[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Therapeutic Targeting of Cancer Cells
Era of Molecularly Targeted Agents
Shivaani Kummar, Anthony J. Murgo, Joseph E. Tomaszewski and James H. Doroshow
• Molecularly targeted anticancer agents (MTAs) are those that selectively target specific molecular features of cancer cells such as aberrations in genes, proteins, or pathways that regulate tumor growth, progression, and survival.
• Molecular targets include the following: products of activating mutations and translocations, growth factors and receptors, aberrant signal transduction and apoptotic pathways, factors that control tumor angiogenesis and microenvironment, dysregulated proteins, DNA repair machinery, and aberrant epigenetic mechanisms.
• The development of MTAs requires innovative strategies that differ from those traditionally applied to nontargeted conventional chemotherapy.
• Successful development of an MTA depends largely on the importance of the target in controlling tumor cell proliferation and survival and effective modulation of the target in the tumor at clinically achievable concentrations.
• Primary objectives of clinical trials of MTAs differ from those used in trials of conventional chemotherapy. An important objective in phase 1 trials of MTAs should be to determine a phase 2 dose based on optimal target modulation (i.e., a biologically effective dose) rather than on maximum tolerated dose. In addition, objective tumor response may not be an adequate end point for efficacy evaluations of MTAs that have a primarily cytostatic effect. Alternate end points, such as progression-free survival, may be more appropriate.
• Functional and molecular imaging plays an increasingly important role in the development of MTAs.
Molecular Targets
The increasing number and assortment of molecular targets can be broadly categorized according to genetic or functional properties, including products of activating gene mutations and translocations; growth factors and receptors; aberrant signal transduction and apoptotic pathways; factors that control tumor angiogenesis and microenvironment; dysregulated proteins; DNA repair machinery; and aberrant epigenetic mechanisms (Tables 28-1 and 28-2).
Table 28-1
U.S. Food and Drug Administration–Approved Molecularly Targeted Agents
| FDA Approved Agents(s) | Target | Disease Indication |
| Imatinib mesylate | BCR-ABL, KIT, PDGFR-β | Ph+ CML, Ph+ ALL, chronic monomyelocytic leukemia, dermatofibrosarcoma protuberans |
| Dasatinib | BCR-ABL, Src family kinases | Ph+ CML |
| Nilotinib hydrochloride monohydrate | BCR-ABL | Ph+ CML |
| All-trans retinoic acid | PML-RAR | Acute promyelocytic leukemia |
| Sunitinib malate | VEGFR, KIT, PDGFR-α | GIST, pancreatic neuroendocrine tumors, kidney cancer |
| Vemurafenib | BRAFV600E mutation | BRAFV600E-positive melanoma |
| Crizotinib | ALK tyrosine kinase | ALK-positive non–small cell lung cancer |
| Erlotinib | EGFR | Non–small cell lung cancer |
| Panitumumab | EGFR | Colorectal cancer |
| Cetuximab | EGFR | Head and neck cancer, colorectal cancer |
| Trastuzumab | ErbB-2 | HER2 overexpressing breast cancer |
| Lapatinib | ErbB-2 | HER2 overexpressing breast cancer |
| Vismodegib | Hedgehog pathway | Advanced/metastatic basal cell carcinoma |
| Brentuximab vedotin | CD30 | Hodgkin lymphoma, anaplastic large cell lymphoma |
| Ofatumumab | CD20 | CLL |
| Ibritumomab | Non-Hodgkin lymphoma | |
| Rituximab | B-cell non-Hodgkin lymphoma | |
| Alemtuzumab | CD52 | B-cell chronic lymphocytic leukemia |
| Bevacizumab | VEGF | Colorectal cancer, non–small cell lung cancer, renal cell carcinoma |
| Vandetanib | VEGFR | Medullary thyroid cancer |
| Sorafenib | VEGFR, B-Raf kinase | Kidney cancer, hepatocellular cancer |
| Pazopanib | VEGFR | Soft tissue sarcomas, kidney cancer |
| Axitinib | VEGFR | Kidney cancer |
| Temsirolimus | mTOR | Renal cell carcinoma |
| Everolimus | mTOR | Renal cell carcinoma, advanced pancreatic neuroendocrine tumors |
| Azacitidine | DNA methyltransferase | Myelodysplastic syndrome |
| Decitabine | DNA methyltransferase | Myelodysplastic syndrome |
| Vorinostat | Histone deacetylase | Cutaneous T-cell lymphoma |
| Romidepsin | Histone deacetylase | Cutaneous T-cell lymphoma |
| Bortezomib | Proteosome | Multiple myeloma |
| Ipilimumab | Cytotoxic T-lymphocyte–associated antigen 4 | Metastatic melanoma |
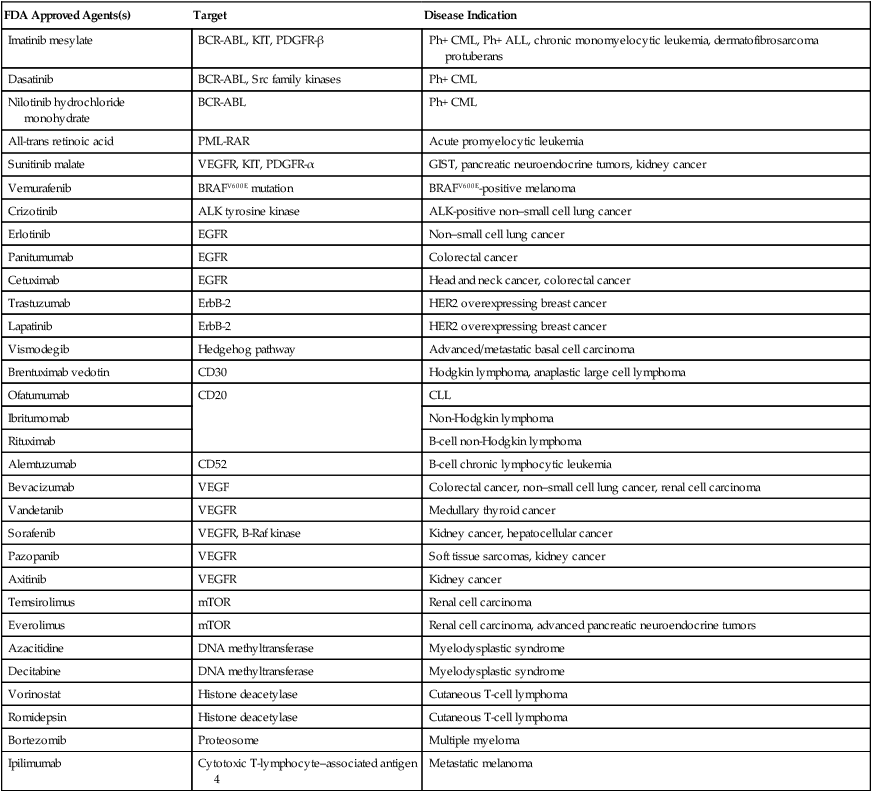
Table 28-2
Classes of Molecularly Targeted Anticancer Agents under Clinical Development
| Target | Role of Target | Disease Indication | References |
| FMS-like tyrosine kinase-3 (FLT-3) | Regulates cell cycle progression, proliferation, and survival | Acute myeloid leukemia | 61 |
| Poly (ADP-ribose) polymerase (PARP) | Single-strand DNA break repair | Various solid tumors, BRCA-positive breast and ovarian cancer | 62 |
| O6-Alkylguanine DNA alkyltransferase | Prevents intrastrand DNA cross-links | 63 | |
| Insulin-like growth factor-1 receptor (IGF-1R) | Regulates cell proliferation, differentiation and survival | Various adult and pediatric tumors | 64 |
| Insulin-like growth factor binding protein-3 (IGFBP3) | Regulates cell proliferation, differentiation and survival | Various adult and pediatric tumors | 64 |
| Hypoxia-inducible factor-1α (HIF-1α) | Regulates tumor cell response to oxygen deprivation | HIF-1α–expressing solid tumors | 65 |
| αvβ3 integrin receptor | Involved in cell adhesion | Glioma and various solid tumors | 66, 67 |
| Syk | Inhibits differentiation and induces growth factor–independent proliferation of pre–B cells | B-cell lymphomas | 68 |
| MET | Regulates cell growth, anti-apoptosis, altered cytoskeletal function | Solid tumors | 69 |
| MEK1/2 | Regulates tumor cell proliferation and survival | Various solid tumors, melanoma | 70, 71 |
| RAS | Promote cell proliferation and survival | Acute myeloid leukemia | 72 |
| RAF | Regulates tumor cell proliferation and survival | Melanoma and other solid tumors | 73 |
| RET | Regulates tumor cell proliferation and survival | Medullary thyroid cancer | 74 |
| Akt kinase | Regulator of cell cycle and apoptotic pathway | Variety of solid and hematologic cancer | 75 |
| Clusterin | Promotes cell survival | Variety of solid tumors | 76 |
| HSP-90 | Chaperone for several oncogenic proteins and growth factors | Myeloid leukemia and solid tumors | 77 |
| Bcl-2 | Antiapoptotic | Lymphomas, solid tumors | 78 |
| Tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) | Apoptotic mechanism | Various types of cancer | 79 |
| Angiopoietin 1 and 2 | Reduces blood flow to tumor | Solid tumors | 80 |
| DNA methyltransferase inhibitors | Reexpression of genes silenced by methylation | Myelodysplastic syndrome, acute myeloid leukemia, various solid tumors in combination with chemotherapy or radiation | 58, 59 |
| Src inhibitors | Src family kinases that interact with other growth, proliferation, and angiogenic pathways | Solid tumors | 81 |
| VEGFR, VEGF | Reduces blood flow to tumor | Solid tumors | 11 |
| CD105 (Endoglin) | Reduces blood flow to tumor | Solid tumors | 82 |
| HDAC | Reexpression of silenced genes | T-cell lymphoproliferative disorders, adenoid cystic cancer, solid tumors | 83 |
| Retinoid receptor | Increase in reactive oxygen species and dihyceramide levels resulting in cytotoxicity | T-cell lymphoproliferative disorders | 84, 85 |
| Wee 1 | DNA repair and cell cycle (abrogates the G2 cell cycle checkpoint) | Solid tumors | 86 |
| BTK inhibitor | Bruton tyrosine kinase | Lymphoma, chronic lymphocytic leukemia | 87 |
| Gamma secretase inhibitor | Notch pathway | Solid tumor | 88 |
| CDK inhibitors | Cyclin-dependent kinases (cell cycle, proliferation) | Solid tumors | 89 |
| CHK1 inhibitors | Checkpoint kinases (cell cycle) | Solid tumors | 90 |
| DNA minor groove | DNA damage | Solid tumors | 91 |
| SMAC mimetic, IAP inhibitor | Induction of apoptosis | Solid tumors | 92 |
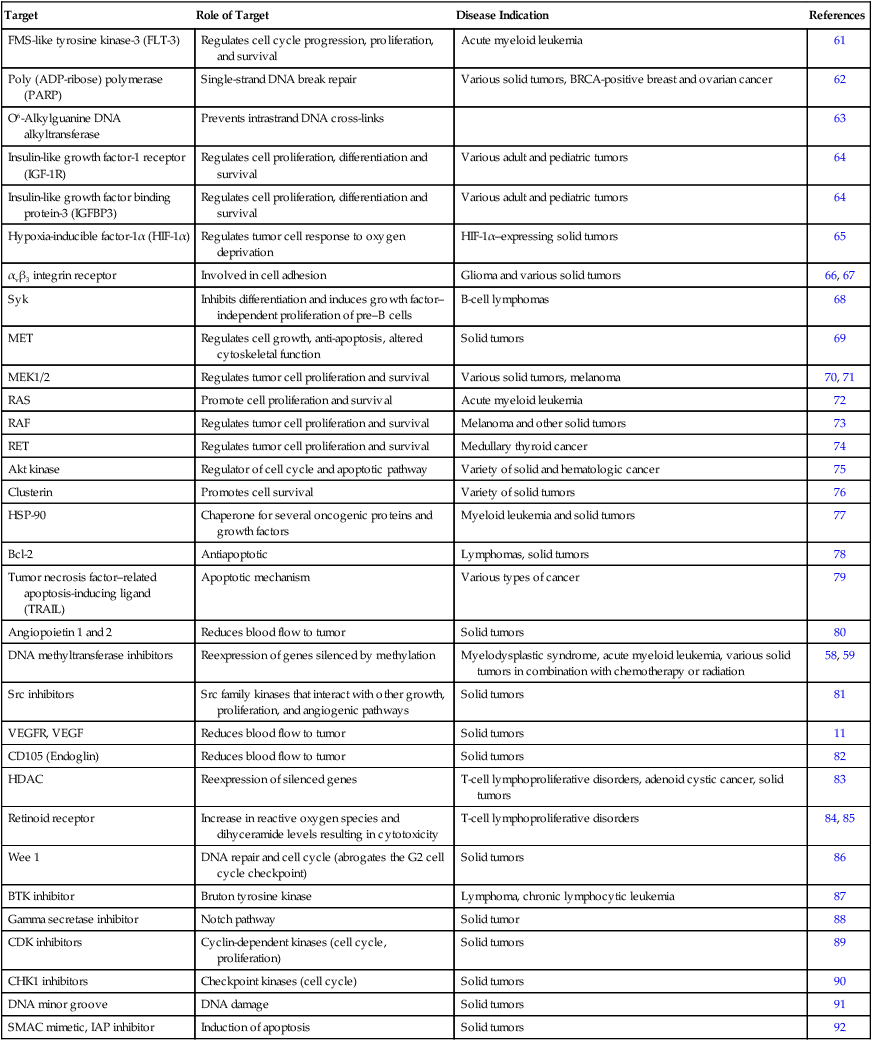
The most promising molecular targets are those solely responsible for sustaining tumor growth and survival because agents that potently and selectively inhibit these critical targets are likely to have a major clinical impact. Probably the best example of a critical target is BCR-ABL in patients with chronic myelogenous leukemia (CML). BCR-ABL is a fusion protein formed by the reciprocal translocation of chromosomes 9 and 22. Knowledge that this dysregulated tyrosine kinase played a causal role in the pathogenesis of essentially all cases of CML spurred preclinical studies, which led to the development of a potent and selective ABL tyrosine kinase inhibitor (TKI), imatinib mesylate (previously known as STI571).1 Subsequent clinical trials established imatinib mesylate as the first highly effective molecularly targeted therapy for CML and a prototype for the development of others in the class. Imatinib mesylate is also a potent inhibitor of other tyrosine kinases including PDGFR and KIT, and it is highly effective in the treatment of gastrointestinal stromal tumors (GISTs) bearing activating c-KIT mutations and in some GISTs bearing activating PDGFR mutations.1,2
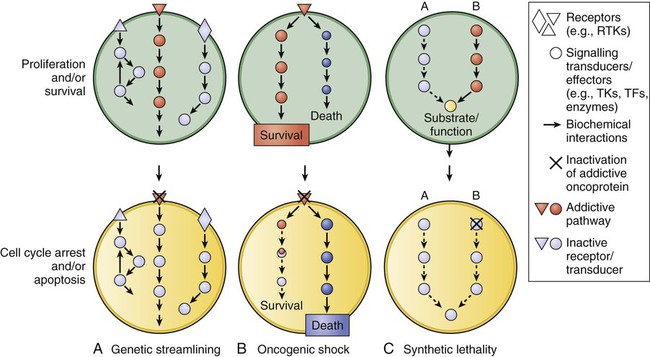
Unfortunately, most human tumors, including the most common types, are genetically complex and do not have a single critical target, and the relative critical importance of a target in different tumors may vary. Most tumor types have various genetic and molecular abnormalities driving their growth and survival. The existence of multiple abnormalities in one or more molecular pathways helps explain resistance to molecularly targeted therapy and provides a rationale for treatment strategies combining two or more targeted agents.3,4 However, cancer cells may become “addicted” or physiologically dependent on the sustained activity of specific oncogenes for maintenance of a malignant phenotype and for survival. This dependence mechanism, termed oncogene addiction7–7 (Fig. 28-1), is associated with differential attenuation rates of prosurvival and proapoptotic signals stemming from the oncoprotein, with predominant apoptotic signals resulting in cell killing. The latter process, termed “oncogenic shock,”8 could explain the remarkably rapid clinical responses to TKIs in some patients with solid tumors, including those typically having complex molecular abnormalities. Other possible factors controlling sensitivity or resistance to molecularly targeted therapy include increased expression of the target due to gene amplification or transcription, emergence of resistant target gene mutations, and overexpression of multidrug transporter membrane proteins.4,7
Interest in molecular therapy directed at factors controlling angiogenesis has increased since the United States Food and Drug Administration (FDA) approved several agents that target vascular endothelial growth factor (VEGF) and its receptor (VEGFR). The VEGF pathway, involving the VEGF family of proteins and their receptors, is an important regulator of both physiological and pathological angiogenesis. Through its signaling pathways, VEGF/VEGFR activation contributes to increased vascular permeability, mobilization of bone marrow–derived endothelial cell precursors, degradation of the extracellular matrix, and endothelial cell division, differentiation, migration, and survival.9,10 VEGF overexpression occurs in most types of cancers, including colorectal, renal, gastric, pancreatic, liver, lung, breast, thyroid, and genitourinary; it also occurs in glioma and other intracranial tumors, as well as in hematologic malignancies, and it is associated with tumor growth and a worse clinical outcome in a number of these tumor types.10,11
Potential therapeutic strategies to inhibit signaling through VEGF and VEGFR pathway activation include monoclonal antibodies directed against VEGF or VEGFR, TKIs, and antisense strategies (antisense oligodeoxynucleotides, antisense RNA, and small interfering RNAs). In 2004, the FDA approved bevacizumab, a humanized murine monoclonal antibody directed against VEGF, for treating metastatic colorectal cancer in combination with fluorouracil-based chemotherapy. The FDA subsequently approved sunitinib, sorafenib, and axitinib, three small-molecule TKIs with activity against VEGFR, for the treatment of advanced renal cancer.11 In addition, sorafenib is effective for treating hepatocellular carcinoma, a tumor against which standard cytotoxic chemotherapy has little or no activity.11,12 The experience with these agents has established the VEGF/VEGFR pathway as a valid target for cancer therapeutics.11
Mammalian target of rapamycin (mTOR) has also emerged as a validated target with the demonstration that the small-molecule mammalian target of rapamycin inhibitors temsirolimus and everolimus are effective in the treatment of renal cell carcinoma.13 This activity of temsirolimus is attributed to the downregulation of factors that control cell growth and angiogenesis.13
Preclinical Development of MTAs
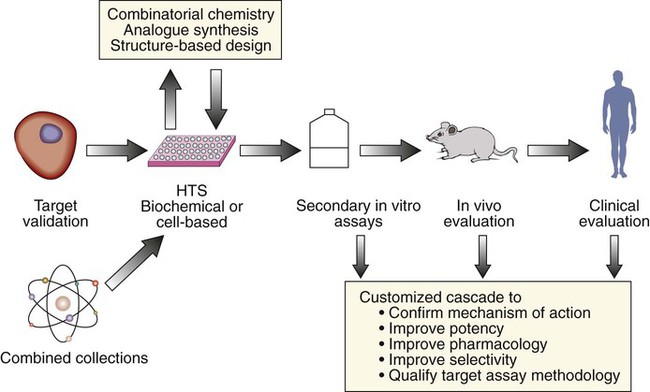
The discovery and development of molecularly targeted therapies requires closely aligned laboratory and clinical research, integrating drug discovery, development, and clinical investigation. In such a cooperative setting, researchers can effectively take rational and iterative steps from target identification to clinical evaluation (Box 28-1) (Fig. 28-2). A crucial early step in developing a molecularly targeted therapy is target validation, defined as experimental evaluation of the role of a given gene or protein.14,15 The target validation process involves a variety of preclinical approaches, including genetic, cell-based, and animal models.16 Validation and prioritization of molecular targets for therapeutic development depends on a variety of criteria, taking into consideration chemical, biological, clinical, and practical factors15 (Box 28-2). The fundamental goal is to provide evidence that the target is valid (i.e., that affecting the target inhibits tumor growth, progression, or survival) and that making drugs that hit the target is feasible. The next major step in the development of molecularly targeted therapy is finding/synthesizing compounds directed against that target.
Empirical approaches traditionally used to screen for cytotoxic agents are not optimal for MTAs. Rather, screening for MTAs should be target-based. The increasing number of potential targets and the availability of sophisticated high-throughput screening technology provide tremendous opportunities to screen an enormous set of diverse small molecular compounds for promising therapeutics.15 Furthermore, the availability of genetically engineered mouse models provide the opportunity to better screen compounds in vivo.16 A more in-depth discussion of these important drug discovery tools and their implications for the molecularly targeted drug development process is beyond the scope of this chapter.
Optimal development of an MTA requires careful assessment of pharmacokinetic (PK) and pharmacodynamic (PD) effects in relevant nonclinical models before initiating clinical trials. Preclinical in vivo pharmacologic and toxicologic testing is necessary to establish the starting dose and schedule for clinical trials, to evaluate effects on normal host tissue, and to help make predictions about serum and tissue levels required for target modulation, as well as effects on tumor growth. Currently, most in vivo efficacy studies involve human tumor cell murine xenograft models.17 However, xenograft models have limitations, including the requirement for an immunocompromised host, and they are not ideal for simulating the complex relationship between tumor and microenvironment, such as angiogenesis. Moreover, xenograft models have not been very predictive of drug efficacy in patients with cancer; activity in a particular histology in a xenograft tumor model does not closely correlate with activity in the same human cancer histology.18 However, agents that have activity against a broad range of tumor types in xenografts have a better chance of clinical activity than do agents that do not have activity against a broad range of tumor types. Major variables to consider in designing xenograft studies include the molecular characteristics of the tumor, site of implantation, size of the tumor at commencement of drug treatment, and dose and schedule of administration. Clearly, future development of MTAs would benefit from more predictive in vivo models. Although limitations exist, prudent use of genetically engineered mouse models in conjunction with traditional xenograft models holds promise for accelerating targeted drug development.17
Clinical Development of MTAs
Successful development of MTAs may require different strategies than those traditionally used for the development of conventional cytotoxic chemotherapeutic agents (Table 28-3).19 Consequently, the discovery of an increasing number of molecularly targeted compounds has created new challenges in drug development, both for dose determination and efficacy evaluation. In animal models, some of these newer agents are cytostatic in that they exhibit growth inhibitory and antimetastatic effects without causing impressive tumor regression. Although the specific effects of each class are dependent on the target or targets inhibited, in patients, compared with cytotoxic agents, MTAs are more likely to result in tumor growth inhibition than tumor regression. Pivotal clinical trials of sorafenib in patients with renal cell and hepatocellular carcinoma, for example, substantiate the cytostatic properties of this small-molecule VEGFR inhibitor, with results showing marked improvement in progression-free survival (PFS) with minimal objective tumor response by response evaluation criteria in solid tumors (RECIST) criteria.12,20
Table 28-3
Properties and Development of Nontargeted versus Molecularly Targeted Anticancer Agents
| Nontargeted Chemotherapy | Molecularly Targeted | |
| Drug discovery | Empirical approaches; screening for compounds that inhibit tumor growth/survival | Target-based approaches; screening for compounds that hit tumor target |
| Antitumor effects | Cytotoxic; tumor shrinkage | May be cytostatic; growth inhibition |
| Host toxicity | Nonselective, toxic to multiple normal organ systems | Selective; depends on target(s) specificity; “off-target” effects |
| Therapeutic window | Usually narrow | Usually wider |
| Phase 1 primary end points | DLT and MTD; PK | Target inhibition and optimal biological dose; PK |
| Dosing | Intermittent | Intermittent or continuous |
| Phase 2 efficacy trial end points | Objective tumor response (tumor shrinkage) | Tumor response or progression-free survival |
| Phase 2 dose | Based on MTD | Optimal biological dose |
| Patient selection | Histology | Target expression, if possible |
| Interval to clinical response | Relatively short (e.g., 4-8 wk) | Variable |
DLT, Dose-limiting toxicity; MTD, maximum tolerated dose; PK, pharmacokinetic.
Dose Determination
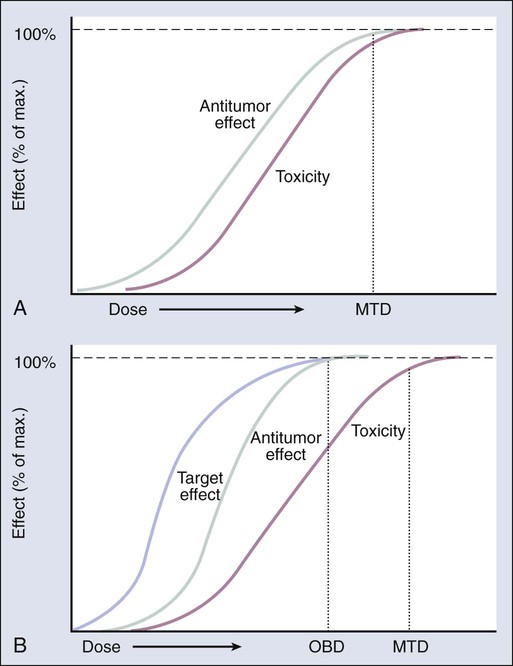
The primary objective of phase 1 trials of conventional cytotoxic agents ordinarily is to define maximum tolerated dose (MTD), under the assumption that the higher the dose, the greater the antitumor activity (Fig. 28-3, A). Thus the dose recommended for phase 2 efficacy trials is typically the MTD. However, MTAs are generally less toxic and, because of their selectivity, may be effective at doses much lower than the MTD (Fig. 28-3, B). MTAs act on specific targets differentially expressed or activated in cancer cells, and thus they tend to have a wider therapeutic index than traditional cytotoxics. Therefore increasing the dose in a traditional phase 1 trial to the point of normal tissue tolerance (MTD) may be an irrelevant or nonachievable end point with an MTA. Rather, target inhibition may be a better end point, with the primary objective of clinical trials being the determination of an “optimal biological dose,” the lowest dose that maximally inhibits the relevant target or pathway.19,21
Phase 1 trials intended primarily to determine a dose that optimally inhibits a target in tumor tissue usually involve pre- and posttreatment tumor tissue acquisition. However, the requirement for tissue biopsies is costly and hampers clinical trial enrollment because only patients with tumors accessible for biopsy are eligible, and not all otherwise eligible patients consent to the procedure. Therefore in lieu of tumor biopsies, one strategy for evaluating target modulation and determining a biologically active dose is to use less-invasive procedures involving target or biomarker assessment in surrogate tissues expressing the target, such as circulating tumor cells (CTCs).22,23 For example, demonstrating a “biologically effective dose” in CTCs may serve as a guide in dose-finding trials. However, before an effect on target in surrogate tissue can be used for dose determination, it must be established that it is a true surrogate for drug effects in patients’ tumors. This need was exemplified in trials conducted with O6-benzylguanine, an inhibitor of O6-alkylguanine DNA alkyltransferase (AGT), as a modulator of resistance, in combination with bis-chloroethylnitrosourea in melanoma and other malignancies.24 No difference was found in the efficacy of the combination compared with single-agent bis-chloroethylnitrosourea, although inhibition of AGT levels was demonstrated in peripheral blood mononuclear cells at the doses tested; however, tumor samples were not obtained to show that AGT was actually inhibited at the dose and schedule of O6-benzylguanine tested. The differential effect of O6-benzylguanine on AGT levels in tumor versus peripheral blood mononuclear cells was only established in a separate trial.25
To define a biologically effective dose, it is essential to have a validated target (defined previously) and a reliable, reproducible assay to evaluate target inhibition. This requires having a clear understanding of the dynamics and consequences of target inhibition—specifically, the degree of target effect required for tumor growth inhibition, whether the target needs to be inhibited continuously or intermittently, and the target’s function in normal cells. Thus, optimal clinical development of MTAs requires extensive preclinical studies, including animal models that are predictive of eventual clinical effect and are applicable to thorough assay interrogation.26 After the appropriate preclinical development of a reliable pharmacodynamic assay, the drug and its effect on tumor (as evaluated by the assay) should optimally be evaluated in first-in-human pre–phase 1 or “phase 0” studies before definitive phase 1 trials are initiated.19,21,27
Because these pilot pre–phase 1 studies involve a limited number of subjects and very limited drug exposure with no therapeutic intent, they may require less than the usual amount of preclinical toxicity data in accordance with the FDA guidance for Exploratory Investigational New Drug (IND) studies.28 Several types of studies can potentially be conducted under the purview of an Exploratory IND designation, including:
1. Studies that provide important PK information (e.g., oral bioavailability);
2. Studies designed to select the most promising lead agent from several candidates;
3. Studies to determine whether a mechanism of action (e.g., target inhibition) observed in animal models can be observed in humans; and
4. Studies to evaluate in vivo binding properties of novel imaging agents using extremely small doses of the test agent (“microdosing”).
Another approach to determine an optimal dose of an MTA is to conduct a randomized phase 2 trial with a clinical response end point comparing two or more doses. This approach may be necessary if dose selection cannot rely on a biological marker. However, for this design to be effective, the agent must have some degree of clinical activity, yielding objective responses or affecting PFS. Under these circumstances, the randomized phase 2 design is useful in selecting a maximally efficacious dose with the least amount of toxicity that is applicable to subsequent phase 3 trials.29
Efficacy Evaluation
Because of the expanding number of anticancer agents under development with novel mechanisms of action, there is an increased need to develop novel “best fit” paradigms for demonstrating efficacy. A primary end point of objective response rate, considered standard for most phase 2 trials of traditional cytotoxic agents, may not be adequate for targeted agents that produce growth inhibition without tumor regression (Table 28-3).19 Furthermore, evidence of a therapeutic response to cytotoxic drugs is expected to occur relatively early in the course of treatment (e.g., after one to two cycles), whereas MTAs may require dosing for longer periods at low doses to be efficacious and may have maximum clinical effects in combination rather than as single agents. These considerations are exemplified by the clinical development of sorafenib. The single-agent rate of objective response using RECIST criteria with this multi-targeted TKI is minimal (2% to 3%) in patients with advanced renal cell carcinoma. Yet a phase 3 trial of the agent in this disease resulted in a marked improvement in PFS, forming the basis for FDA approval.20 If the main decision factor for further development had been objective tumor response, the true effectiveness of sorafenib may not have been established. For this reason, exploratory efficacy trials of molecularly targeted cytostatic agents increasingly include PFS as a primary end point.
However, molecular heterogeneity and differences in patient and tumor characteristics make evaluation of PFS difficult to interpret, particularly in relatively small exploratory studies without concomitant comparator controls. One trial design proposed to overcome some of these concerns is the randomized discontinuation design, which is considered a type of enrichment trial.30 With one variant of this design, all eligible patients receive a study agent for a defined period, followed by restaging; patients with a predefined degree of tumor shrinkage continue to take the drug, and those with progressive disease are removed from the study. Only the patients who show evidence of stable disease undergo randomization to receive either the study drug or placebo. Thus the randomized population is enriched with sensitive patients because only the patients who do not experience early drug failure continue to receive therapy. The fact that all patients have the opportunity to receive the investigational drug is considered a major attraction of this study design.30
The application of the randomized discontinuation design or other types of novel screening designs in exploratory efficacy studies may be particularly useful in the early development of a molecularly targeted cytostatic agent for which a dependable assay to select patients based on target expression is not available.29,31 Furthermore, randomized phase 2 designs, in general, are applicable to exploratory trials evaluating two or more doses or schedules.32,33 These studies should be considered before embarking on more definitive phase 3 trials, particularly when uncertainty exists about the optimal dosing regimen.
Sometimes the approach taken to evaluate the efficacy (e.g., PFS) of an MTA is to conduct a trial comparing the results of treatment with the targeted agent in combination with a standard agent or regimen to that with the standard regimen alone. The intent of such studies is to show superior efficacy with the combination. The “add-on” approach is useful for the evaluation of agents expected to have little efficacy when used alone, and it is particularly attractive because all patients receive an active agent or regimen. For example, these designs were critical to the successful development of trastuzumab in breast cancer and bevacizumab in colorectal cancer.36–36
This approach, however, is not without significant risk of failure, as was the case with the randomized trials of gefitinib and erlotinib combined with chemotherapy in persons with non–small cell lung cancer (NSCLC).37,38 Reasons for failure include suboptimal dose of the MTA, antagonism between the MTA and the chemotherapy, lack of synergistic or additive antitumor effects, or dilution of the study population with patients having insensitive tumors.39 Such designs should only be considered if there is a reasonable estimate of the single-agent activity of the MTA in the specific patient population to be studied, the agent is administered at an optimally efficacious dose, and the combination is supported by a strong scientific rationale. Furthermore, for agents with targets expressed in only a small proportion of patients, the study should be enriched with patients who have tumors that express the target or have another marker predictive of activity.
Use of Pharmacodynamic Markers
Trial designs for exploratory studies of MTAs may need to rely more on PD end points as markers or surrogates for antitumor effect than on toxicity or objective tumor response. This situation requires the development of PD assays that reliably measure drug-target effect. To do so, the assay must be accurate, precise, reproducible, sensitive, robust, and have a dynamic range tight enough to detect differences between baseline and posttreatment values21 (Box 28-3). Furthermore, the assay methodology assessing drug effect on the target should be developed and authenticated first in in vivo animal models and then preferably in phase 0 clinical trials.
The development of mechanism-based biomarker assays can significantly expedite drug development, because such assays aid in determining early on if a drug modulates the target. This approach could also help select the lead agent from a group of compounds, help determine dose, guide patient selection and assessment of response, and provide the basis for combination trials.19,40,41 However, the development of a reliable assay to measure the target is required, as exemplified by the comparative value of the measurement of HER2/neu by immunohistochemistry versus fluorescence in situ hybridization before selection for trastuzumab therapy.34,42
Rational Use of Functional and Molecular Imaging
Interest in utilizing novel imaging technologies for the development of MTAs is increasing.40,43 Molecular imaging has the potential to monitor both normal and abnormal biochemical and physiological parameters in individual patients. Unlike anatomic imaging, molecular imaging displays biochemical and physiological abnormalities underlying disease, rather than the structural consequences of these abnormalities. Functional and molecular imaging can be helpful in phase 1 studies for dose determination, as discussed earlier, and may be applicable as noninvasive tools to detect drug-target effect, as well as to provide early evidence of efficacy. It may also prove useful in patient selection.
Detecting and monitoring responsiveness to a cytostatic agent using functional imaging, such as [18F]-fluorodeoxyglucose (FDG) positron emission tomography (PET), has been evaluated in detail. Uptake of the glucose analogue FDG by tumor cells is a marker of glucose metabolism, number of viable cells, and the level of cell proliferation in a tumor mass. Thus treatment that causes metabolic changes associated with inhibition of tumor cell proliferation or death should have a relatively rapid effect on the degree of FDG uptake. Decrease in the maximum standardized uptake value on FDG-PET was found to be an early predictor of response to imatinib therapy for patients with advanced GIST.44 These changes can occur as early as 24 to 48 hours after initiating imatinib and predict for objective response or stabilization of disease as determined subsequently by computed tomography scanning. Conversely, failure to achieve a decrease in FDG-PET uptake after treatment with imatinib is predictive of a poor clinical response for patients with GIST.
Another area of mounting interest is the use of PET and novel imaging probes to visualize cancer targets and processes in vivo.41 These probes include a wide variety of small molecules, peptides, and antibodies to image processes, including cell proliferation, angiogenesis, apoptosis, hypoxia, PK, and multidrug resistance. In addition, probes exist or are in development to image tumor cell receptors such as epidermal growth factor receptor (EGFR), human epidermal growth factor receptor-2, and bombesin, to name a few.45 The investigational radiopharmaceutical agent 16 alpha-[18F]-fluoro-17 beta-estradiol (FES), which is synthesized by adding the radioactive element, 18F, in the D ring of estradiol, has been used to assess the pharmacodynamic effect of tamoxifen, fulvestrant, and aromatase inhibitors,46 and the level of uptake of FES has been correlated with tumor response to tamoxifen.47
3′-[18F]Fluoro-3′-deoxythymidine PET is particularly useful as an in vivo marker of tumor cell proliferation and for monitoring response to antiproliferative therapy.48 Finally, interest in the use of optical imaging by spectral fluorescence enhancement is increasing because of its sensitivity and ability to detect very small clusters of cancer cells.49 However, optical imaging has the disadvantage of being limited to settings where direct imaging is possible (e.g., intraoperative or endoscopic), because whole-body imaging is not yet feasible. Progress in the clinical application of molecular imaging depends on the development of probes that have better sensitivity and specificity, as well as increases in the signal-to-noise or spatial resolution of molecular imaging devices. Unquestionably, noninvasive imaging will play an increasingly important role in the development and application of molecularly targeted therapy.
Patient Selection
An important consideration in the enrollment of patients on exploratory efficacy trials of MTAs is the use of patient selection to optimize the chance of a positive effect. This optimization is not feasible unless the patients enrolled have tumors that express or have a high probability of expressing the target. Until recently, essentially all phase 2 trials have evaluated a drug in a specific histologic type (e.g., breast cancer or NSCLC) on the presumption that a given histologic type represents a relatively uniform disease entity. However, an increasing amount of evidence shows that tumors are heterogeneous at the molecular level within any given histologic type and that genetic variations in the tumor may significantly affect drug sensitivity. Clearly, patient selection beyond histologic type is becoming increasingly important in drug development, especially for agents likely to benefit specific patient subsets. For example, the efficacy of crizotinib in patients with advanced NSCLC whose tumors harbor a specific type of alteration in the anaplastic lymphoma receptor tyrosine kinase (ALK) gene,50 and the efficacy of vemurafenib in patients with metastatic melanoma carrying the V600E BRAF mutation,51 might have been overlooked in trials of these agents in larger, unselected groups of patients with NSCLC or melanoma.
Some mutations may confer sensitivity to a particular agent, whereas other mutations confer resistance, such as exon 11 mutations of c-Kit in GIST52 and T315I BCR-ABL mutations in CML, respectively, in the case of imatinib. On the other hand, the effect of inhibiting the same mutation may be context dependent based on disease histology, as evidenced by the >50% overall response rate to vemurafenib in patients with advanced melanoma who carried the BRAFV600E mutation as opposed to a <5% overall response rate in patients with advanced colorectal carrying the same mutation.53,54 It has been shown that vemurafenib treatment, with inhibition of the BRAFV600E oncoprotein, results in feedback activation of EGFR, which is present on a majority of colorectal cancer cells, resulting in resistance.54 This finding might explain the poor response of patients with colorectal cancer to vemurafenib treatment. In addition, targeting BRAF mutation in tumors bearing other types of mutations such as in RAS or wild-type BRAF can lead to excessive signaling of the RAF-MEK-ERK pathway with unintended clinical consequences, such as the development of keratoacanthomas and squamous cell carcinomas.55
The development and FDA approval of vemurafenib for the treatment of melanoma is limited to patients with BRAFV600-mutation–positive tumors. To select patients for vemurafenib treatment, the FDA concurrently approved the Cobas 4800 BRAFV600 mutation test. The approved indication for vemurafenib specifically includes the limitation that treatment of patients with wild-type melanoma is not recommended because essentially no safety or efficacy data are available for these patients. The decision to limit the development of vemurafenib for melanoma to patients with BRAF mutations was reasonable given what was known about the drug’s mechanism of action and existing nonclinical data. However, in general, it is useful to obtain at least some information about how a drug affects target (test)-negative as well as target (test)-positive patients; the data may be important for the validation of the biomarker assay and to make certain the drug is not effective in test-negative patients. Crizotinib was approved by the FDA for the treatment of patients with ALK-rearrangement–positive NSCLC based on two single-arm trials demonstrating unprecedented overall response rates of 50% to 60%. However, preliminary data from a small cohort of ALK-negative patients revealed a potentially promising response rate of about 20%, suggesting that further evaluation of crizotinib in patients with ALK-negative tumors is warranted.56 The response in ALK-test–negative patients could be due to activity against other crizotinib targets such as MET or ROS1, independent of the drug’s effect on ALK.
Rational Approaches to Combination Therapy
An increasing number of MTAs in development have some clinical activity against selected types of cancer. However, with few exceptions, such as imatinib for the treatment of CML and GIST, MTAs have limited effectiveness as single agents, and any clinical benefit is restricted to a relatively small proportion of patients. Because molecular abnormalities in cancer are usually complex, and multiple processes and pathways are responsible for tumor proliferation and survival, the likelihood that engaging any one specific target will have a major antitumor impact is relatively low. Consequently, there is considerable interest in combining MTAs that affect two or more critical targets with the expectation that such regimens will have more pronounced antitumor activity.3 Because of the large number of targets and MTAs in development, the number of potential combinations is almost limitless. Thus the design of clinical trials evaluating MTAs in combination should not be empirical but based on a sound scientific rationale. Various strategies are being used to devise rational drug combinations that optimize antitumor efficacy while limiting toxicity3 (Table 28-4).
Table 28-4
Strategies Combining Molecularly Targeted Anticancer Agents with Other Modalities
| Combination Strategies | Potential Mechanism |
| MTAs plus cytotoxic therapy Combination with chemotherapy Combination with radiotherapy |
Modulate targets involved in tumor sensitivity/resistance or repair mechanisms |
| Two or more MTAs | |
| Antibody directed at ligand or receptor plus a small-molecule TKI directed at the same receptor (e.g., anti-VEGF antibody plus VEGFR TKI) | Maximizes target inhibition |
| Agents that inhibit two or more signal transduction molecules in the same pathway (e.g., EGFR TKI plus RAF inhibitor: MEK and RAF inhibitor) | Maximizes pathway inhibition |
| Agents that inhibit parallel pathways (EGFR inhibitor plus HER2 inhibitor) | Maximize tumor inhibition by effecting multiple cellular mechanisms |
| Agents that inhibit a target and a compensatory feedback loop (e.g., mTOR inhibitor plus an Akt inhibitor) | Maximize tumor response by inhibiting compensatory resistance mechanisms |
| Agents that target the tumor microenvironment/vasculature plus an agent directed against tumor cell proliferation | Maximize clinical benefit by inhibiting tumor cell proliferation, invasion, and metastasis |
| Epigenetic remodeling agents used in combination or in concert with agents directed against a reexpressed target | Enhance restoration of expression of tumor suppressor genes, apoptotic mechanisms, or tumor antigens |
| MTA plus surgery | |
| Administer MTA following complete anatomic resection (adjuvant) | Maximize benefit by targeting minimal residual disease |
| Administer MTA before surgical resection (neoadjuvant) | Evaluate for complete pathological response and target engagement |
As noted previously, the use of MTAs in combination with standard chemotherapy has met with mixed success. Among the successes are trials demonstrating improved survival with the addition of trastuzumab in persons with breast cancer34 and bevacizumab in persons with colorectal cancer35,36 and NSCLC.57 Among the failures are randomized trials of gefitinib and erlotinib combined with chemotherapy in persons with NSCLC.37,38 The reasons these latter combination trials failed to show superiority over chemotherapy alone are not clear, but they include the possibility of suboptimal dosing (as may have been the case for gefitinib), antagonism with the chemotherapy agents, or dilution of the study population with patients harboring insensitive tumors.39 Combining MTAs can also cause chronic toxicities that limit dosing and compromise efficacy. Consequently, there is a need to better elucidate these variables by using reliable predictive preclinical models for efficacy and toxicity and appropriately designing early-phase clinical trials to address PK, PD, efficacy, and toxicity considerations in advance of large phase 2 and phase 3 clinical trials.
Rational combinations of MTAs that have been evaluated clinically maximize inhibition of a single target or pathway, inhibit two or more parallel cellular processes or pathways, inhibit tumor cell processes and the microenvironment in concert, and use epigenetic remodeling agents to increase the expression of a target (Table 28-4). For example, inhibitors of DNA methyltransferase and histone deacetylase are being studied alone and in combination to restore the expression of tumor suppressor genes and other genes silenced by DNA methylation, such as hormone receptors and tumor antigens, to make tumor cells more sensitive to targeted hormonal and vaccine therapy, respectively.58,59
The potential advantages of using two or more MTAs in combination are clear. Another approach is to design a single agent with specificity against multiple targets of interest. One successful example is the small-molecule human epidermal growth factor receptor-2/EGFR inhibitor lapatinib, which recently received FDA approval for the treatment of advanced breast cancer.60 Avoiding the complex trial designs that may be necessary to isolate the effects of each agent, as well as the potentially complicated intellectual property negotiations with another company needed to perform combination MTA trials, are incentives for pharmaceutical companies to take this approach. Regardless of whether developing single agents with multiple targets or combinations of agents that focus on a limited number of targets is the drug development strategy employed, it is important to minimize the risk of clinical toxicity by avoiding overlapping or off-target toxicities.
Conclusions and Future Directions
The development of MTAs requires different strategies than those traditionally applied to conventional, nontargeted chemotherapy. Empirical methods for the development of standard agents are mostly inadequate for the development of MTAs. Rather, optimal MTA development from discovery through clinical testing requires the integration of innovative methods that are primarily target-based. Furthermore, novel clinical trial designs have evolved with the introduction of MTAs. Most phase 1 trials of MTAs should have as a primary objective the determination of a recommended phase 2 dose based on optimal target modulation, because escalation to MTD may not be needed or feasible; however, accomplishing this goal successfully requires reliable procedures for tissue acquisition and handling in conjunction with authenticated assay methodologies to measure target modulation. These target assay procedures are best developed and performed in preclinical in vitro and in vivo models and, if feasible, in phase 0 clinical studies in a limited number of patients in advance of large, more definitive trials.21
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Therapeutic Targeting of Cancer Cells
Era of Molecularly Targeted Agents
Shivaani Kummar, Anthony J. Murgo, Joseph E. Tomaszewski and James H. Doroshow
• Molecularly targeted anticancer agents (MTAs) are those that selectively target specific molecular features of cancer cells such as aberrations in genes, proteins, or pathways that regulate tumor growth, progression, and survival.
• Molecular targets include the following: products of activating mutations and translocations, growth factors and receptors, aberrant signal transduction and apoptotic pathways, factors that control tumor angiogenesis and microenvironment, dysregulated proteins, DNA repair machinery, and aberrant epigenetic mechanisms.
• The development of MTAs requires innovative strategies that differ from those traditionally applied to nontargeted conventional chemotherapy.
• Successful development of an MTA depends largely on the importance of the target in controlling tumor cell proliferation and survival and effective modulation of the target in the tumor at clinically achievable concentrations.
• Primary objectives of clinical trials of MTAs differ from those used in trials of conventional chemotherapy. An important objective in phase 1 trials of MTAs should be to determine a phase 2 dose based on optimal target modulation (i.e., a biologically effective dose) rather than on maximum tolerated dose. In addition, objective tumor response may not be an adequate end point for efficacy evaluations of MTAs that have a primarily cytostatic effect. Alternate end points, such as progression-free survival, may be more appropriate.
• Functional and molecular imaging plays an increasingly important role in the development of MTAs.
Molecular Targets
The increasing number and assortment of molecular targets can be broadly categorized according to genetic or functional properties, including products of activating gene mutations and translocations; growth factors and receptors; aberrant signal transduction and apoptotic pathways; factors that control tumor angiogenesis and microenvironment; dysregulated proteins; DNA repair machinery; and aberrant epigenetic mechanisms (Tables 28-1 and 28-2).
Table 28-1
U.S. Food and Drug Administration–Approved Molecularly Targeted Agents
| FDA Approved Agents(s) | Target | Disease Indication |
| Imatinib mesylate | BCR-ABL, KIT, PDGFR-β | Ph+ CML, Ph+ ALL, chronic monomyelocytic leukemia, dermatofibrosarcoma protuberans |
| Dasatinib | BCR-ABL, Src family kinases | Ph+ CML |
| Nilotinib hydrochloride monohydrate | BCR-ABL | Ph+ CML |
| All-trans retinoic acid | PML-RAR | Acute promyelocytic leukemia |
| Sunitinib malate | VEGFR, KIT, PDGFR-α | GIST, pancreatic neuroendocrine tumors, kidney cancer |
| Vemurafenib | BRAFV600E mutation | BRAFV600E-positive melanoma |
| Crizotinib | ALK tyrosine kinase | ALK-positive non–small cell lung cancer |
| Erlotinib | EGFR | Non–small cell lung cancer |
| Panitumumab | EGFR | Colorectal cancer |
| Cetuximab | EGFR | Head and neck cancer, colorectal cancer |
| Trastuzumab | ErbB-2 | HER2 overexpressing breast cancer |
| Lapatinib | ErbB-2 | HER2 overexpressing breast cancer |
| Vismodegib | Hedgehog pathway | Advanced/metastatic basal cell carcinoma |
| Brentuximab vedotin | CD30 | Hodgkin lymphoma, anaplastic large cell lymphoma |
| Ofatumumab | CD20 | CLL |
| Ibritumomab | Non-Hodgkin lymphoma | |
| Rituximab | B-cell non-Hodgkin lymphoma | |
| Alemtuzumab | CD52 | B-cell chronic lymphocytic leukemia |
| Bevacizumab | VEGF | Colorectal cancer, non–small cell lung cancer, renal cell carcinoma |
| Vandetanib | VEGFR | Medullary thyroid cancer |
| Sorafenib | VEGFR, B-Raf kinase | Kidney cancer, hepatocellular cancer |
| Pazopanib | VEGFR | Soft tissue sarcomas, kidney cancer |
| Axitinib | VEGFR | Kidney cancer |
| Temsirolimus | mTOR | Renal cell carcinoma |
| Everolimus | mTOR | Renal cell carcinoma, advanced pancreatic neuroendocrine tumors |
| Azacitidine | DNA methyltransferase | Myelodysplastic syndrome |
| Decitabine | DNA methyltransferase | Myelodysplastic syndrome |
| Vorinostat | Histone deacetylase | Cutaneous T-cell lymphoma |
| Romidepsin | Histone deacetylase | Cutaneous T-cell lymphoma |
| Bortezomib | Proteosome | Multiple myeloma |
| Ipilimumab | Cytotoxic T-lymphocyte–associated antigen 4 | Metastatic melanoma |
Table 28-2
Classes of Molecularly Targeted Anticancer Agents under Clinical Development
| Target | Role of Target | Disease Indication | References |
| FMS-like tyrosine kinase-3 (FLT-3) | Regulates cell cycle progression, proliferation, and survival | Acute myeloid leukemia | 61 |
| Poly (ADP-ribose) polymerase (PARP) | Single-strand DNA break repair | Various solid tumors, BRCA-positive breast and ovarian cancer | 62 |
| O6-Alkylguanine DNA alkyltransferase | Prevents intrastrand DNA cross-links | 63 | |
| Insulin-like growth factor-1 receptor (IGF-1R) | Regulates cell proliferation, differentiation and survival | Various adult and pediatric tumors | 64 |
| Insulin-like growth factor binding protein-3 (IGFBP3) | Regulates cell proliferation, differentiation and survival | Various adult and pediatric tumors | 64 |
| Hypoxia-inducible factor-1α (HIF-1α) | Regulates tumor cell response to oxygen deprivation | HIF-1α–expressing solid tumors | 65 |
| αvβ3 integrin receptor | Involved in cell adhesion | Glioma and various solid tumors | 66, 67 |
| Syk | Inhibits differentiation and induces growth factor–independent proliferation of pre–B cells | B-cell lymphomas | 68 |
| MET | Regulates cell growth, anti-apoptosis, altered cytoskeletal function | Solid tumors | 69 |
| MEK1/2 | Regulates tumor cell proliferation and survival | Various solid tumors, melanoma | 70, 71 |
| RAS | Promote cell proliferation and survival | Acute myeloid leukemia | 72 |
| RAF | Regulates tumor cell proliferation and survival | Melanoma and other solid tumors | 73 |
| RET | Regulates tumor cell proliferation and survival | Medullary thyroid cancer | 74 |
| Akt kinase | Regulator of cell cycle and apoptotic pathway | Variety of solid and hematologic cancer | 75 |
| Clusterin | Promotes cell survival | Variety of solid tumors | 76 |
| HSP-90 | Chaperone for several oncogenic proteins and growth factors | Myeloid leukemia and solid tumors | 77 |
| Bcl-2 | Antiapoptotic | Lymphomas, solid tumors | 78 |
| Tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) | Apoptotic mechanism | Various types of cancer | 79 |
| Angiopoietin 1 and 2 | Reduces blood flow to tumor | Solid tumors | 80 |
| DNA methyltransferase inhibitors | Reexpression of genes silenced by methylation | Myelodysplastic syndrome, acute myeloid leukemia, various solid tumors in combination with chemotherapy or radiation | 58, 59 |
| Src inhibitors | Src family kinases that interact with other growth, proliferation, and angiogenic pathways | Solid tumors | 81 |
| VEGFR, VEGF | Reduces blood flow to tumor | Solid tumors | 11 |
| CD105 (Endoglin) | Reduces blood flow to tumor | Solid tumors | 82 |
| HDAC | Reexpression of silenced genes | T-cell lymphoproliferative disorders, adenoid cystic cancer, solid tumors | 83 |
| Retinoid receptor | Increase in reactive oxygen species and dihyceramide levels resulting in cytotoxicity | T-cell lymphoproliferative disorders | 84, 85 |
| Wee 1 | DNA repair and cell cycle (abrogates the G2 cell cycle checkpoint) | Solid tumors | 86 |
| BTK inhibitor | Bruton tyrosine kinase | Lymphoma, chronic lymphocytic leukemia | 87 |
| Gamma secretase inhibitor | Notch pathway | Solid tumor | 88 |
| CDK inhibitors | Cyclin-dependent kinases (cell cycle, proliferation) | Solid tumors | 89 |
| CHK1 inhibitors | Checkpoint kinases (cell cycle) | Solid tumors | 90 |
| DNA minor groove | DNA damage | Solid tumors | 91 |
| SMAC mimetic, IAP inhibitor | Induction of apoptosis | Solid tumors | 92 |
The most promising molecular targets are those solely responsible for sustaining tumor growth and survival because agents that potently and selectively inhibit these critical targets are likely to have a major clinical impact. Probably the best example of a critical target is BCR-ABL in patients with chronic myelogenous leukemia (CML). BCR-ABL is a fusion protein formed by the reciprocal translocation of chromosomes 9 and 22. Knowledge that this dysregulated tyrosine kinase played a causal role in the pathogenesis of essentially all cases of CML spurred preclinical studies, which led to the development of a potent and selective ABL tyrosine kinase inhibitor (TKI), imatinib mesylate (previously known as STI571).1 Subsequent clinical trials established imatinib mesylate as the first highly effective molecularly targeted therapy for CML and a prototype for the development of others in the class. Imatinib mesylate is also a potent inhibitor of other tyrosine kinases including PDGFR and KIT, and it is highly effective in the treatment of gastrointestinal stromal tumors (GISTs) bearing activating c-KIT mutations and in some GISTs bearing activating PDGFR mutations.1,2
Unfortunately, most human tumors, including the most common types, are genetically complex and do not have a single critical target, and the relative critical importance of a target in different tumors may vary. Most tumor types have various genetic and molecular abnormalities driving their growth and survival. The existence of multiple abnormalities in one or more molecular pathways helps explain resistance to molecularly targeted therapy and provides a rationale for treatment strategies combining two or more targeted agents.3,4 However, cancer cells may become “addicted” or physiologically dependent on the sustained activity of specific oncogenes for maintenance of a malignant phenotype and for survival. This dependence mechanism, termed oncogene addiction7–7 (Fig. 28-1), is associated with differential attenuation rates of prosurvival and proapoptotic signals stemming from the oncoprotein, with predominant apoptotic signals resulting in cell killing. The latter process, termed “oncogenic shock,”8 could explain the remarkably rapid clinical responses to TKIs in some patients with solid tumors, including those typically having complex molecular abnormalities. Other possible factors controlling sensitivity or resistance to molecularly targeted therapy include increased expression of the target due to gene amplification or transcription, emergence of resistant target gene mutations, and overexpression of multidrug transporter membrane proteins.4,7
Interest in molecular therapy directed at factors controlling angiogenesis has increased since the United States Food and Drug Administration (FDA) approved several agents that target vascular endothelial growth factor (VEGF) and its receptor (VEGFR). The VEGF pathway, involving the VEGF family of proteins and their receptors, is an important regulator of both physiological and pathological angiogenesis. Through its signaling pathways, VEGF/VEGFR activation contributes to increased vascular permeability, mobilization of bone marrow–derived endothelial cell precursors, degradation of the extracellular matrix, and endothelial cell division, differentiation, migration, and survival.9,10 VEGF overexpression occurs in most types of cancers, including colorectal, renal, gastric, pancreatic, liver, lung, breast, thyroid, and genitourinary; it also occurs in glioma and other intracranial tumors, as well as in hematologic malignancies, and it is associated with tumor growth and a worse clinical outcome in a number of these tumor types.10,11
Potential therapeutic strategies to inhibit signaling through VEGF and VEGFR pathway activation include monoclonal antibodies directed against VEGF or VEGFR, TKIs, and antisense strategies (antisense oligodeoxynucleotides, antisense RNA, and small interfering RNAs). In 2004, the FDA approved bevacizumab, a humanized murine monoclonal antibody directed against VEGF, for treating metastatic colorectal cancer in combination with fluorouracil-based chemotherapy. The FDA subsequently approved sunitinib, sorafenib, and axitinib, three small-molecule TKIs with activity against VEGFR, for the treatment of advanced renal cancer.11 In addition, sorafenib is effective for treating hepatocellular carcinoma, a tumor against which standard cytotoxic chemotherapy has little or no activity.11,12 The experience with these agents has established the VEGF/VEGFR pathway as a valid target for cancer therapeutics.11
Mammalian target of rapamycin (mTOR) has also emerged as a validated target with the demonstration that the small-molecule mammalian target of rapamycin inhibitors temsirolimus and everolimus are effective in the treatment of renal cell carcinoma.13 This activity of temsirolimus is attributed to the downregulation of factors that control cell growth and angiogenesis.13
Preclinical Development of MTAs
The discovery and development of molecularly targeted therapies requires closely aligned laboratory and clinical research, integrating drug discovery, development, and clinical investigation. In such a cooperative setting, researchers can effectively take rational and iterative steps from target identification to clinical evaluation (Box 28-1) (Fig. 28-2). A crucial early step in developing a molecularly targeted therapy is target validation, defined as experimental evaluation of the role of a given gene or protein.14,15 The target validation process involves a variety of preclinical approaches, including genetic, cell-based, and animal models.16 Validation and prioritization of molecular targets for therapeutic development depends on a variety of criteria, taking into consideration chemical, biological, clinical, and practical factors15 (Box 28-2). The fundamental goal is to provide evidence that the target is valid (i.e., that affecting the target inhibits tumor growth, progression, or survival) and that making drugs that hit the target is feasible. The next major step in the development of molecularly targeted therapy is finding/synthesizing compounds directed against that target.
