Chapter 10 Cytokines in liver, biliary, and pancreatic disease
Microbiologic Recognition and Toll-Like Receptors
In mounting an immune response, the challenge to the host is to identify the presence of an infectious process early and to coordinate its considerable resources to eradicate the pathogen, while minimizing tissue damage and chronic inflammation. The recognition of microbial pathogens and the ability to distinguish self from nonself relies heavily on the innate immunity and cell surface receptors present on antigen-presenting cells in the liver (Akira & Hemmi, 2003). Antigen-rich blood from the portal circulation has the potential to activate either the innate and adaptive immune responses against infections or to maintain immunologic tolerance of harmless antigens (Tacke et al, 2009). In addition, the infiltration of monocytes during liver injury is an important adaptive mechanism leading to chronic infammation (Karlmark et al, 2009). The immune response to a biologic insult is a highly organized process that involves pattern-recognition receptors (PRRs) that identify preserved structures of different pathogens. Toll-like receptors (TLRs) are an important family of at least 10 PRRs that are ubiquitously expressed in humans (Akira & Takeda, 2004). Current evidence demonstrates crucial roles for TLRs in alcoholic liver disease, nonalcoholic steatoheaptitis, hepatitis B, hepatitis C, hepatic fibrosis and cirrhosis, hepatocellular carcinoma, primary biliary cirrhosis, acetaminophen-induced hepatotoxicity, and ischemia-reperfusion (I/R) liver injury, all of which are potential therapeutic targets (Pimental-Nunes et al, 2010; Table 10.1).
Table 10.1 Toll-like Receptors and Their Potential Targets for Gastrointestinal Diseases
| Organ | Disease | Target |
|---|---|---|
| Stomach | Helicobacter pylori infection | 2, 4, 9 |
| Adenocarcinoma | 2, 4, 9 | |
| Pancreas | Acute pancreatitis | 2, 4, 9 |
| Chronic pancreatitis | 3 | |
| Pancreatic cancer | 2, 3, 6, 9 | |
| Liver | Alcoholic liver disease | 4 |
| Nonalcoholic steatohepatitis | 4 | |
| Hepatitis B | 3, 7, 8, 9 | |
| Hepatitis C | 3, 7, 9 | |
| Fibrosis/cirrhosis | 4, 9 | |
| Hepatocellular carcinoma | 3, 4 | |
| Ischemia/reperfusion | 2, 4 | |
| Colon | Inflammatory bowel disease | 2, 4, 5, 9 |
| Adenocarcinoma | 2, 4, 5, 9 |
Endotoxin Structure and Biologic Activity
Lipopolysaccharides (LPSs) from gram-negative bacteria have a wide range of harmful biologic activities and are therefore also considered endotoxins (Dinarello, 2004). Research over the past decade has elucidated the mechanisms by which the host recognizes endotoxin and the innate immune response is initiated. Much more is known about the cell surface receptors and signaling pathways involved in the host response to endotoxin than is known about several other microbial products and TLRs. Although endotoxin has been implicated in the beneficial and injurious host responses to gram-negative infections, the exact role of endotoxin in the pathogenesis of human disease remains unclear. Similarities in host responses to various microbial products, including LPSs from gram-negative bacteria and the glycoconjugates of gram-positive bacterial and fungal infections, suggest that the host innate immune system has evolved to respond consistently to microbial invasion, regardless of its antigenic source (Fearon & Locksley, 1996).
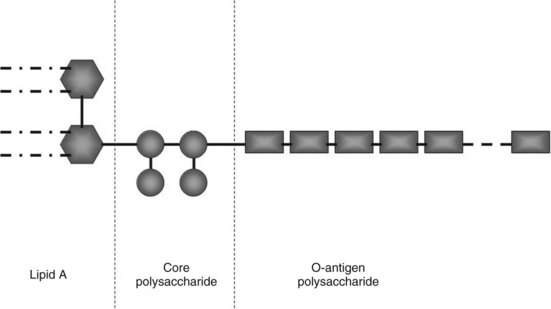
Endotoxin is the major constituent of the outer cell wall of gram-negative bacteria and can comprise 65% by weight of the total bacterium. Endotoxin is shed spontaneously from the cell walls of living bacteria and is released in copious amounts upon cell death and cell lysis. Endotoxin comprises three major components: an inner lipid A, an intermediate R-core oligosaccharide, and an outer O-polysaccharide (Fig. 10.1). The inner lipid A portion and the R-core oligosaccharide generally are conserved, whereas the structure of the O-polysaccharide is unique for each strain of bacteria. The R-core oligosaccharide is exposed in rough forms of gram-negative bacteria. The internal lipid A moiety is poorly antigenic but seems to be a primary factor in inducing most of the biologic effects of LPS. Binding of endotoxin to serum proteins and its activation of host immunity through its cellular receptor are mediated predominantly via the lipid A moiety; the O-antigen is primarily immunogenic (Ulevitch & Tobias, 1999).
The host response to endotoxin is immediate (within minutes), is dose dependent, and affects all organ and tissue systems (Table 10.2). High-dose endotoxin exposure in rodents and primates reproduces the toxic effects of gram-negative bacteremia, characterized by hemodynamic collapse, shock, organ failure, and death (Beutler et al, 1985). Low-dose administration of endotoxin to human volunteers (Fong et al, 1990; Michie et al, 1988) produces a variety of constitutional symptoms consistent with a milder infectious process, including fever, myalgia, tachycardia, occasional hypotension, transient leukopenia followed by neutrophilia, and a hepatic acute-phase response (Table 10.3).
| Fever |
Secondary to sympathetic neurogenic activity
Indirectly caused by catecholamine and prostaglandin release
Secondary to tissue hypoperfusion caused by vasoconstriction, endothelial injury, and reduced cardiac output
Indirectly caused by complement activation, neutrophil degranulation, prostacyclin and prostaglandin release, nitric oxide production
Produces pooling of blood in pulmonary and splanchnic vascular beds with hypotensive shock
Increased resting energy expenditure
Increased plasma growth hormone, corticotropin, and cortisol
Increased lipolysis with raised plasma free fatty acid levels
Alterations in carbohydrate metabolism, increased gluconeogenesis, and glycogenolysis
Decreased albumin synthesis, increased acute-phase response protein production mediated by IL-6 and, to a lesser extent, TNF-α and IL-1
IL, interleukin; TNF, tumor necrosis factor; CSF, colony-stimulating factor; PAF, platelet activating factor
Table 10.3 Biologic Responses to Low-Dose Endotoxin in Human Volunteers
| Constitutional Responses* |
| Cytokine/Hormone Responses |
| Hemodynamic Responses |
| Leukocyte Responses |
| Metabolic Responses |
TNF-α, tumor necrosis factor α; IL, interleukin; s-TNFR, soluble TNF receptor
Data from Fong et al, 1990: The acute splanchnic and peripheral tissue metabolic response to endotoxin in humans. J Clin Invest 85:1896-1904.
Although the host responses to endotoxin are likely to be dose dependent, there is now general acceptance that the clinical sequelae associated with systemic endotoxin administration are probably secondary to the host innate immune response rather than a result of direct interactions between endotoxin and cell membranes or secretory proteins (Fearon & Locksley, 1996; Ulevitch & Tobias, 1999). At high doses, endotoxin can produce direct endothelial injury, but most host responses to endotoxin are mediated by complement activation and the release of humoral factors, including proinflammatory cytokines, nitric oxide (NO), and prostaglandins.
Immune Regulation and Response to Endotoxin (SEE CHAPTER 9)
The host defense against many gram-negative bacteria is dependent on the innate immune recognition of endotoxin, and this response must be highly sensitive and self-limited. The sensitivity of this system is crucial for prompt mobilization to combat infection, but a risk of severe immune-mediated pathology exists if this response not self-limited. Optimal sensitivity is achieved by the strictly ordered interactions of endotoxin with different extracellular and cell surface proteins, including lipopolysaccharide (LPS)-binding protein (LBP), cluster of differentiation (CD)14, MD-2, and Toll-like receptor 4 (TLR-4) (Gioannini & Weiss, 2007). The presence of endotoxin in the plasma and lymphatic system is initially recognized by serum proteins and lipoproteins, and this recognition initiates the activation of the innate immune response (Beutler et al, 2003). Redundancy is the hallmark of this activation process, as proteins, glycoproteins, lipids, and nucleic acids of prokaryotic origins are recognized by different TLRs and other components of innate immunity. For the most part, the innate immune system relies on cell surface receptors and hepatic secretory proteins, primarily opsonins, to recognize carbohydrate and lipid, protein, and DNA structures indicative of a microbial infection.
Different constituents of specific microbial products, including endotoxin, are recognized by different aspects of the innate immune system (Ulevitch & Tobias, 1999). The first step in the recognition of endotoxin by the innate immune system is its binding to a hepatic secretory protein, LBP, to form the LPS-LBP complex (Fig. 10.2). LBP is a member of a group of homologous lipid-binding proteins that function as lipid-transfer proteins (Tobias et al, 1986, 1989; Wright et al, 1989). Secreted by the liver and found in human blood in 2 to 20 mg/mL concentrations, LBP binds avidly to the lipid A moiety of endotoxin and mediates its transfer to the cell surface endotoxin receptor, CD14. Although CD14 functions as a ligand-binding protein for endotoxin-LBP complexes, CD14 is bound to the cell membrane through a glycosyl–phosphatidylinositol anchor, and it lacks an intracellular domain capable of transducing a signal. LPS-LBP complex binding to CD14 alone is insufficient to transduce a signal.
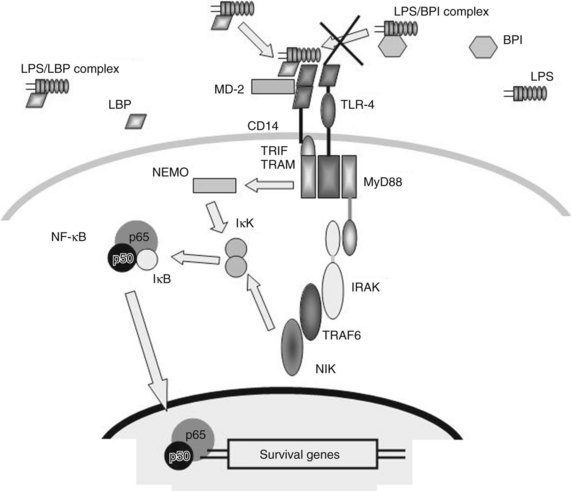
The signal-transducing component of the CD14 bipartite complex protein has been identified as a member of the family of TLRs, specifically TLR-4 (Beutler, 2002). Although TLR-4 is expressed in several cells of healthy liver, signs of inflammation are absent because of low expression levels of TLR-4 and modulation of TLR-4 signaling. Mounting evidence suggests that altered LPS–TLR-4 signaling plays a role in the pathogenesis of chronic liver disease (Soares, 2010). TLR-4 seems to interact with CD14, the LBP-endotoxin complex, and a third protein, MD-2 (Viriyakosol et al, 2001).
Defining the complex role of CD14 in endotoxin-induced activation of TLR-4 is a challenge, and whether this role is obligatory remains unclear (Beutler et al, 2006; Jiang et al, 2005). However, the activation of TLR-4 by endotoxin does appear to require the simultaneous binding of endotoxin and TLR-4 by MD-2 (Gioannini et al, 2004). TLR-4 signaling can occur via a Toll-like and interleukin (IL)-1 receptor (TIR) domain and the formation of a scaffold composed of members of myeloid differentiation factor 88 (MyD88) adaptor proteins. At the same time, TLR-4 signaling also occurs through a MyD88-independent pathway that involves the adaptor protein TIR domain–containing adaptor inducing interferon-β (TRIF). This latter pathway seems to be more essential for the expression of type I interferons (IFNs) and IFN-dependent proteins in response to endotoxin exposure (Yamamoto et al, 2003). Although it is presently unknown how CD14-dependent signaling is integrated from the cell surface via TLR-4, the confirmation of TLR-4 as the signaling complex is important, because it identifies a signal transduction pathway and potential therapeutic targets.
High-density lipoproteins can bind endotoxin in serum and may play a role in protecting against endotoxemia (Cue et al, 1994). An additional endogenous mechanism to suppress endotoxin responsiveness is through the release of an LBP-homologous protein, bactericidal/permeability-increasing protein (BPI; see Fig. 10.2). BPI is a 55- to 60-kD protein structurally similar to LBP, which binds endotoxin through its lipid A moiety. However, the resulting BPI-endotoxin complexes do not bind to CD14 or transduce a signal (Elsbach, 1998). BPI acts as an endogenous endotoxin inhibitor and has shown some modest beneficial effects in patients with meningococcal sepsis (Levin et al, 2000). In addition, BPI released from activated neutrophils can bind directly to endotoxin being expressed on the surface of gram-negative bacteria, and it is growth arresting and cytolytic for the bacteria (von der Mohlen et al, 1996).
BPI is stored in the granules of neutrophils and is released on neutrophil activation and degranulation. In a healthy adult, BPI levels in the serum are very low (15 to 50 ng/mL), but concentrations increase in response to an inflammatory or endotoxemic challenge (Calvano et al, 1994). In contrast, LBP concentrations are several logs higher in patients in the surgical ICU (2 to 20 mg/mL) and increase only modestly in response to inflammation. During infection, the ratio of LBP to BPI concentrations in the plasma approaches 1000:1, and this ratio favors endotoxin signaling. In closed-space infections infiltrated with neutrophils, however, BPI exceeds LBP concentrations, and BPI concentrations are directly proportional to neutrophil counts. These findings suggest that in the normal healthy adult and in patients with sepsis, the relationship between plasma LBP and BPI concentrations favors the recognition of endotoxin and activation of the innate immune response. In local infectious sites, where neutrophil and inflammatory cell recruitment has occurred, the increased BPI response is presumably compensatory and aimed at the continued activation of the immune system and reduction of bacterial growth.
Endotoxin signaling via the CD14 receptor and TLR-4 involves a novel IL-1 receptor-associated kinase, which activates a secondary kinase, NF-κB–inducing kinase (NIK) (Yang et al, 1999). Activation of this cascade rapidly induces the phosphorylation of additional kinases, which results in NF-κB translocation and the transcription of NF-κB–dependent genes (see Fig. 10.2). Genes containing NF-κB response elements are numerous and include most of the proinflammatory cytokines, including tumor necrosis factor (TNF)-α, IL-1, IL-6, IL-8, IL-12, IL-18, and IFN-γ (Blackwell & Christman, 1997).
Most of the systemic inflammatory responses to endotoxin are mediated by the release of proinflammatory cytokines and other humoral factors, predominantly TNF-α, IL-1, and IL-6 and, to a lesser extent, Fas ligand (FasL) (Suffredini et al, 1999). These proinflammatory cytokines not only globally regulate the inflammatory response to endotoxin, they also play a crucial role in reprogramming the metabolic and protein synthetic responses by the liver. As we will discuss in greater detail, TNF-α and IL-6 in particular play unique roles in regulating not only the hepatocyte acute-phase response to endotoxin but also hepatocyte proliferation versus apoptosis in liver regeneration (Michalopoulos & DeFrances, 1997; see Chapter 5), viral hepatitis (Hayashi & Mita, 1997; see Chapter 64), and toxic liver injury (Batey et al, 1999; Bradham et al, 1998; McClain et al, 1999). In contrast, FasL is a potent inducer of hepatocyte apoptosis and has been implicated in the pathogenesis of viral hepatitis (Kondo et al, 1997; Hayashi & Mita, 1997).
Tumor Necrosis Factor Superfamily
Since the discovery of TNF-α 35 years ago, the TNF superfamily has grown to comprise at least 20 related proteins that signal through greater than 30 receptors (Grewal, 2009). Members of the TNF superfamily are primarily homotrimeric proteins, with the exception of lymphotoxin, and they exist primarily in a membrane-associated form (Bazzoni & Beutler, 1996). As a general rule, members of the TNF family are primarily involved in the regulation of cell proliferation and apoptosis, although several of the members—including TNF-α, TNF-β, FasL, CD30L, and CD40L—also have proinflammatory properties.
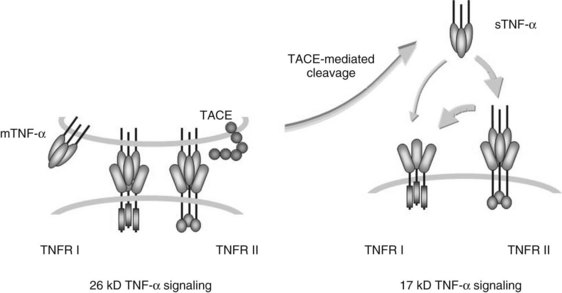
TNF-α is synthesized as a bioactive cell-associated protein that is primarily involved in juxtacrine signaling of cytotoxicity. This 26-kD intermediate is enzymatically cleaved by a matrix metalloproteinase to a 17-kD secreted form that acts in a paracrine or endocrine fashion (Fig. 10.3). TNF-α originally was characterized as a factor that produced necrosis of solid tumors in vivo (Carswell et al, 1975). It subsequently was recognized that TNF-α modulates growth, differentiation, and metabolism in a variety of cell types; it can produce cachexia by stimulating lipolysis, inhibiting lipoprotein lipase activity in adipocytes, and stimulating hepatic lipogenesis; and it can initiate apoptosis in hepatocytes and lymphoid cells (Table 10.4; Ksontini et al, 1998).
| Immune Cells | Nonimmune Cells | In Vivo |
|---|---|---|
FSH, follicle-stimulating hormone; G-CSF, granulocyte colony-stimulating factor; GH, growth hormone; GM-CSF, granulocyte-macrophage colony-stimulating factor; ICAM, intercellular adhesion molecule; IL, interleukin; LIF, leukemia inhibitory factor; TSH, thyroid-stimulating hormone; VCAM, vascular cell adhesion molecule
TNF-α is a powerful inducer of the inflammatory response both directly and through stimulation of numerous downstream proinflammatory mediators. Secondary mediators that are known to be induced by systemically administered TNF-α include cytokines (IL-1, IL-2, IL-4, IL-6, IL-10, IL-12, IL-18, IL-23, type I and type II IFN, transforming growth factor-β, leukemia inhibitory factor [LIF], and macrophage migration inhibitory factor [MIF]), hormones (cortisol, epinephrine, glucagon, insulin, and norepinephrine), and assorted other molecules (acute-phase proteins, IL-1 receptor antagonist [IL-1Ra], leukotrienes, oxygen free radicals, NO, platelet-activating factor, and prostaglandins; Tracey & Cerami, 1994). Other principal biologic effects of TNF-α are listed in Table 10.4 (see Chapter 9).
Not only is TNF-α involved in tissue inflammation, but there is a growing recognition that it is also a prominent ligand for the activation of programmed cell death. Apoptosis occurs naturally during growth and development, but it may also result from certain pathologic conditions in which local and systemic production of TNF-α is increased. Hepatocytes are particularly sensitive to TNF-α–induced apoptosis, especially during simultaneous transcriptional inhibition (Leist et al, 1994).
TNF-α has been administered systemically in low doses to human volunteers and regionally in higher doses to sarcomas and melanomas. The physiologic responses to low-dose (50 mg/m2) TNF-α administration were remarkably similar to the responses seen with low-dose endotoxin administration: fever and constitutional symptoms of pain, headache, myalgia, and nausea. Hematologically, the volunteers developed a rapid leukopenia, followed by neutrophilia with sustained lymphopenia and monocytopenia (van der Poll et al, 1992). Plasma IL-6 levels increased 40-fold, and the volunteers developed an acute-phase protein response. Prostaglandin (6-keto prostaglandin [PG] F1a) production also was markedly increased. Metabolically, the patients exhibited increased lipolysis and glucose turnover (van der Poll et al, 1991b). There also were significant effects on the vascular endothelium. Within 1 hour, TNF-α administration induced activation of the fibrinolytic pathway, followed by activation of the coagulation cascade (van der Poll et al, 1991a).
At higher doses of TNF-α, as achieved with regional perfusion of limbs, the systemic release of large quantities of protein occasionally occurs, and the effects on the vascular endothelium are profound. Under these conditions, activation of the endothelium is evident by increased release of soluble selectins and integrins, and hemodynamic instability is common (Aderka et al, 1998; Zwaveling et al, 1996); therefore continuous monitoring for systemic leakage is required.
Interleukin-1 Family
The IL-1 family of ligands and cytokines are similar to the TNF superfamily in that they are also primarily associated with inflammation. IL-1 possesses several biologic properties that result in increased expression of downstream proinflammatory genes. The most salient and relevant is the ability of IL-1 to initiate and sustain the expression of cyclooxygenase type 2 and inducible NO synthase (Dinarello, 1996). This property accounts for the large amount of PGE2 and NO produced by cells exposed to IL-1 or in subjects injected with IL-1. Other important proinflammatory properties of IL-1 are its ability to increase IL-8 synthesis and to express adhesion molecules on endothelial cell surfaces, which accounts for the infiltration of inflammatory cells into the extravascular space. IL-1 also potentiates the biologic activities of TNF-α, and modest doses of the two in combination can be lethal in experimental animals.
The original IL-1 superfamily consisted of two members: IL-1α and IL-1β. Currently, the IL-1 superfamily also contains IL-1Ra, IL-16, IL-17, IL-18, IL-33, and at least six other homologues in the IL-1 family (Schmitz et al, 2005). IL-1α and IL-1β are agonists, and IL-1Ra is a specific receptor antagonist for the IL-1 receptor family. The naturally occurring IL-1Ra seems to be unique in cytokine biology (Arend, 1993). The intron–exon organization of three primary IL-1 genes suggests the duplication of a common gene some 350 million years ago. Processing of IL-1α or IL-1β to “mature” forms requires specific cellular proteases. In contrast, IL-1Ra evolved with a signal peptide and is readily transported out of the cell and is termed secreted IL-1Ra.
Even under conditions of cell stimulation, human blood monocytes do not process or readily secrete mature IL-1α, which is probably only released by dying cells, as it is a key factor released by necrotic cells that promotes inflammation (Chen, 2007). The IL-1α precursor proIL-1α is synthesized in association with cytoskeletal structures (microtubules), which is in contrast to most proteins translated in the endoplasmic reticulum. ProIL-1α is fully active as a precursor and remains intracellular. The opposite is the case with the IL-1β precursor proIL-1β, which is not fully active, and a considerable amount is secreted after cleavage by a specific, intracellular cysteine protease, IL-1β–converting enzyme, or caspase-1. Caspase-1 plays a central role in the biology of IL-1β, IL-18, and IL-33 and has become a therapeutic target for modulating IL-1–based diseases (Martinon & Tschopp, 2004). IL-33 is an activator of Th2 cells and has recently been shown to play a protective role in the development of atherosclerosis (Miller et al, 2008; Sanada et al, 2007) and is thought to be released by cell death (Carriere et al, 2007).
Although animal experiments revealed that IL-1 was proinflammatory, a great deal of information has been gathered from studies in which humans have been injected with recombinant IL-1α or IL-1β. Humans have received IL-1α infusions as part of clinical trials following bone marrow transplantation because of its hematopoietic properties. Although the duration of leukopenia and thrombocytopenia were reduced, patients had serious signs of systemic inflammation that included fever, hypotension, and flulike symptoms, similar to endotoxemia (Smith et al, 1993). Interestingly, the exquisite sensitivity of humans to IL-1 given systemically was not appreciated from animal studies.
Interleukin-6
IL-6 is another pleiotropic cytokine produced by a wide variety of cells, including T cells, B cells, endothelial cells, fibroblasts, monocytes, and macrophages. The biologic activities of IL-6 include immune regulation, hematopoiesis, inflammation, and oncogenesis (see Chapter 9). The IL-6 gene has been mapped to chromosome 7, and its product varies from 21 to 28 KD, depending on posttranslational modifications. IL-6 belongs to a much larger superfamily of related cytokines, including leukemia inhibitory factor, oncostatin M, ciliary neurotrophic factor, cardiotrophin-1, and IL-11. All members of this superfamily share a modest degree of structural homology, but more importantly, all these related cytokines use a common signal transduction pathway through heterocomplexes composed of gp130. Each ligand has its own receptor, which combines in a duplex to transduce its signal through the JAK/Stat signaling cascade.
Although microbial products are potent inducers of IL-6, the synthesis of IL-6 seems to depend on the release of more proximal cytokines, primarily TNF-α and IL-1. The biologic activities of IL-6 are numerous and can be divided into those involved in the regulation of the hematopoietic system and those involved in activation of the innate immunity. Recent studies have elucidated a crucial role for IL-6 in regulating the balance between T-regulatory cells and a new subset of T-helper cells that produce IL-17 (Kimura & Kishimoto, 2010). In the liver, IL-6 is secreted by Kupffer cells and is an important regulator of innate immune response and a stimulator of hepatocyte proliferation. Although IL-6 shares several biologic responses with TNF-α and IL-1, IL-6 is the predominant regulator of the hepatic acute-phase response and induces hepatocyte proliferation during liver regeneration (Table 10.5).

Fas Ligand
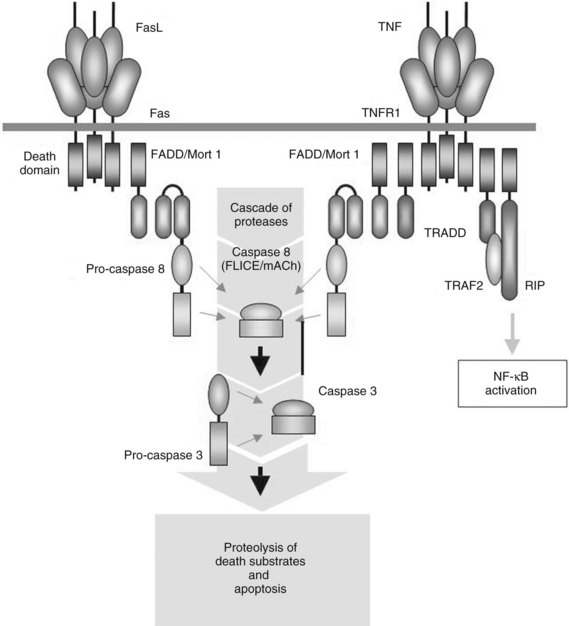
FasL is another member of the TNF superfamily. Similar to TNF-α, FasL is synthesized and expressed first as a 40-kD membrane–associated protein and is then processed further by matrix metalloproteinases to a homotrimeric 26-kD secreted form (Nagata & Golstein, 1995). Historically, FasL was presumed to have predominantly proapoptotic properties, especially for hepatocytes, and these were associated primarily with its membrane-associated form. Although it was noted that secreted FasL formed trimeric structures and retained its ability to ligate the Fas receptor, it was unclear whether soluble FasL could induce apoptosis (Schneider et al, 1998). A model of Fas activation has recently been proposed that distinguishes five separate stages (Lee et al, 2006). FasL occurs in a membrane-bound found and in a soluble form that is the result of splicing of the membrane-bound form. Studies in mice in particular suggested that the soluble forms of FasL may be receptor antagonists of Fas, which inhibit cell-associated FasL-mediated apoptosis.
The hypothesis that soluble FasL is strictly an inducer of apoptosis has been dispelled by several groups, who reported that soluble FasL also has proinflammatory properties. FasL can induce NF-κB through NF-κB–inducing kinase (NIK) signaling (Malanin et al, 1997). Ottonello and colleagues (1999) showed that soluble FasL in humans is endowed with potent chemotactic properties for neutrophils at concentrations incapable of producing apoptosis. FasL does not seem to activate neutrophils, only to recruit them; neutrophil calcium flux, superoxide production, and degranulation are not affected. Other investigators have come to similar conclusions (Chen et al, 1998; Seino et al, 1998). In addition, Park and colleagues (2003) suggested that FasL can activate blood monocytes. These authors showed that these inflammatory properties are separate and distinct from the proapoptotic properties of FasL. This observation is significant in part because it suggests that FasL, similar to TNF-α and other members of the TNF superfamily, may contribute to neutrophil and monocyte infiltration and recruitment as part of the host inflammatory response. This observation is particularly relevant in viral hepatitis, in which increased FasL expression and neutrophil infiltration have been observed.
Although FasL was originally thought to be expressed only by cells of the lymphoid or myeloid lineage—including predominantly T and B cells, phagocytes, and natural killer cells—it is now recognized that FasL frequently is expressed by nonlymphoid cells (Kiener et al, 1997; Liles et al, 1996). The Fas receptor, also called CD95 or Apo1, is widely expressed on a variety of cell types, including hepatocytes. The Fas receptor shares structural homology with the TNF receptor I (Fig. 10.4), and Fas signaling can also lead to proinflammatory events through the activation of NF-κB–dependent pathways, which may be responsible for some its chemotactic properties.
Transforming Growth Factor-β
TGF-β is another ubiquitously expressed cytokine that plays a role in several cellular functions, including apoptosis, cell cycle regulation, immune regulation, and inflammation (Massague, 2008; Bierie & Moses, 2006). TGF-β is produced in an inactive form that requires activation by proteases such as matrix metalloproteinase (MMP)2 and MMP9. Interestingly, TGF-β and its proteases are present at high levels in the periphery of certain tumors (Reiss & Barcellos-Hoff, 1997), and TGF-β also seems to be a potent inducer of fibrosis (see Chapter 6) and eventually of carcinogensis of the liver (see Chapter 8C). TGF-β signaling is via phosphorylation of Smad3, but the location of phosphorylation determines whether the effect will be oncogenic or tumor suppressive. Phosphorylation at the C-terminal region of Smad3 transmits a tumor-suppressor signal, and phosphorylation at the linker region initiates oncogenic sequelae. Therefore TGF-β signaling appears to play a role in chronic liver disease progressing to cancer. In addition, mutations in the genes encoding the receptors for TGF-β and decreased expression of downstream Smad proteins have been reported in pancreatic cancer (Derynck et al, 2001).
Cytokines in the Liver and Pancreas
Regulation of Cytokine Expression in the Liver
Cytokine production by resident cell populations in the liver contributes significantly, not only to local cytokine appearance and organ homeostasis but also to the cytokines’ emergence into the systemic circulation. Fong and colleagues (1990) cannulated the hepatic vein of human volunteers and examined the efflux of TNF-α and IL-6 from the splanchnic bed after intravenous endotoxin administration. Almost 40% of the TNF-α that appeared in the systemic circulation was derived from the splanchnic bed. Not only are Kupffer cells a potential source of proinflammatory cytokines in the liver, biliary epithelial cells and venous endothelial cells can also make significant quantities of cytokines, especially TNF-α, during liver regeneration (Bradham et al, 1998). In addition, resident and infiltrating T cells and natural killer cells can contribute significantly to TNF-α and FasL production, especially during viral-induced hepatic injury (Hayashi & Mita, 1997). There is some evidence that virally infected hepatocytes themselves can express TNF-α directly (Gonzalez-Amaro et al, 1994). TNF-α, IL-1, IL-6, and FasL expression is increased in the liver in response to intravenous endotoxin administration, generalized peritonitis, hepatic I/R injury, infected burn injury, and concanavalin A–induced hepatitis and during liver regeneration (Solorzano et al, 1997; Tannahill et al, 1999).
Cytokines and the Hepatic Acute-Phase Response
The local and systemic release of TNF-α, IL-1, and IL-6 provide anabolic signals to the liver that direct the increased synthesis of hepatic acute-phase reactant proteins (Table 10.6). IL-1 and IL-6 in particular act on the hypothalamic–pituitary axis to stimulate the release of corticotropin-releasing hormone, corticotropin, and cortisol, and these serve to facilitate the mobilization of free amino acids from skeletal muscle, connective tissue, and gut to the liver, where amino acid uptake is enhanced. These free amino acids become the precursors for increased protein synthesis and the altered protein synthetic pattern seen in patients with chronic inflammation and mediated directly by IL-6, IL-1, and to a lesser extent TNF-α.
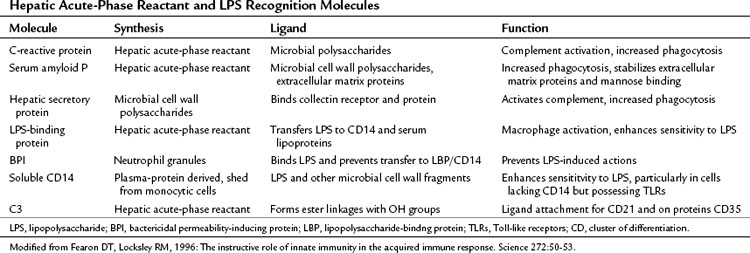
These three cytokines transcriptionally suppress albumin synthesis, the primary protein secretory product of the liver in the healthy adult. Decreased albumin synthesis and hypoalbuminemia are well-described consequences of chronic liver disease, and the patient’s plasma albumin concentration often has been predictive of adverse outcome in a variety of hospitalized patients. In addition, IL-6, IL-1, and TNF-α induce the transcription of a class of heavily glycosylated secretory proteins, termed acute-phase reactants. The number of acute-phase reactants synthesized by the liver in response to these cytokines is quite large and has already been reviewed (Gabay & Kushner, 1999). As a class, the acute-phase proteins have diverse biologic functions (Table 10.7), but many of these proteins are recognition molecules of the innate immune system, including C-reactive protein, MBP (collectin), LBP, amyloid A, and amyloid P (Fearon & Locksley, 1996). These proteins bind microbial polysaccharides or LPS and either activate complement or enhance phagocytosis. Other acute-phase reactants—such as α1-antitrypsin, α1-antichymotrypsin, and tissue inhibitors of metalloproteinases—are protease inhibitors, whereas some, such as haptoglobin and hemopexin, are antioxidants. The function of these latter two groups of acute-phase reactants is to reduce local tissue damage secondary to activation of the innate immune response.
| Acute-Phase Proteins Whose Plasma Concentrations Increase |
| Complement System |
LPS, lipopolysaccharide; IL, interleukin.
Modified from Gabay C, Kushner I, 1999: Acute-phase proteins and other systemic responses to inflammation. N Engl J Med 340:448-454.
Cytokines and the Pancreas
Many pathways that regulate pancreatic development have already been identified and include TGF-β, Notch, Hedgehog, fibroblast growth factor, and epidermal growth factor (EGF) (Rane, 2006; see Chapter 1A). Dysregulation of these pathways appears to be important in many pancreatic disease states.
Much of the current data pertaining to the role of cytokines in the pancreas comes from the diabetes, pancreatitis, and adenocarcinoma literature. It is known that islet cells produce a variety of cytokines in response to both physiologic and pathologic exposure to nutrients (Donath et al, 2010). The inflammatory mediators of cytokine production are produced by α, β, endothelial, ductal, and immune cells. A chronic state of metabolic stress is believed to induce an inflammatory state that leads to islet cell dysfunction and insulin secretion failure. A significant role for TGF-β in the progression of type 1 diabetes has been demonstrated in mice (Baxter et al, 1995).
TGF-β proteins are regulators of pancreatic cell function and appear to have key roles in the development of nondiabetic pancreatic disease as well. The first model of pancreatic adenocarcinoma was generated with overexpression of TGF-β (Greten et al, 2001). These mice developed fibrosis and then pancreatic adenocarcinoma. Additionally, TGF-β2 subset levels appear to be associated with advanced tumor stage (Friess et al, 1993). Patients with long-standing pancreatitis are thought to be at increased risk of developing adenocarcinoma. TGF-β levels are elevated in patients with chronic pancreatitis, and this appears to correlate with both fibrosis and hyperglycemia (Fogar et al, 1998).
Cytokines and Apoptosis
TNF-α seems to be a prominent ligand for the activation of cellular apoptosis, but how TNF-α induces apoptosis in hepatocytes remains unclear. Although hepatocytes are particularly sensitive to TNF-α–mediated apoptosis, the cells first must be exposed to transcriptional inhibition. Incubating cultured hepatocytes with TNF-α or administering TNF-α does not produce hepatocyte apoptosis unless D-galactosamine or actinomycin D is coadministered. The requirement for transcriptional inhibition is likely explained by TNF-α simultaneously activating signaling pathways in hepatocytes that both induce and antagonize apoptosis, suggesting a delicate balance among TNF-α signaling pathways (Fig. 10.5; Beg & Baltimore, 1996; Van Antwerp et al, 1996). Induction of the proinflammatory properties of TNF-α, induced in part through activation of NF-κB–dependent “survival genes,” seems to inhibit apoptosis directly (Baker & Reddy, 1998). NF-κB is a ubiquitous transcription factor that plays a crucial role in the cellular response to signal transduction by TNF-α and IL-6. All members of this family (NF-κB) share a conserved region of approximately 300 amino acids important for their dimerization, nuclear translocation, and DNA binding (Baeuerle, 1998). These NF-κB proteins form homodimers and heterodimers, and their activity is modulated by interactions with inhibitory proteins of the I-κB family.
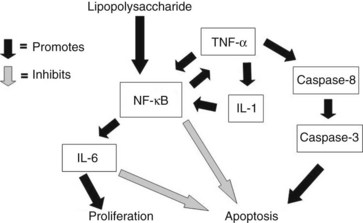
These findings have several clinical implications. The role of NF-κB in the development of type 1 diabetes and its effect on β cells is still unclear, but both antiapoptotic and proapoptotic effects have been described (Donath et al, 2008). Recent studies suggest that enhanced activation of the NF-κB pathway via NIK overexpression may exist in pancreatic adenocarcinoma (Nishina et al, 2009). Robin and colleagues (2005) showed that alcohol abuse increases the sensitivity of hepatocytes to TNF-α–mediated apoptosis and hepatotoxicity. This increased sensitivity seems to be secondary to the failure of TNF-α to induce NF-κB activation in the livers of animals after alcohol abuse. Similarly, radiation to hepatocytes prevents induction of survival genes and increases the apoptotic response to TNF-α (Christiansen et al, 2004). In contrast, ischemic preconditioning seems to protect livers from subsequent apoptotic cell death secondary to induction of NF-κB and IL-6 (Iwasaki et al, 2002). In contrast, activation of TNF-α signaling of caspase-8 and caspase-3 via tumor necrosis factor receptor type 1–associated death domin/Fas-associated death domain (TRADD/FADD) leads to apoptosis through pathways shared by FasL signaling. The balance between these two pathways ultimately determines whether a cell responds to TNF-α by proliferating or undergoing apoptosis (Beg & Baltimore, 1996; Van Antwerp et al, 1996).
The role that specific cytokines play in apoptotic liver injury secondary to endotoxin administration has been well characterized. Similar to high-dose TNF-α in the absence of transcriptional inhibitors, high-dose endotoxin administration results in significant liver injury secondary to hepatocyte necrosis, although hepatocyte apoptosis is minimal (Bohlinger et al, 1996). Simultaneous inhibition of transcription, either by depleting hepatocyte nucleotides with D-galactosamine or by blocking transcription with actinomycin D, results in a 1000-fold increased sensitivity to endotoxin-induced lethality, and the hepatic injury is characterized by massive liver apoptosis (Solorzano et al, 1997). The liver injury seems to depend primarily on TNF-α expression, although FasL, interferon-γ, IL-12, IL-18, and IL-23 probably also contribute. Mice lacking either TNF-α or the TNF type I receptor are completely protected from endotoxin and D-galactosamine–induced hepatocyte apoptosis and mortality, whereas they are still susceptible to high-dose endotoxin-induced lethality. Fas agonists differ from TNF-α in that they apparently can induce massive hepatocyte apoptosis in the absence of transcriptional inhibition. The differential response to apoptosis mediated by TNF-α and FasL seems to be related to the observation that FasL is a significantly weaker inducer of NF-κB than is TNF-α. IL-1 and NO protect hepatocytes from TNF-α–mediated apoptosis, presumably by stimulating NF-κB activation and inhibiting caspase activities (Bohlinger et al, 1996). Caspases play central roles as the “suicide effectors” of hepatocyte apoptosis, demonstrated by the treatment of mice with broad-acting caspase inhibitors to prevent apoptotic liver injury and mortality (Kunstle et al, 1997; Mignon et al, 1999).
Nitric Oxide
Oxidative stress has been implicated in the pathogenesis of apoptosis, cancer, diabetes, hypertension, and inflammation. This implies that it may be a final common pathway by which numerous diseases exert their harmful effects. The contribution of NO to liver injury and apoptosis is complex. NO is a lipophilic, short-lived molecule (with a half-life of seconds) that physiologically seems to have roles in vasodilation, immune response, apoptosis, and protection of cells from oxidative killing (Wink et al, 1999). NO derived from NO synthase originating in endothelial cells and other nonparenchymal liver cells seems to be hepatoprotective in most cases (Ou et al, 1997). NO from inducible NO synthase (iNOS) in hepatic parenchymal cells can either damage or protect hepatocytes. Upregulation of iNOS occurs after stimulation by endotoxin and by cytokines such as IL-1b (Kitade et al, 1996; Zamora et al, 2000). Depending on the level of redox stress, iNOS can cause detrimental effects, such as direct cytotoxicity and suppression of hepatic protein synthesis, in a manner similar to the septic state (Taylor et al, 1998; Chen et al, 2003). After resuscitation from profound hemorrhagic shock, iNOS can also mediate activation of NF-κB and increases in IL-6 expression, which leads to acute liver injury followed by recovery (Hierholzer et al, 1998).
Alternatively, with lower redox states, NO plays a cytoprotective role by suppressing apoptosis in hepatocytes and reducing mitochondrial dysfunction to limit liver injury (Brookes et al, 2000; Wang et al, 2002; Chen et al, 2003). NO exerts its cytoprotective effect during acute inflammation by suppressing caspase activation, inducing heat shock proteins (Kim et al, 1997), upregulating hemoxygenase-1 (Zamora et al, 2000), and activating the cyclic guanosine monophosphate signal pathway to interrupt apoptosis (Kim et al, 1997; Wang et al, 2002; Chen et al, 2003). NO can act as an electron acceptor for S-nitrosylation, which can alter the catalytic site of caspases and render them inactive (Li et al, 1997). NO also can induce the expression of heat shock proteins 70 and 32 (Kim et al, 1995; Li et al, 2000). These proteins may function by refolding damaged proteins in the liver, modulating caspase activation, and regulating expression of hemoxygenase-1, an inducible enzyme that catalyzes the degradation of heme into biliverdin, releasing carbon monoxide as a by product, which is thought to confer local antiinflammatory effects (Otterbein et al, 2000). NO also can bind soluble guanylyl cyclase, which can increase intracellular cyclic guanosine monophosphate, suppressing apoptosis and caspase activity (Lucas et al, 2000). Which particular mechanism predominates likely depends on the specific circumstances (Chen et al, 2003). To delineate further the effect of NO on hepatocytes, Wang and others (2002) are continuing to evaluate genetic changes using DNA microarray and large-scale proteomic analysis.
Liver Regeneration
The liver’s ability to regenerate is a unique quality that is essential for its functions of controlling metabolism and detoxification (see Chapter 5). This capability to fully regenerate after insult differs significantly from other organs, which heal with scar (Fausto et al, 2006; Michalopoulos et al, 2007). However, in certain chronic liver injury states, the liver’s regenerative capacity can be insufficient, leading to the replacement of functional epithelial tissue with connective tissue and to development of cirrhosis. A thorough understanding of the mechanisms and factors by which liver regeneration takes place is important in developing therapies to enhance regenerative capacity and prevent cirrhosis.
It has been shown that humoral factors are required, because hepatectomy in a rodent joined in parabiotic circulation results in proliferation and regeneration of the intact liver from the other member of the pair (Moolten & Bucher, 1967). This work provides convincing evidence that some mitogenic factor or factors are released into the peritoneal or systemic circulation during liver regeneration. Studies with recombinant proteins, cytokine inhibitors, and transgenic mice have convincingly shown that at least three cytokines—TNF-α, IL-6, and hepatocyte growth factor—play crucial roles in this regenerative process (Michalopoulos & DeFrances, 1997). The regenerative process of the liver can be divided into three phases: 1) initiation, 2) expansion, and 3) termination (Zimmermann et al, 2004) and is discussed elsewhere (Chapter 5). However, cytokines and growth factors play a significant role. The initiation phase is regulated by cytokines such as IL-6 and TNF-α, the expansion stage is regulated by mitogenic factors such as hepatocyte growth factor, and the termination phase is where mitosis stops, and sometimes apoptosis begins to control overgrowth. Additional cytokines, TGF-β and EGF, also may have roles. The TGF-β superfamily of cytokines appears to play a significant role in maintaining embryonic stem cell identity, and the differentiation of pancreas and liver progenitor cells is restricted by this pathway (Wandzioch & Zaret, 2009).
Several converging lines of evidence suggest that TNF-α, and particularly its signaling through the type I receptor, is essential for liver regeneration. Treatment of animals with neutralizing antibodies against TNF-α delays regeneration and decreases DNA synthesis (Yamada et al, 1997). DNA synthesis is also impaired after partial hepatectomy in TNF-receptor type I knockout mice. Whether these defects are due directly to the effects of TNF-α or are mediated through TNF-α–dependent expression of other mediators, including IL-6, is not fully understood, because treating TNF-receptor type I knockout mice with exogenous IL-6 corrects this delay in regeneration.
The role of IL-6 in hepatic regeneration also has been established (Drucker et al, 2010). Plasma IL-6 concentrations increase after partial hepatectomy, peaking within 24 to 48 hours (Matsunami et al, 1992; Rai et al, 1996). Although it is controversial whether IL-6 is directly mitogenic for hepatocytes in culture, it is clear that IL-6 is mitogenic for biliary epithelial cells (Matsumoto et al, 1994), and the absence of IL-6 delays regeneration in vivo (Cressman et al, 1996). Hepatocyte DNA synthesis after a partial hepatectomy is suppressed in IL-6 knockout mice. A recent randomized trial suggested a beneficial effect of pentoxifylline, a TNF-α inhibitor, on liver regeneration that seems to be IL-6 mediated (Petrowsky et al, 2010). IL-6 expression required for a normal regenerative response seems to depend on NF-κB translocation. NF-κB–dependent pathways are required to protect hepatocytes from TNF-α–mediated apoptosis. Studies from several groups have shown that inhibition of TNF-α–mediated NF-κB activation exaggerates apoptotic pathways initiated by TNF-α and FasL. Xu and colleagues (1998) noted that blocking NF-κB in RALA hepatocytes resulted in TNF-α–mediated apoptosis. Actinomycin D pretreatment of the same cells also promoted TNF-α apoptosis but did not prevent NF-κB translocation. Actinomycin D prevented the transcription of NF-κB–induced genes in response to TNF-α. In addition, inhibition of NF-κB using an inhibitory adenoviral vector in the livers of mice after hepatic resection produces massive apoptosis and liver failure (Iimuro et al, 1998). Taken together, these findings suggest that blockade of NF-κB–dependent transcription of certain genes in hepatocytes during liver regeneration results in TNF-α–mediated apoptosis.
Pathogenesis of Endotoxin and Cytokines in Liver, Biliary, and Pancreatic Disease
Endotoxemia and Bacterial Translocation
Bacterial translocation has been defined as the process by which viable enteric bacteria cross the intestinal mucosal barrier to mesenteric lymph nodes and remote organs and tissues (Lemaire et al, 1997; MacFie, 1997). Because the bowel contains massive amounts of viable, pathogenic bacteria and endotoxin, several physiologic mechanisms must be disrupted for bacterial translocation to occur.
Endotoxemia itself can further promote bacterial translocation, possibly suggesting an amplification cascade. Endotoxemia frequently produces hypoperfusion of the splanchnic bed, leading to decreased perfusion of the distal ileum and cecum and increased intestinal permeability. In mice, administration of endotoxin induced bacterial translocation, which could be blocked by inhibitors of xanthine oxidase activity (Deitch et al, 1989a, 1989b, 1989c). In humans, low-dose endotoxin administration increases urinary excretion of orally administered D-mannitol, suggesting a reduction in gut barrier function (O’Dwyer et al, 1988).
Bacterial translocation has been proposed to occur in acute necrotizing pancreatitis (Fritz et al, 2010) and numerous biliary diseases, including obstructive jaundice, cirrhosis, portal hypertension, and alcoholic hepatitis (Nehez & Andersson, 2002; Wiest & Garcia-Tsao, 2005). Although the measurement of endotoxin in the blood of patients using current assays is problematic, increased appearance of endotoxin in the systemic circulation is a reproducible event. Several investigators have postulated that endotoxemia and bacterial translocation are the “engines” that drive the systemic inflammatory response syndrome in liver disease (Szabo et al, 2002).
Systemic endotoxemia rarely occurs in patients without liver disease or sepsis. In patients with cirrhosis, alcoholic hepatitis, or obstructive jaundice, systemic appearance of endotoxin has been reported in as many as 70% of patients. Increased levels are also associated with adverse outcomes (Hanck et al, 1998; Kimmings et al, 1995). One hypothesis for obstructive jaundice is that the absence of bile secretion leads to endotoxemia, because enteric biliary drainage of bile salts reduces the absorption of endotoxin (Kimmings et al, 1995, 2000).
The liver is the primary site of endotoxin uptake and detoxification from the blood. Clearance of endotoxin by the liver is greater from the portal enteric drainage than from the hepatic artery. Considerable experimental and clinical evidence suggests that the increased systemic appearance of endotoxin seen in patients with liver and biliary tract disease is the result of increased enteric absorption and reduced clearance from the hepatic reticuloendothelial system (Kimmings et al, 1995). Increased endotoxemia has been reported in patients with cirrhosis, acute hepatic failure, severe hepatitis B and C infections, and obstructive jaundice. In many cases, the appearance of endotoxemia in fulminant hepatic failure correlates strongly with liver disease severity, coexisting renal failure, and mortality. Endotoxin is detected at a higher frequency in the circulation of patients with cirrhosis who exhibit ascites, renal dysfunction, and portocaval shunting, and endotoxemia is a predictor of adverse outcome (Kimmings et al, 1995), although endotoxin can bypass the liver directly via the lymphatics. In rats subjected to a cecal ligation and puncture, endotoxin appearance in the thoracic lymph duct was higher by a factor of several hundred than in the portal blood, suggesting that lymphatic flow may represent a significant source for the systemic circulation (Olofsson et al, 1986).
Pathogenesis of Proinflammatory Cytokines
Convincing circumstantial evidence shows that increased TNF-α–mediated and FasL-mediated apoptosis contributes to hepatocyte injury in chronic hepatitis C infections and in alcoholic hepatitis. Increased TNF-α and FasL expression has been detected in liver biopsy specimens from patients with hepatitis C infections (Fukuda et al, 1995; Larrea et al, 1996), presumably localized to infiltrating inflammatory cells (Kupffer, T, and natural killer cells). In addition, patients with chronic hepatitis B and C often have elevated plasma TNF-α concentrations and occasionally have increased soluble Fas and FasL concentrations. Plasma concentrations of the shed TNF receptors are commonly elevated in chronic hepatitis B and C infections, and levels frequently correlate with the extent of inflammation and hepatocyte death.
Increased hepatocyte apoptosis also has been detected in livers from patients with chronic hepatitis C (Bantel et al, 2004; McPartland et al, 2005). Anti–TNF-α agents administered to mice infected with chronic hepatitis C appear to be antiviral by promoting apoptosis and preventing regeneration of hepatocytes (Brenndorfer et al, 2010). Similarly to plasma TNF-α levels, IL-6 and endotoxin levels are both increased in patients with alcoholic hepatitis, and concentrations often correlate with mortality (Bird et al, 1990; Khoruts et al, 1991; Naveau et al, 1998; Sheron et al, 1991). Peripheral blood monocytes from patients with alcoholic cirrhosis spontaneously release more TNF-α and IL-6 than monocytes from healthy adults, and they do so in response to ex vivo endotoxin stimulation (Schafer et al, 1995). One explanation for this increased endotoxemia and TNF-α expression is that ethanol consumption increases the permeability of the gut to bacterial products such as endotoxin, potentially inducing TNF-α and FasL expression in the liver.
Several reports have shown elevated levels of TNF-α, its shed receptors, and IL-6 in animals and patients with obstructive jaundice (Kimmings et al, 1995). In animal models of experimental biliary obstruction, TNF mRNA expression in the liver is increased, and plasma concentrations rise (Beierle et al, 1996). These animals produce more TNF-α after a surgical injury than do healthy mice, and the increased production correlates with an adverse outcome (Bemelmans et al, 1992, 1993). In patients with obstructive jaundice, elevated TNF-α production by peripheral blood mononuclear cells also has been detected (Kimura et al, 1998; Puntis & Jiang, 1996).
There is considerable controversy regarding the increased level of circulating TNF-α and whether it is biologically active, because many assays only detect total TNF-α. The shed receptors of TNF-α are natural inhibitors of TNF-α bioactivity, and their concentrations also increase in patients with obstructive jaundice. Shedding of the TNF receptors may represent a protective mechanism to reduce the pathologic responses to TNF-α by binding and blocking their interaction with cellular receptors, and binding of TNF-α to its shed receptor may facilitate its clearance by the kidneys. In mice with jaundice, the concentrations of the shed receptors are increased (Bemelmans et al, 1996), and similar findings are seen in patients with obstructive liver disease (Kimmings et al, 1995).
Serum IL-6 levels are significantly higher in patients with biliary obstruction compared with control subjects (Akiyama et al, 1998; Kimura et al, 1999; Puntis & Jiang, 1996; Yamashiki et al, 1998). In one study, serum IL-6 levels decreased significantly after drainage (Akiyama et al, 1998). Patients with elevated serum IL-6 levels had lower serum levels of total protein and albumin, higher mean age, and more frequent positive cultures of bile compared with patients with lower serum IL-6 levels.
Cytokines in Ischemia-Reperfusion Injury
Visceral ischemia and reperfusion are frequent occurrences during hepatobiliary surgery, liver transplantation, trauma surgery, aortic surgery, and mesenteric vascular surgery and can lead to pulmonary and hepatic injury and multiorgan failure. Elective hepatic resection in particular is often performed using portal inflow occlusion with intermittent periods of reperfusion, which potentially reduces blood loss during the resection but can result in hepatocellular damage. I/R injury is associated with release of TNF-α and other proinflammatory cytokines (Colletti et al, 1996; Liu et al, 2000). In rats, intestinal ischemia of 120 minutes followed by reperfusion for 30 minutes was associated with 10-fold increases in circulating TNF-α over controls, which were transient and cleared by 60 minutes of reperfusion (Caty et al, 1990). Blocking TNF-α or IL-1 could mitigate the degree of organ damage (Welborn et al, 1996). After ischemia, gut-derived endotoxin was detected in the portal vein, which preceded the appearance of TNF-α in systemic plasma (Caty et al, 1990). I/R injury is also associated with activation of the oxidant-sensitive TNF transcription factor, NF-κB (Donnahoo et al, 2000; Yoshidome et al, 1999). Studies in mice demonstrate that reperfusion of ischemic liver tissue causes a series of events that include superoxide anion generation and NF-κB–mediated expression of injurious and protective mediators that favor injury (Abe et al, 2009). IL-10 seems to protect against acute lung injury that can occur after hepatic I/R injury (Huber et al, 2000; Welborn et al, 2003; Yoshidome et al, 1999). Hemoxygenase-1 also has been shown to reduce I/R injury in an ex vivo liver transplant model in rats (Amersi et al, 1999), and low-dose carbon monoxide has been shown to be hepatoprotective (Amersi et al, 2002).
In humans, thoracoabdominal aortic aneurysm repair necessitates complete occlusion of the thoracic aorta above the celiac artery for 20 minutes to more than 1 hour, which is associated with pulmonary and renal dysfunction, consumptive coagulopathies, and mortality of 15% (Derrow et al, 2001; Rectenwald et al, 2002). For patients undergoing thoracoabdominal aortic aneurysm repair, visceral I/R is also associated with proinflammatory cytokine release (Welborn et al, 2000). Compared with patients undergoing infrarenal abdominal aortic aneurysm repair, in which the viscera remain perfused, patients undergoing thoracoabdominal aortic aneurysm repair had elevated levels of TNF-α, IL-6, IL-8, and IL-10 and the TNF receptors p55 and p75 (Welborn et al, 2000). The postoperative cytokine response was associated with the duration of visceral ischemia, and when left atrial femoral bypass was used to reduce ischemia time, the systemic TNF-α, p75, and IL-10 responses were attenuated. Patients with early and sustained peaks of IL-6 and TNF-α were more likely to develop multisystem organ failure.
Microarray technology and multiplex protein analyses were used on blood leukocytes and plasma in 10 patients to identify adverse clinical outcomes after thoracoabdominal aortic aneurysm repair with its requisite I/R injury (Feezor et al, 2004). From roughly 30,000 genes, changes in 146 correlated with an adverse clinical outcome. Several—such as monocyte-to-macrophage differentiation-associated factor, matrix metalloproteinase-8, and platelet factor 4—are involved in the innate immune response. Others are associated with a general stress response and cytoprotection, such as the upregulation of heat shock proteins; several genes involved with major histocompatability complex (MHC) class I were downregulated. Many of the differences seen in patients who eventually would develop multiorgan dysfunction were evident preoperatively, suggesting a preexisting proinflammatory state in these patients with diseased vasculature (Feezor et al, 2004).
Cytokines in Ablation of Hepatic Tumors (See Chapter 83, Chapter 85A, Chapter 85B, Chapter 85C, Chapter 85D )
For resectable primary and metastatic tumors, resection remains the standard of care (Blumgart & Fong, 1995; DeMatteo et al, 1999). However, several ablative techniques have been described, which by definition leave tumor debris in situ and can elicit a complex immunologic response. Cryoablation entails the use of metallic probes cooled with liquid nitrogen and placed into the tumor or on the liver surface (Chapman et al, 2000). Freezing of the tumor tissue results in obliteration of small vessels and disruption of hepatocyte plasma membranes (Chapman et al, 2000). In early clinical series, approximately 1% of patients treated with cryoablation developed “cryoshock,” a syndrome characterized by acute respiratory distress syndrome, multiorgan failure, and disseminated intravascular coagulation; it results in death in nearly 20% of patients who develop it (Seifert & Morris, 1999). This complication, now rarely seen, was noted to be dose dependent, such that the risk increases proportionately with the volume of treated liver, and especially when more than 30% of the total hepatic volume is cryoablated (Chapman et al, 2000; Sarantou et al, 1998). This process seems to be mediated by NF-κB, which becomes activated, and transcription of TNF-α and other cytokines is increased (Barnes & Karin, 1997; Blackwell et al, 1999; Chapman et al, 2000; Seifert et al, 2002).
Radiofrequency ablation (RFA) involves the dispersal of heat around a needle passed into a tumor to cause coagulative necrosis. The technology behind this technique is limited by a smaller zone of ablation, which prevents its use in larger tumors (Chapman et al, 2000). In contrast to cryoablation, after treatment with RFA, liver cell organelles undergo thermal coagulative destruction, but the plasma membrane stays intact (Chapman et al, 2000). In rodent models in which the size of the ablation was controlled, rats treated with RFA had markedly diminished lung inflammation and lower TNF-α and macrophage inhibitory protein-2 levels (analogous to human IL-8) compared with rats treated with cryoablation (Chapman et al, 2000; Ng et al, 2004). In a study of 17 patients undergoing RFA, production of cytokines (TNF-α, IL-1b, IL-1Ra, IL-6, IL-8, and IL-10) and the TNF cytokine receptors (p55 and p75) was not altered at any time within the first 48 hours after the procedure (Schell et al, 2002). When stimulated with endotoxin (LPS), blood samples from the same patients showed normal ability to elaborate cytokines, suggesting that the absence of cytokine production after RFA was not related to the patients’ underlying disease or inability to produce cytokines but rather was due to thermal coagulation of hepatocytes (Schell et al, 2002).
Microwave ablation is a more recently developed technology that is being used with increasing frequency to treat liver tumors. Microwave ablation involves application of microwaves directly to liver tissue to produce rapid temperature elevations and cause coagulative necrosis. Zhang and colleagues (2002) demonstrated a significant increase in T cells, NK cells, and monocytes in microwave-ablated hepatocellular patients. This response was maximal at day 3 but persisted to day 30. Interestingly, patients with a higher degree of immune cell infiltration had lower recurrence rates, similar to RFA patients (Hansler et al, 2006).
Future Directions
Although it is clearly premature to celebrate the widespread use of cytokine or anticytokine therapies for patients with liver, biliary, or pancreatic disease, research is certainly heading in that direction. Several treatment modalities that offer the opportunity to interrupt endotoxin and the proinflammatory cytokine responses in liver disease remain of considerable interest. Antiendotoxin therapies are of particular interest in obstructive jaundice and acute alcoholic hepatitis. In chronic viral hepatitis, in which increased TNF-α–mediated and FasL-mediated apoptosis are presumed to occur, enthusiasm remains for the opportunity to intervene with either immunoadhesins or antibodies. The clinical use of antiinflammatory cytokines, such as IL-10, is likely to occur at some point in the near future, although the risk of added immunosuppression remains a concern. The pegylated form of IFN-α, which has greater stability and in vivo activity than IFN-α alone, has substantially improved viral response rates in hepatitis C patients (see Chapter 64). As a result, it is now considered the standard of care for treatment of hepatitis C and may also have some efficacy in hepatitis B patients.
Antiendotoxin antibodies have been evaluated in patients with sepsis, generally with disappointing results. Although concerns have been raised about the neutralizing capacity and specificity of some of these antibodies, the lack of efficacy could be explained in part by the inadequate selection of patients and the timing of administration. Only a fraction of the patients receiving antiendotoxin therapies were actually endotoxemic, and the administration of the antibody usually occurred 12 to 24 hours after the onset of symptoms. Recently, focus has been placed on the development of antiendotoxin vaccines, which likely will be tested in Phase I human trials soon (Cross, 2010). An alternative approach is recombinant BPI. In human volunteers treated with low-dose endotoxin, recombinant BPI administration reduced the proinflammatory cytokine responses and activation of the fibrinolytic and coagulation pathways (von der Mohlen et al, 1995). Recombinant BPI continues to be under U.S. Food and Drug Administration (FDA) investigation for its potential use in children with meningococcal sepsis (Levin et al, 2000). Although the results of this trial indicated a trend toward a survival benefit, this study was underpowered to detect significant differences in mortality. In rats subjected to a partial hepatectomy, recombinant BPI reduced liver injury and the magnitude of the inflammatory response (Kimmings et al, 1999).
Although most of the cytokine and anticytokine therapies currently in use are not specifically indicated for liver, biliary, or pancreatic disease, much information about their effects will be learned in the coming years, and some of these therapies may well emerge as effective therapies in patients with inflammatory, neoplastic, or other hepatopancreatobiliary disorders. A recent phase I/II trial of intratumoral injections of TLR-2 and TLR-6 antagonists for R2 resected pancreatic adenocarcinoma shows a possible increased survival, but larger trials will be needed (Schmidt et al, 2007). Preclinical studies indicate that TLR-3 antagonists may delay the development of pancreatic cancer (Schwartz et al, 2009). The now common use of TNF-α and pegylated IFN demonstrate the potential application of cytokine therapy to liver disease. The liver’s complex interplay of metabolism, immunology, cytokine productivity, and regenerative capacity make it ideally suited for the development of cytokine-directed therapies.
Abe Y, et al. Mouse model of liver ischemia and reperfusion injury: method for studying reactive oxygen and nitrogen metabolism in vivo. Free Radic Biol Med. 2009;46:1-7.
Aderka D, et al. Shedding kinetics of soluble tumor necrosis factor (TNF) receptors after systemic TNF leaking during isolated limb perfusion: relevance to the pathophysiology of septic shock. J Clin Invest. 1998;101:650-659.
Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85-95.
Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499-511.
Akiyama T, et al. Serum and bile interleukin 6 after percutaneous transhepatic cholangio-drainage. Hepatogastroenterology. 1998;45:665-671.
Amersi F, et al. Upregulation of heme oxygenase-1 protects genetically fat Zucker rat livers from ischemia/reperfusion injury. J Clin Invest. 1999;104:1631-1639.
Amersi F, et al. Ex vivo exposure to carbon monoxide prevents hepatic ischemia/reperfusion injury through p38 MAP kinase pathway. Hepatology. 2002;35:815-823.
Arend WP. Interleukin-1 receptor antagonist. Adv Immunol. 1993;54:167-227.
Baeuerle PA. IkappaB-NF-kappaB structures: at the interface of inflammation control. Cell. 1998;95:729-731.
Baker SJ, Reddy EP. Modulation of life and death by the TNF receptor superfamily. Oncogene. 1998;17:3261-3270.
Bantel H, et al. Detection of apoptotic caspase activation in sera from patients with chronic HCV infection is associated with fibrotic liver injury. Hepatology. 2004;40:1078-1087.
Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066-1071.
Batey RG, et al. Alcoholic hepatitis as a T-cell mediated disorder: an hypothesis. Alcohol Clin Exp Res. 1999;23:1207-1209.
Baxter AG, et al. The genetics of the NOD mouse. Diabetes Metab Rev. 1995;11:315-335.
Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. N Engl J Med. 1996;334:1717-1725.
Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha–induced cell death. Science. 1996;274:782-784.
Beierle EA, et al. Hepatic tumor necrosis factor-alpha production and distant organ dysfunction in a murine model of obstructive jaundice. Am J Surg. 1996;171:202-206.
Bemelmans MH, et al. Cytokines tumor necrosis factor and interleukin-6 in experimental biliary obstruction in mice. Hepatology. 1992;15:1132-1136.
Bemelmans MH, et al. Effect of antitumour necrosis factor treatment on circulating tumour necrosis factor levels and mortality after surgery in jaundiced mice. Br J Surg. 1993;80:1055-1058.
Bemelmans MH, et al. Increased concentrations of tumour necrosis factor (TNF) and soluble TNF receptors in biliary obstruction in mice: soluble TNF receptors as prognostic factors for mortality. Gut. 1996;38:447-453.
Beutler B. TLR4 as the mammalian endotoxin sensor. Curr Top Microbiol Immunol. 2002;270:109-120.
Beutler B, et al. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229:869-871.
Beutler B, et al. How we detect microbes and respond to them: the Toll-like receptors and their transducers. J Leukoc Biol. 2003;74:479-485.
Beutler B, et al. Genetic analysis of host resistence: Toll-like receptor signaling and immunity at large. Annu Rev Immunol. 2006;24:353-389.
Bierie B, Moses HL. Tumor microenvironment: TGFbeta—the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506-520.
Bird GL, et al. Increased plasma tumor necrosis factor in severe alcoholic hepatitis. Ann Intern Med. 1990;112:917-920.
Blackwell TS, Christman JW. The role of nuclear factor-kappa B in cytokine gene regulation. Am J Respir Cell Mol Biol. 1997;17:3-9.
Blackwell TS, et al. Acute lung injury after hepatic cryoablation: correlation with NF-kappa B activation and cytokine production. Surgery. 1999;126:518-526.
Blumgart LH, Fong Y. Surgical options in the treatment of hepatic metastasis from colorectal cancer. Curr Probl Surg. 1995;32:333-421.
Bohlinger I, et al. DNA fragmentation in mouse organs during endotoxic shock. Am J Pathol. 1996;149:1381-1393.
Bradham CA, et al. Mechanisms of hepatic toxicity: I. TNF-induced liver injury. Am J Physiol. 1998;275:G387-G392.
Brenndorfer ED, et al. Anti-tumor necrosis factor alpha promotes apoptosis and prevents regeneration in a transgenic mouse model of hepatitis C. Hepatology. 2010;52:1553-1563.
Brookes PS, et al. Concentration-dependent effects of nitric oxide on mitochondrial permeability transition and cytochrome c release. J Biol Chem. 2000;275:20474-20479.
Calvano SE, et al. Changes in polymorphonuclear leukocyte surface and plasma bactericidal/permeability-increasing protein and plasma lipopolysaccharide-binding protein during endotoxemia or sepsis. Arch Surg. 1994;129:220-226.
Carriere V, et al. IL-33, the IL-1–like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A. 2007;104:282-287.
Carswell EA, et al. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975;72:3666-3670.
Caty MG, et al. Evidence for tumor necrosis factor–induced pulmonary microvascular injury after intestinal ischemia-reperfusion injury. Ann Surg. 1990;212:694-700.
Chapman WC, et al. Hepatic cryoablation, but not radiofrequency ablation, results in lung inflammation. Ann Surg. 2000;231:752-761.
Chen CJ, et al. Identification of a key pathway required for sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851-856.
Chen JJ, et al. Regulation of the proinflammatory effects of Fas ligand (CD95L). Science. 1998;282:1714-1717.
Chen T, et al. Role of nitric oxide in liver injury. Curr Mol Med. 2003;3:519-526.
Christiansen H, et al. Irradiation leads to susceptibility of hepatocytes to TNF-alpha–mediated apoptosis. Radiother Oncol. 2004;72:291-296.
Colletti LM, et al. The role of cytokine networks in the local liver injury following hepatic ischemia/reperfusion in the rat. Hepatology. 1996;23:506-514.
Cressman DE, et al. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274:1379-1383.
Cross AS. Development of an anti-endotoxin vaccine for sepsis. Subcell Biochem. 2010;53:285-302.
Cue JI, et al. Reconstituted high-density lipoprotein inhibits physiologic and tumor necrosis factor alpha responses to lipopolysaccharide in rabbits. Arch Surg. 1994;129:193-197.
Deitch EA, et al. Inhibition of endotoxin-induced bacterial translocation in mice. J Clin Invest. 1989;84:36-42.
Deitch EA, et al. Endotoxin-induced bacterial translocation: a study of mechanisms. Surgery. 1989;106:292-300.
Deitch EA, et al. Endotoxin induces bacterial translocation and increases xanthine oxidase activity. J Trauma. 1989;29:1679-1683.
DeMatteo RP, et al. Surgical treatment of malignant liver tumours. Baillieres Best Pract Res Clin Gastroenterol. 1999;13:557-574.
Derrow AE, et al. The outcome in the United States after thoracoabdominal aortic aneurysm repair, renal artery bypass, and mesenteric revascularization. J Vasc Surg. 2001;34:54-61.
Derynck R, et al. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117-129.
Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095-2147.
Dinarello CA. Infection, fever, and exogenous and endogenous pyrogens: some concepts have changed. J Endotoxin Res. 2004;10:201-222.
Donath MY, et al. Cytokines and beta-cell biology: from concept to clinical translation. Endocr Rev. 2008;29:334-350.
Donath MY, et al. Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends Endocrinol Metab. 2010;21:261-267.
Donnahoo KK, et al. Early renal ischemia, with or without reperfusion, activates NFkappaB and increases TNF-alpha bioactivity in the kidney. J Urol. 2000;163:1328-1332.
Drucker C, et al. Impact of interleukin-6 classic- and trans-signaling on liver damage and regeneration. J Autoimmunity. 2010;34:29-37.
Elsbach P. The bactericidal/permeability-increasing protein (BPI) in antibacterial host defense. J Leukoc Biol. 1998;64:14-18.
Fausto N, et al. Liver regeneration. Hepatology. 2006;43:S45-S53.
Fearon DT, Locksley RM. The instructive role of innate immunity in the acquired immune response. Science. 1996;272:50-53.
Feezor RJ, et al. Genomic and proteomic determinants of outcome in patients undergoing thoracoabdominal aortic aneurysm repair. J Immunol. 2004;172:7103-7109.
Fogar P, et al. Transforming growth factor beta, fibrogenesis and hyperglycemia in patients with chronic pancreatitis. J Med. 1998;29:277-287.
Fong YM, et al. The acute splanchnic and peripheral tissue metabolic response to endotoxin in humans. J Clin Invest. 1990;85:1896-1904.
Friess H, et al. Enhanced expression of the type II transforming growth factor beta receptor in human pancreatic cancer correlates with decreased survival. Gastroenterology. 1993;105:1846-1856.
Fritz S, et al. Bacterial translocation and infected pancreatic necrosis in acute necrotizing pancreatitis derives from small bowel rather than from colon. Am J Surg. 2010;200:111-117.
Fukuda R, et al. Expression rate of cytokine mRNA in the liver of chronic hepatitis C: comparison with chronic hepatitis B. J Gastroenterol. 1995;30:41-47.
Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448-454.
Gioannini TL, Weiss JP. Regulation of interactions of Gram-negative bacterial endotoxins with mammalian cells. Immunol Res. 2007;39:249-260.
Gioannini TL, et al. Isolation of an endotoxin–MD-2 complex that produces Toll-like receptor 4–dependent cell activation at picomolar concentrations. Proc Natl Acad Sci. 2004;101:4186-41891.
Gonzalez-Amaro R, et al. Induction of tumor necrosis factor alpha production by human hepatocytes in chronic viral hepatitis. J Exp Med. 1994;179:841-848.
Greten FR, et al. A model of pancreatic cancer development. Pancreatology. 2001;1:363-368.
Grewal IS. Overview of TNF superfamily: a chest full of potential therapeutic targets. Adv Exp Med Biol. 2009;647:1-7.
Hanck C, et al. Presence of plasma endotoxin is correlated with tumour necrosis factor receptor levels and disease activity in alcoholic cirrhosis. Alcohol Alcohol. 1998;33:606-608.
Hansler J, et al. Activation and dramatically increased cytolytic activity of tumor-specific T lymphocytes after radio-frequency ablation in patients with hepatocellular carcinoma and colorectal liver metastasis. World J Gastroenterol. 2006;12:3716-3721.
Hayashi N, Mita E. Fas system and apoptosis in viral hepatitis. J Gastroenterol Hepatol. 1997;12:S223-S226.
Hierholzer C, et al. Essential role of induced nitric oxide in the initiation of the inflammatory response after hemorrhagic shock. J Exp Med. 1998;187:917-928.
Huber TS, et al. Anticytokine therapies for acute inflammation and the systemic inflammatory response syndrome: IL-10 and ischemia/reperfusion injury as a new paradigm. Shock. 2000;13:425-434.
Iimuro Y, et al. NFkappaB prevents apoptosis and liver dysfunction during liver regeneration. J Clin Invest. 1998;101:802-811.
Iwasaki Y, et al. Protective effect of ischemic preconditioning against intermittent warm ischemia–induced liver injury. J Surg Res. 2002;107:82-92.
Jiang Z, et al. CD14 is required for MyD88-independent LPS signaling. Nat Immunol. 2005;6:565-571.
Karlmark KR, et al. Hepatic recruitment of inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50:261-274.
Khoruts A, et al. Circulating tumor necrosis factor, interleukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology. 1991;13:267-276.
Kiener PA, et al. Human monocytic cells contain high levels of intracellular Fas ligand: rapid release following cellular activation. J Immunol. 1997;159:1594-1598.
Kim YM, et al. Nitrogen oxide–induced autoprotection in isolated rat hepatocytes. FEBS Lett. 1995;374:228-232.
Kim YM, et al. Nitric oxide inhibits apoptosis by preventing increases in caspase-3–like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138-31148.
Kimmings AN, et al. Inflammatory and immunologic effects of obstructive jaundice: pathogenesis and treatment. J Am Coll Surg. 1995;181:567-581.
Kimmings AN, et al. Treatment with recombinant bactericidal/permeability-increasing protein to prevent endotoxin-induced mortality in bile duct–ligated rats. J Am Coll Surg. 1999;189:374-379.
Kimmings AN, et al. Systemic inflammatory response in acute cholangitis and after subsequent treatment. Eur J Surg. 2000;166:700-705.
Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. 2010;40:1830-1835.
Kimura F, et al. Hyperactive cytokine response after partial hepatectomy in patients with biliary obstruction. Eur Surg Res. 1998;30:259-267.
Kimura F, et al. Serum interleukin-6 levels in patients with biliary obstruction. Hepatogastroenterology. 1999;46:1613-1617.
Kitade H, et al. Interleukin 1 beta markedly stimulates nitric oxide formation in the absence of other cytokines or lipopolysaccharide in primary cultured rat hepatocytes but not in Kupffer cells. Hepatology. 1996;23:797-802.
Kondo T, et al. Essential roles of the Fas ligand in the development of hepatitis. Nat Med. 1997;3:409-413.
Ksontini R, et al. Revisiting the role of tumor necrosis factor alpha and the response to surgical injury and inflammation. Arch Surg. 1998;133:558-567.
Kunstle G, et al. ICE-protease inhibitors block murine liver injury and apoptosis caused by CD95 or by TNF-alpha. Immunol Lett. 1997;55:5-10.
Larrea E, et al. Tumor necrosis factor alpha gene expression and the response to interferon in chronic hepatitis C. Hepatology. 1996;23:210-217.
Lee KH, et al. The role of receptor internalization in CD95 signaling. EMBO J. 2006;25:1009-1023.
Leist M, et al. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-alpha requires transcriptional arrest. J Immunol. 1994;153:1778-1788.
Lemaire LC, et al. Bacterial translocation in multiple organ failure: cause or epiphenomenon still unproven. Br J Surg. 1997;84:1340-1350.
Levin M, et al. Recombinant bactericidal/permeability-increasing protein (rBPI21) as adjunctive treatment for children with severe meningococcal sepsis: a randomised trial. rBPI21 Meningococcal Sepsis Study Group. Lancet. 2000;356:961-967.
Li CY, et al. Heat shock protein 70 inhibits apoptosis downstream of cytochrome c release and upstream of caspase-3 activation. J Biol Chem. 2000;275:25665-25671.
Li J, et al. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem Biophys Res Commun. 1997;240:419-424.
Liles WC, et al. Differential expression of Fas (CD95) and Fas ligand on normal human phagocytes: implications for the regulation of apoptosis in neutrophils. J Exp Med. 1996;184:429-440.
Liu P, et al. Role of endogenous nitric oxide in TNF-alpha and IL-1beta generation in hepatic ischemia-reperfusion. Shock. 2000;13:217-223.
Lucas KA, et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375-414.
MacFie J. Bacterial translocation in surgical patients. Ann R Coll Surg Engl. 1997;79:183-189.
Malanin NL, et al. MAP3K-related kinase involved in NF-kappaB induction by TNF, CD95 and IL-1. Nature. 1997;385:540-544.
Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-574.
Massague J. TGFbeta in cancer. Cell. 2008;134:215-230.
Matsumoto K, et al. Human biliary epithelial cells secrete and respond to cytokines and hepatocyte growth factors in vitro: interleukin-6, hepatocyte growth factor and epidermal growth factor promote DNA synthesis in vitro. Hepatology. 1994;20:376-382.
Matsunami H, et al. Serial changes of h-HGF and IL-6 in living-related donor liver transplantation with special reference to their relationship to intraoperative portal blood flow. Transplant Proc. 1992;24:1971-1972.
McClain CJ, et al. Cytokines in alcoholic liver disease. Semin Liver Dis. 1999;19:205-219.
McPartland JL, et al. Apoptosis in chronic viral hepatitis parallels histological activity: an immunohistochemical investigation using anti-activated caspase-3 and M30 cytodeath antibody. Int J Exp Pathol. 2005;86:19-24.
Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60-66.
Michalopoulos GK, et al. Liver regeneration. J Cell Physiol. 2007;213:286-300.
Michie HR, et al. Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med. 1988;318:1481-1486.
Mignon A, et al. LPS challenge in D-galactosamine-sensitized mice accounts for caspase-dependent fulminant hepatitis, not for septic shock. Am J Respir Crit Care Med. 1999;159:1308-1315.
Miller AM, et al. IL-33 reduces the development of atherosclerosis. J Exp Med. 2008;205:339-346.
Moolten FL, Bucher NL. Regeneration of rat liver: transfer of humoral agent by cross circulation. Science. 1967;158:272-274.
Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449-1456.
Naveau S, et al. Plasma levels of soluble tumor necrosis factor receptors p55 and p75 in patients with alcoholic liver disease of increasing severity. J Hepatol. 1998;28:778-784.
Nehez L, Andersson R. Compromise of immune function in obstructive jaundice. Eur J Surg. 2002;168:315-328.
Ng KK, et al. Comparison of systemic responses of radiofrequency ablation, cryotherapy, and surgical resection in a porcine liver model. Ann Surg Oncol. 2004;11:650-657.
Nishina T, et al. NIK is involved in constitutive activation of the alternative NF-kappaB pathway and proliferation of pancreatic cancer cells. Biochem Biophys Res Commun. 2009;388:96-101.
O’Dwyer ST, et al. A single dose of endotoxin increases intestinal permeability in healthy humans. Arch Surg. 1988;123:1459-1464.
Olofsson P, et al. Endotoxin: routes of transport in experimental peritonitis. Am J Surg. 1986;151:443-446.
Otterbein LE, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422-428.
Ottonello L, et al. Soluble Fas ligand is chemotactic for human neutrophilic polymorphonuclear leukocytes. J Immunol. 1999;162:3601-3606.
Ou J, et al. Differential effects of nonselective nitric oxide synthase (NOS) and selective inducible NOS inhibition on hepatic necrosis, apoptosis, ICAM-1 expression, and neutrophil accumulation during endotoxemia. Nitric Oxide. 1997;1:404-416.
Park DR, et al. Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J Immunol. 2003;170:6209-6216.
Petrowsky H, et al. Effects of pentoxifylline on liver regeneration: a double-blinded, randomized, controlled trial in 101 patients undergoing major liver resection. Ann Surg. 2010;252:813-822.
Pimental-Nunes P, et al. Toll-like receptors as therapeutic targets in gastrointestinal diseases. Expert Opin Ther Targets. 2010;14:347-368.
Puntis MC, Jiang WG. Plasma cytokine levels and monocyte activation in patients with obstructive jaundice. J Gastroenterol Hepatol. 1996;11:7-13.
Rai RM, et al. Kupffer cell depletion by gadolinium chloride enhances liver regeneration after partial hepatectomy in rats. Am J Physiol. 1996;270:G909-G918.
Rane SG, et al. Transforming growth factor-beta pathway: role in pancreas development and pancreatic disease. Cytokine Growth Factor Rev. 2006;17:107-119.
Reiss M, Barcellos-Hoff MH. Transforming growth factor-beta in breast cancer: a working hypothesis. Breast Cancer Res Treat. 1997;45:81-95.
Rectenwald JE, et al. Functional outcome after thoracoabdominal aortic aneurysm repair. J Vasc Surg. 2002;35:640-647.
Robin MA, et al. Alcohol increases tumor necrosis factor alpha and decreases nuclear factor-kappa B to activate hepatic apoptosis in genetically obese mice. Hepatology. 2005;42:1280-1290.
Sanada S, et al. IL-33 and ST2 comprise a critical biochemically induced and cardioprotective signaling system. J Clin Invest. 2007;117:1538-1549.
Sarantou T, et al. Complications of hepatic cryosurgery. Semin Surg Oncol. 1998;14:156-162.
Schafer C, et al. Tumor necrosis factor and interleukin-6 response of peripheral blood monocytes to low concentrations of lipopolysaccharide in patients with alcoholic liver disease. Z Gastroenterol. 1995;33:503-508.
Schell SR, et al. Pro- and antiinflammatory cytokine production after radiofrequency ablation of unresectable hepatic tumors. J Am Coll Surg. 2002;195:774-781.
Schmidt J, et al. Intratumoral injection of the Toll-like receptor-2/6 agonist “macrophage-activating lipopeptide-2” in patients with pancreatic carcinoma: a phase I/II trial. Br J Cancer. 2007;97:598-604.
Schmitz J, et al. IL-33, an interleukin-1–like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2–associated cytokines. Immunity. 2005;23:479-490.
Schneider P, et al. Conversion of membrane-bound Fas (CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med. 1998;187:1205-1213.
Schwartz AL, et al. Phenylmethimazole decreases Toll-like receptor 3 and noncanonical Wnt5a expression in pancreatic cancer and melanoma together with tumor cell growth and migration. Clin Cancer Res. 2009;15:4114-4122.
Seifert JK, Morris DL. World survey on the complications of hepatic and prostate cryotherapy. World J Surg. 1999;23:109-114.
Seifert JK, et al. Large volume hepatic freezing: association with significant release of the cytokines interleukin-6 and tumor necrosis factor α in a rat model. World J Surg. 2002;26:1333-1341.
Seino K, et al. Chemotactic activity of soluble Fas ligand against phagocytes. J Immunol. 1998;161:4484-4488.
Sheron N, et al. Elevated plasma interleukin-6 and increased severity and mortality in alcoholic hepatitis. Clin Exp Immunol. 1991;84:449-453.
Smith JW2nd, et al. The effects of treatment with interleukin-1 alpha on platelet recovery after high-dose carboplatin. N Engl J Med. 1993;328:756-761.
Soares JB, et al. The role of lipopolysaccharide/Toll-like receptor 4 signaling in chronic liver disease. Hepatol Int. 2010;4:659-672.
Solorzano CC, et al. Involvement of 26-KD cell-associated TNF-alpha in experimental hepatitis and exacerbation of liver injury with a matrix metalloproteinase inhibitor. J Immunol. 1997;158:414-419.
Suffredini AF, et al. New insights into the biology of the acute phase response. J Clin Immunol. 1999;19:203-214.
Szabo G, et al. Liver in sepsis and systemic inflammatory response syndrome. Clin Liver Dis. 2002;6:1045-1066.
Tacke F, et al. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36:4-12.
Tannahill CL, et al. Discordant tumor necrosis factor-alpha superfamily gene expression in bacterial peritonitis and endotoxemic shock. Surgery. 1999;126:349-357.
Taylor BS, et al. Inducible nitric oxide synthase in the liver: regulation and function. Biochemistry (Mosc). 1998;63:766-781.
Tobias PS, et al. Isolation of a lipopolysaccharide-binding acute phase reactant from rabbit serum. J Exp Med. 1986;164:777-793.
Tobias PS, et al. Identification of a lipid A binding site in the acute phase reactant lipopolysaccharide binding protein. J Biol Chem. 1989;264:10867-10871.
Tracey KJ, Cerami A. Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Annu Rev Med. 1994;45:491-503.
Ulevitch RJ, Tobias PS. Recognition of gram-negative bacteria and endotoxin by the innate immune system. Curr Opin Immunol. 1999;11:19-22.
Van Antwerp DJ, et al. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787-789.
van der Poll T, et al. Fibrinolytic response to tumor necrosis factor in healthy subjects. J Exp Med. 1991;174:729-732.
van der Poll T, et al. Tumor necrosis factor mimics the metabolic response to acute infection in healthy humans. Am J Physiol. 1991;261:E457-E465.
van der Poll T, et al. Effects on leukocytes after injection of tumor necrosis factor into healthy humans. Blood. 1992;79:693-698.
Viriyakosol S, et al. MD-2 binds to bacterial lipopolysaccharide. J Biol Chem. 2001;276:38044-38051.
von der Mohlen MA, et al. Inhibition of endotoxin-induced cytokine release and neutrophil activation in humans by use of recombinant bactericidal/permeability-increasing protein. J Infect Dis. 1995;172:144-151.
von der Mohlen MA, et al. Release of bactericidal/permeability-increasing protein in experimental endotoxemia and clinical sepsis: role of tumor necrosis factor. J Immunol. 1996;156:4969-4973.
Wandzioch E, Zaret KS. Dynamic signaling network for the specification of embryonic pancreas and liver progenitors. Science. 2009;324:1701-1710.
Wang Y, et al. Mechanisms of hepatoprotection by nitric oxide. Ann N Y Acad Sci. 2002;962:415-422.
Welborn MB3rd, et al. Visceral ischemia-reperfusion injury promotes tumor necrosis factor (TNF) and interleukin-1 (IL-1) dependent organ injury in the mouse. Shock. 1996;6:171-176.
Welborn MB3rd, et al. The relationship between visceral ischemia, proinflammatory cytokines, and organ injury in patients undergoing thoracoabdominal aortic aneurysm repair. Crit Care Med. 2000;28:3191-3197.
Welborn MB3rd, et al. Role of endogenous interleukin-10 in local and distant organ injury after visceral ischemia-reperfusion. Shock. 2003;20:35-40.
Wiest R, Garcia-Tsao G. Bacterial translocation (BT) in cirrhosis. Hepatology. 2005;41:422-433.
Wink DA, et al. Antioxidant effects of nitric oxide. Methods Enzymol. 1999;301:413-424.
Wright SD, et al. Lipopolysaccharide (LPS) binding protein opsonizes LPS-bearing particles for recognition by a novel receptor on macrophages. J Exp Med. 1989;170:1231-1241.
Xu Y, et al. NF-kappaB inactivation converts a hepatocyte cell line TNF-alpha response from proliferation to apoptosis. Am J Physiol. 1998;275:C1058-C1066.
Yamada Y, et al. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci U S A. 1997;94:1441-1446.
Yamamoto M, et al. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science. 2003;301:640-643.
Yamashiki M, et al. Analysis of serum cytokine levels in primary biliary cirrhosis patients and healthy adults. J Clin Lab Anal. 1998;12:77-82.
Yang RB, et al. Signaling events induced by lipopolysaccharide-activated Toll-like receptor 2. J Immunol. 1999;163:639-643.
Yoshidome H, et al. Interleukin-10 suppresses hepatic ischemia/reperfusion injury in mice: implications of a central role for nuclear factor kappaB. Hepatology. 1999;30:203-208.
Zamora R, et al. Inducible nitric oxide synthase and inflammatory diseases. Mol Med. 2000;6:347-373.
Zhang J, et al. Significance of changes in local immunity in patients with hepatocellular carcinoma after percutaneous microwave coagulation therapy. Chin Med J (Eng). 2002;115:1367-1371.
Zimmermann A, et al. Regulation of liver regeneration. Nephrol Dial Transplant. 2004;4:6-10.
Zwaveling JH, et al. High plasma tumor necrosis factor (TNF)-alpha concentrations and a sepsis-like syndrome in patients undergoing hyperthermic isolated limb perfusion with recombinant TNF-alpha, interferon-gamma, and melphalan. Crit Care Med. 1996;24:765-770.