[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Diseases of the Pleura and Mediastinum
Benjamin E. Haithcock, Timothy M. Zagar, Longzhen Zhang and Thomas E. Stinchcombe
Malignant Pleural Mesothelioma (MPM)
• MPM is a rare disease with 2,000 to 3,000 cases annually in the United States.
• It is associated with prior occupational asbestos exposure, but some patients without known asbestos exposure will develop MPM.
• Four subtypes exist: epithelioid, sarcomatous, biphasic epithelial (also referred to as mixed), and desmoplastic.
• Epithelioid is the most common and may be associated with a better prognosis.
• Diagnosis must be differentiated from metastatic adenocarcinoma or non–small cell lung cancer with adenocarcinoma histology. No single test is diagnostic of MPM, and it frequently requires a panel of immunohistochemistry markers to confirm the diagnosis.
• Cytologic analysis of pleural fluid is not reliable to exclude MPM, and many times a thoracoscopic biopsy may be required for definitive diagnosis. Evidence of invasion into subpleural adipose tissue is the most reliable indicator of malignancy.
• Computed tomographic (CT) scan is the initial staging procedure.
• Positron emission tomography (PET) or PET-CT scan has the ability to detect extrathoracic disease.
• Cervical mediastinoscopy may be useful for detecting mediastinal involvement.
• Peritoneal lavage or laparoscopy is indicated if peritoneal involvement is suspected.
• For patients with operable disease without significant co-morbidities, surgical options include extrapleural pneumonectomy or pleurectomy and decortication.
• Single-institution and phase II trials have demonstrated the feasibility of multimodality therapy.
• The benefit of post- or preoperative radiation and chemotherapy is undefined.
• For patients with unresectable disease or metastatic disease, without significant treatment co-morbidities, and preserved performance status, treatment with a platinum agent (cisplatin or carboplatin) and antifolate (pemetrexed or raltitrexed) is the standard therapy.
• A variety of agents have shown activity in the second-line setting.
• Radiation therapy provides palliation of symptoms, and postoperative radiation therapy may reduce the rate of local and port site recurrence.
• Most common of all mediastinal tumors, it comprises approximately 20% of the tumors of the mediastinum.
• Differential includes thymoma, thymic carcinoma, and thymic carcinoid.
• Approximately 50% of patients with thymoma are asymptomatic at the time of diagnosis.
• Thymomas may be associated with parathymic syndromes (e.g., myasthenia gravis, pure red blood cell aplasia, and hypogammaglobulinemia)
• Thymomas are classified according to the World Health Organization (WHO) classification, which is associated with 10-year overall survival rates.
• Primary therapy is complete surgical resection.
• Postoperative radiation therapy should be considered for patients with stage IIB disease, close surgical margins, WHO grade B type, and tumor adherent to the pericardium.
• Chemotherapy with cisplatin or anthracycline-based therapy is an option for patients with unresectable or metastatic disease.
• Patients with malignant pleural effusions have a poor prognosis, and pleural effusion is considered metastatic disease.
• Common symptoms include dyspnea on exertion, shortness of breath, and cough.
• Most common causes of malignant pleural effusion are lung cancer, breast cancer, lymphoma, and cancer of unknown primary.
• Cytology can be diagnostic of type of malignancy, and patients with exudative effusion and known metastatic cancer should be considered as having a malignant pleural effusion.
• Thoracentesis may be diagnostic and provide symptomatic relief for the patient.
• Management strategies include intermittent thoracentesis, indwelling pleural catheters, and talc pleurodesis. The optimal treatment strategy may depend on the patient’s prognosis, performance status, and type of malignancy.
Primary Tumors of the Pleura: Mesothelioma
Primary benign and malignant tumors of the pleura are rare, and they include solitary fibrous tumor, adenomatoid tumor, pleural desmoid tumors, calcifying fibrous pseudotumor, and malignant pleural mesothelioma (MPM).1,2 For clinicians, the most frequent differential diagnosis is solitary fibrous tumor versus MPM. Solitary fibrous tumors of the pleura are significantly less common than MPM and are not associated with asbestos exposure.3 Solitary fibrous tumors are generally asymptomatic at the time of diagnosis, and the radiographic features are a well-circumscribed, peripheral mass that abuts the pleural surface, frequently attached by a pedicle.4 Approximately 10% to 20% of solitary fibrous tumors are classified as malignant; malignant tumors are characterized by mitoses, necrosis, atypia, and hypercellularity.4 Radiographically malignant solitary fibrous tumors compared to benign tumors tend to be larger and are associated with increased likelihood of being positive on positron emission tomography (PET).5,6 Patients with malignant solitary fibrous tumors tend to have a higher recurrence rate and worse survival.7–10 The primary treatment for solitary fibrous tumors is complete surgical resection if feasible.12–12 Because of the rarity of solitary fibrous tumors, it is difficult to define the natural history and the optimal management, but postoperative radiation and chemotherapy are not standard therapies. The primary focus of this section will be on MPM because that is the most common pleura malignancy.
Epidemiology
The association between MPM and asbestos exposure has been well established for decades; in the mid-20th-century, commercial uses for asbestos were developed, which led to increased occupational exposure of asbestos. Common sites of occupational exposure were in asbestos mines, shipyards, cement factories, or work with insulation. Asbestos refers to six fibrous silicate minerals found widely throughout the world, and is divided into two categories based on the chemical composition and crystalline structure13,14: a serpentine form (ie, chrysotile) and a thin, rodlike form (ie, amphiboles, including crocidolite, amosite, anthophyllite, tremolite, and actinolyte).15 The association of the amphibole form and MPM is well established, but the association between the serpentine form and MPM is a matter of debate.13 The long latency period between asbestos exposure and the development of MPM can make identification of the type, amount, and duration of asbestos exposure difficult. There does not appear to be a linear relationship between exposure and risk of developing MPM, and it appears that prolonged exposure is required in order to develop MPM. This is important for workers who had sporadic or limited exposure.13 A higher rate of MPM has been observed among family members of workers with occupational exposure caused by secondary exposure from clothes and close contact.16,17 Some patients with MPM will not report a known asbestos exposure, so the absence of a history of asbestos exposure should not eliminate MPM from the differential diagnosis.18 Other less common etiologic agents associated with MPM include prior radiation therapy, diagnostic use of thorium dioxide (Thorotrast), mineral fibers with similar properties (e.g., erionite), and potentially simian virus 40.19 In the United States, the incidence of MPM is estimated to be 2000 to 3000 cases per year and it appears to be rising, which is related to not only increased asbestos exposure a generation ago but also improved recognition.20,21
Despite the clear association between asbestos exposure and MPM, only a minority of people with significant exposure develop MPM (~5%), and the identification of MPM clusters within certain families raises the question about influence of genetics in carcinogenesis.14 A study of a MPM epidemic in Cappadocia, Turkey, and the United States revealed that the risk of developing MPM is transmitted in certain high-risk families, suggesting a genetic predisposition to erionite carcinogenesis.22 A high incidence of MPM in some families in the United States has been linked to germline mutations of the BAP1 gene.23 BAP1 somatic mutations have been identified in 25% of sporadic MPM as well.23,24 BAP1 appears to regulate deubiquitination during the DNA damage response and the cell cycle, and mutations that affect the deubiquitination of BAP1 or the nuclear localization reduce the tumor suppressor activity of BAP1.25 Other potential mechanisms of carcinogenesis include direct mechanical interference of asbestos fibers with chromosome segregation during mitosis or the generation of oxidants by macrophages attempting to digest asbestos fibers.28–28 The presence of asbestos causes inflammation, and the chronic inflammation associated with the asbestos fibers contributes to the process of carcinogenesis.14,29,30 The mechanism of carcinogenesis from chronic inflammation appears to be related to the cytokines tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β).30,31
Clinical Presentation
The most common presenting symptoms for patients with MPM are shortness of breath, dyspnea on exertion, and chest wall pain or discomfort. Other less common symptoms include fever of unknown origin, sweats, weight loss, and a decline in performance status.32 Many times patients have symptoms for months prior to presenting to a medical professional. On physical examination, decreased breath sounds on auscultation or dullness to percussion may be present. Frequently, a chest x-ray is performed and this reveals a pleural effusion and/or opacification in one hemithorax. There are no specific laboratory abnormalities or tumor markers diagnostic of MPM, although it has been associated with thrombocytosis.33
A thoracentesis is frequently performed to alleviate the patient’s symptoms, and it is part of the initial evaluation. A cytologic analysis of the pleural fluid is frequently performed; however, pleural effusion cytology is not reliable. Diagnostic cytologic criteria have not been established and invasion cannot be assessed on the specimen.36–36 Evidence of invasion into the subpleural adipose tissue is the most reliable indicator of malignancy.34,37 Thus, if there is clinical suspicion of MPM, a pleural effusion cytology negative for malignancy is not sufficient to eliminate MPM from the differential. Many times, a thoracoscopic biopsy is required, and if the tumor is surgically resectable the thoracoscopic port should be placed within the line of a potential thoracotomy so the area can be excised at the time of thoracotomy; this is to help prevent seeding/recurrence at the port site, which has been reported.
A number of prognostic markers have been identified including performance status, histology, presence or absence of chest pain, younger age, and normal platelet count.32,33,38–41 Many of these factors were identified from retrospective analyses that included small numbers of patients. The European Organization for Research and Treatment of Cancer (EORTC) performed a multivariate analysis and found poor prognosis to be associated with poor performance status, a high white blood cell count, a probable/possible histologic diagnosis of MPM (compared to definite diagnosis), male gender, and sarcomatous histologic subtype.42 On the basis of these five factors, patients were divided into a good prognosis group (1-year survival rate of 40%, 95% confidence interval [CI] = 30% to 50%), and a poor prognosis group (1-year survival rate of 12%, 95% CI = 4% to 20%). This model was validated based on independent clinical trials.43 An analysis of a phase III trial also validated the EORTC prognostic index and identified pain and appetite loss as independent prognostic factors.44 The prognostic index is useful to assist in the interpretation of phase II trials and may assist the clinician in estimating the prognosis for clinical decision making.
Pathology
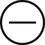
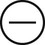
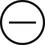
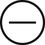
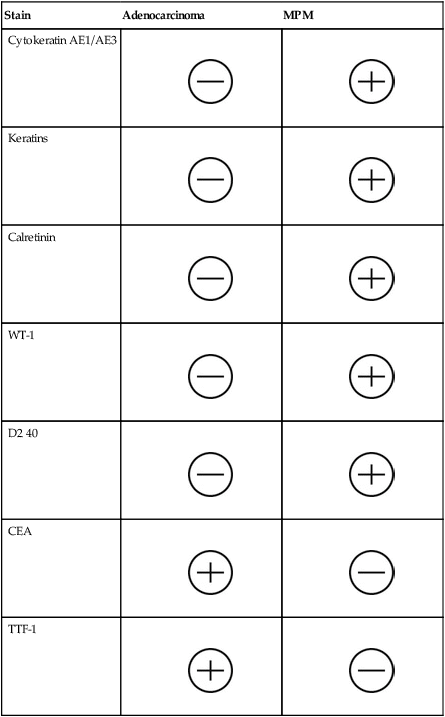
The four subtypes of MPM according to the World Health Organization (WHO) are as follows: epithelioid, sarcomatous, biphasic epithelial (also referred to as mixed), and desmoplastic.45 The epithelioid type is the most common and has a better prognosis compared to the other types. It can be difficult to distinguish MPM from reactive mesothelial hyperplasia, non–small cell lung cancer (NSCLC) with adenocarcinoma histology, and metastatic adenocarcinoma. Unfortunately, no single immunohistochemistry (IHC) stain can differentiate MPM from NSCLC with adenocarcinoma histology, and a panel of IHC stains is frequently used. One practice is to test for two markers that are positive in MPM (e.g., cytokeratin AE1/AE3, keratins [e.g., CK 5/6, CK 7], calretinin, Wilms tumor 1 [WT-1; nuclear staining], D2-40) as well as two that are negative for MPM (e.g., carcinoembryonic antigen [CEA] and thyroid transcription factor 1 [TTF-1]) (Table 73-1).46
Table 73-1
Histochemistry Studies to Distinguish NSCLC and MPM46
| Stain | Adenocarcinoma | MPM |
| Cytokeratin AE1/AE3 |
 |
 |
| Keratins |
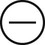 |
 |
| Calretinin |
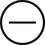 |
 |
| WT-1 |
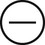 |
 |
| D2 40 |
 |
 |
| CEA |
 |
 |
| TTF-1 |
 |
 |
Staging
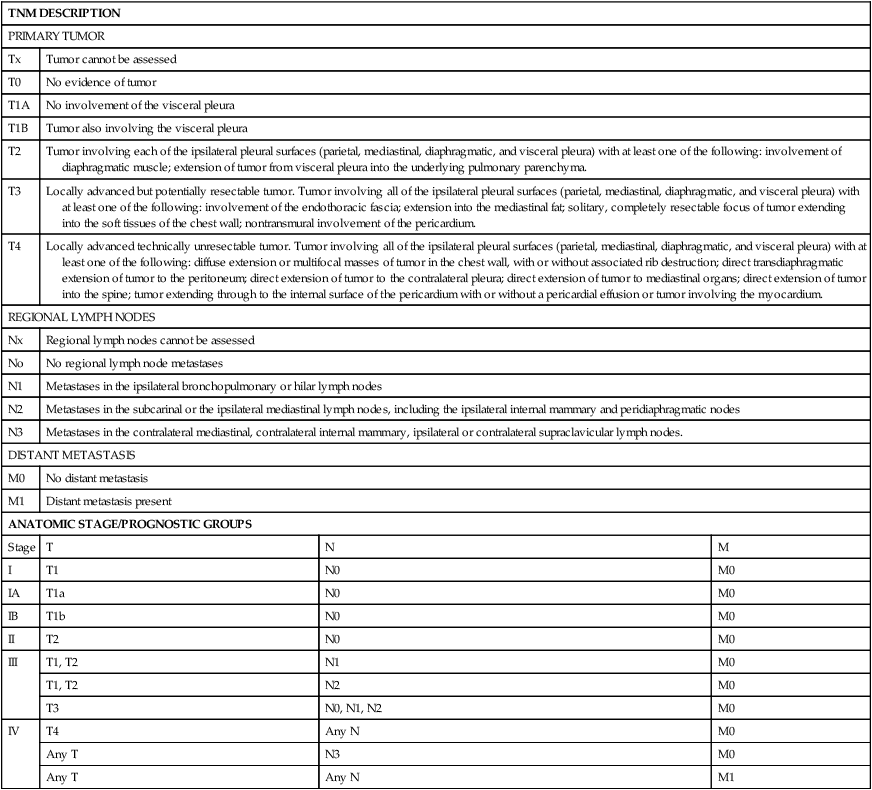
The natural history of MPM is growth along the pleural surfaces of the thoracic cavity and invasion of the surrounding lung tissue, followed by transdiaphragmatic extension, peritoneal spread, and metastatic disease. The staging system provides an estimate of the prognosis, and an assessment if the tumor is potentially resectable. A number of different staging systems have been used,47,48 and there has been a lack of a consensus on the optimal staging system. The tumor, nodal, and metastasis (TNM) staging system is often used (Table 73-2).49 Computed tomography (CT) is the standard staging procedure. An assessment of the mediastinal lymph nodes is critical for assessment of staging and for potential resection, and the staging tests frequently used are CT, fluorodeoxyglucose positron emission tomography (FDG-PET), CT-PET, and mediastinoscopy. A recent systemic review of the imaging modalities revealed that FDG-PET was superior to CT scan, but inferior to CT/FDG-PET, in terms diagnostic specificity, sensitivity, and staging.50 The mean standardized uptake value was statistically higher in malignant compared to benign disease, and PET-CT had a higher sensitivity for detection of lymph node involvement. PET or PET-CT also has the ability to detect extrathoracic disease, and approximately 10% to 25% of patients will have distant disease detected with PET scanning.51,52 Transdiaphragmatic extension is a contraindication to surgical resection, and some centers perform peritoneal lavage or laparoscopy. In one study, the use of peritoneal lavage prior to surgical resection demonstrated transdiaphragmatic invasion of the peritoneal cavity or peritoneal metastases in approximately 10% of patients.53 The role of cervical mediastinoscopy is undetermined, but may be of value for patients being considered for surgical resection. A recent retrospective review revealed that patients without mediastinal involvement had a significantly better survival compared to patients with mediastinal disease.54 Noninvasive tests improve the staging, in particular, to increase the detection of patients with metastatic disease and to assist in the selection of the optimal site to biopsy; however, definitive staging requires surgical resection and pathological examination of the specimen.
Table 73-2
| TNM DESCRIPTION | |||
| PRIMARY TUMOR | |||
| Tx | Tumor cannot be assessed | ||
| T0 | No evidence of tumor | ||
| T1A | No involvement of the visceral pleura | ||
| T1B | Tumor also involving the visceral pleura | ||
| T2 | Tumor involving each of the ipsilateral pleural surfaces (parietal, mediastinal, diaphragmatic, and visceral pleura) with at least one of the following: involvement of diaphragmatic muscle; extension of tumor from visceral pleura into the underlying pulmonary parenchyma. | ||
| T3 | Locally advanced but potentially resectable tumor. Tumor involving all of the ipsilateral pleural surfaces (parietal, mediastinal, diaphragmatic, and visceral pleura) with at least one of the following: involvement of the endothoracic fascia; extension into the mediastinal fat; solitary, completely resectable focus of tumor extending into the soft tissues of the chest wall; nontransmural involvement of the pericardium. | ||
| T4 | Locally advanced technically unresectable tumor. Tumor involving all of the ipsilateral pleural surfaces (parietal, mediastinal, diaphragmatic, and visceral pleura) with at least one of the following: diffuse extension or multifocal masses of tumor in the chest wall, with or without associated rib destruction; direct transdiaphragmatic extension of tumor to the peritoneum; direct extension of tumor to the contralateral pleura; direct extension of tumor to mediastinal organs; direct extension of tumor into the spine; tumor extending through to the internal surface of the pericardium with or without a pericardial effusion or tumor involving the myocardium. | ||
| REGIONAL LYMPH NODES | |||
| Nx | Regional lymph nodes cannot be assessed | ||
| No | No regional lymph node metastases | ||
| N1 | Metastases in the ipsilateral bronchopulmonary or hilar lymph nodes | ||
| N2 | Metastases in the subcarinal or the ipsilateral mediastinal lymph nodes, including the ipsilateral internal mammary and peridiaphragmatic nodes | ||
| N3 | Metastases in the contralateral mediastinal, contralateral internal mammary, ipsilateral or contralateral supraclavicular lymph nodes. | ||
| DISTANT METASTASIS | |||
| M0 | No distant metastasis | ||
| M1 | Distant metastasis present | ||
| ANATOMIC STAGE/PROGNOSTIC GROUPS | |||
| Stage | T | N | M |
| I | T1 | N0 | M0 |
| IA | T1a | N0 | M0 |
| IB | T1b | N0 | M0 |
| II | T2 | N0 | M0 |
| III | T1, T2 | N1 | M0 |
| T1, T2 | N2 | M0 | |
| T3 | N0, N1, N2 | M0 | |
| IV | T4 | Any N | M0 |
| Any T | N3 | M0 | |
| Any T | Any N | M1 | |
Surgical Evaluation and Resection
As mentioned previously, the majority of patients present with a large pleural effusion and, on imaging, pleural studding.55,56 In these patients, thoracentesis and pleural biopsy is the initial step in making a diagnosis. Thoracentesis is able to provide a diagnosis of MPM in 33% to 84% of cases. Other modalities to assist in establishing a diagnosis includes the use of a Cope or Abrams needle. This is effective in 30% to 50% of cases.57
Thoracoscopy is an essential tool to aid in the diagnosis and management of MPM. This is especially helpful in patients with large pleural effusions with no appreciable mass on imaging. With thoracoscopy, the surgeon is able to directly visualize the entire thorax space. This includes evaluation of the visceral and parietal pleura and chest wall. In addition, mediastinal structures including pericardium and mediastinal lymph nodes can be directly evaluated to aid in determining the extent of future resection. Lastly, the diaphragm can be inspected from the thoracic side to determine the extent of disease. If on imaging or during thoracoscopy there is diaphragmatic involvement, laparoscopy must be strongly considered.58 At the time of thoracoscopy, ample biopsies of abnormal pleura can be performed directly. In addition, if contralateral thoracic involvement of MPM is suspected, thoracoscopic approaches can be used to aid in confirming the diagnosis.
After determining the extent of disease, the patient must be evaluated medically to determine suitability for resection and to help guide the type of resection required. An echocardiogram is performed to assess cardiac function and, in particular, to evaluate if there is any right heart dysfunction. If extrapleural pneumonectomy (EPP) is performed, this creates pulmonary hypertension and some degree of right heart strain. Preoperative cardiac imaging will help assess whether the patient can tolerate this insult on the heart. In addition, duplex imaging of the lower extremities has been found to be of assistance in identifying patients with deep vein thrombosis (DVT) to prevent the often lethal complication of pulmonary embolism in a pneumonectomy patient.59 For patients identified to have a DVT preoperatively, treatment with anticoagulation and, if appropriate, placement of an inferior vena cava (IVC) filter is highly recommended.
Patients fit for surgery must have a Karnofsky performance status greater than 70 and have normal kidney and hepatic function. In addition, their room air Pco2 must be less than 45 mm Hg, Po2 greater than 65 mm Hg, and an ejection fraction of 45% or greater. In addition, in accordance with the American College of Chest Physicians (ACCP) guidelines, patients should have a forced expiratory volume in the first second (FEV1) greater than 2 L or have a predicted postoperative (PPO) FEV1 of greater than 800 mL. Those patients with an FEV1 less than 2 L should undergo quantitative perfusion scan to determine their exact PPO FEV1.60 Patients with PPO FEV1 of less than 800 mL may be candidates for pleurectomy and decortication (P/D) rather than EPP.
The primary goal of surgery in the multimodality treatment of MPM is to achieve maximum cytoreduction of the tumor. With MPM, this is defined as an R1 resection.61 Despite the effectiveness of chemotherapy and radiotherapy, surgical therapy remains the foundation of potential curative treatment for MPM. The secondary objective of surgery is to improve symptoms. This includes evacuation of the pleural effusion and pulmonary decortication of an entrapped lung, which improves pain related to chest wall invasion of the MPM. There are three operations that are typically used in the treatment of MPM. These include EPP, P/D, and palliative limited pleurectomy.
EPP involves en bloc resection of the pleura, lung, pericardium, and diaphragm. The steps involved with this procedure have been described previously62 and include
3. Division of the pulmonary artery and vein
4. Division of the main stem bronchus
5. Mediastinal lymphadenectomy
With careful selection of patients and operations performed by experienced thoracic surgeons in centers knowledgeable of the treatment of MPM, operative mortality is approximately 5%.
P/D involves attempts at complete resection of gross pleural disease without resection of the lung parenchyma. During this procedure, resection of the pericardium and diaphragm may be required and therefore require reconstruction, similar to the techniques used for EPP. Mediastinal lymphadenectomy is also performed. Again, the main difference between P/D and EPP involved the pulmonary resection. The mortality for this procedure is 1.8% to 4%,63,64 yet there is less morbidity as a result of preservation of the pulmonary parenchyma.
Chemotherapy
For a prolonged period of time, there was doubt about the efficacy of chemotherapy in unresectable MPM and it is only recently that palliative chemotherapy has been accepted. The goals of chemotherapy are to reduce disease related symptoms, maintain or improve quality of life, and extend overall survival (OS). In general, for patients to be considered chemotherapy candidates, they should be ambulatory (ie, an Eastern Cooperative Oncology Group [ECOG] performance status [PS] of 0 to 2 or a Karnofsky PS of ≥70), have adequate organ function, and not have significant co-morbidities. One challenge when attempting to assess the activity of chemotherapy is that assessment of radiographic response by the commonly used Response Evaluation Criteria in Solid Tumors (RECIST) is difficult given the radiographic appearance and growth pattern of MPM.65 Consequently, many physicians use a modified RECIST using the sum of six unidimensional measurements of pleural thickness in order to reduce intra- and interobserver variability.66 The modified RECIST may be particularly useful when assessing the activity of novel agents.
The standard first-line therapy for MPM is a platinum agent (e.g., cisplatin or carboplatin) with an antifolate agent (e.g., pemetrexed or raltitrexed), based on two phase III clinical trials.67,68 A phase III trial by Vogelzang et al. compared cisplatin alone and with pemetrexed in chemotherapy-naïve patients who were not eligible for surgical resection (n = 456).67 Patients assigned to the combination arm compared to the cisplatin alone arm experienced a superior overall response rate (ORR), median time to progression, and OS (Table 73-3). A phase III trial of the EORTC compared cisplatin alone to cisplatin and raltitrexed in chemotherapy-naïve patients (n = 250).68 Patients assigned to the combination experienced a numerically higher ORR and longer progression-free survival (PFS), and a statistically significant longer OS. No difference in health-related quality of life was observed between the two arms.
Table 73-3
Select Studies of First-Line Treatments for MPM
| First Author | Comparison | No. of Patients | Overall Response Rate | Median Time to Progression or Progression-Free Survivor | Overall Survival |
| Vogelzang67 | Cisplatin vs cisplatin/pemetrexed | 456 | 16.7% | 3.9 mo | 9.3 mo |
| 41.3% | 5.7 mo | 12.1 mo | |||
| P < 0.0001 | P = 0.001 | P = 0.02 | |||
| Van Meerbeeck68 | Cisplatin vs cisplatin/raltitrexed | 250 | 13.6 % | 4.0 | 8.8 mo |
| 23.6% | 5.3 | 11.4 mo | |||
| P = 0.056 | HR = 0.78, P = 0.059 | HR = 0.76, P = 0.048 | |||
| Santoro69 | Cisplatin/pemetrexed vs carboplatin/pemetrexed | 1704 | 26.3% | 7 mo | NAa |
| 21.7% | 6.9 mo | NA |
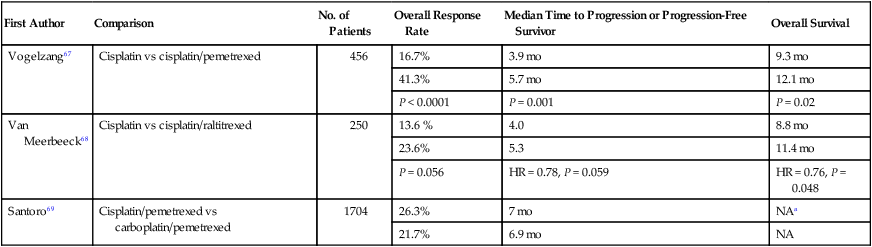
NA, not available; HR, hazard ratio
aThe 1-year survival rate is the cisplatin/pemetrexed and carboplatin/pemetrexed groups was 63.1% and 64%, respectively.
One clinical question is whether carboplatin can be substituted for cisplatin, but data from randomized trials investigating this question are not available. However, retrospective data from a large expanded access program is available; the selection of carboplatin or cisplatin was based on physician’s selection. The International Extended Access Program was a multicenter, nonrandomized study that enrolled chemotherapy-naïve patients; patients could receive pemetrexed with cisplatin (n = 843) or carboplatin (n = 861).69 The ORR, median time to progression, and 1-year survival rates were similar (Table 73-3). Regarding safety, patients receiving cisplatin/pemetrexed experienced a higher rate of grade 3 or 4 neutropenia (36.1% vs 23.9%) and a lower rate of grade 3 or 4 anemia (7.2% vs 14.3%) and thrombocytopenia (5.0% vs 14.3%). Other phase I or II trials have revealed a similar efficacy to carboplatin and pemetrexed.70,71 These data suggest that the cisplatin or carboplatin in combination with pemetrexed have similar efficacy, and carboplatin may be substituted for cisplatin in patients who have a relative or absolute contraindication to cisplatin.
A phase III trial compared active symptom control (ASC) alone, defined as treatment with steroids, analgesic drugs, bronchodilators, palliative radiotherapy, to ASC plus mitomycin, vinblastine, and cisplatin (MVP), or ASC plus weekly vinorelbine.72 The primary end point was OS, and the trial was designed to enroll 840 patients to detect a 3-month difference in OS. The trial design was changed because of slow accrual, and the ASC was compared to ASC plus chemotherapy in a two-arm design with the ability to detect an improvement from a median OS of 9 months with ASC to 12 months with ASC plus chemotherapy (5% significance level, 76% power, and 420 total patients). A significant difference in ASC compared to ASC plus chemotherapy was not detected (hazard ratio [HR] = 0.89, 95% confidence interval [CI] = 0.72 to 1.10, P = 0.29, median OS = 7.6 and 8.5 months, respectively). An exploratory analysis suggested a survival benefit with the addition of vinorelbine, but there was no evidence of survival benefit with MVP. These results are provocative, but interpretation is difficult because survival in the chemotherapy arm was poor, the changes in the study design and the chemotherapy used does not reflect current standards. The OS of the ASC provides an estimated survival for patients who elect not to pursue chemotherapy.
All patients experience disease progression, and many patients will be candidates for further chemotherapy. Currently, there is no defined standard of care for second-line therapy, and numerous single-arm, phase II trials have been performed of single agent or combination in the second-line setting.73 Some of these trials have demonstrated activity in terms of response, but none have been promising enough to warrant a phase III trial. Single agent vinorelbine and in combination with cisplatin has demonstrated activity, and would be considered a reasonable choice.74,75 The combination of cisplatin or carboplatin and gemcitabine may be a reasonable choice in the appropriate patient, and outside the context of a clinical trial.76,77 The activity of single agent gemcitabine appears to be limited,78,79 and the combination of gemcitabine and pemetrexed does not appear to have greater activity than single agent pemetrexed.80
In many cancers, the development of targeted therapies has revolutionized treatment. Several targeted therapies have been investigated in MPM but these results have been disappointing. The epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors have been investigated in phase II trials, and the results do not reveal significant activity.83–83 This is probably due to the absence of EGFR mutations in MPM.84 Patients with MPM have elevated vascular endothelial growth factor (VEGF) levels, and thus there is preclinical rationale for investigating bevacizumab, a monoclonal antibody against VEGF. However, a randomized phase II trial of cisplatin and gemcitabine alone and with bevacizumab in chemotherapy-naïve patients did not reveal an improvement in the primary end point of PFS or OS with the addition of bevacizumab.85 A randomized phase II/III trial of cisplatin and pemetrexed alone and with bevacizumab is ongoing, and data are available from the phase II component of the trial. The disease control rate was numerically higher in the bevacizumab-containing arm compared to the chemotherapy alone arm, and the rate of grade 3 or 4 toxicities were similar in the two arms. The criteria to proceed to the phase III component of the trial were met.86 Phase II trials of multitargeted tyrosine kinase inhibitors have not revealed significant activity and it is unlikely these agents will have a role in MPM.87–90 A phase III trial compared thalidomide maintenance therapy to observation in patients who did not experience disease progression after four cycles of carboplatin or cisplatin with pemetrexed; a similar PFS and OS were observed in the two arms.91 Imatinib has demonstrated limited efficacy in MPM as well.92,93 The indiscriminate investigation of targeted therapy and lack of a well-identified marker to enrich trial enrollment have probably contributed to the lack of success of targeted therapy in MPM.
Chemotherapy for Patients With Resectable MPM
Several phase studies have investigated preoperative or neoadjuvant chemotherapy in patients with resectable MPM.94 One single-arm phase II trial investigated four cycles of preoperative cisplatin and pemetrexed followed by EPP and hemithoracic radiation. The ORR was 32.5% (95% CI = 22.2 to 44.1), and patients experiencing a radiologic complete or partial response experienced a median survival of 26.0 months compared to 13.9 months among patients with stable or progressive disease (P = 0.05). Several studies have investigated preoperative chemotherapy with cisplatin or carboplatin and gemcitabine, and the response rate observed with this combination is 25% to 35%97–97; 40% to 80% of patients enrolled underwent EPP in these studies. In general, these studies indicate that this approach is feasible with reasonable toxicity in carefully selected patients, but it is difficult to estimate the additional benefit of the preoperative therapy in terms of OS or increased rate of surgical resection. Although a definitive conclusion cannot be made, preoperative chemotherapy is a reasonable approach in select patients. Our practice has been to use four cycles of cisplatin and pemetrexed because that is the combination that has demonstrated benefit in advanced disease in a phase III trial.
Radiotherapy
MPM is difficult to treat because of its locally aggressive behavior, and surgical resection has been the cornerstone of curative treatment. Unfortunately, after EPP or P/D, the risk for local recurrence is high, ranging from 30% to 60%.64,98–100 Because the most common site of treatment failure is the ipsilateral hemithorax, the need to optimize local control has provided impetus for evaluation of adjuvant radiotherapy (RT). In fact, in a series of retrospective studies, the use of adjuvant hemithoracic RT after EPP or P/D to the chest cavity has been shown to improve local control and survival.103–103 Although some believe that MPM is relatively radiation “resistant,” there is in vivo data to suggest that the opposite is true and that MPM cells are actually quite sensitive; however, it is difficult to deliver effective tumoricidal doses of radiation to the pleura without significant toxicity, including death. The target volume is the preoperative extent of the pleural space, which is large, irregular, and close to radiosensitive normal structures and several critical structures, including the lungs, heart, and liver.104
Radiotherapy as a Component of Radical Treatment
Extrapleural Pneumonectomy and Adjuvant Radiotherapy
Much of the data regarding adjuvant RT after EPP is from single institutions and is retrospective in nature, which makes generalization/creation of a standard of care difficult. What is clear from all of the data is that outcomes of patients with this disease are far from optimal and newer combinations of therapy must be studied. Gordon et al. reported on eight patients treated with EPP, RT, and doxorubicin-containing chemotherapy.105 One patient remained disease free for 2 years, but the other 7 patients recurred locally by 18 months, highlighting the aggressive course of disease.
Rusch et al. retrospectively analyzed 105 patients, looking for factors contributing to poor local control.101 They postulated that the main cause of local failure after surgery and radiation therapy was insufficient radiation dose. As a result, they conducted a phase II trial evaluating an increased dose of RT (54 Gy) following resection (70% had EPP).102 Of the 54 patients who had an EPP and postoperative RT, 7 patients had a locoregional failure, but only 2 of these patients did not also synchronously have distant metastases. One grade 4 esophageal fistula was observed, but otherwise toxicity was manageable. Chemotherapy was not used in this trial and outcomes may have been better if it had been.
Intensity-Modulated Radiotherapy After EPP
Further attempts at improving local control with radiotherapy after EPP have focused on the use of intensity-modulated radiotherapy (IMRT); IMRT has the flexibility to deliver dose distributions that conform to complicated convex and concave target volumes, while minimizing dose to critical structures in proximity.106 What limits our ability to give higher doses and perhaps achieve higher control rates are the surrounding normal tissues, most often the remaining lung, heart, and liver.
In 2007, Rice et al. reported on 63 patients who underwent EPP and received IMRT (median dose 45 Gy) at the MD Anderson Cancer Center.107 Median OS was poor (10.2 months), but only 5% patients had recurrence within the irradiated field. Buduhan and colleagues analyzed 46 patients who received induction chemotherapy, followed by EPP and adjuvant radiotherapy (median dose 50.4 Gy); 24 patients were treated with conventional external beam radiotherapy (EBRT) and 14 with IMRT.108 Survival was somewhat higher than historical controls (median 24 months). The incidence of local recurrence was 14.3% with IMRT versus 41.7% with EBRT (P = 0.03), suggesting that adjuvant IMRT might be more effective in terms of local control than EBRT. However, IMRT tends to give low doses to larger volumes of normal tissue and has been shown to increase treatment-related morbidity and mortality.
In a series of 13 patients treated with IMRT (median dose 54 Gy) at Harvard, 6 patients died secondary to radiation pneumonitis.109 These patients had a significantly higher mean volume of lung that received radiation. Unfortunately, there are no accepted dose-limit parameters that have been established in this setting, and often attempts are made to limit the low-dose volume as much as possible.
A more modern series was reported from Duke University.110 Thirty patients underwent EPP and received adjuvant IMRT. Two-year local control, DFS, and OS rates were 47%, 34%, and 50%, respectively. Four patients experienced radiation-related pulmonary toxicity (3 grade 3 or 4, 1 grade 5), though the authors pointed out that these 4 patients were treated early on in their experience with IMRT, and that none of the most recently treated 15 patients developed pneumonitis. They suggested minimizing the mean dose to the remaining healthy lung, as well as limiting the dose of that lung that receives 5 Gy or more to less than 50%.
Pleurectomy/Decortication and Adjuvant Radiotherapy
P/D involves resection of all gross tumor without removing the lung, and as one might expect, local control remains the primary issue. Various strategies have been investigated to improve local control, including intrapleural chemotherapy, photodynamic therapy, brachytherapy, and EBRT. Reports have varied regarding the efficacy of radiation in this setting. P/D and adjuvant RT yields an OS of 9 to 18 months, depending on the series cited.113–113
An analysis from Memorial Sloan Kettering Cancer Center summarized the results of 123 patients treated with P/D followed by hemithoracic RT (median dose 42.5 Gy), with or without brachytherapy.114 The median and 2-year OS was 13.5 months and 23%, respectively. Multivariate analysis for OS revealed radiation doses <40 Gy, nonepithelioid histology, left-sided disease, and the use of an implant all to be unfavorable prognostic factors (P < 0.02). This study suggests that P/D and adjuvant hemithoracic RT is not an effective treatment option for MPM. As a result of poor local control with conventional RT techniques after P/D, IMRT may have the potential to improve these results.
Radiotherapy for Palliation of Symptoms
Radiotherapy has been reported to produce relief of chest pain and objective tumor responses, though data are limited. The first prospective assessment of the role of RT in palliation was performed in Glasgow in the 1980s.115 Twenty-two patients received 30 Gy in 10 fractions to the whole hemithorax for pain; 13 had less pain at 1 month, but this had fallen to 3 patients by 3 months and 1 patient by 5 months; median survival was only 4 months. In a second study in Sweden, 47 patients received 40 Gy in 20 fractions to the hemithorax; 16 subsequently received doxorubicin and cyclophosphamide.116 Only 1 of 31 (3%) exhibited a partial response, and the authors reported no favorable effect on chest pain, weight loss, or improvement in performance status. It is the practice at our institution to utilize localized RT to sites of pleural disease that may be eroding bone or causing focal pain. We do not recommend hemithoracic RT for palliation.
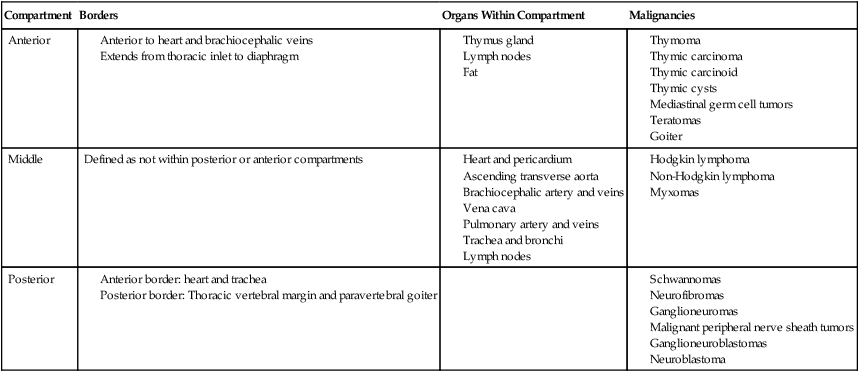
Diseases of the Mediastinum
The mediastinum is located in the central part of the chest, and the borders are considered the two pleural cavities, the diaphragm and the thoracic inlet.117 The mediastinum is further divided into the anterior, middle, and posterior compartments based on anatomic structures to assist in the development of a differential diagnosis (Table 73-4).118,119 Primary tumors of the mediastinum include neoplastic, congenital, and inflammatory conditions; approximately two-thirds of all mediastinal tumors are benign.118,120 The differential diagnosis of a mediastinal mass includes thymoma, thymic carcinoma, non-Hodgkin lymphomas (NHLs), Hodgkin lymphoma, teratomas, primary mediastinal germ cell tumors, thymic cysts, lymphangioma, and intrathoracic goiter. Masses in the anterior mediastinum are more likely to be malignant; and of the masses identified in anterior, middle, and posterior mediastinum, 59%, 29%, and 16%, respectively are malignant.121
Table 73-4
Mediastinal Compartments and Organs118, 119
| Compartment | Borders | Organs Within Compartment | Malignancies |
| Anterior |
The potential diagnoses of non-Hodgkin lymphoma (NHL), Hodgkin disease (HD) and mediastinal germ cell tumor require special consideration. Many NHL involve the mediastinum; however, there is frequently disease outside the mediastinum at the time of diagnosis. Hodgkin’s lymphoma is the most common mediastinal lymphoma and frequently presents with a bulky anterior mediastinal mass as the primary site of disease. Lymphoblastic lymphomas occur in the mediastinum, generally in adolescents and young adults, and can grow rapidly and cause airway or superior vena cava compression. The diagnosis and management of these lymphomas are covered in other chapters of this book (see Chapter 97). If a primary mediastinal germ cell is suspected based on the clinical and radiographic presentation, then a testicular exam and laboratory exam of α-fetoprotein, β-human chorionic gonadotropin and lactate dehydrogenase (LDH) should be part of the evaluation. Mature teratomas are the most common mediastinal germ cell tumors, and the radiographic appearance of teratomas on CT can assist in the identification of these tumors.118 The management of mediastinal germ cell tumors is similar to the standard management of other germ cell tumors, and is covered in other chapters in this book (see Chapter 86). The focus of this section will be the management of primary malignancies of the mediastinum.
Anterior Mediastinal Mass
Thymoma
The most common tumor of the anterior mediastinum is thymoma, and it is estimated that thymomas represent 20% of tumors of the mediastinum. Most patients are older than 40 years and they occur with equal frequency in men and women.118 Approximately 50% of patients with thymoma are asymptomatic at the time of diagnosis, and the mass is incidentally detected on an imaging study. Among patients who are symptomatic at the time of diagnosis, the most common associated symptoms are chest pain, cough, and dyspnea related to compression of adjacent structures.118,122 Radiographically, thymoma is an anterior-superior mediastinal mass that is well defined, encapsulated, and is generally homogeneous.123 It may become heterogeneous if there are areas of hemorrhage, necrosis, or cyst formation. Assessing invasion on CT scan can be challenging, but characteristic features of invasion are vascular compromise and irregular interface with the surrounding lung.123 Thymoma can spread circumferentially around the pleura, and the CT should extend through the abdomen to evaluate for potential transdiaphragmatic extension.
Approximately half of patients will have a parathymic syndrome such as myasthenia gravis, pure red cell aplasia, or hypogammaglobulinemia. Myasthenia gravis occurs in 30% to 50% of patients with thymoma, but only 15% of patients with myasthenia gravis will have thymoma.118,124 Pure red cell aplasia occurs in only 5% of patients with thymoma, but 50% of patients with pure red cell aplasia will have a thymoma.118 Thus, patients with suspected thymoma should undergo evaluation for these autoimmune disorders, and patients with these autoimmune disorders should be evaluated for thymomas as part of the initial workup. Patients with myasthenia gravis may have a better prognosis, possibly because of more indolent disease and/or earlier detection because of the symptoms of myasthenia.125 Thymectomy will result in resolution of the parathymic syndrome in patients with pure red blood cell aplasia and improvement in myasthenia gravis; all thymic tissue should be resected to reduce the rate of recurrence of parathymic syndromes.126
Thymomas are pathologically classified as epithelial neoplasms and are characterized by a combination of epithelial cells and mature lymphocytes. Thymic carcinoids and thymic carcinomas are tumors of the thymic epithelial cells, but are considered distinct and different clinical entities. Although the thymic carcinoids are histopathologically distinct, the distinction between thymomas and thymic carcinomas can be more difficult. Thymic carcinomas have malignant cytologic features whereas thymomas are generally considered cytologically benign.127 Thymomas are generally classified based on the World Health Organization (WHO) classification (Table 73-5),45 and the most commonly used staging system is the Masaoka clinical staging system (Table 73-6).128 The traditional tumor, node, and metastasis staging system is less applicable to thymomas because they tend to spread locally and metastases are generally intrathoracic (ie, pleural, pericardium, or diaphragm). This WHO pathologic system has been correlated with 10-year OS rates.129
Table 73-5
WHO Classification of Thymoma45
| WHO Type | Histologic Terminology |
| A | Spindle cell, medullary |
| AB | Mixed |
| B1 | |
| B2 | Cortical |
| B3 | |
| C |
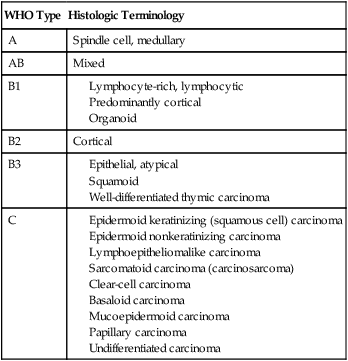
Table 73-6
Masaoka Clinical staging of Thymoma128
| Stage | Description |
| I | Macroscopically and microscopically completely encapsulated (tumor can invade into but not through the capsule) |
| IIA | Microscopic invasion through capsule |
| IIB | Macroscopic invasion into surrounding fatty tissue or mediastinal pleura |
| IIIA | Macroscopic invasion into neighboring organs (eg, pericardium or lung) |
| IIIB | Macroscopic invasion of great vessels |
| IVA | Pleural or pericardial dissemination |
| IVB | Lymphogenous or hematogenous metastases |
The rarity of thymoma has made it difficult to determine the optimal management, and many of the publications in the medical literature are retrospective analyses from single institutions. The disease course of thymoma can be very variable with late relapses, which complicate the evaluation of disease-free or relapse-free survival. In order to develop a standard approach to the treatment of patients with thymoma, a systemic review and the development of practice guidelines were developed by Cancer Care Ontario’s Program in evidence-based care.127 The clinical approach to a patient with thymoma is outlined in Fig. 73-1. The primary treatment for patients with thymoma is surgical resection if technically feasible and if the patient does not have significant co-morbidities. For patients with completely resected stage I disease, postoperative radiotherapy or systemic therapy is not recommended, as the recurrence rates after surgery alone are low. There is one randomized trial looking at the use of postoperative radiotherapy and numerous retrospective series suggesting the lack of benefit of adjuvant radiotherapy in completely resected stage I tumors (Table 73-7).130–138
Table 73-7
Masaoka Stage I Results From Several Institutions Using Postoperative Radiation or No Adjuvant Therapy
| Author, Year | Case No. | Years of Study | Radiation Dose (Gy) | Recurrence Rate | Survival Rate | |
| Surgery Alone | With RT | |||||
| Monden, 1985130 | 38 | 30-50 | 1/12 | 0/26 | ||
| Curran, 1988131 | 43 | 1960-1985 | 50 (32-60) | 0/42 | 0/1 | 67% (5-y) |
| Fujimura, 1987132 | 31 | 1965– | 0/31 | 74.3% (10-y) | ||
| Pollack, 1992133 | 11 | 1962-1987 | 50 | 1/6 | 1/5 | 74% (DFS, 5-y) |
| Quintanilla-Martinez, 1994134 | 52 | 1939-1990 | 0/52 | 100% (5-y) | ||
| Blumberg, 1995138 | 25 | 1949-1993 | 45 (5.23-116) | 1/25 | — | 95% (5-y) 86% (10-y) |
| Zhang, 1999135 | 29 | 1981-1996 Prospective |
Lymphocytic, 50 epithelial, 60 |
0/13 | 0/16 | 92% surgery, 88% with RT (10-y) |
| Ogawa, 2002136 | 17 | 1997-1998 Multiinstitute |
30-60 IF/WM |
— | 0/17 | 100% (10-y) |
| Singhal, 2003137 | 30 | 1992-2002 | 45-55 IF/EF |
1/27 | 0/3 | 94% (5-y) |
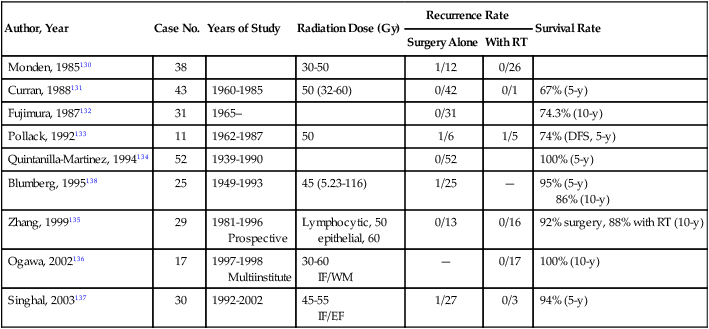
RT, radiotherapy; DFS, disease-free survival; WM, whole mediastinum.
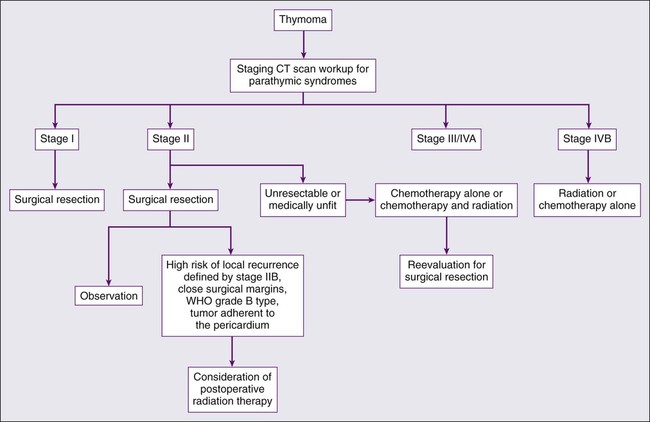
For patients with stage II disease, the surgical approach remains the same, and postoperative systemic therapy is not recommended. Radiation therapy should be considered for patients with stage II disease with a high risk of recurrence, often defined as invasion through the capsule (stage IIB), close surgical margins, WHO grade B type, and tumor adherent to the pericardium. Although the acute toxicities are generally limited and manageable, the long-term toxicities, specifically the risk for coronary artery disease and second malignancies, need to be considered.139,140 Preoperative chemotherapy or radiation therapy is not recommended for patients with stage I or II disease.
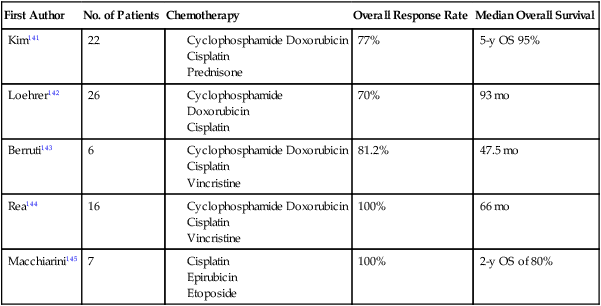
The optimal management strategy for patients with stage III disease is unclear, and a variety of multimodality treatment approaches are employed, including preoperative chemotherapy alone and with radiation, and surgical resection with postoperative chemotherapy or radiation therapy. The diagnosis of thymoma should be pathologically confirmed prior to initiating any preoperative therapy. The goals of preoperative therapy are to reduce the tumor size and render the marginally resectable or unresectable tumor resectable; therefore, radiographic response is frequently used as a trial end point. Most of the chemotherapy regimens contain a platinum or an anthracycline agent and have demonstrated response rates of 70% to 100% (Table 73-8).141–145 Interpretation of the long-term survival benefit of preoperative chemotherapy is not possible because of the small size of the trials and the variation in the use and timing of surgical resection, chemotherapy, and radiation. Patients with stage III and IVA thymoma were enrolled in these trials as well. These trials suggest that preoperative chemotherapy is feasible with acceptable acute toxicity. When using preoperative therapy, our approach has been to use chemotherapy alone or sequential chemotherapy and radiation, but concurrent chemoradiotherapy is a reasonable approach provided the treatment volumes are reasonable.127 Postoperative radiation therapy is commonly used in this situation; adjuvant chemotherapy may be considered but is not routine practice.
Table 73-8
Trials of Preoperative Therapy in Advanced Thymoma
| First Author | No. of Patients | Chemotherapy | Overall Response Rate | Median Overall Survival |
| Kim141 | 22 | 77% | 5-y OS 95% | |
| Loehrer142 | 26 | 70% | 93 mo | |
| Berruti143 | 6 | 81.2% | 47.5 mo | |
| Rea144 | 16 | 100% | 66 mo | |
| Macchiarini145 | 7 | 100% | 2-y OS of 80% |
Patients who experience a relapse after therapy for early stage or locally advanced disease should be evaluated for surgical resection if the relapse is isolated, and there has been a reasonable disease-free interval. Unfortunately, there are no specific criteria for a second resection and the decision is based on the individual patient and clinical judgment. For patients with an isolated relapse that is not amendable to surgical resection, radiation therapy is a reasonable option. For patients with stage IVB or disease not suitable for surgical resection or radiotherapy, systemic therapy is the standard of care, but the superiority of one systemic therapy has not been established. A number of different combinations with anthracycline and/or platinum agents have demonstrated activity in this setting, and there is not a defined standard of care.146–149 For patients with cardiac dysfunction anthracycline-based regimens should be avoided, and for patients with renal insufficiency cisplatin-based therapy should be avoided. The combination of octreotide alone and with prednisone has demonstrated activity. The advantage of this combination is that it does not contain a cytotoxic agent. There is some evidence that patients who have octreotide-avid disease on octreoscan derive greater benefit from this combination.150,151 Unfortunately, trials with targeted therapy such as belinostat, imatinib, dasatinib, sunitinib, sorafenib, gefitinib, and erlotinib have not revealed promising response rates or have consisted solely of case reports with responses.152–159
Radiation Therapy for Thymoma
Radiotherapy is given in five once-daily fractions per week for approximately 5 weeks. It is our policy to use standard fractions of 1.8 to 2.0 Gy to a total dose of 45 to 50.4 Gy. There are conflicting data on whether there is a dose response,136,160 but most agree that positive margins or gross residual disease require a dose of at least 50 Gy.161 Standard three-dimensional conformal radiotherapy (e.g., a wedged pair) is often sufficient to encompass the target volume, which includes the tumor bed and any gross residual disease. The presence of metallic surgical clips can help the radiation oncologist with target delineation. If a tumor grossly invades the adjacent lung and is resected, we do not routinely extend our fields to cover the area in lung that was resected. IMRT is sometimes necessary if the volume of critical structures (most often heart and/or lungs) is too large. A practical problem with the use of IMRT is the low dose spillage of dose that occurs, which may increase the risk of radiation-induced pneumonitis. Patients are immobilized with their arms above their head and radiation therapy planning is CT scan based. The anterior mediastinum often does not move much during the respiratory cycle, but if there is concern for motion, a four-dimensional scan can be performed to account for this.
Thymic Carcinoma
Thymic carcinomas are rare epithelial malignancies that have a more aggressive clinical course than thymomas. Thymic carcinomas comprise approximately 15% of thymic tumors, and have a 29% 10-year OS rate, which is significantly less than thymomas.129 Squamous cell carcinoma, lymphoepitheliomalike carcinoma, and undifferentiated are the most common histologic cell types.162,163 Thymic carcinoma is frequently locally invasive and can metastasize to regional lymph nodes and distant sites.163 The radiographic appearance is of a large, poorly defined and infiltrative anterior mediastinal mass; pleural and pericardial effusions are common.164 There is a paucity of data on the management of thymic carcinomas, but they are generally treated with multimodality therapy and with similar paradigms to thymomas. Surgical resection, when feasible, is the preferred therapy. There is a greater inclination to use postoperative radiotherapy and chemotherapy because of the higher rate of local and distant relapses; however, the impact of these modalities on clinical outcome is uncertain. When unresectable, our practice is to treat with chemoradiotherapy in fit patients. After approximately 50 Gy of radiation is delivered, we assess if the patient’s disease has become resectable—if it has not, we often administer an additional 10 to 16 Gy (total dose to gross disease 60 to 66 Gy).
Thymic Carcinoid
Thymic carcinoids are histologically different from thymomas and thymic carcinomas, and resemble other carcinoid tumors. They most commonly affect men in their 30s to 50s, and patients may be asymptomatic or have symptoms related to compression of adjacent structures.165 The radiographic appearance is often a large, lobulated, and invasive anterior mediastinal mass.165 Patients generally do not experience the classic carcinoid syndrome, but approximately 50% of patients have Cushing syndrome from ectopic production of adrenocorticotropin hormone or multiple endocrine neoplasia syndrome.165,166 Regional and distant metastases occur in up to 73% of patients, and bone metastases can be osteoblastic. The primary treatment is surgical resection, and radiotherapy for symptomatic local and distant metastases. These tumors are generally resistant to cytotoxic chemotherapy, but recent trials of targeted therapies have shown activity in low- to intermediate-grade pancreatic neuroendocrine tumors, and suggest possible activity in locally advanced or metastatic thymic carcinoids.167,168 Patients with Cushing syndrome need proper management of their paraneoplastic syndrome, and may require a referral to an endocrinologist for optimal management.
Nonmalignant Thymic Tumors
Thymolipoma is a rare, benign, slow-growing tumor of thymus that tends to affect young adults; women and men are affected equally.169 This tumor often presents as a large, encapsulated mass of adipose tissue and thymic tissue.170 CT scans reveal the combination of fat and soft tissue with attachment to the thymus, and conformation to adjacent structures.118 Surgical resection is curative. Congenital thymic cysts are thought to be remnants of the thymopharyngeal duct, and can be found from the neck to the anterior mediastinum.171 Inflammatory thymic cysts are potentially related to inflammation from a neoplasm (e.g., Hodgkin lymphoma, seminoma, thymic carcinoma).162 Congenital cysts tend to be <6 cm, are usually rounded, uniloculated or multiloculated, and have thin walls without inflammatory change. In contrast, acquired cysts are usually larger, multiloculated, and have variable wall thickness. The primary therapy is surgical resection, and inflammatory cysts should be carefully examined for an underlying neoplasm. Cervical goiters can extend into the mediastinum, and will have characteristic radiologic appearance; the treatment of choice for the symptomatic patient is surgical resection.
Tumors of the Middle Mediastinum
Primary malignant tumors of the heart are rare, and often benign. Myxomas are the most common benign tumor, and tend to be located in the left atrium. Malignant tumors may involve the heart through direct extension or metastases. The types of malignant tumors reported to metastasize to the heart include angiosarcoma, rhabdomyosarcoma, MPM, fibrosarcoma, lymphoma, extraskeletal osteosarcoma, neurogenic sarcoma, malignant teratoma, thymoma, leiomyosarcoma, liposarcoma, and synovial cell sarcoma.172 Angiosarcoma and rhabdomyosarcoma are the most common types of malignant tumors. The clinical symptoms of a cardiac tumor vary depending on the tumor location and the status of distant disease, but can include embolization. Pericardial involvement or metastases can occur with a variety of tumors. Excision is the primary treatment modality, but many times surgical excision is unsuccessful and patients are left with residual disease.173,174 Radiation and/or chemotherapy are options, but the benefit is limited. The prognosis for patients with malignant cardiac tumors is poor because tumors are frequently metastatic at the time of diagnosis, experience complications of the cardiac metastases, and many of the tumors are not particularly responsive to chemotherapy or radiotherapy. In addition, the radiation tolerance of the heart and adjacent critical structures (e.g., lung, esophagus) is limited, and oftentimes administering large doses of radiation results in significant morbidity with a small chance of long-term disease control.
Pericardial involvement can result in pericarditis, pericardial effusions, and potentially pericardial tamponade. The standard workup includes a chest CT, cardiac echocardiography, and an electrocardiogram. Patients with a cardiac tamponade physiology require immediate percutaneous drainage of the pericardial fluid. MPM is a very rare tumor in this location, and frequently rapidly fatal.175,176 Patients may present with a mediastinal mass, pericardial effusion and/or tamponade, and congestive heart failure.175–179 In the majority of the cases, complete surgical resection is not feasible, and the role and benefit of chemotherapy and radiation is unknown.
Posterior Mediastinal Tumors
Neurogenic tumors are the most common cause of posterior mediastinal mass, and they account for 75% of primary posterior mediastinal malignancies.119,180 Neurogenic tumors are classified according to the tissue of origin. Schwannoma, neurofibroma, and malignant tumors of peripheral nerve sheath arise from the peripheral nerves, and ganglioneuroma, ganglioneuroblastoma, and neuroblastoma arise from the sympathetic ganglia.181 Schwannomas are benign, slow-growing neoplasms that frequently arise from a spinal nerve root. The majority of patients are asymptomatic, but some patients will experience paresthesia or pain from compression of adjacent structures or extensions of the tumor into the spinal canal.182 Neurofibromas occur at a higher rate among patients with neurofibromatosis, and frequently patients with this disorder present with multiple neurofibromas or a single plexiform neurofibroma.183 Neurofibromatosis is not associated with multiple Schwannomas. Both Schwannomas and neurofibromas have similar radiographic appearance and classically are sharply marginated, spherical, and lobulated paraspinal masses.119 Patients whose tumors have suspected extension into the spinal canal should undergo a magnetic resonance imaging scan before pursuing surgical resection. The primary treatment is surgical resection, and tumors with extension into the spinal canal frequently require a combined neurosurgical and thoracic surgery approach.
Malignant peripheral nerve sheath tumors are the malignant variety of Schwannomas and neurofibromas and are also referred to as malignant Schwannomas, malignant neurofibromas, and neurogenic fibrosarcomas.119 These are pleomorphic, cellular malignancies, and about half of patients have neurofibromatosis. Malignant peripheral nerve sheath tumors may occur spontaneously or after radiation exposure.184 The most frequent symptoms are pain and/or neurologic deficits and symptoms; an enlarging mass is frequently associated with the onset of symptoms. These malignancies have the potential for hematogenous dissemination, and patients frequently develop pulmonary metastases. The preferred treatment is complete surgical excision; however, many times that is not feasible because of anatomic considerations. If complete surgical resection is not feasible then simple excision or subtotal resection with postradiation therapy is an alternative.185,186 Radiation doses of at least 50 to 60 Gy are required to obtain local control. Local recurrence is frequent, and adverse prognostic factors include large tumor size, incomplete resection, and neurofibromatosis.185,187
The sympathetic ganglia and adrenal glands can give rise to ganglioneuromas, ganglioneuroblastomas, and neuroblastomas; up to a third of neuroblastomas will originate in the mediastinum.119 Ganglioneuromas are benign and well differentiated as opposed to neuroblastomas, which are aggressive and undifferentiated malignant tumors. Ganglioneuromas typically affect adolescents and young adults, and most patients are asymptomatic. When symptoms are present, it is usually related to compression of adjacent structures or extension of the tumor into the spinal canal. The standard treatment is surgical resection. Ganglioneuroblastomas exhibit features of ganglioneuroma and neuroblastoma188; patients are generally young children (age < 10 years).189 Patients present with symptoms related to the large tumor size, compression of adjacent structures, or distant metastases. Neuroblastoma is an aggressive tumor, characteristically a tumor of young children. The treatment of these two malignancies is covered in other sections of this book (see Chapter 95).
Pleural Effusions
The incidence of malignant effusions is approximately 150,000 per year in the United States. The most common tumors associated with malignant pleural effusions are lung cancer (37.5%), breast cancer (16.8%), lymphoma (11.5%), and unknown primary (10.7%); lung cancer is the most common cause of pleural effusion among men and breast cancer is the most common cause among women.190 The presence of a malignant pleural effusion represents metastatic disease, and the prognosis is poor, with median survival times of 5 to 15 months.190 The variability in the prognosis may reflect the variability in the efficacy of treatments for the primary tumor and at which point patients develop pleural effusions in the natural history of the disease. The majority of patients who present with a pleural effusion are symptomatic. The most common symptom is dyspnea due to reduced compliance of the chest wall, depression of the ipsilateral diaphragm, reduction in lung volume, and mediastinal shift.191 Other common symptoms include cough and chest wall discomfort, and patients have constitutional symptoms frequently associated with advanced malignancy such as weight loss, fatigue, anorexia. Physical examination findings include decreased breath sounds on the affected side or dullness to percussion.
Management of Malignant Pleural Effusions
A pleural effusion occurs when there is an accumulation of pleural fluid due to an imbalance in hydrostatic pressure and oncotic pressure or an increase in permeability within the pleural space; the fluid accumulates between the parietal and visceral pleura. This space has a small amount of fluid the purpose of which is to lubricate the pleura to assist with lung mechanics. Typically, the rate of pleural fluid production is 0.01 mL/kg/h. This fluid is typically absorbed through parietal pleural lymphatics at a rate 28 times faster than the production.192 There are several circumstances that result in an increase in pleural fluid and include193
• increase in hydrostatic pressure (congestive heart failure),
• decrease in oncotic pressure (malnourished patients),
• increase in the space available for pleural fluid accumulation (lung collapse),
• increase in the permeability of the microvascular circulation secondary to inflammatory mediators,
• inadequate removal of pleural fluid through lymphatics secondary to obstruction from tumor or fibrosis, and
• transdiaphragmatic movement of ascitic fluid into the pleural space.
Imaging studies are helpful in the initial evaluation of pleural effusion. With plain chest radiographs, there is typically blunting of the costophrenic angle with a meniscus. This usually corresponds to at least 500 mL of pleural fluid.194 The pleural effusion in conjunction with other radiographic findings can help with determining the etiology of the pleural effusion. Ultrasonography is helpful at detecting pleural effusions as small as 50 mL. Ultrasonography can also be helpful in localization of the pleural fluid for thoracentesis. CT is helpful in determining the cause of the pleural effusion, assessing the characteristics of the fluid (loculated), and assisting with treatment planning.
As mentioned previously, thoracentesis is the next key step in the evaluation of the pleural effusion, and can be both therapeutic and diagnostic (Fig. 73-2). Complete drainage of the effusions allows for the clinician to determine if the effusion was the cause of the patient’s symptoms and it allows for adequate testing of the fluid to occur. It will also help the clinician to determine if the lung completely reexpands to fill the thoracic cavity. There are no absolute contraindications to thoracentesis. Complications related to thoracentesis include pneumothorax (3% to 20%),195 reexpansion pulmonary edema (1% to 2%),196 subcutaneous hematoma, and pleural infections.
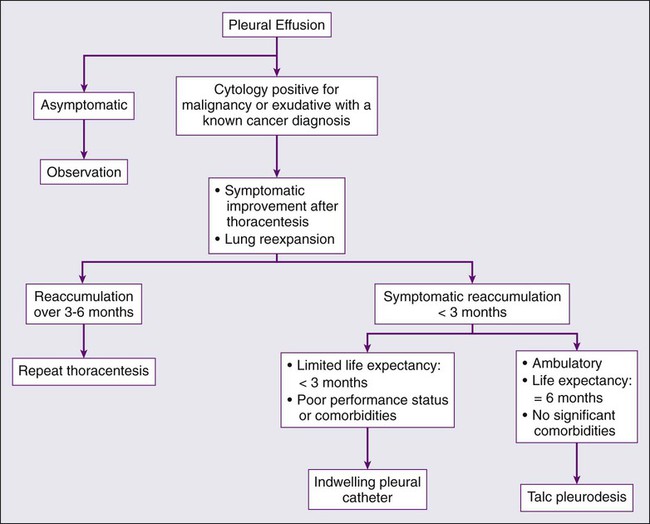
The pleural effusion should undergo analysis to determine the etiology of the effusion. Light’s criteria are used to determine whether an effusion is exudative versus transudative.197 This is performed by measuring the serum and pleural fluid LDH and protein levels. Exudative effusions include one or more of the following: pleural fluid/serum LDH greater than 0.6; pleural fluid/serum protein greater than 0.5; and pleural fluid LDH greater than two-thirds of the upper normal limits for serum.
The differential diagnosis for exudative etiologies for effusions is significantly larger and therefore other studies are helpful in determining the cause of the exudative pleural effusions. Low pH (<7.20) is the result of increased acid production by either cells or organisms and is typically related to an infectious etiology such as empyema or esophageal rupture or malignancy. Low glucose (<60 mg/dL) is due to increased utilization by polymorphonuclear leukocytes, malignant cells, and bacteria.198 A lymphocyte-predominant pleural effusion is typically seen in lymphoma-related effusions. Basophils are typically seen in association with leukemia, and plasma cell–predominant effusions are seen in myeloma associated conditions. Cytologic evaluation of the effusion is helpful in determining the etiology of a pleural effusion in 33% to 84% of cases. If thoracentesis is nondiagnostic, the next procedure of choice is thoracoscopy with biopsy. The clinical etiology to malignant pleural effusion includes most malignancies, with lung and breast most common.199
After thoracentesis or complete removal of the pleural effusion, if the patient does not see an improvement in his of her symptoms then no further therapy is warranted. If after thoracentesis or complete removal of the pleural effusion, there is improvement in symptoms, then the clinical decision revolves around whether there is complete expansion of the lung after drainage of the effusion. If the lung completely reexpands, then pleurodesis should be considered. Pleurodesis involves the sclerosis of the parietal and visceral pleura to prevent pleural fluid reaccumulation and to cause a pleural symphysis. Various agents such as bleomycin and tetracycline have been used in the past but sterilized talc is the sclerosing agent of choice for thoracic pleural symphysis.199 The sterilized talc is instilled by one of two methods; the patient may be taken to the operating room and thoracoscopy performed. Under these conditions, the appropriate diagnosis is confirmed and the lung parenchyma is inspected to confirm that the lung reexpands after removal of the pleural effusion. Aerosolized talc is then instilled in the chest cavity (approximately 4 to 5 g). A tube thoracostomy is then inserted, remains in place for 48 hours, and is then removed. The second method involves placement of a tube thoracostomy at the bedside. After confirmation of complete pulmonary expansion on chest x-ray, 4 to 5 g of sterile talc is suspended in 120 mL of sterile saline and inserted into the tube thoracostomy. The tube is clamped for 4 hours as the patient changes positions every 30 minutes. The tube is then unclamped, remains in place for 48 hours, and is then removed. The disadvantages to this approach are the hospitalization of the patient, small but possible incident of a talc reaction, and possible reexpansion pulmonary edema.200
If the lung does not reexpand, and the patient’s symptoms improve after thoracentesis, then the clinician should consider placement of an indwelling pleural catheter. This is a special catheter that is placed in an outpatient setting, which drains the pleural effusion on a regular basis. As the drainage decreases, the frequency of drainage should decrease such that if no fluid remains after 2 weeks of minimal drainage, the catheter is removed. This process is effective in that it is safe and effective in managing malignant pleural effusions in patients with entrapped lungs. There is a 25% chance of reexpansion of the lung with chronic frequent drainage of the catheter.201 The disadvantage of this process includes use of a chronic catheter for weeks to months and small risk of infection.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Diseases of the Pleura and Mediastinum
Benjamin E. Haithcock, Timothy M. Zagar, Longzhen Zhang and Thomas E. Stinchcombe
Malignant Pleural Mesothelioma (MPM)
• MPM is a rare disease with 2,000 to 3,000 cases annually in the United States.
• It is associated with prior occupational asbestos exposure, but some patients without known asbestos exposure will develop MPM.
• Four subtypes exist: epithelioid, sarcomatous, biphasic epithelial (also referred to as mixed), and desmoplastic.
• Epithelioid is the most common and may be associated with a better prognosis.
• Diagnosis must be differentiated from metastatic adenocarcinoma or non–small cell lung cancer with adenocarcinoma histology. No single test is diagnostic of MPM, and it frequently requires a panel of immunohistochemistry markers to confirm the diagnosis.
• Cytologic analysis of pleural fluid is not reliable to exclude MPM, and many times a thoracoscopic biopsy may be required for definitive diagnosis. Evidence of invasion into subpleural adipose tissue is the most reliable indicator of malignancy.
• Computed tomographic (CT) scan is the initial staging procedure.
• Positron emission tomography (PET) or PET-CT scan has the ability to detect extrathoracic disease.
• Cervical mediastinoscopy may be useful for detecting mediastinal involvement.
• Peritoneal lavage or laparoscopy is indicated if peritoneal involvement is suspected.
• For patients with operable disease without significant co-morbidities, surgical options include extrapleural pneumonectomy or pleurectomy and decortication.
• Single-institution and phase II trials have demonstrated the feasibility of multimodality therapy.
• The benefit of post- or preoperative radiation and chemotherapy is undefined.
• For patients with unresectable disease or metastatic disease, without significant treatment co-morbidities, and preserved performance status, treatment with a platinum agent (cisplatin or carboplatin) and antifolate (pemetrexed or raltitrexed) is the standard therapy.
• A variety of agents have shown activity in the second-line setting.
• Radiation therapy provides palliation of symptoms, and postoperative radiation therapy may reduce the rate of local and port site recurrence.
• Most common of all mediastinal tumors, it comprises approximately 20% of the tumors of the mediastinum.
• Differential includes thymoma, thymic carcinoma, and thymic carcinoid.
• Approximately 50% of patients with thymoma are asymptomatic at the time of diagnosis.
• Thymomas may be associated with parathymic syndromes (e.g., myasthenia gravis, pure red blood cell aplasia, and hypogammaglobulinemia)
• Thymomas are classified according to the World Health Organization (WHO) classification, which is associated with 10-year overall survival rates.
• Primary therapy is complete surgical resection.
• Postoperative radiation therapy should be considered for patients with stage IIB disease, close surgical margins, WHO grade B type, and tumor adherent to the pericardium.
• Chemotherapy with cisplatin or anthracycline-based therapy is an option for patients with unresectable or metastatic disease.
• Patients with malignant pleural effusions have a poor prognosis, and pleural effusion is considered metastatic disease.
• Common symptoms include dyspnea on exertion, shortness of breath, and cough.
• Most common causes of malignant pleural effusion are lung cancer, breast cancer, lymphoma, and cancer of unknown primary.
• Cytology can be diagnostic of type of malignancy, and patients with exudative effusion and known metastatic cancer should be considered as having a malignant pleural effusion.
• Thoracentesis may be diagnostic and provide symptomatic relief for the patient.
• Management strategies include intermittent thoracentesis, indwelling pleural catheters, and talc pleurodesis. The optimal treatment strategy may depend on the patient’s prognosis, performance status, and type of malignancy.
Primary Tumors of the Pleura: Mesothelioma
Primary benign and malignant tumors of the pleura are rare, and they include solitary fibrous tumor, adenomatoid tumor, pleural desmoid tumors, calcifying fibrous pseudotumor, and malignant pleural mesothelioma (MPM).1,2 For clinicians, the most frequent differential diagnosis is solitary fibrous tumor versus MPM. Solitary fibrous tumors of the pleura are significantly less common than MPM and are not associated with asbestos exposure.3 Solitary fibrous tumors are generally asymptomatic at the time of diagnosis, and the radiographic features are a well-circumscribed, peripheral mass that abuts the pleural surface, frequently attached by a pedicle.4 Approximately 10% to 20% of solitary fibrous tumors are classified as malignant; malignant tumors are characterized by mitoses, necrosis, atypia, and hypercellularity.4 Radiographically malignant solitary fibrous tumors compared to benign tumors tend to be larger and are associated with increased likelihood of being positive on positron emission tomography (PET).5,6 Patients with malignant solitary fibrous tumors tend to have a higher recurrence rate and worse survival.7–10 The primary treatment for solitary fibrous tumors is complete surgical resection if feasible.12–12 Because of the rarity of solitary fibrous tumors, it is difficult to define the natural history and the optimal management, but postoperative radiation and chemotherapy are not standard therapies. The primary focus of this section will be on MPM because that is the most common pleura malignancy.
Epidemiology
The association between MPM and asbestos exposure has been well established for decades; in the mid-20th-century, commercial uses for asbestos were developed, which led to increased occupational exposure of asbestos. Common sites of occupational exposure were in asbestos mines, shipyards, cement factories, or work with insulation. Asbestos refers to six fibrous silicate minerals found widely throughout the world, and is divided into two categories based on the chemical composition and crystalline structure13,14: a serpentine form (ie, chrysotile) and a thin, rodlike form (ie, amphiboles, including crocidolite, amosite, anthophyllite, tremolite, and actinolyte).15 The association of the amphibole form and MPM is well established, but the association between the serpentine form and MPM is a matter of debate.13 The long latency period between asbestos exposure and the development of MPM can make identification of the type, amount, and duration of asbestos exposure difficult. There does not appear to be a linear relationship between exposure and risk of developing MPM, and it appears that prolonged exposure is required in order to develop MPM. This is important for workers who had sporadic or limited exposure.13 A higher rate of MPM has been observed among family members of workers with occupational exposure caused by secondary exposure from clothes and close contact.16,17 Some patients with MPM will not report a known asbestos exposure, so the absence of a history of asbestos exposure should not eliminate MPM from the differential diagnosis.18 Other less common etiologic agents associated with MPM include prior radiation therapy, diagnostic use of thorium dioxide (Thorotrast), mineral fibers with similar properties (e.g., erionite), and potentially simian virus 40.19 In the United States, the incidence of MPM is estimated to be 2000 to 3000 cases per year and it appears to be rising, which is related to not only increased asbestos exposure a generation ago but also improved recognition.20,21
Despite the clear association between asbestos exposure and MPM, only a minority of people with significant exposure develop MPM (~5%), and the identification of MPM clusters within certain families raises the question about influence of genetics in carcinogenesis.14 A study of a MPM epidemic in Cappadocia, Turkey, and the United States revealed that the risk of developing MPM is transmitted in certain high-risk families, suggesting a genetic predisposition to erionite carcinogenesis.22 A high incidence of MPM in some families in the United States has been linked to germline mutations of the BAP1 gene.23 BAP1 somatic mutations have been identified in 25% of sporadic MPM as well.23,24 BAP1 appears to regulate deubiquitination during the DNA damage response and the cell cycle, and mutations that affect the deubiquitination of BAP1 or the nuclear localization reduce the tumor suppressor activity of BAP1.25 Other potential mechanisms of carcinogenesis include direct mechanical interference of asbestos fibers with chromosome segregation during mitosis or the generation of oxidants by macrophages attempting to digest asbestos fibers.28–28 The presence of asbestos causes inflammation, and the chronic inflammation associated with the asbestos fibers contributes to the process of carcinogenesis.14,29,30 The mechanism of carcinogenesis from chronic inflammation appears to be related to the cytokines tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β).30,31
Clinical Presentation
The most common presenting symptoms for patients with MPM are shortness of breath, dyspnea on exertion, and chest wall pain or discomfort. Other less common symptoms include fever of unknown origin, sweats, weight loss, and a decline in performance status.32 Many times patients have symptoms for months prior to presenting to a medical professional. On physical examination, decreased breath sounds on auscultation or dullness to percussion may be present. Frequently, a chest x-ray is performed and this reveals a pleural effusion and/or opacification in one hemithorax. There are no specific laboratory abnormalities or tumor markers diagnostic of MPM, although it has been associated with thrombocytosis.33
A thoracentesis is frequently performed to alleviate the patient’s symptoms, and it is part of the initial evaluation. A cytologic analysis of the pleural fluid is frequently performed; however, pleural effusion cytology is not reliable. Diagnostic cytologic criteria have not been established and invasion cannot be assessed on the specimen.36–36 Evidence of invasion into the subpleural adipose tissue is the most reliable indicator of malignancy.34,37 Thus, if there is clinical suspicion of MPM, a pleural effusion cytology negative for malignancy is not sufficient to eliminate MPM from the differential. Many times, a thoracoscopic biopsy is required, and if the tumor is surgically resectable the thoracoscopic port should be placed within the line of a potential thoracotomy so the area can be excised at the time of thoracotomy; this is to help prevent seeding/recurrence at the port site, which has been reported.
A number of prognostic markers have been identified including performance status, histology, presence or absence of chest pain, younger age, and normal platelet count.32,33,38–41 Many of these factors were identified from retrospective analyses that included small numbers of patients. The European Organization for Research and Treatment of Cancer (EORTC) performed a multivariate analysis and found poor prognosis to be associated with poor performance status, a high white blood cell count, a probable/possible histologic diagnosis of MPM (compared to definite diagnosis), male gender, and sarcomatous histologic subtype.42 On the basis of these five factors, patients were divided into a good prognosis group (1-year survival rate of 40%, 95% confidence interval [CI] = 30% to 50%), and a poor prognosis group (1-year survival rate of 12%, 95% CI = 4% to 20%). This model was validated based on independent clinical trials.43 An analysis of a phase III trial also validated the EORTC prognostic index and identified pain and appetite loss as independent prognostic factors.44 The prognostic index is useful to assist in the interpretation of phase II trials and may assist the clinician in estimating the prognosis for clinical decision making.
Pathology
The four subtypes of MPM according to the World Health Organization (WHO) are as follows: epithelioid, sarcomatous, biphasic epithelial (also referred to as mixed), and desmoplastic.45 The epithelioid type is the most common and has a better prognosis compared to the other types. It can be difficult to distinguish MPM from reactive mesothelial hyperplasia, non–small cell lung cancer (NSCLC) with adenocarcinoma histology, and metastatic adenocarcinoma. Unfortunately, no single immunohistochemistry (IHC) stain can differentiate MPM from NSCLC with adenocarcinoma histology, and a panel of IHC stains is frequently used. One practice is to test for two markers that are positive in MPM (e.g., cytokeratin AE1/AE3, keratins [e.g., CK 5/6, CK 7], calretinin, Wilms tumor 1 [WT-1; nuclear staining], D2-40) as well as two that are negative for MPM (e.g., carcinoembryonic antigen [CEA] and thyroid transcription factor 1 [TTF-1]) (Table 73-1).46
Table 73-1
Histochemistry Studies to Distinguish NSCLC and MPM46
| Stain | Adenocarcinoma | MPM |
| Cytokeratin AE1/AE3 |
|
|
| Keratins |
|
|
| Calretinin |
|
|
| WT-1 |
|
|
| D2 40 |
|
|
| CEA |
|
|
| TTF-1 |
|
|
Staging
The natural history of MPM is growth along the pleural surfaces of the thoracic cavity and invasion of the surrounding lung tissue, followed by transdiaphragmatic extension, peritoneal spread, and metastatic disease. The staging system provides an estimate of the prognosis, and an assessment if the tumor is potentially resectable. A number of different staging systems have been used,47,48 and there has been a lack of a consensus on the optimal staging system. The tumor, nodal, and metastasis (TNM) staging system is often used (Table 73-2).49 Computed tomography (CT) is the standard staging procedure. An assessment of the mediastinal lymph nodes is critical for assessment of staging and for potential resection, and the staging tests frequently used are CT, fluorodeoxyglucose positron emission tomography (FDG-PET), CT-PET, and mediastinoscopy. A recent systemic review of the imaging modalities revealed that FDG-PET was superior to CT scan, but inferior to CT/FDG-PET, in terms diagnostic specificity, sensitivity, and staging.50 The mean standardized uptake value was statistically higher in malignant compared to benign disease, and PET-CT had a higher sensitivity for detection of lymph node involvement. PET or PET-CT also has the ability to detect extrathoracic disease, and approximately 10% to 25% of patients will have distant disease detected with PET scanning.51,52 Transdiaphragmatic extension is a contraindication to surgical resection, and some centers perform peritoneal lavage or laparoscopy. In one study, the use of peritoneal lavage prior to surgical resection demonstrated transdiaphragmatic invasion of the peritoneal cavity or peritoneal metastases in approximately 10% of patients.53 The role of cervical mediastinoscopy is undetermined, but may be of value for patients being considered for surgical resection. A recent retrospective review revealed that patients without mediastinal involvement had a significantly better survival compared to patients with mediastinal disease.54 Noninvasive tests improve the staging, in particular, to increase the detection of patients with metastatic disease and to assist in the selection of the optimal site to biopsy; however, definitive staging requires surgical resection and pathological examination of the specimen.
Table 73-2
| TNM DESCRIPTION | |||
| PRIMARY TUMOR | |||
| Tx | Tumor cannot be assessed | ||
| T0 | No evidence of tumor | ||
| T1A | No involvement of the visceral pleura | ||
| T1B | Tumor also involving the visceral pleura | ||
| T2 | Tumor involving each of the ipsilateral pleural surfaces (parietal, mediastinal, diaphragmatic, and visceral pleura) with at least one of the following: involvement of diaphragmatic muscle; extension of tumor from visceral pleura into the underlying pulmonary parenchyma. | ||
| T3 | Locally advanced but potentially resectable tumor. Tumor involving all of the ipsilateral pleural surfaces (parietal, mediastinal, diaphragmatic, and visceral pleura) with at least one of the following: involvement of the endothoracic fascia; extension into the mediastinal fat; solitary, completely resectable focus of tumor extending into the soft tissues of the chest wall; nontransmural involvement of the pericardium. | ||
| T4 | Locally advanced technically unresectable tumor. Tumor involving all of the ipsilateral pleural surfaces (parietal, mediastinal, diaphragmatic, and visceral pleura) with at least one of the following: diffuse extension or multifocal masses of tumor in the chest wall, with or without associated rib destruction; direct transdiaphragmatic extension of tumor to the peritoneum; direct extension of tumor to the contralateral pleura; direct extension of tumor to mediastinal organs; direct extension of tumor into the spine; tumor extending through to the internal surface of the pericardium with or without a pericardial effusion or tumor involving the myocardium. | ||
| REGIONAL LYMPH NODES | |||
| Nx | Regional lymph nodes cannot be assessed | ||
| No | No regional lymph node metastases | ||
| N1 | Metastases in the ipsilateral bronchopulmonary or hilar lymph nodes | ||
| N2 | Metastases in the subcarinal or the ipsilateral mediastinal lymph nodes, including the ipsilateral internal mammary and peridiaphragmatic nodes | ||
| N3 | Metastases in the contralateral mediastinal, contralateral internal mammary, ipsilateral or contralateral supraclavicular lymph nodes. | ||
| DISTANT METASTASIS | |||
| M0 | No distant metastasis | ||
| M1 | Distant metastasis present | ||
| ANATOMIC STAGE/PROGNOSTIC GROUPS | |||
| Stage | T | N | M |
| I | T1 | N0 | M0 |
| IA | T1a | N0 | M0 |
| IB | T1b | N0 | M0 |
| II | T2 | N0 | M0 |
| III | T1, T2 | N1 | M0 |
| T1, T2 | N2 | M0 | |
| T3 | N0, N1, N2 | M0 | |
| IV | T4 | Any N | M0 |
| Any T | N3 | M0 | |
| Any T | Any N | M1 | |
Surgical Evaluation and Resection
As mentioned previously, the majority of patients present with a large pleural effusion and, on imaging, pleural studding.55,56 In these patients, thoracentesis and pleural biopsy is the initial step in making a diagnosis. Thoracentesis is able to provide a diagnosis of MPM in 33% to 84% of cases. Other modalities to assist in establishing a diagnosis includes the use of a Cope or Abrams needle. This is effective in 30% to 50% of cases.57
Thoracoscopy is an essential tool to aid in the diagnosis and management of MPM. This is especially helpful in patients with large pleural effusions with no appreciable mass on imaging. With thoracoscopy, the surgeon is able to directly visualize the entire thorax space. This includes evaluation of the visceral and parietal pleura and chest wall. In addition, mediastinal structures including pericardium and mediastinal lymph nodes can be directly evaluated to aid in determining the extent of future resection. Lastly, the diaphragm can be inspected from the thoracic side to determine the extent of disease. If on imaging or during thoracoscopy there is diaphragmatic involvement, laparoscopy must be strongly considered.58 At the time of thoracoscopy, ample biopsies of abnormal pleura can be performed directly. In addition, if contralateral thoracic involvement of MPM is suspected, thoracoscopic approaches can be used to aid in confirming the diagnosis.
After determining the extent of disease, the patient must be evaluated medically to determine suitability for resection and to help guide the type of resection required. An echocardiogram is performed to assess cardiac function and, in particular, to evaluate if there is any right heart dysfunction. If extrapleural pneumonectomy (EPP) is performed, this creates pulmonary hypertension and some degree of right heart strain. Preoperative cardiac imaging will help assess whether the patient can tolerate this insult on the heart. In addition, duplex imaging of the lower extremities has been found to be of assistance in identifying patients with deep vein thrombosis (DVT) to prevent the often lethal complication of pulmonary embolism in a pneumonectomy patient.59 For patients identified to have a DVT preoperatively, treatment with anticoagulation and, if appropriate, placement of an inferior vena cava (IVC) filter is highly recommended.
Patients fit for surgery must have a Karnofsky performance status greater than 70 and have normal kidney and hepatic function. In addition, their room air Pco2 must be less than 45 mm Hg, Po2 greater than 65 mm Hg, and an ejection fraction of 45% or greater. In addition, in accordance with the American College of Chest Physicians (ACCP) guidelines, patients should have a forced expiratory volume in the first second (FEV1) greater than 2 L or have a predicted postoperative (PPO) FEV1 of greater than 800 mL. Those patients with an FEV1 less than 2 L should undergo quantitative perfusion scan to determine their exact PPO FEV1.60 Patients with PPO FEV1 of less than 800 mL may be candidates for pleurectomy and decortication (P/D) rather than EPP.
The primary goal of surgery in the multimodality treatment of MPM is to achieve maximum cytoreduction of the tumor. With MPM, this is defined as an R1 resection.61 Despite the effectiveness of chemotherapy and radiotherapy, surgical therapy remains the foundation of potential curative treatment for MPM. The secondary objective of surgery is to improve symptoms. This includes evacuation of the pleural effusion and pulmonary decortication of an entrapped lung, which improves pain related to chest wall invasion of the MPM. There are three operations that are typically used in the treatment of MPM. These include EPP, P/D, and palliative limited pleurectomy.
EPP involves en bloc resection of the pleura, lung, pericardium, and diaphragm. The steps involved with this procedure have been described previously62 and include
3. Division of the pulmonary artery and vein
4. Division of the main stem bronchus
5. Mediastinal lymphadenectomy
With careful selection of patients and operations performed by experienced thoracic surgeons in centers knowledgeable of the treatment of MPM, operative mortality is approximately 5%.
P/D involves attempts at complete resection of gross pleural disease without resection of the lung parenchyma. During this procedure, resection of the pericardium and diaphragm may be required and therefore require reconstruction, similar to the techniques used for EPP. Mediastinal lymphadenectomy is also performed. Again, the main difference between P/D and EPP involved the pulmonary resection. The mortality for this procedure is 1.8% to 4%,63,64 yet there is less morbidity as a result of preservation of the pulmonary parenchyma.
Chemotherapy
For a prolonged period of time, there was doubt about the efficacy of chemotherapy in unresectable MPM and it is only recently that palliative chemotherapy has been accepted. The goals of chemotherapy are to reduce disease related symptoms, maintain or improve quality of life, and extend overall survival (OS). In general, for patients to be considered chemotherapy candidates, they should be ambulatory (ie, an Eastern Cooperative Oncology Group [ECOG] performance status [PS] of 0 to 2 or a Karnofsky PS of ≥70), have adequate organ function, and not have significant co-morbidities. One challenge when attempting to assess the activity of chemotherapy is that assessment of radiographic response by the commonly used Response Evaluation Criteria in Solid Tumors (RECIST) is difficult given the radiographic appearance and growth pattern of MPM.65 Consequently, many physicians use a modified RECIST using the sum of six unidimensional measurements of pleural thickness in order to reduce intra- and interobserver variability.66 The modified RECIST may be particularly useful when assessing the activity of novel agents.
The standard first-line therapy for MPM is a platinum agent (e.g., cisplatin or carboplatin) with an antifolate agent (e.g., pemetrexed or raltitrexed), based on two phase III clinical trials.67,68 A phase III trial by Vogelzang et al. compared cisplatin alone and with pemetrexed in chemotherapy-naïve patients who were not eligible for surgical resection (n = 456).67 Patients assigned to the combination arm compared to the cisplatin alone arm experienced a superior overall response rate (ORR), median time to progression, and OS (Table 73-3). A phase III trial of the EORTC compared cisplatin alone to cisplatin and raltitrexed in chemotherapy-naïve patients (n = 250).68 Patients assigned to the combination experienced a numerically higher ORR and longer progression-free survival (PFS), and a statistically significant longer OS. No difference in health-related quality of life was observed between the two arms.
Table 73-3
Select Studies of First-Line Treatments for MPM
| First Author | Comparison | No. of Patients | Overall Response Rate | Median Time to Progression or Progression-Free Survivor | Overall Survival |
| Vogelzang67 | Cisplatin vs cisplatin/pemetrexed | 456 | 16.7% | 3.9 mo | 9.3 mo |
| 41.3% | 5.7 mo | 12.1 mo | |||
| P < 0.0001 | P = 0.001 | P = 0.02 | |||
| Van Meerbeeck68 | Cisplatin vs cisplatin/raltitrexed | 250 | 13.6 % | 4.0 | 8.8 mo |
| 23.6% | 5.3 | 11.4 mo | |||
| P = 0.056 | HR = 0.78, P = 0.059 | HR = 0.76, P = 0.048 | |||
| Santoro69 | Cisplatin/pemetrexed vs carboplatin/pemetrexed | 1704 | 26.3% | 7 mo | NAa |
| 21.7% | 6.9 mo | NA |
NA, not available; HR, hazard ratio
aThe 1-year survival rate is the cisplatin/pemetrexed and carboplatin/pemetrexed groups was 63.1% and 64%, respectively.
One clinical question is whether carboplatin can be substituted for cisplatin, but data from randomized trials investigating this question are not available. However, retrospective data from a large expanded access program is available; the selection of carboplatin or cisplatin was based on physician’s selection. The International Extended Access Program was a multicenter, nonrandomized study that enrolled chemotherapy-naïve patients; patients could receive pemetrexed with cisplatin (n = 843) or carboplatin (n = 861).69 The ORR, median time to progression, and 1-year survival rates were similar (Table 73-3). Regarding safety, patients receiving cisplatin/pemetrexed experienced a higher rate of grade 3 or 4 neutropenia (36.1% vs 23.9%) and a lower rate of grade 3 or 4 anemia (7.2% vs 14.3%) and thrombocytopenia (5.0% vs 14.3%). Other phase I or II trials have revealed a similar efficacy to carboplatin and pemetrexed.70,71 These data suggest that the cisplatin or carboplatin in combination with pemetrexed have similar efficacy, and carboplatin may be substituted for cisplatin in patients who have a relative or absolute contraindication to cisplatin.
A phase III trial compared active symptom control (ASC) alone, defined as treatment with steroids, analgesic drugs, bronchodilators, palliative radiotherapy, to ASC plus mitomycin, vinblastine, and cisplatin (MVP), or ASC plus weekly vinorelbine.72 The primary end point was OS, and the trial was designed to enroll 840 patients to detect a 3-month difference in OS. The trial design was changed because of slow accrual, and the ASC was compared to ASC plus chemotherapy in a two-arm design with the ability to detect an improvement from a median OS of 9 months with ASC to 12 months with ASC plus chemotherapy (5% significance level, 76% power, and 420 total patients). A significant difference in ASC compared to ASC plus chemotherapy was not detected (hazard ratio [HR] = 0.89, 95% confidence interval [CI] = 0.72 to 1.10, P = 0.29, median OS = 7.6 and 8.5 months, respectively). An exploratory analysis suggested a survival benefit with the addition of vinorelbine, but there was no evidence of survival benefit with MVP. These results are provocative, but interpretation is difficult because survival in the chemotherapy arm was poor, the changes in the study design and the chemotherapy used does not reflect current standards. The OS of the ASC provides an estimated survival for patients who elect not to pursue chemotherapy.
All patients experience disease progression, and many patients will be candidates for further chemotherapy. Currently, there is no defined standard of care for second-line therapy, and numerous single-arm, phase II trials have been performed of single agent or combination in the second-line setting.73 Some of these trials have demonstrated activity in terms of response, but none have been promising enough to warrant a phase III trial. Single agent vinorelbine and in combination with cisplatin has demonstrated activity, and would be considered a reasonable choice.74,75 The combination of cisplatin or carboplatin and gemcitabine may be a reasonable choice in the appropriate patient, and outside the context of a clinical trial.76,77 The activity of single agent gemcitabine appears to be limited,78,79 and the combination of gemcitabine and pemetrexed does not appear to have greater activity than single agent pemetrexed.80
In many cancers, the development of targeted therapies has revolutionized treatment. Several targeted therapies have been investigated in MPM but these results have been disappointing. The epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors have been investigated in phase II trials, and the results do not reveal significant activity.83–83 This is probably due to the absence of EGFR mutations in MPM.84 Patients with MPM have elevated vascular endothelial growth factor (VEGF) levels, and thus there is preclinical rationale for investigating bevacizumab, a monoclonal antibody against VEGF. However, a randomized phase II trial of cisplatin and gemcitabine alone and with bevacizumab in chemotherapy-naïve patients did not reveal an improvement in the primary end point of PFS or OS with the addition of bevacizumab.85 A randomized phase II/III trial of cisplatin and pemetrexed alone and with bevacizumab is ongoing, and data are available from the phase II component of the trial. The disease control rate was numerically higher in the bevacizumab-containing arm compared to the chemotherapy alone arm, and the rate of grade 3 or 4 toxicities were similar in the two arms. The criteria to proceed to the phase III component of the trial were met.86 Phase II trials of multitargeted tyrosine kinase inhibitors have not revealed significant activity and it is unlikely these agents will have a role in MPM.87–90 A phase III trial compared thalidomide maintenance therapy to observation in patients who did not experience disease progression after four cycles of carboplatin or cisplatin with pemetrexed; a similar PFS and OS were observed in the two arms.91 Imatinib has demonstrated limited efficacy in MPM as well.92,93 The indiscriminate investigation of targeted therapy and lack of a well-identified marker to enrich trial enrollment have probably contributed to the lack of success of targeted therapy in MPM.
Chemotherapy for Patients With Resectable MPM
Several phase studies have investigated preoperative or neoadjuvant chemotherapy in patients with resectable MPM.94 One single-arm phase II trial investigated four cycles of preoperative cisplatin and pemetrexed followed by EPP and hemithoracic radiation. The ORR was 32.5% (95% CI = 22.2 to 44.1), and patients experiencing a radiologic complete or partial response experienced a median survival of 26.0 months compared to 13.9 months among patients with stable or progressive disease (P = 0.05). Several studies have investigated preoperative chemotherapy with cisplatin or carboplatin and gemcitabine, and the response rate observed with this combination is 25% to 35%97–97; 40% to 80% of patients enrolled underwent EPP in these studies. In general, these studies indicate that this approach is feasible with reasonable toxicity in carefully selected patients, but it is difficult to estimate the additional benefit of the preoperative therapy in terms of OS or increased rate of surgical resection. Although a definitive conclusion cannot be made, preoperative chemotherapy is a reasonable approach in select patients. Our practice has been to use four cycles of cisplatin and pemetrexed because that is the combination that has demonstrated benefit in advanced disease in a phase III trial.
Radiotherapy
MPM is difficult to treat because of its locally aggressive behavior, and surgical resection has been the cornerstone of curative treatment. Unfortunately, after EPP or P/D, the risk for local recurrence is high, ranging from 30% to 60%.64,98–100 Because the most common site of treatment failure is the ipsilateral hemithorax, the need to optimize local control has provided impetus for evaluation of adjuvant radiotherapy (RT). In fact, in a series of retrospective studies, the use of adjuvant hemithoracic RT after EPP or P/D to the chest cavity has been shown to improve local control and survival.103–103 Although some believe that MPM is relatively radiation “resistant,” there is in vivo data to suggest that the opposite is true and that MPM cells are actually quite sensitive; however, it is difficult to deliver effective tumoricidal doses of radiation to the pleura without significant toxicity, including death. The target volume is the preoperative extent of the pleural space, which is large, irregular, and close to radiosensitive normal structures and several critical structures, including the lungs, heart, and liver.104
Radiotherapy as a Component of Radical Treatment
Extrapleural Pneumonectomy and Adjuvant Radiotherapy
Much of the data regarding adjuvant RT after EPP is from single institutions and is retrospective in nature, which makes generalization/creation of a standard of care difficult. What is clear from all of the data is that outcomes of patients with this disease are far from optimal and newer combinations of therapy must be studied. Gordon et al. reported on eight patients treated with EPP, RT, and doxorubicin-containing chemotherapy.105 One patient remained disease free for 2 years, but the other 7 patients recurred locally by 18 months, highlighting the aggressive course of disease.
Rusch et al. retrospectively analyzed 105 patients, looking for factors contributing to poor local control.101 They postulated that the main cause of local failure after surgery and radiation therapy was insufficient radiation dose. As a result, they conducted a phase II trial evaluating an increased dose of RT (54 Gy) following resection (70% had EPP).102 Of the 54 patients who had an EPP and postoperative RT, 7 patients had a locoregional failure, but only 2 of these patients did not also synchronously have distant metastases. One grade 4 esophageal fistula was observed, but otherwise toxicity was manageable. Chemotherapy was not used in this trial and outcomes may have been better if it had been.
Intensity-Modulated Radiotherapy After EPP
Further attempts at improving local control with radiotherapy after EPP have focused on the use of intensity-modulated radiotherapy (IMRT); IMRT has the flexibility to deliver dose distributions that conform to complicated convex and concave target volumes, while minimizing dose to critical structures in proximity.106
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
