Cancers Arising in the Ovary
Mark Morgan, Jeff Boyd, Ronny Drapkin and Michael V. Seiden
• Ninety percent of ovarian cancers represent a collection of “epithelial tumors” that increase in incidence with age, with patients diagnosed at a median age of 63 years. Stromal tumors represent less than 10% of malignant tumors arising in the ovary and typically present with symptoms related to sex hormone production. The remaining tumors are germ cell tumors that occur in adolescents and young adults.
• Lifetime risk of epithelial ovarian cancer is approximately 1 in 85. Risks are lower in multiparous women and those who have taken oral contraceptives. Risks are significantly higher in women with germline mutations in BRCA1 or BRCA2 or other DNA mismatch repair genes.
• Epithelial and stromal tumors spread primarily by exfoliation of cells into the peritoneal cavity whereas germ cell tumors tend to spread by lymphatics or hematogenously.
• Patients should usually undergo surgical resection of ovarian tumors with comprehensive surgical staging. Most women with epithelial ovarian cancer present with advanced disease and aggressive surgical cytoreduction is recommended. Approximately 10% of these patients will also present with malignant pleural effusions. Comprehensive surgical staging is recommended for stromal and germ cell tumors although the role of lymphadenectomy is unclear when there is no gross lymphadenopathy.
• Epithelial tumors are typically treated with carboplatin and paclitaxel as well as maximal surgical cytoreduction. A high percentage of women will experience a complete clinical remission; however, the majority who present with advanced disease will relapse within a few years. Median survivals for women with advanced disease are approaching 5 years. Germ cell tumors typically are treated with bleomycin, etoposide, and cisplatin in regimens initially described in the management of testicular carcinoma with the majority of women cured of disease.
• Women with early-stage disease typically receive adjuvant therapy with regimens similar to those used for advanced disease.
Management of Recurrent Disease
• Most women with epithelial cancer will develop recurrent intraperitoneal tumor. Tumors recurring greater than 6 to 12 months after the prior exposure to platinum are often retreated with platinum-based therapy. Patients with rapid recurrence after platinum therapy may be palliated with a variety of drugs, including liposomal doxorubicin, topotecan, and gemcitabine. In addition, bevacizumab has significant activity in the management of recurrent disease.
• Most women with recurrent epithelial cancer will eventually have significant bowel dysfunction. Optimal management often requires a multidisciplinary approach.
Introduction
Ovarian cancer is the leading cause of gynecologic cancer–related deaths and the fifth overall cause of cancer deaths among American women.1 Globally, it afflicts more than 200,000 women each year and claims the lives of over 125,000 annually.2 The high case-to-fatality ratio is in part due to the fact that there is currently no effective screening tool for the early detection of this disease. The vast majority of cases are diagnosed at late stage (International Federation of Gynecology and Obstetrics [FIGO] stage III and IV),3 for which the 5-year survival rate is less than 30%.4
Ovarian cancer is a large collection of tumors that arise or present in a relatively small organ. However, each of these tumor types has very different histologic features and more importantly biological and genetic features that define each of a myriad of malignancies. Tumors are typically divided into epithelial malignancies, stromal tumors, and germ cell tumors. Epithelial malignancy, the most common and lethal of the family of ovarian malignancies, is a misnomer because the ovarian surface epithelium is a mesothelium yet primary ovarian mesotheliomas are extraordinarily rare. Instead, it is likely that epithelial tumors are the result of some still poorly defined and oncogenically induced mesenchymal–epithelial transformation.5 Alternatively, they may represent a metastatic site from the epithelium that initially resided in an extraovarian müllerian site such as the uterus or fallopian tube.
Epidemiology
Risk factors for ovarian cancer can be divided into those that confer markedly elevated risk compared with those that confer moderate or, more commonly, very modest increased risk of ovarian cancer. In the general population, the lifetime risk of ovarian cancer in a woman is approximately 1.25% or 1 case per 80 female births.1 Globally, ovarian cancer is found at slightly higher incident rates in individuals of western and central European descent with age-adjusted rates of approximately 11 cases/100,000 year. Africans and south central Asians may have rates a few times lower, although it is possible some of these differences are from underreporting.6
Table 89-1 reviews the risk factors associated with ovarian cancer. The highest risk is associated with germline mutations in BRCA1 that confer a lifetime risk of ovarian cancer of 25% to 50%.7–10 Significant elevations in risk are also seen in individuals with germline mutations in BRCA2.10–10 Although less wellstudied, mutation in the mismatch repair genes (MLH1, MSH2, PSM2, MSH6) as seen in hereditary nonpolyposis colorectal cancer (HNPCC) or Lynch syndrome also confer significant risk, which has been estimated in a few studies to be as high as 1% per year for women in their 40s or 50s.11 With the use of more comprehensive yet targeted sequencing of genes associated with DNA repair, it appears there is a heightened risk of ovarian cancer in a variety of individuals with germline mutations in genes such as Check1, Rad51, p53, as well as others.12 Although these germline mutations confer markedly elevated risk, there are several lifestyle or environmental factors associated with moderate or modest impact on risk. Age is a major risk factor, with rates of ovarian cancer rising sharply in perimenopausal years and peaking well past menopause. Other risk factors include increased body mass index, a history of polycystic ovarian syndrome, and perineal talc exposure.13–17 Some risk factors such as endometriosis may confer risk for specific types of ovarian cancer, namely clear cell and endometrioid types.18–19
Table 89-1
Genetic, Lifestyle, and Environmental Factors Associated with Epithelial Ovarian Cancer Risk
| Risk Factor | Magnitude of Risk |
| BRCA1 mutation | ↑↑↑ |
| BRCA2 mutation | ↑↑ |
| HNPCC | ↑↑ |
| Endometriosis | ↑ |
| Nulliparity | ↑ |
| High BMI | ↑ |
| Estrogen around menopause | ↑ |
| Early menarche and/or late menopause | ↑ |
| Perineal exposure to talc | ↑ |
| Protective Factors | Magnitude of Protection |
| Oral contraceptives | ↓↓ |
| IUD | ↓ |
| Tubal ligation | ↓ |
| Multiple births | ↓ |
| Late menarche and/or early menopause | ↓ |
| History of mumps | ↓ |
| History of bone fracture | ↓ |
BMI, Body mass index; HNPCC, hereditary nonpolyposis colorectal cancer; IUD, intrauterine device.
There are several protective factors. For example, late menarche, early menopause, early first birth, and breast feeding are associated with a decreased risk of ovarian cancer.14,20–22 Likewise, there is an incremental risk reduction per each year of oral contraceptive use.23 All of these factors are associated with a decrease in the number of ovulatory cycles. However, the reduction in risk may not be explained solely by changes in ovulatory cycles because tubal ligation and intrauterine device (IUD) use are also strongly protective.23,24 Curiously, a history of bone fracture and mumps may be protective and are associated with a higher likelihood of circulating anti-MUC 1 autoantibodies.27–27 Whether this relates to a mechanism of immune surveillance is unclear. Other behaviors may have complex effects, such as cigarette smoking, which increases the risk of mucinous cancer (particularly borderline tumors), decreases the risk of clear cell and endometrioid carcinoma, and leaves the incidence of serous cancer unchanged.28,29
Biological and Molecular Genetic Characteristics
Ovarian cancer is an extraordinarily complex set of malignancies, each characterized by unique histogenesis, early natural history, and molecular genetic features. Although this is evident when epithelial ovarian carcinoma (EOC) is compared with nonepithelial ovarian cancers (e.g., sex cord-stromal tumors), new insights into the etiology and biological characteristics of the more common EOCs are rapidly emerging. Historically, there was wide acceptance of the concept that the epithelial component of the ovary gives rise to four common histologic variants. There was controversy, however, about whether tumorigenesis occurs in the single-cell layer of the surface epithelium or in architectural variations such as cystic inclusions. This concept is problematic from the perspective of embryologic development because EOCs are of serous, endometrioid, mucinous, or clear cell histologies, like their gynecologic counterparts of müllerian origin—the fallopian tubes, uterus, and upper vagina. However, the ovary is not derived from the müllerian duct; it develops separately on the urogenital ridge, the surface epithelium of which is a modified mesothelium, contiguous with, and morphologically resembling, the peritoneal mesothelial lining. Although metaplasia during malignant transformation is a possibility, the question persists as to whether EOC invariably arises from the ovary or perhaps in some cases from müllerian remnants, for example, rete ovarii, lesions such as endometriosis, or other müllerian-derived components of the reproductive tract.30 This section will address various theories and classification schemes for the etiology of EOCs, their fundamental biological features, and the molecular genetic features of EOCs and nonepithelial ovarian cancers.
In addition to the well-recognized molecular and biological heterogeneity among EOC histologic variants, a more sophisticated model is emerging for the classification of EOCs based on histology, tumor grade, and early natural history.31 One such model posits that a “type I” tumor category consists of low-grade serous, low-grade endometrioid, clear cell, mucinous, and transitional (Brenner) carcinomas. In the case of low-grade serous tumors and mucinous tumors, they are suggested to arise from corresponding benign cystic epithelium, often through a borderline (low malignant potential) tumor, supporting the classical paradigm of stepwise morphologic progression during tumorigenesis. Type I tumors are generally indolent, of early stage at diagnosis, and genetically stable. Ironically, these tumors are relatively chemoresistant, and thus the prognosis may be poor when metastasis occurs, although survival times are typically long. Clear cell and low-grade endometrioid cancers may be considered a subclass of type I tumors, some proportion of which arise from the malignant transformation of endometriotic lesions that implant on the ovary. In contrast, “type II” tumors are aggressive, generally chemosensitive initially, often presenting at an advanced stage, and include high-grade serous (and other high-grade histologic type) carcinomas, undifferentiated carcinomas, and carcinosarcomas (Fig. 89-1). Type II tumors are considerably more common than type I tumors; the etiology and biological characteristics of high-grade serous tumors, the most common EOC, will be discussed in greater detail.
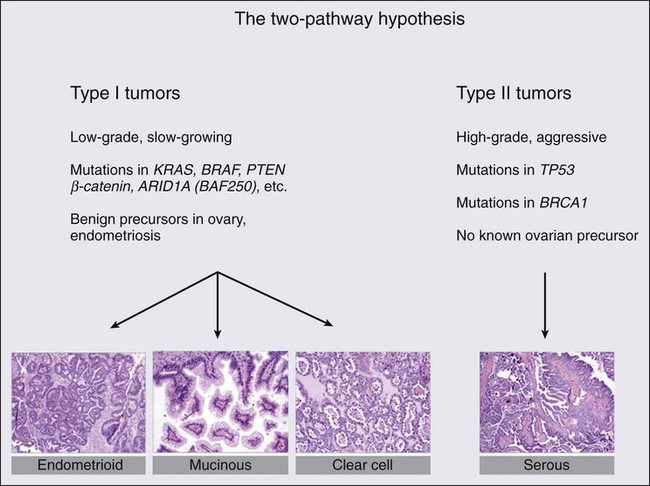
Molecular Genetics of Type I Tumors
The most remarkable distinction between type I and type II tumors is the absence of mutations in the TP53 gene and low chromosomal instability in the former, and the near ubiquitous presence of TP53 mutations and high chromosomal instability in the latter class of tumors.32,33 In terms of well-characterized candidate genetic alterations in type I tumors, low-grade serous tumors display mutually exclusive KRAS or BRAF mutations, but the proportion of affected cases is now debated.34 Recent data from a whole exome analysis of low-grade serous tumors indicate the presence of very few point mutations; 64 distinct genes were mutated in 8 tumors.35 However, in a validation set, only KRAS and BRAF mutations were reproducibly found. These data support the concepts of relative genetic stability of low-grade serous tumors and that KRAS and BRAF mutations are likely important in their pathogenesis. Other types of genetic alterations, such as translocations and epigenetic alterations, have yet to be adequately explored, however, and clearly, at least 50% of low-grade serous tumors appear not to harbor KRAS or BRAF mutations. Interestingly, KRAS mutations are present in approximately 50% of mucinous carcinomas, of both the gastrointestinal and müllerian subtypes, but BRAF mutations are uniformly absent.36 These molecular characteristics of low-grade serous cancers and mucinous cancers are common in borderline serous and mucinous tumors as well, supporting the hypothesized pathogenic continuum of these two tumor types.37
A common molecular genetic alteration also exists in clear cell and low-grade endometrioid cancers; the ARID1A gene, encoding a key component of the SWI-SNF chromatin remodeling complex, is a tumor suppressor gene mutated in approximately half of clear cell EOCs and in approximately one-third of endometrioid EOCs.38,39 In two patients, the same ARID1A mutations were evident in cancers adjacent to atypical endometriosis, providing strong genetic evidence for a link between endometriosis and these two cases of type I cancers.38 Low-grade endometrioid and clear cell cancers are also characterized by mutations in genes encoding components of the Wnt signaling pathway, including CTTNB1, PTEN, and PIK3CA.40 Finally, the PPP2R1A gene, encoding a subunit of protein phosphatase 2A, is mutated in a small fraction of all four histologic variants of type I EOCs, but mutations are invariably absent in type II tumors.41
Molecular Genetics of Type II Tumors
An enormous advance in our understanding of the molecular genetic landscape of type II EOCs was facilitated by the Cancer Genome Atlas Project, which analyzed more than 300 high-grade serous EOCs.33 In addition to whole exome, genome, and comprehensive gene expression analyses, gene methylation and copy number alterations were also examined. As noted above, TP53 mutation and a high level of chromosomal instability are present in essentially all high-grade serous tumors. Germline mutation of the BRCA1 or BRCA2 genes was observed in 17% of cases, with somatic mutations in an additional 3% of tumors. Interestingly, mutations in only seven additional genes were observed in as many as 2% to 4% of cases. However, a consideration of other types of genetic aberrations, such as gene amplification, and upregulation or downregulation of gene expression, indicates the presence of oncogenic alterations in the RB pathway in 67% of cancers, alterations in the PI3K/RAS pathway in 45% of cases, and in the NOTCH pathway in 22% of cases. Additionally, defective homologous recombination-mediated DNA repair, as influenced by BRCA1/2 mutation or genetic or epigenetic alterations in additional genes with a role in this process, is present in 51% of cases. Finally, the FOXM1 signaling pathway, affecting cell cycle progression or DNA repair, is altered in 84% of high-grade serous tumors. It is likely that this type of multifaceted approach to the detection of pathogenic alterations in critical pathways, when applied to type I tumors, will yield similar insights.
Pathogenesis of High-Grade Serous Carcinoma
The most enduring theory of ovarian carcinogenesis holds that ovarian carcinomas arise from müllerian metaplasia of the ovarian surface epithelium or subcortical epithelial inclusions and develop as a function of genotoxic stimuli introduced to this epithelium during reproductive years (Fig. 89-2).42 Although this model supports the genesis of type I tumors, very few high-grade serous carcinomas have been encountered at a stage at which their ovarian origin can be determined with confidence and there is scant evidence for the existence of a precursor lesion in the ovarian surface epithelium or circulating immune complexes. Insight into the pathogenesis of high-grade serous carcinomas came from investigating the prevalence of occult ovarian and fallopian tube cancers in women with germline BRCA gene mutations. Inherited mutations in BRCA1 or BRCA2 are associated with familial ovarian and breast cancer syndromes and account for approximately 11% to 15% of ovarian carcinomas.3,4,43,44 Mutations in either gene confer a 15% to 40% lifetime risk of developing ovarian cancer.45 Many women with germline BRCA mutations elect to undergo risk-reducing bilateral salpingo-oophorectomy, after which the ovaries are thoroughly examined for evidence of occult cancer. Until recently, the fallopian tubes were not systematically examined in these cases. Consequently, early-stage tubal cancers have been rarely detected and severely underreported in BRCA mutation carriers. Recent studies suggest that the fallopian tube may harbor a cell-of-origin, the fallopian tube secretory epithelial cell (FTSEC), for high-grade serous carcinomas of the ovary. Supportive evidence for this hypothesis includes (1) ~5% to 10% of BRCA1 mutation carriers undergoing prophylactic surgery will have an early lesion, termed serous tubal intraepithelial carcinoma (STIC) in their fallopian tube fimbria; (2) >50% of women with stage III/IV pelvic serous cancer also harbor a STIC; (3) identical TP53 mutations have been identified in STICs and corresponding serous carcinomas12; (4) nonneoplastic FTSEC and serous carcinoma share similar morphological and immunophenotypic features; and (5) a candidate precursor lesion (the p53 signature) composed of benign-appearing FTSECs that harbor DNA damage and TP53 mutations, has been described in the fallopian tube (Fig. 89-3).45–50 These observations suggest that pelvic serous carcinomas previously assigned to different sites of origin (ovary, fallopian tube, or peritoneum), share a common carcinogenic pathway not previously appreciated, which originates in the FTSEC.
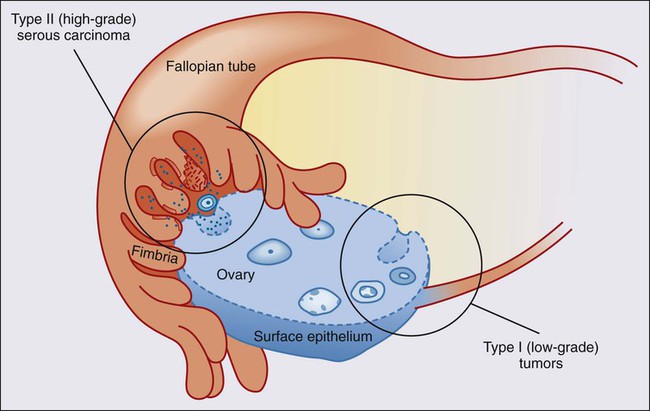
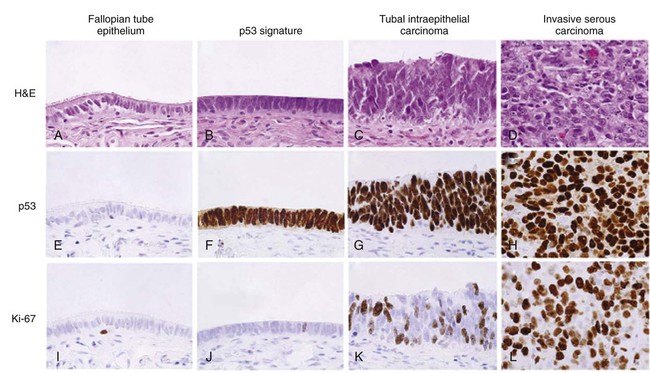
This new model of pathogenesis is compelling and also entirely consistent with longstanding epidemiological observations. It has long been documented that lifetime ovulation is positively correlated with high-grade serous carcinoma, and that factors such as parity and birth control, which would decrease lifetime ovulation, have a protective effect.51 During ovulation, the follicular fluid surrounding the ovum is released and bathes the surrounding tissue, including the ovarian surface epithelium and fallopian tube fimbria proximal to the ovary. The composition of this fluid, known to play a critical, albeit poorly understood, role in the development of the follicle, is ill defined but contains hormones, fatty acids, reactive oxygen species, and growth factors that can have mutagenic effects on the surrounding epithelium with each successive ovulatory cycle. In fact, a recent study in a mouse model showed that ovulation causes DNA damage to tubal epithelial cells, suggesting that the distal fallopian tube is susceptible to double-strand DNA breakage during ovulation.52
The shifting model of serous cancer pathogenesis is starting to impact clinical care. Sectioning and extensive examination of the fimbriated end of the fallopian tube is becoming common practice in academic tertiary care centers. Encouragingly, this has resulted in independent validation and support for the fallopian tube model of serous pathogenesis.53–56 The model is also stirring discussion about whether prophylactic surgery should be limited to the fallopian tubes, or whether one of both ovaries may be spared in BRCA mutation carriers. Salpingectomy alone carries an obvious decrease in early menopause–related morbidity and improvement in quality of life, including preservation of fertility, but evidence supporting this approach for the management of high-risk women is still very limited. A related question is whether fimbriectomy is a viable alternative to tubal ligation as a sterilization technique.57 Arguably, sterilization practices that target the fimbria may maximize the protective effect of tubal sterilization on serous cancer prevention. However, the value of such a practice must be evaluated in the context of a clinical trial that carefully evaluates the safety and feasibility of fimbriectomy with long-term follow-up to ensure that it is effective to reducing the risk of “ovarian cancer” in an at-risk population. Nevertheless, barring the emergence of a successful chemopreventive agent or early detection strategy, removing fimbrial tissue is a provocative, albeit yet unproven, surgical approach to reducing serous malignancies.
In addition to the aforementioned clinical issues that can now be addressed, this new model has sparked the development of novel fallopian tube–based experimental platforms for the discovery of biomarkers and therapeutic targets.25 Coupled with a recent report from The Cancer Genome Atlas (TCGA), which provided a comprehensive, panoramic view of the genomic complexity of serous carcinomas, we are now poised to leverage these new insights for better patient management and care.
Sex Cord-Stromal Tumors
The sex cord-stromal tumors account for approximately 7% of all ovarian neoplasms, and consist of two broad categories, granulosa-stromal cell tumors, and Sertoli-stromal cell tumors.58 The majority of these tumors are relatively indolent and are associated with a favorable long-term prognosis. A substantial proportion of sex cord-stromal tumors is diagnosed in patients under 40 years of age; these tumors have the potential to produce steroid hormones. Remarkably, a single somatic missense mutation of the FOXL2 gene is present in the great majority of all adult-type granulosa cell tumors, rare in other granulosa cell tumors, and largely absent in EOCs or other tumor types.59 The FOXL2 gene encodes a transcription factor known to be critical for granulosa cell development, and is likely pathognomonic for adult granulosa cell tumors. Clues to the pathogenesis of Sertoli-stromal cell tumors derive from the observation that germline truncating mutations in DICER1, which encodes an endoribonuclease in the RNase III family essential for processing microRNAs, are observed in families with the pleuropulmonary blastoma-family tumor and dysplasia syndrome, which includes nonepithelial ovarian tumors, including sex cord-stromal tumors.60 Somatic missense mutations of DICER1 are found in about one-third of nonepithelial ovarian tumors, including 60% of Sertoli-Leydig cell tumors. This finding suggests a novel mechanism through which perturbation of microRNA processing may be oncogenic. Finally, women affected by the rare Peutz-Jeghers hamartomatous polyp syndrome, associated with germline mutation of the STK11 (LKB1) serine-threonine kinase gene, are susceptible to sex cord tumors with annular tubules.61
Germ Cell Tumors
Ovarian germ cell tumors are composed of seven major histologic tumor types derived from the primitive germ cells of the embryonic gonad, and account for approximately 3% of all ovarian neoplasms. Although distinct immunohistochemical features typically distinguish the various histologic types of germ cell tumors, specific genetic alterations have been described only for dysgerminomas, the most common malignant ovarian germ cell neoplasm occurring in pure form. They tend to be tetraploid, with the occasional presence of a small isochromosome, i(12p). Copy number alterations have been identified affecting gains on 12p, 12q, 21q, 22q, and loss on 13q.62 Missense mutation of codon 816 of the KIT oncogene occurs in approximately one-third of dysgerminomas, and is associated with advanced stage at presentation. As such, this mutation represents a potential therapeutic target.63
Breast and Ovarian Cancer Syndrome
The existence of a familial breast and ovarian cancer syndrome was formally recognized as early as 1978 by Lynch and colleagues,64 and the BOC syndrome is now a well-accepted clinical entity. The great majority of individuals affected by the BOC syndrome are BRCA1 or BRCA2 mutation carriers. A site-specific manifestation of familial ovarian carcinoma in which an excess of ovarian cancer but not breast or other cancers occurs was also recognized at one time, but genetic linkage analyses failed to demonstrate association of these kindreds to any locus other than the BOC susceptibility gene BRCA1.65 These families are most appropriately considered as affected by a variant manifestation of the BOC syndrome in which breast cancer is rare or undocumented. Significant risks of cancers other than breast and ovarian associated with BRCA1/2 appear to be limited to uveal melanoma, pancreatic, prostate, and male breast cancers in BRCA2 mutation carriers, although the lifetime risks of these cancers are low compared with female breast and ovarian cancers.66 The Society of Gynecologic Oncology developed guidelines to assist in identifying individuals at risk for BOC syndrome,67 and these were also endorsed by the American College of Obstetricians and Gynecologists.68
Genetics of the BOC Syndrome
Following the original reports of genetic linkage of early-onset breast cancer families and some breast and ovarian cancer families to the BRCA1 locus on chromosome 17q,69 the BRCA1 gene was cloned and characterized in 1994.70 Shortly thereafter, the BRCA2 locus on chromosome 13q was defined,71 and the gene was identified in 1995.72 Based on a large volume of literature, a reasonable estimate is that 55% of hereditary ovarian cancers are linked to BRCA1 and 25% to BRCA2 in the context of the BOC syndrome, and 15% to the Lynch syndrome genes. Several additional genes, including RAD51C, RAD51D, and BRIP1, account for up to 5% of hereditary ovarian cancers.73
The BRCA genes share remarkable similarity in both structure and function. Mutations occur throughout both genes, with more than 80% being nonsense or frameshift alterations leading to a truncated protein product; the remainder are missense and other “variants of uncertain significance,” and a very small proportion consist of large-scale gene rearrangements. Well over 1000 distinct inherited mutations in each gene have been described. The prevalence of BRCA mutations in the general population has been estimated to be as high as one in 400. This figure varies considerably among distinct populations, however. Of note, some ethnic or geographically isolated populations carry specific BRCA mutations at a high frequency, generally as a consequence of a founder effect.74 This phenomenon is most pronounced in the Ashkenazi Jewish population, in which approximately 2.5% of individuals carry one of two distinct mutations in BRCA1 (185delAG or 5382insC) or the 6174delT mutation in BRCA2.75 In a search for genotype-phenotype correlations, the study of BOC families revealed the presence of an “ovarian cancer cluster region” in the very large exon 11 gene of BRCA2. Emerging data suggest that a similar region exists in BRCA1, but the mechanism for these effects are not clear. Mutations of the BRCA genes are inherited in an autosomal dominant fashion; somatic loss of heterozygosity at the BRCA loci in ovarian cancers invariably affects the wild-type allele, consistent with their function as classic tumor suppressor genes.
Given the very large size and multiple functional domains within the BRCA proteins, it is not surprising that both interact physically with numerous additional proteins and participate in multiple cellular processes. Over the years, a major challenge had been to distinguish among the various functions of BRCA1/2 insofar as they relate directly to tumor suppression.77 Based on a wealth of data from model systems, there is now strong consensus that the primary role of both proteins in this regard is in the repair of DNA double strand breaks through the process of homologous recombination; loss of this function leads to genomic instability and tumorigenesis.80–80 The BRCA1 protein is substantially more diverse with respect to protein–protein interactions, and involvement in protein supercomplexes, playing a role in DNA replication, cell cycle checkpoint control, apoptosis, regulation of transcription, chromatin unfolding, and protein ubiquitination. Although the BRCA2 protein probably also plays a role in cell cycle checkpoint control and regulation of mitosis, its key function appears to involve serving as a scaffold for RAD51 in the repair of DNA double strand breaks, as for BRCA1, through homology-directed DNA repair.81
Ovarian Cancer Associated with the BOC Syndrome
In carriers of mutant BRCA1 or BRCA2, penetrance is high but incomplete for ovarian cancer. Numerous studies using multiple designs have proposed the lifetime risk of ovarian cancer associated with germline BRCA mutation, with estimates converging around 40% for BRCA1 and 20% for BRCA2. There is considerable evidence that these risks are affected by other genetic and environmental factors clustering in families. Data from large collaborative genomewide association studies suggest that several common alleles modify ovarian cancer risk associated with BRCA mutation carriers.82
The clinical and pathological characteristics of BRCA-linked compared with sporadic ovarian cancer are distinct. The age at diagnosis for BRCA1-linked ovarian cancer patients (age 45 to 50 years) is considerably younger than for those in the general population (age 61 years). In contrast, BRCA2-linked ovarian cancer patients are diagnosed at a significantly older age (55 to 60 years) than BRCA1-linked patients.83,84 Notably, in contrast with hereditary breast cancer, BRCA-linked ovarian cancers do not occur in women under 30 years. These data have considerable implications for clinical intervention in unaffected mutation carriers. The histopathological characteristics of BRCA-associated ovarian cancers differ from the spectrum associated with their sporadic counterparts. In the BRCA population, tumors of high-grade serous histology are overrepresented, although endometrioid and clear cell tumors are also observed. Tumors of mucinous histology and borderline (low malignant potential) tumors do not occur in association with BRCA mutation. In addition, low-grade serous ovarian cancers are the result of a distinct pathogenic process, and do not occur in the BRCA population. Finally, there does not appear to be a difference between the histopathological spectrum of tumors in the BRCA1 compared with the BRCA2 population.85
The clinical course of BRCA-linked ovarian cancer patients is dramatically different from that of sporadic ovarian cancer patients matched for all other clinical and pathological criteria of prognostic significance. Specifically, progression-free survival and overall survival are significantly improved in BRCA mutation carriers. This prognostic difference has been unequivocally demonstrated in both the Ashkenazi Jewish founder population83,86 as well as the general population,87,88 when comparing BRCA-linked tumors with sporadic tumors. Furthermore, the improved outcome appears to be greater for BRCA2 mutation carriers compared with BRCA1 mutation carriers. The biological basis for this phenomenon is also reasonably well established based on work in vitro and in model systems in vivo. Platinum-based combination chemotherapy, the standard first-line treatment for epithelial ovarian carcinoma, is an “accidental” targeted or personalized therapy in this context. Cis(carbo)platin is known to create interstrand DNA cross links, which are repaired by the cell through creation of a double-strand break at the crosslink, which is then repaired through homologous recombination. Cells deficient in homologous recombination-mediated DNA repair are thus hypersensitive to the cytotoxic effects of platinum-based therapy (as well as other agents that act through similar mechanisms, e.g., mitomycin C).89
Screening, Surveillance, and Risk Reduction in the High-Risk Population
The screening of individuals from the general population for hereditary predisposition to the BOC syndrome is a complex and controversial topic. Several groups have made consensus recommendations on this topic, all referencing different criteria, including the American College of Medical Genetics, the National Comprehensive Cancer Network, the American Society of Clinical Oncology, and the American College of Obstetricians and Gynecologists. In 2005, the U.S. Preventive Services Task Force published its recommendation statement. To summarize, the recommendation is against routine referral for genetic counseling or BRCA genetic testing for women whose family history is not associated with an increased risk for a deleterious BRCA mutation (grade D recommendation), whereas the recommendation for women whose family history is associated with an increased risk for deleterious BRCA mutation be referred for genetic counseling and evaluation for BRCA testing (grade B recommendation).90
Although it is generally accepted that women with germline BRCA mutations should be routinely screened for ovarian cancers, there are no data to suggest that ovarian cancer screening leads to improved survival in the high-risk population. An evaluation of 13 ovarian cancer screening studies of high-risk women found that of 70 screen-detected tumors, only 24% were early-stage, similar to the incidence in the general population. This finding suggests that there is likely to be little difference in survival between screened and unscreened populations.91
In the context of prevention, essentially all published retrospective and prospective studies of the effect of risk-reducing bilateral salpingo-oophorectomy (RRSO) in BRCA mutation carriers demonstrate a significant reduction in the risk of ovarian cancer and, generally, breast cancer as well. Typical is the largest and most recent prospective, multicenter cohort study of 2482 women with BRCA mutations.92 In women undergoing RRSO, the procedure was associated with a substantially lower risk of ovarian cancer, first diagnosis of breast cancer, all-cause mortality, breast cancer–specific mortality, and ovarian cancer–specific mortality. These and a wealth of similar data from the literature contribute to consensus recommendations from several professional organizations and experts in the field, including a review of the literature by the Society of Gynecologic Oncology Clinical Practice Committee, which recommends RRSO in the BRCA population.93 The frequent finding of occult ovarian and tubal carcinomas following RRSO reinforce this recommendation, as well as the importance of careful pathological examination of the entire specimen following surgery (see previous section on Etiology and Biological Characteristics—High-Grade Serous Cancer). Hysterectomy is not recommended in this context because there is no evidence for an increased risk of uterine cancer in the BRCA population; however, the use of tamoxifen or estrogen replacement therapy in some patients may affect this decision. Factors such as current age, child-bearing considerations, and BRCA1 versus BRCA2 mutation status contribute to the timing of the procedure with respect to patient age at the time of RRSO, but the consensus recommendation is to perform the procedure no later than age 40 years. Cost- and comparative-effectiveness studies clearly support this recommendation in the context of cancer prevention in BRCA mutation carriers.94,95
Also well established is the protective effect of oral contraceptives on ovarian cancer risk in the BRCA population. In a large representative study, duration of oral contraceptive use increasingly reduced the risk of ovarian cancer by 50% to 60% with up to 5 years of use.96 Historically, use of this chemoprevention strategy has been tempered by conflicting data on an association with increased risk of breast cancer in BRCA mutation carriers. However, a recent meta-analysis of available data concludes that there is no evidence that recent oral contraceptive formulations increase breast cancer risk in BRCA carriers.97 Nevertheless, as with most medical interventions, a discussion between women and their physicians of the benefit–risk ratio is strongly indicated.
Lynch Syndrome
A history of the recognition and clinical definition of this syndrome is instructive with respect to implications for risk assessment, screening, and prevention. In 1913, Alfred Warthin described “cancer family G,” observed to have an excess of gastric and uterine cancers, but after several generations of observation, members were also documented to suffer from excessive colorectal and extraintestinal tumors.98 Subsequent studies of similar families by Henry Lynch and colleagues lead to a description of the “cancer family syndrome.”99,100 In 1984, the terms Lynch syndrome I and Lynch syndrome II were proposed by Boland and Troncale as corresponding to site-specific familial colorectal cancer and the cancer family syndrome, respectively, without antecedent polyposis.101 Recognizing that Lynch syndromes I and II are manifestations of the same polygenic-based cancer predisposition syndrome, corresponding to the cloning and characterization of the genes responsible for this syndrome in the early 1990s (see later), several collaborative groups, the first meeting in Amsterdam, began proposing criteria designed to provide a uniform basis for diagnosis of a single clinical entity that came to be known as hereditary nonpolyposis colorectal cancer (HNPCC) syndrome.102 The so-called Amsterdam criteria (and later the revised Amsterdam II criteria)103 are very specific for identifying likely gene mutation carriers but are relatively insensitive. They are therefore criticized for being overly exclusive when used as a guide for referring individuals for genetic consultation. Recognizing that there was little consensus with respect to the criteria or threshold for recommending genetic testing for HNPCC, the National Cancer Institute Workshop on HNPCC Syndrome created a set of criteria that, when met, warrant genetic screening.104 These criteria have become known as the Bethesda Guidelines, which appear to be substantially more sensitive but less specific than the Amsterdam criteria as identifying HNPCC kindreds with pathogenic mutations (Box 89-1). This test, described below, is relatively simple and inexpensive, and a negative result translates into a very low probability of the existence of a pathogenic mutation predisposing to what, coming full circle, is now widely referred to simply as Lynch syndrome.
Genetics of Lynch Syndrome
Clues to the genetic basis of Lynch syndrome first emerged in 1993, with several independent observations of somatic hypermutability of a class of DNA repetitive elements, known as microsatellites, in sporadic and familial colorectal tumors.105 This observation was accompanied by reports of the genetic linkage of Lynch syndrome kindreds to two loci, one on chromosome 2p and another on chromosome 3p. Identification and cloning of the relevant genes at these loci, MSH2 on chromosome 2106 and MLH1 on chromosome 3107 quickly followed, together with the realization that these genes encoded human orthologs of yeast DNA mismatch repair proteins. The microsatellite instability phenotype previously observed in colorectal and other cancer types associated with the Lynch syndrome could be explained by loss of function of these DNA mismatch repair genes. In Lynch syndrome, one of these genes is inherited in a mutant form through the germline, whereas in sporadic colorectal, endometrial, and gastric carcinomas affected by microsatellite instability, somatic silencing of MLH1 through promoter hypermethylation is the primary pathogenic mechanism.108 Most patients (90%) with Lynch syndrome carry a mutation in either MSH2 or MLH1, with approximately 10% of cases attributable to MSH6 mutation and a very small number to PMS2 mutation. The DNA mismatch repair genes responsible for Lynch syndrome are classical tumor suppressor genes that sustain loss of function mutations; they are inherited in an autosomal dominant fashion with somatic loss of the wild-type allele required for tumorigenesis. The basis for incomplete and variable tissue-specific penetrance of these genes remains unknown.
In women, Lynch syndrome is most frequently associated with colorectal and endometrial cancers, with a 40% to 60% lifetime risk of each.109 Other cancers, especially of the gastrointestinal and genitourinary tracts, are common.110 The lifetime risk estimates for EOC associated with Lynch syndrome range from 6% to 20%, and the clinicopathological features of these cancers appear to be distinct compared to ovarian cancer in the general population. Although data are limited, in the two largest studies to date, the mean age at diagnosis is mid-40s, most cancers are well or moderately differentiated endometrioid, clear cell histologic types are overrepresented, and early-stage cancers are relatively common.111,112 Additionally, synchronous endometrial cancers are observed in approximately 20% of cases. There is some evidence that MSH6 may account for a higher proportion of gynecologic, including ovarian cancers.
Screening, Surveillance, and Risk Reduction in the Lynch Syndrome Population
The prevalence of DNA mismatch repair gene mutations in the general population is estimated at 1 in 500 to 1 in 1000,85 and the proportion of all ovarian cancers arising in the context of Lynch syndrome is estimated at 1% to 4%.113 With respect to screening tumor tissues for the possible presence of Lynch syndrome, it is necessary to incorporate the risk of endometrial cancer into guidelines for females, and numerous consensus recommendations are published. The Bethesda Guidelines are sensitive but have a low specificity, and are now less commonly used in the clinical setting. Another approach uses algorithms that incorporate the types of cancer in a family, the age of occurrence, and the relationships of family members with cancer to estimate the likelihood of an individual having Lynch syndrome; several of these computerized algorithms are available online.114 Recently, cost-effectiveness and other considerations have led to an emerging consensus that reflex testing of all incident colorectal, and perhaps endometrial, cancer cases, using immunohistochemistry to assess mismatch repair gene expression, is the most appropriate initial screen for Lynch syndrome.116–116 Of note, 92% of ovarian cancers associated with germline mismatch repair gene mutation demonstrate loss of gene expression in the corresponding gene as assessed by immunohistochemistry.112
Although consensus recommendations uniformly endorse increased surveillance for colorectal cancer in this population, based on the resultant decreased mortality, evidence is lacking for an effective methodology for screening women for gynecologic malignancies. There is, however, evidence for the efficacy of prophylactic, or risk-reducing, surgery. In a retrospective cohort of 315 women with mismatch repair gene mutations, in which 61 had prophylactic hysterectomy with or without bilateral salpingo-oophorectomy, matched to women without surgery and followed for approximately 10 years, no ovarian cancers developed in those who had surgery, whereas 12 (5.5%) who did not developed ovarian cancer.117 Using a different approach, data from a decision analytic model that considered 10,000 theoretical women with a mismatch repair gene mutation indicated that only 28 prophylactic surgeries would be needed to prevent one ovarian cancer in this population.118 Risk-reducing surgery, compared with surveillance, is also the most cost-effective option from a societal healthcare cost perspective.119 Taken together, available data would suggest that prophylactic hysterectomy and bilateral salpingo-oophorectomy be offered to all women genetically susceptible to Lynch syndrome at the age of 35 years, or once child-bearing is complete, following a careful discussion of the risks, benefits, and limitations of this procedure.
Screening in the General Population
Approximately 70% of women with epithelial ovarian cancer are seen with advanced-stage disease where lethality is high, with approximately 90% of these women eventually experiencing a recurrence of their disease and most eventually succumbing to platinum-resistant disease. In contrast, the 30% of women who present with early-stage disease enjoy significantly better short- and long-term outcomes, with some reports suggesting 60% to 70% of these women are cured.120 Thus, it would seem intuitive that an effective screening test that detects disease confined to the ovary should produce a higher portion of patients cured of their disease and a corresponding decrease in ovarian cancer–associated mortality. In addition, soon after the discovery of CA-125 in the 1980s, it was appreciated that the majority of women with advanced-stage disease had elevations of this biomarker and subsequent studies identified that 40% to 70% of women with early-stage disease had an elevation of this marker as well.120–125 Thus for the last 15 years there has been a concerted effort to determine whether pelvic ultrasound, CA-125, or the combination might spare women from presenting with advanced-stage disease. In subsequent years, the discovery of additional biomarkers such as human epididymis-4 (HE-4) as well as panels of biomarkers or even more complex proteomic profiles have at first glance offered the hope of early detection.
Despite these challenging statistics, a clinical trial reported in the 1990s suggested that large-scale screening trials might be feasible and CA-125 might meet that requirement. In particular, Jacobs and colleagues reported a randomized trial involving over 20,000 women with half randomly assigned to annual blood draws on 3 successive years for CA-125 determination and the remaining half randomly assigned to standard care.126 In the screened group, approximately 4% of the women had a CA-125 of greater than 30, triggering a pelvic ultrasound. Twenty-nine women ultimately underwent surgical exploration to detect six ovarian cancers (positive predictive value of 20%). Over the next 8 years, 10 additional women were found to have ovarian cancer between screens. Ovarian cancer developed in 19 women in the unscreened group, with more high-grade tumors in this unscreened group. Although there were 9 and 18 deaths in the screened versus unscreened populations, this did not reach statistical significance.126 In addition, two large follow-up studies have been conducted.
In the United States, the Prostate Lung-Colon-Ovary (PLCO) screening trial invited 78,000 women aged 55 to 74 years to participate. Women were randomly assigned one to one, with 39,000 women assigned to six annual CA-125 serum determinations and four annual transvaginal ultrasound examinations. The remaining 39,000 women were not screened. As might be expected, the screened group had more ovary cancers detected (212) compared with the unscreened group (176).127 Stage and grade of tumors at diagnosis were similar in both groups. Screening generated a significant consumption of resources. Although 212 women in the screened group had a diagnosis of cancer, there were 3285 false positives, with 1080 surgical procedures, and 163 significant complications resulting from screen-associated procedures. Most sobering was the fact that 118 women died of ovarian cancer in the screened group as compared with 100 women in the unscreened group.127 Subset analysis of a portion of these serum specimens using a larger collection of serum biomarkers failed to demonstrate that any panel of biomarkers outperformed CA-125 in early detection. It is possible that HE-4 or some secondary collection of biomarkers might prove to be a superior second-stage screening test in individuals with an elevated CA-125 as compared with transvaginal ultrasound.
A second study, based in the United Kingdom under the acronym of the UKCTOCS trial, compared an ultrasound-screened population (n = 51,000) with a CA-125-screened population (n = 51,000) and an unscreened control population (n = 100,000). Unlike, earlier trials, the group of women assigned to CA-125 screening were triaged not only on their CA-125 value but also the rate of change in CA-125 as observed over time using a Risk of Ovarian Cancer Algorithm (ROCA). Individuals whose ROCA analysis crossed a specific threshold were referred for ultrasound in a second round of screening (this arm was referred to as the multimodality screening arm). Although the final survival data will not be available for several years, the results of the prevalence screen have been reported and demonstrated that ultrasound alone was associated with an unacceptable false-positive rate. The multimodal screening rate demonstrated sensitivity, specificity, and positive predictive values of 89.4%, 99.8%, and 43.3%, respectively.128 Ovarian cancer death rates, arguably the only meaningful value, will be available in a few years. Until that time, none of the major medical societies recommend screening in average-risk asymptomatic women.
Clinical Manifestations, Patient Evaluation, Staging, and Debulking
Early-stage ovarian cancer is usually found during the evaluation of a pelvic mass detected either by pelvic examination or radiologic imaging (most commonly ultrasound). Occasionally, it is found incidentally after radiologic evaluation for other medical indications. Although ovarian cancer has been thought of as a “silent disease,” presenting with minimal symptoms until advanced stages, recent evidence suggests that clinical symptoms frequently predate the diagnosis by 3 to 6 months. A “symptom index” developed by Goff based on abdominal bloating, increased abdominal size, difficulty eating, and early satiety identified 57% of women with early disease and 80% of women with advanced disease in a case-control study.129 Although these symptoms are common in women presenting to primary care clinics, the frequency, duration, and severity were worse in the women with ovarian cancer. Other studies have not confirmed the usefulness of the symptom index,130 but studies in this area are ongoing and hope to at least raise the awareness of women and primary care providers to potential symptoms that should be evaluated further.131 If a mass suspicious for ovarian cancer is detected on physical examination or ultrasound, then further imaging such as computed tomography (CT) may be indicated.
A serum CA-125 may also be helpful, although it is less specific in premenopausal women where conditions such as endometriosis, pelvic inflammatory disease, or leiomyomas may cause it to be elevated. The CA-125 can also be elevated in pregnancy. Although elevated in about 80% of women with epithelial ovarian cancer, it is also normal in approximately 50% of women with early ovarian cancer.132 Recently, other tests have been evaluated to increase the sensitivity and decrease the false positives associated with the CA-125, especially in premenopausal women. None of these tests to date increase the positive predictive value enough to be used in screening, but they may be useful in prompting referral to surgeons experienced in the management of gynecologic malignancies when a suspicious mass is detected. Several studies have shown that evaluation and treatment outcome is superior for women treated primarily by gynecologic oncologists. However, in the United States, only about one-third of patients are referred to gynecologic oncologists for their primary surgery.133 Currently, a multiple marker assay that includes CA-125-II is available and FDA approved (OVA-1) for use in women with a pelvic mass for whom surgery is planned. This test has a high negative predictive value (96%), allowing a clinician to feel confident that if normal, the mass is unlikely to be cancer and referral to a specialist may not be required. However, it should not be used as a screening test or to determine if surgery should be performed.134 Another blood test, an immunoassay for the HE-4 protein, has been approved for monitoring for recurrence or progression of ovarian cancer. This test is also not sensitive or specific enough alone to be used as a screening test but may prove useful in eliminating some of the false positives associated with the CA-125 (such as endometriosis or pelvic inflammation) when evaluating a pelvic mass.135,136
An initial surgical approach is important for diagnosis and staging of presumed early disease and cytoreduction or debulking of advanced disease. The FIGO staging system for ovarian cancer is most commonly used and is based on the findings at surgery (Table 89-2). Traditionally, a large vertical midline incision has been recommended for both staging and debulking. However, this recommendation is being challenged by some who argue that advances in preoperative imaging and minimally invasive surgical capabilities can make alternative surgical approaches, such as laparoscopic or robotic-assisted surgery, viable alternatives in selected cases.139–139 This is especially true with the more frequent use of neoadjuvant chemotherapy when large bulky tumors may be effectively cytoreduced by treatment prior to surgery.
Table 89-2
International Federation of Gynecology and Obstetrics Staging System for Ovarian Carcinoma
| Stage | Description |
| I | Growth limited to the ovaries |
| IA | One ovary; no ascites; capsule intact; no tumor on external surface |
| IB | Two ovaries; no ascites; capsule intact; no tumor on external surface |
| IC | One or both ovaries with either surface tumor; ruptured capsule; or ascites or peritoneal washings with malignant cells |
| II | Pelvic extension |
| IIA | Involvement of uterus and/or tubes |
| IIB | Involvement of other pelvic tissues |
| IIC | Stage IIA or IIB with factors as in stage IC |
| III | Peritoneal implants outside pelvis and/or positive retroperitoneal or inguinal nodes |
| IIIA | Grossly limited to true pelvis; negative nodes; microscopic seeding of abdominal peritoneum |
| IIIB | Implants of abdominal peritoneum ≤2 cm; nodes negative |
| IIIC | Abdominal implants >2 cm and/or positive retroperitoneal or inguinal nodes |
| IV | Distant metastases |
Data from the New FIGO stage grouping.7
Ovarian cancer spreads primarily by exfoliation into the peritoneal cavity, direct spread, or lymphatic dissemination. Subclinical metastatic disease can be found in approximately 30% of women with an apparent localized cancer, with retroperitoneal lymph nodes being involved about 10% of the time.140 Therefore, a systematic approach to surgical staging is recommended. This involves obtaining washings for peritoneal cytology, omentectomy, careful inspection of the entire peritoneal cavity, random biopsies of pelvic and abdominal peritoneal surfaces, bilateral pelvic and paraaortic lymph node sampling, and in most cases a total hysterectomy and removal of both fallopian tubes and ovaries. Frozen section should be obtained and for young women desiring childbearing, the uterus and contralateral ovary can almost always be preserved when a germ cell or sex cord-stromal tumor is found because they are rarely bilateral. This is also true for low malignant potential (borderline) epithelial tumors and low-grade serous tumors on frozen section. Invasive cancer is found in 20% to 30% of these cases on final pathology. Therefore, complete staging is still indicated for these tumors. In the absence of enlarged lymph nodes, lymphadenectomy may be omitted in sex cord-stromal tumors and possibly even germ cell tumors.141 Although lymph node metastasis are more common in germ cell tumors (especially dysgerminoma), adjuvant treatment decisions can usually be made based on histology.
More than two-thirds of patients with epithelial ovarian cancer will present with disseminated disease (stage III or greater). In these cases, women almost always present with weeks or months of increasing gastrointestinal or urologic symptoms, often prompting evaluation from multiple specialists including gastroenterologists, urologists, gynecologists, and sometimes even psychiatrists to consider such diagnoses as irritable bowel syndrome, gastritis, interstitial cystitis, menopause, or depression. In these patients, preoperative evaluation typically demonstrates a complex adnexal mass with cystic and solid components as well as ascites, peritoneal deposits, and a pleural effusion (particularly on the right side). CA-125 is typically elevated in women with serous carcinoma, the most common histologic subtype. As described above, surgical exploration to confirm the diagnosis and proceed with an attempt at maximal surgical cytoreduction is frequently the primary step in the management of these patients. Griffiths in 1975 demonstrated that the amount of residual disease after surgery was a significant prognostic factor in advanced ovarian cancer, and this observation has been confirmed in multiple subsequent studies.142,143 Retrospective review of large GOG randomized trials and the Scottish Randomized Trial in Ovarian Cancer (SCOTROC-1) has suggested that a large part of the benefit in their trials was due to the inclusion of subsets of patients who either started with minimal extrapelvic disease or who did not require extensive surgery to obtain “optimal” status.144,145 Optimal debulking surgery has generally been defined either as no residual tumor nodule >2 cm in diameter, or more recently, no tumor nodule >1 cm in diameter. More recent data suggest that even patients with extensive initial metastatic disease requiring radical surgery of the upper abdomen and diaphragm can obtain improved outcomes only if all gross disease is resected.146,147 This is understandable because (1) there is significant interobserver difference in clinical measurement and (2) the designation of ≤1 or ≤2 cm generally implies diffuse small volume residual disease or carcinomatosis. It would be unusual to be considered “optimally debulked” if there were only a few small nodules remaining. If that were the case, these few nodules would usually be resected. A meta-analysis of 6885 patients with stage III and IV ovarian cancer by Bristow in 2002 demonstrated that in any given cohort of patients, each 10% increase in the proportion of patients undergoing maximal cytoreduction was associated with a 5.5% increase in median cohort survival time.148 Further, Aletti147 found that surgeons at the Mayo Clinic who were more willing to perform radical debulking procedures had improved survivals compared with surgeons less likely to do those procedures for the same disease burden. In reviewing this study and the available data of five other single-institution series and eight clinical trials published since 2003, Chang and Bristow concluded that (1) complete cytoreduction to no gross residual disease is associated with significantly longer overall survival, (2) radical cytoreductive surgery is able to at least partly counteract the effect of tumor burden, and (3) the survival outcome of patients with advanced ovarian cancer is strongly influenced by the individual surgeons ability and willingness to undertake radical surgical procedures to achieve minimal residual disease.149
Three randomized trials have been completed addressing the issue of debulking surgery in the primary management of epithelial ovarian cancer. Two trials, one European and the other American, evaluated patients who had a very unsuccessful primary debulking procedure and explored the value of a second surgical procedure after several cycles of chemotherapy. This strategy of interval debulking surgery was based on the hypothesis that chemotherapy would chemically debulk patients, making surgical cytoreduction possible. Both trials randomly assigned patients to either six cycles of chemotherapy or “interval debulking surgery” after three cycles of cisplatin-based chemotherapy. Both trials gave an additional three cycles of chemotherapy to the patients randomly assigned to interval debulking surgery. The European trial, conducted by the European Organization for Research and Treatment of Cancer (EORTC) and using cisplatin and cyclophosphamide, showed a statistically significant 6-month improvement in median survival for patients undergoing interim cytoreduction.150 The American Gynecologic Oncology Group (GOG) trial, using cisplatin and paclitaxel, showed no significant difference in progression-free and median survival (Table 89-3).151 It is possible that the use of paclitaxel in the GOG trial eliminated the benefit of interval debulking surgery. Also, the initial surgery in the GOG trial was more often performed by gynecologic oncologists than by general gynecologists as in the European trial. This approach resulted in patients starting the GOG trial with less disease and therefore on average they did not receive as large a benefit in the subsequent debulking procedure. In the GOG trial, only 36% of patients were converted from suboptimal to optimal versus 45% of patients in the EORTC trial. Both arms of the GOG trial had a superior median survival than the interval debulking arm of the EORTC trial.
Table 89-3
Results of Two Studies of Interval Surgical Cytoreduction
| Parameter | All | IDS | No IDS |
| EORTC STUDY55 | |||
| Patients | 408 | 150 | 149 |
| Response after three cycles | |||
| Complete response rate | 17% | ||
| Partial response rate | 55% | ||
| Progression-free survival (mo) | 15 | 12.5 | |
| Survival (mo) | 27 | 19 | |
| GOG STUDY56 | |||
| Patients | 425 | 216 | 209 |
| Progression-free survival (mo) | 10.5 | 10.8 | |
| Survival (mo) | 32 | 33 |
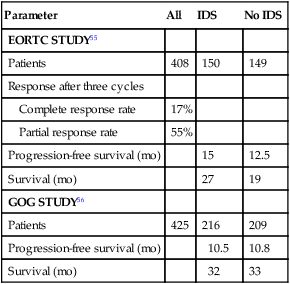
The third trial conducted by the EORTC and the National Cancer Institute of Canada (NCIC) randomly assigned patients to neoadjuvant chemotherapy followed by interval cytoreduction and chemotherapy or primary cytoreduction followed by chemotherapy. The patients all had advanced-stage IIIC or IV disease, with more than 50% having >5 cm extraovarian metastasis. There was no difference in median survival between the 2 groups (30 months for neoadjuvant chemotherapy and 29 months for primary cytoreduction). Absence of residual tumor after surgery was the strongest independent predictor of survival on multivariate analysis (P < 0.001).The morbidity after interval cytoreduction was less than with primary surgery. The authors concluded that neoadjuvant chemotherapy is not inferior to primary surgery for stage IIIC and IV ovarian cancer and that neoadjuvant chemotherapy, followed by interval debulking surgery, is a reasonable option when medical comorbidities preclude an aggressive surgical approach or that the chance of resection of all gross disease is small.152 Vergote, the author of this study, recommends that primary debulking surgery may still be preferable for patients assessed preoperatively to have <5 cm extraovarian metastasis. He has also proposed criteria for neoadjuvant chemotherapy (Box 89-2).153 Others have argued that the survival and optimal debulking rate in the EORTC-NCIC study is inferior to that seen in other trials and single-institutional series for similar patients.154 Therefore, it is argued that until a confirmatory study is done, primary debulking should still be the standard. However these cross-study comparisons have limited validity because of different entry criteria in other trials and biases involved in a nonrandomized case series.
Primary Therapy of Early-Stage Epithelial Ovarian Cancer
Approximately 25% to 30% of epithelial ovarian cancers present as stage I or II disease with an overrepresentation of nonserous histologies. The incidence varies depending upon the extent of staging because approximately 30% of patients will be upstaged when comprehensive staging surgery is performed.155 Tumor grade and rupture before and during surgery are important factors in prognosis and determining whether early disease warrants adjuvant therapy. There is controversy over the prognostic importance of histologic type, with some studies but not all studies suggesting clear cell carcinoma has been overrepresented in recurrences in chemotherapy trials of early-stage disease.156,157 In contrast, early-stage mucinous tumors likely have a slightly better prognosis.
Studies evaluating survival for observation alone after surgery for patients with stage IA or B, well-differentiated tumors have shown that the 5-year survival rate is more than 90%.158 Survival approaches 100% for patients who have had comprehensive surgical staging, and no further therapy is recommended for these patients.159 Patients with early disease who are considered at high risk and have been included in adjuvant trials are shown in Table 89-4.
Table 89-4
Risk Groups for Limited Ovarian Carcinoma
| Group | Characteristics |
| Low risk | Grade I or II disease |
| Intact capsule | |
| No tumor on external surface | |
| Negative peritoneal cytology | |
| No ascites | |
| Growth confined to ovaries | |
| High risk* | Grade III disease |
| Ruptured capsule | |
| Tumor on external surface | |
| Positive peritoneal cytology | |
| Ascites | |
| Growth outside ovaries |
*If any high-risk factors are present, the patient is considered high risk.
Data from Young RC, Walton L, Ellenberg SS, et al. Adjuvant therapy in stage I and stage II epithelial ovarian cancer: results of two prospective randomized trials. N Engl J Med 1990;322:1021-1027.
Although both whole pelvic and whole abdominal radiotherapy have been used in the past as adjuvant therapy for early ovarian cancer, several small studies failed to show superiority to radiation over chemotherapy.160,161 Concerns regarding short- and long-term toxicity, especially radiation-induced bowel damage in patients who have undergone abdominal surgery, has caused this modality to fall out of favor. Intraperitoneal radioactive colloid has also been used in the past. Four randomized trials comparing intraperitoneal radioactive phosphorous (32P) with chemotherapy in early ovarian cancer have been completed. None showed an overall survival advantage to 32P; however, frequent and sometimes serious bowel complications have also resulted in this approach being largely abandoned.158,162–164
There have been four randomized trials evaluating observation versus platinum-based chemotherapy in early ovarian cancer. Two small studies with limited power showed no significant difference in overall survival for patients with stage I, grade 2 or 3 disease using carboplatin or cisplatin for six cycles.163,165 A combined analysis of two other large European randomized trials (ACTION and ICON-1) showed a significant improvement in disease-free and overall survival for adjuvant platinum-based chemotherapy (Table 89-5).166,167 However, a subset analysis with limited power failed to show a survival benefit in patients in the ACTION trial who underwent comprehensive surgical staging.167 The duration of treatment that is optimal for patients with high-risk early ovarian cancer is uncertain. The GOG randomly assigned 427 patients with unfavorable early ovarian cancer to three or six cycles of carboplatin and paclitaxel. Unfavorable was defined as patients with stage IA or IB disease if their histology was grade 3 or clear cell, stage IC, and completely resected stage II. The carboplatin dose was given at an area under the curve (AUC) of 7.5 and the paclitaxel was given at 175 mg/m2 over 3 hours. The 5-year survival rate was 84% for stage I and 73% for stage II and there was no significant difference between the two arms. There was a 25% lower recurrence rate for the patients receiving six cycles but the study was powered to identify only a larger difference (50%) and therefore this difference also was not statistically significant. Toxicity, in particular neurotoxicity, was expectedly greater with six courses.168 The GOG has recently completed a trial randomly assigning the same patient population to three cycles of carboplatin and paclitaxel, this time at a carboplatin AUC of 6, with or without an additional 24 weeks of weekly paclitaxel at 40 mg/m2. The 5-year survival for stage I disease was 88.8% and for stage II disease was 78.9%. There was no difference in recurrence or survival between regimens. As expected, there was a significant increase in toxicity, especially neuropathy, with the prolonged regimen.169
Table 89-5
| Observation | Chemotherapy | Relative Risk | |
| Patients | 460 | 465 | |
| Recurrence free at 5 y | 65% | 76% | 0.64† |
| Alive at 5 y | 74% | 82% | 0.67‡ |
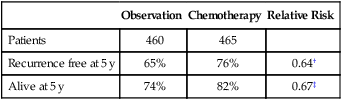
*Randomized trial of patients at high risk of recurrence.
Data from Vergote I, Trimbos B, Guthrie D, et al. Results of a randomized trial in 923 patients with high-risk early ovarian cancer, comparing adjuvant chemotherapy with no further treatment following surgery [abstract]. Proc ASCO 2001;20:201a.
Primary Therapy of Advanced-Stage Epithelial Ovarian Cancer
The initial approach to patients with presumed epithelial ovarian, fallopian tube, and primary peritoneal cancer usually involves cytoreductive surgery followed by platinum and taxane–based chemotherapy. Several controversies exist regarding the timing of initial surgery in relation to chemotherapy, the role of “interval cytoreduction” after suboptimal debulking surgery, the role of intraperitoneal chemotherapy, the importance of dose-intensity, and whether to incorporate newer targeted drugs such as bevacizumab into frontline therapy.170
In 1986, Omura et al. reported the results of a GOG phase III randomized controlled trial establishing the superiority of adding cisplatin to doxorubicin and cyclophosphamide.171 Subsequent GOG trials failed to show a benefit to keeping doxorubicin in the combination, or to doubling the dose intensity of cisplatin and cyclophosphamide.172,173 Eight cycles of cisplatin and cyclophosphamide at 50 and 500 mg/m2, respectively, did not improve response or survival compared with the same drugs given for four cycles at 100 and 1000 mg/m2, respectively. However, a Scottish trial that doubled the dose intensity of cisplatin from 50 to 100 mg/m2 while keeping the cyclophosphamide dose stable at 750 mg/m2 did show an improvement in response and survival when both regimens were given for six cycles.174 This suggests that the total dose may be important once a certain dose intensity is achieved. Subsequently paclitaxel was tested in ovarian cancer and found to have significant activity in platinum-resistant disease. GOG 111, reported by Maguire et al. in 1996, demonstrated the superiority of cisplatin and paclitaxel over cisplatin and cyclophosphamide in patients with advanced, suboptimally debulked ovarian cancer. In this trial, cisplatin was given at 75 mg/m2 and paclitaxel at 135 mg/m2 over 24 hours. The prolonged infusion of paclitaxel reduces the incidence of hypersensitivity reactions and the severity of neurotoxicity. This is especially important when given with cisplatin. The median survival for the patients in the paclitaxel arm was 37 months versus 25 months in the cyclophosphamide group.175 These results were confirmed by a European-Canadian trial (OV-10) in patients with stage IIB-IV disease. Neurotoxicity was significantly higher in that trial, however, because it used a higher dose of paclitaxel (175 mg/m2) given over 3 hours.176
Contrasting these studies are two large randomized trials that did not show superiority of the platinum combination therapy over single-agent platinum chemotherapy. GOG 132 randomized patients with suboptimally debulked ovarian cancer to cisplatin at 100 mg/m2 versus cisplatin/paclitaxel (75 and 135 mg/m2/24 h) versus paclitaxel (200 mg/m2/24 h). The two platinum-containing regimens had comparable survival (26 months) and were superior to single-agent paclitaxel. In the platinum arm, there was a high crossover to paclitaxel before progression, suggesting that sequential therapy may explain the equivalent survival.177 ICON-3 was a large multinational trial that randomly assigned patients with stage I–IV disease to carboplatin/paclitaxel versus cisplatin/doxorubicin/cyclophosphamide or single-agent carboplatin (chosen prior to randomization). This showed no difference in progression-free or overall survival.178
Since then, a series of randomized trials performed by the GOG and in Europe have established carboplatin, given at an AUC of 6 to 7.5, with paclitaxel (over 3 hours) or docetaxel as the standard treatment for advanced-stage epithelial ovarian cancer. GOG 158, which randomly assigned optimally debulked patients with stage III disease to carboplatin/paclitaxel versus cisplatin/paclitaxel (24 hour) showed no difference in median progression-free or overall survival, but a more favorable toxicity profile with the carboplatin-containing regimen. Grade 4 neutropenia, gastrointestinal, renal, and metabolic toxicities were more common in the cisplatin arm. The median progression-free and overall survival for the carboplatin/paclitaxel arm was 20.7 and 57.4 months, respectively.179 Because the carboplatin-containing regimen can be given on an outpatient basis, this has become the preferred regimen used today. Two European studies showed similar results with different patient populations, but reported increased neurotoxicity with the cisplatin/paclitaxel regimen, likely because they used 3-hour infusions of paclitaxel. These trials also gave carboplatin at an AUC of 5 or 6.180,181 Based on these studies, carboplatin (AUC 6) and paclitaxel 175 mg/m2 over 3 hours given every 21 days has become the standard chemotherapy regimen for both early and advanced disease. Substituting docetaxel for paclitaxel may reduce the severity of neurotoxicity without compromising efficacy.182 However, duration of drug exposure may be critical for paclitaxel cytotoxicity. A randomized phase III trial from Japan reported a significant progression-free and 3-year survival advantage using weekly paclitaxel (80 mg/m2) and every-21-day carboplatin (AUC 6). This “dose-dense regimen” has demonstrated similar benefit in breast cancer but is associated with more myelosuppression and neurotoxicity. In the Japanese trial in ovarian cancer, fewer than half of the patients assigned to the dose-dense regimen completed the treatment according to protocol.183 A GOG trial attempting to confirm the Japanese dose-dense study has recently been completed and the results are pending. This trial also allowed concurrent and maintenance bevacizumab.
Because there are multiple drugs with activity in ovarian cancer (Table 89-6), it is reasonable to consider adding additional agents to standard therapy. However, there does not seem to be an advantage to adding a third chemotherapeutic agent to platinum/paclitaxel based therapy. Three international randomized trials failed to demonstrate an improvement in survival with the addition of an anthracycline (doxorubicin or epirubicin) to platinum-based therapy.184,185 Bookman et al. reported on a large international five-arm randomized trial looking at the addition of pegylated liposomal doxorubicin (PLD), topotecan, or gemcitabine to carboplatin and paclitaxel and found no difference in progression-free and overall survival (Table 89-7). The trial, led by the GOG, enrolled 4312 patients with advanced-stage ovarian cancer and included patients who were both optimally and suboptimally debulked. For patients with >1 cm residual, gross-residual ≤1cm, or microscopic residual, the median progression-free survival rates were 13, 16, and 29 months, respectively, and the median overall survival times were 33, 40, and 68 months, respectively. There was no benefit seen for the addition of a third agent to any of these subgroups.186
Table 89-6
Active Agents in the Management of Ovarian Cancer
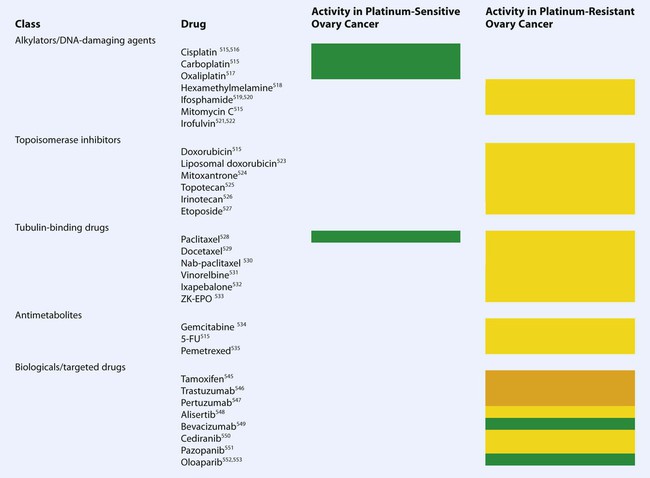 |

Green: high activity; yellow: modest activity; orange: very limited activity as single agent.
Table 89-7
Schema for GOG Protocol 182: An International Trial of the Addition of a Third Agent to Front-Line Therapy
| Regimen I | Paclitaxel plus carboplatin × 8 cycles |
| Regimen II | Paclitaxel plus carboplatin plus gemcitabine × 8 cycles |
| Regimen III | Paclitaxel plus carboplatin plus PLD × 8 cycles (PLD given every other cycle) |
| Regimen IV | Gemcitabine plus carboplatin × 4 cycles followed by paclitaxel plus carboplatin × 4 cycles |
| Regimen V | Topotecan plus carboplatin × 4 cycles followed by paclitaxel plus carboplatin × 4 cycles |
GOG, American Gynecologic Oncology Group; PLD, Pegylated liposomal doxorubicin.
Maintenance Therapy
Only one trial has shown a potential benefit of maintenance therapy after complete remission. A Southwest Oncology Group (SWOG)/GOG trial randomly assigned patients to either 3 or 12 cycles of intravenous paclitaxel at 175 mg/m2 every 4 weeks. The trial was halted after a planned interim efficacy analysis showed an improvement in progression-free survival (28 vs. 21 months, one sided P = 0.0035). A survival benefit could not be determined because the study was halted, at which time crossover to the paclitaxel maintenance arm occurred. Because of neurotoxicity, the dose of paclitaxel was reduced to 135 mg/m2 during the trial.187
Other European trials using intraperitoneal cisplatin, high-dose chemotherapy, topotecan, or epirubicin also failed to demonstrate a survival benefit when used as consolidation or maintenance therapy.188–192 The GOG is currently running a randomized trial of no further therapy versus paclitaxel (135 mg/m2) or Xyotax (paclitaxel polyglumex) every 4 weeks for 12 cycles for patients in complete remission, with the primary end point being overall survival.
Intraperitoneal (IP) Therapy
Ovarian cancer remains clinically confined to the peritoneal cavity for a large part of its clinical course. Therefore, it is logical to consider that direct administration of chemotherapy to the peritoneal cavity may produce a therapeutic advantage. Because many agents that are active in ovarian cancer have a pharmacologic advantage when given intraperitoneally, it is also possible to reach dose-intensity levels not possible with conventional administration routes. For example, both carboplatin and cisplatin have a tenfold to twentyfold higher local concentration when given intraperitoneally. In addition, because of absorption through peritoneal surfaces, systemic levels of these drugs are comparable when the drugs are given intravenously or IP (AUC although not Cmax). Paclitaxel has a 1000-fold increase in intraperitoneal exposure when given intraperitoneally due to a long intraperitoneal residence time with systemic levels (AUC) approximately 50% of that seen with intravenous dosing. Because intraperitoneal chemotherapy generally penetrates tumor nodules for only 1 to 2 mm, its use has been largely limited to patients with small-volume residual disease after primary cytoreduction.193 In January 2006, the National Cancer Institute (NCI) of the United States issued a clinical advisory, suggesting that a combination of IV and IP chemotherapy should be considered in the front-line treatment of patients with optimally debulked ovarian cancer. This was based on a meta-analysis of eight trials that showed an average 21.6% decrease in the risk of death when IP chemotherapy was used.194 Three U.S. studies were particularly influential in the analysis (Table 89-8).197–197 Unfortunately, only one used equivalent doses of IP and IV therapy, leaving some doubt about the influence of the IP route as the most significant variable. Regardless, the most recent trial by Armstrong, using a more intensive IP regimen, reported an impressive improvement in overall survival (66 vs. 50 months, P = 0.03) for the IP arm. This improvement occurred despite the fact that only 42% of patients on the IP arm received the planned six cycles. The increased toxicity of the IP regimen used in this trial, combined with the difficulty of administering IP chemotherapy, has limited the adoption of this regimen as standard therapy.
Table 89-8
GOG Randomized Trials of Intraperitoneal Chemotherapy Trials
| Study | IP Regimen | IV Regimen | Survival IP (mo) | Survival IV (mo) | P Value |
| Alberts 1996 | Cisplatin 100 mg/m2 Cyclophosphamide 600 mg/m2 |
Cisplatin 100 mg/m2 Cyclophosphamide 600 mg/m2 |
49 | 41 | 0.02 |
| Markman 2001 | IV carboplatin AUC 9 IP cisplatin 100 mg/m2 IV paclitaxel 135 mg/m2 |
Cisplatin 75 mg/m2 Paclitaxel 135 mg/m2 |
67 | 51 | 0.05 |
| Armstrong 2006 | IV paclitaxel 135 mg/m2 IP cisplatin 100 mg/m2 IP paclitaxel 60 mg/m2 |
Cisplatin 75 mg/m2 Paclitaxel 135 mg/m2 |
66 | 50 | 0.03 |

To make the regimen used in the Armstrong study more tolerable, several modifications have been proposed. In addition, data from Fujiwara in Japan suggest that appropriately dosed IP carboplatin may be a suitable alternative to IP cisplatin.198 The GOG has recently completed a phase III trial of IV carboplatin (AUC 6) with IV dose-dense paclitaxel (80 mg/m2, days 1, 8, 15), versus IP carboplatin (AUC 6) with IV dose-dense paclitaxel, versus IP cisplatin (75 mg/m2), IV paclitaxel (135 mg/m2 over 3 hours) and IP paclitaxel (60 mg/m2, day 8). Each regimen was given on 21 day cycles for six cycles. Bevacizumab (15 mg/kg) was also given IV starting on the second cycle of each treatment and continued for 16 cycles after completing chemotherapy. It is hoped that this study will answer whether carboplatin and paclitaxel is more effective when carboplatin is given IP at equivalent doses or if a more tolerable modification of the IP regimen used in the Armstrong study is superior to either carboplatin-based regimen.
Biological Agents in Front-Line Therapy
Tumor angiogenesis appears to be an important process in epithelial ovarian cancer development. Bevacizumab is a monoclonal antibody that can neutralize vascular endothelial growth factor (VEGF), a promoter of the initiation phase of angiogenesis. Two phase III trials have been reported that have added bevacizumab to standard therapy with six cycles of carboplatin and paclitaxel. GOG 0218 and ICON 7 both showed an improved progression-free survival when bevacizumab was given concurrently and as maintenance therapy for a total of 18 cycles in ICON 7 and 22 cycles in GOG 0218. There were some differences in patient populations, with the GOG trial only including patients with stage III and IV disease and the ICON 7 allowing patients with high-risk, early disease (stage I and II). Bevacizumab was given at 15 mg/kg in the GOG study and 7.5 mg/kg in ICON 7.199–200 In GOG 0218, the progression-free survival was extended by 3.8 months (14.1 vs. 10.3 months, P < 0.001) and in ICON 7 by 1.7 months ( 24.1 vs. 22.4 months, P = 0.04). Although the final planned end point of both trials was progression-free survival, GOG 0218 was originally powered to detect a survival difference. However, during the trial it became apparent that patient and doctor request for unblinding at the time of progression, with the subsequent treatment of patients randomly assigned to placebo with bevacizumab, would make evaluation of survival as an end point infeasible. With the final survival analysis pending at the time of the reports, there was no overall survival advantage in either study. However, a preplanned analysis of ICON 7 demonstrated an overall survival benefit of almost 8 months in a high-risk group with stage IV or >1 cm residual disease (36.6 vs. 28.8 months, P = 0.002). Hypertension was the most common complication in both trials and required treatment in 20% to 25% of patients. The rate of gastrointestinal perforation was approximately double with bevacizumab, but still less than 3%.
Germ Cell Tumors
Malignant germ cell tumors account for approximately 3% to 5% of ovarian malignancies in Western Countries. They occur primarily in teens and young adults with a peak age in the early 20s. Because they are so rare, treatment of malignant germ cell tumors is based largely on the experience with the more common testicular germ cell tumors. These tumors typically exhibit rapid growth and patients are seen with abdominal pain, sometimes leading to emergent surgery because of rupture or hemorrhage. The most common type is the dysgerminoma (analogous to testicular seminoma) that may be associated with an elevated serum LDH or a low-level elevation of the serum hCG when nests of syncytiotrophoblastic cells are present. Bilaterality is more common (10% to 15%) with dysgerminoma than with other types. They may also be associated with dysgenetic gonads (46,XY) and a gonadoblastoma. Dysgerminomas are very radiosensitive, but radiation therapy is rarely used today because of its adverse effect on fertility. Nondysgerminomatous tumors include yolk sac tumors, immature teratomas, embryonal carcinoma, choriocarcinoma, polyembryoma, and mixed germ cell tumors. These tumors are often associated with the production of AFP or hCG (Table 89-9). The serum CA-125 can also be nonspecifically elevated. Prior to modern chemotherapy, they were almost always fatal.
Table 89-9
Serum Tumor Markers for Ovarian Germ Cell Tumors
| Histologic Subtype | hCG | AFP | LDH |
| Dysgerminoma | +/− | − | + |
| Yolk sac tumor | − | + | − |
| Immature teratoma | − | +/− | − |
| Embryonal carcinoma | + | +/− | +/− |
| Choriocarcinoma | + | − | +/− |
| Polyembryoma | + | +/− | − |
| Mixed germ cell | +/− | +/− | +/− |
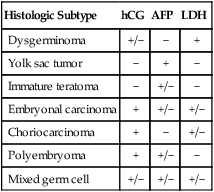
AFP, α-Fetoprotein; hCG; human chorionic gonadotropin; LDH, serum lactate dehydrogenase.
Because germ cell tumors are often large and solid, the initial surgical approach is usually via laparotomy with a vertical incision. Fertility-sparing, unilateral salpingo-oophorectomy is almost always indicated, even with advanced disease, except in the case of dysgenetic gonads. Complete staging or debulking as for an epithelial ovarian cancer is recommended. However, the role of lymphadenectomy and staging when there is no visible disease or enlarged lymph nodes has been questioned recently.201 That is because all tumor types except stage IA dysgerminomas and low-grade stage IA immature teratomas will require adjuvant chemotherapy anyway and the salvage rate for untreated patients with recurrence is very high. If no adjuvant therapy for these early tumors is chosen, careful serologic and radiologic surveillance is required, and most recurrences occur in the first 2 years. Investigators at the Charing Cross Hospital in London have suggested that all stage I germ cell tumors can be observed. In their series of 37 patients, 22% of the dysgerminomas and 36% of the nondysgerminomas recurred within 13 months. However, there was a high salvage rate with chemotherapy, and the overall disease-specific survival rate was 94%.202
Postoperative chemotherapy with bleomycin, etoposide, and cisplatin (BEP) has replaced earlier regimens that included vincristine, dactinomycin, and cyclophosphamide (VAC). This approach was developed for testicular germ cell tumors and has been confirmed to be effective in ovarian germ cell tumors. Both 5- and 3-day BEP regimens have been reported with very high cure rates but as of yet not directly compared in ovarian germ cell tumors. Completely resected ovarian germ cell tumors (stages I, II, and III) can expect cure rates of more than 90% with three cycles of BEP. Incompletely resected and stage IV disease should receive four cycles. Resection of residual masses after chemotherapy for metastatic immature teratoma and mixed germ cell tumors should be considered. For other types, serologic and radiologic follow-up is indicated.203,204 Positron emission tomography may be especially useful in this setting. Recurrent disease after platinum-based therapy may be salvaged with high-dose chemotherapy and stem cell rescue.
Sex Cord-Stromal Tumors
Fertility-sparing surgery with a unilateral salpingo-oophorectomy is appropriate for premenopausal women desiring childbearing. Although staging procedures similar to epithelial ovarian cancer has been recommended in the past, recent evidence suggests that lymph node involvement is rare and lymphadenectomy may not be necessary in apparent early disease.205–206 Nodal recurrences have been reported however. Adjuvant therapy is not indicated for early, resected disease and is generally used for metastatic or recurrent disease. Historically, BEP has been used based on a GOG phase II trial and case series.207 However, this regimen is toxic and it is not clear that a regimen typically used for germ cell tumors is optimal for stromal tumors. In small case series, taxanes and aromatase inhibitors have been found to have some activity.208 Also, serum vascular endothelial growth factor A has been found to be elevated in patients with granulosa cell tumors and bevacizumab has demonstrated activity in vitro and in small numbers of patients.209
Management of Women with Recurrent Epithelial Ovarian Cancer
Women with evidence of tumor response followed by subsequent tumor progression within 6 months of completing platinum-containing therapy are considered to have platinum-resistant tumors. This is a relative term in that some patients with rising CA-125 occurring 6 months after platinum-based therapy will often respond well to retreatment with a platinum regimen, whereas those who have evidence of bulky recurrence weeks after completing platinum essentially never respond. Women with evidence of progressive disease more than 6 months after completing therapy are considered to have tumors that are potentially platinum sensitive. As with platinum-resistant tumors, the group of women with platinum sensitive disease have variable response rates to retreatment with platinum ranging from 30% for women with a median platinum-free interval of 6 to 12 months to >70% for women with a platinum-free interval of greater than 2 years.212–212 In addition, recurrent serous tumors and smaller tumors may demonstrate greater response to therapy at relapse compared with bulky tumors (>5 cm) or tumors of nonserous histology.213
Women with platinum-refractory disease typically are symptomatic and require either prompt treatment with a nonplatinum agent or transition to best support care. For women with platinum-resistant or platinum-sensitive recurrent disease, the first question to be asked is whether the patients should have immediate therapy versus a course of careful observation until a time that tumor symptoms or bulk warrant therapy. In particular, the routine use of monitoring of patients with serial CA-125 values will often detect the recurrence of disease months to occasionally a year or more prior to the development of clinical symptoms.214–215 Furthermore, the increasing use of sophisticated imaging techniques including positron emission tomography–computed tomography (PET-CT) scans has successfully identified women with very early recurrences of disease.216
Recently, Rustin and colleagues reported a study evaluating the role of CA-125 monitoring of patients in a first clinical remission after platinum-based therapy of advanced-stage ovarian cancer.217 Patients had CA-125s evaluated every 3 months but half the patients (and their treating physicians) were blinded to those results and hence were not aware of a rising CA-125 (and by extension progression of subclinical recurrent disease). Women assigned to early treatment initiated treatment within 28 days of the time in which their CA-125 reached twice normal whereas those assigned to late treatment remained blinded to CA-125 and initiated therapy at clinical or symptomatic relapse. As anticipated, physicians and their patients acted on a rising CA-125 alone initiated therapy on average 4.8 months earlier than those blinded to the value. The patients and physicians randomly assigned to CA-125 disclosure received more chemotherapy but did not enjoy a longer survival (early treatment overall survival = 25.7 months vs. late treatment 27.1 months). The study has been criticized because of the limited use of paclitaxel and the failure to prescribe specific therapies in each arm at the time of relapse. Nevertheless, there is a lack of compelling data that early initiation of chemotherapy in asymptomatic patients with recurrent disease prolongs survival. Likewise, the early initiation of palliative chemotherapy in women with asymptomatic recurrent disease is unlikely to improve quality of life in the short term. Many investigators have concluded that careful observation is an appropriate strategy in asymptomatic patients with a rising CA-125.
In patients who will not accept observation, who have symptomatic disease, or who demonstrate significant tumor bulk that risks impending gastrointestinal, pulmonary, or urologic dysfunction, treatment is warranted. Figure 89-4 demonstrates a treatment approach supported by available, albeit somewhat limited, collection of randomized trials in this patient population.
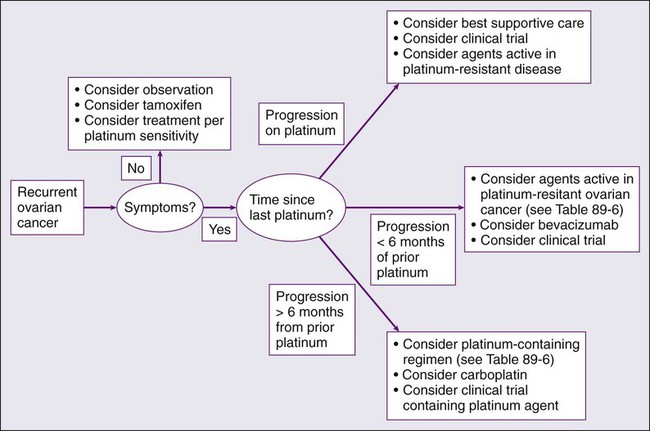
Management of Platinum-Sensitive Recurrent Disease
Randomized studies looking at the value of treatment with platinum versus non–platinum-containing regimen demonstrates much improved responses and duration of response with a platinum-containing regimen.218 In the study by Cantu, women with long platinum-free intervals were randomly assigned to single-agent paclitaxel versus cyclophosphamide, Adriamycin, and cisplatin. Despite the fact that the study randomly assigned less than 100 women, there was a statistically significant improvement in overall survival in women randomized to the platinum-containing regimen. Likewise, there are several randomized trials comparing platinum plus a second agent (either paclitaxel, epirubicin, PLD, or gemcitabine) versus single-agent platinum.219–223 Only a few of the studies accrued large enough numbers of patients to have reasonable power to detect a modest difference in survival. The largest study, ICON 4, compared the use of either single-agent cisplatin or carboplatin versus the same drug with paclitaxel. This study, as well as the GEICO study, demonstrated superior outcomes with the paclitaxel regimen.221–222 In ICON 4, the doublet had a superior response rate (66% compared with 54%; P = 0.06), progression-free survival at 1 year (50% compared with 40%; P = 0.001) and overall survival at 2 years (57% vs 50%; P = 0.023) compared with women assigned to single-agent cisplatin.221 Likewise, similar results were seen in the randomized trial of carboplatin versus gemcitabine and carboplatin (Table 89-10).223 This modest-sized randomized trial of 356 patients with platinum-sensitive first recurrence of disease demonstrated that women treated with carboplatin (AUC 4) and gemcitabine (1000 mg/m2 days 1 and 8) had a response rate of 47% compared with 31% in the women assigned to carboplatin dosed at an AUC = 5 (P = 0.0016). Progression-free survival likewise was superior in the doublet arm at 8.6 months compared with 5.8 months for the women on single-agent carboplatin (P = 0.0031).223 The study was not powered to detect a survival difference, and the overall survival in the two groups was not statistically different. Randomized trials have also explored a comparison of PLD alone with PLD and the novel marine product trabectedin. In a large trial that included women with platinum-sensitive and platinum-resistant disease, the group of women with platinum-sensitive disease had improved progression-free survival with the trabectedin containing doublet compared with PLD.226–226 There was not a statistically significant improvement in survival. Because there was not a platinum-containing comparator arm, it is difficult to evaluate what precise role this combination might have in the management of recurrent and platinum-sensitive ovarian cancer.
Table 89-10
Randomized Trials of Single Agents Versus Combination Therapy in Recurrent and Platinum Sensitive Ovary Cancer
| N | Control | Experimental | PFS (C/E) | OS (C/E) | |
| Cantu | 97 | Paclitaxel | Cyclophosphamide Adriamycin Cisplatin |
9 mo 15.7 mo P = 0.038 |
25.8 mo 34.7 mo P = 0.043 |
| Monk | 417 | PLD | PLD Trabectedin |
7.5 mo 9.2 mo P = 0.017 |
23 mo 26 mo P = 0.11 |
| Bolis | 300 | Carboplatin | Carboplatin Epirubicin |
16 mo 20 mo (NS) |
NS |
| Parmar | 802 | Platinum | Carboplatin Paclitaxel |
10 mo 13 mo P = 0.0004 |
24 mo 29 mo P = 0.02 |
| Gonzales-Martin | 81 | Carboplatin | Carboplatin Paclitaxel |
8 mo 11.2 mo P = 0.02 |
17 mo NRY* P = 0.0021 |
| Pfisterer | 356 | Carboplatin | Carboplatin Gemcitabine |
5.8 mo 8.6 mo P = 0.0032 |
NR NS |
| Markman | 61 | Carboplatin | Carboplatin PLD |
8 mo 12 mo P = 0.02 |
18 mo 31 mo NS |
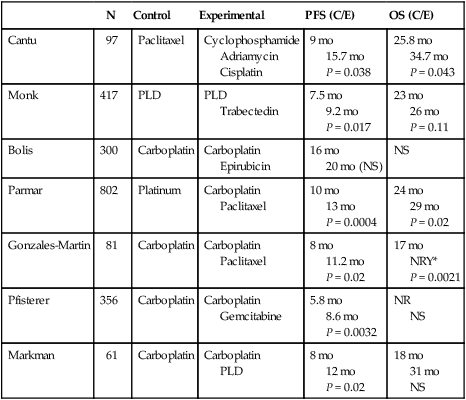
Only a few randomized trials have been reported comparing platinum doublets (Table 89-11).229–229 Of note, the recently published Calypso trial compared PLD with carboplatin to paclitaxel with platinum. In this study, neuropathy, alopecia, and allergy to platinum were more common in the paclitaxel-treated population whereas skin, mucosal, and hematologic toxicity was more common in the PLD-containing arm. Progression-free survival, but not overall survival, was slightly longer in the PLD-treated group. The remarkably lower rate of platinum allergy was also seen in the randomized trial of single carboplatin versus PLD with platinum.220,230
Table 89-11
Randomized Trials of Combination Therapy in Platinum Sensitive Ovarian Cancer
| N | Control | Experimental | PFS (C/E) | OS (C/E) | |
| Bafaloukos | 189 | Carboplatin Paclitaxel |
Carboplatin PLD |
10.8 mo 11.8 mo NS |
29.4 mo 24.7 mo NS |
| Martin | 976 | Carboplatin Paclitaxel |
Carboplatin PLD |
9.4 mo 11.3 mo P = 0.005 |
33.0 mo 30.7 mo NS |
| Secord | 150 | Carboplatin Docetaxel |
Carboplatin Docetaxel |
13.4 mo 8.4 mo P = 0.02 |
33.2 mo 30.1 mo NS |
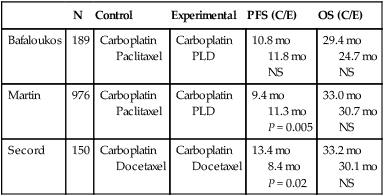
A single randomized phase II of concurrent versus sequential therapy has also been reported. In the study reported by Secord and colleagues, patients were randomly assigned between docetaxel (days 1 and 8) and concurrent carboplatin versus docetaxel on days 1 and 8 of a 21-day cycle for six cycles followed by subsequent carboplatin as a single agent for six cycles. The sequential therapy demonstrated a 62% higher rate of tumor progression (hazard ratio [HR] 1.62, P = 0.02). Overall survival between the two groups appeared similar (33 months for concurrent therapy vs. 30 months for sequential therapy; not significant).231
The addition of molecularly targeted agents to the treatment of women with recurrent and platinum-sensitive disease has been recently reported in the OCEANS (Ovarian Cancer Study Comparing Efficacy and Safety of Chemotherapy and Anti-Angiogenic Therapy in Platinum-Sensitive Recurrent Disease) study,232 which evaluated the benefit of adding bevacizumab to carboplatin and gemcitabine in women with platinum-sensitive recurrent disease. In this moderate-size study of 484 patients, the non bevacizumab arm received a placebo to minimize bias and either placebo or bevacizumab was continued postchemotherapy in patients with a durable response. The objective response rate was 79% in bevacizumab arm and 57% in the placebo arm (P < 0.0001). Bevacizumab-based therapy also extended progression-free survival from 7.4 to 10.4 months (HR 0.534, 95% confidence interval [CI] 0.41 to 0.7). Although mature survival analysis for this randomized trial is still not available, to date the overall survival for the two groups is similar (median 35.2 months in the placebo arm and 33.3 months in the bevacizumab arm).
Recently, investigation in basic biological mechanisms of DNA repair have demonstrated that cells deficient in homologous recombination have defects in the repair of double-strand DNA breaks and are dependent on alternative mechanism of repair. In particular, alternative mechanisms of base excision repair require poly(ADP-ribose) polymerase (PARP) to locate at sites of DNA damage as an integral part of the DNA repair. Tumors deficient in BRCA1 or BRCA2 have defects in homologous recombination and thus are dependent on PARP and by extension are exquisitely sensitive to PARP inhibition.235–235
Several PARP inhibitors are currently in clinical trials, with the greatest experience currently reported with olaparib. Early phase I and II studies of this agent demonstrated activity in both platinum-sensitive and platinum-resistant disease.234,236 In addition, armed with the knowledge that a significant portion of women with high-grade serous cancer have defects in the homologous DNA end joining and rely on the base excision repair pathway, two groups have conducted randomized phase II studies evaluating the value of a PARP inhibitor as a maintenance strategy in women entering second partial or complete response after retreatment with platinum-based therapy in the setting of recurrent and potentially platinum-sensitive disease.237 The “study 19” trial evaluated olaparib versus placebo as maintenance therapy in women with high-grade serous carcinoma who had just successfully achieved at least a partial response to platinum-based therapy. Women assigned to olaparib had an 8.4-month progression-free survival compared with 4.8 months for women on placebo (HR 0.35, 95% CI, 0.25 to 0.49, P < 0.00001). Early survival data reveal no difference although the study was modest in size and these data are not mature.51 It is not yet known what portion of these patients had germline or somatic mutations in BRCA1 or BRCA2 and whether that subgroup experienced greater benefit than women without the BRCA mutation.
The second study had similar eligibility with a slightly different design: the PARP inhibitor was added to carboplatin and paclitaxel during treatment for women assigned to the active treatment arm. The women on the experimental treatment arm received carboplatin at an AUC of 4 compared with those on the control arm who received carboplatin at an AUC of 6. Women on the experimental treatment arm continued on single-agent olaparib as maintenance at the conclusion of their paclitaxel and carboplatin therapy if they had demonstrated a tumor response. Progression-free survival, the primary end point of the study, was prolonged on the olaparib arm at 12.2 versus 9.6 months for the control arm (HR 0.51, P = 0.0012). Survival data are not yet available.238 From these studies, it is clear that olaparib has activity in women in high-grade serous carcinoma. Much but not all of this activity is likely in the population with germline or somatic mutations in BRCA 1 or 2. Other individuals may also have a BRCA-deficient phenotype because of alteration in expression or structure of the many BRCA-related proteins. A cell’s ability to perform homologous repair of DNA damage has recently been referred to as BRCAness.239,240 Determining predictive markers for BRCAness remains an area of intense research interest. Currently, there are no FDA-approved PARP inhibitors, and bevacizumab is not approved for the treatment of ovarian cancer at this time.
Treatment of Recurrent Platinum Resistant Epithelial Ovarian Cancer
Women who have clinical evidence of progressive ovarian cancer within 6 months of their last dose of platinum or platinum-based combination therapy are typically defined as having platinum-resistant disease and retreatment with platinum is expected to have a less than 10% chance of having a significant and clinically meaningful response. A variety of drugs with variable mechanisms of action have demonstrated activity in ovarian cancer, including taxanes (paclitaxel, docetaxel, nab-paclitaxel), topoisomerase 1 and 2 inhibitors (topotecan and liposomal doxorubicin and etoposide), epothilones (ixabepilone and ZK-EPO), alkylators (hexamethymelamine, ifosfamide), and antimetabolites (pemetrexed, gemcitabine) (see Table 89-6).241–261
Typically, the management of women with recurrent disease includes the delivery of single agents in sequence. Essentially all the chemotherapeutic agents listed above have response rates in range of 10% to 20%, with time to progression typically 3 to 4 months. Because almost all these studies are single-arm studies, it is difficult to determine the relative activity for these agents.241–261 Randomized comparisons of one palliative strategy versus an alternative strategy are limited in platinum-resistant disease and are reviewed in Table 89-12. For example, a comparison of every-3-week paclitaxel to standard topotecan resulted in similar response rates and progression-free survival.262 Likewise, randomized comparisons of topotecan to PLD demonstrated no significant differences in women with platinum-resistant disease.263 A similar study comparing gemcitabine with PLD demonstrated no significant difference in response rate or progression-free survival although there was a borderline improvement in overall survival favoring the PLD arm; however, it is notable that some of these patients would be classified as platinum-sensitive recurrent disease.264 In addition, schedules have also been evaluated such as day 1 through 5 topotecan versus weekly topotecan (5-day topotecan modestly superior)265,266 or every 3-week paclitaxel versus weekly paclitaxel (equivalent).267
Table 89-12
Randomized Trials in Platinum-Resistant Ovarian Cancer
| N | Control | Experimental | PFS (Control/Experimental) | OS | |
| Rosenberg | 208 | Paclitaxel (3w) | Paclitaxel (w) | 8.1 m 6.1 m ns |
14.7m* 13.6 ns |
| Sehouli | 194 | Topotecan d1-5 | Topotecan (w) | 3.0m 4.4m ns |
9.3m 9.6m ns |
| ten Bokkel Huinink | 226 | Paclitaxel (3w) | Topotecan d1-5 | 5.2m 3.4m ns |
NR |
| Gordon | 474 | Topotecan | PLD | 3.3m 2.4m ns |
8m 9.8m ns |
| Ferrandina | 153 | PLD | Gemcitabine | 3.9m 4.8m ns |
13m 11.8m ns |
| Lortholary | 165 | Paclitaxel(w) | Paclitaxel (w) Topotecan (w) Or Paclitaxel (w) Carboplatin (w) |
3.7 m 5 m (ns) |
NR |
| Vergote | 461 | PLD or Topotecan |
Canfosfamide | NR | 13.5m 8.5m p<.01 |
| Monk | 228 | PLD | PLD Trabectedin |
3.7 m 4.0 m ns |
12m 13m ns |
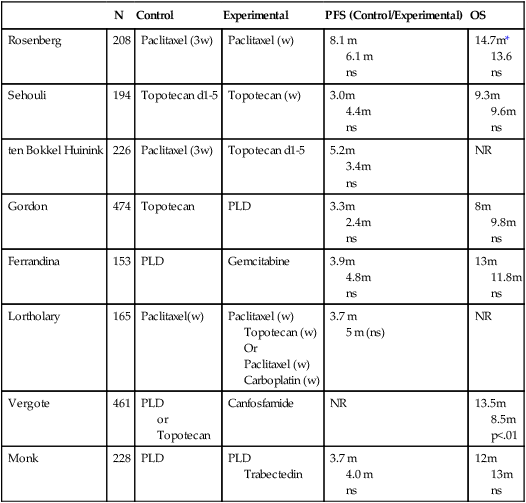
Studies of doublet versus single-agent chemotherapy demonstrated greater toxicity with doublets but no improvement in survival.225,226,268–270 For example, trabectedin and PLD versus PLD and, in a separate study, canfosfamide and liposomal doxorubicin versus liposomal doxorubicin, failed to demonstrate superior clinical outcome with the doublet.49 Trials of topotecan either as a single agent or compared with combinations with either etoposide or gemcitabine demonstrated no additional benefit to addition of the second agent, and the doublets increased the risk of thrombocytopenia.
Biological or targeted therapies date back to tamoxifen therapy to target estrogen receptors, but in the last decade a collection of new targeted therapies have been explored,271–285 including EGFR inhibitors (gefitinib, erlotinib, trastuzumab, EMD7200, lapatinib), VEGF inhibitors (bevacizumab, pazopanib, cediranib), and a host of tyrosine kinase inhibitors (dasatinib, imatinib, sunitinib, sorafenib). A partial list of active agents are listed in Table 89-6. Although not yet FDA approved for the treatment of women with ovarian cancer, a large body of literature is accumulating on the efficacy of bevacizumab in platinum-resistant ovarian cancer.275,286,287 Several studies have demonstrated that a large portion of ovarian tumors are rich in VEGF expression, and high levels of VEGF are typically found in the serum and especially in malignant ascites. In a study by Burger, single-agent bevacizumab demonstrated a response rate of 21% with a 6-month progression-free survival rate of 40%.275 Notable in this and other phase II studies was the dramatic resolution of ascites and/or pleural effusions within weeks of initiating therapy. Similar data have also been seen with aflibercept (a VEGF decoy receptor) and catumaxomab (a trifunctional antibody that binds T cells and EpCAM).286,287 The drug has been well tolerated, with the principal toxicity being manageable hypertension, especially in women on prolonged therapy. More dire complications, such as bowel perforation, have been reported in as many as 10% of patients with bulky recurrent disease and/or evidence of partial bowel obstruction. Similar toxicities have been reported with aflibercept, strongly suggesting it is a class effect of VEGF inhibition.286,288 Perforation rates are much lower in women with small-volume disease and on primary treatment.
There have been numerous studies evaluating the combination of bevacizumab with a variety of standard cytotoxic agents including platinum, paclitaxel, cyclophosphamide, topotecan, and liposomal doxorubicin.284,285 In a variety of phase II trials, the addition of bevacizumab has generated both significant response rates and progression-free survival that seemed to be superior to the single-agent drug. This led to the recently reported Aurelia study that compared bevacizumab and standard therapy with a variety of single agents in women with platinum-resistant ovarian cancer. In this trial, women with recurrent and platinum-resistant disease were randomly assigned to bevacizumab with chemotherapy or just chemotherapy. Physicians were allowed to select paclitaxel, liposomal doxorubicin, or topotecan as the palliative chemotherapy arm. In the randomized phase II study, the time to progression on the bevacizumab plus chemotherapy arm was 6.7 months as compared with 3.4 months (HR 0.48, P < 0.001).289 Overall survival data are not yet available. Perforation rates were 1.7% in the bevacizumab arm compared with 0% in the nonbevacizumab arm.
Controversies, Problems, Challenges, Future Possibilities, and Clinical Trials
Role of Surgery in the Management of Recurrent Platinum-Resistant Disease
There is literature that reports retrospective experiences from high-volume surgical centers that demonstrate that patients who undergo aggressive surgical cytoreduction at the time of disease recurrence do better than those who do not have surgical procedures.292–292 In particular patients with platinum-sensitive disease with a limited number of discrete lesions who undergo successful complete surgical cytoreduction of tumor have a superior survival rate compared with those who have partial resections or, alternatively, are not offered surgical exploration. The major flaw in these studies is the significant risk of selection bias because often only the healthiest patients with modest burden of disease are offered surgery, and surgical resectability may be a predictive marker for more favorable biology or chemosensitivity. A randomized trial looking at the value of surgery and chemotherapy versus chemotherapy alone is currently open and accruing patients slowly. Until the time that we have better-quality data, it is appropriate to counsel patients on the uncertain benefits of surgical resection in this clinic setting.
Management of Low-Grade Serous Tumors
Recurrent invasive borderline tumors or more commonly low-grade serous carcinomas prove to be a major therapeutic challenge.293,294 Although their clinical progression tends to be slow (often measured over years), they tend to be resistant to chemotherapies that prove to be active in high-grade malignancies. As discussed above, the genomic abnormalities that underlie this malignancy are distinct from those of high-grade serous carcinoma.295,296 Large, single-institution experience suggests that cytotoxic therapy response rates are fewer than 10% with similar or perhaps slightly higher responses to antihormonal agents such as tamoxifen.297 A recent phase II study has suggested significant activity of a small molecule that preferentially inhibits the progrowth kinase, MEK, which is regulated in part by Ras and Raf, both of which are often mutated in low-grade serous tumors. A randomized trial of an MEK inhibitor versus standard therapies is currently being planned.
Management of Nonserous “Epithelial” Ovarian Tumors
There is now overwhelming molecular evidence that mucinous, clear cell, and many endometrioid tumors are molecularly different from serous tumors.31 Thus, it is not surprising that carboplatin and paclitaxel is not the ideal regimen for these malignancies. Unfortunately, until very recently, these histologies have been lumped in with serous carcinoma in large randomized trials. Although their low frequency has prevented significant subset analysis, even in large randomized trials, there is ample evidence that both metastatic clear cell and mucinous tumors do poorly after treatment with carboplatin and paclitaxel as compared with tumors with serous histology. Because of its histologic and to a lesser extent molecular similarity to mucinous tumors of the gastrointestinal (GI) tract, mucinous tumors are occasionally treated with GI regimens such as 5-fluorouracil and oxaliplatin (FOLFOX) with bevacizumab with anecdotal responses.298,299 Curiously, approximately 20% of these tumors demonstrate Her-2-neu amplification and responses to trastuzumab have been described.300 An international randomized trial of carboplatin and paclitaxel versus oxaliplatin and capecitabine is currently underway but will take many years to accrue. Likewise, clear cell tumors have been treated with alternative regimens. Particularly in Japan where this type of ovarian neoplasm represents up to 30% of the overall ovary cancer incidence regimens other than paclitaxel and carboplatin are used although it is unclear if these generate superior outcomes.301,302 Anecdotal evidence suggests that, like clear cell tumors of the kidney, some of the women with these tumors have demonstrated very durable responses to antiangiogenic agents.303,304 As described above, many of these tumors have loss of ARID1A tumor suppressor,38,39 a molecule involved in epigenetic regulation through its participation in multiprotein complexes that influence chromatin structure. Translating this information into a rational therapeutic strategy is not yet available.
Drug Resistance and Biomarker Panels
Many companies now offer panels that use techniques such as in vitro tissue culture, immunohistochemistry, quantitative RT-PCR, or now genomic sequencing to evaluate the tumor and provide this information to patients and their physicians for selecting therapy, particularly in the setting of recurrent disease. A significant body of literature exists that demonstrates that in vitro drug resistance assays have a good ability to identify drugs that likely will generate little clinic benefit; however, they tend to be much less accurate in selecting the most active drug.305,306 Clinical trials that randomly assign patients between assay-selected therapy and physician choice are difficult to design and no large trials exist to definitely answer their role in the management of women with newly diagnosed or recurrent disease. The role of comprehensive molecular evaluation, including deep sequencing, is rapidly expanding and is available through a variety of companies. Despite the availability of this technology, the identification of validation of predictive biomarkers is a large unmet need in this malignancy and thus the routine clinical application of these assays is as yet unproven. Ideally, the use of such assays should be coordinated with the collection of fresh or recent tumor tissue and in the context of a clinical trial.