[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Vascular and Interstitial Biology of Tumors
Rakesh K. Jain, Vikash P. Chauhan and Dan G. Duda
• A solid tumor is an organ composed of neoplastic cells and stromal cells nourished by a vasculature made of endothelial cells—all embedded in an extracellular matrix. The interactions among these cells and between these cells, their surrounding matrix, and their local microenvironment control the expression of various genes. The products encoded by these genes, in turn, control the pathophysiological characteristics of the tumor. Tumor pathophysiology governs not only tumor growth, invasion, and metastasis but also the response to various therapies.
• Tumor vasculature is made of host vessels co-opted by cancer cells and by new vessels formed by the processes of vasculogenesis and angiogenesis. A constellation of positive and negative regulators of angiogenesis governs the process of neovascularization.
• Tumor vessels are abnormal in terms of their organization, structure, and function. These abnormalities contribute to heterogeneity in vascular permeability, blood flow, and the microenvironment.
• Tumor interstitial matrix is formed of proteins secreted by stromal and cancer cells and by those leaked from the nascent blood vessels.
• Tumor interstitium is heterogeneous, with some regions fairly permeable and others difficult to penetrate. Modification of collagen and hyaluronan in the matrix can improve penetration of large-molecular-weight therapeutics.
• Solid components of tumors—cancer cells, stromal cells, and matrix molecules—mechanically compress blood and lymphatic vessels, resulting in reduced perfusion that limits oxygen and drug supply. Depleting these constituents decompresses blood vessels to enhance perfusion and drug delivery.
• Interstitial hypertension is a hallmark of solid tumors and results from vessel leakiness, lack of functional lymphatics, and compression of vessels. This elevated fluid pressure contributes to blood flow heterogeneity and directly hinders the penetration of large-molecular-weight therapeutics. Alleviating interstitial hypertension improves oxygen and drug delivery to tumors.
• Judicious application of angiogenic therapy can normalize the tumor vessels and make them more efficient for delivery of oxygen (a known radiosensitizer) and drugs. Antiangiogenic agents can prune tumor vessels, induce cancer cell apoptosis, and lower interstitial hypertension in tumors.
• Thus far, eight antiangiogenic agents have been approved for patients with certain types of cancer. Based on these successes, antiangiogenic therapy is expected to make a difference in many other tumor types. Two main hurdles to further development of antiangiogenic agents are the better understanding of the mechanisms of action of these agents and the development of biomarkers to select patients for these drugs and to predict and monitor their effects.
Introduction
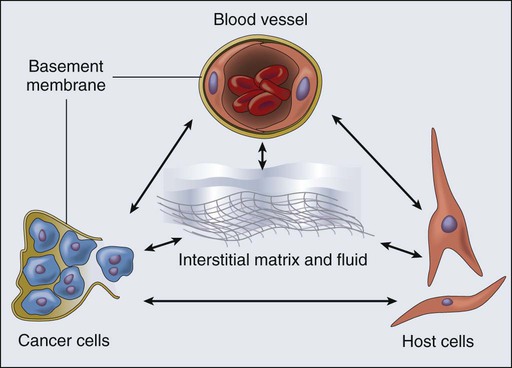
A solid tumor is an organ composed of neoplastic cells and host stromal cells nourished by a vasculature made of endothelial cells—all embedded in an extracellular matrix (Fig. 8-1). The interactions among these cells and between these cells, their surrounding matrix, and their local microenvironment control the expression of various genes. The products encoded by these genes, in turn, control the pathophysiological characteristics of the tumor. Tumor pathophysiology governs not only tumor growth, invasion, and metastasis but also the response to various therapies. In this chapter we will discuss various pathophysiological parameters that characterize the vascular and extravascular compartments of a tumor and the mechanisms governing the formation and function of these compartments.
Vascular Compartment
New Vessel Formation
It has been known for nearly a century that the vascular system is associated with tumor growth in animals and humans.1 Powerful insights into the neovascularization of transplanted tumors using transparent window techniques were developed in the 1940s.2–5 The possibility that tumors produce an “angiogenic” substance was suggested in 1968.6 The hypothesis that blocking angiogenesis should block tumor growth and metastasis was proposed shortly thereafter in 1971.7 The concept that a tissue acquires angiogenic capacity during neoplastic transformation—and, by extension, that antiangiogenesis could be used to prevent cancer—was put forward in 1978.8 The first antiangiogenic agent approved for patients with cancer was bevacizumab, an antibody specific to vascular endothelial growth factor (VEGF), on the basis of the increased survival seen in patients with metastatic colorectal cancer with the combination of bevacizumab and standard chemotherapy in a pivotal randomized placebo-controlled phase III trial.9 At present, various antiangiogenesis and proangiogenesis strategies are being evaluated clinically to prevent or treat a large number of diseases, including cancer.12–12 Both normal and pathological angiogenic processes are governed by the net balance between proangiogenic and antiangiogenic factors.13,14 This balance is spatially and temporally regulated under physiological conditions, so that the “angiogenic switch” is “on” when needed (e.g., during embryonic development, wound healing, and formation of the corpus luteum) and “off” at other times. During neoplastic transformation and tumor progression, this regulation is deranged, and blood vessels form ectopically to support a growing tumor mass.
Cellular Mechanisms
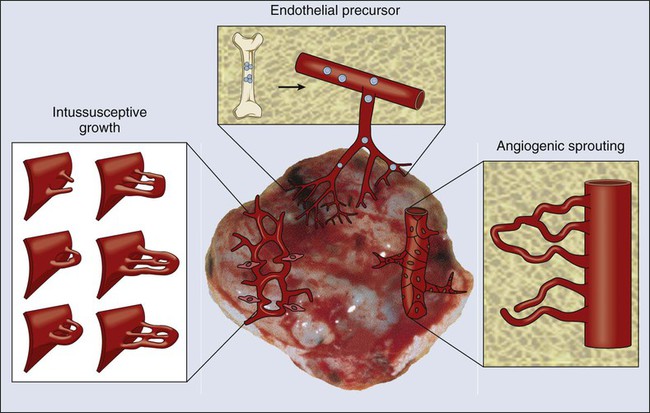
At least four cellular mechanisms are involved in the vascularization of tumors: co-option, intussusception, sprouting (angiogenesis), and vasculogenesis (Fig. 8-2).11 Tumor cells can co-opt and grow around existing vessels to form “perivascular” cuffs. However, as stated earlier, these cuffs cannot grow beyond the diffusion limit of critical nutrients and may actually cause the collapse of the vessels because of growth pressure (referred to as “solid stress”). Alternatively, an existing vessel may enlarge in response to the growth factors released by tumors, and an interstitial tissue column may grow in the enlarged lumen and partition the lumen to form an expanded vascular network. This mode of intussusceptive microvascular growth has been observed during tumor growth, wound healing, and gene therapy.15–18 “Sprouting” angiogenesis is perhaps the most widely studied mechanism of vessel formation. During sprouting angiogenesis, the existing vessels become leaky in response to growth factors released by normal cells or cancer cells; the basement membrane and the interstitial matrix dissolve; pericytes dissociate from the vessel; endothelial cells (ECs) migrate and proliferate to form an array/sprout; a lumen is formed in the sprout (a process referred to as canalization); branches and loops are formed by confluence and anastomosis of sprouts to permit blood flow; and finally, these immature vessels are invested in basement membrane and pericytes. During physiological angiogenesis, these vessels differentiate into mature arterioles, capillaries, and venules, whereas in tumors they remain largely immature.5,11,12,19 During embryonic development, a primitive vascular plexus is formed from endothelial precursor cells (EPCs, also known as angioblasts) by a process referred to as vasculogenesis. In adults, EPCs—mobilized from bone marrow niches into the peripheral blood circulation—also can contribute to neovascularization (a process referred to as “postnatal” vasculogenesis) in tumors and other tissues.22–22 In addition, recent reports demonstrated that a fraction of endothelial cells in tumor vessels may be derived via transdifferentiation from cancer cells,23 or from cancer stemlike cells (e.g., in glioblastomas).11,24–26 In this larger context, the term “vasculogenesis” might be invoked to describe neovascularization in tumors from tumor stem cells.27 Tumor vasculogenesis also may occur from EPCs of non–bone marrow origin that are recruited from adjacent tissues and/or circulation.28,29 The current challenge is to discern the relative contribution of each of the these mechanisms of neovascularization during tumor growth and during and after treatment of tumors.27,30
Molecular Mechanisms
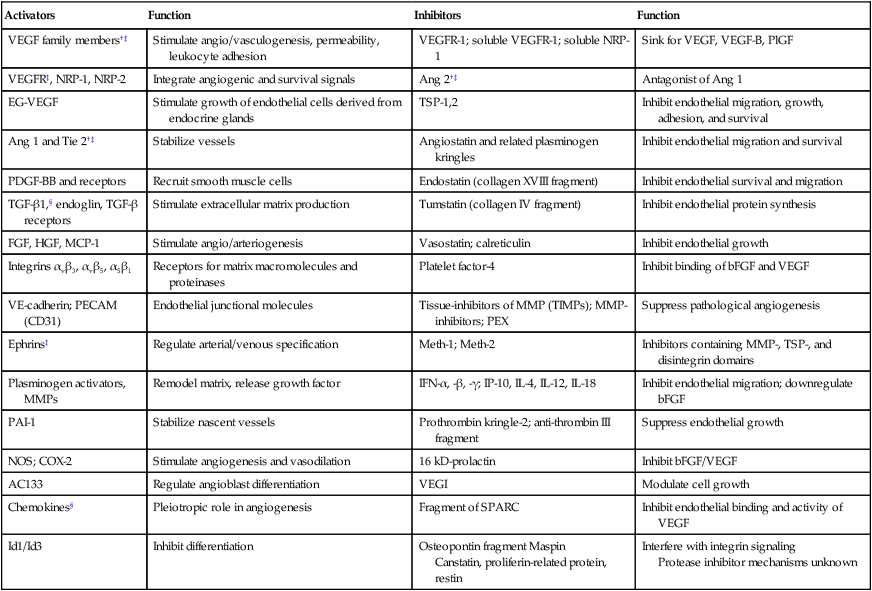
Various proangiogenic and antiangiogenic molecules that orchestrate different steps in vessel formation, along with their functions, are listed in Table 8-1. VEGF currently is considered the most critical proangiogenic molecule. Originally discovered in 1983 as the vascular permeability factor and cloned in 1989, VEGF increases vascular permeability, promotes migration and proliferation of ECs, serves as an EC survival factor, can mobilize EPC populations from the bone marrow, and is known to upregulate leukocyte adhesion molecules on ECs.15,22,31–33 During tumor progression, or with treatment, the number of distinct angiogenic molecules produced by a tumor can increase.36–36 Thus after VEGF signaling is blocked, a tumor might rely on other, alternative angiogenic molecules (e.g., basic fibroblast growth factor [bFGF], stromal-derived factor 1α [SDF1α], placental growth factor [PlGF], or interleukin-8 [IL-8]).37 Other positive regulators of angiogenesis include the angiopoietins that are involved in stabilizing vessels and controlling vascular permeability; various proteases involved in dissolving/remodeling matrix and releasing growth factors; and recently discovered organ-specific angiogenic stimulators (e.g., endocrine gland–derived VEGF).19,38,39 Angiogenesis inhibitors include endogenous soluble receptors of various proangiogenic ligands (e.g., sVEGFR1/sFLT1) and molecules that downregulate the expression of stimulators (e.g., interferons) or that interfere with the release of the stimulators or binding with their receptors (e.g., platelet factor 4). Thrombospondins are among the first and most well-characterized endogenous inhibitors that interfere with the growth, adhesion, migration, and survival of ECs.13 Other endogenous inhibitors include fragments of various plasma or matrix proteins (e.g., angiostatin, a fragment of plasminogen; endostatin, a fragment of collagen XVIII; and tumstatin, a fragment of collagen IV).42–42 Neither the mechanisms of action of the matrix-derived inhibitors nor their physiological role are well understood.43 The generation of proangiogenic and antiangiogenic molecules can be triggered by metabolic stress (e.g., low po2, low pH, or hypoglycemia), mechanical stress (e.g., shear stress or solid stress), immune/inflammatory cells that have infiltrated the tissue, and genetic mutations (e.g., activation of oncogenes or deletion of suppressor genes that control the production of angiogenesis regulators).13,14,44–47 These molecules can emanate from cancer cells, endothelial cells, stromal cells, blood, and extracellular matrix (Fig. 8-3).48–51 Because the normal host cells differ among organs, the detailed mechanisms of angiogenesis might depend on the specific host-tumor interactions operating within a given tissue.52–59 Furthermore, because the tumor microenvironment is likely to change during tumor growth, regression, and relapse, profiles of proangiogenic and antiangiogenic molecules are likely to change with time and space.60,61 The challenge currently is to develop a unified conceptual framework to describe the temporal and spatial profiles of this increasingly diverse array of angiogenesis regulators with the aim of developing effective therapeutic strategies.37,62–64
Table 8-1
Angiogenesis Activators and Inhibitors*
| Activators | Function | Inhibitors | Function |
| VEGF family members†‡ | Stimulate angio/vasculogenesis, permeability, leukocyte adhesion | VEGFR-1; soluble VEGFR-1; soluble NRP-1 | Sink for VEGF, VEGF-B, PlGF |
| VEGFR‡, NRP-1, NRP-2 | Integrate angiogenic and survival signals | Ang 2†‡ | Antagonist of Ang 1 |
| EG-VEGF | Stimulate growth of endothelial cells derived from endocrine glands | TSP-1,2 | Inhibit endothelial migration, growth, adhesion, and survival |
| Ang 1 and Tie 2†‡ | Stabilize vessels | Angiostatin and related plasminogen kringles | Inhibit endothelial migration and survival |
| PDGF-BB and receptors | Recruit smooth muscle cells | Endostatin (collagen XVIII fragment) | Inhibit endothelial survival and migration |
| TGF-β1,§ endoglin, TGF-β receptors | Stimulate extracellular matrix production | Tumstatin (collagen IV fragment) | Inhibit endothelial protein synthesis |
| FGF, HGF, MCP-1 | Stimulate angio/arteriogenesis | Vasostatin; calreticulin | Inhibit endothelial growth |
| Integrins αvβ3, αvβ5, α5β1 | Receptors for matrix macromolecules and proteinases | Platelet factor-4 | Inhibit binding of bFGF and VEGF |
| VE-cadherin; PECAM (CD31) | Endothelial junctional molecules | Tissue-inhibitors of MMP (TIMPs); MMP-inhibitors; PEX | Suppress pathological angiogenesis |
| Ephrins‡ | Regulate arterial/venous specification | Meth-1; Meth-2 | Inhibitors containing MMP-, TSP-, and disintegrin domains |
| Plasminogen activators, MMPs | Remodel matrix, release growth factor | IFN-α, -β, -γ; IP-10, IL-4, IL-12, IL-18 | Inhibit endothelial migration; downregulate bFGF |
| PAI-1 | Stabilize nascent vessels | Prothrombin kringle-2; anti-thrombin III fragment | Suppress endothelial growth |
| NOS; COX-2 | Stimulate angiogenesis and vasodilation | 16 kD-prolactin | Inhibit bFGF/VEGF |
| AC133 | Regulate angioblast differentiation | VEGI | Modulate cell growth |
| Chemokines§ | Pleiotropic role in angiogenesis | Fragment of SPARC | Inhibit endothelial binding and activity of VEGF |
| Id1/Id3 | Inhibit differentiation | Osteopontin fragment Maspin Canstatin, proliferin-related protein, restin |
Interfere with integrin signaling Protease inhibitor mechanisms unknown |
*Selected list updated from reference 11; for complete function and references, see supplementary information (http://steele.mgh.harvard.edu).
Vascular Architecture
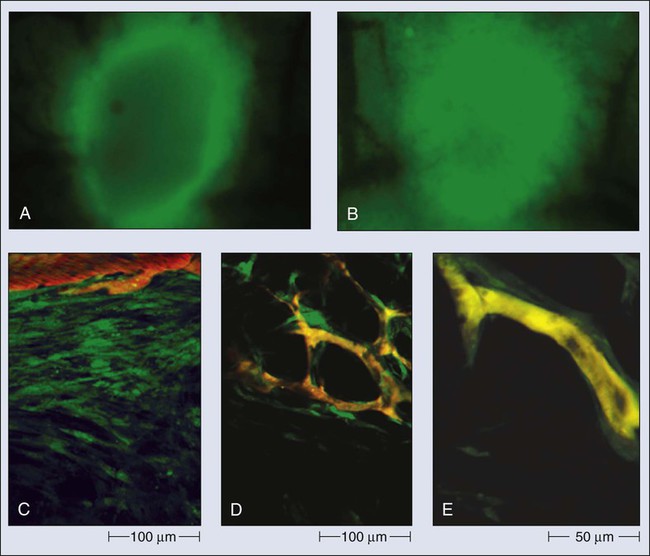
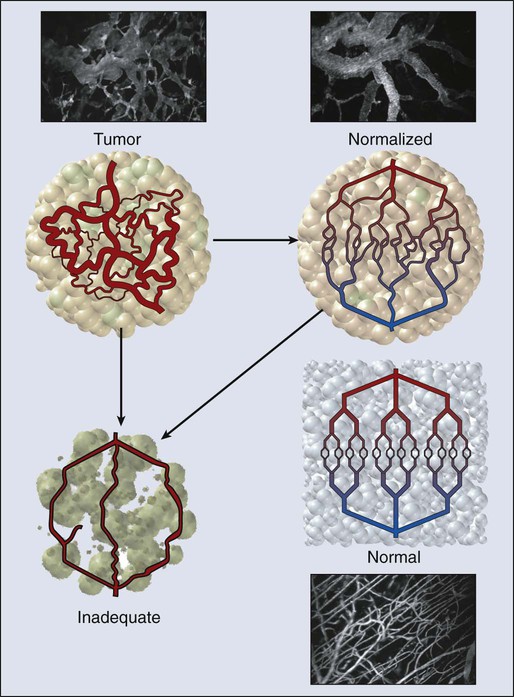
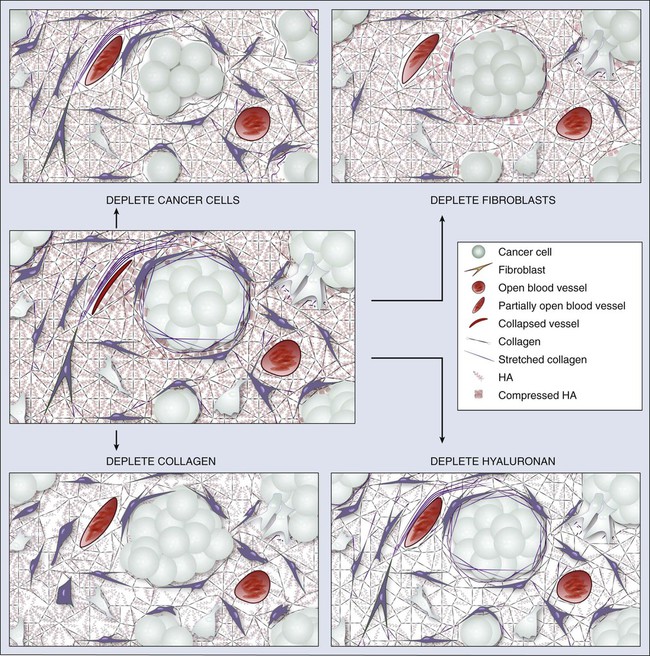
In normal tissue, blood flows from an artery to arterioles to capillaries to venules to a vein. Although the tumor vasculature originates from these host vessels and the mechanisms of angiogenesis are similar, its organization may differ dramatically, depending on the tumor type, its location, and whether it is growing, regressing, or relapsing.15,65–68 In general, tumor vessels are dilated, saccular, tortuous, and chaotic in their patterns of interconnection.69 For example, whereas normal vasculature is characterized by dichotomous branching, tumor vasculature has many trifurcations and branches with uneven diameters.70,71 The fractal dimensions and minimum path lengths of tumor vasculature are different from those of normal host vasculature.67–67 The molecular mechanisms of this abnormal vascular architecture are not understood, but it seems reasonable to hypothesize that the imbalance of VEGF and angiopoietins is a key contributor.19,72 In mice, “normalization” of the tumor vasculature observed during therapies that reduce VEGF (e.g., hormone withdrawal from a hormone-dependent tumor), interfere with VEGF signaling (e.g., treatment with anti-VEGF or anti-VEGFR2 antibody; Fig. 8-4), or mimic an antiangiogenic cocktail (e.g., trastuzumab [Herceptin] treatment of a HER2-overexpressing tumor) is in concert with this molecular hypothesis.61,63,73–76 Mechanical solid stress (i.e., physical forces exerted by solid tissue components) generated by proliferating cancer and stromal cells and their excessive interstitial matrix production also can lead to the partially compressed or totally collapsed vessels often found in tumors (Fig. 8-5).64,77,78 The decompression of blood vessels observed after induction of apoptosis in perivascular cells or stromal cells, or enzymatic degradation of matrix hyaluronan, supports this mechanical hypothesis.78–81 Perhaps the combination of both molecular and mechanical factors renders the tumor vasculature abnormal, and thus both types of factors must be taken into account when designing novel strategies for cancer treatment.
Blood Flow and Microcirculation
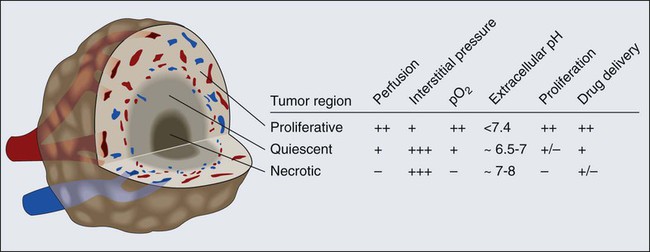
Blood flow in a vascular network, whether normal or abnormal, is governed by the arteriovenous pressure difference and flow resistance. Flow resistance is a function of the vascular architecture (referred to as geometric resistance) and of the blood viscosity (rheology, referred to as viscous resistance).69 Abnormalities in both vasculature and viscosity increase the resistance to blood flow in tumors.71,82–84 As a result, overall perfusion rates (i.e., blood flow rate per unit volume) in tumors are lower than in many normal tissues.85,86 Both macroscopically and microscopically, tumor blood flow is temporally and spatially chaotic. Macroscopically, four spatial regions can be recognized in a tumor (Fig. 8-6):
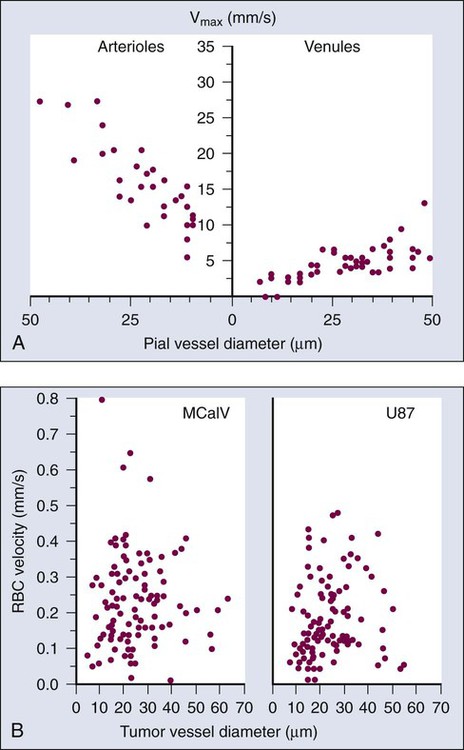
At the microscopic level, in normal tissues, erythrocyte velocity is dependent on vessel diameter, but there is no such dependence in most tumors.54,59,89 Furthermore, the average erythrocyte velocity can be an order of magnitude lower in some tumors compared with that of normal host tissue (Fig. 8-7).59 In a given vessel within a tumor, blood flow fluctuates with time and can reverse its direction.87,89,90 In addition to the elevated geometric and viscous (rheologic) resistance, other molecular and mechanical factors contribute to this spatial and temporal heterogeneity. These factors include imbalance between proangiogenic and antiangiogenic molecules, “solid stress” generated by proliferating tumor cells and matrix production, vascular remodeling by intussusception, and coupling between luminal and interstitial fluid pressure via hyperpermeability of tumor vessels.6,16,64,65,77–79,91–93 This heterogeneity contributes to both acute and chronic hypoxia in tumors—a major cause of resistance to radiation and other therapies. Considerable effort has gone into increasing tumor blood flow for improving radiation therapy, or decreasing tumor perfusion in the case of hyperthermia. Increased perfusion also correlates with a positive response to radio/chemotherapy and patient survival.94,95 Increased perfusion has been difficult to achieve reproducibly, because tumor vasculature consists of both vessels co-opted from the preexisting host vasculature and vessels resulting from the angiogenic response of host vessels to cancer cells. The former are invested in normal contractile perivascular cells, whereas the latter lack these perivascular cells or these cells are abnormal.48,69,96 Presumably as a result, efforts to increase tumor blood flow by pharmacologic or physical agents have not always been reproducible or successful.48,69,85,96 On the other hand, the strategy of decreasing or shutting down tumor blood flow by “stealing” blood away from the “passive component” of the tumor vasculature by vasodilators, by vascular targeting, or by intravascular coagulation has shown promise in experimental systems.85,86,97–99 It also appears that judiciously applied antiangiogenic therapy could “normalize” the abnormal tumor microcirculation by pruning the immature vessels (see Fig. 8-4), thus rendering the remaining vasculature more responsive to vasoactive agents.100
Vascular Permeability
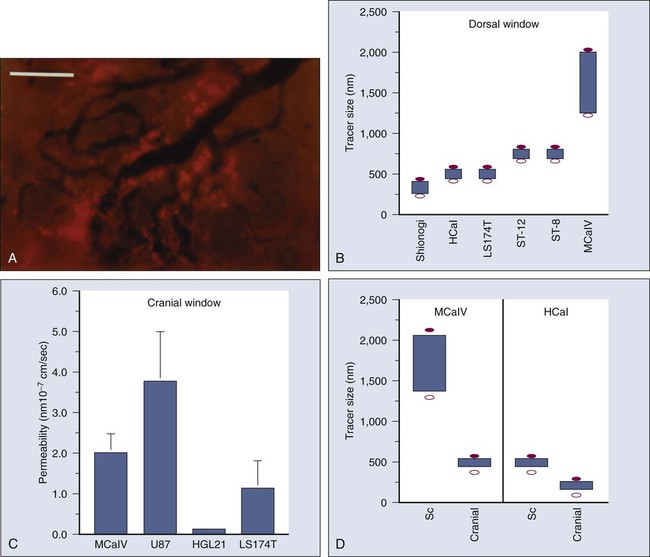
Once a blood-borne molecule has reached an exchange vessel, its extravasation occurs by diffusion, convection, and, to some extent, presumably by transcytosis.100,101 The effective permeability, P, of a molecule depends on the size, shape, charge, and flexibility of the molecule and on the size, shape, charge, and dynamics of the transvascular transport pathway. In normal vessels, these pathways include diffusion along the EC membrane (for lipophilic solutes), trans-EC diffusion, interendothelial junctions (<7 nm), open or closed fenestrations (<10 nm), and transendothelial channels (including vesicles or vesicovacuolar channels).15,102 Some of these anatomic pathways may be lined with glycocalyx on ECs, thus effectively reducing the size of the pathway. A basement membrane may further retard the movement of molecules. Ultrastructure studies show widened interendothelial junctions, increased numbers of fenestrations, vesicles, and vesicovacuolar channels in tumor vessels, and a lack of normal basement membrane and pericytes.15,51,96,102,103 In concert with these ultrastructural findings, both vascular permeability and hydraulic conductivity (a measure of water movement by pressure gradient) of tumors, in general, are significantly higher than those for various normal tissues.59,104–108 Furthermore, unlike normal vessels in the surrounding tissue, tumor vessels partially lose molecular size–based selectivity in permeability to different molecules.109 Despite increased overall permeability, not all blood vessels of a tumor are leaky (Fig. 8-8, A). Even the leaky vessels have a finite pore size that is tumor dependent (Fig. 8-8, B), and ultrastructural studies show that the larger pore size in tumors represents wide interendothelial junctions.56,103 Thus permeability is inversely related to molecular size due to steric and hydrodynamic hindrance from these pores, and very large molecules, such as nanoparticles, do not penetrate tumors efficiently.110,111 Molecular shape also influences penetration of these pores, such that rod-shaped molecules have a higher permeability than do spherical molecules.112 Similarly, positively charged molecules have a higher affinity for the negatively charged glycocalyx in the pores of these angiogenic tumor vessels, reducing this hindrance and giving them a higher permeability.113–116 Not only do the vascular permeability and pore size vary from one tumor to the next (see Fig. 8-8, A-C), but within the same tumor they vary spatially and temporally, as they do during tumor growth, regression, and relapse.56,60,61 The local microenvironment plays an important role in controlling vascular permeability (Fig. 8-8, D). For example, a human glioma (HGL21) has fairly leaky vessels when grown subcutaneously in immunodeficient mice, but it exhibits blood-brain barrier properties in the brain.59,72 Such site-dependent differences for other tumors have been observed in other orthotopic sites.54,57,58 One possible explanation is that the host-tumor interactions control the production and secretion of cytokines associated with permeability increase (e.g., VEGF) and decrease (e.g., angiopoietin 1).19,39,72,117,118 A better understanding of the molecular mechanisms of permeability regulation in tumors is likely to yield strategies for improved delivery of molecular medicine to tumors.
Movement of Cells Across Vessel Walls
Both cancer cells and immune cells frequently move across the walls of blood vessels—the former in the process of metastasis, and the latter during immune response or cell-based immunotherapy. Both transendothelial pathways (through ECs) and periendothelial pathways (between ECs) have been proposed as a route for intravasation and extravasation of cells. Very little is known about intravasation except that tumors might shed more than a million cells per gram per day and most of these are not clonogenic.119–122 Recently, it was demonstrated that metastatic cells could bring their own stroma from the primary tumor in the form of hereotypic cell clumps.123 More is known about the molecular and cellular mechanisms of extravasation.124 When a cell enters a blood vessel, it can continue to move with the flowing blood, collide with the vessel wall, adhere transiently or stably, and finally extravasate. These interactions are governed both by local hydrodynamic forces and adhesive forces. The former are determined by the vessel diameter and fluid velocity and the latter by the expression, strength, and kinetics of bond formation between adhesion molecules and by the surface area of contact.125–129 Deformability of cells affects both types of forces.130 In addition, cancer cells may grow intravascularly at a distant site (e.g., in the lungs).131
Rolling of endogenous leukocytes is generally low in tumor vessels, whereas stable adhesion (≥30 sec) is comparable between normal vessels and tumor vessels.132 On the other hand, both rolling and stable adhesion are nearly zero in angiogenic vessels induced in collagen gels by bFGF or VEGF, two of the most potent angiogenic factors.133 Whether this observation is due to a low flux of leukocytes into angiogenic vessels and/or to downregulation of adhesion molecules in these immature vessels currently is not known. The age of the animal also plays an important role in leukocyte-endothelial interactions.134 Further insight into the types of cells that adhere to tumor vessels comes from studies on the localization of IL-2-activated natural killer (A-NK) cells in normal and tumor tissues in mice using positron emission tomography.135,136 After systemic injection, these cells localized primarily in the lungs immediately after injection and could not be detected in the tumor.135 Increased rigidity caused by IL-2 activation might contribute to the mechanical entrapment of these cells in the lung microcirculation.137,138 Constitutive expression of certain adhesion molecules in the lung vasculature also might facilitate their retention in the lungs.124 One approach to reducing lung entrapment is to reduce the rigidity of these cells.112,117 Alternatively, the lung can be circumvented by injecting A-NK cells directly into the blood supply of tumors. In this case, A-NK cells, both xenogenic and syngeneic, adhered to some blood vessels in three different tumor models via CD18 and very large antigen-4 (VLA-4) on the A-NK cells and intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin on the activated endothelium of angiogenic vessels.33,136,139–142 These molecules can be upregulated by tumor necrosis factor–α (TNF-α) and a protein of 90 kd molecular weight (p90) that is secreted by some neoplastic cells and downregulated by transforming growth factor–β (TGF-β)—also, presumably, secreted by cancer cells.55,125,143–147 Surprisingly, the proangiogenic VEGF also can upregulate these molecules, whereas another proangiogenic molecule, bFGF, can downregulate these molecules.33,51,61,76,148 Inflammatory cells such as monocyte/macrophages or neutrophils also are recruited by VEGF and may play important roles in promoting matrix remodeling and angiogenesis.149 The challenge currently is to decrease nonspecific entrapment of immune cells in normal vessels and to increase their delivery to tumor vessels to improve various cell-based therapies, including gene therapy.
Extravascular Compartment
Composition and Origin
The extravascular compartment of a solid tumor consists of neoplastic cells (parenchyma) and host cells (e.g., inflammatory cells and fibroblasts) residing in an interstitial matrix bathed by the interstitial fluid (see Fig. 8-1). Depending on the tumor type and its stage of differentiation, neoplastic cells might be dispersed in the matrix as individual cells (e.g., lymphomas and melanomas) or as clumps, sheets, or nests (e.g., carcinomas). More than 80% of tumors are carcinomas arising from epithelial cells. The remaining cancers include sarcomas arising from mesenchymal cells (e.g., bone or muscle cells), lymphomas arising from lymphoid tissue, leukemias arising from hematopoietic cells, and hemangiomas arising from ECs. In a poorly differentiated carcinoma, the cancer cells might be packed loosely in clumps, whereas in a well-differentiated carcinoma, the cells might be connected with intercellular junctions and tightly packed in a nest enveloped by a basement membrane. With tumor progression, cancer cells can invade the basement membrane and spread to other regions.15 These various types of normal host cells must migrate into the tumor from normal tissue. Inflammatory cells might enter the tumor via blood vessels or might infiltrate from the adjacent tissue or lymphatics.124 Other host cells, such as fibroblasts, might proliferate and migrate from the adjacent connective tissue.19,48,150 The interstitial subcompartment of a tumor is bounded by the walls of the blood vessels on one side and by the membranes of cancer and stromal cells on the other. In normal tissues, the blood vessels are surrounded by a basement membrane, which, as previously discussed, is defective in tumors.19 In addition, functional lymphatics might be confined to the tumor margin.151,152 The interstitial space of tumors, like that of normal tissues, is composed of a collagen and elastin fiber network that provides structural support to the tissue. Interdispersed in this cross-linked structure are the interstitial fluid and macromolecular constituents (polysaccharides, hyaluronan, and proteoglycans), which form a hydrophilic gel. Compared with current understanding of blood vessel formation, understanding of stroma generation is minimal. Dvorak and coworkers have proposed that the extravasated plasma protein fibrinogen, a key component of the tumor interstitial fluid, clots to form fibrin, which serves as a major component of the provisional stroma. This provisional stroma eventually is replaced by more mature connective tissue stroma. The tumor interstitial fluid also contains several other Arg-Gly-Asp (RGD)-containing proteins, including fibronectin, vitronectin, osteopontin, thrombospondin, decorin, and tenacin.15 These proteins are present in both free and bound forms. Their Arg-Gly-Asp sequence provides a binding site for adhesion that assists in the migration of various cells, including stromal cells. In addition to extravasating from the leaky tumor vessels, these proteins, along with collagen and various proteoglycans, also are synthesized by the stromal cells, albeit in a form that differs from that in the plasma or normal tissues.15 Tumor interstitial fluid also can contain various growth factors that facilitate stroma formation. For example, in vitro studies suggest that platelet-derived growth factor-BB (PDGF-BB) is involved in the recruitment of fibroblasts to tumors, and TGF-β induces the production of collagen and other matrix molecules in tumors.19,153 With the increasing interest in using the fragments of matrix constituents for controlling angiogenesis, increased understanding of the molecular and cellular mechanisms of stroma generation in tumors will increase.43
Interstitial Transport
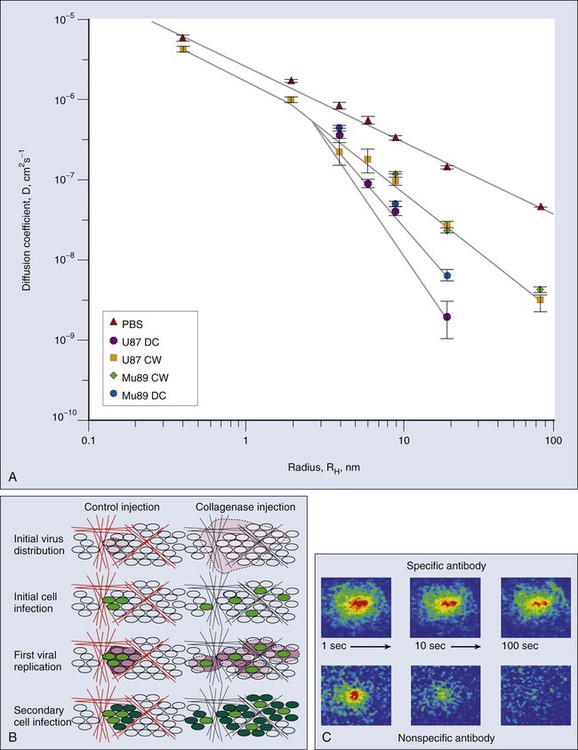
Once a molecule has extravasated, its movement through the interstitial space occurs by diffusion and convection.101,154 Diffusion is proportional to the concentration gradient in the interstitium, and convection is proportional to the interstitial fluid velocity, which, in turn, is proportional to the fluid pressure gradient in the interstitium. Just as the interstitial diffusion coefficient D (cm2/s) relates the diffusive flux to the concentration gradient, the interstitial hydraulic conductivity K (cm2/mmHg/sec) relates the interstitial velocity to the pressure gradient.134 Values of these transport coefficients are governed by the structure and composition of the interstitial compartment and by the physicochemical properties of the solute molecule.155–165 The value of K for a human colon carcinoma xenograft (LS174T), measured using two different methods, was found to be higher than that of a hepatoma, which, in turn, was higher than that of the normal liver.167–167 Using fluorescence recovery after photobleaching, D of various molecules in tumors was found to be about one third that in water and higher than the values in the host tissue (Fig. 8-9, A).157,168 Collagen content and structure have a significant effect on D in tumors.159,163,166,169–173 This finding is surprising because hyaluronan and proteoglycans, not collagen, account for most of the resistance to transport in normal tissues. Because collagen is produced by host cells (e.g., fibroblasts), the penetrability into a tumor depends on the host-tumor interaction (see Fig. 8-9, A). Thus agents that interfere with collagen synthesis and/or organization (e.g., relaxin, losartan, and bacterial collagenase) might increase interstitial transport in tumors156,169,170 (Fig. 8-9, B). The time constant for a molecule with diffusion coefficient D to diffuse across a distance L is approximately L2/4D. For diffusion of immunoglobulin G (IgG) in tumors, this time constant is on the order of 1 hour for a 100-µm distance, days for a 1-mm distance, and months for a 1-cm distance. Thus for a 1-mm distance in tumor, diffusional transport would take days, and for a 1-cm distance in tumor, it would take months. Furthermore, the path through the interstitium is tortuous over large length scales, greatly increasing the distance a molecule must travel.158 If the central vessels have collapsed completely as a result of cellular proliferation and interstitial matrix rearrangement, the reduced delivery of macromolecules by blood flow would make diffusion the primary mechanism of delivery to this necrotic center.6,79 Binding of a low- or high-molecular-weight drug to plasma proteins and various tissue components could further retard their transport in tumors.168,174–180 The role of binding is clearly illustrated in Figure 8-9, C, which compares the rate of fluorescence recovery of a photobleached spot in tumor tissue injected with a nonspecific versus a specific IgG. In addition to the heterogeneity of D in tumors, the most unexpected result of these photobleaching studies was the large extent (30% to 40%) of nonspecific binding.168 These results collectively suggest that the interstitial compartment of a tumor can be a formidable barrier to the uniform delivery of therapeutic macromolecules (e.g., antibodies and genes) in tumors, and strategies are needed to modify this barrier.
Lymphangiogenesis and Lymphatic Transport
In most normal tissues, extravasated plasma and macromolecules are taken up by the lymphatics and returned to the blood circulation. It is widely accepted that lymphatic vessels are present in the tumor margin and the peritumoral tissue (Fig. 8-10, A). Indeed, invasion of peritumoral lymphatics is considered to be a poor prognostic factor for several tumors (e.g., breast, colorectal, and endometrial cancers), and lymphatic metastasis is a major cause of morbidity and mortality for others (e.g., melanoma, head and neck cancer, lung cancer, and cervical cancer). The hotly debated issue for nearly a century has been whether anatomically defined lymphatic vessels are present within solid tumors and, if so, whether they function (see Fig. 8-10, A).181,182 Currently available immunohistochemical markers stain for structures in some tumors that resemble lymphatic vessels. Because many of these markers lack specificity, however, it is not clear whether they stain functional lymphatic vessels, ECs from remnant lymphatic vessels, or some other structures (e.g., preferential fluid channels).152,166,183,184 Mechanical “solid stress” induced by proliferating tumor cells and matrix production compresses and impairs lymphatic vessels that are co-opted or formed within a tumor.6,78,80 The impaired lymphatic vessels, in turn, contribute to the interstitial hypertension characteristic of animal and human tumors (to be discussed shortly). Embryonic lymphatic vessels originate primarily from blood vessels according to the following process (Fig. 8-10, B).187–187
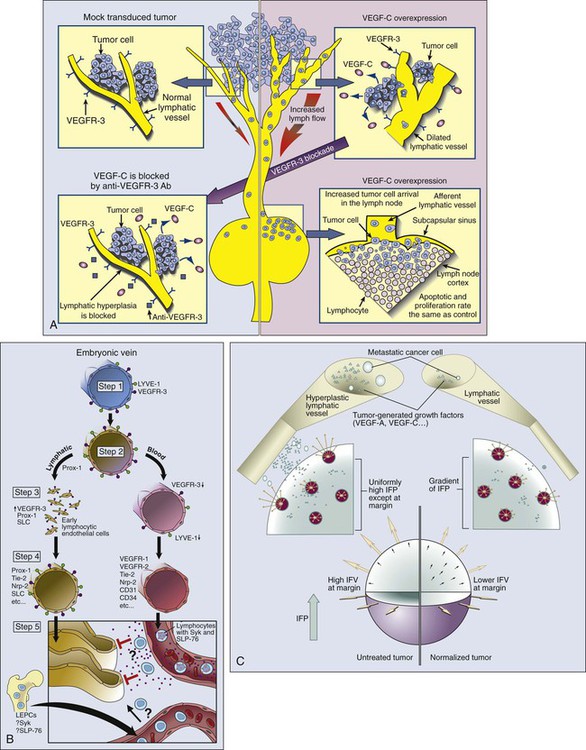
1. In the early embryo, endothelial cells of the cardinal vein express lymphatic vascular endothelial receptor-1 (LYVE-1) and VEGFR3, molecules observed primarily (but not exclusively) on lymphatic vessels in normal adult tissues.
2. A yet unknown signal triggers the expression of the homeobox gene Prox-1 so that the protein is displayed in a polarized fashion in the endothelial cells of the cardinal vein. This action marks the first stage of commitment to the lymphatic lineage.
3. These LYVE-1+VEGFR3+Prox-1+ cells then start to bud, again in a polarized fashion.
4. At this stage, these early lymphatic endothelial cells start expressing secondary lymphoid chemokine and increased levels of VEGFR3, markers of mature lymphatic ECs.
Members of the angiopoietin family (Ang-2) and its receptor (Tie-2), as well as podoplanin, presumably are involved in the maturation and patterning of these nascent lymphatic vessels.188 The hematopoietic signaling pathway, Syk/SLP-76, contributes to the separation of lymphatics from blood vessels. The molecules involved in angiogenesis also are involved in lymphangiogenesis. For example, VEGF-C and VEGF-D can induce both angiogenesis and lymphangiogenesis and are associated with lymphogenic metastasis in a variety of tumors.151 Their receptor VEGFR3 is present in both lymphatic and selected vascular endothelium. As is the case with vascular angiogenesis, other positive and negative regulators (e.g., angiopoietins) and other receptors (e.g., chemokine receptors and neuropilins) could be involved in lymphangiogenesis, and as previously discussed, mechanisms analogous to co-option, intussusception, sprouting, and vasculogenesis might operate in lymphatic growth.11,188 Similar to the recently discovered organ-specific angiogenic molecule (EG-VEGF) and endothelial precursor cells, there could be organ-specific lymphangiogenic molecules and lymphatic EPCs that contribute to tumor-associated lymphangiogenesis.22,38,186 Moreover, the proteolytic processing of lymphangiogenic molecules and the phenotype and function of the resulting lymphatics might depend not only on the tumor type but also on the host organ in which the tumor is growing.76,184–186
The mechanical and/or molecular signals that could trigger the lymphangiogenic switch are unknown. Because lymphatic vessels help maintain the balance of fluid in tissues, hydrostatic pressure is a probable trigger. Whether the hyperplasia and the increased density of lymphatic vessels seen in the tumor margins are a response to elevated hydrostatic pressure in tumors and whether the newly formed lymphatics are able to remain open and carry cancer cells are open questions. Techniques such as microlymphangiography, reagents that block signaling of VEGF-C and VEGF-D, and lymphangiogenic factors yet to be discovered will allow us to answer these important questions.4,184,186,189–197
Interstitial Hypertension
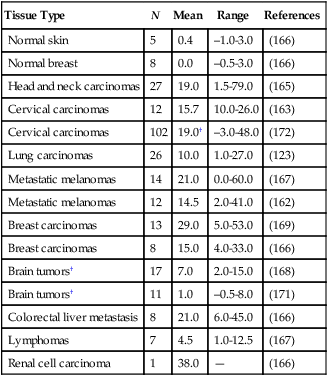
Unlike normal tissues, in which the interstitial fluid pressure (IFP) is around 0 mm Hg, both animal and human tumors exhibit interstitial hypertension (Table 8-2).151,154,167,198–209 The tumor IFP begins to increase as soon as the host vessels become leaky in response to permeability factors such as VEGF, and thus IFP can be lowered by antibodies against VEGF or VEGFR2.212–212 The IFP increases with tumor size in some tumors and remains independent of tumor size in others.198–200,202,203 Three mechanisms contribute to interstitial hypertension in tumors. In normal tissues, the lymphatics maintain the fluid homeostasis; thus the lack of functional lymphatics in tumors is a key contributor. Indeed, DiResta and colleagues have been able to lower the IFP by placing “artificial lymphatics” in tumors.213 The second contributor is the high permeability of tumor vessels. As a result, the hydrostatic and oncotic (colloid osmotic) pressures become almost equal between the intravascular and extravascular spaces.201,214 At least two pieces of evidence support this hypothesis. First, reducing permeability by blocking VEGF signaling lowers IFP.211,212,215–218 Second, IFP increases and decreases with the microvascular pressure within seconds.221–221 The two mechanisms described thus far can only explain interstitial hypertension up to 20 to 30 mm Hg, the microvascular pressure of most exchange vessels (the hydrostatic pressure within the lumina of capillaries) in our bodies, but IFPs as high as 94 mm Hg have been measured in human tumors.200 Because microvascular pressure (MVP) is the driving force for IFP in tumors, these tumors must have a high MVP. Indeed, this is the case.201 Two possible explanations exist for elevated MVP in tumors: the tumor vessels have reduced arterial resistance so that the MVP becomes closer to arterial pressure, and/or the tumor vessels have increased venous resistance as a result of compression and tortuosity so that the entire intratumor vascular network is under hypertension. Indirect evidence for the latter explanation comes from the decrease in IFP after decompression of tumor vessels by taxol-induced apoptosis of perivascular cells.79
Table 8-2
Interstitial Fluid Pressure (mm Hg) in Normal and Neoplastic Tissues in Patients*
| Tissue Type | N | Mean | Range | References |
| Normal skin | 5 | 0.4 | –1.0-3.0 | (166) |
| Normal breast | 8 | 0.0 | –0.5-3.0 | (166) |
| Head and neck carcinomas | 27 | 19.0 | 1.5-79.0 | (165) |
| Cervical carcinomas | 12 | 15.7 | 10.0-26.0 | (163) |
| Cervical carcinomas | 102 | 19.0† | –3.0-48.0 | (172) |
| Lung carcinomas | 26 | 10.0 | 1.0-27.0 | (123) |
| Metastatic melanomas | 14 | 21.0 | 0.0-60.0 | (167) |
| Metastatic melanomas | 12 | 14.5 | 2.0-41.0 | (162) |
| Breast carcinomas | 13 | 29.0 | 5.0-53.0 | (169) |
| Breast carcinomas | 8 | 15.0 | 4.0-33.0 | (166) |
| Brain tumors† | 17 | 7.0 | 2.0-15.0 | (168) |
| Brain tumors† | 11 | 1.0 | –0.5-8.0 | (171) |
| Colorectal liver metastasis | 8 | 21.0 | 6.0-45.0 | (166) |
| Lymphomas | 7 | 4.5 | 1.0-12.5 | (167) |
| Renal cell carcinoma | 1 | 38.0 | — | (166) |
1. Reduced transmural pressure gradients due to equilibrium between MVP and IFP reduce convection across tumor vessel walls and thus compromise the transport of macromolecules.201,220
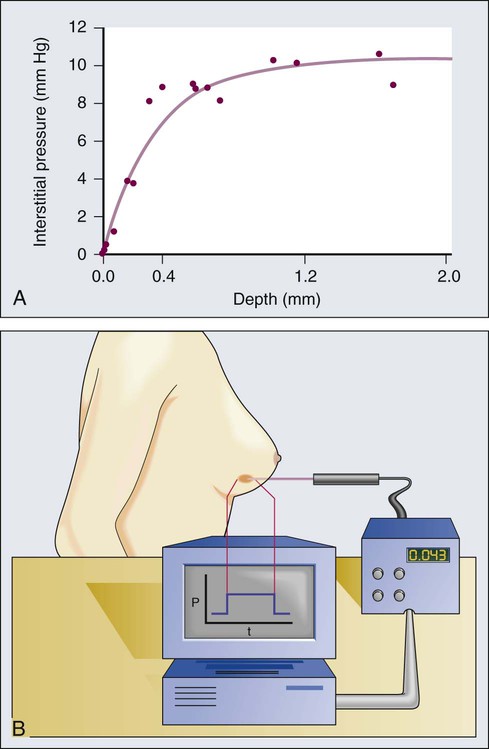
2. Because IFP is nearly uniform throughout a tumor and drops precipitously in the tumor margin, the interstitial fluid oozes out of the tumor into the surrounding normal tissue, carrying macromolecules with it (Fig. 8-11).198,222
3. Finally, transmural coupling between IFP and MVP due to high permeability of tumor vessels can lead to blood flow stasis in tumors without physically occluding the vessels.93–93
The enhanced permeability also can facilitate lymphatic metastasis (Fig. 8-10, C). Thus decreasing vascular permeability might restore the transmural pressure gradients and potentially resume/reestablish blood flow in the nonperfused regions of tumors. Some direct and indirect antiangiogenic therapies might “normalize” the tumor vasculature through this mechanism (see Fig. 8-4).100 This normalization also can reduce the number of cells shed into peritumor lymphatics and alleviate peritumor edema (see Fig. 8-10, C).73,223
Metabolic Environment
Hypoxia
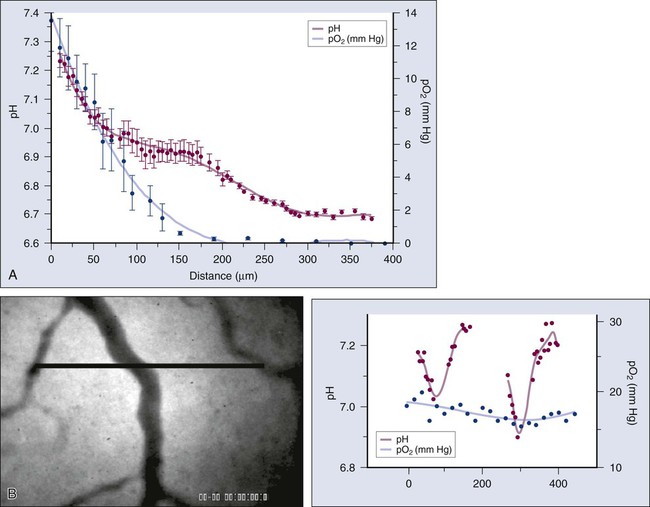
A key function of the vasculature is to provide adequate levels of nutrients and oxygen to the parenchymal cells and to remove waste products. Based on the anatomy of the capillary bed and a mathematical model of oxygen diffusion and consumption, the Nobel laureate August Krogh introduced the concept of a diffusion limit for oxygen of 100 to 200 µm nearly a century ago.224 This unit of tissue—a single capillary surrounded by a 100- to 200-µm-radius cylinder—is referred to as a “Krogh cylinder” in physiology. Nearly 50 years later, Thomlinson and Gray identified similar “cords” in human lung cancer and found necrotic cells beyond 180 mm away from blood vessels, presumably as a result of lack of oxygen.225 This is referred to as “chronic hypoxia” or “diffusion-limited” hypoxia. Although various hypoxia markers and microelectrodes have suggested these gradients, the first direct measurements of these perivascular gradients, along with pO2 and blood flow rate of the same vessels, became possible only recently with the development of phosphorescence quenching microscopy (Fig. 8-12, A).226,227 As discussed previously, blood flow in tumor vessels is intermittent, and thus some regions of a tumor are starved for oxygen periodically. The resulting hypoxia is referred to as “acute hypoxia” or “perfusion-limited hypoxia.”228,229 A necessary consequence of intermittent blood flow is the resumption of blood flow after shutdown, and the resulting production of free radicals can lead to “reperfusion injury” or “reoxygenation injury,” applying additional selection pressure on cancer cells.
Low pH
Another consequence of the abnormal microcirculation of the tumor is low extracellular pH. There are at least two sources of H+ ions in tumors—lactic acid and carbonic acid.230 The former results from glycolysis and the latter from conversion of CO2 and H2O via carbonic anhydrase. The intracellular pH of cancer cells remains neutral or alkaline (pH 7.4), however, despite the acidic extracellular pH. Because carbonic anhydrase-9 (CAIX), various glucose transporters (GLUT1, GLUT3), and enzymes in the glycolytic pathway are upregulated by hypoxia, one would expect low extracellular pH and hypoxia to track each other and to colocalize with regions of low blood flow.231 Surprisingly, there is a lack of spatial correlation among these parameters (Fig. 8-12, B), a discovery made possible by recent developments in optical techniques that permit the simultaneous high-resolution mapping of multiple physiologic parameters.226 A potential explanation for this lack of concordance is that some perfused tumor vessels carry hypoxic blood.226 Thus although they might not be able to deliver adequate oxygen to the surrounding cells, they may be able to carry away the waste products (e.g., lactic acid).
Molecular, Cellular, and Therapeutic Consequences
The presence of oxygen during irradiation makes the damage to DNA produced by radiation-induced free radicals permanent, whereas such damage can be repaired under hypoxic conditions.232 Therefore hypoxia in solid tumors significantly reduces their radiation sensitivity. Tumor hypoxia also is associated with resistance to some chemotherapeutics.232 Similarly, low extracellular pH can affect the cellular uptake and cytotoxicity of some therapeutics adversely or favorably.235–235 As a result, for nearly half a century, considerable preclinical and clinical efforts have been focused on alleviating hypoxia by improving tumor perfusion with various therapies, including the following:
• Increasing oxygen content of the blood (via hyperbaric oxygenation, for example)
• Increasing hemoglobin/hematocrit (via erythropoietin, for example)
1. Use hypoxia and/or low pH to activate drugs or to attract anaerobic bacteria.
2. Dissect hypoxia-induced pathways to identify novel targets for drug development.
The first strategy has led to the development of agents such as tirapazamine and to the rejuvenation of interest in bacteriolytic therapy; both approaches are still being tested in clinical trials.88,232 The second strategy has revealed several molecular determinants of the physiological and pathophysiological responses to hypoxia.231 The balance between hypoxia-induced apoptosis/necrosis on one hand, and the increased resistance to cell death mediated by various hypoxia-induced pathways on the other, determines whether a tumor can survive and even grow under hypoxic conditions. Ultimately, hypoxia might select for tumor cells that are more malignant, more invasive and genetically unstable, and less susceptible to apoptosis, thus rendering them resistant to various therapies. Therefore several molecules in the hypoxia-induced pathways are now being targeted in the development of diagnostic and therapeutic agents. Finally, normalization of tumor vessels by antiangiogenic agents may reduce hypoxia within tumor environment and synergize with radiation therapy.237
Hypoxia-induced pathways include genes involved in the following processes:
• Oxygen delivery (e.g., heme oxygenase 1 and erythropoietin)
• Glycolysis and glucose uptake (e.g., GLUT1 and GLUT3 and hexokinase-1 and hexokinase-2)
• pH control (e.g., carbonic anhydrase-9 and carbonic anhydrase-12)
• Stress-response pathways (e.g., growth arrest– and DNA damage–induced gene GADD153)
• Growth factor signaling (e.g., IL-6, IL-8, and insulin-like growth factor-2 [IGF-2])
• Angiogenesis (e.g., VEGF-A, VEGFR1, Ang-2, Tie-2, FGF-3, TGF-β, nitric oxide synthase [NOS], cyclooxygenase-2 [COX-2], and hepatocyte growth factor [HGF])
• Transcription (e.g., HIF1α and HIF2α, JUN, FOS, and nuclear factor–κB [NF-–κB])
• Apoptosis (e.g., BCL-interacting killer [BIK], annexin V, 19-kd interacting protein-3 [NIP3], and NIP3-like protein X [NIX])
• Growth inhibition signaling factors (e.g., p21, p27, and GADD153)
• Invasion and metastasis (e.g., metalloproteinases [MMPs], CXCL12/CXCR4, and plasminogen activator inhibitor-1 [PAI-1])231
Of the various molecules involved in sensing and responding to hypoxia, HIF1α has received the most attention. This transcription factor is upregulated in several human tumors.231 Regulated by a proline hydroxylase, HIF1α can activate the genes for angiogenesis, vasodilation, glycolysis, and erythrocyte production by binding to the hypoxia-response element.238,239 Surprisingly, teratomas arising from HIF1α–/– embryonic stem cells grow more rapidly despite lower levels of VEGF and angiogenesis.51,240 This counterintuitive finding could be a result of the ability of HIF1α–/– cells to survive under hypoxic conditions instead of undergoing apoptosis.48 Interestingly, other HIF1α–/– cancer cells lead to slowly growing tumors. As a result, molecular therapies that target HIF1α or hypoxia-response element are under intensive investigation for cancer detection and treatment.241
Antiangiogenic Agents in the Clinic
Two major problems currently plague the nonsurgical treatment of malignant solid tumors. First, physiological barriers within tumors impede the delivery of therapeutics and oxygen (a key sensitizer to ionizing radiation) at effective concentrations to all cancer cells.242,243 Second, inherent or acquired resistance resulting from genetic and epigenetic mechanisms reduces the effectiveness of both conventional and novel therapies.244 Can we take advantage of the unique pathophysiology of tumors to overcome these problems for better management of cancer?
Mechanisms of Action
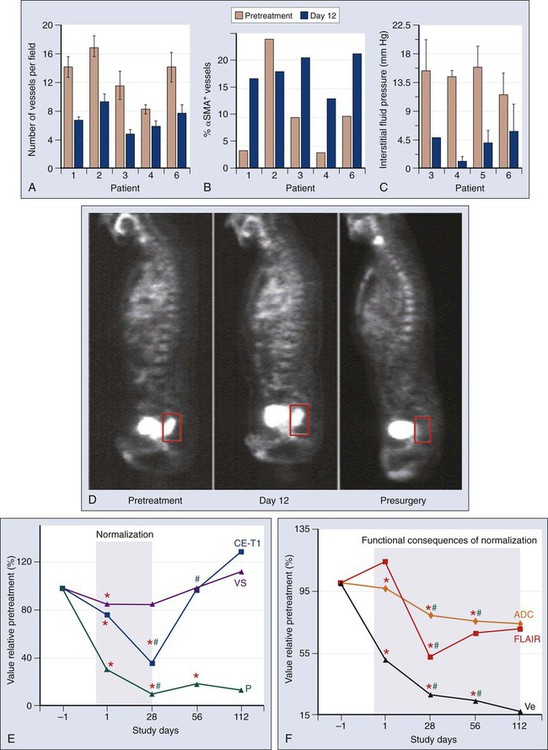
Recent clinical data offer hope, with seven U.S. Food and Drug Administration–approved VEGF-blocking antiangiogenic agents (bevacizumab, sorafenib, sunitinib, pazopanib, axitinib, vandetanib, regorafenib, and aflibercept) already in clinical use.11,245 Studies in patients with rectal cancer and glioblastoma have largely confirmed the hypotheses that anti-VEGF therapy has antivascular effects and can induce vascular normalization in patients with cancer.216,218,245–249 In brief, bevacizumab alone reduced the tumor tissue vascular density by approximately half at day 12 after the first infusion and reduced significantly the tumor blood flow evaluated on computed tomography scans and the number of viable circulating endothelial cells and progenitor cells. Clinically, both the low-dose-bevacizumab infusion and the high-dose-bevacizumab infusion showed a less hyperemic/hemorrhagic appearance but no significant tumor regression after bevacizumab treatment upon flexible sigmoidoscopy.216,218 Although the significant pruning of tumor vasculature led to a significant increase in cancer cell apoptosis, it also led to a more mature (pericyte-covered) tumor vasculature and a stable or increased cancer cell proliferation (Fig. 8-13, A-D).218–218 Whether the increase in tumor cell apoptosis was due to a direct or indirect effect of bevacizumab is currently unclear. Similarly, the role of VEGF blockade on immune or stromal cells in patients is not yet understood. After bevacizumab therapy alone, the tumor IFP was consistently decreased, particularly in the patients with high baseline values.218–218 This finding suggested that in human tumors, similar to mouse models, the tumor microenvironment was normalized by the reduction of the excessive vascularization and potentially sensitized the tumor to the subsequent cytotoxic therapy within the “normalization window.”212,237 Imaging studies yielded more supportive data for the normalization hypothesis: despite the significant reduction in vessel density and blood flow, the fluorodeoxyglucose uptake measured on positron emission tomography scans (a measure of tumor metabolic activity) and the P • S product (proportional to the penetration of tracer in tumor) evaluated on computed tomography scans did not significantly change at day 12.216,218 Antiangiogenic therapy with cediranib in recurrent glioblastoma demonstrated the existence of a window of vascular normalization in patients with cancer (Fig. 8-13, E and F).247,248 Moreover, two recent studies of cediranib in glioblastoma showed that some patients respond with increased perfusion and that these patients survive longer than do nonresponders.95
Biomarkers
Similar to many targeted therapies, the survival benefit after antiangiogenic therapies remains modest. Unfortunately, unlike many anticancer targeted therapies, there are no validated biomarkers of patient selection, response, or resistance for antiangiogenic agents.37 If we could we find biomarkers to identify patients more likely to benefit from these drugs, the survival benefit in patients taking anti-VEGF drugs would be improved. Unlike preclinical models, the phase III bevacizumab experience in patients with metastatic colorectal cancer did not identify P53, KRAS, or BRAF status, VEGF or thrombospondin-2 (TSP2) expression, or microvascular density at baseline as predictive markers of response.250,251 Similarly, circulating VEGF levels—although of potential biomarker value—have unclear predictive biomarker value.37,252 In collaborative studies at Massachusetts General Hospital, these questions have been examined in more than 20 multidisciplinary trials by measuring tissue, circulating and imaging biomarkers at various time points during and after treatment and correlating these with treatment outcomes. Several promising biomarkers have emerged. One is the level of circulating sVEGFR1/sFLT1 (an endogenous blocker of VEGF and PlGF) as a biomarker of intrinsic resistance to anti-VEGF therapy. It was reported that patients with locally advanced rectal carcinoma who had high plasma sVEGFR1 levels before treatment were less likely to benefit—or experience toxicities—from bevacizumab therapy combined with chemoradiation.218,253 The same association was found for newly diagnosed patients with glioblastoma multiforme (GBM), triple-negative breast cancer, hepatocellular carcinoma (HCC), and metastatic colorectal carcinoma who were treated with anti-VEGF agents.254–257 Of interest, a retrospective analysis in two randomized phase III trials recently has shown that a genetic variation in VEGFR1 gene correlates with increased VEGFR1 expression and poor outcome of bevacizumab treatment in patients with metastatic renal cell carcinoma and pancreatic ductal adenocarcinoma.238 A second potential biomarker is the SDF1α/CXCR4 axis as a potential evasive pathway. It was found that circulating levels of the chemokine SDF1α increase in patients at the time of evasion from anti-VEGF therapies. This was the case in patients with rectal carcinoma who were treated with bevacizumab, in patients with GBM who were treated with cediranib, in patients with HCC who were treated with sunitinib, and in patients with sarcoma after sorafenib.215,247,256,258,259 Interestingly, the sources of SDF1α are different in each of these diseases, and so is the role of the SDF1α/CXCR4 pathway.260 For example, in persons with GBM this pathway appears to facilitate invasion of cancer cells and co-option of host vessels by invading cancer cells. These biomarker candidates need to be tested prospectively. Of note, based on these findings, Dr. Patrick Wen (Dana Farber Cancer Institute, Boston, MA) has started a clinical trial with AMD3100 (an anti-CXCR4 drug) plus bevacizumab in patients with recurrent GBM (ClinicalTrials.gov Identifier: NCT01339039).
Toxicity
Experimental studies have shown that, in 11 of the 17 healthy organs studied, VEGF blockade can significantly decrease the number of normal capillaries.261 In patients with cancer, most anti-VEGF agents often induce proteinuria, hypertension, thyroid-stimulating hormone elevation, and gastrointestinal toxicity, but agent-specific toxicities also have been reported.10,262 In addition, the long-term effects of antiangiogenic therapies in patients with less advanced lesions remain to be established.
Conclusion
The recent successes of the anti-VEGF agents have raised great hope and have demonstrated important lessons about the significance of the target, timing, and dosage of each agent37,75:
• According to the results of the phase III trials completed to date, bevacizumab can increase median overall survival in certain cancers when combined with standard chemotherapy.
• Anti-VEGF therapy with bevacizumab can increase overall survival and/or progression-free survival in patients with advanced colorectal carcinoma, non–small cell lung carcinoma, breast carcinoma, and renal cell carcinoma when combined with cytotoxic agents or immunotherapy. Recently, aflibercept (or “VEGF-Trap,” an sFLT1 chimera) with chemotherapy showed improved survival in second line in persons with metastatic colorectal cancer.
• Improved survival has been observed with broad-spectrum multitargeted tyrosine kinase inhibitors (e.g. sorafenib, sunitinib, pazopanib, axitinib, and vandetanib) when used in monotherapy. Thus far, VEGF receptor–selective tyrosine kinase inhibitors have failed to confer the same survival advantage as bevacizumab when combined with chemotherapy.
• Anti-VEGF therapy can prune and “normalize” tumor vascular structure and function.
• An urgent need exists to identify biomarkers to guide anti-VEGF therapy and combination therapies using anti-VEGF agents.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Vascular and Interstitial Biology of Tumors
Rakesh K. Jain, Vikash P. Chauhan and Dan G. Duda
• A solid tumor is an organ composed of neoplastic cells and stromal cells nourished by a vasculature made of endothelial cells—all embedded in an extracellular matrix. The interactions among these cells and between these cells, their surrounding matrix, and their local microenvironment control the expression of various genes. The products encoded by these genes, in turn, control the pathophysiological characteristics of the tumor. Tumor pathophysiology governs not only tumor growth, invasion, and metastasis but also the response to various therapies.
• Tumor vasculature is made of host vessels co-opted by cancer cells and by new vessels formed by the processes of vasculogenesis and angiogenesis. A constellation of positive and negative regulators of angiogenesis governs the process of neovascularization.
• Tumor vessels are abnormal in terms of their organization, structure, and function. These abnormalities contribute to heterogeneity in vascular permeability, blood flow, and the microenvironment.
• Tumor interstitial matrix is formed of proteins secreted by stromal and cancer cells and by those leaked from the nascent blood vessels.
• Tumor interstitium is heterogeneous, with some regions fairly permeable and others difficult to penetrate. Modification of collagen and hyaluronan in the matrix can improve penetration of large-molecular-weight therapeutics.
• Solid components of tumors—cancer cells, stromal cells, and matrix molecules—mechanically compress blood and lymphatic vessels, resulting in reduced perfusion that limits oxygen and drug supply. Depleting these constituents decompresses blood vessels to enhance perfusion and drug delivery.
• Interstitial hypertension is a hallmark of solid tumors and results from vessel leakiness, lack of functional lymphatics, and compression of vessels. This elevated fluid pressure contributes to blood flow heterogeneity and directly hinders the penetration of large-molecular-weight therapeutics. Alleviating interstitial hypertension improves oxygen and drug delivery to tumors.
• Judicious application of angiogenic therapy can normalize the tumor vessels and make them more efficient for delivery of oxygen (a known radiosensitizer) and drugs. Antiangiogenic agents can prune tumor vessels, induce cancer cell apoptosis, and lower interstitial hypertension in tumors.
• Thus far, eight antiangiogenic agents have been approved for patients with certain types of cancer. Based on these successes, antiangiogenic therapy is expected to make a difference in many other tumor types. Two main hurdles to further development of antiangiogenic agents are the better understanding of the mechanisms of action of these agents and the development of biomarkers to select patients for these drugs and to predict and monitor their effects.
Introduction
A solid tumor is an organ composed of neoplastic cells and host stromal cells nourished by a vasculature made of endothelial cells—all embedded in an extracellular matrix (Fig. 8-1). The interactions among these cells and between these cells, their surrounding matrix, and their local microenvironment control the expression of various genes. The products encoded by these genes, in turn, control the pathophysiological characteristics of the tumor. Tumor pathophysiology governs not only tumor growth, invasion, and metastasis but also the response to various therapies. In this chapter we will discuss various pathophysiological parameters that characterize the vascular and extravascular compartments of a tumor and the mechanisms governing the formation and function of these compartments.
Vascular Compartment
New Vessel Formation
It has been known for nearly a century that the vascular system is associated with tumor growth in animals and humans.1 Powerful insights into the neovascularization of transplanted tumors using transparent window techniques were developed in the 1940s.2–5 The possibility that tumors produce an “angiogenic” substance was suggested in 1968.6 The hypothesis that blocking angiogenesis should block tumor growth and metastasis was proposed shortly thereafter in 1971.7 The concept that a tissue acquires angiogenic capacity during neoplastic transformation—and, by extension, that antiangiogenesis could be used to prevent cancer—was put forward in 1978.8 The first antiangiogenic agent approved for patients with cancer was bevacizumab, an antibody specific to vascular endothelial growth factor (VEGF), on the basis of the increased survival seen in patients with metastatic colorectal cancer with the combination of bevacizumab and standard chemotherapy in a pivotal randomized placebo-controlled phase III trial.9 At present, various antiangiogenesis and proangiogenesis strategies are being evaluated clinically to prevent or treat a large number of diseases, including cancer.12–12 Both normal and pathological angiogenic processes are governed by the net balance between proangiogenic and antiangiogenic factors.13,14 This balance is spatially and temporally regulated under physiological conditions, so that the “angiogenic switch” is “on” when needed (e.g., during embryonic development, wound healing, and formation of the corpus luteum) and “off” at other times. During neoplastic transformation and tumor progression, this regulation is deranged, and blood vessels form ectopically to support a growing tumor mass.
Cellular Mechanisms
At least four cellular mechanisms are involved in the vascularization of tumors: co-option, intussusception, sprouting (angiogenesis), and vasculogenesis (Fig. 8-2).11 Tumor cells can co-opt and grow around existing vessels to form “perivascular” cuffs. However, as stated earlier, these cuffs cannot grow beyond the diffusion limit of critical nutrients and may actually cause the collapse of the vessels because of growth pressure (referred to as “solid stress”). Alternatively, an existing vessel may enlarge in response to the growth factors released by tumors, and an interstitial tissue column may grow in the enlarged lumen and partition the lumen to form an expanded vascular network. This mode of intussusceptive microvascular growth has been observed during tumor growth, wound healing, and gene therapy.15–18 “Sprouting” angiogenesis is perhaps the most widely studied mechanism of vessel formation. During sprouting angiogenesis, the existing vessels become leaky in response to growth factors released by normal cells or cancer cells; the basement membrane and the interstitial matrix dissolve; pericytes dissociate from the vessel; endothelial cells (ECs) migrate and proliferate to form an array/sprout; a lumen is formed in the sprout (a process referred to as canalization); branches and loops are formed by confluence and anastomosis of sprouts to permit blood flow; and finally, these immature vessels are invested in basement membrane and pericytes. During physiological angiogenesis, these vessels differentiate into mature arterioles, capillaries, and venules, whereas in tumors they remain largely immature.5,11,12,19 During embryonic development, a primitive vascular plexus is formed from endothelial precursor cells (EPCs, also known as angioblasts) by a process referred to as vasculogenesis. In adults, EPCs—mobilized from bone marrow niches into the peripheral blood circulation—also can contribute to neovascularization (a process referred to as “postnatal” vasculogenesis) in tumors and other tissues.22–22 In addition, recent reports demonstrated that a fraction of endothelial cells in tumor vessels may be derived via transdifferentiation from cancer cells,23 or from cancer stemlike cells (e.g., in glioblastomas).11,24–26 In this larger context, the term “vasculogenesis” might be invoked to describe neovascularization in tumors from tumor stem cells.27 Tumor vasculogenesis also may occur from EPCs of non–bone marrow origin that are recruited from adjacent tissues and/or circulation.28,29 The current challenge is to discern the relative contribution of each of the these mechanisms of neovascularization during tumor growth and during and after treatment of tumors.27,30
Molecular Mechanisms
Various proangiogenic and antiangiogenic molecules that orchestrate different steps in vessel formation, along with their functions, are listed in Table 8-1. VEGF currently is considered the most critical proangiogenic molecule. Originally discovered in 1983 as the vascular permeability factor and cloned in 1989, VEGF increases vascular permeability, promotes migration and proliferation of ECs, serves as an EC survival factor, can mobilize EPC populations from the bone marrow, and is known to upregulate leukocyte adhesion molecules on ECs.15,22,31–33 During tumor progression, or with treatment, the number of distinct angiogenic molecules produced by a tumor can increase.36–36 Thus after VEGF signaling is blocked, a tumor might rely on other, alternative angiogenic molecules (e.g., basic fibroblast growth factor [bFGF], stromal-derived factor 1α [SDF1α], placental growth factor [PlGF], or interleukin-8 [IL-8]).37 Other positive regulators of angiogenesis include the angiopoietins that are involved in stabilizing vessels and controlling vascular permeability; various proteases involved in dissolving/remodeling matrix and releasing growth factors; and recently discovered organ-specific angiogenic stimulators (e.g., endocrine gland–derived VEGF).19,38,39 Angiogenesis inhibitors include endogenous soluble receptors of various proangiogenic ligands (e.g., sVEGFR1/sFLT1) and molecules that downregulate the expression of stimulators (e.g., interferons) or that interfere with the release of the stimulators or binding with their receptors (e.g., platelet factor 4). Thrombospondins are among the first and most well-characterized endogenous inhibitors that interfere with the growth, adhesion, migration, and survival of ECs.13 Other endogenous inhibitors include fragments of various plasma or matrix proteins (e.g., angiostatin, a fragment of plasminogen; endostatin, a fragment of collagen XVIII; and tumstatin, a fragment of collagen IV).42–42 Neither the mechanisms of action of the matrix-derived inhibitors nor their physiological role are well understood.43 The generation of proangiogenic and antiangiogenic molecules can be triggered by metabolic stress (e.g., low po2, low pH, or hypoglycemia), mechanical stress (e.g., shear stress or solid stress), immune/inflammatory cells that have infiltrated the tissue, and genetic mutations (e.g., activation of oncogenes or deletion of suppressor genes that control the production of angiogenesis regulators).13,14,44–47 These molecules can emanate from cancer cells, endothelial cells, stromal cells, blood, and extracellular matrix (Fig. 8-3).48–51 Because the normal host cells differ among organs, the detailed mechanisms of angiogenesis might depend on the specific host-tumor interactions operating within a given tissue.52–59 Furthermore, because the tumor microenvironment is likely to change during tumor growth, regression, and relapse, profiles of proangiogenic and antiangiogenic molecules are likely to change with time and space.60,61 The challenge currently is to develop a unified conceptual framework to describe the temporal and spatial profiles of this increasingly diverse array of angiogenesis regulators with the aim of developing effective therapeutic strategies.37,62–64
Table 8-1
Angiogenesis Activators and Inhibitors*
| Activators | Function | Inhibitors | Function |
| VEGF family members†‡ | Stimulate angio/vasculogenesis, permeability, leukocyte adhesion | VEGFR-1; soluble VEGFR-1; soluble NRP-1 | Sink for VEGF, VEGF-B, PlGF |
| VEGFR‡, NRP-1, NRP-2 | Integrate angiogenic and survival signals | Ang 2†‡ | Antagonist of Ang 1 |
| EG-VEGF | Stimulate growth of endothelial cells derived from endocrine glands | TSP-1,2 | Inhibit endothelial migration, growth, adhesion, and survival |
| Ang 1 and Tie 2†‡ | Stabilize vessels | Angiostatin and related plasminogen kringles | Inhibit endothelial migration and survival |
| PDGF-BB and receptors | Recruit smooth muscle cells | Endostatin (collagen XVIII fragment) | Inhibit endothelial survival and migration |
| TGF-β1,§ endoglin, TGF-β receptors | Stimulate extracellular matrix production | Tumstatin (collagen IV fragment) | Inhibit endothelial protein synthesis |
| FGF, HGF, MCP-1 | Stimulate angio/arteriogenesis | Vasostatin; calreticulin | Inhibit endothelial growth |
| Integrins αvβ3, αvβ5, α5β1 | Receptors for matrix macromolecules and proteinases | Platelet factor-4 | Inhibit binding of bFGF and VEGF |
| VE-cadherin; PECAM (CD31) | Endothelial junctional molecules | Tissue-inhibitors of MMP (TIMPs); MMP-inhibitors; PEX | Suppress pathological angiogenesis |
| Ephrins‡ | Regulate arterial/venous specification | Meth-1; Meth-2 | Inhibitors containing MMP-, TSP-, and disintegrin domains |
| Plasminogen activators, MMPs | Remodel matrix, release growth factor | IFN-α, -β, -γ; IP-10, IL-4, IL-12, IL-18 | Inhibit endothelial migration; downregulate bFGF |
| PAI-1 | Stabilize nascent vessels | Prothrombin kringle-2; anti-thrombin III fragment | Suppress endothelial growth |
| NOS; COX-2 | Stimulate angiogenesis and vasodilation | 16 kD-prolactin | Inhibit bFGF/VEGF |
| AC133 | Regulate angioblast differentiation | VEGI | Modulate cell growth |
| Chemokines§ | Pleiotropic role in angiogenesis | Fragment of SPARC | Inhibit endothelial binding and activity of VEGF |
| Id1/Id3 | Inhibit differentiation | Osteopontin fragment Maspin Canstatin, proliferin-related protein, restin |
Interfere with integrin signaling Protease inhibitor mechanisms unknown |
*Selected list updated from reference 11; for complete function and references, see supplementary information (http://steele.mgh.harvard.edu).
Vascular Architecture
In normal tissue, blood flows from an artery to arterioles to capillaries to venules to a vein. Although the tumor vasculature originates from these host vessels and the mechanisms of angiogenesis are similar, its organization may differ dramatically, depending on the tumor type, its location, and whether it is growing, regressing, or relapsing.15,65–68 In general, tumor vessels are dilated, saccular, tortuous, and chaotic in their patterns of interconnection.69 For example, whereas normal vasculature is characterized by dichotomous branching, tumor vasculature has many trifurcations and branches with uneven diameters.70,71 The fractal dimensions and minimum path lengths of tumor vasculature are different from those of normal host vasculature.67–67 The molecular mechanisms of this abnormal vascular architecture are not understood, but it seems reasonable to hypothesize that the imbalance of VEGF and angiopoietins is a key contributor.19,72 In mice, “normalization” of the tumor vasculature observed during therapies that reduce VEGF (e.g., hormone withdrawal from a hormone-dependent tumor), interfere with VEGF signaling (e.g., treatment with anti-VEGF or anti-VEGFR2 antibody; Fig. 8-4), or mimic an antiangiogenic cocktail (e.g., trastuzumab [Herceptin] treatment of a HER2-overexpressing tumor) is in concert with this molecular hypothesis.61,63,73–76 Mechanical solid stress (i.e., physical forces exerted by solid tissue components) generated by proliferating cancer and stromal cells and their excessive interstitial matrix production also can lead to the partially compressed or totally collapsed vessels often found in tumors (Fig. 8-5).64,77,78 The decompression of blood vessels observed after induction of apoptosis in perivascular cells or stromal cells, or enzymatic degradation of matrix hyaluronan, supports this mechanical hypothesis.78–81 Perhaps the combination of both molecular and mechanical factors renders the tumor vasculature abnormal, and thus both types of factors must be taken into account when designing novel strategies for cancer treatment.
Blood Flow and Microcirculation
Blood flow in a vascular network, whether normal or abnormal, is governed by the arteriovenous pressure difference and flow resistance. Flow resistance is a function of the vascular architecture (referred to as geometric resistance) and of the blood viscosity (rheology, referred to as viscous resistance).69 Abnormalities in both vasculature and viscosity increase the resistance to blood flow in tumors.71,82–84 As a result, overall perfusion rates (i.e., blood flow rate per unit volume) in tumors are lower than in many normal tissues.85,86 Both macroscopically and microscopically, tumor blood flow is temporally and spatially chaotic. Macroscopically, four spatial regions can be recognized in a tumor (Fig. 8-6):
At the microscopic level, in normal tissues, erythrocyte velocity is dependent on vessel diameter, but there is no such dependence in most tumors.54,59,89 Furthermore, the average erythrocyte velocity can be an order of magnitude lower in some tumors compared with that of normal host tissue (Fig. 8-7).59 In a given vessel within a tumor, blood flow fluctuates with time and can reverse its direction.87,89,90 In addition to the elevated geometric and viscous (rheologic) resistance, other molecular and mechanical factors contribute to this spatial and temporal heterogeneity. These factors include imbalance between proangiogenic and antiangiogenic molecules, “solid stress” generated by proliferating tumor cells and matrix production, vascular remodeling by intussusception, and coupling between luminal and interstitial fluid pressure via hyperpermeability of tumor vessels.6,16,64,65,77–79,91–93 This heterogeneity contributes to both acute and chronic hypoxia in tumors—a major cause of resistance to radiation and other therapies. Considerable effort has gone into increasing tumor blood flow for improving radiation therapy, or decreasing tumor perfusion in the case of hyperthermia. Increased perfusion also correlates with a positive response to radio/chemotherapy and patient survival.94,95 Increased perfusion has been difficult to achieve reproducibly, because tumor vasculature consists of both vessels co-opted from the preexisting host vasculature and vessels resulting from the angiogenic response of host vessels to cancer cells. The former are invested in normal contractile perivascular cells, whereas the latter lack these perivascular cells or these cells are abnormal.48,69,96 Presumably as a result, efforts to increase tumor blood flow by pharmacologic or physical agents have not always been reproducible or successful.48,69,85,96 On the other hand, the strategy of decreasing or shutting down tumor blood flow by “stealing” blood away from the “passive component” of the tumor vasculature by vasodilators, by vascular targeting, or by intravascular coagulation has shown promise in experimental systems.85,86,97–99 It also appears that judiciously applied antiangiogenic therapy could “normalize” the abnormal tumor microcirculation by pruning the immature vessels (see Fig. 8-4), thus rendering the remaining vasculature more responsive to vasoactive agents.100
Vascular Permeability
Once a blood-borne molecule has reached an exchange vessel, its extravasation occurs by diffusion, convection, and, to some extent, presumably by transcytosis.100,101 The effective permeability, P, of a molecule depends on the size, shape, charge, and flexibility of the molecule and on the size, shape, charge, and dynamics of the transvascular transport pathway. In normal vessels, these pathways include diffusion along the EC membrane (for lipophilic solutes), trans-EC diffusion, interendothelial junctions (<7 nm), open or closed fenestrations (<10 nm), and transendothelial channels (including vesicles or vesicovacuolar channels).15,102 Some of these anatomic pathways may be lined with glycocalyx on ECs, thus effectively reducing the size of the pathway. A basement membrane may further retard the movement of molecules. Ultrastructure studies show widened interendothelial junctions, increased numbers of fenestrations, vesicles, and vesicovacuolar channels in tumor vessels, and a lack of normal basement membrane and pericytes.15,51,96,102,103 In concert with these ultrastructural findings, both vascular permeability and hydraulic conductivity (a measure of water movement by pressure gradient) of tumors, in general, are significantly higher than those for various normal tissues.59,104–108 Furthermore, unlike normal vessels in the surrounding tissue, tumor vessels partially lose molecular size–based selectivity in permeability to different molecules.109 Despite increased overall permeability, not all blood vessels of a tumor are leaky (Fig. 8-8, A). Even the leaky vessels have a finite pore size that is tumor dependent (Fig. 8-8, B), and ultrastructural studies show that the larger pore size in tumors represents wide interendothelial junctions.56,103 Thus permeability is inversely related to molecular size due to steric and hydrodynamic hindrance from these pores, and very large molecules, such as nanoparticles, do not penetrate tumors efficiently.110,111 Molecular shape also influences penetration of these pores, such that rod-shaped molecules have a higher permeability than do spherical molecules.112 Similarly, positively charged molecules have a higher affinity for the negatively charged glycocalyx in the pores of these angiogenic tumor vessels, reducing this hindrance and giving them a higher permeability.113–116 Not only do the vascular permeability and pore size vary from one tumor to the next (see Fig. 8-8, A-C), but within the same tumor they vary spatially and temporally, as they do during tumor growth, regression, and relapse.56,60,61 The local microenvironment plays an important role in controlling vascular permeability (Fig. 8-8, D). For example, a human glioma (HGL21) has fairly leaky vessels when grown subcutaneously in immunodeficient mice, but it exhibits blood-brain barrier properties in the brain.59,72 Such site-dependent differences for other tumors have been observed in other orthotopic sites.54,57,58 One possible explanation is that the host-tumor interactions control the production and secretion of cytokines associated with permeability increase (e.g., VEGF) and decrease (e.g., angiopoietin 1).19,39,72,117,118 A better understanding of the molecular mechanisms of permeability regulation in tumors is likely to yield strategies for improved delivery of molecular medicine to tumors.
Movement of Cells Across Vessel Walls
Both cancer cells and immune cells frequently move across the walls of blood vessels—the former in the process of metastasis, and the latter during immune response or cell-based immunotherapy. Both transendothelial pathways (through ECs) and periendothelial pathways (between ECs) have been proposed as a route for intravasation and extravasation of cells. Very little is known about intravasation except that tumors might shed more than a million cells per gram per day and most of these are not clonogenic.119–122 Recently, it was demonstrated that metastatic cells could bring their own stroma from the primary tumor in the form of hereotypic cell clumps.123 More is known about the molecular and cellular mechanisms of extravasation.124 When a cell enters a blood vessel, it can continue to move with the flowing blood, collide with the vessel wall, adhere transiently or stably, and finally extravasate. These interactions are governed both by local hydrodynamic forces and adhesive forces. The former are determined by the vessel diameter and fluid velocity and the latter by the expression, strength, and kinetics of bond formation between adhesion molecules and by the surface area of contact.125–129 Deformability of cells affects both types of forces.130 In addition, cancer cells may grow intravascularly at a distant site (e.g., in the lungs).131
Rolling of endogenous leukocytes is generally low in tumor vessels, whereas stable adhesion (≥30 sec) is comparable between normal vessels and tumor vessels.132 On the other hand, both rolling and stable adhesion are nearly zero in angiogenic vessels induced in collagen gels by bFGF or VEGF, two of the most potent angiogenic factors.133 Whether this observation is due to a low flux of leukocytes into angiogenic vessels and/or to downregulation of adhesion molecules in these immature vessels currently is not known. The age of the animal also plays an important role in leukocyte-endothelial interactions.134 Further insight into the types of cells that adhere to tumor vessels comes from studies on the localization of IL-2-activated natural killer (A-NK) cells in normal and tumor tissues in mice using positron emission tomography.135,136 After systemic injection, these cells localized primarily in the lungs immediately after injection and could not be detected in the tumor.135 Increased rigidity caused by IL-2 activation might contribute to the mechanical entrapment of these cells in the lung microcirculation.137,138 Constitutive expression of certain adhesion molecules in the lung vasculature also might facilitate their retention in the lungs.124 One approach to reducing lung entrapment is to reduce the rigidity of these cells.112,117 Alternatively, the lung can be circumvented by injecting A-NK cells directly into the blood supply of tumors. In this case, A-NK cells, both xenogenic and syngeneic, adhered to some blood vessels in three different tumor models via CD18 and very large antigen-4 (VLA-4) on the A-NK cells and intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin on the activated endothelium of angiogenic vessels.33,136,139–142 These molecules can be upregulated by tumor necrosis factor–α (TNF-α) and a protein of 90 kd molecular weight (p90) that is secreted by some neoplastic cells and downregulated by transforming growth factor–β (TGF-β)—also, presumably, secreted by cancer cells.55,125,143–147 Surprisingly, the proangiogenic VEGF also can upregulate these molecules, whereas another proangiogenic molecule, bFGF, can downregulate these molecules.33,51,61,76,148 Inflammatory cells such as monocyte/macrophages or neutrophils also are recruited by VEGF and may play important roles in promoting matrix remodeling and angiogenesis.149 The challenge currently is to decrease nonspecific entrapment of immune cells in normal vessels and to increase their delivery to tumor vessels to improve various cell-based therapies, including gene therapy.
Extravascular Compartment
Composition and Origin
The extravascular compartment of a solid tumor consists of neoplastic cells (parenchyma) and host cells (e.g., inflammatory cells and fibroblasts) residing in an interstitial matrix bathed by the interstitial fluid (see Fig. 8-1). Depending on the tumor type and its stage of differentiation, neoplastic cells might be dispersed in the matrix as individual cells (e.g., lymphomas and melanomas) or as clumps, sheets, or nests (e.g., carcinomas). More than 80% of tumors are carcinomas arising from epithelial cells. The remaining cancers include sarcomas arising from mesenchymal cells (e.g., bone or muscle cells), lymphomas arising from lymphoid tissue, leukemias arising from hematopoietic cells, and hemangiomas arising from ECs. In a poorly differentiated carcinoma, the cancer cells might be packed loosely in clumps, whereas in a well-differentiated carcinoma, the cells might be connected with intercellular junctions and tightly packed in a nest enveloped by a basement membrane. With tumor progression, cancer cells can invade the basement membrane and spread to other regions.15 These various types of normal host cells must migrate into the tumor from normal tissue. Inflammatory cells might enter the tumor via blood vessels or might infiltrate from the adjacent tissue or lymphatics.124 Other host cells, such as fibroblasts, might proliferate and migrate from the adjacent connective tissue.19,48,150 The interstitial subcompartment of a tumor is bounded by the walls of the blood vessels on one side and by the membranes of cancer and stromal cells on the other. In normal tissues, the blood vessels are surrounded by a basement membrane, which, as previously discussed, is defective in tumors.19 In addition, functional lymphatics might be confined to the tumor margin.151,152 The interstitial space of tumors, like that of normal tissues, is composed of a collagen and elastin fiber network that provides structural support to the tissue. Interdispersed in this cross-linked structure are the interstitial fluid and macromolecular constituents (polysaccharides, hyaluronan, and proteoglycans), which form a hydrophilic gel. Compared with current understanding of blood vessel formation, understanding of stroma generation is minimal. Dvorak and coworkers have proposed that the extravasated plasma protein fibrinogen, a key component of the tumor interstitial fluid, clots to form fibrin, which serves as a major component of the provisional stroma. This provisional stroma eventually is replaced by more mature connective tissue stroma. The tumor interstitial fluid also contains several other Arg-Gly-Asp (RGD)-containing proteins, including fibronectin, vitronectin, osteopontin, thrombospondin, decorin, and tenacin.15 These proteins are present in both free and bound forms. Their Arg-Gly-Asp sequence provides a binding site for adhesion that assists in the migration of various cells, including stromal cells. In addition to extravasating from the leaky tumor vessels, these proteins, along with collagen and various proteoglycans, also are synthesized by the stromal cells, albeit in a form that differs from that in the plasma or normal tissues.15 Tumor interstitial fluid also can contain various growth factors that facilitate stroma formation. For example, in vitro studies suggest that platelet-derived growth factor-BB (PDGF-BB) is involved in the recruitment of fibroblasts to tumors, and TGF-β induces the production of collagen and other matrix molecules in tumors.19,153 With the increasing interest in using the fragments of matrix constituents for controlling angiogenesis, increased understanding of the molecular and cellular mechanisms of stroma generation in tumors will increase.43
Interstitial Transport
Once a molecule has extravasated, its movement through the interstitial space occurs by diffusion and convection.101,154 Diffusion is proportional to the concentration gradient in the interstitium, and convection is proportional to the interstitial fluid velocity, which, in turn, is proportional to the fluid pressure gradient in the interstitium. Just as the interstitial diffusion coefficient D (cm2/s) relates the diffusive flux to the concentration gradient, the interstitial hydraulic conductivity K (cm2/mmHg/sec) relates the interstitial velocity to the pressure gradient.134 Values of these transport coefficients are governed by the structure and composition of the interstitial compartment and by the physicochemical properties of the solute molecule.155–165 The value of K for a human colon carcinoma xenograft (LS174T), measured using two different methods, was found to be higher than that of a hepatoma, which, in turn, was higher than that of the normal liver.167–167 Using fluorescence recovery after photobleaching, D of various molecules in tumors was found to be about one third that in water and higher than the values in the host tissue (Fig. 8-9, A).157,168 Collagen content and structure have a significant effect on D in tumors.159,163,166,169–173 This finding is surprising because hyaluronan and proteoglycans, not collagen, account for most of the resistance to transport in normal tissues. Because collagen is produced by host cells (e.g., fibroblasts), the penetrability into a tumor depends on the host-tumor interaction (see Fig. 8-9, A). Thus agents that interfere with collagen synthesis and/or organization (e.g., relaxin, losartan, and bacterial collagenase) might increase interstitial transport in tumors156,169,170 (Fig. 8-9, B). The time constant for a molecule with diffusion coefficient D to diffuse across a distance L is approximately L2/4D. For diffusion of immunoglobulin G (IgG) in tumors, this time constant is on the order of 1 hour for a 100-µm distance, days for a 1-mm distance, and months for a 1-cm distance. Thus for a 1-mm distance in tumor, diffusional transport would take days, and for a 1-cm distance in tumor, it would take months. Furthermore, the path through the interstitium is tortuous over large length scales, greatly increasing the distance a molecule must travel.158 If the central vessels have collapsed completely as a result of cellular proliferation and interstitial matrix rearrangement, the reduced delivery of macromolecules by blood flow would make diffusion the primary mechanism of delivery to this necrotic center.6,79 Binding of a low- or high-molecular-weight drug to plasma proteins and various tissue components could further retard their transport in tumors.168,174–180 The role of binding is clearly illustrated in Figure 8-9, C, which compares the rate of fluorescence recovery of a photobleached spot in tumor tissue injected with a nonspecific versus a specific IgG. In addition to the heterogeneity of D in tumors, the most unexpected result of these photobleaching studies was the large extent (30% to 40%) of nonspecific binding.168
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
