Hairy Cell Leukemia
Jae H. Park and Martin S. Tallman
• Hairy cell leukemia (HCL) is an uncommon clonal B-cell lymphoproliferative disorder.
• BRAF V600E mutation is present in nearly all cases of HCL but absent in other B-cell lymphoproliferative disorders and represents a disease-defining genetic event in HCL.
• Physical findings generally are confined to splenomegaly.
• The purine analogs are the therapeutic agents of choice.
• Most patients who receive treatment with cladribine or pentostatin have prolonged survival.
• BL22 or HA22, an immunoconjugate of an anti-CD22 antibody linked to a truncated Pseudomonas exotoxin A, is a novel agent that has been very effective in the management of relapsed and refractory HCL.
• BRAF inhibitors represent the first molecularly targeted therapy in HCL and will soon be explored in clinical studies.
Introduction
Hairy cell leukemia (HCL) is a rare chronic B-cell lymphoproliferative disorder with unique morphologic features and an excellent prognosis. Bouroncle and colleagues described 26 patients in 1958 with what then was called “leukemic reticuloendotheliosis” and are credited with the initial description of HCL as a distinct clinical entity.1 The disease is characterized by splenomegaly, pancytopenia, and infiltration of the bone marrow with lymphocytes that have irregular cytoplasmic projections when identified in the peripheral blood.2,3 Although the origin of the malignant cell was unclear for some time, immunoglobulin gene rearrangements confirm that the disease is a clonal B-cell malignancy.6–6 The pattern of expression of B-cell–associated surface antigens (see later discussion) reflects a degree of differentiation between the mature B cell of chronic lymphocytic leukemia and the plasma cell of multiple myeloma.7,8 A majority of patients have few symptoms at the time of diagnosis. However, occasionally patients are seen with life-threatening pancytopenia, symptomatic splenomegaly, serious infections, or constitutional signs and symptoms justifying treatment, or such problems may develop later in the disease course.9,10
Treatment strategies have evolved relatively rapidly during the past 50 years. Splenectomy was the first effective treatment described and remained the therapeutic modality of choice for many years. Although the mechanism of benefit is not clear, removing the spleen leads to normalization of the peripheral blood counts in approximately one half of all patients.11–14 Interferon-α induces a high overall response rate; however, most responses are partial.14–21 Remarkable progress has occurred with the introduction of the two purine analogs, 2′-deoxycoformycin (2′-DCF; pentostatin)22–33 and 2-chlorodeoxyadenosine (2-CdA; cladribine).24,34–44 Most patients with both previously treated and untreated HCL achieve durable complete remission with either of these agents. With use of molecular techniques, however, minimal residual disease (MRD) can be identified in most if not all patients, suggesting that most patients are not actually cured of their disease. Nevertheless, with either purine analog, a majority of patients enjoy prolonged periods of progression-free and overall survival.
Recently, whole-exome gene sequencing of HCL identified the presence of BRAF V600E mutation in nearly all patients with this mutation absent in other B-cell lymphoid malignancies.45 This discovery of the highly specific and recurrent mutation in HCL identified the BRAF V600E mutation as the disease-defining genetic event in HCL and has generated great interest and excitement for novel diagnostic possibilities and therapeutic targeting of the BRAF pathway.
Epidemiology
Relatively little is known about the epidemiology of HCL, partly because of its rarity. In the United States, HCL represents 2% of adult leukemias, with only approximately 600 to 800 new cases diagnosed each year.46,47 Although anecdotal reports of familial HCL have been published, no clear genetic predisposition has been recognized.48–54 The median age at diagnosis is 52 years; and for unclear reasons the disease occurs in men more often than in women, with a sex ratio of approximately 4 : 1.55 Although reported incidence rates are similar in the United States and Great Britain,46,47 classic HCL is rare in Japan, where a distinct variant form has been described.58–58
Etiology and Pathogenesis
The etiology of HCL has not been determined. An association with exposure to benzene,59,60 organophosphorus insecticides,61 or other solvents62 has been suggested but has not been confirmed.63 Exposure to radiation,64 agricultural chemicals,60 or wood dust47 and a previous history of infectious mononucleosis62 also have been suggested as potential associations. However, among a majority of patients, no such exposures can be identified. Cyclin D1, an important cell cycle regulator, may play a role in the molecular pathogenesis of HCL. Overexpression of the cyclin D1 protein has been described in HCL patients.65,66 Unlike in mantle cell lymphoma, 11q13 rearrangements are not detected in most patients with HCL, suggesting other mechanisms of gene deregulation.66
The hairy cell may be derived from the memory B cell because the genome-wide expression profile is close to that of postgerminal center B cells.67 Evidence suggests that hairy cells may come from B cells of the splenic marginal zone because hairy cells have a splenic expression signature that reflects tissue components, such as the marginal zone, that are not well represented in lymph node tissue.68 However, expansion of the red pulp of the spleen, so characteristic of HCL, is in contrast to expansion of the white pulp, which involves the splenic marginal zone. Hairy cells express B-cell CLL/lymphoma 2 (BCL2), a universal inhibitor of apoptosis.69 Survival of hairy cells may be further promoted by overexpression of Fms-like tyrosine kinase 3 (FLT3), which activates the phosphatidylinositol-3-kinase (PI3K) pathway that activates anti-apoptotic signals.67,70 Hairy cells produce basic fibroblast growth factor (bFGF) and overexpress bFGF tyrosine kinase receptor,67,71 which can activate the PI3K-Akt cascade.72
The characteristic appearance of the hairy cells is due to expression of β-actin, which is polymerized to F-actin, located in the cortical cytoskeleton, in the peripheral part of the cell, that serves to support the hairlike projections.67 The actual hairlike projections probably are due to increased expression of pp52, a leukocyte-specific intracellular phosphoprotein, that binds to F-actin.73,74
More recently, whole-exome gene sequencing of HCL identified BRAF V600E mutation in the entire tumor-cell clone of nearly all patients with HCL, which is absent in other B-cell lymphoid malignancies.45 This discovery implicates the BRAF V600E mutation in the pathogenesis of HCL. The concept that the BRAF V600E mutation may represent a key driver mutation in HCL is further supported by a preliminary evidence of striking clinical activity of the specific BRAF inhibitor vemurafenib in a patient with refractory HCL.75 However, further characterization of the BRAF pathway in HCL and formal clinical studies of BRAF inhibitors in this disease will be required to confirm the implicated pathogenic mechanisms of the BRAF pathway in HCL.
Clinical Presentation
At the time of diagnosis, most patients have symptoms attributable to anemia, neutropenia, thrombocytopenia, or splenomegaly. Approximately 25% of patients have fatigue or weakness and 25% have infection; another 25% come to medical attention because of incidental discovery of splenomegaly or an abnormal peripheral blood cell count.55
Most patients are relatively well at the time of diagnosis. The most common and invariably the only physical finding is splenomegaly, which occurs in approximately 80% of patients.3,55 The spleen is palpable 5 cm below the left costal margin in approximately 60% of patients. Hepatomegaly occurs in approximately 20% of patients. Unlike in many other chronic lymphoproliferative disorders, peripheral adenopathy is uncommon at diagnosis, with less than 10% of patients having peripheral nodes larger than 2 cm. Although adenopathy is not common at diagnosis, internal adenopathy may develop after a prolonged disease course76,77 and is present in 75% of patients at autopsy.78 The characteristic distribution in HCL is probably caused by expression of the integrin receptor α4β1 by the hairy cells and its interaction with the vascular cell adhesion molecule-1 (VCAM-1) found on splenic and hepatic endothelia and in bone marrow and splenic stroma.79
Patients with HCL are susceptible to both gram-positive and gram-negative bacterial infections.80 In addition, susceptibility to atypical mycobacterial infections,81 particularly those caused by Mycobacterium kansasii, as well as to invasive fungal infections, also has been documented.80 Other opportunistic infections that have been reported include legionnaires’ disease,82 toxoplasmosis,83 and Listeria monocytogenes infection.84 The milieu specific for this increased susceptibility to infections comprises granulocytopenia, monocytopenia, poor granulocyte reserve and abnormal mobilization,85 T-cell dysfunction,86 and decreased numbers of dendritic cells and antigen-presenting cells.87
Rarely, patients with HCL may have associated systemic immunologic disorders,88 including scleroderma and polymyositis89 and polyarteritis nodosa.90 HCL has been associated with other cutaneous lesions such as erythematous maculopapules91 and pyoderma gangrenosum.92,93 An associated coagulopathy manifested by factor VIII antibodies has been reported.94 Osseous involvement also has been described, primarily lytic lesions in the axial skeleton, usually the proximal femur.95,96 Rarely, osteolytic lesions may be associated with paraproteinemia.97 A rare case of HCL occurring with systemic mast cell disease has been reported.98
Laboratory Evaluation
Pancytopenia is present in approximately 50% of patients with HCL at diagnosis; most other patients have suppression of one or two cell lines.3,55 Most patients with HCL have leukopenia, although 10% to 20% of patients exhibit a “leukemic phase” with a white blood cell count higher than 10,000 to 20,000/µL. Monocytopenia is a characteristic but often overlooked finding.1,3,55 Other laboratory findings include abnormal hepatic transaminase levels (19%), azotemia (27%), and hypergammaglobulinemia (18%), which rarely is monoclonal.3,97,99 Unlike in chronic lymphocytic leukemia, hypogammaglobulinemia is uncommon.
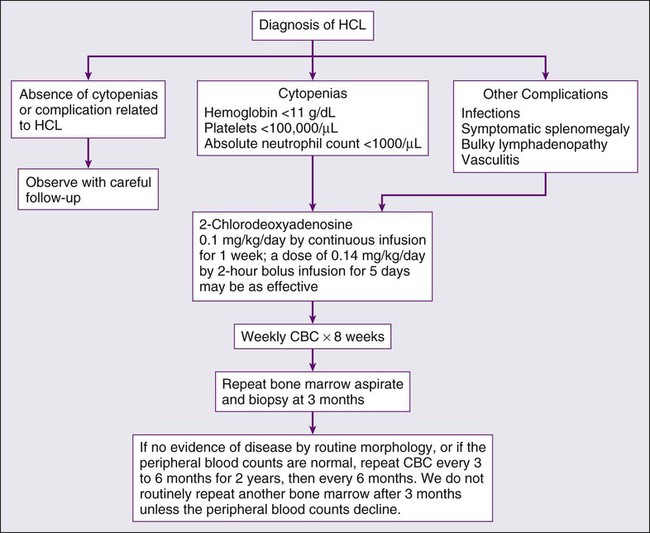
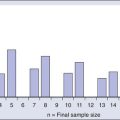
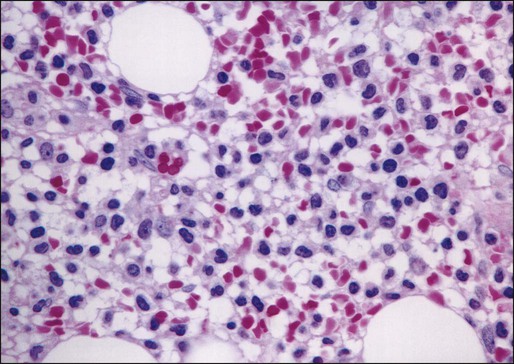
The morphologic features of hairy cells are distinctive (Fig. 103-1). The neoplastic cells are one to two times the size of a small lymphocyte. The nuclei are round, oval, indented, or monocytoid; rarely, they appear convoluted.100 The nuclei are located in a central or eccentric position. The chromatin pattern is netlike, and nucleoli are indistinct or absent. The amount of cytoplasm varies, ranging from scant to abundant; a pale blue-gray color is characteristic. The cytoplasmic borders are irregular and exhibit fine, hairlike projections or ruffled borders. Occasionally, cytoplasmic granules are present. Rarely, the cytoplasm exhibits basophilia, or rod-shaped inclusions that correspond to ribosomal lamellar complexes, observed on ultrastructural examination in approximately 40% of cases.101
Examination of the bone marrow core biopsy specimen is critical in the diagnosis of HCL because of its characteristic histopathological appearance102–105 (Figs. 103-2 and 103-3). The bone marrow usually is hypercellular in most patients, but this finding may be variable. Hairy cell infiltration may be diffuse, patchy, or interstitial or a combination of these patterns. In patients with diffuse involvement, large areas of the bone marrow are replaced by hairy cells, with complete effacement of marrow in some patients. With patchy infiltration, small subtle clusters of hairy cells are present focally or scattered throughout the bone marrow. Unlike in lymphomas, the hairy cells do not form well-defined, discrete aggregates; instead, they merge subtly with the surrounding residual hematopoietic tissue. In the interstitial pattern of involvement, variable numbers of hairy cells infiltrate between normal hematopoietic cells and fat, with preservation of the overall bone marrow architecture. Hairy cell nuclei in biopsy sections are round, oval, or indented and widely separated from each other by abundant clear or lightly eosinophilic cytoplasm; rarely, the cells are convoluted or spindle shaped. The nuclear chromatin is lightly condensed, nucleoli are inconspicuous, and mitotic figures are rare or absent. Extravasated red blood cells often are seen, and blood lakes, similar to those observed in the spleen, also may be observed. Reticulin stains of the bone marrow trephine biopsy specimen in HCL show a moderate to marked increase in reticulin fibers.
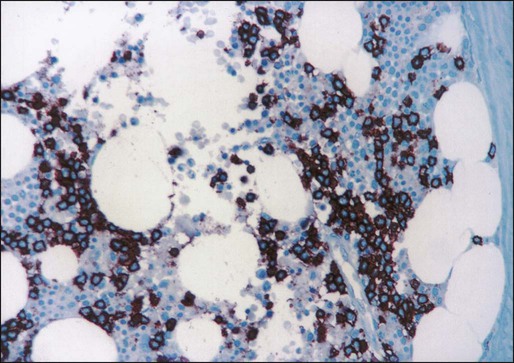
Normal hematopoietic cells usually are decreased in HCL; the number of granulocytes typically is more severely reduced than are erythroid precursors and megakaryocytes. In 10% to 20% of patients with HCL, the bone marrow is hypocellular. The hypocellularity may be severe,106 with a marrow appearance resembling that in patients with aplastic anemia.
Historically, cytochemical demonstration of tartrate-resistant acid phosphatase (TRAP) activity has been used to confirm the diagnosis of HCL.107 TRAP-positive cells are found in most cases of HCL at diagnosis, although the percentage of positive cells varies greatly among patients. A positive TRAP stain in conjunction with characteristic histopathology is essentially diagnostic of HCL. Today, however, the routine use of immunophenotyping by flow cytometry for the diagnosis of chronic lymphoproliferative disorders has made reliance on the TRAP stain less important.
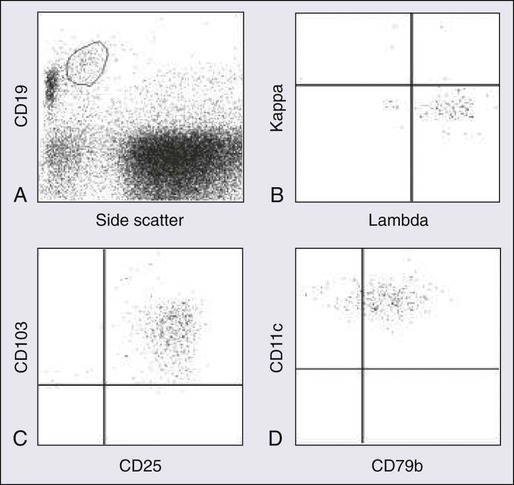
Flow cytometric immunophenotyping is an essential part of the diagnostic evaluation, both to identify the characteristic immunophenotypic profile of HCL and to distinguish it from other chronic B-cell and T-cell lymphoproliferative disorders. Because hairy cells exhibit distinctive light scatter characteristics and immunophenotype, they can be identified even when present in very low levels (less than 1% of lymphocytes) in either the peripheral blood or bone marrow aspirate.108 This property is useful not only at the time of diagnosis but also after therapy to assess for residual disease.109
Hairy cells show bright CD45 expression with increased forward and side scatter resembling that of large lymphocytes or monocytes. They exhibit a mature B-cell phenotype and express one or more heavy chains and monotypic light chains. The numbers of cases with expression of kappa or of lambda light chains are approximately equal. Surface immunoglobulin is of moderate to bright intensity. Hairy cells strongly express pan–B-cell antigens, including CD19, CD20, CD22, and CD79b. They usually stain negatively for CD5, CD10, and CD23. They strongly express CD11c, CD25, and FMC7. CD103, an antigen expressed on mucosal T cells and some activated T cells, is expressed in a majority of cases of HCL.108,110 Approximately 35% of patients have a variant immunophenotype, but the hairy cells from such patients have the characteristic morphologic features of classic hairy cells and these patients have the same excellent response to treatment with purine analogs.111 Several B-cell–associated antibodies, including CD20, CD79a, and DBA.44, react with hairy cells in fixed, routinely processed tissue sections. Although these antibodies are not specific for HCL, they are useful in documenting the B-cell nature of the infiltrate and highlighting the extent of bone marrow infiltration at the time of diagnosis and after therapy.112–115
Clonal cytogenetic abnormalities are present in approximately two thirds of patients with HCL. Karyotype analysis is rarely required or useful to establish the diagnosis, however. The most frequently involved chromosomes include chromosomes 1, 2, 5, 6, 11, 14, 19, and 20. In particular, chromosome 5 is altered in 40% of patients, most commonly as trisomy 5, pericentric inversions, or interstitial deletions involving band 5q13.118–118
In rare cases, the diagnosis of HCL is made by histologic analysis of splenic tissue after splenectomy or splenic needle core biopsy. Splenic involvement in HCL is characterized by diffuse infiltration of the red pulp cords and sinuses, with atrophy or replacement of the white pulp. Blood-filled sinuses, lined by hairy cells, often are present but are not pathognomonic for HCL; they have been referred to as “pseudosinuses.”119 The liver shows both sinusoidal and portal infiltration by hairy cells. Involved lymph nodes commonly exhibit partial effacement, with hairy cells infiltrating the paracortex and medulla in a leukemic pattern. The leukemic cells often surround residual lymphoid follicles and extend through the capsule.
A recent whole-exome sequencing study of HCL cells in parallel with normal cells has identified the presence of the BRAF V600E mutation in all of 48 cases of HCL.45 This finding has been extended and corroborated in larger cohorts of patients from different institutions using allele-specific or quantitative real-time polymerase chain reaction (PCR) assays. The BRAF mutation has now been reported in more than 400 cases of HCL and was not evident in more than 700 cases of other B-cell lymphoproliferative disorders.45,120–125 These data suggest that the BRAF V600E mutation represents a reliable molecular marker for diagnosis of HCL, and the BRAF mutational analysis will likely become part of the diagnostic armamentarium in HCL. However, a recent study reported the absence of the BRAF mutation in a subset of classic HCL, particularly HCL cases expressing immunoglobulin heavy variable 4-34 (IGHV4-34) immunoglobulin rearrangement.124 Therefore, as more information becomes available on the prevalence and functional role of BRAF mutations in HCL, the BRAF mutation analysis should be used in combination with cytomorphology and immunophenotyping to further improve and strengthen the diagnosis of patients with HCL.
Differential Diagnosis
Considerations in the differential diagnosis for HCL include other chronic B-cell lymphoproliferative disorders associated with splenomegaly, including prolymphocytic leukemia, splenic marginal zone lymphoma, and hairy cell variant (Box 103-1). Patients with prolymphocytic leukemia typically have splenomegaly, but this disorder usually can be distinguished from HCL by the marked leukocytosis, the characteristic morphologic appearance of the prolymphocytes, and an immunophenotypic profile different from that for HCL.126–129 Splenic marginal zone lymphoma exhibits some clinical and morphologic features similar to those of HCL, but, in contrast, the bone marrow infiltrates are sharply demarcated from the surrounding normal tissue and intrasinusoidal infiltration often is prominent. In addition, the immunophenotypic profile differs from that for HCL, including negative staining for CD103.130–133 Hairy cell variant exhibits morphologic features that are intermediate between those of HCL and prolymphocytic leukemia. Unlike HCL, hairy cell variant is associated with prominent leukocytosis, lack of monocytopenia, and absence of CD25 expression.134–138 Finally, infiltrates of systemic mastocytosis in the bone marrow may resemble those seen in HCL. Immunohistochemistry studies, however, show the mast cells, unlike hairy cells, to be negative for B-cell antigens and positive for tryptase.139 Although cytomorphologic and immunophenotypic analysis along with clinical presentation can usually secure the diagnosis of HCL, the molecular analysis for the BRAF V600E mutation can be used to further improve the discrimination of HCL from HCL variant cases and other chronic B-cell lymphoproliferative disorders.
Treatment
Indications
HCL almost always has an indolent course, with some patients surviving 10 years without need for therapy.140 In a majority of patients, however, progressive disease eventually leads to complications resulting from anemia, bleeding, splenomegaly, or recurrent infections. Therapy is indicated when the patient has significant cytopenias; symptomatic organomegaly or adenopathy; repeated infections; or constitutional signs and symptoms such as fever, night sweats, or fatigue. Persistent blood cell counts showing an absolute neutrophil count less than 1000/µL, a hemoglobin value less than 11.0 g/dL, or a platelet count below 100,000/µL are guidelines that can serve as indications for therapy.
Role of Splenectomy
Splenectomy was the first effective therapy for HCL and remained the initial treatment of choice until approximately 2 decades ago.11–14 After splenectomy, counts for all three cell lines return to normal in 40% to 70% of patients.13,141 This response is maintained for a median of 20 months in approximately two thirds of patients, and the overall 5-year survival rate is approximately 70%.141 No correlation has been found between spleen size and response to splenectomy. Splenectomy may have a possible role in an occasional patient to establish the diagnosis, in rare cases of splenic rupture, or in patients with life-threatening thrombocytopenia or a significant bleeding diathesis, because emergency splenectomy can lead to a rapid rise in the platelet count. Except in these very unusual circumstances, however, little, if any, role for splenectomy is recognized since the introduction of the purine analogs.
Chemotherapeutic Approaches
Cytotoxic chemotherapy was given for the treatment of HCL before the advent of more effective therapies in the early 1980s. A variety of agents, including anthracyclines,142 alkylating agents (chlorambucil),143 and high-dose methotrexate,144 demonstrate activity. Combination chemotherapy, such as the CHOP regimen (cyclophosphamide, hydroxy doxorubicin [Adriamycin], vincristine [Oncovin], prednisone), produces long-lasting normalization of peripheral blood counts.145 In one report in the literature of successful syngeneic (identical twin) bone marrow transplantation, the patient remained free of disease at least 15 years later.146 Although HCL is sensitive to chemotherapy, significant myelosuppression and toxicity are associated with use of this modality. Therefore, conventional chemotherapy is now a therapy of only historical interest.
Interferon
Interferon was first reported in 1984 to be an effective therapy for patients with HCL15; since then, many large studies have confirmed its activity.15–21 The precise mechanism of action of interferon is not known, but its activity may be attributable to a decrease in the production of cytokines such as granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, interleukin-3, and interleukin-6, perhaps related to the characteristic monocytopenia associated with interferon treatment.147 Studies suggest that interferon-α results in apoptotic death of hairy cells, mediated by tumor necrosis factor-α.148 Despite a high overall response rate of 75% to 90%, most patients achieve only partial remission (i.e., they have a partial response [PR], defined as normalization of all peripheral blood counts).18,19 Interferon commonly is administered subcutaneously at a dose of 3 million IU/m2 three times a week for 12 to 18 months. During the first 2 months of treatment, the white blood cell count and hemoglobin level often decrease, occasionally precipitating transfusion. The platelets normalize earliest in responding patients, followed by the hemoglobin and the white blood cell count. An absolute neutrophil count greater than 1500/µL is achieved after a median of 5 months of therapy. The most common manifestations of toxicity include flulike symptoms, anorexia and fatigue, nausea and vomiting, diarrhea, dry skin, peripheral neuropathies, and central nervous system dysfunction, usually manifested as depression or memory loss. Other than myelosuppression, the most common laboratory abnormality is elevation of hepatic transaminase levels. The median failure-free survival period after discontinuing interferon therapy ranges from 6 to 25 months in different series.18–20,149 Patients with more than 30% hairy cells in the marrow or a platelet count less than 160,000/µL at the end of treatment have a higher risk of early relapse.20–20 In addition, patients who express the CD5 antigen appear to respond poorly to interferon.150 Patients can be maintained on long-term interferon therapy at a dose of 3 million IU subcutaneously given three times a week with minimal toxicity. Sixty percent of patients have sustained their initial response for a median of 5 years, 9% discontinued therapy early because of unexpected neurologic toxicity, and only 13% stopped therapy because of progressive disease.21 A recently updated study by Benz and colleagues confirmed the tolerability and efficacy of long-term therapy with interferon.151 In this study, 9% discontinued therapy because of progressive disease and 17% stopped therapy because of side effects. Although treatment of HCL with interferon is effective, complete responses are uncommon, and failure-free survival usually is short after discontinuation of treatment. Furthermore, the purine analogs have completely supplanted interferon for the treatment of newly diagnosed HCL.
Purine Analogs
As early as the 1960s, Giblett and colleagues observed that 30% of children with severe combined immunodeficiency syndrome lacked the enzyme adenosine deaminase (ADA).152 It appeared that the accumulation of the triphosphorylated form of deoxyadenosine was responsible for lymphocyte depletion.153 Therefore, the deliberate inhibition of ADA was recognized as a potentially useful antileukemic strategy. The methods to accomplish this therapeutic effect included the development of agents to bind irreversibly to ADA or to resist the action of the enzyme. These agents affect both dividing and nondividing cells.154 After purine analog therapy, accumulation of deoxyadenosine triphosphates leads to DNA strand breaks and inhibition of DNA repair, which ultimately results in cell apoptosis.
Pentostatin (2′-Deoxycoformycin)
Pentostatin, or 2′-DCF, was the first agent to induce a significant number of complete responses in HCL.22,155 This drug binds to ADA, resulting in irreversible inhibition of the enzyme,156 which is found in all lymphoid cells and is important in purine metabolism. In most studies, complete response is defined by disappearance of hairy cells in the blood and bone marrow, complete normalization of peripheral counts (hemoglobin concentration > 12.0 g/dL, platelet count > 100,000/µL, and absolute neutrophil count > 1500/µL), and resolution of splenomegaly and lymphadenopathy. A partial response requires normalization of blood cell counts, greater than 50% reduction in hairy cells in the bone marrow, and greater than 50% reduction in splenomegaly.
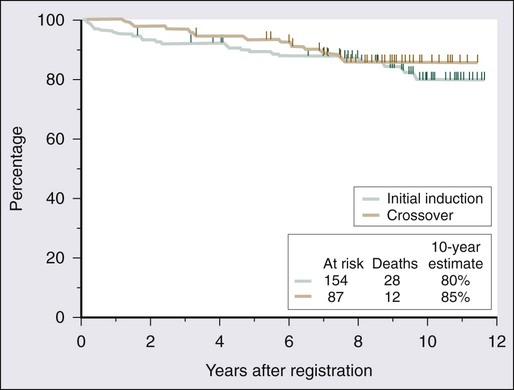
A number of studies demonstrate the efficacy of 2′-DCF in patients with HCL (Table 103-1).23–33 A large prospective, randomized study showed that the complete response rate and relapse-free survival rate are significantly better with 2′-DCF than with interferon.31 Various dosing schedules have been reported in early studies, but the current dose is 4 mg/m2 given by intravenous infusion every 2 weeks until maximum response. The median number of cycles required by patients until best response has ranged from 6 to 12 cycles.24,27,29,33 In one of the earlier studies conducted by the Eastern Cooperative Oncology Group, most patients achieved maximal response within the first 6 months.27 Therapy is relatively well tolerated; neutropenia, fever, and infections are the most frequent toxicities.25,27,32 One of the largest published series providing long-term evaluation was reported by the Southwest Oncology Group.25 A total of 241 patients received 2′-DCF on a phase III trial comparing interferon and 2′-DCF; 154 were randomly assigned to receive 2′-DCF initially, with crossover of 87 additional patients from the interferon group after it failed to produce improvement. Seventy-two percent of patients achieved a complete response with 2′-DCF. Long-term survival was not statistically different based on initial treatment; in both groups, overall survival was 90% at 5 years and 81% at 10 years (Fig. 103-4). With longer follow-up, 2′-DCF does not appear to be curative in all patients. Studies with follow-up periods longer than 5 years after treatment report relapses in 15% to 48% of patients.23–25,30
Table 103-1
Activity of 2′-Deoxycoformycin in Hairy Cell Leukemia
| Study | No. of Patients | Previously Untreated | Median No. (of Cycles) | CR (%) | PR (%) | NR (%) | Median Follow-up (mo) | Relapse (%) | Median Time to Relapse (mo) |
| Johnston et al.26 | 28 | 18 | NA | 89 | 11 | 0 | 14 | 4 | 13.8 |
| Ho et al.28 | 33 | 0 | NA | 33.3 | 45.5 | 0 | 14.5 | 8 | 10 |
| Rafel et al.32 | 78 | 35 | 7 | 72 | 16 | 1 | 31 | 33 | 29 |
| Cassileth et al.27 | 50 | 19 | 6 | 64 | 20 | 16 | 39 | 14 | CR: 13; PR: 31 |
| Catovsky et al.29 | 148 | 23 | 9 | 74.3 | 22.3 | 3.4 | 42 | 8 | 22 |
| Ribeiro et al.33 | 50 | 18 | 12 | 44 | 52 | 0 | 47 | 10 | 22 |
| Grever et al.25,31 | 154 | 154 | NA | 76 | 3 | 5* | 57 | 9 | NA |
| Maloisel et al.23 | 238 | 67 | 9 | 83 | 16.6 | 6 | 63.5 | 15 | NA |
| Kraut et al.30 | 24 | NA | NA | 100 | 0 | 0 | 82 | 48 | 30 |
| Dearden et al.24 | 220 | 188 | 10 | 82 | 14 | 4 | 192 | 44 | 51.5 |
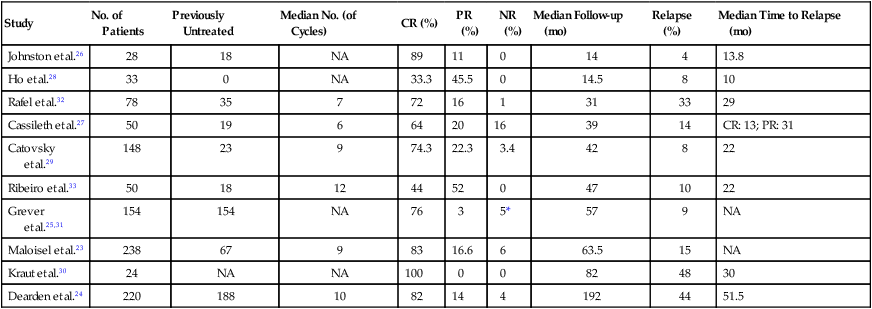
CR, complete response; NA, not available; NR, no response; PR, partial response.
Cladribine (2-Chlorodeoxyadenosine)
Cladribine, or 2-CdA, is a purine analog that is resistant to the action of ADA. This agent accumulates in the lymphoid cells, possibly because they are rich in the enzyme deoxycytidine kinase.153 This enzyme phosphorylates 2-CdA to the active 5′-triphosphate form, creating a deoxynucleotide that cannot readily exit the cell. This compound inhibits ribonucleotide reductase, which results in decreased synthesis of deoxynucleotides. Both DNA synthesis and repair are impaired.
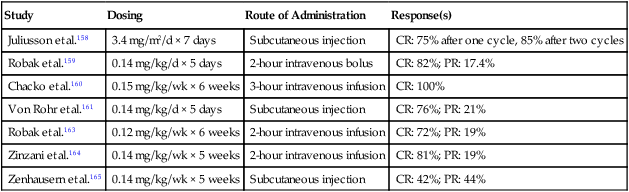
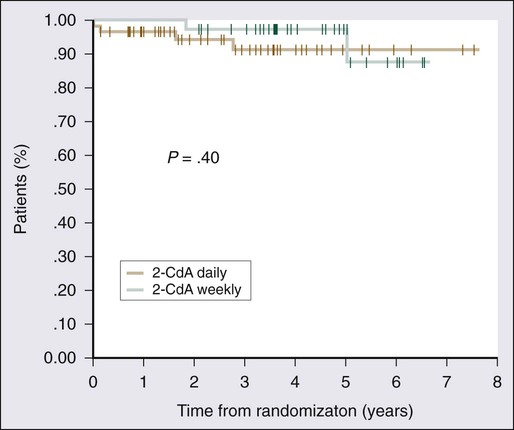
2-CdA was first reported to be effective in HCL by Piro and colleagues in 1990.34 Twelve patients were given a single cycle of 2-CdA at a dose of 0.1 mg/kg/d by continuous infusion for 7 days; a complete response was obtained in 11 of the 12 patients within 8 weeks of treatment. A number of subsequent studies have shown similar remarkable efficacy (Table 103-2).24,34–44 Clear orders for use of the 7-day infusion pump are critical because poor response to therapy has been attributed to underdosing of the drug when 1 day’s dose was administered over 7 days.157 Excellent results with alternative dosing schedules and routes of administration have been reported (Table 103-3).158–165 In the study conducted by Von Rohr and colleagues, 2-CdA was administered as a subcutaneous bolus injection of 0.14 mg/kg/d for 5 days to 62 patients.161 The results were similar to those of previous studies, with 76% of patients achieving a complete response and an overall response rate of 97%. Robak and colleagues conducted a prospective randomized trial comparing a standard 5-day schedule with a 6-week schedule, which had been reported to be potentially less toxic.162,163 These investigators observed similar complete response rates, overall response rates, and progression-free and overall survival rates, with no less toxicity163 (Fig. 103-5).
Table 103-2
Activity of 2-Chlorodeoxyadenosine in Hairy Cell Leukemia*
| Study | No. of Patients | Previously Untreated | CR (%) | PR (%) | NR (%) | Median Follow-up (mo) | Relapse (%) | Median Time to Relapse (mo) |
| Estey et al.40 | 46 | 27 | 78 | 11 | 11 | 9 | 2 | 17.8 |
| Juliusson and Liliemark41 | 16 | 3 | 75 | 0 | 13 | 12 | 0 | |
| Tallman et al.42 | 20 | 12 | 80 | 20 | 0 | 12 | 5 | NA |
| Piro et al.44 | 144 | 69 | 85 | 12 | 2 | 14 | 3 | 36 |
| Piro et al.34 | 12 | 3 | 92 | 8 | 0 | 15.5 | 0 | |
| Seymour et al.35 | 46 | 27 | 78 | 11 | 11 | 30 | 20 | 16 |
| Jehn et al.37 | 42 | 32 | 98 | 2 | 0 | 33 | 14 | 29 |
| Von Rohr et al.161 | 62 | 33 | 76 | 21 | 3 | 46 | 24 | 38 |
| Hoffman et al.43 | 49 | 21 | 76 | 24 | 0 | 55 | 24 | NA |
| Saven et al.38 | 349 | 179 | 91 | 7 | 2 | 58 | 26 | CR: 30; PR: 24 |
| Goodman et al.39 | 207 | 119 | 95 | 5 | 0 | 108 | 37 | 42 |
| Chadha et al.36 | 86 | 60 | 79 | 21 | 0 | 116 | 36 | 35 |
| Dearden et al.24 | 45 | 12 | 84 | 16 | 0 | 45 | 29 | 23.5 |
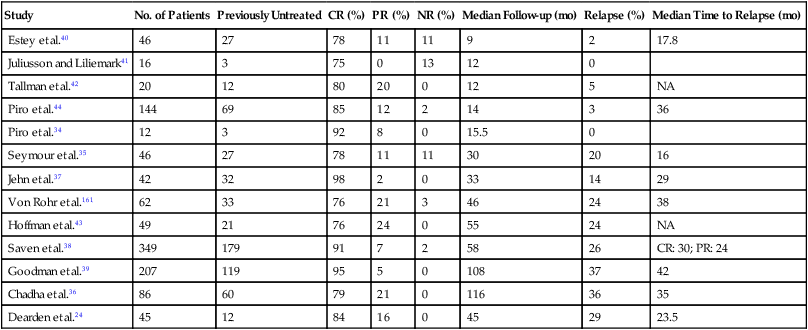
CR, complete response; NA, not available; NR, no response; PR, partial response.
Table 103-3
Alternate Schedules of 2-Chlorodeoxyadenosine Therapy for Treatment of Hairy Cell Leukemia
| Study | Dosing | Route of Administration | Response(s) |
| Juliusson et al.158 | 3.4 mg/m2/d × 7 days | Subcutaneous injection | CR: 75% after one cycle, 85% after two cycles |
| Robak et al.159 | 0.14 mg/kg/d × 5 days | 2-hour intravenous bolus | CR: 82%; PR: 17.4% |
| Chacko et al.160 | 0.15 mg/kg/wk × 6 weeks | 3-hour intravenous infusion | CR: 100% |
| Von Rohr et al.161 | 0.14 mg/kg/d × 5 days | Subcutaneous injection | CR: 76%; PR: 21% |
| Robak et al.163 | 0.12 mg/kg/wk × 6 weeks | 2-hour intravenous infusion | CR: 72%; PR: 19% |
| Zinzani et al.164 | 0.14 mg/kg/wk × 5 weeks | 2-hour intravenous infusion | CR: 81%; PR: 19% |
| Zenhausern et al.165 | 0.14 mg/kg/wk × 5 weeks | Subcutaneous injection | CR: 42%; PR: 44% |
The most common toxicities associated with 2-CdA treatment are neutropenia and fever. Investigators at the Scripps Clinic reported results for the largest collection of patients with HCL treated with 2-CdA. Of 349 patients who received a single cycle of 2-CdA, 87% had grade 3 or 4 neutropenia, 42% had a neutropenic fever, but only 13% had documented infections, none of which was opportunistic.38 Most of the fevers seen with administration of 2-CdA do not appear to represent infection but may be due to release of cytokines. Because of the high incidence of neutropenic fever, these investigators conducted a prospective trial examining the effect of filgrastim in 35 patients receiving 2-CdA.166 Filgrastim was administered on days −3 through −1, and again after completion of 2-CdA therapy until the absolute neutrophil count was more than 2000/µL on 2 consecutive days. When compared with historical controls, patients in the filgrastim treatment group more rapidly achieved an absolute neutrophil count greater than 1000/µL, in 9 days versus 22 days. The incidence of fever and need for hospital admission, however, did not differ between the two groups. Therefore, the routine use of prophylactic filgrastim is not indicated in this setting. Such a strategy may be useful in the rare patient who has a life-threatening infection and requires therapy with a purine analog.
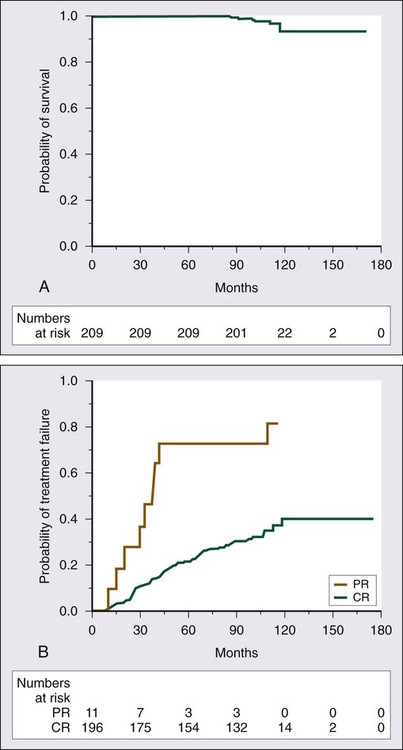
Although response rates are very high with 2-CdA, they are not sustained in all patients. The experience with 2-CdA is similar to that with 2′-DCF in that relapses have appeared with longer follow-up (see Table 103-2). In a report from the Scripps Clinic, after a median follow-up period of 108 months, 37% of patients experienced relapse, with a median time to relapse of 42 months.39 Initial PR is associated with a shorter duration of remission (Fig. 103-6). Of the patients who experienced relapse, 79% received re-treatment with 2-CdA; the overall response rate was 92%, including 75% with a complete remission.39 One retrospective study has suggested that durability of response is greater with 2′-DCF than with 2-CdA. At 45 months of follow-up for patients treated with 2′-DCF and patients treated with 2-CDA, relapse rates were 9.7% and 29%, respectively.24 To date, no randomized trials of the two agents have been conducted to resolve this issue. Thus far, no plateau has been reached with either agent, and relapses continue to occur after initial treatment.
Immunosuppression with Purine Analogs
Treatment with both 2′-DCF and 2-CdA results in prolonged immunosuppression.41,167–169 A decrease in the total lymphocyte count occurs with 2′-DCF, with a greater reduction in T cells than in B cells or natural killer cells.168 The levels of CD4+ and CD8+ cells decrease to fewer than 200 cells/µL for at least 6 months after 2′-DCF treatment is discontinued. In a series of 15 patients with HCL treated with 2′-DCF with long-term follow-up, the median time to recovery of CD4+ lymphocyte counts to normal was 54 months.169 Treatment with 2-CdA induces similar suppression of CD4+ cell counts.35 The median time to recovery of CD4+ cell counts to normal after completion of 2-CdA therapy was 40 months. Treatment with 2-CdA affects one distinct subset of CD4+ T cells. The CD4+/CD45RA+ subset is significantly reduced for as long as 5 years; the CD4+/CD45RO+ T cells, which secrete cytokines and enhance B cell function, are not suppressed.170 In addition, recovery of CD8 and NK cells is more rapid; they normalize within 3 months of treatment with 2-CdA.171 This finding may explain why opportunistic infections, other than an occasional case of herpes zoster, are surprisingly uncommon.33,38,41,170
Prognosis
Before the routine introduction of interferon therapy, survival at 4 years was reported to be 68%.172 With the use of purine analogs, durable remissions are obtained; and even after relapse, re-treatment with a purine analog results in good responses. The 5-year survival rates are greater than 85%.24,25,29,33,38,39 Flinn and colleagues reported long-term results for 241 patients with HCL treated with 2′-DCF; the overall survival rate was 90% at 5 years and 81% at 10 years (see Fig. 103-4).25 The leading causes of death were second malignancies and infection. Only 2 of 40 deaths in this series were attributable to HCL.
A series of 209 patients with HCL treated with 2-CdA at the Scripps Clinic have been followed for at least 7 years.39 The overall survival rate at 9 years is 97% (see Fig. 103-6A). At least three other large long-term follow-up studies of 2′-DCF or 2-CdA have been published.173,174 After a median follow-up period of 8.5 years, Jehn and associates reported a relapse rate of 39%, but the overall survival rate at 12 years was 79%.173 At Northwestern University, among 86 patients, the overall survival rate at 12 years was 87%.174 After a median follow-up period of 9.7 years, 36% of patients had experienced relapse. Else and colleagues reported a study with one of the longest follow-ups of patients who received a purine analog, 2-CdA in 45 patients and 2′-DCF in 188, with a median follow-up period of 16 years.174 Although an earlier report from the same group of investigators suggested a lower relapse rate associated with use of 2′-DCF, in this more recent report, the response, relapse, and overall survival rates at 10 years were similar. These studies have also suggested that the quality of the initial response can be predictive of the outcome, with a longer disease-free survival and a delayed time to treatment failure in patients achieving a complete response after initial therapy compared with those with lesser responses, but overall survival did not differ significantly between these two groups of patients.39,174
Two recent studies have examined potential predictors of outcome in HCL and have identified subsets at higher risk of treatment failure. In a study from the National Institutes of Health, patients with IGHV4-34 rearrangement had significantly lower response rates to initial therapy with 2-CdA (none in 6 patients with V4-34+ HCL vs. 36 of 40 patients with V4-34− HCL; P <.001) and shorter median overall survival (8.63 years in V4-34+ HCL vs. 26.22 years in V4-34− HCL; P < .001).175 In another study, 6 of 58 newly diagnosed HCL patients were identified to have unmutated IGHV4-34, which was associated with a significantly higher rate of failure after treatment with 2-CdA: 5 of 6 patients with unmutated IGHV4-34 failed to respond to initial 2-CdA treatment, whereas all 52 patients with mutated IGHV4-34 benefited from treatment (P < .001).176 Although the prognostic value of these molecular parameters needs to be validated in a larger cohort, the suboptimal response to initial therapy with 2-CdA suggests a need for alternative treatment approaches in these patients.
Other Considerations in Management
Evaluation of Minimal Residual Disease
The remarkable activity of the purine analogs has led to the examination of post-treatment bone marrow biopsy specimens to detect MRD in patients otherwise in complete remission. Immunohistochemistry studies using anti-CD20, DBA.44, and anti-CD45RO antibodies in paraffin-embedded biopsy specimens have been used most frequently.111–114,177–179 Newer techniques for MRD detection include flow cytometric immunophenotyping, consensus primer PCR assay,109 and patient-specific relative quantitative (RQ) PCR assay.180 Depending on the criteria used, 13% to 51% of patients in apparent complete remission have evidence of MRD111–113,181 (Fig. 103-7). In one study, no statistically significant difference in incidence of MRD was found between patients receiving 2′-DCF and those given 2-CdA.181 The presence of MRD appears to predict relapse.113,179 In one study evaluating MRD in 66 patients, 50% of those with MRD experienced relapse whereas only 6% of patients without MRD did so.181 Two trials have suggested that administration of the anti-CD20 monoclonal antibody rituximab after 2-CdA therapy is effective in eradicating MRD.182,183 However, a study by Sigal and colleagues suggests that HCL patients with detectable MRD and even morphologically evident disease can live many years after initial therapy without experiencing overt relapse.184 Therefore, it remains unknown whether MRD eradication should be the goal of therapy in HCL and whether such a strategy prolongs overall survival. Furthermore, the definition of what constitutes MRD varies, and the optimal time to evaluate the maximal benefit of purine analogs remains unclear.185
Treatment of Relapse
Relapse often is detected on bone marrow biopsy alone, and immediate re-treatment is not necessary. In a study by Kraut and colleagues, relapse was detected at a median of 30 months from achievement of remission with 2′-DCF but re-treatment was initiated at a median of 60 months after first remission.30 Re-treatment with purine analogs resulted in complete remission in 5 of 7 patients. Patients with an initial response to 2-CdA also respond well to re-treatment with a purine analog. In the Scripps Clinic series, 76 of 207 patients experienced relapse and 79% received re-treatment with 2-CdA. The overall response rate was 92%, including 75% with a complete response.39 The median duration of the second response was 35 months, which is comparable with the 42-month duration of first response. Responses continue to be seen even with a third cycle of 2-CdA in 80% of patients receiving re-treatment. In patients who experience relapse after purine analog therapy, re-treatment may be with either 2-CdA or 2′-DCF. Alternative agents for treatment of relapsed or treatment-refractory disease include HA22, rituximab, and BRAF inhibitors (see New Therapies).
Risk of Second Malignancies
An association has been noted between HCL and second malignancies, although such a relationship is difficult to determine with certainty. Malignancies that have been observed in patients with HCL include melanoma, prostate cancer, gastrointestinal cancers, non-Hodgkin lymphoma, and nonmelanomatous skin cancers. It is not clear whether HCL itself increases risk or whether the type of therapy may play a role. Kampmeier and colleagues reported a significantly increased incidence of second malignancies in patients with HCL treated with interferon.186 The British Columbia Cancer Agency reported data from a 20-year follow-up study of 117 patients: a second malignancy developed in 31%, and 30% of these malignancies were diagnosed before the diagnosis of HCL.187 The risk was elevated regardless of the type of therapy. This association has not been uniformly observed, however.188,189 Investigators at the M.D. Anderson Cancer Center reported no excess of second malignancies among 350 patients who received either interferon, 2-CdA, or 2′-DCF.190 The immunosuppression due to the purine analogs may play a role in the increased malignancy incidence, but the evidence is not clear. Long-term follow-up studies of patients with HCL treated with 2′-DCF have not demonstrated a statistically significant increased risk of second malignancies.23,25 Other studies have suggested that treatment with 2-CdA is associated with an increased cancer risk.38,39,191 In a review of the Scripps Clinic experience with 349 patients, 8% of patients had a second malignancy that developed at a median time of 62 months after the diagnosis of HCL and 21 months after treatment with 2-CdA.38 Of note, 11% of the patients in this study had a diagnosis of malignancy before the diagnosis of HCL. It is not clear, therefore, that therapy increases risk; HCL itself may be associated with an inherent predisposition to malignancy.
New Therapies
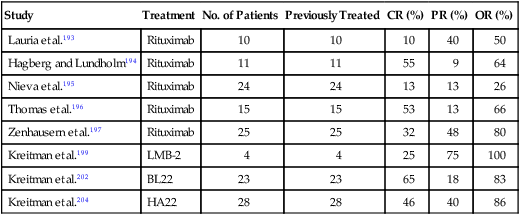
The anti–CD-20 monoclonal antibody rituximab has been tested in patients with HCL refractory to other treatments.192,193 Two different regimens of rituximab have been used in patients with previously treated HCL: 375 mg/m2/wk for 4 doses and 375 mg/m2/wk for 8 doses. With the use of these regimens, overall response rates ranging from 26% to 80% have been reported (Table 103-4).193–197 This wide range of response rates may be due to a heterogeneous patient population and different dosing schedules of rituximab. Review of pretreatment characteristics in these studies suggests a lower response rate in patients with higher number of prior treatments and extensive pretreatment HCL bone marrow infiltration. Using eight weekly doses of rituximab, Thomas and colleagues196 observed an overall response rate of 60% in 15 patients with relapsed or refractory HCL and suggested that four weekly doses may be inadequate for some patients. Although the responses obtained from the rituximab therapy appeared durable (median duration of remission 14 to 32 months), relapses were observed in 33% to 42% responding patients in the studies with extended follow-ups.195,196 It appears that rituximab has activity in HCL, but its optimal dose and schedule and potential synergy with other agents need to be explored further.
Table 103-4
Antibody and Immunoconjugate Therapy in Treatment of Hairy Cell Leukemia
| Study | Treatment | No. of Patients | Previously Treated | CR (%) | PR (%) | OR (%) |
| Lauria et al.193 | Rituximab | 10 | 10 | 10 | 40 | 50 |
| Hagberg and Lundholm194 | Rituximab | 11 | 11 | 55 | 9 | 64 |
| Nieva et al.195 | Rituximab | 24 | 24 | 13 | 13 | 26 |
| Thomas et al.196 | Rituximab | 15 | 15 | 53 | 13 | 66 |
| Zenhausern et al.197 | Rituximab | 25 | 25 | 32 | 48 | 80 |
| Kreitman et al.199 | LMB-2 | 4 | 4 | 25 | 75 | 100 |
| Kreitman et al.202 | BL22 | 23 | 23 | 65 | 18 | 83 |
| Kreitman et al.204 | HA22 | 28 | 28 | 46 | 40 | 86 |
CR, complete response; OR, overall response; PR, partial response.
CD-25, also known as Tac, is the α subunit of the interleukin-2 receptor and is expressed in 80% of patients with HCL.198 LMB-2, anti-Tac(Fv)-PE38, is an immunotoxin that contains the variable heavy domain of anti-Tac fused to the amino terminus of a 38-kD truncated form of the Pseudomonas exotoxin.199 Tac has demonstrated some efficacy in patients with CD25+ hematologic malignancies. After binding to CD25, the compound is internalized, leading to apoptosis and cell death. Of four patients with HCL refractory to standard therapies, including 2-CDA and interferon regimens, all demonstrated a response to LMB-2, with one complete response.199 LMB-2 is well tolerated and appears to have no hematologic toxicity. Larger studies need to be conducted to better determine the safety and efficacy of this agent.
Another promising immunotoxin under investigation is recombinant immunotoxin containing anti-CD22 monoclonal antibody and PE38, the Pseudomonas exotoxin. CD22 is expressed by normal B cells and B-cell leukemias and lymphomas, including HCL, but is not found on stem cells.200 Kreitman and colleagues recently updated their original study201 and reported the results of a phase II trial of 36 patients with HCL treated with the anti-CD22 recombinant immunotoxin called BL22.202 The overall response rate was 72%, with 47% of patients showing a complete response. Of the 17 complete responders, only 3 had MRD, as determined by immunohistochemistry studies of bone marrow. The median complete response duration was not reached in the study, and only 4 of 17 patients with complete responses experienced relapse at a median follow-up of 26 months. This therapy appears to be well tolerated with transient mild vascular leak syndrome in most patients and reversible hemolytic-uremic syndrome in 8%. No other hematologic toxicity or decrease in T-cell count was observed.202 Kreitman and colleagues further modified BL22 to increase the affinity for CD22 by using hot-spot mutagenesis. The resulting protein, called HA22, which differs from BL22 by two amino acids, has a 14-fold increased binding affinity for CD22 and a larger increase in cytotoxicity.203 A recent report of the phase I clinical trial with HA22 in 28 patients with relapsed or refractory HCL demonstrated the overall response rate of 86% with 46% complete response, most of which were MRD negative assessed by immunohistochemistry.204 At a median follow-up of 29 months, 10 of 13 patients (80%) remain in complete response. Based on the encouraging results of this study, a randomized study of HA22 versus best alternative therapy (i.e., rituximab or 2-CdA) is underway in patients with HCL and multiple relapses.
One of the new exciting areas of research in HCL is the investigation of clinical activity of BRAF inhibitors based on the specific and high prevalence of the BRAF V600E mutation in HCL.45 The concept that the BRAF V600E mutation may represent a key driver mutation in HCL is supported by a preliminary evidence of striking clinical activity of the specific BRAF inhibitor vemurafenib in a patient with refractory HCL.75 A group of investigators in Germany recently reported that a patient with HCL and massive splenomegaly and pancytopenia, who was refractory to three lines of purine analog-based treatment, achieved a complete response by day 43 of therapy with the specific BRAF inhibitor vemurafenib.75 This observation substantiates the BRAF mutation as a potential therapeutic target in HCL, and a multicenter clinical trial with the BRAF inhibitor in relapsed and refractory HCL is scheduled to open soon in the United States.
Lastly, increased angiogenesis has been reported in patients with HCL.207–207 Microvessel density is higher in the marrow of patients with active HCL than in normal control subjects. Treatment with both interferon-α and 2-CdA is associated with a decrease in microvessel density.207–207 These observations suggest a potential role for anti-angiogenesis agents.