Fetal and Neonatal Neurologic Injury
Tania F. Esakoff MD, Sarah J. Kilpatrick MD, PhD, Arvind Palanisamy MBBS, MD, FRCA
Chapter Outline
The detection and diagnosis of fetal and neonatal brain injury have been advanced by improvements in functional imaging and the identification of potential biochemical markers. Evidence indicates that inflammatory mediators play an important role in the pathophysiology of fetal brain injury. Evidence also suggests that maternal administration of magnesium sulfate before anticipated early preterm birth reduces the risk for cerebral palsy in surviving infants. New data suggest that induced hypothermia is beneficial for the treatment of neonatal hypoxic ischemic encephalopathy. Of specific concern to anesthesia providers is rodent and nonhuman primate data that suggest that fetal exposure to anesthetic agents may have harmful effects on neurogenesis and synapse formation in the developing brain. Overall, however, little progress has been made in reducing the incidence of neonatal brain injury and cerebral palsy.
Fetal Brain Development
Generation of the various cell types that populate the developing brain, and the subsequent layering and organization, is a precisely regulated process encoded by genetic programs and modified by epigenetic influences.1–4 Contrary to previous dogma, it is now well established that the brain continuously evolves during ontogeny and that these processes are susceptible to subtle changes in the internal and external milieu. Although such neurodevelopmental processes occur throughout the human lifespan, the process is most robust and dynamic during the perinatal period.5 Much of our understanding of the processes that drive fetal brain development comes from studies in rodents and nonhuman primates.6 With recent advances in neuroimaging, it is now possible to study brain anatomy and assess neurobehavioral changes in the human fetus.7
When pathways leading to orderly brain development are deconstructed, three major events appear critical to the establishment of functional synapses. Neuronal proliferation, migration, and cellular differentiation occur in a preordained fashion to establish early neural circuitry. These processes often overlap and occur at different rates in different brain regions. Neurogenesis, a term that encompasses both neuronal proliferation and subsequent survival, begins with neural stem/progenitor cells in neurogenic niches such as the subventricular zone and the subgranular zone of the dentate gyrus. These neural progenitor cells undergo mitosis to generate immature neurons that migrate in a radial fashion and laminate the cortex in an “inside-out” fashion.8 Interneurons, which comprise 10% to 15% of the total neuronal cells in the brain, originate from the ganglionic eminences in the developing brain.9 These newly generated interneurons, which play an indispensable role in circuit inhibition, migrate in a tangential manner to populate distinct brain areas. Both forms of migration are guided by cell-intrinsic mechanisms as well as by structural scaffolds and humoral mediators such as gamma-aminobutyric acid (GABA) and glutamate.10,11
In humans, neurogenesis starts and peaks at 5 and 25 weeks’ gestation, respectively, while neuronal migration is completed between 30 and 36 weeks’ gestation.12 Between 20 and 40 weeks’ gestation, these processes are followed by the generation of an array of supporting glial cells, such as astrocytes and oligodendrocytes.12 Concurrently, synapse formation begins as early as the 10th week of gestation and continues to increase gradually at a rate of approximately 4% per week until the end of the second trimester. After this phase, a robust and exponential increase in synapse formation (almost 40,000 synapses/min) occurs between 28 weeks’ and term gestation.13 These processes, in conjunction with the onset of myelination, result in a fivefold increase in brain volume and the appearance of morphologic features of the mature brain such as sulci and gyri. By 24 weeks’ gestation the fetus has all the neural machinery necessary to perceive pain.14 Many clinicians recommend that appropriate measures should be taken to provide fetal analgesia during fetal surgical procedures from this point onward.15
Although the ontogeny of neurotransmitter systems is less well studied, a wealth of animal and human data indicates that these systems appear very early in life, before the phase of active synaptogenesis.12 The presence of these neuromodulatory substances before synapse formation lends credence to the view that they serve a trophic role during early brain development, a role that is distinct from their predominant role of facilitating synaptic neurotransmission in the mature brain. Among these neurotransmitters, GABA remains the most widely studied (Figure 10-1).16 Although GABA has an inhibitory action in the mature brain, GABA serves an excitatory role during fetal brain development. The major mechanism for this role reversal is the differential expression of chloride ion transporters NKCC1 and KCC2; these transporters increase the intracellular concentration of chloride in developing neurons.17 On stimulation of GABA receptors that are expressed in neural progenitor cells and immature neurons, chloride ions are actively extruded, causing membrane depolarization rather than the hyperpolarization seen in mature neurons. This depolarizing effect of GABA decreases DNA synthesis and inhibits proliferation of neural progenitor cells,18 causes concentration- and time-dependent effects on neuronal migration,11 and plays a major role in activity-dependent synapse formation.19
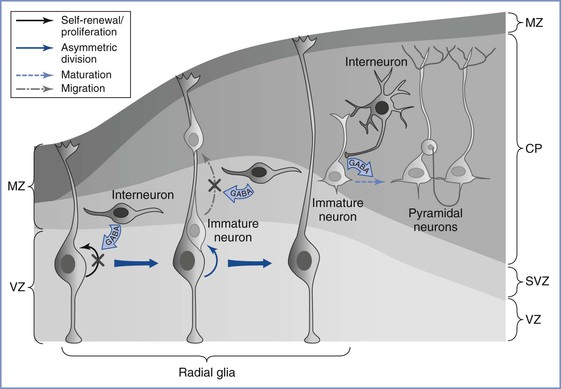
FIGURE 10-1 Gamma–aminobutyric acid’s (GABA) role in regulating embryonic cortical development. During corticogenesis, interneurons migrating in the subventricular zone (SVZ) can release GABA and activate GABAergic receptors on the radial glia, depolarizing these progenitors and decreasing their proliferation. Radial glia generate immature pyramidal neurons through asymmetric division, and the migration of these immature neurons along the radial fibers is decreased by GABA signaling. As young neurons assume their position in the cortex and begin to mature, GABA-mediated depolarization by the interneurons is required for the development of dendritic arbors and excitatory synaptic inputs from other pyramidal neurons. MZ, marginal zone; VZ, ventricular zone; SVZ, subventricular zone; CP, cortical plate. (From Wang DD, Kriegstein AR. Defining the role of GABA in cortical development. J Physiol 2009; 587:1873-9.)
The N-methyl-D-aspartate (NMDA)-subtype glutamate receptors originate later than the GABA receptors and remain functionally silent because of magnesium ion–induced channel blockade; thus, they play a limited role during early brain development. Dopaminergic, cholinergic, and serotonergic systems develop concomitantly and appear fully functional by the second trimester.12 Pharmacologic interventions (e.g., ethanol, antiepileptic drugs) that act directly or indirectly on these powerful neuromodulator systems induce long-lasting impairment of fetal brain development, mainly owing to impaired neurogenesis and/or altered neuronal migration.20–22 Alteration of this excitation-inhibition balance is purported to be responsible for an array of childhood neurodevelopmental disorders.
Experimental studies reveal that the fetal blood-brain barrier is morphologically well developed and functionally competent at term.23 Convincing evidence confirms that the endothelial tight junctions of the blood-brain barrier are as effective in the term fetus as in the adult, although the exact time that blood-brain barrier competency is established in the human fetus is unknown. In rodents, data suggest that the fetal blood-brain barrier is established between embryonic days 11 and 17 (term gestation is 22 days), a time period that corresponds to approximately the late second and early third trimesters in humans.24
Cerebral Palsy
History, Definitions, and Significance
In 1861, John Little, an orthopedic surgeon, first described cerebral palsy in a report to the Obstetrical Society of London. Described as a neonatal neurologic disorder associated with difficult labor or birth trauma, the disorder was known as Little’s disease until William Osler coined the term cerebral palsy in 1888.25 A precise definition and classification of cerebral palsy has proved elusive. In the forward to the “Report on the Definition and Classification of Cerebral Palsy,” published in 2007 in Developmental Medicine and Child Neurology, Peter Baxter25 wrote, “This [supplement] illustrates the difficulties inherent in trying to agree what we mean by the terms we use and that a classification that suits one purpose, such as a diagnostic approach, may not always be ideal for others, such as therapy issues. Defining and classifying cerebral palsy is far from easy. We do need a consensus that can be used in all aspects of day-to-day care and for future research on cerebral palsy.”
Today, cerebral palsy is defined as a nonprogressive disorder of the central nervous system (CNS) present since birth that includes some impairment of motor function or posture.25 Intellectual disability (formerly known as mental retardation) may be present but is not an essential diagnostic criterion. Various forms of cerebral palsy exist, with differences in pathology, pathophysiology, and potential relationships with intrapartum events. The literature on cerebral palsy is difficult to review and understand. Data from individual studies are difficult to compare because of variations in the duration of follow-up, birth weight classifications, inclusion criteria for congenital abnormalities, and exclusion criteria for various causes of death. Terms such as hypoxic-ischemic encephalopathy of the newborn, newborn asphyxia, birth asphyxia, and asphyxia neonatorum are difficult to distinguish. Some authorities, including the American College of Obstetricians and Gynecologists (ACOG), have argued that the term birth asphyxia should be abandoned.26
Intrapartum events continue to receive the blame for some cases of cerebral palsy. It is a widely believed theory that an intrapartum reduction in fetal oxygen delivery may cause cerebral palsy, and early reports in primates demonstrated that perinatal asphyxia could cause brain injury.27 Continuous electronic fetal heart rate (FHR) monitoring, which has largely replaced intermittent FHR auscultation during labor, is believed to prompt the delivery of at-risk fetuses and thus reduce asphyxial events. However, despite a higher incidence of cesarean delivery, no reduction in the incidence of cerebral palsy has been observed since the widespread implementation of continuous electronic FHR monitoring during labor.28,29 Further, among patients with new-onset late FHR decelerations, an estimated 99% of tracings would be false positive “if used as an indicator for subsequent development of cerebral palsy.”30 This information is probably surprising to the lay public and trial lawyers as well as to obstetricians; a survey of maternal-fetal medicine fellows showed that they, too, have greatly overestimated the diagnostic accuracy of FHR monitoring.30
Large randomized trials have not demonstrated better fetal and neonatal outcomes with continuous electronic FHR monitoring than with intermittent FHR auscultation.31,32 In an editorial citing observations made by Schifrin and Dame,33 Friedman34 opined, “The absence of either suggestive or overtly ominous fetal heart rate patterns is reliably reassuring.” Unfortunately, there is little objective evidence that reassuring FHR tracings exclude the subsequent occurrence of cerebral palsy. In a 1993 review of published FHR monitoring studies, Rosen and Dickinson35 could not identify FHR patterns that were consistently associated with neurologic injuries. Moreover, no consistent FHR pattern was observed in a subset of 55 brain-damaged infants. These investigators concluded, “We do not advocate the abandonment of the use of electronic fetal monitoring, but we do believe that it is yet to be proved to be of value in predicting or preventing neurologic morbidity.” A more focused application of FHR monitoring may ultimately be found useful. For example, fetal inflammatory changes, which can be associated with neurologic injury, may be associated with characteristic FHR findings.36 Thus, electronic FHR monitoring provides incomplete data that should be evaluated in the clinical context in which is it used.
Despite significant limitations in the use of intrapartum electronic FHR monitoring, there is no doubt that it will continue to be used for the foreseeable future. In a review of medicolegal issues in FHR monitoring, Schifrin and Cohen37 noted that despite its limitations, “Monitoring deserves credit for reducing intrapartum death, one of the original rationales for its development.”
A 2008 workshop (sponsored by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the ACOG, and the Society for Maternal-Fetal Medicine [SMFM]) updated the definitions of various types of FHR tracings to simplify interpretation for providers.38 As a result, a three-category system was developed. Category I FHR tracings provide the most reassuring evidence of fetal well-being and strongly predict normal fetal acid-base status at the time of observation. Category III tracings are the most ominous; these tracings predict abnormal fetal acid-base status at the time of observation and require prompt evaluation. Most FHR tracings are category II (indeterminate). Category II tracings are not predictive of abnormal fetal acid-base status, but they do not provide sufficient evidence to be classified as either normal or abnormal; these tracings require continued surveillance and reevaluation (see Chapter 8).
Intrapartum events are responsible for some cases of cerebral palsy39; however, these cases are few. After exclusion of infants with significant congenital anomalies, intrapartum events—including asphyxial insults—likely account for only 5% to 8% of all cases of cerebral palsy at all gestational ages.40,41 In 1999, the International Task Force on Cerebral Palsy published a consensus statement summarizing criteria that are necessary and suggestive of an intrapartum etiology for neurologic abnormalities (Box 10-1).42 In 2010, a proposed evidence-based neonatal workup to confirm or refute allegations of intrapartum asphyxia was published.43
Epidemiology and Etiology
The causes of cerebral palsy are not known, but the varying forms suggest a multifactorial etiology. The Collaborative Perinatal Project still represents one of the largest studies of the antecedent factors associated with cerebral palsy. The investigators in this study evaluated the outcomes of 54,000 pregnancies among patients who delivered at 12 university hospitals between 1959 and 1966. They evaluated more than 400 variables in a univariate analysis,44 which identified potential risk factors that were then subjected to a more rigorous multivariate analysis.45 Maternal age, parity, socioeconomic status, smoking history, maternal diabetes, duration of labor, or use of anesthesia was not associated with cerebral palsy in the univariate analysis. The factors most strongly associated with cerebral palsy in the multivariate analysis were (1) maternal mental retardation, (2) birth weight of 2000 g or less, and (3) fetal malformations. Other factors associated with cerebral palsy included (1) breech presentation (but not vaginal breech delivery), (2) severe proteinuria (more than 5 g/24 h) during the second half of pregnancy, (3) third-trimester bleeding, and (4) a gestational age of 32 weeks or less. There was a slight association between cerebral palsy and fetal bradycardia, chorioamnionitis, and low placental weight. However, only 37% of the cases of cerebral palsy occurred in patients with one or more of these identified risk factors.
Rosen and Dickinson46 reviewed studies from Europe, Australia, and the United States that were published between 1985 and 1990 and included data from 1959 to 1982. The incidence of cerebral palsy ranged from 1.8 to 4.9 (composite rate of 2.7) cases per 1000 live births. The incidence of certain conditions in infants with cerebral palsy was as follows: birth weight less than 2500 g, 26%; diplegia, 34%; hemiplegia, 30%; quadriplegia, 20%; and extrapyramidal forms, 16%.
Two more recent studies from Australia reexamined the risk factors for cerebral palsy. A large epidemiologic study from 1998 noted an incidence of neonatal encephalopathy of 3.8 per 1000 term births.47 The investigators identified preconception and antepartum factors that were associated with neonatal encephalopathy (Box 10-2). In the second study from 2011, the greatest risks for cerebral palsy included (1) preterm birth, (2) fetal growth restriction (also known as intrauterine growth restriction), (3) perinatal infection, and (4) multiple gestation.48 Upper respiratory tract and gastrointestinal infections during pregnancy and instrumental (forceps or vacuum) vaginal delivery were not associated with cerebral palsy.48 Evidence suggests that intrapartum factors alone are associated with neonatal encephalopathy in less than 5% of cases.47,49 These data, along with the recognition that most patients with identified risk factors do not have children with cerebral palsy, have led the majority of investigators to agree that most cases of cerebral palsy cannot be predicted and that the identification of pregnancy-related conditions contributes minimally to the identification of patients at risk for having a child with cerebral palsy.
In 2000, the ACOG and the American Academy of Pediatrics (AAP) convened the Neonatal Encephalopathy and Cerebral Palsy Task Force. The resulting landmark report,49 which was released in 2003, was reviewed and endorsed by such groups as the U.S. Department of Health, the National Institutes of Health, the SMFM, the Child Neurology Society, the March of Dimes Birth Defects Foundation, the Society of Obstetricians and Gynaecologists of Canada, and the Royal Australian and New Zealand College of Obstetricians and Gynaecologists. The Task Force extended the earlier international consensus statement regarding the requirements for establishing a causal relationship between intrapartum events and cerebral palsy (see Box 10-1).42 The consensus statement led to several medicolegal conclusions42,49:
Phelan et al.50 subsequently confirmed that fetuses that experienced a sudden and sustained deterioration of the FHR, and that subsequently were found to have cerebral palsy, demonstrated characteristics consistent with the ACOG/AAP Task Force criteria50 for intrapartum asphyxial injury.
Peripartum Asphyxia and Cerebral Palsy
Asphyxia may be defined as insufficient exchange of respiratory gases.51 Although accurate, this definition does not include an index of severity or have any predictive value. Unfortunately, most studies have not used a uniform definition of birth asphyxia.52–54
In 1953, Dr. Virginia Apgar, an anesthesiologist, introduced her scoring system to identify newborn infants in need of resuscitation and to assess the adequacy of subsequent resuscitation efforts.55 Although the Apgar score has also been used to identify infants at risk for cerebral palsy, only a weak association has been found.56,57 In the Collaborative Perinatal Project, only 1.7% of children with a 1-minute Apgar score of 3 or less developed cerebral palsy.58 Among infants who weighed more than 2500 g at delivery, the incidence of cerebral palsy was 4.7% if the 5-minute Apgar score was 0 to 3 and 0.2% if the 5-minute Apgar score was at least 7. Among infants who weighed less than 2500 g with the same 5-minute Apgar scores, the incidence of cerebral palsy was 6.7% and 0.8%, respectively. Among all infants, a higher incidence of cerebral palsy was observed if the Apgar score remained 3 or less for longer than 5 minutes. The incidence of early neonatal death increased among those infants with prolonged neonatal depression.
Most infants who subsequently manifest evidence of cerebral palsy have a normal 5-minute Apgar score. In the Collaborative Perinatal Project, only 15% of the infants in whom cerebral palsy later developed had a 5-minute Apgar score of 3 or less, approximately 12% had a score of 4 to 6, and the remaining 73% had a score of at least 7.58 It must also be noted that preterm delivery is independently associated with a low Apgar score.
Although most cases of cerebral palsy are not attributed to intrapartum insults, intrapartum asphyxia does occur and can have serious consequences. However, the degree of asphyxia necessary to produce irreversible CNS injury is unclear. In some cases, an intrapartum insult that might have otherwise been innocuous might be superimposed on subclinical chronic fetal compromise and result in permanent injury.
Umbilical cord blood gas measurements are often used to diagnose suspected asphyxia. However, the definition of normal umbilical cord blood gas and pH measurements remains unclear.51 In one study of 15,073 vigorous neonates (arbitrarily defined as having a 5-minute Apgar score of 7 or more) conducted between 1977 and 1993, the median umbilical arterial blood gas measurements (with the 2.5th percentile in parentheses) were as follows: pH 7.26 (7.10), PO2 17 (6) mm Hg, PCO2 52 (74) mm Hg, and base excess −4 (−11) mEq/L.51 Only small differences in median pH and other measurements were present when infants were grouped according to gestational age. These data suggest that umbilical arterial blood pH in vigorous neonates can be as low as 7.10 and base excess may be as low as −11 mEq/L.
Although intrapartum events are most likely associated with a minority of cerebral palsy cases, clinical studies have attempted to define the associated extent and duration of perinatal asphyxia. Fee et al.59 defined asphyxia as an umbilical arterial blood pH of less than 7.05 with a base deficit greater than 10 mEq/L; they concluded that this threshold was a poor predictor of adverse neurologic outcomes. Goodwin et al.60 defined asphyxia as an umbilical arterial blood pH of less than 7.00; with the use of this definition, hypoxic ischemic encephalopathy and abnormal neurologic outcome were associated with acidemia. Goldaber et al.61 also observed greater neonatal morbidity and mortality among term infants (birth weight > 2500 g) with an umbilical arterial blood pH of less than 7.00.
Low et al.62,63 also studied complications of intrapartum asphyxia in term and preterm infants. They developed a complication score that expressed the magnitude of neonatal complications. Among term infants, the frequency and severity of newborn complications increased with the severity and duration of metabolic acidosis at birth. Importantly, respiratory acidosis at birth did not predict complications in newborns. Similar results were noted for preterm infants delivered between 32 and 36 weeks’ gestation. In contrast, in infants delivered before 32 weeks’ gestation, complications were similar in the control and asphyxia (defined as umbilical arterial blood buffer base < 30 mmol/L) groups. When this scoring system was used in term infants, the threshold for moderate or severe newborn complications was an umbilical arterial blood base deficit of 12 mmol/L.63
Relatively few studies have followed neurodevelopmental examinations for a sufficient duration to make meaningful conclusions about peripartum predictors of neurologic injury. Nagel et al.64 performed such examinations in 30 children in whom umbilical arterial blood pH was less than 7.00 at delivery, 28 of whom survived the neonatal period. Evaluation at 1 to 3 years of age detected 3 children who had experienced an episode of hypertonia. The majority of children exhibited no major problems, with only 1 child displaying mild motor developmental delay. Another study examined neonatal complications (neonatal death, grade 3 or 4 intraventricular hemorrhage, gastrointestinal dysfunction, and neonatal seizures) in 35 newborns with an umbilical arterial blood pH of less than 7.00 at delivery, 3 of whom died during the neonatal period.65 An umbilical arterial blood base deficit greater than or equal to 16 mmol/L and a 5-minute Apgar score less than 7 had a sensitivity and specificity for predicting adverse neonatal outcomes of 79% and 81%, respectively. These investigators did not perform any follow-up neurologic examinations after the neonatal period.65
Because metabolic acidosis may be a predictor of complications in newborns, the severity of intrapartum acidosis could be an important variable. Gull et al.66 studied a small cohort of 27 patients with terminal bradycardia who were delivered vaginally. Not surprisingly, the umbilical arterial blood base deficit was greater in infants with end-stage bradycardia than in controls. The loss of short-term FHR variability for more than 4 minutes during terminal bradycardia correlated with the development of metabolic acidosis.
The relationship between umbilical arterial blood base excess values and the timing of hypoxic injury has been estimated in human and animal studies.67 In addition, in a 2010 systematic review and meta-analysis, an umbilical cord arterial blood pH less than 7.00 was significantly associated with important, biologically plausible, adverse neonatal outcomes (i.e., neonatal mortality, hypoxic ischemic encephalopathy, intraventricular hemorrhage, periventricular leukomalacia, cerebral palsy).68 Unfortunately, this relationship does not consider the role of previous or repetitive hypoxic episodes before the episode in question and therefore cannot accurately pinpoint the time of injury. Fortunately, the human fetus is quite robust, and episodes of intrauterine asphyxia usually yield a normal neonate. A much smaller number of fetuses experiencing such episodes die in utero. Blumenthal69 concluded that there is a fine threshold between normality and death from asphyxia.
The increased presence of nucleated red blood cells in the umbilical circulation at delivery has been proposed as a marker of the occurrence and timing of intrauterine asphyxia.50,70,71 However, data from these investigations demonstrated considerable variability and were influenced by birth weight and gestational age.72 Furthermore, the timing of the injury may be difficult to determine with confidence, because multiple episodes of asphyxia may have occurred. In such cases, the nucleated red blood cell count may reflect and implicate only the most recent, but possibly least important, event. Both nucleated red blood cell and lymphocyte counts appear to undergo more sustained elevations in cases of antepartum asphyxia than in cases of intrapartum asphyxia.49
Chorioamnionitis, Fever, and Cerebral Palsy
An association between cerebral palsy and chorioamnionitis has been demonstrated in preterm infants and term infants of normal birth weight.73,74 An elevated maternal temperature is one sign of chorioamnionitis, but alone it is insufficient for the diagnosis. Other signs include, but are not limited to, maternal and fetal tachycardia, foul-smelling amniotic fluid, uterine tenderness, and maternal leukocytosis. The diagnosis remains unproven until confirmed by placental culture or histologic examination.
The mechanism by which chorioamnionitis is associated with cerebral palsy is unclear; however, inflammatory cytokines may play a role (see later discussion).75–77 A landmark meta-analysis published in 2000 reported that both clinical and histologic chorioamnionitis were strongly associated with an increased risk for cerebral palsy and periventricular leukomalacia in both preterm and term infants.78 In a 2010 meta-analysis, both histologic (pooled odds ratio [OR], 1.83; 95% confidence interval [CI], 1.17 to 2.89) and clinical chorioamnionitis (OR, 2.42; 95% CI, 1.52 to 3.84) were again found to be significantly associated with cerebral palsy.79
Several studies have demonstrated a tendency for maternal temperature to rise after administration of epidural analgesia during labor (see Chapter 37).80 Although the mechanism of epidural analgesia–associated maternal pyrexia remains unclear, fever due to epidural analgesia alone is not associated with cerebral palsy.81 Epidural analgesia has been blamed for the common obstetric practice of antibiotic administration to mothers with fever but no other evidence of chorioamnionitis. This practice may lead to unnecessary neonatal sepsis evaluations and antibiotic exposure.82 Rather than treat all women with pyrexia for presumed chorioamnionitis, Mayer et al.83 correctly noted that physicians should make an effort to differentiate true chorioamnionitis from incidental maternal fever. These investigators found that additional signs of chorioamnionitis were present in all cases in which the diagnosis was later confirmed by culture or pathologic examination. Neuraxial anesthesia is not a risk factor for cerebral palsy.44
The mode of delivery has been examined as an independent risk factor for periventricular leukomalacia, the most common form of ischemic brain injury in surviving preterm infants in whom cerebral palsy later develops. In 99 women with chorioamnionitis who delivered between 25 and 32 weeks’ gestation, vaginal delivery had a significant association with periventricular leukomalacia.84 Because the cesarean delivery group likely included a substantial number of infants with nonreassuring FHR tracings, the better outcomes in these infants is striking. Prospective trials are needed to confirm these retrospective observations.
Pathophysiology of Fetal Asphyxia
Intrauterine Hypoxemia and the Fetal Brain
The fetus is exclusively dependent on the placenta for oxygen and nutrients; thus, acute and chronic conditions that affect the placenta or the umbilical cord can deprive the fetus of one or more of these vital resources. Recent evidence from experimental animal models and humans suggest that both hypoxemic and inflammatory pathways interact and augment fetal brain damage.
The spectrum of neurologic injury in neonates depends on the duration and gestational age at hypoxemic-ischemic insult. Acute hypoxemia during the early- to mid-gestational period in sheep affects the predominant neurodevelopmental events such as neurogenesis and neuronal migration. Such hypoxemia causes the death of cerebellar Purkinje cells and hippocampal pyramidal neurons, as well as impaired neuronal migration.85 In contrast, acute hypoxemia in late gestation appears to spare the hippocampus and cerebellum but causes neuronal death in the cerebral cortex and striatum.86 Furthermore, acute perinatal anoxia causes long-term changes in dendritic arborization and synaptic connectivity.87,88
Experimental models of chronic hypoxemia, based on restriction of placental mass or blood flow, demonstrate an array of completely different effects on the fetal brain. Chronic placental insufficiency relatively spares the fetal brain compared with other organ systems, although it results in reduced fetal brain weight. Overall, neurons appear to survive chronic and mild hypoxemia; even minor behavioral changes appear to resolve fully by adulthood in animal models. It is not known whether these effects are mediated by hypoxemia per se, or by other accompanying conditions such as chronic reduction of fetal nutrient supply or altered maternal-fetal endocrine status.
Dysregulation of neuronal calcium transport appears to be the initial pathway by which cerebral hypoxemia causes perinatal neuronal injury.89 Hypoxia-induced changes in the NMDA receptor increases cellular permeability to calcium; such increases in intracellular calcium trigger a variety of downstream effects, ultimately resulting in generation of free radicals, peroxidation of lipid membranes, and nuclear fragmentation. It has long been recognized that developing oligodendroglia are highly vulnerable to excitotoxic injury in preterm infants.90 Altered maturation or premature oligodendrocyte death can occur in areas of severe hypoxia-ischemia as a result of up-regulation of inflammatory cytokines by activated microglia, elevated glutamate levels, or depleted levels of the antioxidant glutathione. It is highly likely that a combination of these mechanisms, modified by the nature and duration of insults and gestational age, determine the ultimate neurobehavioral phenotype.
Maternal Inflammation and Fetal Brain Injury
Although the development of the fetal brain is encoded by genetic programming, such programs remain highly susceptible to environmentally induced epigenetic modifications and appear closely intertwined with maternal immune and endocrine systems. Recent experimental and epidemiologic studies reveal that maternal infection and inflammation early in pregnancy can cause an array of neurodevelopmental abnormalities in the offspring such as schizophrenia and autism.91–94 Among maternal infections, chorioamnionitis is the best characterized and thoroughly investigated model of perinatal neuroinflammation. Although the exact contribution of maternal inflammation to perinatal brain injury is obscured because of the association of chorioamnionitis with preterm delivery and hypoxic-ischemic encephalopathy, inflammatory experimental models have revealed much information on cytokine induction, their transport across the placenta and amniotic fluid, and subsequent activation of the fetal immune system.
The exact mechanism by which maternal inflammation triggers a fetal immune response is likely multifactorial (Figure 10-2). Despite the presence of circulating immune cells as early as 7 weeks’ gestation in humans, antigen presentation is suboptimal because of reduced expression of the major histocompatibility complex class II on antigen-presenting cells. Furthermore, the T cells are relatively immature. Therefore, maternally derived humoral mediators seem credible candidates to initiate and perpetuate an inflammatory cascade across the placenta. This idea has gained traction with the identification of maternal interleukin-6 (IL-6) in the fetal circulation as early as the second trimester, suggesting the possibility of transplacental transfer of proinflammatory cytokines.95 Proinflammatory mediators such as IL-6 cause significant impairment of placental blood flow and fetal hypoxemia in animal models, dysregulate the barrier function of both the placenta and the immature fetal blood-brain barrier, trigger production of acute-phase proteins from the fetal liver, promote T-cell entry into the immature brain parenchyma, and disrupt the orderly patterning of the fetal cerebral cortex.91,93,96–99 The role of inflammatory mediators in this phenomenon is reinforced by the direct correlation that exists between plasma levels of IL-6 and the severity of functional deficits in offspring.100,101 In addition to IL-6, cytokines such as IL-1β, IL-7, and IL-13 are up-regulated in the fetal brain after a prenatal immune insult, a phenomenon that suggests collective activation of the innate fetal immune response.102
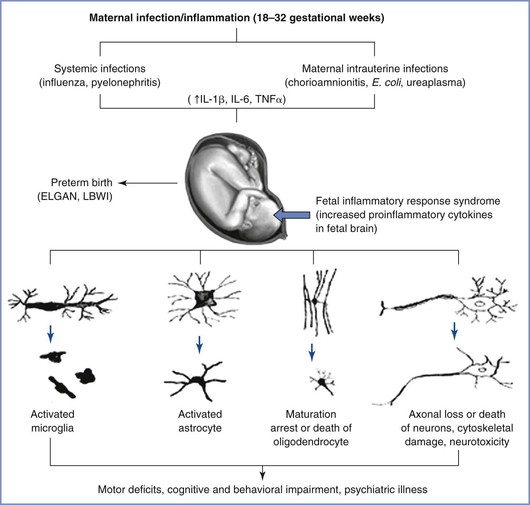
FIGURE 10-2 Probable mechanisms of fetal brain injury with in utero exposure to maternal inflammation. IL, interleukin; TNF, tissue necrosis factor; LBWI, low birth weight infant; ELGAN, extremely low gestational age neonate. (From Burd I, Balakrishnan B, Kannan S. Models of fetal brain injury, intrauterine inflammation, and preterm birth. Am J Reprod Immunol 2012; 67:287-94.)
Both microglia (the major resident macrophages in the developing brain) and the complement system have been implicated as amplifiers of this immune response. During normal fetal development, microglia invade and colonize the fetal brain during the first and second trimesters103 and are readily activated by proinflammatory mediators such as IL-1β. Activated microglial cells either cause a direct cytotoxic effect on oligodendrocytes and impair myelination or produce long-lasting alterations in neuronal-glial crosstalk, resulting in impaired synaptic function and subsequent neurodevelopmental disorders.3,91,104
At the cellular level, numerous mechanisms are involved in propagating the prenatal immune response. Collectively, robust experimental evidence suggests that prenatal inflammation alters fetal brain development at the molecular, cellular, and circuit levels. Epidemiologic studies have shown a strong correlation between maternal infection/inflammation and neurodevelopmental disorders such as schizophrenia and autism.92,105,106
Animal Models of Fetal Asphyxia
Much of our knowledge of the fetal response to insufficient exchange of respiratory gases has been gained through the use of animal models. However, the limitations of these models must be acknowledged. Raju107 reviewed the various animal models of fetal brain injury. At birth, sheep and guinea pig brains are much closer to maturity than the human brain. In this regard, rat pup and human brains are more similar to each other because they both undergo significant extrauterine development (Figure 10-3).108 Nonetheless, the importance of this distinction has been challenged. Previously, investigators relied mainly on morphologic milestones (e.g., the brain growth spurt) to compare species at different stages of development. A computerized method attempted to more accurately compare observations among 10 species (including humans) by evaluating the mathematical relationships of more than 100 developmental events and factors (e.g., evolutionary, genetic, neurochemical, neuroanatomic).109 Although all events have not been catalogued for any one species, the iterative process allows information to be added to improve the theoretic model and is freely available online.* This method is not completely understood or accepted but may explain some of the variability observed among various models of developmental brain injury.
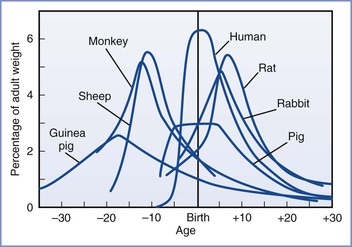
FIGURE 10-3 Brain growth spurts of seven mammalian species expressed as first-order velocity curves of the increase in weight with age. The units of time for each species are as follows: guinea pig (days); rhesus monkey (4 days); sheep (5 days); pig (weeks); human (months); rabbit (2 days); rat (days). Rates are expressed as a percentage of adult weight for each unit of time. (From Penning DH. Fetal and neonatal neurologic injury. In Chestnut DH, Polley LS, Tsen LC, Wong CA, editors. Chestnut’s Obstetric Anesthesia: Principles and Practice. 4th edition. Philadelphia, Mosby, 2009; modified from Dobbing J, Sands S. Comparative aspects of the brain growth spurt. Early Hum Dev 1979; 3:79-83.)
One advantage of the chronically instrumented fetal lamb is that it is similar in size to the human fetus, thus facilitating the placement of electrodes and vascular catheters in both the fetus and the mother. Investigators may obtain measurements while the mother (and fetus) remains anesthetized or from awake animals that have recovered from surgery. Studies of animals with continuous instrumentation allow the assessment of fetal breathing movements, gross body movements, brain electrical activity (electroencephalogram), and blood gas and pH measurements. Blood concentrations of glucose, lactate, and various hormones can also be determined. Microdialysis techniques have been used to evaluate neurotransmitter release within the fetal brain in vivo in acute, exteriorized, and chronic preparations.110–112 Other studies have measured fetal cerebral blood flow in vivo during episodes of hypoxemia113 and during maternal infusion of ethanol.114 Together, these studies have enhanced the understanding of the fetal brain response to pathophysiologic insults in utero. Ultimately, these insights may lead to improved diagnoses, treatment, and prevention of fetal brain injury.
Studies have used a variety of methods to produce fetal hypoxemia and acidemia in fetal lambs. Each method attempts to mimic one or more clinically relevant situation(s), including (1) decreased concentration of maternal inspired oxygen for several hours115 or days116; (2) decreased uterine blood flow, which may be accomplished by placement of an adjustable clamp on the common iliac artery117; (3) decreased umbilical blood flow, either by total obstruction118 or by means of a slow, progressive obstruction119; (4) selective uteroplacental embolization120; (5) maternal hemorrhage121; and (6) a combination of two insults, such as hypoxemia plus hypotension.122
Care must be exercised in the application of knowledge gained from hypoxia-ischemia studies conducted on nonfetal models (e.g., rat pups) to the problem of insufficient intrauterine gas exchange. The fetus and the fetal brain exist in a relatively hypoxemic environment. Despite preferential streaming of the most highly oxygenated blood to the brain and heart, the average PO2 measured in the carotid artery of fetal lambs at term is approximately 22 mm Hg.123 Further, unlike adult conditions in which global anoxia (i.e., cardiac arrest) or focal ischemia (i.e., stroke) is the clinical correlate, fetal asphyxia typically involves diminution, but not absence, of delivery of oxygen, with variable degrees of respiratory or metabolic acidosis. A complete loss of cerebral blood flow rarely occurs, except as a terminal event. Of course, prolonged hypoxemia and decreased oxygen delivery can lead to acidemia and myocardial failure, followed by ischemia and rapid fetal demise. Fetal hypoxemia may result from the compromise of any or all of the steps involved in maternal-fetal oxygen transport (Box 10-3).124 The impact of repeated hypoxic-ischemic insults should not be underestimated, and numerous clinical scenarios can be envisioned whereby this might occur (e.g., repetitive umbilical cord occlusion, chronic abruption). Moreover, brief insults that may be harmless could cause damage if repeated, as has been demonstrated in adult rats125 and in fetal lambs.126
The neuropathology of intrauterine asphyxia depends, to some extent, on gestational age. In fetal lambs exposed to sustained hypoxemia with developing acidemia, immature fetuses demonstrated a predominantly periventricular injury, whereas mature fetuses had a primarily cortical injury, although there was some overlap (Figure 10-4).127 This finding is consistent with injury patterns in humans. It is not surprising that the biophysical and biochemical responses to hypoxemia vary between preterm and term fetuses. Matsuda et al.115 observed that the development of metabolic acidemia, reduced fetal breathing and body movements, and an altered sleep state were much less pronounced in mid-gestational fetal lambs subjected to hypoxemia than in fetal lambs at term.
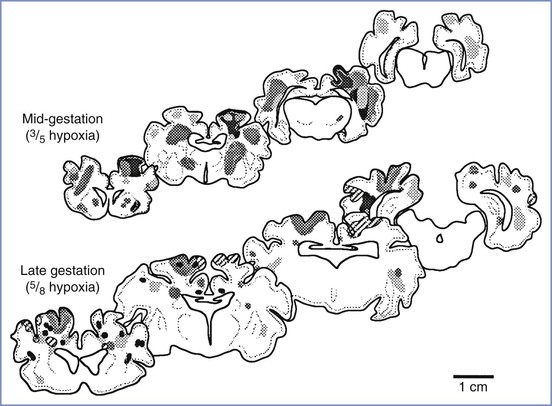
FIGURE 10-4 Composite diagram showing distribution of hypoxic injury in mid-gestational (top) and near-term (bottom) fetal lambs at 3 days after 8 hours of arterial hypoxemia. Hypoxemia was produced by placing the pregnant ewe in a chamber with reduced ambient oxygen. Each shading pattern represents an individual animal. The severity of injury is not indicated in this diagram. (From Penning DH, Grafe MR, Hammond R, et al. Neuropathology of the near-term and midgestation ovine fetal brain after sustained in utero hypoxemia. Am J Obstet Gynecol 1994; 170:1425-32.)
Neuropathology of Fetal Asphyxia
The mechanism and timing of an asphyxial insult can affect the resulting fetal or neonatal pathology. Acute, complete asphyxia must be distinguished from incomplete, brief, or intermittent asphyxia or chronic hypoxemia. Complete asphyxia may occur in the setting of a total placental abruption or umbilical cord occlusion (as may occur with a uterine rupture or umbilical cord prolapse), which if unrecognized and not treated rapidly leads to fetal demise. Incomplete asphyxia may occur in any setting in which oxygen delivery to the fetus is inadequate to meet all of its needs (e.g., brief and/or repeated episodes of partial umbilical cord occlusion, placental embolization, or incomplete placental abruption). This latter category of asphyxia presumably contributes to the largest proportion of cases of cerebral palsy attributed to antepartum events. In these cases, the insult is not severe enough to lead to immediate fetal death but can profoundly affect fetal brain growth and development. Ongoing studies are attempting to determine whether there is a period of time in utero when the fetus is especially vulnerable to neurologic injury.
Using a primate model to perform seminal research on the subject of perinatal brain injury, Myers27 identified two patterns of injury based on whether the fetus suffered complete or partial asphyxia. True complete asphyxia was demonstrated in fetal monkeys at term subjected to varying durations (0 to 25 minutes) of complete asphyxia. These fetuses were resuscitated when possible, a procedure that often required the use of cardiac massage and epinephrine, and postmortem examinations revealed extensive pathology in brainstem areas. In humans, such a severe intrauterine insult would most likely be incompatible with extrauterine survival. If survival did occur, the infant would show obvious encephalopathy and multiorgan system dysfunction at birth. The second pattern (i.e., partial asphyxia) is more relevant to the discussion of human cerebral palsy. In studies of fetal monkeys subjected to partial asphyxia,128 some animals demonstrated cortical necrosis, subcortical white matter damage, and basal ganglia damage. Although these two studies form the core of our knowledge of perinatal brain injury in primates, there were relatively few animals in each experimental group, and considerable variation in response occurred. Some animals suffered no injury, whereas others could not be resuscitated.
Several investigators have attempted to summarize the neuropathology of fetal and neonatal asphyxia.128–130 Volpe131 emphasized that the variation in neuropathology after intrauterine asphyxia depends on the fetal gestational age. Volpe also proposed a framework for these variations. The principal sites of injury in preterm fetuses are the white matter (especially periventricular white matter) and the basal ganglia, whereas older fetuses demonstrate injury primarily in the gray matter of the cortex and cerebellum.
Periventricular leukomalacia is the most common pathologic finding in preterm infants with brain injury.131 This lesion is characterized by coagulative necrosis of the white matter adjacent to the lateral ventricles and around the foramen of Monro, especially at the external angle of the lateral ventricles and the optic radiation.132 With long-term survival, the lesion may progress to a widening of the ventricles and hydrocephalus ex vacuo. Clinically, periventricular leukomalacia may not be apparent at birth.129 Developing hydrocephalus may be detected on computed tomography or ultrasonographic examination. In more subtle cases, magnetic resonance imaging (MRI) may show decreased myelination (see later discussion).133
The pathophysiology of periventricular leukomalacia is unclear. Conventional wisdom has held that periventricular leukomalacia is an ischemic lesion unique to preterm infants.134 The insult is thought to occur in an arterial border zone perfused by end-arterial branches of the middle and posterior cerebral arteries. This border zone has been identified by DeReuck,134 who demonstrated periventricular arborizations between vessels penetrating to the ventricles (i.e., ventriculopedal vessels) and between vessels arising from the ventriculochoroidal arteries (i.e., ventriculofugal vessels). Others have challenged DeReuck’s anatomic findings and have questioned whether periventricular leukomalacia is a purely ischemic lesion.135,136
White matter in the immature fetal brain may be at increased risk for hypoxic-ischemic injury because of a limited ability of its vessels to vasodilate.131 If this were true, autoregulation would be precluded in situations of hypotension. However, at least one study has shown that blood flow to white matter actually may increase (relative to gray matter) during fetal asphyxia.137 Fetal white matter may be more metabolically active than gray matter because of large numbers of actively myelinating cells.131 In situations of marginal oxygen supply, glia are subsequently at greater risk for injury. One study has suggested that immature astrocytes are more susceptible to ischemic death than mature astrocytes.138 Studies in fetal lambs have successfully produced pathologic changes similar to those present in infants with periventricular leukomalacia.127 These models may help clarify the mechanism of this common pathologic correlate of cerebral palsy in the preterm infant.
Fetal Adaptive Responses
The fetus takes advantage of several adaptive responses for survival and growth in the relatively hypoxemic intrauterine environment; these adaptive changes to intrauterine hypoxemia vary between immature and mature fetuses. Fetal responses to asphyxia may be categorized as an alteration of fetal metabolism or maximization of fetal oxygen transport (Box 10-4).124 Richardson139 defined the oxygen margin of safety as the extent to which fractional oxygen extraction can increase and fetal arterial PO2 can decrease before tissue oxygen supplies are inadequate. Regardless of the etiology of decreased oxygen delivery to the fetus, fetal oxygen consumption is maintained by increasing oxygen extraction until oxygen delivery is approximately 50% of normal.140 Lower levels of tissue oxygen tension result in progressive metabolic acidemia and a terminal decrease in oxygen consumption.139
Alterations in substrate use may affect the fetal response to insufficient exchange of respiratory gases. Unlike the adult brain, the fetal brain can use ketone bodies and lactate as alternative energy sources.141 In gravid ewes, a reduction in uterine blood flow results in reduced fetal glucose consumption.141 Current opinion holds that hyperglycemia should be avoided in adult humans at risk for ischemia.142 Hyperglycemia may exacerbate metabolic acidosis by providing substrate for anaerobic metabolism, which increases lactic acid production. However, Vannucci and Mujsce,143 citing experiments in neonatal rat pups, suggested that the immature brain may respond differently and that glucose administration may actually reduce hypoxic-ischemic brain injury. These investigators did not consider earlier work by Blomstrand et al.,144 who studied the effects of hypoxia in the anesthetized, exteriorized fetal lamb. In that study, hyperglycemia accelerated the loss of somatosensory evoked potentials, the onset of metabolic acidosis, and the reduction of cerebral oxygen consumption. Until these different observations are reconciled, the maintenance of normoglycemia in utero appears prudent.
During chronic hypoxemia, the fetus may also restrict the use of energy derived from oxidative metabolism to maintain essential cellular processes. This may lead to decreased somatic growth and fetal growth restriction. Using an ovine model of asphyxia, Hooper145 detected decreased incorporation of tritiated [3H]-thymidine (which reflects decreased DNA turnover and, presumably, decreased cell division) in fetal tissue. The decrease in incorporation of tritiated [3H]-thymidine was not uniform in all tissues. The rates of DNA synthesis were maintained in most fetal tissues (including the fetal brain) but were greatly reduced in the lung, the skeletal muscle, and the thymus gland.
The fetus can conserve additional energy by decreasing breathing and gross body movements. Rurak and Gruber146 demonstrated a 17% reduction in oxygen consumption in fetal lambs that were paralyzed by a neuromuscular blocking agent. Perceptible fetal movements represent an index of fetal health. Many obstetricians instruct their patients to count episodes of fetal activity for specified periods and to consult them if fetal movements are decreased or absent (see Chapter 6). Fetal hypoxemia results in decreases in both activity and rapid eye movement (REM) sleep in fetal lambs. REM sleep states are associated with an increased cerebral metabolic rate for oxygen (CMRO2).113 Thus, during periods of fetal stress, reductions in fetal body movements or REM sleep lead to a significant decline in fetal energy expenditure.
Oxygen deprivation typically results in a change in and/or redistribution of fetal cardiac output.147 The magnitude of these changes depends on the mechanism and severity of oxygen deprivation. Sheldon et al.148 demonstrated that experimental fetal hypoxemia (produced by the administration of a decreased maternal-inspired concentration of oxygen) resulted in greater blood flow to the brain, myocardium, and adrenal glands. In fetal lambs, a brief (4-minute) complete arrest of uterine and ovarian blood flow resulted in a decrease in blood flow to all organs except the myocardium and adrenal glands.149
Fetal and Neonatal Assessment
Fetal Neurobehavioral Assessment
With recent advances in the understanding of prenatal brain development and imaging technology, there is considerable interest in monitoring and codifying fetal neurologic development and behavior to predict postnatal neurodevelopment.150 The driving principle is that fetal behavioral patterns reflect complex interactions between the maternal environment and primitive neuronal network generators in the developing brain. There is an overwhelming convergence of opinion that most neurodevelopmental disorders have an intrauterine origin and that there is extensive neurobehavioral continuity from the fetal to the neonatal period.151
Although the assessment of high-risk pregnancies has included an analysis of certain aspects of fetal behavior, until recently there have been no unified scales for assessment of fetal neurobehavior. Current fetal neurobehavioral scales assess a variety of behaviors that can be categorized into the four main domains described by DiPietro150: (1) heart rate, (2) motor activity, (3) existing behavioral state, and (4) responsiveness to external stimuli. The Fetal Neurobehavioral Coding System (FENS) incorporates most elements of fetal behavior and is a direct extension of the NICU Network Neurobehavioral Scale (NNNS) used for neonatal assessment.152 Using ultrasonography, FENS analysis can identify specific behaviors in fetuses with growth restriction; compared with normally developing fetuses, fetuses with growth restriction demonstrate a delayed appearance of behavioral states, longer behavioral state transitions, and disorganized behavioral patterns. These tests have been validated in other paradigms, including pregnancies that were complicated by maternal diabetes, substance abuse, and cigarette smoking.
A recently developed, more comprehensive scale is the Kurjak Antenatal Neurodevelopmental Test (KANET), which includes an assessment of eight fetal parameters related to fetal behavior, general movements, and other physical signs (e.g., head circumference, presence or absence of overlapping cranial sutures, finger movements).153 However, these fetal assessment studies are time-consuming and require specific training to codify behaviors. In addition, because the brain structures driving such behaviors have not been clearly identified, it is difficult to understand the significance of differences in behavior, if any.
Fetal Neuroimaging Assessment
In vivo MRI provides details of the architecture of the developing brain beginning in the eighteenth gestational week and can quantify brain growth and structural abnormalities.154 The use of more sophisticated techniques such as MR tractography and functional fetal MRI is likely to enhance our understanding of normal brain development and thus facilitate identification of abnormal development. Until controlled trials demonstrate adequate sensitivity, specificity, and positive predictive power for these tests, their clinical potential is limited. These potential advantages will need to be balanced against the potential detrimental effects of ultrasonography and MRI on fetal neuronal development and migration.155
Neonatal Radiologic Diagnosis of Cerebral Injury
MRI is a useful tool in the diagnosis of neonatal brain injury.156,157 MRI can assist in the diagnosis of hypoxic ischemic encephalopathy in newborn infants,158 provide three-dimensional evaluations to determine the volume of gray matter and the extent of white matter myelination (thus providing valuable insights into normal and abnormal brain development),159 and estimate the timing of the brain injury in patients with cerebral palsy.160 The presence of cerebral edema confirms recent-onset brain injury; edema develops 6 to 12 hours after injury and resolves within 4 days.161 Unfortunately, the changes are subtle and the time frame of interest may extend before or after the intrapartum period. Nonetheless, the information can be quite helpful, and early imaging should be performed in cases of suspected brain injury. There is a strong correlation between anatomic brain lesions detected on MRI and specific types of cerebral palsy.161 MRI is particularly sensitive in the detection of periventricular leukomalacia, although many children with this MRI abnormality have clinically normal neurologic development.162
New imaging techniques, such as diffusion tensor imaging and magnetic resonance spectroscopy, may offer advantages over conventional MRI when performed early (i.e., hours) after a hypoxic-ischemic insult. Diffusion tensor imaging detects the microscopic movement of water particles in brain tissue. Magnetic resonance spectroscopy analyzes the signal of protons attached to molecules such as glutamate, glutamine, and lactate, among others.163 These methods, developed in rabbit164 and sheep165 models of intrauterine hypoxia, detect acute chemical changes in brain tissue and may accurately predict motor outcome in preterm infants.166 Injury patterns detected with these methods are present for several days and resolve over the next week, at which point the chronic injury becomes visible with conventional MRI. Identification of injuries shortly after birth with these newer techniques can support the hypothesis that an injury occurred within days of delivery.163 Thus, magnetic resonance spectroscopy and diffusion tensor imaging are powerful new tools for timing the occurrence and understanding the pathophysiology of perinatal brain injury.163 Cerebral ultrasonography remains a useful technique in the early neurologic neonatal assessment,167 especially for the critically ill infant who might not be a candidate for transfer to an MRI facility.
Anesthesia and Brain Injury
Anesthetic agents have profound effects on brain metabolism and synaptic transmission. These effects may be direct or indirect and protective or harmful.
Labor Analgesia and the Fetal Brain
Labor analgesia usually entails administration of lower concentrations of analgesic/anesthetic agents for a longer duration than occurs during administration of anesthesia for surgical procedures. Despite widespread use of analgesic and sedative drugs during labor, little attention has been paid to the neurodevelopmental consequences of antepartum and intrapartum fetal exposure to these drugs. Because neurodevelopmental events at term are quite different from those that occur during the second trimester, there is a need to design experimental studies to investigate the effects of analgesic techniques and drugs administered during the third trimester of pregnancy.
Parenteral Opioids
Among systemic opioids used for labor analgesia, meperidine remains the most widely studied. Although it is well recognized that opioids cross the placenta and enter the fetal circulation,168 the long-term effects of peripartum opioid exposure on the infant’s neurodevelopmental trajectory are unclear. Only a few preclinical studies have addressed this question.169,170 Endogenous opioid systems are active in the fetal brain, and the presence of their cognate receptors at critical sites during this period suggests that these systems are intricately linked to early neurodevelopment.171–174 Preclinical evidence suggests that opioid mechanisms play an important role in both early and adult neurogenesis by modulating neuronal progenitor proliferation and differentiation.175–177 Of concern, fetal rat exposure to morphine during the entire second trimester alters offspring hippocampal development.178 However, animal studies of opioid abuse in pregnancy should not be extrapolated to peripartum opioid use in humans because of differences in the gestational age as well as differences in drug dose and duration of administration. Only focused studies will reveal the true consequences of intravenous opioid administration for labor analgesia at term gestation.
Neuraxial Techniques
Studies of neuraxial analgesia in labor usually focus on analgesic quality and obstetric and short-term neonatal outcomes. To date, no randomized trials have evaluated the long-term effects of neuraxial analgesia on brain development in offspring. Epidurally administered local anesthetics cross the placenta and enter the fetal circulation. Golub179 randomized nonlaboring pregnant rhesus monkeys at term to receive epidural bupivacaine (total dose of 1.2 mg/kg) or saline.179 No differences in specific cognitive deficits were identified between groups; however, exposed offspring demonstrated a prolonged increase in motor disturbance behaviors at 10 to 12 months of age, suggesting that perinatal interventions can alter postnatal behavioral ontogeny. In the only human evidence to date, investigators examined the association between the use of neuraxial labor analgesia and the incidence of childhood learning disabilities in a population-based birth cohort of children from Olmsted County, Minnesota.180 The incidence of childhood learning disabilities was not associated with the use of neuraxial labor analgesia (adjusted hazard ratio, 1.05).180
Inhalational Agents
The use of inhalational anesthetic agents during labor and delivery became popular after the successful use of chloroform by John Snow during Queen Victoria’s delivery of Prince Leopold in 1853. Other inhalational agents were subsequently introduced, including nitrous oxide, trichloroethylene, cyclopropane, and methoxyflurane. Although the use of halogenated agents has been supplanted by widespread adoption of neuraxial techniques for labor analgesia, nitrous oxide is still widely used in many countries. Typically it is administered as 50% nitrous oxide in oxygen using a blender device (e.g., Nitronox in the United States) or premixed in a single cylinder (e.g., Entonox in the United Kingdom) (see Chapter 22).
Scientific studies of the fetal and neonatal effects of inhalational analgesics are generally of limited quality.181,182 Available evidence suggests that inhalational anesthetic agents, including nitrous oxide, have minimal or no effect on Apgar and neurobehavioral adaptation scores immediately after delivery.183,184 However, none of these studies has evaluated long-term neurodevelopmental outcomes. This knowledge gap is critical, because robust evidence suggests that early-life neural reprogramming, following pharmacologic and inflammatory insults, affects behavioral development later in life. Of concern is compelling animal evidence that anesthetic agents, when administered during a critical period of brain development, cause widespread neurodegeneration with subsequent learning, memory, and behavioral problems (see later discussion).185,186 Nitrous oxide, in particular, is now known to be a potent developmental neurotoxin in animal models, yet its effects (if any) on human neurodevelopment are unclear.
Epidemiologic evidence from young children receiving anesthetic agents appears to support the possibility that anesthesia and surgery are associated with learning disabilities and attention deficit disorders later in life.187 Thus, although the pattern of nitrous oxide administration during labor is unlike its administration for surgical anesthesia, the administration of nitrous oxide for labor analgesia merits closer scientific scrutiny.
Maternal Anesthesia and the Fetal Brain
Despite the popularity of neuraxial techniques in obstetric anesthesia, many pregnant women continue to require general anesthesia for either pregnancy-related or nonobstetric surgical procedures. The commonly used anesthetic agents freely cross the placenta and reach the fetal brain, causing fetal sleep or sedation. Obstetric anesthesia research has focused primarily on the teratogenic effects of anesthetic agents administered during the first trimester (see Chapter 17) and the effects of anesthetic agents on neonatal behavior when administered during cesarean delivery. Historically, the second trimester has been assumed to be a safe period for surgery and anesthesia, primarily because of a lack of targeted studies. However, during the past decade, extensive animal research has shown that anesthetic agents, when administered during the phase of synaptogenesis, can induce a profound neurodegenerative response in the developing brain and cause functional impairment in offspring.185,188
Human epidemiologic studies appear to support an association between early childhood exposure to anesthetic agents and subsequent functional impairment. However, it is unclear whether these adverse outcomes result from the underlying disease, surgery, or anesthesia or a combination of these factors.187,189 A recent study found evidence that repeated anesthesia exposures or a cumulative anesthesia exposure time greater than 2 hours during early childhood was associated with a nearly twofold increase in the incidence of learning disabilities and attention deficit hyperactivity disorder.189 Because human synaptogenesis appears to begin during the third trimester, there is now serious concern that intrauterine fetal exposure to these anesthetic agents may result in similar functional impairment. Because there is no precise way to monitor human fetal brain development in utero, the potential long-term effects of maternal anesthesia on the fetal brain must be investigated in animal models. However, given the considerable differences in neural maturation among species (Figure 10-5; see also Figure 10-3), and the duration of anesthesia exposure in relation to the lifespan of the organism, these results should be interpreted with caution.190,191
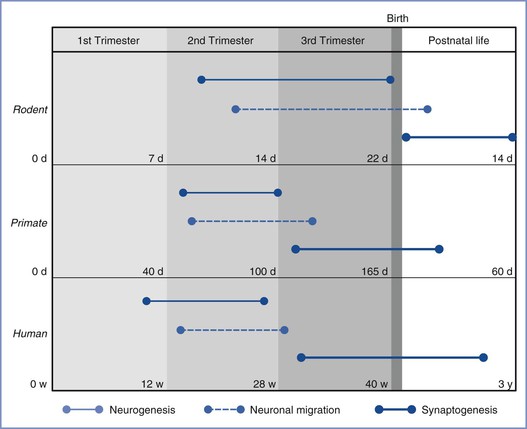
FIGURE 10-5 Time lines of major neurodevelopmental events in utero in rodents, nonhuman primates, and humans. Events are as marked in the figure legend (d, days; w, weeks; y, years). Synaptogenesis is predominantly a postnatal event in rodents, unlike that in primates and humans. (From Palanisamy A. Maternal anesthesia and fetal neurodevelopment. Int J Obstet Anesth 2012; 21:152-62.)
The exact mechanisms by which anesthetic agents impair early brain development are still under active investigation.192 Anesthetic agent interactions with GABA and the NMDA-subtype of glutamate receptors decrease activity-dependent synapse formation and cause apoptotic neurodegeneration in multiple areas of the developing brain. These histopathologic changes have been well investigated, especially in the hippocampal formation, which is an area that is crucial for memory. Early exposure to anesthetic agents affects long-term potentiation in the hippocampus and affects spatial working memory in animal models.185 These changes do not appear to be caused by direct cytotoxicity193 but rather by a combination of effects on both neuronal and non-neuronal cells in the developing brain (Box 10-5).
Of specific concern are the effects of anesthetic agents on neurogenesis and synapse formation in the fetal brain. Human neural ontogeny suggests that the second trimester is a period of active fetal brain development, with neuroblast proliferation peaking between the 5th and 25th postmenstrual weeks. Because GABA and glutamate play a crucial role in these processes, there is concern that prolonged and nonphysiologic modulation of the fetal GABA and glutamatergic systems, as might occur during second-trimester maternal anesthesia, might affect neurogenesis, neuronal migration, and/or synapse formation.
In one of the first animal studies to simulate a clinically relevant scenario,194 a single exposure to 1.4% isoflurane (1 MAC [minimum anesthetic concentration]) for 4 hours during the second trimester caused long-lasting impairment of spatial working memory in rodent offspring. Although the exact mechanisms behind these behavioral disturbances are unclear, other studies suggest that mid-gestational exposure to isoflurane up-regulates the proapoptotic protein caspase-12, decreases overall synapse numbers in the fetal hippocampus, and down-regulates the plasticity-associated protein GAP-43.195,196 Similar results have been reported in pregnant guinea pigs and macaques, suggesting that the fetal brain remains highly susceptible to maternal mid-trimester anesthesia.197,198 Furthermore, isoflurane suppresses neurogenesis in rodents both in vitro as well as in vivo,199–201 causing a depletion of the neural stem cell pool. At least in vitro, this phenomenon appears to be dose dependent.201 At the present time, the impact of reduced neurogenesis on behavioral deficits and the effect of anesthetic agents on neuronal migration remain unknown.
When these studies are extended to the third trimester, the results are mixed. In one rodent study, maternal administration of 1.3% isoflurane for 6 hours during the third trimester had no effect on offspring neurodevelopment.202 However, these investigators performed another dose-response study in term rodents and found that maternal administration of 3% isoflurane, but not 1.3% isoflurane, for 1 hour caused fetal brain hippocampal neurodegeneration.203 No neurodegenerative changes were observed after third-trimester exposure in guinea pigs.197 Thus, collectively, it appears that the fetal brain is less vulnerable to the adverse effects of anesthetic agents during the third trimester. This phenomenon could be due to the stage of neurodevelopment, or more likely, to an increase in the levels of neuroprotective hormones such as estrogen, progesterone, neurosteroids, and oxytocin during the third trimester.
The relative fetal/neonatal safety of third-trimester maternal anesthesia was supported by a robust, population-based birth cohort study (in Olmsted County, Minnesota) that sought to determine the incidence of learning disabilities in children after maternal administration of general or neuraxial anesthesia during cesarean or vaginal delivery.204 Children exposed to general anesthesia during cesarean delivery were not more likely to develop learning disabilities compared with those born vaginally with no exposure to general anesthesia. Although the study was retrospective and used data from 1976 to 1982, it is reassuring that even the children whose mothers required emergency general anesthesia (presumably secondary to presumed fetal compromise) did not have a higher incidence of learning disability. Further epidemiologic work is required to ascertain the effects of maternal anesthesia during nonobstetric surgery in the second trimester and that of nitrous oxide analgesia during labor.
Meanwhile, the potential for anesthesia-related neurotoxicity will undoubtedly undergo continued scrutiny. In a comprehensive editorial,205 McGowan and Davis made the following conclusions: Additional animal studies are needed to define molecular mechanisms, risks, and potential treatments for anesthetic-related neurotoxicity in the developing brain; future studies should be relevant to human development and clinical practice; human studies of adequate statistical power are needed to identify any evidence of injury from intrauterine exposure to anesthetic agents; future studies should take advantage of advances in genomics and proteomics and should target the identification of sensitive and specific biomarkers for neurocognitive injury; some forms of developmental brain injury might be prevented or ameliorated by periprocedural therapy (e.g., anti-apoptotic agents such as melatonin).205,206
McGowan and Davis205 affirmed the U.S. Food and Drug Administration’s (FDA) conclusion207 that currently, “there [is] no scientific basis to recommend changes in clinical [anesthesia] practice.” They noted that the “real enemy” includes “known and understood causes of brain injury and death,” such as hypoxia and cardiovascular collapse. They cautioned, “[W]e must not let our enthusiasm for understanding the possible neurocognitive risks of anesthetics … obscure our awareness of this enemy or prevent us from alleviating pain.”205 The FDA’s position was reaffirmed in March 2011.208
Fetal Neuroprotection
Throughout gestation, the fetus remains concealed, protected, and nourished by a combination of maternal anatomic and physiologic factors. For example, the amniotic fluid cushions the fetus against trauma, and the placenta serves as a conduit to ensure a continuous supply of maternal nutrients to the developing fetus. Despite these inbuilt protective mechanisms, the fetus remains vulnerable to maternal insults such as infection and fever, drugs, and acute changes in placental physiology. Among all organ systems, the developing central nervous system appears to be most susceptible to such insults. Understanding the developmental aspects of neuroprotective mechanisms will therefore enable generation of targeted neuroprotective therapies.
Role of the Placenta
One of the fundamental neuroprotective mechanisms is the barrier function of the placenta. The placenta serves as a conduit for chemical communication between the mother and the developing fetus; endocrine signals, growth factors, and cytokines freely traverse the placenta, which dynamically adapts to chronic changes in the maternal-fetal environment to preserve fetal growth and viability.209,210 However, this function also allows transplacental transfer of an array of pharmacologically active molecules either by passive diffusion or active transport.211 By virtue of its enzymatic machinery, the placenta is capable of detoxification of some of these potentially harmful chemicals, making it the first line of defense against potentially harmful environmental agents.
Although passive diffusion along a concentration gradient is the most widely studied placental transport mechanism, recent studies have elucidated the roles of two important active transport molecules in the syncytiotrophoblast, which actively extrudes xenobiotics: phospho-glycoprotein (P-gp) and breast cancer resistance protein (BRCP).212 The activity of these transporters varies with gestational age and certain pathophysiologic conditions (e.g., preeclampsia, intrauterine infection) and is influenced by the steroid hormones of pregnancy. Thus, it is possible that placental permeability to certain drugs could depend on a complex interplay of several factors. In addition to this barrier function, the human placenta secretes estrogen and progesterone in very high concentrations; these hormones, as well as others, eventually enter the fetal circulation, where they serve as substrates for de novo neurosteroid synthesis in the fetal brain.213 In particular, allopregnanolone has been shown to exert neuroprotective effects in the fetal brain (see later discussion).214
Humoral Mechanisms
Published studies have extensively investigated the intricate and symbiotic relationship between the fetus and maternal hormones throughout pregnancy. Much of our understanding comes from elaborate murine and primate research models in which changes in maternal levels of hormones closely parallel changes in the fetal plasma and/or brain. Throughout pregnancy, there is a gradual rise in the levels of many maternal hormones such as progesterone, estradiol, and oxytocin.215–217 At term or during labor the levels of these hormones are 40- to 100-fold higher than in the nonpregnant state.
Many of these hormones freely cross the placenta and are transported to the fetal brain, where they profoundly influence neurodevelopment. For example, estradiol and progesterone influence neural stem cell proliferation, modulate apoptosis and synaptogenesis in a region-specific manner, alter subcellular signaling mechanisms, and promote dendritic growth and spinogenesis through specific receptor mechanisms.215,216 Estradiol, in particular, prevents cell death in both neuronal and non-neuronal cell lines.
Maternal plasma oxytocin levels gradually increase during pregnancy and reach a peak during the second stage of labor. Oxytocin is of particular importance because it has significant effects on GABAergic signaling in fetal neurons. In a series of elegant experiments, investigators showed that oxytocin transiently switched the action of GABA on immature rodent fetal neurons from depolarizing to hyperpolarizing at term gestation.218 This finding raises the possibility that oxytocin protects the fetal brain during the stressful process of labor and delivery.219
Neuroprotective Therapies
Magnesium Sulfate and Cerebral Palsy
Until recently there was considerable controversy regarding the role of magnesium sulfate in preventing or possibly exacerbating fetal brain injury. Although some controversy remains, the publication of several large randomized studies of the effect of antenatal maternal magnesium sulfate administration on offspring outcome has dramatically altered practice guidelines and clinical practice.220–222 Although none of these studies demonstrated significant improvement in the primary outcome, all showed reduced cognitive morbidity and none showed any increase in pediatric morbidity or mortality associated with magnesium sulfate use for neuroprotection.
In a placebo-controlled trial of women who were thought likely to deliver within 24 hours and before 30 weeks’ gestation in New Zealand and Australia, Crowther et al.220 reported a lower incidence of substantial gross motor dysfunction (3.4% versus 6.6%; relative risk [RR], 0.51; 95% CI, 0.29 to 0.91) and combined death or substantial gross motor dysfunction (17% versus 22.7%; RR, 0.75; 95% CI, 0.59 to 0.96) in children whose mothers were randomized to receive antenatal magnesium sulfate treatment. In another large trial from France, which included women in preterm labor before 33 weeks’ gestation, a significant reduction in death and/or gross motor dysfunction was again identified in the children whose mothers received magnesium sulfate (25.6% versus 30.8%; OR, 0.62; 95% CI, 0.41 to 0.99).221 A reduction in death and/or motor or cognitive dysfunction (34.9% versus 40.5%; OR, 0.68; 95% CI, 0.47 to 0.99) was observed in the magnesium-exposed offspring at 2 years of age.221 Finally, a randomized, controlled multicenter trial in the United States found that fetal exposure to magnesium sulfate within 24 hours of preterm delivery (between 24 and 32 completed weeks’ gestational age) did not reduce the combined risk for moderate or severe cerebral palsy or death. However, fetal exposure to magnesium sulfate reduced the risk for moderate or severe cerebral palsy among survivors (1.9% versus 3.5%; RR, 0.55; 95% CI, 0.32 to 0.95) and was associated with a decreased overall rate of cerebral palsy (4.2% versus 7.3%; P = .004).222
Although the results are optimistic, it is difficult to compare these trials owing to differences in inclusion criteria, study interventions/dosages, and outcomes. Nonetheless, after the publications of these trials, it has been concluded from meta-analyses that fetal exposure to magnesium sulfate may reduce the risk for cerebral palsy without increasing the risk for neonatal death.223,224
In 2010, the ACOG and the SMFM released a joint committee opinion that supported antenatal maternal magnesium sulfate administration for fetal neuroprotection, stating that the available evidence suggests that magnesium sulfate administered before anticipated early preterm birth reduces the risk for cerebral palsy in surviving infants.225 Physicians electing to use magnesium sulfate for fetal neuroprotection should develop specific guidelines regarding inclusion criteria, treatment regimens, concurrent tocolysis, and monitoring. The ACOG and the SMFM have concluded that it is reasonable to use a protocol based on one of the large randomized trials220–222; magnesium sulfate should be offered to women at high risk for anticipated preterm delivery (< 28 to 32 weeks’ gestational age) within 24 hours.220–222 A loading dose of magnesium sulfate 4 to 6 g should be administered, followed by a maintenance infusion of 1 to 2 g/h for 12 to 24 hours, at which point the risk for impending preterm delivery should be reassessed. If there is no longer a concern for impending delivery, the magnesium sulfate should be discontinued and restarted with active labor or when delivery is again thought to be imminent.
Hypothermia
Some investigators have described improved outcomes after the use of hypothermia in neonates at risk for hypoxic ischemic encephalopathy. One group of investigators has described an experimental model of severe intrauterine hypoxia in preterm fetal sheep, in which asphyxia was produced by 25 minutes of complete umbilical cord occlusion.226 Cerebral hypothermia (fetal extradural temperature reduced from 39° to 29° C) decreased the loss of striatal neurons and oligodendroglia. This finding was associated with improved basal ganglia function after ischemia.
These and other experimental results prompted a randomized clinical trial of whole-body hypothermia for neonates with hypoxic ischemic encephalopathy.227 Eligible neonates were older than 36 weeks’ gestational age, had moderate or severe encephalopathy, and were admitted to the neonatal intensive care unit within 6 hours of birth. Body temperature was lowered to 33.5° C for 72 hours in neonates randomized to hypothermia treatment. Death or moderate to severe disability at 18 to 22 months of age occurred in 44% of 102 infants in the hypothermia group, compared with 62% of 106 infants in the control group (risk ratio, 0.72; 95% CI, 0.54 to 0.95; P = .01). Although encouraging, these results are at odds with those from another large multicenter randomized trial.228 In an editorial attempting to reconcile these opposing results, several possible explanations were suggested.229 Importantly, in the study that demonstrated no benefit, cooling began later and more time was required to achieve complete cooling, because head (not total body) cooling was employed.228 Moreover, the study that showed no benefit with hypothermia may have included infants who were so severely affected that no therapy would have been beneficial.228 This possibility highlights the importance of patient selection in these clinical trials.
In 2012, the whole-body hypothermia investigators published the results of follow-up evaluations of the original study subjects at 6 to 7 years of age. There was no difference in the combined primary outcome of death or an IQ score less than 70 (47% versus 62%, P = .06) between the hypothermia group and the control group.230 The hypothermia group had a lower incidence of death or severe disability (41% versus 60%, P = .03), but there was no difference in moderate or severe disability (35% versus 38%, P = .87). Attention-executive dysfunction occurred in 4% of the hypothermia group versus 13% of the usual care group (P = .19), and visuospatial dysfunction occurred in 4% of the hypothermia group versus 3% of the usual care group (P = .80).230 Thus, although there was no significant difference in the primary outcome, whole-body hypothermia decreased the incidence of death and did not increase the rate of a low IQ score or severe disability among survivors.
A 2013 meta-analysis of 11 randomized controlled trials of hypothermia therapy, which included 1505 term and late preterm infants, concluded that the benefits of cooling on survival and neurodevelopment outweigh the short-term adverse effects.231 The authors advised that hypothermia should be instituted in term and late preterm infants with moderate-to-severe hypoxic ischemic encephalopathy if identified before 6 hours of age. Further trials are necessary to identify appropriate cooling techniques and to refine patient selection.
Experimental Neuroprotection
Perlman232 has reviewed various strategies for treating hypoxic-ischemic neonatal injuries, including administration of inflammatory mediator modulators, excitatory amino acid receptor agonists and antagonists, free radical scavengers, and platelet-activating factor antagonists. These emerging therapeutic strategies stem from basic neuroscience research on brain development and the pathophysiology of ischemic injury.
The role of white matter in the attenuation of hypoxic-ischemic brain damage (e.g., through uptake of excitatory amino acids or sequestration of potassium and hydrogen ions) is underappreciated,233–235 and drugs that inhibit the release of excitatory amino acids or antagonize their receptors may be of benefit.236 Multiple strategies may be necessary to inhibit the deleterious pathways initiated by brain ischemia and hypoxia.237 The combination of oxygen free-radical scavengers and calcium entry–blocking agents appears to have some efficacy in limiting postasphyxial injury in neonatal sheep.238 A “brain cocktail,” consisting of free radical scavengers, modifiers of nitric oxide activity, metabolic inhibitors, calcium and iron chelators, and drugs that affect the excitatory amino acid systems, may someday be administered to fetuses and neonates at risk for brain injury. Additional compounds that may inhibit CNS necrosis or apoptosis, either in utero or in the neonatal period, include agents that interrupt the inflammatory cascade, progesterone, and other steroids.
Among the pharmacologic candidates for neuroprotection, erythropoietin (EPO) appears promising239; EPO is known to have wide-ranging actions, including anti-apoptotic and neurotrophic effects. In a preterm fetal sheep model, EPO was shown to reduce axonal damage and decrease the astrocytic and microglial response to maternally administered lipopolysaccharide (which leads to fetal brain damage).239 In addition, EPO was also shown to confer protection against placental and fetal liver damage, suggesting that EPO could potentially buttress the placental barrier and prevent fetal injury. In the first human clinical study, EPO administration in full-term neonates was associated with an almost 50% reduction in death and disability at 18 months when the hypoxic-ischemic injury was moderate but not severe.240 Although neonatal EPO therapy does not appear to increase the risk for thrombotic effects, the safety and efficacy of EPO administration to pregnant mothers, and its role in fetal neuroprotection, remains to be investigated.240
Another agent with potential for fetal neuroprotection is melatonin, a highly effective antioxidant with reliable transplacental transfer and a wide therapeutic index. Administration of melatonin to fetal sheep compromised by experimental umbilical cord occlusion prevents oxidative stress, reduces lipid peroxidation, modulates microglial activation, and decreases the extent of brain damage.241 The translational potential of other agents such as N-acetylcysteine (NAC), allopurinol, neurosteroids such as allopregnanolone, anesthetic agents such as xenon, and creatine appears limited.
The ability to accurately predict which fetuses are at risk for neurologic injury, and when, is still rudimentary, because the most vulnerable periods of fetal development are still unknown, and noninvasive fetal surveillance techniques are not very advanced. The ability to identify these “at risk” infants in utero is a necessary step in designing effective therapeutic regimens that interfere minimally with the normal trophic activities of the developing brain.
References
1. Caviness VS Jr, Nowakowski RS, Bhide PG. Neocortical neurogenesis: morphogenetic gradients and beyond. Trends Neurosci. 2009;32:443–450.
2. Mitsuhashi T, Takahashi T. Genetic regulation of proliferation/differentiation characteristics of neural progenitor cells in the developing neocortex. Brain Dev. 2009;31:553–557.
3. Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705.
4. MacDonald JL, Roskams AJ. Epigenetic regulation of nervous system development by DNA methylation and histone deacetylation. Prog Neurobiol. 2009;88:170–183.
5. Amrein I, Isler K, Lipp HP. Comparing adult hippocampal neurogenesis in mammalian species and orders: influence of chronological age and life history stage. Eur J Neurosci. 2011;34:978–987.
6. Molnár Z, Clowry G. Cerebral cortical development in rodents and primates. Prog Brain Res. 2012;195:45–70.
7. Huppi PS. Cortical development in the fetus and the newborn: advanced MR techniques. Top Magn Reson Imaging. 2011;22:33–38.
8. Ayala R, Shu T, Tsai LH. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128:29–43.
9. Pleasure SJ, Anderson S, Hevner R, et al. Cell migration from the ganglionic eminences is required for the development of hippocampal GABAergic interneurons. Neuron. 2000;28:727–740.
10. Belvindrah R, Lazarini F, Lledo PM. Postnatal neurogenesis: from neuroblast migration to neuronal integration. Rev Neurosci. 2009;20:331–346.
11. Heng JI, Moonen G, Nguyen L. Neurotransmitters regulate cell migration in the telencephalon. Eur J Neurosci. 2007;26:537–546.
12. de Graaf-Peters VB, Hadders-Algra M. Ontogeny of the human central nervous system: what is happening when? Early Hum Dev. 2006;82:257–266.
13. Levitt P. Structural and functional maturation of the developing primate brain. J Pediatr. 2003;143(4 Suppl):S35–S45.
14. Lowery CL, Hardman MP, Manning N, et al. Neurodevelopmental changes of fetal pain. Semin Perinatol. 2007;31:275–282.
15. Ferschl M, Ball R, Lee H, Rollins MD. Anesthesia for in utero repair of myelomeningocele. Anesthesiology. 2013;118:1211–1223.
16. Wang DD, Kriegstein AR. Defining the role of GABA in cortical development. J Physiol. 2009;587:1873–1879.
17. Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739.
18. LoTurco JJ, Owens DF, Heath MJ, et al. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron. 1995;15:1287–1298.
19. Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284.
20. Cuzon VC, Yeh PW, Yanagawa Y, et al. Ethanol consumption during early pregnancy alters the disposition of tangentially migrating GABAergic interneurons in the fetal cortex. J Neurosci. 2008;28:1854–1864.
21. Uban KA, Sliwowska JH, Lieblich S, et al. Prenatal alcohol exposure reduces the proportion of newly produced neurons and glia in the dentate gyrus of the hippocampus in female rats. Horm Behav. 2010;58:835–843.
22. Manent JB, Jorquera I, Mazzucchelli I, et al. Fetal exposure to GABA-acting antiepileptic drugs generates hippocampal and cortical dysplasias. Epilepsia. 2007;48:684–693.
23. Saunders NR, Liddelow SA, Dziegielewska KM. Barrier mechanisms in the developing brain. Front Pharmacol. 2012;3:46.
24. Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13.
25. The definition and classification of cerebral palsy. Dev Med Child Neurol. 2007;49(Suppl 109):1–44.
26. American College of Obstetricians and Gynecologists. Inappropriate use of the terms fetal distress and birth asphyxia. ACOG Committee Opinion No. 326. December 2005. Obstet Gynecol. 2005;106:1469–1470.
27. Myers RE. Two patterns of perinatal brain damage and their conditions of occurrence. Am J Obstet Gynecol. 1972;112:246–276.
28. Freeman R. Intrapartum fetal monitoring—a disappointing story. N Engl J Med. 1990;322:624–626.
29. Grant A, O’Brien N, Joy MT, et al. Cerebral palsy among children born during the Dublin randomised trial of intrapartum monitoring. Lancet. 1989;2:1233–1236.
30. Hankins GD, Erickson K, Zinberg S, et al. Neonatal encephalopathy and cerebral palsy: a knowledge survey of Fellows of The American College of Obstetricians and Gynecologists. Obstet Gynecol. 2003;101:11–17.
31. Leveno KJ, Cunningham FG, Nelson S, et al. A prospective comparison of selective and universal electronic fetal monitoring in 34,995 pregnancies. N Engl J Med. 1986;315:615–619.
32. MacDonald D, Grant A, Sheridan-Pereira M, et al. The Dublin randomized controlled trial of intrapartum fetal heart rate monitoring. Am J Obstet Gynecol. 1985;152:524–539.
33. Schifrin BS, Dame L. Fetal heart rate patterns. Prediction of Apgar score. JAMA. 1972;219:1322–1325.
34. Friedman EA. The obstetrician’s dilemma: how much fetal monitoring and cesarean section is enough? N Engl J Med. 1986;315:641–643.
35. Rosen MG, Dickinson JC. The paradox of electronic fetal monitoring: more data may not enable us to predict or prevent infant neurologic morbidity. Am J Obstet Gynecol. 1993;168:745–751.
36. Freeman RK. Problems with intrapartum fetal heart rate monitoring interpretation and patient management. Obstet Gynecol. 2002;100:813–826.
37. Schifrin BS, Cohen WR. Medical legal issues in fetal monitoring. Clin Perinatol. 2007;34:329–343.
38. Macones GA, Hankins GD, Spong CY, et al. The 2008 National Institute of Child Health and Human Development workshop report on electronic fetal monitoring: update on definitions, interpretation, and research guidelines. Obstet Gynecol. 2008;112:661–666.
39. Bakketeig LS. Only a minor part of cerebral palsy cases begin in labour: but still room for controversial childbirth issues in court. BMJ. 1999;319:1016–1017.
40. Blair E, Stanley FJ. Intrapartum asphyxia: a rare cause of cerebral palsy. [Erratum appears in J Pediatr 1988 Aug;113(2):420]. J Pediatr. 1988;112:515–519.
41. Espinoza MI, Parer JT. Mechanisms of asphyxial brain damage, and possible pharmacologic interventions, in the fetus. Am J Obstet Gynecol. 1991;164:1582–1589.
42. MacLennan A. A template for defining a causal relation between acute intrapartum events and cerebral palsy: international consensus statement. BMJ. 1999;319:1054–1059.
43. Muraskas JK, Morrison JC, Muraskas JK, Morrison JC. A proposed evidence-based neonatal work-up to confirm or refute allegations of intrapartum asphyxia. Obstet Gynecol. 2010;116:261–268.
44. Nelson KB, Ellenberg JH. Antecedents of cerebral palsy. I. Univariate analysis of risks. Am J Dis Child. 1985;139:1031–1038.
45. Nelson KB, Ellenberg JH. Antecedents of cerebral palsy. Multivariate analysis of risk. N Engl J Med. 1986;315:81–86.
46. Rosen MG, Dickinson JC. The incidence of cerebral palsy. Am J Obstet Gynecol. 1992;167:417–423.
47. Badawi N, Kurinczuk JJ, Keogh JM, et al. Intrapartum risk factors for newborn encephalopathy: the Western Australian case-control study. BMJ. 1998;317:1554–1558.
48. O’Callaghan ME, MacLennan AH, Gibson CS, et al. Epidemiologic associations with cerebral palsy. Obstet Gynecol. 2011;118:576–582.
49. American College of Obstetricians Gynecologists Taskforce on Neonatal Encephalopathy and Cerebral Palsy, The American Academy of Pediatrics. Neonatal Encephalopathy and Cerebral Palsy: Defining the Pathogenesis and Pathophysiology. American College of Obstetricians and Gynecologists: Washington, DC; 2003.
50. Phelan JP, Korst LM, Martin GI, et al. Application of criteria developed by the task force on neonatal encephalopathy and cerebral palsy to acutely asphyxiated neonates. Obstet Gynecol. 2012;119:1056–1057.
52. Chiswick M. Birth asphyxia and cerebral palsy. Int J Obstet Anesth. 1992;1:178–179.
53. Hull J, Dodd K. What is birth asphyxia? Br J Obstet Gynaecol. 1991;98:953–955.
54. Gilstrap LC 3rd, Leveno KJ, Burris J, et al. Diagnosis of birth asphyxia on the basis of fetal pH, Apgar score, and newborn cerebral dysfunction. Am J Obstet Gynecol. 1989;161:825–830.
55. Apgar V. A proposal for a new method of evaluation of the newborn infant. Curr Res Anesth Analg. 1953;32:260–267.
56. Marrin M, Paes BA. Birth asphyxia: does the Apgar score have diagnostic value? Obstet Gynecol. 1988;72:120–123.
57. Sykes GS, Molloy PM, Johnson P, et al. Do Apgar scores indicate asphyxia? Lancet. 1982;1:494–496.
58. Nelson KB, Ellenberg JH. Apgar scores as predictors of chronic neurologic disability. Pediatrics. 1981;68:36–44.
59. Fee SC, Malee K, Deddish R, et al. Severe acidosis and subsequent neurologic status. Am J Obstet Gynecol. 1990;162:802–806.
60. Goodwin TM, Belai I, Hernandez P, et al. Asphyxial complications in the term newborn with severe umbilical acidemia. Am J Obstet Gynecol. 1992;167:1506–1512.
61. Goldaber KG, Gilstrap LC 3rd, Leveno KJ, et al. Pathologic fetal acidemia. Obstet Gynecol. 1991;78:1103–1107.
62. Low JA, Lindsay BG, Derrick EJ. Threshold of metabolic acidosis associated with newborn complications. Am J Obstet Gynecol. 1997;177:1391–1394.
63. Low JA, Panagiotopoulos C, Derrick EJ. Newborn complications after intrapartum asphyxia with metabolic acidosis in the term fetus. Am J Obstet Gynecol. 1994;170:1081–1087.
64. Nagel HT, Vandenbussche FP, Oepkes D, et al. Follow-up of children born with an umbilical arterial blood pH < 7. Am J Obstet Gynecol. 1995;173:1758–1764.
65. Sehdev HM, Stamilio DM, Macones GA, et al. Predictive factors for neonatal morbidity in neonates with an umbilical arterial cord pH less than 7.00. Am J Obstet Gynecol. 1997;177:1030–1034.
66. Gull I, Jaffa AJ, Oren M, et al. Acid accumulation during end-stage bradycardia in term fetuses: how long is too long? Br J Obstet Gynaecol. 1996;103:1096–1101.
67. Ross MG, Gala R, Ross MG, Gala R. Use of umbilical artery base excess: algorithm for the timing of hypoxic injury. Am J Obstet Gynecol. 2002;187:1–9.
68. Malin GL, Morris RK, Khan KS, et al. Strength of association between umbilical cord pH and perinatal and long term outcomes: systematic review and meta-analysis. BMJ. 2010;340:c1471.
69. Blumenthal I. Cerebral palsy—medicolegal aspects. J R Soc Med. 2001;94:624–627.
70. Phelan JP, Korst LM, Ahn MO, Martin GI. Neonatal nucleated red blood cell and lymphocyte counts in fetal brain injury. Obstet Gynecol. 1998;91:485–489.
71. Korst LM, Phelan JP, Ahn MO, Martin GI. Nucleated red blood cells: an update on the marker for fetal asphyxia. Am J Obstet Gynecol. 1996;175:843–846.
72. Leikin E, Verma U, Klein S, Tejani N. Relationship between neonatal nucleated red blood cell counts and hypoxic-ischemic injury. Obstet Gynecol. 1996;87:439–443.
73. Grether JK, Nelson KB. Maternal infection and cerebral palsy in infants of normal birth weight. [Erratum appears in JAMA 1998 Jan 14;279(2):118]. JAMA. 1997;278:207–211.
74. Murphy DJ, Johnson A. Placental infection and risk of cerebral palsy in very low birth weight infants. J Pediatr. 1996;129:776–778.
75. Silverstein FS, Barks JD, Hagan P, et al. Cytokines and perinatal brain injury. Neurochem Int. 1997;30:375–383.
76. Dammann O, Leviton A. Does prepregnancy bacterial vaginosis increase a mother’s risk of having a preterm infant with cerebral palsy? Dev Med Child Neurol. 1997;39:836–840.
77. Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. 1997;42:1–8.
78. Wu YW, Colford JM Jr. Chorioamnionitis as a risk factor for cerebral palsy: a meta-analysis. JAMA. 2000;284:1417–1424.
79. Shatrov JG, Birch SC, Lam LT, et al. Chorioamnionitis and cerebral palsy: a meta-analysis. Obstet Gynecol. 2010;116:387–392.
80. Segal S. Labor epidural analgesia and maternal fever. Anesth Analg. 2010;111:1467–1475.
81. Gray D, Finucane BT. Epidurals and fever: association or cause? CMAJ. 1997;157:511–512.
82. Lieberman E, Lang JM, Frigoletto F Jr, et al. Epidural analgesia, intrapartum fever, and neonatal sepsis evaluation. Pediatrics. 1997;99:415–419.
83. Mayer DC, Chescheir NC, Spielman FJ. Increased intrapartum antibiotic administration associated with epidural analgesia in labor. Am J Perinatol. 1997;14:83–86.
84. Baud O, Ville Y, Zupan V, et al. Are neonatal brain lesions due to intrauterine infection related to mode of delivery? Br J Obstet Gynaecol. 1998;105:121–124.
85. Rees S, Breen S, Loeliger M, et al. Hypoxemia near mid-gestation has long-term effects on fetal brain development. J Neuropathol Exp Neurol. 1999;58:932–945.
86. Rees S, Inder T. Fetal and neonatal origins of altered brain development. Early Hum Dev. 2005;81:753–761.
87. Saraceno GE, Castilla R, Barreto GE, et al. Hippocampal dendritic spines modifications induced by perinatal asphyxia. Neural Plast. 2012;2012:873532.
88. Chen WF, Chang H, Wong CS, et al. Impaired expression of postsynaptic density proteins in the hippocampal CA1 region of rats following perinatal hypoxia. Exp Neurol. 2007;204:400–410.
89. McLean C, Ferriero D. Mechanisms of hypoxic-ischemic injury in the term infant. Semin Perinatol. 2004;28:425–432.
90. Back SA, Han BH, Luo NL, et al. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463.
91. Burd I, Balakrishnan B, Kannan S. Models of fetal brain injury, intrauterine inflammation, and preterm birth. Am J Reprod Immunol. 2012;67:287–294.
92. Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol. 2012;71:444–457.
93. Boksa P. Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav Immun. 2010;24:881–897.
94. Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med. 2011;17:389–394.
95. Dahlgren J, Samuelsson AM, Jansson T, Holmang A. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr Res. 2006;60:147–151.
96. Smith SE, Li J, Garbett K, et al. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702.
97. Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64:61–78.
98. Boles JL, Ross MG, Beloosesky R, et al. Placental-mediated increased cytokine response to lipopolysaccharides: a potential mechanism for enhanced inflammation susceptibility of the preterm fetus. J Inflamm Res. 2012;5:67–75.
99. Garay PA, Hsiao EY, Patterson PH, McAllister AK. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav Immun. 2013;31:54–68.
100. Chiesa C, Pellegrini G, Panero A, et al. Umbilical cord interleukin-6 levels are elevated in term neonates with perinatal asphyxia. Eur J Clin Invest. 2003;33:352–358.
101. Samuelsson AM, Jennische E, Hansson HA, Holmang A. Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1345–R1356.
102. Arrode-Bruses G, Bruses JL. Maternal immune activation by poly(I:C) induces expression of cytokines IL-1beta and IL-13, chemokine MCP-1 and colony stimulating factor VEGF in fetal mouse brain. J Neuroinflammation. 2012;9:83.
103. Monier A, Evrard P, Gressens P, Verney C. Distribution and differentiation of microglia in the human encephalon during the first two trimesters of gestation. J Comp Neurol. 2006;499:565–582.
104. Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458.
106. Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol. 2012;72:1272–1276.
107. Raju TN. Some animal models for the study of perinatal asphyxia. Biol Neonate. 1992;62:202–214.
108. Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83.
109. Clancy B, Finlay BL, Darlington RB, et al. Extrapolating brain development from experimental species to humans. Neurotoxicology. 2007;28:931–937.
110. Hagberg H, Andersson P, Kjellmer I, et al. Extracellular overflow of glutamate, aspartate, GABA and taurine in the cortex and basal ganglia of fetal lambs during hypoxia-ischemia. Neurosci Lett. 1987;78:311–317.
111. Penning DH, Chestnut DH, Dexter F, et al. Glutamate release from the ovine fetal brain during maternal hemorrhage: a study using chronic in utero cerebral microdialysis. Anesthesiology. 1995;82:521–530.
112. Reynolds JD, Penning DH, Dexter F, et al. Dose-dependent effects of acute in vivo ethanol exposure on extracellular glutamate concentration in the cerebral cortex of the near-term fetal sheep. Alcohol Clin Exp Res. 1995;19:1447–1453.
113. Richardson BS, Patrick JE, Abduljabbar H. Cerebral oxidative metabolism in the fetal lamb: relationship to electrocortical state. Am J Obstet Gynecol. 1985;153:426–431.
114. Richardson BS, Patrick JE, Bousquet J, et al. Cerebral metabolism in fetal lamb after maternal infusion of ethanol. Am J Physiol. 1985;249:R505–R509.
115. Matsuda Y, Patrick J, Carmichael L, et al. Effects of sustained hypoxemia on the sheep fetus at midgestation: endocrine, cardiovascular, and biophysical responses. Am J Obstet Gynecol. 1992;167:531–540.
116. Richardson BS, Carmichael L, Homan J, Patrick JE. Electrocortical activity, electroocular activity, and breathing movements in fetal sheep with prolonged and graded hypoxemia. Am J Obstet Gynecol. 1992;167:553–558.
117. Henderson JL, Reynolds JD, Dexter F, et al. Chronic hypoxemia causes extracellular glutamate concentration to increase in the cerebral cortex of the near-term fetal sheep. Brain Res Dev Brain Res. 1998;105:287–293.
118. Mallard EC, Gunn AJ, Williams CE, et al. Transient umbilical cord occlusion causes hippocampal damage in the fetal sheep. Am J Obstet Gynecol. 1992;167:1423–1430.
119. Clapp JF, Peress NS, Wesley M, Mann LI. Brain damage after intermittent partial cord occlusion in the chronically instrumented fetal lamb. Am J Obstet Gynecol. 1988;159:504–509.
120. Clapp JF 3rd, Mann LI, Peress NS, Szeto HH. Neuropathology in the chronic fetal lamb preparation: structure-function correlates under different environmental conditions. Am J Obstet Gynecol. 1981;141:973–986.
121. Reynolds JD, Chestnut DH, Dexter F, et al. Magnesium sulfate adversely affects fetal lamb survival and blocks fetal cerebral blood flow response during maternal hemorrhage. Anesth Analg. 1996;83:493–499.
122. Hohimer AR, Chao CR, Bissonnette JM. The effect of combined hypoxemia and cephalic hypotension on fetal cerebral blood flow and metabolism. J Cereb Blood Flow Metab. 1991;11:99–105.
123. Robillard JE, Weitzman RE, Burmeister L, Smith FG Jr. Developmental aspects of the renal response to hypoxemia in the lamb fetus. Circ Res. 1981;48:128–138.
124. Richardson BS. The fetal brain: metabolic and circulatory responses to asphyxia. Clin Invest Med. 1993;16:103–114.
125. Lin B, Globus M, Dietrich W, et al. Differing neurochemical and morphological sequelae of global ischemia: comparison of single-multiple and multiple-insult paradigms. J Neurochem. 1992;2213–2223.
126. Mallard EC, Williams CE, Gunn AJ, et al. Frequent episodes of brief ischemia sensitize the fetal sheep brain to neuronal loss and induce striatal injury. Pediatr Res. 1993;33:61–65.
127. Penning DH, Grafe MR, Hammond R, et al. Neuropathology of the near-term and midgestation ovine fetal brain after sustained in utero hypoxemia. Am J Obstet Gynecol. 1994;170:1425–1432.
128. Brann AW Jr, Myers RE. Central nervous system findings in the newborn monkey following severe in utero partial asphyxia. Neurology. 1975;25:327–338.
129. Larroche JC. Fetal and perinatal brain damage. Wigglesworth JS, Singer DB. Textbook of Fetal and Perinatal Pathology. Blackwell Scientific: Boston; 1991:807–838.
130. Allan WC, Riviello JJ Jr. Perinatal cerebrovascular disease in the neonate: parenchymal ischemic lesions in term and preterm infants. Pediatr Clin North Am. 1992;39:621–650.
131. Volpe JJ. Brain injury in the premature infant—current concepts of pathogenesis and prevention. Biol Neonate. 1992;62:231–242.
132. Banker BQ, Larroche JC. Periventricular leukomalacia of infancy: a form of neonatal anoxic encephalopathy. Arch Neurol. 1962;7:386–410.
133. Dubowitz LM, Bydder GM, Mushin J. Developmental sequence of periventricular leukomalacia: correlation of ultrasound, clinical, and nuclear magnetic resonance functions. Arch Dis Child. 1985;60:349–355.
134. De Reuck J. The human periventricular arterial blood supply and the anatomy of cerebral infarctions. Eur Neurol. 1971;5:321–334.
135. Nelson MD Jr, Gonzalez-Gomez I, Gilles FH. Dyke Award. The search for human telencephalic ventriculofugal arteries. AJNR Am J Neuroradiol. 1991;12:215–222.
136. Mayer PL, Kier EL. The controversy of the periventricular white matter circulation: a review of the anatomic literature. AJNR Am J Neuroradiol. 1991;12:223–228.
137. Ashwal S, Dale PS, Longo LD. Regional cerebral blood flow: studies in the fetal lamb during hypoxia, hypercapnia, acidosis, and hypotension. Pediatr Res. 1984;18:1309–1316.
138. Juurlink BH, Hertz L, Yager JY. Astrocyte maturation and susceptibility to ischaemia or substrate deprivation. Neuroreport. 1992;3:1135–1137.
139. Richardson BS. Fetal adaptive responses to asphyxia. Clin Perinatol. 1989;16:595–611.
140. Edelstone DI. Fetal compensatory responses to reduced oxygen delivery. Semin Perinatol. 1984;8:184–191.
141. Gu W, Jones CT, Parer JT. Metabolic and cardiovascular effects on fetal sheep of sustained reduction of uterine blood flow. J Physiol (Lond). 1985;368:109–129.
142. Prakash A, Matta BF. Hyperglycaemia and neurological injury. Curr Opin Anaesthesiol. 2008;21:565–569.
143. Vannucci RC, Mujsce DJ. Effect of glucose on perinatal hypoxic-ischemic brain damage. Biol Neonate. 1992;62:215–224.
144. Blomstrand S, Hrbek A, Karlsson K, et al. Does glucose administration affect the cerebral response to fetal asphyxia? Acta Obstet Gynecol Scand. 1984;63:345–353.
145. Hooper SB, Bocking AD, White S, et al. DNA synthesis is reduced in selected fetal tissues during prolonged hypoxemia. Am J Physiol. 1991;261:R508–R514.
146. Rurak DW, Gruber NC. The effect of neuromuscular blockade on oxygen consumption and blood gases in the fetal lamb. Am J Obstet Gynecol. 1983;145:258–262.
147. Jensen A, Berger R. Fetal circulatory responses to oxygen lack. J Dev Physiol. 1991;16:181–207.
148. Sheldon RE, Peeters LL, Jones MD Jr, et al. Redistribution of cardiac output and oxygen delivery in the hypoxemic fetal lamb. Am J Obstet Gynecol. 1979;135:1071–1078.
149. Jensen A, Hohmann M, Kunzel W. Dynamic changes in organ blood flow and oxygen consumption during acute asphyxia in fetal sheep. J Dev Physiol. 1987;9:543–559.
150. DiPietro JA. Neurobehavioral assessment before birth. Ment Retard Dev Disabil Res Rev. 2005;11:4–13.
151. Stanojevic M, Zaputovic S, Bosnjak AP. Continuity between fetal and neonatal neurobehavior. Semin Fetal Neonatal Med. 2012;17:324–329.
152. Salisbury AL, Fallone MD, Lester B. Neurobehavioral assessment from fetus to infant: the NICU Network Neurobehavioral Scale and the Fetal Neurobehavior Coding Scale. Ment Retard Dev Disabil Res Rev. 2005;11:14–20.
153. Honemeyer U, Talic A, Therwat A, et al. The clinical value of KANET in studying fetal neurobehavior in normal and at-risk pregnancies. J Perinat Med. 2013;41:187–197.
154. Prayer D, Kasprian G, Krampl E, et al. MRI of normal fetal brain development. Eur J Radiol. 2006;57:199–216.
156. Barkovich AJ, Westmark K, Partridge C, et al. Perinatal asphyxia: MR findings in the first 10 days. AJNR Am J Neuroradiol. 1995;16:427–438.
157. Martin E, Barkovich AJ. Magnetic resonance imaging in perinatal asphyxia. Arch Dis Child Fetal Neonatal Ed. 1995;72:F62–F70.
158. Younkin DP. Magnetic resonance spectroscopy in hypoxic-ischemic encephalopathy. Clin Invest Med. 1993;16:115–121.
159. Huppi PS, Warfield S, Kikinis R, et al. Quantitative magnetic resonance imaging of brain development in premature and mature newborns. Ann Neurol. 1998;43:224–235.
160. Sugimoto T, Woo M, Nishida N, et al. When do brain abnormalities in cerebral palsy occur? An MRI study. Dev Med Child Neurol. 1995;37:285–292.
161. Okumura A, Kato T, Kuno K, et al. MRI findings in patients with spastic cerebral palsy. II. Correlation with type of cerebral palsy. Dev Med Child Neurol. 1997;39:369–372.
162. Olsen P, Paakko E, Vainionpaa L, et al. Magnetic resonance imaging of periventricular leukomalacia and its clinical correlation in children. Ann Neurol. 1997;41:754–761.
163. Panigrahy A, Blüml S. Advances in magnetic resonance neuroimaging techniques in the evaluation of neonatal encephalopathy. Top Magn Reson Imaging. 2007;18:3–29.
164. Drobyshevsky A, Derrick M, Prasad PV, et al. Fetal brain magnetic resonance imaging response acutely to hypoxia-ischemia predicts postnatal outcome. Ann Neurol. 2007;61:307–314.
165. Fraser M, Bennet L, Helliwell R, et al. Regional specificity of magnetic resonance imaging and histopathology following cerebral ischemia in preterm fetal sheep. Reprod Sci. 2007;14:182–191.
166. Nanba Y, Matsui K, Aida N, et al. Magnetic resonance imaging regional T1 abnormalities at term accurately predict motor outcome in preterm infants. Pediatrics. 2007;120:e10–e19.
167. Eken P, Jansen GH, Groenendaal F, et al. Intracranial lesions in the fullterm infant with hypoxic ischaemic encephalopathy: ultrasound and autopsy correlation. Neuropediatrics. 1994;25:301–307.
168. Kuhnert BR, Kuhnert PM, Philipson EH, Syracuse CD. Disposition of meperidine and normeperidine following multiple doses during labor. II. Fetus and neonate. Am J Obstet Gynecol. 1985;151:410–415.
169. Golub MS, Donald JM. Effect of intrapartum meperidine on behavior of 3- to 12-month-old infant rhesus monkeys. Biol Neonate. 1995;67:140–148.
170. Golub MS, Eisele JH Jr, Donald JM. Obstetric analgesia and infant outcome in monkeys: infant development after intrapartum exposure to meperidine or alfentanil. Am J Obstet Gynecol. 1988;159:1280–1286.
171. Tripathi A, Khurshid N, Kumar P, Iyengar S. Expression of delta- and mu-opioid receptors in the ventricular and subventricular zones of the developing human neocortex. Neurosci Res. 2008;61:257–270.
172. Sheng WS, Hu S, Herr G, et al. Human neural precursor cells express functional kappa-opioid receptors. J Pharmacol Exp Ther. 2007;322:957–963.
173. Stiene-Martin A, Zhou R, Hauser KF. Regional, developmental, and cell cycle-dependent differences in mu, delta, and kappa-opioid receptor expression among cultured mouse astrocytes. Glia. 1998;22:249–259.
174. Sargeant TJ, Day DJ, Mrkusich EM, et al. Mu opioid receptors are expressed on radial glia but not migrating neuroblasts in the late embryonic mouse brain. Brain Res. 2007;1175:28–38.
175. Kim E, Clark AL, Kiss A, et al. Mu- and kappa-opioids induce the differentiation of embryonic stem cells to neural progenitors. J Biol Chem. 2006;281:33749–33760.
176. Hauser KF, Houdi AA, Turbek CS, et al. Opioids intrinsically inhibit the genesis of mouse cerebellar granule neuron precursors in vitro: differential impact of mu and delta receptor activation on proliferation and neurite elongation. Eur J Neurosci. 2000;12:1281–1293.
177. Sargeant TJ, Day DJ, Miller JH, Steel RW. Acute in utero morphine exposure slows G2/M phase transition in radial glial and basal progenitor cells in the dorsal telencephalon of the E15.5 embryonic mouse. Eur J Neurosci. 2008;28:1060–1067.
178. Niu L, Cao B, Zhu H, et al. Impaired in vivo synaptic plasticity in dentate gyrus and spatial memory in juvenile rats induced by prenatal morphine exposure. Hippocampus. 2009;19:649–657.
179. Golub MS. Labor analgesia and infant brain development. Pharmacol Biochem Behav. 1996;55:619–628.
180. Flick RP, Lee K, Hofer RE, et al. Neuraxial labor analgesia for vaginal delivery and its effects on childhood learning disabilities. Anesth Analg. 2011;112:1424–1431.
181. Likis FE, Andrews JC, Collins MR, et al. Nitrous oxide for the management of labor pain. AHRQ Comparative Effectiveness Reviews 2012, Report No. 12-EHC071-EF. Agency for Healthcare Research and Quality (US): Rockville, MD; 2012.
182. Klomp T, van Poppel M, Jones L, et al. Inhaled analgesia for pain management in labour. Cochrane Database Syst Rev. 2012;(9).
183. Reynolds F. The effects of maternal labour analgesia on the fetus. Best Pract Res Clin Obstet Gynaecol. 2010;24:289–302.
184. Stefani SJ, Hughes SC, Schnider SM, et al. Neonatal neurobehavioral effects of inhalation analgesia for vaginal delivery. Anesthesiology. 1982;56:351–355.
185. Jevtovic-Todorovic V, Hartman RE, Izumi Y, et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–882.
186. Brambrink AM, Evers AS, Avidan MS, et al. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology. 2010;112:834–841.
187. Wilder RT, Flick RP, Sprung J, et al. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;110:796–804.
188. Satomoto M, Satoh Y, Terui K, et al. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology. 2009;110:628–637.
189. Sprung J, Flick RP, Katusic SK, et al. Attention-deficit/hyperactivity disorder after early exposure to procedures requiring general anesthesia. Mayo Clin Proc. 2012;87:120–129.
190. Glass NL, Malviya S. Anesthesia in children—limitations of the data on neurotoxicity. N Engl J Med. 2011;364:1466–1467.
191. Palanisamy A. Maternal anesthesia and fetal neurodevelopment. Int J Obstet Anesth. 2012;21:152–162.
192. Patel P, Sun L. Update on neonatal anesthetic neurotoxicity: insight into molecular mechanisms and relevance to humans. Anesthesiology. 2009;110:703–708.
193. Campbell LL, Tyson JA, Stackpole EE, et al. Assessment of general anaesthetic cytotoxicity in murine cortical neurones in dissociated culture. Toxicology. 2011;283:1–7.
194. Palanisamy A, Baxter MG, Keel PK, et al. Rats exposed to isoflurane in utero during early gestation are behaviorally abnormal as adults. Anesthesiology. 2011;114:521–528.
195. Kong FJ, Tang YW, Lou AF, et al. Effects of isoflurane exposure during pregnancy on postnatal memory and learning in offspring rats. Mol Biol Rep. 2012;39:4849–4855.
196. Kong F, Xu L, He D, et al. Effects of gestational isoflurane exposure on postnatal memory and learning in rats. Eur J Pharmacol. 2011;670:168–174.
197. Rizzi S, Carter LB, Ori C, Jevtovic-Todorovic V. Clinical anesthesia causes permanent damage to the fetal guinea pig brain. Brain Pathol. 2008;18:198–210.
198. Slikker W Jr, Zou X, Hotchkiss CE, et al. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol Sci. 2007;98:145–158.
199. Sall JW, Stratmann G, Leong J, et al. Isoflurane inhibits growth but does not cause cell death in hippocampal neural precursor cells grown in culture. Anesthesiology. 2009;110:826–833.
200. Zhu C, Gao J, Karlsson N, et al. Isoflurane anesthesia induced persistent, progressive memory impairment, caused a loss of neural stem cells, and reduced neurogenesis in young, but not adult, rodents. J Cereb Blood Flow Metab. 2010;30:1017–1030.
201. Culley DJ, Boyd JD, Palanisamy A, et al. Isoflurane decreases self-renewal of rat cultured neural stem cells. Anesthesiology. 2011;115:754–763.
202. Li Y, Liang G, Wang S, et al. Effects of fetal exposure to isoflurane on postnatal memory and learning in rats. Neuropharmacology. 2007;53:942–950.
204. Sprung J, Flick RP, Wilder RT, et al. Anesthesia for cesarean delivery and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;111:302–310.
205. McGowan FX Jr, Davis PJ. Anesthetic-related neurotoxicity in the developing infant: of mice, rats, monkeys and, possibly, humans. Anesth Analg. 2008;106:1599–1602.
206. Yon JH, Carter LB, Reiter RJ, et al. Melatonin reduces the severity of anesthesia-induced apoptotic neurodegeneration in the developing rat brain. Neurobiol Dis. 2006;21:522–530.
207. Mellon RD, Simone AF, Rappaport BA, et al. Use of anesthetic agents in neonates and young children. Anesth Analg. 2007;104:509–520.
208. Kuehn BM. FDA considers data on potential risks of anesthesia use in infants, children. JAMA. 2011;305:1749–1750.
209. Petraglia F, Imperatore A, Challis JR. Neuroendocrine mechanisms in pregnancy and parturition. Endocr Rev. 2010;31:783–816.
210. Sibley CP, Brownbill P, Dilworth M, Glazier JD. Review: adaptation in placental nutrient supply to meet fetal growth demand: implications for programming. Placenta. 2010;31(Suppl):S70–S74.
211. Eshkoli T, Sheiner E, Ben-Zvi Z, Holcberg G. Drug transport across the placenta. Curr Pharm Biotechnol. 2011;12:707–714.
212. Hutson JR, Koren G, Matthews SG. Placental P-glycoprotein and breast cancer resistance protein: influence of polymorphisms on fetal drug exposure and physiology. Placenta. 2010;31:351–357.
213. Schubert K, Schade K. Placental steroid hormones. J Steroid Biochem. 1977;8:359–365.
214. Hirst JJ, Palliser HK, Yates DM, et al. Neurosteroids in the fetus and neonate: potential protective role in compromised pregnancies. Neurochem Int. 2008;52:602–610.
215. Tsutsui K. Progesterone biosynthesis and action in the developing neuron. Endocrinology. 2008;149:2757–2761.
216. McCarthy MM. Estradiol and the developing brain. Physiol Rev. 2008;88:91–124.
217. de Geest K, Thiery M, Piron-Possuyt G, Vanden Driessche R. Plasma oxytocin in human pregnancy and parturition. J Perinat Med. 1985;13:3–13.
218. Tyzio R, Cossart R, Khalilov I, et al. Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science. 2006;314:1788–1792.
219. Khazipov R, Tyzio R, Ben-Ari Y. Effects of oxytocin on GABA signalling in the foetal brain during delivery. Prog Brain Res. 2008;170:243–257.
220. Crowther CA, Hiller JE, Doyle LW, et al. Effect of magnesium sulfate given for neuroprotection before preterm birth: a randomized controlled trial. JAMA. 2003;290:2669–2676.
221. Marret S, Marpeau L, Zupan-Simunek V, et al. Magnesium sulphate given before very-preterm birth to protect infant brain: the randomised controlled PREMAG trial*. BJOG. 2007;114:310–318.
222. Rouse DJ, Hirtz DG, Thom E, et al. A randomized, controlled trial of magnesium sulfate for the prevention of cerebral palsy. N Engl J Med. 2008;359:895–905.
223. Doyle LW, Crowther CA, Middleton P, et al. Magnesium sulphate for women at risk of preterm birth for neuroprotection of the fetus. Cochrane Database Syst Rev. 2009;(1).
224. Wolf HT, Hegaard HK, Greisen G, et al. Treatment with magnesium sulphate in pre-term birth: a systematic review and meta-analysis of observational studies. J Obstet Gynaecol. 2012;32:135–140.
225. American College of Obstetricians and Gynecologists. Committee Opinion No. 455. Magnesium sulfate before anticipated preterm birth for neuroprotection. Obstet Gynecol. 2010;115:669–671.
226. George S, Scotter J, Dean JM, et al. Induced cerebral hypothermia reduces post-hypoxic loss of phenotypic striatal neurons in preterm fetal sheep. Exp Neurol. 2007;203:137–147.
227. Shankaran S, Laptook AR, Ehrenkranz RA, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353:1574–1584.
228. Gluckman PD, Wyatt JS, Azzopardi D, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005;365:663–670.
229. Papile LA, Papile L-A. Systemic hypothermia—a “cool” therapy for neonatal hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353:1619–1620.
230. Shankaran S, Pappas A, McDonald SA, et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N Engl J Med. 2012;366:2085–2092.
231. Jacobs SE, Berg M, Hunt R, et al. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev. 2013;(1).
232. Perlman JM, Perlman JM. Intervention strategies for neonatal hypoxic-ischemic cerebral injury. Clin Ther. 2006;28:1353–1365.
233. Swanson RA. Astrocyte glutamate uptake during chemical hypoxia in vitro. Neurosci Lett. 1992;147:143–146.
234. Swanson RA, Choi DW. Glial glycogen stores affect neuronal survival during glucose deprivation in vitro. J Cereb Blood Flow Metab. 1993;13:162–169.
235. Ransom BR, Stys PK, Waxman SG. The pathophysiology of anoxic injury in central nervous system white matter. Stroke. 1990;21(11 Suppl):III52–III57.
236. Graham SH, Chen J, Sharp FR, Simon RP. Limiting ischemic injury by inhibition of excitatory amino acid release. J Cereb Blood Flow Metab. 1993;13:88–97.
237. Volpe JJ. Brain injury in the premature infant—from pathogenesis to prevention. Brain Dev. 1997;19:519–534.
238. Thiringer K, Hrbek A, Karlsson K, et al. Postasphyxial cerebral survival in newborn sheep after treatment with oxygen free radical scavengers and a calcium antagonist. Pediatr Res. 1987;22:62–66.
239. Juul S. Neuroprotective role of erythropoietin in neonates. J Matern Fetal Neonatal Med. 2012;25(Suppl 4):105–107.
240. Rees S, Harding R, Walker D. The biological basis of injury and neuroprotection in the fetal and neonatal brain. Int J Dev Neurosci. 2011;29:551–563.
241. Balduini W, Carloni S, Perrone S, et al. The use of melatonin in hypoxic-ischemic brain damage: an experimental study. J Matern Fetal Neonatal Med. 2012;25(Suppl 1):119–124.
* Translating Time across Developing Mammalian Brains: http://www.translatingtime.net/.