Chapter 8B Molecular pathogenesis of biliary tract cancer
Cholangiocarcinoma
Cholangiocarcinoma, or biliary tract adenocarcinoma, was first described by Durand-Fardel in 1840 (Olnes & Erlich, 2004). This malignancy arises from the ductal epithelium of the biliary tree, either within the liver (intrahepatic cholangiocarcinoma) or from the extrahepatic bile ducts (extrahepatic biliary adenocarcinoma) or gallbladder. It is a rare disease, and because of its infrequent occurrence, relatively little is known about the underlying biology of these tumors. In addition, this disease is fatal in most cases because of its late clinical presentation and the lack of effective nonsurgical therapeutic modalities (see Chapter 49, Chapter 50A, Chapter 50B, Chapter 50C, Chapter 50D ) (Ishak et al, 1994). However, in recent years, important work has been done toward understanding the natural history and biology of these tumors and establishing patterns upon which future treatment regimens can be based.
Classification
More than 90% of cholangiocarcinomas are adenocarcinomas (Nakeeb et al, 1996), and they most commonly arise at or near the hepatic duct confluence. These fall under the general category of extrahepatic cholangiocarcinomas (see Chapter 50B) but are further subclassified as hilar cholangiocarcinoma, or Klatskin tumors, after the Yale University pathologist who was one of the first to report a series of patients with this condition (Nakeeb et al, 1996). Bismuth further subclassified hilar cholangiocarcinomas by precise location with reference to the biliary bifurcation and the lobar hepatic ducts (Bismuth et al, 1992). This classification may be useful for descriptive purposes and for operative planning. The remaining extrahepatic cholangiocarcinomas occur in the lower bile duct (periampullary) and are further subclassified as distal cholangiocarcinomas. These account for a relatively small fraction of all bile duct tumors. Mid–bile duct tumors are exceedingly rare but do occur in very small numbers, and even less common is disease that diffusely involves the biliary tree.
Cholangiocarcinoma may also arise from the intrahepatic ducts peripherally in the liver, giving rise to the subgroup known as intrahepatic or peripheral cholangiocarcinoma (see Chapter 50A) (Liver Cancer Study Group of Japan, 2000). Intrahepatic cholangiocarcinomas were poorly understood until recently. These are usually grouped by International Classification of Disease (ICD) codes with hepatocellular carcinoma as primary liver tumors (Khan et al 2002a, 2002b). Furthermore, many cases previously referred to as liver adenocarcinoma of unknown primary were likely unrecognized intrahepatic cholangiocarcinomas. Worldwide, intrahepatic cholangiocarcinoma is increasing in incidence and mortality. Intrahepatic tumors have been subcategorized by their growth characteristics into mass-forming, periductal-infiltrating, or intraductal-growing types (Liver Cancer Study Group of Japan, 2000).
Epidemiology
Although rare, cholangiocarcinoma has a distinctly higher incidence in certain demographic groups and geographic areas. The peak age for cholangiocarcinoma is the seventh decade, with a slightly higher male preponderance (Olmes & Erlich, 2004). The reported incidence in the United States is 1 to 2 cases per 100,000 (3500 new cases per year) with no clear racial predisposition. Cholangiocarcinoma incidence rates vary markedly worldwide, presumably reflecting differences in either local environmental risk factors or genetics. The highest disease incidence rates are in northeast Thailand (96 per 100,000 men), where it occurs approximately 100 times more often than in the West.
Epidemiologic studies have shown that, globally, the mortality and incidence of intrahepatic biliary tract neoplasms are rising, but those of extrahepatic tumors are static or falling. World Health Organization (WHO) mortality data were examined for the United States, United Kingdom, France, Italy, Japan, and Australia from 1979 to 1998 (Ishak et al, 1994). Mortality rates for intrahepatic cholangiocarcinoma were increased in both sexes in all countries except among Japanese women.
A U.S. study examined trends in intrahepatic cholangiocarcinoma incidence using data from the Surveillance, Epidemiology, and End Results (SEER) program, which represents over 10% of the total U.S. population. Data from 1976 to 2000 were analyzed by age, gender, and ethnicity. The incidence of intrahepatic cholangiocarcinoma increased by 165%, from 0.32 per 100,000 from 1975 through 1979 to 0.85 per 100,000 from 1995 through 1999. This increase was reflected in all groups but was highest in black men (139%), followed by white men (124%) and white women (111%). The increased incidence may be attributable in part to increased detection (Olnes & Erlich, 2004). However, increased detection of a tumor is usually associated with an increase in the proportion of patients with early-stage disease or smaller lesions. The rise in intrahepatic cancer was not associated with a significant change in the proportion of early stage cancers, histologically confirmed tumors, or smaller lesions (Broome et al, 1996). Furthermore, the incidence does not seem to be plateauing, as would be expected if the increase were due to an improvement in diagnostic modalities, such as magnetic resonance imaging (MRI) and computed tomography (CT), which have become established practice in the past several years (Khan et al, 2002a; Shaib & El-Serag, 2004; Taylor-Robinson et al, 2001). Conversely, the incidence of extrahepatic cholangiocarcinoma has shown no such change and may be declining (Tullo et al, 2000). According to the SEER data, U.S. age-standardized mortality rates for extrahepatic tumors fell from 0.6 per 100,000 in 1979 to 0.3 per 100,000 in 1998; and age-standardized incidence rates decreased from 1.08 per 100,000 to 0.82 per 100,000 in the same period.
Most patients have unresectable disease when they are first seen and typically die within 12 months of diagnosis. Sepsis from cholangitis, frequently related to interventions performed for biliary obstruction and progressive liver failure, contribute to the high mortality (Shaib & El-Serag, 2004). Survival is poor, even in resected patients, with less than 5% of patients diagnosed with cholangiocarcinoma surviving to 5 years, a rate which has not changed significantly over the past 30 years. Although cholangiocarcinoma is a relatively rare tumor with an overall dismal prognosis, interest in the biology of this disease has grown in recent years (Khan et al, 2002a; Patel, 2001, 2002; Taylor-Robinson et al, 2001).
Clinical Risk Factors for Cholangiocarcinoma
Primary Sclerosing Cholangitis
Primary sclerosing cholangitis (PSC; see Chapter 41) is the most common condition predisposing to cancer of the biliary tree. Cholangiocarcinoma rates of 8% to 40% have been reported in patients with primary sclerosing cholangitis in follow-up studies and explant specimens (Khan et al, 2002a). Cholangiocarcinoma in such patients tends to present earlier in life, most commonly in the 30- to 50-year age groups, than in sporadic cases (Pitt et al, 1995; Bergquist et al, 1998). Approximately one third of patients with primary sclerosing cholangitis who develop cholangiocarcinoma do so within 2 years of diagnosis, and the risk of carcinogenesis appears to be unrelated to the duration of the inflammatory disease (Pitt et al, 1995; Watanapa & Watanapa, 2002). Two thirds of patients with primary sclerosing cholangitis have associated inflammatory bowel disease, usually ulcerative colitis (Pitt et al, 1995; Watanapa & Watanapa, 2002); however, no association has been found between the risk of cholangiocarcinoma and the severity of inflammatory bowel disease in this cohort of patients (Pitt et al, 1995; Watanapa & Watanapa, 2002).
To date, no reliable factors have been described that reliably stratify cholangiocarcinoma risk in PSC patients, and the clinical diagnosis of cholangiocarcinoma in this setting is difficult. Therefore, molecular markers of cholangiocarcinogenesis, such as KRAS or other mutations, may improve the early diagnosis. In a study by Kubicka and colleagues (2001), KRAS mutations were analyzed by enriched polymerase chain reaction/restriction fragment length polymorphism in the bile fluid of 56 PSC patients and 20 patients with other cholestatic diseases. To assess the value of KRAS mutations as a risk factor for cholangiocarcinogenesis, patients were prospectively investigated over a mean period of 31.5 months. Seventeen patients (30%) with PSC harbored KRAS mutations in bile fluid. In contrast to the group of PSC patients with wild-type KRAS, four cholangiocarcinomas and two cases of bile duct dysplasia were diagnosed in the group of patients with the mutation during the follow-up investigation. Their results indicate that KRAS mutations in bile fluid of PSC patients may represent an early event in cholangiocarcinogenesis; however, most of the PSC patients with KRAS mutations remained tumor free after a long follow-up investigation, consistent with the notion that these mutations are not specific for malignancy but may also occur in normal bile duct mucosa or in dysplasias. Therefore, analysis of KRAS mutations in bile specimens is not useful to make a definitive diagnosis of cholangiocarcinoma in PSC patients, but it is considered a marker of increased risk.
Parasitic Infection
A large body of experimental and epidemiologic data suggest a pathogenic association between cholangiocarcinoma and liver fluke infestation, especially Opisthorcis viverrini and less definitively Clonorchis sinensis (see Chapter 45) (Watanapa, 1996). Most epidemiologic data are from Thailand, which has the highest incidence of cholangiocarcinoma worldwide—87 per 100 000 population—and where an estimated 7 million people are infected with opisthorchiasis (Parkin et al, 1991). Human beings become infected by eating undercooked fish, whereupon adult worms inhabit and lay eggs in the biliary system. In addition to a strongly positive correlation between liver flukes and cholangiocarcinoma in case-control studies (Thamavit et al, 1978), malignant change in the biliary epithelium of Syrian hamsters has been shown after infection with Opisthorcis viverrini, especially when fed nitrosamines (Thamavit, 1988), which are produced by bacteria in fish and other foods and are thought to act as cofactors in carcinogenesis.
Fibrotic and Cystic Liver Hepatobiliary Diseases
Congenital abnormalities of the biliary tree associated with Caroli syndrome, congenital hepatic fibrosis, and choledochal cysts (cystic dilatations of the bile ducts) carry a 15% risk of malignant change after the second decade, at an average age of 34 years (see Chapter 46) (Scott et al, 1980). The overall incidence of cholangiocarcinoma in patients with untreated cysts is up to 28% (Lipsett et al, 1994; Obtsuka et al, 2001). The mechanism of carcinogenesis is unclear but appears to be related to biliary stasis; reflux of pancreatic juice, causing chronic inflammation; activation of bile acids (van Mil et al, 2001); and deconjugation of carcinogens. Bile duct adenomas and biliary papillomatosis are also associated with the development of cholangiocarcinoma.
Bile Salt Transporter Protein Polymorphisms
Bile salt transporter protein polymorphisms in BSEP, ATP8B1, and ABCB4 genes can lead to unstable bile content and deconjugation of xenobiotics previously conjugated in the liver (Jacquemin, 2001; Thompson & Strautnieks, 2001; Meier et al, 2004). In the background of congenital bile duct abnormalities, this process can lead to the development of cholangiocarcinoma at an early stage in life (Kubo et al, 1995). Individuals who are heterozygous for bile salt transporter polymorphisms are thought to have an increased predisposition to cholangiocarcinoma as adults after exposure to cofactors that result in chronic inflammation in the biliary tree (Kubo et al, 1995).
Intrahepatic Biliary Stones
Hepatolithiasis, the presence of interhepatic biliary stones (see Chapters 39 and 44), is rare in the West but relatively common in parts of Asia, and it is associated particularly with peripheral intrahepatic cholangiocarcinoma (Shaib & El-Serag, 2004). Up to 10% of patients with hepatolithiasis develop cholangiocarcinoma (Chen, 1999). In Taiwan, up to 70% of patients with cholangiocarcinoma undergoing resection reportedly have intrahepatic biliary stones; in Japan this figure is 6% to 18% (Okuda et al, 2002; Khan et al, 2003). Biliary stones are thought to cause bile stasis, predisposing to recurrent bacterial infections and subsequent inflammation, a potential cofactor for carcinogenesis (Khan et al, 2003).
Chemical Carcinogen Exposure
Several chemical toxins have been associated with cholangiocarcinoma. Promutagenic DNA adducts have been identified in cholangiocarcinoma tissue, indicating exposure to DNA-damaging agents (Sahani, 2003). Thorotrast, a radiologic contrast agent banned in the 1960s for its carcinogenic properties, has been strongly associated with the development of cholangiocarcinoma many years after exposure and has been reported to increase the risk of cholangiocarcinogenesis to 300 times that of the general population (Shaib & El-Serag, 2004; Hardell et al, 1984). Associations have also been made with exposure to byproducts from the rubber and chemical industries, including dioxins and nitrosamines (Sorensen et al, 1998), and with alcohol and smoking (Chalasani et al, 2000), but results have been conflicting, and no firm conclusions can as yet be drawn.
Viral Hepatitis
Chronic liver disease from any cause has also been associated with cholangiocarcinoma (Shin et al, 1996). A cohort study of over 11,000 patients with cirrhosis, followed up over 6 years, showed a tenfold risk compared with the general population (Shin et al, 1996). More specifically, an association has been shown with chronic hepatitis B and C virus infection. A case-control study from Korea reported that 12.5% of patients with cholangiocarcinoma tested positive for hepatitis C virus (HCV) and 13.8% for hepatitis B virus surface antigen (HBsAg) compared with 3.5% and 2.3% of controls, respectively (Donato et al, 2001). In a second case-control study from Italy, 23% of patients with cholangiocarcinoma were positive for anti-HCV, and 11.5% were HBsAg positive compared with 6% and 5.5% of controls, respectively (Kobayashi et al, 2000). A prospective, controlled study from Japan reported the risk of developing cholangiocarcinoma in patients with cirrhosis related to HCV as 3.5% at 10 years—1000 times greater than the risk in the general population (Yin & Chen, 1998). HCV is an established risk factor for hepatocellular carcinoma, and both hepatocytes and cholangiocytes have the same progenitor cell, supporting a role for the virus in cholangiocarcinogenesis. Furthermore, RNA from HCV has been identified in cholangiocarcinoma tissue (Berthiaume & Wands, 2004). A U.S. case-controlled study of risk factors for intrahepatic cholangiocarcinoma showed adjusted odds ratios (ORs) of 6.1 for HCV carriers and 5.9 for HIV-infected individuals. Diabetes was also prevalent in patients with intrahepatic tumors, with an adjusted OR of 2.0; the pathogenic mechanisms are unclear in diabetics but may be related to an associated increased risk of steatohepatitis (Tulio et al, 2000).
Molecular Pathogenesis
Genome-wide Analysis
Investigation into the molecular pathogenesis of biliary tract tumors has not been an area of vigorous investigation, but slow progress is being made in the understanding of the genetic basis of this disease. A recent study investigated genome-wide mutations in carcinomas of the biliary tree, including intrahepatic and extrahepatic cholangiocarcinoma and gallbladder cancer, using frozen specimens from patients who underwent surgical resection (Miller et al, 2009). Unsupervised hierarchical clustering analysis revealed that cancers from these different sites did not cluster separately, implying that there was no difference in the global gene expression patterns between the biliary cancer subgroups. Figure 8B.1 depicts the top 40 upregulated and downregulated genes for all cancers combined versus normal biliary epithelia control specimens. Although the individual cancer subtypes did not cluster separately, unique differential expression of many genes was seen, compared with normal biliary epithelium in each of the tumor types.

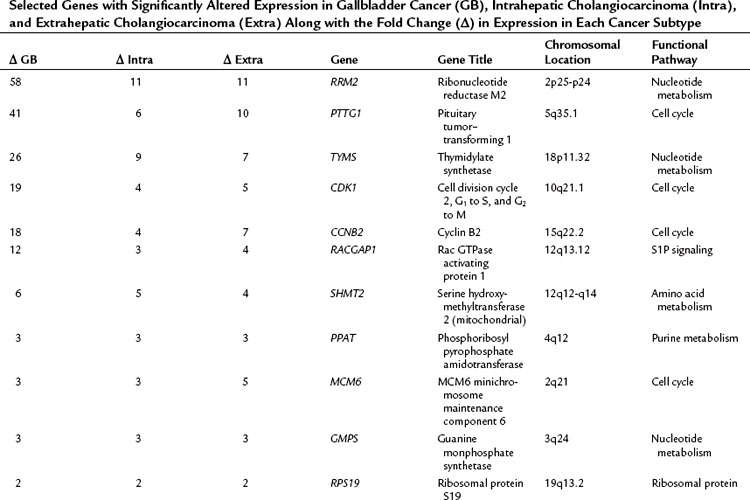
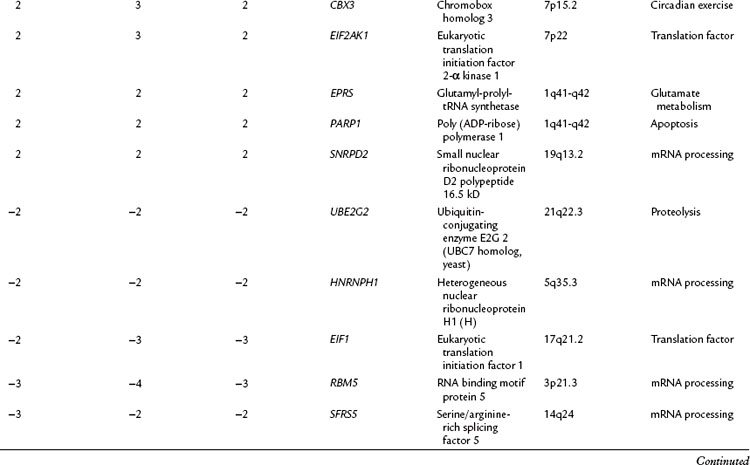
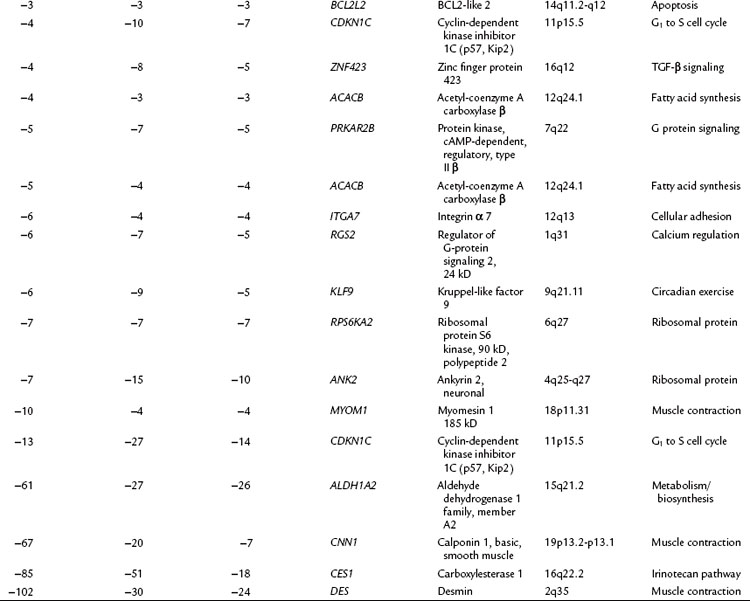
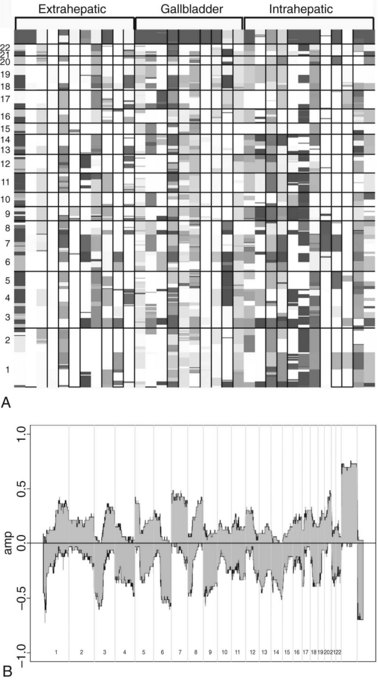
The relationship of gene transcriptional changes among the three biliary cancer subtypes is depicted in a Venn diagram (Fig. 8B.2). Overall, 165 probe sets were commonly differentially expressed in all three cancer types. Selected commonly differentially expressed genes are listed in Table 8B.1. This study also investigated alterations in gene copy number in biliary tract cancers using array-based comparative genome hybridization (CGH) analysis. Considerable heterogeneity in the extent of chromosomal instability between patients was found, even within the same subtype. For example, some patients had mutations in nearly every chromosomal arm, and other patients with the same diagnosis had structural genomic changes (Fig. 8B.3A). But despite the heterogeneity, segments of chromosomes 3p, 6q, 8p, 9p, and 14q were commonly deleted across subtypes of biliary cancers. Commonly amplified regions across cancer types include segments of 1q, 3q, 5p, 7p, 7q, 8q, and 20q (Fig. 8B.3B).
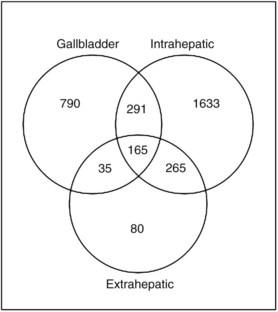
Table 8B.1 Selected Genes with Significantly Altered Expression in Gallbladder Cancer (GB), Intrahepatic Cholangiocarcinoma (Intra), and Extrahepatic Cholangiocarcinoma (Extra) Along with the Fold Change (Δ) in Expression in Each Cancer Subtype
Oncogenes and Tumor Suppressor Genes
Several studies have shown abnormal expression of the KRAS oncogene in 21% to 100% of cases of cholangiocarcinoma and of the TP53 tumor suppressor gene in up to 37% of archival specimens (Isa et al, 2002). These genetic alterations are associated with a more aggressive phenotype in biliary tract tumors (Isa et al, 2002). KRAS and TP53 mutations have also been identified in bile and pancreatic juice of affected patients (Isa et al, 2002; Itoi et al, 1999), but neither KRAS nor TP53 mutational analysis have been shown to be superior to conventional cytopathology in the diagnosis of pancreaticobiliary tumors, but combined analysis of both can increase sensitivity in tissue biopsy and bile specimens (Isa et al, 2002; Itoi et al, 1999; Aishima et al, 2002). Increased expression of c-MET and c-ERBB2 protooncogenes have been shown in cholangiocarcinoma specimens, and it has been suggested that they participate in the metastatic transformation of intrahepatic tumors (Aishima et al, 2002). Point mutations leading to promoter methylation of cell-cycle regulators p16INK4a and p14ARF have also been identified in cholangiocarcinoma related to PSC. Overexpression of the protooncogene BCL2 has been reported to reduce apoptosis in cholangiocarcinoma cell lines (Okaro et al, 2001), and although human cholangiocarcinoma does not express Bcl-2, other antiapoptotic proteins, Mcl-1 and Bcl-xl, are expressed (Taniai et al, 2002).
Genetic Polymorphism at P450
Similar to the development of other tumors, cholangiocarcinogenesis is thought to be a multistep process dependent on the interaction between environmental factors and host genetic factors. Most of the putative environmental risk factors for cholangiocarcinoma cause chronic biliary irritation and can contribute to a promotional stage of carcinogenesis. Genetic polymorphisms in the cytochrome P450 enzymes or in the bile salt transporter proteins are thought to lead to alterations in the efficiency with which environmental toxins (xenobiotics) are handled by the liver. The development of cholangiocarcinoma probably needs a “second hit” to deconjugate such xenobiotics and to expose the bile duct epithelium to damage. Putative secondary hits include chronic inflammation, viral hepatitis, parasitic infestation, and recurrent cholangitis (Khan et al, 2005).
MRP2/ABCC2, MUTYH, NEIL1, and AID
Hoblinger and colleagues (2009) reported that multidrug resistance–associated protein 2 (MRP2/ABCC2) is one of the ATP-binding cassette (ABC) transporters expressed on the apical membrane of hepatocytes and cholangiocytes. ABCC2 plays an important role in the biliary clearance of endogenous and exogenous toxic compounds. The ABCC2 variant c.3972C>T in exon 28 has been shown to be associated with the risk of carcinogenesis. Forsbring and colleagues (2009) recently found that the human h-MUTYH and NEIL1 genes encode DNA glycosylases involved in repair of oxidative base damage, and mutations in these genes are associated with cholangiocarcinoma.
Other work has also suggested that chronic inflammation can play a critical role leading up to cholangiocarcinogenesis. Komori and colleagues (2008) from Japan found that the proinflammatory cytokine–induced aberrant production of activation-induced cytidine deaminase (AID), a member of the DNA/RNA-editing enzyme family, might link bile duct inflammation to an enhanced genetic susceptibility to mutagenesis, leading to cholangiocarcinoma. Ectopic AID production is induced in response to TNF-α stimulation via an NF-κB–dependent pathway. Aberrant expression of AID in biliary cells results in the generation of somatic mutations in tumor-related genes, including TP53, c-MYC, and the promoter region of the CDK2NA sequences. In human tissue specimens, reverse transcription polymerase chain reaction (RT-PCR) analyses revealed that AID was increased significantly in 28 of 30 cholangiocarcinoma tissues (93%), whereas only trace amounts of AID were detected in the normal liver. Immunohistochemistry showed that all of the cholangiocarcinoma tissue samples examined showed overproduction of endogenous AID protein in cancer cells. Moreover, immunostaining for AID was detectable in 16 of 20 bile epithelia in PSC.
Human CYP1A2 and Arylamine N-Acteyltransferases (NAT1 and NAT2)
The CYP1A2, NAT1, and NAT2 genes have been shown to be potential modifiers of an individual’s susceptibility to certain types of cancers. Prawan and colleagues (2005) evaluated the relationship between CYP1A2, NAT1, and NAT2 polymorphisms and cholangiocarcinoma in Thailand. A total of 216 cholangiocarcinoma patients and 233 control subjects were genotyped using PCR. Two CYP1A2 alleles (CYP1A2*1A wild type and *1F), six NAT1 alleles (NAT1*4 wild type, *3, *10, *11, *14A, and *14B), and seven NAT2 alleles (NAT2*4 wild type, *5, *6A, *6B, *7A, *7B, and *13), were analyzed. The CYP1A2*1A/*1A genotype conferred a decreased risk of the cancer (adjusted OR, 0.28; 95% confidence interval [CI], 0.08 to 0.94), compared with CYP1A2*1F/1*F. Frequency distributions of rapid NAT2*13 and two slow alleles, *6B and *7A, were associated with lower cholangiocarcinoma risk. This study suggested that the NAT2 polymorphism may be a modifier of individual risk of cholangiocarcinoma.
Trefoil Factor Family (TFF1)
Trefoil factor family 1 (TFF1) is critical for mucosal protection and tumor suppression in the stomach. To examine its role in cholangiocarcinogenesis, specimens with varying degrees of dysplasia were examined: intrahepatic cholangiocarcinoma (ICC) as a result of hepatolithiasis, biliary epithelial dysplasia with hepatolithiasis, hepatolithiasis without dysplasia or carcinoma, cholangiocarcinoma without hepatolithiasis, and control normal livers (Sasaki et al, 2003). TFF1 expression in the biliary epithelium was increased in hepatolithiasis compared with control livers (P < .01). In biliary epithelial dysplasia and noninvasive intrahepatic cholangiocarcinoma with hepatolithiasis, TFF1 was extensively expressed, and MUC5AC gastric mucin was usually colocalized with TFF1. However, TFF1 expression was significantly decreased in invasive intrahepatic cholangiocarcinoma, despite preserved expression of MUC5AC. A total of four missense mutations were detected: three were found in two noninvasive intrahepatic cholangiocarcinomas with hepatolithiasis (29%) and one in invasive ICC (11%). Loss of heterozygosity of the TFF1 gene was not detectable. The decreased expression of TFF1 in invasive intrahepatic cholangiocarcinoma may be explained by the methylation of the TFF1 promoter region. Upregulation of TFF1 coupled with MUC5AC in biliary epithelium in hepatolithiasis, biliary epithelial dysplasia, and noninvasive intrahepatic cholangiocarcinoma may reflect gastric metaplasia and early neoplastic lesion. Under such conditions, decreased TFF1 expression may lead to increased cell proliferation and then to the invasive character of intrahepatic cholangiocarcinoma.
Aishima SI, et al. c-erbB-2 and c-Met expression relates to cholangiocarcinogenesis and progression of intrahepatic cholangiocarcinoma. Histopathology. 2002;40:269-278.
Bergquist A, et al. Risk factors and clinical presentation of hepatobiliary carcinoma in patients with primary sclerosing cholangitis: a case-control study. Hepatology. 1998;27:311-316.
Berthiaume EP, Wands J. The molecular pathogenesis of cholangiocarcinoma. Semin Liver Dis. 2004;24:127-137.
Bismuth H, Nakache R, Diamond T. Management strategies in resection for hilar cholangiocarcinoma. Ann Surg. 1992;215:31.
Broome U, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut. 1996;38:610-615.
Chalasani N, et al. Cholangiocarcinoma in patients with primary sclerosing cholangitis: a multicenter case-control study. Hepatology. 2000;31:7-11.
Chen MF. Peripheral cholangiocarcinoma (cholangiocellular carcinoma): clinical features, diagnosis and treatment. J Gastroenterol Hepatol. 1999;14:1144-1149.
Donato F, et al. Intrahepatic cholangiocarcinoma and hepatitis C and B virus infection, alcohol intake, and hepatolithiasis: a case-control study in Italy. Cancer Causes Control. 2001;12:959-964.
Forsbring M, et al. Catalytically impaired hMYH and NEIL1 mutant proteins identified in patients with primary sclerosing cholangitis and cholangiocarcinoma. Carcinogenesis. 2009;30(7):1147-1154. Epub 2009 May 14
Hardell L, et al. Aetiological aspects on primary liver cancer with special regard to alcohol, organic solvents and acute intermittent porphyria: an epidemiological investigation. Br J Cancer. 1984;50:389-397.
Hoblinger A, et al. Association of the c.3972C>T variant of the multidrug resistance–associated protein 2 gene (MRP2/ABCC2) with susceptibility to bile duct cancer. Digestion. 2009;80(1):36-39. Epub 2009 May 19
Ishak KK, Anthony PP, Sobin LH. Histological Typing of Tumours of the Liver: WHO International Histological Classification of Tumours. Berlin: Springer-Verlag; 1994.
Isa T, et al. Analysis of microsatellite instability, K-ras gene mutation and p53 protein overexpression in intrahepatic cholangiocarcinoma. Hepatogastroenterol. 2002;49:604-608.
Itoi T, et al. K-ras codon 12 and p53 mutations in biopsy specimens and bile from biliary tract cancers. Pathol Int. 1999;49:30-37.
Jacquemin E. Role of multidrug resistance 3 deficiency in pediatric and adult liver disease: one gene for three diseases. Semin Liver Dis. 2001;21:551-562.
Khan SA, et al. Changing international trends in mortality rates for liver, biliary and pancreatic tumours. J Hepatol. 2002;37:806-813.
Khan SA, et al. Guidelines for the diagnosis and management of cholangiocarcinoma. Gut. 2002;51(Suppl 6):VI1-VI9.
Khan SA, et al. DNA adducts, detected by 32P postlabelling, in human cholangiocarcinoma. Gut. 2003;52:586-591.
Khan SA, et al. p53 Mutations in human cholangiocarcinoma: a review. Liver Int. 2005;25(4):704-716.
Kobayashi M, et al. Incidence of primary cholangiocellular carcinoma of the liver in Japanese patients with hepatitis C virus–related cirrhosis. Cancer. 2000;88:2471-2477.
Komori J, et al. Activation-induced cytidine deaminase links bile duct inflammation to human cholangiocarcinoma. Hepatology. 2008;47(3):888-896.
Kubicka S, et al. K-ras mutations in the bile of patients with primary sclerosing cholangitis. Gut. 2001;48(3):403-408.
Kubo S, et al. Hepatolithiasis associated with cholangiocarcinoma. World J Surg. 1995;19:637-641.
Lipsett PA, et al. Choledochal cyst disease: a changing pattern of presentation. Ann Surg. 1994;220:644-652.
Liver Cancer Study Group of Japan. The general rules for the clinical and pathological study of primary liver cancer, 4th ed. Tokyo: Kanehra; 2000.
Meier Y, et al. Hepatocellular malignancy in ABCB11/BStP disease (progressive familial intrahepatic cholestasis, type 2): four patients. Hepatology. 2004;40(Supp1 1):471A.
Miller G, et al. Genome wide analysis and clinical correlation of chromosomal and transcriptional mutations in cancers of the biliary tract. J Exp Clin Cancer Res. 2009;28:62.
Nakeeb A, et al. Cholangiocarcinoma: a spectrum of intrahepatic, perihilar, and distal tumors. Ann Surg. 1996;224:463-473.
Ohtsuka T, et al. Carcinoma arising in choledochocele. Endoscopy. 2001;33:614-619.
Okaro A, et al. The expression of anti-apoptotic proteins (Bcl-2, Bcl-xl, Mcl-1) by benign, dysplastic and malignant biliary epithelium. J Clin Pathol. 2001;54:927-932.
Okuda K, Nakanuma Y, Miyazaki M. Cholangiocarcinoma: recent progress. Part 1: epidemiology and etiology. J Gastroenterol Hepatol. 2002;17:1049-1055.
Olnes MJ, Erlich R. A review and update on cholangiocarcinoma. Oncology. 2004;66:167-179.
Parkin DM, et al. Liver cancer in Thailand I: a case-control study of cholangiocarcinoma. Int J Cancer. 1991;48:323-328.
Patel T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology. 2001;33:1353-1357.
Patel T. Worldwide trends in mortality from biliary tract malignancies. BMC Cancer. 2002;2:10.
Pitt HA, et al. Malignancies of the biliary tree. Curr Probl Surg. 1995;32:1-90.
Prawan A, et al. Association between genetic polymorphisms of CYP1A2, arylamine N-acetyltransferase 1 and 2 and susceptibility to cholangiocarcinoma. Eur J Cancer Prev. 2005;14(3):245-250.
Sahani D, et al. Thorotrast-induced cholangiocarcinoma: case report. Abdom Imaging. 2003;28:72-74.
Scott J, et al. Bile duct carcinoma: a late complication of congenital hepatic fibrosis: case report and review of literature. Am J Gastroenterol. 1980;73:113-119.
Sasaki M, Tsuneyama K, Nakanuma Y. Aberrant expression of trefoil factor family 1 in biliary epithelium in hepatolithiasis and cholangiocarcinoma. Lab Invest. 2003;83(10):1403-1413.
Shaib Y, El-Serag HB. The epidemiology of cholangiocarcinoma. Semin Liver Dis. 2004;24:115-125.
Shin HR, et al. Hepatitis B and C virus, Clonorchis sinensis for the risk of liver cancer: a case-control study in Pusan, Korea. Int J Epidemiol. 1996;25:933-940.
Sorensen HT, et al. Risk of liver and other types of cancer in patients with cirrhosis: a nationwide cohort study in Denmark. Hepatology. 1998;28:921-925.
Taniai M, et al. p16INK4a promoter mutations are frequent in primary sclerosing cholangitis (PSC) and PSC-associated cholangiocarcinoma. Gastroenterol. 2002;123:1090-1098.
Taylor-Robinson SD, et al. Increase in mortality rates from intrahepatic cholangiocarcinoma in England and Wales 1968-1998. Gut. 2001;48:816-820.
Thamavit W, et al. Effects of dimethylnitrosamine on induction of cholangiocarcinoma in Opisthorchis viverrini–infected Syrian golden hamsters. Cancer Res. 1978;38:4634-4639.
Thamavit W, et al. Enhancement of DHPN induced hepatocellular, cholangiocellular and pancreatic carcinogenesis by Opisthorchis viverrini infestation in Syrian golden hamsters. Carcinogenesis. 1988;9(6):1095-1098. 1988 Jun
Thompson R, Strautnieks S. BSEP: function and role in progressive familial intrahepatic cholestasis. Semin Liver Dis. 2001;21:545-550.
van Mil SW, et al. FIC1 disease: a spectrum of intrahepatic cholestatic disorders. Semin Liver Dis. 2001;21:535-544.
Watanapa P. Cholangiocarcinoma in patients with opisthorchiasis. Br J Surg. 1996;83:1062-1064.
Yin F, Chen B. Detection of hepatitis C virus RNA sequences in hepatic portal cholangiocarcinoma tissue by reverse transcription polymerase chain reaction. Chin Med J (Engl). 1998;111:1068-1070.