[level-membership-for-surgery-category]
Chapter 1A Embryologic development of the liver, biliary tract, and pancreas
Overview of Liver and Biliary Tract Development
Developmental biologists have often marveled at the enormous regenerative capacity of the liver after injury, and such growth represents one of the fastest by mammalian tissues. Presumably this process is based on the recapitulation of embryonic signals in the liver, but our understanding of the mechanisms of liver development is relatively poor. The liver is the largest internal organ and consists of diverse cell types that arise from various embryologic origins. The two principle cell types of the liver are the hepatocytes, which comprise nearly 70% of the mass of the adult organ, and the cholangiocytes. Both cells are derived from the embryonic endoderm. Other cell types of the liver include hematopoietic, Kuppfer, stromal, and stellate cells, all of which are of mesodermal origin (Fig. 1A.1). Despite their homogeneous appearance, hepatocytes do not all function identically; they perform various roles, depending on their physical location within a hepatic lobule, the primary functional unit of the liver. Thus, liver development entails not only hepatocyte and cholangiocyte differentiation but also further cellular differentiation within the hepatocyte population (Fig. 1A.1).
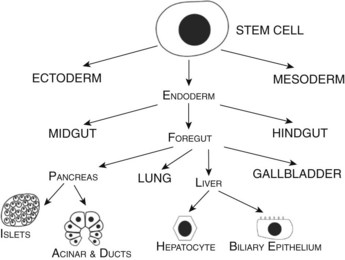
FIGURE 1A.1 Cell lineage schematic for hepatic and pancreatic development from a multipotent progenitor stem cell.
During the third week of gestation, the liver primordium first appears as an outgrowth of the ventral foregut endoderm at the caudal end of the foregut. The proliferation of the epithelial cells in this liver bud leads to its growth and branching with resulting expansion of the growing bud into the surrounding mesenchyme, giving rise to the liver and intrahepatic biliary tree. As it grows caudally, traversing the septum transversum, the persistent connection between the branching epithelium and the foregut develops into the extrahepatic bile ducts and gallbladder (Sadler & Langman, 2006). The hepatoblasts eventually differentiate into hepatocytes and cholangiocytes. The final liver structure continues to develop through the postnatal period. In addition to giving rise to the liver and biliary tract, the proximal endoderm (foregut) also gives rise to respiratory epithelium and the glandular cells of the thyroid, thymus, and pancreas.
Endodermal Patterning
Embryologically, the liver, biliary tract, and pancreas develop through a series of reciprocal interactions between the endoderm and the surrounding mesenchyme. The primitive gut tube, which is derived from the endodermal germ layer during gastrulation, is divided into the foregut, midgut, and hindgut domains, each of which gives rise to specialized regions (Grapin-Botton, 2005). This specialization is initiated by transcription factors expressed in different regions, such as pancreatic and duodenal homeobox gene 1 (PDX1) and pancreas-specific transcription factor 1a (PTF1A), whose coexpression in the endoderm gives rise to the pancreas (Fig. 1A.2) (Grapin-Botton, 2005; Moore-Scott et al, 2007). The definitive endoderm is formed at the ventral side of the vertebrate embryo during gastrulation. Evagination of the endoderm at the anterior end of the embryo generates the ventral foregut, which will eventually give rise to the liver, lung, thyroid, and the ventral pancreas. The dorsal region of the definitive endoderm develops into the intestines and the dorsal pancreas (see Fig. 1A.2).
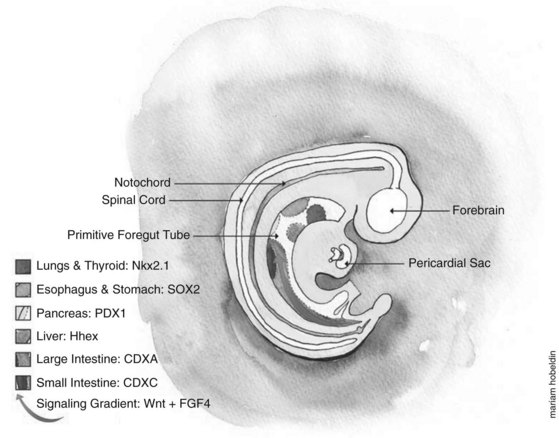
This complex dialogue between the endoderm and mesoderm appears to be critical for the final patterning of the gut tube, where several signaling pathways have been implicated in the regulation of foregut endoderm development, promoting specification along its anterior–posterior axis for organs such as the thyroid, lung, liver, and pancreas. The plasticity of the endoderm was demonstrated by experimentally recombining the posterior mesoderm with early foregut endoderm, leading to repression of liver and pancreatic development in favor of intestinal development (Wells & Melton, 2000; Kumar et al, 2003).
Specific signals from the surrounding mesoderm in the foregut region lead to hepatic specification and subsequent morphogenesis (Guadi et al, 1996). The molecular pathway linking endodermal patterning to the initiation of liver and pancreatic development has been partially elucidated by recent studies in the Xenopus model, supporting a role for FGF4 and WNT signaling from the posterior mesoderm in inhibiting foregut fate and promoting hindgut formation; these signals are inhibited in the anterior endoderm to allow foregut development (McLin et al, 2007). β-Catenin repression in the anterior endoderm is specifically necessary to initiate liver and pancreas development; β-catenin repression in the endoderm then initiates reciprocal signaling with the mesoderm, leading to hepatic induction. To illustrate β-catenin’s role in this process, forced β-catenin expression in the anterior endoderm led to downregulation of the hematopoietically expressed homeobox gene (HHEX) and inhibition of liver formation, whereas repressing β-catenin in the posterior endoderm—future hindgut that normally expresses β-catenin—induced ectopic HHEX expression and ectopic liver bud initiation (McLin et al, 2007). HHEX is a direct target of β-catenin and is one of the earliest foregut markers (Thomas et al, 1998) that is essential for ectopic and normal liver and ventral pancreas development in mice (Keng et al, 2000; Bort et al, 2004). In addition to repressing HHEX, β-catenin also downregulates other foregut markers for liver (for1) (Seo et al, 2002), pancreas (PDX1), lung/thyroid (NKX2-1) (Goss et al, 2009), and intestine (Endocut) (Costa et al, 2003). Similarly, inhibiting β-catenin in the posterior mesoderm led to ectopic expression of other liver and pancreas markers (for1, PDX1, elastase, and amylase) along with inhibition of the intestinal marker Endocut (McLin et al, 2007).
Studies in mouse and chick models demonstrated the need for an FGF2 signal from the cardiogenic mesoderm and bone morphogenetic proteins (BMPs) from the septum transversum mesenchyme (Rossi et al, 2001; Zhang et al, 2004) for liver development. In addition, WNT2B has also been shown to be an important signal for liver specification in Zebrafish (Ober et al, 2006).
Other examples of transcription factors implicated in foregut patterning include the HOX genes. HOXD13 is expressed in the hindgut of mouse and chick embryos (Yokouchi et al, 1995; Roberts et al, 1995; Dolle et al, 1991). Knockout mice homozygous and null mutant for HOXD13 develop with hindgut defects (Kondo et al, 1996). Similarly, when HOXD13 is misexpressed in chick embryo midgut mesoderm, hindgut features are seen in the midgut epithelium (Roberts et al, 1998). The exact mechanism of how the HOX genes induce endodermal patterning remains unknown, as the exact downstream targets are not clear. In addition to HOX genes, retinoic acid signaling has been implicated in this foregut patterning, probably through regulating HOX gene expression. Specifically, retinoic acid signaling appears to specify the position of the pancreatic domain in the foregut of Xenopus, Zebrafish, and mice (Huang et al, 1998; Chen et al, 2004; Martin et al, 2005; Stafford et al, 2004). Goss and colleagues (Goss et al, 2009) demonstrated that WNT2 and WNT2B play an essential role in specifying lung endoderm without affecting the specification of other foregut-derived tissues. WNT2/2B double-knockout (DKO) mutants display complete lung agenesis. NKX2-1, the earliest known transcription factor marker for the developing lung, was also lost from the anterior foregut, confirming the loss of trachea and lung development.
Hepatic Competence
Hepatic competence, or the ability to form liver from the foregut endoderm, is considered the first step in a two-step process for the specification of the liver in vertebrates. Competence is a prerequisite for the endoderm to respond to specific signals, such as Fgfs from the surrounding mesoderm, which then leads to the second step, the induction of liver specific genes such as ALB (albumin). This “competence” is facilitated through the FOXA gene transcription factor family that includes FOXA1 and FOXA2 (forkhead box proteins A1 and A2). FOXA gene expression precedes the induction of the hepatic program by FGF signals in the endoderm, and FOXA2 binding revises α-fetoprotein (AFP) gene transcription in vitro (Crowe et al, 1999). Deletion of FOXA1 and FOXA2 led to no liver bud formation with the loss of AFP expression in the ventral foregut, which would indicate that hepatic specification had failed to initiate (Lee et al, 2005). The forkhead box (FOX) proteins are an extensive family of transcription factors that share homology with the winged helix/forkhead DNA binding domains (Kaestner et al, 2000) that play important roles in regulating expression of genes involved in cellular differentiation, proliferation, transformation, and metabolic homeostasis (Wang et al, 2001; Zaret et al, 1999; Duncan, 2000). In vitro explant cultures demonstrated that Fgf was sufficient to induce ventral foregut endodermal cells to differentiate into hepatoblasts (Guadi et al, 1996). However, when FOXA1/FOXA2-deficient endoderm was cultured with exogenous Fgf2, no liver expression was seen (Lee et al, 2005). This result would indicate the need for FOXA1 and FOXA2 for the onset of hepatogenesis. In vivo DNA-binding studies revealed that the liver-specific albumin gene ALB has an important upstream regulatory binding site for FOXA factors (Guadi et al, 1996; Bossard & Zaret, 1998). Prior to FOXA or GATA4 binding, the ALB gene is transcriptionally silent with a closed chromatin. After binding with FOXA and GATA4, the chromatin domain is thought to become exposed (Cirillo et al, 2002), thus increasing the ability of the gene to be activated. By the E9.5 gestation stage, other transcription factors—CCAAT/enhancer binding protein-β (CEBPB) and NF1, nuclear factor 1—bind to sites adjacent to the FOXA site, and as a result, the albumin gene becomes active (Zaret, 2002). The critical binding of FOXA to chromatin increases the ability of the gene expression to be activated by allowing other transcription factors to bind, hence the hepatic competence.
Hepatic Induction
Fgf from the cardiogenic mesoderm and Bmps from the septum transversum mesenchyme have been implicated in the induction of liver fate in mouse and chick embryonic endoderm. In vitro studies were performed in which the ventral foregut and cardiogenic mesoderm were cocultured in the presence of fibroblast growth factor inhibitors, resulting in the inhibition of liver induction (Jung et al, 1999). Culturing foregut endoderm without the cardiogenic mesoderm in the presence of a low concentration of exogenous FGF2 (2 to 5 ng/mL) rescued hepatic gene expression (Jung et al, 1999) and simultaneously suppressed the expression of the pancreas program (PDX1 expression) (Deutsch et al, 2001), whereas a high concentration of FGF2 (10 to 500 ng/mL) induced NKX2-1, an early marker for respiratory epithelium but not albumin gene expression (Serls et al, 2005). Ventral foregut endoderm cocultured with noggin (Nog), a Bmp inhibitor, inhibited hepatic gene induction, an effect that was rescued when Bmp2 or Bmp4 were added. Despite these results, however, embryos that were homozygous mutant for BMP4 still exhibited normal hepatic gene induction (Rossi et al, 2001; Smith & Harland, 1992). These data indicate that the cardiac mesoderm, a source for FGF signaling, is crucial for the hepatic induction from the ventral foregut endoderm and subsequent morphogenesis, whereas Bmp aids in the process as well (Fig. 1A.3).
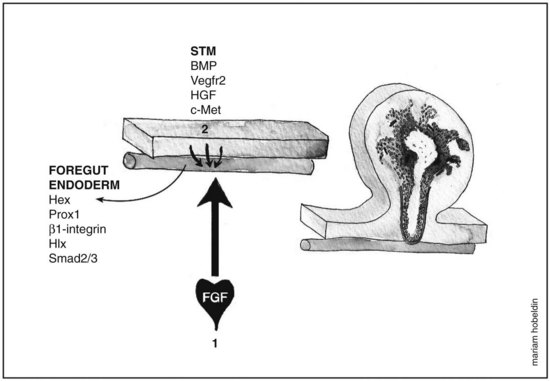
Morphogenesis of the Hepatic Bud
Following hepatic specification (E8.5 to E9.0), the liver starts to express liver-specific genes (ALB, AFP, HNF4A) that eventually form the liver bud. Hepatic bud morphogenesis is facilitated by two transcription factors, HHEX (hematopoietically expressed homeobox, discussed above) (Crompton et al, 1992) and PROX1 (prospero-related homeobox 1) (Oliver et al, 1993). HHEX is expressed in the anterior endoderm at E7.0, eventually giving rise to the ventral pancreas and liver. HHEX-null embryos grow without a liver, thyroid and develop forebrain defects at E11.5; however, evidence of endodermal epithelial thickening suggest a possible defect in differentiation as evidence of possible hepatic induction (Martinez-Barbera et al, 2000).
In a separate study on HHEX-null embryos, liver genes ALB and PROX1 were expressed in the ventral foregut endoderm at around E8.5, with the thickening of the hepatic endoderm region at E9.0 being smaller compared with heterozygote embryos. In addition, a significantly lower proliferation rate was seen in the prospective hepatic domain compared with controls, as demonstrated by bromodeoxyuridine (BrdU) staining, with no evidence of apoptosis (Bort et al, 2004). The basal membrane layer, which is rich in laminin, surrounds the hepatic endoderm and degrades around E9.0 to E9.5, so that the hepatocytes start migrating into the septum transversum mesenchyme (STM) to form the liver bud. This degradation is facilitated by the hepatoblasts, which under normal conditions downregulate E-cadherin. However, in PROX1-null mutant embryos, the progenitor liver cells fail to migrate into the STM as a result of excess E-cadherin and basement membrane proteins laminin and collagen 4. The basal lamina fails to degrade, and the cells remain trapped in the hepatic diverticulum with an overall reduction in liver size (Sosa-Pineda et al, 2000). The bulk of the liver lobe lacks hepatocytes, suggesting that the mesenchymal component contributes most of the liver mass in PROX1-null mutant embryos. A similar phenotype is seen with ONECUT1 and ONECUT2 double mutants, which are required for basal lamina degradation (Margagliotti et al, 2007). Furthermore, pharmacologic inhibition of matrix metalloproteinases (MMPs), extracellular matrix remodeling enzymes usually expressed by hepatoblasts and STM cells, inhibits hepatoblast migration in culture (Margagliotti, 2008). To further illustrate the importance of the extracellular matrix in hepatic bud morphogenesis, hepatoblasts deficient in laminin receptor β1-integrin fail to colonize the liver bud (Fassler & Meyer, 1995). These β1-integrins are among those that act as receptors for extracellular matrix proteins, such as laminins and collagens (Hynes, 1992).
The role of endothelial cells in liver organogenesis has also been studied, with the liver vasculature providing a vital source for hematopoiesis in early life. Endothelial cells are positioned as a loose chain of cells interceding between the thickening hepatic epithelium and the STM (Matsumoto et al, 2001). Embryos that were null mutant for VEGFR2 (also known as KDR) had failure of delamination and subsequent migration by the hepatoblasts (Matsumoto et al, 2001). These results imply that the endothelial cells interact with nascent hepatic cells and aid in liver bud outgrowth.
Liver Bud Growth
The STM cells are closely related to the ventral endoderm and contribute to hepatic induction and growth. This epithelial–mesenchymal interaction is essential for the formation of the liver bud as well as for further expansion and differentiation. During this phase (E9.5 to E15), the liver bud undergoes a tremendous amount of growth and also becomes an important site for hematopoiesis. Factors and signals regulating this critical stage arise from both the hepatic mesenchyme and the STM. These signals include Fgf and Bmp, which promote liver growth in addition to their role in hepatic specification. Bmp4 is strongly expressed in the STM and continues to be expressed at E9.0, during which the liver bud migrates into the STM (Rossi et al, 2001). BMP4-null mutant embryos were developed to assess for a role in liver organogenesis. These null-mutant mice had a delay in the growth of the liver bud, which would indicate that Bmp constitutes an important growth signal for the liver (Rossi et al, 2001).
Another factor that has been implicated in liver growth is HLX, which is expressed prominently in the visceral mesenchyme (STM), into which the liver will expand (Hentsch et al, 1996). HLX-null embryos exhibited severe liver hypoplasia without affecting liver specification (Hentsch et al, 1996). A similar finding was observed in embryos that were null mutant for hepatocyte growth factor (Hgf) (Schmidt et al, 1995). In contrast to HLX−/− embryos, apoptosis was the underlying cause for the severe reduction in liver size. Hgf is produced by the cells lining the sinusoids, which are of mesenchymal origin, mediating its effects through the c-Met tyrosine kinase receptor produced by hepatocytes (Schmidt, 1995). TGF-β signaling is also involved in liver outgrowth. It mediates its signals through SMAD2 and SMAD3, which after phosphorylation translocate to the nucleus to either upregulate or downregulate gene expression. Embryos that were heterozygous mutants for both SMAD2 and SMAD3 exhibited severe liver hypoplasia as a result of a decrease in β1-integrin expression (Weinstein et al, 2001). Intriguingly, this phenotype was rescued when HGF, a potent hepatotrophic growth factor, was added to the culture medium. Presumably, this was a result of the restoration of β1-integrin expression (Kagami et al, 1996), which plays an important role in hepatocytic adhesion to the extracellular matrix; TGF-β and HGF are known to induce the gene (Kagami et al, 1996; Kawakami-Kimura et al, 1997).
Overview of Hepatoblast Differentiation
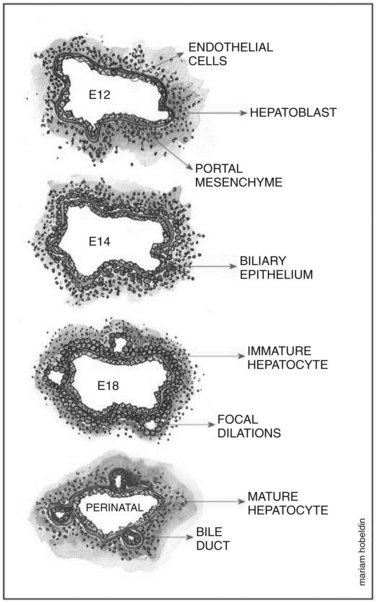
Hepatoblasts begin to differentiate into mature hepatocytes and biliary epithelial cells at around E13.5. Prior to differentiation, the hepatoblasts express genes for adult hepatocytes (ALB, HNF4A), biliary epithelial cells (KRT19), and fetal liver (AFP) (Lemaigre, 2003; Shiojiri et al, 1991). Hepatoblasts in proximity to the portal vein form a bilayer and eventually differentiate into biliary epithelial cells, upregulating biliary-specific cytokeratin-19 (KRT19) and downregulating the other hepatic genes in the process. This bilayer around the portal vein begins to form focal dilations incorporated into the portal mesenchyme to form intrahepatic biliary ducts at E17.0 until birth. Those parts of the ductal bilayer plate not involved in the formation of the ducts progressively regress, and the rest of the hepatoblasts differentiate into mature hepatocytes, arranged in hepatic cords with the bile canaliculi on the apical surfaces (Lemaigre et al, 2003). The bile is produced by the hepatocytes and is secreted into the canaliculi, which are connected to the network of intrahepatic biliary ducts. The bile then flows to the hepatic ducts, transits through the cystic duct, and is stored in the gallbladder; eventually, the bile is excreted into the bowel via the common bile duct. The biliary epithelial cells delineate the lumen of the intrahepatic and extrahepatic biliary tree (hepatic, cystic, and common bile ducts) and the gallbladder (Fig. 1A.4).
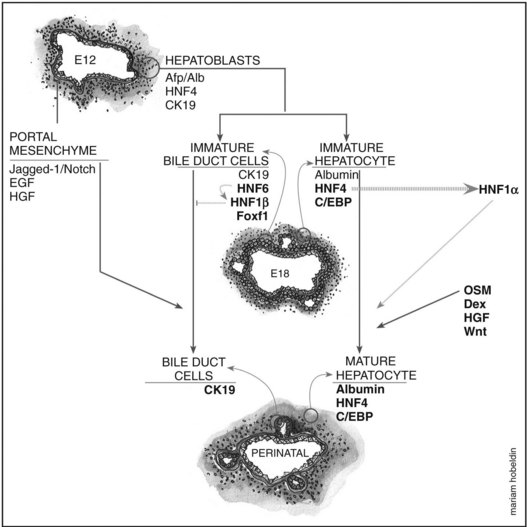
Biliary Epithelial Cell Differentiation and Formation of the Ductal Plate
The exact origin of the biliary epithelial cells has been greatly debated; however, the popular school of thought is that they are derived from bipotential hepatoblasts that can differentiate into either hepatocytes or biliary epithelial cells. This theory is based on the observation that immature hepatoblasts coexpress markers of both hepatocytes (ALB) and biliary epithelial cells (KRT19). The biliary-specific marker KRT19 becomes strongly expressed at a later gestational age, as the cells become ductal cells, whereas other cells transiently express the hepatocyte markers ALB and AFP as they develop into mature hepatocytes (Shiojiri et al, 1991). This theory was further supported when embryonic liver was transplanted into the testes of syngeneic animals, before the emergence of intrahepatic biliary ducts, as it subsequently gave rise to both hepatocytes and typical bile ducts (Shiojiri et al, 1991). However, this finding does not exclude the possibility that ductal cells may be derived from other embryonic cell types. Suzuki and colleagues (Suzuki et al, 2002) used in vitro studies on hepatic “stem” cells from E13 embryonic livers identified as having self-renewing capability and multilineage differentiation potential, to show that these cells could form differentiated hepatocytes, biliary epithelial cells, pancreatic, and intestinal cells.
The first step in triggering the initiation of the transition from a hepatoblast to a biliary epithelial cell is thought to be facilitated through the ONECUT transcription factor hepatocyte nuclear factor 6 (HNF6), which is expressed in the biliary epithelial cells of the developing intrahepatic biliary ducts and in hepatoblasts, gallbladder primordium, and the extrahepatic bile ducts (Landry et al, 1997; Rausa et al, 1997). HNF6−/− embryos displayed severe biliary anomalies that resulted in an absent gallbladder and the extrahepatic bile duct network being replaced with an enlarged structure connecting the liver to the duodenum. In the intrahepatic biliary ducts, however, it caused abnormal differentiation of biliary epithelial cells, resulting in cholestasis. Closer histologic examination revealed an increased number of KRT19-positive cells compared with controls at E13.5, with the development of abnormal large cysts at E15.5 to E16.5 that contained an epithelium of KRT19-expressing cells. These abnormal cysts are similar to those seen in Caroli disease, a disorder with ductal plate malformation and ectasias. Although HNF6 might play a role in the disease, not all of the malformations seen in HNF6−/− mice are manifested in Caroli disease, such as the absence of the gallbladder. The abnormal increase in KRT19-positive cells at E13.5 lacked any proliferative marker, suggesting that they are postmitotic, resulting from an increased number of hepatoblasts that have differentiated toward a biliary lineage prematurely (Clotman et al, 2002). In addition, the excess KRT19-positive biliary epithelial cells formed cordlike extensions within the liver parenchyma, rather than being restricted to the vicinity of the portal vein, as seen in the controls. This observation supports the role of HNF6 in controlling the differentiation of hepatoblasts into biliary epithelial cells and the morphogenesis of the intrahepatic biliary ducts, confining biliary epithelial cells to the periportal area. A similar morphologic defect of intrahepatic biliary ducts was observed in HNF1B−/− embryos. These findings suggest that HNF6 controls intrahepatic biliary duct development via HNF1B (Clotman et al, 2002).
With regard to mesenchymal–epithelial induction of liver primordium and gallbladder, studies have suggested that the mesenchyme contributes to biliary epithelial cell differentiation, where differentiation of hepatoblasts into biliary epithelial cells was stimulated when cocultured with hepatic or lung mesenchyme (Shiojiri & Koike, 1997). It was also noted in the HNF6−/− study that biliary epithelial cell differentiation occurred at the interface between the portal mesenchyme and the liver parenchyma (Clotman et al, 2002). A recent study revealed that the forkhead box f1 (FOXF1) transcription factor may play an important role in mesenchymal–epithelial signaling, an interface required for the development of organs derived from foregut endoderm, such as the pancreas, liver, gallbladder, and lung (Kalinichenko et al, 2002). FOXF1 expression is restricted to the gallbladder mesenchyme and STM. In FOXF1+/−, the gallbladder develops severe structural abnormalities with significant reduction in size and reduced mesenchymal cell numbers, an absent biliary epithelial cell layer, and a deficient external smooth muscle layer. The reduction in mesenchymal cell numbers was attributed to the reduction in the vascular cell-adhesion molecule (VCAM1) and cell-adhesion α5 integrin (ITGA5) genes, both of which are essential for mesodermal formation (Mahlapuu et al, 2001; Yang et al, 1993). Defective smooth muscle layer formation was attributed to diminished levels of platelet-derived growth factor receptor α (PDGFRA), which is essential for smooth muscle cell differentiation (Jacob et al, 1994). FOXF1 is not expressed in intrahepatic biliary duct mesenchyme in wild-type mice, and no defects were seen in the intrahepatic biliary ducts of FOXF1+/− mice; however, FOXF1 mRNA levels in the liver were increased, suggesting that it may be a compensatory mechanism to prevent defects in the liver (Kalinichenko et al, 2002). All these data suggest that FOXF1 is crucial for the development of the extrahepatic bile ducts and gallbladder, with the mesenchyme playing an essential role in biliary epithelial cell differentiation. The interaction between the biliary epithelial cells and the extracellular matrix is also thought to contribute to biliary epithelial cell differentiation.
Integrins are membrane receptors for extracellular matrix proteins that play an important role in mediating the interaction between differentiating cells and the extracellular matrix (Hynes, 1992; Couvelard et al, 1998). Hepatoblasts express integrin heterodimers containing the β1 subunit (α1β1, α5β1, α6β1, and α9β1), and when hepatoblasts differentiate into immature intrahepatic biliary epithelial cells when in contact with the mesenchyme, the morphology of the integrins changes. The primitive intrahepatic biliary epithelial cells upregulate α6β1 expression and lose α1β1, while acquiring several other integrin dimers not previously expressed on hepatoblasts, including α2β1, α3β1, α5β1 and α6β4 (Couvelard et al, 1998). This change is likely due to the different pericellular environment. Intrahepatic biliary epithelial cells are contacted by a basement membrane containing collagen, entactin, and laminin (Desmet, 1985); however, hepatocytes are surrounded by the perisinusoidal matrix, which is devoid of laminin and entactin (Schuppan, 1990). The increase in α6β1 expression—along with the acquisition of biliary-specific expression of α2β1, α3β1, and α6β4, which are integrin receptors for laminin—correlated with the deposition of laminin at the contact points of the portal mesenchyme with the ductal plate (Couvelard et al, 1998).
Remodeling of the Ductal Plate
As mentioned earlier, the double-layered ductal plate around the portal vein begins to form focal dilations incorporated into the portal mesenchyme to form intrahepatic biliary ducts, while the parts of the ductal bilayer plate not involved in the formation of the ducts progressively regresses. This mechanism of regression is thought to be carried out by apoptosis (Terada & Nakanuma, 1995; Sergi et al, 2000). Cell-matrix interactions are also thought to contribute to the remodeling process. Tenascin was found to be expressed in the mesenchyme around the biliary epithelial cells of primitive ducts migrating into the mesenchyme. In contrast, tenascin was absent in the mesenchyme of peripheral ducts (Terada & Nakanuma, 1994).
Tubulogenesis of ductal cells is thought to be controlled by soluble factors secreted from hepatocytes or biliary epithelial cells. When coculturing human biliary epithelial cells with hepatocytes, a marked ductular morphogenic response was induced, and the biliary epithelial cells formed well-organized luminal ducts. This result was reproduced when biliary epithelial cells were grown in a conditioned medium from previous hepatocyte and biliary epithelial cell coculturing (Auth et al, 2001); however, it remains to be seen whether soluble factors do contribute to tubulogenesis in in vivo studies (Fig. 1A.5).
Developmental Relationship Between the Ducts, Vessels, and Mesenchyme of the Portal Tract
A functional relationship appears to exist between the contents of the portal tract (bile duct, hepatic artery, portal vein) based on observations of a number of human diseases termed ductal plate malformations. These diseases include biliary atresia, Caroli disease, and Meckel and Alagille syndromes, in which abnormal biliary ducts are associated with anomalies of the portal mesenchyme and of the portal blood vessels (Lemaigre, 2003). This association was also demonstrated in studies on Notch pathway defects. In Alagille syndrome, an autosomal dominant disease, bile ducts are absent in the portal tract associated with an increased number of arteries and fibrosis. Haploinsufficiency of JAG1, a Notch receptor ligand, is associated with Alagille syndrome, where JAG1 is persistently expressed in the ductal epithelium in humans (Li et al, 1997; Louis et al, 1999). It is also expressed in the endothelial cells of the developing portal vasculature (Crosnier et al, 2000). The animal model for Alagille syndrome was replicated in double-heterozygous mice for mutations in the JAG1 and NOTCH2 genes (J1N2+/−). JAG1 protein was expressed in the hepatic vasculature, and NOTCH2 was expressed in a subset of hepatoblasts surrounding the portal vein, hepatic artery, and bile ducts. Interestingly, although neither JAG1 nor NOTCH2 protein was expressed in the bile duct epithelium of these mice, JAG1 is expressed in bile duct epithelium in humans (Louis et al, 1999; McCright et al, 2002). The differences in JAG1 expression between human and mouse most likely reflects species specificity rather than technical artifacts, as mice that are heterozygous or homozygous for the JAG1 gene did not show the liver symptoms of Alagille syndrome (Xue et al, 1999). Similar biliary abnormalities were observed in NOTCH2-null mutant mice (McCright et al, 2002).
Hepatocyte Differentiation
Other factors that have been shown to promote hepatocyte differentiation include oncostatin M (OSM), an interleukin-6 family cytokine, Hgf, and Wnt (Michalopoulos et al, 2003; Tan et al, 2008). Although as discussed earlier in the chapter, the repression of Wnt signaling in the foregut endoderm is necessary for hepatic specification, its role is reversed at later gestational stages to promoting hepatocyte differentiation. β-Catenin, a central component of the canonical Wnt pathway, is essential for normal development; its aberrant activation in liver was associated with tumors, including hepatic adenomas and hepatocellular cancers (Peifer & Polakis, 2000; de La Coste et al, 1998; Zucman-Rossi et al, 2006). Livers that were deficient in β-catenin displayed decreased numbers of hepatocytes with hepatoblasts that lacked maturation, proliferation, and function (Tan et al, 2008). In addition, CEBPα, a fundamental regulator of hepatocyte differation and maturation (Tan et al, 2008), was decreased. Also noted in these livers was a complete absence of CK-19-positive intrahepatic biliary ducts, suggesting that β-catenin may play a role in biliary differentiation (Tan et al, 2008). In vitro studies showed that OSM, produced by hematopoietic cells in midfetal liver, and Hgf induce hepatic differentiation in the presence of dexamethasone, both working through different pathways (Kamiya et al, 2001). It was demonstrated that embryonic livers at E14.5 expressed G6Pase, tyrosine amino transferase (TAT), and accumulated glycogen, all signs of a mature and differentiated liver when cultured with Hgf or Osm. This finding was further supported when TAT levels, as well as glycogen storage, was significantly reduced in livers derived from mice null mutant for gp130, the common receptor subunit of the IL-6 family cytokines (Kamiya et al, 2001). CEBPα is also critical for the acquisition and maintenance of hepatocyte differention, with knockout mice exhibiting defects in liver growth and architecture as well as an increase in cell proliferation (Flodby et al, 1996). CEBPα is also thought to be a key factor in controlling the switch in the differentiation of bipotential hepatoblasts to become either biliary epithelial cells or hepatocytes. CEBPα starts to be expressed in the endodermal liver primordium at E9.5, and its expression in the nuclei of hepatoblasts and hepatocytes becomes stronger with development. During biliary cell differentiation, CEBPα expression was suppressed in periportal biliary cell progenitors, suggesting that its suppression may be a prerequisite to biliary cell differentiation from hepatoblasts (Shiojiri et al, 2004). Lineage tracing experiments have recently shown that Sox9+ biliary epithelial cells themselves differentiate into mature hepatocytes under normal physiologic conditions (Furuyama et al, 2011). Hepatocyte differentiation from the lineage tagged Sox9+ bile duct cells increased during the hepatic regenerative process, suggesting that the biliary and pancreatic ductal tree (Sox9-expressing domains) contains a previously unappreciated pool of progenitor cells.
Overview of Pancreatic Development
The endocrine pancreas is organized into islets of Langerhans, which consist of five cell subtypes—α, β, δ, ε, and PP cells—that secrete glucagon, insulin, somatostatin, ghrelin, and pancreatic polypeptide, respectively. All totaled these cells occupy only 2% of the adult pancreatic mass (Gittes, 2009). The exocrine pancreas, on the other hand, produces digestive enzymes, is composed of acinar and ductal epithelial cells, and accounts for nearly 98% of the adult pancreatic mass.
Basic Pancreatic Embryology
During early development, the pancreas is formed by the dorsal and ventral bud, each originating from the endodermal lining of the caudal part of the primitive foregut tube after regional specification with transcription factor Pdx1 followed by Ptf1a (Sadler & Langman, 2006; Grapin-Botton, 2005; Moore-Scott et al, 2007). The first morphologic evidence of pancreas development is in the form of mesenchymal condensation overlying the dorsal aspect of the primitive foregut tube at the level of the duodenal anlagen, distal to the stomach. Shortly after, at around E9.5 gestation in mice and day 26 of gestation in humans, the dorsal bud begins to evaginate into the overlying mesenchyme (Kallman & Grobstein, 1964; Picte et al, 1972). Approximately 12 hours later in mice, and 6 days after dorsal bud evagination in humans, the ventral bud begins to arise.
The morphologic development of the ventral pancreatic bud is similar to the dorsal bud; however, it is markedly different at the molecular level. After this initial bud evagination, the pancreatic bud then undergoes elongation of the stalk region, a precursor to the main pancreatic duct and branching morphogenesis of the apical region of the bud. Unlike growth patterns seen in the developing lung, kidney, and salivary glands, in which the branching morphogenesis undergoes a typical 90-degree pattern, the pancreas grows in an acute-angled branching pattern, which can lead to the exclusion, or “squeezing out,” of mesenchyme from between the closely apposed branches of epithelium. This exclusion of mesenchyme may influence epithelial–mesenchymal interactions and lineage selection (Fig. 1A.6). The ventral pancreatic bud, which arises from the base of the hepatic diverticulum, rotates dorsally and eventually fuses with the dorsal pancreatic bud (6 to 7 weeks gestation in humans, E12 to E13 in mice), contributing to the formation of the uncinate process and inferior part of the head of the pancreas, with the rest of the pancreas arising from the dorsal pancreatic bud. The entire ventral pancreatic duct and the distal part of the dorsal pancreatic duct fuse together to form the main pancreatic duct of Wirsung. The remaining proximal part of the dorsal pancreatic duct is either obliterated or persists as a small accessory pancreatic duct, the duct of Santorini (Sadler & Langman, 2006).
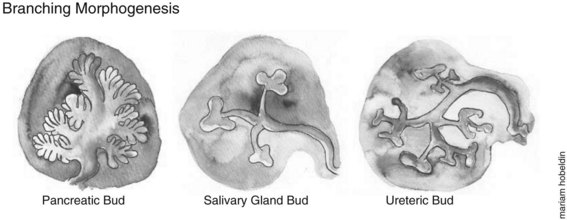
During development the pancreas undergoes a major amplification of the endocrine cell population through two waves of differentiation: an early primary transition (pre-E13.5 in mice) followed by the secondary transition (E13.5 to E16.5 in mice) (Picte et al, 1972; Jensen, 2004). In a similar gestational window, the exocrine pancreatic precursors undergo rapid branching morphogenesis and acinar cell differentiation (see Fig. 1A.6).
Endodermal Patterning of the Pancreas
As mentioned earlier, the pancreas develops through epithelial–mesenchymal interactions. Prior to this, the dorsal foregut became competent to receive propancreatic signals from the notochord (FGFs) (Wells & Melton, 2000) and from the anterior foregut (retinoic acids and BMPs) (Stafford & Prince, 2002; Tiso et al, 2002). The prepancreatic region of the definitive foregut comes into contact with the notochord, whereas the ventral pancreas contacts the lateral plate mesoderm (Kumar et al, 2003).
Dorsal and Ventral Pancreatic Bud Development
The notochord is key to the development of the dorsal pancreas, where it remains in contact with the dorsal prepancreatic endoderm until the paired dorsal aortae fuse in the midline (∼E8.0). The dorsal pancreas failed to form when the notochord was removed from early chick embryos in vitro (Kim et al, 1997), but no effect was seen on ventral pancreas development, further emphasizing the different molecular controls for dorsal versus ventral pancreas development. The ventral pancreas develops under the control of signals from the overlying cardiogenic mesenchyme, which also produces prohepatic signals (FGFs) to induce liver formation. Lack of prohepatic FGF signaling in regions of the cardiogenic mesenchyme will lead to the endoderm by “default” differentiating into ventral pancreas (Deutsch et al, 2001). Sonic Hedgehog (SHH) in the prospective pancreatic endoderm is suppressed by the notochord, which is necessary for dorsal pancreatic development, and SHH is otherwise expressed along the entire primitive gut tube epithelium. When the notochord was deleted in chick embryo cultures, ectopic SHH was seen in the pancreatic region of the foregut endoderm, and the pancreas failed to develop (Hebrok et al, 1998).
Pancreatic Mesenchyme
The pancreatic epithelium becomes enveloped by mesenchyme, which contains important factors that are pivotal for pancreatic morphogenesis. The mesenchyme has a general proexocrine effect on the pancreas through cell contact and through proendocrine effect, mediated by diffusible factors secreted from the mesenchyme (Li et al, 2004). Mesenchymal contact with the epithelium both enhances notch signaling (HES1), which favors the acinar lineage and also inhibits neurogenin 3 expression (NEUROG3), leading to the suppression of endocrine differentiation (Duvillie et al, 2006). The “default” differentiation of the pancreatic epithelium in the absence of mesenchyme is endocrine (Gittes et al, 1996). Thus the mesenchyme has both an inductive effect on the exocrine tissue development and a repressive effect on the development of endocrine cells.
The interaction between the mesenchyme and the pancreatic epithelium is regulated by several factors, the most important of which appears to be the FGFs, which are expressed in several epithelial–mesenchymal interface regions of the pancreas and are particularly important in regulating branching morphogenesis (Hogan, 1999). FGFs 1, 7, and 10 mediate their effect through FGF receptor 2B (FGFR2B) to induce epithelial proliferation, favoring exocrine differentiation (Elghazi et al, 2002; Celli et al, 1998; Dichmann et al, 2003). Null mutation for FGF10 or FGFR2B leads to blunting of branching pancreatic morphogenesis with a lack of proliferation of endocrine progenitor cells, indicating that FGF10 induces proliferation of epithelial cells and prevents endocrine differentation (Bhushan et al, 2001; Pulkkinen et al, 2003). These roles for FGFs are primarily for ongoing development of the buds, in contrast to the role of FGF from the cardiogenic mesenchyme, which inhibits ventral pancreatic bud formation in favor of liver development (Deutsch et al, 2001).
TGF-β Signaling
The TGF-β superfamily has been implicated in early pancreatic development. TGF-β isoforms (TGF-β1, -β2, and -β3) are localized to the pancreatic embryonic epithelium as early as E12.5 and progressively become focused to the acinar cells (Crisera et al, 1999). The ligands mediate their actions through TGF-β receptor type II (TBR-II), a heterodimer receptor; however, in late gestation it localizes to the pancreatic ducts (Tulachan et al, 2007). TBR-II inhibition has been shown to recruit an inappropriately high number of ductal cells to become endocrine progenitor cells, suggesting that TBR-II signaling is important for regulating the flow of endocrine progenitors out of ducts (Sanvito et al, 1995; Lee et al, 1995).
Activins, part of the TGF-β superfamily, also play a role in pancreatic development. When exogenous activin was added to embryonic pancreas explant cultures, it inhibited branching morphogenesis, a process associated with ductal and acinar differentiation (Ritvos et al, 1995). This observation was further supported when follistatin, an activin inhibitor expressed in the pancreatic mesenchyme by E12.5, was added to the pancreatic epithelium; it mirrored the mesenchymal effect, promoting exocrine and repressing endocrine development (Miralles et al, 1998). These observations would support a role for activin in promoting and regulating endocrine differentiation and suppressing exocrine differentiation in the early pancreas. Along similar lines, transgenic mice that express a dominant-negative activin receptor display reduced levels of differentiated islet cells (Kim et al, 2000; Shiozaki et al, 1999; Yamaoka et al, 1998). In vitro in cell lines, activin A favors β-cell differentiation.
SMADs are intracellular signals for the TGF superfamily and have gained interest among developmental biologists, since SMAD4 was specifically identified to be mutated in 50% of pancreatic cancers (Hahn et al, 1996). However, a key role for SMAD4 has not yet been identified in pancreatic development (Simeone et al, 2006; Bardeesy et al, 2006). SMAD7 on the other hand, which acts as a general TGF signaling inhibitor when expressed under the PDX1 promoter, results in a dramatic decrease in β cells at birth—by around 90%—and replacement with glucagon cells (Smart et al, 2006).
Notch Signaling
A key step in pancreatic development is the lineage decision by pancreatic progenitor cells between the endocrine and exocrine lineage. Notch signaling has been identified as a master regulator of this fate decision switch (Apelqvist et al, 1999). However, the exact mechanism for Notch signaling in pancreatic lineage selection remains elusive, and ambiguity still surrounds the exact role of Notch signaling through pancreatic development. It appears that the downstream factors of Notch targets are variably expressed at certain times in development of the pancreas. The early pattern of downstream molecules is a key mediator of Notch signaling in determining endocrine versus exocrine differentiation of progenitor cells and in maintaining a stem cell state in some progenitor cells. Later in gestation, the downstream molecules for Notch signaling change to foster cell maturation and differentiation beyond this initial lineage decision between endocrine and exocrine differentiation.
Hedgehog Signaling
As mentioned earlier, Shh suppression in the prospective pancreatic endoderm is a prerequisite for pancreas formation, however, Hedgehog signaling in pancreatic development appears to be complex. SHH-null mutant mice do not exhibit an expanded pancreatic field; in contrast, Indian Hedgehog (IHH) null mutant mice that develop with a small pancreas indicate a propancreatic role for IHH (Hebrok et al, 2000). However, when combining SHH-null mutation with heterozygous mutation for IHH, an annular pancreas develops. Mice that are null mutant for the Hedgehog Patched receptor Ptc lack Pdx1 and glucagon expression at E9.5, supporting the need to suppress Shh for early pancreatic development (Hebrok et al, 2000). Similarly, transgenic overexpression of Shh or Ihh under the Pax4 promoter led to a significant decrease in endocrine and exocrine tissue mass (Smith et al, 2000).
WNT
As mentioned earlier in the chapter, β-catenin repression in the anterior endoderm is necessary to initiate liver and pancreas development. Consistent with this, PDX1–WNT1 and PDX1–WNT5A transgenic mice developed pancreatic agenesis and severe hypoplasia, respectively, confirming that early Wnt signaling is detrimental for pancreatic development (Heller et al, 2002). Similar to Notch signaling, the role of Wnt signaling beyond endodermal patterning is complex and depends on the time and place of Wnt signaling. β-catenin, which is mostly expressed in the mesenchyme (Heller et al, 2002), when ablated from Pdx1-positive cells resulted in the loss of acinar tissue at birth, confirming earlier studies that exocrine tissue development is mesenchyme dependent (Murtaugh et al, 2005; Wells et al, 2007). Others found a role for Wnt signaling in promoting postnatal pancreatic growth (Heiser et al, 2006), illustrating the complex and multiple roles that Wnts play in pancreatic development.
Endothelial Cells
Recently, an important role for endothelial cells in pancreatic endocrine development has been demonstrated. As mentioned earlier, the notochord separates from the gut endoderm by the fusion of the dorsal aortas around E8.0. Lammert and colleagues (2001) demonstrated that the removal of the dorsal aorta from Xenopus embryos led to the absence of pancreatic endocrine development. In addition, aortic endothelial cells were able to induce pancreatic bud formation and subsequent insulin cell differentiation in the adjacent endoderm. Importantly, ectopic VEGFA in the intestine and stomach led to ectopic formation of islets in the stomach (Lammert et al, 2001). Interestingly, it appears that ventral pancreas development seems to be not dependent on the endothelium despite its close proximity to the vitelline veins (Yoshitomi & Zaret, 2004).
Glucagon
The first detectable endocrine cells during pancreatic development are the glucagon-containing cells at around E9 in the mouse, and recent studies have shown that glucagon signaling is necessary for early differentiation of insulin-expressing cells, which appear around E13.0 (Picte et al, 1972; Prasadan et al, 2002). Glucagon hormone is generated from proglucagon by the action of prohormone convertase 2 (PC2). When either PC2 or the glucagon receptor is knocked out, mutant mice are generated without glucagon, displaying a delay in islet cell differentiation and maturation but still retaining the large amplification of insulin-positive cells (“secondary wave”) later in gestation (Vincent et al, 2003). Exogenous addition of exendin-4, a glucagon-like peptide-1 (GLP-1) analog, was able to rescue the delay in early insulin differentiation (Prasadan et al, 2002). Exendin-4 was also shown also to convert AR42J cells (Zhou et al, 1999; Yew et al, 2004) and ARIP cells (Hui et al, 2001) into insulin-expressing cells. Together these studies strongly support the role of glucagon signaling, through its receptor, in initiating early insulin differentiation.
Extracellular Matrix
The pancreatic epithelium is contained within a continuous sheath of basement membrane, which constitutes the epithelial-mesenchymal interface and plays an important role in guiding pancreatic development (Hisaoka et al, 1993). Basement membrane has also been shown to play an important role in regulating branching morphogenesis in many other organs. Laminin-1 was found to induce duct formation in isolated E11 mouse pancreatic epithelium (Gittes et al, 1996) and to mediate the proexocrine inductive effect of the mesenchyme and the pro–β-cell role later in gestation (Li et al, 2004; Jiang et al, 2001). Cadherins—calcium-dependent adhesion molecules, critical for cell–cell adhesions—play an essential role in migration and differentiation of pancreatic endocrine progenitor cells. E-cadherin and R-cadherin are initially localized to the ducts, but they become downregulated as cells move out of the ducts to form islets (Dahl et al, 1996; Sjodin et al, 1995). N-CAM, another cell-adhesion molecule expressed in mature α and PP cells (Cirulli et al, 1994), when knocked out led to improper aggregation of endocrine cells within the islet (Esni et al, 1999). Thus, cell-adhesion molecules regulate the adhesive properties of endocrine cells, and these adhesive properties are essential for endocrine cell aggregation into islets.
Transcription Factors
The role of transcription factors has been extensively studied in pancreatic development. The most heavily studied is Pdx1, an early marker for pancreatic progenitors that later restricts to the β cells. It is expressed in the prepancreatic region of the primitive foregut tube at E8.5 then expands to be expressed in distal stomach, common bile duct, and duodenum by E10.5 to 11.5 (Offield et al 1996; Guz et al, 1995). Pdx1 is initially expressed throughout the epithelium; however, its expression becomes suppressed in cells as they commit to the endocrine (Jensen et al, 2000) lineage or ducts (Gu et al, 2002). Its expression reappears as cells differentiate to the insulin-positive β-cell lineage. PDX1-null mutant mice and humans developed pancreatic agenesis (Jonsson et al, 1994; Yee et al, 2001; Stoffers et al, 1997a; Stoffers et al, 1997b). Delayed Pdx1 inhibition using a tetracycline-regulatable transgenic knock-in system demonstrated severe blunting of pancreatic development with complete absence of acini and β cells (Holland et al, 2002), indicating that Pdx1 continues to play a role in pancreas development beyond regional specification during endodermal patterning.
Ptf1α
Pancreas-specific transcription factor 1α (Ptf1α) is an early marker of pancreatic progenitor cells, expressed slightly later than Pdx1 at around E9.5; by E18.5, it becomes progressively restricted to acinar cells (Cras-Meneur et al, 2009). PTF1A-null mutant mice develop a phenotype similar to that seen with PDX1-null mutants, with severe pancreatic hypoplasia and absent acini and ducts; however, endocrine cells still develop; interestingly, the endocrine cells migrate out through the pancreatic mesenchyme to populate the spleen (Kawaguchi et al, 2002; Krapp et al, 1998). A PTF1A nonfunctioning mutation has been seen in humans born without a pancreas (Sellick et al, 2004). Furthermore, PTF1A has complex interactions with downstream targets of Notch to control exocrine lineage selection.
NEUROG3
NEUROG3 is one of the earliest markers specific to the pancreatic endocrine lineage, and it is a key driver of endocrine differentiation. It is first expressed at E9.0, and it peaks at around E15.5; it is thought to be antagonized by Notch signaling in cells with an acinar fate (Jensen et al, 2000; Gradwohl et al, 2000). NEUROG3 cells then proliferate, giving rise to postmitotic cells expressing transcription factors NeuroD, Nkx6.1, and Pax6. NEUROG3 shuts off at around E17.5 (Jensen et al, 2000; Gu et al, 2002), and forced overexpression of NEUROG3 leads to cells prematurely committing to an endocrine lineage, with the development of only glucagon-positive clusters (Johansson et al, 2007). Overexpressing NEUROG3 at different times later in gestation (E11 to E12) resulted in the formation of insulin-positive and pancreatic polypeptide–positive cells (Johansson et al, 2007). However, when NEUROG3 is deleted from cells, no pancreatic endocrine cells form (Gradwohl et al, 2000); thus it appears to be a critical and essential factor for pancreatic endocrine differentiation.
PAX6
PAX6 is also another marker of endocrine lineage, but unlike NGN3, it is not absolutely necessary for endocrine formation because mice null mutant for Pax6 still formed endocrine cells, albeit at a reduced rate (St-Onge et al, 1997). Pax6 expression is retained in cells committed to the endocrine lineage as early as E9.5 in glucagon-positive cells or in insulin-positive cells at E12.5, thereafter beginning hormone expression (St-Onge et al, 1997). Transgenic expression of PAX6 under the PDX1 promoter led to islet and ductal hyperplasia, indicating that PAX6 may have roles in the formation of ducts and in islet growth (Yamaoka et al, 2000). However, when PAX6 was ectopically expressed in β cells under the insulin promoter, it led to β-cell apoptosis, indicating that PAX6 promotes proliferation only in pancreatic progenitors, not in mature cells.
PAX4/ARX
PAX4 is another endocrine cell marker and is expressed as early as E9.5; it then peaks at around E13-15, coinciding with the secondary transition of insulin-positive cells (Wang et al, 2004). Its importance was further delineated when β cells and δ cells failed to develop in PAX4-null mutant mice. Early embryonic insulin-positive cells were present in these mice, indicating that PAX4 is only needed for the formation of mature β cells (Wang et al, 2004; Sosa-Pineda et al, 1997). In these mice, the β and δ cells seem to have been replaced by cells coexpressing glucagons and ghrelin, indicating that PAX4 may act as a transcriptional inhibitor for this expression (Wang et al, 2008). PAX4 seems also to inhibit the expression of ARX, a homeobox-containing gene that enhances glucagon-positive cell differentiation (Collombat et al, 2005). ARX, which is downstream of NEUROG3 and is absent in NEUROG3-null mutant mice, when overexpressed in PDX1-positive progenitor cells diverted most β- and δ-cell precursors instead toward α- and PP-cell lineages, without a change in the overall number of endocrine cells. When ARX was knocked out, no α cells were seen (Collombat et al, 2003). These findings further emphasize the opposing roles for PAX4 and ARX in α and PP cells versus β and δ cells (Collombat et al, 2007).
NKX2-2
NKX2-2 is expressed as early as E9.5, is coexpressed with PDX1 as a marker of multipotent pancreatic progenitor cells, and eventually becomes restricted to NGN3-positive cells, persisting in all endocrine lineages except for δ cells (Sussel et al 1998; Chiang & Melton, 2003). NKX2-2–null mutant mice develop with no β cells, reduced PP cells, an 80% reduction in α cells, and no effect on δ cells. However, a large number of ghrelin-positive ε cells were found with no glucagon coexpression, indicating that NKX2-2 normally induces insulin-positive differentiation and represses ε cell formation (Sussel et al, 1998).
NKX6-1 and NKX6-2
Single- and double-null mutant studies for NKX6-1 and NKX6-2 have been done and suggest that both molecules, which are expressed early in pancreatic endodermal cells, play central roles in pancreatic development, especially in β-cell development (Henseleit et al, 2005). NKX6-1, which is downstream of PDX1, becomes restricted to insulin-positive cells; whereas NKX6-2 is restricted to glucagon-positive and amylase-positive cells, after E15.5, it disappears (Sander et al, 2000). In NKX6-1–null mutant mice, an 85% reduction in β cells is seen, whereas NKX6-1 and NKX6-2 double-null mutants developed a 92% and 65% reduction in β cells and α cells, respectively, suggesting that NKX6-1 has an important role in generating β cells. NKX6-2 plays an important role in β-cell formation and can compensate for NKX6-1 absence in β-cell formation (Henseleit et al, 2005; Sander et al, 2000). β-cell loss was rescued with transgenic expression of NKX6-1 or NKX6-2 under the PDX1 promoter (Nelson et al, 2007).
MAFA and MAFB
The transcription factor MAFA is a critical regulator of the insulin gene, and it also regulates glucose-responsive expression of insulin (Matsuoka et al, 2003). MAFA is first expressed in insulin-positive cells during the “secondary transition” wave of insulin-cell differentiation; however, it is not absolutely necessary for β-cell formation, because MAFA-null mutant mice have a normal proportion of insulin-positive cells at birth (Nishimura et al, 2009). MAFB is expressed early in pancreatic endocrine development and, as the pancreas develops, it switches off in insulin-positive forming cells as they form mature β cells that then express MAFA (Nishimura et al, 2006). It was demonstrated that early expression of MAFA under the PDX1 promoter was detrimental to pancreatic development, with a severe reduction in pancreatic mass and in the proliferation of progenitors, suggesting that MAFA is a key regulator for the maturation of pancreatic β cells rather than β-cell specification (Nishimura et al, 2009). The same conclusion was drawn in MAFB-null mutants mice, in which a delay was seen in the development of early insulin- and glucagon-positive cells, with a reduction by almost half of their total mass (Artner et al, 2007).
HNF Cascade
Three HNF factors—HNF6/ONECUT1, HNF1B, and HNF3B (renamed FOXA2)—appear to have an interwoven, complex set of interactions that all impact on pancreatic development, starting from the earliest pancreatic specification all the way through to mature pancreatic cell differentiation and function. They are all expressed in the pancreatic epithelium at around E9.0 to E10.0 with HNF6 appearing to be a key determinant of pancreatic specification (Maestro et al, 2003). This conclusion comes from observations that HNF6−/− embryos developed ventral pancreatic agenesis, with an atrophic dorsal pancreas, along with severe biliary anomalies as described earlier (Jacquemin et al, 2003; Haumaitre et al, 2005). HNF6 is expressed in the foregut–midgut endoderm junction at E8.0 under the control of HNF1B (Poll et al, 2006). HNF6 in turn regulates the expression of FOXA2, which is essential for gut formation (Weinstein et al, 1994). FOXA2, however, is not necessary for pancreas formation, because its deletion did not affect pancreatic development (Lee et al, 2005). As discussed earlier in the bile duct formation section, HNF6 also acts upstream of HNF1B, where HNF6 activates HNF1B to initiate the formation of endocrine progenitors (Clotman et al, 2002).
SOX9
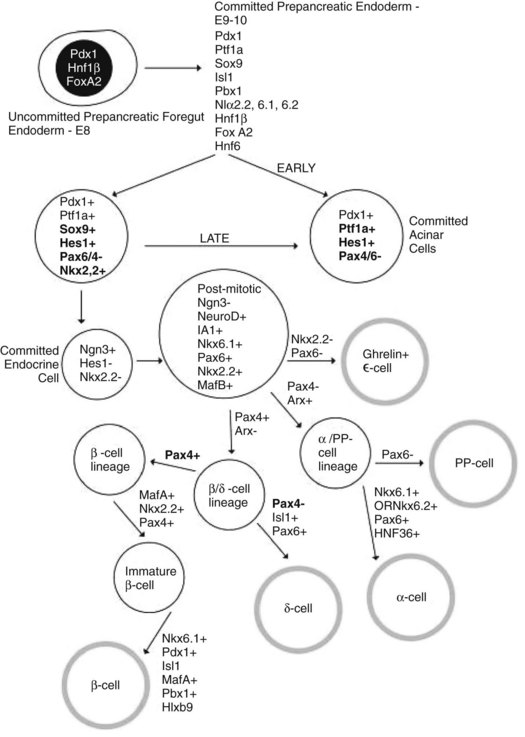
SOX9 acts as a marker for pancreatic progenitor cells that can give rise to all pancreatic cell types, and it is necessary for maintaining cells in a progenitor state (Akiyama et al, 2005). SOX9 mutants display pancreatic hypoplasia as a result of depletion of the pancreatic progenitor pool (Seymour et al, 2007). It has also been suggested that SOX9 mediates HNF6 and the control of NEUROG3-cell formation, because conditional deletion of SOX9 led to the formation of cystic structures similar to those seen in HNF6−/− embryos, associated with absent pancreatic endocrine cells (Seymour et al, 2007). Furthermore, it was found that SOX9 is transiently coexpressed with NEUROG3 and frequently colocalizes with HES1, which acts as a precursor to NEUROG3-positive cells. It was suggested that SOX9 helps mediate the transition of endocrine progenitors from the nonendocrine HES1-positive, NEUROG3-negative state to an HES1-negative, NEUROG3-positive endocrine-committed state (Seymour et al, 2007). Thus, SOX9 acts as a key mediator of the commitment of NEUROG3-positive endocrine progenitors (Fig. 1A.7).
Akiyama H, et al. Osteo-chondroprogenitor cells are derived from Sox9-expressing precursors. Proc Natl Acad Sci U S A. 2005;102(41):14665-14670.
Apelqvist A, et al. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400(6747):877-881.
Artner I, et al. MafB is required for islet beta cell maturation. Proc Natl Acad Sci U S A. 2007;104(10):3853-3858.
Auth MK, et al. Morphogenesis of primary human biliary epithelial cells: induction in high-density culture or by coculture with autologous human hepatocytes. Hepatology. 2001;33(3):519-529.
Bardeesy N, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006;20(22):3130-3146.
Bhushan A, et al. Fgf10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Development. 2001;128(24):5109-5117.
Bort R, et al. Hex homeobox gene-dependent tissue positioning is required for organogenesis of the ventral pancreas. Development. 2004;131(4):797-806.
Bossard P, Zaret KS. GATA transcription factors as potentiators of gut endoderm differentiation. Development. 1998;125(24):4909-4917.
Celli G, et al. Soluble dominant-negative receptor uncovers essential roles for fibroblast growth factors in multi-organ induction and patterning. Embo J. 1998;17(6):1642-1655.
Chen Y, et al. Retinoic acid signaling is essential for pancreas development and promotes endocrine at the expense of exocrine cell differentiation in Xenopus. Dev Biol. 2004;271(1):144-160.
Chiang MK, Melton DA. Single-cell transcript analysis of pancreas development. Dev Cell. 2003;4(3):383-393.
Cirillo LA, et al. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell. 2002;9(2):279-289.
Cirulli V, et al. Expression of neural cell adhesion molecule (N-CAM) in rat islets and its role in islet cell type segregation. J Cell Sci. 1994;107(Pt 6):1429-1436.
Clotman F, et al. The onecut transcription factor HNF6 is required for normal development of the biliary tract. Development. 2002;129(8):1819-1828.
Collombat P, et al. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003;17(20):2591-2603.
Collombat P, et al. The simultaneous loss of Arx and Pax4 genes promotes a somatostatin-producing cell fate specification at the expense of the alpha- and beta-cell lineages in the mouse endocrine pancreas. Development. 2005;132(13):2969-2980.
Collombat P, et al. Embryonic endocrine pancreas and mature beta cells acquire alpha and PP cell phenotypes upon Arx misexpression. J Clin Invest. 2007;117(4):961-970.
Costa RM, et al. Novel gene expression domains reveal early patterning of the Xenopus endoderm. Gene Expr Patterns. 2003;3(4):509-519.
Couvelard A, et al. Expression of integrins during liver organogenesis in humans. Hepatology. 1998;27(3):839-847.
Cras-Meneur C, et al. Presenilins, Notch dose control the fate of pancreatic endocrine progenitors during a narrow developmental window. Genes Dev. 2009;23(17):2088-2101.
Crisera CA, et al. The ontogeny of TGF-beta1, -beta2, -beta3, and TGF-beta receptor-II expression in the pancreas: implications for regulation of growth and differentiation. J Pediatr Surg. 1999;34(5):689-693. discussion 693-684
Crompton MR, et al. Identification of a novel vertebrate homeobox gene expressed in haematopoietic cells. Nucleic Acids Res. 1992;20(21):5661-5667.
Crosnier C, et al. JAGGED1 gene expression during human embryogenesis elucidates the wide phenotypic spectrum of Alagille syndrome. Hepatology. 2000;32(3):574-581.
Crowe AJ, et al. Hepatocyte nuclear factor 3 relieves chromatin-mediated repression of the alpha-fetoprotein gene. J Biol Chem. 1999;274(35):25113-25120.
Dahl U, Sjodin A, Semb H. Cadherins regulate aggregation of pancreatic beta cells in vivo. Development. 1996;122(9):2895-2902.
de La Coste A, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95(15):8847-8851.
Desmet VJ. Intrahepatic bile ducts under the lens. J Hepatol. 1985;1(5):545-559.
Deutsch G, et al. A bipotential precursor population for pancreas and liver within the embryonic endoderm. Development. 2001;128(6):871-881.
Dichmann DS, et al. Expression and misexpression of members of the FGF and TGFbeta families of growth factors in the developing mouse pancreas. Dev Dyn. 2003;226(4):663-674.
Dolle P, et al. The Hox-4.8 gene is localized at the 5′ extremity of the Hox-4 complex and is expressed in the most posterior parts of the body during development. Mech Dev. 1991;36(1-2):3-13.
Duncan SA. Transcriptional regulation of liver development. Dev Dyn. 2000;219(2):131-142.
Duvillie B, et al. The mesenchyme controls the timing of pancreatic beta-cell differentiation. Diabetes. 2006;55(3):582-589.
Elghazi L, et al. Role for FGFR2IIIb-mediated signals in controlling pancreatic endocrine progenitor cell proliferation. Proc Natl Acad Sci U S A. 2002;99(6):3884-3889.
Esni F, et al. Neural cell adhesion molecule (N-CAM) is required for cell type segregation and normal ultrastructure in pancreatic islets. J Cell Biol. 1999;144(2):325-337.
Fassler R, Meyer M. Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev. 1995;9(15):1896-1908.
Flodby P, et al. Increased hepatic cell proliferation and lung abnormalities in mice deficient in CCAAT/enhancer binding protein alpha. J Biol Chem. 1996;271(40):24753-24760.
Furuyama, et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas, and intestine. Nat Genet. 2011;43:34-41.
Gittes GK, et al. Lineage-specific morphogenesis in the developing pancreas: role of mesenchymal factors. Development. 1996;122(2):439-447.
Gittes GK. Developmental biology of the pancreas: a comprehensive review. Dev Biol. 2009;326(1):4-35.
Goss AM, et al. Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev Cell. 2009;17(2):290-298.
Gradwohl G, et al. Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97(4):1607-1611.
Grapin-Botton A. Antero-posterior patterning of the vertebrate digestive tract: 40 years after Nicole Le Douarin’s PhD thesis. Int J Dev Biol. 2005;49(2-3):335-347.
Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447-2457.
Guadi R, et al. Hepatic specification of the gut endoderm in vitro: cell signaling and transcriptional control. Genes Dev. 1996;10(13):1670-1682.
Guz Y, et al. Expression of murine STF-1, a putative insulin gene transcription factor, in beta cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development. 1995;121(1):11-18.
Hahn SA, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271(5247):350-353.
Haumaitre C, et al. Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. Proc Natl Acad Sci U S A. 2005;102(5):1490-1495.
Hebrok M, Kim SK, Melton DA. Notochord repression of endodermal Sonic hedgehog permits pancreas development. Genes Dev. 1998;12(11):1705-1713.
Hebrok M, et al. Regulation of pancreas development by hedgehog signaling. Development. 2000;127(22):4905-4913.
Heiser PW, et al. Stabilization of beta-catenin impacts pancreas growth. Development. 2006;133(10):2023-2032.
Heller RS, et al. Expression patterns of Wnts, Frizzleds, sFRPs, and misexpression in transgenic mice suggesting a role for Wnts in pancreas and foregut pattern formation. Dev Dyn. 2002;225(3):260-270.
Henseleit KD, et al. NKX6 transcription factor activity is required for alpha- and beta-cell development in the pancreas. Development. 2005;132(13):3139-3149.
Hentsch B, et al. Hlx homeo box gene is essential for an inductive tissue interaction that drives expansion of embryonic liver and gut. Genes Dev. 1996;10(1):70-79.
Hisaoka M, Haratake J, Hashimoto H. Pancreatic morphogenesis and extracellular matrix organization during rat development. Differentiation. 1993;53(3):163-172.
Hogan BL. Morphogenesis. Cell. 1999;96(2):225-233.
Holland AM, et al. Experimental control of pancreatic development and maintenance. Proc Natl Acad Sci U S A. 2002;99(19):12236-12241.
Huang D, et al. A conserved retinoic acid responsive element in the murine Hoxb-1 gene is required for expression in the developing gut. Development. 1998;125(16):3235-3246. 1998
Hui H, Wright C, Perfetti R. Glucagon-like peptide 1 induces differentiation of islet duodenal homeobox-1–positive pancreatic ductal cells into insulin-secreting cells. Diabetes. 2001;50(4):785-796.
Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69(1):11-25.
Jacob A, et al. Retinoic acid–mediated activation of HNF-3 alpha during EC stem cell differentiation. Nucleic Acids Res. 1994;22(11):2126-2133.
Jacquemin P, Lemaigre FP, Rousseau GG. The Onecut transcription factor HNF-6 (OC-1) is required for timely specification of the pancreas and acts upstream of Pdx-1 in the specification cascade. Dev Biol. 2003;258(1):105-116.
Jensen J, et al. Independent development of pancreatic alpha and beta cells from neurogenin3-expressing precursors: a role for the notch pathway in repression of premature differentiation. Diabetes. 2000;49(2):163-176.
Jensen J. Gene regulatory factors in pancreatic development. Dev Dyn. 2004;229(1):176-200.
Jiang FX, Georges-Labouesse E, Harrison LC. Regulation of laminin 1–induced pancreatic beta-cell differentiation by alpha6 integrin and alpha-dystroglycan. Mol Med. 2001;7(2):107-114.
Johansson KA, et al. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev Cell. 2007;12(3):457-465.
Jonsson J, et al. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371(6498):606-609.
Jung J, et al. Initiation of mammalian liver development from endoderm by fibroblast growth factors. Science. 1999;284(5422):1998-2003.
Kaestner KH, Knochel W, Martinez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000;14(2):142-146.
Kagami S, et al. Transforming growth factor-beta (TGF-beta) stimulates the expression of beta1 integrins and adhesion by rat mesangial cells. Exp Cell Res. 1996;229(1):1-6.
Kalinichenko VV, et al. Haploinsufficiency of the mouse Forkhead Box f1 gene causes defects in gallbladder development. J Biol Chem. 2002;277(14):12369-12374.
Kallman F, Grobstein C. Fine structure of differentiating mouse pancreatic exocrine cells in transfilter culture. J Cell Biol. 1964;20:399-413.
Kamiya A, Kinoshita T, Miyajima A. Oncostatin M and hepatocyte growth factor induce hepatic maturation via distinct signaling pathways. FEBS Lett. 2001;492(1-2):90-94.
Kawaguchi Y, et al. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32(1):128-134.
Kawakami-Kimura N, et al. Involvement of hepatocyte growth factor in increased integrin expression on HepG2 cells triggered by adhesion to endothelial cells. Br J Cancer. 1997;75(1):47-53.
Keng VW, et al. Homeobox gene Hex is essential for onset of mouse embryonic liver development and differentiation of the monocyte lineage. Biochem Biophys Res Commun. 2000;276(3):1155-1161.
Kim SK, Hebrok M, Melton DA. Notochord to endoderm signaling is required for pancreas development. Development. 1997;124(21):4243-4252.
Kim SK, et al. Activin receptor patterning of foregut organogenesis. Genes Dev. 2000;14(15):1866-1871.
Kondo T, et al. Function of posterior HoxD genes in the morphogenesis of the anal sphincter. Development. 1996;122(9):2651-2659.
Krapp A, et al. The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev. 1998;12(23):3752-3763.
Kumar M, et al. Signals from lateral plate mesoderm instruct endoderm toward a pancreatic fate. Dev Biol. 2003;259(1):109-122.
Lammert E, Cleaver O, Melton D. Induction of pancreatic differentiation by signals from blood vessels. Science. 2001;294(5542):564-567.
Landry C, et al. HNF-6 is expressed in endoderm derivatives and nervous system of the mouse embryo and participates to the cross-regulatory network of liver-enriched transcription factors. Dev Biol. 1997;192(2):247-257.
Lee CS, et al. The initiation of liver development is dependent on Foxa transcription factors. Nature. 2005;435(7044):944-947.
Lee CS, et al. Foxa2 is required for the differentiation of pancreatic alpha-cells. Dev Biol. 2005;278(2):484-495.
Lee MS, et al. Accumulation of extracellular matrix and developmental dysregulation in the pancreas by transgenic production of transforming growth factor-beta 1. Am J Pathol. 1995;147(1):42-52.
Lemaigre FP. Development of the biliary tract. Mech Dev. 2003;120(1):81-87.
Li L, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16(3):243-251.
Li Z, et al. Multifaceted pancreatic mesenchymal control of epithelial lineage selection. Dev Biol. 2004;269(1):252-263.
Louis AA, et al. Hepatic jagged1 expression studies. Hepatology. 1999;30(5):1269-1275.
Maestro MA, et al. Hnf6 and Tcf2 (MODY5) are linked in a gene network operating in a precursor cell domain of the embryonic pancreas. Hum Mol Genet. 2003;12(24):3307-3314.
Mahlapuu M, et al. The forkhead transcription factor Foxf1 is required for differentiation of extra embryonic and lateral plate mesoderm. Development. 2001;128(2):155-166.
Margagliotti S, et al. The Onecut transcription factors HNF-6/OC-1 and OC-2 regulate early liver expansion by controlling hepatoblast migration. Dev Biol. 2007;311(2):579-589.
Margagliotti S, et al. Role of metalloproteinases at the onset of liver development. Dev Growth Differ. 2008;50(5):331-338.
Martin M, et al. Dorsal pancreas agenesis in retinoic acid-deficient Raldh2 mutant mice. Dev Biol. 2005;284(2):399-411.
Martinez-Barbera JP, et al. The homeobox gene Hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development. 2000;127(11):2433-2445.
Matsumoto K, et al. Liver organogenesis promoted by endothelial cells prior to vascular function. Science. 2001;294(5542):559-563.
Matsuoka TA, et al. Members of the large Maf transcription family regulate insulin gene transcription in islet beta cells. Mol Cell Biol. 2003;23(17):6049-6062.
McCright B, Lozier J, Gridley T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development. 2002;129(4):1075-1082.
McLin VA, Rankin SA, Zorn AM. Repression of Wnt/beta-catenin signaling in the anterior endoderm is essential for liver and pancreas development. Development. 2007;134(12):2207-2217.
Michalopoulos GK, et al. HGF-, EGF-, and dexamethasone-induced gene expression patterns during formation of tissue in hepatic organoid cultures. Gene Expr. 2003;11(2):55-75.
Miralles F, Czernichow P, Scharfmann R. Follistatin regulates the relative proportions of endocrine versus exocrine tissue during pancreatic development. Development. 1998;125(6):1017-1024.
Moore-Scott BA, et al. Identification of molecular markers that are expressed in discrete anterior posterior domains of the endoderm from the gastrula stage to mid-gestation. Dev Dyn. 2007;236(7):1997-2003.
Murtaugh LC, et al. Beta-catenin is essential for pancreatic acinar but not islet development. Development. 2005;132(21):4663-4674.
Nelson SB, Schaffer AE, Sander M. The transcription factors Nkx6.1 and Nkx6.2 possess equivalent activities in promoting beta-cell fate specification in Pdx1+ pancreatic progenitor cells. Development. 2007;134:2491-2500.
Nishimura W, Bonner-Weir S, Sharma A. Expression of MafA in pancreatic progenitors is detrimental for pancreatic development. Dev Biol. 2009;333(1):108-120.
Nishimura W, et al. A switch from MafB to MafA expression accompanies differentiation to pancreatic beta cells. Dev Biol. 2006;293(2):526-539.
Ober EA, et al. Mesodermal Wnt2b signaling positively regulates liver specification. Nature. 2006;442(7103):688-691.
Offield MF, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122(3):983-995.
Oliver G, et al. Prox 1, a prospero-related homeobox gene expressed during mouse development. Mech Dev. 1993;44(1):3-16.
Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis—a look outside the nucleus. Science. 2000;287(5458):1606-1609.
Picte RL, et al. An ultrastructural analysis of the developing embryonic pancreas. Dev Biol. 1972;29(4):436-467.
Poll AV, et al. A vHNF1/TCF2-HNF6 cascade regulates the transcription factor network that controls generation of pancreatic precursor cells. Diabetes. 2006;55(1):61-69.
Prasadan K, et al. Glucagon is required for early insulin-positive differentiation in the developing mouse pancreas. Diabetes. 2002;51(11):3229-3236.
Pulkkinen MA, et al. The IIIb isoform of fibroblast growth factor receptor 2 is required for proper growth and branching of pancreatic ductal epithelium but not for differentiation of exocrine or endocrine cells. Mech Dev. 2003;120(2):167-175.
Rausa F, et al. The cut-homeodomain transcriptional activator HNF-6 is coexpressed with its target gene HNF-3 beta in the developing murine liver and pancreas. Dev Biol. 1997;192(2):228-246.
Ritvos O, et al. Activin disrupts epithelial branching morphogenesis in developing glandular organs of the mouse. Mech Dev. 1995;50(2-3):229-245.
Roberts DJ, et al. Sonic hedgehog is an endodermal signal inducing Bmp-4 and Hox genes during induction and regionalization of the chick hindgut. Development. 1995;121(10):3163-3174.
Roberts DJ, et al. Epithelial-mesenchymal signaling during the regionalization of the chick gut. Development. 1998;125(15):2791-2801.
Rossi JM, et al. Distinct mesodermal signals, including BMPs from the septum transversum mesenchyme, are required in combination for hepatogenesis from the endoderm. Genes Dev. 2001;15(15):1998-2009.
Sadler TW, Langman J. Langman’s Medical Embryology, 10th ed. Philadelphia: Lippincott, Williams & Wilkins; 2006.
Sander M, et al. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development. 2000;127(24):5533-5540.
Sanvito F, et al. TGF-beta 1 overexpression in murine pancreas induces chronic pancreatitis and, together with TNF-alpha, triggers insulin-dependent diabetes. Biochem Biophys Res Commun. 1995;217(3):1279-1286.
Schmidt C, et al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373(6516):699-702.
Schuppan D. Structure of the extracellular matrix in normal and fibrotic liver: collagens and glycoproteins. Semin Liver Dis. 1990;10(1):1-10.
Sellick GS, et al. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet. 2004;36(12):1301-1305.
Seo YW, et al. FOR, a novel orphan nuclear receptor related to farnesoid X receptor. J Biol Chem. 2002;277(20):17836-17844.
Sergi C, Kahl P, Otto HF. Contribution of apoptosis and apoptosis-related proteins to the malformation of the primitive intrahepatic biliary system in Meckel syndrome. Am J Pathol. 2000;156(5):1589-1598.
Serls AE, et al. Different thresholds of fibroblast growth factors pattern the ventral foregut into liver and lung. Development. 2005;132(1):35-47.
Seymour PA, et al. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci U S A. 2007;104(6):1865-1870.
Shiojiri N, Koike T. Differentiation of biliary epithelial cells from the mouse hepatic endodermal cells cultured in vitro. Tohoku J Exp Med. 1997;181(1):1-8.
Shiojiri N, Lemire JM, Fausto N. Cell lineages and oval cell progenitors in rat liver development. Cancer Res. 1991;51(10):2611-2620.
Shiojiri N, et al. Suppression of C/EBP alpha expression in biliary cell differentiation from hepatoblasts during mouse liver development. J Hepatol. 2004;41(5):790-798.
Shiozaki S, et al. Impaired differentiation of endocrine and exocrine cells of the pancreas in transgenic mouse expressing the truncated type II activin receptor. Biochim Biophys Acta. 1999;1450(1):1-11.
Simeone DM, et al. Islet hypertrophy following pancreatic disruption of Smad4 signaling. Am J Physiol Endocrinol Metab. 2006;291(6):E1305-E1316.
Sjodin A, Dahl U, Semb H. Mouse R-cadherin: expression during the organogenesis of pancreas and gastrointestinal tract. Exp Cell Res. 1995;221(2):413-425.
Smart NG, et al. Conditional expression of Smad7 in pancreatic beta cells disrupts TGF-beta signaling and induces reversible diabetes mellitus. PLoS Biol. 2006;4(2):e39.
Smith SB, et al. Autoregulation and maturity onset diabetes of the young transcription factors control the human PAX4 promoter. J Biol Chem. 2000;275(47):36910-36919.
Smith WC, Harland RM. Expression cloning of noggin, a new dorsalizing factor localized to the Spemann organizer in Xenopus embryos. Cell. 1992;70(5):829-840.
Sosa-Pineda B, et al. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature. 1997;386(6623):399-402.
Sosa-Pineda B, Wigle JT, Oliver G. Hepatocyte migration during liver development requires Prox1. Nat Genet. 2000;25(3):254-255.
Stafford D, Prince VE. Retinoic acid signaling is required for a critical early step in zebrafish pancreatic development. Curr Biol. 2002;12(14):1215-1220.
Stafford D, et al. A conserved role for retinoid signaling in vertebrate pancreas development. Dev Genes Evol. 2004;214(9):432-441.
Stoffers DA, et al. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet. 1997;17(2):138-139.
Stoffers DA, et al. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15(1):106-110.
St-Onge L, et al. Pax6 is required for differentiation of glucagon-producing alpha cells in mouse pancreas. Nature. 1997;387(6631):406-409.
Sussel L, et al. Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development. 1998;125(12):2213-2221.
Suzuki A, et al. Clonal identification and characterization of self-renewing pluripotent stem cells in the developing liver. J Cell Biol. 2002;156(1):173-184.
Tan X, et al. Beta-catenin deletion in hepatoblasts disrupts hepatic morphogenesis and survival during mouse development. Hepatology. 2008;47(5):1667-1679.
Terada T, Nakanuma Y. Expression of tenascin, type IV collagen and laminin during human intrahepatic bile duct development and in intrahepatic cholangiocarcinoma. Histopathology. 1994;25(2):143-150.
Terada T, Nakanuma Y. Detection of apoptosis and expression of apoptosis-related proteins during human intrahepatic bile duct development. Am J Pathol. 1995;146(1):67-74.
Thomas PQ, Brown A, Beddington RS. Hex: a homeobox gene revealing peri-implantation asymmetry in the mouse embryo and an early transient marker of endothelial cell precursors. Development. 1998;125(1):85-94.
Tiso N, et al. BMP signalling regulates anteroposterior endoderm patterning in zebrafish. Mech Dev. 2002;118(1-2):29-37.
Tulachan SS, et al. TGF-beta isoform signaling regulates secondary transition and mesenchymal-induced endocrine development in the embryonic mouse pancreas. Dev Biol. 2007;305(2):508-521.
Vincent M, et al. Abrogation of protein convertase 2 activity results in delayed islet cell differentiation and maturation, increased alpha-cell proliferation, and islet neogenesis. Endocrinology. 2003;144(9):4061-4069.
Wang J, et al. The concerted activities of Pax4 and Nkx2.2 are essential to initiate pancreatic beta-cell differentiation. Dev Biol. 2004;266(1):178-189.
Wang Q, et al. Ghrelin is a novel target of Pax4 in endocrine progenitors of the pancreas and duodenum. Dev Dyn. 2008;237(1):51-61.
Wang X, et al. Increased levels of forkhead box M1B transcription factor in transgenic mouse hepatocytes prevent age-related proliferation defects in regenerating liver. Proc Natl Acad Sci U S A. 2001;98(20):11468-11473.
Weinstein DC, et al. The winged-helix transcription factor HNF-3 beta is required for notochord development in the mouse embryo. Cell. 1994;78(4):575-588.
Weinstein M, et al. Smad proteins and hepatocyte growth factor control parallel regulatory pathways that converge on beta1-integrin to promote normal liver development. Mol Cell Biol. 2001;21(15):5122-5131.
Wells JM, Melton DA. Early mouse endoderm is patterned by soluble factors from adjacent germ layers. Development. 2000;127(8):1563-1572.
Wells JM, et al. Wnt/beta-catenin signaling is required for development of the exocrine pancreas. BMC Dev Biol. 2007;7:4.
Xue Y, et al. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet. 1999;8(5):723-730.
Yamaoka T, et al. Hypoplasia of pancreatic islets in transgenic mice expressing activin receptor mutants. J Clin Invest. 1998;102(2):294-301.
Yamaoka T, et al. Diabetes and pancreatic tumours in transgenic mice expressing Pax6. Diabetologia. 2000;43(3):332-339.
Yang JT, Rayburn H, Hynes RO. Embryonic mesodermal defects in alpha 5 integrin-deficient mice. Development. 1993;119(4):1093-1105.
Yee NS, Yusuff S, Pack M. Zebrafish pdx1 morphant displays defects in pancreas development and digestive organ chirality and potentially identifies a multipotent pancreas progenitor cell. Genesis. 2001;30(3):137-140.
Yew KH, et al. Interplay of glucagon-like peptide-1 and transforming growth factor-beta signaling in insulin-positive differentiation of AR42J cells. Diabetes. 2004;53(11):2824-2835.
Yokouchi Y, Sakiyama J, Kuroiwa A. Coordinated expression of Abd-B subfamily genes of the HoxA cluster in the developing digestive tract of chick embryo. Dev Biol. 1995;169(1):76-89.
Yoshitomi H, Zaret KS. Endothelial cell interactions initiate dorsal pancreas development by selectively inducing the transcription factor Ptf1a. Development. 2004;131(4):807-817.
Zaret K. Developmental competence of the gut endoderm: genetic potentiation by GATA and HNF3/fork head proteins. Dev Biol. 1999;209(1):1-10.
Zaret KS. Regulatory phases of early liver development: paradigms of organogenesis. Nat Rev Genet. 2002;3(7):499-512.
Zhang W, et al. Regulation of Hex gene expression and initial stages of avian hepatogenesis by Bmp and Fgf signaling. Dev Biol. 2004;268(2):312-326.
Zhou J, et al. Glucagon-like peptide 1 and exendin-4 convert pancreatic AR42J cells into glucagon- and insulin-producing cells. Diabetes. 1999;48(12):2358-2366.
Zucman-Rossi J, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology. 2006;43(3):515-524.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 1A Embryologic development of the liver, biliary tract, and pancreas
Overview of Liver and Biliary Tract Development
Developmental biologists have often marveled at the enormous regenerative capacity of the liver after injury, and such growth represents one of the fastest by mammalian tissues. Presumably this process is based on the recapitulation of embryonic signals in the liver, but our understanding of the mechanisms of liver development is relatively poor. The liver is the largest internal organ and consists of diverse cell types that arise from various embryologic origins. The two principle cell types of the liver are the hepatocytes, which comprise nearly 70% of the mass of the adult organ, and the cholangiocytes. Both cells are derived from the embryonic endoderm. Other cell types of the liver include hematopoietic, Kuppfer, stromal, and stellate cells, all of which are of mesodermal origin (Fig. 1A.1). Despite their homogeneous appearance, hepatocytes do not all function identically; they perform various roles, depending on their physical location within a hepatic lobule, the primary functional unit of the liver. Thus, liver development entails not only hepatocyte and cholangiocyte differentiation but also further cellular differentiation within the hepatocyte population (Fig. 1A.1).
FIGURE 1A.1 Cell lineage schematic for hepatic and pancreatic development from a multipotent progenitor stem cell.
During the third week of gestation, the liver primordium first appears as an outgrowth of the ventral foregut endoderm at the caudal end of the foregut. The proliferation of the epithelial cells in this liver bud leads to its growth and branching with resulting expansion of the growing bud into the surrounding mesenchyme, giving rise to the liver and intrahepatic biliary tree. As it grows caudally, traversing the septum transversum, the persistent connection between the branching epithelium and the foregut develops into the extrahepatic bile ducts and gallbladder (Sadler & Langman, 2006). The hepatoblasts eventually differentiate into hepatocytes and cholangiocytes. The final liver structure continues to develop through the postnatal period. In addition to giving rise to the liver and biliary tract, the proximal endoderm (foregut) also gives rise to respiratory epithelium and the glandular cells of the thyroid, thymus, and pancreas.
Endodermal Patterning
Embryologically, the liver, biliary tract, and pancreas develop through a series of reciprocal interactions between the endoderm and the surrounding mesenchyme. The primitive gut tube, which is derived from the endodermal germ layer during gastrulation, is divided into the foregut, midgut, and hindgut domains, each of which gives rise to specialized regions (Grapin-Botton, 2005). This specialization is initiated by transcription factors expressed in different regions, such as pancreatic and duodenal homeobox gene 1 (PDX1) and pancreas-specific transcription factor 1a (PTF1A), whose coexpression in the endoderm gives rise to the pancreas (Fig. 1A.2) (Grapin-Botton, 2005; Moore-Scott et al, 2007). The definitive endoderm is formed at the ventral side of the vertebrate embryo during gastrulation. Evagination of the endoderm at the anterior end of the embryo generates the ventral foregut, which will eventually give rise to the liver, lung, thyroid, and the ventral pancreas. The dorsal region of the definitive endoderm develops into the intestines and the dorsal pancreas (see Fig. 1A.2).
This complex dialogue between the endoderm and mesoderm appears to be critical for the final patterning of the gut tube, where several signaling pathways have been implicated in the regulation of foregut endoderm development, promoting specification along its anterior–posterior axis for organs such as the thyroid, lung, liver, and pancreas. The plasticity of the endoderm was demonstrated by experimentally recombining the posterior mesoderm with early foregut endoderm, leading to repression of liver and pancreatic development in favor of intestinal development (Wells & Melton, 2000; Kumar et al, 2003).
Specific signals from the surrounding mesoderm in the foregut region lead to hepatic specification and subsequent morphogenesis (Guadi et al, 1996). The molecular pathway linking endodermal patterning to the initiation of liver and pancreatic development has been partially elucidated by recent studies in the Xenopus model, supporting a role for FGF4 and WNT signaling from the posterior mesoderm in inhibiting foregut fate and promoting hindgut formation; these signals are inhibited in the anterior endoderm to allow foregut development (McLin et al, 2007). β-Catenin repression in the anterior endoderm is specifically necessary to initiate liver and pancreas development; β-catenin repression in the endoderm then initiates reciprocal signaling with the mesoderm, leading to hepatic induction. To illustrate β-catenin’s role in this process, forced β-catenin expression in the anterior endoderm led to downregulation of the hematopoietically expressed homeobox gene (HHEX) and inhibition of liver formation, whereas repressing β-catenin in the posterior endoderm—future hindgut that normally expresses β-catenin—induced ectopic HHEX expression and ectopic liver bud initiation (McLin et al, 2007). HHEX is a direct target of β-catenin and is one of the earliest foregut markers (Thomas et al, 1998) that is essential for ectopic and normal liver and ventral pancreas development in mice (Keng et al, 2000; Bort et al, 2004). In addition to repressing HHEX, β-catenin also downregulates other foregut markers for liver (for1) (Seo et al, 2002), pancreas (PDX1), lung/thyroid (NKX2-1) (Goss et al, 2009), and intestine (Endocut) (Costa et al, 2003). Similarly, inhibiting β-catenin in the posterior mesoderm led to ectopic expression of other liver and pancreas markers (for1, PDX1, elastase, and amylase) along with inhibition of the intestinal marker Endocut (McLin et al, 2007).
Studies in mouse and chick models demonstrated the need for an FGF2 signal from the cardiogenic mesoderm and bone morphogenetic proteins (BMPs) from the septum transversum mesenchyme (Rossi et al, 2001; Zhang et al, 2004) for liver development. In addition, WNT2B has also been shown to be an important signal for liver specification in Zebrafish (Ober et al, 2006).
Other examples of transcription factors implicated in foregut patterning include the HOX genes. HOXD13 is expressed in the hindgut of mouse and chick embryos (Yokouchi et al, 1995; Roberts et al, 1995; Dolle et al, 1991). Knockout mice homozygous and null mutant for HOXD13 develop with hindgut defects (Kondo et al, 1996). Similarly, when HOXD13 is misexpressed in chick embryo midgut mesoderm, hindgut features are seen in the midgut epithelium (Roberts et al, 1998). The exact mechanism of how the HOX genes induce endodermal patterning remains unknown, as the exact downstream targets are not clear. In addition to HOX genes, retinoic acid signaling has been implicated in this foregut patterning, probably through regulating HOX gene expression. Specifically, retinoic acid signaling appears to specify the position of the pancreatic domain in the foregut of Xenopus, Zebrafish, and mice (Huang et al, 1998; Chen et al, 2004; Martin et al, 2005; Stafford et al, 2004). Goss and colleagues (Goss et al, 2009) demonstrated that WNT2 and WNT2B play an essential role in specifying lung endoderm without affecting the specification of other foregut-derived tissues. WNT2/2B double-knockout (DKO) mutants display complete lung agenesis. NKX2-1, the earliest known transcription factor marker for the developing lung, was also lost from the anterior foregut, confirming the loss of trachea and lung development.
Hepatic Competence
Hepatic competence, or the ability to form liver from the foregut endoderm, is considered the first step in a two-step process for the specification of the liver in vertebrates. Competence is a prerequisite for the endoderm to respond to specific signals, such as Fgfs from the surrounding mesoderm, which then leads to the second step, the induction of liver specific genes such as ALB (albumin). This “competence” is facilitated through the FOXA gene transcription factor family that includes FOXA1 and FOXA2 (forkhead box proteins A1 and A2). FOXA gene expression precedes the induction of the hepatic program by FGF signals in the endoderm, and FOXA2 binding revises α-fetoprotein (AFP) gene transcription in vitro (Crowe et al, 1999). Deletion of FOXA1 and FOXA2 led to no liver bud formation with the loss of AFP expression in the ventral foregut, which would indicate that hepatic specification had failed to initiate (Lee et al, 2005). The forkhead box (FOX) proteins are an extensive family of transcription factors that share homology with the winged helix/forkhead DNA binding domains (Kaestner et al, 2000) that play important roles in regulating expression of genes involved in cellular differentiation, proliferation, transformation, and metabolic homeostasis (Wang et al, 2001; Zaret et al, 1999; Duncan, 2000). In vitro explant cultures demonstrated that Fgf was sufficient to induce ventral foregut endodermal cells to differentiate into hepatoblasts (Guadi et al, 1996). However, when FOXA1/FOXA2-deficient endoderm was cultured with exogenous Fgf2, no liver expression was seen (Lee et al, 2005). This result would indicate the need for FOXA1 and FOXA2 for the onset of hepatogenesis. In vivo DNA-binding studies revealed that the liver-specific albumin gene ALB has an important upstream regulatory binding site for FOXA factors (Guadi et al, 1996; Bossard & Zaret, 1998). Prior to FOXA or GATA4 binding, the ALB gene is transcriptionally silent with a closed chromatin. After binding with FOXA and GATA4, the chromatin domain is thought to become exposed (Cirillo et al, 2002), thus increasing the ability of the gene to be activated. By the E9.5 gestation stage, other transcription factors—CCAAT/enhancer binding protein-β (CEBPB) and NF1, nuclear factor 1—bind to sites adjacent to the FOXA site, and as a result, the albumin gene becomes active (Zaret, 2002). The critical binding of FOXA to chromatin increases the ability of the gene expression to be activated by allowing other transcription factors to bind, hence the hepatic competence.
Hepatic Induction
Fgf from the cardiogenic mesoderm and Bmps from the septum transversum mesenchyme have been implicated in the induction of liver fate in mouse and chick embryonic endoderm. In vitro studies were performed in which the ventral foregut and cardiogenic mesoderm were cocultured in the presence of fibroblast growth factor inhibitors, resulting in the inhibition of liver induction (Jung et al, 1999). Culturing foregut endoderm without the cardiogenic mesoderm in the presence of a low concentration of exogenous FGF2 (2 to 5 ng/mL) rescued hepatic gene expression (Jung et al, 1999) and simultaneously suppressed the expression of the pancreas program (PDX1 expression) (Deutsch et al, 2001), whereas a high concentration of FGF2 (10 to 500 ng/mL) induced NKX2-1, an early marker for respiratory epithelium but not albumin gene expression (Serls et al, 2005). Ventral foregut endoderm cocultured with noggin (Nog), a Bmp inhibitor, inhibited hepatic gene induction, an effect that was rescued when Bmp2 or Bmp4 were added. Despite these results, however, embryos that were homozygous mutant for BMP4 still exhibited normal hepatic gene induction (Rossi et al, 2001; Smith & Harland, 1992). These data indicate that the cardiac mesoderm, a source for FGF signaling, is crucial for the hepatic induction from the ventral foregut endoderm and subsequent morphogenesis, whereas Bmp aids in the process as well (Fig. 1A.3).
Morphogenesis of the Hepatic Bud
Following hepatic specification (E8.5 to E9.0), the liver starts to express liver-specific genes (ALB, AFP, HNF4A) that eventually form the liver bud. Hepatic bud morphogenesis is facilitated by two transcription factors, HHEX (hematopoietically expressed homeobox, discussed above) (Crompton et al, 1992) and PROX1 (prospero-related homeobox 1) (Oliver et al, 1993). HHEX is expressed in the anterior endoderm at E7.0, eventually giving rise to the ventral pancreas and liver. HHEX-null embryos grow without a liver, thyroid and develop forebrain defects at E11.5; however, evidence of endodermal epithelial thickening suggest a possible defect in differentiation as evidence of possible hepatic induction (Martinez-Barbera et al, 2000).
In a separate study on HHEX-null embryos, liver genes ALB and PROX1 were expressed in the ventral foregut endoderm at around E8.5, with the thickening of the hepatic endoderm region at E9.0 being smaller compared with heterozygote embryos. In addition, a significantly lower proliferation rate was seen in the prospective hepatic domain compared with controls, as demonstrated by bromodeoxyuridine (BrdU) staining, with no evidence of apoptosis (Bort et al, 2004). The basal membrane layer, which is rich in laminin, surrounds the hepatic endoderm and degrades around E9.0 to E9.5, so that the hepatocytes start migrating into the septum transversum mesenchyme (STM) to form the liver bud. This degradation is facilitated by the hepatoblasts, which under normal conditions downregulate E-cadherin. However, in PROX1-null mutant embryos, the progenitor liver cells fail to migrate into the STM as a result of excess E-cadherin and basement membrane proteins laminin and collagen 4. The basal lamina fails to degrade, and the cells remain trapped in the hepatic diverticulum with an overall reduction in liver size (Sosa-Pineda et al, 2000). The bulk of the liver lobe lacks hepatocytes, suggesting that the mesenchymal component contributes most of the liver mass in PROX1-null mutant embryos. A similar phenotype is seen with ONECUT1 and ONECUT2 double mutants, which are required for basal lamina degradation (Margagliotti et al, 2007). Furthermore, pharmacologic inhibition of matrix metalloproteinases (MMPs), extracellular matrix remodeling enzymes usually expressed by hepatoblasts and STM cells, inhibits hepatoblast migration in culture (Margagliotti, 2008). To further illustrate the importance of the extracellular matrix in hepatic bud morphogenesis, hepatoblasts deficient in laminin receptor β1-integrin fail to colonize the liver bud (Fassler & Meyer, 1995). These β1-integrins are among those that act as receptors for extracellular matrix proteins, such as laminins and collagens (Hynes, 1992).
The role of endothelial cells in liver organogenesis has also been studied, with the liver vasculature providing a vital source for hematopoiesis in early life. Endothelial cells are positioned as a loose chain of cells interceding between the thickening hepatic epithelium and the STM (Matsumoto et al, 2001). Embryos that were null mutant for VEGFR2 (also known as KDR) had failure of delamination and subsequent migration by the hepatoblasts (Matsumoto et al, 2001). These results imply that the endothelial cells interact with nascent hepatic cells and aid in liver bud outgrowth.
Liver Bud Growth
The STM cells are closely related to the ventral endoderm and contribute to hepatic induction and growth. This epithelial–mesenchymal interaction is essential for the formation of the liver bud as well as for further expansion and differentiation. During this phase (E9.5 to E15), the liver bud undergoes a tremendous amount of growth and also becomes an important site for hematopoiesis. Factors and signals regulating this critical stage arise from both the hepatic mesenchyme and the STM. These signals include Fgf and Bmp, which promote liver growth in addition to their role in hepatic specification. Bmp4 is strongly expressed in the STM and continues to be expressed at E9.0, during which the liver bud migrates into the STM (Rossi et al, 2001). BMP4-null mutant embryos were developed to assess for a role in liver organogenesis. These null-mutant mice had a delay in the growth of the liver bud, which would indicate that Bmp constitutes an important growth signal for the liver (Rossi et al, 2001).
Another factor that has been implicated in liver growth is HLX, which is expressed prominently in the visceral mesenchyme (STM), into which the liver will expand (Hentsch et al, 1996). HLX-null embryos exhibited severe liver hypoplasia without affecting liver specification (Hentsch et al, 1996). A similar finding was observed in embryos that were null mutant for hepatocyte growth factor (Hgf) (Schmidt et al, 1995). In contrast to HLX−/− embryos, apoptosis was the underlying cause for the severe reduction in liver size. Hgf is produced by the cells lining the sinusoids, which are of mesenchymal origin, mediating its effects through the c-Met tyrosine kinase receptor produced by hepatocytes (Schmidt, 1995). TGF-β signaling is also involved in liver outgrowth. It mediates its signals through SMAD2 and SMAD3, which after phosphorylation translocate to the nucleus to either upregulate or downregulate gene expression. Embryos that were heterozygous mutants for both SMAD2 and SMAD3 exhibited severe liver hypoplasia as a result of a decrease in β1-integrin expression (Weinstein et al, 2001). Intriguingly, this phenotype was rescued when HGF, a potent hepatotrophic growth factor, was added to the culture medium. Presumably, this was a result of the restoration of β1-integrin expression (Kagami et al, 1996), which plays an important role in hepatocytic adhesion to the extracellular matrix; TGF-β and HGF are known to induce the gene (Kagami et al, 1996; Kawakami-Kimura et al, 1997).
Overview of Hepatoblast Differentiation
Hepatoblasts begin to differentiate into mature hepatocytes and biliary epithelial cells at around E13.5. Prior to differentiation, the hepatoblasts express genes for adult hepatocytes (ALB, HNF4A), biliary epithelial cells (KRT19), and fetal liver (AFP) (Lemaigre, 2003; Shiojiri et al, 1991). Hepatoblasts in proximity to the portal vein form a bilayer and eventually differentiate into biliary epithelial cells, upregulating biliary-specific cytokeratin-19 (KRT19) and downregulating the other hepatic genes in the process. This bilayer around the portal vein begins to form focal dilations incorporated into the portal mesenchyme to form intrahepatic biliary ducts at E17.0 until birth. Those parts of the ductal bilayer plate not involved in the formation of the ducts progressively regress, and the rest of the hepatoblasts differentiate into mature hepatocytes, arranged in hepatic cords with the bile canaliculi on the apical surfaces (Lemaigre et al, 2003). The bile is produced by the hepatocytes and is secreted into the canaliculi, which are connected to the network of intrahepatic biliary ducts. The bile then flows to the hepatic ducts, transits through the cystic duct, and is stored in the gallbladder; eventually, the bile is excreted into the bowel via the common bile duct. The biliary epithelial cells delineate the lumen of the intrahepatic and extrahepatic biliary tree (hepatic, cystic, and common bile ducts) and the gallbladder (Fig. 1A.4).
Biliary Epithelial Cell Differentiation and Formation of the Ductal Plate
The exact origin of the biliary epithelial cells has been greatly debated; however, the popular school of thought is that they are derived from bipotential hepatoblasts that can differentiate into either hepatocytes or biliary epithelial cells. This theory is based on the observation that immature hepatoblasts coexpress markers of both hepatocytes (ALB) and biliary epithelial cells (KRT19). The biliary-specific marker KRT19 becomes strongly expressed at a later gestational age, as the cells become ductal cells, whereas other cells transiently express the hepatocyte markers ALB and AFP as they develop into mature hepatocytes (Shiojiri et al, 1991). This theory was further supported when embryonic liver was transplanted into the testes of syngeneic animals, before the emergence of intrahepatic biliary ducts, as it subsequently gave rise to both hepatocytes and typical bile ducts (Shiojiri et al, 1991). However, this finding does not exclude the possibility that ductal cells may be derived from other embryonic cell types. Suzuki and colleagues (Suzuki et al, 2002) used in vitro studies on hepatic “stem” cells from E13 embryonic livers identified as having self-renewing capability and multilineage differentiation potential, to show that these cells could form differentiated hepatocytes, biliary epithelial cells, pancreatic, and intestinal cells.
The first step in triggering the initiation of the transition from a hepatoblast to a biliary epithelial cell is thought to be facilitated through the ONECUT transcription factor hepatocyte nuclear factor 6 (HNF6), which is expressed in the biliary epithelial cells of the developing intrahepatic biliary ducts and in hepatoblasts, gallbladder primordium, and the extrahepatic bile ducts (Landry et al, 1997; Rausa et al, 1997). HNF6−/− embryos displayed severe biliary anomalies that resulted in an absent gallbladder and the extrahepatic bile duct network being replaced with an enlarged structure connecting the liver to the duodenum. In the intrahepatic biliary ducts, however, it caused abnormal differentiation of biliary epithelial cells, resulting in cholestasis. Closer histologic examination revealed an increased number of KRT19-positive cells compared with controls at E13.5, with the development of abnormal large cysts at E15.5 to E16.5 that contained an epithelium of KRT19-expressing cells. These abnormal cysts are similar to those seen in Caroli disease, a disorder with ductal plate malformation and ectasias. Although HNF6 might play a role in the disease, not all of the malformations seen in HNF6−/− mice are manifested in Caroli disease, such as the absence of the gallbladder. The abnormal increase in KRT19-positive cells at E13.5 lacked any proliferative marker, suggesting that they are postmitotic, resulting from an increased number of hepatoblasts that have differentiated toward a biliary lineage prematurely (Clotman et al, 2002). In addition, the excess KRT19-positive biliary epithelial cells formed cordlike extensions within the liver parenchyma, rather than being restricted to the vicinity of the portal vein, as seen in the controls. This observation supports the role of HNF6 in controlling the differentiation of hepatoblasts into biliary epithelial cells and the morphogenesis of the intrahepatic biliary ducts, confining biliary epithelial cells to the periportal area. A similar morphologic defect of intrahepatic biliary ducts was observed in HNF1B−/− embryos. These findings suggest that HNF6 controls intrahepatic biliary duct development via HNF1B (Clotman et al, 2002).
With regard to mesenchymal–epithelial induction of liver primordium and gallbladder, studies have suggested that the mesenchyme contributes to biliary epithelial cell differentiation, where differentiation of hepatoblasts into biliary epithelial cells was stimulated when cocultured with hepatic or lung mesenchyme (Shiojiri & Koike, 1997). It was also noted in the HNF6−/− study that biliary epithelial cell differentiation occurred at the interface between the portal mesenchyme and the liver parenchyma (Clotman et al, 2002). A recent study revealed that the forkhead box f1 (FOXF1) transcription factor may play an important role in mesenchymal–epithelial signaling, an interface required for the development of organs derived from foregut endoderm, such as the pancreas, liver, gallbladder, and lung (Kalinichenko et al, 2002). FOXF1 expression is restricted to the gallbladder mesenchyme and STM. In FOXF1+/−, the gallbladder develops severe structural abnormalities with significant reduction in size and reduced mesenchymal cell numbers, an absent biliary epithelial cell layer, and a deficient external smooth muscle layer. The reduction in mesenchymal cell numbers was attributed to the reduction in the vascular cell-adhesion molecule (VCAM1) and cell-adhesion α5 integrin (ITGA5) genes, both of which are essential for mesodermal formation (Mahlapuu et al, 2001; Yang et al, 1993). Defective smooth muscle layer formation was attributed to diminished levels of platelet-derived growth factor receptor α (PDGFRA), which is essential for smooth muscle cell differentiation (Jacob et al, 1994). FOXF1 is not expressed in intrahepatic biliary duct mesenchyme in wild-type mice, and no defects were seen in the intrahepatic biliary ducts of FOXF1+/− mice; however, FOXF1 mRNA levels in the liver were increased, suggesting that it may be a compensatory mechanism to prevent defects in the liver (Kalinichenko et al, 2002). All these data suggest that FOXF1 is crucial for the development of the extrahepatic bile ducts and gallbladder, with the mesenchyme playing an essential role in biliary epithelial cell differentiation. The interaction between the biliary epithelial cells and the extracellular matrix is also thought to contribute to biliary epithelial cell differentiation.
Integrins are membrane receptors for extracellular matrix proteins that play an important role in mediating the interaction between differentiating cells and the extracellular matrix (Hynes, 1992; Couvelard et al, 1998). Hepatoblasts express integrin heterodimers containing the β1 subunit (α1β1, α5β1, α6β1, and α9β1), and when hepatoblasts differentiate into immature intrahepatic biliary epithelial cells when in contact with the mesenchyme, the morphology of the integrins changes. The primitive intrahepatic biliary epithelial cells upregulate α6β1 expression and lose α1β1, while acquiring several other integrin dimers not previously expressed on hepatoblasts, including α2β1, α3β1, α5β1 and α6β4 (Couvelard et al, 1998). This change is likely due to the different pericellular environment. Intrahepatic biliary epithelial cells are contacted by a basement membrane containing collagen, entactin, and laminin (Desmet, 1985); however, hepatocytes are surrounded by the perisinusoidal matrix, which is devoid of laminin and entactin (Schuppan, 1990). The increase in α6β1 expression—along with the acquisition of biliary-specific expression of α2β1, α3β1, and α6β4, which are integrin receptors for laminin—correlated with the deposition of laminin at the contact points of the portal mesenchyme with the ductal plate (Couvelard et al, 1998).
Remodeling of the Ductal Plate
As mentioned earlier, the double-layered ductal plate around the portal vein begins to form focal dilations incorporated into the portal mesenchyme to form intrahepatic biliary ducts, while the parts of the ductal bilayer plate not involved in the formation of the ducts progressively regresses. This mechanism of regression is thought to be carried out by apoptosis (Terada & Nakanuma, 1995; Sergi et al, 2000). Cell-matrix interactions are also thought to contribute to the remodeling process. Tenascin was found to be expressed in the mesenchyme around the biliary epithelial cells of primitive ducts migrating into the mesenchyme. In contrast, tenascin was absent in the mesenchyme of peripheral ducts (Terada & Nakanuma, 1994).
Tubulogenesis of ductal cells is thought to be controlled by soluble factors secreted from hepatocytes or biliary epithelial cells. When coculturing human biliary epithelial cells with hepatocytes, a marked ductular morphogenic response was induced, and the biliary epithelial cells formed well-organized luminal ducts. This result was reproduced when biliary epithelial cells were grown in a conditioned medium from previous hepatocyte and biliary epithelial cell coculturing (Auth et al, 2001); however, it remains to be seen whether soluble factors do contribute to tubulogenesis in in vivo studies (Fig. 1A.5).
Developmental Relationship Between the Ducts, Vessels, and Mesenchyme of the Portal Tract
A functional relationship appears to exist between the contents of the portal tract (bile duct, hepatic artery, portal vein) based on observations of a number of human diseases termed ductal plate malformations. These diseases include biliary atresia, Caroli disease, and Meckel and Alagille syndromes, in which abnormal biliary ducts are associated with anomalies of the portal mesenchyme and of the portal blood vessels (Lemaigre, 2003). This association was also demonstrated in studies on Notch pathway defects. In Alagille syndrome, an autosomal dominant disease, bile ducts are absent in the portal tract associated with an increased number of arteries and fibrosis. Haploinsufficiency of JAG1, a Notch receptor ligand, is associated with Alagille syndrome, where JAG1 is persistently expressed in the ductal epithelium in humans (Li et al, 1997; Louis et al, 1999). It is also expressed in the endothelial cells of the developing portal vasculature (Crosnier et al, 2000). The animal model for Alagille syndrome was replicated in double-heterozygous mice for mutations in the JAG1 and NOTCH2
[/not-level-membership-for-surgery-category]
