Myelodysplastic Syndromes
David P. Steensma and Richard M. Stone
• The myelodysplastic syndromes (MDS) are a group of clonal marrow failure syndromes originating in a hematopoietic progenitor or stem cell. Approximately 25% to 30% of MDS cases progress to acute myeloid leukemia (AML), which is defined by 20% or more marrow blasts.
• Approximately 50% of MDS cases are associated with a karyotypic abnormality, usually chromosomal aneuploidy.
• Recurrent MDS-associated somatic mutations are now recognized in more than 25 genes. Some of these mutations have prognostic value independent of existing risk stratification tools.
• The most important risk factor for MDS is aging. The risk of developing MDS increases greatly after age 65 years. The median age at diagnosis in the United States is approximately 71 years.
• Approximately 10% to 15% of MDS cases arise as a consequence of therapeutic or environmental exposure to a DNA damaging agent; these are termed secondary or therapy-related MDS. In patients who had been exposed to ionizing radiation or alkylating agents, abnormalities of chromosomes 5 and 7 are common. Patients treated with topoisomerase II inhibitors may develop rearrangements of the MLL gene at 11q23 or the MECOM (MDS1/EVI1) locus on chromosome 3q21q26.
• Patients who develop MDS before age 40 years without a recognized toxin exposure may have a germline DNA repair defect or congenital marrow failure syndrome, such as Fanconi anemia or dyskeratosis congenita. Chromosome breakage analysis and telomere length analysis is appropriate in such patients.
• The 2008 World Health Organization (WHO) classification of MDS, refined from the 2001 WHO and 1982 French–American–British (FAB) MDS classifications, defines several subtypes of MDS with varying risk of AML progression or death. The specific diagnosis depends on the proportion of marrow blasts, the number of cell lineages involved by disease, and the presence or absence of ring sideroblasts or chromosome 5q deletion.
• Minimal diagnostic criteria for MDS (after exclusion of other causes of cytopenias) include either: 10% or more dysplastic cells in one or more myeloid lineages, increased marrow blasts (≥5%), or the presence of a cytogenetic abnormality or another marker of clonal hematopoiesis.
• The incidence of MDS is at least 4 cases per 100,000 persons per year in the United States. Most patients have lower-risk disease at the time of initial diagnosis.
• Not all marrow dysplasia represents MDS. Other causes of cytopenias and abnormal cell morphology must be considered in the initial evaluation of patients, such as nutritional deficiency (e.g., vitamin B12, folate, copper), inflammation, excessive alcohol use, human immunodeficiency virus infection, and non-MDS neoplasms.
• In ambiguous cases, the presence of an MDS-associated cytogenetic abnormality, such as deletion of chromosome 5q, can confirm the diagnosis.
• Several prognostic tools, including the 2012 Revised International Prognostic Scoring System (IPSS-R), are in widespread use and aid in risk stratification and therapy choice. In general, older patients with higher blast proportion, more severe cytopenias, and higher-risk cytogenetic results have a poorer outlook.
• Complications of peripheral blood cytopenias and functional cell defects, including infection and hemorrhage, are the most common causes of death in MDS. Progression to AML following MDS occurs in 25% to 30% of cases and is usually fatal.
• In lower-risk cases, supportive care alone may be appropriate. Epoetin or darbepoetin may alleviate MDS-associated anemia.
• Anemia of MDS associated with chromosome 5q deletion frequently responds to lenalidomide. Thrombocytopenic patients respond less well.
• Chronic red blood cell transfusion can lead to iron overload and is an adverse prognostic marker. The appropriate use of iron chelation therapy in MDS is controversial.
• Azacitidine has been demonstrated to improve survival compared with supportive care in higher-risk MDS. Another DNA methyltransferase inhibitor, decitabine, can also induce hematologic and cytogenetic responses.
• Allogeneic stem cell transplantation is the only potentially curative therapy, and should be considered for patients with higher-risk MDS who are younger than age 75 years and have a matched donor.
Second- or Third-Line Therapies
• Once azacitidine or decitabine fail the patient, or if relapse occurs after a stem cell transplantation, the outlook is very poor, with a median survival less than 6 months. There is currently no established second-line therapy for MDS. Clinical trial enrollment is especially important for this patient group.
Introduction
The term myelodysplastic syndromes (MDS) describes a heterogeneous group of acquired bone marrow failure syndromes that are collectively characterized by clonally restricted and ineffective hematopoiesis.1 MDS-associated ineffective hematopoiesis results in peripheral blood cytopenias, despite a bone marrow that is frequently normocellular or hypercellular when compared with age-matched controls. In addition, MDS carry a variable risk of progression to acute myeloid leukemia (AML), which is currently defined by the World Health Organization (WHO) as the presence in the marrow of 20% or greater blast cells with myeloid lineage markers—that is, immature, neoplastic cells that express myeloid-associated cell surface antigens—that are positive cytochemically for myeloperoxidase, Sudan black, or nonspecific esterase, or which express AML-associated chromosomal or molecular abnormalities.2
History and Terminology
MDS are a relatively recently identified group of medical conditions, and the terminology used to describe these disorders has historically been varied and at times confusing, including nonspecific eponyms such as “di Guglielmo syndrome,” which was once used to describe any erythroid-dominant myeloproliferation.3,4 A detailed history of the evolving concept of MDS and key discoveries contributing to the current state of understanding was recently published.5
In 1938, Rhoads and Barker in New York described 100 patients with anemia who failed to improve during therapy with hematinics such as iron salts or liver extract.6 These patients were described as having “refractory anemia”; a few of them may have had MDS. The term refractory anemia, although coined more than 7 decades ago and in a different context, persists to the present day, even though the anemia of MDS is, fortunately, not always “refractory” to contemporary therapies. A link between a cytopenic prodrome and subsequent development of overt AML was established by French investigators, including Chevalier in the early 1940s and this association was highlighted by Block and Jacobsen in Chicago in their landmark description of “preleukemic acute human leukemia” in 1953.7,8 Björkman in Sweden described acquired sideroblastic leukemia in 1956, which would later become understood as another form of MDS, while by the 1960s chronic myelomonocytic leukemia (CMML) was recognized as a potential precursor state for acute leukemias with monocytic differentiation.9,10
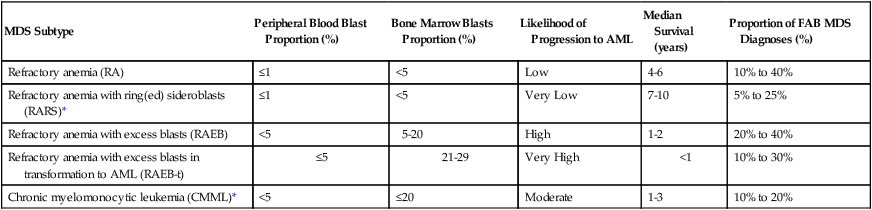
In 1973, Saarni and Linman published the first case series of 143 patients with MDS, which was then termed “preleukemia.”11 By the time of the first formal classification of the acute leukemias by the French–American–British (FAB) Co-Operative Group (in 1976, 3 years after Saarni and Linman’s report), the term “preleukemia” had begun to fall out of favor, as most patients with “preleukemia” never went on to develop leukemia, yet were still at risk for death from complications of disease-associated cytopenias and functional neutrophil or platelet defects.12,13 After a brief vogue for the term “dysmyelopoietic syndromes,” the seminal 1982 FAB classification (Table 99-1), described further below, used the term “MDS,” a moniker that has been with us ever since.14
Table 99-1
French–American–British (FAB) Classification of Myelodysplastic Syndromes (1982)
| MDS Subtype | Peripheral Blood Blast Proportion (%) | Bone Marrow Blasts Proportion (%) | Likelihood of Progression to AML | Median Survival (years) | Proportion of FAB MDS Diagnoses (%) |
| Refractory anemia (RA) | ≤1 | <5 | Low | 4-6 | 10% to 40% |
| Refractory anemia with ring(ed) sideroblasts (RARS)* | ≤1 | <5 | Very Low | 7-10 | 5% to 25% |
| Refractory anemia with excess blasts (RAEB) | <5 | 5-20 | High | 1-2 | 20% to 40% |
| Refractory anemia with excess blasts in transformation to AML (RAEB-t) | ≤5 | 21-29 | Very High | <1 | 10% to 30% |
| Chronic myelomonocytic leukemia (CMML)* | <5 | ≤20 | Moderate | 1-3 | 10% to 20% |
AML, acute myeloid leukemia; MDS, myelodysplastic syndrome.
*In the FAB classification, RARS is additionally defined by ≥15% ring sideroblasts among erythroid precursors, and CMML is additionally defined by ≥1 × 109/L peripheral blood monocytes.
Adapted from Bennett JM, Catovsky D, Daniel M-T, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol 1982;51:189–199.
Although the terminology used by clinicians and pathologists to describe MDS is more consistent than it was 30 years ago, confusion about the precise biological nature of MDS continues. Even though MDS are clearly neoplastic—numerous AML-associated acquired genetic mutations are present in MDS cells, and most of the hematopoietic cells are clonal at the time of MDS diagnosis, long before progression to leukemia15,16—some doctors continue to describe MDS to their patients as peculiar forms of anemia or as marrow failure syndromes without using the word “cancer,” and cancer-specific insurance policies routinely exclude coverage for MDS diagnoses.17,18 The Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute and its associated cancer registries began formally tracking MDS only in 2001.19,20
Diagnosis of MDS can be difficult; choice of therapy, even more so.21 Patient outcomes also vary widely, with the highest-risk patients with MDS having a median survival of only a few months, while those afflicted with the most indolent forms of disease are likely to live for a decade or more.22 (Increasing recognition of this diversity in clinical course and accompanying biological heterogeneity accounts for why the singular usage “myelodysplastic syndrome” is steadily shifting to the plural term, “syndromes.”) Therefore diagnosis and subsequent care of the patient require coordination among a pathologist with morphologic expertise who can diagnose the condition with confidence if characteristic cellular changes are present; a cytogeneticist or molecular pathologist who can reliably identify typical subcellular and genetic abnormalities associated with MDS; a clinical hematologist-oncologist able to assess the patient’s risk and individualize therapy; and, in some cases, a multidisciplinary team experienced in bone marrow and stem cell transplantation.
Epidemiology and Etiology
General Observations
The incidence of MDS has proven difficult to estimate accurately.23 In the United States, early SEER data following initial inclusion of MDS in the program in 2001 suggested an incidence of approximately 3.3 to 3.6 cases per 100,000 people per year in the general population, and 20 cases per 100,000 persons in individuals older than age 65 years.19,20 These SEER data also indicated that the incidence of MDS may be increasing over time, either because of greater recognition of these diseases or a genuine increase in disease frequency. Based on these United States registry data, it has been estimated that approximately 10,000 to 15,000 patients are diagnosed with MDS in the United States each year.19,20
However, a study of Medicare claims data (Medicare is a U.S. government health care payer) suggested a much higher incidence of MDS, approximately 75 cases per 100,000 persons older than age 65 years.24 Although this analysis may be confounded by the use of MDS codes on claims in order to justify use of certain therapies for incompletely evaluated cytopenic patients, if the Medicare data are accurate, more than 45,000 patients in the United States are diagnosed with MDS each year, and the prevalence of these diseases in the United States exceeds 100,000 persons, placing MDS among the most common hematologic malignancies.25 The incidence of MDS rises sharply after the age of 70 years, and physician surveys indicate that the median age at diagnosis in the United States is approximately 70 to 72 years.26
The United States data described above are consistent with reports from other Western countries, including Germany, Sweden, the United Kingdom, and France.29–29 In contrast, Eastern European and Asian patients are significantly younger at the time of MDS diagnosis, and also present with different distributions of MDS subtypes, compared to patients in Western countries.30–33 This observation suggests that the natural history of MDS varies in different regions, either because of background genetics of the population or distinct environmental exposures.
MDS are more common in men than in women, possibly related to past patterns of occupational exposures.19,20 However, a specific subtype of MDS associated with isolated deletion of the long arm of chromosome 5 [del(5q)] and marrow morphology, including hypolobated megakaryocytes, the so-called “5q− syndrome,” is more common in women than in men.34 Aging is by far the most important risk factor for development of MDS, likely because of the progressive accumulation of somatic mutations of hematopoietic stem cells across the human life span.35 Eventually, as such somatic mosaicism emerges, certain patterns of mutation may give hematopoietic cells a growth and survival advantage, and the expanded clone of cells is then at risk for acquiring additional mutations that augment its malignant potential.36
Environmental and Occupational Exposures
The risk of developing MDS is increased after exposure to organic solvents and hydrocarbons, including benzene, likely as a result of acceleration of DNA mutagenesis by such agents.39–39 Even low-level occupational benzene exposure has been associated with hematologic toxicity.38 Polymorphisms in DNA repair enzymes or of the detoxification enzyme NADPH : quinine oxidoreductase (NQO1) may alter patients’ sensitivity to DNA damaging chemicals.40,41
Other exposures that are encountered more commonly by the general population than benzene may also influence MDS risk.42 For instance, case-control studies suggest an increased risk of MDS in association with family history of hematopoietic cancer, tobacco smoking, and exposure to potentially harmful chemicals (i.e., agricultural chemicals, petroleum products, hair dyes or solvents), while higher educational attainment or moderate consumption of wine may be protective.43–46 Compared with de novo MDS, secondary MDS related to an occupational or environmental exposure is more closely associated with an abnormal karyotype.46
Familial Myelodysplastic Syndromes
In general, families of patients with MDS can be reassured that their risk for developing MDS is minimally increased compared to the general population. However, rare cases of familial MDS and AML have been reported,47 particularly in patients with germline RUNX1 mutations, who usually have a prodrome of thrombocytopenia.48 A rare granulocyte colony-stimulating factor (G-CSF) receptor polymorphism (CSF3R gene) also may predispose affected persons to high-risk MDS.49 Germline mutations in CEBPA are another cause of hereditary AML (albeit usually without an MDS prodrome), underscoring the genetic heterogeneity of familial MDS and AML.50,51 In a few families, MDS or AML with monosomy 7 has been reported in multiple individuals in the absence of either phenotypic abnormalities or any history of hematologic disorders. The genetic cause for this is unknown, although childhood monosomy 7 mosaic syndrome predisposes to MDS.52 Other families with recurrent MDS/AML and still-mysterious genetics exhibit a distinct gene expression pattern in affected versus unaffected family members, particularly in genes encoding factors involved in signal transduction.53 Recently, germline mutations in the GATA2 transcription factor were described in association with MDS, monocytopenia, and a risk of mycobacterial infection or lymphedema (“MonoMAC syndrome”).54,55
Therapy-Related Myelodysplastic Syndromes
MDS may be idiopathic and arise de novo, or may instead be secondary to a recognized exposure to a DNA-damaging agent.56–60 So-called secondary or therapy-related MDS (t-MDS) represents approximately 10% to 15% of MDS cases overall, and has a similar poor prognosis to secondary AML (t-AML), such that the WHO classes t-MDS and t-AML together as a single disorder.61
t-MDS/AML has been linked to several classes of exposure, which trigger disease via distinct genetic pathways.62 Common causes of t-MDS/AML include treatment with drugs that alkylate DNA bases (e.g., chlorambucil, melphalan, cyclophosphamide), therapeutic or accidental exposure to ionizing radiation, treatment with inhibitors of topoisomerase II (e.g., etoposide, or anthracyclines such as doxorubicin), or environmental or occupational exposure to other miscellaneous DNA toxins such as kerosene or benzene.46 Combined exposures, such as the combination of radiation therapy with chemotherapy (particularly with alkylating agents), increases the risk of t-MDS/AML compared with single exposures. MDS has also has been reported after exposure to fludarabine.63,64
MDS may rarely evolve from another hematologic disorder, particularly polycythemia vera after treatment with radiophosphorus or busulfan.65 Patients’ sensitivity to DNA damaging agents and inherent risk for t-MDS/AML after an exposure may vary. For instance, specific polymorphisms in the methylene tetrahydrofolate reductase (MTHFR) gene, encoding a protein critical for DNA synthesis, are associated with a higher risk of t-MDS/AML after treatment with cyclophosphamide, particularly in patients with breast cancer.66
The median latency period for t-MDS/AML after exposure to alkylating agents is 5 to 7 years, and the risk appears to be dose-dependent.67,68 In general, t-MDS/AML after alkylating agent or radiation exposure is characterized by deletions or unbalanced translocations, commonly involving chromosome 5 (particularly 5q31) or chromosome 7.69 By contrast, t-MDS or AML after exposure to topoisomerase inhibitors is distinct from that following alkylating agents in several ways: these cases are much less common than alkylating agent or radiation-induced t-MDS/AML; the latency period is shorter (median: 1 to 3 years); disease more frequently manifests with overt leukemia without a preceding MDS phase; a balanced translocation, often involving the MLL gene at 11q23 or the MECOM locus at 3q21q26, is more likely than chromosomal deletion or aneuploidy; and the complete response rate to antileukemia therapies may be higher, although the long-term prognosis is still poor.70,71
It can be challenging in some cases to prove a connection between a suspect exposure and subsequent development of MDS or AML, but the presence of a complex karyotype (i.e., three or more acquired chromosome abnormalities) or abnormalities of chromosomes 5 and 7 or 11q23 suggests t-MDS/AML. The predisposing exposure does not need to be recent. Even more than 50 years after the 1945 atomic weapon discharge over Nagasaki, for instance, the incidence of MDS continued to be higher in exposed persons than in unexposed controls.33
Stem Cell Transplantation and Granulocyte Colony-Stimulating Factor Contributing to Myelodysplastic Syndromes
High-dose chemotherapy and autologous stem cell transplantation (SCT) represent a strong risk factor for subsequent development of t-MDS/AML. For instance, after autologous SCT for malignant lymphoma, the risk of t-MDS/AML at long-term follow-up in some series exceeds 10%, although the use of non–total-body irradiation-containing conditioning regimens may reduce this risk to some degree.72–76 The nature of pretransplantation therapy influences the risk of t-MDS/AML after autologous SCT.77,78 In one study, the risk of MDS after radioimmunotherapy with 131I tositumomab for lymphoma did not appear to be increased above the risk for those who had not received radioimmunotherapy.79
There is also a substantial risk of MDS after allogeneic SCT, but because the most common indications for allogeneic SCT in the United States are MDS and AML and related myeloid neoplasms, the contribution of the procedure itself to subsequent development of “t-MDS/AML” is often difficult to determine. However, cases of posttransplant “relapse” of MDS have been reported with different karyotypes from pretransplant disease, suggesting that they may represent a different (and in some cases donor-derived) dominant clone from the original disease.80
The use of G-CSF in children with congenital neutropenia may increase the incidence of secondary MDS and AML, but this remains controversial, because congenital neutropenia carries an inherent risk of MDS, and without the use of G-CSF many children would die of infection early in life and would not live long enough to develop MDS.83–83 Similarly, an increased incidence of t-MDS/AML was reported in studies of long-term maintenance therapy with lenalidomide for multiple myeloma, but again the causality is unclear, and lenalidomide may improve myeloma survival allowing more time for secondary neoplasms to develop.84 t-MDS/AML has also been reported as a complication following dose dense breast cancer therapy that includes G-CSF support.59
Pediatric Myelodysplastic Syndromes
MDS are rare in children and represent only approximately 5% of hematologic malignancies in patients younger than age 18 years.85,86 When MDS do arise in children, they are usually either secondary to prior treatment of a neoplasm, or are associated with a congenital marrow failure syndrome or germline DNA repair defect, such as Li-Fraumeni syndrome (TP53 germline mutation), Shwachman-Bodian-Diamond syndrome (SBDS germline mutation), or Bloom syndrome (BLM1 germline mutation).87–91 Children with the marrow failure syndrome Fanconi anemia (>13 genes responsible) are at markedly increased risk of developing MDS, and because up to one-third of Fanconi anemia patients have no physical stigmata, the possibility of Fanconi anemia should be considered in all patients diagnosed with MDS before age 40 years without an obvious predisposing cause.89 Dyskeratosis congenita, a congenital marrow failure syndrome caused by germline mutations in genes encoding proteins responsible for telomere maintenance (e.g., DKC1, TERT and TERC), carries a higher risk of aplastic anemia than MDS, though acquired MDS often have altered telomere dynamics.90,92
Children who develop MDS without excess blasts but who appear to lack a predisposing congenital syndrome are provisionally classified by the WHO (see “Therapy: Immunosuppressive Therapy”) as having “refractory cytopenia of childhood” (MDS-RCC).93 Prognostic tools and classification systems useful for adult MDS often perform poorly in pediatric subgroups.96–96
The bone marrow in pediatric MDS is often hypocellular, rather than the hypercellular marrow typical in adults, and there is also a high incidence of unfavorable biological features such as monosomy 7 in childhood MDS.85 The MDS subtypes refractory anemia with ring sideroblasts (RARS) and 5q− syndrome are rare in children, but several types of congenital sideroblastic anemia can potentially be confused with MDS. These congenital syndromes include sideroblastic anemia caused by germline mutations of the X-linked gene ALAS2, which encodes the enzyme responsible for the first and rate-limiting step in heme biosynthesis. Germline ALAS2 mutations can also present late in life as a result of acquired skewing of X chromosome inactivation patterns and can be confused with RARS, but they do not carry a risk of progression to AML.97,98
Pathogenesis and Biology
Although historically there has been considerable confusion about the fundamental nature of MDS, since the WHO revised its classification of neoplastic disease in 1999, MDS have been clearly recognized as malignancies, that is, as forms of cancer.17,99 Hematopoiesis is clonal in MDS, and while clonality alone is not equivalent to malignancy, survival in patients with MDS is diminished compared with healthy controls, even in the lowest-risk cohorts.100
DNA damaging agents can predispose to MDS by accelerating mutation acquisition, and evolving idiopathic somatic mosaicism also puts patients at risk as described above, but the underlying cause of MDS is unknown in most cases, and there is no single unifying pathophysiology. Historically, MDS has been difficult to model in the laboratory, because there are no truly representative MDS cell lines and only a limited number of preclinical models—just a few recently constructed murine models, such as an NUP98-HOXD13 transgenic mouse and an interesting “stromal dysfunction” mouse that evolves an MDS-like syndrome after knockout of Dicer1 in osteoprogenitor cells rather than hematopoietic progenitors.101–104 Only in the present era of high-throughput resequencing has a clear picture of MDS pathogenesis and clonal architecture begun to emerge.16,105
Stem Cell Origin and Microenvironment
Despite the limitations of a lack of cell line or preclinical models, a functional model of MDS molecular pathogenesis is suggested by the clinical behavior of the disease—namely, the progressive transformation of a committed myeloid hematopoietic stem cell with acquisition of successive genetic abnormalities, and clonal expansion, leading to the “classic” clinical phenotype (i.e., hypercellular bone marrow with increased angiogenesis and apoptosis, abnormal cytogenetics, cytopenias, and transformation to higher-risk forms of disease including frank AML).108–108 Intramedullary apoptosis mediated by death receptors and their ligands, such as tumor necrosis factor-α, Fas, or TRAIL (tumor necrosis factor-related apoptosis-inducing ligand), may help explain the “paradox of MDS,” in which a hypercellular marrow is associated with peripheral blood cytopenias.111–111
In the subset of patients with 5q− syndrome, global gene expression profiling and other experiments indicate a stem cell origin for the disease, and the clonal “leukemic” stem cells (sortable via their CD34+, CD38 absent or low, CD90+ expression pattern) persist during therapy with lenalidomide and serve as a nidus for relapse.112,113 It seems highly likely that a stem cell origin is also applicable for other MDS subtypes. The marrow microenvironment may harbor such stem cells in a protective niche and support perpetuation of disease, and the stromal cells may themselves be defective or mutated.102,114,115
Expression Profiling and Point Mutations
Gene expression profiling experiments using complementary DNA microarray technology were the first high-throughput technique to demonstrate biological heterogeneity matching the clinical diversity of patients with MDS, but specific pathophysiological insights from gene expression profiling have been limited because of the difficulty in separating clones and in distinguishing primary “driver” and reactive “passenger” changes in gene expression.116–119 In patients with lower-risk MDS, array-based single-nucleotide polymorphism (SNP) analysis has demonstrated uniparental disomy and copy number changes, and the presence of such abnormalities in patients with a normal karyotype may portend a poorer prognosis.120–123 More recently, whole-genome DNA sequencing efforts focused on the architecture of hematopoiesis in MDS demonstrate that more than 80% of cells are clonal at the time of diagnosis and that in a darwinian process, clones compete for dominance in the marrow, with the hardiest clones surviving and ultimately contributing to leukemia.16
Most patients with MDS are now known to harbor multiple point mutations in coding regions of genes expressed in CD34+ cells at the time of diagnosis, and it seems likely that several mutations with complementary pathobiological function are required for the full clinical picture to emerge.15,28,124 Detection of the presence of these mutations can aid in clinical diagnosis in ambiguous cases.21
The first mutations described in MDS primary cells in 1987 were in the RAS G-protein–coupled signaling pathway.125,126 As of 2012, a quarter-century later, more than 25 different recurrent mutations are recognized, and several of these have prognostic significance independent of existing models.28 Common recurrent mutations (Table 99-2) involve factors important for cell proliferation such as receptor tyrosine kinases and their downstream mediators (e.g., NRAS, KRAS, BRAF, JAK2), pre–messenger RNA splicing (e.g., SF3B1, SRSF2, U2AF1), transcription and cell differentiation (e.g., RUNX1, WT1, PHF6), DNA damage repair (e.g., TP53), and especially epigenetic patterning (e.g., TET2, EZH2, DNMT3A, IDH1/2, ATRX).127–134 The functional consequences and details of interactions between these factors are only just beginning to be understood, and may depend on sequence of mutation acquisition and allele burden. Although a model of inactivation or mutation of tumor suppressor genes has been proposed as a contributor to MDS biology, and may account for the clinical response to DNA hypomethylating agents as described below, the principal tumor suppressors responsible for MDS, if they exist, have not yet been identified.135
Table 99-2
Somatic Mutations in Single Genes Detectable in MDS
| Recurrently Mutated Genes | Mutation Frequency in MDS | Prognostic Impact | Additional Notes |
| PRE-MRNA SPLICING FACTORS (COMPONENTS OF THE SPLICEOSOME) | |||
| SF3B1 | 15% to 20% | Neutral to favorable | Strongly associated with the presence of ring sideroblasts (75% to 80% of RARS cases); often co-occurs with DNMT3A mutations |
| U2AF1 | 10% to 15% | Neutral to adverse | Often co-occurs with del(20q) |
| SRSF2 | 10% to 15% | Adverse | More frequent in CMML (25% to 30%), often co-occurs with RUNX1 or ASXL1 mutations |
| ZRSR2 | 5% to 10% | Neutral to adverse | |
| U2AF2, SFRA1, PRPF40B, SF1 | ~5% | ? | |
| EPIGENETIC MODIFIERS (ALTER DNA METHYLATION OR HISTONE/CHROMATIN MODIFICATIONS) | |||
| TET2 | 20% | Neutral | More frequently observed in in CMML (~40%) |
| DNMT3A | 10% to 15% | Adverse | In AML, is associated with lack of chemoresponsiveness |
| IDH1, IDH2 | <5% | Variable | More frequent in AML than in MDS; when seen with NPM1 in AML, better prognosis |
| ASXL1 | 10% to 20% | Adverse | More frequent in CMML (~40%) |
| EZH2 | 6% | Adverse | More frequent in CMML (~12%) |
| KDM6A (UTX) | <1% | ? | Rare in MDS, more frequent in CMML |
| PHF6 | ? | ? | |
| ATRX | Rare | ? | Associated with acquired α−thalassemia and hemoglobin H inclusion bodies |
| TRANSCRIPTION FACTORS | |||
| RUNX1 | 10% to 15% | Adverse | Germline mutations cause familial platelet disorder with propensity to AML (FPD-AML); somatic mutations are associated with thrombocytopenia, more common in t-MDS/AML |
| ETV6 | <5% | Adverse | In AML, is part of t(12;21) translocation |
| MYBL2 | <1% | ? | |
| GATA2 | Rare | ? | Germline mutations cause familial MDS and other myeloid disorders |
| CEBPA | ? | ? | More frequently mutated in AML, germline mutations cause familial AML (typically without preceding dysplasia) |
| WT1 | ? | ? | More frequently mutated in AML |
| TP53 | 5% to 10% | Adverse | |
| TYROSINE KINASE SIGNALING | As a group, these genes are more often mutated in myeloid malignancies with a proliferative component | ||
| NRAS, KRAS, BRAF | 5% to 10% | Neutral or adverse | NRAS mutations are more common in CMML and AML |
| JAK2 | <5% | Neutral | More frequent in RARS-T (50%) |
| CBL, CBLB | <5% | Adverse | More frequently mutated in CMML |
| FLT3, MPL, KIT | <1% | ? | MPL mutated in (5%) of RARS-T, FLT3, and KIT mutations are rare in MDS and much more common in AML |
| NF1 | ? | ? | |
| PTPN11 | <1% | ? | Rare in MDS, more frequent in JMML (30%) |
| OTHERS | |||
| NPM1 | <2% | ? | Much more frequently mutated in AML where it is associated with favorable prognosis |
| GNAS | <1% | ? | Mutations are known to cause constitutive activation |
| BCOR, BCORL1 | <1% | ? | |
| UMODL1 | ? | ? | |
| ZSWIM4 | ? | ? | |
| Cohesins (RAD21, STAG1, STAG2, SMC3, SMC1A) | ? | ? | |
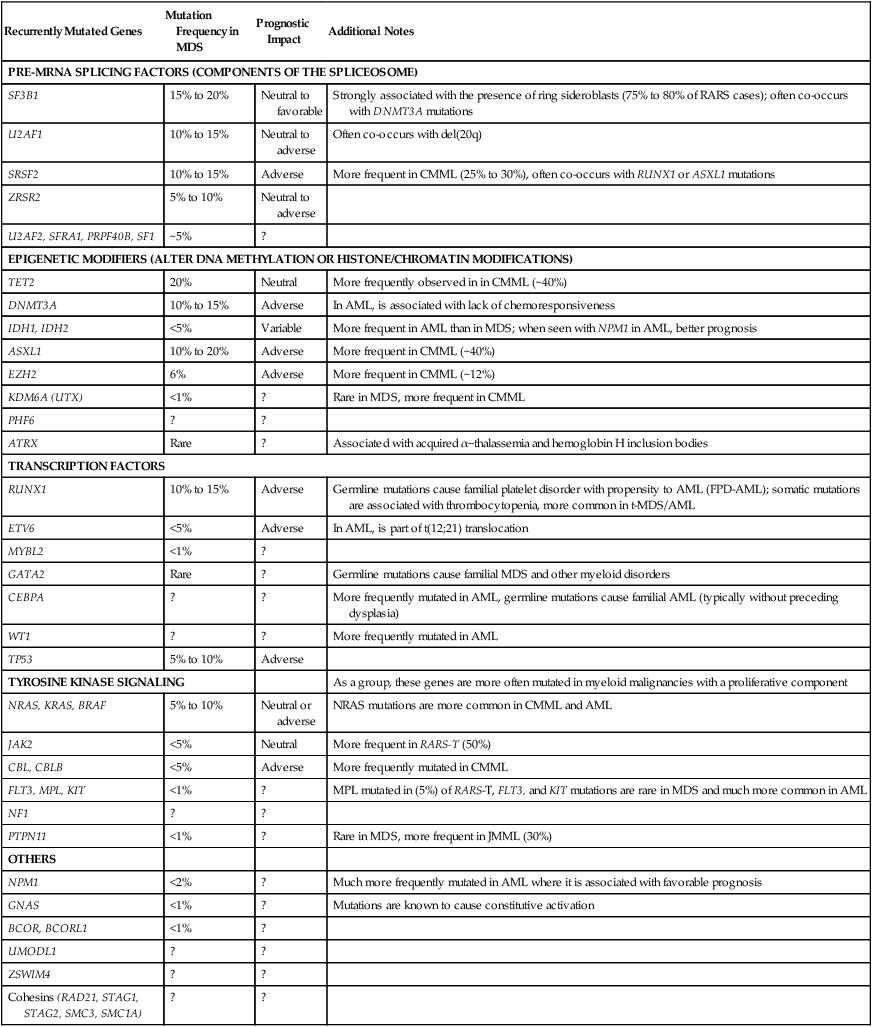
Adapted (modified and updated) from Bejar R, Levine R, Ebert BL. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J Clin Oncol 2011;29:504-515; and Bejar R, Steensma D. Molecular genetics of MDS. eLS online resource, August 2012 http://www.els.net/WileyCDA/ElsArticle/refId-a0023872.html.
Despite the high frequency of clonal cytogenetic alterations in MDS, insights from karyotyping into disease biology have been limited, primarily because balanced translocations are rare, and the more typical megabase pair deletions or aneuploidy are less fertile ground for genome discovery. Much attention has focused on the distal of the two commonly deleted regions on the long arm of chromosome 5 (del5q) in MDS, a genomic region that contains more than 40 genes, many of them expressed in CD34+ cells.136 Elegant knockdown experiments in 2008 demonstrated that haploinsufficiency of ribosomal subunit protein RPS14 contributes to anemia, but cannot explain the clonal advantage or the thrombocytosis common in del(5q) MDS.137 Altered expression of microRNAs mi-145 and mi-146a and other factors in or near the commonly deleted regions, including α-catenin (CTNNA1), SPARC, RIL, NPM1, CDC25C, and EGR1, may also play a role in 5q− syndrome biology.138–143
In addition to the now-numerous point mutations, gene silencing through aberrant promoter methylation also appears to be important in the pathophysiology of MDS, both early in the course of MDS (including protein kinases and some factors involved in signal transduction or stress response, such as DAPK, RAS oncogene family members, BLU, and SOCS1) and later in the disease course (in genes involved in cell-cycle regulation, such as P15INK4B) in patients with excess blasts.144–150 Similarly to a hypothesis about pathologically silenced tumor suppressors, these observations offer insight into both the development and progression of MDS and serve as the rationale for the therapeutic use of hypomethylating agents (DNA methyltransferase inhibitors) in MDS, but disappointingly, no methylation signature defined to date has predicted therapeutic response to this class of drugs.153–153
Immune Dysfunction
Immune modulation appears to play a role in the pathophysiology and possibly also in the progression of MDS in some cases.154,155 An increase in B-lymphocyte apoptosis (but not in T lymphocytes) has been observed, B-cell lineage–affiliated gene expression is markedly reduced in early stage MDS, and mutations in TET2 may be present in both B cells and myeloid cells (but again, not in T cells).158–158 Furthermore, clonal T-cell expansion is common, and natural killer cell function is reduced in MDS, particularly in cases with higher-risk features.159–162 A similar reduction in regulatory T cells (i.e., CD4+, CD25high, Foxp3+ “Tregs”) in lower-risk MDS also may contribute to the emergence of autoreactive T-cell clones that suppress hematopoietic progenitors.163 A gene expression pattern similar to that seen after γ-interferon stimulation also has been observed in MDS.118,164 These observations, coupled with the clinical overlap between MDS and aplastic anemia, have provided a rationale for immunosuppressive treatment of MDS, as described under “Therapy: Immunosuppressive Therapy.”165,166
Clinical Presentation
Patients with MDS may be incidentally discovered when a blood count is performed for a reason unrelated to suspected marrow failure (e.g., during pregnancy167,168), or may present with symptoms related to cytopenias or functional cell abnormalities. Normocytic or macrocytic anemia is typical, and other nonneoplastic causes of anemia (e.g., vitamin B12 with or without folic acid deficiency, medication or alcohol use, chronic liver disease) must be ruled out. Microcytic anemia in MDS is rarely associated with acquired thalassemia, but iron deficiency, renal insufficiency and the anemia of chronic inflammation also are substantially more common causes of anemia in older adults.130,169,170 However, in a study of elderly patients with unexplained macrocytic anemia, most had at least one molecular or cytogenetic marker of clonality suggesting the possibility of “early MDS” (this could also be explained by restricted clonality of hematopoiesis with aging).171 In another study in a geriatric ward of an Israeli hospital, 246 (7.5%) of 3275 admitted patients over a 3-year period had unexplained cytopenias or macrocytosis, and 37 (15%) of those ultimately were shown to have evidence of MDS.172 Many such cases will be inconsequential; for example, mild cytopenias detected in frail elderly patients who will die with MDS, rather than from MDS.
In some cases, a definitive cause for the cytopenias may not be established despite intensive investigation. The designation “idiopathic cytopenia(s) of undetermined significance” (ICUS) has been proposed to describe such patients, but unlike the more familiar monoclonal gammopathy of undetermined significance, ICUS is by definition not yet known to be a clonal disorder.173,174 The natural history of ICUS is unclear, but there is some risk of MDS and close follow-up is recommended. Cytogenetic studies may be helpful in diagnosing of MDS in the absence of clear morphologic dysplasia, and detection of a common MDS-associated chromosomal anomaly allows the diagnosis of “unclassifiable MDS” even when morphology is unrevealing.175 Additionally, studies of novel molecular diagnostic markers, such as SNP arrays or panels of point mutations, to distinguish MDS from other causes of cytopenias are ongoing.176 A transfusion history to identify use of packed red blood cells is valuable in determining the likelihood of response to antithymocyte globulin or erythropoiesis-stimulating agent therapy.176,177
Neutropenia also is a common feature of MDS, present in about one-third of patients at diagnosis and becoming more common with disease progression.178 In addition, MDS-associated neutrophil dysfunction has been described, and patients may present with suppurative infections in the absence of neutropenia.181–181 Infection is by far the most common cause of death in MDS and correlates only in general terms with the neutrophil count; neutropenia and infection are also common adverse effect of MDS therapy.182 Therefore suspected infections in patients with MDS should be treated aggressively. Opportunistic infection rarely also has been reported in MDS (e.g., disseminated Mycobacterium avium-intracellulare infection), presumably related to complex disease-related immunodeficiency.183
Thrombocytopenia is a growing clinical problem in MDS, present in 40% to 65% of patients at diagnosis and a contributing cause to death in approximately 20%.184 MDS uncommonly manifests as isolated thrombocytopenia, and when it does so, it can be difficult to distinguish from immune-mediated thrombocytopenia, which can coexist with MDS.187–187 In contrast, MDS with del(5q) or rearrangements of the MECOM locus can manifest with thrombocytosis.190–190 In addition, a subtype of RARS associated with thrombocytosis (RARS-T) frequently harbors JAK2 or MPL mutations and is classified as an MDS/myeloproliferative neoplasm overlap syndrome.191,192 No close association has been found between thrombocytopenia and serum thrombopoietin levels in patients with MDS.193 In addition to numerical abnormalities, various forms of platelet dysfunction (e.g., loss of granules or cell-surface receptors) have been described in MDS and may contribute to mucosal bleeding at higher platelet counts where hemorrhage usually does not occur (>50 × 109/L).194,195
Uncommonly, MDS presents with unusual immunologic features or accompanying paraneoplastic syndromes.196 These include Sweet syndrome (neutrophilic dermatosis),197,198 Behçet disease,199 inflammatory arthritis and synovitis,200 vasculitis,201 Crohn disease,202 and other immunologic phenomena including a higher-than-expected incidence of monoclonal gammopathy of undetermined significance.205–205 MDS-associated paraneoplastic manifestations do not appear to have an impact on the prognosis or disease course, but can cause bothersome symptoms.206,207
Laboratory Evaluation and Pathology
The diagnosis of MDS requires at least one persistent cytopenia (i.e., anemia, neutropenia, or thrombocytopenia), demonstration of morphologic dysplasia in an appropriate clinical context, and exclusion of other clonal and nonclonal disorders (Box 99-1).21 Box 99-2 outlines a suggested diagnostic evaluation. Bone marrow aspiration and trephine biopsy should be performed in all patients for whom the diagnosis is not apparent based on medical history and basic laboratory testing such as vitamin B12 and folate levels.210–210 Although some clinicians continue to perform marrow aspiration without core biopsy, especially outside of the United States, biopsy is extremely safe and provides valuable information about marrow cellularity and architecture, including the presence of prognostically relevant fibrosis or abnormal localization of immature precursors (central clustering of immature cells, which may correlate with expression of vascular endothelial growth factor and neoangiogenesis).211–214
Characteristic peripheral blood findings in MDS patients include oval macrocytic erythrocytes, anisopoikilocytosis, neutrophil hypogranularity or hypolobation, giant or hypogranular platelets, monocytosis, and circulating immature myeloid cells or blasts. In the erythroid series, common marrow aspirate findings suggestive of MDS include megaloblastoid erythroid maturation, nuclear karyorrhexis, multinucleated erythroid precursors, and ring sideroblasts (defined as erythroid precursors with at least five granules, representing mitochondria laden with mitochondrial ferritin, encircling at least one-third of the nucleus 215,216). Typical marrow findings in other lineages indicative of MDS include hypogranular granulocytes, left-shifted myeloid maturation, abnormal myeloid-to-erythroid ratio, an excess of immature monocytes, dual esterase staining (i.e., simultaneous α-naphthyl butyrate “nonspecific” esterase and chloroacetate “specific” esterase) cells, micromegakaryocytes, and megakaryocyte nuclear lobation abnormalities.217 Some common dysplastic findings in the blood and aspirate that suggest MDS are depicted in Figures 99-1 through 99-8.
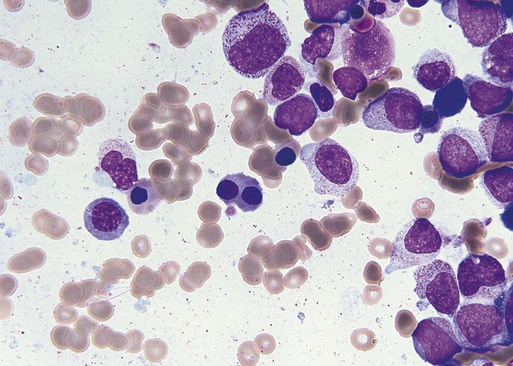
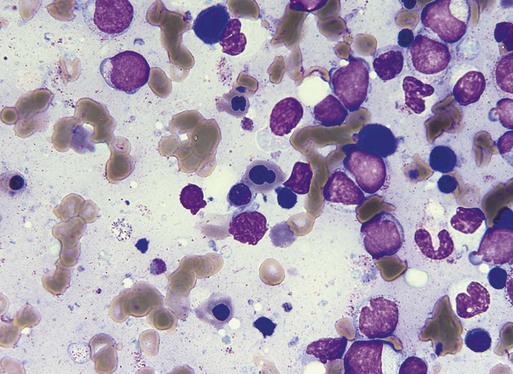
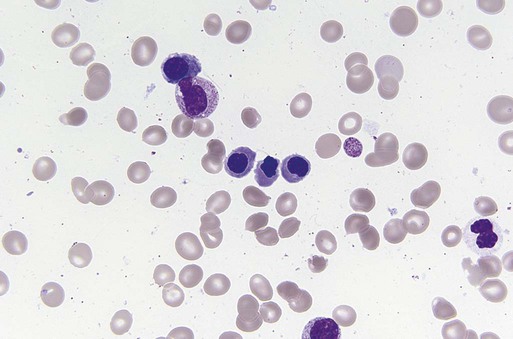
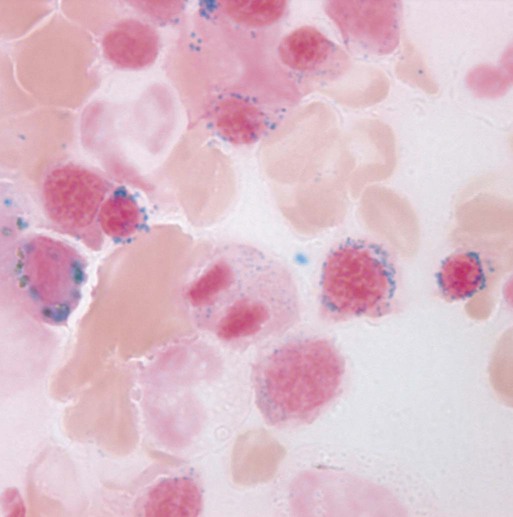
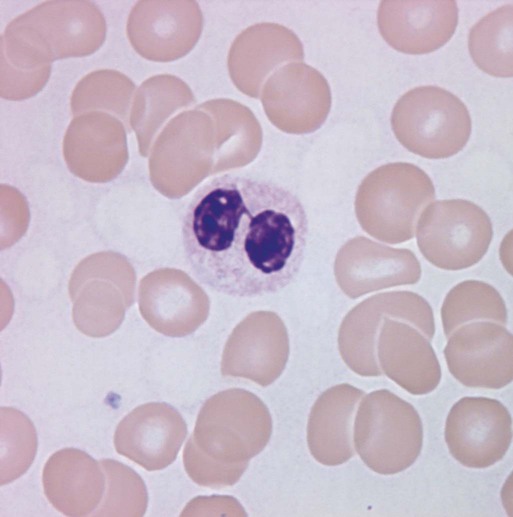
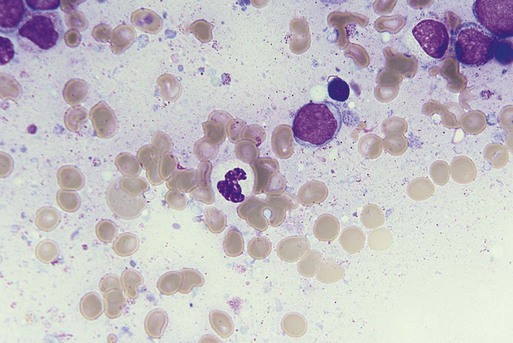
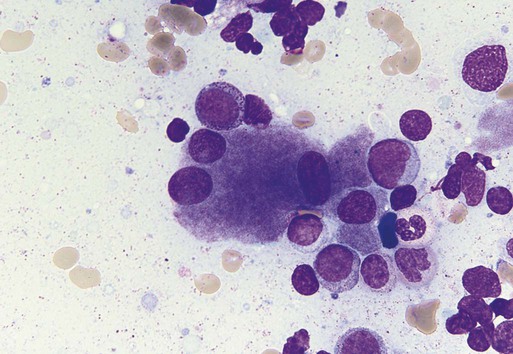
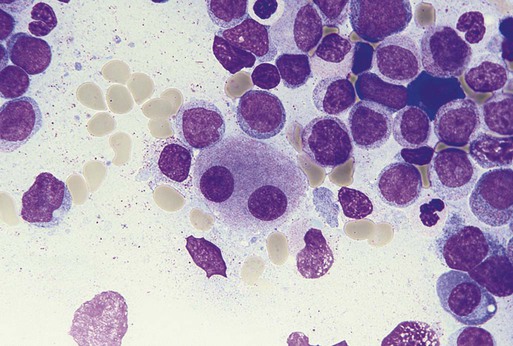
AML is now defined by ≥20% blasts (see “Disease Classification: World Health Organization Classifications”); thus all patients with MDS have <20% marrow blasts, by definition. Minimal diagnostic criteria for MDS were proposed by an international working group in 2006 (see Box 99-1).218,219 In these criteria, there is a requirement for meaningful cytopenias and exclusion of other diagnoses, plus either dysplastic morphologic abnormalities in >10% of cells in at least one of the three major myeloid lineages, ≥5% marrow blasts, or an MDS-associated karyotypic abnormality or other marker of clonal hematopoiesis (e.g., an MDS-associated somatic point mutation). Loss of the Y chromosome, trisomy 8, and deletion of the long arm of chromosome 20 [del(20q)] by themselves are not considered specific enough to define MDS.2,220 Refinements in diagnostic techniques for evaluation of MDS in the near future may include routine incorporation of assays for panels of MDS-associated point mutations or high-resolution SNP array technology.
Cytogenetic evaluation is mandatory whenever MDS is a diagnostic possibility, and may be particularly helpful in establishing the diagnosis in patients with indeterminate morphology.175 The role of interphase fluorescence in situ hybridization (FISH) with probe sets for common MDS-associated abnormalities (e.g., del5q, del7q, +8, del20q, −Y, etc.) has not been established. However, FISH may complement standard cytogenetics, especially when metaphase karyotyping fails or in patients who are unwilling to undergo marrow biopsy.221,222 Some clinicians perform FISH for del(5q) in patients with a normal karyotype when considering therapy with lenalidomide, but it is not clear that patients with occult clones detected in this way respond as well as those with del(5q) clones detected by G-banded karyotyping.223 Because of the potential response to imatinib, patients with CMML should be evaluated for 5q33/platelet-derived growth factor receptor-β translocation in the correct clinical setting, but this translocation is exceptionally rare and not usually karyotypically occult.224
The bone marrow in MDS is usually normocellular or hypercellular for age, but 10% to 20% of cases are accompanied by a hypocellular marrow, and such cases may be difficult to distinguish from aplastic anemia, as described earlier.165,225 MDS may coexist with paroxysmal nocturnal hemoglobinuria clones, although these clones are usually clinically silent and small (<10% of cells). However, in hypocellular cases or when hemolysis is present, peripheral blood flow cytometry for CD55 and CD59 expression and fluorescent aerolysin antigen should be considered to evaluate for paroxysmal nocturnal hemoglobinuria.226
Flow cytometry is increasingly playing a role in MDS diagnosis, and international efforts are ongoing to develop consensus standards for the use of flow cytometry in evaluation of suspected MDS cases.229–229 Flow cytometry may assist in diagnosis, and certain aberrant antigen patterns appear to have prognostic significance.228 Although flow cytometry is often used as for estimating blast cell proportion, morphologic assessment remains the “gold standard” for blast calculation, as flow counts are subject to processing artifacts and to variability in placement of gates by the cytometry operator. Bone marrow flow cytometry studies may be helpful if concurrent T-cell large granular lymphocyte disease is suspected.230
Dysplastic morphology is not specific for MDS and has been reported in various other settings, including recovery following chemotherapy or SCT, hemolysis, and even in the marrow of healthy subjects (where it is usually mild, involving <10% of cells, and is most common in the erythroid lineage).231,232 The presence of bone marrow dysplasia at presentation in patients with de novo AML (i.e., with no antecedent history of MDS) suggests a history of unrecognized MDS, but this finding does not appear to have independent prognostic implications.235–235 Other causes of cytopenias accompanied by dysplasia include vitamin B12 deficiency, folate deficiency, and copper deficiency, the latter of which is often associated with peripheral neuropathy and is becoming increasingly common as bariatric surgery proliferates and as zinc supplements are widely used by health faddists.236,237 Reversible dysplastic changes also have been noted rarely after immunosuppressive medications, and ring sideroblasts can result from medications such as antituberculosis drugs (e.g., isoniazid, pyrazinamide) or from vitamin B6 or copper deficiency, so unilineage erythroid dysplasia with a normal karyotype should prompt some concern about the security of the MDS diagnosis.238,239
A baseline reticulocyte count, iron studies (ferritin, serum iron, total iron-binding capacity) and serum erythropoietin level are recommended for patients with anemia, particularly for those with lower-risk disease who may be candidates for blood transfusion and treatment with recombinant erythropoietin. Baseline human leukocyte antigen (HLA) typing should be performed in any patient who is a potential candidate for allogeneic SCT, and possibly for HLA-DR15 status, which may predict a moderately higher likelihood of response to antithymocyte globulin (ATG).240 HIV testing also should be considered in patients with risk HIV factors.241
Disease Classification
French–American–British Classification
Two morphologic classification systems are commonly used to distinguish MDS subtypes. The FAB system (see Table 99-1), proposed in 1982, could be considered to be largely of historical interest now more than 3 decades later. Yet the FAB system is still commonly found in published papers, is part of the label of all three FDA-approved drugs for MDS-related indications in the United States, and served as the basis upon which the later WHO MDS classification was crafted, so it is worth briefly discussing.14,242 The FAB classification is a morphology-based system that predicts rates of survival and of transformation to AML (AML was defined by the FAB in 1976 as ≥30% blasts12) by dividing MDS into five subgroups, based on the presence or absence of ringed sideroblasts (later called “ring” sideroblasts by the WHO, for etymologic reasons), peripheral blood monocytosis, and myeloblast percentage (see Table 99-1). The five MDS categories recognized in the FAB system are refractory anemia (RA); RARS; RA with excess blasts (RAEB); RA with excess blasts in transformation to AML (RAEB-t); and CMML. CMML is a heterogeneous “overlap syndrome” that can include features of a myelodysplastic syndrome, a myeloproliferative neoplasm (e.g., leukocytosis and organomegaly), or both.243
Both RA and RARS are typified by a hypercellular bone marrow and dysplasia of the erythroid cell lines, with or without dysgranulopoiesis or dysmegakaryopoiesis, in the setting of less than 5% bone marrow blasts and 1% or less peripheral blood myeloblasts. The diagnosis of RARS also requires 15% or more ring sideroblasts; the precise proportion does not have prognostic significance.244 A diagnosis of RAEB requires these foregoing bone marrow abnormalities, along with 5% to 20% bone marrow (or up to 4% peripheral blood) myeloblasts. If Auer rods are present in the myeloblasts, or if the myeloblast percentage in the bone marrow is between 21% and 29% (or more than 4% in the peripheral blood), the diagnosis becomes RAEB-t. CMML is defined by a peripheral blood monocytosis with an absolute monocyte count greater than 1 × 109/L; the degree of marrow monocytosis is not a factor.243 In 1994, the FAB group proposed dividing CMML into a “proliferative” subtype, with a white count >13 × 109/L, and a “dysplastic” subtype with a lower total white count.245 Because peripheral monocytosis often fluctuates near the (arbitrary) CMML threshold, this leads some patients to have FAB CMML at one visit and another form of FAB MDS at another. More generally, wide variation in outcomes within each of the FAB diagnostic categories prompted prognostic models and eventually a revised classification system.
World Health Organization Classifications
In 1997, the WHO began an effort to update its classification of all neoplasms. The third revision of the WHO system for hematologic cancers (the first two WHO cancer classifications had been published before 1981, and were never widely used by clinicians or pathologists) was published in draft form in 1999 (the MDS section is summarized in Table 99-3), with full publication of the WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues following in 2001.99,246
Table 99-3
2008 WHO Classification of Myelodysplastic Syndromes/Neoplasms
| MDS Subtype | Abbreviation | Peripheral Blood: Key Features | Bone Marrow: Key Features | WHO-Estimated % of Patients with MDS |
| Refractory cytopenias with unilineage dysplasia (RCUD): | ||||
| Refractory anemia | RA | Anemia; <1% peripheral blood blasts | Unilineage erythroid dysplasia (in >10% of cells); <5% blasts | 10%-20% |
| Refractory neutropenia | RN | Neutropenia; <1% peripheral blood blasts | Unilineage granulocytic dysplasia; <5% blasts | <1% |
| Refractory thrombocytopenia | RT | Thrombocytopenia; <1% blasts | Unilineage megakaryocytic dysplasia; <5% blasts | <1% |
| Refractory anemia with ring sideroblasts | RARS | Anemia; no blasts | Unilineage erythroid dysplasia; ≥15% of erythroid precursors are ring sideroblasts; 5% blasts | 3%-11% |
| Refractory cytopenias with multilineage dysplasia | RCMD | Cytopenia(s); <1% blasts; no Auer rods | Multilineage dysplasia ± ring sideroblasts; <5% blasts; no Auer rods | 30% |
| Refractory anemia with excess blasts, type 1 | RAEB-1 | Cytopenia(s); <5% blasts; no Auer rods | Unilineage or multilineage dysplasia; 5% to 9% blasts; no Auer rods | 40%* |
| Refractory anemia with excess blasts, type 2 | RAEB-2 | Cytopenia(s); 5%-19% blasts; ± Auer rods | Unilineage or multilineage dysplasia; 10% to 19% blasts; ±Auer rods | |
| Myelodysplastic syndrome (MDS) associated with isolated del(5q) | Del(5q) | Anemia; normal or high platelet count; <1% blasts | Isolated 5q31 chromosome deletion; anemia, hypolobated megakaryocytes | 5% |
| Childhood MDS, including refractory cytopenia of childhood (provisional) | RCC | Pancytopenia | <5% Marrow blasts for RCC; marrow usually hypocellular | <1% |
| MDS, unclassifiable | MDS-U | Cytopenias; ≤1% blasts | Does not fit other categories; dysplasia; <5% blasts; if no dysplasia, MDS-associated karyotype | ? |
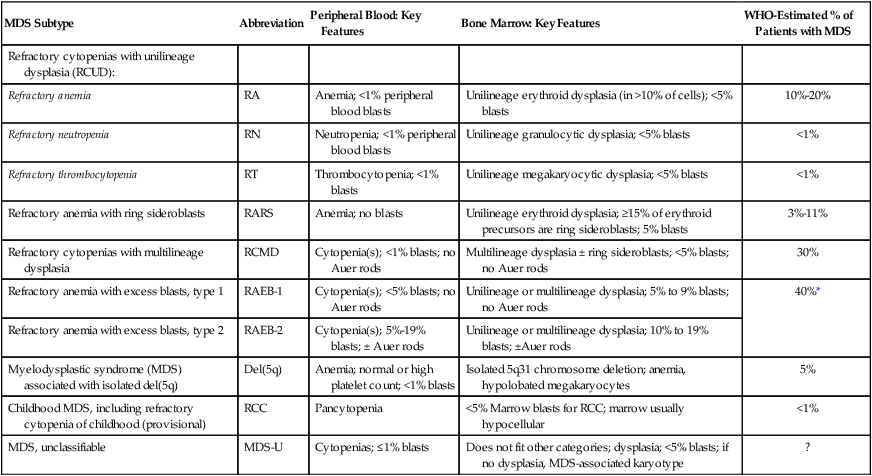
*This 40% estimate represents the proportion of patients with RAEB-1 or RAEB-2, collectively.
Adapted from Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC Press; 2008. pp. 87–107.
There are a number of differences between the FAB and WHO classifications, including the inclusion of a limited number of karyotypes in the WHO classification, separation of several FAB groupings into multiple subtypes, and presentation of specific threshold for defining a cell lineage as involved by “dysplasia” (i.e., >10% dysplastic cells). The most notable change was that the WHO lowered the blast threshold for making a diagnosis of AML from 30% to 20% blasts in the blood or bone marrow, thus eliminating the FAB RAEB-t category, a change that not all investigators welcomed.242,247 CMML was also formally separated from the MDSs into a new overlap category of “myelodysplastic/myeloproliferative disorders” (MDS/MPD) that also included juvenile myelomonocytic leukemia and RARS-T. A cytogenetically defined MDS subgroup, “MDS with deletion 5q,” was added.
In addition, the WHO committee separated RAEB into two categories based on blast proportion, RAEB-1 (5% to 9% blasts) and RAEB-2 (11% to 19% blasts), as the prognosis varied widely within the FAB RAEB category. The WHO committee recognized that patients with anemia only and erythroid-restricted dysplasia have a distinct and favorable outcome compared with those with multiple cytopenias and multilineage dysplasia, and introduced a new category, “refractory cytopenia with multilineage dysplasia” (RCMD) to describe the latter, separating those with multilineage dysplasia and ring sideroblasts (RCMD-RS) from those without (RCMD). Finally, treatment-related MDS/AML was given a separate diagnostic category, and a new “unclassifiable” category was introduced (MDS-U) for cases not fitting other categories. This classification was quickly independently validated,248 although specific criticisms were levied, such as the unnecessary retention of an Auer-rod based classification and a high proportion of MDS-U.242,247,249
In 2008, the WHO published the fourth edition of Classification of Tumours of Haematopoietic and Lymphoid Tissues.2 From the MDS standpoint, this update represented only a minor change from the previous 2001 WHO classification.250 In the 2008 version, the WHO included a token pediatric MDS category (MDS-RCC, described above), merged RCMD-RS and RCMD as the prognosis for these was identical and not affected by the presence of rings,251 and clarified that the diagnosis of MDS-U can be made without morphologic abnormalities if a convincing MDS-associated karyotype is present. A new general category of “refractory cytopenia with unilineage dysplasia” (RCUD) was also introduced, consisting of familiar RA together with two rarer entities, refractory neutropenia and refractory thrombocytopenia, which are associated with single cytopenias other than anemia.
Prognosis
Because patients with MDS vary so widely in their risk of death or leukemia progression, a number of risk stratification systems have been developed in recent decades (the first, the Bournemouth scoring system, was proposed in 1985252), in order to more accurately estimate outcomes and aid clinical decision making.253 Key prognostic factors incorporated into existing models include the age of the patient, number and degree of cytopenias, karyotypic findings, marrow and blood blast proportion, and morphologic subtype of disease.22
The pathological classification, whether the FAB or the WHO system is used, does offer some insight into prognosis. Lower-risk forms of MDS are less likely to evolve to AML and in general are associated with less severe cytopenias; as a consequence, affected patients are more likely to live longer than those with high-risk MDS. Using the FAB classification system, the lower-risk MDS category includes patients with RA and RARS, in whom median survival ranges from approximately 3 to 8 years and risk of AML transformation is approximately 10% to 35%.256–256 In the WHO classification system, the more benign subtypes include RCUD (RA, refractory neutropenia, refractory thrombocytopenia), RARS, RCMD, some types of MDS-U, and MDS with an isolated deletion of 5q. In these patients, median survival ranges from approximately 2 to 8 years, and risk of AML transformation is about as low as for FAB RA/RARS.248,257,258 Patients with RCUD and RARS enjoy a longer survival than those with RCMD, whereas those with the 5q− syndrome, or with an isolated del(5q) cytogenetic abnormality without the associated syndromic morphologic changes but with less than 5% blasts, have the best overall survival and lowest likelihood of AML transformation.248 Outcomes for subtypes vary widely depending on karyotype. For patients with higher-risk disease (i.e., those with an increased percentage of marrow blasts—using the FAB system, this includes those with RAEB, RAEB-t, and some CMML patients; in the WHO system, it is RAEB-1 and -2), median survival ranges from 0.5 to 1.5 years and AML progression is almost guaranteed, unless the patient dies from complications of cytopenias first.258,259 Prognostic estimates worsen with increasing age and with the presence of comorbid conditions.262–262
International Prognostic Scoring System
Beginning in 1994, an International MDS Risk Assessment Workshop (IMRAW) formed and subsequently analyzed seven combined international MDS databases; these efforts resulted in the landmark International Prognostic Scoring System (IPSS), published in 1997 (Table 99-4).178 The IPSS analyzed 816 largely untreated patients with de novo MDS (only 5% had received oral chemotherapy, and 2% had received hematopoietic growth factors) and found that a combined score based on three factors—the number of cytopenias, karyotype, and percentage of bone marrow blasts—accurately predicted survival and the risk of AML transformation (Tables 99-5 and 99-6). Patients were divided into four IPSS subgroups: low (a score of 0); intermediate-1 (a score of 0.5 to 1.0); intermediate-2 (a score of 1.5 to 2.0); and high (a score of 2.5 or greater; see Table 99-6). IPSS low and intermediate-1 subgroups are sometimes lumped together as “lower-risk” MDS, in contrast to “higher-risk” (intermediate-2 and high subgroups). This system has further usefulness within lower-risk MDS groups when patients are analyzed by age cohorts, for example, younger or older than age 60 years, or younger or older than age 70 years, but age has little bearing on the outcomes in IPSS higher-risk MDS.
Table 99-4
The 1997 International Prognostic Scoring System for Myelodysplastic Syndromes: Scoring Summary
| Prognostic Factor | Category Score (Sum All 3 Subscores to Calculate the Overall IPSS Score) | ||||
| 0 (Best) | 0.5 | 1 | 1.5 | 2.0 (Worst) | |
| Marrow blast proportion (%) | <5 | 5-10 | — | 11-20 | 21-30* |
| Karyotype | Good risk [normal, isolated −Y, isolated del(5q), or isolated del(20q)] | Intermediate (all karyotypes not defined as good or poor) | Poor (abnormal chromosome 7 or a complex karyotype (defined as ≥3 anomalies)) | — | — |
| Peripheral blood cytopenias† | 0 or 1 | 2 or 3 | — | — | — |
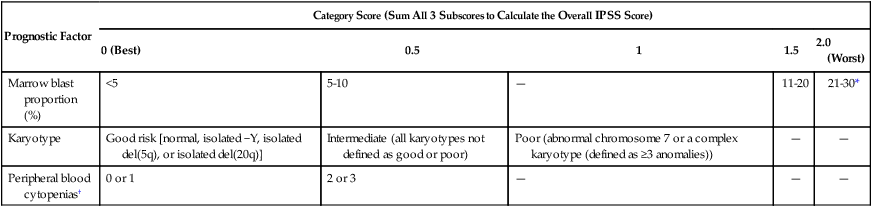
*Patients with ≥20% blasts are no longer considered myelodysplastic syndrome and were redefined as acute myeloid leukemia by the World Health Organization in 2001.
†The IPSS definition of peripheral blood cytopenias is as follows: hemoglobin <10 g/dL; absolute neutrophil count <1800/mm3; and platelet count <100 × 109/L.
Adapted from Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997;89:2079–2088.
Table 99-5
The 1997 International Prognostic Scoring System MDS Risk Stratification System
| Risk Category | Total Score | Median Survival (Years) | Median Survival (Years) for Patients <60 Years Old (n = 205) |
Median Survival (Years) for Patients ≥60 Years Old (n = 611) |
Time Until 25% of Surviving Patients in Category Developed Leukemia (Years) |
| Low risk | 0 | 5.7 | 11.8 | 4.8 | 9.4 |
| Intermediate-1 (INT-1) | 0.5 or 1.0 | 3.5 | 5.2 | 2.7 | 3.3 |
| Intermediate-2 (INT-2) | 1.5 or 2.0 | 1.2 | 1.8 | 1.1 | 1.1 |
| High | ≥2.5 | 0.4 | 0.3 | 0.5 | 0.2 |

Four risk categories were defined for the 1997 IPSS, based on 816 patients with de novo disease.
Data are from Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997;89:2079–2088.
Table 99-6
Recurrent Acquired Chromosome (Karyotypic) Abnormalities in MDS and Possible Associated Genes
| Recurrent MDS-Associated Chromosomal Abnormalities | Frequency Observed | Potentially Involved Genes |
| VERY GOOD PROGNOSIS | ||
| −Y | 2.2% | Unknown |
| del(11q) | 0.7% | Unknown, MLL? |
| GOOD PROGNOSIS | ||
| None (normal karyotype) | 55.1% | Various |
| del(5q) ± 1 other abnormality | 8.0% | RPS14, HSPA9, EGR1, APC, NPM1, miR-145/146, CTNNA1, others |
| del(20q) | 1.7% | MYBL2, L3MBTL1 |
| del(12p) | 0.6% | ETV6 |
| INTERMEDIATE PROGNOSIS | ||
| +8 | 4.7% | MYC |
| del(7q) | 0.5% | EZH2 |
| i(17q) | 0.4% | TP53, NF1 |
| +19 | 0.4% | Unknown |
| Above + 1 other abnormality | 3.4% | Various |
| Any other single abnormality | 9.0% | Various |
| POOR PROGNOSIS | ||
| −7 ± del(7q) | 2.8% | EZH2 |
| inv(3)/t(3q)/del(3q) | 0.4% | MECOM (MDS1–EVI1) |
| 3 separate abnormalities | 2.1% | Various |
| VERY POOR PROGNOSIS | ||
| More than 3 abnormalities | 7.0% | Various |
International Prognostic Scoring System Criticisms
Although the IPSS has been criticized for being developed from data that preceded the current treatment era, and is also limited in its applicability only to patients with newly diagnosed de novo MDS (i.e., not previously treated patients, and not t-MDS/AML), it remained the most widely used predictive tool for 15 years after its publication, determining eligibility for clinical trials and even being incorporated into FDA labeling for lenalidomide. Additional criticisms of the IPSS include its insensitivity to the degree of cytopenias (for example, the IPSS treats patients with platelet counts of 90 × 109/L the same as those with 9 × 109/L, even though multiple series have demonstrated that the risk for the latter is much greater184,263), the wide variation in outcomes within each of the four IPSS risk groups (25% to 30% of patients with low-risk MDS die within 2 years of diagnosis, which is not exactly low risk),100 the small number of included karyotypes,264 overemphasis of blasts at the expense of cytogenetics,264 use of an outdated 30% AML blast cutoff dating back to the 1982 FAB classification, and lack of applicability to children (a separate IPSS has been proposed for pediatric use94).
Furthermore, the IPSS fails to incorporate numerous clinical and biological markers that have emerged as having prognostic importance since 1997.253 Chief among these is transfusion dependence—a greater number of transfusions is associated with worse outcomes—and elevated serum ferritin levels >1000 ng/mL also appear to carry an independent risk for poorer outcome,265–268 although there are dissenters on this point.269 Other potential IPSS-independent high-risk prognostic variables obtainable during routine testing include elevated β2-microglobulin,270 low absolute lymphocyte count,271 adverse flow cytometry score,228 and high lactate dehydrogenase level.272
Neither the IPSS nor any of the newer prognostic systems described below incorporate any molecular markers in their risk stratification models. Novel molecular patterns that have been reported to predict outcomes range from changes in expression of specific genes (e.g., BMI1, WT1273,274) and an abnormal SNP karyotype (especially for patients with normal G-banded metaphase cytogenetic results123) to the presence of specific mutations such as DNMT3A, ASXL1, EZH2, TP53, NRAS, SRSF2, and RUNX1.15,28,127,275 In patients with sideroblastic anemia, one group reported that SF3B1 mutations are associated with favorable outcome, but other groups found that these mutations are neutral, once histology is taken into account.132,133,276,277 In the future, these molecular changes will likely be incorporated into more robust prognostic scoring systems than any of the existing models, once the complexities of allele burden, polyclonality, and co-existing mutations are accounted for.
World Health Organization Classification-Based Prognostic Scoring System and MD Anderson Risk Models
Within a few years of publication of the IPSS, various groups began to express concerns about its limitation, and several investigators subsequently proposed new models. In 2007, for instance, Malcovati and colleagues in Europe proposed the WHO classification-based Prognostic Scoring System (WPSS), which has many similarities to the IPSS (Table 99-7).278
Table 99-7
Revised Version of the World Health Organization Classification-Based Prognostic Scoring System
| Score | ||||
| Prognostic Variable | 0 | 1 | 2 | 3 |
| WHO Classification | RCUD, RARS, MDS with Isolated Deletion (5q) | RCMD | RAEB-1 | RAEB-2 |
| Karyotype class* | Good | Intermediate | Poor | |
| Severe anemia† | No | Yes | ||
| Risk Group | Points | Median Survival, Months | Median Time Until AML Evolution, Months |
| Very low | 0 | 139 | Not reached |
| Low | 1 | 112 | 176 |
| Intermediate | 2 | 68 | 93 |
| High | 3 to 4 | 21 | 21 |
| Very high | 5 to 6 | 13 | 12 |
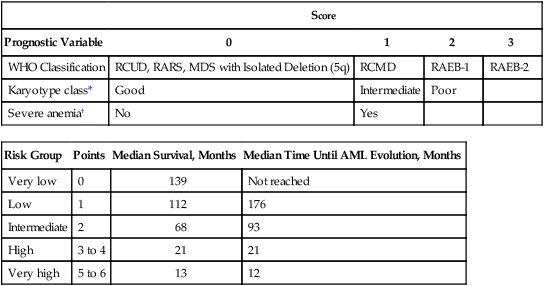
*Karyotypes: Same as for IPSS.
†Severe anemia: Hemoglobin <9.0 g/dL in males, <8.0 g/dL in females.
Adapted and modified from Malcovati L, Della Porta MG, Strupp C, et al. Impact of the degree of anemia on the outcome of patients with myelodysplastic syndrome and its integration into the WHO classification-based Prognostic Scoring System (WPSS). Haematologica 2011;96:1433–1440.
The WPSS is based on data from a learning cohort of 467 de novo MDS patients assessed in Italy from 1992 to 2002, and a validation cohort of 620 MDS patients from Germany seen from 1982 to 2003. Patients are given a score based on their WHO classification (instead of blast proportion in the IPSS), cytogenetic risk score (using the same IPSS scoring scheme), and transfusion requirement (instead of number of cytopenias in the IPSS). Based on this cumulative score, patients are divided into one of five WPSS risk groups: very low, low, intermediate, high, and very high. After the initial publication, the investigators modified the WPSS by incorporating an increased risk group for marrow fibrosis,211 and later they substituted hemoglobin level for transfusion dependence,279 because transfusion needs are often difficult to assess immediately after MDS diagnosis.
In 2008, investigators at the MD Anderson Cancer Center in Houston developed both a general MDS risk model (Tables 99-8 and 99-9) and a specific model for patients with IPSS lower-risk disease.100,280 The general risk model is more complex than any of the other prognostic systems, but has the advantage of being dynamic (such that it can be applied to previously treated patients or patients diagnosed a year or more earlier), sensitive to the degree of thrombocytopenia, and applicable to CMML. The MD Anderson general risk model has been independently validated by several other groups.281,282 The lower-risk model was designed to assess which patients with IPSS lower-risk disease are actually at high risk for disease progression, and divides patients into three subgroups. It, too, has been independently validated and correlates well with molecular findings, even though it is a clinicopathologic model.28
TABLE 99-8
MD Anderson Cancer Center General Risk Model for MDS and CMML
| Prognostic Factor | Points (Sum, 0-17) |
| Performance Status: ≥2 | 2 |
| Age: 60 to 64 years | 1 |
| Age: ≥65 years | 2 |
| Platelets: <30 × 109/L | 3 |
| Platelets: 30 to 49 × 109/L | 2 |
| Platelets: 50 to 199 × 109/L | 1 |
| Hemoglobin: <12 g/dL | 2 |
| Bone marrow blasts: 5% to 10% | 1 |
| Bone marrow blasts: ≥11% | 2 |
| White blood count: ≥20 × 109/L | 2 |
| Karyotype: abnormal chromosome 7 or complex (≥3 abnormalities) | 3 |
| Prior transfusion | 1 |
Adapted and modified from Kantarian H, O’Brien S, Ravandi F, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer 2008;113:1351–1361.
Table 99-9
Median Survival in Three Series Using MD Anderson Cancer Center General Risk Model for MDS and CMML
| Series | |||
| Risk Group | MD Anderson (Kantarjian 2008) n = 1915 |
Mayo Clinic (Hugo 2009) n = 1503 |
H. Lee Moffit Cancer Center (Komrokji 2012) n = 844 |
| Median Survival (Months) | |||
| Low (0-4 points) | 54 | 51 | 61 to 92* |
| Intermediate 1 (5-6 points) | 25 | 21 | 28 to 49* |
| Intermediate 2 (7-8 points) | 14 | 13 | 15 to 27* |
| High (≥9 points) | 6 | 5 | 8 to 14* |

*In Moffit series, first value is median survival from time of referral to tertiary center; second value is for survival from time of diagnosis.
Adapted and modified from Kantarian H, O’Brien S, Ravandi F, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer 2008;113:1351–1361; Hugo SE, Bundrick SC, Hanson CA, Steensma DP. Independent validation of the MD Anderson Cancer Center risk model for myelodysplastic syn (MDS), and comparison to the InternaPrognostic Scoring System (IPSS) and the World Health Organization-Based Prognostic Scoring System (WPSS). ASH Annual Meeting Abstracts 2009;114(22):3814. Komrokji RS, Corrales-Yepez M, Al Ali N, et al. Validation of the MD Anderson Prognostic Risk Model for patients with myelodysplastic syndrome. Cancer 2012;118:2659–2664.
Revised International Prognostic Scoring System
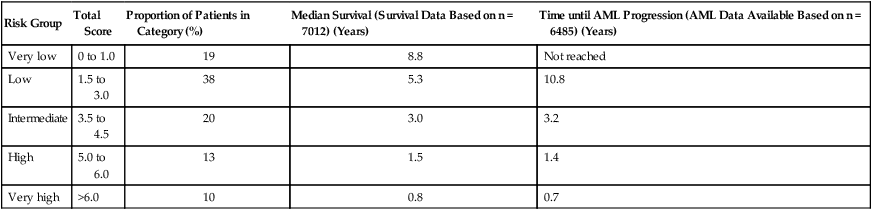
In 2012, an expanded IMRAW revised the IPSS (IPSS-R), based on an analysis of more than 7000 patients from 11 countries (see Tables 99-6, 99-10, and 99-11).22 Although still only applicable to untreated patients with de novo MDS and useful only at the time of diagnosis, the IPSS-R offers several important advantages over the 1997 IPSS. The largest of these is inclusion of a broader set of chromosome abnormalities than the original (see Table 99-6), and IPSS-R also weights poor-risk chromosome changes more heavily compared to marrow blast proportion than did the IPSS.264 The IPSS-R has five risk groups, whereas the original IPSS had four, and it is sensitive to the degree of cytopenias to a limited extent. The IPSS-R also includes different marrow blast subgroups than the IPSS, including two different cohorts with less than 5% blasts (as 4% blasts is not really normal). Using the IPSS-R, 27% of patients with IPSS lower-risk disease will be “upstaged” to a higher risk score, while 18% of IPSS higher-risk disease will be “downstaged” to a lower risk score and better prognosis.22
Table 99-10
2012 Revised International Prognostic Scoring System for MDS (IPSS-R)
| Parameter | Categories and Associated Scores | ||||
| Cytogenic Risk Group | Very good | Good | Intermediate | Poor | Very Poor |
| 0 | 1 | 2 | 3 | 4 | |
| Marrow blast proportion | ≤2% | 2% to <5% | 5% to 10% | >10% | |
| 0 | 1 | 2 | 3 | ||
| Hemoglobin | ≥10 g/dL | 8 to <10 g/dL | <8 g/dL | ||
| 0 | 1 | 1.5 | |||
| Absolute neutrophil count | ≥0.8 × 109/L | <0.8 × 109/L | |||
| 0 | 0.5 | ||||
| Platelet count | ≥100 × 109/L | 50 to 100 × 109/L | <50 × 109/L | ||
| 0 | 0.5 | 1 | |||
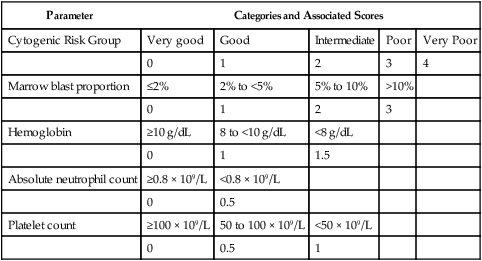
Possible ranges of summed scores: 0 to 10.
Greenberg PL, Tuechler H, Schanz J, et al. Revised International Prognostic Scoring System (IPSS-R) for myelodysplastic syndromes. Blood 2012;120(12):2454-65.
Table 99-11
Risk Groups and Outcomes Defined by the 2012 IPSS-R
| Risk Group | Total Score | Proportion of Patients in Category (%) | Median Survival (Survival Data Based on n = 7012) (Years) | Time until AML Progression (AML Data Available Based on n = 6485) (Years) |
| Very low | 0 to 1.0 | 19 | 8.8 | Not reached |
| Low | 1.5 to 3.0 | 38 | 5.3 | 10.8 |
| Intermediate | 3.5 to 4.5 | 20 | 3.0 | 3.2 |
| High | 5.0 to 6.0 | 13 | 1.5 | 1.4 |
| Very high | >6.0 | 10 | 0.8 | 0.7 |
Adapted from Greenberg PL, Tuechler H, Schanz J, et al. Revised International Prognostic Scoring System (IPSS-R) for myelodysplastic syndromes. Blood 2012;120(12):2454-65.