Local Anesthetics and Opioids
Brenda A. Bucklin MD, Alan C. Santos MD, MPH
Chapter Outline
Local anesthetics and opioids are often used for pain relief in obstetric practice. Local anesthetics may be used for infiltration anesthesia, peripheral (pudendal) nerve block, or neuraxial block, whereas opioids are administered both systemically and neuraxially. The physiologic changes that occur during pregnancy may affect the pharmacology of both local anesthetics and opioids. In turn, these analgesic drugs may have effects on the mother and the fetus.
Local Anesthetics
Molecular Structure
All local anesthetic molecules except cocaine contain a desaturated carbon ring (aromatic portion) and a tertiary amine connected by an alkyl chain (Figure 13-1). The intermediate alkyl chain, by virtue of its ester or amide linkage, is the basis for the classification of local anesthetics as amino-esters (which are hydrolyzed by pseudocholinesterase) and amino-amides (which undergo hepatic microsomal metabolism) (Table 13-1). The aromatic ring of the esters, which renders the molecule lipid soluble, is a derivative of benzoic acid. The amide aromatic ring is a homologue of aniline. The tertiary-amine portion acts as a proton acceptor; thus, local anesthetics behave as weak bases. In its quaternary (i.e., “protonated”) form, the terminal amine is the water-soluble portion. The Henderson-Hasselbalch equation predicts the relative proportions of local anesthetic that exist in the ionized and un-ionized form. The higher the pKB (base dissociation constant) relative to physiologic pH, the smaller the proportion of drug that exists in the un-ionized form. All amide local anesthetics (with the exception of lidocaine) exist as stereoisomers because of the presence of an asymmetric carbon adjacent to the terminal amine.
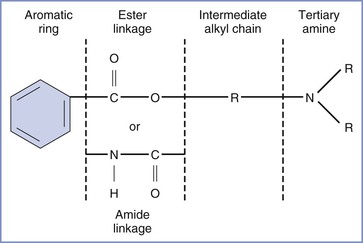
FIGURE 13-1 Structure of the molecule of a local anesthetic. R, alkyl group. (Modified from Santos AC, Pedersen H. Local anesthetics in obstetrics. In Petrie RH, editor. Perinatal Pharmacology. Cambridge, MA, Blackwell Scientific, 1989:373.)
TABLE 13-1
Physicochemical Characteristics and Fetal-to-Maternal (F/M) Blood Concentration Ratios at Delivery for Commonly Used Local Anesthetic Agents
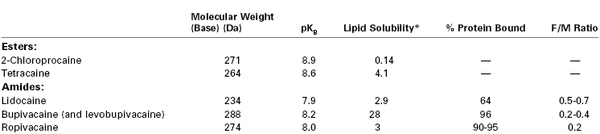
* N-heptane/pH = 7.4 buffer.
Modified from Santos AC, Pedersen H. Local anesthetics in obstetrics. In Petrie RH, editor. Perinatal Pharmacology. Cambridge, MA, Blackwell Scientific, 1989:375.
Clinical formulations of local anesthetics are prepared as hydrochloride salts to increase their solubility in water. These solutions are usually acidic (i.e., pH of 4 to 6) to enhance formation of the water-soluble quaternary amine and to prevent oxidation of the epinephrine present in epinephrine-containing solutions.
Chirality
With the exception of lidocaine, amide local anesthetics are known as chiral compounds because they have a single asymmetric carbon adjacent to the amino group and thus exist in isomeric forms that are mirror images of each other. The direction in which the isomers rotate polarized light distinguishes them as either dextrorotary (D) or levorotary (L) isomers. This distinction is important, because individual isomers of the same drug may have different biologic effects. As a rule, the levorotary isomer of a drug has greater vasoconstrictor activity and a longer duration of action but less potential for systemic toxicity than the dextrorotary form.1
In the past, single-isomer formulations were costly to produce; and for that reason, local anesthetics used clinically (e.g., bupivacaine) have contained a racemic mixture of both the dextrorotary and levorotary forms of the drug. However, with improved techniques of selective extraction, two commercially available single-isomer formulations of local anesthetic are now available, ropivacaine and levobupivacaine. Levobupivacaine is the levorotary isomer of bupivacaine; it is currently not marketed in the United States. Ropivacaine is a homologue of mepivacaine and bupivacaine, but, unlike these other local anesthetics, it is formulated as a single levorotary isomer rather than as a racemic mixture. A propyl group on the pipechol ring distinguishes ropivacaine from bupivacaine (which has a butyl group) and mepivacaine (which has a methyl group).2 Thus, it is not surprising that the physicochemical characteristics of ropivacaine are intermediate between those of mepivacaine and bupivacaine.
The reduction in systemic toxicity observed with administration of the levorotary isomers may be both drug and concentration dependent. For example, one study in isolated guinea pig hearts noted that bupivacaine isomers lengthened atrioventricular conduction time more than ropivacaine isomers did. In contrast to other measured variables, “atrioventricular conduction time showed evident stereoselectivity” for bupivacaine at the lowest concentration studied (0.5 µM) but only at much higher concentrations for ropivacaine (> 30 µM).3
Mechanism of Action
At rest, the interior of a nerve cell is negatively charged in relation to its exterior. This resting potential of 60 to 90 mV exists because the concentration of sodium in the extracellular space greatly exceeds that in the intracellular space. The converse is true for potassium. Excitation results in the opening of membrane channels, which allows sodium ions to flow freely down their concentration gradient into the cell interior. Thus, the electrical potential within the nerve cell becomes less negative until, at the critical threshold, rapid depolarization occurs. This depolarization is needed to initiate the same sequence of events in adjacent membrane segments and for propagation of the action potential. Thereafter, sodium channels close and the membrane once again becomes impermeable to the influx of sodium. The negative resting membrane potential is reestablished as sodium is removed from the cell by active transport. At the same time, potassium passively accumulates within the resting cell.
Interference with sodium-ion conductance appears to be the mechanism by which local anesthetics reversibly inhibit the propagation of the action potential. Four major theories attempt to explain this effect. The most prominent hypothesis is that the local anesthetic interacts with receptors in the nerve cell membrane that control channels involved in sodium conductance.4 There may be more than one site at which local anesthetics bind to sodium-channel receptors (Figure 13-2).
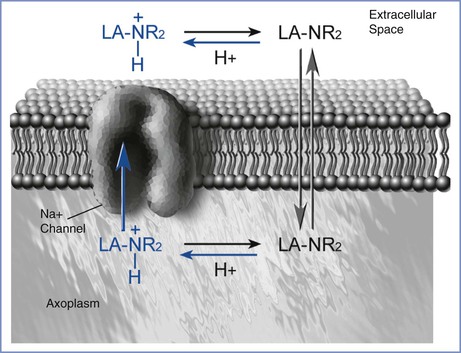
FIGURE 13-2 Local anesthetic access to the sodium channel. The uncharged molecule (LA-NR2) diffuses most easily across the lipid membrane and interacts with the sodium channel at an intramembranous site. The charged molecule (LA-NR2H+) gains access to a specific receptor on the sodium channel in the intracellular space. (Drawing by Naveen Nathan, MD, Northwestern University Feinberg School of Medicine, Chicago, IL.)
The Meyer-Overton theory offers a second explanation for local anesthetic action. This hypothesis suggests that the lipid-soluble portion of the local anesthetic molecule expands the cell membrane and interferes with rapid sodium conductance. A third possibility is that local anesthetics may alter the membrane surface charge, a change that would inhibit propagation of the action potential. Fourth, local anesthetics may displace calcium from sites that control sodium conductance.
Both the un-ionized and ionized forms of a local anesthetic are involved in pharmacologic activity. The un-ionized base, which is lipid soluble, diffuses through the cell membrane, whereas the charged form is much more active in blocking the sodium channel.
Pharmacodynamics
Pregnant women typically require smaller doses of local anesthetic compared with nonpregnant women for neuraxial blockade. This effect may be evident as early as the second trimester.5,6 This difference has been attributed to enhanced spread of local anesthetic due to epidural venous engorgement. However, mechanical effects alone do not account for the observation that the spread of spinal and epidural analgesia in early pregnancy is similar to that in pregnant women at term.5–7 In fact, pregnancy may also enhance neuronal sensitivity to local anesthetics. For example, pregnancy increases median nerve susceptibility to lidocaine.8 In vitro studies demonstrated that the onset of neural blockade was faster, and lower concentrations of bupivacaine were required to block vagal fibers, in pregnant rabbits than in nonpregnant rabbits.9
Hormonal and biochemical changes may be responsible for the greater susceptibility to neural blockade during pregnancy. For example, one study demonstrated an enhanced effect of bupivacaine in isolated vagus fibers from nonpregnant, ovariectomized rabbits who had received long-term (4 days) but not short-term exposure to progesterone.10 A higher pH and lower bicarbonate and total carbon dioxide content have been demonstrated in cerebrospinal fluid (CSF) from women undergoing cesarean delivery than in CSF from age-matched nonpregnant controls. A higher pH increases the proportion of local anesthetic that exists as the base form and facilitates diffusion of the drug across nerve membranes.7
Pharmacokinetics
Pregnancy is associated with progressive physiologic adaptations that may influence drug disposition (see Chapter 2). However, it is difficult to predict with certainty the effects of pregnancy on the pharmacokinetics of an individual drug.
2-Chloroprocaine
2-Chloroprocaine is hydrolyzed rapidly by plasma pseudocholinesterase to chloroaminobenzoic acid and H2O. The in vitro half-life of 2-chloroprocaine in sera from men is less than 15 seconds.11 Although pregnancy is associated with a 30% to 40% decrease in pseudocholinesterase activity, the half-life of 2-chloroprocaine in maternal plasma in vitro is 11 to 21 seconds. After epidural injection, the half-life of 2-chloroprocaine in the mother ranges from 1.5 to 6.4 minutes.12 The longer half-life after epidural administration results from continued absorption of the drug from the injection site. Administration of 2-chloroprocaine to patients with low pseudocholinesterase activity may result in prolonged local anesthetic effect and a greater potential for systemic toxicity.13
Lidocaine
The volume of the central compartment and the volume of distribution are greater in pregnant ewes than in nonpregnant ewes.14,15 Bloedow et al.15 observed that the total body clearance of lidocaine was similar in the two groups of animals. They concluded that the elimination half-life of lidocaine, which depends on the balance between volume of distribution and clearance, was longer in pregnant ewes.15 In contrast, Santos et al.14 concluded that the elimination half-life of lidocaine was similar in the two groups of sheep because the total body clearance of the drug was greater in pregnant animals than in nonpregnant animals. This discrepancy could result from differences in the complexity of the surgical preparation and the allowed recovery period. In pregnant women, the elimination half-life of lidocaine after epidural injection is approximately 114 minutes.16
Lidocaine is metabolized to two active compounds, monoethylglycinexylidide (MEGX) and glycinexylidide (GX). Monoethylglycinexylidide can be detected in maternal plasma within 10 to 20 minutes after neuraxial injection of lidocaine, whereas glycinexylidide can be detected within 1 hour of epidural injection but rarely after subarachnoid injection.17,18 Urinary excretion of unchanged lidocaine is negligible in sheep (i.e., < 2% of the administered dose) and is not affected by pregnancy.14
The physiologic changes that occur during pregnancy are progressive. However, little information is available about the pharmacokinetics of local anesthetics before term. In one study, total clearance of lidocaine was similar at 119 and 138 days’ gestation in gravid ewes (term is 148 days).19
Lidocaine is predominantly bound to alpha1-acid glycoprotein (AAG) in plasma.20 Pregnancy leads to a decreased concentration of AAG; thus, the free plasma fraction of lidocaine is higher in term pregnant women than in nonpregnant controls.20 The increase in the free fraction of lidocaine occurs early in gestation and is progressive.21
Bupivacaine
At least two studies compared the pharmacokinetics of bupivacaine after epidural administration in pregnant and nonpregnant women.22,23 The absorption rate, the area under the concentration-time curve, and the elimination half-life (12 to 13 hours) were similar in the two groups. The elimination half-life of bupivacaine after epidural administration is much longer than that reported after intravenous injection, largely because the drug is continuously absorbed over time from the epidural space.
After intravenous injection, the volume of distribution of bupivacaine is lower in pregnant sheep than in nonpregnant sheep.24 In contrast, ovine pregnancy is associated with a greater volume of distribution of lidocaine.14,15 The differences in gestational effects on the volume of distribution of the two local anesthetics may result from the greater binding of bupivacaine to plasma proteins during gestation (whereas the converse occurs with lidocaine).24 In one study, urinary excretion of unchanged bupivacaine was not affected by pregnancy and was less than 1% of the administered dose.22 Nonetheless, low concentrations of bupivacaine may be detected in the urine of pregnant women for as long as 3 days after delivery.25
Bupivacaine undergoes dealkylation in the liver to 2,6-pipecolyxylidide (PPX). After epidural injection of bupivacaine for cesarean delivery, PPX was detected in maternal plasma within 5 minutes and remained detectable for as long as 24 hours.25 With the lower doses required for labor analgesia, PPX was found only if the block was maintained with multiple reinjections during a period that exceeded 4 hours.26 Pregnancy may affect metabolism of bupivacaine.22 For example, pregnant women have higher serum PPX concentrations, but the unconjugated 4-hydroxy metabolite is not produced in significant amounts. The reason for this finding is unclear but may be related to the effects of hormonal changes on hepatic enzyme systems. Both progesterone and estradiol are competitive inhibitors of microsomal oxidases, whereas reductive enzymes are induced by progesterone.24 Bupivacaine is bound extensively to AAG and albumin.27 This protein binding is reduced during late pregnancy in humans.28
Long-acting pipechol amide local anesthetics, such as bupivacaine, are beneficial for neuraxial labor analgesia because they produce a relative motor-sparing block as compared with other local anesthetics. The effective dose in 50% of cases (ED50) for motor block after intrathecally administered bupivacaine was lower in pregnant than in nonpregnant women (3.96 mg and 4.14 mg, respectively).29
Ropivacaine
Pregnant sheep have a smaller volume of distribution and a slower clearance of ropivacaine than nonpregnant animals.24 However, the relationship between volume of distribution and clearance is such that the elimination half-life is similar in pregnant and nonpregnant animals.
After intravenous injection in laboratory animals, the elimination half-life of ropivacaine is shorter than that of bupivacaine.24,30 Similar findings have been described after intravenous injection in nonpregnant human volunteers.31 The shorter elimination half-life of ropivacaine has been attributed to a faster clearance and a shorter mean residence time than for bupivacaine.24
Peak plasma concentration (Cmax) after epidural administration of 0.5% ropivacaine and 0.5% bupivacaine for cesarean delivery are similar (1.3 µg/mL and 1.1 µg/mL, respectively).32 The elimination half-life of ropivacaine is 5.2 ± 0.6 hours, which is shorter than that for bupivacaine, at 10.9 ± 1.1 hours. No difference in clearance between the two drugs has been noted.
Like bupivacaine, ropivacaine is metabolized by hepatic microsomal cytochrome P450. The major metabolite is PPX, and minor metabolites are 3′- and 4′-hydroxy-ropivacaine.33
Ropivacaine is highly bound (approximately 92%) to plasma proteins but less so than bupivacaine (96%).34 Indeed, at plasma concentrations occurring during epidural anesthesia for cesarean delivery, the free fraction of ropivacaine is almost twice that of bupivacaine.32 In sheep, pregnancy is associated with a greater binding of ropivacaine (and bupivacaine) to plasma proteins.24 In pregnant women undergoing epidural analgesia, the free fraction of ropivacaine decreases as the concentration of AAG increases, up to the point at which the receptors are saturated.35 However, there is little correlation between the free fraction and umbilical cord blood levels of ropivacaine at delivery.35
Effect of Histamine (H2)-Receptor Antagonists
Histamine (H2)-receptor antagonists (e.g., cimetidine, ranitidine, famotidine) are administered to increase gastric pH and reduce the parturient’s risk for aspiration pneumonitis. Drug disposition may be affected by binding to hepatic cytochrome P450, thereby reducing hepatic blood flow and renal clearance, especially with cimetidine. However, short-term administration of H2-receptor antagonists does not alter the pharmacokinetics of amide local anesthetics in pregnant women.36,37
Effects of Preeclampsia
Pathophysiologic changes associated with preeclampsia (e.g., reduced hepatic blood flow, abnormal liver function, decreased intravascular volume) may also affect maternal blood concentrations of local anesthetics (see Chapter 36). For example, Ramanathan et al.38 found that total body clearance of lidocaine after epidural injection was significantly lower in preeclamptic women than in normotensive women; however, the elimination half-life of lidocaine was similar in the two groups. Nonetheless, decreased clearance may result in greater drug accumulation with repeated injections of lidocaine in women with preeclampsia. In contrast, long-acting amides have a relatively low hepatic extraction, and changes in liver blood flow with preeclampsia may have less effect on the metabolic clearance.
Effect of Diurnal Variation
Pain may exhibit temporal variation in intensity due to diurnal neuroendocrine or external factors. Further, the pharmacokinetics and pharmacodynamics of local anesthetics may exhibit temporal patterns (i.e., chronobiology). In one study, the duration of action of epidural bupivacaine was approximately 25% longer when it was administered between 7:00 AM and 7:00 PM than between 7:00 PM and 7:00 AM.39 In contrast, another study found no diurnal variation with intrathecal bupivacaine administered for labor analgesia.40 The authors suggested that observed temporal differences in duration of analgesia may be explained by external influences such as shift changes for nurses and anesthesiologists.40
Toxicity
Systemic absorption or intravascular injection of a local anesthetic may result in local anesthetic systemic toxicity (LAST). Toxicity most often involves the central nervous system (CNS), but cardiovascular toxicity also may occur. Less common are tissue toxicity and hypersensitivity reactions.
Central Nervous System Toxicity
The severity of CNS effects is proportional to the blood concentration of local anesthetic. This relationship is well described for lidocaine (Figure 13-3). Initially, the patient may complain of numbness of the tongue, tinnitus, or lightheadedness. At high plasma concentrations, convulsions occur because of a selective blockade of central inhibitory neurons that leads to increased CNS excitation.41 At still higher concentrations, generalized CNS depression or coma may result from reversible blockade of both inhibitory and excitatory neuronal pathways. Finally, depression of the brainstem and cardiorespiratory centers may occur.
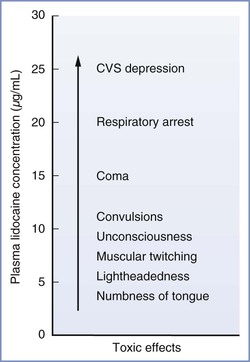
FIGURE 13-3 Signs and symptoms of systemic toxicity with increasing lidocaine concentrations. CVS, cardiovascular system. (Modified from Carpenter RL, Mackey DC. Local anesthetics. In Barash PG, Cullen BF, Stoelting RK, editors. Clinical Anesthesia. Philadelphia, Lippincott, 1992:527.)
The relative toxicity of a local anesthetic correlates with its potency. For lidocaine, etidocaine, and bupivacaine, the ratio of the mean cumulative doses that cause convulsions in dogs is approximately 4 : 2 : 1, which is similar to their relative anesthetic potencies.42 The same relative toxicity was demonstrated in human volunteers.43 Local anesthetics may be ranked in order of decreasing CNS toxicity as follows: bupivacaine, ropivacaine, levobupivacaine, lidocaine, and 2-chloroprocaine.44 Tetracaine, etidocaine, and mepivacaine are used rarely in obstetric anesthesia practice.
Other factors (e.g., the speed of injection) may affect CNS toxicity. In humans, the mean dose of etidocaine that elicited signs of CNS toxicity was lower during a 20-mg/min infusion than during a 10-mg/min infusion.43 The seizure threshold also may be affected by metabolic factors. For example, in cats, an increase in PaCO2 or a decrease in pH results in a reduction in the seizure-dose threshold for local anesthetics. Respiratory acidosis may result in delivery of more drug to the brain; alternatively, respiratory acidosis may result in “ion trapping” of the local anesthetic and/or an increase in the unbound fraction of drug available for pharmacologic effect.45–47
Cardiovascular Toxicity
The cardiovascular system is much more resistant than the CNS to the toxic effects of local anesthetics. Severe, direct cardiovascular depression is rare, especially in association with the use of lidocaine. Prompt administration of oxygen and, if necessary, initiation of ventilatory and circulatory support usually prevent cardiac arrest after unintentional intravenous injection of lidocaine.48 Progressive depression of myocardial function and profound vasodilation occur only at extremely high plasma concentrations.48 In contrast, the more potent amide local anesthetics (i.e., bupivacaine) have a more narrow margin of safety, expressed as the ratio between the dose (or plasma concentration) required to produce cardiovascular collapse and the dose (or plasma concentration) required to produce convulsions.48 A partial explanation is the fact that supraconvulsant doses of bupivacaine (but not of lidocaine) precipitate lethal ventricular arrhythmias.49–51 These arrhythmias may be caused by exaggerated electrophysiologic effects (e.g., depression of ventricular conduction) out of proportion to bupivacaine’s anesthetic potency.52
Two theories have been proposed to explain why malignant ventricular arrhythmias occur with bupivacaine but not with lidocaine. Both bupivacaine and lidocaine rapidly block cardiac sodium channels during systole, but bupivacaine dissociates from these channels during diastole at a much slower rate than lidocaine.52 Thus, at physiologic heart rates, the diastolic period is of sufficient duration for lidocaine to dissociate from sodium channels, whereas a bupivacaine block becomes intensified. This difference makes bupivacaine much more potent than lidocaine in depressing conduction and inducing reentrant-type ventricular arrhythmias. Alternatively, other investigators have suggested that high concentrations of local anesthetic in the brainstem may lead to systemic hypotension, bradycardia, and ventricular arrhythmias.53 These effects occur more commonly with bupivacaine because of its high lipid solubility, which facilitates transfer across the blood-brain barrier. An echocardiographic study in anesthetized dogs suggested that bolus injection of bupivacaine results in systolic dysfunction, especially involving the right ventricle, which precedes the occurrence of arrhythmias.54
Systemic Toxicity of Ropivacaine and Levobupivacaine
In perfused preparations of myocardium, ropivacaine is intermediate between bupivacaine and lidocaine in its depressant effect on cardiac excitation and conduction as well as in its potential to induce reentrant-type ventricular arrhythmias.34 In dogs, the margin of safety between convulsive or lethal doses and plasma concentrations of drug is greater for ropivacaine than for bupivacaine but less than that for lidocaine.55 The arrhythmogenicity of ropivacaine in pigs also is intermediate between that of lidocaine and bupivacaine.56 In sheep, the ratio of fatal doses of bupivacaine, ropivacaine, and lidocaine is 1 : 2 : 9.57 Ropivacaine was found to cause fewer CNS symptoms and was 25% less toxic than bupivacaine (as defined by the doses and plasma concentrations that were tolerated) when administered to healthy male volunteers.58
Most studies comparing the systemic toxicity of ropivacaine and bupivacaine have used equal doses of each, and, therefore, cannot resolve the controversy as to whether ropivacaine truly is less cardiotoxic or merely less potent than bupivacaine. This issue would be of concern only if larger doses of ropivacaine than bupivacaine were required to produce comparable regional blocks. Indeed, several studies in laboring women suggest that ropivacaine is 25% to 40% less potent than bupivacaine.59–61 Thus, the need for a larger dose of ropivacaine may negate the expected benefits of its apparently wider margin of safety. However, results from one laboratory study showed that ropivacaine produces less cardiotoxicity than bupivacaine, even when given at equipotent doses.62
Long-acting amide local anesthetics—even the newer drugs—are very potent and may cause cardiac arrest with a misplaced injection or relative overdose. Indeed, several cardiac arrests have been reported with the use of ropivacaine,63,64 including one in a woman undergoing a cesarean delivery with epidural anesthesia.65 In contrast to that induced by bupivacaine,66 resuscitation from a cardiac arrest induced by ropivacaine may be successful more often than not.63–65
Evidence suggests that levobupivacaine causes fewer arrhythmias than the racemic drug. Valenzuela et al.67 demonstrated that levobupivacaine caused less inhibition of inactivated sodium channels than either the dextrorotary or racemic drug. In comparison with dextrorotary and racemic bupivacaine, levobupivacaine resulted in less QRS widening and a lower frequency of malignant ventricular arrhythmias in isolated, perfused rabbit hearts.68 Similarly, levobupivacaine produced less second-degree heart block and atrioventricular conduction delay than the other two forms of the drug in isolated perfused guinea pig hearts.3
In laboratory animals, the systemic toxicity of levobupivacaine is intermediate between that of bupivacaine and ropivacaine.69 Potency ratio data for epidural bupivacaine, ropivacaine, and levobupivacaine in laboring women are inconsistent, but studies suggest that levobupivacaine is equipotent or less potent than bupivacaine (see Chapter 23).70,71 Altogether, published data and clinical experience suggest that any benefits from the reduction in risk for systemic toxicity with levobupivacaine are not obtained at the expense of efficacy. Like ropivacaine, levobupivacaine may cause cardiac arrest but is associated with a better response to resuscitation than racemic bupivacaine.72
Effects of Pregnancy on Systemic Toxicity
Central Nervous System Toxicity.
It is unclear whether pregnancy lowers the seizure threshold for amide local anesthetic agents. In one study, seizures occurred at lower doses of bupivacaine, levobupivacaine, and ropivacaine in pregnant ewes than in nonpregnant ewes.69 However, the difference was small (10% to 15%) and probably of negligible clinical significance. In studies in sheep and rats, pregnancy did not reduce the doses required to cause convulsions after intravenous administration of mepivacaine, bupivacaine, or lidocaine.51,73 Magnesium sulfate, which is frequently used in obstetric practice, does not affect the seizure-dose threshold of lidocaine.74
Cardiovascular Toxicity.
In 1979, Albright66 alerted anesthesiologists to several cases of sudden cardiovascular collapse after unintentional intravascular injection of bupivacaine and etidocaine in pregnant women. The fact that cardiac arrest occurred concurrently with or shortly after the onset of convulsions was especially disconcerting. Most of these cases were fatal, and subsequent controversy centered on whether resuscitation was instituted promptly and effectively. Nonetheless, the U.S. Food and Drug Administration (FDA) restricted the use of the highest concentration (0.75%) of bupivacaine in pregnant women.
Several physiologic changes that occur during pregnancy place the parturient at higher risk for refractory cardiac arrest than the nonpregnant patient. First, reduced functional residual capacity and a higher metabolic rate increase the risk for and hasten the onset of hypoxemia during periods of hypoventilation or apnea. Second, aortocaval compression decreases the efficacy of closed-chest cardiac massage in the supine position.75 Third, a large bolus of drug injected into an epidural vein might reach the heart rapidly through a dilated azygous system. However, none of these factors adequately explains why cardiac arrest and difficult resuscitation are very rare in parturients intoxicated with lidocaine or mepivacaine.66,76
Results of laboratory studies of the effects of pregnancy on bupivacaine cardiotoxicity have been contradictory. Pregnancy-related hormones enhance the cardiotoxicity and arrhythmogenicity of bupivacaine in vitro.77,78 For example, the magnitude and severity of bupivacaine-induced electrophysiologic changes are greater in myocardium obtained from nonpregnant rabbits treated with progesterone or beta-estradiol than in myocardium from untreated controls.77,78 The electrophysiologic effects of lidocaine are less pronounced than those of bupivacaine, even in hormonally treated animals. Studies conducted in vivo have been less conclusive. In earlier investigations, significantly lower doses and plasma concentrations of bupivacaine, but not of mepivacaine or lidocaine, were required to produce circulatory collapse in pregnant sheep than in nonpregnant sheep.49–51 However, a study involving a larger number of sheep and more rigorous methods (e.g., randomization, blinding) failed to confirm that pregnancy enhances the cardiotoxicity of bupivacaine.69
Progesterone does not increase myocardial sensitivity to ropivacaine.79 Likewise, pregnancy does not enhance the systemic toxicity of ropivacaine or levobupivacaine in sheep.69
Extrapolation of results of animal studies to obstetric anesthesia practice is difficult, for several reasons. First, in the aforementioned sheep studies, the drug was administered by constant-rate intravenous infusion. In contrast, in pregnant women intoxicated with bupivacaine, cardiac arrest occurred after unintended intravascular injection of a large bolus of drug. Second, a potential for bias existed in the animal studies because randomization and blinding were not used in all studies and some relied on historical controls.49–51 Third, it is unclear whether resuscitation in the reported clinical cases was accompanied by prompt and effective relief of aortocaval compression.75
Nonetheless, bupivacaine remains a popular local anesthetic for obstetric anesthesia. In current practice, heightened vigilance, use of an appropriate test dose, and fractionation of the therapeutic dose have made epidural anesthesia a safe technique for use in obstetric patients (see Chapter 12). In a study of anesthesia-related maternal mortality, Hawkins et al.80 noted that the number of maternal deaths resulting from local anesthetic toxicity decreased after 1984, the year that the FDA withdrew approval for the epidural administration of 0.75% bupivacaine in obstetric patients. However, LAST has been recognized for decades as an important potential cause of maternal mortality.81 In our judgment, adherence to the aforementioned clinical precautions—rather than the proscription against the epidural administration of 0.75% bupivacaine—has been responsible for the lower number of maternal deaths due to LAST. Anesthesia providers should be aware that intravenous injection of 0.25% and 0.5% bupivacaine can also cause LAST.
The availability of single levorotary isomers of a local anesthetic may be advantageous because these drugs have a greater margin of safety than bupivacaine, with similar blocking properties, although at a higher cost. From the standpoint of systemic toxicity, the use of these isoforms may be more beneficial in parturients undergoing cesarean delivery, who require higher doses than administered for analgesia during labor. Nonetheless, a greater margin of safety with these new drugs should not be a substitute for proper technique.
Treatment of Systemic Toxicity
Meticulous attention to good technique and adherence to guidelines for maximum recommended dose are mandatory. (The use of a test dose to identify misplaced injections is discussed in Chapter 12.) Incremental injection of the therapeutic dose, careful observation of the patient, and monitoring of vital signs usually provide early warning of an impending reaction. In mild cases, discontinuation of the administration of drug, administration of supplemental oxygen, and maintenance of normal ventilation often limit the severity of the reaction. In 2012, the American Society of Regional Anesthesia and Pain Medicine developed a checklist for managing LAST (Box 13-1). In patients who show signs of CNS excitation, a small dose of an intravenous sedative-hypnotic drug with strong anticonvulsant properties such as a benzodiazepine (diazepam up to 5 mg or midazolam 1 to 2 mg) or propofol (10 to 20 mg) may prevent progression to convulsions.82 In one study, prophylactic administration of a benzodiazepine reduced the incidence of both convulsions and mortality in mice intoxicated with amide local anesthetics.83 However, propofol should be avoided in patients with cardiovascular instability because of its cardiovascular depressant properties.82
If convulsions should occur, oxygenation and ventilation must be maintained to prevent hypoxemia, hypercarbia, and acidosis.45,46,82,84 Patency of the airway must be restored. It may be necessary to suction the airway first in some patients. Management should consist of administration of 100% oxygen and tracheal intubation, if required. Convulsions may be terminated quickly with a small dose of a benzodiazepine. Maternal circulation should be supported by maintenance of left uterine displacement and administration of a vasopressor as needed. Because a high plasma concentration of local anesthetic may cause myocardial depression and vasodilation, a mixed alpha- and beta-adrenergic agonist (e.g., ephedrine) may be preferable to a pure alpha-adrenergic agonist. Vasopressin, calcium entry-blocking agents, beta-adrenergic blocking agents, and local anesthetics should be avoided.82 Fortunately, convulsions induced by intravenous injection of a relatively small dose of local anesthetic are usually self-limited because of rapid redistribution of the drug.
Persistent hypotension and bradycardia may require administration of epinephrine. However, individual epinephrine doses should not exceed 1 µg/kg.82 In a dog study of bupivacaine toxicity,85 amrinone was superior to epinephrine in improving cardiac contractility depressed by bupivacaine. Bupivacaine-induced ventricular arrhythmias should not be treated with lidocaine, because local anesthetic toxicity is additive.
Cardiac arrest should be treated according to the American Heart Association’s Advanced Cardiac Life Support (ACLS) guidelines, modified for pregnancy (see Chapter 55).86 The pelvis should be tilted leftward to prevent or relieve aortocaval compression, which renders cardiac massage ineffective. Prompt cesarean delivery of the infant may be necessary to relieve aortocaval compression (venous return) and restore maternal circulation. Prolonged resuscitation may be needed until myocardial washout of bupivacaine has occurred.52
Lipid emulsion therapy has been incorporated into guidelines for the treatment of LAST.82,86 The salutary effect of lipid emulsion therapy may be related to the greater affinity of bupivacaine for the lipid and the dissociation of bupivacaine from the cardiac sodium channels or to the binding of plasma bupivacaine by the lipid fraction in the blood. Alternatively, others have proposed that bupivacaine poisons the normal energy transport mechanisms in the mitochondria and that lipid emulsion bypasses those mechanisms to provide energy substrate.87 In a suspected case of bupivacaine intoxication in a parturient, manifested by facial and limb twitching and unconsciousness, prophylactic administration of 100 mL of lipid emulsion prevented progression to full cardiovascular collapse.88 At least one case report has described the successful use of lipid emulsion to treat refractory cardiac arrest due to LAST.89
The timing of lipid emulsion administration is controversial.82 Early treatment may prevent cardiovascular collapse, but only a few patients progress to cardiovascular collapse. The decision to administer lipid emulsion should be based on “clinical severity and rate of progression of LAST.”82 Propofol should not be used to treat LAST; its lipid content is inadequate, and the cardiodepressant effects of the drug are detrimental during resuscitation from LAST. A protocol for treatment of LAST, including the administration of lipid emulsion, is presented in Box 13-1.
After maternal recovery, fetal condition should be assessed promptly. In theory, a delay in delivery may allow back-diffusion of local anesthetic from the fetus to the mother, which may be of benefit to the neonate by decreasing neonatal plasma bupivacaine levels. Laboratory studies have demonstrated this phenomenon after the administration of bupivacaine90 but not lidocaine.91
Tissue Toxicity
Neurologic complications of neuraxial anesthesia are rare and result mostly from direct neural trauma, infection, injection of toxic doses of local anesthetic, or the injection of the wrong drug.
In 1980, several cases of prolonged or permanent sensory and motor deficits after subarachnoid injection of a large dose of 2-chloroprocaine intended for epidural block were described.92 Studies comparing the neurotoxicity of 2-chloroprocaine with that of other local anesthetics have yielded conflicting results, most likely related to the use of different methodologies and different species.93,94 It has been suggested that neurotoxicity was caused by sodium metabisulfite, an antioxidant present in the commercial formulation used in the reported cases.93 The pH of this formulation was between 2.7 and 4.0. In CSF rendered more acidic by 2-chloroprocaine, metabisulfite generates sulfur dioxide, which is lipid soluble and can diffuse into the nerve cells.95 Intracellular hydration of sulfur dioxide generates sulfurous acid, which may cause profound intracellular acidosis and irreversible damage.
Subsequently, the manufacturer released another preparation of 2-chloroprocaine, which was free of bisulfite but contained ethylenediaminetetraacetic acid (EDTA). This was followed by several reports of severe, incapacitating paralumbar pain and spasm associated with epidural injection of large volumes of drug.96 The etiology is unclear, although chelation of calcium by disodium EDTA may result in a reduced tissue calcium concentration and local tetany of the affected muscles.
In a 2004 study, Taniguchi et al.97 suggested that sodium bisulfite was the “scapegoat” for 2-chloroprocaine neurotoxicity. They concluded that neurologic deficits associated with unintentional intrathecal injection of 2-chloroprocaine likely resulted from a direct effect of the 2-chloroprocaine, not the sodium bisulfite.
The current preparation of 2-chloroprocaine that is marketed for epidural administration does not contain EDTA or other preservatives. It is packaged in colored vials to reduce the oxidation of the 2-chloroprocaine. Low-dose, preservative-free 2-chloroprocaine (30 to 60 mg) is now being studied as a possible alternative to lidocaine for spinal anesthesia.98
Lidocaine has been used for spinal anesthesia for more than 50 years, in thousands upon thousands of patients, with apparent safety. However, cauda equina syndrome, sacral nerve root deficits, or transient neurologic toxicity can occur after subarachnoid injection of lidocaine.99,100 Neurotoxicity of local anesthetics is concentration dependent101 and is not unique to lidocaine.102,103 Some investigators have speculated that slow injection of local anesthetic through a spinal microcatheter results in maldistribution and pooling of high concentrations of hyperbaric lidocaine in the cauda equina area, resulting in neurotoxicity and cauda equina syndrome.99,100
Milder manifestations of neurotoxicity also may occur. As early as 1954, mild, transient neurologic symptoms were reported after spinal anesthesia with lidocaine.104 Transient neurologic symptoms (TNS) (dysesthesia or low back pain radiating to the buttocks, thighs, and calves) have been observed in surgical patients even after conventional (i.e., single-shot) spinal anesthesia with hyperbaric 5% lidocaine (see Chapter 32).100 In response to concerns that intrathecal injection of hyperbaric 5% lidocaine might be associated with TNS, in 1994 the FDA Advisory Committee on Anesthetic Drugs recommended that the injected drug concentration be reduced by dilution with an equal volume of either preservative-free saline or CSF. However, Pollock et al.105 reported that there was no difference in the incidence of TNS when spinal lidocaine 50 mg was diluted to 2%, 1%, or 0.5% solutions before administration and that the overall incidence of TNS did not differ from that of historic controls given 5% lidocaine.
Interestingly, the exposure of frog sciatic nerve to lidocaine results in a progressive, irreversible loss of impulse activity beginning at a concentration of 1%.101 The investigators in this study noted that “the range of lidocaine that produces such changes in mammalian nerve awaits determination.”101 Meanwhile, it seems prudent to take the following precautions99,106:
1. Dilute the commercial 5% lidocaine for intrathecal injection as recommended by the FDA.
Generally, if pencil-point, side-hole spinal needles are used, it is recommended that the injection port should be directed cephalad. However, an epidemiologic survey did not implicate dose and needle bevel direction as factors that affect the risk for TNS.106 A meta-analysis of randomized controlled trials comparing spinal lidocaine with other local anesthetics (bupivacaine, prilocaine, procaine, and mepivacaine) found that the relative risk (RR) for development of TNS was higher with lidocaine than with the other local anesthetic agents (RR, 4.35; 95% confidence interval [CI], 1.98 to 9.54).107 It has not been conclusively proven that TNS are manifestations of neurotoxicity.
Pregnancy may be associated with a reduced risk for TNS. Studies suggest that the incidence of TNS after spinal anesthesia with lidocaine or bupivacaine is equally low (< 3%) in women having cesarean delivery and those undergoing postpartum tubal ligation.108,109
Allergic Reactions
True allergy to a local anesthetic is rare.110 Further, anaphylactic and anaphylactoid reactions may be the result of additives such as methylparaben and metabisulfite.110,111 Clinical criteria are important in the diagnosis because there is often a delay in obtaining confirmatory laboratory data. The alleged allergy to a local anesthetic can be substantiated in only 15% of patients by a history of urticaria, bronchospasm, facial edema, and/or cardiovascular instability.112 Adverse reactions (e.g., CNS and cardiovascular symptoms) may mimic hypersensitivity but may not actually be a result of hypersensitivity. These symptoms may be caused by hyperventilation or vasovagal syncope during injection of the drug, sympathetic stimulation (e.g., palpitations, tachycardia) from epinephrine, or edema related to the injection itself (Box 13-2).
Some pregnant women claim to be allergic to “Novocaine” or “the caine” drugs. Obstetricians should refer such patients to an allergist and an anesthesiologist for appropriate evaluation well before the expected date of delivery. In many cases, a carefully obtained history excludes true hypersensitivity. If IgE-mediated hypersensitivity is suspected, patients should be referred to an allergist for further evaluation. Phillips et al.111 recommended testing with skin prick or intradermal testing using appropriate positive (diluted histamine) and negative (normal saline) controls. Intradermal testing is more sensitive but is associated with a false-positive rate of 8% to 15%.110 If the skin testing is negative, subcutaneous provocative dose testing is a useful method to confirm that the drug is safe to use clinically.111 Alternatively, if skin testing is positive, the testing sequence (skin testing followed by provocative subcutaneous testing) should be repeated with an alternative agent.
The subcutaneous provocative test can be performed by any physician qualified to manage hypersensitivity reactions. Appropriate emergency equipment and drugs (e.g., epinephrine, H1– and H2-receptor antagonists) should be immediately available for resuscitation. Although not mentioned in many protocols, establishing intravenous access before testing seems prudent. The back and the ventral aspects of the forearm are the preferred sites for testing. Areas with abnormal skin coloration or dermographia should be avoided. A history of recent treatment with antihistamines, salicylates, or corticosteroids may alter the test results.113
The following protocol has been proposed by Chandler et al.114 (Table 13-2) and has been used successfully in at least one published case.115 After a negative needle-prick test, increasing volumes of undiluted local anesthetic (typically 1% concentration) are injected subcutaneously at 15-minute intervals. In patients with an especially strong history of a severe reaction, the series may be preceded by injection of diluted solutions (e.g., a 1 : 100 solution, followed by a 1 : 10 solution). A fresh syringe and a 30-gauge needle should be used for each subsequent injection. Additional refinements may consist of the use of both a negative control and a positive control injection. A local anesthetic that is not in the same class as the drug in question should be tested; if an ester is suspected as the offending agent, testing should be performed with an amide agent, and vice versa. If possible, the drug tested should be suitable for local infiltration and for epidural and subarachnoid block.
TABLE 13-2
A Protocol for Provocative Dose Testing with Local Anesthetics
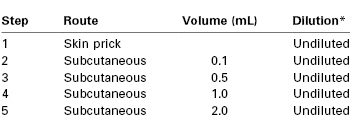
* See text for initial dilution suggestions for patients with a history of severe allergy.
From Chandler MJ, Grammer LC, Patterson R. Provocative challenge with local anesthetics in patients with a prior history of reaction. J Allergy Clin Immunol 1987; 79:885.
The test is considered positive if there is a change in the patient’s clinical status or if a skin wheal more than 10 mm in diameter, with or without a flare, arises within 10 minutes of injection and persists for at least 30 minutes.113 If provocative dose testing is completed without a reaction, the local anesthetic used and the final dose given should be recorded; the patient (and the referring physician) should be informed that her risk for an adverse reaction to subsequent administration of that drug and dose is no greater than that for the general population.114,115
Management of an Allergic Reaction.
Pharmacologic therapy of a severe allergic reaction involves (1) inhibition of mediator synthesis and release, (2) reversal of the effects of these mediators on target organs, and (3) prevention of the recruitment of other inflammatory processes. In general, catecholamines, phosphodiesterase inhibitors, antihistamines, and corticosteroids have been used for this purpose (Box 13-3).116 Higher doses of catecholamines may be required in a patient who has received sympathetic blockade. In addition, pregnancy itself decreases responsiveness to catecholamines.117 Despite its potential adverse effect on uterine blood flow, epinephrine remains the cornerstone of therapy for allergic reactions. In one reported case, a mother was treated successfully with epinephrine 100 µg without any apparent adverse effects on the newborn.118
Effects on the Uterus and Placenta
Uterine Blood Flow
The association of paracervical block anesthesia with fetal bradycardia has been attributed to the high concentration of local anesthetic deposited in the vicinity of the uterine arteries (see Chapter 24). Human uterine artery segments obtained at the time of cesarean hysterectomy constrict when exposed to high concentrations of lidocaine,119 mepivacaine,119 or bupivacaine.120
These findings also have been confirmed in laboratory animals. Fishburne et al.121 observed a dose-related decrease in uterine blood flow during uterine arterial infusion of 2-chloroprocaine, lidocaine, or bupivacaine in gravid ewes. A 25% reduction in uterine blood flow occurred at the following calculated plasma concentrations of local anesthetic: bupivacaine, 7 µg/mL; 2-chloroprocaine, 11.5 µg/mL; and lidocaine, 19.5 µg/mL. However, when plasma local anesthetic concentrations mimic those that occur in ordinary clinical practice, local anesthetics have no adverse effect on uterine blood flow.122–124 In pregnant ewes, uterine blood flow remained unchanged during an intravenous infusion of lidocaine or bupivacaine that resulted in plasma concentrations of 0.81 to 4.60 and 1.5 to 2.0 µg/mL, respectively.122,124 Similarly, intravenous injection of 2-chloroprocaine, 0.67 and 1.34 mg/kg, did not reduce uterine blood flow velocity in pregnant guinea pigs.123
Pregnancy may enhance uterine vascular reactivity to local anesthetic agents. Isolated human uterine artery segments obtained from term parturients constrict at a lower lidocaine concentration than uterine artery segments from nonpregnant patients.119,125 Uterine artery sensitivity to local anesthetics increases as early as the second trimester of pregnancy and may be related to an increase in estrogen levels.119,121 However, these studies were performed before the recognition of the importance of intact vascular endothelium in the in vitro assessment of vascular tone.
The exact mechanism by which high concentrations of local anesthetics cause uterine artery vasoconstriction (while causing dilation in other vascular beds) is unclear. This vasoconstriction may result from modulation of calcium-channel regulation because verapamil and nifedipine ablate the response.125 Alternatively, local anesthetics may affect cyclic nucleotides and alter the ionic content and contractility of uterine vascular smooth muscle.126 Clinical experience with the use of local anesthetics supports the view that clinical concentrations of these drugs do not adversely affect the uterine vasculature (see Chapter 3).127,128
All local anesthetics can reduce uterine blood flow at plasma concentrations that greatly exceed those occurring during the routine practice of obstetric anesthesia.121 There has been an added concern that the levorotary isomers of local anesthetics, which produce vasoconstriction at clinical doses,129 may reduce uteroplacental perfusion and adversely affect fetal well-being. It is reassuring to note that ropivacaine, even at plasma concentrations that are almost two times greater than would be expected to occur during clinical use, does not reduce uterine blood flow or affect fetal heart rate (FHR), blood pressure, or acid-base measurements in pregnant sheep.122 In humans, Doppler velocimetry studies have shown that ropivacaine has little effect on the uteroplacental or fetal circulation when it is administered to provide epidural anesthesia for cesarean delivery.127 Similarly, clinically relevant plasma concentrations of levobupivacaine had no adverse effect on uterine blood flow.122
Umbilical Blood Flow
Fetal well-being also depends on the adequacy of fetal perfusion of the placenta. The regulatory mechanisms that control flow through the umbilical vessels are poorly understood. Lidocaine does not affect spiral strips obtained from human umbilical artery segments at concentrations up to 5 µg/mL, but it produces relaxation in concentrations from 30 to 900 µg/mL.130 Bupivacaine also does not constrict umbilical artery segments at clinically relevant concentrations of 0.3 and 1 µg/mL.130 At higher concentrations, the effect of bupivacaine appears to be biphasic. Constriction occurs at concentrations of 5 to 25 µg/mL, and relaxation occurs at concentrations greater than 125 µg/mL.130,131 Hypercarbia but not hypoxemia lessens the contractile response of umbilical vessels to bupivacaine in vitro.132
Decreases in umbilical blood flow of as much as 43% accompany intravenous administration of lidocaine 4 mg/kg in pregnant sheep.133 However, plasma concentrations of the drug were higher than would be expected with clinical use, and all ewes exhibited signs of CNS toxicity, which may reduce umbilical blood flow.
Advances in noninvasive Doppler imaging have facilitated clinical assessment of umbilical cord blood flow velocity. The ratio of the systolic (S) peak to the diastolic (D) trough of the umbilical artery waveform is used as a measure of vascular resistance. The S/D ratio in the umbilical artery decreases during normal pregnancy, and high ratios usually are associated with fetal compromise (see Chapter 6). Local anesthetics administered for epidural anesthesia do not adversely affect the umbilical artery S/D ratio.134,135 In fact, labor epidural analgesia with 1.5% lidocaine or 2% 2-chloroprocaine resulted in a decrease in the S/D ratio.134,135 This favorable change may have resulted from pain relief. Other investigators have noted no appreciable change or a slight decrease in the S/D ratio after the epidural administration of amide local anesthetics for elective cesarean delivery.127,128,136
Uterine Tone and Contractility
Changes in uterine tone and contractility may affect uteroplacental perfusion. Local anesthetics exert direct effects on uterine smooth muscle. One study reported that exposure to high concentrations of local anesthetic in vitro led to contraction of human myometrial segments obtained at the time of cesarean delivery.137 These findings have been corroborated in laboratory animals.138 Further, Belitzky et al.139 observed that direct intramyometrial injection of 1% procaine resulted in uterine hyperstimulation and fetal compromise in pregnant women. In all of these reports, the myometrium was exposed to higher than normal concentrations of the drug. In other studies, however, intravenous infusion of lidocaine or bupivacaine that resulted in clinically relevant plasma concentrations did not affect uterine tone or uterine activity in pregnant ewes.122,124 In a recent study using electrohysterogram monitoring, levobupivacaine caused less uterine muscle relaxation after intramyometrial injection in rats than did bupivacaine.140
Drug Interactions with 2-Chloroprocaine and Lidocaine
Epidural 2-chloroprocaine may affect the efficacy of other drugs administered in the neuraxis. Previous administration of 2-chloroprocaine (even a test dose) may reduce the quality and duration of analgesia produced by subsequent epidural injection of morphine or fentanyl.141,142 Several hypotheses have been proposed for this antagonism. The low pH of the 2-chloroprocaine solution may result in acidification of the epidural space and thus may favor formation of the poorly diffusible, charged form of the opioid. Second, it has been suggested that 2-chloroprocaine (or its metabolite, chloroaminobenzoic acid) may act as a specific μ-opioid receptor antagonist because a κ-opioid receptor agonist (e.g., butorphanol) is not antagonized by 2-chloroprocaine.141 However, using an in vitro hippocampal slice model, Coda et al.143 concluded that 2-chloroprocaine opioid antagonism did not appear to act through a μ-opioid receptor. Third, a “window” may be caused by the rapid regression of 2-chloroprocaine before the onset of analgesia with epidural morphine.144 This mechanism is supported by the results of a study145 in which women who received spinal bupivacaine anesthesia for cesarean delivery were randomly assigned to receive either epidural morphine with 2-chloroprocaine or epidural morphine with saline-placebo. There was no difference in post–cesarean delivery epidural morphine analgesia between the two groups; presumably the spinal bupivacaine provided adequate analgesia until the onset of epidural morphine analgesia.145
2-Chloroprocaine also reduces the subsequent efficacy of bupivacaine.146 Corke et al.147 suggested that chloroaminobenzoic acid is responsible for this effect. Administration of buffered 2-chloroprocaine does not prevent the antagonism of epidural bupivacaine.148
The use of neuraxial opioids alone or in combination with local anesthetics has become ubiquitous in obstetric anesthesia for enhancing analgesia during labor or for providing effective pain relief after cesarean delivery. Lidocaine is a frequently used drug for epidural anesthesia during cesarean delivery. In a recent study,149 epidural administration of 20 to 35 mL of epidural 2% lidocaine with fentanyl 1 hour before administration of extended-release epidural morphine increased the peak plasma concentration(Cmax) of morphine when compared with similar women who received a combined spinal-epidural (CSE) technique (intrathecal bupivacaine and fentanyl) with no epidural medication for cesarean delivery (see Chapter 28).
Potency of Bupivacaine, Ropivacaine, and Levobupivacaine
The levorotary compounds ropivacaine and levobupivacaine were developed because of the concerns about the safety of high doses of bupivacaine. Many studies have addressed the question of relative potency among the three drugs. Ropivacaine is approximately 10 times less lipid soluble (N-heptane/buffer) than bupivacaine, a difference that is important for two reasons.2 First, ropivacaine may penetrate more slowly into the large, heavily myelinated motor neurons, resulting in less motor block than occurs with bupivacaine. Second, the issue raises questions as to whether ropivacaine is equipotent to bupivacaine. Indeed, a higher dose of ropivacaine is required to produce a sensory and motor block comparable with that produced by bupivacaine after spinal injection.150,151 Similarly, the EC50 (the local anesthetic concentration at which 50% of women have pain relief, also known as the minimum local anesthetic concentration [MLAC]) of epidural ropivacaine is almost twice as great as that of epidural bupivacaine in laboring women.59 Critics of the use of EC50 data to compare potency argue that it provides no information on the shape and slope of the dose-effect relationship, which can vary with drug concentration, and further, that it provides no information on the effective clinical dose (ED95 [effective dose in 95% of cases]).152
Studies of the EC50 of epidural levobupivacaine are conflicting; one study found that levobupivacaine was essentially equipotent to bupivacaine,70 whereas others suggest that ropivacaine and levobupivacaine have similar potency.153,154 Levobupivacaine may have a greater motor-sparing effect than bupivacaine when given for the initial intrathecal injection. For example, in one study, none of 37 women who received intrathecal levobupivacaine 2.5 mg (with sufentanil and epinephrine) had evidence of motor block.155 In contrast, 13 of 38 (34%) women given intrathecal bupivacaine 2.5 mg demonstrated a Bromage grade 1 motor block.
In obstetric anesthesia practice, the clinical effects of epidural levobupivacaine and ropivacaine are indistinguishable from those of epidural bupivacaine for labor analgesia.156 The choice of bupivacaine, levobupivacaine, or ropivacaine does not affect the method of delivery or neonatal condition.156 For cesarean delivery, epidural levobupivacaine 0.5% is virtually identical to epidural bupivacaine 0.5%.157 The levorotary isomers (ropivacaine and levobupivacaine) may provide a greater margin of safety when large volumes of a concentrated solution of local anesthetic are required (e.g., epidural anesthesia for cesarean delivery). However, there may be little advantage to using levobupivacaine or ropivacaine when dilute solutions are used for epidural labor analgesia or when a small dose is used for spinal anesthesia.
Placental Transfer
Most drugs, including local anesthetics, cross the placenta. The factors that influence the placental transfer of a drug include (1) the physicochemical characteristics of the drug, (2) the concentration of free drug in the maternal blood, (3) the permeability of the placenta, and (4) the hemodynamic events occurring within the fetal-maternal unit.
Local anesthetics cross placental membranes by a process of simple (i.e., passive) diffusion. The rate of transfer (not necessarily the amount) of a particular drug is described by the Fick equation, as follows:
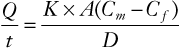
where Q/t is the rate of diffusion; K is the diffusion constant for the drug; A is the surface area available for transfer; Cm is free drug concentration in the maternal blood; Cf is the free drug concentration in the fetal blood; and D is the thickness of the trophoblastic epithelium. In general, K is affected by molecular size, lipid solubility, and the degree of ionization.
Molecular Size
Compounds with a molecular weight of less than 500 Da cross the placenta easily, whereas drugs like digoxin, which have a molecular weight higher than 500 Da, have a slower rate of diffusion.158 Molecular weights of local anesthetics range from 234 to 288 Da (see Table 13-1). These small differences in molecular weight should not affect the rate of placental transfer because the diffusion constant (K) is inversely proportional to the square root of the molecular weight.159
Ionization and Lipid Solubility
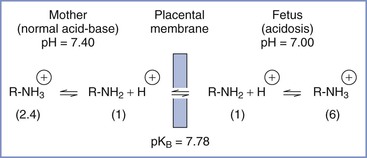
Local anesthetics are weak bases; they have a relatively low degree of ionization and considerable lipid solubility at physiologic pH. The basic un-ionized local anesthetic molecule is more lipid soluble than the ionized moiety and determines placental transfer in a protein-free perfusate.160
The relationship between pH and pKB may affect drug accumulation in the fetus. For the amide local anesthetics, pKB values are close enough to physiologic pH that changes in fetal pH may alter the balance between ionized and un-ionized drug. In the acidotic fetus, a greater proportion of drug in the ionized form results in a larger total amount of local anesthetic in fetal plasma, because of “ion trapping” (Figure 13-4).161–163 Elimination of lidocaine from fetal blood is slower in the asphyxiated fetus than in the nonasphyxiated fetus.133 Accumulation of lidocaine may be greater in fetal tissues, where the pH is even lower than that in fetal blood.163
Protein Binding
Perhaps most confusing and least understood are the effects of protein binding on placental transfer. Amide local anesthetics are bound predominantly to AAG and to a much lesser extent to albumin.20 The extent of protein binding varies among the local anesthetic agents (see Table 13-1). For a given local anesthetic, the proportion of free drug increases as blood concentration increases because of the saturation of binding sites. Binding of local anesthetics in the fetal plasma is approximately half that in the mother.90,91
The fetal-to-maternal (F/M) blood concentration ratios of amide local anesthetic agents are listed in Table 13-1. The lower F/M blood concentration ratios of highly protein-bound drugs (e.g., bupivacaine) have been attributed to their more restricted placental transfer compared with less protein-bound drugs (e.g., lidocaine). Indeed, the rate of bupivacaine transfer across rabbit placenta perfused in situ is lower than that of lidocaine transfer.164,165 Some investigators have suggested that protein binding in the maternal plasma should not affect the diffusion of drugs across the placenta because the dissociation from plasma proteins is essentially instantaneous.159,166 In more recent studies, the relatively low umbilical vein–to–maternal vein blood concentration ratio for bupivacaine has been attributed to differences in protein binding between maternal plasma and fetal plasma (Figure 13-5).90,91,167,168 Let us assume that the total concentration of lidocaine or bupivacaine in the maternal plasma is 2 mg/L. Lidocaine and bupivacaine are approximately 50% and 90% bound to maternal plasma proteins, respectively. Thus, the free concentrations of drug available for placental transfer are 1.0 and 0.2 mg/L, respectively. At equilibrium, the concentration of free drug is equal on the two sides of the placenta. In the fetus, however, lidocaine and bupivacaine are approximately 25% and 50% bound to fetal plasma proteins, respectively. Thus, the total lidocaine concentration in fetal plasma is 1.33 mg/L, resulting in an F/M ratio of 0.67; for bupivacaine, the corresponding values are 0.4 mg/L and 0.2.
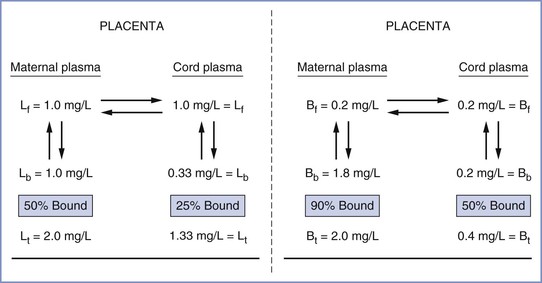
FIGURE 13-5 Demonstration of how distribution of local anesthetics across the placenta may be predicted from differences in drug protein binding in maternal and fetal plasma. Left, lidocaine (L); f, b, t, free, bound, and total drug concentrations, respectively. Right, bupivacaine (B). Lidocaine umbilical cord–to–maternal plasma ratio (F/M) = 0.67; bupivacaine F/M = 0.20. (From Tucker GT, Mather LE. Properties, absorption, and disposition of local anesthetic agents. In Cousins MJ, Bridenbaugh PO, editors. Neural Blockade in Clinical Anesthesia and Management of Pain. 2nd edition. Philadelphia, Lippincott, 1988:95.)
Substantial accumulation of bupivacaine occurred in human fetuses whose mothers received the drug for epidural anesthesia.25 After delivery, measurable plasma and urine concentrations persisted for as long as 3 days.25 In vitro studies using a perfused human placental model have found that the placental transfer of ropivacaine is similar to that of bupivacaine.169 Intravenous infusion of ropivacaine or bupivacaine to pregnant sheep results in steady-state maternal plasma concentrations of 1.5 to 1.6 µg/mL and fetal concentrations of approximately 0.28 µg/mL.122 Tissue concentrations of ropivacaine in fetal heart, brain, liver, lung, kidneys, and adrenal glands were similar to those of bupivacaine.122 Datta et al.32 noted that the free fraction of ropivacaine at delivery was approximately twice that of bupivacaine in neonates whose mothers received the drug for epidural anesthesia during labor or cesarean delivery.
Maternal Blood Concentration of Drug
The maternal blood concentration of local anesthetic is determined by (1) the dose, (2) the site of administration, (3) metabolism and excretion, and (4) the effects of adjuvants such as epinephrine (see later discussion). For a given local anesthetic, the maternal blood concentration determines fetal drug exposure and is the only variable of the Fick equation that may be influenced by the clinician.
Dose.
In general, higher doses result in higher maternal and fetal blood concentrations. For example, Kuhnert et al.18 found that doubling the mean (± SD) dose of epidural lidocaine from 300 ± 195 mg to 595 ± 127 mg almost doubled the concentration in umbilical cord blood. The elimination half-life of amide local anesthetics is relatively long; thus, repeated epidural injection or continuous infusion of the drug may lead to accumulation in the maternal plasma. This statement does not apply to 2-chloroprocaine, however, which is rapidly hydrolyzed by pseudocholinesterase.12
Site of Administration.
The rates of absorption and peak plasma concentrations depend on the vascularity at the site of administration. The peak plasma concentration of lidocaine is achieved within 9 to 10 minutes after paracervical block. In contrast, absorption from the lumbar epidural space, which is less vascular, occurs at a slower rate; the peak plasma concentration is not achieved until 25 to 40 minutes after administration.18,170 Injection of local anesthetic into the caudal rather than the lumbar epidural space may result in higher blood levels because of the need for a higher drug volume to provide comparable anesthesia to that provided by lumbar epidural injection.171
In the past, it was thought that subarachnoid administration of a local anesthetic resulted in less systemic absorption than epidural administration. However, peak blood concentrations of lidocaine have been reported to be similar after subarachnoid and epidural administration.172 In another study, subarachnoid administration of lidocaine 75 mg for cesarean delivery resulted in low but measurable fetal plasma concentrations of the drug.17
Placenta
Maturation of the placenta may affect the rate of drug transfer. In pregnant mice, diazepam and its metabolites cross the placenta more rapidly in late pregnancy.173 Uptake and metabolism of drugs by the placenta would be expected to reduce transfer to the fetus. However, placental drug uptake of local anesthetics is limited, and it is unlikely that this organ metabolizes the amide local anesthetic agents.174 This may not be true for the ester local anesthetics. For example, cocaine is biotransformed when it is incubated with human placental microsomal fraction, presumably because of cholinesterase activity within the placenta.175 Placental metabolism of para-aminobenzoic acid also has been demonstrated.176
Teratogenicity
The teratogenicity of anesthetics is not an issue during parturition, but local anesthetics often are used for procedures during the first trimester of pregnancy. In vitro studies have suggested that local anesthetics may have some adverse developmental effects. Even at low concentrations, these agents have caused reversible reduction of cell division in tissue culture.177–182 However, structural anomalies have not been observed in intact animals.183–185 Mid-pregnancy administration of lidocaine or mepivacaine in rats has been associated with behavioral changes in the offspring.186,187
Extrapolation of laboratory findings to humans is tenuous for several reasons. First, a drug may be teratogenic in one species but not in others. Second, a 1-hour drug exposure in a pregnant rat (with a gestation of 21 days) is excessive and not analogous to several hours of clinical anesthesia during human pregnancy. Third, the doses of local anesthetics used in animal studies greatly exceed those administered for clinical anesthesia. Indeed, a large, multicenter study demonstrated that the risk for congenital anomalies in humans was not increased by the administration of benzocaine, procaine, tetracaine, or lidocaine during early pregnancy.188 However, a twofold increase in the incidence of congenital anomalies was noted in infants whose mothers had received mepivacaine. The small number of patients who received mepivacaine in this study (n = 82) and the fact that no adverse effects occurred with the use of other amide agents have raised doubts about the validity of this observation.189
Fetal and Neonatal Effects
Pharmacokinetics
Local anesthetics, once transferred across the placenta, are distributed in the fetus. Factors that influence tissue uptake of the drug include (1) fetal plasma protein binding, (2) lipid solubility, (3) the degree of ionization of the drug, and (4) hemodynamic changes that affect the distribution of fetal cardiac output. The fetal plasma protein-binding capacity of local anesthetics is approximately 50% that of maternal plasma.90,91,190 Thus, at any given total plasma concentration of local anesthetic, there is greater availability of free drug in the fetus than in the mother.90,91,190–192 Studies have examined the distribution of lidocaine in fetal tissues after an intravenous injection of the drug to animals.19,193 The higher concentration of lidocaine in the liver, myocardium, and brain (compared with other fetal tissues) reflects rapid distribution of the drug to highly perfused tissues. The only organ in which lidocaine concentrations in the fetus have been found to exceed those in the mother is the liver. This finding is not surprising, given the high lipid content of the fetal liver and the fact that it receives most of the blood returning from the placenta by means of the umbilical vein.193 Fetal acidosis and hypoxemia result in circulatory adaptations that increase blood flow to vital organs (e.g., brain, heart, adrenal glands).194 The concentration of lidocaine in these organs is higher in asphyxiated fetuses than in healthy fetuses.163,194
Any drug that reaches the fetus undergoes metabolism and excretion. The term newborn has the hepatic enzymes necessary to metabolize local anesthetics.17,18,195–197 Nonetheless, the elimination half-life of these drugs is longer in the neonate than in the adult.196,197 The use of mepivacaine in obstetric epidural analgesia fell into disfavor after a report indicating that the elimination half-life of the drug in the neonate was approximately 9 hours, or three times as long as the neonatal half-life for lidocaine.198 It is ironic that it was later discovered that the neonatal elimination half-life for bupivacaine may be as long as 14 hours.199
Morishima et al.197 compared the pharmacokinetics of lidocaine among adult ewes and fetal and neonatal lambs. The metabolic (hepatic) clearance in the lambs was similar to that in adults, and renal clearance was greater than that in adults. Nonetheless, the elimination half-life was more prolonged in the lambs. This latter finding has been attributed to a greater volume of distribution in the lamb. Thus, at any given time, a smaller fraction of lidocaine accumulated in the body is available for clearance by hepatic metabolism. The greater renal clearance noted in neonates is a result of decreased protein binding, which increases the proportion of drug available to the kidneys for excretion.
The elimination half-life of local anesthetics in the fetus is similar to that in the adult because, unlike the newborn, the fetus can excrete drug across the placenta back to the mother.90,197 With bupivacaine, this transfer may occur even though the total plasma drug concentration in the mother may exceed that in the fetus.90
Systemic Toxicity
In general, the neonate is more sensitive than the adult to the depressant effects of drugs. However, the seizure threshold for local anesthetics in the neonate appears to be similar to that in the adult.200
Morishima et al.201 compared the relative CNS toxicity and cardiovascular toxicity of lidocaine in adult ewes and fetal and neonatal lambs. Greater doses (when calculated on a milligram-per-kilogram basis) were required to elicit toxic manifestations in the fetus and neonatal lamb than in the adult. However, the plasma concentrations of the drug associated with toxic manifestations were similar in the three groups of animals. The greater dose tolerated by fetuses than by neonates and adults was attributed to placental clearance of drug back to the mother and better maintenance of blood gas tensions during convulsions. In the neonate, a large volume of distribution is most likely responsible for the high doses of local anesthetic required to have toxic effects.
Studies of bupivacaine cardiotoxicity are inconsistent. In vitro, the sinoatrial node of neonatal guinea pigs was found to be more sensitive than that of adults to the cardiodepressant effect of bupivacaine.202 In contrast, 2-day-old piglets demonstrated greater resistance than older animals to the arrhythmogenic and CNS effects of bupivacaine.203
Fetal Heart Rate
Changes in FHR after administration of local anesthetics are most often related to indirect effects such as maternal hypotension and uterine tachysystole (see Chapter 23). Local anesthetics probably have little direct effect on FHR, except perhaps after paracervical block. Rather, labor itself may be the single most important factor that alters FHR patterns.204 Transient changes in FHR variability and an increase in the incidence of periodic decelerations have been observed during administration of neuraxial analgesia in laboring women.205,206 In contrast, in the absence of labor, FHR patterns are not affected even by the larger doses of local anesthetics required during administration of epidural anesthesia for cesarean delivery.204 The FHR changes noted in laboring women were transient and did not affect the condition of their newborns.205,206 In a recent study, investigators found no significant difference in the number or type of fetal electrocardiographic ST-segment changes (ST-waveform analysis [STAN] events) in women with a high-risk singleton gestation who received epidural analgesia for labor when compared with a control group of women who did not receive epidural analgesia.207