Therapeutic Antibodies and Immunologic Conjugates
• Because of their tumor selectivity, monoclonal antibodies offer exceptional opportunities for targeted therapy.
• When naked, monoclonal antibodies can kill tumors by receptor blockade and by actively inducing apoptosis.
• Tumor cytotoxicity is mediated through white cells by activating antibody-dependent cell-mediated cytotoxicity, and in the presence of serum, by complement-mediated cytotoxicity.
• Bispecific or multifunctional constructs can greatly enhance the antitumor effect of antibodies.
• Antibodies can deliver effector molecules in the form of drug conjugates, radioimmunoconjugates, immunocytokines, immunotoxins, immunoenzymes, immunoliposomes, and retargeted killer cells.
• Naked antibodies generally do not have overlapping toxicity profiles with chemotherapy and radiation therapies, and dose-limiting toxicities of immunoconjugates vary depending on the cytotoxic moiety (e.g., myelosuppression in radioimmunoconjugates) being used.
• Antibodies are likely to be most beneficial at the time of minimal residual disease, especially when used in conjunction with standard therapy.
• The following antibodies are licensed by the Food and Drug Administration and are in use for the cancers listed (target molecule in bold):
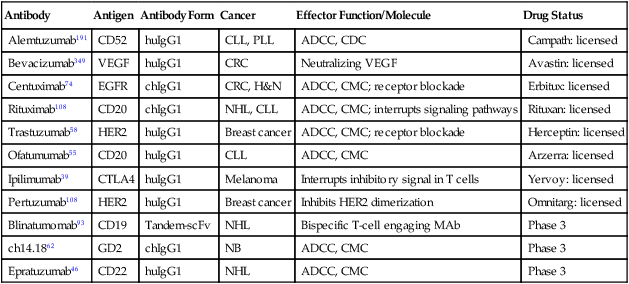
 Alemtuzumab (Campath): B-chronic lymphocytic leukemia (CD52)
Alemtuzumab (Campath): B-chronic lymphocytic leukemia (CD52)
 Bevacizumab (Avastin): colorectal cancer (VEGF)
Bevacizumab (Avastin): colorectal cancer (VEGF)
 Cetuximab (Erbitux): colorectal cancer, head and neck cancer (EGFR)
Cetuximab (Erbitux): colorectal cancer, head and neck cancer (EGFR)
 Ibritumomab (Zevalin): non-Hodgkin lymphoma (Yttrium-90, CD20)
Ibritumomab (Zevalin): non-Hodgkin lymphoma (Yttrium-90, CD20)
 Rituximab (Rituxan): non-Hodgkin lymphoma (CD20)
Rituximab (Rituxan): non-Hodgkin lymphoma (CD20)
 Trastuzumab (Herceptin): breast cancer (HER2)
Trastuzumab (Herceptin): breast cancer (HER2)
 Ipilimumab (Yervoy): melanoma (CTLA4)
Ipilimumab (Yervoy): melanoma (CTLA4)
 Ofatumumab (Arzerra): CLL (CD20)
Ofatumumab (Arzerra): CLL (CD20)

 Pertuzumab (Perjeta): breast cancer (HER2)
Pertuzumab (Perjeta): breast cancer (HER2)
• In the coming decade, other monoclonal antibodies currently in various phases of clinical trial, as well as those approved for nononcologic indications, may be added to the list. The prospects for further innovation in this established cancer treatment modality are highly favorable.
Introduction
The clinical development of antibody therapy was accelerated by the introduction of the hybridoma technique in 1975 and the emergence of recombinant technology.1 Through these innovations, individual plasma cells can be immortalized, and cloning of heavy and light chain repertoires from animals and humans is now routinely performed. In the past three decades, monoclonal antibodies (MAbs) have evolved from research tools to inclusion in a rapidly increasing list of licensed pharmaceutical agents. They have generated excitement on many fronts and will likely play a pivotal role in the history of cancer medicine (Box 32-1). The clinical utility of MAbs for in vitro diagnosis and ex vivo manipulation of blood or stem cells is well recognized. Their role in the treatment and prophylaxis of graft-versus-host disease is detailed in Chapter 33. The use of B-cell idiotype and anti-idiotypic antibodies as tumor vaccines is described in Chapter 6. This chapter will focus on the application of naked therapeutic antitumor MAbs and immunologic conjugates in cancer therapy.
Effector Mechanisms of Monoclonal Antibodies
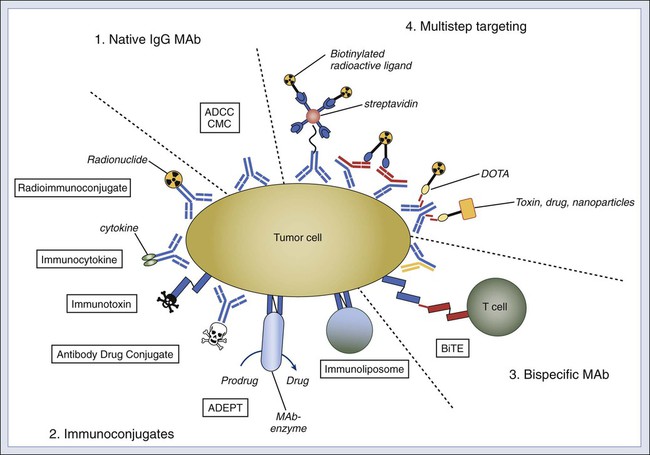
Antitumor MAbs can mediate highly effective tumoricidal functions both in vitro and in vivo (Fig. 32-1). These functions include signaling through receptor binding, antibody-dependent cell-mediated cytotoxicity (ADCC), and complement-mediated cytotoxicity (CMC).
Signaling by Receptor Cross-Linking and Receptor Blockade
When the antigen is a cell surface receptor, its clustering by multivalent MAbs can induce apoptosis.2 Apoptosis increases with hyper cross-linking (e.g., CD20 target on lymphoma cells).5–5 Both caspase dependent and independent programmed cell death pathways appear to be involved.6 In AIDS-related lymphoma, anti-CD20 MAb diminishes p38MAPK signaling and Bcl-2 expression, whereas in non–AIDS-related lymphoma, signaling through CD20 inhibits AP-1 and nuclear factor (NF)-κB, leading to downregulation of Bcl-xL, thereby sensitizing lymphoma cells to chemotherapy.7 Direct receptor blockade by MAbs has also been reported for epidermal growth factor receptor-1 EGF-R18 and human epidermal growth factor receptor-2 (HER-2),9 leading to upregulation of the BH3-only protein Bnip3L, thereby sensitizing tumor cells to chemotherapy.10 MAb inhibition of vascular endothelial growth factor receptor-1 (VEGFR-1)11 or VEGFR-212 can also enhance the efficacy of chemotherapy.
Cytophilic MAb and ADCC
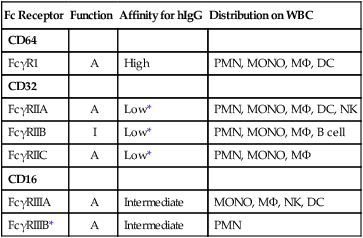
The Fc region of immunoglobulin G (IgG) MAb interacts with both activating and inhibitory Fc receptors (FcγR).13 Humans have four activating FcγRs: FcγRI (CD64) is a high-affinity FcγR, whereas FcγRIIA (CD32A), FcγRIIIA (CD16A), and FcγRIIIB (CD16B) are low-affinity FcγRs. FcγRIIB (CD32B) is the only known inhibitory FcγR (Table 32-1). All FcγRs are transmembrane glycoproteins except for FcγRIIIB, which is anchored on neutrophils by glycosyl phosphatidylinositol. FcγRI also has an extracellular portion composed of three Ig-like domains, whereas FcγRII and FcγRIII have only two domains. This phenomenon allows FcγRI activation by a single IgG molecule (or monomer), whereas the latter two Fcγ receptors must bind multiple IgG molecules within an immune complex to be activated. FcγRIIA carries the immunoreceptor tyrosine–based activation motif (ITAM) in its intracellular tail for activation. Other FcγRs do not have an ITAM but interact with an adaptor protein called the accessory γ chain (Fcγ subunit), which carries a cytoplasmic ITAM. ITAM becomes tyrosine phosphorylated by members of the Src-kinase family, with subsequent recruitment of kinases containing SH2. These events lead to the activation of phosphatidylinositol 3-kinase and phospholipase-Cγ, followed by protein kinase C activation and sustained calcium elevation.13 These biochemical cascades trigger phagocytosis, degranulation, cytokine release, and ADCC. In sharp contrast to activating FcγRs, FcγRIIB is a single-chain receptor that carries the immunoreceptor tyrosine–based inhibitory motif in its cytoplasmic domain. Engagement of this inhibitory receptor activates phosphatase SHP-1 to remove phosphate groups from tyrosine residues, leading to downregulation of both biochemical and cellular functions. The ratio of activating to inhibitory FcγRs on immune cells, such as dendritic cells (DCs), macrophages, and neutrophils, can influence the antitumor properties of MAbs.
Table 32-1
Properties of IgG Fc Receptors13
| Fc Receptor | Function | Affinity for hIgG | Distribution on WBC |
| CD64 | |||
| FcγR1 | A | High | PMN, MONO, MΦ, DC |
| CD32 | |||
| FcγRIIA | A | Low* | PMN, MONO, MΦ, DC, NK |
| FcγRIIB | I | Low* | PMN, MONO, MΦ, B cell |
| FcγRIIC | A | Low* | PMN, MONO, MΦ |
| CD16 | |||
| FcγRIIIA | A | Intermediate | MONO, MΦ, NK, DC |
| FcγRIIIB* | A | Intermediate | PMN |
Inflammatory mediators (interferon-γ or C5a) increase activating FcγRs and downregulate inhibitory FcγRIIB, whereas interleukin-4 (IL-4), IL-10, and tumor growth factor-β upregulate FcγRIIB, thereby raising the thresholds for cell activation. Removing the inhibitory signals by FcγRIIB-blocking antibodies has shown efficacy in preclinical models.13 This finding may be relevant for cross-presentation of antigens, which is acquired endocytically through Fc receptors on DCs during the induction of tumor-specific T-cell responses.14 In addition to these FcγRs, a unique class of Fc receptor called FcRn (neonatal) is found on endothelial cells and regulates antibody catabolism.15 FcRn is similar in structure to major histocompatibility complex class I antigen. It binds IgG at an acidic pH of 6.0 to 6.5 but not at neutral or higher pH. When serum IgG is internalized by endothelial cells through pinocytosis, it becomes FcRn-bound in the acidic endosomes and escapes lysosomal degradation by recycling to the cell surface for release back into blood, where the pH is neutral. This regulation of serum half-life by FcRn binding can be exploited in antibody engineering. Although most therapeutic antibodies have been primarily IgGs, both IgA1 and IgA2 can also mediate efficient ADCC by binding to FcαRI (CD89) on human neutrophils and monocytes/macrophages.16
Certain cancer cells such as colon carcinoma, lymphoma, leukemia, neuroblastoma, and melanoma are effectively killed by natural killer (NK) cells, neutrophils, and activated monocytes in vitro in the presence of specific MAbs. Depending on the affinity of the MAb for the individual FcγR, both NK cells (carrying FcγRII and FcγRIII) and neutrophils (bearing all three FcγR) can mediate efficient ADCC. Because of its high affinity, FcγRI is generally occupied by monomeric IgG in human plasma. Human IgG subclasses (IgG1, IgG2, IgG3, and IgG4) have differential affinities for FcγRII and FcγRIII. Chimeric or humanized IgG1 antibodies (e.g., Lym-1 specific for the human leukocyte antigen–DR subregion, and ch14.18 or hu3F8 for GD2) can exploit FcγRIII for lymphocyte ADCC while using FcγRII for myeloid ADCC.19–19 Among the four IgG subclasses, IgG2 has the lowest affinity for the inhibitory receptor FcγRIIB.13 Mouse IgG3 (e.g., 3F8 specific for GD2) can engage both FcγRII and FcγRIII in ADCC,20 despite the low affinity of the monomer for human FcγRs. Polymorphic FcR alleles (FCGR2A23–23 and FCGR3A22,24) with higher affinity for human IgG1 mediated more effective ADCC in vitro and were associated with superior clinical responses to rituxan, cetuximab, or Herceptin. In addition to FcγRs, adhesion molecules are critical for MAb-mediated ADCC. These molecules include CR3 (CD11b/Cd18)17,18,20 and CD66b17 for neutrophil-mediated ADCC and LFA-1 (CD11a/CD18) for NK-mediated ADCC.25 Because cytokines can increase the expression of adhesion molecules, granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon-γ,18,26–28 and IL-229,30 have been used to activate either myeloid- or NK-mediated effector models. Furthermore, because both GM-CSF and IL-2 expand the effector cell pools, they enhance the effector to target ratio, a critical determinant of both in vitro and preclinical models of antibody-based therapy. Optimal combinations of MAbs and cytokines in the appropriate clinical setting are being explored.31–34
Signaling by Agonistic and Antagonistic Antibodies
Activating signaling receptors on human cytotoxic lymphocyte35 and specifically NK cells36 can enhance ADCC functions. Although most relevant for CD8+ T-cell–mediated tumor lysis (e.g., CD137 [4-1BB], CD134 [OX40], and GITR), agonistic antibodies to CD137 have now been shown to potentiate NK-ADCC in anti-CD20 and trastuzumab therapy.37,38 Conversely, use of antagonistic antibodies to remove immune checkpoint blockade allows T cells to perform more effective surveillance. The first successful example of such an immune approach was the anti-CTLA4 MAb ipilimumab,39 which was approved by the Food and Drug Administration (FDA) for melanoma. An equally exciting strategy targets the programmed death–1 (PD-1) T-cell coreceptor or its ligand B7-H1/PD-L1 and B7-DC/PD-L2, a pathway that maintains an immunosuppressive tumor microenvironment.40 Phase 1 and 2 clinical trials of anti–PD-141 or anti–PD-L142 have shown tumor responses in melanoma, non–small cell lung cancer (NSCLC), renal cell carcinoma, and ovarian cancer. Besides turning on T cells, removing inhibitory signals on NK cells could also have clinical potential for both hematologic and solid malignancies.45–45 Blocking MAbs (e.g., anti-CD47) have also been effective in unleashing macrophages to phagocytose tumor cells in the absence or presence of MAb,46 both in vitro and in vivo, in leukemia/lymphoma47–51 and in solid tumor models.49,51 In addition, binding of MAb to certain tumor cell surface receptors can be directly cytotoxic, independent of ADCC or CMC. For example, induction of cell death through death receptor family–like TRAIL receptors on tumor cells may have potential for cancer therapy.52,53
Complement Activation
IgG initiates the classic complement cascade by binding C1q to its CH2 domain. C1q is more avid for human IgG1 and IgG3 than for IgG2 and has no affinity for IgG4.54 CMC potency of individual IgG MAb is also dependent on antibody affinity,55 antigen density, and nature of the target or the tumor cell. Although some tumor cell lines (e.g., lymphoma and neuroblastoma) are sensitive to CMC, many are resistant to complement because of anticomplement surface proteins such as decay accelerating factor (DAF; CD55),58–58 homologous restriction factor (CD59),56,59,60 and membrane cofactor protein (CD46).57–59,61 The effect of complement extends beyond direct tumor lysis. After complement activation, tumor-bound C3b is cleaved rapidly by plasma protease factor I to iC3b. Through the iC3b receptors CR3 (Mac-1 or alphaMbeta2-integrin) and CR4 (CD11c/CD18, alphaXbeta2-integrin) on leukocytes, tumor cells are opsonized.62 C3a and C5a, byproducts of complement activation, are also potent mediators of inflammation63 and are chemotactic for phagocytic leukocytes, drawing them to the tumor sites. C5a can also downregulate the inhibitory receptor FcγRIIB13 or induce secondary cytokines to increase vascular permeability for both MAbs and effector cells. Preclinical models of anti-CD20 immunotherapy showed that CMC could interfere with ADCC,64 and the adverse clinical outcome correlations with C1qA levels supported this observation.65
Clinical Application of Naked MAb Directed at Cancer Cells
Lymphoma and Leukemia
In 1997, the anti-CD20 chimeric antibody rituximab became the first MAb approved by the FDA for the treatment of cancer (Table 32-2). It was based on a single-arm multicenter study of 166 patients with relapsed or refractory, low-grade, or follicular non-Hodgkin lymphoma (NHL).66 Chemosensitization of rituximab when combined with cyclophosphamide, doxorubicin, vincristine, and prednisone was later demonstrated in diffuse large B-cell NHL.67,68 The addition of rituximab to induction chemotherapy improved survival of patients with follicular lymphoma compared with induction chemotherapy alone, but most patients were not cured and experienced a relapse after a median of 4 years.69 Rituximab maintenance improved progression-free survival (PFS)70 and overall survival (OS) in a meta-analysis.71
Table 32-2
Naked Monoclonal Antibody for Cancer Therapy
| Antibody | Antigen | Antibody Form | Cancer | Effector Function/Molecule | Drug Status |
| Alemtuzumab191 | CD52 | huIgG1 | CLL, PLL | ADCC, CDC | Campath: licensed |
| Bevacizumab349 | VEGF | huIgG1 | CRC | Neutralizing VEGF | Avastin: licensed |
| Centuximab74 | EGFR | chIgG1 | CRC, H&N | ADCC, CMC; receptor blockade | Erbitux: licensed |
| Rituximab108 | CD20 | chIgG1 | NHL, CLL | ADCC, CMC; interrupts signaling pathways | Rituxan: licensed |
| Trastuzumab58 | HER2 | huIgG1 | Breast cancer | ADCC, CMC; receptor blockade | Herceptin: licensed |
| Ofatumumab55 | CD20 | huIgG1 | CLL | ADCC, CMC | Arzerra: licensed |
| Ipilimumab39 | CTLA4 | huIgG1 | Melanoma | Interrupts inhibitory signal in T cells | Yervoy: licensed |
| Pertuzumab108 | HER2 | huIgG1 | Breast cancer | Inhibits HER2 dimerization | Omnitarg: licensed |
| Blinatumomab93 | CD19 | Tandem-scFv | NHL | Bispecific T-cell engaging MAb | Phase 3 |
| ch14.1862 | GD2 | chIgG1 | NB | ADCC, CMC | Phase 3 |
| Epratuzumab46 | CD22 | huIgG1 | NHL | ADCC, CMC | Phase 3 |
For most patients, rituximab was well tolerated.72 Severe adverse events thought to be a result of complement activation often occurred with the first infusion,73 especially if the patient had high numbers of circulating tumor cells. These infusion-related reactions usually appeared 30 to 120 minutes after MAb injection, and they typically were associated with severe cardiopulmonary events with deaths (<0.1%) occurring within 24 hours. B-cell depletion occurred in most patients, although hypogammaglobulinemia appeared in only 14% of all cases, and it was not associated with clinical morbidity.66 Severe mucocutaneous reactions occurred rarely (0.07%), resulting in some fatalities. Late-onset neutropenia after rituximab therapy has been reported in 5% to 27% of patients and has been correlated with high-affinity FcγRIIIA polymorphism.74
Poor tumor response to rituximab occurs when a low-affinity FcγRIII polymorphic allele is found in the patient’s white blood cells and when the patient has a low density of CD20 molecules or the presence of complement inhibitory molecules (CD55 and CD59) on tumor cells.75 Ofatumumab is a next-generation humanized anti-CD20 antibody with more potent cytolytic potential partly because of membrane proximity of its epitope,55 and it is now approved for fludarabine-refractory chronic lymphocytic leukemia that is resistant to alemtuzumab or for bulky lymphadenopathy. Ocrelizumab, Veltuzumab, GA101, AME-133v, and PRO131921 are all humanized anti-CD20 antibodies with enhanced binding to low-affinity FcR that currently are undergoing clinical testing.75
Campath-1H (Alemtuzumab), a humanized rat IgG1 anti-CD52 MAb, demonstrated antitumor activity against patients with recurrent B-cell chronic lymphocytic leukemia who had not responded to fludarabine therapy in an international phase 2 trial involving 21 centers (n = 93 patients), and it was approved by the FDA.76,77 Grade 3 or 4 infections were reported in 26.9% of patients.78 Opportunistic infections including bacterial sepsis and viral infections, as well as marrow aplasia, have since been reported.77,79 Its utility for eradicating minimal residual disease (MRD) and T-cell malignancies are being explored.80,81
Epratuzumab is a humanized IgG1 antibody directed at CD22, a cell surface antigen expressed in B-precursor acute lymphoblastic leukemia (ALL)84–84; it shows modest activity among patients with NHL, as a single agent85,86 or in combination with rituximab.87,88 Most clinical efforts have been focused on its immunotoxins.89 SGN-30 (chIgG1 anti-CD30), XmAb2513 (humanized SGN-30), and MDX-060 were developed for patients with Hodgkin disease,90 with modest clinical activity as a naked MAb.
Blinatumomab, a tandem single-chain variable fragment (scFv) bispecific antibody (bispecific T-cell engaging antibody), retargets cytolytic T cells onto tumor cells by transiently inducing a cytolytic synapse between a cytotoxic T cell and the cancer target cell. Granules containing granzymes and the pore-forming protein perforin fuse with the T-cell membrane and discharge their toxic content, leading to target cell apoptosis. At extremely low doses of 60 µg/m2/day × 4 to 8 weeks given as continuous intravenous injection, major responses among 82% of patients with mantle cell lymphoma, follicular lymphoma, and diffuse large B-cell lymphoma,91,92 as well as molecular complete remission (CR) in 80% of patients with adult B-ALL,92,93 were observed. Together with CD20, CD22, and CD52, the possibilities for antibody-based immunotherapy of ALL have now greatly expanded, although details for the optimal combination, such as the required level of antigen expression, timing, schedule, dosage, and stage of disease, still need to be defined.94 In patients with myeloid leukemia, the addition of lintuzumab (huM195, IgG1, and anti-CD33) to salvage induction chemotherapy was found to be safe, but it did not result in a statistically significant improvement in response rate or survival in patients with refractory/relapsed acute myeloid leukemia (AML).95
Solid Tumors
Trastuzumab (Herceptin) is a humanized MAb against the receptor tyrosine kinase ERBB2 (also known as HER2/NEU) on breast cancer cells. Along with inducing CMC and ADCC, it can induce HER2 protein downregulation, prevent HER2-containing heterodimer formation, initiate G1 arrest, induce p27, prevent HER2 cleavage, and inhibit angiogenesis.96 Up to 25% of breast cancers are positive for HER2, which is associated with aggressiveness and poor prognosis.97,98 Based on its synergy with chemotherapy in vitro,99,100 trastuzumab was tested in a large phase 3 trial of 469 patients with breast cancer, where its combination with chemotherapy produced a longer median response duration (9.1 vs. 6.1 months), higher overall response (OR) (50% vs. 32%), and a lower death rate at 1 year (22% vs. 33%)101 than did chemotherapy alone. On the basis of this trial, the FDA approved the use of trastuzumab and paclitaxel as a first-line treatment of HER2-overexpressing metastatic breast cancer. Subsequent studies showed that 1 year of treatment with trastuzumab after adjuvant or neoadjuvant chemotherapy significantly improved disease-free survival among women with HER2-positive breast cancer.102,103 A 0.8% incidence of severe congestive heart failure and a 3.6% incidence of significant left ventricular ejection fraction decreases were found in the trastuzumab treatment group compared with 0% and 0.6%, respectively, in the observation group,104 although both symptomatic and asymptomatic events were largely manageable and reversible.105 Recent studies also suggest that synergy between trastuzumab and chemotherapy may still be effective beyond disease progression.106 The combination of trastuzumab and small molecules targeting HER2 in the adjuvant and neoadjuvant settings is being actively pursued.107 Despite the clinical benefit of trastuzumab, disease in patients with metastatic breast cancer eventually progresses. Pertuzumab, a humanized MAb that inhibits HER2 dimerization, produced an 18% to 24% response when combined with trastuzumab among patients with HER2-positive metastatic breast cancer. In the phase 3 randomized study (n = 808 patients), the combination of pertuzumab + docetaxel + trastuzumab versus docetaxel + trastuzumab as first-line treatment for HER2-positive breast cancer showed significant prolongation of PFS (18.5 vs. 12.4 months) with similar toxicity profiles except for febrile neutropenia and diarrhea.108 On the basis of this study, pertuzumab (Perjeta) was approved by the FDA. In advanced HER2-positive gastric cancer, a phase 3 trial of trastuzumab plus platinum-based chemotherapy versus chemotherapy alone showed an OS advantage of 2.7 months.109 However, its benefit when added to standard chemotherapy was uncertain for NSCLC110,111 or ovarian cancer.112
Cetuximab (chIgG1 anti-EGFR) is an FDA-approved chimeric antibody designed to induce receptor blockade; it reverses resistance to topoisomerase I inhibitors in addition to having modest monotherapy activity in patients with colorectal cancer (CRC),113,114 with improvements in PFS and OS.115 First-line treatment with cetuximab plus FOLFIRI, compared with FOLFIRI alone, reduced the risk of progression of metastatic CRC, although no significant difference was found in OS.116 An acneiform rash occurred in 82.9% of patients and a grade 3 rash was observed in 4.9%; it was exacerbated by sunlight or secondarily infected by Staphylococcus aureus, which was sometimes preventable by prophylactic topical antibiotics. Serious hypersensitivity-hypomagnesemia reactions requiring H1 antihistamines and steroids developed in only a minority of patients (~1%).113 Response and survival correlated strongly with the severity of the rash. In contrast, clinical benefit did not correlate with EGFR immunostaining of the tumor.117 Neither EGFR kinase domain mutations nor EGFR gene amplification appeared to be essential for response to cetuximab. Although response to cetuximab was strongly associated with wild-type KRAS,113 not all KRAS-mutated tumors were resistant to cetuximab.118 Expression of epiregulin and amphiregulin were biomarkers for efficacy.119 Panitumumab, a fully human anti-EGFR antibody generated using human IgG-transgenic mouse technology,120 was much less immunogenic, stimulating binding antibodies in only 1.8% and neutralizing antibodies in 0.2% of patients.121 As first-line treatment in previously untreated patients with metastatic CRC, panitumumab plus FOLFOX4 (fluorouracil, leucovorin, and oxaliplatin) improved median PFS significantly from 8 to 9.6 months for patients with wild-type KRAS tumors in a phase 3 randomized trial, but did not improve their OS.122 As second-line treatment, panitumumab plus fluorouracil, leucovorin, and irinotecan (FOLFIRI) was better than FOLFIRI alone in prolonging median PFS (from 3.9 to 5.9 months) and median OS (from 12.5 to 14.6 months).123 The potential role of anti-EGFR antibody in patients with gastroesophageal cancer is under investigation.124
When combined with radiotherapy for locally advanced squamous-cell carcinoma of the head and neck, cetuximab improved the duration of locoregional control from 14.9 to 24.4 months, the median OS from 29.3 to 49 months, and 5-year OS from 36.4% to 45.6%, with Hazard Ratio 0.49 among patients with ≥grade 2 acneiform rash.125 As first-line treatment of recurrent or metastatic head and neck cancer, addition of cetuximab to platinum + fluorouracil in a randomized phase 3 trial increased the response rate from 20% to 36%, the prolonged median PFS from 3.3 to 5.6 months, and the median OS from 7.4 to 10.1 months.126 Two large randomized phase 3 trials (FLEX and BMS099) demonstrated a small benefit (~1 month) in OS, but not in PFS, with the addition of cetuximab to first-line chemotherapy in patients with advanced NSCLC.127,128 Subgroup analyses suggested a significant association between early occurrence of skin rash129 and intensity of EGFR staining130 with efficacy.
Despite initial enthusiasm,131 adjuvant therapy with edrecolomab (17-1A, Panorex), a mouse IgG2a antibody specific for epithelial cell adhesion molecule (EpCAM) on malignant and normal epithelial cells, did not improve disease-free survival or OS in subsequent phase 3 studies in patients with CRC.132,133 The story of edrecolomab provided a case study for the clinical failure of MAb on a major scale.134 Despite the lack of clinical efficacy, anti-idiotype network and T-cell responses against antibody-modified tumors were found.135,136 Adecatumumab, the fully human MAb, is undergoing testing in gynecologic cancers.137 Catumaxomab (Removab), a rat-mouse bispecific antibody engaging CD3 on T cells and EpCAM on tumor cells, was recently approved in Europe for malignant ascites.138,139 Oregovomab (MAb B43.13, anti-CA125), a murine IgG1 antibody for ovarian cancer, did not improve time to relapse in a phase 3 randomized trial of 145 patients.140 A correlation of improved survival and human anti-mouse antibody (HAMA)/idiotype network response was of biological interest.141
Among the ganglioside antigens on neuroectodermal tumors, GD3 (MAb R24 for melanoma)142 and GD2 (MAb 3F8 and ch14.18 for neuroblastoma)34,143 have been studied clinically. GD2 is present on a variety of solid tumors in addition to neuroblastoma, including osteosarcoma, retinoblastoma, some soft tissue sarcomas, brain tumors, mesenchymal stem cells,144 and cancer stem cells.145 The clinical effectiveness of anti-GD2 MAb for large, soft tissue masses was minimal. Its utility was more suitable for microscopic marrow disease in the presence of GM-CSF and/or IL-2.146,147 Activation of CD11b by GM-CSF on myeloid cells correlated with patient PFS.33 Missing ligands for killer immunoglobulin-like receptors on NK cells significantly affected both PFS and OS.45,143 Failure to achieve early MRD remission was the strongest predictor of clinical outcome. HAMA response was a strong positive predictor of OS and was associated with the induction of Ab2 and Ab3 through the idiotype network, implicating a potential role of the host immune response in maintaining clinical remission.148,149 With anti-GD2 therapy, disease relapse was primarily at isolated sites, and isolated central nervous system relapse, previously rare, was salvageable by combining surgical resection, radiation, and intrathecal iodine-131 MAb.150 Clinical development of anti-GD2 MAb was limited by its adverse effect of pain, thus precluding dose escalation. Humanized forms of 3F8 (hu3F8)19 and 14.18 (hu14.18)151 are currently undergoing active clinical investigations.
Complications and Contraindications
Toxicities of MAbs are in general manageable and self-limited. Common acute reactions include fever, chills, headache, nausea, fatigue, angioedema, urticaria, pruritus, blood pressure fluctuations, and bronchospasm. Lethal or irreversible adverse effects unique to some MAbs include cytokine release (anti-lymphocyte MAb) and complement activation (anti-CD20) syndromes,73 immune suppression (anti-CD20 and anti-CD52) and associated viral reactivation/opportunistic infections/progressive multifocal leukoencephalopathy,77,79,152 preexisting IgE responses to galactose-α-1,3-galactose (cetuximab),153 cardiotoxicity (anti-HER2),101 and late-onset neutropenia (rituximab).74 The severe cytokine release syndrome associated with antilymphocyte (e.g., anti-CD28, TGN1412) MAb is extremely rare,154 although TGN1412 provided a case study for potential tragic complications for any first-in-human biological agent. A unique, severe, self-limited acute adverse effect is the pain syndrome from cross-reactivity of anti-GD2 MAb with peripheral pain fibers.155,156 Murine MAb induces HAMA responses, which can alter the pharmacokinetics and pharmacodynamic properties of repeat MAb injections. The murine-derived N-glyconeuraminic acid (e.g., cetuximab) that is introduced when MAb is produced in mouse cell lines can react with natural antibodies in humans.157 HAMA is primarily directed to the murine Fc portion of the antibody, although anti-idiotypic responses have also been reported.158 With chimeric, humanized, primatized, and human antibodies, the incidence of neutralizing antibody decreases,159 but their frequency varies with individual MAb from minimal (0.2%)121 to substantial (49%).160
Immunoconjugates
The clinical utility of naked MAb can be limited by both host (number and activity of effector cells, FcR polymorphism, and interference by inhibitory receptors) and tumor factors (antigen heterogeneity and complement regulatory proteins). Although the CMC and ADCC functions of naked MAb (see Fig. 32-1) can be improved by altering the Fc protein structure161 or by modifying Fc-glycosylation,162 substantial gains in clinical potentials of MAb can derive from immunoconjugates. These include (1) radioimmunoconjugates to deliver β and α emitters,163,164 (2) immunotoxins165 and antibody drug conjugates,168–168 (3) bispecific MAb (specific for soluble ligands, cell surface antigens, and receptors) to enhance signal blockade or to cross-link effector cells selectively to tumors,169,170 (4) immunocytokines to deliver cytokines to tumor sites while minimizing systemic toxicities,171 (5) antibody-directed enzyme prodrug therapy (ADEPT) to pretarget enzymes to tumor sites for prodrug activation so that high local concentrations of active drugs can be released without triggering systemic toxicities,172 and (6) immunoliposomes to deliver drugs, isotopes, DNA, or toxins.173,174
Radioimmunoconjugates
MAbs have the potential to target and ablate tumors in radioimmunotherapy (RIT).175 Radioimaging can map the biodistribution of MAbs and quantify the relative amounts of MAb deposited in various tissues and organs, thus allowing more precise radiation dose estimates in therapeutic studies. With the advent of single photon emission computed tomography and positron emission tomography, accurate dosimetry is readily achievable. In preclinical models, ablation of established xenografts is possible, although radiation damage to the marrow remains dose-limiting. For patients with lymphoma and leukemia, antitumor activity of RIT is highly reproducible, but major responses in solid tumors are rare. Unlike naked antibodies, the bystander effect of RIT from cross-firing of the radioisotopes accounts for most of the toxicities of radioimmunoconjugates, hence limiting their efficacy.
Most clinical applications of RIT utilize β-emitting radioimmunoconjugates (Table 32-3). β Particles have a relatively long range (0.8 to 5 mm) and low linear energy transfer (approximately 0.2 keV/µm). This long range results in the delivery of radiation not only to the antigen-positive tumor cells but also to surrounding antigen-negative tumor cells, as well as to the neighboring normal tissues. Thus β emitters can treat bulky diseases effectively but are not optimal for eradicating single cells or micrometastasis. Most early human studies of RIT used 131I, a long-lived β-particle emitter. Because of its γ-particle emission, it is also suitable for dosimetry studies. However, this γ-particle emission poses a radio hazard at high treatment doses, necessitating patient isolation. In vivo dehalogenation of iodinated antibodies can compromise tumor dose with subsequent thyroid damage from the released iodide. Yttrium-90 (90Y) is a pure β emitter; its lack of γ radiation allows outpatient treatment. However, 90Y has its limitations, including deposition in bone when dissociated from the MAb complex and its lack of γ emissions, thereby requiring the use of indium-111 to estimate biodistribution and dosimetry. Besides 90Y, other β emitters recently explored include rhenium-186, rhenium-188, copper-67, and lutetium-177, each with its own limitations.
Table 32-3
Choice of Radioisotopes for Radioimmunotherapy
| Isotope | Particle(s) Emitted | Half-Life Hours | Maximum Energy (KeV) | Mean range of α- or β- particle emission (mm) |
| Iodine-131 (131I) | β, γ | 193 | 610 | 0.8 |
| Yttrium-90 (90Y) | β | 64 | 2280 | 2.7 |
| Copper-67 (67Cu) | β | 62 | 577 | 1.8 |
| Lutetium-177 (177Lu) | β | 161 | 496 | 1.5 |
| Rhenium-188 (188Re) | β, γ | 17 | 2120 | 2.4 |
| Actinium-225 (225Ac) | α | 240 | 5935 | 0.05-0.08 |
| Astatine-211 (211At) | α | 7.2 | 7450 | 0.05-0.08 |
| Bismuth-213 (213Bi) | α | 0.77 | 5982 | 0.05-0.08 |
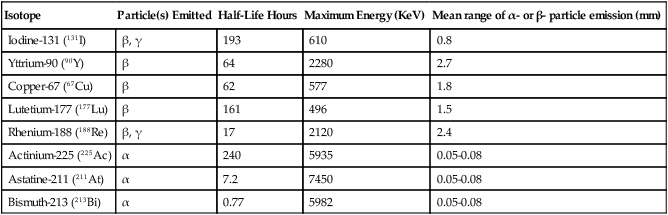
Alpha particles are helium nuclei; when compared with β particles, they have a shorter range (50 to 80 µm) and a higher linear energy transfer (approximately 100 keV/µm).176 As few as one or two α particles can destroy a target cell. RIT using α emitters should result in less nonspecific toxicity to normal bystanders, as well as more efficient single-cell killing. This modality is ideal for controlling MRD. Isotopes that emit α particles such as astatine-211 and bismuth-213 have been tested in clinical trials.213 Bi-HuM195 (anti-CD33) administered intravenously for AML177 and 211At conjugated to MAb 8C16 and administered intraventricularly or intrathecally for gliomas178 have been well tolerated and have produced clinical responses. The relative lack of extramedullary toxicities should encourage further development of this targeting technique for micrometastases or neoplasms on the surface of body compartments, such as peritoneal and leptomeningeal metastasis.181–181
Radiolabeled MAbs for Lymphoma
Despite the potential of RIT, only two antibodies have been licensed thus far, namely, Zevalin and Bexxar (Table 32-4).182 Because sequestration of MAbs in the liver or spleen compromises tumor delivery, a large dose of naked anti-CD20 antibody is often used during RIT to reduce liver or spleen uptake. At 0.2 to 0.4 mCi/kg (7.4 to 15 MBq/kg) of 90Y-ibritumomab (mouse IgG1 anti-CD20), dosimetry showed median radiation-absorbed doses of 7.4 Gy to spleen, 4.5 Gy to liver, 2.1 Gy to lung, 0.23 Gy to kidney, 0.62 Gy (blood-derived method) and 0.97 Gy (sacral image–derived method) to red marrow, and 0.57 Gy to the total body, with a median effective blood half-life of 27 hours.183 Grade 4 neutropenia, thrombocytopenia, and anemia occurred in 30% to 35%, 10% to 14%, and 3% to 8% of patients, respectively. Serious grade 3 and 4 toxicities occurred in 3% of patients, and life-threatening events occurred in 1% to 5% of patients. Four weeks after therapy, no circulating B cells could be detected, and recovery began around 12 weeks after therapy, usually achieving normal limits by 9 months. Serum IgG and IgA remained unchanged throughout, whereas IgM dropped below normal before recovering by 6 months. In 3.8% of patients, HAMA or human antichimeric antibody developed. RIT-related myelodysplasia and acute myelogenous leukemia were reported in 1% of patients 8 to 34 months after treatment.184 In a phase 3 randomized study, a single dose of 0.4 mCi/kg 90Y-ibritumomab was more effective than rituximab at 375 mg/m2/week for 4 weeks in patients with relapsed or refractory low-grade, follicular, or transformed NHL.185,186 This difference was statistically significant for OR (80% vs. 56%), CR (30% vs. 16%), and the probability of ≥6-month durable responses (64% vs. 47%), respectively. In a phase 3 randomized trial in patients with advanced follicular lymphoma in first remission, rituximab at 250 mg/m2 (day 7 and day 0) was used to clear peripheral B cells. When compared with control, the addition on day 0 of 90Y-ibritumomab tiuxetan (14.8 MBq/kg) converted 77% of PR to CR and prolonged PFS from 13.3 months to 36.5 months.187 In other studies, increasing the dose or dose intensity of rituximab has not produced a meaningful improvement in outcome.
Table 32-4
Radiolabeled Monoclonal Antibody and Drug Conjugates
| Antibody | Antigen | Antibody Form | Cancer | Isotope/Drug | Drug Status |
| Tositumomab191 | CD20 | muIgG2a | NHL | Iodine-131 | Bexxar: licensed* |
| Ibritumomab183 | CD20 | muIgG1 | NHL | Yttrium-90 | Zevalin: licensed |
| Gemtuzumab ozogamicin257 | CD33 | huIgG1 | AML | Calicheamicin | Mylotarg: licensed* |
| Brentuximab vedotin262 | CD30 | chIgG1 | Hodgkin disease, ALCL | Auristatin | Adcetris: licensed |
| Trastuzumab emtansine (T-DM1)273 | HER2 | huIgG1 | Breast cancer | Mertansine | Kadcyla: licensed |
| Inotuzumab ozogamicin166 | CD22 | huIgG1 | NHL | Calicheamicin | Phase 3 (terminated) |
| Epratuzumab tetraxetan199 | CD22 | huIgG1 | NHL | Yttrium-90 | LymphoCide: phase 3 |
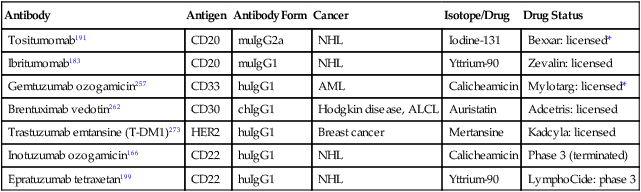
131I-tositumomab (Bexxar; mouse IgG2a anti-CD20) achieved 71% OR (34% CR) in a phase 2 trial (n = 59) of chemotherapy-refractory/relapsed patients with NHL. In 17% of patients, HAMA was induced.188 Using myeloablative doses of 280 to 785 mCi (calculated to deliver 25 to 72 Gy to critical organs), 30 of 36 patients (83%) achieved durable CR, with OS of 68% and PFS of 42% (median follow-up, 42 months).189,190 Hypothyroidism developed in 60% of patients 6 to 12 months after therapy. Secondary myelodysplastic syndrome (MDS)/AML was reported in 2% to 5% of patients.190,191 Similarly, myeloablative doses (10 to 31 Gy) of 131I-anti-CD37 MAb produced 84% CR and 11% PR in patients with NHL, with eight patients in continual remission 46 to 95 months after therapy. Extramedullary toxicities were mild at doses of less than 23 Gy, beyond which cardiopulmonary toxicity became dose-limiting.192 131I- tositumomab193,194 or 90Y-ibritumomab tiuxetan195 was also explored as part of autologous hematopoietic cell transplant conditioning regimens. At myeloablative doses, MDS/therapy-related AML developed in 8.3% at 5 years, which was comparable with patients receiving chemotherapy alone.196 Despite FDA approval, patient referral for RIT was limited because of alternative nonradioactive options, as well as logistics and economic concerns.197
Because the B-cell antigen CD22 is internalized, the anti-CD22 antibody is ideal for RIT. 131I-epratuzumab (hLL2, humanized anti-CD22),198 90Y-epratuzumab,199,200 and 186Re-epratuzumab201 have all shown antitumor activity against B-cell lymphoma. In addition, unlike other anti-CD20 antibodies, optimal biodistribution was achieved (hLL2) without the need for preinjection of unlabeled antibody. Other promising antigen systems for RIT include CD19202 and HLA-DR,203 CR2,204 CD37,205 CD30,206 and B-cell idiotypes.207 In one study, HAMA response to the MAb Lym-1 appeared to correlate with improved survival.208
Radiolabeled MAb for Leukemia
Radiolabeled MAb targeted to lineage-specific antigens have been safely administered to patients with leukemia.209 90Y-anti-CD25 was active in acute T-cell leukemia210; myelosuppression was the primary toxicity. 131I-anti-CD33 (for AML, MDS, and myeloblastic chronic myeloid/myelogenous leukemia [CML]),211,212 90Y-anti-CD33,209 131I-anti-CD45 (for AML, ALL, and MDS),213 and 188Re-anti-CD66c (for AML, ALL, and CML)214 all delivered significant radiation doses to the bone marrow and are particularly effective as part of a conditioning regimen for hematopoietic stem cell transplantation. Radioconjugates that emit α particles (213Bi-anti-CD33 and actinium-225-anti-CD33) may be better suited for the treatment of small-volume disease.215,216
Radioimmunotherapy of Solid Tumors
The antitumor activity of RIT in solid tumors is less encouraging (Table 32-4).217 A number of radiolabeled MAbs given intravenously have been tested clinically with modest clinical benefit: 131I-hMN-14 (humanized anti–carcinoembryonic antigen [CEA] IgG at 60 mCi/m2) for CRC in remission or with small-volume metastasis,175 131I-huA33 for CRC,218 90Y-anti-CEA for carcinoma,219 131I-cG250 (chimeric anticarbonic anhydrase IX),220 and 177Lu-cG250 for renal cell carcinoma.221 Intravenous anti-GD2 131I-3F8 was tested in children with metastatic neuroblastoma at high doses (6 to 28 mCi/kg).222 Although responses were seen in both soft tissue masses and bone marrow, the use of myeloablative 131I-3F8 (20 mCi/kg) did not add survival benefit compared with naked 3F8 among patients with high-risk metastatic stage 4 neuroblastoma in first remission.143,146
Compartmental RIT
90Y-CC49 (a second-generation murine antibody of B72.3, ≤24 mCi/m2),223 and 90Y-HMFG1 (mouse anti-MUC1, 666 MBq/m2)224 were tolerated intraperitoneally in patients with recurrent ovarian cancer, although no evidence of survival gain was found. Intrathecal and intraventricular administration for leptomeningeal carcinomatosis and intratumoral RIT of malignant brain tumors using 131I-81C6 (antitenascin MAb) have produced objective responses and prolonged patient survival.225,226 211At-81C6 is an example of α-particle therapy for MRD in patients with malignant glioma.227 Intraventricular 131I-3F8228 and 131I-8H9150 are also being tested in RIT for leptomeningeal cancers in both children and adults, with highly favorable radiation dose ratios of cerebrospinal fluid to blood. Among children with recurrent neuroblastoma metastatic to the central nervous system, long-term remissions have been achieved.150
Multistep Targeting or Pretargeting
To improve tumor uptake and reduce systemic toxicity, a multistep procedure that pretargets the antibody before the binding of the cytotoxic ligand to the tumor has been used successfully.229 Generally, a tumor-specific antibody is conjugated to a ligand binder, such as streptavidin (with high affinity for biotin) or ligand-specific antibody (binding to metal chelators, such as diethylene triamine pentaacetic acid or 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid [DOTA]).232–232 In the first step, these bispecific antibodies (172 to 200 kD) are allowed to localize to tumors in vivo, and any excess is cleared from the blood with or without a clearing agent (e.g. DOTA-dextran or biotinylated dendrimers). The small radiolabeled ligand (DOTA-radiometal or its biotinylated form) is then injected intravenously. The ligand penetrates tissues rapidly, and by virtue of the high-affinity interaction, binds tightly to the antibody-conjugate at the tumor site. Unbound ligand is quickly excreted through the kidneys. Because of the short transit time of the toxic ligand (radionuclides or toxins), a substantial improvement in the therapeutic ratio is achievable without sacrificing the percent injected dose per gram in tumor.233 Pretargeting using antibodies to CEA234 was tested in CRC,235 small cell lung cancer,236 and medullary thyroid carcinoma,234,237 along with NT-LU-10 in CRC,238 CC49 in gastrointestinal cancer,239 and anti-CD20 MAb in NHL,240,241 all with variable success. A three-step approach that used biotinylated MAb, followed by avidin/streptavidin, and then biotinylated radiometal chelate, was also applied to glioma with encouraging results.242,243 However, immunogenicity of avidin/streptavidin will limit repeated injections.
Immunotoxins and Antibody Drug Conjugates
Chemotherapeutic drugs have been conjugated to MAbs for selective tumor delivery. Doxorubicin, melphalan, methotrexate, and vinca alkaloids conjugated to MAbs have limited clinical success. BR96-doxorubicin directed at Lewis Y antigen has shown no clinical benefit in phase 2 trials in patients with breast cancer244 or gastric cancer.245
Agents such as ribosome-inactivating toxins can be potent cancer drugs. One major limitation is the lack of tumor selectivity.246 Two-chain toxins (e.g., ricin and diphtheria toxin [DT]) utilize their B chain for cell binding and their A chain for inhibition of protein synthesis; other toxins (e.g., Pseudomonas exotoxin [PE], Pokeweed antiviral protein, gelonin) have a built-in receptor for cell attachment. When conjugated to MAbs, they become immunotoxins. These toxins can be genetically modified for MAb conjugation and for an improved safety profile.165 In recombinant toxins (e.g., PE40, PE38, or DT DAB486), the cell-binding domains were replaced by scFv.165,246
MAbs conjugated to different toxins have been actively explored for cancer therapy246: ricin toxin A-chain (RTA) (RTA to anti-CD7, anti-CD22, and anti-CD25); DT (anti-IL2R); and PE (anti-CD25, anti-CD22,247,248 anti-Lewis Y,249 and anti-HER2). A common toxicity is the vascular leak syndrome, characterized by marked fluid overload, dyspnea, and sensorimotor neuropathies.250 Deglycosylated RTA devoid of mannose and fucose has reduced hepatic sequestration, allowing longer serum half-life. Anti-CD22-dsFv-PE (RFB4[dsFv]-PE38, BL22) demonstrated activity for NHL251 and cladribine-resistant hairy cell leukemia.252 Adverse effects include transient hypoalbuminemia and elevated aminotransferase levels; serious but reversible hemolytic-uremic syndrome has also been reported.
An expanding collection of toxic natural compounds, such as calicheamicins253 and maytansinoids,254 are now candidates for antibody drug conjugates. Gemtuzumab ozogamicin (Mylotarg) is an anti-CD33 antibody conjugated to calicheamicin. Acting like a prodrug, calicheamicin is released from the antibody after internalization, forming a diradical that induces double-strand DNA breaks. Gemtuzumab ozogamicin was active in childhood refractory AML255 and achieved a 30% response rate among patients with refractory AML who were 60 years or older.256 When tested as a low-fractionated dose regimen in addition to frontline chemotherapy in a phase 3 randomized trial, 2-year event-free survival was improved from 17.1% to 40.8% and 2-year OS was improved from 41.9% to 53.2%.257 Toxicities included reversible myelosuppression (especially neutropenia and thrombocytopenia), abnormal liver function tests, and bilirubinemia. The incidence of hepatic toxicity including venoocclusive disease was ~2%.166 These safety concerns led to U.S. marketing withdrawal. Inotuzumab ozogamicin (CMC-544, anti-CD22 calicheamicin conjugate) has showed activity in NHL,258 and phase 3 results are pending.166 MAb-calicheamicin conjugates (e.g., anti-Lewis Y259 and anti-Mucin260) have not been successful to date in solid tumors. With most immunotoxins, immunogenicity has been a major constraint, although pegylation may reduce immunogenicity.261 Brentuximab Vedotin (Adcetris, SGN-35) is a conjugate of chimeric anti-CD30 MAb SGN-30 and monomethyl auristatin E, which disrupts microtubule when internalized. Monomethyl auristatin E is linked to SGN-30 through a protease-cleavable covalent linker cAC10. Because of its activity and safety profile, it gained FDA approval rapidly for refractory or relapsed Hodgkin disease262,263 and anaplastic large cell lymphoma.264,265 Auristatin conjugated through cysteine residues (MEDI-547, human anti-EphA2 MAb) was too toxic,266 and when conjugated to MAb specific for carbonic anhydrase showed early promise267; however, clinical development was halted by Bayer Healthcare. Auristatin conjugated to human anti-GPNMB antibody (Glembatumumab vedotin) is undergoing early-phase clinical trials in patients with breast cancer and melanoma.268,269 Trastuzumab emtansine (T-DM1) is an antibody conjugate comprising trastuzumab and DM1, a microtubule polymerization inhibitor.270 Among patients with HER2-positive metastatic breast cancer, objective responses were seen with minimal toxicity in a phase 1271 and phase 2 study.272 In a randomized phase 2 first-line study, T-DM1 showed at least equivalent efficacy when compared with trastuzumab plus docetaxel.273 Human antidrug antibody was detected in 4.5% of patients.274
Cellular Immunoconjugates with Bispecific Antibodies
Tumor-selective MAbs can be rendered cytophilic by conjugation with MAbs specific for the trigger molecules on T lymphocytes, NK cells, and granulocytes.169,275,276 These molecules include CD3, CD28, Fc receptors (CD64, CD16) and FcαRI (CD89).170 Bispecific antibodies (BiAb), which redirect polyclonal T-cells to tumors, seem to be most promising. Here, one binding site of the BiAb engages CD3 on T cells, whereas the other binding site determines tumor specificity, for example, B-NHL (CD19),91,93 breast cancer (HER2),279–279 Hodgkin lymphoma (CD30), malignant ascites (EpCAM),138,139 CRC (EpCAM),280 melanoma (MCSP),281 prostate (PSMA),282 gastrointestinal and CRC (EGFR283 or CEA284). Similar successes have been reported for the trigger molecule CD28 for ALL (CD19 and CD20),285,286 melanoma (MCSP),287 and Hodgkin disease (CD30),288 although severe cytokine release syndrome associated with anti-CD28 MAb TGN1412154 may discourage their final clinical translation. BiAb targeting FcγRIII (CD16) has also been explored for Hodgkin disease (CD30),289 CD19,290 CD33,291 EGFR,292 and HER2,293 whereas FcγRI was exploited for CD30,294 EGFR,295,296 and HER2.297 BiAb has also been made selective for epitopes outside the Fc-binding domain of FcγR to bypass competition with serum IgG.
Immunocytokines
Cell-mediated cytotoxicity has been highly effective against tumors in vitro and in animal models. Immunocytokines171,298 have shown remarkable success in activating and redirecting effectors to human tumors. Most of these studies have focused on NK, natural killer T cells (NKT), or T cells171 and granulocytes.18 Antibody-IL-2 immunocytokine can eradicate metastatic murine neuroblastoma while inducing long-term antitumor immunity.171,298 After initial successes with IL-2 immunocytokine,299 constructs containing other cytokines have been explored,171 including IL-12, tumor necrosis factor, and lymphotoxin. This emerging technology has been successfully applied to a number of antigens and clinical models,300 including GD2299,301 and fibronectin.302,303 Whereas measurable tumor responses were uncommon in clinical trials, microscopic marrow tumor responses have been reported.299 More recently, the combination of a plasmid DNA vaccine and IL-2 immunocytokine in the mouse model was shown to be more effective than when either one was administered alone.304
Immunoenzymes for ADEPT
Another novel approach (ADEPT) uses MAbs to deliver a covalently conjugated enzyme to the tumor, which can then activate a nontoxic prodrug.172 Despite preclinical successes, ADEPT has been difficult to translate into clinical benefit. Significant impediments to broaden clinical implementation include immunogenicity of antibody-enzyme conjugate, as well as the presence of endogenous enzymes or endogenous substrates and endogenous inhibitors of these enzymes within the tumors.
Immunoliposomes
With advances in liposome technology, several liposomal agents have been licensed for use in patients with cancer.305,306 When coated with polyethylene-glycol, uptake by the reticuloendothelial system is inhibited, thereby prolonging residence time in the blood. Concurrent developments in drug-loading technology have improved the efficiency and stability of drug entrapment in liposomes. Although there is passive accumulation of liposomes in tumors through enhanced permeability and retention, their uptake can be enhanced when engrafted with surface antibodies or their derivatives. For example, scFv or Fab can target liposomes for uptake into tumors bearing CD19,307 HER2,308 EGFR,309 and GD2.310 When liposomes fuse with the tumor targets, their contents can be efficiently delivered intracellularly. Although their potential is high, the clinical benefit of MAb-targeted liposomes in cancer therapy remains to be proven.
Improving the Efficacy of Antibody-Based Cancer Therapies
MAbs have been chimerized and humanized to reduce immunogenicity, enhance their binding to human FcR, and prolong their serum half-life (see Fig. 32-2).311 Chimeric MAbs are made by joining the antigen-combining variable domains of a mouse MAb to human constant domains: mouse VL to human CL and mouse VH to human CH1–CH2–CH3.161 In humanized MAb, the antigen-binding loops (known as complementarity-determining regions) from a mouse MAb are grafted into a human IgG.312 Human antibodies can also be derived from scFv or Fab phage display libraries,313,314 which is particularly useful for poorly immunogenic or self antigens.315 Alternatively, human MAb can be made from hIgG-transgenic mice.316
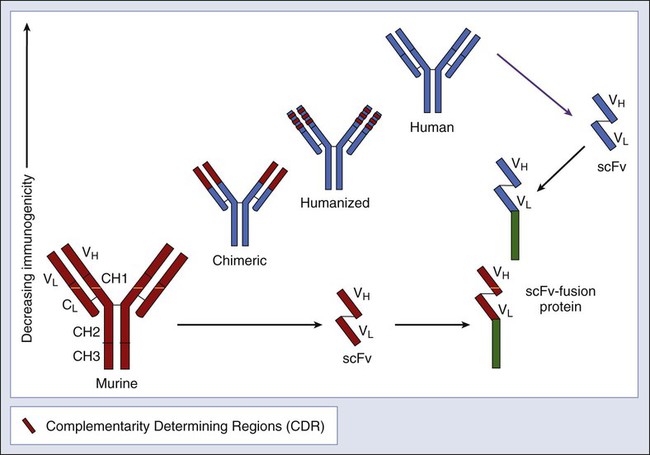
Because Fc is necessary for antitumor effect, chimerizing or humanizing mouse MAb with the human IgG1 or IgG3 Fc regions can improve ADCC and CMC functions. Similarly, removing FcγRIIB inhibitory receptor recognition also can enhance antitumor activity.317 Point mutations in the Fc region have increased its affinity for the activating Fc receptors, or decreased its affinity for FcγRIIB.318 Glycosylation of IgG at Asn297 stabilizes the tertiary structure of the CH2 domain, which is critical for effector functions, including ADCC and CMC.319 Glycosylation depends on the cell line that produces the MAb; increasing the bisected complex oligosaccharides in the Fc region320 or defucosylation can greatly improve ADCC properties.321,322 CMC can also be improved by Fc region mutations to increase C1q binding,323 or decreased by the K322A mutation.324
The antigen-binding affinity, molecular architecture, and oligomerization states of MAb can be reengineered to enhance tumor binding and uptake.325 For example, affinity can be increased using phage display,326 ribosome display,327 DNA shuffling,328 or yeast display.329 However, because the “binding-site” barrier can impede tumor penetration if the MAb has very high affinity,330 the optimal MAb may indeed be a medium-affinity IgG, especially for surface antigens expressed at high density. In addition, the size of the MAb is critical. ScFv are small (25 kDa) and rapidly cleared by the kidney. On the other hand, MAb forms with molecular weight in the range of 100 to 200 kDa should be ideal for tumor targeting.331 Besides increasing avidity, oligomerization can increase antitumor activity through a multitude of mechanisms including CMC/ADCC, induction of apoptosis, growth arrest, and synergy with chemotherapy or immunotoxins.4 The construction of dual or multispecific MAbs will further expand options.169 While scFv is a powerful building block for polymeric forms or novel fusion proteins,332,333 single-domain antibodies may further expand the possibilities of antibody-based cancer therapies.334
Alternative Targets for Anticancer Antibodies
Besides the ability to block receptors from interaction with their natural ligand, MAbs can inhibit receptor dimerization or receptor interaction with coreceptors.335 HER2 is a ligandless member of the ErbB receptor family that functions as a coreceptor with HER1/EGFR, HER3, and HER4. MAb 2C4 hinders the recruitment of HER2 into HER ligand complexes sterically and inhibits in vitro and in vivo growth of breast and prostate tumors. Pertuzumab, its humanized version, has produced highly favorable clinical responses and was recently approved by the FDA.108 Another new class of tumor-specific antigens are the T-cell epitopes, normally recognized by T-cell receptors. These epitopes are tumor-derived peptides presented on specific HLA alleles. Using human scFv phage libraries, highly specific antibodies have been made against these epitopes and exploited in cancer therapeutic strategies.336–342
Most of the MAb targeting effort has been focused on individual tumor cells, but alternative strategies directed at tumor neovasculature343,344 tumor stroma,345 fibronectin,302,303 tumor stem cells,145,346 tumor infiltrating macrophages,347 or tumor infiltrating T cells348 are promising approaches. Bevacizumab (Avastin), a humanized IgG1 specific for VEGF, was effective against metastatic renal cancer,349,350 and when combined with chemotherapy, it was effective against NSCLC,351 metastatic CRC,352 and metastatic breast cancer.353 Furthermore, MAb can be made to inhibit homing of angiogenic progenitors, e.g., anti-VLA4 (Natalizumab),354 anti-VEGF-R1,355 or anti VEGF-R2/KDR.356,357 Targeting tumor vasculature may have significant advantages compared with direct tumor targeting,358 in that endothelial cells, unlike tumor cells, are less likely to acquire resistance. Another angiogenesis target is αVβ3 integrin, which initiates endothelial proliferation, migration, and matrix remodeling.359 In a phase 1 trial, chimeric IgG1 (MEDI-522) specific for αVβ3360 was well tolerated and tumor perfusion was possibly modified.