Cancer of the Kidney
Roberto Pili, Eric Kauffman and Ronald Rodriguez
• Renal cell carcinoma (RCC) accounts for 3% of malignancies in adults.
• Cigarette smoking (in more than 20% of cases) and obesity (in more than 30%) are established causal factors for RCC.
• Four percent of cases of RCC arise from hereditary syndromes.
• Different subtypes of RCC are characterized by distinct clinical behavior, genetic abnormalities, and molecular signatures.
• Clear cell RCC is the most common histologic subtype, representing approximately 70% of all sporadic RCCs.
• The von Hippel-Lindau tumor suppressor gene is genetically and epigenetically altered in more than 75% of sporadic cases of clear cell RCC.
• Prognosis for RCC is dependent on tumor histologic type, grade, and stage.
• Nephron-sparing surgery has become the gold standard, when feasible.
• Follow-up guidelines for resected RCC include history, physical examination, periodic metabolic panels, and abdominal and chest computed tomography (CT) studies 4 to 6 months after surgery.
• High-dose bolus interleukin-2 (IL-2), though toxic and of limited use in selected patients, remains a therapeutic option for clear cell RCC because of its potential for durable complete response. Additional immunotherapeutic approaches under development and promising results have been reported with the immunocheckpoint inhibitor PD-1 antibody. Identification of predictors of response to immunotherapies is undergoing.
• Antiangiogenesis drugs have become the new standard of care in the first-line setting for clear cell RCC. Clinical benefit has also been shown with vascular endothelial growth factor and mammalian target of rapamycin inhibitors in subsequent lines of therapies. Novel targets for therapeutic interventions have been identified and are being exploited in clinical testing.
• Optimal treatment for non–clear cell RCC remains a challenge because of the genetic differences and little knowledge of the dysregulated molecular biology driving these cancers. Rational preclinical and clinical testing is needed.
Incidence and Risk Factors for Sporadic Renal Cell Adenocarcinoma
The estimated new cases of kidney and renal pelvis tumors for 2012 was 64,770.1 A predominance of cases in male patients has been reported, with an estimated 40,250 men developing disease in 2012, compared with 24,520 women. The estimated number of deaths in 2013 is 13,570 for both sexes (8,650 in men and 4,920 in women). The increasing incidence of RCC observed in the past had been attributed to increased detection as a result of the widespread use of imaging modalities such as computed tomography (CT), ultrasonography, and magnetic resonance imaging (MRI).2,3 Although a decrease in the size of diagnosed renal cell tumors over time has been noted, an increasing incidence of large and late-stage RCC has also been observed, and partly accounts for the overall increase in incidence.3 In the United States, increases in incidence have been more rapid among women than among men and among African Americans than among whites, leading to a shift in excess from among whites to among African Americans.4
Cigarette smoking and obesity are the most consistently established causal risk factors, accounting for more than 20% and 30% of renal cell cancers, respectively.4,5 Hypertension, rather than antihypertensive drugs, appears to influence renal cell cancer development, although the mechanism is unknown.4,5 This was the conclusion of a large study of 363,992 Swedish men who received at least one physical examination between 1971 and 1992 and who were followed up until death or until the end of 1995.5 The relative risk for RCC was 1.3 for former smokers and 1.6 for current smokers. The relative risk for renal pelvis cancer was even higher at 1.6 for former smokers and 3.5 for current smokers. With regard to obesity, patients with a body mass index in the highest one-eighth of the cohort had a relative risk of 1.9 when compared with patients in the leanest subgroup. Hypertension was confirmed as a third risk factor for RCC. A study tested whether smoking is associated with mutations in the von Hippel-Lindau tumor suppressor gene (VHL) in 337 cases of sporadic RCC among 120,852 people over a mean follow-up period of 11.3 years; the findings suggest that smoking causes RCC independently of VHL mutations.6
Patients with end-stage renal disease have an increased incidence of RCC when compared with the general population. Patients receiving prolonged dialysis tend to develop acquired renal cystic disease, possibly as a result of disordered proliferation within the native kidney. In these patients, the tumors often are bilateral and multifocal, with a papillary histology.7 Accordingly, these patients should be monitored regularly with renal ultrasound examinations or noncontrast MRI. If the patient is on dialysis, then nephrectomy is typically preferred, even when the tumor is smaller than 4 cm, providing the risk of surgery is reasonable.
Additional evidences suggest a potential role in RCC for alcohol consumption, occupational exposure to trichloroethylene, and high parity among women. However, further research is needed into the potential causal effects of genetic factors and their interaction with environmental exposures. Large studies employing genome-wide scanning technology are in progress to provide novel discoveries in renal carcinogenesis.8
Pathology
Kidney tumors usually are unilateral but may be bilateral in 2% to 4% of cases.9 These tumors tend to grow into the renal vein and may form a tumor thrombus that extends into the vena cava and even the right atrium. Vascular involvement is present in 4% to 10% of patients at the time of presentation.10 From a pathological and surgical prospective, it is important to distinguish a tumor thrombus form a positive margin at the vascular surface, as a true positive margin (with actual invasion into the wall of the vessel) portends a poor prognosis.
RCC is a clinically and pathologically heterogeneous disease.11 The 2004 World Health Organization (WHO) classification for renal neoplasms recognizes several distinct histologic subtypes of RCC (Table 82-1). These subtypes include clear cell RCC, papillary RCC, chromophobe RCC, hereditary cancer syndromes, multilocular cystic RCC, collecting duct carcinoma, medullary carcinoma, mucinous tubular and spindle cell carcinoma, neuroblastoma-associated RCC, Xp11.2 translocation–TFE3 carcinoma, and unclassified lesions.12,13 Clear cell RCC is the most common adult RCC, representing 70% of all RCCs. Papillary type I and type II RCC account for 10% to 15%, chromophobe RCC for 4% to 6%, collecting duct carcinoma for less than 1%, and unclassified lesions for 4% to 5% of RCCs. Tumors may be composed of mixed histologic subtypes, and each subtype may feature high-grade sarcomatoid characteristics. Histologic differentiation of most subtypes of RCC can be accomplished with hematoxylin-and-eosin staining techniques. The conventional histologic pattern is the most common, characterized by large clear cells with abundant cytoplasm. The chromophobe pattern is granular with abundant mitochondria. The papillary or tubulopapillary variant may represent a different type of tumor, because they tend to be smaller with fewer anaplastic features. The most widely used grading system for RCC is the nuclear grading system developed by Fuhrman and colleagues.14 This system assigns a grade from I to IV, based on nuclear size, roundness, and other morphologic features, such as the prominence of nucleoli and the presence or absence of clumped chromatin. Patients with tumors of high Fuhrman grade tend to have poorer clinical outcomes. However, many pathologists omit Fuhrman grade when the apparent aggressiveness of the histology is not related to prognosis (e.g., chromophobe carcinoma, which tends to have a favorable prognosis even when the cellular characteristics appear aggressive).
Table 82-1
Histologic Classification of Renal Cell Carcinoma
| Sporadic RCC 2004 WHO Classification | ||
| Histologic Tumor Type | Prevalence (%) | Cytogenetic Findings |
| Clear cell RCC | 70 | 3p25-26 (VHL) |
| Papillary RCC | 10-15 | Trisomy of chromosomes 7 and 17, loss of Y chromosome, 7q34 (c-Met) |
| Chromophobe RCC | 4-6 | Loss of multiple chromosomes: 1, 2, 6, 10, 13, 17, 21 |
| Multilocular cystic RCC | <1 | Extracellular matrix gene |
| Collecting duct carcinoma | <1 | Loss of multiple chromosomes: 1, 6, 8, 13, 14 |
| Medullary carcinoma | <1 | Sickle cell trait |
| Mucinous tubular and spindle cell carcinoma | <1 | |
| Neuroblastoma-associated RCC | <1 | |
| Xp11.2 translocation–TFE3 carcinoma | 1-2 | Translocations involving Xp11.2 (TFE3) |
| Unclassified lesions | 4-5 | |
| Hereditary RCC: Syndromes and Histologic Tumor Types | |||
| Hereditary Syndrome | Chromosome (Gene) Abnormality | Histologic Type of Renal Tumor | Systemic Manifestations |
| von Hippel-Lindau | 3p26 (VHL) | Clear cell RCC | Retinal angiomas, central nervous system hemangioblastomas, pheochromocytoma |
| Hereditary papillary RCC | 7q34 (Met) | Type 1 papillary RCC | None |
| Hereditary leiomyoma RCC | 1q42–43 (FH) | Type 2 papillary RCC | Cutaneous and uterine leiomyomas |
| Birt-Hogg-Dube syndrome | 17p11.2 (BHD) | Chromophobe RCC, oncocytoma, hybrid tumors | Skin lesions, lung cysts |
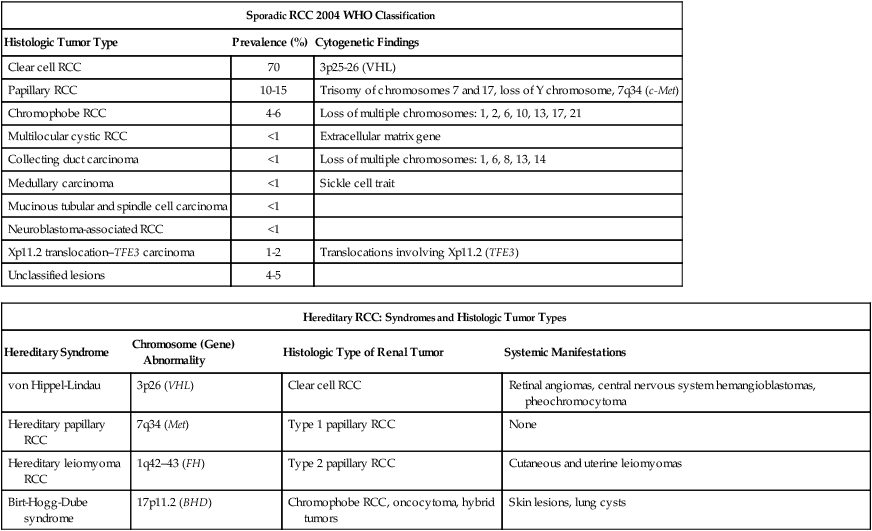
RCC, renal cell carcinoma; WHO, World Health Organization.
Adapted from Prasad SR, Humphrey PA, Catena JR, et al. Common and uncommon histologic subtypes of renal cell carcinoma: imaging spectrum with pathologic correlation. RadioGraphics 2006;26:1795–1806.
Genetics and Epigenetics of Renal Cell Carcinoma
Until recently, RCC was thought to represent a monomorphic disease arising from a probable common precursor cell but with different histologic and clinical manifestations. Genetic characterization based on cytogenetics and molecular biology has established that different subtypes of RCCs are characterized by distinct genetic abnormalities and molecular signatures reflecting the differences in the cell type, biology, and underlying molecular mechanisms.15 Additional tumor metabolic pathways may explain the biological diversity of RCC.
Sporadic Renal Cell Carcinoma
Clear Cell Renal Cell Carcinoma
A common genetic feature signature of sporadic clear cell RCC is the loss of chromosome 3p, suggesting the presence of one or more RCC tumor suppressor genes at this site. The von Hippel-Lindau tumor suppressor gene (VHL), which resides on chromosome 3p25, is mutated or silenced in greater than 50% of sporadic clear cell RCCs.16,17 Germline VHL mutations give rise to von Hippel-Lindau syndrome, which is characterized by an increased risk of blood vessel tumors (hemangioblastomas), endocrine tumors and RCC. The VHL gene product, pVHL, is the substrate recognition module of an E3 ubiquitin ligase that targets the hypoxia-inducible factor (HIF) α transcription factors (HIF1α, HIF2α, and HIF3α) for destruction in the presence of oxygen. Hypoxic cells, or cells lacking pVHL (“pseudohypoxic”), accumulate high levels of HIF, which activates the transcription of a variety of genes, including vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF-3) B, and transforming growth factor alpha (TGF-2). Restoration of pVHL function in VHL−/− mutant renal carcinoma cells suppresses their ability to form tumors experimentally by reducing HIFα levels.18,19 Inhibition of HIFα is necessary and sufficient for tumor suppression by pVHL in RCC nude mouse xenograft assays. This provides a rationale for treating VHL−/− RCC with inhibitors of HIFα or its downstream targets. While the HIF1α isoform was initially believed to be more important, increasing literature supports HIF2α as the more important HIFα member in mediating tumorigenesis.18,19 Although most investigation has focused on the role of HIFα isoforms, the pVHL protein also has several other targets in addition to HIFα, postulated by some to contribute to tumorigenesis. Elucidating these targets will lead to further knowledge of how pVHL suppresses tumor growth. Analysis of mutations in exon 3 of the VHL gene may be useful in refining the diagnostic criteria for conventional RCC versus chromophobe RCC with clear cell features.20
A recent work from Dr. Rathmell’s group have generated gene expression microarray data using software that implements iterative unsupervised consensus clustering algorithms to identify the optimal molecular subclasses. A Consensus Cluster analysis identified two distinct subtypes of clear cell RCC within the training set, designated clear cell type A (ccA) and B (ccB). In each subtype the analysis of data defined a small, highly predictive gene set. A validation data set of 177 tumors was analyzed. Tumors designated ccA had improved disease-specific survival compared with ccB (median survival of 8.6 vs. 2.0 years). These preliminary data suggest that a cluster subtype classification is possible in RCC as performed in other diseases such as breast cancer. Prospective clinical studies will be necessary to validate these important findings.21
The genetic studies in familial RCC have led to the identification of specific molecular signatures in non–clear cell histotypes as well, such as Met hyperactivation in papillary type I, fumarate hydratase mutation in papillary type II, and c-Kit overexpression in chromophobe RCC (see “Papillary Types I and II Renal Cell Carcinoma”). Preliminary results from ongoing genetic studies have shown the possibility of clustering different histotypes based, for example, on kinase expression (Fig. 82-1A).22
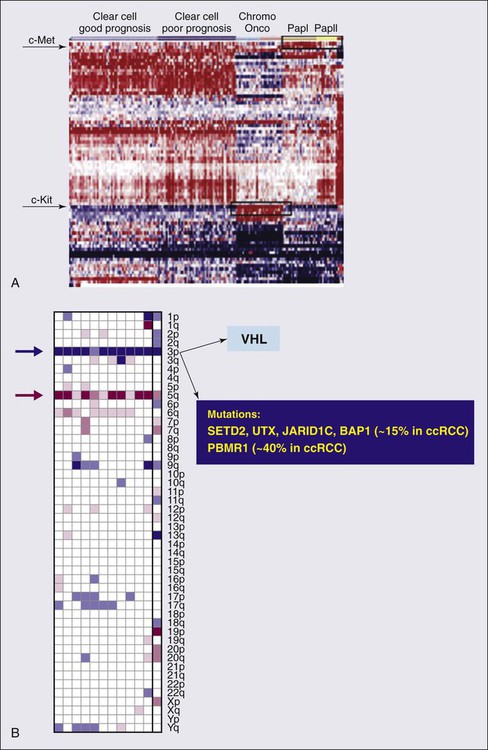
DNA, histones, and nonhistone proteins are condensed into a highly complex nucleoprotein structure known as chromatin that acts as a prototype for all of the genetic information. Chromatin conformation can be in a heterochromatin form, a highly compact structure, associated primarily with transcriptionally inactive genes, or a euchromatin form, consisting of a more open or relaxed structure, associated with transcriptionally active genes.23 Epigenetic mechanisms mediated by histone deacetylases (HDACs), histone acetyltransferases, and histone methyltransferases play critical roles in various biological and cellular processes, including cell proliferation, angiogenesis, hypoxia-related effects, and cell-cycle regulation. Histones on the N-terminal are altered by acetylation, methylation, phosphorylation, ubiquitination, sumoylation, deamination, or adenosine diphosphate ribosylation.24,25 The different histone residues and their modifications result in either transcriptionally active or repressive marks. For example, histone H3 lysine 4 (designated as H3K4), H3K36, and H3K79 are associated with active marks, whereas H3K9, H3K27, and H4K20 are associated with repressive marks.26
Several studies have recently reported SETD2, a histone methyltransferase, as a tumor suppressor gene in RCC.27,28 An initial study consisting of a large-scale next-generation sequencing of primary RCC genomes identified somatic truncating mutations in SETD2 (approximately 3%).28 These mutations are associated with a decrease in H3K36 trimethylation levels in several clear cell renal carcinoma cell lines, VHL mutations and/or hypoxic phenotype (determined by a panel of hypoxia-related genes) in clinical samples, as well as a chromatin remodeling complex protein PBRM1.29 It is also important to note that SETD2 and PBRM1 are located on the short arm of the human chromosome 3 (3p21.31), in close proximity to the VHL gene and the 3p region is frequently altered in clear cell RCCs30 (see Fig. 82-1B). Although mutations in enzymes governing chromatin remodeling are often found in advanced stage disease, these mutations have not been correlated to survival.31 The H3K27 methyltransferase EZH2 (which is associated with aggressiveness in breast cancer and overexpressed in metastatic prostate cancer)32 is overexpressed in renal tumor patient samples compared with their normal adjacent.33 Conversely, histone demethylases governing H3K27 methylation status UTX and JMJD3 are overexpressed in clear cell RCCs compared with the adjacent nontumor tissue, which corresponds to the decreased H3K27 methylation in clear cell RCC.34 These preliminary evidences suggest that alterations in histone-modifying genes may be associated with RCC and could be exploited for therapeutic interventions.
More recently, the BAP1 protein, a nuclear deubiquitinase, has been reported to be inactivated in 15% of clear cell RCCs. BAP1 mediates suppression of cell proliferation, ubiquitinates the histone 2A lysine 119, and its loss sensitizes RCC cells in vitro to genotoxic stress. Initial evidences suggest that BAP1 loss is associated with high tumor grade and poor prognosis. Interestingly, mutations in BAP1 and PBRM1 appear to be mutually exclusive. These preliminary results provide a rationale for future integrated pathological and molecular genetic classification of RCC that will likely lead to subtype specific treatments.35
Intratumor heterogeneity has always been considered a potential major clinical hurdle to develop personalized-medicine strategies that depend on results from single-tumor biopsy samples. A recent report has now shown very elegantly that intratumor heterogeneity is a reality.36 Different techniques, including exome sequencing, chromosome aberration analysis, and ploidy profiling on multiple spatially separated samples obtained from primary renal carcinomas and associated metastatic sites, determined that 63% to 69% of all somatic mutations were not detectable across every tumor region. Mutational intratumor heterogeneity was seen for multiple tumor suppressor genes and chromatin remodeling genes, including SETD2, PTEN, and KDM5C. Gene-expression signatures associated with good and poor prognosis were detected in different regions of the same tumor. These new intriguing findings, combined with the complex genomic landscape, make us rethink the common assumption that a single genomic test might guide therapy. The timing and location of the tumor sample acquisition remain a challenge, but there is still an opportunity to make personalized medicine a goal to achieve in the future for the treatment of cancer, and RCC in particular.
Papillary Types I and II Renal Cell Carcinoma
As with clear cell RCC, genetic studies in familial RCC have led to the identification of genes responsible for non–clear cell histotypes as well. However, unlike clear cell RCC, gene mutations identified in hereditary non–clear cell RCC are absent in the vast majority of sporadic cases. Activating mutations in the Met oncogene responsible for hereditary papillary RCC (see “Familial Renal Cell Carcinoma”) are found in only approximately 10% of sporadic papillary type I RCC cases.37 The Met tyrosine kinase receptor localizes to the cell membrane where it binds its extracellular ligand, hepatocyte growth factor, triggering intracellular activation of the Akt, Rac, and MAP kinase signaling pathways, promoting cell proliferation and migration. Hyperactivation of Met signaling is believed to promote tumorigenesis by upregulation of these downstream pathways. Both Met and hepatocyte growth factor localize to chromosome 7, which is commonly amplified in sporadic papillary type I RCC.
The fumarate hydratase gene encoding a Krebs cycle enzyme and mutated in hereditary papillary type II RCC (as part of hereditary leiomyomatosis and RCC syndrome; see “Familial Renal Cell Carcinoma”) has not been identified in sporadic papillary type II.38 However, increased activity of the NRF2 transcription factor resulting from fumarate hydratase loss in hereditary papillary type II has also been demonstrated in sporadic papillary type II renal cancers.39,40
Chromophobe Renal Cell Carcinoma
As with papillary types of RCC, the genetic mutations underlying sporadic chromophobe RCC tumorigenesis remain to be elucidated, and appear to have little mutational overlap with hereditary chromophobe RCC. The folliculin gene mutated in the most common type of hereditary chromophobe RCC (Birt-Hogg-Dubé; see “Familial Renal Cell Carcinoma”) is rarely mutated (0% to 10%) in sporadic chromophobe RCC tumors.41 While PTEN has been implicated in a rarer type of hereditary chromophobe RCC, its mutation is yet to be identified in the sporadic disease.42
TFE3-Fusion Renal Cell Carcinoma
Also known as Xp11 translocation kidney cancer, TFE3-fusion RCC represents <1% of all sporadic renal cell cancers. It is the most recently designated histologic subtype of RCC by the WHO. TFE3-fusion RCC occurs in younger patients and is the most common mutation in pediatric RCC tumors. These tumors are clinically aggressive and commonly present with metastasis, particularly to regional lymph nodes. These tumors harbor a pathognomonic fusion between the TFE3 gene of chromosome Xp11.2 and one of a number of possible fusion partners on various chromosomes, most commonly PRCC, ASPRC1, and SFPQ. The TFE3 gene encodes a transcription factor involved in the regulation of many proteins implicated in carcinogenesis, including TGF, Met, Rb, Folliculin, Ets, and E-cadherin. It is believed that the fusion promotes tumorigenesis by causing dysregulated transcriptional TFE3 activity. Immunohistochemical detection of nuclear TFE3 expression is suggestive of the underlying fusion mutation43; however, definitive diagnosis requires genetic confirmation by karyotype, fluorescence in situ hybridization, or polymerase chain reaction (PCR).
Rarely, fusions between the related transcription factor gene, TFEB, and the MALAT1/Alpha gene also are found in renal cancers. Less than 30 cases have thus far been reported. The histology of these tumors appears to be distinct from TFE3-fusion tumors. TFEB-fusion cancers similarly occur in younger patients, but in contrast to TFE3-fusion cancers, appear to confer an excellent prognosis. The function of the TFEB transcription factor is unknown, but a central role in lysosome biogenesis and autophagy regulation has been suggested.44
Familial Renal Cell Carcinoma
In a small percentage (5%) of cases, RCC is a feature of one of several hereditary syndromes.15,45 Such syndromes are associated with distinct histologic subtypes of RCC, and in each case patients have increased risk of multifocal tumor development.15,45 Management is dependent on preservation of renal function. Close surveillance and minimization of surgical procedures constitute the mainstay of treatment.
von Hippel-Lindau syndrome is a disorder of autosomal dominant inheritance that occurs in 1 in 40,000 births. The mean age at onset is in the fourth decade of life. The syndrome is inherited as a result of a germline mutation in a single allele of the VHL gene tumor suppressor gene located on chromosomal band 3p25–26.15,45,46 Sporadic loss of the remaining wild-type VHL allele provides the “second hit” necessary for tumorigenesis, most commonly via chromosome 3p deletion. Multifocal tumorigenesis is observed in multiple organ systems, with each tumor harboring an independent second VHL mutation. Renal manifestations include cysts and clear cell RCC tumors. Both tend to be multifocal and bilateral, and are found in the majority of patients with von Hippel-Lindau disease.15,45,46 Hundreds of independent clear cell cancers may be present in a single kidney, including dozens of macroscopic tumors. VHL syndrome patients are at high risk for chronic renal insufficiency because of the lifelong risk of multifocal RCC tumor development and need for repeat renal surgeries. As a result, VHL patients should undergo active surveillance until the largest tumor reaches 3 cm, at which time attempts may be made to resect all tumors in that kidney. Resection by enucleation without clamping of the main renal artery is recommended to maximize nephron sparing. Surgical candidates, particularly those with numerous tumors, are counseled as to the high possibility of local recurrence from de novo tumor formation and future ipsilateral surgery. The discovery of the VHL gene in hereditary clear cell RCC enabled the subsequent identification of a VHL mutation in sporadic clear cell RCC tumors (see above).47
Birt-Hogg-Dubé syndrome is a disorder of autosomal dominant inheritance. Signs and symptoms usually manifest in the fifth decade of life and include renal tumors and cysts, benign skin tumors (fibrofolliculomas) and pulmonary cysts, which can lead to spontaneous pneumothorax. The renal neoplasms may be multifocal and bilateral tumors and most often have pure chromophobe histology or a “hybrid” mixture of chromophobe and oncocytoma; infrequently, pure oncocytoma tumors may be present. Patients can present with several different tumor types within the same kidney and the presence of benign (oncocytoma) and malignant tumors within the same kidney should immediately prompt the suspicion of Birt-Hogg-Dubé (BHD) syndrome. The BHD gene mutated in this syndrome encodes the protein Folliculin and is located on chromosome 17p11.2.15,45,46,48,49 The BHD gene appears to have the characteristics of a loss-of-function tumor suppressor gene.50 Folliculin has unknown function but is found in complexes with adenosine monophosphate-activated protein kinase, the major sensor of cell energy and a negative regulator of the mammalian target of rapamycin (mTOR) pathway. Recently, multiple studies have implicated Folliculin in adherens junction formation and signaling.51,52
Tuberous sclerosis is a syndrome of autosomal dominant inheritance, with two genes identified, TSC1, located on 9q34, and TSC2, located on 16p13.3. It affects 1 in 6000 people and is usually diagnosed at birth.15,45,46 This syndrome encompasses multiple organ systems, including dermatologic, cardiac, pulmonary, and renal. Skins lesions include facial angiofibromas, periungual fibroma, shagreen patches, and hypopigmented macules. Patients also develop cardiac rhabdomyomas, pulmonary lymphangioleiomyomatosis, retinal hamartomas, subependymal nodules, and giant cell astrocytomas. The renal manifestations include bilateral and multifocal angiomyolipomas (AMLs) and less commonly clear cell renal carcinoma. In contrast with spontaneous AML, AML in this setting can be associated with a low risk of occult RCC (1%). The TSC1 and TSC2 gene products inhibit activation of mTOR signaling, a major promoter of protein synthesis and cell growth.
Hereditary papillary renal cell carcinoma (HPRCC), inherited as an autosomal dominant trait, is caused by mutations in Met protooncogene on chromosomal band 7q31–34.15,46,53 It is characterized primarily by bilateral, multifocal papillary type I RCC. These tumors are not aggressive and rarely metastasize. Age at onset is around the fifth decade. The Met oncogene encodes a membrane tyrosine kinase that, in HPRCC, harbors an activating mutation in the kinase domain. Hyperactivation of the Met oncoprotein leads to upregulation of several intracellular signaling pathways involving Akt, Rac, and MAP kinase.
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) is a disease of autosomal dominant inheritance. The gene for this disorder localizes to chromosomal band 1q42.3–43 and has been identified as fumarate hydratase (FH).15,45,46 Age at onset is between the third and fourth decades of life. The syndrome consists of cutaneous leiomyomas, uterine leiomyomas (fibroids), and papillary type II RCC tumors with high metastatic potential, even when small in size. Unlike the VHL patients, in whom delay in surgical treatment is usually the rule until the largest tumor reaches 3 cm, there should be no delay in treatment of solid renal lesions in these patients. Cystic lesions in these patients should be followed with close surveillance and early surgical intervention for any radiographic development of a potentially solid component. Thirty percent of patients have solitary and unilateral renal tumors.45,46
The FH gene functions as a tumor suppressor, with loss of the second allele detected in kidney tumors. The wild-type gene encodes an enzyme in the Krebs cycle catalyzing fumarate conversion into malate. Loss of the FH enzyme leads to accumulation of fumarate, which has been suggested to promote carcinogenesis through indirect stabilization of transcription factors HIFα and NRF2.39,40,54
Distinct from HPRCC and HLRCC, Malchoff and colleagues have described a three-generation family with five cases of papillary thyroid carcinoma and two cases with papillary renal neoplasia.53 With the use of linkage analysis, these investigators demonstrated that the fPTC/PRN phenotype was linked to 1q21.53 They characterized a distinct inherited tumor syndrome that may establish a link between papillary RCC and familial papillary thyroid carcinoma.53
Hereditary renal cell cancer associated with melanoma has been recently described. The TFE3 gene mutated in sporadic RCC (see “Diagnosis of Renal Cell Carcinoma”) is one of four members of the MiT family of transcription factors; although TFE3 mutations have not been identified in hereditary RCC syndromes, the related MiT member, MiTF, was shown to have a specific amino acid substitution associated with hereditary RCC tumors associated with melanoma.55 This substitution confers hyperactivation of MiTF transcriptional activity by preventing its sumoylation and degradation. Histologic features of these MiTF renal cancers are yet to be characterized.
Diagnosis of Renal Cell Carcinoma
As the use of imaging methods has become more widespread, the frequency of incidental detection of RCC has increased. Patients with RCC typically present with a mass involving the kidney that is suggestive of the diagnosis. Nephrectomy is the most effective therapy for RCC that is confined to the kidney and should be used both diagnostically and therapeutically in most patients who are suitable surgical candidates. In certain clinical settings, percutaneous biopsy of a renal mass should be considered. In a retrospective study of 115 consecutive percutaneous biopsies performed on renal masses in 113 patients, investigators found percutaneous biopsy to be of high sensitivity in three clinical groups: patients with a known malignancy (N = 55), patients with no known malignancy and suspected unresectable tumor (N = 36), and nonsurgical patients with a mass suspected to be a resectable RCC (N = 8).57 Percutaneous biopsy of renal masses appears to be safe, carrying only a minimal risk of tumor spread. Urologists should consider increasing the indications for renal biopsy of small renal masses that appear to be RCC, especially in elderly and surgically unfit patients. The standardization of a sheathed biopsy technique by interventional radiology has alleviated fears of tumor seeding through the biopsy tract. With more experience and follow-up preoperative biopsy, this strategy has the potential to decrease unnecessary treatment, because up to a third of small renal masses are now reported to be benign at surgery. Percutaneous biopsy also may allow better selection of renal tumors for active surveillance and minimally invasive ablative therapies. However, there are certain histologic subtypes that cannot be easily distinguished by percutaneous biopsy. Oncocytoma for instance can only be diagnosed by resection, as rarely clear cell carcinoma may harbor regions of oncocytic cells, which are indistinguishable from oncocytoma with a single-needle core. In cases where oncocytoma may be suspected (e.g., in a patient with prior multifocal oncocytomas in the contralateral kidney), several staged biopsies of the mass can be performed to increase the confidence in the diagnosis. A RCC is unlikely to have three separate biopsies all positive for oncocytic cells only without any clear cell components. Finally, initial therapy for mRCC may potentially be stratified by histologic subtype and, in the future, molecular characteristics.
Staging Systems for Renal Cell Carcinoma
The tumor–node–metastasis (TNM) system is a dynamic staging method that continually changes on the basis of new evidence from clinical studies (Table 82-2).58 This staging system is a method of stratifying patients with cancer and is based on data from large multicenter studies with large numbers of patients and a good level of evidence. Despite continual revisions to the methodology to incorporate new clinical evidence, however, the optimal RCC patient stratification using the TNM staging system remains controversial, and further revisions probably will be needed. Revision of the TNM staging system for RCC is likely to result in the simultaneous update of the integrated prognostic systems currently used with this traditional method of staging.
Table 82-2
Tumor–Node–Metastasis (TNM) Staging System for Renal Cell Carcinoma
| Staging | Classification | 1987 | 1997 | 2002 |
| Tumor | T1 | Tumor ≤2.5 cm, limited to kidney | Tumor ≤7 cm, limited to kidney | NA |
| T1a | NA | NA | Tumor ≤4 cm, limited to kidney | |
| T1b | NA | NA | Tumor >4 cm and ≤7 cm, limited to kidney | |
| T2 | Tumor >2.5 cm, limited to kidney | Tumor >7 cm, limited to kidney | Tumor >7 cm, limited to kidney | |
| T3 | Tumor extends into major veins or invades adrenal or perinephric tissues, but not beyond Gerota fascia | Tumor extends into major veins or invades adrenal or perinephric tissues, but not beyond Gerota fascia | Tumor extends into major veins or invades adrenal or perinephric tissues, but not beyond Gerota’ fascia | |
| T3a | Perinephric or adrenal extension | Perinephric or adrenal extension | Perinephric or sinus fat or adrenal extension | |
| T3b | Renal vein involvement | Renal vein or vena cava involvement below diaphragm | Renal vein or vena cava involvement below diaphragm | |
| T3c | Vena cava involvement below diaphragm | Vena cava involvement above diaphragm | Vena cava involvement above diaphragm | |
| T4 | Outside Gerota’s fascia | Outside Gerota fascia | Outside Gerota’ fascia | |
| T4a | Vena cava involvement above diaphragm | NA | NA | |
| T4b | NA | NA | NA | |
| Node | Nx | Regional lymph nodes cannot be assessed | Regional lymph nodes cannot be assessed | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis | No regional lymph node metastasis | No regional lymph node metastasis | |
| N1 | Metastases in one lymph node ≤2 cm in greatest dimension | Metastases in one regional lymph node | Metastases in one regional lymph node | |
| N2 | Metastases in one lymph node >2 cm, but not >5 cm in greatest dimension | Metastases in more than one regional lymph node | Metastases in more than one regional lymph node | |
| N3 | Metastases in one lymph node >5 cm in greatest dimension | NA | NA | |
| Metastasis | Mx | Distant metastasis cannot be assessed | Distant metastasis cannot be assessed | Distant metastasis cannot be assessed |
| M0 | No distant metastases | No distant metastases | No distant metastases | |
| M1 | Distant metastases | Distant metastases | Distant metastases |
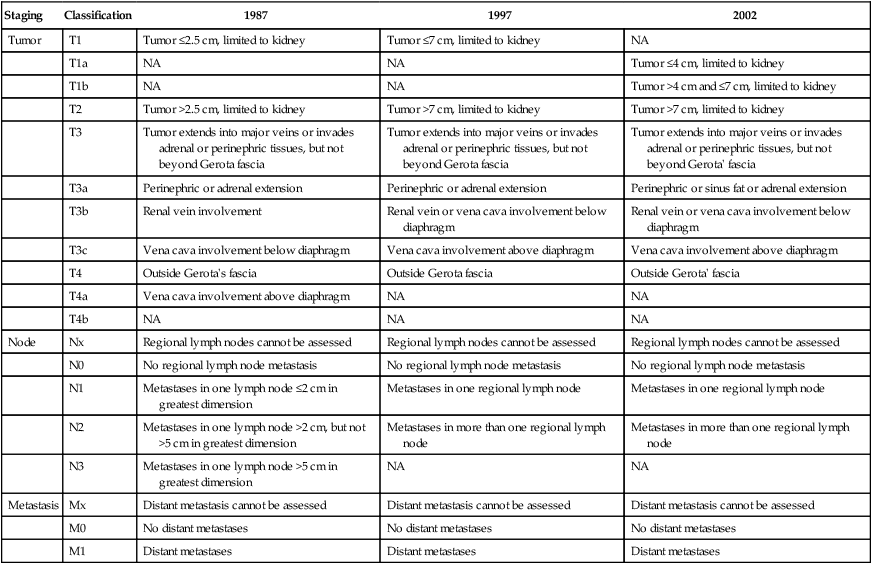
From Ficarra V, Galfano A, Mancini M, et al: TNM staging system for renal-cell carcinoma: current status and future perspectives. Lancet Oncol 2007;8:554–558.
The first TNM staging system for RCC was developed in 1978.59 In this system, tumors are characterized on the basis of the degree of local extension of the tumor at the primary site (T), the involvement of regional lymph nodes (N), and the presence or absence of distant metastases (M). The addition of numbers to each of the TNM components allows one to more precisely categorize the extent of malignant disease at the primary, lymph node, and metastatic sites, respectively. The classification may be clinical (cTNM) or histopathological (pTNM). In the case of RCC, the regional lymph nodes are defined as the hilar, abdominal paraaortic, and paracaval nodes. Laterality does not affect N categories. A retrospective study (N = 252) conducted to validate this system found that survival was most dependent on the local extent of the primary tumor, with 5-year overall survival rates of 100% for pT1, 91% for pT2, 58% for pT3, and 25% for pT4.60 The system also resulted in poor stratification of patients, with few patients in the pT1 (N = 7) and pT4 (N = 9) categories.
In the 1987 revision of the TNM system, a value of 2.5 cm was chosen to distinguish pT1 from pT2 tumors, and an entirely new set of criteria was introduced to evaluate lymph node status. Perhaps most important, comprehensive stage groupings were introduced to define more precisely the impact of TNM characteristics on survival. Again, however, very few patients were stratified to stage I, with only 11 of 872 patients classified in this category in one study.60,61 Furthermore, a study of 337 RCC tumors confined to the kidney found no survival difference between stage I and stage II tumors at the 2.5-cm breakpoint, although a significant difference was identified at the 7.5-cm breakpoint. In 1997, the American Joint Committee on Cancer (AJCC)62 and the International Union Against Cancer (UICC)63 published an updated version of the TNM system. The result of this international collaboration was a more uniform categorization of RCC based on improved clinical evaluation and management. In this version, the breakpoint between pT1 and pT2 was increased from 2.5 cm to 7.0 cm. In addition, the requirement for the T3a classification was more precisely defined as invasion of the adrenal gland by direct extension of the tumor or invasion of perinephric fat. The subcategories of venous tumor extension (pT3) were revised so that venous tumor extension above the diaphragm was classified as T3c, whereas venous tumor extension limited to the vena cava below the diaphragm was classified as T3b. This reflected the decreased adverse prognostic significance of venous tumor extension alone. The classification of lymph node metastasis also was simplified to include involvement of a single lymph node (N1) or multiple lymph nodes (N2), with the stipulation that four to eight nodes should be analyzed before assigning the pN0 classification.64
In the most recent (2010) 7th edition to the AJCC staging for RCC,65 T2 lesions are now subclassified as T2a (>7 cm up to 10 cm) and T2b (>10 cm), while ipsilateral adrenal involvement is now staged based on type of invasion: T4 if contiguous, M1 if not contiguous. In addition, renal vein involvement is reclassified as T3a, and nodal involvement is simplified to either N0 or N1, with the N2 stage dropped. In the previous (2002) edition of the manual, the key change was the subdivision of T1 lesions into T1a and T1b. The rationale was based on evidence from studies of patients undergoing partial nephrectomy, a procedure commonly used for tumors that are 4 cm or smaller. It has been reported that patients who undergo partial nephrectomy for RCC tumors smaller than 4 cm have equivalent survival to those undergoing radical nephrectomy.66 In a separate study of 485 patients undergoing nephron-sparing surgery for RCC with a mean follow-up period of 47 months, patients were divided into four groups based on the size of the primary.67 Patients in group 1 (tumors less than 2.5 cm in diameter) and those in group 2 (tumors 2.5 to 4.0 cm) had equivalent survival, although survival was significantly greater for groups 1 and 2 than for group 3 (tumors 4 to 7 cm) and group 4 (tumors greater than 7 cm). These findings were similar to those previously published in a separate series of 394 patients.68
Since the last revision of the system in 2002, data from several studies have provided issues for discussion in the next revision of the system.58 New data are available from large multicenter studies that recruited adequate numbers of patients with level 2 or 3 evidence.71–71 Prognostic systems include not only anatomical features but also other clinical and pathological variables, but will also eventually provide more accurate information for patient counseling, follow-up planning, patient selection for clinical trials, and adequate assessment of the results.58 Integrated systems are less widely used than the TNM system, because they are difficult to apply and contain several inadequately defined clinical and histopathological variables. The TNM system will become a more refined and advanced instrument used by all clinicians and researchers involved in the management of RCC.
Prognostic Factors for Renal Cell Carcinoma
Although TNM stage, Fuhrman grade, and Eastern Cooperative Oncology Group (ECOG) performance status are the most widely recognized prognostic factors in RCC, research continues to determine strong and easily available prognostic parameters that may help to classify patients in groups with different risks for death from renal cancer. The prognosis for patients with RCC is dependent primarily on disease stage. Patients with histopathological stage pT1 or pT2 (organ confined) disease have the best prognosis, with 5-year cancer-specific survival rates after nephrectomy ranging from 71% to 97%.72 For patients with locally advanced tumors, 5-year cancer-specific survival rates after nephrectomy decrease by 20% to 53%, and once RCC has metastasized, the 5-year survival rate is less than 10%.
Numerous models exist to predict disease recurrence after nephrectomy for all histologic types and specifically for clear cell RCC.73 The natural history and risk group stratification have also been evaluated in those with newly diagnosed mRCC and in patients with previously treated metastatic disease. Currently, RCC histologic subtypes are classified according to the UICC and AJCC recommendations.74 These recommendations are based on the Heidelberg classification system,75 which categorizes RCCs as clear cell, papillary, chromophobe, collecting duct, and unclassified RCC subtypes. Recent studies have suggested that stratification by histologic subtype could lend prognostic value.76,77
The University of California at Los Angeles (UCLA) Integrated Staging System (UISS) was developed for the purpose of improving the prognostic accuracy of the 1997 TNM staging system by incorporating clinical variables.72 The study was based on the analysis of data for patients with RCC treated at a single institution between 1989 and 1999 (N = 661). All patients underwent radical or partial nephrectomy, and most patients with metastatic disease received recombinant IL-2-based immunotherapy within the context of 11 clinical trials. The median follow-up period was 37 months. Patients with papillary tumors (N = 42) showed a trend toward improved prognosis that did not achieve statistical significance, whereas patients with sarcomatoid (N = 45) and collecting duct tumors (N = 3) had worse prognosis. Survival rates for patients with clear cell and chromophobe histologic patterns were similar. The UISS uses five stratification groups (I through V) that incorporate variables commonly used in clinical practice, including 1997 TNM stage, ECOG performance status, and Fuhrman grade. The projected 5-year survival rates published by the UISS group were as follows: 94% for group I, 67% for group II, 39% for group III, 23% for group IV, and 0% for group V. The original UCLA integrated staging system has since been modified so that patients are grouped into two general categories: those with nonmetastatic disease at the time of diagnosis and those with metastatic disease.78 Each category is then divided into high-, intermediate-, and low-risk subcategories, based on the 1997 TNM staging system, the Fuhrman grade, and the ECOG performance status.
To confirm the ability of the UISS to stratify patients with localized and mRCC into risk groups, an international multicenter study was conducted.73 A total of 4202 patients from eight academic centers were classified according to the UISS. The UISS stratified both localized and mRCC cases into three different risk groups. For localized RCC, the 5-year survival rates were 92%, 67%, and 44% for low-, intermediate-, and high-risk groups, respectively. A trend toward a higher risk of death was observed in all centers for increasing UISS risk category. For mRCC, the 3-year survival rates were 37%, 23%, and 12% for low-, intermediate-, and high-risk groups, respectively. In six of eight centers, a trend toward a higher risk of death was observed for increasing UISS risk category. A greater variability in survival rates among centers was observed for high-risk patients. These results suggest that the UISS is an accurate predictor of survival for patients with localized RCC, applicable to external databases. Although the UISS may be useful for patients with mRCC, it may be less accurate in this subset of patients because of the heterogeneity of patients and treatments.
A retrospective, single-institution review of 24 consecutive clinical trials conducted at Memorial Sloan-Kettering Cancer Center using cytokines or chemotherapy for the treatment of advanced RCC (N = 670) identified a small subgroup of patients (N = 30) who were long-term survivors after nephrectomy and treatment with interferon-α, interleukin (IL)-2, or surgical resection of metastasis.79 The five most prominent negative prognostic factors that were identified by multivariate analysis included low Karnofsky performance status (less than 80%), elevated lactate dehydrogenase (greater than 1.5 times the upper limit of normal), low serum hemoglobin (below the lower limit of normal), high corrected serum calcium (greater than 10 mg/dL), and absence of nephrectomy. Patients with zero risk factors were assigned a favorable-risk status; those with one or two risk factors, an intermediate-risk status; and those with three or more risk factors, a poor-risk status. All long-term survivors in this study were in either good- or intermediate-risk groups.
In an additional study from the Memorial Sloan-Kettering Cancer Center, the relationship between pretreatment clinical features and survival was assessed in 251 patients with advanced RCC treated during 29 consecutive clinical trials between 1975 and 2002.80 Clinical features were first examined in univariate analyses, and then a stepwise modeling approach based on Cox regression was used to form a multivariate model. The median survival time for the 251 patients was 10.2 months and differed according to year of treatment, with patients whose tumor was treated after 1990 showing longer survival. In this group, the median overall survival time was 12.7 months. Because the purpose of this analysis was to establish prognostic factors for present-day clinical trial design, prognostic factor analysis was performed on data for these patients. Pretreatment features associated with a shorter survival in the multivariate analysis were low Karnofsky performance status, low hemoglobin level, and high corrected serum calcium. These features were used as risk factors to categorize patients into three different groups. The median time to death in patients with zero risk factors was 22 months. The median survival time in patients with one of these prognostic factors was 11.9 months. Patients with two or three risk factors had a median survival time of 5.4 months. These results suggest that the Memorial Sloan-Kettering Cancer Center risk categories (proposed by Motzer and coworkers80) can be used in clinical trial design and interpretation.
The natural history and risk group stratification also have been evaluated in patients with newly diagnosed mRCC. For patients diagnosed with disease recurrence, no specific risk stratification tools have been available at the time of recurrence. A retrospective study sought to evaluate the usefulness of the prognostic score suggested by Motzer and coworkers.80 From January 1989 to July 2005, patients with localized RCC treated by nephrectomy in whom recurrent disease subsequently developed were identified. Each patient was given a total risk score of 0 to 5, with 1 point for each of five prognostic variables (recurrence at less than 12 months after nephrectomy, serum calcium concentration greater than 10 mg/dL, hemoglobin concentration less than the lower limit of normal, lactate dehydrogenase level greater than 1.5 times the upper limit of normal, and Karnofsky performance status less than 80%). Patients were categorized into low- (score = 0), intermediate- (score = 1 to 2), and high-risk subgroups (score = 3 to 5). The final cohort included 118 patients, with a median survival time of 21 months from the time of recurrence. Median duration of follow-up for survivors was 27 months. Overall survival was associated with risk group category. Low-risk, intermediate-risk, and high-risk criteria were fulfilled in 34%, 50%, and 16% of patients, respectively. Median survival times for low-risk, intermediate-risk, and high-risk patients were 76, 25, and 6 months, respectively. Two-year overall survival rates for low-risk, intermediate-risk, and high-risk patients were 88%, 51%, and 11%, respectively. These additional data support the use of a scoring system based on objective clinical and laboratory data to achieve meaningful risk stratification for both patient counseling and clinical trial entry.
The Memorial Sloan-Kettering Cancer Center and UCLA prognostic systems are similar in several ways. First, both systems are based on a series of consecutive clinical trials for RCC conducted at single institutions. Second, good performance status was found to correlate with lower risk in both systems. Third, cytoreductive nephrectomy for metastatic disease before immunotherapy was found to confer a more favorable prognosis (Memorial Sloan-Kettering Cancer Center) or was used as standard therapy (UCLA) in both systems. In 2007, Karakiewicz and associates proposed a nomogram for prediction of RCC-specific survival in nonmetastatic and metastatic disease, using a development cohort of 2530 patients and an external validation cohort of 1422 patients.81 The nomogram differs from the UISS with respect to one predictor. The symptom classification replaces the ECOG performance system within the nomogram. Moreover, the nomogram provides individual estimation of RCC-specific survival, instead of grouping patients within discrete strata. Direct comparison of the nomogram with the UISS strata, within an external validation cohort, demonstrated higher accuracy for the nomogram, with 89% and 87%, respectively, at 2 and 5 years, compared with 86% and 82% for UISS. A recent publication reports the results from an international, comprehensive database of more than 3500 patients that includes previously reported clinical prognostic factors. Three risk groups were identified and the model was validated using an independent data set of 645 patients treated with tyrosine kinase inhibitor (TKI) therapies. The results show that median survival in the favorable-, intermediate-, and poor-risk groups was 26.9 months, 11.5 months, and 4.2 months, respectively. Performance status, number of metastatic sites, time from diagnosis to treatment, and pretreatment hemoglobin, white blood count, lactate dehydrogenase, alkaline phosphatase, and serum calcium remain important prognostic factors. The model showed good concordance when tested among patients treated with TKI therapies.82
Heng et al. reported a novel model that validates components of the Memorial Sloan-Kettering Cancer Center model with the addition of platelet and neutrophil counts to establish a prognosticator algorithm for overall survival in patients with mRCC treated with vascular endothelial growth factor (VEGF) inhibitors.83 In their latest report, the authors conducted an external validation and comparison with existing databases in RCC patients treated with VEGF inhibitors in the first-line setting at 13 institutions members of the Consortium’s database.84 They compared the Database Consortium model with the Cleveland Clinic Foundation, the International Kidney Cancer Working Group, the French, and the Memorial Sloan-Kettering Cancer Center models. A total of 1028 patients were assessed, with the majority having complete data. Median overall survival was 18.8 months. The previously defined prognostic factors (anemia, thrombocytosis, neutrophilia, hypercalcemia, Karnofsky performance status <80%, and <1 year from diagnosis to treatment) were independent predictors of reduced survival in this external validation set. The results showed that median overall survival was 43.2 months in the favorable-risk group (no risk factors), 22.5 months in the intermediate-risk group (one to two risk factors), and 7.8 months in the poor-risk group (three or more risk factors). The concordance index of the Database Consortium with the other models ranged between 0.636 and 0.687. Now that this Database Consortium model has been externally validated, it can be applied to stratify patients by risk in clinical trials involving anti-VEGF therapies and to counsel patients about prognosis.
Management Options for Localized Disease
Radical Nephrectomy
Radical nephrectomy is the traditional gold-standard treatment for localized RCC. Long-term cancer-specific survival for pT1a, pT1b, pT2, and pT3 lesions can be approximated at greater than 90%, 80%, 70%, and 60%, respectively. Historically, a radical nephrectomy has included resection of the ipsilateral adrenal gland. However, adrenal resection increases the risk of life-threatening adrenal insufficiency (addisonian crisis) should the contralateral adrenal gland require resection or become otherwise functionally compromised. Today, it is common practice to limit adrenalectomy to the setting of large or upper pole renal tumors, given a low risk of adrenal involvement with smaller RCC tumors. Even in this setting, ipsilateral adrenalectomy has been challenged by recent studies suggesting that negative CT imaging of the ipsilateral adrenal gland can effectively rule out RCC involvement.85 Although still controversial, in the setting of negative cross-sectional imaging, adrenal gland preservation should be considered to avoid long-term risk of addisonian crisis.
The role of regional lymphadenectomy at the time of radical nephrectomy is also controversial. The most common regional landing sites for RCC are the paracaval, interaortocaval, and retrocaval nodes for the right kidney, and the paraaortic, interaortocaval, and preaortic nodes for the left kidney. In the presence of radiographic or palpable lymphadenopathy, a lymph node dissection is justified. In the absence of such findings, the role of lymphadenectomy is prognostic only, with no level 1 evidence to support a therapeutic benefit. Some series suggest a therapeutic role for lymphadenectomy specifically in patients without lymphadenopathy if their primary tumors are otherwise high risk, but these series are limited by retrospective nature and sample size. The European Organization for Research and Treatment of Cancer (EORTC) 30881 trial has provided the only prospective randomized trial investigating radical nephrectomy (N = 772) with or without lymphadenectomy, but showed no benefit with regional lymphadenectomy at a median follow up of 12.6 years.86 This trial has been criticized for its inclusion of patients with predominantly low tumor stage and thus a low rate of nodal positivity (4%), suggesting insufficient power to detect a therapeutic benefit. In the absence of level 1 evidence supporting such a role, most urologists continue to avoid lymphadenectomy at nephrectomy, given the significant risk for hemorrhage during dissection along the aorta and vena cava.
Nephron-Sparing Surgery
Radical nephrectomy has been shown to cause significant decreases in renal function, raising the risk of chronic renal insufficiency and dialysis, and chronic renal insufficiency, in turn, raises the risk of cardiovascular events, hospitalization, and overall mortality.87 Acknowledging the detrimental impact of chronic renal insufficiency on overall health, urologists have increasingly strived over the past 2 decades to preserve renal function in RCC patients. With increasing sensitivity and use of imaging studies leading to detection of smaller renal tumors, partial nephrectomy has become an effective alternative to radical nephrectomy for patients with localized RCC, resulting in low morbidity and good oncologic outcomes.
Traditional indications for partial nephrectomy have included conditions in which radical resection would leave the patient anephric (bilateral RCC, a horseshoe kidney, or a solitary kidney), requiring immediate dialysis; and unilateral RCC with a contralateral kidney at risk for compromised function, as in diabetes, hypertensive nephrosclerosis, renal artery stenosis, renal calculi, and chronic pyelonephritis. Initial outcomes under such indications supported operative feasibility and oncologic safety of a nephron-sparing approach. Patient selection quickly expanded to include localized primary tumors with diameter of 4 cm or less and exophytic location, regardless of renal function.88 Although the risk of local recurrence is necessarily increased with a nephron-sparing approach, risk of cancer-specific mortality appears similar to a radical approach. Perioperative complications are, however, more common with partial nephrectomy, due largely to increased bleeding/transfusion and urinary leaks related to collecting system injury.
Indications for partial nephrectomy have expanded to include select larger tumors.89 Several studies have now retrospectively compared partial and radical nephrectomy for larger tumors up to 7 cm, demonstrating similar oncologic outcomes.90,91 Leibovich and associates compared outcomes in 91 patients managed with nephron-sparing surgery and 841 patients who underwent radical nephrectomy for 4- to 7-cm tumors.90 These investigators found no statistical difference in cancer-specific survival and distant metastases between the two groups. Becker similarly compared 196 patients undergoing partial (N = 45) or radical nephrectomy (N = 151) for clear cell RCC tumors of 4 to 7 cm. They observed no differences in oncologic outcomes between the two groups, but noted better postoperative renal function at 3 months for patients undergoing nephron sparing.91 With regard to feasibility of partial nephrectomy, tumor location (exophytic vs. endophytic) may be a better determinant than tumor size. With either larger size or more endophytic location, ischemic time can be expected to increase, along with complications related to bleeding and collecting system injury (urinary leak).94–94 The indications for nephron-sparing surgery are partly dependent on the experience and skill of the surgeon.
The primary oncologic concern with nephron-sparing surgery is the risk or recurrence in the same kidney, avoided with radical nephrectomy. Ipsilateral intrarenal recurrence occurs in 1% to 6% of patients after partial nephrectomy, and may result from either primary tumor multifocality (including de novo tumor formation) or positive surgical margins.95 However, it remains controversial whether positive surgical margins during partial nephrectomy increase the risk of RCC recurrence or have no prognostic significance. Many series support good local recurrence-free survival for patients with positive margins and intermediate-term follow up. Furthermore, immediate or delayed radical nephrectomy most often fails to reveal any residual disease in these patients. In a recent description of a large single-institutional series, Kwon et al. found no relation between margin status after partial nephrectomy and primary tumor risk (metastatic potential).96 However, local recurrence occurred in 4% of patients with a positive compared to only 0.5% of patients with negative surgical margins, and all cases of local recurrence in both groups were from high-risk primary tumors. The authors concluded that local recurrence may be more likely when positive surgical margins occur in the specific setting of high-risk primary tumor histology, whereas positive margins with low-risk primary tumors may have no significance. Current practice generally assumes no prognostic significance of positive surgical margins.
Given the benefit of nephron preservation and potential insignificance of positive margins, the necessity of a 1-cm resection margin proposed by Vermooten in 1950 has come under question.97 Margin width during partial nephrectomy has thus been increasingly reduced over time.98,99 Several recent partial nephrectomy series show no association between disease recurrence and margin size.100–100 Some urologists now advocate a zero-margin tumor “enucleation,” taking advantage of a natural plane between the renal parenchyma and tumor pseudocapsule, a histologically confirmed structure ranging from 0.04 mm to 0.79 mm in width.101 In a series of 232 patients undergoing enucleation of tumors smaller than 4 cm, Carini et al. reported 97% and 95% 5- and 10-year cancer-specific survival rates, respectively, with no cases of positive margins or recurrences in the resection bed.102 According to these investigators, even though pseudocapsular microinfiltration is frequent (33%) in enucleated tumors, positive margins are still avoided because of a surrounding chronic inflammatory layer that is, on average, 1 mm in width.101 In addition to optimizing renal preservation, tumor enucleation enables the surgeon to work in a relatively avascular plane and may reduce blood loss during resection and risk of collecting system injury. The reduction in bleeding may allow resection without clamping of the main renal artery, avoiding renal ischemia.103
A large volume of existing retrospective literature now suggests survival benefits of partial nephrectomy over radical nephrectomy, but suffers from an uncertain selection bias. In a meta-analysis of more than 40,000 patients undergoing radical or partial nephrectomy, the latter was associated with a significant benefit in both survival and renal function; however, the quality of pooled studies was judged to be low.104 Recently, a large multiinstitutional randomized trial from EORTC comparing partial and radical nephrectomy for tumors 5 cm or smaller has challenged current thinking on the role of nephron sparing.105 A total of 541 patients were enrolled in this study and median follow-up was more than 9 years. Based on an intention-to-treat analysis, the radical nephrectomy group cohort showed better overall survival compared with patients assigned to partial nephrectomy. Because there were very few cancer-specific deaths in either cohort, the benefit in overall survival for radical nephrectomy was attributed to a reduction in non–cancer-related deaths. This unexpected outcome has come under intense scrutiny and the study has been heavily criticized because of underaccrual and its intention-to-treat analysis with frequent study arm crossover. Perhaps unduly, these findings have been largely dismissed by the urology community.
Surgical Approach
For cancers confined to the kidney requiring radical nephrectomy, a laparoscopic approach is generally preferred based on its tendency for lower blood loss, faster inpatient and outpatient recovery, and similar oncologic outcomes relative to an open approach, although these differences are inferred largely from retrospective comparisons.106–110 In a prospective randomized comparison between these two approaches among patients with tumors up to 8 cm, Burgess et al. found that laparoscopic radical nephrectomy achieved significantly better postoperative pain scores and convalescence, but no reduction in hospitalization.111
While laparoscopy is suitable for most radical nephrectomies, large renal tumors often are best approached in open fashion. In some instances, however, even large tumors can be approached laparoscopically if the local anatomy is favorable, although intraoperative mobilization of the kidney may become more difficult. In a retrospective comparison of open and laparoscopic radical nephrectomy for T2 tumors with a mean size of 10 cm, Hemal et al. observed lower blood loss, less narcotic use, and quicker recovery with laparoscopy, at the expense of a longer mean operative time.112 In another retrospective study involving tumors larger than 7 cm, complications were more common with laparoscopic than open radical nephrectomy.103 Conversion from laparoscopy to open can be expected at higher frequency with large tumors. Gong et al. reported open conversion in 12% of laparoscopic nephrectomies for T2 tumors compared with only 1% for T1 tumors, although postoperative recovery was no different.114
In contrast to radical nephrectomy, the gold standard approach to partial nephrectomy remains an open incision. However, with increasing usage of minimally invasive therapies and enhanced dexterity afforded by robotic assistance, the indications for laparoscopy are expanding to include partial nephrectomy in select patients. Today, the choice between open and robotic or laparoscopic partial nephrectomy depends on anatomic location of the tumor, body habitus, and ability to tolerate pneumoperitoneum. The optimal tumor for laparoscopic partial nephrectomy is small (<4 cm) and peripheral/exophytic, although experienced laparoscopists may take on larger and more centrally located lesions. In a study from a single high-volume laparoscopic surgeon, similar operative outcomes were observed for laparoscopic resection of tumors larger than 4 cm (N = 58) compared to smaller tumors (N = 367), with the exception of a 6- to 8-minute increase in ischemic time.115 Central tumors resected laparoscopically were associated with increases in both ischemic and overall operative times, but not bleeding. An experienced surgeon can thus perform technically challenging laparoscopy with good surgical and oncologic outcomes.116
The advent of the daVinci robot has simplified the most difficult technical portion of the operation, namely intracorporeal suturing and reconstruction of the resection defect. This has translated to improvements in several key operative outcomes for minimally invasive partial nephrectomy, in addition to a quicker learning curve. In one experienced minimally invasive surgeon’s series of 492 laparoscopic (N = 231) or robotic (N = 261) partial nephrectomies, the latter approach correlated with a significant reduction in operative time, ischemia time, positive margins, and perioperative complications, despite greater patient comorbidity and tumor complexity.117 Other retrospective series indicate reduced blood loss with the robotic approach as well.118,119
The single remaining concern for laparoscopic and robotic nephron-sparing surgery is the amount of warm ischemic time and resulting loss of renal function. Precooling of the kidney prior to renal arterial clamping during open partial nephrectomy is believed to reduce ischemic injury but has thus far not been feasible with a laparoscopic approach. Warm ischemic times during laparoscopy in excess of 20 minutes have been suggested to result in irreversible injury. For this reason, complex resections, including large, endophytic or multifocal tumors, may require alternative methods, including either open resection using cold ischemia, or resection of the tumor without main renal artery clamping. The latter approach increases blood loss and the likelihood of transfusion, but optimizes preservation of renal function. Recently, renal hypoperfusion by pharmacologically induced hypotension has been proposed to avoid main renal artery clamping, but the safety and efficacy requires further study.120 Newer methods include superselective segmental renal artery clamping, with or without administration of near infrared dyes to visualize clamping impact. Such strategies may allow resection without putting the entire kidney through an ischemic insult.121,122 However, immediate and long-term efficacy of these technical innovations are still under active investigation.
The common benefits of cosmesis and pain control with minimally invasive surgery is partly compromised during laparoscopic radical nephrectomy by the substantial skin/fascial incision needed to extract the kidney and surrounding Gerota’s fat en bloc. Some laparoscopists have accordingly advocated morcellation of the specimen as an alternative to removing the kidney as a whole. An intact extraction has the benefit of maintaining kidney and tumor integrity for more precise histopathological evaluation, decreases the chance of tumor seeding of the port sites or renal fossa, and requires shorter surgery time.123 Pautler and colleagues compared the histopathological diagnosis of their operative needle biopsy before morcellation with the morcellated specimen.124 They found adequate histopathologic material for diagnosis but were unable to stage most cases.124 Although it has been argued that lack of pathological tumor staging information does not affect management of the patient, the tumor stage is in fact the major determinant of current risk-based postoperative surveillance regimens.125 Hence, although there was initially great enthusiasm among the laparoscopic surgeons to remove the kidneys and tumors through morcellation, this practice has been abandoned and nearly all urologic surgeons now prefer intact extraction of the kidney and mass for accurate histopathological diagnosis and staging.
Thermal Ablation for Renal Cell Carcinoma
Urologists pioneered minimally invasive surgery and have been constantly searching for less-invasive techniques. With the advent of energy-based ablative alternatives to open and laparoscopic surgeries in selected patients, it is now possible to achieve cancer-specific survival with decreased surgical morbidity. Two types of ablation are currently performed, cryoablation and radiofrequency ablation. Although either can be performed laparoscopically under direct vision, ablation is most often performed percutaneously under ultrasound or CT guidance. While the usefulness of ablative therapies is still being evaluated by many groups, the American Urological Association consensus guidelines considers the efficacy of ablative techniques to be slightly less than surgical resection and should be reserved primarily for patients who decline surgery or who are not good surgical candidates but who desire treatment.126
Renal cryoablation has been developed for the treatment of small renal tumors as an alternative to nephron-sparing surgery, with the goal of reducing operative morbidity. Use of standard cryoprobes of 3 to 8 mm can cause rupture of the renal capsule and parenchyma, resulting in significant bleeding. The use of multiple 2.4-mm cryoprobes under intraoperative real-time ultrasound guidance may decrease the risk of bleeding and make this technique a more feasible approach.127 Increasing evidence suggests that cryoablation is an acceptable surgical alternative to traditional open and laparoscopic nephrectomy.128–132 In addition to shorter hospital stay and less morbidity, cryoablation has been shown to be an effective nephron-sparing cancer therapy.132 Patients selected for this option tend to have peripheral lesions 5 cm or less in diameter.128–132 Minimally invasive therapy without the sequelae of open or laparoscopic surgery may be reasonable in these situations, particularly in morbid or elderly patients who are poor surgical candidates. Cryoablation surgery has been described in the literature with open, laparoscopic, and, more recently, imaging-guided percutaneous approaches.133,134
Multiple theories have been proposed for the mechanism of action of cryoablative surgery. The most accepted theory postulates direct cellular injury leading to coagulative necrosis.130,131 Injury to the cancer cells occurs as a result of intracellular ice crystal formation during the freezing phase of the treatment.130,131 The freezing process causes protein damage, which injures the cell membrane and essential enzymatic processes.130 Ice crystals that form within the cell disrupt intracellular organelles and membranes. Additionally, indirect ischemic injury caused by occlusion of the microvasculature during the thaw phase creates stasis within blood vessels, leading to end-tissue infarction. These ensuing insults to renal tissue lead to liquefactive necrosis.130
In most cases, the cryoprobes used are 2.4 mm in diameter. The usual procedure is percutaneous CT guidance cryotherapy, in which each lesion is treated with a double freeze–thaw cycle, with active freeze at 100% efficiency for a minimum of 10 minutes and a passive thaw of 8 minutes.128 These cycle times consistently deliver the lethal freeze of −4° C within 1 cm of the ice ball edge.128 The critical threshold for necrosis has been reported as between −19.4° C and −40° C.128 The number of probes used is specific to the size and shape of the lesion, with freeze margins of 0.5 to 1 cm of the lesion.
Numerous studies of cryoablation support adequate oncologic efficacy with short- and intermediate-term patient follow-up. However, there are at present sparse long-term data available. Gill and colleagues recently provided the first 10-year oncologic outcomes for cryotherapy using a laparoscopic approach. The mean tumor size was 2.3 cm and cancer specific survival at 10 years was 83%, comparable but slightly lower than would be expected for surgical resection of renal cancers of this size.135 Because the renal tumor is not excised and histopathological margins are unknown, whether the entire tumor has been extirpated remains uncertain.128 This challenge forces frequent CT and MRI follow-up studies on patients who receive this therapy.
Potential complications of RFA are similar to cryoablation, and include urinary leak/fistula, ureteral stricture or obstruction as a result of tissue sloughing, and injury to adjacent organs. Bleeding complications are infrequently encountered with RFA and make this procedure an attractive option for poor surgical candidates on blood thinning medication. Allaf and colleagues reported increased pain with RFA compared to cryoablation, attributing this to a potential analgesic effect of the latter.136
As with cryotherapy, numerous studies now support effective short-to-intermediate–term oncologic efficacy of RFA; however, longer-term outcomes data are, at present, sparse.137–140 Psutka and colleagues documented oncologic outcomes for 185 RFA patients with a median tumor size of 3 cm and a median follow-up of 6.3 years. Only 2.2% and 1.6% of patients developed metastasis and died, respectively, although 8% and 24% of T1a and T1b patients, respectively, were not disease-free.141 The low rate of metastasis and death is promising, and validation of these longer-term outcomes is now awaited.
Active Surveillance
The precise risk of metastasis with active surveillance is unclear, but appears to be around 1% to 2% based on current literature.142,143 However, follow up in these studies is mostly short- or intermediate-term only, thus this long-term rate may be an underestimate, particularly given the 5% to 10% rate of metastasis expected after surgical resection of pT1a tumors. In addition, reporting bias may further underestimate the rate of metastasis on active surveillance. During patient selection for active surveillance, the metastatic risk must be weighed in light of both the patient’s willingness to undergo treatment and the patient’s operative risk, including general health/performance status, renal function, and tumor complexity. Percutaneous tumor biopsy at present does not play a routine role during patient selection, probably because of concerns of common undergrading and a historically high nondiagnostic rate, although accuracy of 80% to 90% is described in most contemporary biopsy series.
Presently, there are no standardized guidelines for surveillance regimens and thresholds for implementation of delayed treatment. A number of studies have attempted to identify clinical predictors of metastasis for patients on active surveillance, but are limited by low metastatic case number. In many cases of metastasis on active surveillance, the primary tumor has progressed to greater than 4 cm. Accordingly, tumor size may provide a useful trigger for intervention. Increasingly, tumor growth rate is also being used as a trigger. Although growth rate of small renal masses does not differ between malignant and benign tumors (0.2 to 0.3 cm/year), high-grade and particularly metastatic primary tumors appear to grow faster, with the latter ranging from 1.3 to 2.9 cm/year.142,144–147 A growth rate of more than 0.5 cm/year has accordingly been implemented by some urologists as a threshold for intervention. This approach has been challenged by the fact that oncocytomas commonly demonstrate rapid growth rates.148
Surveillance after Treatment of Localized Renal Cell Carcinoma
Sporadic Renal Cell Carcinoma
A need has been recognized for standardization of surveillance protocols after nephrectomy for sporadic RCC. Multiple surveillance protocols have been proposed.71,149–151 An important consideration in determining the appropriate frequency of surveillance imaging after surgery is the ability to provide curative or palliative salvage therapy. Because improved survival can be achieved for certain subsets of patients with recurrent disease, an active approach to surveillance is warranted. For instance, patients with solitary metachronous metastases treated aggressively with surgical resection have a 5-year survival rate of 20% to 44%.152–155 Extended survival (21 to 136 months) also can be achieved in as many as 33% of patients who undergo resection of an isolated local recurrence in the retroperitoneum.156,157 Follow-up imaging after curative therapy has been challenged on the basis of survival benefit and cost-effectiveness.158
No current randomized trials exist, so little evidence on the appropriate follow-up after surgery is available. Too much surveillance places a financial and psychological burden on the patient, whereas too little surveillance may increase morbidity and mortality. Patients are risk stratified, and a follow-up schedule is determined on the basis of stage and grade of the disease. Most recurrence occurs within 3 to 5 years after nephrectomy.159 Ljungberg and colleagues prospectively reviewed data for 187 patients with pT1 to pT3 disease who underwent radical nephrectomy at their center.151 These investigators found that 80% of metastatic cancers were diagnosed within 3 years after surgery.151 It has been documented that Furman nuclear grade, TNM stage, ECOG status, and DNA ploidy are good predictors of tumor progression and tumor recurrence.159 T1 tumors recur between 38 and 45 months; T3 tumors recur between 17 and 28 months.159 Furman grades 1 through 4 tumors are associated with risk of metastasis of 9%, 61%, 79%, and 87%, respectively. Diploid tumors carry a decreased risk of recurrence compared with aneuploid tumors.151
Imaging for surveillance is based largely on risk of metastasis by anatomic site. The major metastatic sites, in order of frequency, include lung (3% to 16%), bone (2% to 8%), regional lymph nodes, liver (1% to 7%), ipsilateral adrenal, contralateral kidney, and brain (2% to 4%). Currently, a history and physical examination (H&P), serum studies (calcium level, alkaline phosphatase level, and liver transaminases), and imaging studies (chest plain radiographs or chest CT scan and abdominal CT scan) done at time points when disease recurrence is likely are used for surveillance.160 The lung has the greatest incidence of metastasis, so early diagnosis is pertinent.160 Chest radiography has been shown to detect up to 90% of lung metastases.159 Although chest CT is more sensitive than chest radiography, it yields more false positives, and its role in surveillance is still unknown.159 In the case of bone metastasis, however, only palliative therapies are available, therefore screening bone scintigraphy and radiographs are not warranted.160 Abdominal CT and liver transaminase determination are an integral part of surveillance because of treatment options available for local recurrence or liver recurrence.160
Levy and coworkers have suggested a surveillance protocol based on TNM staging.150 For T1 disease, a 6-month H&P and serum studies are performed annually for 3 years. For T2 disease, 3-month H&P and semiannual chest radiographs and serum studies are performed; at 24 and 60 months an abdominal CT is recommended. For T3 disease, 3-month follow-up visits that include serum studies and chest x-ray are suggested. Then at 24 and 60 months, a CT scan of the abdomen is recommended. Close follow-up is suggested for T4 disease, including H&P, CT of the abdomen, chest radiographs, and serum studies every 3 months for 2 years and every 4 months for the third and fourth years. Complete follow-up is indicated semiannually in year 5 and then annually.
Zisman and coworkers, in an attempt to standardize RCC staging and management protocol, developed the UISS, described earlier.161 This system incorporates histologic grade, ECOG status, and TNM stage to improve prognostication for RCC.161 With this system, they created a surveillance protocol for patients grouped according to recurrence risk. For the low-risk group, these investigators recommend H&P, serum laboratory studies, and chest CT scan annually. At 2 and 4 years after surgery, the patient should have an abdominal CT scan. The intermediate-risk group is recommended to have H&P, serum laboratory studies, and chest CT scan semiannually for 3 years and then annually for 10 years. They should also have an abdominal CT annually for 2 years and then every 2 years up to 10 years postsurgery. The high-risk group should have H&P, serum studies, and chest CT semiannually for 3 years and then annually for 10 years. These patients also should have an abdominal CT scan semiannually for 2 years and then annually for 5 years and then every 2 years up until 10 years. Throughout the protocol, after 3 years of surveillance, the chest CT can alternate with chest radiography.
Lifelong surveillance is necessary for patients with RCC. Late recurrence is arbitrarily defined as a recurrence more than 10 to 20 years after nephrectomy. In sporadic RCC, recurrences have been documented as long as 45 years after initial surgical resection.162 The appropriate intensity of follow-up after 5 years remains to be established. Approximately 85% of recurrences, however, will be detected in the first 3 years after resection of the primary.163
von Hippel-Lindau Disease and Other Familial Renal Cell Carcinomas
Patients with von Hippel-Lindau RCC or other familial forms of RCC are at high risk for local recurrence after nephron-sparing surgery and require close lifelong surveillance. More than 80% of patients with von Hippel-Lindau disease treated with nephron-sparing surgical resection will have a recurrence in the ipsilateral kidney within 10 years, and lesions will develop in the contralateral kidney, if they have not done so already.164 This is because multiple microscopic lesions are present throughout the kidneys, despite the grossly normal appearance of the parenchyma.165 A diagnosis of von Hippel-Lindau disease should be considered in any patient with early onset or multifocal RCC or RCC in conjunction with the following: a history of visual or neurological symptoms; a family history of blindness, central nervous system (CNS) tumors, or RCC; or coexisting pancreatic cysts, epididymal lesions, or inner ear tumors.166,167 Standard recommendations for surveillance in patients with von Hippel-Lindau disease include (a) CT of the abdomen and pelvis every 6 months in patients with solid renal lesions and every 12 months in patients without solid renal lesions, with alternating MRI and CT in younger patients to avoid secondary malignancies associated with prolonged use of the former, (b) annual physical and ophthalmologic evaluations, (c) estimation of urinary catecholamines every 1 to 2 years, (d) MRI of the CNS every 2 years, and (e) periodic auditory examinations. Molecular genetic and clinical screening should be offered to appropriate family members based on an autosomal-dominant pattern of inheritance.164,167,168
Perioperative Medical Therapies
Neoadjuvant Therapies
Presurgical approaches in RCC are currently under investigation based on the evidence that therapies targeting the VEGF axis have shown clinical benefit in the metastatic setting.169 The rationale for the use of VEGF inhibitors in this setting is to downsize locally advanced or nonresectable tumor and offer either organ-sparing surgery or radical nephrectomy, respectively. The use of antiangiogenic therapies perioperatively would also have a potential role in controlling early micrometastatic disease. To date, small, single-institution studies have shown that this approach appears feasible in terms of safety and may achieve meaningful tumor response.170,171
The presurgical approach is also being tested in the setting of cytoreductive nephrectomy to assess the response to antiangiogenics and determine the optimal candidates for debulking surgery. For example, a recent study assessed the safety and efficacy of presurgical bevacizumab in patients with mRCC. In this single-arm, phase II trial, patients received bevacizumab plus erlotinib (23 patients) or bevacizumab alone (27 patients) for 8 weeks followed by restaging. Forty-two patients underwent nephrectomy. Median progression-free survival and overall survival were 11 and 25.4 months, respectively. The conclusions from this study were that presurgical treatment with bevacizumab therapy yields clinical outcomes comparable with post-surgical treatment with antiangiogenic therapy in patients with mRCC, but it may result in wound-healing delays.172 At this time, a neoadjuvant therapeutic approach should only be considered in a clinical trial setting.
Adjuvant Therapies
A role for adjuvant therapy after nephrectomy remains to be established, and to date, observation remains the standard of care outside a clinical trial. Clinical studies with immunotherapies have failed to demonstrate a clinical benefit. One randomized, multicenter, prospective study has reported on the efficacy of adjuvant interferon alfa-2b given after nephrectomy to patients with Robson stages II and III RCC.173 Patients randomized to receive interferon alfa-2b were given a dose of 6 million IU intramuscularly 3 times per week for 6 months, starting within 1 month of surgery. Patients in the observation-only group who relapsed were given interferon alfa-2b, 10 million IU intramuscularly, 3 times per week, or the best available treatment at the time of relapse. Treatment groups were well matched at baseline for patient and tumor characteristics. No significant differences were observed in overall or event-free survival for patients randomized to management by observation (N = 124) and patients receiving interferon alfa-2b (N = 123). This study did not support the use of adjuvant interferon alfa-2b after nephrectomy. An analyzed and reported phase III study of interferon-α as adjuvant treatment for resectable RCC by the ECOG/Intergroup showed that adjuvant treatment with interferon did not contribute to better overall survival or relapse-free survival.174
Cytoreductive Nephrectomy for Patients with Metastatic Renal Cell Carcinoma
Approximately one third of patients diagnosed with RCC present with metastatic disease.175 The role of nephrectomy in patients with metastatic disease at the time of diagnosis has long been the subject of debate. A general consensus exists that noncurative nephrectomy is appropriate when symptoms produced by the primary tumor require palliation, when the metastatic tumor burden is less than that of the primary tumor, and when the patient wishes to receive treatment with high-dose IL-2. With the approval of multiple systemic targeted therapies, nephrectomy is now considered the standard of care in patients who are of good performance status with a resectable primary lesion and who are able to receive small molecule inhibitors for RCC.
Cytoreductive surgery with systemic therapy has been shown to induce clinical benefit.175 The Southwest Oncology Group (SWOG) conducted a randomized study in which patients who were acceptable candidates for nephrectomy either underwent radical nephrectomy followed by therapy with interferon alfa-2b (N = 120) or received interferon alfa-2b alone (N = 121). The primary end point was survival, with objective response (OR) as the secondary end point. The patients were stratified by SWOG performance status (0 or 1), the presence or absence of lung metastases only, and the presence or absence of at least one measurable metastatic lesion in the region not to be resected. After randomization, patients either underwent immediate radical nephrectomy followed by interferon alfa-2b or were immediately given interferon alfa-2b without surgery. Interferon alfa-2b was then continued until disease progression. The median survival time was 11.1 months for patients who received nephrectomy plus interferon alfa-2b, and 8.1 months for patients who received interferon alfa-2b alone (P = 0.05). Nonetheless, interpretation of these data is difficult in light of the poor median survival seen in both arms of this study.176
The patients benefiting the most from cytoreductive surgery appear to be those who have a good ECOG performance status (0 or 1) and preferably with metastasis limited to the lungs.177 Risk and benefits, however, need to be weighed in the decision to perform cytoreductive surgery. Patients with poor performance status and a large metastatic disease burden (greater than the tumor in the kidney) are less likely to derive benefit from surgery. Frequently, patients with metastatic disease can undergo resection laparoscopically. This is always preferred when technically feasible, because it allows a shorter recovery interval until starting systemic therapy.
Mickisch and colleagues, in the EORTC randomized trial, compared radical nephrectomy plus immunotherapy with immunotherapy alone.178 These investigators found that nephrectomy plus immunotherapy delayed time to progression of the disease and improved survival. Bromwich and associates found a mean survival benefit of 4 to 10 months.179 Of the 268 patients selected for the study, only 20 (7%) were selected for cytoreductive nephrectomy. Owing to the associated risks of the surgery, recovery time, and adverse reactions of the immunotherapy, they concluded after careful patient selection that cytoreductive surgery may only be beneficial to a small segment of renal cell patients.
Rabets and coworkers compared outcomes data for laparoscopic and for open cytoreductive nephrectomy.180 The selection criteria for laparoscopic surgery were presence of lesions 15 cm or less in diameter and confined to the kidney. These investigators found no statistically significant survival benefits between the surgical modalities. Laparoscopic nephrectomy was found to have fewer morbidities, including less operative blood loss and shorter hospital stay. They concluded that even with advanced disease, laparoscopic nephrectomy is a safe alternative to open nephrectomy in metastatic disease. With proper patient selection patients with mRCC may still benefit from laparoscopic surgery.175,177,180,181
A recent retrospective study reviewed baseline characteristics and outcomes of 314 patients with mRCC to assess the impact of cytoreductive nephrectomy on overall survival. (The impact of cytoreductive nephrectomy on survival of patients with mRCC receiving VEGF-targeted therapy.182 The authors concluded that cytoreductive nephrectomy was independently associated with prolonged overall survival (19.8 months vs. 9.4 months) favoring the patients who underwent surgery and received subsequent anti-VEGF therapies. An ongoing randomized trial (CARMINA study) is randomizing patients with newly diagnosed mRCC to either undergo surgery and then sunitinib treatment or directly to sunitinib and it will hopefully answer the question whether cytoreductive nephrectomy should remain the standard of care in the TKI’s era.
Resection of Metastases in Renal Cell Carcinoma
Resection of solitary metastases from RCC is associated with improved survival, although the selection criteria have been poorly defined. This issue was addressed in a retrospective, single-institution analysis of patients with recurrent RCC (N = 278) at the Memorial Sloan-Kettering Cancer Center.183 Recurrent disease was defined as solitary if it recurred within the resected renal bed or if one organ system or site was involved. Multiple unilateral lesions in the lung were considered to represent a solitary site of metastasis. Bilateral lung involvement or recurrence in two or more sites was considered to constitute multiple sites. Surgery for metastatic disease was considered curative if metastases were curatively resected and noncurative if gross tumor was left behind. Of the 278 patients who underwent initial curative nephrectomy, recurrence was solitary in 155 patients and multiple in 123 patients, with a median time to first recurrence of 25 months. The overall 5-year survival rate of patients who underwent curative resection for the first recurrence was 44% (N = 141), 14% for those who received noncurative resection (N = 70), and 11% for those who were treated nonsurgically (N = 67). Favorable predictors of survival by multivariate analysis included a single site of first recurrence, curative resection of the first metastasis, a disease-free interval of more than 12 months, and a metachronous presentation with recurrence. Curative resection of isolated metastases to glandular tissue (thyroid, salivary gland, pancreas, adrenal, ovary) was associated with the best 5-year overall survival rate (63%), followed by resection of isolated lung metastases (54%). In this study, resection of solitary brain metastases, however, was associated with poor outcome, with an 18% 5-year overall rate. The 5-year overall survival rates of 46% and 44%, respectively, for patients who underwent second (N = 62) or third curative resection (N = 22) of subsequent metastases after initial curative metastasectomy were not significantly different from those for patients who received only initial curative metastasectomy (44%; N = 141). Thus patients who undergo complete resection of metastatic disease in a solitary site after a disease-free interval of longer than 12 months may experience long-term survival. As with previous reports, complete resection is the important factor, rather than the number of sites resected, even in patients who have undergone prior metastectomy.184,185
Brain metastases from RCC raise specific therapeutic problems because they are relatively unresponsive to whole-brain radiation therapy and tend to bleed. Stereotactically guided high-precision irradiation (radiosurgery) has shown promising results in selected patients with brain metastases from RCC.186 Radiosurgery appears attractive owing to its low risk of toxicity and minimal invasiveness. Multiple lesions can be treated at the same time, and retreatment can be performed for local or distant recurrences.
Lymphadenectomy in Locally Advanced Disease
The role of lymphadenectomy in RCC remains controversial. Nodal disease has been found to be a predictor of poor prognosis even with M0 disease, although, there is very little evidence to support the value of lymph node dissection.189–189 The decision for lymph node dissection should be based on multiple factors. The need for accurate staging, decreased local recurrence rates, and improved survival should be weighed against risk of morbidity and mortality.187,190 One argument against lymph node dissection is that the kidney is drained hematologically and lymphatically through multiple pathways.187 Owing to the unpredictable pathways of spread accurate staging may not be possible. From 58% to 95% of patients with lymph node disease also have synchronous metastasis.191 consequently, the likelihood of finding patients with localized disease is low at 2% to 9%. Canfield and associates did a retrospective study supporting the need for aggressive nodal resection in the presence of positive lymph nodes without evidence of metastasis.191 Blom and coworkers of the EORTC Genitourinary Group conducted a large randomized trial comparing radical nephrectomy with lymph node dissection and radical nephrectomy alone.189 At 5-year follow-up evaluation, these workers found no significant difference in morbidity and mortality between the two groups after surgery. They also found no significant difference in survival. A small subset of patients may benefit from lymph node dissection. Pantuck and associates have shown improved survival in patients found to have disease-positive lymph nodes who undergo cytoreductive nephrectomy and postoperative immunotherapy.190 The literature is variable on the benefits of lymph node dissection in patients with renal neoplasms. Overall, lymphadenectomy provides little staging information and confers no clear benefit in decreasing risk of recurrence or in survival.
Immunotherapies for Advanced Disease
High-Dose Interleukin 2
Inpatient high-dose bolus IL-2 received FDA approval for treatment for patients with stage IV RCC in 1992 based on data presented on 255 patients who were entered into seven phase II clinical trials.192,193 In these studies, patients received 600,000 to 720,000 IU/kg of recombinant human IL-2 by 15-minute infusion every 8 hours during two 5-day courses (maximum: 14 doses per course) separated by 5 to 9 days of rest. Stable or responding patients received 2 to 5 courses of therapy at 8- to 12-week intervals and then were observed while not receiving any additional therapy. ORs were seen in 37 (15%) of the 255 patients, including 17 complete responses (CRs; 7%) and 20 partial responses (PRs; 8%). The median duration of response was 54 months for all of the responders and 20 months for partial responders; the median has not yet been reached for complete responders. The median survival was 16 months for all 255 patients. Most patients who achieved a CR that lasted longer than 30 months and those with PRs after resection resulting in “no evidence of disease” after a response to high-dose IL-2 were unlikely to experience disease progression and may actually be cured.
Although the inpatient high-dose bolus IL-2 regimen produces favorable outcomes in a handful of patients, it also is associated with significant toxic effects and cost and is not universally available. Low-dose IL-2 regimens (with or without interferon-α) have produced similar response rates and survival in nonrandomized phase II trials, but responses appeared to be less durable than those seen with high-dose IL-2.194–197
In an effort to determine the value of outpatient subcutaneous IL-2 and interferon-α relative to high-dose intravenous IL-2, the Cytokine Working Group did a phase III trial in which patients were randomized to receive either outpatient IL-2 and interferon-α every 6 weeks or standard high-dose inpatient IL-2 every 12 weeks.198,199 Of the 193 patients who were enrolled, 192 were evaluable for toxicity and tumor response. The response rate for high-dose IL-2 was 23%, versus 10% for IL-2 and interferon-α. Eight patients achieved a CR while taking high-dose IL-2, versus only three patients taking low-dose IL-2 and interferon-α. The median response durations were 24 months for high-dose IL-2 and 15 months for IL-2 and interferon-α. Median overall survival times were 17.5 and 13 months, favoring high-dose IL-2. Ten patients (nine major responders) who received high-dose IL-2 were progression free at 3 years, versus three patients (two major responders) who received IL-2 and interferon-α. Of note, responses to high-dose IL-2 were seen with equal frequency across the stratification criteria, whereas low-dose IL-2 and interferon-α seemed to produce fewer responses in patients with liver or bone metastases and in those who had not undergone prior nephrectomy to remove the primary tumor. For patients with bone or liver metastases, or with unresected primary tumor, survival was superior with high-dose IL-2 compared with IL-2 and interferon-α, whereas no significant survival differences between the two treatments were noted for patients who had undergone prior nephrectomy or who were without bone or liver metastases.
Taken together, these studies suggest that high-dose intravenous bolus IL-2 is superior in terms of response rate and possibly response quality to regimens that involve either low-dose IL-2 and interferon-α, intermediate- or low-dose IL-2 alone, or interferon-α alone.199 The superiority of high-dose IL-2 is particularly apparent in patients with tumor metastases in immune-sequestered sites, such as liver or bone, or whose primary tumor has not been resected, or who fall into the intermediate-risk or poor-risk group defined by the French Immunotherapy Group. Consequently, although low-dose cytokine therapy has a limited role in mRCC, it must be concluded that high-dose intravenous IL-2 should remain the preferred therapy for appropriately selected patients with access to such therapy. In view of the toxicity and limited efficacy of high-dose intravenous IL-2 therapy, however, additional efforts should be directed at better defining the patient population for whom this therapy is appropriate.200
Use of interferon-α as a single agent has resulted in only a modest survival benefit in the randomized setting, although combination of this agent with low-dose IL-2 (and perhaps with fluorouracil) may lead to an improved response rate, but evidence of survival benefit in a prospective randomized trial is lacking.203–203 Although interferon-α has been the outpatient treatment of choice in most of Europe and the United States until recently, benefit again appears to be restricted to patients with good prognostic risk.204
Predictors of Response to Cytokine Therapies
Responses to immunotherapy most frequently are seen in patients with RCC of clear cell histology.205–208 This observation was detailed in a retrospective analysis of pathology specimens obtained from 231 patients (163 primary and 68 metastatic tumor specimens) who had received IL-2 therapy in Cytokine Working Group clinical trials.208 For patients with primary tumor specimens available for review, the response rate to IL-2 was 21% (30 of 146) for patients with clear cell histology primary tumors, compared with 6% for patients with non-clear cell histology (1 responder in 17 patients). Among the patients with clear cell carcinoma, response to IL-2 was also associated with the presence of alveolar features and the absence of papillary or granular features. The response rate in patients whose primary tumors had “good” predictive features (e.g., greater than 50% alveolar and no granular or papillary features) was 39% (14 of 36). Patients with tumors that contained “poor” predictive features (e.g., greater than 50% granular or any papillary features) had a response rate of 3% (1 of 33). When this model was then applied to the 68 patients with specimens from metastatic sites, those patients who were treated without resection of their primary tumors, five tumor responses were seen in the 20 patients with “good” predictive features, whereas no tumor responses were seen in the 16 patients in the “poor” predictive group, thus supporting the validity of the model developed from the primary kidney tumor specimens. As a result of these data, it may be appropriate for patients whose primary tumor is of non–clear cell histologic type, or of clear cell histologic type but with “poor” predictive features, to forgo IL-2-based treatment altogether. Even in the most favorable predictive group, however, more than 50% of patients failed to respond to IL-2 therapy, so additional investigations to identify tumor-associated predictors of responsiveness to IL-2 are still necessary.205
Some investigators have begun to examine tumor tissue to identify immunohistochemical markers that may predict the outcomes for patients with RCC. Carbonic anhydrase IX (CAIX) has been identified as one potential marker. Bui and colleagues used a monoclonal antibody designed to detect CAIX expression to perform an immunohistochemical analysis of paraffin-embedded RCC specimens. These investigators showed that greater than 90% of RCCs express CAIX, and that its expression decreases with advancing stage.209 In their analysis, high CAIX expression in primary tumors was seen in 79% of patients and was associated with improved survival and possibly improved response to IL-2-based therapy. Building on this work, Atkins and coworkers performed a nested case-control study within the larger cohort of patients for whom the histopathological findings were analyzed.210 CAIX expression levels were correlated with response to IL-2, pathological risk categorization, and survival. Median survival times were 3 years and 1 year for high and low CAIX expressors, respectively. Although tumor response was seen in six patients with low CAIX staining, survival beyond 5 years was seen only in the patients with high-CAIX–expressing tumors. High-CAIX staining was associated with better histopathological features but remained an independent predictor of response. Additional studies to explain these preliminary observations and correlate results with previously described clinical features are necessary.205
The Cytokine Working Group has completed the high-dose IL-2 “Select” Trial. The primary objective of this study was to determine, in a prospective fashion, if the predictive model proposed by Atkins and associates can identify a group of patients with advanced RCC who are significantly more likely to respond to high-dose IL-2-based therapy (“good” risk) than a historical, unselected patient population. New factors (including baseline immune function, immunohistochemical markers, and gene expression patterns) that might be associated with response to high-dose IL-2 therapy have been explored in an attempt to more narrowly limit the application of IL-2 to those patients most likely to benefit.205
In this multicenter prospective study, patients with histologically confirmed RCC that was metastatic or unresectable, ECOG PS 0 to 1 and adequate organ function received high-dose IL-2 (600,000 U/kg/dose intravenously every 8 hours on days 1 through 5 and 15 to 19 [maximum 28 doses] every 12 weeks). The primary end point of the study was to determine the major response rate and associated “good” predictive features. One hundred twenty eligible patients were accrued between 2007 and 2009. The majority of patients (approximately 70%) had an ECOG performance status of 0 and were Memorial Sloan-Kettering Cancer Center intermediate risk. At the time of the analysis the investigator assessed, relative risk was 30% in clear cell RCC with six complete remissions. Response to IL-2 was not associated with any pretreatment clinical factor and was not seen in patients with non–clear cell histology. Preliminary analysis of CAIX staining did not show correlation with outcome. Additional analyses are still ongoing.211