeratotic cells with hyperchromatic nuclei and eosinophilic cytoplasm are seen in the epidermis. Atypical cells can invade follicular and ductal structures. The histology of invasive SCC shows masses of epidermal cells proliferating into the dermis. Atypical squamous cells and mitotic figures are seen (Fig. 70-6). The cells have abundant eosinophilic cytoplasm and large nuclei. Horn pearls, which are a result of keratinization of squamous cells, are seen. The dermis may show a marked inflammatory reaction. Spindle cell SCC is a rare variant composed of mainly spindle cells, but some squamous differentiation may be seen. Spindle cells have large vesicular nuclei and scant cytoplasm and intermingle with the collagenous stroma. Spindle cell SCC is poorly differentiated with numerous mitoses and deep invasion and requires immunohistochemistry to differentiate it from other spindle cell tumors. Another uncommon histologic variant of SCC is the acantholytic or adenoid SCC, seen more on the face and neck. There are nests of tumor cells with dyskeratotic cells and central acantholysis forming pseudoglandular structures.
Clinical Manifestations/Patient Evaluation/Staging
SCC occurs more on the head and neck area in whites, but in blacks there is no predilection for sun-exposed areas of the body.112 SCC in situ can arise de novo or from premalignant lesions, such as actinic keratosis or arsenical keratosis. In fact, in whites, the majority of SCCs arise from actinic keratoses. These can eventually spread beyond the epidermis and become invasive. Bowen disease is a SCC in situ arising de novo. If Bowen disease occurs on the glans penis or rarely vulva it is referred as erythroplasia of Queyrat. Bowen disease often appears as a well-defined single red plaque with dry surface scaling. Lesions are slow growing and asymptomatic, thus ignored by many patients. The physician might initially treat it as psoriasis or nummular eczema with no response. There is a 3% to 5% chance that Bowen disease can progress into invasive SCC.113 Clinically erythroplasia of Queyrat resembles Bowen disease, but lacks the dry superficial scale. Instead, the surface is moist and smooth.
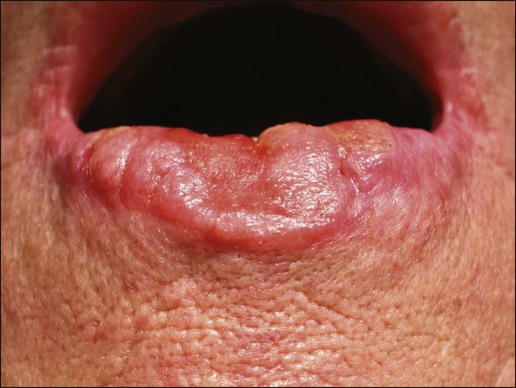
Invasive SCC can also arise from normal skin. It starts as a small, firm, dull-red nodule, which can undergo central ulceration. Sometimes if SCC arises from solar keratosis an adherent keratotic scale can be seen. If ignored, the lesion grows horizontally and vertically and may become fixed to the underlying tissue. The surface might become ulcerated with bleeding, malodorous exudate, or crust. The borders are usually elevated and firm. Occasionally a fungoid lesion without ulceration can be seen. SCC of the lower lip often arises from previously solar-damaged areas (actinic cheilitis). Initially local thickening of the vermilion border occurs that then progresses into a noduloulcerative lesion (Fig. 70-7). Persistent subungual erythema with pain and swelling should alert the physician for SCC of the nail region. This form of SCC may be confused with warts (Fig. 70-8). As mentioned earlier, SCCs can arise from chronic sinuses, scars, and chronic thermal damage.
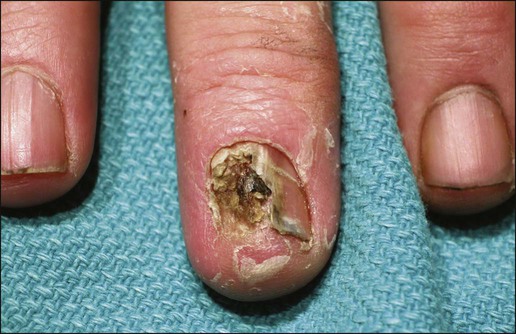
The risk of metastasis of SCC is variable, depending on the site and tumor characteristics. The deeper and larger the tumor, the higher is the chance of metastasis. Recurrent tumors are at high risk for metastasis. Lesions with perineural involvement have a 35% metastatic rate.114 Among the different histologic types, desmoplastic SCCs are more likely to metastasize.115 The risk of metastasis of SCC derived from actinic keratosis is low (0.5% to 3.7%) compared with SCC arising in radiation-induced SCC and chronic osteomyelitis (20% and 31%, respectively).118–118 High-risk areas for metastasis include tumors arising from the dorsal hands, lips, ears, and penis. For example, SCC of the lower lip has a 15% risk of metastasis.119 Bowen disease and erythroplasia of Queyrat have a low chance of metastasis, but once they become invasive, the risk of metastasis increases significantly.
Evaluation for patients with SCC is similar to that of BCC, and stratification of SCC into high-risk or low-risk tumors is practical to guide treatment. However, physicians must be reminded that SCCs in general have a greater potential for recurrence and metastasis than BCCs. The first step is to estimate the peripheral and vertical extension of tumor cells. The new seventh edition of the AJCC staging system for noneyelid skin cancer, including cutaneous SCC, incorporates tumor-specific (T) staging features, such as thickness greater than 2 mm, Clark level greater than IV, perineural invasion, anatomic site, and degree of histologic differentiation (see Table 70-1).120,121 Other reported high-risk features that may result in local and distant metastasis are size greater than 2 cm, recurrent status, and immunocompromised host.122 Next, the patient should be evaluated for metastasis to the lymph nodes and other organs. Note that eyelid skin cancers are staged differently, using a separate system that is described in the section on sebaceous carcinoma. For SCC patients, closer followup intervals are recommended, such as every 3 to 6 months for the first several years, depending on the location, size, aggressiveness, and node status of the primary tumor. The exception would be small SCCs arising from actinic keratoses on sun-exposed surfaces, where the rate of metastasis is very small (0.5%). In these patients, routine followup every 6 to 12 months is all that is required.
Primary Therapy
The treatment options for SCC are similar to BCC treatment, with some differences (Fig. 70-9).
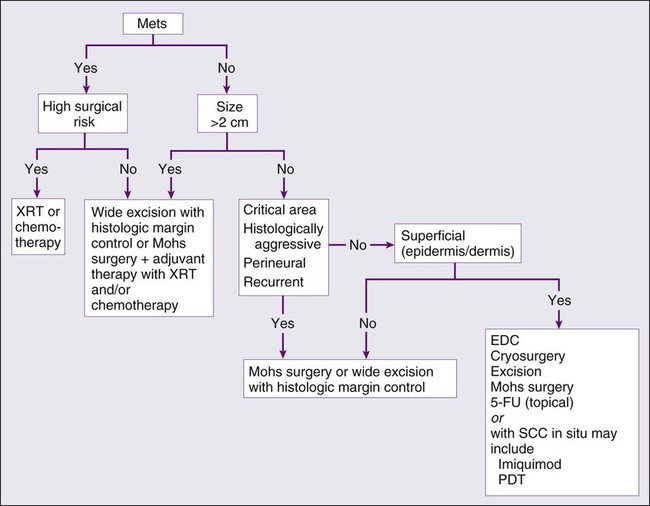
Destructive methods or surgical excision can treat Bowen disease effectively. When cancer cells invade hair follicles or ducts, treatment by excision is preferred. Small uncomplicated SCCs (<1 cm) in low-risk locations are frequently treated with EDC. Cryotherapy is an alternative treatment. Simple surgical excision with margin control is commonly used for smaller tumors and those on the trunk and extremities. According to the newest NCCN guidelines, the recommended margin for low-risk SCC is 4 to 6 mm, and for high-risk tumor is 10 mm, if possible.123 The excision should include the subcutaneous fat. Mohs micrographic surgery is the treatment of choice for high-risk SCCs and for tissue preservation where tissue sparing is cosmetically or functionally vital.124
Radiation can also be used as a primary choice of treatment in the elderly at high risk for surgical complications or patients with contraindications to anesthesia or surgery. Radiation can be useful to treat tumors on the eyelids, nose, ears, and lips when surgery is not practical. The disadvantages are the cost, blind margin control, and prolonged treatment duration. The cure rate for tumors larger than 2 cm is 85% to 95%.125 Radiation therapy is best avoided in treating verrucous carcinomas and younger patients because of poor long-term cosmetic results. SCCs that have arisen from ulcers, scars, and irradiated sites are better removed by margin-controlled surgery than by radiation therapy.
Treatment of Locally Advanced and Metastatic Disease
Aggressive or deeply invasive SCCs are treated with deep surgical excision with margin control when practical. Mohs surgery may be helpful in delineating critical areas in massive tumors or tumors with ill-defined borders. SCC invading surrounding structures such as bone and cartilage must also be excised. Tumors with neural or perineural involvement that are at high risk of recurrence should be completely excised preferably with the Mohs technique. High-risk tumors may require adjuvant therapy with radiation therapy126 or chemotherapy.127,128
A recent review article summarized treatment modalities for locally advanced nonmetastatic cutaneous SCC.129 Commonly used traditional chemotherapy includes oral capecitabine and intravenous platinum-based regimens. Biological response modifiers refer to agents that augment host antitumor immune responses, examples of which include oral isotretinoin and interferons. Because in vitro studies demonstrated synergism between retinoids and interferons, these agents have been used in combination with success.130,131 Newer targeted agents are promising, including epidermal growth factor receptor antagonists, small-molecule tyrosine kinase inhibitors, and proteasome inhibitors.
Sentinel lymph node biopsy may be considered in patients with high-risk SCC.132,133 If lymph node metastasis has occurred, treatment is wide surgical resection of tumor with regional lymph node dissection with or without adjunctive radiation. However, elective lymph node dissection is not commonly performed in the absence of evidence for regional spread.
Keratoacanthoma
Keratoacanthomas (KAs) are common cutaneous neoplasms that are thought to derive from the infundibular portion of hair follicles. They most often present as solitary rapidly growing, pink or flesh-colored, dome-shaped or crateriform nodules. They occur most commonly on sun-exposed areas in fair-skinned elderly individuals.134 Clinically, KAs are characterized by their spontaneous involution over several months with residual atrophic scarring and local tissue destruction.135
Epidemiology
Although most cases of KA are solitary, subsets of individuals with multiple KAs have been described. These include the familial Ferguson-Smith type, comprising multiple KAs at an early age, and the generalized eruption of thousands of small KAs as described by Grzybowski.136 KAs frequently occur in individuals with MTS, XP, and solid-organ transplantation.137 They also occur in areas of scarring from prior trauma, thermal burns, and surgical treatment, and in association with many benign conditions such as stasis dermatitis, lichen planus, lichen simplex chronicus, psoriasis, and discoid lupus erythematosus.134
Etiology and Biological Characteristics
Debate exists as to whether KAs are a distinct clinical entity or a subtype of well-differentiated SCC. Significant efforts have been made to find practical, reliable criteria to distinguish KA from SCC.138,139 Similarities include expression of oncogenetic and cell cycle–regulating proteins including p53; a subset of these tumors also share trisomy 7 anomaly.140–143 On the other hand, studies on apoptotic marker P2X7, on antiapoptotic Bcl-xL protein, on loss of heterozygosity, on expression of angiotensin type 1 receptor and of desmogleins 1 and 2, on adhesion molecules vascular cellular adhesion molecule and intercellular adhesion molecule, on telomerase activity, and on chromosomal aberrations assessed by comparative genomic hybridization suggest that KA and SCC possess distinct genetic aberrations that result in separate developmental pathways.144–152 One study localized recurrent aberrations to chromosomes 17, 19, 20, and X in one-third of the KAs studied. Recurrent aberrations on chromosomes 7, 8, 10, and 13 were characteristic of SCC, not of KA, perhaps accounting for the progressive growth and lack of regression of SCC.153 Microsatellite instability and loss of heterozygosity are seen uncommonly in KA unless associated with MTS.
Prevention and Early Detection
Because UV exposure is a key component of etiology, sun-protective clothing, sunblock use, and avoidance of UV exposure are the primary means of prevention. Timely diagnosis and appropriate treatment of chronic scarring conditions could reduce incidence in the affected population. Self skin-examination or examination by a dermatologist to identify the characteristic lesion is the only means of early detection.135
Pathology and Pathways of Spread
Histologically, a mature KA has a distinctive architecture characterized by a keratin-filled crater lined by a proliferating squamous epithelium with abundant pale eosinophilic “glassy” cytoplasm, epithelial lipping, and multiple keratin horn pearls with central orthokeratosis. Cytology is often indistinguishable from that of a well-differentiated cystic SCC. At the base of the tumor, islands and strands of atypical cells may invade the dermis. Frequently there is a heavy inflammatory infiltrate at the base of the lesion comprising lymphocytes, plasma cells, neutrophils, and eosinophils. Atypical mitoses, as well as perineural and perivascular invasion, also may be present, particularly in tumors involving the head and neck.154,155
Clinical Manifestations/Patient Evaluation/Staging
Patients will usually describe rapid onset and growth of a firm nodule with central depression filled with keratinous crustlike material over sun-exposed sites. The lesion will involute spontaneously leaving an atrophic scar over the course of several weeks. If evaluated early, a biopsy extending into the dermis is warranted to secure the diagnosis and to assess for SCC. Palpation of regional lymph nodes is indicated if there is suspicion for SCC, a cutaneous carcinoma known to metastasize.134
Primary Therapy
Although not thought to metastasize in general, several cases of highly aggressive and metastatic KAs have been reported, leaving some to question whether these were truly KAs or rather KA-like SCCs.136 However, given reported cases of local destruction and metastatic disease, it is recommended that KAs be treated in the same manner as SCCs. Complete conservative excision or Mohs micrographic surgery is the mainstay of therapy.156 Radiotherapy, photodynamic therapy, electrodessication and curettage, systemic immunosuppressive medications, systemic retinoids, and topical and intralesional immunosuppressive and immunomodulating modalities have been reported with varying success.157–163
Nonmelanoma Skin Cancer in Immunocompromised Hosts
Patients receiving chronic immunosuppression are at significantly increased risk for development of NMSC. Additionally, they are at increased risk of morbidity and mortality as a result of development of multiple primary tumors and more aggressive subtypes of skin carcinoma. Although increased risk is associated with immunosuppression intended to treat many ailments, studies of solid organ transplant recipients offer the clearest data at this time. As available immunosuppressive drugs and care for the chronically immunosuppressed population improve, the longevity of this population also is increasing. With increased longevity, the burden of NMSC in this group has grown significantly.164
Epidemiology
Reports on the incidence of NMSC varies in organ transplant populations studied from different parts of the world and also varies with the specific transplanted organ. Within 10 years from solid organ transplantation, 15% to 43% of recipients will develop NMSC. The highest risk is associated with heart and lung transplantation, the next with kidney transplantation, and the least with liver transplantation.164 This corresponds to the overall degree of immunosuppression required to prevent rejection of the specific organ in each population. The most significant risk factors for the development of NMSC in organ transplant recipients include advanced age, cumulative UV exposure pretransplantation, degree and duration of immunosuppression, fair skin type, diagnosis of NMSC prior to transplantation, pretransplantation disease requiring prior immunosuppression (e.g., systemic lupus), and diagnosis of lymphoma before or after transplantation.99,165
In addition to the organ transplant population, there is mounting evidence for concern regarding increased risk of NMSC in other immunosuppressed populations. There is significantly increased risk for patients with rheumatologic disease and psoriasis receiving tumor necrosis factor inhibitors, methotrexate and prednisone.166,167 Immune dysregulation from chronic lymphocytic leukemia elevates risk of aggressive SCC and subsequent mortality.168 Highly active antiretroviral therapy has dramatically increased the longevity of the HIV-infected populations and with that has evolved a significant increase in BCCs and SCCs.169 The use of thiopurines to treat inflammatory bowel disease has been shown to substantially increase the risk of SCC development.170
Prevention and Early Detection
Appropriate dermatologic care for organ transplant recipients (and presumably others with significant chronic immunosuppression, although these guidelines are less-well established) should be based on risk stratification. All individuals should receive baseline screening and risk assessment by a dermatologist and education regarding skin cancer risk, photoprotection, and self-screening. Many transplantation centers have dermatologists with specific expertise in the care of these patients. Those determined to be at increased risk should be screened every 6 to 12 months, or more frequently if they develop multiple or aggressive skin cancers.99
Primary Therapy
Treatment of NMSC in organ transplant recipients and similarly immunosuppressed patients should be tailored to the individual’s needs. Standard modalities, including excision, electrodessication and curettage, curettage and cryotherapy, and Mohs micrographic surgery, are appropriate in most cases. Aggressive treatment of precancerous lesions with standard modalities, such as cryotherapy, topical retinoids, imiquimod, topical 5-fluorouracil, and photodynamic therapy, is beneficial. Patients with extensive photodamage amounting to areas of “field cancerization” should be identified and treated aggressively with standard modalities, then monitored closely to reduce the risk of developing numerous or aggressive tumors in these areas.100
Locally Advanced Disease and Palliation
Organ transplant recipients developing multiple new primary skin cancers (5 to 10 per year) or skin cancers with aggressive histologic subtypes despite appropriate monitoring and therapy are candidates for chemopreventive therapy. Acitretin can be used at doses from 10 to 25 mg daily with significant reduction in the number of new NMSC for many patients. Dosage is limited by side effects. Liver function, kidney function, and blood lipids must be monitored while on therapy. The chemopreventive effect of acitretin lasts only for the duration of therapy, and a rebound of SCC may occur upon discontinuation of acitretin.99,100
Revision of immunosuppression may be appropriate for patients who develop multiple or histologically aggressive NMSC. Consultation should take place between the patient and the patient’s transplant physicians regarding possible reduction of current medication or a switch to a comparable immunosuppressive agent with a better side-effect profile for skin cancer. When graft function is stable and surgical wounds have healed, sirolimus or everolimus may be considered for their antineoplastic properties. Sirolimus is associated with a significant reduction in malignancies posttransplantation. Azathioprine should be discontinued if possible because of its photocarcinogenic properties. Tacrolimus is preferable to cyclosporine as a first-line calcineurin inhibitor because of an associated lower risk of NMSC. Although triple-drug immunosuppressive therapy remains the standard treatment after organ transplantation at most centers, dual therapy and calcineurin inhibitor monotherapy have been shown to reduce NMSC without significant increase in organ rejection in patients more than 6 years posttransplantation.165
Treatment of Metastatic Disease
Patients with aggressive, large, recurrent, numerous or metastatic NMSC should be referred to a provider with expertise in the evaluation of their metastatic potential and management. A role for sentinel lymph node biopsy has been established in select aggressive SCC. Adjuvant radiation therapy is helpful in selected cases of SCC or BCC that are aggressive or have high-risk features. Involvement of a multidisciplinary transplant team that includes transplant physicians, dermatologists, oncologists, radiation oncologists, otorhinolaryngologists, and plastic surgeons, may benefit those patients who are extensively afflicted with NMSC.165
Future Possibilities and Clinical Trials
Current efforts to improve outcomes for organ transplant recipients with nonmelanoma skin cancers are centered on modification of current immunosuppressive regimens and adaptation of immunosuppressive regimens using newer agents with more favorable side-effect profiles regarding skin cancer development, such as sirolimus and everolimus. Transplantation centers continue to investigate regimens that minimize immunosuppression at induction and later in the course of transplantation for appropriate patient candidates. For example, calcineurin monotherapy (compared to two- or three-agent regimens) was associated with a higher graft survival rate and a lower skin cancer rate in selected patients 6 to 12 years posttransplantation.171 Ongoing studies are needed to further understand appropriate strategies for optimization of graft survival weighed against the risk of carcinogenesis.
Sebaceous Carcinoma
Sebaceous carcinoma is a rare, potentially devastating tumor that derives from the adnexal epithelium of sebaceous glands. Principally, the tumor occurs in two different clinical scenarios: ocular sebaceous carcinoma (OSC) and extraocular sebaceous carcinoma (ESC).172,173
Epidemiology
OSC accounts for 0.2% to 0.8% of all eyelid tumors and 1% to 1.5% of all malignant tumors of the eyelid.174 OSC affects women 1.5 times more frequently than men; however, a slight male preponderance occurs in cases of ESC.175–179 The mean age at diagnosis is 65 years (range: 40 to 60 years), but OSC has been reported in adults in their nineties and in a 3-year-old child. Sebaceous carcinoma seems to have a higher incidence in the Asian population.180–183
Etiology and Biological Characteristics
This tumor is associated with MTS, an autosomal-dominant disorder of mismatch repair genes characterized by sebaceous tumors (benign and malignant), as well as other visceral malignant tumors.184 Although sebaceous carcinoma with microsatellite instability has been found in several renal organ transplant recipients,184 only a subset of these tumors possess microsatellite instability. Mutations in p53 are significantly associated with sebaceous carcinoma and are independent of microsatellite instability, therefore suggesting a divergence in molecular pathogenesis.185 Epigenetic inactivation of E-cadherin, leading to loss of the membrane bound molecule, is associated with reduced disease-free survival in OSC.186
There is evidence that the chronic inflammation caused by chalazion may be causative in OSC. The unsaturated 8-carbon fatty acid oleic acid has been shown to cause dysplasia in glandular structures.172
Risk factors associated with poor prognosis include duration longer than 6 months; vascular or lymphatic involvement; orbital extension; poorly differentiated tumor morphology; multicentricity; pagetoid spread into the conjunctival epithelium, cornea, or skin; upper and lower lid involvement; and history of radiation exposure.180,187–189 In the past, 14% to 25% of patients reportedly had metastasis, most frequently involving regional nodal basins, followed by spread to the liver, lung, brain, and bones.175,177–180 Because of increased index of suspicion, physician education, and earlier recognition with biopsy, the metastatic rate has decreased to 10% in some series.189 Among industrialized countries, periocular mortality is reportedly 9% to 15%. Systemic disease is associated with a grave prognosis.174
Prevention and Early Detection
The diagnosis is often elusive, because OSC is notorious for masquerading as more common ocular disorders, such as chalazion, keratoconjunctivitis, blepharoconjunctivitis, SCC, BCC, granulomatous disorders, and benign neoplasms.175,188,190–192 Initial recognition is 50% among ophthalmologists and only 18.6% among general practitioners.194–194 Often there is a delay in diagnosis of 6 months to a year, and in some series up to 2.9 years, which contributes to increased morbidity and mortality.175,189 Although UV radiation does not seem to play a role in the pathogenesis of OSC, ionizing radiation is associated with it. No other means of prevention are known.174
Pathology and Pathways of Spread
Microscopic examination shows that OSC and ESC invade surrounding tissue by direct extension into the dermis or the overlying epithelium.189 The tumor demonstrates epitheliotropic spread in 37% to 80% of cases. Atypical sebaceous cells have a pagetoid distribution within the conjunctival epithelium.187,189 As much as 18% of cases of OSC may have multifocal regions of tumor involvement.195
There are basically two broad classification schemes with regard to light microscopic findings. Font196 described three classes of tumor based on degree of differentiation: well differentiated, moderately well differentiated, and poorly differentiated. Others prefer to classify the tumor on the basis of growth pattern: lobular, comedocarcinoma, papillary, and mixed.178 The intraepithelial neoplasia growth pattern and pagetoid spread are not addressed in this classification scheme. Recognition of this particular biphasic nature of OSC is critical to appreciating the biological behavior of this recalcitrant tumor. The histologic features include anaplastic cells with varying degrees of differentiation. Squamoid or basaloid features may be present.175 Lobulated aggregates of basaloid cells usually contain irregular, hyperchromatic nuclei and vacuolated cytoplasm.188,195,197,198 In cases of intraepithelial neoplasia, the epithelial pagetoid cells lack intercellular bridges and contain large hyperchromatic nuclei, prominent nucleoli, and abundant, pale-staining cytoplasm. Special stains, such as oil red O and Sudan black, often are helpful in delineating the atypical sebocytes; however, poorly differentiated tumors may not stain positively for lipids.189,190 In addition, undifferentiated sebocytes often lack the foamy cytoplasm of their more differentiated counterparts and may resemble atypical SCC with standard hematoxylin-and-eosin staining.175,188,197
Both OSC and ESC are aggressive tumors that recur locally if inadequately managed and readily metastasize. Metastasis may occur through lymphatic vessels, and in the case of OSC, it is postulated to occur through the lacrimal and excretory systems.175
Clinical Manifestations/Patient Evaluation/Staging
OSC typically manifests as a painless, slowly growing, yellowish papule (Fig. 70-10). The upper lid is affected two to three times more frequently than the lower lid. The tumor usually originates from the meibomian gland (sebaceous gland in the tarsus) or from a sebaceous gland of the eyelash (glands of Zeis).176,179,187 Clinically, the signs can be subtle with only moderate lid thickening, eyelash alopecia, pale corneal changes, conjunctival injection, or thickening along the temporal or inferior temporal limbus.175,177,190,199 There are case reports of ipsilateral upper and lower eyelid involvement and bilateral upper and lower lid involvement.188,191,195 With regard to the rarer ESC, initial diagnosis can be elusive because the clinical appearance often is pleomorphic and nonspecific and varies from a banal-appearing nodule to large ulcerating masses.200–203 This variant most often occurs on the head and neck region, reflecting the abundance of sebaceous glands there.200
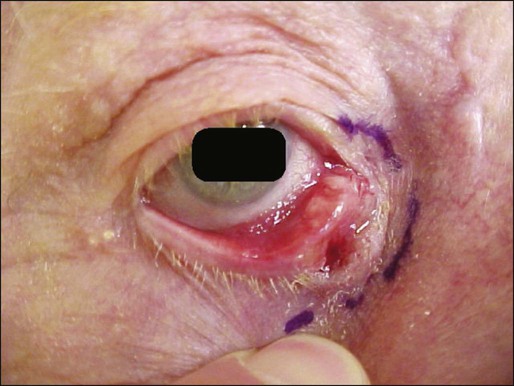
Evaluation of patients with known or suspected sebaceous gland carcinoma includes complete history with review of systems, family history and consideration of MTS, complete skin examination, and palpation of nodal basins and structures adjacent to the lesion. A chest radiograph, complete blood cell count, liver function tests, and electrolytes are recommended. If MTS is a consideration, a colonoscopy is warranted.175
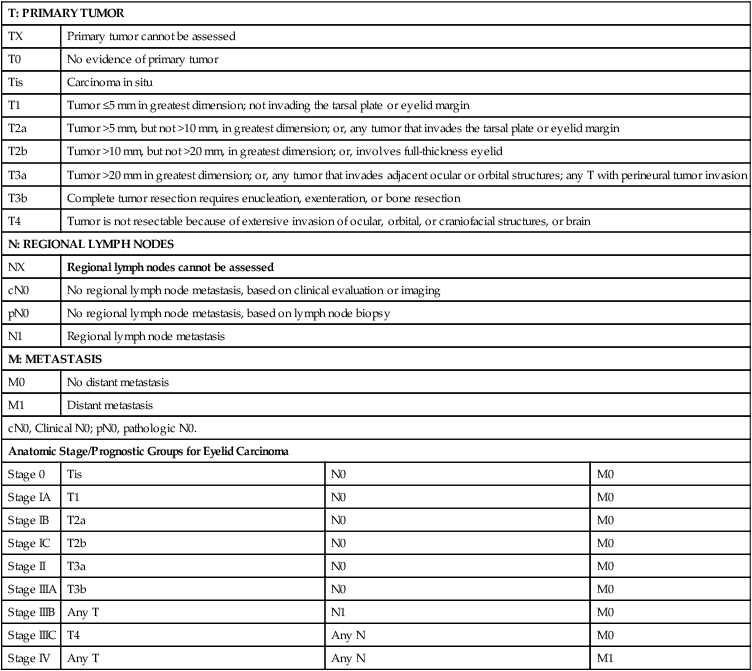
If suspicious lesions are identified, a full-thickness biopsy is required to ensure adequate tissue sampling. For OSC, it has been recommended that as many as nine mucosal biopsy specimens be obtained and mapped from the medial and lateral portions of the bulbar and palpebral conjunctiva of the upper and lower lids given the prevalence of pagetoid spread.204,205 Table 70-2 provides TNM staging for eyelid cancer.
Table 70-2
TNM Staging for Eyelid Carcinoma
| T: PRIMARY TUMOR | |||
| TX | Primary tumor cannot be assessed | ||
| T0 | No evidence of primary tumor | ||
| Tis | Carcinoma in situ | ||
| T1 | Tumor ≤5 mm in greatest dimension; not invading the tarsal plate or eyelid margin | ||
| T2a | Tumor >5 mm, but not >10 mm, in greatest dimension; or, any tumor that invades the tarsal plate or eyelid margin | ||
| T2b | Tumor >10 mm, but not >20 mm, in greatest dimension; or, involves full-thickness eyelid | ||
| T3a | Tumor >20 mm in greatest dimension; or, any tumor that invades adjacent ocular or orbital structures; any T with perineural tumor invasion | ||
| T3b | Complete tumor resection requires enucleation, exenteration, or bone resection | ||
| T4 | Tumor is not resectable because of extensive invasion of ocular, orbital, or craniofacial structures, or brain | ||
| N: REGIONAL LYMPH NODES | |||
| NX | Regional lymph nodes cannot be assessed | ||
| cN0 | No regional lymph node metastasis, based on clinical evaluation or imaging | ||
| pN0 | No regional lymph node metastasis, based on lymph node biopsy | ||
| N1 | Regional lymph node metastasis | ||
| M: METASTASIS | |||
| M0 | No distant metastasis | ||
| M1 | Distant metastasis | ||
| cN0, Clinical N0; pN0, pathologic N0. | |||
| Anatomic Stage/Prognostic Groups for Eyelid Carcinoma | |||
| Stage 0 | Tis | N0 | M0 |
| Stage IA | T1 | N0 | M0 |
| Stage IB | T2a | N0 | M0 |
| Stage IC | T2b | N0 | M0 |
| Stage II | T3a | N0 | M0 |
| Stage IIIA | T3b | N0 | M0 |
| Stage IIIB | Any T | N1 | M0 |
| Stage IIIC | T4 | Any N | M0 |
| Stage IV | Any T | Any N | M1 |
From American Joint Committee on Cancer Staging Manual, Seventh Edition, 2010.
Primary Therapy
Management of sebaceous gland carcinoma is primarily surgical, although there are a few reports of tumors managed primarily with radiation.206 Given the paucity of cases, no randomized, controlled studies have been conducted to evaluate therapy. Traditionally, wide local excision with a margin of 4 to 6 mm of clinically normal tissue has been used.175 Exenteration is recommended for extensive bulbar conjunctival involvement or evidence of orbital invasion.179 In his personal series of 60 consecutive patients with a mean followup of 41 months, Shields and colleagues reported a local recurrence rate of 18%, exenteration of 13%, and metastasis of 8%. 189 Some experts consider Mohs micrographic surgical technique the treatment of choice, because it preserves the maximum amount of healthy tissue. This quality is especially crucial in the treatment of ocular tissue.188,189,207–210 In addition, it allows for microscopic examination of all margins of the obtained tissue to ensure complete tumor eradication. In 2002, Snow et al reported results of nine cases treated by Mohs micrographic surgery, achieving a 90% success rate with 3-year followup.202
One group is recommending sentinel lymph node biopsy versus strict regional lymph node surveillance for OSCs T2b (AJCC TNM staging); in other words, where the tumor is larger than 10 mm but not larger than 20 mm in greatest dimension or involves a full-thickness eyelid.211
Treatment of Locally Advanced and Metastatic Disease
Local recurrence usually is the result of inadequate management of the primary tumor.175 If the recurrent lesion is small enough, treatment can be repeated with Mohs micrographic surgery or local excision.189 More extensive OSC tumor involvement usually necessitates exenteration.194,198 Ophthalmologists have considered cryotherapy a useful modality in the management of epibulbar disease in a small subset of patients, and this therapy may be an alternative to exenteration.182,212,213 Regional lymph node metastasis is managed by surgical resection and adjuvant radiation.175
Merkel Cell Carcinoma
Merkel cell carcinoma (MCC) is a rare and aggressive primary cutaneous neuroendocrine carcinoma. Since first described in 1972 by Toker,214 MCC has had many synonyms, including primary small cell carcinoma of the skin, trabecular carcinoma, APUDoma, neuroendocrine carcinoma, endocrine carcinoma, and primary undifferentiated tumor of the skin.217–217 The identification of electron-dense neurosecretory granules in MCC suggests the Merkel cell, which is a specialized receptor cell of touch localized in the basal layer of the epidermis, as the most probable cell of origin.220–220 However, the presence of epithelial and sarcomatous characteristics in MCC also suggests that MCC may arise from primitive totipotent epidermal stem cells.221
Epidemiology
SEER (Surveillance, Epidemiology, and End Results) data indicate age-adjusted incidence rates of 0.18 to 0.41 per 100,000 persons in the United States.222 The incidence tripled from 1986 to 2001, and approximately 1500 cases of MCC are diagnosed each year.225–225 MCC is rare in blacks; most patients are white (94.9%).222 Ninety percent of cases reported occur in patients older than 50 years, with a mean of 69 years old at diagnosis.222,226,227 Males are affected more commonly than females (61% male vs. 39% female).222,228
Etiology and Biological Characteristics
Our knowledge of the etiology and biological behavior of MCC remains limited.223,229–231 The major new discovery is the identification of Merkel cell polyomavirus (MCPyV), which has been confirmed positive in approximately 80% of MCC cases, with clonal integration into the tumor genome.232–235 Further studies are necessary to understand the role of MCPyV in MCC pathogenesis, and its impact on the course of disease and prognosis.236 Immunosuppression is a well-recognized risk factor for MCC,231,237 indicated by the increased prevalence of MCC in organ transplant recipients,240–240 human immunodeficiency virus carriers,241 patients with autoimmune diseases,244–244 and patients with hematologic neoplasias, particularly chronic lymphocytic leukemia.237,245,246 Other reported associations include the presence of BCC and SCC,247 skin radiation exposure,248,249 arsenic exposure,250 and advancing age.223
Prevention and Early Detection
A high index of clinical suspicion and skin biopsies for pathological and immunohistochemical evaluation are encouraged to achieve early detection of MCC, as the disease stage is the best predictor of survival.222 Risk factors associated with MCC need to be minimized whenever possible.
Pathology and Pathways of Spread
Histologically, MCCs are dermal-based tumors typically composed of small, uniform, basophilic, and ovoid cells measuring up to 15 μm in diameter. These cells have frequent mitotic figures, apoptotic bodies, scanty cytoplasm, vesicular nuclei with finely dispersed chromatin and inconspicuous nucleoli, giving a so-called salt-and-pepper appearance.216,251–253 The tumor may extend from the dermis into the subcutaneous fat or deeper, but usually spares the epidermis. Up to 10% of MCC may have a pagetoid pattern of epidermal infiltration,256–256 which can mimic other tumors with intraepidermal involvement. MCC also may have both squamous and adnexal differentiation.217 Nearly 40% of MCC are associated with adjacent or overlying Bowen disease or squamous cell carcinoma,257 and less frequently, with BCC or eccrine tumors. There are three main architectural subtypes of MCC: intermediate cell, small cell, and trabecular.251,253 The most common type, intermediate cell type, is characterized by large sheets or clusters of uniform, less compact cells with foci of necrosis, occasionally arising near adnexal structures and connecting with epidermis, and often with surrounding lymphocytic infiltrate.215,255,258–260 The small cell type arises in the dermis and appears as sheets of cells interrupted by strands of connective tissue. Glandular or pseudoglandular structures are absent. The cells are round and small and may demonstrate “crush” artifact.259 The least common type, trabecular pattern, is arranged in cords admixed among a fibrovascular background, occasionally with pseudoglandular structures and involvement of adnexal structures, such as hair follicles.215,216,258–260 Whether these subtypes have prognostic significance is yet to be determined.220 Lymphatic invasion is frequently encountered and has negative prognostic implications.216,258,259,261,262
MCC bears similarity to other small cell neoplasms and must be differentiated from cutaneous metastatic lesions such as carcinoid or small cell undifferentiated carcinoma of the lung via immunohistochemical studies.216,251–253 Cytokeratin 20 (CK20), an intermediate filament protein, is the most important marker that stains approximately 80% to 90% of MCCs in a characteristic paranuclear dotlike pattern.251,259,263–265 In contrast, small cell carcinoma of the lung stains positively for thyroid transcription factor-1 (TTF-1) but negatively for CK20.251 In addition, MCC stains positively for a panel of neuroendocrine markers with variable sensitivity, including neuron-specific enolase, which lacks specificity for MCC, and chromogranin A and synaptophysin, which lack sensitivity for MCC.251 MCC also stains positively for epithelial membrane antigen and Ber-EP4.264 Recently, CM2B4, an antibody that recognizes the large T (LT) antigen of the MCPyV, has been detected in approximately 70% of MCC.266 Differential diagnoses for small, round blue cell tumors that may mimic MCC also include blastic lymphomas like Burkitt lymphoma and cutaneous Ewing sarcoma/peripheral primitive neuroectodermal tumors. Lymphomas can be differentiated from MCC by expression of CD45 (leukocyte common antigen) and other T- or B-cell markers via immunohistochemical staining. Detection of EWS/FLI-1, a fusion transcript caused by chromosomal translocation t(11;22)(q24;q12), by reverse transcription polymerase chain reaction (RT-PCR) or fluorescence in situ hybridization (FISH) confirms the diagnosis of cutaneous Ewing sarcoma/peripheral primitive neuroectodermal tumors.267
Clinical Manifestations/Patient Evaluation/Staging
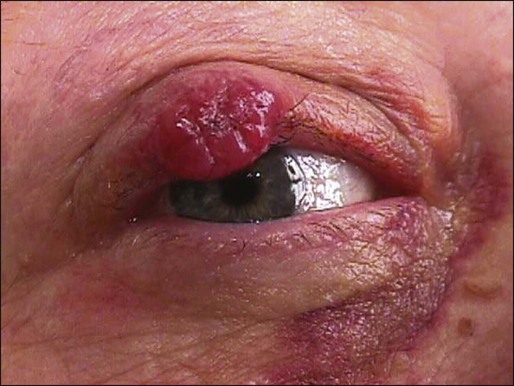
MCC often presents as a painless, indurated, erythematous to violaceous nodule with a smooth, shiny surface on sun-damaged skin, and, less commonly, as a plaque with satellite metastases. Surface ulcerations are rare.215–217,226,228,268 Heath et al summarized clinical features of MCC in an acronym: AEIOU (asymptomatic/lack of tenderness, expanding rapidly, immune suppression, older than 50 years, and ultraviolet-exposed site on a person with fair skin),237 suggesting clinical appearance of MCC is rather nondescript. MCC occurs predominantly in the head and neck (41% to 50%), followed by the extremities (32% to 38%) and trunk (12% to 14%).268 Ten percent of these tumors are in the periocular areas (Fig. 70-11).269 MCC also has been reported in extracutaneous sites, such as the vulva, endocervix, penis, esophagus, bladder, and calvarium.242,270–273 The salivary glands, nasal cavity, lip, lymph nodes, vulva, vagina and esophagus are the most common extracutaneous sites.222 The clinical differential diagnosis includes BCC, SCC, amelanotic melanoma, lymphoma, and metastatic disease. Histologic and immunohistochemical studies are the key to confirm the diagnosis. At the time of presentation, approximately 66% cases of MCC are localized disease, 27% with nodal involvement and 7% with distant metastatic disease.228
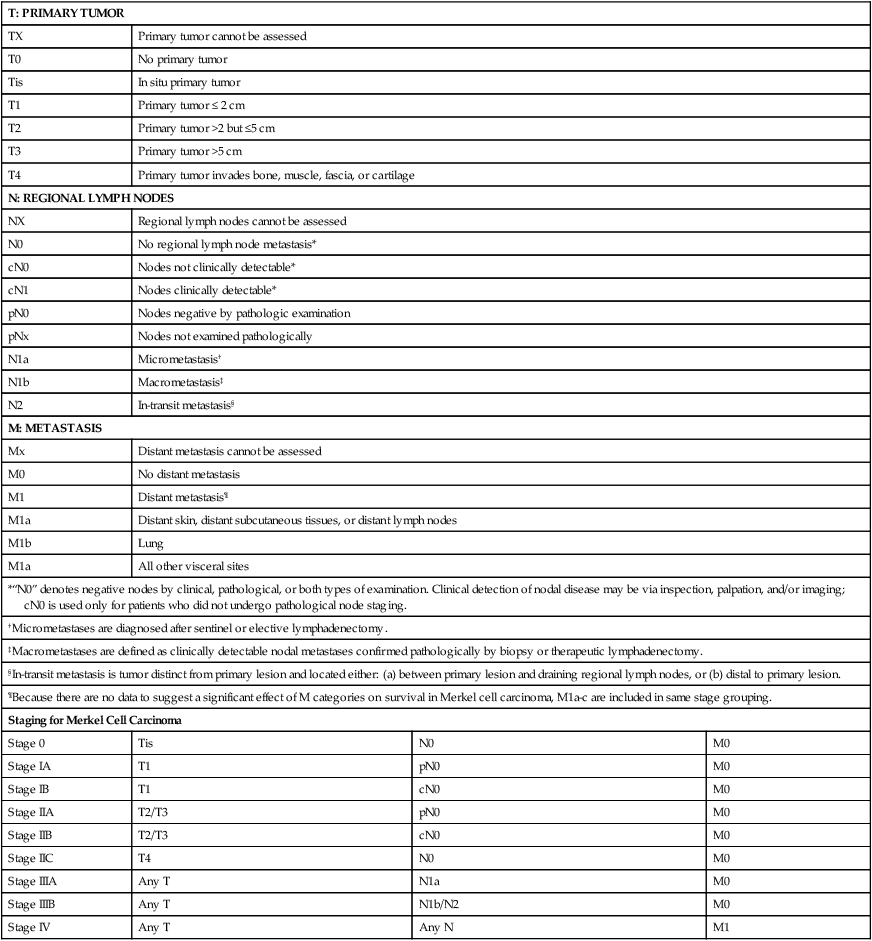
Patients diagnosed with MCC are commonly evaluated by a detailed history, a physical examination performed with emphasis on the skin and lymph nodes, a basic workup, such as complete blood cell count and hepatic and renal function tests, and appropriate imaging tests.274 In the absence of clinically positive lymph nodes, sentinel lymph node biopsy (SLNB) is generally recommended in MCC patients to obtain accurate nodal staging, especially to detect microscopic nodal disease.275–278 SLNB revealed nodal positivity in approximately one-third of patients who were otherwise negative by physical examination.275,279–281 Computed tomography (CT) evaluation of the chest, abdomen, and pelvis may be useful for initial staging, although CT scans reportedly have low sensitivity (20%) for detecting nodal involvement and low specificity for distant disease.275 In selected patients, high-resolution ultrasound has been used for evaluation of regional lymph node basins preoperatively and in followup visits. When distant metastases are expected, 18F-fluorodeoxyglucose positron emission tomography (PET)/CT fusion scanning has an emerging role for whole-body screening.274 Tumor staging is performed to guide management. The classification and staging of MCC had been challenging, with five different staging systems being proposed in the past 20 years.228 In 2010, the AJCC released the first consensus staging system for MCC based on tumor–nodes–metastases (TNM) criteria (Table 70-3).220,229 To facilitate patient care and research in MCC, the Centers for Disease Control and Prevention (CDC) granted seven MCC-specific codes that became active as of October 1, 2009.220,230
Table 70-3
TNM Criteria and Staging Groups for Merkel Cell Carcinoma
| T: PRIMARY TUMOR | |||
| TX | Primary tumor cannot be assessed | ||
| T0 | No primary tumor | ||
| Tis | In situ primary tumor | ||
| T1 | Primary tumor ≤ 2 cm | ||
| T2 | Primary tumor >2 but ≤5 cm | ||
| T3 | Primary tumor >5 cm | ||
| T4 | Primary tumor invades bone, muscle, fascia, or cartilage | ||
| N: REGIONAL LYMPH NODES | |||
| NX | Regional lymph nodes cannot be assessed | ||
| N0 | No regional lymph node metastasis* | ||
| cN0 | Nodes not clinically detectable* | ||
| cN1 | Nodes clinically detectable* | ||
| pN0 | Nodes negative by pathologic examination | ||
| pNx | Nodes not examined pathologically | ||
| N1a | Micrometastasis† | ||
| N1b | Macrometastasis‡ | ||
| N2 | In-transit metastasis§ | ||
| M: METASTASIS | |||
| Mx | Distant metastasis cannot be assessed | ||
| M0 | No distant metastasis | ||
| M1 | Distant metastasis¶ | ||
| M1a | Distant skin, distant subcutaneous tissues, or distant lymph nodes | ||
| M1b | Lung | ||
| M1a | All other visceral sites | ||
| *“N0” denotes negative nodes by clinical, pathological, or both types of examination. Clinical detection of nodal disease may be via inspection, palpation, and/or imaging; cN0 is used only for patients who did not undergo pathological node staging. | |||
| †Micrometastases are diagnosed after sentinel or elective lymphadenectomy. | |||
| ‡Macrometastases are defined as clinically detectable nodal metastases confirmed pathologically by biopsy or therapeutic lymphadenectomy. | |||
| §In-transit metastasis is tumor distinct from primary lesion and located either: (a) between primary lesion and draining regional lymph nodes, or (b) distal to primary lesion. | |||
| ¶Because there are no data to suggest a significant effect of M categories on survival in Merkel cell carcinoma, M1a-c are included in same stage grouping. | |||
| Staging for Merkel Cell Carcinoma | |||
| Stage 0 | Tis | N0 | M0 |
| Stage IA | T1 | pN0 | M0 |
| Stage IB | T1 | cN0 | M0 |
| Stage IIA | T2/T3 | pN0 | M0 |
| Stage IIB | T2/T3 | cN0 | M0 |
| Stage IIC | T4 | N0 | M0 |
| Stage IIIA | Any T | N1a | M0 |
| Stage IIIB | Any T | N1b/N2 | M0 |
| Stage IV | Any T | Any N | M1 |
From American Joint Committee on Cancer Staging Manual, Seventh Edition, 2010; Wang TS, Byrne PJ, Jacobs LK, Taube JM. Merkel cell carcinoma: update and review. Semin Cutan Med Surg 2011;30:48-56.
Primary Therapy
Treatment of MCC remains challenging. The overall 5-year relative survival in patients with local, nodal positive, and distant MCC is 64%, 39%, and 18%, respectively.228 Because of its rare incidence, there is currently a lack of standard therapeutic procedures for the management of MCC. Most cases are managed by wide local excision along with sentinel lymph node biopsy, followed by adjuvant radiotherapy to the primary site and the regional lymph node draining system,220,229–231,268 and are best managed in a multidisciplinary approach.282 Surgical excision remains the standard treatment for localized MCC without any evidence of regional or distant metastases.220,229–231,268 Wide local excision of 2.5 to 3.0 cm margins, made deep into the muscle fascia, has been the historical standard for MCC of the trunk or extremity. When tissue conservation is required, such as lesions on the face, the use of Mohs micrographic surgery followed by adjuvant radiotherapy is particularly suitable.283 MCCs are usually highly sensitive to ionizing radiation therapy. The use of adjuvant radiotherapy has been associated with improved survival,284 and is routinely administered in MCC.285,286 Radiation can be used as primary therapy for MCC lesions that are not resectable in selected patients.286
Treatment of Locally Advanced Disease and Palliation
After a positive lymph node is detected, it remains unclear whether patients benefit most from a complete node dissection, radiotherapy only, or both.268,287 Radical lymphadenectomy should be considered the first-line treatment for sentinel node positive patients. However, elderly, poor surgical candidates with micrometastases to lymph nodes may receive radiotherapy alone. In general, patients with larger tumors, macrometastases, or extranodal invasion require combined therapy. For patients with less than optimal margins, especially in the head-neck region, where adjuvant radiotherapy is planned, conservative excision of lymph nodes close to the positive node basin without performing a radical lymphadenectomy might be considered.268 Adjuvant use of chemotherapy in patients with regional disease is currently not supported by evidence to suggest improved survival.288
Treatment of Metastatic Disease
Systemic chemotherapy is used as a palliative measure in the setting of metastatic MCC, occasionally combined with radiation therapy. There is no evidence-based standardized chemotherapy and no survival benefit yet demonstrated. Many drugs have been used as monotherapy, or more often in combination, including cisplatin, etoposide, doxorubicin, vincristine, cyclophosphamide, methotrexate, and 5-fluorouracil.229,230,268,289 Selected cases with in-transit metastasis may be successfully treated with hyperthermic isolated limb perfusion of chemotherapeutic agents such as melphalan.290
Controversies/Problems/Challenges/Future Possibilities and Clinical Trials
Studies to better elucidate the molecular pathogenesis, such as understanding the functional role of MCPyV in MCC, are vital to discover potential molecular targets for novel therapies. Well-designed, prospective clinical trials are needed to unify and guide management for MCC.289
Dermatofibrosarcoma Protuberans
DFSP, recognized as a distinctive entity since 1920s,291,292 is a low- to intermediate-grade soft-tissue sarcoma that arises from the dermis. Approximately 85% to 90% of DFSPs are low grade, while the remainder contain a high-grade fibrosarcoma component (DFSP-FS), and are therefore considered as intermediate grade.293 Although distant metastasis has been reported, DFSP has a high propensity for local recurrence even after wide local excision.
Epidemiology
DFSP constitutes less than 1% of all malignant tumors, and accounts for 2% to 6% of all soft-tissue sarcoma in the United States.294 The estimated incidence is 0.8 to 4.5 cases per million persons per year.295–299 The tumor commonly occurs between 20 and 50 years of age, with only rare cases occurring in young children.300 In one large series, children accounted for 6% of all DFSP cases.301 There are approximately 166 pediatric cases reported in the literature, and these are divided into congenital and acquired types (38 and 128 cases, respectively).302 Giant cell fibroblastoma, which occurs more commonly in young children, is thought to be on a clinical and histopathologic spectrum with DFSP.303 Although most of the studies showed that the sexes are affected equally, some series suggested a slight male or female preponderance.295–297,300,304,305 Congenital DFSP, however, is most commonly reported in females.302 Although most reported cases of DFSP occur in whites, the condition has been reported in all races, including African Americans and Asians.295–297,306
Etiology and Biological Characteristics
The cause of DFSP is unknown; however, approximately 10% to 20% of patients report a history of antecedent trauma.295,300,307–309 There have been reports of DFSP arising in multiple immunization sites, burn scars, and surgical scars.306,310,311 A characteristic molecular feature of DFSP is the presence of cytogenetic abnormality as a result of formation of a supernumerary chromosomal ring or linear chromosomal translocation t (17;22) involving chromosomes 17 and 22.312 Such genetic alteration results in a unique fusion product in which the gene for platelet-derived growth factor B (PDGF-B) is fused with the highly expressed collagen 1A1 (COL1A1) gene. This leads to constitutive activation of the PDGF receptor followed by constitutive activation of cell growth, therefore essential to the development of DFSP.313–317 The detection of the COL1A1-PDGF-B fusion product by RT-PCR has been shown in more than 90% cases of DFSP in both adults and children,312 thereby demonstrating its diagnostic value. The test may be especially useful in atypical cases where diagnosis is uncertain based on histology.
Characteristically, DFSP manifests as a solitary nodule. However, there may be multicentric primary lesions. There is a link between multiple eruptive DFSP and immunodeficiency states. Eight cases of DFSP associated with adenosine deaminase (ADA) deficiency have been reported, six of which had multicentric lesions.318 The authors postulate that a DNA repair defect may lead to the (17;22)(q22;q13) translocation causing overexpression of PDGF-B and subsequent formation of fibrotic tumors.
Pathology and Pathways of Spread
DFSP is characterized by a proliferation of monomorphic spindle-shaped cells with little cytologic atypia, arranged in fascicles that create the typical storiform, cartwheel, or pinwheel pattern. These cells often infiltrate the dermis, dissecting between the native collagen bundles, as well as the subcutaneous fat.322–322 Fascia, muscle, and underlying bone may be involved in long-standing lesions.297,300,306,313 The tumor cells intersect in tight right angles around central vessels. The overlying epidermis usually is normal appearing and a tumor-free zone, known as a “Grenz zone,” may be present between tumor and epidermis in early lesions. The peripheral margins may have a deceptively bland appearance resembling normal collagen and making interpretation of tumor margins difficult.297 Congenital DFSP also has typical histologic features of DFSP, however lesions from younger patients can be difficult to interpret.321,323,324 Histologic variants of DFSP include those that contain interstitial mucin (myxoid DFSP) or pigmented dendritic cells, also known as Bednar tumor.325–328 This variant tends to occur more often in dark-skinned persons and accounts for approximately 1% to 5% of all cases of DFSPs.325 Despite its characteristic features on hematoxylin and eosin (H&E), immunohistochemical staining may be necessary to help differentiate DFSP from other fibrohistiocytic proliferations. CD34 is a surface antigen often expressed on normal hematopoietic stem cells, interstitial dendritic cells in the dermis, as well as peri-eccrine and perifollicular cells. Spindle cells in DFSP usually stain positive for CD34. The sensitivity of this marker in diagnosing DFSP ranges from 84% to 100%.329,330 DFSP stains negative for factor XIIIa, a key feature differentiating it from dermatofibroma. In addition, it stains negative for S100, which can be used to distinguish it from nerve tumors and melanoma. Although CD34 is a useful diagnostic marker, it is worth noting that in the fibrosarcomatous variant of DFSP, there can be loss of CD34 positivity in approximately 50% of cases.331
Clinical Manifestations/Patient Evaluation/Staging
DFSP usually manifests as a flesh-colored, firm, asymptomatic nodule or plaque (Fig. 70-12). The tumors can also be violaceous or red-brown, especially along the margins at an early stage. As the tumor enlarges, it becomes firm to palpation and nodular. The nodular component is fixed to the dermis initially, but progressive fixation to the deeper structure can be observed late in the course of the disease.332 The majority of DFSPs exhibits an indolent growth pattern and are often diagnosed after being present for years. The tumor size at diagnosis typically ranges from 1 to 5 cm, but neglected lesions can be as large as 20 cm in diameter.295,297,306 Untreated DFSP can enlarge in a relentless manner with accelerated growth that results in nodularity or multiple outcroppings or protuberances. Ulceration occurs when the tumor erodes through the epidermis. Atypical clinical manifestations include sclerotic plaques with a morpheaform appearance.213 Lesions associated with pigment are known as Bednar tumors.325
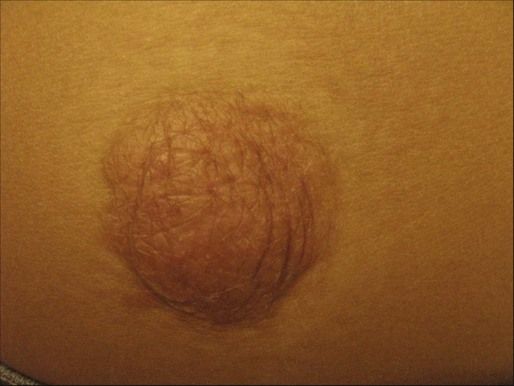
Approximately 50% to 60% of DFSP lesions occur on the trunk, 25% of those occurring on the chest and shoulders.296,297,300,306 Approximately 20% arise on the proximal aspects of the upper or lower extremities,297–300 and 10% to 15% occur on the head and neck, with the scalp being affected less than 5% of the time.296,297,300,306 In rare instances, acral sites have been reported, mostly in young children and adolescents.296,300,301,326 Often there is a delay in diagnosis given the indolent growth pattern, rarity and nonspecific features of the tumor. DFSP often is mistaken for lipoma, deep-seated epidermal cyst, scar, hypertrophic scar, keloid, dermatofibroma, nodular fasciitis, as well as infantile fibromatosis and vascular tumor in young children. The diagnosis is usually made via histologic evaluation of biopsied tissue.
Once the diagnosis of DFSP is confirmed, a complete history, review of systems, and physical examination with focus on the skin and structures contiguous with the tumor and palpation of lymph node basins should be performed. Because local or systemic metastases are extremely rare, neither prophylactic lymph node dissection nor extensive laboratory or imaging studies are indicated. Magnetic resonance imaging (MRI) is useful to determine the depth of the tumor if it is large and/or unresectable. CT is usually not indicated unless there is concern about underlying bone involvement. Although it is locally aggressive, DFSP rarely metastasizes. When pulmonary metastasis is suspected, a preoperative chest x-ray or chest CT should be considered. The overall rate of distant metastasis is approximately 5% and of regional metastasis is 1%.305 In the past, higher rates of metastasis were partly a result of inclusion of malignant fibrohistiocytoma (MFH) in DFSP case series.335–335 MFH is now understood to be a totally different entity from DFSP.
Similar to other soft-tissue sarcomas, most clinical staging of DFSP or DFSP-FS is based on the staging system established by the American Musculoskeletal Tumor society (MSTS) according to histologic grade and anatomic confinement of the tumor (Table 70-4).336 A more recent classification system in 2010 was developed by the AJCC/International Union Against Cancer (IUAC) based on a staging system for soft-tissue sarcomas.337 The system stratified the primary tumor stage as equal to or smaller than 5 cm versus larger than 5 cm in size, but discounted the possibility of even greater increased risk of recurrence for tumors larger than 10 cm in size.
Table 70-4
DFSP Staging System Based on American Musculoskeletal Tumor Society (MSTS) Staging System
| Stage IA | Low-grade DFSP or intermediate-grade DFSP-FS that is within the cutaneous/subcutaneous compartment and can be managed adequately by a wide excision alone (i.e., dissection outside of the reactive zone) |
| Stage IB | Low-grade DFSP or intermediate-grade DFSP-FS that has extended outside of an anatomic compartment (i.e., into underlying fascia or muscle) |
| Stage II | High-grade histology |
| Stage III | Local lymph node involvement |
From American Joint Committee on Cancer and National Comprehensive Cancer Network have not set forth a staging system specifically for DFSP.
Primary Therapy
The traditional mainstay of therapy for DFSP has been surgical resection, specifically wide local excision with a margin of 2 to 4 cm of normal-appearing skin as recommended by the NCCN.306,338,339 Unfortunately, the biological and histologic features of the tumor coupled with the inability to clinically determine tumor margins account for exceptionally high recurrence rates when using standard surgical therapy. Reported local recurrence rates are as high as 60% after standard surgical excision and 4% to 40% after wide local excision.338,340–342 Approximately 80% of local recurrences occur within 3 years.295,306,314 Local recurrence after 10 years is rare but has been reported.297,300 Although some authors believe that wide excision with histologic microscopic margin clearance may be effective,343 achieving adequate margins may not often be feasible, particularly in children or young adults where functional disability, gross mutilation and unacceptable cosmetic outcomes are more likely. Mohs micrographic surgery (MMS) has been receiving greater recognition as a useful method to manage DFSP.338,339,344–351 The capability of microscopic examination of the entire specimen margins allows the surgeon to localize and excise the tumor in a precise manner. This method also allows for maximum conservation of tissue, a crucial factor in certain anatomic areas, such as the head, neck, distal extremities, and genitalia. A review of several recent published studies showed that more than 50% of cases treated with MMS required surgical margins less than 1.5 cm.340 Results of several studies have suggested that MMS may be beneficial in the management of DFSP; however, few series have provided complete 5-year follow-up data.338,339,344–351 To date, there is no randomized study demonstrating the superiority of MMS over wide local excision. Some argue that tissue processing using frozen sections in standard MMS makes it difficult to accurately define histologic margins. Therefore, modified MMS has been employed by many. The technique involves resection of the tumor followed by processing the specimen using standard permanent fixation and evaluation. This technique allows the use of immunohistochemical stains to help define the tumor margin, as it is often difficult to distinguish the lesion from scar tissue on frozen section.
Locally Advanced Disease and Palliation
In cases when local recurrence occurs, the tumors have significant propensity for deeper invasion into fascia, muscle, or bone, which makes complete resection even more difficult. Therefore, when surgical margins are positive, repeat surgery is usually recommended followed by adjuvant radiotherapy (RT). The therapy, however, should not be used as primary treatment or a substitute for inadequate surgical resection. Recently, imatinib mesylate (Gleevec), a small-molecule tyrosine kinase inhibitor that inhibits platelet-derived growth factor receptors (PDGFRs), has shown promise for unresectable DFSP.334,352 There is evidence suggesting that patients with confirmed t(17;22) gene translocation benefit from imatinib.343,353 Current NCCN guidelines suggest molecular analysis using cytogenetics before consideration of imatinib therapy. Imatinib has been approved in the United States and Europe for use in adult patients who have unresectable, recurrent and/or metastatic DFSP. The broad indication allows imatinib to be used as both neoadjuvant and adjuvant therapy as an alternative to radiotherapy. There is minimal evidence supporting a role for conventional chemotherapy in the treatment of DFSP.
Cutaneous Angiosarcoma
Cutaneous angiosarcoma (CAS) is a rare, extremely malignant tumor originating from endothelial cells of vascular or lymphatic differentiation.354
Epidemiology
CAS accounts for less than 1% of all soft-tissue sarcomas. SEER data show that CAS has an equal distribution between sexes, occurs mostly in whites (88%), with a mean age of 73 years, and seldom in African Americans (4%). CAS arises predominantly in the head and neck areas (62%), followed by the trunk (24%).355
Etiology and Biological Characteristics
The exact etiology of CAS remains largely unclear. In 1948, Stewart-Treves reported an association between angiosarcoma and postmastectomy lymphedema.356 Since then, chronic lymphedema of any origin, such as postsurgery, Milroy disease, or filariasis, has become a well-recognized risk factor for the development of CAS.354 RT is another independent risk factor.357,358 Other proposed associations include a history of trauma,359 herpes zoster,360 chronic osteomyelitis,361 foreign materials,361 and arteriovenous fistula sites in renal transplant recipients.362,363 Chemical carcinogens, such as vinyl chloride and arsenic, are associated with hepatic angiosarcoma.364,365 UV light exposure does not seem to be an inciting factor for CAS. Although angiosarcomas are reported in renal transplant recipients, the role of immunosuppression to the pathogenesis of angiosarcoma is uncertain.354
Efforts have been made to explore the etiology of angiosarcoma at the molecular biology level. Reported findings include overexpression of vascular endothelial growth factor (VEGF) and its receptors, mutations in TP53, overexpression of Wilms tumor-1 and Galectin-3, activation of signaling pathways including RAS and PI3-kinase signaling, and overexpression of the transcription factor ETS1.354
Pathology and Pathways of Spread
Although CAS is highly aggressive, its histologic features can be subtle, making it difficult to differentiate from a benign proliferative or inflammatory lesion under light microscopy.354 In general, tumors appear poorly circumscribed and infiltrative, with a range of differentiation within a single lesion, which can range from a well-differentiated, angiomatous pattern, to a poorly differentiated or even highly anaplastic, solid pattern. The pathognomonic findings are abnormal, pleomorphic, hyperchromatic, malignant endothelial cells that can be rounded, polygonal, or fusiform, and can have an epithelioid appearance.354,366,367 The well-differentiated, angiomatous pattern is characterized by anastomosing, functioning vascular sinusoids continuous with normal vascular channels, dissecting between dermal collagen bundles, around but usually not invading skin appendages, and often with lymphocytic infiltrates. The vessels are irregular and lined by atypical endothelial cells, which can range from a nearly normal, single endothelial-cell lining to more aggressive, multilayered, papillarylike projections into irregular vascular lumens. In the solid pattern, poorly differentiated tumors contain the malignant endothelial cells that are arranged in solid sheets or cords, with areas of hemorrhage and necrosis, and invading the dermis and underlying tissue. Those poorly differentiated or anaplastic tumors can resemble carcinoma or amelanotic melanoma.354,366
Immunohistochemical stains are helpful in diagnosis of CAS. CAS typically expresses endothelial markers including von Willebrand factor, CD34, CD31, Ulex europaeus agglutinin 1, and VEGF. von Willebrand factor, Ulex europaeus agglutinin 1, and CD31 are the most useful markers in poorly differentiated cases.368 Negative cytokeratin staining is generally helpful for differentiating CAS from carcinoma; however, epithelioid angiosarcomas can also express cytokeratins.369,370 The negative staining for melanocytic markers, such as S100 and human melanoma black-45, can help to distinguish CAS from melanoma. It has been shown that high-level amplifications of MYC are seen in postradiation CAS, in contrast to the absence of MYC in other postradiation atypical vascular lesions. Therefore, immunohistochemical stainings for MYC may be useful for mapping of postradiation CAS lesions and for careful tumor margin control.371
Clinical Manifestations/Patient Evaluation/Staging
There are three main clinical variants of CAS: (a) idiopathic CAS of the scalp and face, (b) CAS associated with chronic lymphedema, most commonly following mastectomy (Stewart-Treves syndrome), and (c) postradiation angiosarcoma, which is the least common variant.372,373 CAS of the scalp and face subtype occurs more on the scalp (50%) than the face or neck (30%).374 It usually presents as a painless, rapidly proliferating growth of the subcutaneous tissue plane (Fig. 70-13). The tumor is usually smooth on surface, firm or spongy on palpation, and often shows an ill-defined area of dusky to violaceous erythema resembling a bruise or healing contusion. The tumor infiltrates the surrounding structure in a centrifugal and multifocal fashion, which may result in a much larger subclinical extension than the apparent clinical margins, and multiple foci separated by an isthmus of normal-appearing skin. Long-standing CAS may develop superimposed tumors and nodules. Ulceration and bleeding are not characteristic unless the CAS is larger than 10 cm in diameter. A significant number of patients with CAS are associated with another primary malignant neoplasm either before or after the development of the angiosarcoma.355 CAS can be mistaken for a benign lesion given the subtlety and nondescript nature of the clinical findings. Initial differential diagnoses include cellulitis, nonspecific inflammatory changes, fungal infection, trauma, hemangioma, cutaneous lymphoma, and pseudolymphoma.366,373,375,376 Reported atypical clinical scenarios include rosacealike signs and symptoms, chronic edema of the eyelids, xanthelasmalike lesions, and even recurrent angioedema.378–378
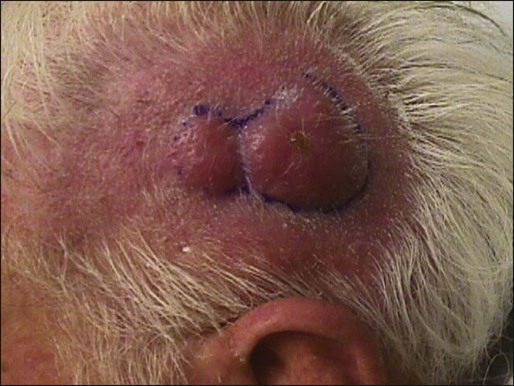
Survival rates of CAS correlate with age, anatomical site, and stage of disease. Patients younger than 50 years with tumors arising in the trunk and localized to the skin have the best prognosis.355 The most important prognostic factors include tumor size and ability to achieve negative margins, with tumor grade also being considered to be a prognostic factor for recurrence and overall survival.381–381 Favorable outcomes are more likely when the tumor (a) has been present for less than 6 months, (b) is less than 5 cm in size, and (c) is amenable to combined surgery and radiation therapy (e.g., CAS of the nose).374
Initial patient evaluation includes biopsy to confirm histology. The peripheral margin of CAS may be partially assessed with random biopsies performed in a gridlike pattern circumferentially around the tumor.374 Ideally, the biopsies should be full thickness of the skin and include the muscle and fascia if possible for complete assessment of the depth of the margin. MRI is often used to delineate the extent of the primary lesion before surgery, however, CT and MRI have not been extremely helpful unless there has been marked bony invasion.382,383 Soft-tissue invasion of the tumor may be partially visualized, but the exact extent of the margins is poorly delineated as a result of the diffuse infiltrating nature of the tumor. Although angiosarcomas can spread through the lymphatic system, the value of sentinel lymph node biopsy has not been established.354
Soft-tissue sarcoma, including deep angiosarcoma, is staged using the IUAC/AJCC system. This is based on the TNM staging system, with an additional notation for histologic grade.67 However, cutaneous angiosarcoma may be difficult to stage using the AJCC system.384
Primary Therapy
Because of its rapidly progressive, infiltrative and multifocal nature, achieving local control and prevention of metastasis in CAS has been a challenge. In some series, survival rates were only 10% to 20% after 5 years.373,375,379,380,385,386 Given the rarity of angiosarcoma, no single institution has enough experience to establish definitive treatment guidelines. Radical surgery attempting for complete resection is considered the primary treatment of choice. Wide excision margins of 3 to 6 cm or more are recommended; however, complete excision is often difficult to achieve because of diffuse tissue infiltration and tumor location.354 Mohs micrographic surgery with adjuvant radiation has been shown to produce a favorable outcome.374,387,388
Postoperative adjuvant RT, with large doses (>50 Gy) and wide treatment fields, is employed routinely as it improves local control and overall survival,375,379,389 although there is still an extremely high rate of recurrence and local failure.381 Radiation treatment alone is generally thought to be inadequate treatment for potentially curable disease, and further RT is usually avoided for radiation-induced angiosarcomas. The role of chemotherapy in the neoadjuvant or adjuvant setting after definitive surgery and RT is not well established and has not been proven to increase survival.381,390,391
Locally Advanced Disease and Palliation
Locally advanced lesions are managed by wide local excision and adjuvant RT. Additional surgery in locally recurrent disease to achieve a pathological complete resection may improve survival.392 Management of regional lymph node metastasis is surgical resection and adjuvant radiation.374 When the lesion is too extensive for surgery or when the patient is a poor surgical candidate, VEGF-A monoclonal antibody, bevacizumab might be considered. There is a single case report of scalp CAS in which remission was achieved after treatment with bevacizumab and RT without surgical intervention.393 Bevacizumab has also been used in conjunction with RT as postsurgical treatment.394
Treatment of Metastatic Disease
Cytotoxic chemotherapy is the primary treatment option for metastatic angiosarcoma, although there is no convincing evidence to show survival benefit. The main drug groups used in CAS are paclitaxel, docetaxel, pegylated liposomal doxorubicin, and liposomal daunorubicin.354,393 The taxanes paclitaxel and docetaxel have antiangiogenic activity and are therefore of particular interest in the management of angiosarcomas.395
Controversies/Problems/Challenges/Future Possibilities and Clinical Trials
Further prospective studies are necessary to clarify treatment strategies for CAS. Antiangiogenic biological molecules are promising candidates used in conjunction with surgery and RT. Examples include VEGF-A monoclonal antibody, bevacizumab; broad-spectrum tyrosine-kinase inhibitors, sorafenib; VEGFR-inhibitor, axitinib; vascular disrupting compound, combretastatin; and thalidomide, owing to its antiangiogenic properties. Immune modulators, interferon-α or interleukin-2 have also been tried in limited cases.354