Chapter 5 Current concepts on the etiology and pathogenesis of Parkinson disease
Anatomical and biochemical pathology of PD
Historical introduction
Parkinson disease (PD) was first described in 1817 with the publication by James Parkinson of a book entitled An Essay on the Shaking Palsy (Parkinson, 1817). In it, he described six individuals with the clinical features that have come to be recognized as a disease entity. One of the people was followed in detail over a long period of time; the other five consisted of brief descriptions, including two of whom he had met walking in the street, and another whom he had observed at a distance. Such distant observations without a medical examination demonstrate how easily distinguishable the condition is. The physical appearance of flexed posture, resting tremor, and shuffling gait are readily recognizable. Parkinson’s opening description has his key essentials: “Involuntary tremulous motion, with lessened muscular power, in parts not in action and even when supported; with a propensity to bend the trunk forward, and to pass from a walking to a running pace: the senses and intellects being uninjured.” In the small monograph, Parkinson provided a detailed description of the symptoms and also discussed the progressive worsening of the disorder, which he called “the shaking palsy” and also its Latin term “paralysis agitans.”
After the publication of Parkinson’s Essay, the disease was widely accepted in the medical community. It took 70 years for the name of the disorder to be referred to as “Parkinson’s disease,” as recommended by the French neurologist Charcot (1879) who argued against the term “paralysis agitans” (see Goetz, 1987, for English translation) and recommended the disorder be named after James Parkinson. Charcot argued that there is no true paralysis, but rather the “lessened muscular power” is what is today called akinesia, hypokinesia, or bradykinesia; all three terms often being used interchangeably by clinicians, although these three terms specifically refer to lack of movement, small movement, and slow movement, respectively. These terms represent a paucity of movement not due to weakness or paralysis. Similarly, Charcot emphasized that tremor need not be present in the disorder, so “agitans” and “shaking” are not appropriate as part of the name of the disorder.
The clinical features of PD and its differential diagnosis are presented in Chapter 4.
Incidence, prevalence, and mortality
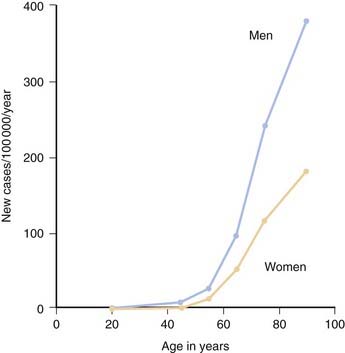
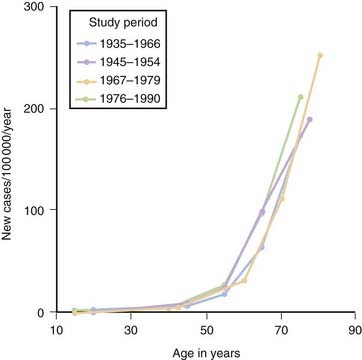
Although PD can develop at any age, it begins most commonly in older adults, with a peak age at onset around 60 years. The likelihood of developing PD increases with age, with a lifetime risk of about 2% (Elbaz et al., 2002). A positive family history doubles the risk of developing PD to about 4%. A summation from seven population-based studies in various European countries found the overall prevalence of PD in people aged over 65 to be 1.8%, with an increase from 0.6% for persons aged 65–69 years to 2.6% for people aged 85–89 years (de Rijk et al., 2000). Twin studies indicate that PD with an onset under the age of 50 years is more likely to have a genetic relationship than for patients with an older age at onset (Tanner et al., 1999). Males have higher prevalence (male-to-female ratio of 3:2) and incidence rates than females (Fig. 5.1) (Bower et al., 2000), but the age-specific incidence rates have not varied over the past 70 years (Fig. 5.2) (Rocca et al., 2001). The incidence rates vary among studies, but average between 11.0 and 13.9/100 000 population per year (Van Den Eeden et al., 2003). In a northern California study, the incidence rates varied among ethnic groups, being highest in Hispanics, then non-Hispanic whites, then Asians, and lowest in African-Americans (Van Den Eeden et al., 2003).
The prevalence rates of PD have varied in different studies and in different countries. The figure of 187 per 100 000 population given by Kurland (1958) is a reasonable estimate in the US. But if the population is restricted to adults older than 39 years of age, the prevalence rate is 347 per 100 000 (Schoenberg et al., 1985) since both prevalence and incidence rates increase with age. At age 70, the prevalence is approximately 550 per 100 000, and the incidence is 120 per 100 000/year. At the present time, approximately 850 000 individuals in the US have PD, although one recent estimate is less than this (Hirtz et al., 2007). The number is expected to grow as the population ages (Dorsey et al., 2007). Advancing age is the single greatest risk factor for developing sporadic PD. Approximately 2% of the population will have PD by the time they reach the age of 80 years.
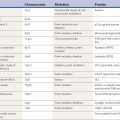
In the pre-levodopa era, mortality was reported to be three-fold greater in patients with PD (Hoehn and Yahr, 1967). The mortality rate was reduced to 1.6-fold greater than age-matched non-PD individuals after the introduction of levodopa (Yahr, 1976; Elbaz et al., 2003). Today, patients with PD can live 20 or more years, depending on the age at onset (Kempster et al., 2007). Death in PD is usually due to some concurrent unrelated illness or due to the effects of decreased mobility, aspiration, or increased falling with subsequent physical injury. The Parkinson-plus syndromes typically progress at a faster rate and often cause death within 9 years. Thus, the diagnosis of PD versus other forms of parkinsonism is of prognostic importance, as well as of therapeutic significance because it almost always responds to at least a moderate degree with levodopa therapy, whereas the Parkinson-plus disorders usually do not.
Anatomical pathology of PD
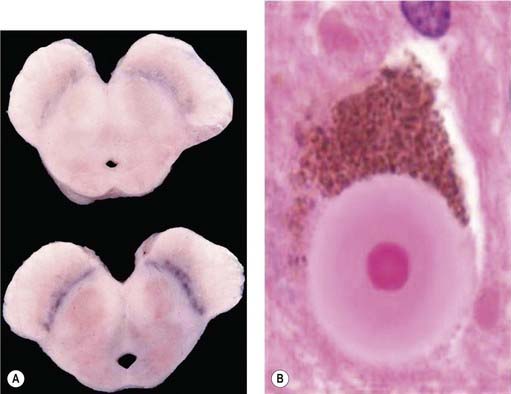
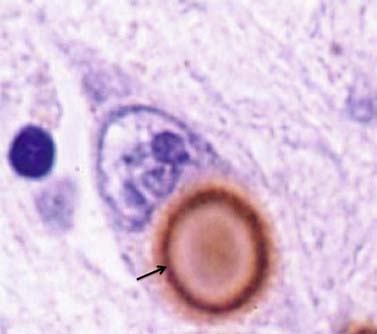
It was many years after Parkinson’s original description before the basal ganglia were recognized by Meynert in 1871 as being involved in disorders of abnormal movements. And it was not until 1895 that the substantia nigra (SN) was suggested to be affected in PD. Brissaud (1895) suggested this on the basis of a report by Blocq and Marinesco (1893) of a tuberculoma in that site that was associated with a contralateral hemiparkinsonian tremor. These authors were careful to point out that the pyramidal tract and the brachium conjunctivum above and below the level of the lesion contained no degenerating fibers. The importance of the SN was emphasized by Tretiakoff in 1919. He studied the SN in nine cases of PD, one case of hemiparkinsonism, and three cases of postencephalitic parkinsonism, finding neuronal loss in this nucleus in all cases. With the hemiparkinsonian case, Tretiakoff found a lesion in the nigra on the opposite side, concluding that the nucleus served the motor activity on the contralateral side of the body. The SN, so named because of its normal content of neuromelanin pigment, was noted to show depigmentation, loss of nerve cells, and gliosis. These findings remain the principal and essential histopathologic features of the disease. In his study, Tretiakoff also found Lewy bodies in the SN, expanding the earlier observation of Lewy (1912, 1914), who had discovered the presence of these cytoplasmic inclusions in the substantia innominata and the dorsal vagus nucleus in PD. Lewy bodies (Fig. 5.3) are now widely recognized as the major pathologic hallmark of the disorder. Lewy bodies have since been seen in autonomic ganglia, the peripheral nervous system and in other regions of the central nervous system, including the cerebral cortex (Jellinger, 2009a).
Foix and Nicolesco (1925) made a detailed study of the pathology of PD in 1925 and found that the most constant and severe lesions are in the substantia nigra. Since then many workers, including Hassler (1938) and Greenfield and Bosanquet (1953), have confirmed these findings and added other observations, including involvement of other brainstem nuclei such as the locus coeruleus and the raphe nuclei. The pigmented cells of the locus coeruleus contain neuromelanin; these cells are also lost in PD, with many of those remaining containing Lewy bodies. The asymmetry of clinical signs in PD is reflected by the asymmetrical and more severe contralateral loss of substantia nigra pars compacta (SNc) neurons. Neuronal loss extends beyond loss in SNc, locus coeruleus, and raphe, with loss of neurons in the dorsal motor vagal nucleus, hypothalamus, the nucleus basalis of Meynert, and sympathetic ganglia (Forno, 1982; Jellinger, 1987). There are also losses of glutamatergic projection neurons from thalamus to the basal ganglia (Henderson et al., 2000) and glutamatergic projection neurons from the presupplementary motor cortex to the premotor cortices (Halliday et al., 2005).
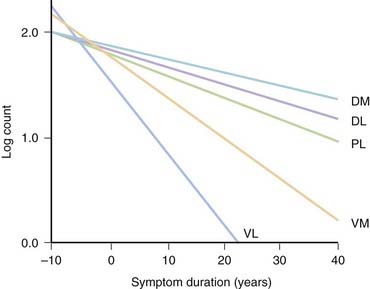
PD and the Parkinson-plus syndromes have in common a degeneration of SNc dopaminergic neurons, with a resulting deficiency of striatal dopamine due to loss of these nigrostriatal neurons. Accompanying this neuronal loss is an increase in glial cells in the nigra and a loss of the neuromelanin normally contained in the dopaminergic neurons. There is a reduction of nigral neurons and striatal dopamine with aging also (Carlsson and Winblad, 1976; McGeer et al., 1977; Fearnley and Lees, 1991) (Fig. 5.4), and although PD is associated with increasing age, the greater rate (Fig. 5.5) and the pattern of cell loss in the SN differs between that in aging and that in PD (Figs 5.6, 5.7) (Fearnley and Lees, 1991). Clinical features begin to emerge when approximately 80% of striatal dopamine content (or 60% of nigral dopaminergic neurons) are lost (Bernheimer et al., 1973). The course of clinical decline is associated with the progressive reduction of striatal dopamine (Riederer and Wuketich, 1976).
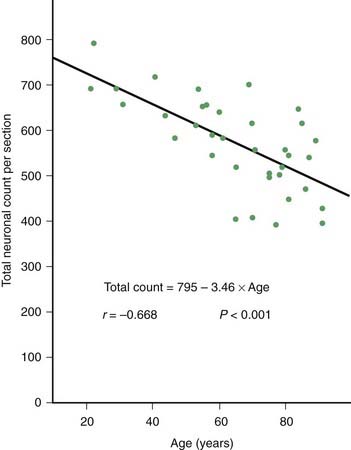
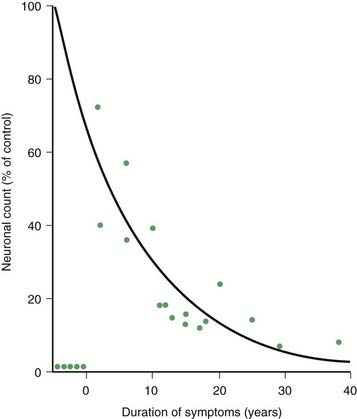
Figure 5.5 Total pigmented nigral neurons with disease duration.
From Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 1991;114:2283–2301.
Pathologically, almost all patients with PD have Lewy bodies in the SN and locus coeruleus. Juvenile PD (Dwork et al., 1993; Hattori et al., 2000), now recognized as being due mainly to homozygous parkin mutations, is a major exception. Another exception is with PD due to the R1441G mutation in the LRRK2 gene (Martí-Massó et al., 2009).The pathology has not yet been described in juvenile PD patients due to homozygous mutations in DJ-1 genes, but in one case of young-onset PD with PINK1 mutation, the autopsy showed the presence of Lewy bodies (Samaranch et al., 2010). There are no Lewy bodies in the Parkinson-plus syndromes or in postencephalitic parkinsonism (Jellinger, 2009b).
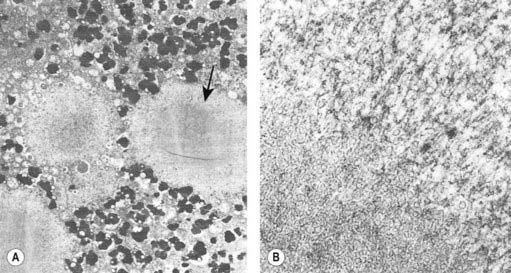
The presence of Lewy bodies in the SNc and the locus coeruleus plus the clinical features of PD (without the characteristic clinical features of some other form of parkinsonism) are usually used to make the pathologic diagnosis of PD, but there is no complete agreement among neuropathologists about the pathologic criteria for the diagnosis of PD (Forno, 1982, 1996). Some patients with clinical PD die with nigral degeneration without Lewy bodies (Rajput et al., 1991; Hughes et al., 1992, 2002). In fact, as mentioned above, patients with juvenile PD usually do not have Lewy bodies, especially those with homozygous parkin mutations (Dwork et al., 1993; Hattori et al., 2000). Thus, Lewy bodies confirm the diagnosis and are a critical pathologic marker to confirm the diagnosis, but they are not necessary and their presence is not pathognomonic for PD (Forno 1982). They are found in 4–6% of routine autopsies (Forno, 1982), the incidence rate increases with age as in PD (Fearnley and Lees, 1991) (Table 5.1), and people dying with such incidental Lewy bodies are considered to have a presymptomatic state of PD. The site of regional nigral neuronal loss in incidental Lewy body disease is the lateral region in the ventral tier, the same as for PD (Fig. 5.6) (Fearnley and Lees, 1991). These brains show a reduction in striatal dopaminergic markers (e.g., tyrosine hydroxylase and vesicular monoamine transporter 2), but not as severe as those with clinical PD (DelleDonne et al., 2008; Dickson et al., 2008). Cortical Lewy bodies in patients with dementia and no parkinsonism could be a separate disease or a variant in the presentation of the same disorder that causes PD. Lewy bodies consist of a dense inner core surrounded by a radiating filamentous outer zone (Fig. 5.8) (Duffy and Tennyson, 1965; Forno and Norville, 1976).
| Age | No. | Percent |
|---|---|---|
| <20 | 0/2 | |
| 20–29 | 0/6 | |
| 30–39 | 0/6 | |
| 40–49 | 1/7 | 3.7 |
| 50–59 | 0/20 | |
| 60–69 | 2/51 | 3.9 |
| 70–79 | 3/56 | 5.4 |
| 80–89 | 6/49 | 12.2 |
| 90–99 | 1/8 | 12.5 |
Data from Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 1991;114:2283–2301.
Biochemical pathology of PD
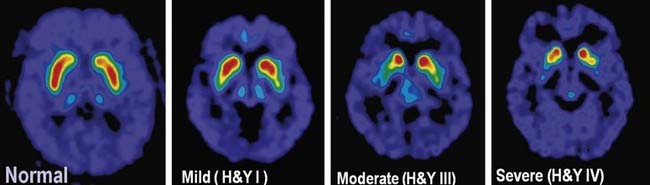
The pigmented neurons of both the SNc and the ventral tegmental area (medial to the SN in the midbrain) contain dopamine. The former neurons project to the neostriatum, the latter to the limbic system and the neocortex. In PD, the mesolimbic and mesocortical neurons are relatively spared, whereas the nigrostriatal neurons are progressively lost. As a result, there is a corresponding decrease of dopamine content in both the nigra and the striatum, with the innervation of the posterior putamen affected first and most severely, as can be detected in FDOPA PET scans (Fig. 5.9). Whereas dopamine is reduced initially in the posterior striatum in PD, over time, as the disease progresses, all striatal subregions are affected to a similar degree (Nandhagopal et al., 2009).
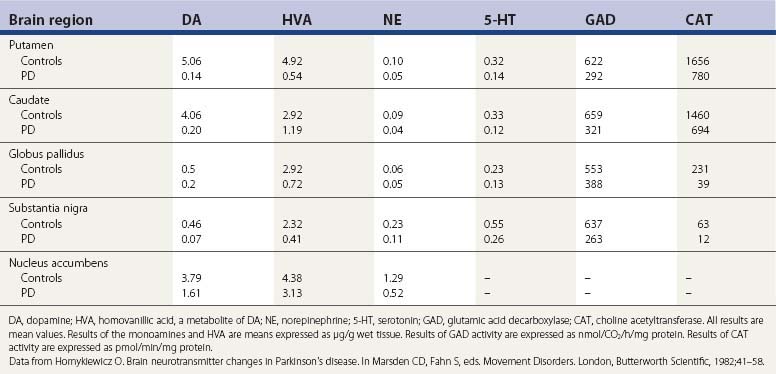
The pigmented neurons of the locus coeruleus contain norepinephrine, and these neurons project widely in the CNS. A third set of monoaminergic neurons are those containing serotonin (5-HT), located in the raphe of the pons and medulla. In PD there is a progressive loss of all three types of monoaminergic cells, particularly the dopaminergic cells. So, in addition to a depletion of striatal dopamine, there is also a reduction in brain norepinephrine and 5-HT (Ehringer and Hornykiewicz, 1960; Hornykiewicz, 1966) (Table 5.2). There is also a reduction in other neurotransmitters (Agid et al., 1987) as well as enzyme activities for the synthesis of other neurotransmitters (Table 5.2), indicating that the biochemical changes in PD extend beyond the loss of only the monoamines.
Table 5.2 Concentration of neurotransmitters, their metabolites, and synthesizing enzymes in selected regions of brain of controls and PD
In addition to the motor features of PD, the symptoms that define the clinical diagnosis, there are also a host of nonmotor symptoms, some occurring before the motor symptoms and some occurring after (see Chapter 8). The early motor symptoms of bradykinesia and rigidity and tremor are associated with monoaminergic cell and chemical loss. The later motor symptoms of flexed posture, loss of postural reflexes, and the freezing phenomenon appear to correlate poorly with dopaminergic deficit. The nonmotor symptoms probably are the result of loss of neuronal function other than dopamine. Catecholamine reduction in PD is seen in the autonomic nervous system and accounts for the reduction in MIBG SPECT scan labeling in the heart in PD due to loss of postganglionic myocardial sympathetic nerve fibers (Rascol and Schelosky, 2009).
Among the neurotransmitter changes is the reduction of brain acetylcholine (Bohnen and Albin, 2010). Acetylcholinesterase (AChE), which serves as a marker for cholinergic neurons, can be measured by PET scanning (Shimada et al., 2009). A reduction of AChE begins early in PD (Bohnen and Albin, 2009). Reduced thalamic AChE activity correlates with falling in PD (Bohnen et al., 2009), and in part represents decreased cholinergic output of the pedunculopontine nucleus (PPN), which appears to be important for gait. Cortical loss of acetylcholine probably contributes to the dementia seen in PD (Shimada et al., 2009; Bohnen and Albin, 2010). Overall, reduced AChE is more widespread and profound both in PD with dementia and in dementia with Lewy bodies (Shimada et al., 2009).
There are compensatory changes, such as supersensitivity of dopamine receptors, so that symptoms of PD are first encountered only when there is about an 80% reduction of dopamine concentration in the putamen (or a loss of 60% of nigral dopaminergic neurons) (Bernheimer et al., 1973). Another compensatory mechanism is an increase in neurotransmitter turnover, as detected by an increased HVA/DA ratio. In a major review, Hornykiewicz (1966) correlated loss of dopamine concentration in the striatum with severity of bradykinesia and rigidity in PD. With further loss of dopamine concentration, parkinsonian bradykinesia becomes more severe. The progressive loss of the dopaminergic nigrostriatal pathway can be detected during life using PET and SPECT scanning (Fig. 5.9); these show a continuing reduction of FDOPA and dopamine transporter ligand binding in the striatum that correlates with the bradykinesia score in the Unified Parkinson’s Disease Rating Scale (Brooks et al., 1990; Snow et al., 1994; Seibyl et al., 1995; Morrish et al., 1996; Vingerhoets et al., 1997; Broussolle et al., 1999; Benamer et al., 2000). Using special statistical techniques, FDG PET also shows a correlation between worsening bradykinesia and increase of lentiform metabolism (Eidelberg et al., 1995). In fact, using FDG PET demonstrates a metabolic network characteristic of PD compared to other forms of parkinsonism (Hirano et al., 2009; Tang et al., 2010). While postsynaptic dopamine receptors have been thought to be preserved in PD, a recent study suggested a downregulation of the D3 receptors in drug-naive PD patients (Boileau et al., 2009).
Interconnected pathogenic mechanisms of PD
The best correlation of symptoms with progressive loss of striatal dopamine are those of bradykinesia and rigidity, which relate to striatal dopamine deficiency and loss of SNc dopaminergic neurons and can be correlated with a progressive decrease of dopaminergic imaging by PET or SPECT (see above). Since these symptoms are two of the cardinal features of the disease, research efforts have concentrated on the pathogenic mechanisms that cause loss of the nigrostriatal dopamine system, and this will be reflected in this review. Similar mechanisms might involve the other monoaminergic systems (noradrenergic and serotonergic). But there is little knowledge of the pathogenesis of neuronal loss for nonaminergic neurons. It seems likely that loss of these other monoamines might be instrumental in the high rate of depression (Mayeux et al., 1984) and anxiety in patients with PD. Little is known about the anatomical or biochemical associations for the other clinical features of the disease, including motoric features of tremor, freezing, flexed posture and loss of postural reflexes, and the multitude of nonmotor features.
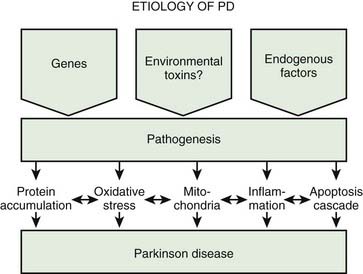
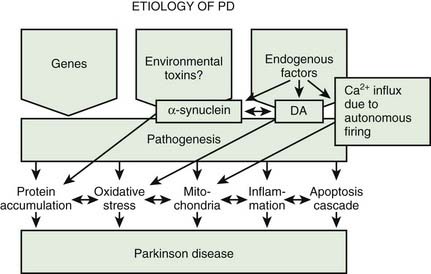
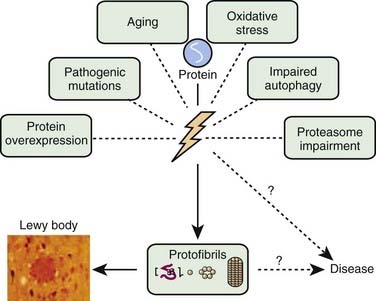
A variety of pathogenic mechanisms have been uncovered for the loss of dopamine neurons, and probably more will be uncovered. The reader is referred to reviews on this topic for details (Dauer and Przedborski, 2003; Jenner and Olanow, 2006; Gupta et al., 2008). With the development of genetic causes of PD, a multiple hit hypothesis was proposed by Sulzer (2007), discussed later in this chapter. Evidence has accumulated over decades from pathologic and biochemical findings that implicate oxidative stress, mitochondrial dysfunction, excitotoxicity, inflammation, and apoptosis as taking place in the SNc (Fig. 5.10). More recently protein aggregation in the form of Lewy bodies and Lewy neurites has placed a major emphasis on the accumulation of toxic protein as perhaps the most important pathogenic factor. Each of these factors cross-interact with the others to add to the pathogenesis of cell death. Toxic proteins accumulate because of insufficient degradation or an excess synthesis that the normal degradation process cannot handle.
Oxidative stress
A key source of oxidative stress in monoaminergic neurons is via monoamine metabolism and auto-oxidation (discussed in more detail below). Since most research in PD neurodegeneration has been applied to dopaminergic neurons, we will focus on dopamine. Antioxidant defenses protect cells, and one of the leading antioxidants is reduced glutathione (GSH), and this is reduced in the SNc of PD patients at postmortem (Perry et al., 1982; Sian et al., 1994). This reduction of GSH is specific to PD brains, and is not seen in atypical parkinsonisms with nigral degeneration. The reduction of GSH likely reflects an excess utilization of this reducing agent, implying a high degree of oxidative stress taking place there. The decrease in GSH in the SNc occurs in incidental Lewy bodies as well as in PD, suggesting that oxidative stress has occurred prior to nerve cell loss. Some evidence suggests that GSH depletion itself may play an active role in PD pathogenesis (Martin and Teismann, 2009). Iron accumulation in the nigra also contributes to oxidative stress (Dexter et al., 1989a). Oxidized products of lipids, DNA, and protein are also seen in the PD nigra (Dexter et al.,1989b; Sanchez-Ramos et al., 1994), providing postmortem evidence of oxidative stress. The formation of neuromelanin in dopamine neurons is derived from the condensation of oxidized dopamine products, and thus represents a protective mechanism by the cell to defend itself against oxidative stress (Sulzer et al., 2000). One of the mutant genes that can cause PD, DJ-1, functions normally to protect against oxidative stress and is discussed below.
It is of interest that an endogenous substance, uric acid, which has antioxidant properties, has been correlated with a reduction in developing PD (Weisskopf et al., 2007). Subsequent studies evaluating urate levels in plasma in the PRECEPT (Schwarzschild et al., 2008) and CSF in the DATATOP (Ascherio et al., 2009) clinical trials found that higher urate levels in men were associated with a slower rate of progression of PD.
Mitochondrial dysfunction
Mitochondria appear to play an important role in the pathogenesis of PD (Banerjee et al., 2009). The finding that 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) intoxication causes parkinsonism (Langston et al., 1983), and the discovery that MPTP selectively destroys dopamine neurons and impairs complex I activity in the mitochondria (Nicklas et al., 1987) led to the study of mitochondria in PD patients. Decreased complex I activity is seen in the SNc of PD brains and not elsewhere, nor in other forms of parkinsonism (Mizuno et al., 1989; Schapira et al., 1990). Another complex I toxin, rotenone, which is a commonly used pesticide, also damages SNc neurons in animals (Betarbet et al., 2000). We will see below that two genes related to PD, namely parkin and PINK1, help maintain the integrity and function of the mitochondria, and loss of function of these genes results in PD. Mitochondrial dysfunction impairs ATP production, which hinders energy-dependent mechanisms of degradation of misfolded proteins by the ubiquitin-proteasomal system. Mitochondrial dysfunction is both a cause and a consequence of oxidative stress. Deregulation of mitochondrial respiration leads to generation of reactive oxygen species, contributing to oxidative stress, while oxidative and nitrosative stress deteriorates mitochondrial function. An early event in MPTP toxicity is oxidative stress, which is the consequence of an inability to transport electrons due to the inhibition of mitochondrial complex I. The accumulating electrons are a source of oxidative stress (Zhou et al., 2008). Besides their role in electron transport and oxidative phosphorylation, mitochondria are a major cellular source of free radicals, affect calcium homeostasis, and instigate cell-death pathways via apoptosis (Henchcliffe and Beal, 2008; Schapira, 2008).
Other mechanisms
Excitotoxicity from excessive glutamatergic activity results in an increase in calcium and can damage mitochondria; this has been implicated in PD (Beal, 1998). Nitrosative stress is induced by nitric oxide forming peroxynitrite, leading to protein nitration, and has also been suggested as a pathogenic factor (Tsang and Chung, 2009). Inflammation is seen in PD SN (McGeer et al., 1988a, 1988b), but is not considered an early event (Zhou et al., 2008). Rather, inflammation appears to augment the continuing degeneration (Tansey et al., 2007; Hirsch and Hunot, 2009; Saijo et al., 2009; Przedborski, 2010). Apoptosis is thought to represent the cellular death mechanism in PD (Mochizuki et al., 1996; Hirsch et al., 1999).
Accumulation of toxic proteins
The accumulation of too much protein in the cell has invoked the concept that these proteins are toxic and can lead to cell death. The concept of accumulated unwanted protein derived from genetic studies with the discovery that the protein α-synuclein is present in Lewy bodies (discussed below). Proteins are commonly damaged by misfolding or some other alteration, so that they are not functioning normally, and the cell attempts to repair them or eliminate them through degradation by either the ubiquitin-proteasomal system or by autophagy via the lysosome. Thus, misfolded, damaged, or altered proteins are either repaired or else removed from the cell, otherwise they would accumulate and act as toxins that would damage the cell. The first step to eliminate such proteins is to try to repair them through chaperone-mediated mechanisms. If this fails, the ubiquitin-proteasomal system attempts to remove the unwanted protein. If this is not capable of removing the protein, then autophagy via the lysosome takes place (Kopito, 2000; Pan et al., 2008). Autophagy appears to play a major role in removing α-synuclein and other unwanted proteins. This mechanism is described later in this chapter.
Genetics as an etiologic factor of PD
Familial PD
Families with PD occurring in several members have been recognized over the years, with approximately 10% of newly diagnosed patients reporting someone else in the family having PD. One of the earliest to describe a family with multiple affected members was Allen (1937). Perhaps the largest pedigree of PD described to date and with many generations affected was that reported by Mjönes (1949) of a Swedish and Swedish-American family. He found an autosomal dominant inheritance pattern in this family with 60% penetrance. But generally, since most PD patients are sporadic without a positive family history, the disease was not thought to have a genetic etiology.
Twin studies
To study the possibility of a genetic cause of PD though, Duvoisin, Eldridge and their colleagues (1981) evaluated twins with PD. They found zero concordance in 12 monozygotic twin pairs and concluded that “genetic factors appear not to play a major role in the etiology of PD.” This group (Ward et al., 1983) continued to analyze twin pairs and now in 43 monozygotic (MZ) and 19 dizygotic (DZ) pairs with the index case having definite PD, the frequency of PD in MZ twins was similar to that expected in an unrelated control group matched for age and sex. The authors again concluded that “the major factors in the etiology of PD are nongenetic.” A Finnish twin study 5 years later (Martilla et al., 1988) also found low concordance in MZ twin pairs and also concluded that PD appears to be “an acquired disease not caused by a hereditary process.” However, Bill Johnson, then at Columbia University, began to question the conclusions of the twin studies (Johnson et al., 1990), saying they were too small to be statistically conclusive and recommended that linkage studies be conducted.
The momentum toward a genetic etiology of PD
In 1990 Golbe and Duvoisin and their colleagues described a large kindred with autosomal dominant PD originating in Contoursi, Italy, with some of the family having emigrated to the US. This led Duvoisin to rethink his previous conclusions that PD is largely nongenetic (Duvoisin and Johnson, 1992). At this time there were also reports that FDOPA PET scans can detect decreased FDOPA uptake in some nonaffected relatives, including twins with an affected co-twin (Brooks, 1991). A large PET study in twins indicated that the concordance for decreased striatal FDOPA uptake in PD twins is greater than previously realized (Burn et al., 1992). With the availability of the Contoursi kindred and with the tools of linkage analysis, the stage was set for finding a gene mutation in familial PD.
Finding the first gene mutation in PD
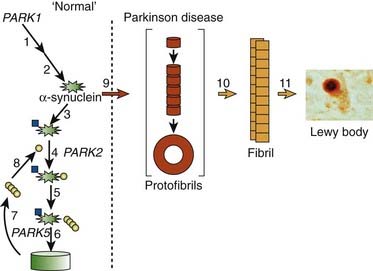
Linkage analysis was carried out on the Contoursi kindred, and after several years of searching, linkage to chromosome 4q21–q23 was found (Polymeropoulos et al., 1996). By the next year, Polymeropoulos and colleagues (1997) identified the mutated gene, SNCA, for the protein α-synuclein. The Contoursi family actually originated in Greece and immigrated to Italy. The mutation (Ala53Thr) was also found in three small, unrelated Greek families. Subsequently, two other mutations were found in SNCA, which also caused autosomal dominant PD, namely Ala30Pro in a German family (Krüger et al., 1998) and Glu46Lys in a Spanish family (Zarranz et al., 2004). Families with SNCA mutations have a younger age at onset (usually in their forties) and a more rapidly worsening course of PD, and also with some early cognitive impairment. So, the gene mutation, on average, causes a more severe form of the disease than the typical adult-onset sporadic case. The SNCA gene was originally labeled PARK1.
Although all these SNCA mutations are rare in causing all worldwide cases of both familial and sporadic PD, the protein α-synuclein has taken on a premier and most important role as being highly likely involved in the disease’s pathogenesis, including the sporadic cases. Immediately after the gene mutation was discovered (Polymeropoulos et al., 1997), Spillantini and colleagues (1997) found that fibrillary α-synuclein is a major component of the Lewy body, a finding confirmed the following year (Mezey et al., 1998). In fact, staining microscopic brain slices with the antibody for α-synuclein has become the pathologist’s tool to detect Lewy bodies, and probably accounts for the recognition that diffuse Lewy body disease (DLBD), also known as dementia with Lewy bodies (DLB), is the second most common cause of dementia after Alzheimer disease. Prior to staining for α-synuclein, ubiquitin antibodies were used because ubiquitin is another protein found within the Lewy body. Ubiquitin is located in the central core and α-synuclein in the surrounding halo (Fig. 5.11). Autopsies on patients with mutated or excess amounts of α-synuclein show an abundance of Lewy bodies (Seidel et al., 2010).
Other gene mutations in PD
After the SNCA gene mutation was found, other investigators began collecting families with PD and conducting linkage analyses on them and then cloning the gene when linkage was obtained. Japanese investigators discovered the second gene to cause PD, which they called parkin and designated as PARK2, with the protein also called parkin (Kitada et al., 1998). The gene mutation was initially reported in a family with autosomal recessive juvenile parkinsonism without Lewy bodies that had been linked to chromosome 6q25.2–q27 (Matsumine et al., 1998). Discovery was aided because many patients had deletions of large sections of chromosome 6, where the gene resides. Parkin mutations have now been identified pan-ethnically and are thought to be the cause of approximately 50% of familial young-onset PD and 15–20% of sporadic young-onset PD (<50 years). Over 70 mutations, including exon rearrangements, point mutations, and deletions, have been identified, many of them recurrent in different populations. The identification of parkin mutations is inversely correlated with age of onset, with the earliest age of onset having the greatest association. However, PARK2 does not appear to be restricted to young-onset PD, and parkin mutations have been identified in individuals over 50 (Lücking et al., 2000). Juvenile parkin-related PD is associated with mutations in both parkin alleles (homozygotes or compound heterozygotes). Some heterozygotes have now been recognized as having adult-onset PD with Lewy bodies (Farrer et al., 2001; Pramstaller et al., 2005). A single parkin mutation (heterozygote) may be responsible for instances of later onset PD (Foroud et al., 2003), and numerous heterozygote cases have now been found (Pramstaller et al., 2005). This is a crucial finding in the search for the cause of idiopathic PD, and it is clear that parkin plays an important role. The frequency and penetrance of parkin mutations have yet to be determined. PARK2, especially in juvenile cases, has been characterized by slow clinical progression, sustained response to levodopa, high likelihood of levodopa-induced dyskinesias, dystonia, sleep benefit, and hyperreflexia. The rate of loss of FDOPA striatal uptake is much slower in symptomatic and asymptomatic parkin carriers, compatible with the slow clinical worsening in this form of PD (Pavese et al., 2009).
The locus for PARK3 has been localized by linkage to chromosome 2p13 in a small group of European families with autosomal dominant PD with incomplete penetrance (Gasser et al., 1998). Affected individuals have clinical and pathologic findings similar to sporadic late-onset PD, including age of onset. The sepiapterin reductase gene is a likely candidate for PARK3 (Karamohamed et al., 2003; Sharma et al., 2006). Sepiapterin reductase is an enzyme in the pathway for tetrahydrobiopterin synthesis, a cofactor for tyrosine hydroxylase. At present PARK3 is considered a susceptibility gene.
The evolution of the gene identification for PARK4 is a fascinating story. Waters and Miller (1994) described a kindred with autosomal dominant Lewy body parkinsonism in four generations, with an early age at onset (PARK4). The kindred was extended (Muenter et al., 1998; Farrer et al., 1999; Gwinn-Hardy et al., 2000), and action tremor similar to essential tremor and dementia were prominent within the family, and widespread Lewy bodies were seen in the cerebral cortex. Linkage was reported to chromosome 4p, but this haplotype also occurred in individuals in the pedigree who did not have clinical Lewy body parkinsonism but rather suffered from postural tremor only (Farrer et al., 1999). A second genome-wide search in this family found linkage on chromosome 4q and the mutation to be a triplication of a region of the chromosome that includes the SNCA gene (Singleton et al., 2003), the same gene that is mutated in PARK1. Instead of a mutation, though, this triplication produces a doubling of the normal, wild-type α-synuclein protein. A second PD family (this one being Swedish-American) with PD dementia with triplication of SNCA has also been reported (Farrer et al., 2004). The large Swedish family reported originally by Mjönes (1949) has been genetically analyzed and found to consist of duplications and triplications of normal SNCA (Fuchs et al., 2007). The remarkable thing about it is that the family has an α-synuclein duplication in one branch, and a triplication in the other, with a correspondingly different clinical picture (late versus early onset, respectively).
PARK5, an autosomal dominant mutation of the gene for ubiquitin-carboxy-terminal-hydrolase L1 (UCHL1), was found in a single family and was mapped to chromosome 4p14 (Leroy et al., 1998). Additional families with this mutation are necessary before concluding that UCHL1 is a definite causative gene. But since this enzyme is part of the ubiquitin-proteasomal system, it implicates this degradative pathway as being important in the pathogenesis of PD (see below). Another potential pathogenic mechanism is the association of UCH-L1 with cellular membranes, including the endoplasmic reticulum (Liu et al., 2009). The membrane-associated form of UCH-L1 has been correlated with the intracellular level of α-synuclein.
Families with autosomal recessive, young-onset PD in southern Europe were mapped to chromosome 1p35–p36, labeled as PARK6 (Valente et al., 2002), and Valente and her colleagues (2004) identified two homozygous mutations in the PTEN-induced putative kinase 1 (PINK1) gene. Novel homozygous mutations in this gene were subsequently found in six unrelated Asian families (Hatano et al., 2004). Other families around the globe have been reported, and heterozygosity appears to increase susceptibility to developing PD (Hedrich et al., 2006). PINK1 is a mitochondrial serine/threonine kinase, and appears important in combating oxidative stress. The patients with PINK1 mutations resemble those with parkin mutations. Excessive parkin can rescue the PINK1 mutant phenotype, indicating that parkin works downstream from PINK1 in a common pathway. One autopsy of a young-onset patient with PD due to PINK1 mutations showed the presence of Lewy bodies (Samaranch et al., 2010).
PARK7, the DJ-1 gene mutation, is another cause of early-onset, autosomal recessive PD and is due to mutations on chromosome 1p36 (Bonifati et al., 2003). The mutations and gene identification were found in a consanguineous Dutch family and families in Italy and Uruguay. PARK7, like parkin and PINK1, is characterized by slow progression and a good response to levodopa. No autopsies have been reported for individuals with DJ-1. The crystal structure of DJ-1 reveals a redox-reactive center. DJ-1 may be activated in the presence of reactive oxygen species under conditions of oxidative stress. DJ-1, therefore, is an important redox-reactive signaling intermediate controlling oxidative stress (Kahle et al., 2009). DJ-1 and PINK1 are both important for mitochondrial function (Cookson, 2010). Knock-out mutants of DJ-1 enhance mitochondrial oxidative stress produced by pacemaking of SNc neurons (Guzman et al., 2010).
The most common PD gene mutation found so far has been PARK8, the gene located on chromosome 12p11.2–q13.1 for the protein leucine-rich repeat kinase 2 (LRRK2) (Zimprich et al., 2004), which is also called dardarin (Paisan-Ruiz et al., 2004). Initially, linkage to chromosome 12 was reported in a large Japanese autosomal dominant family without Lewy bodies (Funayama et al., 2002). But the Zimprich and Paisan-Ruiz families included subjects with Lewy body pathology, tangle pathology, and without Lewy bodies. One LRRK2 mutation, G2019S, is the most common and is seen in more than 2% in sporadic North American and English PD patients (Gilks et al., 2005; Nichols et al., 2005). The highest frequencies are in the Portuguese (Bras et al., 2005), Ashkenazi Jewish (Clark et al., 2006; Ozelius et al., 2006), and North African Arab (Lesage et al., 2006) patients, even in the absence of a family history of PD. The clinical, pathologic, and genetic epidemiology of LRRK2 were reviewed by Haugarvoll and Wszolek (2006) and mixed pathology resembling typical PD, tauopathies (tangles), and multiple system atrophy (MSA) were seen. However, patients with typical MSA were not found to have the LRRK2 G2019S mutation (Ozelius et al., 2007). In the Chinese population the G2019S mutation is rare, but about 9% of PD patients have a heterozygous G2385R mutation (Fung et al., 2006; Farrer et al., 2007). LRRK2 mutations show the typical age at onset and are not prone to be young onset (Clark et al., 2006). Penetrance is not complete and variable rates have been reported. Penetrance increases with age. In one report, it increases from 28% at age 59 years, to 51% at 69 years, and 74% at 79 years (Healy et al., 2008), and in another report the penetrance rate was 24% at the age of 80 (Clark et al., 2006).
How mutations in LRRK2 cause PD is not clear. Increased kinase activity appears to be involved. Enhanced kinase activity (approximately three-fold) has been consistently reported for G2019S; but four disease mutants, R1441C, R1441G, Y1699C, and I2020T, have kinase activity less than two-fold of the wild-type protein (Dauer and Ho, 2010). Inhibitors of LRRK2 kinase have been found to be protective in both in vitro and in vivo models of LRRK2-induced neurodegeneration. These results establish that LRRK2-induced degeneration of neurons in vivo is kinase-dependent (Lee et al., 2010).
PARK9 is actually a Parkinson-plus disorder rather than PD. The autosomal recessive disorder has a mutation for the A form of parkinsonism-plus seen with a mutation of lysosomal type 5 ATPase, ATP13A2, on chromosome 1q36, known as PARK9. There is juvenile onset, and the disorder was originally known as the Kufor-Rakeb syndrome, after the village in Jordan where the affected families reside (Najim al-Din et al., 1994). This syndrome had features of Parkinson-plus with pyramidal signs, dystonia, and supranuclear gaze palsy. The disorder is levodopa-responsive, and some cases show putaminal and caudate iron accumulation, making it another neurodegeneration with brain iron accumulation (NBIA) (Schneider et al., 2010).
A genome-wide screen on 117 Icelandic patients with classic late-onset Parkinson disease and 168 of their unaffected relatives from 51 families resulted in linkage to chromosome 1p32 (Hicks et al., 2002). This susceptibility gene for PD was designated as PARK10.
A genome-wide screen on 150 families with PD resulted in finding linkage for a susceptibility gene on chromosome 2q36–q37, designated as PARK11 (Pankratz et al., 2003a). Mutations in the gene coding for GRB10-interacting GYF protein 2 (GIGYF2) was reported (Lautier et al., 2008), but this has been questioned by other investigators (Guo et al., 2009; Nichols et al., 2009).
A genome-wide search also found a positive LOD score for a locus on chromosome Xq21–q25 (Pankratz et al., 2003b), which has been designated as PARK12.
Using a candidate gene approach based on the phenotype of motor neuron degeneration in mice, a genome search on German PD patients found a novel mutation of a mitochondrial serine protease HTRA2 (high temperature requirement protein A2) in four patients (Strauss et al., 2005; Plun-Favreau et al., 2008). This mutation has been listed as PARK13.
Homozygosity for a missense mutation in the PLA2G6 gene was discovered in a large consanguineous Pakistani family with neurodegeneration with brain iron accumulation (NBIA2), a condition known as infantile neuroaxonal dystrophy (Morgan et al., 2006). Spasticity, cerebellar ataxia, and optic atrophy are common in these infants (Kurian et al., 2008). But the phenotypic spectrum has been found to be much broader. It can cause autosomal recessive dystonia–parkinsonism at all ages, including adults, and without evidence of brain iron accumulation (Paisan-Ruiz et al., 2009). It has been labeled PARK14.
Parkinsonism with spasticity occurring in the first decade of life has been called pallido-pyramidal syndrome (Davison, 1954). It is a recessive disorder that responds well to levodopa therapy (Horowitz and Greenberg, 1975; Bronstein and Vickrey, 1996). It has also been called parkinsonism-pyramidal syndrome, and the mutation was found in the FBXO7 gene coding for the F-box only protein 7 (Di Fonzo et al., 2009). It has been labeled PARK15.
PARK16 was discovered by a large genome-wide association study in the Japanese population on chromosome 1q32 (Satake et al., 2009). This finding was replicated in a large genome-wide association study in the European population (Simón-Sánchez et al., 2009) as did another replication study, which showed an association in ethnic Chinese but no association in caucasians of European ancestry (Vilariño-Güell et al., 2010). The nature of the abnormal gene is unknown.
A mutation in the EIF4G1 gene on chromosome 3q27 was recently found in European and American families, all with the same ancestral haplotype (Chartier-Harlin et al., 2009).
Although not designated with a PARK label, heterozygotic mutations of the lysosomal enzyme, β-glucocerebrosidase (GBA) gene on chromosome 1q21 (homozygotes are the cause of Gaucher disease) have been associated with PD (Goker-Alpan et al., 2004; Aharon-Peretz et al., 2004, 2005; Clark et al., 2005) and in other Lewy body disorders (Goker-Alpan et al., 2006). Mutations of GBA are most common in the Ashkenazi Jewish population. Between 17–30% of the Ashkenazi sporadic PD patients are heterozygotes for the GBA mutation (Aharon-Peretz et al., 2005; Clark et al., 2007). The GBA mutation is associated with onset younger than 50 years compared to the older age onset in sporadic PD (Clark et al., 2007). Heterozygous mutations have been found in patients with PD in all ethnic groups, and has been designated a risk factor for PD. Being a lysosomal enzyme, it implies a link between autophagy malfunction and PD.
Evaluating eight mutations of GBA among 420 patients with PD and 4138 controls of Israeli Ashkenazi origin, the relative frequency of mutation carriers was 18% versus 4% and severe mutations were associated with 13-fold increased risk of PD compared to two-fold risk increase in subjects with mild mutations (Gan-Or et al., 2008). In addition, 14% of the studied patients had the G2019S LRRK2 mutation. Thus more than a third of all PD Ashkenazi Jewish patients carried the GBA or the LRRK2 mutation. In one study involving a Japanese population of 534 patients with PD and 544 controls, 55 patients with PD (9.4%) and only 2 controls (0.37%), odds ratio (OR) 28.0, had at least one of 11 pathogenic heterozygous variants in the GBA gene (Mitsui et al., 2009). In another study, involving 790 British (non-Jewish) PD patients and 257 controls, GBA mutations were found in 33 (4.18%) of patients and 3 (1.17%) of controls (OR 3.7) (Neumann et al., 2009). The GBA-positive patients were younger at onset and 45% had visual hallucinations and 48% had cognitive decline or dementia. Diffuse neocortical Lewy body pathology occurred more frequently in the 17 GBA-positive patients examined at autopsy, than in controls. A large worldwide 16-center collaborative project involving 5691 patients with PD and 4898 controls revealed an OR of 5.43 for any GBA mutation in patients versus controls (Sidransky et al., 2009).
Compared to the general population, parkinsonism in patients with Gaucher disease is more frequent, occurs at an earlier age, responds less well to levodopa, and is more frequently associated with cortical dysfunction (Kraoua et al., 2009). Enzyme replacement therapy and substrate reduction therapy were ineffective. The independently increased risk of GBA mutations in patients with PD, together with the identification of mutations in a lysosomal type 5 P-type ATPase gene responsible for the PARK9 locus, suggests the link between neurodegeneration producing the PD phenotype and impaired autophagic lysosomal mechanisms (Pan et al., 2008). For those with Gaucher disease 5–7% develop PD by age 70, and 9–12% before age 80 years (Rosenbloom et al., 2011).
Families with compound heterozygosity in the DNA polymerase gamma 1 (POLG1) were discovered to have early onset PD in addition to a polyneuropathy (Davidson et al., 2006; Orsucci et al., 2011). This enzyme is involved in the replication of mitochondrial DNA, indicating that impaired mitochondrial function can lead to PD.
Perry syndrome is a rapidly progressive disease with familial parkinsonism, hypoventilation, depression, and weight loss (Perry et al., 1975; Wider and Wszolek, 2008). In addition to severe neuronal loss in the substantia nigra and locus coeruleus, there is also loss of putative respiratory neurons in the ventrolateral medulla and raphe nucleus (Tsuboi et al., 2008). There are neuronal and glial inclusions that stain positive for TAR DNA-binding protein-43 (TDP-43) (Wider et al., 2009), as seen in ALS and frontotemporal dementia. Mutations in the DCTN1 gene, which codes for dynactin 1, has been found (Farrer et al., 2009). The dynactin complex is important for many cellular transport functions including axonal transport of vesicles.
An infantile dystonia–parkinsonism disorder due to homozygous loss-of-function mutations in the SLC6A3 gene encoding the dopamine transporter has been reported (Kurian et al., 2009). Such mutations have been associated with attention deficit hyperactivity disorder. One would expect increased synaptic dopamine, and therefore a hyperkinetic disorder rather than a parkinsonian-dystonia disorder, which is usually associated with a deficiency of striatal dopamine and thus decreased synaptic dopamine reaching the dopamine receptor. The CSF contained an elevated level of homovanillic acid (HVA), the final metabolite of dopamine, indicating a high turnover of dopamine. It would appear that the synaptic dopamine is being rapidly metabolized by monoamine oxidase (MAO) and catechol-O-methyltransferase. Perhaps the dopamine receptor was downregulated. There was a poor response to levodopa therapy.
Other familial or childhood-onset dystonia–parkinsonisms are those due to homozygous mutations of the gene for tyrosine hydroxylase and the heterozygous mutations of the gene for the tyrosine hydroxylase cofactor, GTPCH1. Both of these disorders are discussed in Chapter 12.
There is emerging evidence that some patients with parkinsonism are born with fewer than normal dopaminergic neurons as a result of some in utero or perinatal event or as a result of mutations in genes that code for various transcription factors, such as nuclear receptor-related 1 (NURR1) (Bensinger and Tontonoz, 2009), human achaete-scute homolog 1 (HASH1), and paired-like homeodomain transcription factor 3 (PITX3), and other genes necessary for the development and maintenance of the dopaminergic system (Le et al., 2009).
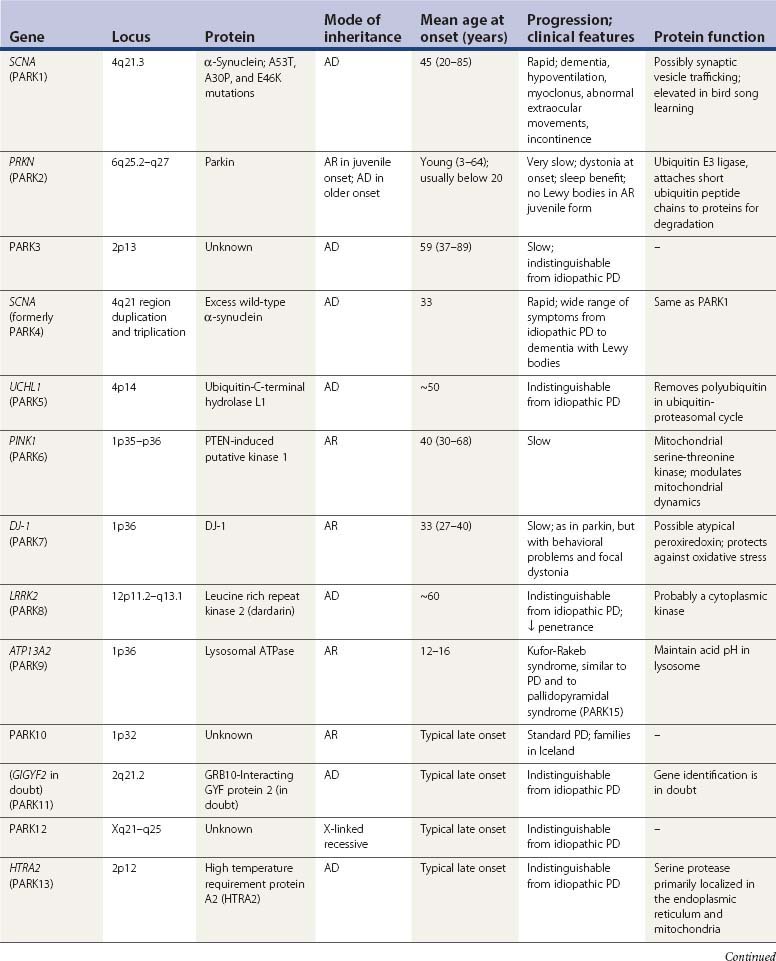
A summary of the genetic alterations described above is presented in Table 5.3.
Environmental factors contributing to etiology of or protection against PD
Environmental toxic factors
A gene–environment interaction is a common theme in the etiology of PD. With the discovery of MPTP as a toxin causing parkinsonism due to dopamine neuron degeneration (Langston et al., 1983), an environmental etiology seemed likely. MPTP is the protoxin; it is oxidized by MAO-B to MPP+, which can be taken up into dopamine nerve terminals via the dopamine transporter. Once there, it inhibits complex I in the mitochondrial respiratory chain, thereby reducing ATP synthesis and increasing superoxide radicals (for review, see Dauer and Przedborski, 2003). But MPTP-induced parkinsonism is not PD; moreover inhibition of MAO-B with selegiline (Parkinson Study Group, 1993) or rasagiline (Parkinson Study Group, 2004) does not halt progression of PD. Rotenone is used to poison unwanted fish in lakes, and like MPP+ inhibits complex I and can cause experimental parkinsonism (Betarbet et al., 2000). Whether it is an important environmental factor is questionable, although the association of PD in men with pesticide exposure is positive whereas exposure to other chemicals is not (Frigerio et al., 2006). Two pesticides in particular, maneb and paraquat, show a high risk for PD, and the risk is even greater if a person is exposed to both (Costello et al., 2009).
There are other pesticides known to cause dopaminergic damage, including ziram, which has been shown to inhibit the ubiquitin/proteasome system in primary ventral mesencephalic cultures (Chou et al., 2008). In an analysis of serum samples of 50 PD patients and 43 controls, beta-hexachlorocyclohexane (β-HCH) was more often detectable in patients with PD (76%) than controls (40%) (Richardson et al., 2009). One study showed the world’s highest prevalence of PD may be among the Amish in the northeast United States. The prevalence of PD was 5703/100 000 (nearly 6%) of people 60 years old or older, more than three times the prevalence for the rest of the United States (Racette et al., 2009). Since normal subjects were more related to each other than were subjects with clinically definite PD, this suggests that environmental factors, such as high use of pesticides, may contribute to the high prevalence of PD in this community. To investigate occupation, specific job tasks or exposures, and risk of parkinsonism and clinical subtypes, a multicenter case-control study comparing lifelong occupation and job-task histories was created to determine associations with parkinsonism. In the study, referred to as SEARCH (Study of Environmental Association and Risk of Parkinsonism using Case-Control Historical Interviews), 519 patients and 511 controls were analyzed. Increased risk of parkinsonism was found with pesticide use (OR 1.9, 95% CI 1.12–3.21), whereas tobacco and greater caffeine intake were inversely associated with PD risk (Tanner et al., 2009).
Other than pesticides, there is little consistent evidence for environmental factors. Other putative risk factors are head trauma, certain occupations, and milk consumption (Tanner, 2010). Against a strong, new environmental factor is the lack of clusters of PD and an unchanging incidence rate over decades (Rocca et al., 2001). With the discovery of individual genes causing PD, a major genetic contribution to the etiology of PD has become predominant. It is likely that there are specific genes that can cause familial PD and susceptibility genes that make the individual with those genes sensitive to triggers to pathogenesis from environmental or endogenous factors, such as oxidative stress. Endogenous factors, such as dopamine in nigral neurons and autonomous cell firing through calcium channels with the aging process, are discussed below under pathogenesis.
Infectious agents remain a possible etiologic factor. For example, the highly pathogenic H5N1 avian influenza viruses enter the CNS and cause activation of microglia and α-synuclein phosphorylation and aggregation; in the SN there is loss of dopaminergic neurons (Jang et al., 2009a). Viruses known to induce parkinsonism include Coxsackie, Japanese encephalitis B, St Louis, West Nile, and HIV viruses (Jang et al., 2009b).
Environmental protective factors
There are a couple of environmental factors that are consistently associated with a reduced risk for developing PD, namely smoking (Ritz et al., 2007) and caffeine consumption. In the huge study (more than 140 000 men and women) in the Cancer Prevention Study II Nutrition Cohort, there was a lower risk of developing PD that correlated with (a) more years smoked, (b) smoking more cigarettes per day, (c) older age at quitting smoking, and (4) fewer years since quitting smoking (Thacker et al., 2007). It was concluded that the dependence of this association on the timing of smoking during life is consistent with a biologic effect.
Coffee consumption was also found to be associated with a lower risk of developing PD. First discovered by Ross and colleagues (2000) in Japanese-American men in Hawaii, this association has held up in subsequent epidemiologic studies. Ascherio and colleagues (2001) evaluated this association in two other large cohorts being followed, and found a similar association with coffee consumption and a mild lowered risk of developing PD in men (the authors speculated that estrogen use in women eliminated the caffeine benefit). Focus centered on caffeine and then speculation about the adenosine A2A receptor since this is affected by caffeine (Checkoway et al., 2002; Hernan et al., 2002; Schwarzschild et al., 2002; Ascherio et al., 2003, 2004).
A higher serum urate level is correlated with a reduction in developing PD (Weisskopf et al., 2007). Subsequent studies evaluating urate levels in plasma in the PRECEPT (Schwarzschild et al., 2008) and CSF in the DATATOP (Ascherio et al., 2009) clinical trials found that higher urate levels in men were associated with a slower rate of progression of PD. Urate has antioxidant properties. Use of anti-inflammatory drugs is another putative protective factor (Gagne and Power, 2010).
Endogenous factors that contribute to the etiology of PD
Dopamine induces oxidative stress
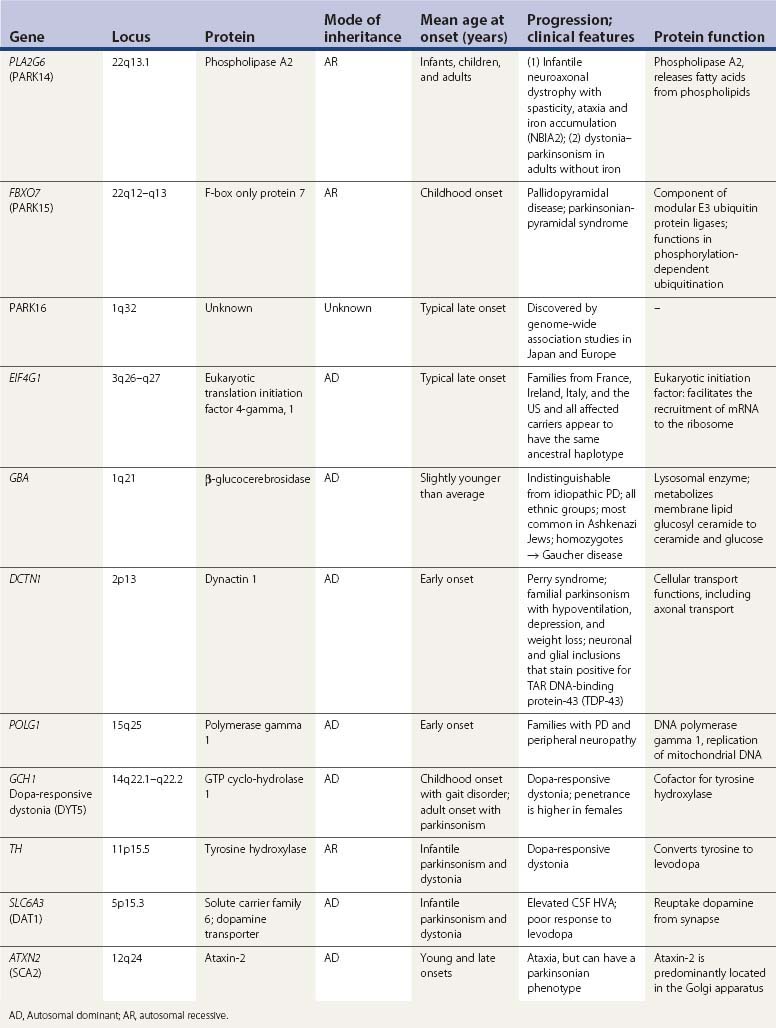
The SNc neurons are dopaminergic, and this monoamine provides a steady source of oxidative stress (Fahn and Cohen, 1992). Levodopa and dopamine can be auto-oxidized to oxyradicals (semiquinones and quinones) and dopamine can be enzymatically oxidized by MAO-A (located in the dopamine nerve terminals) to form the intermediate dihydroxyphenylacetaldehyde (DOPAL) and final metabolite, homovanillic acid (HVA) (Fig. 5.12). For every molecule of dopamine metabolized by MAO, a molecule of hydrogen peroxide is formed, which can be a precursor of more oxyradicals. DOPAL has been found to enhance aggregation of α-synuclein (W.J. Burke et al., 2008). This may be an important mechanism of nigral neuron cell death because injection of DOPAL into rat nigra results in dopamine neuron loss (W.J. Burke et al., 2008).
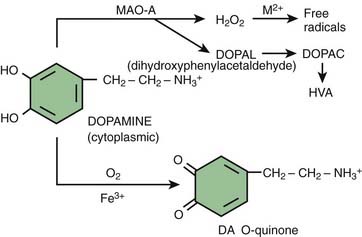
The cell’s defense is to condense the oxidized products and turn them into insoluble neuromelanin (Sulzer et al., 2000) (Fig. 5.13). This could explain why the most susceptible cells are those with the most neuromelanin (Hirsch et al., 1988, 1989); not because neuromelanin itself is toxic, but rather because its presence indicates these are the cells with the most oxidative stress. Cytosolic dopamine quinone can also form adducts with other compounds, including cysteine to make a toxic compound.
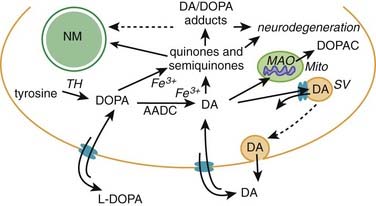
Interaction between dopamine and α-synuclein
In the dopamine neuron, a special situation occurs for wild-type α-synuclein. α-Synuclein is a soluble protein in cytoplasm that is natively unfolded. By virtue of its containing an A2 alpha helix domain, it can associate with vesicle membranes (Clayton and George, 1998). A potential role for binding to synaptic vesicles is suggested by an enhanced synaptic recovery for evoked DA release in the knock-out animal (Maroteaux et al., 1988) and alterations in the distal synaptic vesicle pool in mutants (Abeliovich et al., 2000; Cabin et al., 2002). α-Synuclein can form into oligomers (also called protofibrils), which are beta-sheets that consist of spheres, chains, and rings. These protofibrils permeabilize the synaptic vesicles that store dopamine by forming pore-like assemblies on their surface (Mosharov et al., 2006). This allows leakage of vesicular dopamine into the cytosol, thus enriching cytosolic dopamine to enhance oxidative stress. A vicious cycle is at play because cytosolic dopamine reacts with α-synuclein to form a covalent adduct that slows the conversion of protofibrils to fibrils, thus sustaining the presence of the protofibrils (oligomers) (Conway et al., 2000; Rochet et al., 2004). The presence of the oligomers then leads to more permeabilization of the synaptic vesicles and more cytosolic dopamine (Sulzer, 2010). A diagram of normal soluble α-synuclein forming oligomers, fibrils, aggregates and their deposition in Lewy bodies is seen in Figure 5.14.
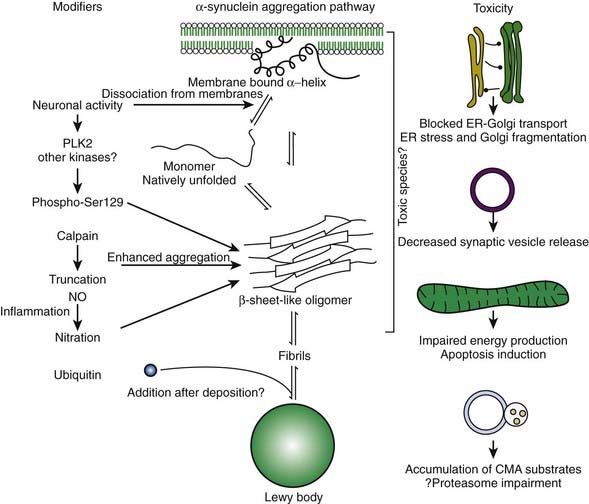
Another critical peculiarity of α-synuclein-dopamine adducts is that they block chaperone-mediated autophagy (CMA) by binding and blocking the receptors on the lysosome from accepting other proteins and from being broken down by the CMA mechanism (Martinez-Vicente et al., 2008). This would lead to accumulation of toxic proteins in the cell, which would lead to the cell’s dysfunction and death. A more complete discussion about the role of autophagy in removing damaged proteins is presented below.
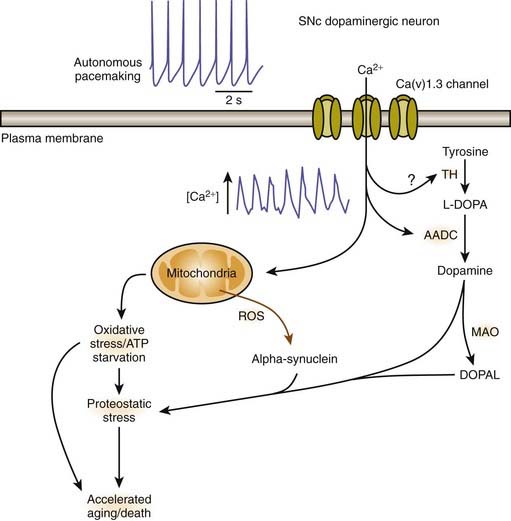
Calcium influx into SNc dopaminergic neurons
A remarkable feature in the pathology of PD is that while SN dopamine neurons, particularly those located in the ventral tier of the SNc, undergo selective death, the neighboring dopamine neurons of the ventral tegmental area (VTA) are generally unaffected. The reason for this selective pattern of dopaminergic neuronal death remains unclear and should be an important clue to pathogenesis. There are differences between the dopaminergic neurons in the SN and the VTA. There is less neuromelanin (apparently indicating less long-term oxidative stress from cytosolic dopamine) and there is more of the calcium-binding protein, calbindin (Iacopino et al., 1992). The expression of the vesicular monoamine transporter (VMAT2) that delivers cytosolic dopamine into the synaptic vesicles may also be somewhat higher in the VTA (Peter et al., 1995). Adult SNc dopaminergic neurons fire autonomously due to calcium ion influx, but VTA dopaminergic neurons use sodium ions (Surmeier, 2007).
A physiologic explanation has been suggested by Surmeier (2007). Unlike most neurons in the brain, SNc dopamine neurons are autonomously active; that is, they generate action potentials at a clock-like 2–4 Hz in the absence of synaptic input. In this respect, they are much like cardiac pacemakers. Juvenile dopamine-containing neurons in the substantia nigra pars compacta use sodium ion influx as the pacemaking mechanism. But these mechanisms remain latent in adulthood (Chan et al., 2007). Instead their adult autonomous activity is generated by Ca2+ ions whereas most neurons use channels that allow Na+ ions across the membrane. The SNc dopamine neurons rely on L-type Ca(v)1.3 Ca2+ channels. With increased intracellular calcium, mitochondrial function can be affected with increased demand on oxidative phosphorylation, leading to increased production of reactive oxygen species and eventually cellular damage. As the cells undergo more stress over time, they thus “age faster.” This would be a link with the risk factor of age (Surmeier, 2007). Blocking Ca(v)1.3 Ca2+ channels in adult neurons induces a reversion to the juvenile form of pacemaking (Chan et al., 2007). Such blocking (“rejuvenation”) protects these neurons in both in-vitro and in-vivo models of PD, pointing to a new strategy that could slow or stop the progression of the disease (Chan et al., 2007; Surmeier, 2007). An intriguing finding is that a case-control analysis within the UK-based General Practice Research Database revealed that calcium-channel blockers when used to treat hypertension are associated with a lower prevalence of PD in those individuals compared to other antihypertensive therapies (Becker et al., 2008). This finding has been substantiated by a similar epidemiologic analysis of the Danish medical database. Subjects prescribed centrally-acting dihydropyridines (calcium channel blockers) prior to 2 years before the index date (onset of PD in affected subjects) were less likely to develop PD (OR 0.73, 95% CI 0.54–0.97) (Ritz et al., 2010). In contrast, risk estimates were close to null for the peripherally-acting dihydropyridine amlodipine and for other antihypertensive medications.
Mosharov and colleagues (2009) found that an interaction between Ca2+, cytosolic dopamine, and α-synuclein appears to underlie the susceptibility of SN neurons in PD. α-Synuclein is critical, for without it, dopaminergic neurons are resistant to levodopa toxicity, and Ca2+ influx through L-type channels elevates dopamine synthesis to potentially toxic levels (Mosharov et al., 2009). Figure 5.15 illustrates how Ca2+ influx influences mitochondria and oxidative stress and may lead to cell death. Figure 5.16 illustrates how all the above endogenous factors influence the pathogenesis of PD.
Where does PD pathology begin? How does it progress? Braak’s staging system
Following the development of Lewy neurites
Braak and his colleagues, using staining techniques to identify α-synuclein in neurons (Lewy neurites), studied 30 autopsied brains that were observed to have incidental Lewy body pathology (Del Tredici et al., 2002). Instead of finding α-synuclein staining fibers in the substantia nigra, they found them in non-catecholaminergic neurons of the dorsal glossopharyngeus–vagus complex, in projection neurons of the intermediate reticular zone, and in specific nerve cell types of the gain-setting system (coeruleus–subcoeruleus complex, caudal raphe nuclei, gigantocellular reticular nucleus), olfactory bulb, olfactory tract, and/or anterior olfactory nucleus, all in the absence of nigral involvement (Fig. 5.17). None of the melanized areas were affected, and the first of these to show Lewy changes was the locus coeruleus. Based on these findings of the location of Lewy neurites, these investigators proposed that PD begins in the lower brainstem and the olfactory apparatus.
Braak followed this study with one investigating Lewy neurites in 41 autopsies that were characteristic of PD clinically and pathologically and in 69 others who had no clinical features but whose autopsies showed Lewy neurites so that he could determine if there was a pattern of progression (Braak et al., 2003, 2004). A pattern of α-synuclein-immunopositive Lewy neurites and Lewy bodies was discerned, in which these features began in the anterior olfactory nucleus and in the lowest part of the brainstem (stage 1) and then progressed rostrally up the brainstem. The raphe and locus coeruleus were the next to be involved (in stage 2). The substantia nigra became involved in stage 3, the mesocortex and thalamus in stage 4, the neocortex association areas and prefrontal cortex in stage 5, and the entire neocortex, but only mildly so in primary motor and sensory areas, in stage 6 (Fig. 5.18). As the stages develop, the sites involved in the earlier stages become more severely affected.
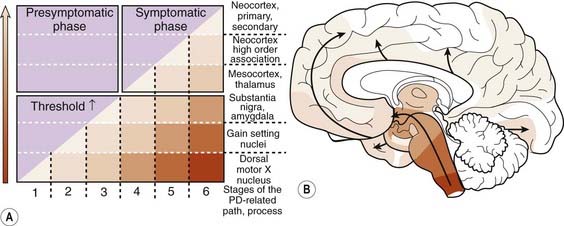
Figure 5.18 Braak staging of PD based on development of Lewy neurites.
From Braak H, Ghebremedhin E, Rub U, et al. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 2004;318(1):121–134.
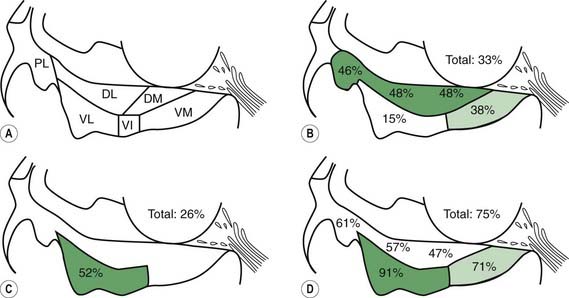
In a subsequent titled lecture, Braak and colleagues (2006) reinforced his staging system and cited support from other neuropathologic reports. They state that their samples did not include cases of diffuse Lewy body disease, which has its Lewy bodies starting in the cerebral cortex. All 110 cases they examined fell into one of six different subgroups based on the location of the brain regions involved (Fig. 5.19). Each of the subgroups displayed newly affected regions and those previously involved were worse. They pointed out that long unmyelinated axons, which the nigrostriatal pathway has, are the susceptible sites for Lewy neurites.
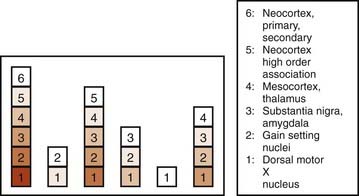
Figure 5.19 All 110 cases of Braak et al. (2006) fall into just one of six categories of locations of Lewy neurites.
From Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 2004;318(1):121–134.
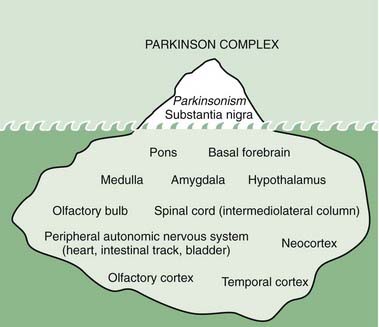
Although α-synuclein accumulation may not start in the nigra, unless the nigra becomes involved, the symptoms of PD do not develop. On the other hand, it is possible that symptoms of decreased sense of smell, depression, decreased assertiveness, impaired spatial sense, and even constipation may be preclinical symptoms of PD (Fig. 5.20) (Langston, 2006). Such a concept is leading researchers to seek clinical and laboratory biomarkers to detect PD before it becomes manifest with motor symptoms.
Are Lewy bodies and Lewy neurites toxic or protective?
Since they were first discovered, most neurologists and neuropathologists assumed that Lewy bodies somehow contribute to the cause or pathogenesis of the disease. Only recently has this assumption come under serious doubt. In fact, there is a large school of scientists who consider the formation of Lewy bodies as a means for the neuron to protect itself. Initiated by Lansbury and colleagues (2002, 2006), the concept has evolved that oligomers (protofibrils) of α-synuclein are toxic, in part because they permeabilize synaptic vesicle membranes. The protofibrils can be fibrillated (aggregated) into amyloid, thereby removing them, and thus protecting the neuron (Figs 5.21, 5.22). These aggregates thus represent a protective mechanism by the cell against oligomers’ toxicity. This view is not universally accepted, and some neuroscientists believe that the fibrils are the toxic element. This remains a controversial point.
Is the Braak staging system for PD established as denoting the progression of pathology in PD?
A comprehensive appraisal of the Braak hypothesis was conducted by R.E. Burke and colleagues (2008). From their analysis, it is clear that the caudal-to-rostral spread of Lewy neurites is not entirely universal. Here, we summarize the arguments they made in concluding that this pattern of abnormal synucleinopathy – from caudal brainstem toward rostral structures over time – is not completely established for PD, but augmented with more recent findings, suggesting that the majority of PD patients might follow this pattern.
1 Unlikely that PD is universally preceded by caudal synucleinopathy
Many brains with incidental Lewy bodies with reduced numbers of nigral dopamine neurons do not show lower brainstem synucleinopathy. In a study by Parkkinen and her colleagues (2005), 16% of subjects with incidental Lewy bodies in the nigra did not have synuclein pathology in the lower brainstem. In another study looking at distribution of incidental Lewy bodies (iLBD), 34 of 235 brains studied had iLBD (14.5%) and all but one could be assigned a Braak PD stage (Frigerio et al., 2009). There were three patterns of distribution of α-synuclein pathology: (1) diffuse cortical and subcortical; (2) no cortical, but a caudal-to-rostral ascending pattern, primarily involving brainstem; and (3) intermediate between these two categories. Also, 6/33 cases failed to follow the pattern of contiguous spread proposed by Braak. These findings suggest dichotomy in the distribution of iLBD: some cases fit the Braak ascending scheme, conceptually consistent with preclinical PD, whereas others displayed prominent cortical involvement that might represent preclinical DLB.
2 Dementia with Lewy bodies (DLB) in the cerebral cortex often does not have brainstem synucleinopathy
Braak and colleagues (2003) excluded DLB in their series; yet many DLB cases have features of PD. In the autopsy series by Parkkinen and colleagues (2008), there were 226 cases of Lewy bodies in either the dorsal motor nucleus of vagus, substantia nigra or basal forebrain; 55% in Braak stage 5 or 6 lacked clinical features of dementia or parkinsonism. Moreover, of cases with dementia, 28% did not fit the Braak scheme of brainstem Lewy neurites being present.
3 Many elderly people have Braak pathology from stages 1 through 6 and die without developing PD
R.E. Burke and colleagues (2008) analyzed cases from the literature and plotted the Braak stages and the age at death, which showed no correlation between these two. Therefore, there is no certainty that those with early Braak stages will develop PD, and not everyone with advanced Braak stages has PD.
4 No correlation between Braak stages and the clinical severity of PD
R.E. Burke and colleagues (2008) plotted the Hoehn and Yahr (H&Y) scores published by Braak and colleagues (2003) and reported that individuals either had no clinical PD or had H&Y scores of 3–5 without a relationship to the Braak stage.
5 REM sleep behavior disorder (RBD) usually does not necessarily precede PD and not everyone with RBD develops PD
RBD is suspected of arising in the pons-medulla area, and therefore the Braak scheme would have RBD occurring before PD, if RBD is a component of PD. In the survey of their PD patients, Scaglione and colleagues (2005) found that only 33% had RBD. Of these, PD preceded RBD in 73%, an average of 8 years after onset of PD. In another study, RBD preceded PD in only 22% (De Cock et al., 2007). Postuma and colleagues (2009) followed patients with idiopathic RBD and found the risk for developing any neurodegenerative disease (PD, DLB, MSA, or Alzheimer disease) is 17.7% by 5 years, 40.6% by 10 years, and 52.4% by 12 years. Only 14 out of 93 developed PD. Subsequent studies reveal that approximately 50% of individuals with idiopathic RBD will develop PD and that the more severe the loss of atonia on baseline polysomnograms, the better the prediction of the development of PD (Postuma et al., 2010).
6 PD due to LRRK2 mutations does not always have Lewy body pathology
In patients with LRRK2 mutations who have PD, nigral degeneration is seen, but many do not have Lewy bodies, even though they may have the identical molecular mutation as those who do have Lewy bodies (Haugarvoll and Wszolek, 2006). Some patients have tau pathology. This is another example, like those described above, that Lewy bodies are not essential to have PD. Because the genetics are the same in these LRRK2 families, one can question the role of α-synuclein in the pathogenesis of all cases of PD.
In addition to the above arguments, there are many other concerns about the Braak hypothesis (Attems and Jellinger, 2008; Halliday et al., 2008; Parkkinen et al., 2008; Kalaitzakis et al., 2009). For example, the progression outlined by Braak is not consistent with the natural history of PD (Lees et al., 2009). A study evaluating 71 PD cases showed that caudorostral spread did not occur in 47% of the cases and that extensive synucleinopathy can occur without overt neurological signs (Kalaitzakis et al., 2008). Some pathologists have, therefore, proposed of a “multicentric” origin of PD (Dickson et al., 2008).
7 Results of neuroimaging do not completely match the pattern of the Braak pattern of progression
Brooks (2010) has analyzed alterations in PET to see if they correlated with the Braak staging. Microglial activation as detected by PET has been seen in the cerebral cortex prior to dementia in early PD, suggesting a Braak level 5 or 6 at that early stage. Braak staging suggests that the serotonergic and noradrenergic systems could become dysfunctional ahead of the dopaminergic system in PD. But PET studies with serotonergic markers show less midbrain pathology than the dopaminergic nigrostriatal system. FDOPA uptake in the locus coeruleus appears to be preserved until late disease, indicating that the loss of noradrenergic function is a late phenomenon in PD, despite the presence of Lewy body pathology in Braak stage 2. Assessment of cholinergic function reveals a significant reduction in parietal and occipital cortex in early PD, although this would represent Braak stage 4 disease. In the heart, MIBG and 18F-dopamine studies suggest early involvement of the sympathetic ganglia in PD, which is in keeping with Braak staging. Even so, in Hoehn and Yahr stage 1 PD, 50% of patients have normal MIBG uptake.
8 Some nonmotor features of PD appear before the motor signs as predicted by the Braak staging hypothesis
Autonomic involvement accounts for constipation seen in PD. A case-control study at the Mayo Clinic matched each incident case of PD (n = 196) by age and gender to the general control population (Savica et al., 2009). Constipation preceding PD was more common in cases than in controls (P = 0.0005). This association of constipation was found to have occurred for 20 or more years before the onset of motor symptoms of PD.
Bowel movement frequency was monitored and compared with postmortem SN neuronal loss in the Honolulu-Asia Aging Study (Petrovitch et al., 2009). There were evaluations in 414 men aged 71–93 years who later had postmortem evaluations. Men with 1 or more bowel movements daily had a significantly higher density of neurons in the SN compared to those with <1 daily. Constipation was associated with low SN neuron density independent of the presence of Lewy bodies. Incidental Lewy bodies were evaluated in these men. For men with <1, 1, and >1 bowel movement/day, corresponding percentages of incidental Lewy bodies were 24.1%, 13.5%, and 6.5% (Abbott et al., 2007), reflecting that fewer bowel movements were associated with more Lewy bodies.
Olfaction dysfunction (hyposmia) also precedes the motor signs of PD. Olfaction was assessed in 2267 men in the Honolulu-Asia Aging Study and followed for up to 8 years (Ross et al., 2008). The odds ratio for developing PD in the lowest quartile of smell sensitivity was 5.2 compared with the top two quartiles. In another prospective study, involving nonaffected relatives of PD patients, 40 hyposmic and 38 normosmic individuals were studied at baseline and 2 years later with clinical evaluation and a dopamine transporter (DAT) scan (Ponsen et al., 2004). By 2 years, 10% of the individuals with hyposmia, who also had strongly reduced DAT binding at baseline, had developed clinical PD as opposed to none of the other relatives in the cohort. In the remaining nonparkinsonian hyposmic relatives, the average rate of decline in DAT binding was significantly higher than in the normosmic relatives. These results indicate that idiopathic olfactory dysfunction is associated with an increased risk of developing PD of at least 10%.
Supporting evidence that peripheral nervous system is affected before CNS Lewy neurites was the finding that cardiac sympathetic denervation – as measured by MIBG scintigraphy – precedes nigrostriatal loss (Tijero et al., 2010).
9 Human autopsy study to determine validity of Braak staging scheme
Dickson and colleagues (2010) studied the density and distribution of Lewy bodies (LBs) in several conditions. They found that the proportion of cases that fitted the PD staging scheme was 67% for incidental LBs (n = 12); 86% for progressive supranuclear palsy (PSP) with incidental LBs (n = 18); 86% for pure Lewy body disease (LBD) with minimal or no Alzheimer type pathology (n = 52); 84% for LBD with concomitant Alzheimer disease (AD) (n = 84); and only 6% for AD with amygdala predominant LBs (n = 64). They conclude that the PD staging scheme of Braak is valid, except in the setting of advanced AD.
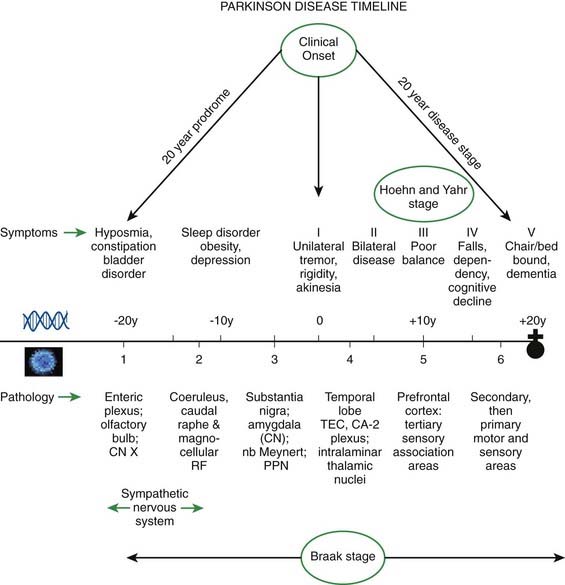
Braak and his colleagues are now estimating that the disease (pathology of Lewy neurites) begins in the olfactory and autonomic system by approximately 20 years before the onset of the motor symptoms of PD, and they have tried to correlate the Braak staging with the Hoehn and Yahr staging of clinical PD (Hawkes et al., 2010) (Fig. 5.23).
Clues on pathogenesis from monogenic PD
PARK1 (SNCA) and PARK4 (SNCA) and α-synuclein accumulation as Lewy neurites and Lewy bodies
Based on the presence of α-synuclein in Lewy bodies and on the pattern of progressive Lewy neurite formation from the caudal to rostral areas of the brain, it is clear that α-synuclein has a special place in the pathogenesis of PD. The normal function of α-synuclein, however, may have little or nothing to do with its role in pathogenesis. Its ability to self-aggregate, its presence in Lewy bodies, the apparent ability of pathogenic mutations or nitrated or DA-reacted proteins to be more prone to aggregation, the ability of protofibrils to disrupt membrane, and the identification of PARK4 as a triplication of the chromosomal region that contains the α-synuclein gene, all seem to point to a toxic function of α-synuclein itself. This notion is also supported by the finding that mice that lack α-synuclein expression are resistant to MPTP toxicity, although neuronal and synaptic vesicle uptake appear unaltered (Dauer et al., 2002). Expression of mutant α-synuclein also causes an apparent increase in cytosolic DA levels, suggesting a link between genetic and oxidative stress pathways (Lotharius et al., 2002). Thus, it may be that the disease stems from inappropriate or dysregulated degradation of this protein. The accumulated, insoluble, aggregated α-synuclein appears to play a vital role in the pathogenesis, leading to the suggestion that impaired protein degradation may be a key problem.
An approach to treating the toxicity from the accumulation of α-synuclein may be by the use of inhibitors of sirtuin 2. Sirtuins are members of the histone deacetylase family of proteins that participate in a variety of cellular functions and play a role in aging. Inhibition of SIRT2 rescued α-synuclein toxicity and modified inclusion morphology in a cellular model of PD and in a Drosophila model of the disease (Outeiro et al., 2007). The results suggest a link between neurodegeneration and aging.
PARK2 (PRKN) and PARK5 (UCHL1) and the ubiquitin-proteasomal system (UBS)
The key mechanisms through which misfolded or toxic proteins are degraded are the ubiquitin-proteasomal system (UBS) and autophagy via the lysosome. The cell initially attempts to restore such proteins using specific chaperones (heat-shock proteins) (Kopito, 2000), but if that is unsuccessful, the cell degrades the protein. The first attempt by the cell to remove these unwanted proteins is via the ubiquitin-proteasomal system. If that is unsuccessful, the process of autophagy is employed, using the lysosomes (Martinez-Vicente and Cuervo, 2007; Pan et al., 2008). Attention was initially directed to UBS because of the normal function of the proteins of PARK 2 (parkin) and PARK5 (UCHL1). The former is an E3 ligase and the latter separates the polyubiquitin chain into its separate components, both of which are active processes in the UBS. More recently, studies indicate that parkin is also related to mitochondria integrity and function (Mortiboys et al., 2008). PINK1 regulates parkin to its localization in the mitochondria by phosphorylating parkin (Kim et al., 2008). Both mutant parkin and PINK1 result in swollen mitochondria. Parkin also stabilizes PINK1 (Shiba et al., 2009) to maintain the mitochondria. When expressed in a Drosophila model of PD, wild-type parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration (Ng et al., 2009). Parkin also promotes macroautophagy of damaged mitochondria (Narendra et al., 2008). This is another example of the important role of the autophagy by the lysosome in PD. Parkin may play a role in sporadic PD because it is inactivated by nitrosative, oxidative, and dopaminergic stress (Dawson and Dawson, 2010).
Protein degradation by autophagy – its importance in PD discovered by studying degradation of mutated α-synuclein
The lysosome degrades cell products by three different mechanisms – macroautophagy, microautophagy, and chaperone-mediated autophagy (Martinez-Vicente and Cuervo, 2007; Pan et al., 2008).
Wild-type α-synuclein is degraded by the ubiquitin-proteasomal system and chaperone-mediated autophagy (Cuervo et al., 2004, 2010). Mutated α-synuclein is unable to enter the lysosome; it becomes trapped at the lysosomal surface and that blocks other material from entering the lysosome and being degraded. Thus, mutated α-synuclein itself is not degraded, and “gums” up the process for other substrates. In addition to mutated α-synuclein being unable to penetrate the lysosome, the same problem exists for altered α-synuclein (phosphorylated, nitrated, or oxidized α-synuclein); these compounds bind to lysosomal membrane with high affinity and are not translocated into the lysosome (Fig. 5.24) (Martinez-Vicente and Cuervo, 2007). As mentioned above in the discussion about endogenous factors leading to selective vulnerability of dopamine neurons, the same problem develops with adducts of α-synuclein and dopamine; these adducts block the translocation of other CMA-attached proteins into the lysosome.
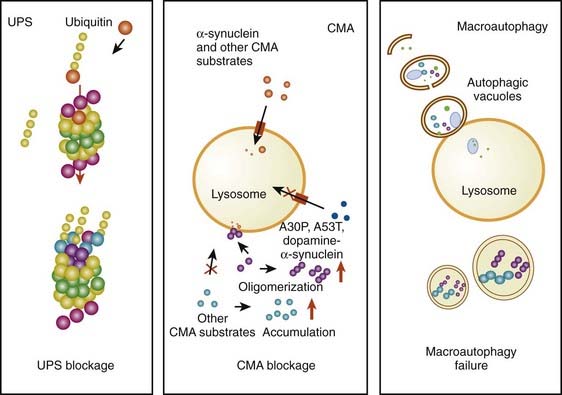
PARK6 (PINK1), PARK7 (DJ-1), and PARK8 (LRRK2)
The PARK8 mutation is the one with the highest prevalence for PD of all the known gene mutations. The protein dardarin (also known as leucine rich repeat kinase 2 (LRRK2) has GTPase and kinase enzymatic domains plus at least two protein–protein interaction domains (Hardy et al., 2006). The common G2019S mutation and an adjacent mutation are located at the N-terminal portion in the kinase domain. This site contains the Mg2+ ion required for kinase function. Mutated LRRK2 is disinhibited and has increased kinase activity and induces a progressive reduction in neurite length and branching both in primary neuronal cultures and in the intact rodent CNS (MacLeod et al., 2006). In contrast, LRRK2 deficiency leads to increased neurite length and branching (MacLeod et al., 2006). Because most patients with PARK8 have Lewy bodies, the biochemical defect appears to be upstream of α-synuclein, but because some patients do not have Lewy bodies and may have other inclusions, such as tau tangles, the exact pathogenesis of PARK8’s effect is still not clear.
PINK1 harbors a mitochondrial targeting motif and a serine/threonine kinase domain (Valente et al., 2004). PINK1 protein appears to accumulate within the intermembrane space of mitochondria (Silvestri et al., 2005). There is genetic interaction between PINK1 and parkin; Drosophila deficient in PINK1 can be restored to health with overexpression of parkin, but not vice versa (Abeliovich and Beal, 2006). This suggests a genetic pathway, with parkin functioning downstream of PINK1.
DJ-1 appears to play a role in protecting against oxidative stress, including that produced by dopamine (Lev et al., 2009). DJ-1 deficiency sensitizes dopamine neurons to oxidative stressors in vitro and in the intact CNS (Abeliovich and Beal, 2006). Over-expression appears to protect against oxidative insults.
Lewy bodies in fetal dopaminergic neurons; infectious protein hypothesis
In the May 2008 issue of Nature Medicine three papers were published about postmortem findings of Lewy bodies in a few of the many surviving fetal dopaminergic neurons that were implanted into subjects with PD more than 11 years earlier. In two of the papers three of the subjects – implanted 11, 14 and 16 years earlier – had rare α-synuclein-containing inclusions resembling Lewy bodies (Li et al., 2008; Kordower et al., 2008). In the third paper, these α-synuclein-containing inclusions were sought out but not found in the brains of five subjects who were implanted 9–14 years earlier (Mendez et al., 2008). A fourth patient with PD who died 14 years posttransplantation also was found to have Lewy bodies in some of the otherwise numerous healthy grafted cells (Kordower et al., 2008).
Clearly, the differences between the studies need to be sorted out. The Lewy body findings, however, suggest a factor in the host striatum that can lead to α-synuclein accumulation and Lewy body formation. It also suggests that some factor(s) in the striatum may play a role in PD pathogenesis. This would imply that dopamine nerve terminals – and not the cell bodies – may be the initial site of pathology for the demise of the dopaminergic nigrostriatal neuron. The findings support the concept that the pathogenic process is chronically active in the PD brain, as McGeer and colleagues (1988a) have pointed out by their observations of continuing inflammation in the striatum. One speculation is that a prion-like mechanism might explain how PD pathology can transfer from the host to the graft (Kordower and Brundin; 2009).
Desplats and colleagues (2009) explored that possibility of neuron-to-neuron transfer of labeled α-synuclein from neuronal cells to neural stem cells in both in-vitro and in-vivo systems. α-Synuclein was transmitted via endocytosis to neighboring neurons and neuronal precursor cells, forming Lewy-like inclusions. With impaired autophagy, there was accumulation of transmitted α-synuclein and inclusion formation. The authors suggest that this mechanism may explain the topographical progression of Lewy pathology in PD suggested by Braak et al. (2003). Olanow and Prusiner (2009) proposed that the topographical spread of α-synuclein would be analogous to the spread of prion protein in prion diseases. Supporting this concept, Hansen and colleagues (2011) showed that exogenous dopaminergic cells can take up α-synuclein grafted into mice with overexpressed α-synuclein.
Multiple hit hypothesis with a central role of α-synuclein
Sulzer (2007) divides the genetic mutations resulting in PD into several categories as follows.
With so many etiologic and pathogenic factors all resulting in the common denominator of loss of dopaminergic SNc cells, it is difficult to grasp a single unifying concept. Sulzer (2007) attempts to do just that with his multiple hit hypothesis. He suggests that “multiple hits” that combine toxic stress, for example, from dopamine oxidation or mitochondrial dysfunction, with an inhibition of a neuroprotective response, such as loss of function of parkin or stress-induced autophagic degradation, underlie selective neuronal death.
Despite the multiple hits that may be necessary for sporadic PD, α-synuclein appears to play a central role. This concept is supported by (1) its presence in Lewy bodies, which are found in all cases except those caused by parkin, some cases of LRRK2, and possibly DJ-1; (2) awareness that excess of wild-type α-synuclein can cause PD, and (3) indications that it may have prion-like properties. This would imply that preventing the accumulation of α-synuclein or finding means to eliminate it could be an effective approach to arrest and even reverse the disease process. A diagram illustrating how some of the genetic and pathogenic factors cause dopaminergic neuronal death is presented in Figure 5.25.
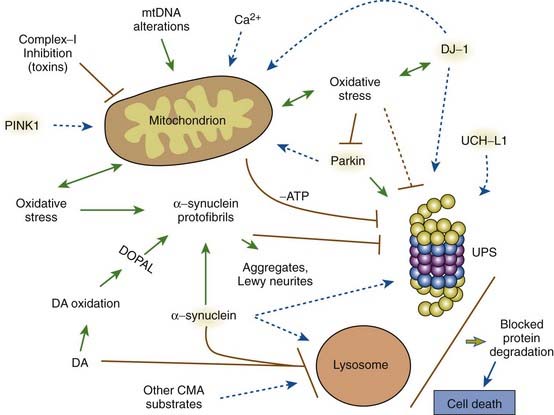
Animal models
A reliable animal model that produces excess α-synuclein and manifests the PD phenotype could prove invaluable. A number of animal models manifesting features of PD have been developed. Perhaps the first was the reserpinized animal. Reserpine, discovered in India as rauwolfia, was known to produce a state of akinesia, i.e., drug-induced parkinsonism. One of the pioneering discoveries was that L-dopa is able to reverse this akinesia (Carlsson et al., 1957). Over the years, other agents were used to damage the dopamine neurons. These include 6-hydroxydopamine, MPTP (Halliday et al., 2009), and rotenone (Pan et al., 2009a). Systemically administered proteasome inhibitors have not been found to consistently produce dopaminergic. When lactacystin, a proteasome inhibitor, is injected in the region of the SN, it does produce pathologic, biochemical, and behavioral changes, suggestive of parkinsonism (Pan et al., 2009b).
The disappointment with the above animal models is that they are good for finding symptomatic therapies, but they have not been successful in helping to find neuroprotective therapies. Drugs that are effective by providing protection in some animal models have not been found to be protective in patients (Rascol et al., 2003; Lang et al., 2006; Olanow et al., 2006; Parkinson Study Group PRECEPT Investigators, 2007). Thus, these models, which are toxin models such as MPTP, have not been proven helpful in finding neuroprotective agents. Perhaps a genetic model would prove useful. Until recently knock-in, knock-out, and transgenic models have resulted in loss of dopaminergic SNc neurons but without the clinical phenotype of PD. More recently Li and colleagues (2009) developed a transgenic mouse model with an altered missense mutation on the LRRK2 gene that produced some aspects of the parkinsonian phenotype, including older age at onset of symptoms and response to levodopa therapy. There was axonal pathology of the nigrostriatal neurons, but no loss of dopaminergic SNc cells. It will be interesting to use this animal model for neuroprotective studies and then see if successful agents will prove beneficial in patients with PD.
Abbott R.D., Ross G.W., Petrovitch H., et al. Bowel movement frequency in late-life and incidental Lewy bodies. Mov Disord. 2007;22(11):1581-1586.
Abeliovich A., Beal M.F. Parkinsonism genes: culprits and clues. J Neurochem. 2006;99(4):1062-1072.
Abeliovich A., Schmitz Y., Farinas I., et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239-252.
Agid Y., Javoy-Agid F., Ruberg M. Biochemistry of neurotransmitters in Parkinson’s disease. In: Marsden C.D., Fahn S., editors. Movement Disorders, vol. 2. London: Butterworths & Co.; 1987:166-230.
Aharon-Peretz J., Badarny S., Rosenbaum H., Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson disease: phenotype-genotype correlation. Neurology. 2005;65(9):1460-1461.
Aharon-Peretz J., Rosenbaum H., Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2004;351(19):1972-1977.
Allen W. Inheritance of the shaking palsy. Arch Intern Med. 1937;60:424-436.
Ascherio A., Chen H., Schwarzschild M.A., et al. Caffeine, postmenopausal estrogen, and risk of Parkinson’s disease. Neurology. 2003;60(5):790-795.
Ascherio A., LeWitt P.A., Xu K., et al. Parkinson Study Group DATATOP Investigators. Urate as a predictor of the rate of clinical decline in Parkinson disease. Arch Neurol. 2009;66(12):1460-1468.
Ascherio A., Weisskopf M.G., O’Reilly E.J., et al. Coffee consumption, gender, and Parkinson’s disease mortality in the cancer prevention study II cohort: the modifying effects of estrogen. Am J Epidemiol. 2004;160(10):977-984.
Ascherio A., Zhang S.M., Hernan M.A., et al. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann Neurol. 2001;50(1):56-63.
Attems J., Jellinger K.A. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease. Neuropathol Appl Neurobiol. 2008;34:466-467.
Banerjee R., Starkov A.A., Beal M.F., Thomas B. Mitochondrial dysfunction in the limelight of Parkinson’s disease pathogenesis. Biochim Biophys Acta. 2009;1792(7):651-653.
Beal M.F. Excitotoxicity and nitric oxide in Parkinson’s disease pathogenesis. Ann Neurol. 1998;44(Suppl 1):S110-S114.
Becker C., Jick S.S., Meier C.R. Use of antihypertensives and the risk of Parkinson disease. Neurology. 2008;70(16 Pt 2):1438-1444.
Benamer H.T.S., Patterson J., Wyper D.J., et al. Correlation of Parkinson’s disease severity and duration with I-123-FP-CIT SPECT striatal uptake. Mov Disord. 2000;15(4):692-698.
Bensinger S.J., Tontonoz P. A Nurr1 pathway for neuroprotection. Cell. 2009;137:26-28.
Bernheimer H., Birkmayer W., Hornykiewicz O., et al. Brain dopamine and the syndromes of Parkinson and Huntington. J Neurol Sci. 1973;20:415-455.
Betarbet R., Sherer T.B., MacKenzie G., et al. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3(12):1301-1306.
Blocq P., Marinesco G. Sur un cas de tremblement Parkinsonien hemiplegique, symptomatique d’une tumeur de peduncule cerebral. CR Soc Biol Paris. 1893;5:105-111.
Bohnen N.I., Albin R.L. Cholinergic denervation occurs early in Parkinson disease. Neurology. 2009;73(4):256-257.
Bohnen N.I., Albin R.L. The cholinergic system and Parkinson disease. Behav Brain Res. 2010. Jan 7 [Epub ahead of print]
Bohnen N.I., Müller M.L., Koeppe R.A., et al. History of falls in Parkinson disease is associated with reduced cholinergic activity. Neurology. 2009;73(20):1670-1676.
Boileau I., Guttman M., Rusjan P., et al. Decreased binding of the D3 dopamine receptor-preferring ligand [11C]-(+)-PHNO in drug-naive Parkinson’s disease. Brain. 2009;132:1366-1375.
Bonifati V., Rizzu P., van Baren M.J., et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299(5604):256-259.
Bower J.H., Maraganore D.M., McDonnell S.K., Rocca W.A. Influence of strict, intermediate, and broad diagnostic criteria on the age- and sex-specific incidence of Parkinson’s disease. Mov Disord. 2000;15(5):819-825.
Braak H., Bohl J.R., Muller C.M., et al. Stanley Fahn Lecture 2005: the staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov Disord. 2006;21:2042-2051.
Braak H., Del Tredici K., Rub U., et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197-211.
Braak H., Ghebremedhin E., Rub U., et al. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318(1):121-134.
Bras J.M., Guerreiro R.J., Ribeiro M.H., et al. G2019S dardarin substitution is a common cause of Parkinson’s disease in a Portuguese cohort. Mov Disord. 2005;20(12):1653-1655.
Brissaud E. Leçons sur les Maladies Nerveuses. Paris: Masson et Cie; 1895.
Bronstein J., Vickrey B. Pallido-pyramidal syndrome. Parkinsonism Relat Disord. 1996;2(2):105-106.
Brooks D.J. Detection of preclinical Parkinson’s disease with PET. Geriatrics. 1991;46(Suppl 1):25-30.
Brooks D.J. Examining Braak’s hypothesis by imaging Parkinson’s disease. Mov Disord. 2010;25(Suppl 1):S83-S88.
Brooks D.J., Ibanez V., Sawle G.V., et al. Differing patterns of striatal F-18-DOPA uptake in Parkinson’s disease, multiple system atrophy, and progressive supranuclear palsy. Ann Neurol. 1990;28:547-555.
Broussolle E., Dentresangle C., Landais P., et al. The relation of putamen and caudate nucleus 18F-Dopa uptake to motor and cognitive performances in Parkinson’s disease. J Neurol Sci. 1999;166(2):141-151.
Burke R.E., Dauer W.T., Vonsattel J.P. A critical evaluation of the Braak staging scheme for Parkinson’s disease. Ann Neurol. 2008;64(5):485-491.
Burke W.J., Kumar V.B., Pandey N., et al. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 2008;115(2):193-203.
Burn D.J., Mark M.H., Playford E.D., et al. Parkinson’s disease in twins studied with 18F-dopa and positron emission tomography. Neurology. 1992;42(10):1894-1900.
Cabin D.E., Shimazu K., Murphy D., et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci. 2002;22(20):8797-8807.
Carlsson A., Lindqvist M., Magnusson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature. 1957;180:1200.
Carlsson A., Winblad B. Influence of age and time interval between death and autopsy on dopamine and 3-methoxytyramine levels in human basal ganglia. J Neural Transm. 1976;38:271-276.
Chan C.S., Guzman J.N., Ilijic E., et al. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature. 2007;447(7148):1081-1086.
Charcot J.M. translated by Sigerson G. second ed. Clinical Lectures on Diseases of the Nervous System, vol. I. Philadelphia: Henry C. Lea. 1879.
Chartier-Harlin M.-C., Dachsel J., Hulihan M., et al. EIF4G1 mutations in familial parkinsonism. Parkinsonism Relat Disord. 2009;13(Suppl 3):S145-S146.
Checkoway H., Powers K., Smith-Weller T., et al. Parkinson’s disease risks associated with cigarette smoking, alcohol consumption, and caffeine intake. Am J Epidemiol. 2002;155(8):732-738.
Chou A.P., Maidment N., Klintenberg R., et al. Ziram causes dopaminergic cell damage by inhibiting e1 ligase of the proteasome. J Biol Chem. 2008;283(50):34696-34703.
Clark L.N., Nicolai A., Afridi S., et al. Pilot association study of the beta-glucocerebrosidase N370S allele and Parkinson’s disease in subjects of Jewish ethnicity. Mov Disord. 2005;20(1):100-103.
Clark L.N., Ross B.M., Wang Y., et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology. 2007;69(12):1270-1277.
Clark L.N., Wang Y., Karlins E., et al. Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology. 2006;67(10):1786-1791.
Clayton D.F., George J.M. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998;21(6):249-254.
Conway K.A., Lee S.J., Rochet J.C., et al. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc Natl Acad Sci USA. 2000;97(2):571-576.
Cookson M.R. alpha-Synuclein and neuronal cell death. Mol Neurodegener. 2009;4:9.
Cookson M.R. DJ-1, PINK1, and their effects on mitochondrial pathways. Mov Disord. 2010;25(Suppl 1):S44-S48.
Costello S., Cockburn M., Bronstein J., et al. Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am J Epidemiol. 2009;169(8):919-926.
Cuervo A.M., Stefanis L., Fredenburg R., et al. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292-1295.
Cuervo A.M., Wong E.S., Martinez-Vicente M. Protein degradation, aggregation, and misfolding. Mov Disord. 2010;25(Suppl 1):S49-S54.
Dauer W., Ho C.C. The biology and pathology of the familial Parkinson’s disease protein LRRK2. Mov Disord. 2010;25(Suppl 1):S40-S43.
Dauer W., Kholodilov N., Vila M., et al. Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci USA. 2002;99(22):14524-14529.
Dauer W., Przedborski S. Parkinson’s Disease: Mechanisms and Models. Neuron. 2003;39:889-909.
Davison C. Pallido-pyramidal disease. J Neuropathol Exp Neurol. 1954;13:50-59.
Davidzon G., Greene P., Mancuso M., et al. Early-onset familial parkinsonism due to POLG mutations. Ann Neurol. 2006;59(5):859-862.
Dawson T.M., Dawson V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov Disord. 2010;25(Suppl 1):S32-S39.
De Cock V.C., Vidailhet M., Leu S., et al. Restoration of normal motor control in Parkinson’s disease during REM sleep. Brain. 2007;130(Pt 2):450-456.
DelleDonne A., Klos K.J., Fujishiro H., et al. Incidental Lewy body disease and preclinical Parkinson disease. Arch Neurol. 2008;65(8):1074-1080.
Del Tredici K., Rub U., De Vos R.A., et al. Where does Parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol. 2002;61(5):413-426.
de Rijk M.C., Launer L.J., Berger K., et al. Prevalence of Parkinson’s disease in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000;54(11 Suppl 5):S21-S23.
Desplats P., Lee H.J., Bae E.J., et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA. 2009;106(31):13010-13015.
Dexter D.T., Carter C.J., Wells F.R., et al. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J Neurochem. 1989;52:381-389.
Dexter D.T., Wells F.R., Lees A.J., et al. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J Neurochem. 1989;52:1830-1836.
Dickson D.W., Fujishiro H., DelleDonne A., et al. Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol. 2008;115(4):437-444.
Dickson D.W., Uchikado H., Fujishiro H., Tsuboi Y. Evidence in favor of Braak staging of Parkinson’s disease. Mov Disord. 2010;25(Suppl 1):S78-S82.
Di Fonzo A., Dekker M.C., Montagna P., et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology. 2009;72(3):240-245.
Dorsey E.R., Constantinescu R., Thompson J.P., et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68(5):384-386.
Duffy P.E., Tennyson V.M. Phase and electron microscopic observations of Lewy bodies and melanin granules in the substantia nigra and locus caeruleus in Parkinson’s disease. J Neuropath Exp Neurol. 1965;24:398-414.
Duvoisin R.C., Johnson W.G. Hereditary Lewy-body parkinsonism and evidence for a genetic etiology of Parkinson’s disease. Brain Pathol. 1992;2(4):309-320.
Duvoisin R.C., Eldridge R., Williams A., et al. Twin study of Parkinson disease. Neurology. 1981;31:77-80.
Dwork A.J., Balmaceda C., Fazzini E.A., et al. Dominantly inherited, early-onset parkinsonism: Neuropathology of a new form. Neurology. 1993;43:69-74.
Ehringer H., Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Klin Wochenschr. 1960;38:1236-1239.
Eidelberg D., Moeller J.R., Isikawa T., et al. Assessment of disease severity in parkinsonism with fluorine-18- fluorodeoxyglucose and PET. J Nucl Med. 1995;36:378-383.
Elbaz A., Bower J.H., Maraganore D.M., et al. Risk tables for parkinsonism and Parkinson’s disease. J Clin Epidemiol. 2002;55(1):25-31.
Elbaz A., Bower J.H., Peterson B.J., et al. Survival study of Parkinson disease in Olmsted county, Minnesota. Arch Neurol. 2003;60(1):91-96.
Fahn S., Cohen G. The oxidant stress hypothesis in Parkinson’s disease: evidence supporting it. Ann Neurol. 1992;32:804-812.
Farrer M., Chan P., Chen R., et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol. 2001;50:293-300.
Farrer M., Gwinn-Hardy K., Muenter M., et al. A chromosome 4p haplotype segregating with Parkinson’s disease and postural tremor. Hum Mol Genet. 1999;8:81-85.
Farrer M.J., Hulihan M.M., Kachergus J.M., et al. DCTN1 mutations in Perry syndrome. Nat Genet. 2009;41(2):163-165.
Farrer M., Kachergus J., Forno L., et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55(2):174-179.
Farrer M.J., Stone J.T., Lin C.H., et al. Lrrk2 G2385R is an ancestral risk factor for Parkinson’s disease in Asia. Parkinsonism Relat Disord. 2007;13(2):89-92.
Fearnley J.M., Lees A.J. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114:2283-2301.
Foix C., Nicolesco I. Anatomie Cerebrale; Les Noyeux Gris Centraux et la Region Mesencephalo-Sous-Opitique, Suive d’un Appendice sur l’Anatomie Pathologique de la Maladie de Parkinson. Paris: Masson et Cie; 1925.
Forno L.S. Pathology of Parkinson’s disease. In: Marsden C.D., Fahn S., editors. Movement Disorders. London: Butterworth Scientific; 1982:25-40.
Forno L.S. Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol. 1996;55:259-272.
Forno L.S., Norville R.L. Ultrastructure of Lewy bodies in the stellate ganglion. Acta Neuropathol. 1976;34(3):183-197.
Foroud T., Uniacke S.K., Liu L., et alParkinson Study Group. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology. 2003;60(5):796-801.
Frigerio R., Fujishiro H., Ahn T.B., et al. Incidental Lewy body disease: Do some cases represent a preclinical stage of dementia with Lewy bodies? Neurobiol Aging. 2009. Jun 26 [Epub ahead of print]
Frigerio R., Sanft K.R., Grossardt B.R., et al. Chemical exposures and Parkinson’s disease: a population-based case-control study. Mov Disord. 2006;21(10):1688-1692.
Fuchs J., Nilsson C., Kachergus J., et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology. 2007;68(12):916-922.
Funayama M., Hasegawa K., Kowa H., et al. A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol. 2002;51(3):296-301.
Fung H.C., Chen C.M., Hardy J., et al. A common genetic factor for Parkinson disease in ethnic Chinese population in Taiwan. BMC Neurol. 2006;6:47.
Gagne J.J., Power M.C. Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology. 2010;74(12):995-1002.
Gan-Or Z., Giladi N., Rozovski U., et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology. 2008;70:2277-2283.
Gasser T., Muller-Myhsok B., Wszolek Z.K., et al. A susceptibility locus for Parkinson’s disease maps to chromosome 2p13. Nat Genet. 1998;18:262-265.
Gilks W.P., Abou-Sleiman P.M., Gandhi S., et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet. 2005;365(9457):415-416.
Goetz C.G. Charcot, the Clinician: The Tuesday Lessons. Excerpts from Nine Case Presentations on General Neurology Delivered at the Salpetriere Hospital in 1887–88 By Jean-Martin Charcot. Translated with Commentary. New York: Raven Press; 1987. 123–124
Goker-Alpan O., Giasson B.I., Eblan M.J., et al. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology. 2006;67(5):908-910.
Goker-Alpan O., Schiffmann R., LaMarca M.E., et al. Parkinsonism among Gaucher disease carriers. J Med Genet. 2004;41(12):937-940.
Golbe L.I., Di Iorio G., Bonavita V., et al. A large kindred with autosomal dominant Parkinson’s disease. Ann Neurol. 1990;27:276-282.
Greenfield J.G., Bosanquet F.D. The brain-stem lesions in parkinsonism. J Neurol Neurosurg Psychiatry. 1953;16:213-226.
Guo Y., Jankovic J., Zhu S., et al. GIGYF2 Asn56Ser and Asn457Thr mutations in Parkinson disease patients. Neurosci Lett. 2009;454:209-211.
Gupta A., Dawson V.L., Dawson T.M. What causes cell death in Parkinson’s disease? Ann Neurol. 2008;64(Suppl 2):S3-S15.
Guzman J.N., Sanchez-Padilla J., Wokosin D., et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468(7324):696-700.
Gwinn-Hardy K., Mehta N.D., Farrer M., et al. Distinctive neuropathology revealed by alpha-synuclein antibodies in hereditary parkinsonism and dementia linked to chromosome 4p. Acta Neuropathol (Berl). 2000;99(6):663-672.
Halliday G., Hely M., Reid W., Morris J. The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol. 2008;115:409-415.
Halliday G., Herrero M.T., Murphy K., et al. No Lewy pathology in monkeys with over 10 years of severe MPTP Parkinsonism. Mov Disord. 2009;24:1519-1523.
Halliday G.M., Macdonald V., Henderson J.M. A comparison of degeneration in motor thalamus and cortex between progressive supranuclear palsy and Parkinson’s disease. Brain. 2005;128(Pt 10):2272-2280.
Hansen C., Angot E., Bergström A.L., et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 2011;121(2):715-725.
Hardy J., Cai H., Cookson M.R., et al. Genetics of Parkinson’s disease and parkinsonism. Ann Neurol. 2006;60(4):389-398.
Hassler R. Zur Pathologie der Paralysis Agitans und des postenzephalitischen Parkinsonismus. J Psychol Neurol. 1938;48:387-476.
Hatano Y., Li Y., Sato K., et al. Novel PINK1 mutations in early-onset parkinsonism. Ann Neurol. 2004;56(3):424-427.
Hattori N., Shimura H., Kubo S., et al. Autosomal recessive juvenile parkinsonism: a key to understanding nigral degeneration in sporadic Parkinson’s disease. Neuropathology. 2000;20(Suppl):S85-S90.
Haugarvoll K., Wszolek Z.K. PARK8 LRRK2 parkinsonism. Curr Neurol Neurosci Rep. 2006;6(4):287-294.
Hawkes C.H., Del Tredici K., Braak H. A timeline for Parkinson’s disease. Parkinsonism Relat Disord. 2010;16(2):79-84.
Healy D.G., Falchi M., O’Sullivan S.S., et al. International LRRK2 Consortium. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7(7):583-590.
Hedrich K., Hagenah J., Djarmati A., et al. Clinical spectrum of homozygous and heterozygous PINK1 mutations in a large German family with Parkinson disease: role of a single hit? Arch Neurol. 2006;63(6):833-838.
Henchcliffe C., Beal M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4(11):600-609.
Henderson J.M., Carpenter K., Cartwright H., Halliday G.M. Degeneration of the thalamic caudal intralaminar nuclei in Parkinson’s disease. Ann Neurol. 2000;47:345-352.
Hernan M.A., Takkouche B., Caamano-Isorna F., Gestal-Otero J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann Neurol. 2002;52(3):276-284.
Hicks A.A., Petursson H., Jonsson T., et al. A susceptibility gene for late-onset idiopathic Parkinson’s disease. Ann Neurol. 2002;52(5):549-555.
Hirano S., Eckert T., Flanagan T., Eidelberg D. Metabolic networks for assessment of therapy and diagnosis in Parkinson’s disease. Mov Disord. 2009;24(Suppl 2):S725-S731.
Hirsch E., Graybiel A.M., Agid Y. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature. 1988;334:345-348.
Hirsch E.C., Graybiel A.M., Agid Y. Selective vulnerability of pigmented dopaminergic neurons in Parkinson’s disease. Acta Neurol Scand. 1989;80(Suppl 126):19-22.
Hirsch E.C., Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382-397.
Hirsch E.C., Hunot S., Faucheux B., et al. Dopaminergic neurons degenerate by apoptosis in Parkinson’s disease. Mov Disord. 1999;14(2):383-385.
Hirtz D., Thurman D.J., Gwinn-Hardy K., et al. How common are the “common” neurologic disorders? Neurology. 2007;68(5):326-337.
Hishikawa N., Hashizume Y., Yoshida M., Sobue G. Clinical and neuropathological correlates of Lewy body disease. Acta Neuropathol. 2003;105(4):341-350.
Hoehn M.M., Yahr M.D. Parkinsonism: Onset, progression and mortality. Neurology. 1967;17:427-442.
Hornykiewicz O. Dopamine (3-hydroxytyramine) and brain function. Pharmacol Rev. 1966;18:925-964.
Hornykiewicz O. Brain neurotransmitter changes in Parkinson’s disease. In: Marsden C.D., Fahn S., editors. Movement Disorders. London: Butterworth Scientific; 1982:41-58.
Horowitz G., Greenberg J. Pallido-pyramidal syndrome treated with levodopa. J Neurol Neurosurg Psychiatry. 1975;38(3):238-240.
Hughes A.J., Daniel S.E., Ben-Shlomo Y., Lees A.J. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125(Pt 4):861-870.
Hughes A.J., Daniel S.E., Kilford L., Lees A.J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease – a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181-184.
Iacopino A., Christakos S., German D., et al. Calbindin-D28K-containing neurons in animal models of neurodegeneration: possible protection from excitotoxicity. Brain Res Mol Brain Res. 1992;13(3):251-261.
Jang H., Boltz D., Sturm-Ramirez K., et al. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci USA. 2009;106(33):14063-14068.
Jang H., Boltz D.A., Webster R.G., Smeyne R.J. Viral parkinsonism. Biochim Biophys Acta. 2009;1792(7):714-721.
Jellinger K. The pathology of parkinsonism. In: Marsden C.D., Fahn S., editors. Movement Disorders, vol. 2. London: Butterworths & Co.; 1987:124-165.
Jellinger K.A. Formation and development of Lewy pathology: a critical update. J Neurol. 2009;256(Suppl 3):270-279.
Jellinger K.A. Absence of alpha-synuclein pathology in postencephalitic parkinsonism. Acta Neuropathol.. 2009;118(3):371-379.
Jenner P., Olanow C.W. The pathogenesis of cell death in Parkinson’s disease. Neurology. 2006;66:S24-S36.
Johnson W.G., Hodge S.E., Duvoisin R. Twin studies and the genetics of Parkinson’s disease: A reappraisal. Mov Disord. 1990;5:187-194.
Kahle P.J., Waak J., Gasser T. DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic Biol Med. 2009;47(10):1354-1361.
Kalaitzakis M.E., Graeber M.B., Gentleman S.M., Pearce R.K. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease: a critical analysis of alpha-synuclein staging. Neuropathol Appl Neurobiol. 2008;34:284-295.
Kalaitzakis M.E., Graeber M.B., Gentleman S.M., Pearce R.K. Evidence against a reliable staging system of alpha-synuclein pathology in Parkinson’s disease. Neuropathol Appl Neurobiol. 2009;35:125-126.
Karamohamed S., DeStefano A.L., Wilk J.B., et alGenePD Study Group. A haplotype at the PARK3 locus influences onset age for Parkinson’s disease: the GenePD study. Neurology. 2003;61(11):1557-1561.
Kempster P.A., Williams D.R., Selikhova M., et al. Patterns of levodopa response in Parkinson’s disease: a clinico-pathological study. Brain. 2007;130(Pt 8):2123-2128.
Kim Y., Park J., Kim S., et al. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377(3):975-980.
Kitada T., Asakawa S., Hattori N., et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605-608.
Kopito R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10(12):524-530.
Kordower J.H., Brundin P. Lewy body pathology in long-term fetal nigral transplants: is Parkinson’s disease transmitted from one neural system to another? Neuropsychopharmacology. 2009;34(1):254.
Kordower J.H., Chu Y., Hauser R.A., et al. Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov Disord. 2008;23(16):2303-2306.
Kraoua I., Stirnemann J., Ribeiro M.J., et al. Parkinsonism in Gaucher’s disease type 1: ten new cases and a review of the literature. Mov Disord. 2009;24:1524-1530.
Krüger R., Kuhn W., Muller T., et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106-108.
Kurian M.A., Morgan N.V., MacPherson L., et al. Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology. 2008;70(18):1623-1629.
Kurian M.A., Zhen J., Cheng S.Y., et al. Homozygous loss-of-function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism-dystonia. J Clin Invest. 2009;119(6):1595-1603.
Kurland L.T. Epidemiology: incidence, geographic distribution and genetic considerations. In: Fields W.S., editor. Pathogenesis and treatment of parkinsonism. Springfield, IL: Charles C Thomas; 1958:5-49.
Lang A.E., Gill S., Patel N.K., et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol. 2006;59(3):459-466.
Langston J.W. The Parkinson’s complex: parkinsonism is just the tip of the iceberg. Ann Neurol. 2006;59(4):591-596.
Langston J.W., Ballard P., Tetrud J.W., Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979-980.
Lansbury P.T.Jr, Brice A. Genetics of Parkinson’s disease and biochemical studies of implicated gene products. Curr Opin Genet Develop. 2002;12:299-306.
Lansbury P.T., Lashuel H.A. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443(7113):774-779.
Lautier C., Goldwurm S., Dürr A., et al. Mutations in the GIGYF2 (TNRC15) gene at the PARK11 locus in familial Parkinson disease. Am J Hum Genet. 2008;82(4):822-833.
Le W., Chen S., Jankovic J. Etiopathogenesis of Parkinson’s disease: A new beginning? Neuroscientist. 2009;15:28-35.
Lee B.D., Shin J.H., Van Kampen J., et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat Med. 2010;16(9):998-1000.
Lees A.J., Hardy J., Revesz T. Parkinson’s disease. Lancet. 2009;373(9680):2055-2066.
Leroy E., Boyer R., Auburger G., et al. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395:451-452.
Lesage S., Dürr A., Tazir M., et al. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med. 2006;354:422-423.
Lev N., Ickowicz D., Barhum Y., et al. DJ-1 protects against dopamine toxicity. J Neural Transm. 2009;116(2):151-160.
Lewy F.H. Paralysis agitans. I. Pathologische Anatomie. In: Lewandowsky M., editor. Handbuch der Neurologie. Berlin: Springer; 1912:920-933.
Lewy F.H. Zur pathologischen Anatomie der Paralysis agitans. Dtsch Z Nervenheilk. 1914;1:50-55.
Li J.Y., Englund E., Holton J.L., et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14(5):501-503.
Li Y., Liu W., Oo T.F., et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat Neurosci. 2009;12(7):826-828.
Liu Z., Meray R.K., Grammatopoulos T.N., et al. Membrane-associated farnesylated UCH-L1 promotes alpha-synuclein neurotoxicity and is a therapeutic target for Parkinson’s disease. Proc Natl Acad Sci USA. 2009;106(12):4635-4640.
Lotharius J., Barg S., Wiekop P., et al. Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J Biol Chem. 2002;277:38884-38894.
Lücking C.B., Durr A., Bonifati V., et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med. 2000;342(21):1560-1567.
MacLeod D., Dowman J., Hammond R., et al. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52(4):587-593.
Maroteaux L., Campanelli J.T., Scheller R.H. Synuclein: a neuron-specific protein localized to the nucleus and presynapic nerve terminal. J Neurosci. 1988;8:2804-2815.
Martí-Massó J.F., Ruiz-Martínez J., Bolaño M.J., et al. Neuropathology of Parkinson’s disease with the R1441G mutation in LRRK2. Mov Disord. 2009;24(13):1998-2001.
Martilla R.J., Kaprio J., Kostenvuo M.D., Rinne U.K. Parkinson’s disease in a nationwide twin cohort. Neurology. 1988;38(1):217-219.
Martin H.L., Teismann P. Glutathione–a review on its role and significance in Parkinson’s disease. FASEB J. 2009;23(10):3263-3272.
Martinez-Vicente M., Cuervo A.M. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6(4):352-361.
Martinez-Vicente M., Talloczy Z., Kaushik S., et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118(2):777-788.
Matsumine H., Yamamura Y., Kobayashi T., et al. Early onset parkinsonism with diurnal fluctuation maps to a locus for juvenile parkinsonism. Neurology. 1998;50:1340-1345.
Mayeux R., Stern Y., Cote L., Williams J.B.W. Altered serotonin metabolism in depressed patients with Parkinson’s disease. Neurology. 1984;34:642-646.
McGeer P.L., Itagaki S., Akiyama H., McGeer E.G. Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol. 1988;24:574-576.
McGeer P.L., Itagaki S., Boyes B.E., McGeer E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285-1291.
McGeer P.L., McGeer E.G., Suzuki J.S. Aging and extrapyramidal function. Arch Neurol. 1977;34:33-35.
Mendez I., Viñuela A., Astradsson A., et al. Dopamine neurons implanted into people with Parkinson’s disease survive without pathology for 14 years. Nat Med. 2008;14(5):507-509.
Meynert T. Ueber Beitrage zur differential Diagnose der paralytischen. Irrsinns. Wiener Med Presse. 1871;11:645-647.
Mezey E., Dehejia A.M., Harta G., et al. Alpha synuclein is present in Lewy bodies in sporadic Parkinson’s disease. Mol Psychiatry. 1998;3(6):493-499.
Mitsui J., Mizuta I., Toyoda A., et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol. 2009;66:571-576.
Mizuno Y., Ohta S., Tanaka M., et al. Deficiencies in complex subunits of the respiratory chain in Parkinson’s disease. Biochem Biophys Res Commun. 1989;163:1450-1455.
Mjönes H. Paralysis agitans. A clinical and genetic study. Acta Psychiatr Neurol. 1949;54:1-195.
Mochizuki H., Goto K., Mori H., Mizuno Y. Histochemical detection of apoptosis in Parkinson’s disease. J Neurol Sci. 1996;137(2):120-123.
Moore D.J., West A.B., Dawson V.L., Dawson T.M. Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci. 2005;28:57-87.
Morgan N.V., Westaway S.K., Morton J.E., et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet. 2006;38(7):752-754.
Morrish P.K., Sawle G.V., Brooks D.J. An (F-18)dopa-PET and clinical study of the rate of progression in Parkinson’s disease. Brain. 1996;119:585-591.
Mortiboys H., Thomas K.J., Koopman W.J., et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol. 2008;64(5):555-565.
Mosharov E.V., Larsen K.E., Kanter E., et al. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62(2):218-229.
Mosharov E.V., Staal R.G., Bove J., et al. Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci. 2006;26(36):9304-9311.
Muenter M.D., Forno L.S., Hornykiewicz O., et al. Hereditary form of parkinsonism-dementia. Ann Neurol. 1998;43:768-781.
Najim al-Din A.S., Wriekat A., Mubaidin A., et al. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol Scand. 1994;89(5):347-352.
Nandhagopal R., Kuramoto L., Schulzer M., et al. Longitudinal progression of sporadic Parkinson’s disease: a multi-tracer positron emission tomography study. Brain. 2009;132(Pt 11):2970-2979.
Narendra D., Tanaka A., Suen D.F., Youle R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795-803.
Neumann J., Bras J., Deas E., et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain. 2009;132(Pt 7):1783-1794.
Ng C.H., Mok S.Z., Koh C., et al. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci. 2009;29(36):11257-11262.
Nichols W.C., Kissell D.K., Pankratz N., et alParkinson Study Group-PROGENI Investigators. Variation in GIGYF2 is not associated with Parkinson disease. Neurology. 2009;72(22):1886-1892.
Nichols W.C., Pankratz N., Hernandez D., et alParkinson Study Group-PROGENI investigators. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet. 2005;365(9457):410-412.
Nicklas W.J., Youngster S.K., Kindt M.V., Heikkila R.E. MPTP, MPP+ and mitochondrial function. Life Sci. 1987;40:721-729.
Olanow C.W., Prusiner S.B. Is Parkinson’s disease a prion disorder? Proc Natl Acad. Sci USA. 2009;106(31):12571-12572.
Olanow C.W., Schapira A.H., LeWitt P.A., et al. TCH346 as a neuroprotective drug in Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol. 2006;5(12):1013-1020.
Orsucci D., Caldarazzo Lenco E., Mancuso M., Siciliano G. POLA1-related and other “Mitochondrial Parkinsonisms”: an overview. J Mol Neurosci. 2011;44(1):17-24.
Outeiro T.F., Kontopoulos E., Altmann S.M., et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317(5837):516-519.
Ozelius L.J., Foroud T., May S., et alNorth American Multiple System Atrophy Study Group. G2019S mutation in the leucine-rich repeat kinase 2 gene is not associated with multiple system atrophy. Mov Disord. 2007;22(4):546-549.
Ozelius L.J., Senthil G., Saunders-Pullman R., et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2006;354(4):424-425.
Paisan-Ruiz C., Bhatia K.P., Li A., et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65(1):19-23.
Paisan-Ruiz C., Jain S., Evans E.W., et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44(4):595-600.
Pan T., Kondo S., Le W., Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 2008;131:1969-1978.
Pan T., Rawal P., Wu Y., et al. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience. 2009;164(2):541-551.
Pan T., Zhu W., Zhao H., et al. Nurr1 deficiency predisposes to lactacystin-induced dopaminergic neuron injury in vitro and in vivo. Brain Research. 2009;1222:222-229.
Pankratz N., Nichols W.C., Uniacke S.K., et alParkinson Study Group. Significant linkage of Parkinson disease to chromosome 2q36–37. Am J Hum Genet. 2003;72(4):1053-1057.
Pankratz N., Nichols W.C., Uniacke S.K., et alParkinson Study Group. Genome-wide linkage analysis and evidence of gene-by-gene interactions in a sample of 362 multiplex Parkinson disease families. Hum Mol Genet.. 2003;12(20):2599-2608.
Parkinson J. An Essay on the Shaking Palsy. London: Sherwood, Neely, and Jones; 1817.
Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early parkinson’s disease. N Engl J Med. 1993;328:176-183.
Parkinson Study Group. A controlled, randomized, delayed-start study of rasagiline in early Parkinson disease. Arch Neurol. 2004;61(4):561-566.
Parkinson Study Group PRECEPT Investigators. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology. 2007;69(15):1480-1490.
Parkkinen L., Kauppinen T., Pirttilä T., et al. Alpha-synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol. 2005;57(1):82-91.
Parkkinen L., Pirttilä T., Alafuzoff I. Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol. 2008;115(4):399-407.
Pavese N., Khan N.L., Scherfler C., et al. Nigrostriatal dysfunction in homozygous and heterozygous parkin gene carriers: an 18F-dopa PET progression study. Mov Disord. 2009;24(15):2260-2266.
Perry T.L., Bratty P.J., Hansen S., et al. Hereditary mental depression and Parkinsonism with taurine deficiency. Arch Neurol. 1975;32:108-113.
Perry T.L., Godin D.V., Hansen S. Parkinson’s disease: a disorder due to nigral glutathione deficiency? Neurosci Lett. 1982;33:305-310.
Peter D., Liu Y., Sternini C., et al. Differential expression of two vesicular monoamine transporters. J. Neurosci. 1995;15:6179-6188.
Petrovitch H., Abbott R.D., Ross G.W., et al. Bowel movement frequency in late-life and substantia nigra neuron density at death. Mov Disord. 2009;24(3):371-376.
Plun-Favreau H., Gandhi S., Wood-Kaczmar A., et al. What have PINK1 and HtrA2 genes told us about the role of mitochondria in Parkinson’s disease? Ann. NY Acad Sci. 2008;1147:30-36.
Polymeropoulos M.H., Higgins J.J., Golbe L.I., et al. Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science. 1996;274:1197-1199.
Polymeropoulos M.H., Lavedan C., Leroy E., et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045-2047.
Ponsen M.M., Stoffers D., Booij J., et al. Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann Neurol. 2004;56(2):173-181.
Postuma R.B., Gagnon J.F., Rompré S., Montplaisir J.Y. Severity of REM atonia loss in idiopathic REM sleep behavior disorder predicts Parkinson disease. Neurology. 2010;74(3):239-244.
Postuma R.B., Gagnon J.F., Vendette M., et al. Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology. 2009;72(15):1296-1300.
Pramstaller P.P., Schlossmacher M.G., Jacques T.S., et al. Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol. 2005;58(3):411-422.
Przedborski S. Inflammation and Parkinson’s disease pathogenesis. Mov Disord. 2010;25(Suppl 1):S55-S57.
Racette B.A., Good L.M., Kissel A.M., et al. A population-based study of parkinsonism in an Amish community. Neuroepidemiology. 2009;33(3):225-230.
Rajput A.H., Rozdilsky B., Rajput A. Accuracy of clinical diagnosis in parkinsonism – a prospective study. Can J Neurol Sci. 1991;18:275-278.
Rascol O., Olanow C.W., Brooks D., et al. Effect of riluzole on Parkinson’s disease progression: A double-blind placebo-controlled study. Neurology. 2003;60(Suppl 1):A288.
Rascol O., Schelosky L. 123I-metaiodobenzylguanidine scintigraphy in Parkinson’s disease and related disorders. Mov Disord. 2009;24(Suppl 2):S732-S741.
Richardson J.R., Shalat S.L., Buckley B. Elevated serum pesticide levels and risk of Parkinson disease. Arch Neurol. 2009;66:870-875.
Riederer P., Wuketich S. Time course of nigrostriatal degeneration in Parkinson’s disease. J Neural Transm. 1976;38:277-301.
Ritz B., Ascherio A., Checkoway H., et al. Pooled analysis of tobacco use and risk of Parkinson disease. Arch Neurol. 2007;64(7):990-997.
Ritz B., Rhodes S.L., Qian L., et al. L-type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol. 2010;67(5):600-606.
Rocca W.A., Bower J.H., McDonnell S.K., et al. Time trends in the incidence of parkinsonism in Olmsted County, Minnesota. Neurology. 2001;57(3):462-467.
Rochet J.C., Outeiro T.F., Conway K.A., et al. Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson’s disease. J Mol Neurosci. 2004;23(1–2):23-34.
Rosenbloom B., Balwani M., Bronstein J.M., et al. The incidence of Parkinsonism in patients with type 1 Gaucher disease: Data from the ICGG Gaucher Registry. Blood Cell Mol Dis. 2011;46(1):95-102.
Ross G.W., Abbott R.D., Petrovitch H., et al. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA. 2000;283(20):2674-2679.
Ross G.W., Petrovitch H., Abbott R.D., et al. Association of olfactory dysfunction with risk for future Parkinson’s disease. Ann Neurol. 2008;63(2):167-173.
Saijo K., Winner B., Carson C.T., et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47-59.
Samaranch L., Lorenzo-Betancor O., Arbelo J.M., et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain. 2010;133(Pt 4):1128-1142.
Sanchez-Ramos J.R., Overvik E., Ames B.N. A marker of oxyradical-mediated DNA damage (8-hydroxy-2′deoxyguanosine) is increased in nigro-striatum of Parkinson’s disease brain. Neurodegeneration. 1994;3:197-204.
Satake W., Nakabayashi Y., Mizuta I., et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41(12):1303-1307.
Savica R., Carlin J.M., Grossardt B.R., et al. Medical records documentation of constipation preceding Parkinson disease: A case-control study. Neurology. 2009;73(21):1752-1758.
Scaglione C., Vignatelli L., Plazzi G., et alBologna, Genova, Parma and Pisa Universities group for the study of REM Sleep Behavior Disorder in Parkinson’s Disease. REM sleep behaviour disorder in Parkinson’s disease: a questionnaire-based study. Neurol Sci. 2005;25(6):316-321.
Schapira A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008;7:97-109.
Schapira A.H.V., Cooper J.M., Dexter D., et al. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem. 1990;54:823-827.
Schneider S.A., Paisan-Ruiz C., Quinn N.P., et al. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord. 2010;25(8):979-984.
Schoenberg B.S., Anderson D.W., Haerer A.F. Prevalence of Parkinson’s disease in the biracial population of Copiah County, Mississippi. Neurology. 1985;35:841-845.
Schwarzschild M.A., Chen J.F., Ascherio A. Caffeinated clues and the promise of adenosine A(2A) antagonists in PD. Neurology. 2002;58(8):1154-1160.
Schwarzschild M.A., Schwid S.R., Marek K., et alParkinson Study Group PRECEPT Investigators. Serum urate as a predictor of clinical and radiographic progression in Parkinson disease. Arch Neurol. 2008;65(6):716-723.
Seibyl J.P., Marek K.L., Quinlan D., et al. Decreased single-photon emission computed tomographic ((123))I beta-CIT striatal uptake correlates with symptom severity in Parkinson’s disease. Ann Neurol. 1995;38:589-598.
Seidel K., Schöls L., Nuber S., et al. First appraisal of brain pathology owing to A30P mutant alpha-synuclein. Ann Neurol. 2010;67(5):684-689.
Sharma M., Mueller J.C., Zimprich A., et alEuropean Consortium on Genetic Susceptibility in Parkinson’s Disease (GSPD). The sepiapterin reductase gene region reveals association in the PARK3 locus:analysis of familial and sporadic Parkinson’s disease in European populations. J Med Genet. 2006;43(7):557-562.
Shiba K., Arai T., Sato S., et al. Parkin stabilizes PINK1 through direct interaction. Biochem Biophys Res Commun. 2009;383(3):331-335.
Shimada H., Hirano S., Shinotoh H., et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology. 2009;73(4):273-278.
Sian J., Dexter D.T., Lees A.J., et al. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348-355.
Sidransky E., Nalls M.A., Aasly J.O., et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361(17):1651-1661.
Silvestri L., Caputo V., Bellacchio E., et al. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet. 2005;14(22):3477-3492.
Simón-Sánchez J., Schulte C., Bras J.M., et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41(12):1308-1312.
Singleton A.B., Farrer M., Johnson J., et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841.
Snow B.J., Lee C.S., Schulzer M., et al. Longitudinal fluorodopa positron emission tomographic studies of the evolution of idiopathic Parkinsonism. Ann Neurol. 1994;36:759-764.
Spillantini M.G., Schmidt M.L., Lee V.M.Y., et al. alpha-Synuclein in Lewy bodies. Nature. 1997;388:839-840.
Strauss K.M., Martins L.M., Plun-Favreau H., et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum Mol Genet. 2005;14(15):2099-2111.
Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci. 2007;30(5):244-250.
Sulzer D. Clues to how alpha-synuclein damages neurons in Parkinson’s disease. Mov Disord. 2010;25(Suppl 1):S27-S31.
Sulzer, D., Bogulavsky, J., Larsen, K.E., Behr, G., Karatekin, E., Kleinman, M.H., et al., 2000. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc Natl Acad Sci USA 97, 11869–11874.
Surmeier D.J. Calcium, ageing, and neuronal vulnerability in Parkinson’s disease. Lancet Neurol. 2007;6(10):933-938.
Surmeier D.J. alpha-Synuclein at the synaptic gate. Neuron. 2010;65(1):3-4.
Tang C.C., Poston K.L., Eckert T., et al. Differential diagnosis of parkinsonism: a metabolic imaging study using pattern analysis. Lancet Neurol. 2010;9(2):149-158.
Tanner C.M. Advances in environmental epidemiology. Mov Disord. 2010;25(Suppl 1):S58-S62.
Tanner C.M., Ottman R., Goldman S.M., et al. Parkinson disease in twins – an etiologic study. JAMA. 1999;281:341-346.
Tanner C.M., Ross G.W., Jewell S., et al. Occupation and risk of parkinsonism: A multicenter case-control study. Arch Neurol. 2009;66(9):1106-1113.
Tansey M.G., McCoy M.K., Frank-Cannon T.C. Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol. 2007;208(1):1-25.
Thacker E.L., O’Reilly E.J., Weisskopf M.G., et al. Temporal relationship between cigarette smoking and risk of Parkinson disease. Neurology. 2007;68(10):764-768.
Tijero B., Gomez-Esteban J.C., Llorens V., et al. Cardiac sympathetic denervation precedes nigrostriatal loss in the E46K mutation of the alpha-synuclein gene (SNCA). Clin Auton Res. 2010;20(4):267-269.
Tretiakoff C. Contribution a l’etude de l’anatomie pathologique du locus niger de Soemmering avec quelques dedutions relatives a la pathogenie des troubles du tonus musculaire et de la maladie de Parkinson. These de Paris; 1919.
Tsang A.H., Chung K.K. Oxidative and nitrosative stress in Parkinson’s disease. Biochim Biophys Acta. 2009;1792(7):643-650.
Tsuboi Y., Dickson D.W., Nabeshima K., et al. Neurodegeneration involving putative respiratory neurons in Perry syndrome. Acta Neuropathol. 2008;115(2):263-268.
Valente E.M., Abou-Sleiman P.M., Caputo V., et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158-1160.
Valente E.M., Brancati F., Ferraris A., et al. PARK6-linked parkinsonism occurs in several European families. Ann Neurol. 2002;51(1):14-18.
Van Den Eeden S.K., Tanner C.M., Bernstein A.L., et al. Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am J Epidemiol. 2003;157(11):1015-1022.
Vilariño-Güell C., Ross O.A., Ansly J.O., et al. An independent replication of PARK 16 in Asian samples. Neurology. 2010;75(24):2248-2249.
Vingerhoets F.J., Schulzer M., Calne D.B., Snow B.J. Which clinical sign of Parkinson’s disease best reflects the nigrostriatal lesion? Ann Neurol. 1997;41(1):58-64.
Ward C.D., Duvoisin R.C., Ince S.E., et al. Parkinson’s disease in 65 pairs of twins and in a set of quadruplets. Neurology. 1983;33:815-824.
Waters C.H., Miller C.A. Autosomal dominant Lewy body parkinsonism in a four-generation family. Ann Neurol. 1994;35:59-64.
Weisskopf M.G., O’Reilly E., Chen H., et al. Plasma urate and risk of Parkinson’s disease. Am J Epidemiol. 2007;166(5):561-567.
Wider C., Dickson D.W., Stoessl A.J., et al. Pallidonigral TDP-43 pathology in Perry syndrome. Parkinsonism Relat Disord. 2009;15(4):281-286.
Wider C., Wszolek Z.K. Rapidly progressive familial parkinsonism with central hypoventilation, depression and weight loss (Perry syndrome) – a literature review. Parkinsonism Relat Disord. 2008;14(1):1-7.
Yahr M.D. Evaluation of long-term therapy in Parkinson’s disease: Mortality and therapeutic efficacy. In: Birkmayer W., Hornykiewicz O., editors. Advances in Parkinsonism. Basle: Editiones Roche; 1976:444-455.
Zarranz J.J., Alegre J., Gomez-Esteban J.C., et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55(2):164-173.
Zhou C., Huang Y., Przedborski S. Oxidative stress in Parkinson’s disease: a mechanism of pathogenic and therapeutic significance. Ann NY Acad Sci. 2008;1147:93-104.
Zimprich A., Biskup S., Leitner P., et al. Mutations in LRRK2 cause autosomal-dominant Parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601-607.