[level-membership-for-neurology-category]
Chapter 19 The tardive syndromes
Phenomenology, concepts on pathophysiology and treatment, and other neuroleptic-induced syndromes
Overview
Fundamentals and definitions
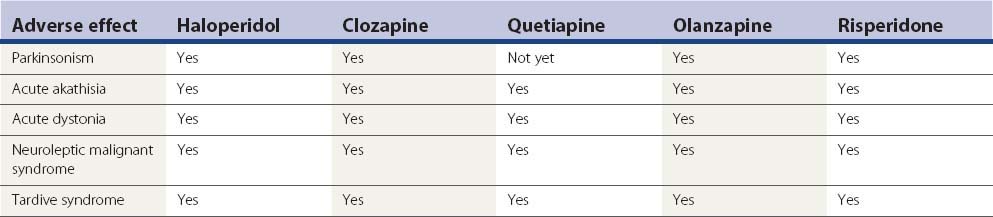
A variety of neurologic adverse effects are seen with drugs that block dopamine D2 receptors. Because these complications are mainly movement disorders and likely relate to the D2 receptors in the striatum and limbic system, they are usually called extrapyramidal reactions. These are listed in Table 19.1 and are covered in the clinical sections that follow.
Table 19.1 Neurological adverse effects of dopamine receptor antagonists
Adapted from Fahn S: The tardive dyskinesias. In Matthews WB, Glaser GH (eds): Recent Advances in Clinical Neurology, vol. 4. Edinburgh: Churchill Livingstone, 1984, pp. 229–260.
The term tardive syndromes refers to a group of disorders that fit all of the following essential criteria: (1) Phenomenologically, the clinical features are that of a movement disorder – i.e., abnormal involuntary movements or a sensation of restlessness that often causes “unvoluntary” movements; (2) the disorder is caused by the patient’s having been exposed to at least one DRBA within 6 months of the onset of symptoms (in exceptional cases, exposure could be up to 12 months); and (3) the disorder persists for at least 1 month after stopping the offending drug (Fahn, 1984a; Stacy and Jankovic, 1991). The question arises as to what to call persistent dyskinesias that are induced by drugs other than DRBAs. For example, Miller and Jankovic (1992) described such a patient who had been exposed to flecainide, a drug not known to be a DRBA. Another drug, buspirone, an azospirone compound, is an anxiolytic that is not known to have any dopamine receptor-blocking activity. Yet there is a report of two patients who had persistent movement disorders after prolonged treatment with this drug (LeWitt et al., 1993). One patient had cervical-cranial dystonia, and the other had an exacerbation of preexisting spasmodic torticollis and TD. It is possible, however, that future laboratory investigation will reveal flecainide and buspirone or one of their metabolites actually to be a DRBA.
Although there are several phenomenologically distinct types of tardive disorders, the collective group is referred to as tardive dyskinesia for historical reasons. Unfortunately, the term tardive dyskinesia is often used also to refer to a phenomenologically specific type of tardive syndrome, so the literature is often confusing; this note of caution is particularly important in trying to understand whether the author of a paper is referring to the tardive syndromes collectively or to a specific tardive syndrome. Since there could be different pathophysiologic mechanisms and treatments for the different forms of tardive syndromes, it is best that they be divided phenomenologically (Table 19.2). In this review, the tardive syndromes as a whole are referred to as tardive dyskinesia, and the specific type that was historically and initially labeled as tardive dyskinesia is referred to as classic tardive dyskinesia. Other names have been used for classic tardive dyskinesia (see Table 19.2). The phenomenologically essential component of classic tardive dyskinesia is the presence of repetitive, almost rhythmic, movements. These are almost always present in the mouth region, and therefore are also called O-B-L dyskinesias.
Table 19.2 Terminology of the tardive syndromes
| Descriptions | Equivalent common names |
|---|---|
| Tardive syndromes as a group | Tardive syndrome |
| Tardive dyskinesia | |
| Repetitive, rhythmic movements, usually in the oral-buccal-lingual region | Classic tardive dyskinesia (TD) |
| Oral-buccal-lingual (O-B-L) dyskinesias | |
| Tardive stereotypy | |
| Rhythmic chorea | |
| Dystonic movements and postures | Tardive dystonia |
| Restlessness and the movements that occur as a result | Tardive akathisia |
| Myoclonus | Tardive myoclonus |
| Tremor | Tardive tremor |
| Tics | Tardive tics |
| Tardive tourettism | |
| Chorea | Withdrawal emergent syndrome |
| Tardive chorea | |
| Oculogyria | Tardive oculogyric crisis |
| Parkinsonism | Tardive parkinsonism (if it exists) |
Tardive dyskinesia was first described in patients who were treated for schizophrenic psychosis with antipsychotics (Schonecker, 1957; Sigwald et al., 1959). These abnormal movements appeared late in the course of treatment, in contrast to acute dystonic reactions and drug-induced parkinsonism, which had previously been recognized as complications from antipsychotics, hence the term tardive. The offending antipsychotics are now known to block the D2 dopamine receptor; i.e., these drugs are DRBAs. Since the first descriptions, TD has also been noted in patients without psychiatric disorders who had other indications for using dopamine receptor antagonists, such as those with gastrointestinal complaints (Casey, 1983), with Gilles de la Tourette syndrome (Riddle et al., 1987), or with dystonia (Greene and Fahn, 1988).
Dopamine receptor antagonists produce many undesirable side effects, most of which occur relatively early in the course of treatment and are reversible on discontinuation of the medication. However, disfiguring and disabling abnormal involuntary movements were also noted to often occur late in the course of treatment and these were often noted to persist, even after discontinuation of the medication. Hence, the term “tardive” was coined, referring to the late and insidious onset (Faurbye et al., 1964). Initially the term tardive dyskinesia was equated with stereotypic repetitive movements of oral, buccal, and lingual distribution (Schonecker, 1957), but subsequently other types of movements have been recognized (Burke et al., 1982; Fahn, 1984a). As such, the concept of tardive dyskinesia has evolved and has been modified considerably since the initial recognition of the syndrome.
The prevalences of drug-induced parkinsonism and the various tardive syndromes have been compared by van Harten and colleagues (1996b) on the island of Curaçao, which has only one psychiatric facility. In 194 inpatients, the prevalence for classic tardive dyskinesia (TD) was 39.7%, that for parkinsonism was 36.1%, that for tardive dystonia was 13.4%, and that for akathisia was 9.3%. Combinations of two or more of these phenomenologies occurred in 30% of patients (van Harten et al., 1997). van Harten and colleagues (2006) continued to follow their patients over 9 years (mean duration of 18 years exposure to first-generation antipsychotic drugs); they found the annual incidence rate for classic TD to be 10.2% and for tardive dystonia to be 0.7%. Severity was associated with age and akathisia but not with drug-induced parkinsonism.
Dopamine receptors and their antagonists
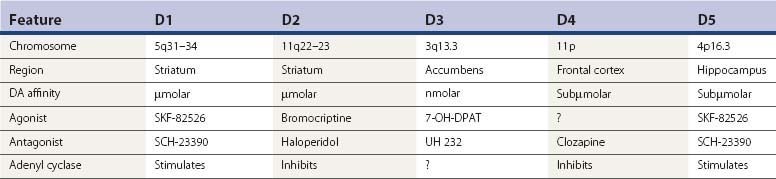
Since TD is an iatrogenic disorder and the most constant feature of the syndrome is the pharmacologic class of the responsible etiologic agent, it is important to understand the nature of the drugs that produce TD (Table 19.3). Although models of abnormal basal ganglia circuitry have been proposed to explain the mechanism of TD (Marchand and Dilda, 2006), the pathophysiology is still poorly understood. Dopamine receptors are classified into five subtypes, based on the genetics of the receptors; they are labeled D1, D2, D3, D4, and D5 (Kebabian and Calne, 1979; Sokoloff et al., 1990). Table 19.4 characterizes the five dopamine receptors. It is the dopamine D2 receptor-blocking action of drugs that has been linked to the tardive syndromes and other neuroleptic drug-induced movement disorders (described later and listed in Table 19.5).
| Class of drug | Examples of drugs in each class |
|---|---|
| 1. Phenothiazines | |
| a. Aliphatic | Chlorpromazine (Thorazine) Triflupromazine (Vesprin) |
| b. Piperidine | Thioridazine (Mellaril) Mesoridazine (Serentil) |
| c. Piperazine | Trifluoperazine (Stelazine) Prochlorperazine (Compazine) Perphenazine (Trilafon) Fluphenazine (Prolixin) Perazine |
| 2. Thioxanthenes | |
| a. Aliphatic | Chlorprothixene (Tarctan) |
| b. Piperazine | Thiothixene (Navane) |
| 3. Butyrophenones | Haloperidol (Haldol) Droperidol (Inapsine) |
| 4. Diphenylbutylpiperidine | Pimozide (Orap) |
| 5. Dibenzazepine | Loxapine (Loxitane) Asenapine (Saphris) |
| 6. Dibenzodiazepine | Clozapine (Clozaril) Quetiapine (Seroquel) |
| 7. Thienobenzodiazepine | Olanzapine (Zyprexa) |
| 8. Pyrimidinone | Risperidone (Risperidal) Paliperidone ER (9-hydroxyrisperidone) |
| 9. Benzisothiazole | Ziprasidone (Geodon) |
| 10. Benzisoxazole | Iloperidone (Zomaril) |
| 11. Substituted benzamides | Metoclopramide (Reglan) Tiapride (Tiapridex) Sulpiride (Meresa) Clebopride Remoxipride Veralipride (Agreal, Agradil) Amisulpride (Solian) Levosulpiride |
| 12. Indolones | Molindone (Moban) |
| 13. Quinolinone | Aripiprazole (Abilify) |
| 14. Tricyclic | Amoxapine (Asendin) |
| 15. Calcium channel blockers | Flunarizine (Sibelium) Cinnarizine (Stugeron) |
| 16. N-acetyl-4-methoxytryptamine | Melatonin |
| 17. Interferon-alpha | Pegylated interferon alpha 2b (IFN-α) (Pegylated = polyethylene glycol (PEG) attached to proteins) |
Recently introduced drugs in Table 19.3, e.g., iloperidone (Jain, 2000), ziprasidone (Hirsch et al., 2002), amisulpride (Curran and Perry, 2002), aripiprazole (Tamminga and Carlsson, 2002), paliperidone ER (Chwieduk and Keating, 2010), levosulpiride (Shin et al., 2009), and asenapine (Kane et al., 2010), need to be in clinical use for several years before their full potential in causing tardive dyskinesia syndromes can be known. The earlier ones in the above sentence have already been found to induce the acute and delayed movement disorders described in this chapter. In Table 10.3, the calcium channel blockers deserve comment. Cinnarizine (1-diphenylmethyl-4-(3-phenyl-2-propenyl) piperazine) and its difluorinated derivative flunarizine can induce parkinsonism (Teive et al., 2004). They are antagonists at the D2 receptors (Belforte et al., 2001), and they also inhibit the MgATP-dependent generation of a transmembrane proton electrochemical gradient and dopamine vesicular uptake (Terland and Flatmark, 1999). Whether either of these latter mechanisms, rather than a proposed DRBA action, is responsible for the neuroleptic activity is uncertain. In regard to melatonin, there is a case report of withdrawal-emergent O-B-L dyskinesias associated with akathisia that occurred with sudden discontinuation of chronic melatonin use (Giladi and Shabtai, 1999). Such withdrawal syndromes are typical of drugs that block dopamine receptors. On resumption of melatonin, the patient’s O-B-L dyskinesia and akathisia cleared. Sudden cessation of the drug again brought on the symptoms; slow taper over 2 months was effective without incident. This case suggests that either the melatonin product the patient was taking was impure and was contaminated with a DRBA or melatonin itself has dopamine receptor antagonist properties. In support of the latter is the result of a blinded crossover study showing some suppression of the tardive dyskinesia with melatonin (Shamir et al., 2001). Pegylated interferon alpha-2b (IFN-α), used in the treatment of hepatitis C virus infection, has been reported to cause parkinsonism, akathisia, and acute dystonic reaction (Quarantini et al., 2007). IFN-α has been shown to decrease dopaminergic activity in mice (Shuto et al., 1997).
Some drugs that block dopamine D2 receptors are promoted for medical problems other than psychosis, but these drugs can cause drug-induced parkinsonism, acute dystonic reactions, tardive syndromes, and NMS, just like the drugs that are promoted for the treatment of psychosis. Metoclopramide (Ganzini et al., 1993) and clebopride (Sempere et al., 1994) are used mainly for dyspepsia and as antiemetic agents. Amoxapine has a tricyclic structure and is marketed as an antidepressant drug, but a metabolite has dopamine receptor-blocking activity and has been implicated in producing TD (Kang et al., 1986; Sa et al., 2001). Veralipride is a substituted benzamide that is used for the treatment of menopausal hot flushes (Masmoudi et al., 1995). Pimozide is marketed for the treatment of Tourette syndrome. Some commercial preparations contain dopamine receptor antagonists in combination with other drugs, and this can lead to inadvertent use of these drugs. A popular combination is that of perphenazine and amitriptyline, marketed as Triavil and Etrafon. Risperidone is commercially promoted with the suggestion that it might have less risk of drug-induced complications, but this appears not to be the case. Parkinsonism and TD have been noted in association with the calcium channel antagonists flunarizine and cinnarizine (Micheli et al., 1987). Both of these medications have mild dopamine receptor antagonist activity, which is thought be the mechanism for their complications (Micheli et al., 1989). Recognition of these drugs is essential not only in making the diagnosis of TD but also in preventing the occurrence of TD by being able to avoid using them.
The D1 and D2 receptors are found mainly in the striatum and nucleus accumbens, as well as in the substantia nigra, amygdala, cingulate cortex, and entorhinal area. The anterior lobe of the pituitary gland has only D2 receptors, and the thalamus and cerebral cortex outside of the cingulate and entorhinal area contain D1 receptors only (De Keyser et al., 1988). D2 receptor affinities of dopamine receptor antagonists correlate closely with antipsychotic and antiemetic properties of the drugs (Creese et al., 1976). Dopamine receptor antagonists are often referred to as neuroleptics or antipsychotics; the former term indicates the effect of drugs in producing parkinsonism, and the latter indicates the effect of controlling psychosis.
“Atypical” antipsychotics
The label “atypical” refers to a lower propensity of the antipsychotic agent to induce parkinsonism or a variety of other movement disorders described later (and listed in Table 19.5), that is these agents have a lower propensity to be neuroleptics. A number of epidemiologic studies have found that the atypicals are less likely to induce tardive dyskinesia and other movement disorder problems (Tarsy and Baldessarini, 2006). Dolder and Jeste (2003) found that the atypicals reduced the incidence of tardive dyskinesia by half.
Dopamine and serotonin (5-HT) receptor antagonism
Although drugs that are labeled as atypical antipsychotic agents block dopamine D2 receptors, they also block serotonin 5-HT2A receptors, and some investigators attribute their antipsychotic effect to this mechanism and recommend research and drug development on newer agents that block other 5-HT receptors (Meltzer, 1999).
How tightly drugs to bind to the D2 receptor (their occupancy rate) is currently considered to be important in the drug having neuroleptic potential in direct proportion. Seeman and Tallerico (1998) found that the atypical antipsychotic drugs bind more loosely than classic antipsychotic drugs (neuroleptics) to the D2 receptors. Kapur and colleagues (1999) measured D2 and 5-HT2 receptor occupancies by positron emission tomography (PET) scan for patients receiving clozapine, risperidone, or olanzapine. Clozapine showed a much lower D2 occupancy (16–68%) than risperidone (63–89%) and olanzapine (43–89%); all three showed greater 5-HT2 occupancy than D2 occupancy at all doses, although the difference was greatest for clozapine. In their PET study, Moresco and colleagues (2004) found that striatal D2 receptor occupancy was significantly higher with olanzapine than with clozapine. All these studies support the relationship between receptor occupancy and the clinical observations that clozapine and quetiapine are the most “atypical,” with the least propensity to cause parkinsonism, tardive syndromes, or other neuroleptic drug reactions.
Despite interest in the 5-HT2A receptor, it is the D2 receptor that is associated with the antipsychotic effect (Seeman, 2010). Antipsychotic clinical doses correlate with their affinities for this receptor. The receptor has high- and low-affinity states. Clozapine and quetiapine do not elicit parkinsonism and rarely result in tardive dyskinesia because they are released from D2 within 12–24 hours. Traditional antipsychotics remain attached to D2 receptors for days, preventing relapse, but allowing accumulation that can lead to tardive dyskinesia. Glutamate agonists that treat schizophrenia have affinity for the dopamine D2(High) receptor and the D3 receptor (Seeman and Guan, 2009).
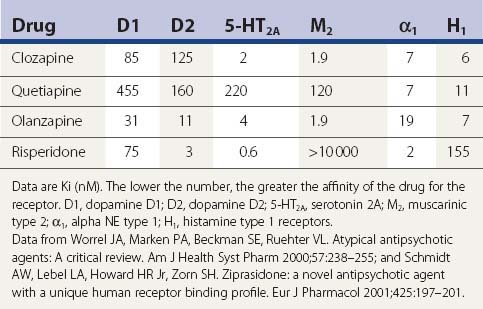
A comparison of the receptor-binding profile of quetiapine, clozapine, olanzapine, and risperidone is presented in Table 19.5. Both quetiapine and clozapine have poor affinity for the dopamine D2 receptor, which probably accounts for the low incidence of inducing parkinsonism and tardive syndromes. Risperidone has the highest affinity for the D2 receptor, resembling haloperidol (see Table 19.6), and is therefore more of a “typical” than an “atypical” antipsychotic.
Table 19.6 A more complete listing of affinities for human receptors and rat transporters by antipsychotic drugs
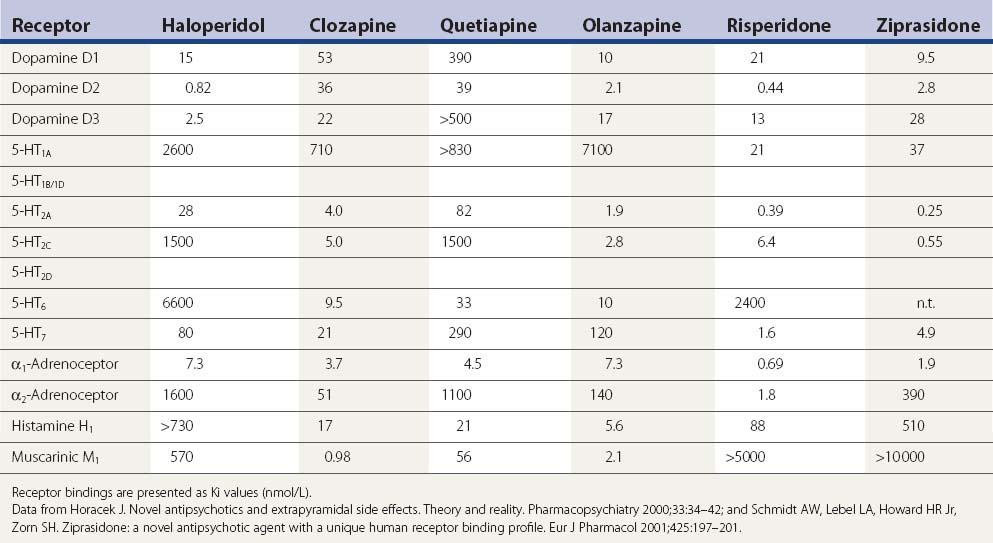
In one PET study striatal D1 and D2 receptor occupancies were evaluated (Tauscher et al., 2004). D1 occupancy ranged from 55% with clozapine to 12% with quetiapine (rank order: clozapine > olanzapine > risperidone > quetiapine). The striatal D2 occupancy ranged from 81% with risperidone to 30% with quetiapine (rank order: risperidone > olanzapine > clozapine > quetiapine). The ratio of striatal D1/D2 occupancy was significantly higher for clozapine (0.88) relative to olanzapine (0.54), quetiapine (0.41), or risperidone (0.31). In an [123I]epidepride single photon emission computed tomography (SPECT) study involving clozapine-, olanzapine-, and haloperidol-treated schizophrenia patients, as well as drug-naive patients and healthy controls, midbrain D(2/3) receptor occupancy was studied. Of those on medication, occupancy was least for those on clozapine (5%), next for those on olanzapine (28%), and greatest for those on haloperidol (40%); no significant differences were observed in the temporal poles (Tuppurainen et al., 2009). All imaging studies are compatible with the clinical results in that clozapine and quetiapine, with the least neuroleptic features, have the lowest D2 receptor occupancy rate.
Clozapine
Clozapine was the first agent to be labeled as an atypical antipsychotic and deservedly so, although rare case reports do exist of drug-induced parkinsonism (Kurz et al., 1995), but it has been reported not to induce rigidity, and it rarely induces parkinsonian tremor (Gerlach and Peacock, 1994). It has also caused rare cases of acute akathisia (Friedman, 1993; Safferman et al., 1993), acute dystonic reaction (Kastrup et al., 1994; Thomas et al., 1994), tardive syndromes (Dave, 1994), tardive dystonia (Bruneau and Stip, 1998; Molho and Factor, 1999a, van Harten et al., 2008b), tardive akathisia (Kyriakos et al., 2005), tardive oculogyria (Uzun and Doruk, 2007), and NMS (Sachdev et al., 1995; Benazzi, 1999; Lara et al., 1999; Gambassi et al., 2006). There are also parkinsonian features that are seen with clozapine. In a prospective study, seven out of 25 patients on clozapine developed TD (Bunker et al., 1996). In a retrospective study, comparison of clozapine and typical antipsychotics showed no lower prevalence of tardive dyskinesia in the clozapine group (Modestin et al., 2000). But it is not clear that the patients in the clozapine group had not been previously exposed to typical antipsychotics and had not developed TD while receiving them. What is clear from this study is that the conversion to using clozapine has not markedly reduced the prevalence of extrapyramidal syndromes; this finding supports the realization that clozapine does not effectively treat TD once it has developed. How effective its use would be in preventing TD in the first place still needs to be verified. For patients with PD, the low propensity to augment existing parkinsonism makes clozapine very useful in treating patients who have dopaminergic drug-induced psychosis (Factor and Friedman, 1997; Friedman et al., 1999).
A SPECT study measuring dopamine D2 receptor binding reveals lower binding with clozapine than with typical neuroleptics (Broich et al., 1998). In animal studies, rats that have been pretreated with haloperidol for 4 weeks develop vacuous chewing movements (VCMs) after treatment with a dopaminergic, whereas rats that have been pretreated with clozapine do not (Ikeda et al., 1999).
A problem with clozapine is that weekly blood counts are required because there is a 1% to 2% incidence of leukopenia, which is reversible if the drug is withdrawn within 1–2 weeks. Granulocyte colony-stimulating factor can be an effective means to treat the agranulocytosis (Sperner-Unterweger et al., 1998). Other common adverse effects are drowsiness, drooling, weight gain, and seizures. An unusual adverse effect with clozapine was a case of myokymia (David and Sharif, 1998). Myocarditis associated with clozapine has been reported (Hill and Harrison-Woolrych, 2008).
Quetiapine
After clozapine, quetiapine is the most favorable in being least likely to induce parkinsonism, NMS (Stanley and Hunter, 2000), or tardive syndromes. In a prospective, head-to-head comparison with risperidone treatment in schizophrenia, quetiapine had far fewer extrapyramidal adverse effects, but more orthostasis (Perez et al., 2008). D2 receptor antagonism is relatively selective for limbic than striatal receptors for clozapine and sertindole, followed by quetiapine, ziprasidone, olanzapine, and remoxipride, whereas risperidone in many respects has a profile that resembles that of haloperidol (Arnt and Skarsfeldt, 1998). In primates, quetiapine and clozapine were equally less likely to induce oral dyskinesias compared to standard antipsychotics (Peacock and Gerlach, 1999). Like clozapine, quetiapine easily induces drowsiness, so when it is used to treat dopa-induced psychosis, it should be taken at bedtime. The major advantage over clozapine is that it does not require blood tests because it does not induce agranulocytosis. However, there may be a rare risk that quetiapine can cause agranulocytosis (Ruhe et al., 2001). Since its introduction, there have been reports of acute dystonic reactions (Jonnalagada and Norton, 2000; Desarker and Sinha, 2006), acute akathisia (Prueter et al., 2003), neuroleptic malignant syndrome (El-Gaaly et al., 2009; Gortney et al., 2009), and also TD (Ghaemi and Ko, 2001). In 367 patients treated for 12 months with quetiapine (mean dose 720 mg/day), parkinsonism was seen in 10%, akathisia in 3%, dystonia in 1.4%, and “hyperkinesia” in 1.7% (Perez et al., 2008).
In a SPECT study using the D2/D3 ligand [I-123]-epidepride the percent occupancy of receptors in the limbic system (temporal lobe) and striatal receptors while patients were receiving quetiapine was 60% and 32%, respectively, which is similar to clozapine (Stephenson et al., 2000). In another SPECT study, quetiapine was shown to occupy 5-HT2A receptors in the frontal and temporal cortex (Jones et al., 2001). A PET study using [18F]fallypride showed similar results (Vernaleken et al., 2010).
A patient with oculogyric crisis, who failed to improve after withdrawal of antipsychotic medication, was successfully treated with quetiapine (Gourzis et al., 2007). Drug-induced parkinsonism and acute akathisia due to other neuroleptics can be reduced by switching patients to quetiapine (Cortese et al., 2008). Drowsiness and weight gain are common; postural hypotension is not infrequent. A rare complication of quetiapine is hepatotoxicity (Shpaner et al., 2008).
Olanzapine
In contrast to clozapine and quetiapine, olanzapine more readily increases parkinsonian symptoms in patients with PD (Jimenez-Jimenez et al., 1998; Molho and Factor, 1999b; Granger and Hanger, 1999), but does so less readily than risperidone and conventional antipsychotics. Drug-induced parkinsonism, including the rabbit syndrome, is seen with olanzapine (Durst et al., 2000), but less than with haloperidol (Peuskens et al., 2009). It can induce acute akathisia (Kurzthaler et al., 1997; Jauss et al., 1998) and TD but less so than haloperidol (Tollefson et al., 1997; Wood 1998). It can also induce NMS (Filice et al., 1998; Moltz and Coeytaux, 1998; Burkhard et al., 1999; Levenson, 1999; Margolese and Chouinard, 1999; Sierra-Biddle et al., 2000; Abu-Kishk et al., 2004; Zaragoza Fernandez et al., 2006), tardive dyskinesia (Herran and Vazquez-Barquero, 1999), and tardive dystonia (Dunayevich and Strakowski, 1999). Acute dystonic reactions also occur (Beasley et al., 1997; Landry and Cournoyer, 1998; Vena et al., 2006).
A direct comparison with chlorpromazine showed similar parkinsonism and acute akathisia for the two drugs (Conley et al., 1998). A double-blind comparison with haloperidol by Beasley and colleagues (1999) showed a much lower risk of developing tardive dyskinesia with olanzapine. After 1 year of exposure, 0.52% of patients developed TD with olanzapine and 7.45% developed TD with haloperidol. An open-label comparison with conventional antipsychotics after a 9-month follow-up after discharge from the hospital favored olanzapine, with TD being present in 2.3% for olanzapine (2/87), and 16.7% (12/72) for the conventional treatment (Mari et al., 2004). There are case reports of agranulocytosis induced by olanzapine (Meissner et al., 1999; Naumann et al., 1999; Tolosa-Vilella et al. 2002), including in a patient who had previously had agranulocytosis with clozapine (Benedetti et al., 1999). This has been attributed to some similar structural and pharmacologic properties of clozapine. A case of restless legs syndrome with periodic movements in sleep has been attributed to olanzapine (Kraus et al., 1999).
Olanzapine has relative regional mesolimbic dopaminergic selectivity and a broad-based binding affinity for serotonin (all 5-HT2 receptor subtypes and the 5-HT6 receptor), dopamine (D2, D3, and D4 receptors), muscarinic, and α1-adrenergic receptors (Bymaster et al., 1999). A PET study in schizophrenic patients being treated with olanzapine revealed that this drug is a potent 5-HT2 blocker, but also a blocker of D2 dopamine receptors similar to risperidone and less so than clozapine (Kapur et al., 1998). Patients on olanzapine were also studied with [123I]iodobenzamide (IBZM) SPECT; the D2 receptor was occupied 60% and 83% of the time at doses of 5 mg/day and 10 mg/day, respectively (Raedler et al., 1999). Such high rates of occupancy probably account for olanzapine’s tendency to worsen parkinsonism and induce DRBA complications, because, as was noted above, D2 receptor occupancy rates are directly related to neuroleptic potential.
Risperidone
With the success of clozapine, there is a commercial advantage for pharmaceutical companies to tout other antipsychotics as “atypical.” Such has been claimed for risperidone, but this drug can readily induce parkinsonism including the rabbit syndrome (Levin and Heresco-Levy, 1999), tardive syndromes (Haberfellner 1997; Silberbauer 1998; Hong et al., 1999; Ananth et al., 2000), and NMS (Newman et al., 1997; Norris et al., 2006). The prevalence of parkinsonism from risperidone is usually considered to be less than that with conventional neuroleptics, but it was observed in 42% compared to 29% in those on haloperidol (Knable et al., 1997). Tardive dyskinesia and tardive dystonia occurred in a patient who was exposed only to risperidone (Bassitt and Garcia, 2000). The annual incidence of TD in patients taking risperidone has been estimated to be 0.3%, compared to an annual incidence in patients taking conventional neuroleptics of 5–10% (Gutierrez-Esteinou and Grebb, 1997). However, in an open-label prospective study of 255 institutionalized patients with dementia who were treated with risperidone, the 1-year cumulative incidence of TD was 2.6% (Jeste et al., 2000).
Like conventional neuroleptics, risperidone induces acute dystonic reactions in marmosets, in contrast to clozapine, which does not (Fukuoka et al., 1997). In a report of an open-label comparison with haloperidol, Rosebush and Mazurek (1999) found the two drugs to have a similar side effect profile. In a prospective follow-up of first-episode schizophrenic patients treated with risperidone, movement disorders developed in more than one-third of these patients, who had previously never been exposed to antipsychotic drugs (Lang et al., 2001). When risperidone is compared with low-potency antipsychotics, such as thioridazine, no difference was discerned in the rates of developing movement disorders (Schillevoort et al., 2001).
In a SPECT study dopamine receptor binding with IBZM showed risperidone to have effects between those of haloperidol and clozapine, with a dose–response curve for risperidone showing greater similarity to that of haloperidol (Dresel et al., 1998). A PET study showed dopamine D2 receptors occupancies of about 70% and 60% in the striatum and extrastriatum, respectively (Ito et al., 2009). Clearly, risperidone is not an “atypical” antipsychotic agent. Risperidone’s occupancy of the 5-HT2 receptors is about 90%, and its occupancy of the D2 receptors is between 50% and 80%, but the latter correlates with the extrapyramidal side effects (Yamada et al., 2002).
Ziprasidone
This benzisothiazole was approved by the Food and Drug Administration in 2001. There are already case reports of acute dystonic reactions (Weinstein et al., 2006; Yumru et al., 2006, Rosenfield et al., 2007), NMS, rhabdomyolysis, and pancreatitis (Murty et al., 2002; Yang and McNeely, 2002; Gray 2004). It has been reported to cause tardive dyskinesia (Mendhekar, 2005) and tardive dystonia (Papapetropoulos et al., 2005; Tsai et al., 2008). Although it is a potent 5-HT2A antagonist (like risperidone, olanzapine, and clozapine), it is also a D2 antagonist in humans as detected by PET scan (Bench et al., 1993, 1996). But in-vitro studies reveal much lower affinity for the D2 receptor than for the 5-HT2A receptor (Seeger et al., 1995), and ziprasidone also binds less tightly to the D2 receptor than dopamine (Seeman, 2002). Ziprasidone has two other unique features compared to other antipsychotic agents: (1) it is a potent 5-HT1A agonist and thus inhibits dorsal raphe serotonergic cell firing (Sprouse et al., 1999) and increases cortical dopamine release (Rollema et al., 2000), and (2) it inhibits neuronal uptake of 5-HT and norepinephrine in a manner comparable to the antidepressant imipramine (Schmidt et al., 2001). What these unique actions might contribute to antipsychotic activity or to propensity for or against extrapyramidal reactions is unclear. Ziprasidone has been reported to induce a case of oculogyric crisis in an adult (Viana Bde et al., 2009).
Aripiprazole
This quinolinone has a novel mechanism of action. Like a number of other “atypical” antipsychotics, it is an antagonist at the 5-HT2A receptors. It is also a 5-HT1A partial agonist (Jordan et al., 2002). What is novel is that it is a partial agonist at the dopamine D2 receptor. It has a higher affinity for the presynaptic autoreceptor than for the postsynaptic receptor. Hence, it reduces dopamine synthesis and release through an agonist action at the dopamine autoreceptor (Tamminga and Carlsson, 2002). Because of its novel action as a partial D2 agonist, it was anticipated that it might cause fewer extrapyramidal adverse effects, and clinical trials reported favorable results (Kane et al., 2002). However, D2 ligand binding PET revealed a dose-related high occupancy state of 71.6% at 2 mg/day to 96.8% at 40 mg/day (Kegeles et al., 2008). With wider use, it has been shown to worsen PD (Friedman et al., 2006; Wickremaratchi et al., 2006), and there are already reports of it causing parkinsonism (López-Torres et al., 2008), NMS (Chakraborty and Johnston, 2004; Hammerman et al., 2006; Palakurthi et al., 2007), acute dystonic reactions (Desarkar et al., 2006; Fountoulakis et al., 2006), and tardive dyskinesia (Zaidi and Faruqui, 2008; Abbasian and Power, 2009; Friedman, 2010).
Amisulpride
Amisulpride, a substituted benzamide, is a highly selective antagonist for dopamine D2 and D3 receptors in the limbic region, which would predict potent antipsychotic activity with a low potential to cause extrapyramidal symptoms (Lecrubier, 2000). It binds less tightly to the D2 receptor than do the typical antipsychotics (Seeman, 2002). Using the full range of recommended amisulpride dosage, striatal occupancies up to 90% can be measured (Vernaleken et al., 2004). It is not yet commercially available in the United States. It has already been reported to induce tardive oculogyric crisis (Mendhekar et al., 2010).
Adverse effects from second-generation antipsychotics
Aside from the D2-blocking effects described above, the five drugs in Table 19.7 have a number of other adverse effects. Sedation is a particular problem seen with each of them, but particularly clozapine and quetiapine. Metabolic side effects are now widely recognized with the second-generation antipsychotics, with weight gain a common problem (Leucht et al., 2009; Patel et al., 2009). Table 19.7 lists the common adverse effects of these drugs, along with haloperidol, for comparison.
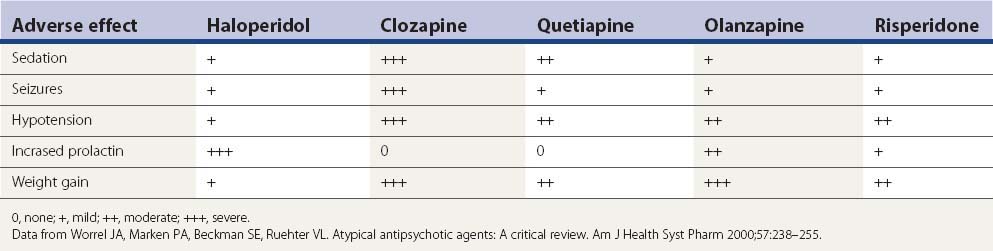
It is not likely that drug-induced tardive syndromes will disappear because use of both typical and atypical agents continues. In a survey in Lombardy (Italy), 35 363 individuals over the age of 65 were prescribed an antipsychotic prescription (2.18 subjects per 100 inhabitants, with two-thirds receiving first-generation agents) (Percudani et al., 2004). Although there may be a lower risk of developing these disorders with “atypicals,” these drugs can induce them (Table 19.8). It is even possible that if physicians consider “atypicals” safe to use, their use will increase, even in situations in which the risk from typicals might have precluded their use. In the United States, the prevalence of atypical antipsychotic use was found to be 267.1 per 100 000 subjects aged 19 years and younger and was more than twice as high for male patients as for female patients (Curtis et al., 2005).
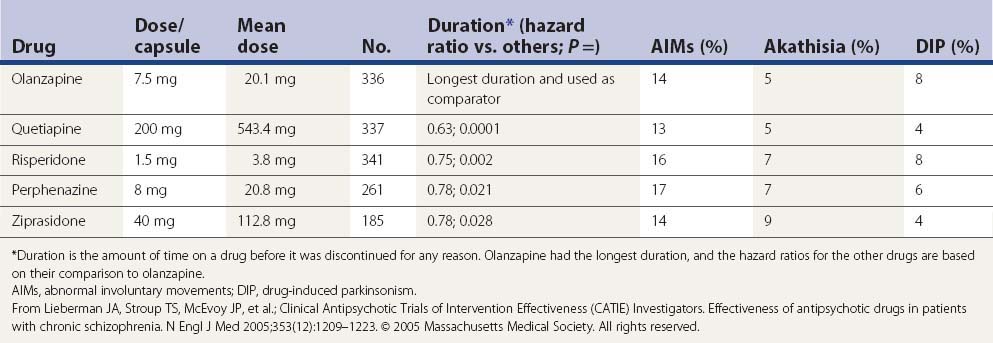
In a large US randomized controlled trial (the CATIE study) comparing second-generation antipsychotics (olanzapine, quetiapine, risperidone, perphenazine, or ziprasidone) with the first-generation perphenazine, only olanzapine had a slightly more favorable outcome of both the patients’ ability to stay on this drug for a longer duration and with slightly less drug-induced parkinsonism, akathisia, and tardive dyskinesia than all other drugs including perphenazine (Table 19.9); perphenazine fared as well as the other so-called ‘atypicals” (Lieberman et al., 2005). A similar outcome was seen in a smaller British study (CUtLASS) (Jones et al., 2006). This was a randomized controlled clinical trial involving 57 centers, with flexible dosing up to 4 capsules/day in 1460 schizophrenic subjects (after excluding 33 from one center, integrity concern). The primary outcome was time to discontinuation, with a maximum observation of 18 months. TD was present in 231 subjects at baseline, and these subjects were excluded from the perphenazine arm. The outcome had multiple comparison, requiring a P value ≤0.017 to be significant.
In a more detailed assessment of extrapyramidal side effects in the CATIE study, there were no significant differences in incidence or change in rating scales for parkinsonism, dystonia, akathisia, or tardive dyskinesia when comparing the second-generation antipsychotics with perphenazine or comparing between second-generation antipsychotics (Miller et al., 2008). Secondary analyses revealed greater rates of concomitant antiparkinsonism medication among individuals on risperidone and lower rates among individuals on quetiapine.
Metoclopramide
After the introduction of metoclopramide for gastrointestinal disorders, reports of acute dystonic reactions in children and tardive dyskinesia in adults began to appear (Cochlin, 1974; Kataria et al., 1978; Lavy et al., 1978). Today, metoclopramide has become a common cause of TD, and the legal profession has found such cases ripe for lawsuits. The annual incidence of TD from metoclopramide has been reported to be between 4.5% and 23% per year (Grimes, 1981; Ganzini et al., 1993), about the same as that for haloperidol ~5.7%/year (Tollefson et al., 1997; Beasley et al., 1999; Csernansky et al., 2002), but higher than those for risperidone ~1.8%/year (Lemmens et al., 1999; Davidson et al., 2000; Jeste et al., 2000; Csernansky et al., 2002) and olanzapine ~1.2%/year (Tollefson et al., 1997; Beasley et al., 1999). Because antipsychotic drugs have an affinity for brain neuromelanin (Seeman, 1988), Chen and colleagues (2010) examined the amount of metoclopramide and antipsychotic drugs binding to postmortem human substantia nigra. They found that in clinical conditions the amount of metoclopramide that would bind to nigra is much higher than the amount of raclopride, haloperidol, or olanzapine that would be expected to be bound and they suggest that this binding might explain the higher annual incidence of TD induced by metoclopramide.
Neurologic side effects of dopamine D2 receptor antagonists
To better understand the tardive syndromes, it is important to recognize the variety of other movement disorders that are induced by the dopamine receptor antagonists at different points in the course of treatment. These movement disorders are often lumped together as extrapyramidal syndromes, but the lumping often hinders the effort to sort out the clinical characteristics and pathophysiology of separate syndromes. It is better to subdivide them into their phenomenologic types (see Table 19.1). Movements that may be seen include acute dystonia, acute akathisia, parkinsonism, tardive syndromes, and NMS. Both dystonia and akathisia also occur as subtypes of tardive syndromes and both subtypes are discussed in more detail in the section on tardive syndromes.
Acute dystonia
The earliest abnormal involuntary movement to appear after initiation of dopamine receptor antagonist therapy is an acute dystonic reaction. In about half of the cases, this reaction occurs within 48 hours, and in 90% of cases, it occurs by 5 days after starting the therapy (Ayd, 1961; Garver et al., 1976). The reaction may occur after the first dose (Marsden et al., 1975).
Dystonic movements are sustained muscle contractions, frequently causing twisting and repetitive movements, or abnormal postures (Fahn, 1988). In a series of 3775 patients, Ayd (1961) found that acute dystonia is the least frequent side effect, affecting about 2–3% of patients, with young males being most susceptible. The incidence rate increases to beyond 50% with highly potent dopamine receptor blockers such as haloperidol (Boyer et al., 1987). In one prospective study, Aguilar and colleagues (1994) reported that 23 of 62 patients developed acute dystonia after haloperidol was introduced, that anticholinergic pretreatment significantly prevented this, and that younger age and severity of psychosis were risk factors. In the prospective study by Kondo and colleagues (1999), 20 (51.3%) of 39 patients placed on nemonapride had dystonic reactions, onset occurring within 3 days after the initiation of treatment in 90%. As in other series, the incidence of acute dystonia was significantly higher in males than in females (77.8% vs. 28.6%, P < 0.05), and younger males (≤30 years) had an extremely high incidence (91.7%). The incidence is much lower with the so-called atypical antipsychotics; Raja and Azzoni (2001) observed only 41 cases out of 1337 newly admitted patients treated with antipsychotics, which included 8 treated with risperidone, 1 with olanzapine, and 1 with quetiapine. A meta-analysis compared intramuscular injections of second-generation antipsychotics (SGAs) to haloperidol injections (Satterthwaite et al., 2008). SGAs were associated with a significantly lower risk of acute dystonia (relative risk = 0.19) and acute akathisia (relative risk = 0.25), compared with haloperidol alone.
All agents that block D2 receptors can induce acute dystonic reactions, including risperidone (Brody, 1996; Simpson and Lindenmayer, 1997) and clozapine (Kastrup et al., 1994). One child developed an acute dystonic reaction after ingestion of a dextromethorphan-containing cough syrup (Graudins and Fern, 1996). Dextromethorphan has several different known pharmacologic actions, but D2 receptor blockade is not one of them. A case of acute dystonia following abrupt withdrawal of bupropion (DA reuptake blocker) was reported (Wang et al., 2007). A case of acute dystonia following abrupt withdrawal of dexamphetamine in a patient taking risperidone was also reported (Keshen and Carandang, 2007). Serotonergic agents have also been reported to induce acute dystonic reactions (Lopez-Alemany et al., 1997; Madhusoodanan and Brenner, 1997; Olivera, 1997). The mechanism could relate to inadequate release of dopamine from the nerve terminals in the striatum owing to the inhibitory effect of serotonin on dopamine neurons in the substantia nigra pars compacta. The opioid σ1 and σ2 receptors have also been implicated (Matsumoto and Pouw, 2000).
Acute dystonic reactions most often affect the ocular muscles (oculogyric crisis), face, jaw, tongue, neck, and trunk, and less often the limbs. Oculogyric crisis has previously been noted to occur as a common feature of postencephalitic parkinsonism (Duvoisin and Yahr, 1965). A typical acute dystonic reaction may consist of head tilt backward or sideways with tongue protrusion and forced opening of the mouth, often with arching of trunk and ocular deviation upward or laterally (Rupniak et al., 1986). The forcefulness of the muscle contractions can be extremely severe and led to auto-amputation of the tongue in one patient (Pantanowitz and Berk, 1999).
Mazurek and Rosebush (1996) studied the timing in the development of an acute dystonic reaction in 200 patients who were taking a neuroleptic medication for the first time. The neuroleptic was given twice daily, and over 80% of the episodes of acute dystonia occurred between 12 noon and 11 p.m.
Reserpine and α-methylparatyrosine, which deplete presynaptic monoamines, have not been associated with acute dystonic reactions (Duvoisin, 1972; Marsden et al., 1975; Walinder et al., 1976). However, another dopamine depletor, tetrabenazine (TBZ), has been reported to induce acute dystonic reactions (Burke et al., 1985). One possible explanation for this difference is that in addition to depleting dopamine, TBZ blocks dopamine receptors (Reches et al., 1983).
It is important to mention the case described by Wolf (1973) of a patient who developed an oral dyskinesia while taking reserpine. Figure 1 in his paper clearly shows a dystonic phenomenon and not the complex, rapid, stereotypic movements of classic tardive dyskinesia. This development appeared too late after initiation of reserpine to be considered an acute dystonic reaction, however. Whether this could be an example of tardive dystonia is not clear, since the phenomenology of tardive dystonia is identical to that of naturally occurring primary dystonia; therefore, the patient could have had a coincidental case of spontaneous, idiopathic oromandibular dystonia (Fahn, 1984b). Thus, there is no absolute evidence that reserpine induces acute dystonic reactions, classic tardive dyskinesia, or tardive dystonia. In fact, symptoms of these three tardive syndromes can be suppressed by reserpine, and eventually reserpine can be withdrawn successfully in many patients without exacerbation of the symptoms (Fahn, 1983). Furthermore, the long time that reserpine has been available without a clear-cut case of tardive syndromes can be compared to the much shorter duration of use of metoclopramide, in which there are already cases of acute dystonic reactions, classic tardive dyskinesia, and tardive dystonia as a consequence of its use (Casteels-Van Daele et al., 1970; Gatrad, 1976; Pinder et al., 1976; Reasbeck and Hossenbocus, 1979; Miller and Jankovic, 1989; Lang, 1990).
Of 452 patients given high-dosage oral metoclopramide to control emesis, Kris and colleagues (1983) observed 14 who developed acute dystonic reactions. However, there was a distinct preponderance of the reactions occurring in children (6/22) compared to adults (8/430). Intravenous metoclopramide is more likely to induce it than oral administration (Pinder et al., 1976). In a study at a Veterans Administration hospital, comparing patients treated with metoclopramide and controls, the relative risk for TD was 1.67, and the relative risk for drug-induced parkinsonism was 4.0 (Ganzini et al., 1993).
The available biochemical explanations for the acute dystonic reaction are unsatisfactory, but several observations contribute to the understanding of the phenomena, relating it to dopamine, muscarinic, and sigma receptors. Acute neuroleptic administration produces sudden increase of dopamine release and increased turnover lasting for 24–48 hours after a single dose (O’Keefe et al., 1970; Marsden and Jenner, 1980). This effect is blocked by anticholinergics consistent with their efficacy in treatment of acute dystonic reactions (O’Keefe et al., 1970). Moreover, in baboons that were pretreated with reserpine and α-methylparatyrosine, which markedly reduces presynaptic dopamine concentration, haloperidol-induced acute dystonic reaction is abolished or greatly reduced (Meldrum et al., 1977). On the other hand, blockade of the postsynaptic receptor fades in about 12 hours after a single dose of antipsychotics, and supersensitivity of receptors begins to develop. Therefore, presynaptic dopaminergic excess in combination with the emerging supersensitive postsynaptic dopamine receptors could result in markedly increased striatal dopaminergic activity at about 20–40 hours after a neuroleptic dose. This period corresponds to the critical time for acute dystonic reactions in human subjects who were given a single dose of butaperazine (Garver et al., 1976). However, extrapolating the data from experimental animals to the clinical situation has its limitations including the fact that rats do not develop acute dystonic reactions. Further data on human cerebrospinal fluid (CSF) dopamine metabolites as an indicator of presynaptic function might prove to be of value in pursuing the hypothesis. Jeanjean and colleagues (1997) suggested that the σ2 receptors could be involved in the acute dystonic reaction. They found a correlation between the clinical incidence of neuroleptic-induced acute dystonia and binding affinity of drugs for the sigma receptor.
Another animal model of acute dystonia is the common marmoset that is treated with haloperidol (Fukuoka et al., 1997). But it takes at least 6 weeks of such treatment to develop this reaction. The dystonia subsides only to reappear when haloperidol treatment is restarted; other neuroleptics, including risperidone, can also make the dystonia reappear, but clozapine was without such an effect. In this animal model, the anticholinergic agent trihexyphenidyl inhibited the induction of acute dystonia.
In patients with acute dystonic reactions, symptoms can be relieved within minutes after parenteral anticholinergics or antihistaminics (Paulson, 1960; Waugh and Metts, 1960; Smith and Miller, 1961). Diphenhydramine 50 mg, benztropine mesylate or biperiden 1–2 mg is given intravenously and can be repeated if the effect is not seen in 30 minutes. Intravenous diazepam has also been shown to be effective and can be used as an alternative therapy (Korczyn and Goldberg, 1972; Gagrat et al., 1978; Rainer-Pope, 1979). If untreated, the majority of cases still resolve spontaneously in 12–48 hours after the last dose of the dopamine receptor antagonists. Dopamine receptor antagonists with high anticholinergic activities have low incidence rates of acute dystonic reactions (Swett, 1975). Therefore, prophylactic use of anticholinergics (Arana et al., 1988) and benztropine (Goff et al., 1991) has been studied and reported to be helpful in reducing the risk of acute dystonic reactions, especially in young patients on high-potency drugs. Three cases of recurrent episodes of acute dystonia and oculogyric crises despite withdrawal of the offending DRBAs (haloperidol in two cases, metoclopramide in one) have been reported, responding to anticholinergics each time (Schneider et al., 2009). The response of acute dystonic reactions to anticholinergics is so characteristic that it is difficult to explain the report of a few cases apparently due to amitriptyline, which has considerable anticholinergic activity and no known DRBA activity (Ornadel et al., 1992).
A case of an acute dystonic reaction occurring in an elderly person with bipolar disorder taking the serotonin uptake inhibitor paroxetine has been reported (Arnone et al., 2002). Speculation about the risk due to previous exposure to neuroleptics was raised. This class of drug can reduce firing rate of nigral dopaminergic neurons owing to their inhibition by serotonin.
Acute akathisia
The term akathisia, from the Greek, meaning unable to sit down, was coined by Haskovec in 1901 (cited by Mohr and Volavka, 2002), long before antipsychotic drugs were introduced. Akathisia was seen in some patients with advanced parkinsonism, and in others, it was frequently thought to be functional. Akathisia refers to an abnormal state of excessive restlessness, a feeling of the need to move about, with relief of this symptom on actually moving. Today, it is most frequently encountered as a side effect of neuroleptic drugs.
Two major issues of akathisia remain in confusion. First, there is no consensus about diagnostic criteria. Some authors consider akathisia to be an abnormal subjective state and regard the movements as an expression of the subjective state but not a necessary feature for the diagnosis (Van Putten, 1975). Others recognize the characteristic patterns of restless movements and consider presence of movements to be sufficient for the diagnosis (Munetz and Cornes, 1982; Barnes et al., 1983; Gibb and Lees, 1986). A second point of confusion is that akathisia occurs not only in an early-onset, self-limited form (acute akathisia), but also as a late-onset, persistent form (tardive akathisia). Much of the literature on akathisia does not distinguish between acute and tardive akathisia, which makes the interpretation of the literature difficult. The recognition of tardive akathisia as a distinct subsyndrome of tardive syndromes has been more recent (Fahn, 1978, 1983; Braude and Barnes, 1983; Weiner and Luby, 1983). For the discussion of the clinical features of akathisia, acute and tardive akathisia are lumped together, since they are similar, but their treatments and most likely their pathophysiologies are distinct.
The subjective aspect of akathisia is characterized by inner tension and aversion to remaining still. Patients complain of vague inner tension, emotional unease, or anxiety with vivid phrases such as “jumping out of my skin” or “about to explode.” Subjective descriptions, however, can be nonspecific. Inner restlessness and inability to remain still can be present in a significant number of psychiatric patients without akathisia and in control subjects without psychiatric problems (Braude et al., 1983). In an attempt to clarify the issue, Braude et al. (1983) systematically surveyed the frequency of various complaints and found that inability to keep the legs still was the most characteristic complaint and was present in over 90% of patients with akathisia in contrast to about 20% of those with other psychiatric disturbances. Others noted more conservative estimation of the frequency of complaints related to the legs from 27% (Gibb and Lees, 1986) to 57% (Burke et al., 1989). Various authors also described atypical features such as “acting out,” suicidal ideation, disruptive behaviors, homicidal violence, sexual torment, terror, and exacerbation of psychosis as akathitic phenomena (Van Putten and Marder, 1987). Evaluation of the subjective aspect also depends on patients’ ability to describe their feelings. Those with psychosis, dementia, or learning disability are often unable to provide useful descriptions for diagnosis. Although akathisia may manifest itself as subjective feeling alone, lack of specific subjective feeling and variable expression by patients pose a diagnostic dilemma. Therefore, the presence of the motor phenomenon is very helpful for the diagnosis.
Akathisia can present as focal pain or burning, usually in the oral or genital region (Ford et al., 1994). The symptom of moaning may be a verbal expression of the subjective feeling of akathisia. Some patients may moan as part of a generalized akathitic state and have other motor evidence of akathisia, such as marching in place, inability to sit still accompanied by associated walking about, inability to lie quietly with writhing and rolling movements, and making stereotypic caressing or rocking movements. The differential diagnosis of moaning includes parkinsonism, akathisia, levodopa usage (Fahn et al., 1996), dementia, pain, and other syndromes of phonations, such as tics, oromandibular dystonia, and Huntington disease, as discussed by Fahn (1991).
The motor aspect of akathisia (akathitic movements) is generally described as excessive movements that are complex, semipurposeful, stereotypic, and repetitive. Braude and colleagues (1983) found that rocking from foot to foot, walking on the spot, and coarse tremor and myoclonic jerks of the feet were characteristic of akathitic movements. Others agree that various leg and feet movements are more common in patients with akathisia than in those with TD (Gibb and Lees, 1986; Burke et al., 1989). However, they also noted that these did not distinguish akathisia from the group that did not meet criteria for akathisia (Gibb and Lees, 1986), and movements involving other parts of the body such as trunk rocking, respiratory grunting, face rubbing, and shifting weight while sitting were also frequent (Burke et al., 1989). Although there are not enough data and consensus on the diagnostic movements of akathisia, these movements seem to be characteristic enough to be recognized by different authors who have independently documented similar phenomena.
Akathisia is seen in patients with Parkinson disease (Lang and Johnson, 1987), in patients abusing cocaine (Daras et al., 1994), and as an adverse effect of selective serotonin uptake inhibitors (Poyurovsky et al., 1995). Acute akathisia occurs not only as an adverse effect of DRBAs but also fairly commonly as an acute adverse effect of the dopamine depletors – reserpine, TBZ, and α-methyl tyrosine (Marsden and Jenner, 1980).
In Ayd’s review (1961), half of the cases of acute akathisia occurred within 25 days of drug treatment and 90% occurred within 73 days. Acute akathisia was the most common side effect of DRBAs, occurring in 21.2% of patients in that study (Ayd, 1961). In a more recent study by Sachdev and Kruk (1994) of 100 consecutive patients placed on neuroleptics, mild akathisia developed in 41% and moderate to severe akathisia in 21%. In a literature review, Sachdev (1995b) reported that incidence rates for acute akathisia with conventional neuroleptics vary from 8% to 76%, with 20% to 30% being a conservative estimate. Sachdev stated that preliminary evidence suggests that the newer atypical antipsychotic drugs are less likely to produce acute akathisia. Using the criterion that both subjective and objective phenomena are required for the diagnosis of acute akathisia, Miller and colleagues (1997) found an incidence rate of 22.4%, 75% of which occurred within the first 3 days of exposure to a neuroleptic. Muscettola and colleagues (1999) found a prevalence rate of 9.4%.
The potency of neuroleptics has been associated with incidence of akathisia, ranging from 0.5% for reserpine (Marsden and Jenner, 1980) to 75% for haloperidol (Van Putten et al., 1984). Other risk factors are neuroleptic dose, the rate of dosage increase, and the development of drug-induced parkinsonism (Sachdev and Kruk, 1994). Akathisia has occurred with all second-generation antipsychotics as well as first-generation ones (Kane et al., 2009).
As with acute dystonic reactions, akathisia has also been induced by serotonergic agents (Chong, 1996; Lopez-Alemany et al., 1997). The mechanism was discussed in the above section on acute dystonia.
As was noted previously, akathisia needs to be distinguished from other conditions such as agitated depression; restless legs syndrome, in which similar subjective sensations may be described by patients but are mainly localized to legs and are present particularly at night (Blom and Ekbom, 1961); or complex motor tics with preceding aura, which show more variety of abnormal movements; and complex vocal tics (Jankovic and Fahn, 1986). Akathisia can also be obscured by other psychiatric disorders, or it could be mistaken for a psychiatric disease. For example, when patients with psychosis develop akathisia after withdrawal from antipsychotic drugs, it may be mistaken for recurrence of psychosis. Paradoxical dystonia (see Chapter 12) in which dystonic movements are relieved by movement can be mistaken for akathisia.
The pathophysiology of acute akathisia remains poorly understood. On the basis of the observation in rats that show increased locomotor activity after blockade of the mesocortical dopamine system (Carter and Pycock, 1978), reduction of this dopaminergic projection was suggested to be responsible for akathisia (Marsden and Jenner, 1980). However, tardive akathisia cannot be explained by this hypothesis because dopamine depletors can ameliorate those symptoms. The observation that acute akathisia can occur with a serotonin uptake inhibitor (Altshuler et al., 1994) indicates that inhibiting dopamine neurons in the substantia nigra by such drugs could link the dopamine system with akathisia. These types of drugs have been reported to increase parkinsonism in patients with PD (Meco et al., 1994). One attractive possibility is that akathisia might reflect an alteration of the dopaminergic mesolimbic system.
Because propranolol has been reported to be beneficial in treating acute akathisia, another suggestion is that acute akathisia results from alterations in the cingulate cortex, the piriform cortex, or area 1 of the parietal cortex based on effects of propranolol in these regions in haloperidol-treated rats (Ohashi et al., 1998).
Ayd (1961) noted that acute akathisia is self-limited, disappearing on discontinuation of neuroleptics, and is well controlled by anticholinergics despite continuation of neuroleptics. Others have noted that only patients with concomitant parkinsonism improve significantly with anticholinergics (Kruse, 1960; Braude et al., 1983). Amantadine may also help, but patients can develop a tolerance (Zubenko et al., 1984a). Beta-blockers at relatively low doses below 80 mg of propranolol per day have been noted to be effective in many studies including one with a double-blind design (Lipinski et al., 1984; Zubenko et al., 1984b; Adler et al., 1986; Dupuis et al., 1987). Nonlipophilic beta-blockers that have poor penetration to the central nervous system (CNS) are not as effective (Lipinski et al., 1984; Dupuis et al., 1987). Selective beta-blockers might not be as effective as nonselective ones (Zubenko et al., 1984b); however, when two equally lipophilic beta-blockers, propranolol and betaxolol, were compared, they were equally effective in treating acute akathisia (Dumon et al., 1992) although the former is a beta-2-blocker and the latter is a beta-1-blocker. In a rat model of acute akathisia (neuroleptic-induced defecation), a lipophilic beta-1-blocker was found to be effective in reducing the phenomenon (Sachdev and Saharov, 1997).
Clonidine also reduces central noradrenergic activity by stimulating central alpha-2 receptors and has been noted to be effective in a small number of studies. The sedating effect is pronounced, however (Adler et al., 1987). Nicotine patches have been reported to reduce akathisia (Anfang and Pope, 1997). Weiss and colleagues (1995) found cyproheptadine, an antiserotonergic agent, to be effective in ameliorating akathisia. In a small placebo-controlled trial, mianserin, a 5-HT2 antagonist, was found to reduce the severity of acute akathisia (Poyurovsky et al., 1999). Trazadone has also been reported to be beneficial (Stryjer et al., 2003). Poyurovsky and Weizman (2001) discuss the potential of serotonin agents in akathisia. A literature search revealed mirtazapine to be effective and superior to propranolol (43.3% vs. 30.0%) (Hieber et al., 2008).
Parkinsonism
Neuroleptic-induced parkinsonism (usually referred to as extrapyramidal syndrome or EPS by psychiatrists and as drug-induced parkinsonism by neurologists) is a dose-related side effect and is indistinguishable phenomenologically from idiopathic PD, including high frequency of tremor and asymmetric signs (Hardie and Lees 1988). SPECT imaging of the dopamine transporter, however, may be helpful in determining whether the neuroleptic-induced parkinsonism is entirely drug-induced or an exacerbation of subclinical PD (Lorberboym et al. 2006). It develops with use of both DRBAs and dopamine-depleting drugs such as reserpine and TBZ. Some authors have noted perioral tremor and termed this rabbit syndrome, which is a localized form of parkinsonian tremor (Decina et al., 1990). The incidence of parkinsonism varies. Korczyn and Goldberg (1976) found 61%, and Muscettola and colleagues (1999) found 19.4%. Women are affected almost twice as frequently as men, which is the reverse of the ratio in idiopathic PD. Neuroleptic-induced parkinsonism also occurs increasingly with advanced age (Ayd, 1961; Hardie and Lees 1988) in parallel with the incidence of idiopathic PD.
Blockade of dopamine receptors by antagonists or depletion of presynaptic monoamines by drugs such as reserpine mimics the deficient dopamine state in PD. All DRBAs can induce parkinsonism, except clozapine (there are only rare reports with clozapine) (Factor and Friedman, 1997). Risperidone can do so (Gwinn and Caviness, 1997; Simpson and Lindenmayer, 1997), as well as olanzapine and only rarely quetiapine. Parkinsonism from neuroleptics is typically reversible when the medication is reduced or discontinued. Sometimes, the reversal can take many months; an interval of up to 18 months has been noted in the literature (Fleming et al., 1970).
Some patients show persisting parkinsonism despite prolonged discontinuation of neuroleptics (Stephen and Williamson, 1984; Hardie and Lees, 1988), giving rise to consideration of a proposed condition of tardive parkinsonism. A study of 8-week exposure of rats to haloperidol found a highly significant 32–46% loss of tyrosine hydroxylase (TH) immunoreactive neurons in the substantia nigra, and 20% contraction of the TH-stained dendritic arbor (Mazurek et al., 1998). Perhaps such pathologic changes account for some cases of prolonged drug-induced parkinsonism in humans. Several cases in the literature had initial resolution of parkinsonism and later reappearance of the symptoms without re-exposure to neuroleptics (Hardie and Lees, 1988). Two cases that had complete resolution of drug-induced parkinsonism after withdrawal of neuroleptics showed evidence of mild PD at autopsy (Rajput et al., 1982). Although one assumes that these patients had subclinical PD, the effect of neuroleptics on the disease progression is unknown. The use of the term “tardive parkinsonism” to refer to cases of persistent parkinsonism remains an enigma. Some are due to concurrent development of progressive PD, and there are no autopsied proven examples of non-PD in any example. Therefore, this suggests that there is as yet no evidence of tardive parkinsonism.
With the introduction of selective serotonin reuptake inhibitors (SSRIs) to treat depression, it has been noticed that these drugs can sometimes worsen parkinsonism in patients with PD (Meco et al., 1994) and occasionally can induce parkinsonism in patients who never had symptoms of PD (Coulter and Pillans, 1995; DiRocco et al., 1998). In an intensive monitoring program in New Zealand of the SSRI drug fluoxetine over a 4-year period, there were 15 reports of parkinsonism in 5555 patients who were exposed to the drug (Coulter and Pillans, 1995). Four of these 15 patients were also on a neuroleptic and one was on metoclopramide. In a literature search, parkinsonism was found with different classes of antidepressants, is not dose related, and can develop with short-term or long-term use (Madhusoodanan et al., 2010). The explanation for inducing or enhancing parkinsonism is that increased serotonergic activity in the substantia nigra will inhibit dopamine-containing neurons, thus causing functional dopamine deficiency in the nigrostriatal pathway (Baldessarini and Marsh, 1992).
The possibility of the existence of tardive parkinsonism comes up from time to time because some patients have continued parkinsonism despite long-term discontinuation of the DRBA (Melamed et al., 1991). However, there is always the possibility that the patient had preclinical Parkinson disease prior to developing drug-induced parkinsonism. Then, when the DRBA is withdrawn, the parkinsonism persists because of progressively worsening PD. One would need to show that there are no Lewy bodies or that the PET scan shows no loss of fluorodopa uptake in patients believed to have tardive parkinsonism.
Anticholinergics can be effective in reducing the severity of the parkinsonism induced by DRBAs, whereas dopaminergic drugs (that activate the dopamine receptors) are ineffective, probably because they are not able to displace the DRBA from its binding to the receptor. Levodopa up to 1000 mg in combination with a peripheral dopa decarboxylase inhibitor had no significant effect (Hardie and Lees, 1988), nor did apomorphine, a dopamine receptor agonist (Merello et al., 1996). On the other hand, levodopa can effectively reverse parkinsonism induced by dopamine depletors, such as reserpine. In fact, the discovery of the dopamine hypothesis for parkinsonism was based on this observation (Carlsson et al., 1957; Carlsson 1959). Treatment is usually initiated with anticholinergics or amantadine (Mindham et al., 1972; Johnson, 1978; Konig et al., 1996).
Neuroleptic malignant syndrome
NMS is an idiosyncratic reaction that can sometimes be life-threatening. The clinical triad consists of (1) hyperthermia, usually with other autonomic dysfunctions such as tachycardia, diaphoresis, and labile blood pressure; (2) extrapyramidal signs, usually increased muscle tone of rigidity or dystonia, often with accompanying elevation of muscle enzymes; and (3) alteration of mental status, such as agitation, inattention, and confusion. Fever is not an essential symptom (Peiris et al., 2000), and it can be delayed (Norris et al., 2006). The syndrome begins abruptly while the patient is on therapeutic, not toxic, dosages of medication. In a review of 340 clinical reports of NMS in the literature (Velamoor et al., 1994), changes in either mental status or rigidity were the initial manifestations of NMS in 82.3% of cases with a single presenting sign. All the symptoms are fully manifest within 24 hours and reach a maximum within 72 hours. There does not seem to be any relationship with the duration of therapy. NMS can develop soon after the first dose or at any time after prolonged treatment. Recovery usually occurs within 1 to several weeks, but can be fatal in 20–30% of cases (Henderson and Wooten, 1981; Gute and Baxter, 1985). Even with awareness of the potential of fatality in modern medicine, death still occurs (van Maidegem et al., 2002). Muscle biopsies have shown swelling and edema, with 10–50% of fibers involved with vacuoles but scanty mononuclear infiltration (Behan et al., 2000).
All agents that block D2 receptors can induce NMS, including risperidone (Raitasuo et al., 1994; Webster and Wijeratne, 1994; Dave, 1995; Singer et al., 1995; Levin et al., 1996), clozapine (Miller et al., 1991; Amore et al., 1997; Dalkilic and Grosch, 1997), amisulpride (Bottlender et al., 2002), olanzapine (Kontaxakis et al., 2002; Kogoj and Velikonja, 2003), and phenothiazines with antihistaminic activity, such as alimemazine (van Maidegem et al., 2002). A case of NMS associated with bupropion has been reported (Kasantikul and Kanchanatawan, 2006). TBZ has been reported to cause NMS; this seems likely to be due to its D2-blocking activity (Reches et al., 1983) rather than to its dopamine-depleting action (by blocking the vesicular dopamine transporter). Reserpine has no known dopamine receptor antagonism, only dopamine-depleting activity (also by blocking the vesicular dopamine transporter), and has not been reported to cause NMS.
In a Japanese study, 10 of 564 (1.8%) patients who received antipsychotics developed NMS (Naganuma and Fujii, 1994), many more than the 12 of 9792 patients (0.1%) reported previously (Deng et al., 1990). Risk factors that were found were psychomotor excitement, refusal of food, weight loss, and oral administration of haloperidol at 15 mg/day or above (Naganuma and Fujii, 1994). Young males appear to be more predisposed to NMS (Gratz and Simpson, 1994), but the reason for this is uncertain. In a case-control study searching for risk factors, Sachdev et al. (1997) found that patients with NMS were more likely to be agitated or dehydrated, often needed restraint or seclusion, had received larger doses of neuroleptics, and more often had previous treatment with electroconvulsive therapy (ECT) before the development of the syndrome.
The pathophysiologic mechanism of NMS is not well understood. Autopsies failed to show any consistent findings (Itoh et al., 1977). A similar syndrome has been reported following abrupt withdrawal of levodopa (Friedman et al., 1985; Hirschorn and Greenberg, 1988; Keyser and Rodnitzky, 1991), suggesting a common mechanism of acute dopamine deficiency (Henderson and Wooten, 1981). IBZM SPECT in one patient showed the dopamine receptor to be blocked in the acute phase of NMS, but the patient had been receiving a D2 blocker, which would be expected to result in this finding (Jauss et al., 1996). There is a report of a patient who developed the NMS syndrome following abrupt withdrawal of the combination of a long-acting neuroleptic and an anticholinergic agent (Spivak et al., 1996). Because it responded to procyclidine administration, it implicates a muscarinic overactivity. There are also reports of an NMS-like syndrome following the sudden withdrawal of amantadine (has dopaminergic and antimuscarinic activity) (Ito et al., 2001), and following withdrawal of baclofen, with recovery after reintroduction of baclofen (Turner and Gainsborough, 2001).
The idiosyncratic nature and rarity of the syndrome remain unexplained. Ram and colleagues (1995) evaluated the structure of the D2 receptor gene in 12 patients who had a history of NMS. One patient was found to have a nucleotide substitution of an exon of the D2 gene. The A1 allele of the TaqI A polymorphism of the dopamine D2 receptor gene appears to occur more commonly in patients who developed NMS (Suzuki et al., 2001). Kishida et al. (2004) found that patients with NMS had a higher association with a polymorphism in the D2 receptor gene.
Treatment of NMS consists of discontinuing the antipsychotic drugs and providing supportive measures. Rapid relief of symptoms has been reported with the use of dantrolene, bromocriptine, or levodopa (Granato et al., 1982; Gute and Baxter, 1985). Nisijima and colleagues (1997) found levodopa to be more effective than dantrolene, but Tsujimoto and colleagues (1998) found intravenous dantrolene plus hemodialysis to be effective. Subcutaneous apomorphine has been found to be effective as a solo treatment (Wang and Hsieh, 2001). Gratz and Simpson (1994) recommended using anticholinergics in an attempt to reverse rigidity prior to utilizing bromocriptine. Carbamazepine was dramatically effective in two patients (with recurrence on withdrawal of the drug) (Thomas et al., 1998). Steroids added to standard therapy have been reported to speed recovery time (Sato et al., 2003). Re-exposure to dopamine receptor antagonists does not necessarily lead to recurrence of NMS (Singh and Albaranzanchi, 1995; Singh and Hambidge, 1998). Residual catatonia that can last weeks to months has been reported, with some patients responding to ECT (Caroff et al., 2000). Hyponatremia can sometimes occur; it has been attributed to inappropriate secretion of antidiuretic hormone and also to cerebral salt-wasting syndrome associated with the NMS (Lenhard et al., 2007). When present, salt replacement is necessary.
Tardive syndromes
The first use of dopamine receptor antagonists for psychiatric disorders was in the early 1950s, and credit for the first report of TD is given to Schonecker (1957), who reported four patients with TD induced by chlorpromazine. Sigwald and colleagues (1959) provided the first detailed descriptions of the syndrome and divided it into acute, subacute, and chronic subtypes. Uhrbrand and Faurbye (1960) published the first systematic review of the complication among 500 psychiatric patients and noted 29 patients with the disorder. Faurbye and colleagues (1964) later coined the term tardive dyskinesia and emphasized the increased incidence of the syndrome with chronic exposure. Despite numerous reports of the classic O-B-L repetitive stereotypic movements, establishment of this disorder as a distinct clinical entity took decades of epidemiologic studies (American Psychiatric Association, 1980; Jeste and Wyatt, 1982a; Kane and Smith, 1982). Confusion arose in part from the difficulty of characterizing and communicating the exact type of movements these patients develop and distinguishing these from the ones that occur spontaneously. It should be noted that these drug-induced movements can be variable in duration; they may be short-lasting and fade slowly after discontinuation of the medication, suppressed by the medication itself, or persistent.
Rigorous epidemiologic data are available only for classic tardive dyskinesia (Jeste and Wyatt, 1982a; Kane and Smith, 1982), but tardive dystonia and tardive akathisia warrant separate recognition beyond their differences in clinical phenomenology because prognosis, at-risk population, and treatment are different. Some authors have noted chronic vocal and motor tics resembling Tourette syndrome (Klawans et al., 1978; Bharucha and Sethi, 1995), and others noted myoclonic movements (Little and Jankovic, 1987; Tominaga et al., 1987) as a chronic persistent problem of neuroleptic therapy, but further studies are necessary to establish them as distinct entities. More recently, a combination of resting, postural, and action tremor has been reported in five patients that persisted despite withdrawal of the offending DRBAs and that improved with treatment with the antidopaminergic TBZ (Stacy and Jankovic, 1992). The tremor was accompanied by other tardive phenomenology, and the authors suggested that this is another tardive syndrome, calling it tardive tremor. Tardive tremor has been reported with metoclopramide (Tarsy and Indorf, 2002).
Withdrawal emergent syndrome
Withdrawal emergent syndrome was first described in children who had been on antipsychotic drugs for a long period of time and then were withdrawn abruptly from their medication (Polizos et al., 1973). The movements are choreic and resemble those of Sydenham disease (Videos 19.1 and 19.2). The abnormal movements are brief and flow from one muscle to another in a seemingly random way. They differ from the movements of classic tardive dyskinesia, which are brief, but stereotypical and repetitive. The movements in withdrawal emergent syndrome involve mainly the limbs, trunk, and neck, and rarely the oral region, which is the most prevalent site in classic tardive dyskinesia. The dyskinetic movements disappear spontaneously within several weeks after withdrawal of the DRBA. For immediate suppression of movements, dopamine receptor antagonists can be reinstituted and withdrawn gradually without recurrence of the withdrawal emergent syndrome (Fahn, 1984a). A withdrawal reaction from melatonin with O-B-L dyskinesia and akathisia was reported by Giladi and Shabtai (1999) and was described earlier in the chapter. 
Withdrawal emergent syndrome is analogous to the classic tardive dyskinesia seen in adults, except that the course is more benign and movements are more generalized, resembling the choreic movements of Sydenham disease. In fact, most cases of tardive dyskinesia that have been reported in children have a benign course and the phenomenology has been reported to be more generalized choreic movements rather than stereotypic repetitive movements of oral, buccal, and lingual distribution. Acute withdrawal of chronic antipsychotic drugs in adults can also lead to transient tardive dyskinesia, which disappears within 3 months. These types of movements have been labeled withdrawal dyskinesia (Gardos et al., 1978; Schooler and Kane, 1982).
On the other hand, acute withdrawal of DRBA can precipitate a persistent akathisia (Poyurovsky et al., 1996; Rosebush et al., 1997) or dyskinesia, i.e., tardive akathisia and other tardive syndromes. Acute withdrawal should be avoided because of the propensity to induce TD, and a slow taper and withdrawal should be substituted. Abrupt withdrawal of risperidone therapy in one elderly person resulted in a near-fatal development of respiratory dyskinesia (Komatsu et al., 2005).
Classic tardive dyskinesia
Dyskinesia is a general term referring to abnormal involuntary movements. The term tardive dyskinesia has been used to refer to abnormal movements that are seen as a complication of long-term dopamine receptor antagonist therapy, mainly the type that presents with rapid, repetitive, stereotypic movements involving the oral, buccal, and lingual areas. However, over the years, other types of movements have been noted as complications of dopamine receptor antagonist therapy. These movements have more specific terminologies, such as dystonia and akathisia. Therefore, some authors refer to the type of movements that were originally described as “classic tardive dyskinesia” for lack of a more specific and distinct name for the movements (Fahn, 1989). Some have called them tardive stereotypy (Stacy and Jankovic, 1991; Jankovic, 1994) because of their repetitive, rather than random, nature. However, the stereotypical movements in classic tardive dyskinesia are so characteristic and resemble those seen in almost all patients with this disorder, in contrast to other types of stereotypies that are seen in patients with learning disability, autism, and psychosis, that the term tardive stereotypy does not convey this uniqueness and therefore seems unsatisfactory. Stereotyped hand clasping appears to be a rare form for the presentation of classic tardive dyskinesia (Kaneko et al., 1993).
On the other hand, some have used the term tardive dyskinesia as equivalent to any oral dyskinesia. It therefore needs to be emphasized that the term “tardive” has become synonymous with chronic neuroleptic complications and should be reserved for these disorders. Sustained dystonic movements of the lower face must be distinguished from classic tardive dyskinesia. Frequently, patients on anticholinergics or other medications develop oral dyskinesia with dryness of mouth. Other movements such as myokymia, myoclonus, and tics must be distinguished. The differential diagnosis of oral dyskinesia is summarized in Table 19.10, whereas Table 19.11 compares clinical features of tardive dyskinesia, Huntington disease, and oromandibular dystonia, the three most common forms of oral dyskinesias.
| 1. Chorea, rhythmical, stereotypical (see also Kurlan and Shoulson, 1988) |
Adapted from Fahn S. The tardive dyskinesias. In Matthews WB, Glaser GH, eds: Recent Advances in Clinical Neurology, vol. 4. Edinburgh: Churchill Livingstone, 1984; pp. 229–260.
Table 19.11 Clinical features of classic tardive dyskinesia (TD), oromandibular dystonia (OMD) and Huntington disease (HD)
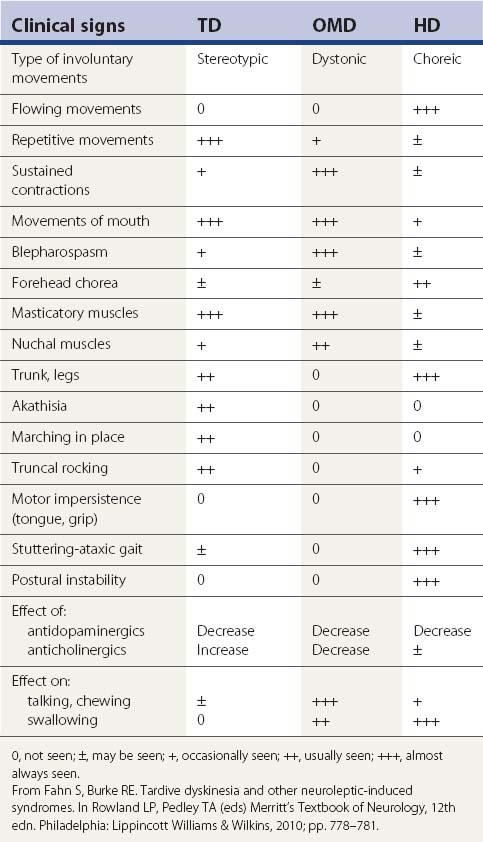
Clinical features of classic tardive dyskinesia
The clinical features of classic TD are quite distinct from the features of other movement disorders (Fahn, 1984a). The principal site is the face, particularly around the mouth, typically called oral-buccal-facial (O-B-L) dyskinesias. The limbs and trunk are affected less often than the mouth. Even when they are involved, it is usually in addition to involvement of the mouth. The forehead and eyebrows are seldom involved unless tardive dystonia is also present; this is in contrast to Huntington disease, in which chorea of the forehead and eyebrows is more common than choreic movements of the oral musculature. In TD, the mouth tends to show a pattern of repetitive, complex chewing motions (Video 19.3; see also Video 1.73), occasionally with smacking and opening of the mouth, tongue protrusion (flycatcher tongue) (Video 19.4), lip pursing, sucking movements, and fishlike lip puckering movements. The rhythmicity and coordinated pattern of movement are striking. This stereotypic pattern is in contrast to the dyskinesias that are seen in Huntington disease, in which the movements are without a predictable pattern. Usually, the limb involvement is limited to the distal part. Like the mouth region, the movements of the distal limbs show a repetitive pattern, earning the label of piano-playing fingers and toes. When the patient is sitting, the legs often move repeatedly, with flexion and extension movements of the toes and foot tapping. When the patient is lying down, flexion and extension of thighs may be seen. Rhythmic truncal rocking can be seen when the patient is lying, sitting, or standing (Video 19.5). The respiratory pattern can be involved with dyskinesia, causing hyperventilation at times and hypoventilation at other times (Yassa and Lai, 1986). In a study of the breathing pattern in patients with TD, patients had an irregular tidal breathing pattern, with a greater variability in both tidal volume and time of the total respiratory cycle (Wilcox et al., 1994). The presence of respiratory dyskinesia never causes a medical problem, although it might look alarming. Esophageal (associated with lingual) dyskinesias have also been reported, resulting in increased intraesophageal pressure and death due to asphyxiation in one patient (Horiguchi et al., 1999). 
Abnormal movements in schizophrenia in the absence of exposure to DRBAs
Tardive dyskinesia as an entity induced by dopamine receptor blockers has been challenged by reports claiming that spontaneous movements are sometimes encountered in patients with schizophrenia who have never been exposed to neuroleptic agents (see the review by Boeti et al., 2003). McCreadie and colleagues (2002) evaluated 37 schizophrenic patients never treated with antipsychotics and followed for 18 months. Nine (24%) had dyskinesia on both occasions, 12 (33%) on one occasion, and 16 (43%) on neither occasion. Twenty-one (57%) had dyskinesia on at least one occasion. Thirteen patients (35%) had parkinsonism on at least one occasion. It is critical that the quality of the dyskinesia be reported, because the classic O-B-L dyskinesias are very distinct and almost specific for tardive dyskinesia, whereas many other types of movements could represent a different disorder. The presence of parkinsonism, though, suggests that the patients had been exposed to neuroleptics, but that the investigators were clueless as to the exposure.
Many authors noted the existence of spontaneous oral dyskinesia occurring in untreated populations and in untreated schizophrenics. McCreadie and colleagues (1996) examined 308 elderly individuals in Madras, India, looking for abnormal movements and found them in 15%. The prevalence of spontaneous dyskinesia has been reported to be as high as 20% in psychiatric or nursing home patients (Brandon et al., 1971), but the average rate is about 5% (Jeste and Wyatt, 1982a; Kane and Smith, 1982). Others noted increasing prevalence of spontaneous dyskinesia with age (Klawans and Barr, 1982). Some studies looking at healthy elderly populations estimate the prevalence rate to be about 1% (Lieberman et al., 1984; D’Alessandro et al., 1986). Blanchet and colleagues (2004) evaluated 1018 (69.3% women) noninstitutionalized, frail elderly subjects attending day care centers to document the prevalence and phenomenology of spontaneous oral dyskinesia. The prevalence rate for spontaneous oral dyskinesia was 3.7% (4.1% for women and 2.9% for men). They reported more frequent ill-fitting dental devices, oral pain, and a lower rate of perception of good oral health compared to nondyskinetic subjects.
The true prevalence rate of spontaneous oral dyskinesia that can mimic TD may be even lower, considering the fact that many other identifiable oral dyskinesias listed in Table 19.10 such as oromandibular dystonia can be difficult to distinguish from classic tardive dyskinesia and may be counted as spontaneous dyskinesia in epidemiologic studies. On the other hand, spontaneous oral dyskinesia resembling the stereotypical O-B-L dyskinesia of classic tardive dyskinesia has been reported in an aged cynomologus monkey (Rupniak et al., 1990). Some patients with oral dyskinesias resembling those of TD and unexposed to DRBAs would seemingly be idiopathic in origin, but careful workup might reveal other etiologies (Table 19.10), including treatment with lithium (Meyer-Lindenberg and Krausnick, 1997) and brainstem infarcts (Fahn et al., 1986).
Reports of oral dyskinesias occurring in schizophrenic patients who have never been exposed to neuroleptics raise the question as to how specifically the O-B-L movements should be attributed to DRBAs and whether or not they could be due to the schizophrenia. Fenn and colleagues (1996) examined 22 never-medicated schizophrenics in Casablanca, Morocco. Three had abnormal movements that are said to be characteristic of TD. Fenton and colleagues (1997) compared the prevalence of spontaneous oral dyskinesias among drug-naive schizophrenics and patients with other psychiatric disorders. They found that dyskinetic movements were more common in the former group. Gervin and colleagues (1998) reported 6 (7.6%) out of 79 first-episode schizophrenics to have spontaneous dyskinesias. Until movement disorder experts can evaluate the movement phenomenology, it is possible that these individuals have some disorder other than TD. Moreover, it is possible that the history of nondrug exposure is faulty. The overwhelming evidence is that the abnormal movements that are seen with DRBAs are due to these drugs.
Epidemiology, risk factors, and natural history
Epidemiologic studies looking into the prevalence of TD have been confounded by the factors that affect the detection of the abnormal involuntary movements as well as the variables that affect the prognosis of the movements. Therefore, it is not surprising to find a wide range of prevalence estimations from 0.5% to 65% in the literature (Jeste and Wyatt, 1982a; Kane and Smith, 1982). The prevalence of TD has been noted to have increased from 5% before 1965 to 25% in the late 1970s (Jeste and Wyatt, 1982a; Kane and Smith, 1982). Mean prevalence rates calculated by two different reviewers, however, agree well at around 20% (Jeste and Wyatt, 1982a; Kane and Smith, 1982).
The prevalence rates of spontaneous dyskinesias and dyskinesias in people who have been exposed to neuroleptics have been compared. In the study by Woerner and colleagues (1991) the overall prevalence of spontaneous dyskinesias was 1.3% in 400 healthy elderly people and 4.8% in elderly inpatients, with a range of 0–2% among psychiatric patients who had never been exposed to neuroleptics. These investigators reported prevalence of TD of 13.3% and 36.1% in voluntary and state psychiatric hospitals, respectively. There was an interplay between age and gender. Among younger patients, men had higher rates; among subjects over age 40 years, rates were higher in women. Van Os and colleagues (1999) also found an increased risk for men in the younger population.
In one prospective study examining development of TD with low-dose haloperidol, the 12-month incidence of probable or persistent tardive dyskinesia was 12.3% (Oosthuizen et al., 2003). In a much larger study, the annual incidence rates range from 5% in a younger population (mean age 28) (Kane et al., 1986) to 12% in an older group (mean age 56) (Barnes et al., 1983). Kane and colleagues’ data (1986) also show that the cumulative incidence of TD increases linearly with increasing duration of neuroleptic exposure at least for the first 4–5 years of such exposure. In a subsequent study by these authors, Chakos and colleagues (1996) studied prospectively 118 patients in their first episode of psychosis who were treatment-naive and were then followed for up to 8.5 years while they were on neuroleptics. The cumulative incidence of presumptive TD was 6.3% after 1 year of follow-up, 11.5% after 2 years, 13.7% after 3 years, and 17.5% after 4 years. Persistent TD had a cumulative incidence of 4.8% after 1 year, 7.2% after 2 years, and 15.6% after 4 years. Thus, the earlier findings of Kane and colleagues (1986) of about 5% a year cumulative incidence seem to have been confirmed.
Jeste et al. (1999) looked at the cumulative incidence of tardive dyskinesia after exposure to neuroleptics in patients over the age of 45 years. The mean cumulative incidence was 3.4%, 5.9%, and 22.3% at 1, 3, and 12 months, respectively. Woerner and his colleagues (1998) studied patients over the age of 54 when first exposed to neuroleptics. The cumulative rates of tardive dyskinesia were 25%, 34%, and 53% after 1, 2, and 3 years of cumulative antipsychotic treatment. A greater risk of tardive dyskinesia was associated with a history of ECT treatment, higher mean daily and cumulative antipsychotic doses, and the presence of extrapyramidal signs early in treatment. From both these studies, the incidence rates for patients beginning treatment with conventional antipsychotics in their fifth decade or later are three to five times what has been found for younger patients, despite treatment with lower doses.
Studies have been carried out in other countries. A prospective study of 11 psychiatric facilities in Japan found the prevalence of TD to be 7.6%, the annual incidence rate to be 3.7%, and an annual remission rate to be 28.7% (Inada et al., 1991). On the other hand, Hayashi and colleagues (1996) reported a prevalence of 22.1% in 258 patients receiving neuroleptics. A study in Austria reported the follow-up of the 270 patients still in a psychiatric hospital after 10 years out of an original population of 861 patients; the prevalence rate of TD was 3.7% in 1982 and 12.7% in 1992; the major risk factor for TD was advanced age (Miller et al., 1995). Jeste and colleagues (1995) followed 266 patients over the age of 45 years to determine incidence and prevalence rates of tardive dyskinesia following exposure to neuroleptics, using electromechanical sensors to detect the presence of movements. Cumulative incidence of TD was 26%, 52%, and 60% after 1, 2, and 3 years, respectively. These rates, which are higher than those found by Kane and his colleagues, might reflect the sensitive sensors used, and these could possibly give false-positive findings. The same group compared the development of abnormal movements in the orofacial and limb-truncal areas in these 266 middle-aged and elderly patients treated with neuroleptics and found that the cumulative incidence of orofacial TD was 38.5% and 65.7% after 1 and 2 years, respectively, whereas that of limb-truncal TD was 18.6% and 32.6% after 1 and 2 years, respectively (Paulsen et al., 1996).
Host and treatment factors affect the development, severity, and persistence of TD, thereby resulting in different prevalence rates. Age of the patient has been the most consistent factor that adversely affects the incidence, prognosis, and severity of tardive dyskinesia (Smith and Baldessarini, 1980; Jeste and Wyatt, 1982a; Kane and Smith, 1982; Kane et al., 1986). Possibly the youngest individual who developed O-B-L tardive dyskinesia was a 2-month-old girl after a 17-day treatment with metoclopramide for gastroesophageal reflux (Mejia and Jankovic, 2005). The movements persisted for at least 9 months after the drug was discontinued. This patient is the first documented case of tardive dyskinesia in an infant.
Female sex has been associated with increased prevalence of TD, especially in the older population (Jeste and Wyatt, 1982a; Kane and Smith, 1982; Kane et al., 1988). Several authors noted increased incidence and prevalence among patients with affective disorders compared to schizophrenia or schizoaffective disorders (Gardos and Casey, 1983). Other host factors such as presence of previous brain damage as noted by increased ventricular size remain controversial (Jeste and Wyatt, 1982a; Kane and Smith, 1982). Some noted poor treatment response of schizophrenia to drug treatment as a risk factor for development of tardive dyskinesia (Chouinard et al., 1986). The parameters of drug exposures such as dose, duration, type of neuroleptics, and drug-free intervals have been very difficult to correlate with risk of TD partly because accurate history concerning the drug treatment is usually not available and the drug itself can affect the detection of TD by masking or uncovering it.
Ethnicity has been found to be an important risk factor in both the development and prognosis of tardive dyskinesia, African-Americans being more susceptible than European-Americans (Wonodi et al., 2004a). In a meta-analysis of the literature, the only statistically significant risk factors for TD were non-white ethnicity and having developed drug-induced parkinsonism; the association with older age was suggestive but inconclusive (Tenback et al., 2009).
Prospective studies have noted that the total cumulative drug exposure correlates with incidence of withdrawal tardive dyskinesia (Kane et al., 1985), and development of TD 5 years later in patients who did not have TD initially (Chouinard et al., 1986). Continued use of DRBAs after the appearance of TD also adversely affects subsequent prognosis (Kane et al., 1986). Drug holidays were once advocated to decrease the risk of TD, but an increased number of drug-free intervals was found to worsen the prognosis after withdrawal (Jeste et al., 1979). The risk of developing TD is three times as great for patients with more than two neuroleptic interruptions as for patients with two or fewer interruptions (van Harten et al., 1998). Other than reduced risk with clozapine, olanzapine, and quetiapine, no other particular type of neuroleptic, including depot preparations, has been clearly identified as a risk factor (Yassa et al., 1988). The effect of other drugs such as anticholinergics on the incidence and prevalence of TD is controversial, although anticholinergics may increase its severity (Yassa, 1988). Lithium may decrease the chance of TD development (Kane et al., 1986; van Harten et al., 2008a). Development of parkinsonism tends to predispose patients to TD (Chouinard et al., 1986; Kane et al., 1986). Substance abuse has also been reported to be a risk factor (Bailey et al., 1997). Lacking the cytochrome P450 enzyme required for metabolism exogenous toxins because of nonfunctional alleles of the CYP2D6 gene appears to be a risk factor for developing both TD and drug-induced parkinsonism (Andreassen et al., 1997). Another genetic risk factor is polymorphism in the ATP13A gene, especially for drug-induced parkinsonism (Kasten et al., 2011).
A review of the literature by Correll and colleagues (2004) found a lower incidence with the newer generation of antipsychotics (Table 19.12). A European consortium of investigators looking at 6-month results came to a similar conclusion (Tenback et al., 2005). In their review of the literature, Tarsy and Baldessarini (2006) also suggest that there may be a declining incidence of TD as the second-generation antipsychotics are being more regularly used. On the other hand, in a survey on elderly patients with dementia treated with older or newer generation antipsychotics, no statistical difference was found in the development of nonparkinsonian movement disorders in relation to treatment class (Lee et al., 2005). Children appear to be increasingly treated with “atypical” antipsychotics and in one study, 9% developed TD (Wonodi et al., 2007).
Table 19.12 Annual incidence of tardive dyskinesia comparing the second generation antipsychotics with haloperidol
| Population | Second-generation antipsychotics | Haloperidol |
|---|---|---|
| Children | 0% | |
| Adults | 0.8% | 5.4% |
| Mixture of adults and elderly | 6.8% | |
| Older than 53 years | 5.3% |
Data from Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry 2004;161(3):414–425.
Although the majority of TD occurs while patients are on chronic treatment with DRBAs or shortly after discontinuing them, many cases have been reported in which the TD occurred after only a short interval of treatment and persisted. Some authors define TD arbitrarily as dyskinesias that occur after a minimum of 3 months of dopamine receptor antagonist therapy (Schooler and Kane, 1982), but it appears that there is no safe low-incidence period right after the initiation of treatment with DRBAs nor is there any particularly high-risk period. The overall risk appears to accumulate as time goes on, although whether it continues to increase linearly even after the first several years of exposure is not clear from available data. Kang and colleagues’ (1986) retrospective data also show that the cumulative number of patients who developed tardive dystonia increased almost linearly from the first few months.
Probably most patients with TD have mild cases that could improve and disappear over time if the offending DRBAs were withdrawn. In an attempt to look at risk factors for the severe forms of TD, Caligiuri and colleagues (1997) conducted a longitudinal prospective study to determine the incidence of severe TD in middle-aged and elderly psychiatric patients. The cumulative incidence of severe TD was 2.5% after 1 year, 12.1% after 2 years, and 22.9% after 3 years. Risk factors for severe TD were higher daily doses of neuroleptics at study entry, greater cumulative amounts of prescribed neuroleptic, and greater severity of worsening negative psychiatric symptoms.
Once established, TD does not frequently become more severe even though the DRBA is continued (Labbate et al., 1997). Fernandez and colleagues (2001) evaluated the dyskinesia of TD and the clinical features of parkinsonism in 53 patients residing in a state psychiatric hospital over a 14-year period. TD improved and parkinsonism worsened in patients who continued to receive neuroleptic drugs. But the natural history of TD is not easy to determine because the DRBAs that cause the movement disorders also tend to suppress the movements, while they can cause drug-induced parkinsonism.
Movements that disappear with increasing dose or resumption of DRBAs have been called masked TD (Schooler and Kane, 1982). Conversely, movements of TD typically increase after discontinuation of the DRBAs. Transient dyskinesias that appear after withdrawal of DRBAs have been called withdrawal dyskinesias (Gardos et al., 1978). The term covert dyskinesia has also been used for dyskinesia that is not detectable during drug administration and is first noted during drug reduction or withdrawal and becomes permanent (Gardos et al., 1978). Therefore, the difference between withdrawal and covert dyskinesia is in duration. In fact, the spectrum of TD ranges from mild transient withdrawal dyskinesia to severe irreversible TD, which can occur during or after discontinuation of the antipsychotic drugs. These examples illustrate the complexity in the detection of TD in the presence of DRBAs. In an attempt to estimate the false-negative rate of detection for TD in patients taking DRBAs, Kane and colleagues (1988) withdrew the drug from patients who showed no evidence of TD while taking DRBAs. Withdrawal TD was seen in 34% of 70 subjects, with persistence beyond 3 months in 7 of them.
Pathophysiology
Anatomical pathology – human and animal models
Postmortem pathologic studies are few and show nonspecific findings in the striatonigral system, which might be due to aging or other uncontrolled factors (Christensen et al., 1970). Jellinger (1977) found swollen large (i.e., cholinergic) neurons in the rostral caudate nucleus in 46% of neuroleptic-treated patients. In a review of the literature Harrison (1999) notes other studies in which cholinergic neurons are affected by these drugs. In a magnetic resonance imaging (MRI) study, 2-year exposure to neuroleptics in schizophrenic patients resulted in an increase in caudate and lenticular nucleus volume, while in those who were similarly exposed to atypical antipsychotics, there was a decrease in volume (Corson et al., 1999).
Since TD is etiologically related to DRBAs and since the clinical pharmacology is most consistent with dopaminergic hyperactivity, many studies have been directed to the dopamine system (Klawans, 1973). Chronic dopamine receptor antagonist treatment in rats shows evidence of cell loss in ventrolateral striatum (Nielson and Lyon, 1978), changes in synaptic patterns in the striatum at the electron microscopic level (Benes et al., 1985), and increases in glutamatergic synapses in the striatum (Meshul et al., 1994). In the accumbens in rats who developed vacuous chewing movements (VCMs), the dendritic surface area is reduced, and dynorphin-positive terminals contact more spines and form more asymmetric specializations than those in animals without the syndrome (Meredith et al., 2000). Andreassen and colleagues (2001) found electron microscopic changes in rats who developed haloperidol-induced VCMs. The nerve terminal area in the striatum was increased but with a lower density of glutamate immunoreactivity. These results suggest that striatal glutamatergic transmission is affected in association with haloperidol-induced VCMs.
Anatomical pathology – in-vitro models
Galili-Mosberg and colleagues (2000) exposed neuronal and PC-12 cultures to haloperidol and its three metabolites. These induced cell death by apoptosis, which was protected by the antioxidants vitamin E and N-acetylcysteine. Marchese and colleagues (2002) found a shrinkage of TH-immunostaining (TH-IM) cell bodies in the substantia nigra pars compacta and reticulata and a reduction of TH-immunostaining in the striatum of haloperidol-treated rats with the arising of VCMs. No differences were observed in TH-IM neurons of the ventral tegmental area and nucleus accumbens versus control rats.
Rodent models – dopamine and other neurotransmitter biochemistry
In experimental animals, a short-term treatment of dopamine receptor antagonist can increase striatal dopamine synthesis and turnover that revert to normal levels within a few days despite continuing treatment (Asper et al., 1973). Receptor binding increases in 2 days and persists for up to a week (Asper et al., 1973; Klawans, 1973; Muller and Seeman, 1977). The change in receptor state correlates well with stereotypic gnawing movements by rats that are induced by a dopamine agonist such as apomorphine (Clow et al., 1980). Chronic treatment for a year in rats produces an increase in receptor binding sites by the sixth month of treatment, overcoming the initial effects of receptor blockade. The increase of receptor binding and functional hypersensitivity takes about 6 months to return to baseline after withdrawal of drug treatment. Increased response of striatal adenylate cyclase to dopamine still persists at 6 months (Clow et al., 1980). The significance of this finding is not clear. Extrapolating from this rodent model of dopaminergic supersensitivity to patients with TD has limitations. Rats do not usually develop spontaneous movements such as tardive dyskinesia, but they do demonstrate increased sensitivity to exogenously administered dopamine agonists and can develop VCMs.
The receptor supersensitivity in rodents develops in all the animals treated relatively early in the course of dopamine antagonist treatment and is completely reversible in 100% of animals shortly after withdrawal. This is clearly at odds with human TD, in which large individual variations and susceptibility factors are important and the disorder often starts late in the course of treatment and becomes persistent. Nonetheless, this model has been most useful for quick screening of potential neuroleptics that are likely to cause TD from those that are not. Calabresi and colleagues (1992) suggested that hypersensitivity of the D2 presynaptic receptors located on terminals of glutamatergic corticostriatal fibers in animals with chronic treatment with haloperidol would inhibit glutamate release in the striatum, thereby reducing the gamma-aminobutyric acid (GABA) output from postsynaptic striatal neurons. Dopamine D2 receptor occupancy appears to be a contributing factor in the development of the rodent model (Turrone et al., 2002). Andreassen and Jorgensen (2000) review the possible role of increased striatal glutamatergic activity in the rat model as an etiologic factor and suggest that excitoxicity is a possible cause of TD. Anatomic changes found by these authors were presented in the previous section.
Rodent models – behavioral changes
A common rodent model is that based on the increased rate of VCMs in rats, which develop after several months of dopamine receptor antagonist treatment (Gunne et al., 1982). The movements persist for several months after neuroleptic withdrawal. The behavioral syndrome is spontaneous in contrast to the stereotypic gnawing movement that requires apomorphine to induce. The pharmacologic response is similar to that seen with TD in that acute haloperidol alleviates the behavioral syndrome. In these aspects, this model is closer to the characteristics of human TD than models that are based on dopamine receptor supersensitivity.
The VCMs that persist after withdrawal of dopamine receptor antagonist do so at the time when the receptor-binding studies show no supersensitivity (Waddington et al., 1983). Blocking N-methyl-D-aspartate (NMDA) receptors with memantine while the rats are receiving treatment with haloperidol allowed these rats to lose the VCMs sooner than those animals who did not receive memantine (Andreassen et al., 1996). This result supports the concept that excessive NMDA receptor stimulation might be a mechanism underlying the development of persistent dyskinesias in humans. Opioid receptor antagonists were also shown to block VCMs in rats, suggesting that increases in dynorphin in the direct striatonigral pathway and enkephalin in the indirect striatopallidal pathway following chronic neuroleptic administration are both likely to contribute to tardive dyskinesia (McCormick and Stoessl, 2002).
In one study of chronic haloperidol treatment in the rat, the animals that developed VCMs were compared with those that did not develop the movements (Shirakawa and Tamminga, 1994). Whereas all animals showed an increase in the dopamine D2 family receptor binding in the striatum and in the nucleus accumbens, and an increase in GABA-A receptors in the substantia nigra pars reticulata (SNr), only those with VCMs had a significant decrease in dopamine D1 receptor density in the SNr. One explanation is that increased dopamine release from dendrites in the SNr downregulates the D1 receptor. But how this would produce dyskinesias is not clear. In dorsal striatal neurons, however, there was no change in D1 receptor mRNA nor was there a change in cell counts in rats with neuroleptic-induced VCMs (Petersen et al., 2000).
Stereotyped behavior was evaluated in rats treated chronically with haloperidol, followed by D1 and D2 agonists. Somatostatin levels were then measured. The results suggested that somatostatin – but not enkephalin-containing striatal neurons – contribute to the expression of haloperidol-induced stereotypies (Marin et al., 1996).
Other neurotransmitters in the striatum may also be involved. Chronic haloperidol treatment in rats increases preproenkephalin messenger RNA in striatal neurons, but fail to do so in rats that develop VCMs (Andreassen et al., 1999a). Some authors noted that decreases of glutamic acid decarboxylase (GAD) and GABA in substantia nigra correlate with increased VCM rates (Gunne and Haggstrom, 1983). Others noted no consistent change in GAD and choline acetyltransferase levels (Mithani et al., 1987). However, Delfs and colleagues (1995) reported that rats treated with haloperidol for up to 12 months showed a decrease of GAD mRNA in the external globus pallidus. This result suggests that there is reduced GABAergic transmission in the projection neurons of the external pallidum. Blocking NMDA receptors with memantine while the rats are receiving treatment with haloperidol allowed these rats to lose the VCMs sooner than animals that did not receive memantine (Andreassen et al., 1996). This result supports the concept that excessive NMDA receptor stimulation could be a mechanism underlying the development of persistent dyskinesias in humans. Lesioning the subthalamic nuclei (and thus its glutamatergic efferents) eliminates VCMs after 1–3 weeks (Stoessl and Rajakumar, 1996). The 5-HT3 receptor antagonist ondansetron reverses haloperidol-induced VCMs in rats (Naidu and Kulkarni, 2001).
As a test for the oxidative stress hypothesis in causing TD, rats were cotreated with coenzyme Q10 (CoQ10) along with haloperidol. Cotreatment with CoQ10 did not attenuate the development of the VCMs (Andreassen et al., 1999b). CoQ10 was absorbed, as detected in the serum, but there was no increase of CoQ10 in the brain.
Primate models with behavioral changes
The model that best resembles human tardive dyskinesia is that induced in nonhuman primates by DRBAs (Klintenberg et al., 2002). Primates show delayed onset of dyskinesia during the course of treatment and only a fraction of the population that is treated is affected by dyskinesia. The behavioral effect is spontaneous and bears resemblance to human TD. The dyskinesia also persists after withdrawal of the DRBAs. Unfortunately, neuropathologic and biochemical data from this model are limited. One group suggested that the best neurochemical correlate of dyskinesia induced by DRBAs has been a decrease in GABA and GAD activity in the substantia nigra, subthalamic nucleus, and medial globus pallidus compared to control animals without drug treatment or animals with drug treatment but without dyskinesia (Gunne et al., 1984). This finding correlates with postmortem biochemical studies on humans with TD (see the next section) and with a recent 2-deoxyglucose study in primates with TD. In the latter study, Mitchell and colleagues (1992) reported that primates with TD had a reduced glucose metabolism in the medial globus pallidus and in the ventral anterior and ventral lateral nuclei of the thalamus. These findings correlate with other similar studies of chorea and ballism and suggest a reduced subthalamopallidal output, so that the medial pallidum is not activated. Such a finding would be seen in the hemiballistic animal.
In a 5-year study of fluphenazine treatment of Cebus apella monkeys, re-exposure following 91 weeks of withdrawal increased dyskinesias and dystonias by 300% (Linn et al., 2001). This observation correlates with earlier clinical studies that found that intermittent treatment with neuroleptics can dramatically increase the incidence of dystonias and dyskinesias.
In a study looking for a proposed mitochondrial dysfunction in TD, baboons were treated for 41 weeks with haloperidol, producing TD; the animals were followed for another 17 weeks following withdrawal of haloperidol, during which the TD persisted (Eyles et al., 2000). Striatal mitochondria were examined by electron microscopy and showed no difference in either size or number between treated and control animals.
Human biochemistry
Postmortem analysis
In a study that was well controlled for the premortem state of patients, a significant decrease in GAD activity was found in the subthalamic nucleus of five tardive dyskinesia patients compared to age-matched controls (Andersson et al., 1989). Dopamine receptor binding is not increased in postmortem brains of tardive dyskinesia patients (Crow et al., 1982; Cross et al., 1985; Kornhuber et al., 1989). One study noted increased homovanillic acid levels in the putamen and nucleus accumbens of the postmortem brain (Cross et al., 1985).
Cerebrospinal fluid
CSF GABA concentration was found to be significantly lower in five patients with chronic schizophrenia and TD than in five patients with chronic schizophrenia without TD who were matched for age, duration of schizophrenia, and duration of neuroleptic therapy (Thaker et al., 1987). CSF cyclic-AMP is thought to reflect the dopamine receptor function, and is not significantly elevated in patients with TD. CSF dopamine metabolite studies do not show significant elevation of homovanillic acid (HVA) (Bowers et al., 1979; Nagao et al., 1979). CSF studies also showed a higher concentration of excitatory amino acid neurotransmitters and markers of oxidative stress in patients with TD, supporting the hypothesis of oxidative stress due to enhanced striatal glutamatergic neurotransmission by blocking presynaptic dopamine receptors (Tsai et al., 1998).
PET and SPECT
PET data on the postsynaptic receptor status have failed to show an elevation of striatal receptor density compared to normal controls, but there was a positive correlation with severity of TD (Blin et al., 1989). No increase in dopamine transporter binding in striatum was found in a SPECT study, indicating no loss on dopaminergic nerve terminals (Lavalaye et al., 2001). A prospective study was carried out by using FDG PET in patients receiving antipsychotic drugs. Those who later developed TD had a relative hypermetabolism in temporolimbic, brainstem, and cerebellar regions and hypometabolism in parietal and cingulate gyrus (Szymanski et al., 1996).
Dopamine release studies using amphetamine following raclopride binding of the D2 receptor showed no differences between subjects with and without TD, indicating that dopamine release does not seem to be a factor (Adler et al., 2002).
Genetics
Steen and colleagues (1997) reported that a specific allelic variation of the dopamine D3 receptor (DRD3) gene is found at a higher frequency (22–24%) of homozygosity among patients with TD compared with the relative under-representation (4–6%) of this genotype in patients with no TD. Subsequent studies (Segman et al., 1999; Liao et al., 2001) supported and extended an association between D3 receptor gene and TD in schizophrenia patients. A serine to glycine polymorphism in the first exon of the DRD3 gene appears to be a risk factor for developing TD (Liao et al., 2001, Ozdemir et al., 2001). Lerer and colleagues (2002) also found the presence of the glycine allele carries a higher risk for developing TD. On the other hand, Chong et al. (2003) found the serine allele to have a higher risk. A meta-analysis of genetic studies supports the view that a serine to glycine polymorphism in the D3 receptor gene has a higher risk (odds ratio = 1.17) for developing TD (Bakker et al., 2006). Other polymorphisms in DRD3 have also been associated with TD (Zai et al., 2009).
Beta-arrestin 2 (ARRB2) is an important mediator between DRD2 and serine-threonine protein kinase (AKT) signal cascade. A case-control study to evaluate the association between a polymorphism (Ser280Ser) and antipsychotic-induced TD was performed amongst 381 patients (TD/non-TD = 228/153) in a Chinese population. This polymorphism was significantly more common in those with TD (P = 0.025) (Liou et al., 2008).
A meta-analysis of genetic studies reported that there appears to be a protective effect against developing TD by being (1) a Val-Met heterozygote or a Met carrier for COMT and (2) Ala-Val and Val carriers for MnSOD, while there is a greater risk for TD by having an A2 variant or being an A2-A2 homozygote for DRD2 (Bakker et al., 2008).
Polymorphisms of the CYP2D6 gene, which encodes the cytochrome P450 enzyme debrisoquine/spartein hydroxylase, has been reported to possibly be associated with an increased susceptibility to developing TD (Kapitany et al., 1998; Ohmori et al., 1998; Vandel et al., 1999).
Searches for polymorphisms in the HTR2A receptor gene did not find any significant difference among patients with or without TD (Basile et al., 2001, Segman et al., 2001), but this has been inconsistent (Tan et al., 2001).
Neuroendocrine
Assessment of tuberoinfundibular dopaminergic sensitivity as a general indicator of dopamine supersensitivity shows lack of hyperresponsiveness of growth hormone and prolactin to dopamine agonist (Tamminga et al., 1977). Although these data on human TD are preliminary and indirect, the evidence for dopaminergic hypersensitivity has not been confirmed in human studies (Jeste and Wyatt, 1981; Fibiger and Lloyd, 1984).
Phenylalanine loading
In a double-blind study, 100 mg/kg of phenylalanine or placebo was given orally to 10 patients with TD (Mosnik et al., 1997); the dyskinesia was exacerbated after ingestion of phenylalanine, confirming earlier reports. Impaired phenylalanine metabolism was found in men but not in women (Richardson et al., 1999)
Human imaging
Results of imaging studies are inconsistent. In one MRI study Buchanan and colleagues (1994) found no difference between schizophrenic patients who had tardive dyskinesia and those who did not when measuring volumes of the caudate, putamen and pallidum. In contrast, Brown and colleagues (1996) reported larger caudate nuclei, particularly the right, in schizophrenic patients with TD compared to those without TD. To confuse this subject more, a computed tomographic (CT) study using special statistical methods found that the left caudate nucleus was smaller and the temporal sulci were enlarged (Dalgalarrondo and Gattaz, 1994). Brain iron was found to be normal in the basal ganglia in an MRI study (Elkashef et al., 1994).
Hoffman and Casey (1991) reviewed the literature on reports on CT evaluation of patients with tardive dyskinesia. They found that there is a trend toward larger cerebral ventricles in TD patients but that the overall difference with controls is small. Mion and colleagues (1991) compared MRI scans in young TD patients versus controls and found that the volumes of the caudate nuclei were significantly smaller in the TD patients than in normal controls and in patients on neuroleptics without TD. But covariate analysis failed to show a significant difference.
Oxidative stress and excitotoxicity
Maurer and Moller (1997) studied the effect on mitochondria of neuroleptics added to normal human brain cortex in vitro. Complex I activity was inhibited by all neuroleptics in the following order: haloperidol > chlorpromazine > risperidone >> clozapine. Haloperidol increases reactive oxygen species (generated from mitochondria) in rat cortical cell lines (Sagara, 1998). Erythrocyte Cu,Zn-superoxide dismutase activity was found to be reduced in patients on neuroleptics with TD compared to those without TD (Yamada et al., 1997). There were no differences in plasma vitamin E levels between TD and non-TD schizophrenic patients treated with neuroleptics (Brown et al., 1998). The CSF results mentioned earlier support the notion that by blocking presynaptic dopamine receptors, antipsychotics increase striatal glutamatergic transmission and subsequently excitotoxic stress (Tsai et al., 1998). Finding striatal changes in glutamate terminals in a rodent model lends support to the concept of excitotoxicity as a possible mechanism for developing TD (Andreassen et al., 2001) (see section entitled “Anatomical pathology – human and animal models”).
Developing TD in the presence of dopamine deficiency states
If dopamine receptor supersensitivity plus active dopamine release from nerve terminals with activation of these supersensitive receptors plays any role in the pathogenesis of TD, then one might anticipate that a state of deficiency of dopamine storage would be incompatible with the development of TD. There appears to be some merit to this concept. Fahn and Mayeux (1980) presented the first report of a patient with PD who developed TD. Their patient had unilateral PD, was treated with a dopamine receptor blocker, and subsequently developed classic TD on the contralateral side of the body. There was no TD on the side with the parkinsonism. In essence, this case supports the notion that a marked deficiency of dopamine release renders the development of TD unlikely, as is seen in the parkinsonian half of the body. The other side probably had partial reduction of dopamine stores and reduced release of dopamine, but it was still capable of developing TD. So perhaps this suggests a quantitative requirement (i.e., a threshold of dopamine availability before an individual could develop TD). This concept is compatible with observations that patients with TD who are treated with dopamine-depleting agents, such as reserpine, can substitute the excessive movements with a parkinsonian state.
The second case was a patient with dopa-responsive dystonia (DRD) who developed TD on long-term haloperidol therapy (de la Fuente-Fernandez, 1998). In DRD, there is a reduction of dopamine stores, but not a complete depletion. So again, if the quantity of dopamine available is the critical factor, a state of DRD seems to be quantitatively sufficient with dopamine.
Pathophysiology – summary
The biochemical basis of TD is still unclear, and the explanation involving dopamine receptor supersensitivity alone does not seem to be sufficient. There is little dispute that the primary and early effect of dopamine receptor antagonist is on the dopaminergic system with presynaptic and postsynaptic hypersensitivity. However, the changes do not seem to distinguish patients who develop TD from those who do not. Gerlach (1991) suggested that TD might be due to an increased ratio of D1/D2 receptor activity. The typical neuroleptics block pre- and postsynaptic D2 receptors, leaving D1 receptors spared. Thus, it is proposed that an increased D1 receptor activation would lead to the dyskinesias.
Oxyradicals have been implicated in the pathogenetic mechanism for the tardive syndromes (Lohr, 1991). This is based on the concept that DRBAs cause an increase in dopamine turnover, resulting in an increased synthesis of hydrogen peroxide, a metabolite of oxidative deamination of dopamine. Hydrogen peroxide, if not rapidly metabolized, will form oxyradicals, which can damage cell membranes and other cellular components. This hypothesis has received support from a study in which simultaneous administration of tocopherol to chronic haloperidol treatment in rats prevented the development of behavioral supersensitivity to apomorphine (Gattaz et al., 1993).
Tardive dystonia
Dystonia is most often an idiopathic disorder and can occur at all ages. Persistent dystonic movement as a complication of DRBA therapy has long been noted (Druckman et al., 1962). However, it had not been studied systematically to show that this represents a distinct syndrome until reported by Burke and colleagues (1982). Tardive dystonia has a different epidemiology and pharmacologic response from those of classic tardive dyskinesia. Although secondary dystonia can be caused by many neurologic disorders (Calne and Lang, 1988), tardive dystonia is one of the most common causes (Kang et al., 1986). A rigorous epidemiologic study is not available for tardive dystonia, but the prevalence of tardive dystonia in chronic psychiatric inpatients has been estimated to be 1.5–2% (Friedman et al., 1986; Yassa et al., 1986). When mild forms of dystonia were evaluated, 27 out of 125 patients on chronic antipsychotic medications had some form of dystonia (Sethi et al., 1990), indicating that the prevalence could be higher than was initially realized. Idiopathic torsion dystonia is a much less common condition, with one of the most generous estimations of prevalence at around 1 per 3000 population (Nutt et al., 1988). Therefore, dystonic movements seem to be much more frequent in patients who have been exposed to DRBAs.
In primary dystonia, patients at younger age of onset tend to develop generalized dystonia and those with onset in adulthood are more likely to have craniocervical focal or segmental dystonia. Kang and colleagues (1986) and Kiriakakis and colleagues (1998) have reviewed large series of cases of tardive dystonia and found a similar correlation between distribution of dystonia and age at onset. They found a correlation between the site and age of onset; the site of onset ascended from the lower limbs to the face as the mean age of onset increased. But tardive dystonia rarely affects legs alone even in youngsters (Kang et al., 1986). Regardless of age at onset, tardive dystonia usually progresses over months or years from a focal onset to become more widespread; only 17% remain focal at the time of maximum severity (Kiriakakis et al., 1998). As in primary dystonia, tardive dystonia in adults tends to remain focal or segmental and tends to involve the craniocervical region.
The onset of tardive dystonia can be from days to years after exposure to a DRBA (Kang et al., 1986). Kiriakakis and colleagues (1998) found the range to extend from 4 days to 23 years of exposure (median 5, mean 6.2 ± 5.1 years) in their series of 107 patients, with a mean (±SD) age at onset of 38.3 ± 13.7 years (range 13–68 years). There is no period that is safe from development of tardive dystonia; one patient in the series by Burke et al. (1982) developed it after a one-day exposure. Men are significantly younger than women at onset of dystonia, and it develops after shorter exposure in men (Kiriakakis et al., 1998). Yassa and colleagues (1989) found that severe tardive dystonia was more common in young men while severe classic tardive dyskinesia was more common in older women.
The phenomenology of tardive dystonia can be indistinguishable from that of idiopathic dystonia, including the improvement with sensory tricks (geste antagoniste), which can be used to advantage in creating mechanical devices to reduce the severity of the dystonia (Krack et al., 1998). Focal dystonias, such as tardive cervical dystonia (Molho et al., 1998) and tardive blepharospasm (Sachdev, 1998), can resemble primary focal dystonias (Video 19.6). Retrocollis is more likely to be due to tardive dystonia (Video 19.7), and less likely head or neck trauma, while primary dystonia is uncommon (Papapetropoulos et al., 2007). A comparison of tardive and primary oromandibular dystonia (OMD) showed similar demographics, both occurring predominantly in women with jaw-closing dystonia being the most common form (Tan and Jankovic, 2000). Primary OMD patients were more likely to have coexistent cervical dystonia, and the two types of dystonia responded equally well to botulinum toxin injections. Limb stereotypies, akathisia, and respiratory dyskinesia were seen only in the tardive OMD. 
One clinical presentation of tardive dystonia is particularly more characteristic of tardive dystonia, the combination of retrocollis, trunk arching backward, internal rotation of the arms, extension of the elbows and flexion of the wrists (Video 19.8) (Kang et al., 1986), whereas patients with idiopathic dystonia more often have lateral torticollis and twisting of the trunk laterally. The presence of lightning-like (myoclonic) movements in association with dystonia may be more common in tardive dystonia than in primary dystonia (Video 19.9). Reduction of dystonic movements with voluntary action such as walking is often seen in tardive dystonia. This is distinctly unusual in idiopathic dystonia in which the dystonic movements are usually exacerbated by voluntary action. It can be severe enough to jeopardize patients by causing life-threatening dysphagia (Hayashi et al., 1997; Samie et al., 1987). 
Tardive dystonia tends to occur in all ages without predilection for any particular age range. The mean age of onset in the literature is about 40 years. This is in contrast to idiopathic dystonia, which shows a bimodal distribution with one early peak in childhood and another later peak in adulthood (Fahn, 1988). Tardive dystonia affects both sexes equally, and men have a younger age of onset than women. Duration of exposure to dopamine receptor antagonists at the onset of tardive dystonia can range from as short as 3 weeks to close to 40 years. The mean duration is 7 years, and as many as 20% of cases develop within a year of the therapy. If the cumulative percentage of patients is plotted against the duration of exposure, the data show a linear line extrapolated to the origin of the graph at zero. This suggests that the risk of developing tardive dystonia starts at the initiation of therapy without any safe minimum period of exposure (Kang et al., 1986).
Wojcik and colleagues (1991) reviewed 32 patients with tardive dystonia and found that most were men, but that women had a shorter exposure time to DRBAs. None of their patients had a complete remission, and the condition causes notable disability. On the other hand, van Harten and colleagues (2008b) reported a remission rate of 80% in mild cases of tardive dystonia; the patients remained on antipsychotic medications, so one cannot be certain that the underlying symptoms would not reappear after withdrawal of the DRBAs.
Many patients with tardive dystonia also have classic tardive dyskinesia at some point in their course (Kang et al., 1986; van Harten et al., 1997). It is not clear why some develop dystonia whereas others develop classic tardive dyskinesia or why some develop both. When patients have both types, dystonic symptoms are usually much more pronounced and disabling (Kang et al., 1986; Gardos et al., 1987).
The dystonia can be so severe that complications can occur. One patient with powerful retrocollis fractured the odontoid process (Konrad et al., 2004).
Tardive akathisia
Originally, akathisia was mainly thought of as an acute to subacute side effect of dopamine receptor antagonists. Various authors, however, noted different variants of akathisia that occurred late in the course of the neuroleptic therapy and/or persisted despite discontinuation of neuroleptic therapy (Braude and Barnes, 1983; Fahn, 1983; Weiner and Luby, 1983). Concomitant or subsequent development of TD was also noted (Munetz and Cornes, 1982; Burke et al., 1989). Barnes and Braude (1985) made a systematic attempt to classify the complex variety of akathisia syndromes. They defined the disorder by the presence of both the subjective and objective features and confirmed that there are acute and chronic variants of akathisia. They also distinguished two types of chronic akathisia; one that occurred early in the course at the time of increasing neuroleptic dose and persisted (acute persistent akathisia) and one that occurred during long-term therapy, sometimes during reduction of their neuroleptic dose (tardive akathisia). Burke and colleagues (1989) reviewed experience with 30 patients with persistent akathisia who met both subjective and objective criteria. They found it difficult to distinguish between acute persistent and tardive akathisia in many cases owing to imprecise information about the onset of the disorder relative to initiation of therapy. It is not clear whether these are distinct syndromes or simply two ends of a continuum. Therefore, some researchers prefer to lump them together as tardive akathisia in line with other persistent movement disorders from dopamine receptor antagonists (Burke et al., 1989). Some attempts to classify akathisia syndromes are complex (Lang, 1994; Sachdev, 1995a); a simple method is to consider persistent neuroleptic-induced akathisia to be the tardive akathisia, and withdrawal, transient akathisia to be acute akathisia, as is done here.
The clinical phenomenology of tardive akathisia is thought to be the same as that of acute akathisia. Moaning (Video 19.10; see also Video 1.36) and focal pain are more common in tardive akathisia than in acute akathisia. In Burke and colleagues’ study (1989), the mean age at onset of tardive akathisia was 58 years with a range from 21 to 82 years, similar to the age range of classic tardive dyskinesia. The mean duration of dopamine receptor antagonist exposure before the onset was 4.5 years with a range from 2 weeks to 22 years. Over half of the patients had onset within 2 years. In tardive akathisia, there is a strong likelihood that there will be accompanying tardive dyskinesia (Video 19.11), or tardive dystonia (Video 19.12) movements. All of the patients with tardive akathisia in the study by Burke and colleagues (1989) also had either tardive dyskinesia (93%) or tardive dystonia (33%) or both (27%) at the same time. But isolated tardive akathisia can exist as well. The pathophysiology of akathisia is not understood. 
Treatment of tardive syndromes
The most important point to remember in the management of tardive syndromes is that they are iatrogenic disorders. One should avoid using a DRBA if possible. Patients should be forewarned of the risk of a tardive dyskinesia syndrome before being placed on the drug. In a survey of 520 psychiatrists, only 54% of them disclose this risk (Kennedy and Sanborn, 1992). A study of the impact of informed consent based on questionnaires showed that patients did retain the information both at 4 weeks and at 2 years (Kleinman et al., 1996). Another study comparing patients’ knowledge of TD by a questionnaire revealed that those who were educated about the disorder had more knowledge about it 6 months later (Chaplin and Kent, 1998).
Once a tardive syndrome has been encountered, removal of the etiologic agent must be seriously considered as the first consideration. If it is to be discontinued, a slow taper appears to be safer than sudden withdrawal; the latter might exacerbate the severity of the syndrome. In classic tardive dyskinesia, prospective data show 33% remission in 2 years following elimination of the DRBA (Kane et al., 1986). In retrospective studies, the remission rates were 12% for tardive dystonia and 8% for tardive akathisia (Kang et al., 1986; Burke et al., 1989). Some of these patients remitted only after at least 5 years of abstinence from DRBAs (Klawans et al. 1984; Kang et al., 1986). Younger age is associated with better chance of remission (Smith and Baldessarini, 1980) and earlier detection and discontinuation of dopamine receptor antagonists were more favorable for remission (Quitkin et al., 1977). In a study involving chronic schizophrenics, only 1 of 49 patients who discontinued the antipsychotic drugs had a lasting recovery, but 10 others had some improvement 1 year later (Glazer et al., 1990).
There are necessary indications for long-term use of DRBAs, such as chronic psychotic disorders (American Psychiatric Association, 1980). When patients are not able to discontinue antipsychotic medications, the concern is whether their TD will inexorably get worse requiring higher and higher doses of antipsychotics to suppress the symptoms, but there are no data to indicate a worsening in most patients (Labbate et al., 1997), and the majority of patients show improvement over time (Gardos et al., 1988). Casey and colleagues (1986) noted that those with decreased neuroleptic doses showed no change in their dyskinesia score, and those with stable doses and increased doses showed a mean decrease in their dyskinesia scores at 3- to 11-year follow-up. Although these data do not answer whether the symptoms were simply masked or whether the disease itself has improved in its natural course, at least it appears that continuing antipsychotics does not necessarily aggravate the symptoms.
Treatment of classic tardive dyskinesia (TD)
Dopamine depletors
Dopamine-depleting drugs have rarely, if ever, been noted to produce TD. In fact, among 17 patients with TD who were treated with reserpine by Fahn (1985), 4 remitted eventually while taking reserpine and were able to come off all treatment. Reserpine depletes catecholamine stores in sympathetic nerve terminals as well as in the CNS. Side effects include parkinsonism, apathy, depression, lethargy, and orthostatic hypotension. Some patients might require fairly high doses; significant improvement was reported with up to 5–8 mg/day (Sato et al., 1971; Fahn, 1985), whereas others who used lower doses reported less dramatic responses. Reserpine has a slow onset and a prolonged duration of action, and this must be taken into consideration when doses are changed. Once control of dyskinesia has been obtained, the dosage might need readjustment because of delayed onset of the catecholamine-depleting effect of reserpine with the induction of drug-induced parkinsonism. It is usually a question of balancing benefit and this adverse effect. Sixty-four percent of 96 patients in the literature had at least 50% improvement (Jeste and Wyatt, 1982b). One of the most notable results was that of a double-blind placebo-controlled study by Huang and colleagues (1980) that showed 50% improvement on reserpine 0.75–1.5 mg/day. Fahn’s long-term study (1985) showed that 13 of 17 patients had moderate to marked benefit on higher doses up to 8 mg/day. Nine of the patients who improved also took alpha-methylparatyrosine. Alpha-methylparatyrosine is a competitive inhibitor of TH, the rate-limiting step in catecholamine synthesis. It is not very effective when used alone, but can be a very powerful antidopaminergic drug when used with other presynaptically acting drugs.
TBZ has a quicker onset and shorter duration of action and has fewer peripheral catecholamine-depleting effects than reserpine. TBZ selectively inhibits vesicular monoamine transporter 2 (VMAT2), which is present in the CNS, whereas reserpine also inhibits VMAT1, found peripherally (Table 19.13). Like reserpine, TBZ has not been implicated in causing TD. However, in contrast to reserpine, TBZ does have some dopamine-receptor blocking activity (Reches et al., 1983), which probably accounts for the few reported cases of acute dystonic reaction that have been encountered clinically (Burke et al., 1985). In contrast to reserpine, remission of TD has not been reported during the treatment of TD with TBZ, although it has been seen in one of our patients (Fahn, personal observation). It would seem, therefore, that reserpine has the theoretical advantage of being more likely to allow for a remission of the TD, compared to TBZ. TBZ’s major advantage is the quicker onset and fewer side effects compared to reserpine. TBZ is rapidly absorbed after oral administration and extensively metabolized during the first pass through the liver and/or the gut. One of the major metabolites, dihydrotetrabenazine, has pharmacologic actions similar to those of TBZ, although its ability to pass through the blood–brain barrier is unclear. Large individual variations are noted, and patients with hepatic dysfunction might expect alteration of pharmacokinetics (Mehvar et al., 1987). The dose has to be clinically titrated for each patient.
Table 19.13 Pharmacology provile of tetrabenazine versus reserpine
| Pharmacological property | Tetrabenazine | Reserpine |
|---|---|---|
| Mechanism of action | Selectively binds hVMAT2 Reversibly binds VMAT2 Binds intravesicular site |
Binds hVMAT1 and hVMAT2 Irreversibly binds VMAT Binds cytoplasmic site |
| Peripheral monoamine depletion | No | Yes |
| Duration of action in humans | Short (approx. 12 hours) | Several days |
| Hypotension in humans | No | Yes |
| Gastrointestinal effects in humans | No | Yes |
TBZ, clinically available in many countries, is now considered the treatment of choice for TD (Kenney and Jankovic, 2006). Improvement with TBZ was noted in 68% of 38 patients in the literature at a mean daily dose of 138 mg (Jeste and Wyatt, 1982a). Fahn (1985) reported improvement in five of six patients at doses of 75–300 mg/day in a long-term study. Kazamatsuri and colleagues (1972) noted that 54% of patients improved by at least 50% in a 6-week trial of TBZ compared to placebo treatment. Jankovic and Beach (1997) reported that 90% of patients had a marked improvement. Ondo and colleagues (1999) blindly rated videotapes taken of patients before and after TBZ treatment (mean duration of 20 weeks) and showed improvement in their dyskinesias. A major problem with TBZ therapy is with long-duration exposure. Most patients initially improve dramatically; then while maintained on the original dose some begin to develop features of parkinsonism. Lowering the dose reduces this unwanted effect, but the TD then is less well controlled. Both reserpine and TBZ can induce acute akathisia and depression, so one needs to monitor for these adverse effects and treat them if they should occur. In some cases, however, depression actually improves after the introduction of TBZ, possibly as a result of abolishment of the involuntary movements (Kenney et al., 2006). Antidepressants, including monoamine oxidase inhibitors, can be used effectively to treat the depression. The selective norepinephrine reuptake inhibitor reboxetine has been found to rapidly reverse TBZ-induced depression (Schreiber et al., 1999).
Atypical antipsychotics
By definition the atypical antipsychotics have a reduced propensity to induce extrapyramidal adverse effects, tardive dyskinesia included. However, a range of adverse effects are encountered from the variety of drugs that have at one time or another been labeled as atypical neuroleptics. Today, the dibenzodiazepines (clozapine and quetiapine) are strong candidates for this labeling, and the thienobenzodiazepine olanzapine is less so. But originally, the “atypical” label was applied to the benzamine derivatives, such as sulpiride, metoclopramide, and tiapride. However, all three compounds have been reported to induce tardive dyskinesia or NMS (Casey, 1983; Achiron et al., 1990; Miller and Jankovic, 1990; Duarte et al., 1996). Similarly, risperidone has been touted as “atypical,” but it more resembles a typical antipsychotic, causing parkinsonism and inducing tardive dyskinesia (Buzan, 1996). The typical antipsychotics, including sulpiride, tiapride, and risperidone, by blocking and occupying dopamine D2 receptors, are effective in reducing the severity of tardive dyskinesia (Chouinard, 1995). But so do even stronger DRBAs, such as phenothiazines and haloperidol. The problem with using typical antipsychotics to treat TD is that they are in the class of the offending drugs and hence will prolong the exposure of the patient to the drugs that cause TD.
The question is whether the true “atypical” antipsychotics, such as clozapine and quetiapine, can reduce the symptoms of TD and still allow the healing process in the brain to proceed to eventually eliminate the pathophysiologic causation of the symptoms. There are reports of clozapine successfully reducing the abnormal movements of tardive dyskinesia and tardive akathisia (Huang et al., 1980; Wirshing et al., 1990; Bassitt and Neto, 1998) and in some patients with tardive dystonia (Lieberman et al., 1989, 1991; Van Putten et al., 1990; Friedman, 1994; Trugman et al., 1994; Wolf and Mosnaim, 1994; Raja et al., 1996; van Harten et al., 1996a; Bassitt and Neto, 1998). But the response rate is lower than that with typical antipsychotics, such as haloperidol. Clozapine permits the dyskinesia to disappear in about half the cases in one report (Gerlach and Peacock, 1994). In another, 8 of the 20 patients with TD improved after an average time of 261 ± 188 days of treatment (Bunker et al., 1996). With an average dose of approximately 400 mg/day, Bassitt and Neto (1998) obtained a 50% lessening of dyskinesia. It is still not clear whether the reduction of dyskinesia is due to the small amount of D2-blocking effect or whether actual healing of the TD can take place in the presence of clozapine. The proof of the latter and preferred category would be the lack of reappearance when clozapine is withdrawn. There has not been a report of such a case. Without that evidence, it is likely that the reductions of tardive dyskinesia and tardive dystonia are due to the small amount of D2-blocking activity by clozapine.
There is a case report of TD improving on quetiapine 600 mg/day (Vesely et al., 2000). The patient was not withdrawn from the drug. A large randomized study on high-dose quetiapine also found quetiapine to be effective (Emsley et al., 2004), but high dosages of the atypical antipsychotics become typical by blocking D2 receptors, so the reduction of TD can be due solely to further D2 receptor blockade.
However, clozapine and quetiapine could substitute for a typical antipsychotic in a patient with a tardive syndrome who also has psychosis, thereby controlling the psychosis and possibly still allowing a chance for a complete remission to occur. Such a remission would not take place in the presence of the typical antipsychotic. Like clozapine, quetiapine needs to be tested. Olanzapine has successfully reduced the symptoms of TD (Littrell et al., 1998). But olanzapine is not a true atypical antipsychotic, and the reduction of the symptoms and signs is probably obtained by blockading D2 receptors. This would convey no advantage over using the more classic and conventional typical antipsychotics.
Dopamine agonists
Some investigators have tried to activate the presynaptic dopamine receptors by using low doses of a dopamine agonist, which in turn would reduce the biosynthesis and release of dopamine. Another approach by Alpert and Friedhoff (1980) was the use of levodopa in an attempt to desensitize the postsynaptic dopamine receptors. This can cause initial worsening of symptoms before eventual improvement is expected after discontinuation of levodopa. Unfortunately, dopaminergic drugs can also lead to overt recurrence of underlying psychosis (Fahn, 1983). This approach has theoretical merit, but is very difficult to carry out in many patients and has not been widely used since the initial reports.
Amantadine has been reported to have some benefit (Angus et al., 1997), but it may be due to its glutamate receptor blocking effect rather than its dopaminergic effect.
Nondopaminergic medications
Although neuroleptics are most effective in controlling the abnormal movements, some patients do not respond to the treatment. Numerous investigators (Jeste and Wyatt, 1982b; Jeste et al., 1988) have attempted nondopaminergic treatments. Agents that enhance GABA transmission have been tried because of GABA’s inhibitory effect on the dopaminergic system and experimental data indicating changes in the GABA system in patients and animals that have been treated with chronic neuroleptics. Use of benzodiazepines, baclofen, valproate, and γ-vinyl GABA has met with limited success, partly owing to tolerance and side effects such as worsening of psychosis. Some improvement was found with clonazepam (Thaker et al., 1990). Propranolol, fusaric acid, and clonidine decrease the noradrenergic activity and have been reported to be useful but require further study to clarify their role in treating tardive dyskinesia. Use of cholinergic drugs was based on the reciprocal dopamine–acetylcholine balance in the basal ganglia. Despite a flurry of reports in the 1970s, this modality has been quite limited, and should be reconsidered now that some adequate cholinergic drugs are available. Anticholinergics, pyridoxine, tryptophan, cyproheptadine, vasopressin, naloxone, morphine, and estrogen were reported to be of no benefit. But in a controlled clinical trial involving 15 patients, pyridoxine was found to reduce the severity of TD (Lerner et al., 2001). Buspirone has been reported to be beneficial (Moss et al., 1993), but it is not clear that this drug does not have dopamine-receptor blocking activity. Calcium channel blockers have been reported to reduce the severity of tardive dyskinesia (Kushnir and Ratner, 1989; Duncan et al., 1990; Suddath et al., 1991), but not in all studies (Loonen et al., 1992). A combination of acetazolamide and thiamine was found to reduce both TD and drug-induced parkinsonism (Cowen et al., 1997). Lithium not only may decrease the chance of TD development (Kane et al., 1986; van Harten et al., 2008a), but also may reduce its severity if applied as a treatment (van Harten et al., 2008a).
A review of randomized clinical trials was presented by Soares and McGrath (1999) with a meta-analysis when more than one randomized clinical trial had been carried out. Meta-analysis showed that baclofen, deanol, and diazepam were no more effective than a placebo. Single randomized clinical trials demonstrated a lack of evidence of any effect for bromocriptine, ceruletide, clonidine, estrogen, gamma-linolenic acid, hydergine, lecithin, lithium, progabide, selegiline, and tetrahydroisoxazolopyridinol. Meta-analysis found that five interventions were effective: levodopa, oxypertine, sodium valproate, tiapride and vitamin E; neuroleptic reduction was marginally significant. Vitamin E is more thoroughly discussed in the next paragraph. Data from single randomized clinical trials revealed that insulin, α-methyldopa, and reserpine were more effective than a placebo. Meta-analysis found that 37.3% of placebo-treated subjects improved.
A role for antioxidants has been raised (Cadet and Lohr, 1989; Behl et al., 1995). Treatment with vitamin E has been found to reduce the severity of tardive dyskinesia (Elkashef et al., 1990; Dabiri et al., 1994; Adler et al., 1998; Sajjad, 1998) or have no effect (Lam et al., 1994). The meta-analysis mentioned earlier showed effectiveness. A small clinical trial of 41 subjects comparing 1200 IU/day of vitamin E and placebo found the former to better reduce severity of abnormal involuntary movements (45.9% vs. 4.3%) (Zhang et al., 2004). However, the largest double-blind study, carried out by the Veterans Administration multicenter, a placebo-controlled clinical trial (Adler et al., 1999), found vitamin E not to be effective. As a potential prophylactic agent, vitamin E (3200 IU/day) was found not to protect against development of drug-induced parkinsonism (Eranti et al., 1998). Nor was vitamin E treatment able to prevent neuroleptic-induced VCMs in rats (Sachdev et al., 1999). In a small open-label trial combining vitamins E and C, improvement in TD was seen (Michael et al., 2003).
There continue to be reports of TD responding to open-label trials. Gabapentin (Hardoy et al., 1999, 2003), pyridoxine (Lerner et al., 1999), and branched-chain amino acids (Richardson et al., 2003) are such compounds. Levetiracetam was helpful in a small trial (Konitsiotis et al., 2006).
Injections of botulinum toxin into the muscles causing oral dyskinesia have been reported to be effective in reducing the movements (Rapaport et al., 2000). This includes tongue protrusion, which has been successfully treated with injections into the genioglossal portion of the tongue (van Harten and Hovestadt, 2006).
Sporadic reports noted efficacy of electroconvulsive therapy in refractory cases of TD (Price and Levin, 1978), but Yassa and colleagues (1990) reported success in only one of nine patients.
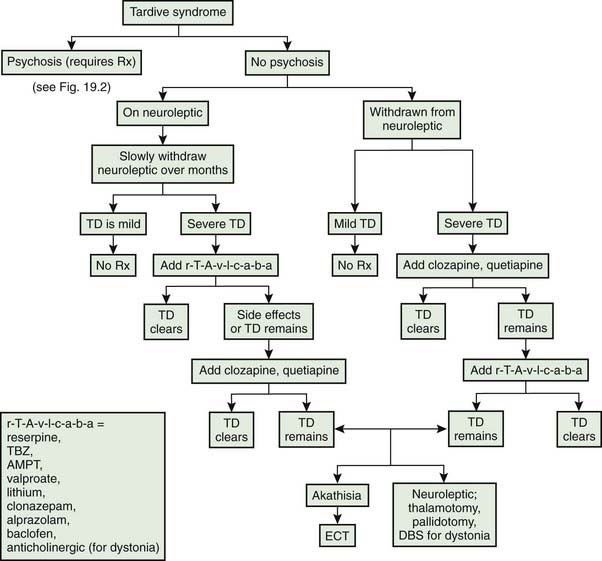
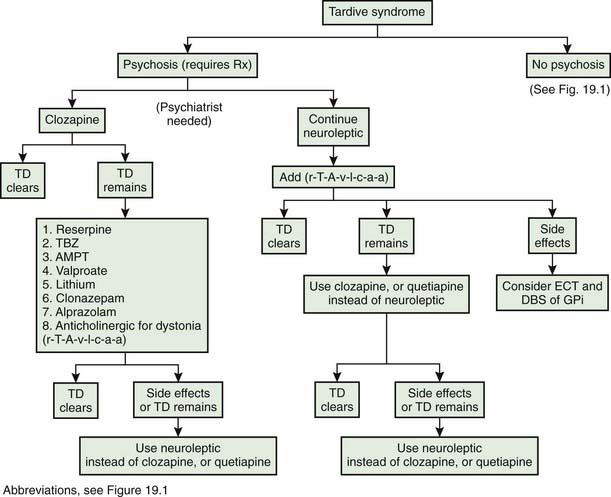
Tardive dystonia
As with classic TD, the most effective medications for tardive dystonia are also antidopaminergic drugs (Kang et al., 1986), but the percentage of patients who improve is smaller. Reserpine and TBZ each produce improvement in about 50% of patients. Some patients who do not respond or have intolerable side effects to one might respond to the other. DRBAs are more effective in suppressing the movements (77%). Symptomatically, those who remained on DRBAs after the onset of tardive dystonia and those who were withdrawn from them do not have a significant difference in their improvement rate. This again is in agreement with the data in classic tardive dyskinesia, in which continued use of DRBAs does not necessarily lead to aggravation of their movements (Casey et al., 1986; Gardos et al., 1988). The atypical antipsychotic clozapine has been helpful in some patients with tardive dystonia (Lieberman et al., 1989, 1991; Van Putten et al., 1990; Friedman, 1994; Trugman et al., 1994; Wolf and Mosnaim, 1994; Raja et al., 1996; van Harten et al., 1996a). There are reports of quetiapine’s effectiveness as well (Gourzis et al., 2005). It is likely that its treatment of tardive dystonia in some situations is due to its D2 receptor-blocking activity resulting in a masking of the symptoms, because withdrawal would exacerbate the dystonia (Krack et al., 1994). The combination of clozapine and clonazepam has been effective in some patients when either drug alone was much less satisfactory (Shapleske et al., 1996).
In tardive dystonia, antimuscarinics are almost as effective as antidopaminergic drugs. This is different from classic tardive dyskinesia, which may get worse with antimuscarinics (Yassa, 1988). Kang and colleagues (1986) reported a 46% improvement rate on antimuscarinics such as trihexyphenidyl and ethopropazine. CNS side effects of the antimuscarinics include forgetfulness, lethargy, psychosis, dysphoria, and personality changes; elderly patients are more susceptible to these. Peripheral side effects include blurred vision, dry mouth, constipation, urinary retention, and orthostatic dizziness. Those who develop side effects to one anticholinergic drug may tolerate another anticholinergic better. Although there is no evidence that one anticholinergic is more efficacious than the others, ethopropazine may produce fewer CNS side effects in elderly patients. Although peripheral pharmacokinetics show relatively short half-lives, their central effects have a very slow onset of action and several weeks are often required before benefit is noticed. Therefore, the medications are started at a low dose, 2.5 mg of trihexyphenidyl or 25 mg of ethopropazine, and increased slowly by 2.5 mg of trihexyphenidyl or 25 mg of ethopropazine weekly until sufficient control of dystonia or intolerable side effects are achieved. As in idiopathic dystonia, many patients respond only to a high dose of anticholinergics. Therefore, every attempt must be made to control side effects so that high-dose anticholinergics may be tried. Kang and colleagues (1986) reported use of a maximum of 450 mg ethopropazine or 32 mg of trihexyphenidyl, and higher doses may be tolerated in young patients if judiciously used. Peripheral side effects are often controlled by peripheral cholinergic drugs, such as oral pyridostigmine and pilocarpine eye drops.
The clinical pharmacology of tardive dystonia indicates two subtypes: one group that responds to antidopaminergic drugs like the other tardive syndromes and one that responds to anticholinergics like idiopathic dystonia. Analysis of clinical characteristics of patients who respond to antidopaminergic drugs and those who respond to anticholinergic drugs has not shown any significant difference (Kang et al., 1986). However, the data from Kang and colleagues (1986) are retrospective and the treatment choice between the two classes of drugs was rather arbitrary, partly based on anticipated side effects. For example, patients who were elderly or who had dementia were treated with dopamine-depleting drugs first, because these patients have greater risk for anticholinergic side effects such as memory loss and confusion. Patients with depression might have relapse of their symptoms on dopamine-depleting drugs and were treated preferentially with anticholinergics. This is a reasonable clinical approach, but characterization of subtypes will require a controlled study with random assignment of the treatment. If either a dopamine depletor or antimuscarinic is ineffective by itself, the combination should be tried.
Benzodiazepines are mainly helpful as adjunctive therapy with dopamine-depleting or anticholinergic drugs, and occasionally can be quite beneficial (Yamamoto et al., 2007). Minimal success with propranolol, levodopa, carbamazepine, and baclofen has been noted. Bromocriptine, deanol, clonidine, lisuride, amantadine, and valproate were reported with mixed results. The calcium channel blocker verapamil was reported to be effective in one patient (Abad and Ovsiew, 1993). Opioids do not have lasting value in suppressing tardive dystonia (Berg et al., 2001). But one study found the combination of naltrexone and clonazepam to offer some benefit (Wonodi et al., 2004b).
If any residual dystonia remains that is localized to one or a few parts of the body, injections of botulinum toxin into the affected parts, such as the orbicularis oculi, masseters, or cervical muscles, might be useful (Chatterjee et al., 1997; Tarsy et al., 1997; Kanovsky et al., 1999).
Electroconvulsive therapy (ECT) might be effective in intractable cases (Yoshida et al., 1996), and deep brain stimulation in the globus pallidus interna is often effective (Franzini et al., 2005; Sako et al., 2008; Gruber et al., 2009), as is intrathecal baclofen (Dressler et al., 1997). Surgery is not always effective and it does pose risks of complications (Trottenberg et al., 2001). However, deep brain stimulation in the pallidum can be safe and effective (Capelle et al., 2010; Chang et al., 2010); it is worth considering when medications fail.
A centrally acting muscle relaxant, eperisone, was successful in treating one patient with tardive dystonia (Nisijima et al., 1998). Eperisone is a beta-aminopropiophenone derivative.
Figures 19.1 and 19.2 depict flowcharts for therapy that can be applied to tardive dystonia, as well as to classic TD.
Tardive akathisia
Tardive akathisia is difficult to treat and does not respond to anticholinergics, which have been reported to help acute akathisia. It and tardive dystonia are the most distressing and disabling features of the tardive symptoms, and their treatment is important. In the study of tardive akathisia by Burke et al. (1989), all of the patients noted that the subjective sensations were distressing. The same study reported that 87% of patients improved on reserpine up to 5 mg/day and 58% on TBZ up to 175 mg/day. In one-third of these patients, the movements were completely suppressed. In this respect the clinical pharmacology is more like that of classic tardive dyskinesia than that of acute akathisia. Opioids were reported to be beneficial (Walters et al., 1986), but the effect has not been persistent (Burke et al., 1989). Electroconvulsive treatment can be effective in those patients whose akathisia has proved to be intractable (Hermesh et al., 1992).
Figures 19.1 and 19.2 depict flowcharts on therapy that can also be applied to tardive akathisia.
Treatment summary
Table 19.14 summarizes a useful approach to treat tardive syndromes in general. The approach to treatment should be earmarked according to whether the patient is psychotic and requires antipsychotic medication, or is not psychotic. For detailed, step-by-step decisions in the treatment of the tardive syndromes, the flowcharts of Figures 19.1 and 19.2 should be helpful.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 19 The tardive syndromes
Phenomenology, concepts on pathophysiology and treatment, and other neuroleptic-induced syndromes
Overview
Fundamentals and definitions
A variety of neurologic adverse effects are seen with drugs that block dopamine D2 receptors. Because these complications are mainly movement disorders and likely relate to the D2 receptors in the striatum and limbic system, they are usually called extrapyramidal reactions. These are listed in Table 19.1 and are covered in the clinical sections that follow.
Table 19.1 Neurological adverse effects of dopamine receptor antagonists
Adapted from Fahn S: The tardive dyskinesias. In Matthews WB, Glaser GH (eds): Recent Advances in Clinical Neurology, vol. 4. Edinburgh: Churchill Livingstone, 1984, pp. 229–260.
The term tardive syndromes refers to a group of disorders that fit all of the following essential criteria: (1) Phenomenologically, the clinical features are that of a movement disorder – i.e., abnormal involuntary movements or a sensation of restlessness that often causes “unvoluntary” movements; (2) the disorder is caused by the patient’s having been exposed to at least one DRBA within 6 months of the onset of symptoms (in exceptional cases, exposure could be up to 12 months); and (3) the disorder persists for at least 1 month after stopping the offending drug (Fahn, 1984a; Stacy and Jankovic, 1991). The question arises as to what to call persistent dyskinesias that are induced by drugs other than DRBAs. For example, Miller and Jankovic (1992) described such a patient who had been exposed to flecainide, a drug not known to be a DRBA. Another drug, buspirone, an azospirone compound, is an anxiolytic that is not known to have any dopamine receptor-blocking activity. Yet there is a report of two patients who had persistent movement disorders after prolonged treatment with this drug (LeWitt et al., 1993). One patient had cervical-cranial dystonia, and the other had an exacerbation of preexisting spasmodic torticollis and TD. It is possible, however, that future laboratory investigation will reveal flecainide and buspirone or one of their metabolites actually to be a DRBA.
Although there are several phenomenologically distinct types of tardive disorders, the collective group is referred to as tardive dyskinesia for historical reasons. Unfortunately, the term tardive dyskinesia is often used also to refer to a phenomenologically specific type of tardive syndrome, so the literature is often confusing; this note of caution is particularly important in trying to understand whether the author of a paper is referring to the tardive syndromes collectively or to a specific tardive syndrome. Since there could be different pathophysiologic mechanisms and treatments for the different forms of tardive syndromes, it is best that they be divided phenomenologically (Table 19.2). In this review, the tardive syndromes as a whole are referred to as tardive dyskinesia, and the specific type that was historically and initially labeled as tardive dyskinesia is referred to as classic tardive dyskinesia. Other names have been used for classic tardive dyskinesia (see Table 19.2). The phenomenologically essential component of classic tardive dyskinesia is the presence of repetitive, almost rhythmic, movements. These are almost always present in the mouth region, and therefore are also called O-B-L dyskinesias.
Table 19.2 Terminology of the tardive syndromes
| Descriptions | Equivalent common names |
|---|---|
| Tardive syndromes as a group | Tardive syndrome |
| Tardive dyskinesia | |
| Repetitive, rhythmic movements, usually in the oral-buccal-lingual region | Classic tardive dyskinesia (TD) |
| Oral-buccal-lingual (O-B-L) dyskinesias | |
| Tardive stereotypy | |
| Rhythmic chorea | |
| Dystonic movements and postures | Tardive dystonia |
| Restlessness and the movements that occur as a result | Tardive akathisia |
| Myoclonus | Tardive myoclonus |
| Tremor | Tardive tremor |
| Tics | Tardive tics |
| Tardive tourettism | |
| Chorea | Withdrawal emergent syndrome |
| Tardive chorea | |
| Oculogyria | Tardive oculogyric crisis |
| Parkinsonism | Tardive parkinsonism (if it exists) |
Tardive dyskinesia was first described in patients who were treated for schizophrenic psychosis with antipsychotics (Schonecker, 1957; Sigwald et al., 1959). These abnormal movements appeared late in the course of treatment, in contrast to acute dystonic reactions and drug-induced parkinsonism, which had previously been recognized as complications from antipsychotics, hence the term tardive. The offending antipsychotics are now known to block the D2 dopamine receptor; i.e., these drugs are DRBAs. Since the first descriptions, TD has also been noted in patients without psychiatric disorders who had other indications for using dopamine receptor antagonists, such as those with gastrointestinal complaints (Casey, 1983), with Gilles de la Tourette syndrome (Riddle et al., 1987), or with dystonia (Greene and Fahn, 1988).
Dopamine receptor antagonists produce many undesirable side effects, most of which occur relatively early in the course of treatment and are reversible on discontinuation of the medication. However, disfiguring and disabling abnormal involuntary movements were also noted to often occur late in the course of treatment and these were often noted to persist, even after discontinuation of the medication. Hence, the term “tardive” was coined, referring to the late and insidious onset (Faurbye et al., 1964). Initially the term tardive dyskinesia was equated with stereotypic repetitive movements of oral, buccal, and lingual distribution (Schonecker, 1957), but subsequently other types of movements have been recognized (Burke et al., 1982; Fahn, 1984a). As such, the concept of tardive dyskinesia has evolved and has been modified considerably since the initial recognition of the syndrome.
The prevalences of drug-induced parkinsonism and the various tardive syndromes have been compared by van Harten and colleagues (1996b) on the island of Curaçao, which has only one psychiatric facility. In 194 inpatients, the prevalence for classic tardive dyskinesia (TD) was 39.7%, that for parkinsonism was 36.1%, that for tardive dystonia was 13.4%, and that for akathisia was 9.3%. Combinations of two or more of these phenomenologies occurred in 30% of patients (van Harten et al., 1997). van Harten and colleagues (2006) continued to follow their patients over 9 years (mean duration of 18 years exposure to first-generation antipsychotic drugs); they found the annual incidence rate for classic TD to be 10.2% and for tardive dystonia to be 0.7%. Severity was associated with age and akathisia but not with drug-induced parkinsonism.
Dopamine receptors and their antagonists
Since TD is an iatrogenic disorder and the most constant feature of the syndrome is the pharmacologic class of the responsible etiologic agent, it is important to understand the nature of the drugs that produce TD (Table 19.3). Although models of abnormal basal ganglia circuitry have been proposed to explain the mechanism of TD (Marchand and Dilda, 2006), the pathophysiology is still poorly understood. Dopamine receptors are classified into five subtypes, based on the genetics of the receptors; they are labeled D1, D2, D3, D4, and D5 (Kebabian and Calne, 1979; Sokoloff et al., 1990). Table 19.4 characterizes the five dopamine receptors. It is the dopamine D2 receptor-blocking action of drugs that has been linked to the tardive syndromes and other neuroleptic drug-induced movement disorders (described later and listed in Table 19.5).
| Class of drug | Examples of drugs in each class |
|---|---|
| 1. Phenothiazines | |
| a. Aliphatic | Chlorpromazine (Thorazine) Triflupromazine (Vesprin) |
| b. Piperidine | Thioridazine (Mellaril) Mesoridazine (Serentil) |
| c. Piperazine | Trifluoperazine (Stelazine) Prochlorperazine (Compazine) Perphenazine (Trilafon) Fluphenazine (Prolixin) Perazine |
| 2. Thioxanthenes | |
| a. Aliphatic | Chlorprothixene (Tarctan) |
| b. Piperazine | Thiothixene (Navane) |
| 3. Butyrophenones | Haloperidol (Haldol) Droperidol (Inapsine) |
| 4. Diphenylbutylpiperidine | Pimozide (Orap) |
| 5. Dibenzazepine | Loxapine (Loxitane) Asenapine (Saphris) |
| 6. Dibenzodiazepine | Clozapine (Clozaril) Quetiapine (Seroquel) |
| 7. Thienobenzodiazepine | Olanzapine (Zyprexa) |
| 8. Pyrimidinone | Risperidone (Risperidal) Paliperidone ER (9-hydroxyrisperidone) |
| 9. Benzisothiazole | Ziprasidone (Geodon) |
| 10. Benzisoxazole | Iloperidone (Zomaril) |
| 11. Substituted benzamides | Metoclopramide (Reglan) Tiapride (Tiapridex) Sulpiride (Meresa) Clebopride Remoxipride Veralipride (Agreal, Agradil) Amisulpride (Solian) Levosulpiride |
| 12. Indolones | Molindone (Moban) |
| 13. Quinolinone | Aripiprazole (Abilify) |
| 14. Tricyclic | Amoxapine (Asendin) |
| 15. Calcium channel blockers | Flunarizine (Sibelium) Cinnarizine (Stugeron) |
| 16. N-acetyl-4-methoxytryptamine | Melatonin |
| 17. Interferon-alpha | Pegylated interferon alpha 2b (IFN-α) (Pegylated = polyethylene glycol (PEG) attached to proteins) |
Recently introduced drugs in Table 19.3, e.g., iloperidone (Jain, 2000), ziprasidone (Hirsch et al., 2002), amisulpride (Curran and Perry, 2002), aripiprazole (Tamminga and Carlsson, 2002), paliperidone ER (Chwieduk and Keating, 2010), levosulpiride (Shin et al., 2009), and asenapine (Kane et al., 2010), need to be in clinical use for several years before their full potential in causing tardive dyskinesia syndromes can be known. The earlier ones in the above sentence have already been found to induce the acute and delayed movement disorders described in this chapter. In Table 10.3, the calcium channel blockers deserve comment. Cinnarizine (1-diphenylmethyl-4-(3-phenyl-2-propenyl) piperazine) and its difluorinated derivative flunarizine can induce parkinsonism (Teive et al., 2004). They are antagonists at the D2 receptors (Belforte et al., 2001), and they also inhibit the MgATP-dependent generation of a transmembrane proton electrochemical gradient and dopamine vesicular uptake (Terland and Flatmark, 1999). Whether either of these latter mechanisms, rather than a proposed DRBA action, is responsible for the neuroleptic activity is uncertain. In regard to melatonin, there is a case report of withdrawal-emergent O-B-L dyskinesias associated with akathisia that occurred with sudden discontinuation of chronic melatonin use (Giladi and Shabtai, 1999). Such withdrawal syndromes are typical of drugs that block dopamine receptors. On resumption of melatonin, the patient’s O-B-L dyskinesia and akathisia cleared. Sudden cessation of the drug again brought on the symptoms; slow taper over 2 months was effective without incident. This case suggests that either the melatonin product the patient was taking was impure and was contaminated with a DRBA or melatonin itself has dopamine receptor antagonist properties. In support of the latter is the result of a blinded crossover study showing some suppression of the tardive dyskinesia with melatonin (Shamir et al., 2001). Pegylated interferon alpha-2b (IFN-α), used in the treatment of hepatitis C virus infection, has been reported to cause parkinsonism, akathisia, and acute dystonic reaction (Quarantini et al., 2007). IFN-α has been shown to decrease dopaminergic activity in mice (Shuto et al., 1997).
Some drugs that block dopamine D2 receptors are promoted for medical problems other than psychosis, but these drugs can cause drug-induced parkinsonism, acute dystonic reactions, tardive syndromes, and NMS, just like the drugs that are promoted for the treatment of psychosis. Metoclopramide (Ganzini et al., 1993) and clebopride (Sempere et al., 1994) are used mainly for dyspepsia and as antiemetic agents. Amoxapine has a tricyclic structure and is marketed as an antidepressant drug, but a metabolite has dopamine receptor-blocking activity and has been implicated in producing TD (Kang et al., 1986; Sa et al., 2001). Veralipride is a substituted benzamide that is used for the treatment of menopausal hot flushes (Masmoudi et al., 1995). Pimozide is marketed for the treatment of Tourette syndrome. Some commercial preparations contain dopamine receptor antagonists in combination with other drugs, and this can lead to inadvertent use of these drugs. A popular combination is that of perphenazine and amitriptyline, marketed as Triavil and Etrafon. Risperidone is commercially promoted with the suggestion that it might have less risk of drug-induced complications, but this appears not to be the case. Parkinsonism and TD have been noted in association with the calcium channel antagonists flunarizine and cinnarizine (Micheli et al., 1987). Both of these medications have mild dopamine receptor antagonist activity, which is thought be the mechanism for their complications (Micheli et al., 1989). Recognition of these drugs is essential not only in making the diagnosis of TD but also in preventing the occurrence of TD by being able to avoid using them.
The D1 and D2 receptors are found mainly in the striatum and nucleus accumbens, as well as in the substantia nigra, amygdala, cingulate cortex, and entorhinal area. The anterior lobe of the pituitary gland has only D2 receptors, and the thalamus and cerebral cortex outside of the cingulate and entorhinal area contain D1 receptors only (De Keyser et al., 1988). D2 receptor affinities of dopamine receptor antagonists correlate closely with antipsychotic and antiemetic properties of the drugs (Creese et al., 1976). Dopamine receptor antagonists are often referred to as neuroleptics or antipsychotics; the former term indicates the effect of drugs in producing parkinsonism, and the latter indicates the effect of controlling psychosis.
“Atypical” antipsychotics
The label “atypical” refers to a lower propensity of the antipsychotic agent to induce parkinsonism or a variety of other movement disorders described later (and listed in Table 19.5), that is these agents have a lower propensity to be neuroleptics. A number of epidemiologic studies have found that the atypicals are less likely to induce tardive dyskinesia and other movement disorder problems (Tarsy and Baldessarini, 2006). Dolder and Jeste (2003) found that the atypicals reduced the incidence of tardive dyskinesia by half.
Dopamine and serotonin (5-HT) receptor antagonism
Although drugs that are labeled as atypical antipsychotic agents block dopamine D2 receptors, they also block serotonin 5-HT2A receptors, and some investigators attribute their antipsychotic effect to this mechanism and recommend research and drug development on newer agents that block other 5-HT receptors (Meltzer, 1999).
How tightly drugs to bind to the D2 receptor (their occupancy rate) is currently considered to be important in the drug having neuroleptic potential in direct proportion. Seeman and Tallerico (1998) found that the atypical antipsychotic drugs bind more loosely than classic antipsychotic drugs (neuroleptics) to the D2 receptors. Kapur and colleagues (1999) measured D2 and 5-HT2 receptor occupancies by positron emission tomography (PET) scan for patients receiving clozapine, risperidone, or olanzapine. Clozapine showed a much lower D2 occupancy (16–68%) than risperidone (63–89%) and olanzapine (43–89%); all three showed greater 5-HT2 occupancy than D2 occupancy at all doses, although the difference was greatest for clozapine. In their PET study, Moresco and colleagues (2004) found that striatal D2 receptor occupancy was significantly higher with olanzapine than with clozapine. All these studies support the relationship between receptor occupancy and the clinical observations that clozapine and quetiapine are the most “atypical,” with the least propensity to cause parkinsonism, tardive syndromes, or other neuroleptic drug reactions.
Despite interest in the 5-HT2A receptor, it is the D2 receptor that is associated with the antipsychotic effect (Seeman, 2010). Antipsychotic clinical doses correlate with their affinities for this receptor. The receptor has high- and low-affinity states. Clozapine and quetiapine do not elicit parkinsonism and rarely result in tardive dyskinesia because they are released from D2 within 12–24 hours. Traditional antipsychotics remain attached to D2 receptors for days, preventing relapse, but allowing accumulation that can lead to tardive dyskinesia. Glutamate agonists that treat schizophrenia have affinity for the dopamine D2(High) receptor and the D3 receptor (Seeman and Guan, 2009).
A comparison of the receptor-binding profile of quetiapine, clozapine, olanzapine, and risperidone is presented in Table 19.5. Both quetiapine and clozapine have poor affinity for the dopamine D2 receptor, which probably accounts for the low incidence of inducing parkinsonism and tardive syndromes. Risperidone has the highest affinity for the D2 receptor, resembling haloperidol (see Table 19.6), and is therefore more of a “typical” than an “atypical” antipsychotic.
Table 19.6 A more complete listing of affinities for human receptors and rat transporters by antipsychotic drugs
In one PET study striatal D1 and D2 receptor occupancies were evaluated (Tauscher et al., 2004). D1 occupancy ranged from 55% with clozapine to 12% with quetiapine (rank order: clozapine > olanzapine > risperidone > quetiapine). The striatal D2 occupancy ranged from 81% with risperidone to 30% with quetiapine (rank order: risperidone > olanzapine > clozapine > quetiapine). The ratio of striatal D1/D2 occupancy was significantly higher for clozapine (0.88) relative to olanzapine (0.54), quetiapine (0.41), or risperidone (0.31). In an [123I]epidepride single photon emission computed tomography (SPECT) study involving clozapine-, olanzapine-, and haloperidol-treated schizophrenia patients, as well as drug-naive patients and healthy controls, midbrain D(2/3) receptor occupancy was studied. Of those on medication, occupancy was least for those on clozapine (5%), next for those on olanzapine (28%), and greatest for those on haloperidol (40%); no significant differences were observed in the temporal poles (Tuppurainen et al., 2009). All imaging studies are compatible with the clinical results in that clozapine and quetiapine, with the least neuroleptic features, have the lowest D2 receptor occupancy rate.
Clozapine
Clozapine was the first agent to be labeled as an atypical antipsychotic and deservedly so, although rare case reports do exist of drug-induced parkinsonism (Kurz et al., 1995), but it has been reported not to induce rigidity, and it rarely induces parkinsonian tremor (Gerlach and Peacock, 1994). It has also caused rare cases of acute akathisia (Friedman, 1993; Safferman et al., 1993), acute dystonic reaction (Kastrup et al., 1994; Thomas et al., 1994), tardive syndromes (Dave, 1994), tardive dystonia (Bruneau and Stip, 1998; Molho and Factor, 1999a, van Harten et al., 2008b), tardive akathisia (Kyriakos et al., 2005), tardive oculogyria (Uzun and Doruk, 2007), and NMS (Sachdev et al., 1995; Benazzi, 1999; Lara et al., 1999; Gambassi et al., 2006). There are also parkinsonian features that are seen with clozapine. In a prospective study, seven out of 25 patients on clozapine developed TD (Bunker et al., 1996). In a retrospective study, comparison of clozapine and typical antipsychotics showed no lower prevalence of tardive dyskinesia in the clozapine group (Modestin et al., 2000). But it is not clear that the patients in the clozapine group had not been previously exposed to typical antipsychotics and had not developed TD while receiving them. What is clear from this study is that the conversion to using clozapine has not markedly reduced the prevalence of extrapyramidal syndromes; this finding supports the realization that clozapine does not effectively treat TD once it has developed. How effective its use would be in preventing TD in the first place still needs to be verified. For patients with PD, the low propensity to augment existing parkinsonism makes clozapine very useful in treating patients who have dopaminergic drug-induced psychosis (Factor and Friedman, 1997; Friedman et al., 1999).
A SPECT study measuring dopamine D2 receptor binding reveals lower binding with clozapine than with typical neuroleptics (Broich et al., 1998). In animal studies, rats that have been pretreated with haloperidol for 4 weeks develop vacuous chewing movements (VCMs) after treatment with a dopaminergic, whereas rats that have been pretreated with clozapine do not (Ikeda et al., 1999).
A problem with clozapine is that weekly blood counts are required because there is a 1% to 2% incidence of leukopenia, which is reversible if the drug is withdrawn within 1–2 weeks. Granulocyte colony-stimulating factor can be an effective means to treat the agranulocytosis (Sperner-Unterweger et al., 1998). Other common adverse effects are drowsiness, drooling, weight gain, and seizures. An unusual adverse effect with clozapine was a case of myokymia (David and Sharif, 1998). Myocarditis associated with clozapine has been reported (Hill and Harrison-Woolrych, 2008).
Quetiapine
After clozapine, quetiapine is the most favorable in being least likely to induce parkinsonism, NMS (Stanley and Hunter, 2000), or tardive syndromes. In a prospective, head-to-head comparison with risperidone treatment in schizophrenia, quetiapine had far fewer extrapyramidal adverse effects, but more orthostasis (Perez et al., 2008). D2 receptor antagonism is relatively selective for limbic than striatal receptors for clozapine and sertindole, followed by quetiapine, ziprasidone, olanzapine, and remoxipride, whereas risperidone in many respects has a profile that resembles that of haloperidol (Arnt and Skarsfeldt, 1998). In primates, quetiapine and clozapine were equally less likely to induce oral dyskinesias compared to standard antipsychotics (Peacock and Gerlach, 1999). Like clozapine, quetiapine easily induces drowsiness, so when it is used to treat dopa-induced psychosis, it should be taken at bedtime. The major advantage over clozapine is that it does not require blood tests because it does not induce agranulocytosis. However, there may be a rare risk that quetiapine can cause agranulocytosis (Ruhe et al., 2001). Since its introduction, there have been reports of acute dystonic reactions (Jonnalagada and Norton, 2000; Desarker and Sinha, 2006), acute akathisia (Prueter et al., 2003), neuroleptic malignant syndrome (El-Gaaly et al., 2009; Gortney et al., 2009), and also TD (Ghaemi and Ko, 2001). In 367 patients treated for 12 months with quetiapine (mean dose 720 mg/day), parkinsonism was seen in 10%, akathisia in 3%, dystonia in 1.4%, and “hyperkinesia” in 1.7% (Perez et al., 2008).
In a SPECT study using the D2/D3 ligand [I-123]-epidepride the percent occupancy of receptors in the limbic system (temporal lobe) and striatal receptors while patients were receiving quetiapine was 60% and 32%, respectively, which is similar to clozapine (Stephenson et al., 2000). In another SPECT study, quetiapine was shown to occupy 5-HT2A receptors in the frontal and temporal cortex (Jones et al., 2001). A PET study using [18F]fallypride showed similar results (Vernaleken et al., 2010).
A patient with oculogyric crisis, who failed to improve after withdrawal of antipsychotic medication, was successfully treated with quetiapine (Gourzis et al., 2007). Drug-induced parkinsonism and acute akathisia due to other neuroleptics can be reduced by switching patients to quetiapine (Cortese et al., 2008). Drowsiness and weight gain are common; postural hypotension is not infrequent. A rare complication of quetiapine is hepatotoxicity (Shpaner et al., 2008).
Olanzapine
In contrast to clozapine and quetiapine, olanzapine more readily increases parkinsonian symptoms in patients with PD (Jimenez-Jimenez et al., 1998; Molho and Factor, 1999b; Granger and Hanger, 1999), but does so less readily than risperidone and conventional antipsychotics. Drug-induced parkinsonism, including the rabbit syndrome, is seen with olanzapine (Durst et al., 2000), but less than with haloperidol (Peuskens et al., 2009). It can induce acute akathisia (Kurzthaler et al., 1997; Jauss et al., 1998) and TD but less so than haloperidol (Tollefson et al., 1997; Wood 1998). It can also induce NMS (Filice et al., 1998; Moltz and Coeytaux, 1998; Burkhard et al., 1999; Levenson, 1999; Margolese and Chouinard, 1999; Sierra-Biddle et al., 2000; Abu-Kishk et al., 2004; Zaragoza Fernandez et al., 2006), tardive dyskinesia (Herran and Vazquez-Barquero, 1999), and tardive dystonia (Dunayevich and Strakowski, 1999). Acute dystonic reactions also occur (Beasley et al., 1997; Landry and Cournoyer, 1998; Vena et al., 2006).
A direct comparison with chlorpromazine showed similar parkinsonism and acute akathisia for the two drugs (Conley et al., 1998). A double-blind comparison with haloperidol by Beasley and colleagues (1999) showed a much lower risk of developing tardive dyskinesia with olanzapine. After 1 year of exposure, 0.52% of patients developed TD with olanzapine and 7.45% developed TD with haloperidol. An open-label comparison with conventional antipsychotics after a 9-month follow-up after discharge from the hospital favored olanzapine, with TD being present in 2.3% for olanzapine (2/87), and 16.7% (12/72) for the conventional treatment (Mari et al., 2004). There are case reports of agranulocytosis induced by olanzapine (Meissner et al., 1999; Naumann et al., 1999; Tolosa-Vilella et al. 2002), including in a patient who had previously had agranulocytosis with clozapine (Benedetti et al., 1999). This has been attributed to some similar structural and pharmacologic properties of clozapine. A case of restless legs syndrome with periodic movements in sleep has been attributed to olanzapine (Kraus et al., 1999).
Olanzapine has relative regional mesolimbic dopaminergic selectivity and a broad-based binding affinity for serotonin (all 5-HT2 receptor subtypes and the 5-HT6 receptor), dopamine (D2, D3, and D4 receptors), muscarinic, and α1-adrenergic receptors (Bymaster et al., 1999). A PET study in schizophrenic patients being treated with olanzapine revealed that this drug is a potent 5-HT2 blocker, but also a blocker of D2 dopamine receptors similar to risperidone and less so than clozapine (Kapur et al., 1998). Patients on olanzapine were also studied with [123I]iodobenzamide (IBZM) SPECT; the D2 receptor was occupied 60% and 83% of the time at doses of 5 mg/day and 10 mg/day, respectively (Raedler et al., 1999). Such high rates of occupancy probably account for olanzapine’s tendency to worsen parkinsonism and induce DRBA complications, because, as was noted above, D2 receptor occupancy rates are directly related to neuroleptic potential.
Risperidone
With the success of clozapine, there is a commercial advantage for pharmaceutical companies to tout other antipsychotics as “atypical.” Such has been claimed for risperidone, but this drug can readily induce parkinsonism including the rabbit syndrome (Levin and Heresco-Levy, 1999), tardive syndromes (Haberfellner 1997; Silberbauer 1998; Hong et al., 1999; Ananth et al., 2000), and NMS (Newman et al., 1997; Norris et al., 2006). The prevalence of parkinsonism from risperidone is usually considered to be less than that with conventional neuroleptics, but it was observed in 42% compared to 29% in those on haloperidol (Knable et al., 1997). Tardive dyskinesia and tardive dystonia occurred in a patient who was exposed only to risperidone (Bassitt and Garcia, 2000). The annual incidence of TD in patients taking risperidone has been estimated to be 0.3%, compared to an annual incidence in patients taking conventional neuroleptics of 5–10% (Gutierrez-Esteinou and Grebb, 1997). However, in an open-label prospective study of 255 institutionalized patients with dementia who were treated with risperidone, the 1-year cumulative incidence of TD was 2.6% (Jeste et al., 2000).
Like conventional neuroleptics, risperidone induces acute dystonic reactions in marmosets, in contrast to clozapine, which does not (Fukuoka et al., 1997). In a report of an open-label comparison with haloperidol, Rosebush and Mazurek (1999) found the two drugs to have a similar side effect profile. In a prospective follow-up of first-episode schizophrenic patients treated with risperidone, movement disorders developed in more than one-third of these patients, who had previously never been exposed to antipsychotic drugs (Lang et al., 2001). When risperidone is compared with low-potency antipsychotics, such as thioridazine, no difference was discerned in the rates of developing movement disorders (Schillevoort et al., 2001).
In a SPECT study dopamine receptor binding with IBZM showed risperidone to have effects between those of haloperidol and clozapine, with a dose–response curve for risperidone showing greater similarity to that of haloperidol (Dresel et al., 1998). A PET study showed dopamine D2 receptors occupancies of about 70% and 60% in the striatum and extrastriatum, respectively (Ito et al., 2009). Clearly, risperidone is not an “atypical” antipsychotic agent. Risperidone’s occupancy of the 5-HT2 receptors is about 90%, and its occupancy of the D2 receptors is between 50% and 80%, but the latter correlates with the extrapyramidal side effects (Yamada et al., 2002).
Ziprasidone
This benzisothiazole was approved by the Food and Drug Administration in 2001. There are already case reports of acute dystonic reactions (Weinstein et al., 2006; Yumru et al., 2006, Rosenfield et al., 2007), NMS, rhabdomyolysis, and pancreatitis (Murty et al., 2002; Yang and McNeely, 2002; Gray 2004). It has been reported to cause tardive dyskinesia (Mendhekar, 2005) and tardive dystonia (Papapetropoulos et al., 2005; Tsai et al., 2008). Although it is a potent 5-HT2A antagonist (like risperidone, olanzapine, and clozapine), it is also a D2 antagonist in humans as detected by PET scan (Bench et al., 1993, 1996). But in-vitro studies reveal much lower affinity for the D2 receptor than for the 5-HT2A receptor (Seeger et al., 1995), and ziprasidone also binds less tightly to the D2 receptor than dopamine (Seeman, 2002). Ziprasidone has two other unique features compared to other antipsychotic agents: (1) it is a potent 5-HT1A agonist and thus inhibits dorsal raphe serotonergic cell firing (Sprouse et al., 1999) and increases cortical dopamine release (Rollema et al., 2000), and (2) it inhibits neuronal uptake of 5-HT and norepinephrine in a manner comparable to the antidepressant imipramine (Schmidt et al., 2001). What these unique actions might contribute to antipsychotic activity or to propensity for or against extrapyramidal reactions is unclear. Ziprasidone has been reported to induce a case of oculogyric crisis in an adult (Viana Bde et al., 2009).
Aripiprazole
This quinolinone has a novel mechanism of action. Like a number of other “atypical” antipsychotics, it is an antagonist at the 5-HT2A receptors. It is also a 5-HT1A partial agonist (Jordan et al., 2002). What is novel is that it is a partial agonist at the dopamine D2 receptor. It has a higher affinity for the presynaptic autoreceptor than for the postsynaptic receptor. Hence, it reduces dopamine synthesis and release through an agonist action at the dopamine autoreceptor (Tamminga and Carlsson, 2002). Because of its novel action as a partial D2 agonist, it was anticipated that it might cause fewer extrapyramidal adverse effects, and clinical trials reported favorable results (Kane et al., 2002). However, D2 ligand binding PET revealed a dose-related high occupancy state of 71.6% at 2 mg/day to 96.8% at 40 mg/day (Kegeles et al., 2008). With wider use, it has been shown to worsen PD (Friedman et al., 2006; Wickremaratchi et al., 2006), and there are already reports of it causing parkinsonism (López-Torres et al., 2008), NMS (Chakraborty and Johnston, 2004; Hammerman et al., 2006; Palakurthi et al., 2007), acute dystonic reactions (Desarkar et al., 2006; Fountoulakis et al., 2006), and tardive dyskinesia (Zaidi and Faruqui, 2008; Abbasian and Power, 2009; Friedman, 2010).
Amisulpride
Amisulpride, a substituted benzamide, is a highly selective antagonist for dopamine D2 and D3 receptors in the limbic region, which would predict potent antipsychotic activity with a low potential to cause extrapyramidal symptoms (Lecrubier, 2000). It binds less tightly to the D2 receptor than do the typical antipsychotics (Seeman, 2002). Using the full range of recommended amisulpride dosage, striatal occupancies up to 90% can be measured (Vernaleken et al., 2004). It is not yet commercially available in the United States. It has already been reported to induce tardive oculogyric crisis (Mendhekar et al., 2010).
Adverse effects from second-generation antipsychotics
Aside from the D2-blocking effects described above, the five drugs in Table 19.7 have a number of other adverse effects. Sedation is a particular problem seen with each of them, but particularly clozapine and quetiapine. Metabolic side effects are now widely recognized with the second-generation antipsychotics, with weight gain a common problem (Leucht et al., 2009; Patel et al., 2009). Table 19.7 lists the common adverse effects of these drugs, along with haloperidol, for comparison.
It is not likely that drug-induced tardive syndromes will disappear because use of both typical and atypical agents continues. In a survey in Lombardy (Italy), 35 363 individuals over the age of 65 were prescribed an antipsychotic prescription (2.18 subjects per 100 inhabitants, with two-thirds receiving first-generation agents) (Percudani et al., 2004). Although there may be a lower risk of developing these disorders with “atypicals,” these drugs can induce them (Table 19.8). It is even possible that if physicians consider “atypicals” safe to use, their use will increase, even in situations in which the risk from typicals might have precluded their use. In the United States, the prevalence of atypical antipsychotic use was found to be 267.1 per 100 000 subjects aged 19 years and younger and was more than twice as high for male patients as for female patients (Curtis et al., 2005).
In a large US randomized controlled trial (the CATIE study) comparing second-generation antipsychotics (olanzapine, quetiapine, risperidone, perphenazine, or ziprasidone) with the first-generation perphenazine, only olanzapine had a slightly more favorable outcome of both the patients’ ability to stay on this drug for a longer duration and with slightly less drug-induced parkinsonism, akathisia, and tardive dyskinesia than all other drugs including perphenazine (Table 19.9); perphenazine fared as well as the other so-called ‘atypicals” (Lieberman et al., 2005). A similar outcome was seen in a smaller British study (CUtLASS) (Jones et al., 2006). This was a randomized controlled clinical trial involving 57 centers, with flexible dosing up to 4 capsules/day in 1460 schizophrenic subjects (after excluding 33 from one center, integrity concern). The primary outcome was time to discontinuation, with a maximum observation of 18 months. TD was present in 231 subjects at baseline, and these subjects were excluded from the perphenazine arm. The outcome had multiple comparison, requiring a P value ≤0.017 to be significant.
In a more detailed assessment of extrapyramidal side effects in the CATIE study, there were no significant differences in incidence or change in rating scales for parkinsonism, dystonia, akathisia, or tardive dyskinesia when comparing the second-generation antipsychotics with perphenazine or comparing between second-generation antipsychotics (Miller et al., 2008). Secondary analyses revealed greater rates of concomitant antiparkinsonism medication among individuals on risperidone and lower rates among individuals on quetiapine.
Metoclopramide
After the introduction of metoclopramide for gastrointestinal disorders, reports of acute dystonic reactions in children and tardive dyskinesia in adults began to appear (Cochlin, 1974; Kataria et al., 1978; Lavy et al., 1978). Today, metoclopramide has become a common cause of TD, and the legal profession has found such cases ripe for lawsuits. The annual incidence of TD from metoclopramide has been reported to be between 4.5% and 23% per year (Grimes, 1981; Ganzini et al., 1993), about the same as that for haloperidol ~5.7%/year (Tollefson et al., 1997; Beasley et al., 1999; Csernansky et al., 2002), but higher than those for risperidone ~1.8%/year (Lemmens et al., 1999; Davidson et al., 2000; Jeste et al., 2000; Csernansky et al., 2002) and olanzapine ~1.2%/year (Tollefson et al., 1997; Beasley et al., 1999). Because antipsychotic drugs have an affinity for brain neuromelanin (Seeman, 1988), Chen and colleagues (2010) examined the amount of metoclopramide and antipsychotic drugs binding to postmortem human substantia nigra. They found that in clinical conditions the amount of metoclopramide that would bind to nigra is much higher than the amount of raclopride, haloperidol, or olanzapine that would be expected to be bound and they suggest that this binding might explain the higher annual incidence of TD induced by metoclopramide.
Neurologic side effects of dopamine D2 receptor antagonists
To better understand the tardive syndromes, it is important to recognize the variety of other movement disorders that are induced by the dopamine receptor antagonists at different points in the course of treatment. These movement disorders are often lumped together as extrapyramidal syndromes, but the lumping often hinders the effort to sort out the clinical characteristics and pathophysiology of separate syndromes. It is better to subdivide them into their phenomenologic types (see Table 19.1). Movements that may be seen include acute dystonia, acute akathisia, parkinsonism, tardive syndromes, and NMS. Both dystonia and akathisia also occur as subtypes of tardive syndromes and both subtypes are discussed in more detail in the section on tardive syndromes.
Acute dystonia
The earliest abnormal involuntary movement to appear after initiation of dopamine receptor antagonist therapy is an acute dystonic reaction. In about half of the cases, this reaction occurs within 48 hours, and in 90% of cases, it occurs by 5 days after starting the therapy (Ayd, 1961; Garver et al., 1976). The reaction may occur after the first dose (Marsden et al., 1975).
Dystonic movements are sustained muscle contractions, frequently causing twisting and repetitive movements, or abnormal postures (Fahn, 1988). In a series of 3775 patients, Ayd (1961) found that acute dystonia is the least frequent side effect, affecting about 2–3% of patients, with young males being most susceptible. The incidence rate increases to beyond 50% with highly potent dopamine receptor blockers such as haloperidol (Boyer et al., 1987). In one prospective study, Aguilar and colleagues (1994) reported that 23 of 62 patients developed acute dystonia after haloperidol was introduced, that anticholinergic pretreatment significantly prevented this, and that younger age and severity of psychosis were risk factors. In the prospective study by Kondo and colleagues (1999), 20 (51.3%) of 39 patients placed on nemonapride had dystonic reactions, onset occurring within 3 days after the initiation of treatment in 90%. As in other series, the incidence of acute dystonia was significantly higher in males than in females (77.8% vs. 28.6%, P < 0.05), and younger males (≤30 years) had an extremely high incidence (91.7%). The incidence is much lower with the so-called atypical antipsychotics; Raja and Azzoni (2001) observed only 41 cases out of 1337 newly admitted patients treated with antipsychotics, which included 8 treated with risperidone, 1 with olanzapine, and 1 with quetiapine. A meta-analysis compared intramuscular injections of second-generation antipsychotics (SGAs) to haloperidol injections (Satterthwaite et al., 2008). SGAs were associated with a significantly lower risk of acute dystonia (relative risk = 0.19) and acute akathisia (relative risk = 0.25), compared with haloperidol alone.
All agents that block D2 receptors can induce acute dystonic reactions, including risperidone (Brody, 1996; Simpson and Lindenmayer, 1997) and clozapine (Kastrup et al., 1994). One child developed an acute dystonic reaction after ingestion of a dextromethorphan-containing cough syrup (Graudins and Fern, 1996). Dextromethorphan has several different known pharmacologic actions, but D2 receptor blockade is not one of them. A case of acute dystonia following abrupt withdrawal of bupropion (DA reuptake blocker) was reported (Wang et al., 2007). A case of acute dystonia following abrupt withdrawal of dexamphetamine in a patient taking risperidone was also reported (Keshen and Carandang, 2007). Serotonergic agents have also been reported to induce acute dystonic reactions (Lopez-Alemany et al., 1997; Madhusoodanan and Brenner, 1997; Olivera, 1997). The mechanism could relate to inadequate release of dopamine from the nerve terminals in the striatum owing to the inhibitory effect of serotonin on dopamine neurons in the substantia nigra pars compacta. The opioid σ1 and σ2 receptors have also been implicated (Matsumoto and Pouw, 2000).
Acute dystonic reactions most often affect the ocular muscles (oculogyric crisis), face, jaw, tongue, neck, and trunk, and less often the limbs. Oculogyric crisis has previously been noted to occur as a common feature of postencephalitic parkinsonism (Duvoisin and Yahr, 1965). A typical acute dystonic reaction may consist of head tilt backward or sideways with tongue protrusion and forced opening of the mouth, often with arching of trunk and ocular deviation upward or laterally (Rupniak et al., 1986). The forcefulness of the muscle contractions can be extremely severe and led to auto-amputation of the tongue in one patient (Pantanowitz and Berk, 1999).
Mazurek and Rosebush (1996) studied the timing in the development of an acute dystonic reaction in 200 patients who were taking a neuroleptic medication for the first time. The neuroleptic was given twice daily, and over 80% of the episodes of acute dystonia occurred between 12 noon and 11 p.m.
Reserpine and α-methylparatyrosine, which deplete presynaptic monoamines, have not been associated with acute dystonic reactions (Duvoisin, 1972; Marsden et al., 1975; Walinder et al., 1976). However, another dopamine depletor, tetrabenazine (TBZ), has been reported to induce acute dystonic reactions (Burke et al., 1985). One possible explanation for this difference is that in addition to depleting dopamine, TBZ blocks dopamine receptors (Reches et al., 1983).
It is important to mention the case described by Wolf (1973) of a patient who developed an oral dyskinesia while taking reserpine. Figure 1 in his paper clearly shows a dystonic phenomenon and not the complex, rapid, stereotypic movements of classic tardive dyskinesia. This development appeared too late after initiation of reserpine to be considered an acute dystonic reaction, however. Whether this could be an example of tardive dystonia is not clear, since the phenomenology of tardive dystonia is identical to that of naturally occurring primary dystonia; therefore, the patient could have had a coincidental case of spontaneous, idiopathic oromandibular dystonia (Fahn, 1984b). Thus, there is no absolute evidence that reserpine induces acute dystonic reactions, classic tardive dyskinesia, or tardive dystonia. In fact, symptoms of these three tardive syndromes can be suppressed by reserpine, and eventually reserpine can be withdrawn successfully in many patients without exacerbation of the symptoms (Fahn, 1983). Furthermore, the long time that reserpine has been available without a clear-cut case of tardive syndromes can be compared to the much shorter duration of use of metoclopramide, in which there are already cases of acute dystonic reactions, classic tardive dyskinesia, and tardive dystonia as a consequence of its use (Casteels-Van Daele et al., 1970; Gatrad, 1976; Pinder et al., 1976; Reasbeck and Hossenbocus, 1979; Miller and Jankovic, 1989; Lang, 1990).
Of 452 patients given high-dosage oral metoclopramide to control emesis, Kris and colleagues (1983) observed 14 who developed acute dystonic reactions. However, there was a distinct preponderance of the reactions occurring in children (6/22) compared to adults (8/430). Intravenous metoclopramide is more likely to induce it than oral administration (Pinder et al., 1976). In a study at a Veterans Administration hospital, comparing patients treated with metoclopramide and controls, the relative risk for TD was 1.67, and the relative risk for drug-induced parkinsonism was 4.0 (Ganzini et al., 1993).
The available biochemical explanations for the acute dystonic reaction are unsatisfactory, but several observations contribute to the understanding of the phenomena, relating it to dopamine, muscarinic, and sigma receptors. Acute neuroleptic administration produces sudden increase of dopamine release and increased turnover lasting for 24–48 hours after a single dose (O’Keefe et al., 1970; Marsden and Jenner, 1980). This effect is blocked by anticholinergics consistent with their efficacy in treatment of acute dystonic reactions (O’Keefe et al., 1970). Moreover, in baboons that were pretreated with reserpine and α-methylparatyrosine, which markedly reduces presynaptic dopamine concentration, haloperidol-induced acute dystonic reaction is abolished or greatly reduced (Meldrum et al., 1977). On the other hand, blockade of the postsynaptic receptor fades in about 12 hours after a single dose of antipsychotics, and supersensitivity of receptors begins to develop. Therefore, presynaptic dopaminergic excess in combination with the emerging supersensitive postsynaptic dopamine receptors could result in markedly increased striatal dopaminergic activity at about 20–40 hours after a neuroleptic dose. This period corresponds to the critical time for acute dystonic reactions in human subjects who were given a single dose of butaperazine (Garver et al., 1976). However, extrapolating the data from experimental animals to the clinical situation has its limitations including the fact that rats do not develop acute dystonic reactions. Further data on human cerebrospinal fluid (CSF) dopamine metabolites as an indicator of presynaptic function might prove to be of value in pursuing the hypothesis. Jeanjean and colleagues (1997) suggested that the σ2 receptors could be involved in the acute dystonic reaction. They found a correlation between the clinical incidence of neuroleptic-induced acute dystonia and binding affinity of drugs for the sigma receptor.
Another animal model of acute dystonia is the common marmoset that is treated with haloperidol (Fukuoka et al., 1997). But it takes at least 6 weeks of such treatment to develop this reaction. The dystonia subsides only to reappear when haloperidol treatment is restarted; other neuroleptics, including risperidone, can also make the dystonia reappear, but clozapine was without such an effect. In this animal model, the anticholinergic agent trihexyphenidyl inhibited the induction of acute dystonia.
[/not-level-membership-for-neurology-category]
