Adult T-Cell Leukemia-Lymphoma
Kunihiro Tsukasaki, Toshiki Watanabe and Kensei Tobinai
• Adult T-cell leukemia-lymphoma (ATL) is a distinct peripheral T-cell malignancy associated with human T-cell leukemia/lymphotropic virus type I (HTLV-I).
• HTLV-I is reverse-transcribed into DNA and randomly integrated into the host cell.
• The HTLV-I genome encodes two unique regulatory proteins—Tax and Rex—responsible for viral expression and cellular transformation. Tax trans-activates viral and cellular genes that could be involved in the pathogenesis of ATL. HTLV-I basic leucine zipper (HTLV-I bZIP; HBZ) is an antisense transcript of HTLV-I, is steadily expressed in ATL cells, and interacts with several host genes and suppresses the activity of Tax.
• A major cluster of HTLV-I–infected individuals and patients with ATL exists on the southwest coast of Japan, where approximately 1.1 million people are infected with the virus.
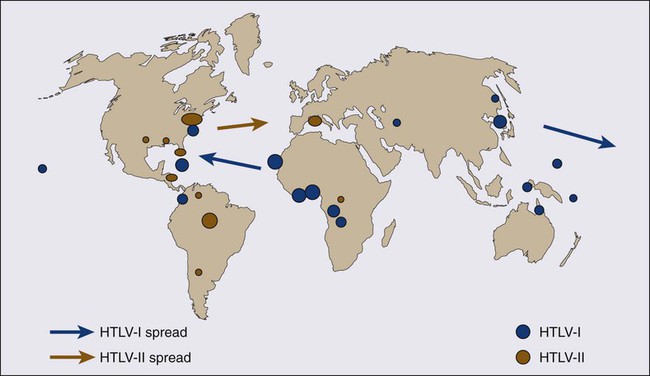
• Other clusters have been noted in the Caribbean islands (African), tropical Africa (African), South America (Mongoloid), and northern Oceania (Melanesian).
• HTLV-I is transmitted from mother to child through breast-feeding, by sexual contact, and by blood-borne transmission.
• The estimated cumulative risk of the development of ATL in HTLV-I–positive individuals is about 3% after transmission from their mothers.
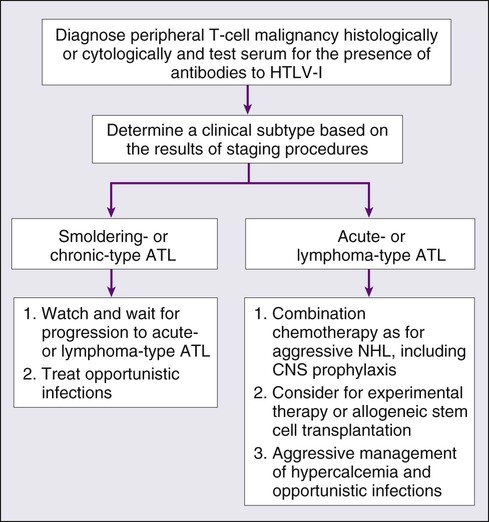
• Patients with ATL show diverse clinical features, and four clinical subtypes have been recognized: acute, lymphoma, chronic, and smoldering.
• The typical manifestations of acute-type ATL include circulating neoplastic cells in the peripheral blood, generalized lymph node swelling, hepatosplenomegaly, skin involvement, opportunistic infections, and hypercalcemia.
• Leukemic cells in the peripheral blood characteristically show markedly polylobated nuclei, so-called flower cells. Their immunophenotypes are CD4/CD25/CCR4+ and CD8− in most cases.
• All histopathological specimens show findings of peripheral T-cell lymphoma of various subtypes.
• ATL is suspected when the aforementioned characteristic clinical manifestations and/or the cytologic findings of leukemic cells in the peripheral blood are recognized.
• Morphologic and immunophenotypic analyses of neoplastic cells in peripheral blood or tumor lesions and a serologic assay against HTLV-I are required for the clinical diagnosis of ATL.
• The demonstration of the monoclonal integration of HTLV-I proviral DNA in the neoplastic cells can lead to a definite diagnosis of ATL.
• An accurate diagnosis of the clinical subtype is vital for appropriate decisions regarding treatment.
• Intensive chemotherapies combining agents used in the treatment of non-Hodgkin lymphoma (NHL) are usually given to patients with the acute or lymphoma subtype of ATL; however, most patients with ATL do not achieve cure with current chemotherapy regimens.
• Further efforts to incorporate promising or new, innovative treatment modalities, such as interferon/zidovudine therapy, new anticancer agents, monoclonal antibody therapy, molecular-targeting therapy, and allogeneic hematopoietic stem cell transplantation (allo-HSCT), are needed for the establishment of risk-adopted therapy.
• Prevention of HTLV-I infections has been achieved in some endemic areas by screening for HTLV-I among blood donors and recommending mothers who are carriers to refrain from breast-feeding. Prevention of ATL among HTLV-I carriers has not been achieved, although several risk factors have been identified.
Introduction
Adult T-cell leukemia-lymphoma (ATL) was first recognized in Japan in 1977.1 The disease was characterized as leukemia of peripheral T cells, generalized lymphadenopathy, hepatosplenomegaly, and skin involvement. Because of its unusual geographic clustering in southwestern Japan, it was postulated that some infectious agent(s) had causative roles. Human T-cell leukemia/lymphotropic virus type I (HTLV-I) was first isolated by Poiesz and associates2 in the United States from cultured cells from one patient with an aggressive variant of mycosis fungoides and from one with Sézary syndrome. Although both patients, who were African Americans, were diagnosed clinically as having cutaneous T-cell lymphoma (CTCL) at the time of reporting, their clinical features were later found to closely resemble those of Japanese patients with ATL.
In 1980, Miyoshi and coworkers3 established the first cell line (MT-1) derived from neoplastic cells in an ATL patient. They co-cultured neoplastic cells from an ATL patient with normal human cord blood leukocytes and established the cell line MT-2 (derived from cord blood lymphocytes), which produced large amounts of type C retrovirus.4 Using the MT-1 cell line, Hinuma and colleagues5 found that patients with ATL had antibodies against the virus-associated antigen in their sera. The “ATL virus” was then isolated and characterized as an RNA retrovirus.6 Because the ATL virus was found to be identical to HTLV by a DNA sequence analysis, it was designated HTLV-I.7
The etiologic association of HTLV-I and ATL is based on the following findings:
• The areas with a high incidence of patients with ATL closely correspond to those with a high prevalence of HTLV-I carriers.8
• HTLV-I immortalizes T cells in vitro.9
• HTLV-I proviral DNA is detected in the neoplastic cells of patients with ATL.10
• Almost all patients with ATL have antibodies against HTLV-I in their sera.
HTLV-I is the first retrovirus found to be associated with a malignant neoplasm in humans.
Virology and Pathogenesis
The RNA genome of HTLV-I is reverse-transcribed into DNA and integrated as a proviral DNA into the host cell. The HTLV-I provirus is 9.0 kilobases long and has structural genes in the order 5′-gag-pol-env-3′. Both ends of the HTLV-I proviral DNA contain repeats called long terminal repeats (LTRs). The HTLV-I gene encodes three structural proteins: group antigen (gag), reverse transcriptase (pol), and envelope (env) proteins. The full-length mRNA is used for the synthesis of gag and pol gene products. The gag protein is synthesized as a precursor polypeptide of 55 kDa that is proteolytically cleaved into the individual gag proteins p19, p24, and p15. The protease is encoded in a different reading frame that spans the 3′ part of the gag region and the 5′ part of the pol region. The pol region encodes the reverse transcriptase, integrase, and RNase H. The env gene encodes two proteins made from a singly spliced mRNA. It is then cleaved intracellularly into an extracellular glycosylated protein (gp46) and a transmembrane (gp21). The pX region at the 3′ end of the genome has the potential to encode essential regulatory proteins (Tax and Rex) and accessory proteins—p8, p12, p13, and p30—that are important for viral infectivity and replication by influencing cellular signaling and gene expression.11–16
The life cycle of a retrovirus begins with the binding of the virus to specific receptors on the cell surface via viral envelope proteins. Three molecules, glucose transporter–1 (GLUT1), neuropilin-1 (NRP1), and heparan sulfate proteoglycans (HSPG) are involved in HTLV-I binding and entry.17–21 HTLV-I transmission is mediated by cell-to-cell contact. A unique structure called the “virologic synapse” was reported to form at the contact site of HTLV-I–infected cells and uninfected T cells, and viral materials are transmitted through it. On the other hand, it was reported that HTLV-I particles move from cell to cell in a complex with numerous extracellular matrix components, forming large surface-associated biofilm-like structures.22 In addition, HTLV-I p12/p8 expression increases T-cell contact through specific adhesion molecules and promotion of cellular conduits appears to enhance cell-to-cell viral transmission.16
Role of Tax
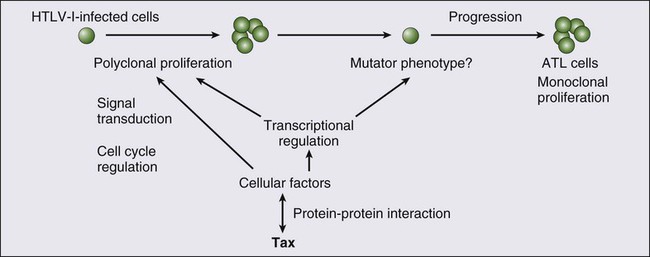
The onset of ATL is preceded by a long period of clinical latency, frequently lasting more than 4 decades. In addition, ATL develops in less than 5% of all individuals infected with HTLV-I. The promoter insertion model was rejected as the leukemogenic mechanism because integration sites of the provirus were random, depending on the patient.11 Consequently, a trans-acting viral factor, Tax, has been shown to be oncogenic, because it transforms and immortalizes rodent fibroblasts and T lymphocytes as well as human T lymphocytes. Tax trans-activates viral transcription through interaction with the cellular basic domain/leucine zipper transcription factors cyclic adenosine monophosphate (cAMP) response element–binding (CREB) factor and cAMP-dependent transcription factor–1 (ATF1). Tax interacts with numerous cellular proteins to reprogram cellular processes, including, but not limited to, transcription, cell cycle regulation, DNA repair, and apoptosis. Tax transcriptionally regulates cellular genes by interaction with enhancer-binding proteins such as CREB, nuclear factor–κB (NF-κB), and serum response factor and by tethering coactivators to the DNA-bound transcription factors. Tax also stimulates cell growth by direct binding to cyclin-dependent kinase holoenzymes and/or inactivating tumor suppressors such as p 53 (tumor protein p53) and DLG1 (discs, large). Furthermore, Tax silences cellular checkpoints, which guard against DNA structural damage and chromosomal missegregation, thereby favoring the manifestation of a mutator phenotype in cells.13,23
Tax interacts and activates specific components of growth factor signal transduction pathways, such as IκB kinase–nuclear factor–κB (IκB kinase [IKK]-NF-κB), RAS/mitogen-activated protein kinase, protein kinase A, and protein kinase C.24,25 Interaction with IKKγ, a component of the IKK complex, results in the constitutive activation of this kinase complex.26 Constitutive activation of the Janus-activated kinase–signal transducer and activator of transcription (JAK-STAT) pathway in HTLV-I–transformed cells has also been reported, although the mechanisms are not well understood.27 Thus, HTLV-I infection results in the aberrant activation of growth-promoting signaling pathways.
The oncogenic capacity of Tax has been reported in various systems; however, cellular transformation by HTLV-I in vivo is a multistage process and viral gene expression is absent in ATL cells in vivo.28,29 Moreover, proviruses integrated in ATL cells are frequently defective, have mutations in the coding region of Tax, and/or are methylated in the 5′ and 5′ LTR regions.32–32 Thus, in addition to promoting growth directly, Tax should endow the infected T cells with capacities that aid the progression to transformed phenotypes in the absence of Tax. In this context, induction of a mutator phenotype by Tax in the infected cells appears to play an important role.33 The role of HTLV-I Tax in the multiple-step leukemogenesis of ATL are illustrated in Figure 108-1.
Presence of Antisense Transcript and Its Function
The expression of antisense strand RNA with the capacity to encode a zinc finger protein (HTLV-I basic leucine zipper [HTLV-I bZIP; HBZ] factor) has opened up a new field of research. HBZ inhibits Tax-dependent viral transcription.34 HBZ RNA is transcribed from the region of the HTLV-I provirus that is spared genomic deletion and has been shown to be expressed in vivo in all ATL patients as well as virus carriers; thus, it might be involved in the growth of ATL cells.35
Role of Chromosomal Abnormalities
Various karyotypic abnormalities have been reported in neoplastic cells of ATL; however, no specific karyotypic abnormality has been found. In general, the chromosomal abnormalities are more complex in the acute type than in the chronic type. Itoyama and colleagues36 reported the results of cytogenetic analysis of 50 cases of ATL and found aneuploidy and multiple breaks more frequently in acute and lymphoma types. Multiple breaks and partial loss of chromosomes correlated with shorter survival. The authors claim that one model of an oncogenic mechanism—activation of a proto-oncogene by translocation of a T-cell receptor (TCR) gene—might not be applicable to the main pathway of development of ATL and that a multiple-step process of leukemogenesis is required.
In a study by Tsukasaki and colleagues,37 64 patients with ATL were analyzed by using comparative genomic hybridization (CGH). The most frequent observations were gains at 14q, 7q, and 3p and losses at 6q and 13q. Chromosomal imbalances, losses, and gains were observed more frequently in acute or lymphoma types. An increased number of chromosomal imbalances was associated with a shorter survival. Paired samples (i.e., samples obtained at different sites) from 4 patients and sequential samples from 13 patients (from 6 during both the chronic phase and acute crisis and from 7 during both acute onset and relapse) were examined by CGH and Southern blotting for HTLV-I. All but two paired samples showed differences on CGH assessment. Two chronic/crisis samples showed distinct results regarding both CGH and HTLV-I integration sites, suggesting clonal changes in ATL at crisis. In 11 patients, the finding of identical HTLV-I sites and clonally related CGH results suggested a common origin of sequential samples. In contrast to chronic/crisis samples, CGH results with all acute/relapse sample pairs showed the presence of clonally related but not evolutional subclones at relapse. It was concluded that clonal diversity is common during the progression of ATL and that CGH alterations are associated with clinical course.
Role of p53 and Other Tumor Suppressor Genes
P53 is a nuclear phosphoprotein that functions as a tumor suppressor. A loss of a normally functioning p53 through mutation or allelic loss has been found in several kinds of malignant neoplasms. Mutations of the p53 gene have also been found in some patients with ATL.38,39 According to a study by Cesarman and coworkers,39 no p53 mutations were detected in samples from 11 patients with the chronic type of ATL, whereas 9 (28%) of 28 samples from patients with the acute type exhibited p53 mutations. In one patient, a tumor sample obtained during the chronic phase did not have a mutation of p53 but the mutation was subsequently detected in a sample that was obtained at crisis. These results suggest that alterations of the p53 gene might contribute to disease progression in a fraction of patients with ATL.
Other putative tumor suppressor genes, p15INK4B and p16INK4A, were reported to be associated with ATL.42–42 Yamada and colleagues41 reported that 28 (25%) of 114 patients with ATL showed homozygous deletions of the p15 and/or p16 genes. These results correlated well with the clinical subtype of ATL. In addition, the patients with deleted p15 and/or p16 genes had significantly shorter survival period than did patients in whom both genes were preserved (P < .0001). Moreover, three of the five patients with chronic-type ATL who progressed to acute-type ATL lost the p16 gene alone or both genes at their exacerbation phase. These results suggest the deletion of p15 and/or p16 to play a key role in the disease progression of some patients with ATL. Uchida and colleagues42 found a point mutation of the p16 gene in 3 (7%) of 44 patients with ATL. It is suggested that the p16 gene is inactivated not only by homozygous deletion but also by point mutation.
Role of HTLV-I Provirus
Several investigators have analyzed the implications of the integration pattern of HTLV-I provirus in the progression of ATL.43,44 It is known that the neoplastic cells of ATL have one complete copy of the HTLV-I provirus per cell in some patients (complete type) whereas others have multiple complete copies per cell (multiple type). The HTLV-I proviruses in the remaining patients do not have the complete genome but rather have a defective genome (defective type). Tsukasaki and colleagues44 found that the median survival times (MSTs) for patients were 7 months, 24 months, and 33 months for defective-type, complete-type, and multiple-type ATL, respectively (P = .006). Among 52 sequentially examined patients, the HTLV-I integration patterns changed in four patients (8%). In three of these four, the rearrangements of the TCRβ gene changed concomitantly, suggesting the appearance of a new ATL clone. The researchers concluded that the frequent clonal change of ATL at crisis reflects the emergence of multiple premalignant clones in viral leukemogenesis.
Tamiya and coworkers43 reported the presence of two types of defective viruses. The type 2 defective virus with a deletion that includes the 5′ LTR was found more frequently in the acute and lymphoma types (39%, 21 of 54) than in the chronic type (6%, 1 of 18). It is postulated that the high frequency of the type 2 defective virus is caused by the genetic instability of the HTLV-I provirus and that this defective virus is selected for because it evades the host’s immune surveillance system.
HTLV-I is an etiologic agent not only in ATL but also in the neurologic disorder known as tropical spastic paraparesis (TSP) or HTLV-I–associated myelopathy (HAM).45,46 In TSP/HAM the HTLV-I provirus remains randomly integrated, whereas in ATL the provirus is monoclonally integrated.
Epidemiology of HTLV-I and Adult T-Cell Leukemia-Lymphoma
Inhabitants of the southwest coast of Japan have the highest prevalence of HTLV-I infections and the highest incidence of ATL in the world.8,46–48 A high prevalence of HTLV-I is also found in inhabitants of the Caribbean islands (African), tropical Africa (African), South America (Mongoloid), and northern Oceania (Melanesian).47–50 Many patients who have been diagnosed with ATL in Western countries are immigrants from the West Indies and tropical Africa. The world map of the distribution of HTLV-I and HTLV-II and the presumed routes of spread is shown in Figure 108-2.48 The geographic clustering of HTLV-I carriers is suggested to be strongly associated with high frequencies of mother-to-child and sexual transmissions of the virus under closed conditions in particular groups.51
Recently, the prevalence of HTLV-I in Japan as determined by screening of blood donors was surveyed.52 The seroprevalence of HTLV-I among 1,196,321 Japanese first-time blood donors from 2006 to 2007 was investigated. A total of 3787 such donors were confirmed to be positive for the anti–HTLV-I antibody. By applying a fitness curve to the age ranges outside the blood donor age range, the present number of HTLV-I carriers covering ages from birth to 99 years was estimated to be at least 1.08 million in Japan; this value was 10% lower than that reported in 1988.53 The adjusted overall prevalence rates were estimated to be 0.66% and 1.02% in men and women, respectively. The peak in carrier numbers was found among individuals in their 70s, which is a shift from the previous peak observed in the 1988 database among individuals in their 50s. As compared with the survey in the 1980s, carriers were distributed not only in the endemic southwestern region of Japan but also throughout the country, particularly in the greater Tokyo metropolitan area. The authors calculated, after applying population projections, that the carrier number will decrease by half in the next two decades; however, the carrier population will age over that interval, meaning that the age of patients with ATL will also be higher.
In endemic areas, there is a marked increase in HTLV-I prevalence with age until 70 years and an increased prevalence among females compared with males. Transmission occurs via sexual and blood-borne routes. A major reason for the increase in seroprevalence with age appears to be the decreasing prevalence of HTLV-I in the population over time, at least in Japan, where it has been most extensively studied. Yamaguchi and coworkers54 reported that the HTLV-I carrier rates among blood donors at Kyusyu in Japan had fallen since 1986 in all age groups younger than age 50 years and in both genders. Recently, the results were reconfirmed in another endemic area in Japan.55 This decrease in HTLV-I carriers among younger blood donors might be explained by improvements in sanitation and general lifestyle changes in recent decades. A shorter duration of breast-feeding, the increasing use of artificial feeding for infants, and decreasing family sizes are also likely to be factors for the recent decline in the vertical transmission rates of HTLV-I.54,55 Overall, there is a slight male predominance of ATL patients, with the male-to-female ratio ranging from 1.1 to 1.5. This ratio contrasts with that of TSP/HAM, which affects females more frequently than males.
1. Mother-to-child-transmission, mainly by HTLV-I–positive lymphocytes in breast milk56
2. Sexual transmission, more commonly from males to females
3. Blood-borne transmission, including blood transfusions and sharing of needles by intravenous drug abusers57,58
The first infection route is vertical transmission from mother to child via HTLV-I–positive lymphocytes in breast milk. The overall infection rate for HTLV-I in children with seropositive mothers has been estimated to be 10% to 30%. HTLV-I-infection has also been reported in about 3% of children not breast-fed, which suggests the possibility of an intrauterine or transvaginal infection.59 However, the intrauterine route was unlikely, given the discordance of HTLV-I DNA in cord blood and the subsequent seroconversion of the infants.60 Several types of intervention have been conducted in HTLV-I–endemic areas in Japan, where seropositive pregnant women are advised not to breast-feed.51 Recently, a nationwide intervention has been initiated in Japan.
The second route is transmission through sexual contact. Transmission of HTLV-I frequently occurs from males to females but rarely from females to males, similar to human immunodeficiency virus (HIV) transmission. HTLV-I has been isolated in semen. It appears likely that the risk of development of ATL after HTLV-I infection by this route of transmission is not high because most of the mothers of ATL patients were seropositive for HTLV-I.61
To prevent HTLV-I transmission through blood transfusions, serologic screening of all blood donors for HTLV-I has been conducted in Japan since November 1986. The present donor screening program for HTLV-I has almost completely prevented transmission by transfusion in Japan. In contrast to red cell and platelet concentrates, fresh-frozen plasma and plasma fractions have never been shown to transmit HTLV-I.57
Approximately 1.1 million HTLV-I–infected individuals reside in Japan, and the annual incidence of ATL in Japan is approximately 1000.52 The annual rate of ATL development among HTLV-I carriers older than 40 years is estimated at 1.5 per 1000 in males and 0.5 per 1000 in females, and the cumulative risk of ATL development among the HTLV-I carriers is estimated to be 2.5% to 5% over the course of a 70-year life span.62 It was postulated that the leukemogenesis of ATL consists of five steps, based on a statistical analysis of ATL development according to age.63
A recent nationwide survey in Japan revealed that compared with previous nationwide studies, the age of ATL patients shifted toward older ages and the mean age gradually increased from 52.7 years in the first survey (cases before 1980) to 61.1 years in the ninth survey (1996 to 1997) and, finally, to 66.0 years in the 10th study (range, 19 to 94; median, 67).64 It has been reported that the age of patients with ATL in areas outside Japan is lower, with an overall mean age in the mid 40s.65
HTLV-I infection early in life, presumably from breast-feeding, is crucial in the development of ATL; 100% of mothers of patients with ATL were HTLV-I carriers as compared with about 30% of mothers of patients with TSP/HAM.61 HTLV-I infection by blood transfusion is associated with a higher risk for the development of TSP/HAM than infection by other routes.66 In contrast, very few cases of ATL after HTLV-I infection by blood transfusion have been reported.67,68 Interestingly, those affected had blood transfusions for a preceding hematologic malignancy and were diagnosed with ATL within 11 years after the HTLV-I infection.
Recently, the development of ATL after kidney, living donor liver, and hematopoietic stem cell transplantations has been reported, possibly associated with the use of immune suppressants.71–71 Other factors reportedly associated with the onset of ATL include HTLV-I infection early in life, increase in age, male sex, family history of ATL, past history of infective dermatitis, smoking of tobacco, serum titers of antibody against HTLV-I, and several human leukocyte antigen (HLA) subtypes.72–77 However, definitive risk factors for the development of ATL among asymptomatic HTLV-I carriers have not been determined. HTLV-I proviral loads have been proposed as an important predictor of the development of ATL, but only a few small prospective studies have been conducted. Recently, Iwanaga and colleagues evaluated 1218 asymptomatic HTLV-I carriers (426 males and 792 females) who were enrolled during 2002 through 2008 for a prospective study on the development of ATL.78 The proviral load at enrollment was significantly higher in males than females (median, 2.10 vs. 1.39 copies/100 peripheral blood mononuclear cells (PBMCs) (P < .0001), in those aged 40 or older, and in those with a family history of ATL. During the follow-up period, 14 participants developed acute ATL. Their baseline proviral loads were high (range, 4.17 to 28.58 copies/100 PBMCs). Multivariate Cox regression analyses indicated that not only a higher proviral load but also advanced age, a family history of ATL, and the first opportunity for HTLV-I testing during treatment for other diseases were independent risk factors for the progression of ATL from a carrier status.
• Viral infection alone is not sufficient for the expression of the malignant phenotype.
• The timing and/or length of viral exposure are critical.
• The long latency period suggests that the disease progression is a multiple-step process. This contrasts with TSP/HAM, which can occur with a shorter latency period, especially among recipients of blood transfusions.
Clinical Manifestations
After HTLV-I was revealed to be associated with ATL, it was found that ATL shows a marked diversity in its clinical manifestations. ATL cases have been subdivided into four distinct clinicopathological entities: acute, lymphoma, chronic, and smoldering types. The recognition of the four clinical subtypes is important in understanding the natural history, clinical features, treatment strategy, and leukemogenesis of ATL. On the basis of a nationwide survey of 854 patients with ATL who were diagnosed between 1983 and 1987 in Japan, the Lymphoma Study Group proposed diagnostic criteria for the four clinical subtypes based on the sites of organ infiltration, presence, absence and degree of leukemic manifestation, a high lactic acid dehydrogenase (LDH) value, and hypercalcemia (Table 108-1)79:
Table 108-1
Diagnostic Criteria for Clinical Subtypes of Adult T-Cell Leukemia-Lymphoma
| Smoldering | Chronic | Lymphoma | Acute | |
| Anti-HTLV-I antibody | + | + | + | + |
| Lymphocyte (×103/µUL) | <4 | ≥4‡ | <4 | * |
| Abnormal T lymphocytes | ≥5%¶ | +§ | ≤1% | +§ |
| Flower cells with T-cell marker | † | † | No | + |
| Lactate dehydrogenase | ≤1.5 N | ≤2 N | * | * |
| Corrected Ca2+ (mEq/L) | <5.5 | <5.5 | * | * |
| Histology-proven lymphadenopathy | No | * | + | * |
| Tumor lesion | ||||
| Skin and/or lung | * | * | * | * |
| Lymph node | No | * | Yes | * |
| Liver | No | * | * | * |
| Spleen | No | * | * | * |
| Central nervous system | No | * | * | * |
| Bone | No | No | * | * |
| Ascites | No | No | * | * |
| Pleural effusion | No | No | * | * |
| Gastrointestinal tract | No | No | * | * |
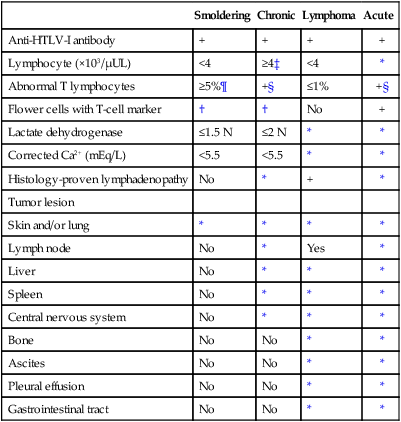
HTLV-I, Human T-cell leukemia/lymphotropic virus I; N, normal upper limit.
*No essential qualification except terms required for other subtype(s).
†Typical “flower cells” may be seen occasionally.
‡Accompanied by T lymphocytosis (3.5 × 103/µL or more).
§If the proportion of abnormal T lymphocytes is less than 5% in peripheral blood, a histologically proven tumor lesion is required.
¶Histologically proven skin and/or pulmonary lesion(s) is required if there are fewer than 5% abnormal T lymphocytes in peripheral blood.
From Shimoyama M and Members of the Lymphoma Study Group (1984-1987). Diagnostic criteria and classification of clinical subtypes of adult T-cell leukemia-lymphoma. Br J Haematol 1991;79:428.
1. The acute type shows a rapidly progressive clinical course and most of the characteristic features of ATL: leukemic manifestation, generalized lymphadenopathy, hepatomegaly, splenomegaly, skin involvement, hypercalcemia, and the infiltration of other organs (central nervous system, gastrointestinal tract, etc.). The symptoms and signs include abdominal pain, diarrhea, ascites, jaundice, pleural effusion, cough, sputum, fever, and unconsciousness because of organ involvement, hypercalcemia, and/or opportunistic infections.
2. The smoldering type shows an indolent clinical course and only a small percentage of leukemic cells, but it also can include skin and lung involvement.
3. The chronic type, lymphocytosis with a high percentage of leukemic cells, is occasionally associated with skin and lung involvement, lymphadenopathy, and hepatosplenomegaly and also shows an indolent clinical course.
4. The lymphoma type includes patients who have the manifestations of nodal NHL without circulating malignant cells in the peripheral blood.
ATL, particularly the aggressive forms (acute and lymphoma types), has been found to infiltrate the stomach and the intestines in 29% and 25% of patients, respectively, at autopsy.80 The involvement may be focal as an isolated gastric lesion or so diffuse as to involve the entire gastrointestinal tract. Extensive infiltration of the intestines can lead to moderate to severe diarrhea and malabsorption. Patients with ATL suffer from a variety of abdominal symptoms (e.g., nausea, vomiting, abdominal fullness, and diarrhea), which might be attributable to infiltration by neoplastic cells, but, because of the associated immunodeficiency, various opportunistic infections such as strongyloidiasis and cytomegalovirus (CMV) gastrocolitis can complicate cases.
Hepatic involvement of ATL cells can be found in up to one fourth of patients with acute and lymphoma subtypes and not infrequently manifests as jaundice and elevated hepatic transaminase levels. Among patients with acute-type or lymphoma-type ATL, palpable hepatomegaly was more frequent and total bilirubin, hepatic transaminase, LDH, and alkaline phosphatase values were higher than among other patients with NHL. Autopsy liver samples disclosed that the portal area was most frequently infiltrated with ATL cells.81
Pulmonary complications, which are common in ATL, are due to leukemic infiltration in one half of patients and to infections with a variety of bacterial and opportunistic organisms in the other half.82 Of 854 Japanese patients with ATL, 26% had active infections at the time of diagnosis.78 The incidence was highest among patients with the chronic and smoldering types (36%) and lower for patients with the acute (27%) and lymphoma (11%) subtypes. The infections that were encountered were bacterial (pneumonia, sepsis, and tuberculosis) in 43%, fungal in 31%, protozoal in 18%, and viral in 8% of patients with ATL (Table 108-2). The immunodeficiency at presentation in ATL can be exacerbated by the neutropenia that is produced by cytotoxic chemotherapy, leading to an extremely high risk of infection throughout the course of therapy. Infections are responsible for the patient’s death in about half of the cases. Pulmonary involvement of ATL cells occurred in 17% of the patients.79
Table 108-2
Infectious Complications at Diagnosis in 818 Japanese Patients with Adult T-Cell Leukemia-Lymphoma
| Type of Infection | No. of Patients* | ||||
| Acute | Lymphoma | Chronic | Smoldering | Total | |
| Bacterial infection | (55) | (9 + 1)† | (25) | (4) | (93) |
| Pneumonia | 35† | 1 | 14 | 4 | 54 |
| Pyoderma | 1 | 1 | 3 | 0 | 5 |
| Septicemia | 6 | 0 | 1 | 0 | 7 |
| Tuberculosis | 7 | 1 | 3 | 0 | 11 |
| Other | 6 | 6 | 4 | 0 | 16 |
| Fungal infection§ | (36 + 2)‡ | (6) | (16) | (8) | (66) |
| Cutaneous | 26 | 5† | 12 | 5 | 48 |
| Oral | 2 | 0 | 0 | 0 | 2 |
| Esophageal | 2 | 0 | 2 | 1 | 5 |
| Pulmonary | 5 | 1 | 1 | 0 | 7 |
| Meningitis | 1 | 0 | 1 | 2 | 4 |
| Protozoal infection§ | (22) | (2) | (10) | (4) | (38) |
| Strongyloidiasis | 13‡ | 2 | 5 | 1 | 21 |
| Giardiasis | 1 | 0 | 0 | 0 | 1 |
| Pneumocystis jiroveci | 8 | 0 | 5 | 3 | 16 |
| Viral infection§ | (13) | (0) | (3) | (0) | (16) |
| Herpes zoster | 7 | 0 | 2 | 0 | 9 |
| CMV pneumonia | 3 | 0 | 0 | 0 | 3 |
| Pneumonitis | 2 | 0 | 1 | 0 | 3 |
| Condyloma acuminatum | 1 | 0 | 0 | 0 | 1 |
| No infection¶ | 339 | 139 | 98 | 29 | 605 |
| Total | 465 | 156 | 152 | 45 | 818 |
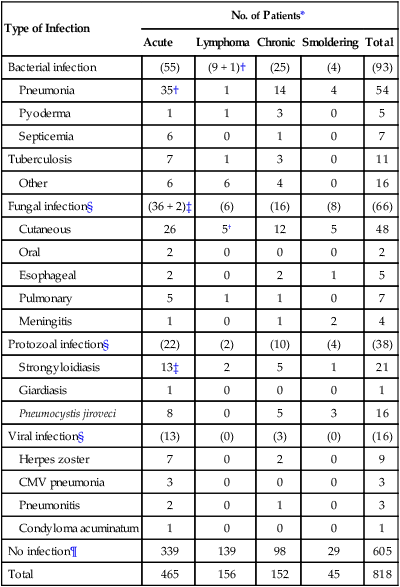
*Values in parentheses indicate the total number of patients in each category.
‡One patient each suffered from oral candidiasis.
From Shimoyama M and Members of the Lymphoma Study Group (1984-1987). Diagnostic criteria and classification of clinical subtypes of adult T-cell leukemia-lymphoma. Br J Haematol 1991;79:42.
Central nervous system (CNS) involvement occurs in approximately 10% of patients with ATL.83 Opportunistic infections of CNS such as cryptococcal meningitis and toxoplasmosis and hypercalcemia can also cause CNS symptoms and signs in ATL patients.
Cutaneous involvement occurs in approximately 40% of patients with ATL. The incidence was highest among patients with the chronic and smoldering types (48%) and lower for patients with the acute (40%) and lymphoma (25%) subtypes.79 A proportion of patients with smoldering ATL have cutaneous involvement without leukemic manifestation.84 Although cutaneous involvement is relatively frequently observed in indolent (smoldering or chronic) ATL, the primary cutaneous tumoral type, although generally included among smoldering ATL, was reported as having a poor prognosis.85,86
Laboratory Findings
Laboratory findings also depend on the clinical subtype of ATL (see Table 108-1).79 Leukocytosis is found among patients with the chronic subtype and a portion of those with the acute and smoldering subtypes at presentation, exhibiting characteristic abnormal lymphoid cells with markedly lobated nuclei, highly indented or lobulated nuclei with condensed chromatin, small or absent nucleoli, and an agranular cytoplasm, termed flower cells. Although not all patients have a leukemic feature, peripheral blood involvement develops in most patients at some time during the course of their disease. Most patients with the acute or lymphoma subtype of ATL have elevated serum LDH levels.
The most striking laboratory finding in patients with ATL is hypercalcemia, which was evident in 32% of Japanese patients with ATL at the time of diagnosis.79 Multiple factors have been suggested to contribute to the development of hypercalcemia. Lytic bone lesions have been described in some patients; however, examinations of bone obtained at autopsy or from bone marrow biopsies usually reveal activated osteoclasts with increased bone resorption; infiltrating neoplastic T cells are rarely found. Patients with ATL have low phosphate levels, hypercalciuria, high levels of nephrogenous cyclic adenosine monophosphate, and low levels of 1,25-dihydroxyvitamin D. This pattern suggests humoral hypercalcemia of malignancy, which was found to be secondary to the production of a parathyroid hormone (PTH)-like molecule by malignant cells. HTLV-I–infected cells were found to produce a protein with PTH-like activity, such as PTH-related protein.87,88 Another suggested contributor to hypercalcemia in patients with ATL is cytokine production by the tumor cells. HTLV-I–infected cell lines and fresh ATL cells from hypercalcemic patients produce tumor necrosis factor (TNF)–α, TNF-β, interleukin (IL)-1α, and IL-1β. Each of these cytokines can enhance osteoclast activity and bone-resorbing activity in animal models. Ishibashi and colleagues89 demonstrated elevated serum levels of TNF-β in 7 of 8 patients with ATL who had complications of hypercalcemia and in none of 28 patients with ATL who had normal serum calcium levels.
Nosaka and coworkers analyzed the expression of various genes that were suggested to regulate serum calcium levels in ATL and reported that the overexpression of the receptor activator of NF-κB (RANK) ligand gene correlated with hypercalcemia.90 ATL cells from patients with hypercalcemia, which highly expressed the transcripts of the RANK ligand (RANKL) gene, induced the differentiation of human hematopoietic precursor cells (HPCs) into osteoclasts in vitro in the presence of macrophage colony-stimulating factor. The authors claime that ATL cells induce the differentiation of hematopoietic precursor cells into osteoclasts through RANKL expressed on their surface, in cooperation with macrophage colony-stimulating factor, and that they ultimately cause hypercalcemia. The etiology of hypercalcemia in ATL is likely to be multifactorial and in individual patients is probably the result of some combination of the factors just described.
Elevated serum levels of soluble IL-2 receptor in patients with ATL, especially in those with the acute or lymphoma subtype, have been noted in several studies.91 The serum level of soluble IL-2 receptor is suggested to be a useful marker for the differential diagnosis of ATL, for evaluating the clinical aggressiveness of the disease, and for monitoring the response to treatment.
Histopathology
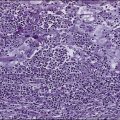
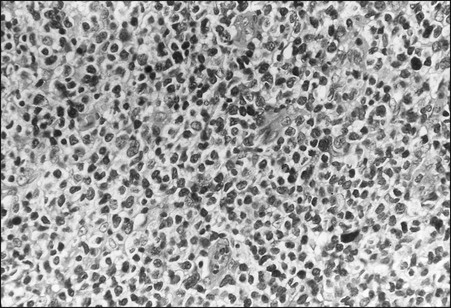
The circulating cells in peripheral blood have markedly polylobated nuclei with homogeneous and condensed chromatin, small or absent nucleoli, and agranular and basophilic cytoplasm—the so-called flower cells that are characteristic of ATL (Fig. 108-3).92 A considerable diversity of morphology among ATL cells has been recognized, however. The morphology of ATL cells in 36 acute cases and 14 chronic cases was investigated. Chronic lymphocytic leukemia–like morphology with round nuclei was more frequent in the chronic type than in the acute type. In contrast, an unusual morphology (lymphoblastic, vacuolated, granular pleomorphic, or large cells) was more frequent in the acute type than in the chronic type. Furthermore, the diversity of cell morphology in ATL was associated with prognostic factors, an aberrant immunophenotype, and a defective HTLV-I genotype.
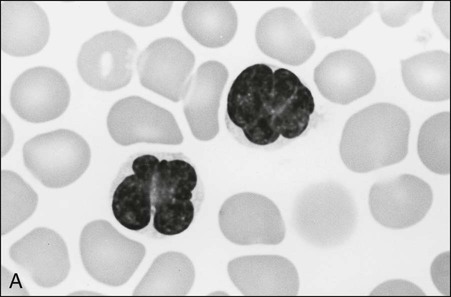
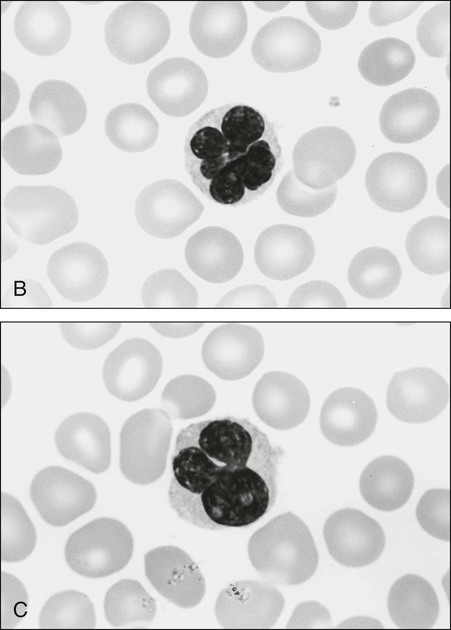
The swollen lymph nodes in patients with ATL show diffuse NHL of various histologic subtypes, including pleomorphic, large cell, mixed cell, or medium-sized cell types.93,94 Figure 108-4 shows the histology of a swollen lymph node obtained from a patient with lymphoma-type ATL. The pleomorphic pattern (i.e., a mixture of various-sized lymphoma cells from small cells to giant cells) and nuclear polymorphism are recognized. Lymph nodes from some patients in the incipient or early neoplastic phase of ATL histologically resemble those that are found in Hodgkin lymphoma.95 Histologic differential diagnosis of ATL according to the World Health Organization (WHO) classification includes peripheral T-cell lymphoma—not otherwise specified (PTCL-NOS), anaplastic large cell lymphoma (ALCL), angioimmunoblastic T-cell lymphoma (AITL), mycoses fungoides/Sézary syndrome (MF/SS), and Hodgkin lymphoma.94,96
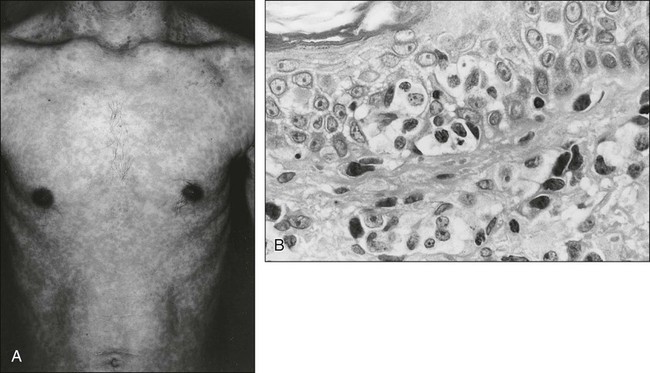
ATL cells frequently involve the skin. Generalized nodular or papulonodular eruptions, as shown in Figure 108-5A, are common; however, tumorous lesions are also recognized in some patients. Erythematous plaque formation and sometimes nodular tumors are other cutaneous manifestations.84–86,94,97 Histologically, diffuse or patchy infiltration of atypical lymphoid cells—usually small or medium in size with pleomorphic nuclear contours in the upper dermis, sometimes with an intraepidermal infiltration—is noted (see Fig. 108-5B). Large nuclear cells with highly irregular or cerebriform features are intermingled in some cases.
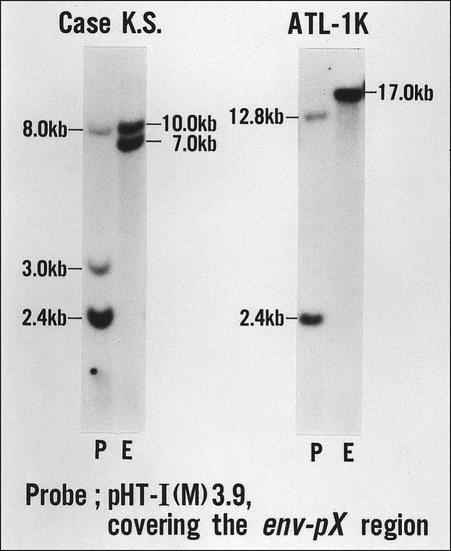
A difficult issue in the diagnosis of ATL is its relationship with other peripheral T-cell malignancies that are not associated with HTLV-I. The clinical diagnosis of ATL is based on the unique combination of its clinical and pathological features. One of the T-cell malignancies likely to be confused with ATL is MF/SS. Because cutaneous involvement is frequent and histologically similar to MF/SS in ATL, the differentiation of smoldering-type ATL from MF/SS is often difficult on the basis of the clinicohistologic manifestations alone. In the differential diagnosis of ATL and other peripheral T-cell malignancies, HTLV-I serology and the molecular detection of the monoclonal integration of HTLV-I proviral DNA are important. Various kinds of serologic assays have been used, including the immunofluorescence assay, the particle agglutination assay, the enzyme-linked immunosorbent assay (ELISA), and the Western blot assay. In general, a particle agglutination assay or ELISA is useful as a screening test and Western blotting is used for confirmation of the presence of serum antibody to HTLV-I. As shown in Figure 108-6, the demonstration of the monoclonal integration of HTLV-I proviral DNA in leukemic/tumor cells by Southern blot analysis can lead to a definitive diagnosis of ATL, although 5% or more ATL cells are required in each sample.
In Western countries, it has been reported that the HTLV-I viral genome was detected in the genomic DNA from patients with MF/SS, and a causal relation between HTLV-I and MF/SS was proposed.98 However, the results were not reconfirmed in subsequent studies by Japanese and international groups.99,100
Several studies have reported the presence of seronegative HTLV-I carriers, and some HTLV-I carriers have been reported to be negative for serum anti-HTLV-I antibodies against viral structural proteins on screening examinations.101 However, subsequent studies with serology/polymerase chain reaction assays suggest that seronegative HTLV-I carriers are extremely rare, although the possibility of their existence remains.102
HTLV-I can infect lymphoid cells of different cell lineages in vitro, but the neoplastic cells in the great majority of ATL cases exhibit the phenotype of mature CD4+ T cells.103 Malignant cells from the peripheral blood or from involved lymph nodes express CD2, CD3, CD4, CD5, the αβ-chains of the TCR, CD25 (IL-2Rα), CD45, CD29, and CCR4 but not CD8, CD7 and CD26.104 It is known that the expression of the CD3/TCR complex is decreased in ATL cells.105 Most acute ATL cells lack CD7, CD26, and HLA-DR with a gradual decline in the positive rate and density for the antigens along with the transformation of chronic ATL cells into acute ATL cells.108–108 Considerable phenotypic heterogeneity has been found in the neoplastic cells of ATL. Although the most common phenotype is CD4+/CD8−, some patients with ATL exhibit a CD4+/CD8+, CD4−/CD8+, or CD4−/CD8− phenotype.109 In addition, some patients with ATL show phenotypic changes throughout the course of their disease. Mature CD4 T cells consist of four helper T cells (Th1, Th17, Th2, and Tfh) and regulatory T cells (Treg). Among them, Th2 and Treg cells are positive for CD25 and CCR4. CCR4, a receptor for thymus and activation regulated chemokine (TARC) and macrophage-derived chemokine (MDC), was expressed in approximately 90% of ATL cases and was associated with poor prognosis and cutaneous involvement.110 Because Treg cells express CD4+ and CD25+ molecules and possess strong immune response suppressive activity, several investigators have analyzed a possible link between ATL cells and Treg cells and found that the forkhead/winged helix transcription factor (FoxP3), a specific marker that is important for the function of Treg cells, was expressed on approximately 60% of ATL cells. These results suggest the origin of ATL cells in most cases to be Treg cells.111,112
One of the remarkable feature of ATL cells (and of most HTLV-I–infected cells) is the expression of IL-2R. Both the α- and β-chains of IL-2R are expressed on the surface of ATL cells. It is postulated that IL-2 and IL-2R are implicated in the pathogenesis of ATL. IL-2R is expected to be an excellent target for monoclonal antibody therapy.113
Clinical Course and Treatment
ATL most often follows the prototypic acute course; however, approximately one fourth of patients show a more indolent course (chronic and smoldering types), with the disease limited predominantly to the peripheral blood and/or skin (and rarely the lung). These patients might experience multiple infections but can remain free of disease progression for many years.79 These indolent diseases frequently progress to full-blown acute- or lymphoma-type ATL, an event that is sometimes called the “crisis.” Some studies have reported that various kinds of infectious episodes might predispose individuals to the transformation from an indolent to an aggressive disease course. Other studies revealed that the presence of genetic alterations such as tumor suppressor abnormalities and aneuploidy in leukemic cells in chronic and smoldering types was associated with a poor prognosis.37,41 At present, however, it is difficult to identify patients at the highest risk of transformation (Fig. 108-7).
Most patients with ATL do not achieve cure with current treatment modalities, even at the early stage of disease. In addition, no treatment has been shown to prevent progression to a more aggressive disease. Therefore, patients with chronic- or smoldering-type ATL have been observed carefully for the development of infectious complications and for signs of progression to acute or lymphoma-type ATL. However, it was recently found that the long-term prognosis of such patients was poorer than expected when compared with that of patients in the early stage of chronic lymphocytic leukemia. In a long-term follow-up study of 78 patients with indolent ATL (favorable chronic- or smoldering-type) with a policy of watchful waiting until disease progression at a single institution, the median survival time was 5.3 years with no plateau in the survival curve. Twelve patients remained alive for more than 10 years, 32 progressed to acute ATL, and 51 died.114 These findings suggest that even “indolent” ATL patients should be carefully observed in clinical practice. Further study is required to establish appropriate management practices for indolent ATL.
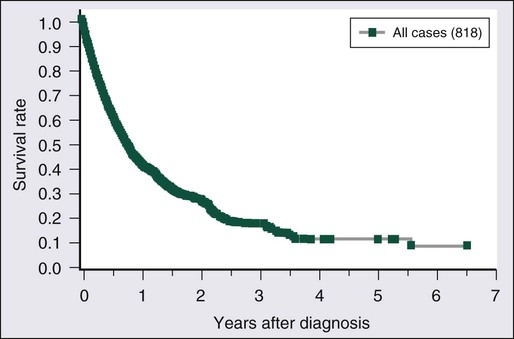
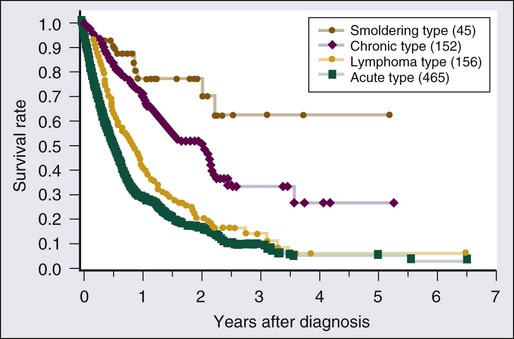
Without treatment, most previously untreated patients with aggressive forms (acute or lymphoma type) of ATL die within weeks or months of diagnosis. The treatment of patients with acute or lymphomatous ATL has not been very successful. Figure 108-8 shows the overall survival (OS) of 818 patients with ATL regardless of disease subtype, and Figure 108-9 presents their survival curves according to the four clinical subtypes in 1980.79 Approximately 85% of the patients received chemotherapy with one of a variety of different regimens. Most of the patients with smoldering-type ATL lived well without chemotherapy for a long period. Approximately two thirds of the chronic-type patients died within about 2.5 years of diagnosis. Patients with lymphoma-type ATL had poor prognoses, with an MST of 10.2 months. The most aggressive type of ATL was the acute type, with an MST of 6.2 months. The projected 4-year survival rates of patients with the lymphoma and acute types were only 5% for each. The clinical subtype clearly determines the prognosis of each patient, suggesting that it can be used as a prognostic indicator and the basis of risk-adopted therapy for patients with ATL.79
The recent International T-Cell Lymphoma Project on 1314 patients revealed that ATL was the fourth most common T-cell lymphoma mainly in Asia (especially Japan), and the prognosis of ATL was the worst among all lymphomas.115 The poor prognosis is associated with the multidrug resistance of ATL cells, high tumor burden with multiple-organ involvement, complicated immunodeficiency and hypercalcemia, and advanced age at onset.116,117 The Lymphoma Study Group of the Japan Clinical Oncology Group (JCOG-LSG) has analyzed prognostic factors for each subtype of ATL.116,118,119 In all patients with ATL, advanced age (≥40 years), poor performance status, high serum LDH level, hypercalcemia, and four or more involved lesions were unfavorable factors. These factors could be used to construct a model for risk grouping. For patients with chronic-type ATL, the major prognostic factors were serum LDH, albumin, and blood urea nitrogen levels. Patients with chronic-type ATL and normal values for the three factors (30% of patients with chronic type disease) had a prognosis as favorable as that of patients with smoldering-type ATL. Thus, patients with the favorable chronic type with normal LDH, albumin, and blood urea nitrogen values need not be treated immediately and can be placed on follow-up without treatment, whereas patients with the unfavorable chronic type who have an abnormal value for at least one of the three factors have an MST of 15 months and are candidates for cytotoxic chemotherapy (Box 108-1).118
Recently, a retrospective review of 807 patients in Japan made a prognostic index for acute- and lymphoma-type ATL based on five prognostic factors: stage, performance status (PS), age, serum albumin level, and soluble IL-2R value.120 In the validation sample, the index was reproducible with MSTs of 3.6, 7.3, and 16.2 months for patients at high, intermediate, and low risk, respectively. The JCOG-LSG conducted a meta-analysis of three consecutive trials exclusively for aggressive ATL (see later discussion).121 OS analysis of a total 276 patients with the acute, lymphoma, or unfavorable chronic types of ATL identified two significant prognostic factors: PS and hypercalcemia. In the validation sample, a proposed prognostic index using the two factors into two strata revealed MSTs of 6.3 and 17.8 months for patients at high and low risk, respectively. In both studies, however, the 5-year OS rate was less than 15% even in the low-risk group, which could not identify the subgroup of patients with sufficient prognosis.
Clinical Trials by the Japan Clinical Oncology Group
Six consecutive chemotherapy trials focusing on ATL have been conducted by JCOG-LSG since 1978.122–130 The first trial, called LSG1 (1978 to 1980), utilized a VEPA regimen, which consisted of vincristine, cyclophosphamide, prednisolone, and doxorubicin. In this study, patients with NHL (including ATL) at an advanced stage were enrolled. The complete remission (CR) rate was lowest (18%) for ATL, intermediate (36%) for peripheral non-ATL T-cell lymphoma (PNTL), and highest (64%) for B-cell lymphoma.122,123 Between 1981 and 1983, the JCOG-LSG conducted a phase III trial that used LSG1-VEPA versus LSG2-VEPA-M (VEPA + methotrexate) against advanced NHL, including ATL.124,125 Patients’ sera were examined for anti–HTLV-I antibody to distinguish ATL from PNTL.131 The CR rate for patients who were given LSG2-VEPA-M for ATL (37%) was higher than that for patients who were given LSG1-VEPA (17%; P = .09). In the LSG1/LSG2 trial, however, the CR rate was significantly lower for ATL than for B-cell lymphoma and PNTL (P < .001). The MST of the 54 patients with ATL treated with LSG1/LSG2 was 6 months, and the estimated 4-year survival rate was only 8%.124,125 These results suggest that CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone)-like chemotherapy of the first generation was not very effective against ATL.
1. VEPA-B: vincristine, cyclophosphamide, prednisolone, doxorubicin, and bleomycin
2. M-FEPA: methotrexate, vindesine, cyclophosphamide, prednisolone, and doxorubicin
3. VEPP-B: vincristine, etoposide, procarbazine, prednisolone, and bleomycin126,127
The CR rate (72%) for the LSG4 protocol among patients with aggressive NHL including ATL was significantly higher than that for the LSG1/LSG2 trial (57%; P < .05). The CR rate for ATL was improved from 28% (LSG1/LSG2) to 43% (LSG4). On the other hand, the CR rate for LSG4 was significantly lower for ATL than for B-cell lymphoma and PNTL (P < .01). The patients with ATL still showed a poor prognosis, with an MST of 8 months and a 4-year survival rate of 12%; however, the continued CR rate was increased to 12% (5 of 43) compared with 4% (2 of 54) in the LSG1/LSG2 trial. A multivariate analysis of the 267 patients with advanced aggressive NHL who were treated with LSG4 demonstrated that the clinical diagnosis of ATL was the most significant unfavorable prognostic factor (relative risk: 3.19; P = .0001) for patients with aggressive NHL in Japan.126,127
The disappointing results with conventional chemotherapies have led to a search for new active agents. Multicenter phase I and II studies of 2′-deoxycoformycin (pentostatin), an irreversible inhibitor of adenosine deaminase) were conducted against ATL in Japan. The phase II study revealed a response rate of 32% (10 of 31) in cases of relapsed or refractory ATL (two with complete response [CR] and eight with partial response [PR]).119 These encouraging results and the proposal of subtype classification of ATL prompted the Japanese investigators to conduct a pentostatin-containing combination phase II trial (JCOG9109; LSG11) as initial chemotherapy exclusively for ATL.128 Sixty-two previously untreated patients with ATL (34 patients with acute, 21 with lymphoma, and 7 with unfavorable chronic subtypes) were enrolled. Vincristine (1 mg/m2 intravenously on days 1 and 8), doxorubicin (40 mg/m2 intravenously on day 1), etoposide (100 mg/m2 intravenously on days 1 through 3), prednisolone (40 mg/m2 orally on days 1 and 2), and pentostatin (5 mg/m2 intravenously on days 8, 15, and 22) were administered every 28 days for 10 cycles unless disease progression or toxic complications occurred. Among the 61 patients who were evaluable for toxicity, 4 patients (7%) died of fatal infections (2 of sepsis and 2 of cytomegalovirus pneumonia). No other fatal nonhematologic toxicities occurred. In the 60 eligible patients, 17 (28%; 95% confidence interval [CI], 19% to 41%) achieved CR, whereas 14 achieved PR (response rate, 52%; 95% CI, 39% to 64%). After a median observation time of 27 months, the MST was 7.4 months and the estimated 2-year survival rate was 17%, findings that were identical to those for the 43 patients with ATL who were treated with the regimen used in the previous LSG4 trial (JCOG8701).126,127 In conclusion, the prognosis of the patients with ATL remained poor even though they were treated with pentostatin-containing combination chemotherapy.
In 1994, the JCOG-LSG initiated a new multiagent combination phase II study (JCOG9303; LSG15). It was a nine-drug regimen comprising vincristine, cyclophosphamide, doxorubicin, prednisolone, ranimustine, vindesine, etoposide, and carboplatin, with the intrathecal administration of methotrexate and prednisolone, for untreated patients with ATL.129 In this study, the elevation of relative dose intensity was attempted with the prophylactic use of granulocyte colony-stimulating factor. In addition, non–cross-resistant agents such as ranimustine and carboplatin were incorporated into the regimens. Ninety-six previously untreated patients with aggressive ATL were enrolled: 58 with acute type, 28 with lymphoma type, and 10 with unfavorable chronic type. Of the 93 eligible patients, 81% responded (75 of 93), with 33 patients (35%) achieving CR and 42 (45%) achieving PR. Patients with lymphoma-type ATL showed a better CR rate (67%, 18 of 27) than patients with acute-type ATL (20%, 11 of 56) and patients with unfavorable chronic-type ATL (40%, 4 of 10). The OS rate of 93 eligible patients at 2 years was 31% (Fig. 108-10). The MST was 13 months, and the median follow-up duration of the 20 surviving patients was 4.2 years. A trend toward better survival for patients with lymphoma type ATL (MST, 20 months) compared with patients with acute-type ATL (MST, 11 months) was recognized (hazard ratio, 1.65). Grade 4 hematologic toxicities of neutropenia and thrombocytopenia were observed in 65% and 53% of the patients, respectively, but grade 4 nonhematologic toxicity was observed in only 1 patient. It was concluded that the regimen used in this LSG15 trial was feasible with mild nonhematologic toxicity and that it improved the clinical outcome of patients with ATL.
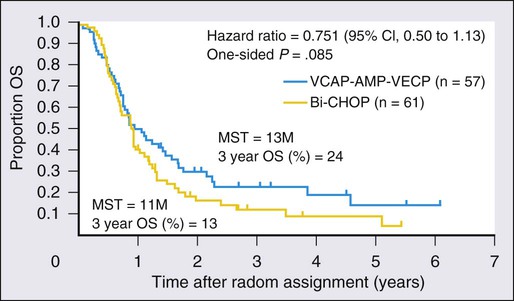
To confirm whether the regimen proposed in the LSG15 study is a new standard for the treatment of aggressive ATL, the JCOG-LSG conducted a phase III study comparing the findings of mLSG15 and CHOP-14 (cyclophosphamide, doxorubicin, vincristine, and prednisolone).130 Previously untreated patients with aggressive ATL were randomly assigned to receive either six courses of the regimen proposed in LSG15 every 4 weeks or eight courses of CHOP-14. Both regimens were supported with granulocyte colony-stimulating factor and intrathecal prophylaxis. The mLSG15 in JCOG9801 was a modified version of the LSG15 in JCOG9303 and consisted of three regimens: VCAP (vincristine, 1 mg/m2 [maximum 2 mg], cyclophosphamide, 350 mg/m2, doxorubicin, 40 mg/m2, prednisolone, 40 mg/m2) on day 1; AMP (doxorubicin 30 mg/m2, ranimustine, 60 mg/m2, prednisolone, 40 mg/m2) on day 8; and VECP (vindesine, 2.4 mg/m2 on day 15; etoposide, 100 mg/m2 on days 15 to 17, carboplatin, 250 mg/m2 on day 15, prednisolone, 40 mg/m2 on days 15 to 17) on days 15 through 17. The next course was to be started on day 29. The modifications in mLSG15 as compared with LSG15 were as follows: (1) the total number of cycles was reduced from seven to six because of progressive cytopenia, especially thrombocytopenia, after repeating the VCAP-AMP-VECP therapy; and (2) cytarabine, 40 mg, was added to methotrexate, 15 mg, and prednisolone, 10 mg, for prophylactic intrathecal administration, at the recovery phases of courses 1, 3, and 5 because of the high frequency of CNS relapse in the JCOG9303 study. One hundred and eighteen patients were randomized. Seventy-two percent of the patients responded, with 23 patients achieving CR (40%) and 18 achieving PR (32%) in the mLSG15 trial. The overall response rate (ORR) was 66%, with 15 patients achieving CR (25%) and 25 achieving PR (41%) in CHOP-14. The CR rate was higher in the mLSG15 arm than the CHOP-14 arm (40% vs. 25%, respectively; P = .020). The median progression-free survival (PFS) time and PFS at 1 year in the former were 7.0 months and 28%, respectively, compared with 5.4 months and 16% in the latter (P = .10). The MST and OS at 3 years were respectively 12.7 months and 24% in the former and 10.9 months and 13% in the latter (P = .085). After the adjustment of patients’ characteristics by Cox regression, the P value for OS became 0.029 because of unbalanced prognostic factors such as bulky lesions and B symptoms. In mLSG15 versus CHOP-14, the percentage of subjects with grade 4 neutropenia, percentage with grade 4 thrombocytopenia, and percentage with grade 3 to 4 infection was 98% versus 83%, 74% versus 17%, and 32% versus 15%, respectively. Three toxic deaths were reported in the former study arm. The longer OS at 3 years and higher CR rate with mLSG15 than CHOP-14 suggest the regimen used in the mLSG15 study arm to be more effective at the expense of greater toxicity, providing a basis for future investigations in the treatment of ATL.130 However, the prognosis is still poor as compared with other hematologic malignancies. Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is now recommended for the treatment of relatively young patients with aggressive ATL (see later discussion). To evaluate the efficacy of allo-HSCT more accurately, especially in view of a comparison with intensive chemotherapy, a prospective multicenter phase II study of the chemotherapeutic regimen used in the mLSG15 study followed by up-front allo-HSCT is ongoing (JCOG0907).
Allogeneic Hematopoietic Stem Cell Transplantation
The results of allo-HSCT for ATL were reported by Japanese investigators.132,133 The authors analyzed 40 patients with the acute and lymphoma types of ATL who were treated with allo-HSCT on Kyusyu island of Japan between 1997 and 2002. All evaluable patients achieved a CR after allo-HSCT, and the median survival time was 9.6 months. The estimated 3-year OS, disease-free survival, and disease relapse rates were 45%, 34%, and 39%, respectively. Among 10 patients with relapsed ATL after allo-HSCT, 5 patients achieved a CR again—3 by the reduction or cessation of immunosuppressive agents, which suggested a graft-versus-ATL effect. These results suggested that allo-HSCT was effective for some patients with aggressive ATL.
Hishizawa and coworkers reported the results of a nationwide retrospective study in 386 patients with ATL who underwent allo-HSCT between 1996 and 2005 with several kinds of conditioning regimens. The 3-year OS for the entire cohort was 33% (95% CI, 28% to 38%).134 Multivariable analysis revealed four recipient factors for a poor prognosis: older age (>50 years), male sex, status other than CR, and use of unrelated cord blood compared with use of HLA-matched related grafts. Treatment-related mortality was higher among patients given cord blood transplants; disease-associated mortality was higher among male recipients or those given transplants not in remission. Among patients who received related transplants, donor HTLV-I seropositivity adversely affected disease-associated mortality. Hishizawa and coworkers concluded that allo-HSCT with currently available graft sources is an effective treatment in selected patients with ATL, although greater effort is warranted to reduce treatment-related mortality. A recent study with the same cohort found that the development of mild-to-moderate acute graft-versus-host-disease (GVHD) confers a lower risk of disease progression and a beneficial influence on survival among allografted patients with ATL.135 More recently, an expanded cohort of the previously mentioned studies was analyzed for a comparison of myeloablative conditioning (MAC) and reduced-intensity conditioning (RIC) for allo-HSCT.136 Although no significant difference in OS between MAC and RIC was observed, there was a trend indicating that RIC contributed to better OS in older patients. Regarding mortality, RIC was significantly associated with ATL-related mortality compared with MAC.
In addition to conventional allo-HSCT, Okamura and associates reported the results of consecutive multicenter feasibility studies of reduced-intensity allo-HSCT against ATL.137 Sixteen patients, all older than 50 years, underwent allo-HSCT from HLA-matched sibling donors after an RIC allo-HSCT consisting of fludarabine (180 mg/m2), busulfan (8 mg/kg), and rabbit antithymocyte globulin (5 mg/kg) in the first trial. The observed regimen-related toxicities and nonhematologic toxicities were acceptable. Disease relapse was the main cause of treatment failure. Three patients who had a relapse subsequently responded to a rapid discontinuation of the immunosuppressive agent and thereafter achieved another remission. After reduced-intensity allo-HSCT, the HTLV-I proviral load became undetectable in eight patients. In their next study, Okamura et al. treated 14 elderly patients with aggressive ATL with reduced intensity stem cell transplantation without antithymocyte globulin and compared the outcomes to the previous study. Engraftment was prompt, and treatment was tolerable. OS and progression-free survival (PFS) at 3 years were 36% and 31%, respectively.138 Compared with the previous study with antithymocyte globulin, complete donor chimerism was significantly delayed. Although the rate of early relapse tended to be decreased, OS or PFS was not improved significantly. Analysis of the combined data from both studies disclosed that grade I to II acute GVHD was the only factor that favorably affected OS and PFS. These results suggested the presence of a graft-versus-ATL effect and the feasibility of a transplant procedure without antithymocyte globulin in elderly ATL patients but could not demonstrate the clinical benefit of incorporating antithymocyte globulin. Reduced intensity allo-HSCT is thus considered to be a feasible treatment for ATL, warranting further investigation.139
Interferon-α and Zidovudine
A small phase II trial in Japan of interferon-α against relapsed/refractory ATL showed a response rate (all PRs) of 33% (8/24), which included 5 of 9 (56%) patients with chronic type ATLs.140 In 1995, Gill and colleagues reported that 11 of 19 patients with acute- or lymphoma-type ATL showed major responses (5 CRs and 6 PRs) to interferon-α and zidovudine.141 The efficacy of this combination was also observed by Hermine’s group; major objective responses were obtained in all 5 patients with ATL (4 with acute type and 1 with smoldering type).142 Although the results of these phase II studies were encouraging, the OS of previously untreated patients with ATL was relatively short (4.8 months) compared with the survival of those in the chemotherapy trials conducted by JCOG-LSG (7 to 8 months) at that time.143 Since then, numerous small phase II studies with interferon-α/zidovudine have shown responses in ATL patients mainly as second-line treatment.144 The therapeutic effect of interferon-α/zidovudine is not due to a direct cytotoxic effect on the leukemic cells.145 Enduring zidovudine treatment of ATL cell lines results in inhibition of the telomerase that reprograms the cells to undergo p53-dependent senescence.146
Recently, the results of a “meta-analysis” on the use of interferon-α/zidovudine for ATL were reported.147 A total of 100 patients received interferon-α and zidovudine as initial treatments. The ORR was 66%, with a 43% CR rate. In this retrospective analysis, the MST was 24 months and the 5-year survival rate was 50% for first-line interferon-α/zidovudine versus 7 months and 20% for 84 patients who received first-line chemotherapy. The median survival time of patients with acute-type ATL treated with first-line interferon-α/zidovudine and chemotherapy was 12 and 9 months, respectively. Patients with lymphoma-type ATL did not benefit from this combination. In addition, first-line interferon-α/zidovudine therapy in chronic- and smoldering-type ATL resulted in a 100% survival rate at a median follow-up time of 5 years. However, because of the retrospective nature of this meta-analysis based on medical records at each hospital, the decision process to select the therapeutic modality for each patient and the possibility of interference with OS by second-line treatment remains unknown. To confirm the promising results of interferon-α/zidovudine in indolent ATL, a prospective multicenter phase III study evaluating the efficacy of interferon-α/zidovudine compared with watchful waiting for indolent ATL is to be initiated in Japan.
Researchers from the United Kingdom reported the results of a retrospective analysis in 73 patients with aggressive ATL (acute-type ATL, 29; lymphoma-type ATL, 44) and suggested that chemotherapy with concurrent/sequential interferon-α/zidovudine as initial treatment might improve survival for both the acute and lymphoma subtypes of ATL compared with chemotherapy alone.148
Response Criteria and Development of New Agents for Treatment of Adult T-Cell Leukemia-Lymphoma
Response Criteria
The complex nature of ATL, often with both leukemic and lymphomatous components, makes assessing responses difficult. A modification of the JCOG response criteria was suggested by an ATL consensus meeting, reflecting criteria for chronic lymphocytic leukemia and NHL that had been published later.151–151 Recently, revised response criteria were proposed for lymphoma. New guidelines were presented that incorporate positron emission tomography (PET), especially for the assessment of CR. It is well known and described in the criteria that several kinds of lymphoma, including PTCLs, are variably fluorodeoxyglucose (FDG) avid.152 Meanwhile, PET or PET combined with computed tomography (CT) is recommended for evaluating responses when the tumorous lesions are FDG-avid at diagnosis.149
Topoisomerase Inhibitors
MST-16 (sobuzoxane), a new orally administered bis(2,6-dioxopiperazine) analog and an inhibitor of topoisomerase II, showed some activity with little cross-resistance toward lymphoid malignancies in vitro and in vivo. MST-16 at 1200 to 2800 mg/day was given orally daily for 7 days, with courses repeated at intervals of 2 to 3 weeks to 24 patients with ATL in a phase I/II study.153 Two CRs and eight PRs occurred in 23 (13 acute, 8 lymphoma, and 2 chronic ATL) evaluable patients. Remission occurred at 7 to 232 (median, 23) days and lasted 43 to 374 (median, 68) days. The major toxic effects were leukopenia (68%), anemia (52%), thrombocytopenia (35%), and gastrointestinal disorders (22%). Although this agent showed promising activity against ATL as a single agent, no further study in combination with other agents has been reported.
Irinotecan Hydrochloride
Irinotecan hydrochloride (CPT-11) is a semisynthetic camptothecin with inhibitory activity against topoisomerase I. Multicenter phase II studies of CPT-11 have been conducted against relapsed or refractory NHL, including ATL, in Japan.154,155 In this study, 9 patients achieved CR, and 17 patients achieved PR (response rate, 38%: 26 of 69) by using a weekly intravenous administration of 40 mg/m2/day for 3 consecutive days. Within this group, 5 of 13 patients with ATL (38%) responded to CPT-11 (1 CR and 4 PR).155 The major toxic effects of CPT-11 were leukopenia, diarrhea, and nausea and/or vomiting. Subsequently, to develop a new effective chemotherapy regimen against NHL and ATL, two phase I/II studies of CPT-11 in combination with carboplatin or etoposide were conducted for relapsed or refractory NHL.156,157 In both studies, however, dose escalation was halted because of hematologic toxicity (in combination with carboplatin) and hepatotoxicity (in combination with etoposide).
Pralatrexate
Pralatrexate (Folotyn) is a novel anti-folate agent with potent preclinical and clinical activity in T-cell malignancies including ATL.158,159 This agent has improved membrane transport and polyglutamylation in tumor cells and high affinity for the reduced folate carrier highly expressed in malignant cells.
Purine Analogs
Several purine analogs have been evaluated for ATL. Among them, pentostatin has been most extensively evaluated as a single agent and in combination with other agents as described earlier.119,128
Other purine analogs clinically studied for ATL are fludarabine and cladribine. Fludarabine is among the standard treatments for B-cell chronic lymphocytic leukemia and other lymphoid malignancies. In a phase I study of fludarabine in Japan, 5 ATL patients and 10 B-cell chronic lymphocytic leukemia patients with refractory or relapsed disease were enrolled.160 Six grade 3 nonhematologic toxicities were only observed in the ATL patients. A PR was achieved only in 1 of the 5 ATL patients, and the duration was short. Cladribine is among the standard treatments for hairy cell leukemia and other lymphoid malignancies. A phase II study of cladribine for relapsed/refractory aggressive ATL in 15 patients revealed only one PR.161 BCX1777 (forodesine), a novel purine nucleoside phosphorylase inhibitor, induces apoptosis, mainly in T cells. In a phase I study, BCX1777 was well tolerated and showed preliminary evidence of activity in relapsed or refractory peripheral T/natural killer-cell malignancies, warranting further investigation.162
Histone Deacetylase Inhibitor
Gene expression governed by epigenetic changes is crucial to the pathogenesis of cancer. Histone deacetylases (HDACs) are enzymes involved in the remodeling of chromatin and play a key role in the epigenetic regulation of gene expression. HDAC inhibitors (HDACis) induce the hyperacetylation of nonhistone proteins as well as nucleosomal histones, resulting in the expression of repressed genes involved in growth arrest, terminal differentiation, and/or apoptosis among cancer cells. Several classes of HDACis have been found to have potent anticancer effects in preclinical studies. HDACis such as vorinostat (suberoylanilide hydroxamic acid), romidepsin (depsipeptide), and panobinostat (LBH589) have also shown promise in preclinical and/or clinical studies against T-cell malignancies, including ATL.163 Vorinostat and romidepsin have been approved for cutaneous T-cell lymphoma (CTCL) by the U.S. Food and Drug Administration. LBH589 has a significant anti-ATL effect in vitro and in mice.164 However, a phase II study for CTCL and indolent ATL was terminated early because of severe infections associated with the shrinkage of skin tumors and formation of ulcers in patients with ATL. Further study is required to evaluate the efficacy of HDACis for PTCL/CTCL, including ATL.
Arsenic Trioxide
Arsenic trioxide (As2O3) is an effective agent for acute promyelocytic leukemia. Ishitsuka and associates165 examined the suppressing effect of As2O3 on the growth of HTLV-I–infected T-cell lines and fresh ATL cells in vitro. Proliferation of four HTLV-I–infected T-cell lines was reduced significantly by As2O3. Ishitsuka’s group claimed that As2O3 has therapeutic potential for the treatment of ATL. Bazarbachi and colleagues166 tested the effects of the combination of As2O3 and interferon-α on cell proliferation, cell cycle phase distribution, and apoptosis in ATL-derived T-cell lines and found a synergistic effect. Recently, a phase II study of the combination of arsenic trioxide, interferon-α, and zidovudine for chronic ATL revealed an impressive response rate and moderate toxicity.167 Although the results appeared promising, the addition of arsenic trioxide to interferon-α/zidovudine, which might be sufficient for the treatment of chronic ATL as described earlier, caused more toxicity and should be evaluated with caution.
Monoclonal Antibodies and Toxin Fusion Proteins
Because most ATL cells express the α-chain of IL-2R (CD25), Waldmann and colleagues treated ATL patients with a MoAb to CD25.113 Anti-Tac (anti-CD25) is a murine monoclonal IgG2a antibody that does not fix human complement, nor does it mediate antibody-dependent cell-mediated cytotoxicity. Anti-Tac has been shown to prevent the growth of certain cell lines in vitro, however, even in the absence of complement, by blocking IL-2 from gaining access to its receptor. Six of 19 patients (32%) who were treated with anti-Tac showed a PR (4 patients) or CR (2 patients) lasting from 9 weeks to more than 3 years.168 One significant impediment to this approach is that a quantity of soluble IL-2R is shed by the tumor cells into the circulation. The soluble IL-2R can bind to anti-Tac and inhibit binding to the tumor cell. Waldmann’s group169 have developed a stable conjugate of anti-Tac with yttrium-90. Among the 16 patients who received 5- to 15-mCi doses, 9 (56%) showed objective responses (2 CRs and 7 PRs). The response lasted longer than that obtained with unconjugated anti-Tac antibody.
LMB-2, composed of the anti-CD25 murine MoAb fused to the truncated form of Pseudomonas toxin, was cytotoxic to CD25-expressing cells including ATL cells in vitro and in mice. Phase I/II trials of this agent showed some effect against hairy cell leukemia, CTCL, and ATL.170 Six of 35 patients in the phase I study had significant levels of neutralizing antibodies after the first cycle. This drug deserves further clinical trials, including its use in combination with cytotoxic agents.
Denileukin diftitox (DAB[389]-IL-2), an IL-2/diphtheria toxin fusion protein targeting IL-2 receptor–expressing malignant T lymphocytes, shows efficacy as a single agent against CTCL and PTCL.171 Also, the combination of this agent with a multiple-agent chemotherapy regimen such as CHOP was promising for PTCL.172 ATL cells frequently and highly express CD25 as just described, and several ATL cases successfully treated with this agent have been reported.173
Yoshie and coworkers reported the frequent expression of CCR4 in ATL and HTLV-I transformed T cells.174 Ishida and coworkers conducted an immunostaining analysis for anti- CCR4 expression in ATL cells obtained from 103 patients with ATL and compared the clinical parameters and OS of the CCR4-positive and CCR4-negative patients. Ninety-one (88%) of the 103 cases were positive for CCR4 staining. Multivariate analysis revealed that CCR4 expression was an independent prognostic factor (P < .05).110 CCR4 is also expressed in about 30% of PTCL and transformed CTCL and 10% to 20% of mycoses fungoides. A novel humanized CCR4 MoAb, mogamulizumab (KW-0761), has been developed, the Fc region of which is defucosylated to enhance antibody-dependent cellular cytotoxicity by increasing its affinity for the Fc receptor on effector cells. A phase I study of dose escalation with four weekly intravenous infusions of mogamulizumab in 16 patients with relapsed CCR4-positive T-cell malignancy (13 ATL and 3 PTCL) revealed that 1 patient, at the maximum dose (1.0 mg/kg), developed grade 3 dose-limiting toxic effects, namely, rashes and febrile neutropenia, and grade 4 neutropenia.175 Other treatment-related grade 3/4 toxic effects were lymphopenia (n = 10), neutropenia (n = 3), leukopenia (n = 2), herpes zoster (n = 1), and acute infusion reaction/cytokine release syndrome (n = 1). Neither the frequency nor the severity of these effects increased with dose escalation or the plasma concentration of the agent. The maximum tolerated dose was not reached. No patients had detectable levels of anti-KW-0761 antibody. The plasma maximum and trough, and the area under the curve for 0 to 7 days of mogamulizumab, tended to increase dependent on dose and frequency. Five patients (31%; 95% CI, 11% to 59%) achieved objective responses: 2 with CRs (0.1; 1.0 mg/kg) and 3 with PRs (0.01; 2 at 1.0 mg/kg). Three of 13 patients with ATL (31%) achieved a response (2 CRs and 1 PR). Responses in each lesion were diverse, that is, good in peripheral blood (6 CRs and 1 PR per 7 evaluable cases), intermediate in skin (3 CRs and 1 PR per 8 evaluable cases), and poor in lymph node (1 CR and 2 PRs per 11 evaluable cases). Mogamulizumab was well tolerated at all the doses tested, demonstrating potential efficacy against relapsed CCR4-positive ATL or PTCL.
A subsequent phase II study of the agent given once per week for 8 weeks at the 1.0 mg/kg to patients with relapsed, aggressive CCR4-positive ATL was conducted.176 Objective responses were noted in 13 of 26 evaluable patients, including eight CRs, with an overall response rate of 50% (95% CI, 30% to 70%). Median PFS and OS were 5.2 and 13.7 months, respectively. The most common adverse events were infusion reactions (89%) and rashes (63%), which were manageable. Based on the results, this agent was approved by the National Health Insurance in Japan. Further investigation of mogamulizumab for treatment of ATL and other T-cell neoplasms is ongoing, including a phase II trial of the VCAP-AMP-VECP regimen combined with mogamulizumab for untreated aggressive ATL.
Some PTCLs, including ATL, as well as most Hodgkin lymphoma and anaplastic large-cell lymphomas, express CD30. To enhance the antitumor activity of CD30-directed therapy, the antitubulin agent monomethyl auristatin E was attached to a CD30-specific MoAb by an enzyme-cleavable linker, producing the antibody-drug conjugate brentuximab vedotin (SGN-35). Brentuximab vedotin induced durable objective responses with acceptable levels of toxicity for most patients with relapsed or refractory CD30-positive lymphomas in several phase I and II studies.177 The promising results warrant further studies of this agent for other CD30-positive PTCLs, including ATL.
CD52 antigen is present on normal and pathological B and T cells. In PTCL, however, CD52 expression varies among patients, with an overall expression rate lower than 50% in one study but not in another.178 ATL cells frequently express CD52 as compared with other PTCLs. The humanized anti-CD52 MoAb alemtuzumab is active against B-cell chronic lymphocytic leukemia and PTCL as a single agent. The combination of alemtuzumab with a CHOP regimen as a first-line treatment for 24 patients with PTCL showed promising results with a CR in 17 (71%) patients and a partial remission in 1, with an overall median duration of response of 11 months; it was associated with mostly manageable infections but included cytomegalovirus reactivation.179 ATL cells express CD52, the target of alemtuzumab, which was active in a preclinical model of ATL and toxic to p53-deficient cells, and several ATL cases successfully treated with this agent have been reported.182–182
Siplizumab is a humanized MoAb targeting CD2 and showed efficacy in a murine ATL model. A phase I dose-escalating study of this agent in 22 patients with several kinds of T cell/natural killer [NK]-cell malignancy revealed six responses (2 CRs in large granular lymphocyte (LGL) leukemia, 3 PRs in ATL, and 1 PR in CTCL). However, 4 patients developed Epstein-Barr virus–associated lymphoproliferative disease.183 The broad specificity of this agent may eliminate both CD4− and CD8+ T cells as well as NK cells without affecting B cells and predispose individuals to the development of Epstein-Barr virus–associated lymphoproliferative syndrome.
Treatment of Complications
Another major obstacle to the successful treatment of ATL is T-cell immunodeficiency. Patients with ATL often have infectious complications at diagnosis. As is shown in Table 108-2, 26% had infections at initial presentation, more than half of which were fungal, protozoal, and viral infections.79 This finding could be due to a profound T-cell immunodeficiency. Other frequently encountered opportunistic infections include Pneumocystis jiroveci infection, tuberculosis, cytomegalovirus infection, and adenovirus infection.
Subclinical immunodeficiency was also evident among healthy carriers of HTLV-I. Strongyloidiasis is frequently associated with smoldering-type ATL and an intermediate state between the healthy carrier state and smoldering-type ATL. With the use of sensitive polymerase chain reaction assays, high incidences of cytomegalovirus, human herpesvirus–6, and Epstein-Barr virus reactivation (51%, 52%, and 22%, respectively) were found in a total of 468 plasma samples from 34 patients receiving cytotoxic chemotherapy for ATL.187 There were differences in the viral reactivation patterns among the three viruses. A high cytomegalovirus load was indicative of subsequent exacerbation of cytomegalovirus reactivation and development of a serious clinical course.
Patients with ATL require some supportive or preventive treatment for fungal, protozoal, and viral infections. A low dose of sulfamethoxazole-trimethoprim and an oral antifungal agent are recommended for use, together with cytotoxic chemotherapy. An anti-Strongyloides agent, such as ivermectin or albendazole, should be considered to avoid systemic infections in patients with a history of exposure to the parasite in the tropics.149
There are case reports of B-cell NHL associated with Epstein-Barr virus and of Kaposi sarcoma in patients with ATL.188,189 The profound immunodeficient state in patients with ATL might allow the emergence of such opportunistic tumors.
Prevention
Two steps should be considered for the prevention of HTLV-I–associated ATL. The first is the prevention of HTLV-I infections. This has been achieved in some endemic areas in Japan by screening for HTLV-I among blood donors and recommending mothers who are carriers to refrain from breast-feeding. For the several decades before initiation of the interventions the prevalence of HTLV-I had declined drastically in endemic areas in Japan, probably because of birth cohort effects.54,55 The elimination of HTLV-I in endemic areas is now considered possible because of the natural decrease in the prevalence as well as intervention of transmission through blood transfusion and breast-feeding.59 The second step is the prevention of ATL among HTLV-I carriers. This goal has not been achieved, in part because the disease develops in only approximately 5% of HTLV-I carriers in their lifetime, although several risk factors have been identified.78 Also, no agent has been found to be effective in preventing the development of ATL among HTLV-I carriers.