Chapter 9 Atypical parkinsonism, parkinsonism-plus syndromes, and secondary parkinsonian disorders
Introduction
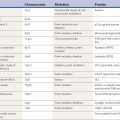
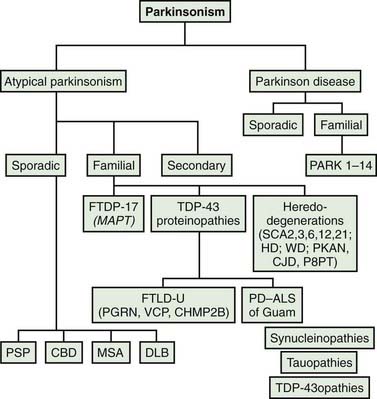
Most patients who are referred to specialized movement disorder clinics with hypokinetic disorders are diagnosed clinically as having Parkinson disease (PD) (Table 9.1) (Jankovic et al., 2000). The second most common group of parkinsonian patients is categorized clinically as having parkinsonism-plus disorders and pathologically as having multiple system degenerations (Fahn, 1977; Jankovic, 1989; Stacy and Jankovic, 1992; Jankovic, 1995b; Jankovic et al., 2000; Litvan et al., 2003; Abdo et al., 2006) (Fig. 9.1). There is, however, a growing body of evidence to support the emerging classification of neurodegenerative disorders according to pathogenetic mechanisms into (1) amyloidoses (e.g., Alzheimer disease, or AD), (2) ubiquitin-proteasome disorders (e.g., PD, parkin PD), (3) synucleinopathies (e.g., PD, multiple system atrophy, or MSA), (4) tauopathies (e.g., frontotemporal dementia (FTD) with parkinsonism, or FTDP; progressive supranuclear palsy, or PSP; corticobasal degeneration, or CBD), (5) polyglutamine expansion diseases (e.g., Huntington disease, spinocerebellar atrophies (SCAs)), and (6) prion diseases (e.g., Creutzfeldt–Jakob disease) (Jankovic, 2008). As our understanding of the mechanisms of these diseases advances, refinements in this classification and new categories of disease will undoubtedly emerge. Besides parkinsonian findings, patients with these disorders exhibit additional (“plus”) features. For example, supranuclear ophthalmoparesis typifies patients with PSP; dysautonomia and ataxia are typically present in MSA; laryngeal stridor occurs in striatonigral degeneration (SND); a combination of apraxia, cortical myoclonus, and “alien hand” occurs in CBD; dementia-parkinsonism occurs not only in PD dementia (PDD), but also in dementia with Lewy bodies (DLB) and AD; and dementia coupled with motor neuron disease occurs in parkinsonism–dementia–amyotrophic lateral sclerosis complex of Guam (Table 9.1). As there are no biologic markers for any of these disorders, the diagnostic criteria are based on the presence of certain clinical features and neuropathologic confirmation (Cummings, 2003; Litvan et al., 2003). While levodopa continues to be the most effective drug for the treatment of motor symptoms associated with PD, a minority (about a third) of patients with atypical parkinsonism also respond to levodopa, although “responsiveness” has not been well defined in the literature (Constantinescu et al., 2007).
| I. Primary (Idiopathic) parkinsonism |
Rarely, psychogenic causes have been implicated in the pathogenesis of parkinsonism (Lang et al., 1995). This chapter focuses only on the sporadic (nongenetic) forms of multisystem degenerations. The secondary and heredodegenerative causes of parkinsonism are covered elsewhere in this book and in other reviews.
Progressive supranuclear palsy
Clinical features and natural history
First described by Steele, Richardson, and Olszewski (Steele et al., 1964; Steele, 1972; Williams et al., 2008) in 1964, progressive supranuclear palsy (PSP) has become a well-characterized, distinct clinical-pathologic entity (Jankovic et al., 1990a; Golbe, 1993; Collins et al., 1995; Litvan, 1998b; Golbe et al., 2007; Golbe and Ohman-Strickland, 2007; Houghton and Litvan, 2007; Azher and Jankovic, 2008). The first volume solely devoted to PSP was published in 1993 (Litvan and Agid, 1993), and a summary of the 1999 “First Brainstorming Conference on PSP” was published (Litvan et al., 2000). The diagnosis of PSP should be considered in any patient with progressive parkinsonism and disturbance of ocular motility (Jankovic, 1984a; Maher and Lees, 1986; Jankovic et al., 1990a; Friedman et al., 1992; Cardoso and Jankovic, 1994).
PSP is considered to be a sporadic disorder, but familial PSP has been reported (Brown et al., 1993; de Yebenes et al., 1995; Tetrud et al., 1996; Rojo et al., 1999; Kaat et al., 2009). The supranuclear ophthalmoparesis was not well documented in the familial cases, and the presence of atypical features, such as early cognitive decline in the family reported by Brown and colleagues (1993) and the relatively early age at onset (53 years) in the de Yebenes and colleagues (1995) kindred, suggested that these families had a neurodegenerative disorder that was distinct from idiopathic PSP. Furthermore, no linkage to the tau gene has been identified in any of these families, although linkage to 1q31.1 has been demonstrated in one large Spanish family (Ros et al., 2005). In a comprehensive epidemiologic-genetic study, 33% of individuals with PSP were found to have at least one first-degree relative with dementia or parkinsonism and 12 families with PSP were identified (5 cases were pathologically confirmed), but the intrafamily phenotype was very variable, the P301L MAPT mutation was identified in only one patient, and there were other limitations in obtaining the family and medical information (Kaat et al., 2009). While it is possible that homozygous mutations occur, such as deletion at codon 296 in MAPT gene identified in one atypical case of PSP (Ferrer et al., 2003), the presence of subhaplotypes overrepresented in individuals with PSP increases the risk of the disease.
In a review of 126 PSP patients, unsteadiness of gait, frequent falling, monotonous speech, loss of eye contact, slowness of movement and of mentation, sloppy eating habits, and nonspecific visual difficulty were the most typical presenting features (Jankovic et al., 1990a). The earliest and most disabling symptom of PSP usually relates to gait and balance impairment, as a result of which patients frequently fall and sustain injuries. The average period from onset of symptoms to the first fall in PSP is 16.8 months, as compared to 108 months in PD, 42 months in MSA, 54 months in dementia with Lewy bodies, and 40.8 months in vascular parkinsonism (Williams et al., 2006). The marked instability is presumably a result of visual-vestibular impairment, axial rigidity, and bradykinesia (Jankovic et al., 1990a). Using computerized posturography, we demonstrated that measures of balance impairment can reliably differentiate between PSP and PD even in early stages of the disease (Ondo et al., 2000). In contrast to the short and shuffling steps, stooped posture, narrow base, and flexed knees that are typically seen in PD, PSP patients have a stiff and broad-based gait, with a tendency to have their knees (and trunk) extended and arms slightly abducted. Instead of turning en bloc, they tend to pivot, which further compromises their balance (Video 9.1). Some PSP patients may present with the syndrome of pure akinesia, also referred to by some as motor blocks (Matsuo et al., 1991; Giladi et al., 1992; Riley et al., 1994) and gait ignition failure (Atchison et al., 1993; Nutt et al., 1993), which is manifested chiefly by akinesia of gait (start hesitation, freezing, motor blocks, festination, disequilibrium with frequent falling), marked impairment of speech (stuttering, stammering, hypophonia), handwriting difficulty (micrographia), and eyelid motor disturbance (blepharospasm, eyelid freezing) without rigidity, tremor, or dementia and without response to levodopa. Primary progressive freezing of gait or pure akinesia with gait freezing may be the initial, main, or the only presentation of PSP (Compta et al., 2007; Williams et al., 2007b; Facheris et al., 2008; Williams and Lees, 2009). Some of the patients with pure freezing may also have PD, pallidonigroluysian degeneration (Ahmed et al., 2008), CBD, and diffuse Lewy body disease (DLBD) (Factor et al., 2006; Williams et al., 2007b). In addition to PSP, frontal gait disorder may be the initial manifestation of AD and CBD (Rossor et al., 1999). Although the PSP may be associated with ataxia and the gait may appear ataxic, the patients usually do not exhibit prominent cerebellar findings. The uncompensated loss of postural reflexes and motor blocks (freezing) especially on turning, coupled with a peculiar lack of insight into the difficulties with equilibrium (possibly secondary to frontal lobe dysfunction), leads to frequent falling. There is, however, also emerging evidence of cerebellar abnormalities, both clinically and pathologically, in PSP (Kanazawa et al., 2009). Abnormal otolith-mediated reflexes may also contribute to the falls of PSP (Liao et al., 2008). Loss of insight, particularly inability to accurately predict performance on future tasks (anticipatory awareness), is a common feature not only in PSP but also in patients with FTD and CBD (O’Keeffe et al., 2007). This is often present even in the early stages of the disease and helps to differentiate PSP from PD (Litvan et al., 1996c). Although the motor subscale of the Unified Parkinson’s Disease Rating Scale (UPDRS) has been found to reliably assess most aspects of PSP (Cubo et al., 2000), the PSP Rating Scale (PSPRS) has been found to be sensitive to disease progression, increasing at a mean rate of about 1 point per month (Golbe and Ohman-Strickland, 2007). PSPRS, along with the Quality of Life Scale (Schrag et al., 2006b), should be used in future clinical trials investigating novel therapeutic interventions. 
Along with postural instability, supranuclear ophthalmoparesis typically manifested by paralysis of downgaze is the most important distinguishing sign of PSP (Litvan et al., 1997c) (Video 9.2). The predictability of these two features with regard to the final pathologic diagnosis was confirmed by a clinical-pathologic study of 24 autopsy-proven cases of PSP (Litvan et al., 1996b). About one-third of PSP patients complain of blurred vision, diplopia, and eye discomfort, but most eventually lose their ability to read or maintain eye contact (Friedman et al., 1992). Involuntary persistence of ocular fixation is a typical, though rarely mentioned, feature of PSP. Other oculomotor abnormalities that are seen in patients with PSP include impairment of saccades, optokinetic nystagmus, and the presence of square wave jerks (Rascol et al., 1991). In early stages of PSP, patients might have only mild limitation of voluntary downgaze and inability to converge, but slowing of horizontal and vertical saccades (also demonstrated by optokinetic nystagmus) appears to be the earliest oculomotor sign of PSP (Video 9.3). Indeed, slowing of vertical saccades, even in the presence of normal vertical saccade amplitude, was found to be an early sign of an autopsy-proven PSP (Hardwick et al., 2009). One study compared saccades, optokinetic nystagmus, and other ophthalmologic signs in six patients with PSP compared to PD and normal control (Garbutt et al., 2004). All PSP patients showed slowing of vertical saccade and quick phase of nystagmus; square wave jerks were more frequent and larger during fixation; vertical optokinetic nystagmus showed impaired slow wave response, and quick phases were slowed and combined with square wave jerks. Deficient generation of the motor command by midbrain burst neurons has been suggested as the primary mechanism for the slow vertical saccades (Bhidayasiri et al., 2001). Slowing of vertical saccades might help to differentiate PSP from other parkinsonian disorders, including PD, MSA, and CBD, although some slowing of vertical saccades can be seen occasionally also in these parkinsonian disorders (Vidailhet et al., 1994; Rivaud-Péchoux et al., 2000; Bhidayasiri et al., 2001). In addition to slow vertical saccades, bilateral impairment of the antisaccade task (the patient is instructed to look in the direction opposite to the visual stimulus) correlates well with frontal lobe dysfunction in PSP (Vidailhet et al., 1994) and other neurodegenerative and frontal lobe disorders (Condy et al., 2004; Munoz and Everling, 2004; Zee and Lasker, 2004). Abnormalities in antisaccades imply a dysfunction of the dorsolateral prefrontal cortex and the superior colliculus (Condy et al., 2004). Later, limitation of vertical and then lateral eye movements follows. The ophthalmoparesis can be overcome by the oculocephalic (doll’s eye) maneuver (Videos 9.1, 9.2, and 9.4), but with disease progression and brainstem involvement, vestibulo-ocular reflexes can be lost, suggesting additional nuclear involvement (Ishino et al., 1974). 
Several clinical-pathologic studies have attempted to establish criteria that separate PSP from other, related disorders. In 60 cases of patients clinically diagnosed with PSP, 47 (78%) of which were pathologically proven, false-positive diagnoses included PD combined with cortical Lewy body pathology or AD, MSA, CBD, Pick disease, motor neuron disease, cerebrovascular disease, and FTD (Osaki et al., 2004). The application of NINDS-SPSP diagnostic criteria (Litvan et al., 1996a) and other criteria improved the accuracy of initial clinical diagnosis only marginally. On the basis of an analysis of 103 pathologically confirmed consecutive cases of PSP, Williams and colleagues (2005) divided PSP into two categories: Richardson syndrome, characterized by the typical features described in the original report, and PSP-P, in which the clinical features overlap with PD and the course is more benign. The latter group, representing about a quarter of all patients with PSP (Williams et al., 2005), has less tau pathology than the classic Richardson syndrome (Williams et al., 2007a, 2008; Williams and Lees, 2009). The mean 4R-tau/3R-tau ratio of the isoform composition of insoluble tangle-tau isolated from the pons was significantly higher in Richardson’s syndrome (2.84) than in PSP-P syndrome (1.63). Further studies are needed to confirm or refute this classification. Another subgroup of PSP that has been identified is the so-called “frontal” PSP, representing about 20% of all PSP patients (Kaat et al., 2007). These patients initially present with behavioral and cognitive symptoms, with or without ophthalmoparesis, and then they evolve into typical PSP.
Pathologically documented cases of PSP without ophthalmoparesis have been reported (Davis et al., 1985; Collins et al., 1995; Daniel et al., 1995). When PSP patients with ophthalmoparesis were compared with those without ophthalmoparesis, no differences in the pathology of the two groups were noted, and there was no correlation between the severity of clinical symptoms and degenerative changes (Daniel et al., 1995). In another pathologic study, brains of patients with PSP who had gaze palsy had two-fold greater loss of neurons in the substantia nigra pars reticulata (SNr) (Halliday et al., 2000). Since SNr projects to the superior colliculi, degeneration of SNr might contribute to the limitation of eye movements. Supranuclear ophthalmoparesis may occur also in DLB (Lewis and Gawel, 1990; Fearnley et al., 1991; De Bruin et al., 1992; Daniel et al., 1995; Brett et al., 2002), CBD (Gibb et al., 1990), postencephalitic parkinsonism, prion disease, Wernicke encephalopathy, dorsal midbrain syndrome, paraneoplastic syndrome, progressive subcortical gliosis, Whipple disease (Jankovic, 1986; Simpson et al., 1995; Averbuch-Heller et al., 1999), Niemann–Pick and Gaucher disease (Shulman et al., 1995; Uc et al., 2000), Kufor-Rakeb syndrome (PARK9; secondary to mutation in ATP13A2 gene on chromosome 1p36) (Hampshire et al., 2001; Schneider et al., 2010), stiff-person syndrome (Oskarsson et al., 2008), primary pallidal degeneration, and other disorders (Calabrese and Hadfield, 1991).
Pseudobulbar symptoms in PSP patients are characterized chiefly by dysarthria, dysphagia, and “emotional incontinence” (Video 9.5). Rigidity, bradykinesia, and hypertonicity of the facial muscles produce deep facial folds and a typical worried or astonished facial expression (Jankovic, 1984b) (Fig. 9.2). The worried appearance is partly due to contraction of the procerus (and possibly corrugator) muscle, the so-called “procerus signs” (Romano and Colosimo, 2001) (Videos 9.1 and 9.2). Speech in PSP is characterized by a spastic, hypernasal, hypokinetic, ataxic, monotonous, low-pitched dysarthria (Kluin et al., 1993) (Videos 9.1, 9.4, and 9.5). The speech rate may be slow or fast, and some patients have severe palilalia and stuttering. An “apraxia of phonation” has been reported in one patient who was aphonic except during periods of excitement or during sleep (Jankovic, 1984b). In contrast, some patients have almost continuous involuntary vocalizations, including loud groaning, moaning, humming, and grunting sounds (Jankovic et al., 1990a). Progressive dysphagia causes most patients to modify their diet, and some eventually need a feeding gastrostomy to maintain adequate nutrition. As a result of chewing difficulties, inability to look down, and poor hand coordination, PSP patients are often described as “sloppy eaters.” 

In a review of dystonia in pathologically proven cases of PD, MSA, and PSP, Rivest and colleagues (1990) found dystonia to be an uncommon feature, noted in only 15 of 118 (13%) cases. They regarded the frequently reported neck extension as a form of axial rigidity rather than dystonia. In another study, the increased neck muscle tone was thought to have features of both dystonia (tonic shortening reaction) and rigidity (antagonist muscle contraction indicative of increased tonic stretch reflex) (Tanigawa et al., 1998). Neck extension, although often noted in published reports, is actually an uncommon sign in PSP. Indeed, neck flexion, usually associated with MSA (Quinn, 1994), can occasionally be seen in PSP (Daniel et al., 1995). In contrast to the typical presence of neck rigidity, truncal muscle tone is only slightly increased, and distal limbs may actually seem hypotonic (Tanigawa et al., 1998). In some patients, however, distal dystonia can be seen (Barclay and Lang, 1997). Although PSP is usually a symmetrical disorder, dystonia represents an occasional exception in that unilateral dystonia may be present, particularly in the more advanced stages of the disease. The most common form of dystonia in PSP is blepharospasm. In one study, 29% of patients had involuntary orbicularis oculi contractions producing blepharospasm, and over one-third had “apraxia” of eyelid opening, eyelid closure, or both (Friedman et al., 1992). Although some (Lepore and Duvoisin, 1985) have hypothesized that these lid abnormalities are due to involuntary supranuclear inhibition of levator palpebrae, others have drawn attention to the similarity of this disorder of eyelid motor control to the parkinsonian phenomenon of sudden transient freezing, hence suggesting the term lid freezing (Jankovic, 1995a). Other terms that are used to describe this condition include pretarsal blepharospasm (Elston, 1992) and focal eyelid dystonia (Krack and Marion, 1994). In one study of 83 patients with PSP, 38 (46%) had some form of dystonia, 22 (24%) had blepharospasm, 22 (27%) had limb dystonia, and 14 (17%) had axial dystonia (Barclay and Lang, 1997). Sometimes, spontaneous arm levitation, a well-recognized sign in CBD, is also seen in patients with PSP and may be wrongly attributed to dystonia (Barclay et al., 1999).
In their original monograph, Steele, Richardson, and Olszewski (1964) indicated that mild dementia was present during early stages of the disease. Though some investigators have reported severe cognitive impairment in this population (Pillon and Dubois, 1993), others have attributed these deficits, at least in part, to poor visual processing (Fisk et al., 1982; Rafal et al., 1988; Jankovic et al., 1990a; Daniel et al., 1995). Despite a relative preservation of short-term memory, cognitive slowing, impairment of executive (goal-directed) functions, and subcortical dementia with deficits in tasks requiring sequential movements, conceptual shifts, and rapid retrieval of verbal knowledge are typically present in patients with PSP (Johnson et al., 1991; Pillon and Dubois, 1993; Litvan et al., 1998e). The memory disturbance that is found in patients with PSP is similar to that of patients with PD and Huntington disease, but it is markedly different from that of patients with AD (Pillon et al., 1994). The apathy, with or without depression, and other hypoactive behaviors that are typically seen in PSP have been attributed to a dysfunction in the frontal cortex and associated circuitry (Litvan et al., 1998e). This is in contrast to Huntington disease, in which behaviors such as agitation, anxiety, and irritability have been related to hyperactivity of the medial and orbitofrontal cortical circuitry. Sparing of olfactory function in PSP, in contrast to that in PD, is another clinical difference between the two neurodegenerative disorders (Doty et al., 1993). Litvan and colleagues (1996d) studied the neuropsychiatric aspects of PSP in 22 patients and found that apathy occurred in 91%, disinhibition in 36%, dysphoria in 18%, anxiety in 18%, and irritability in fewer than 9%. Another sign of frontal lobe dysfunction in PSP is the “applause sign” (signe de l’applaudissement), which probably represents a perseveration of automatic behavior (Dubois et al., 1995, 2005) (Video 9.6). This sign, characteristically present in patients with PSP (but also present in some patients with FTD with parkinsonism and CBD), is manifested by persistence (perseveration) of clapping after the patient is instructed to clap consecutively three times as quickly as possible. In a study of patients with various neurologic disorders evaluated at Baylor College of Medicine, the applause sign was present in 77.8% of 9 patients with CBD, 53.9% of 13 patients with MSA, 52.6% of 19 patients with PSP, 20% of 10 patients with Huntington disease, and 12.5% of 24 patients with PD (Wu et al., 2008). Although the test differentiated patients with CBD from those with PD (P < 0.005) and HD (P < 0.005), it failed to discriminate patients with PSP from other parkinsonian groups, but had a 100% specificity in distinguishing parkinsonian patients from normal subjects. 
After idiopathic PSP, the most common cause of PSP is a multi-infarct state. Multi-infarct or vascular PSP can be difficult to differentiate clinically from the more common idiopathic variety (Dubinsky and Jankovic, 1987; Stern et al., 1989; Winikates and Jankovic, 1994; Rektor et al., 2006). In addition to a much higher frequency of stroke risk factors and abnormal imaging studies, the vascular PSP patients are more likely to have asymmetric and predominantly lower-body involvement, cortical and pseudobulbar signs, dementia, and bowel and bladder incontinence (Winikates and Jankovic, 1994). The concept of vascular PSP is supported by the observation by Ghika and Bogousslavsky (1997), who found that 81% of patients with clinically diagnosed PSP had hypertension. A clinical-pathologic study of four patients who were clinically diagnosed with PSP but found to have vascular PSP at autopsy showed that vascular PSP is characterized by asymmetric signs, falls within 1 year of onset, and vascular lesions on magnetic resonance imaging (MRI) (Josephs et al., 2002). In addition, three of the four patients carried the H2 tau haplotype, whereas 93.7% of patients with idiopathic PSP carry the H1 tau haplotype (78.4% of controls carry this haplotype) (see later). Reported causes of secondary PSP include exposure to organic solvents (McCrank et al., 1989), paraneoplastic syndrome (Jankovic, 1985), mesencephalic tumor (Siderowf et al., 1998), surgery on aorta (Mokri et al., 2004), and other rare and often unsubstantiated causes.
The relentlessly progressive course leads to death, usually from aspiration, within 10 years of onset in the majority of cases. In one clinical-pathologic study of 24 patients with PSP, the median survival from onset was 5.6 years, and this was shorter in men and in patients who experienced falls during the first year of symptoms and with early dysphagia or supranuclear palsy (Litvan et al., 1996c; Litvan, 2003). Overall, the median latency from onset to chairbound state is 5 years and to death is 7 years (Golbe and Ohman-Strickland, 2007).
In another clinical-pathologic study involving 16 cases of PSP, Birdi and colleagues (2002) found the mean survival to be 8.6 years (range: 3–24) years, and the mean age at death was 72.3 years (range: 60–89). The early onset, presence of falls, slowness, and early downward gaze palsy correlated with a rapid progression (Santacruz et al., 1998). In a review of 187 cases, those with early bulbar features had around 5 years less life expectancy than those who had no or late bulbar features (Nath et al., 2003). Similar to other series, the median survival in this study was 5.7 years. Since this figure is based on deceased cases, it might be too pessimistic because slowly progressive cases are still being followed. In another study of 50 PSP patients, Goetz and colleagues (2003) found the median survival from the onset of first symptom to be 7.9 years (6.5 years in the 21 patients who were followed to death). In addition to the short survival, PSP is associated with many symptoms that have a serious impact on the quality of life (Schrag et al., 2006b).
Epidemiology
About 6% of all parkinsonian patients who are evaluated in a specialized clinic fulfill the clinical criteria for PSP. On the basis of a medical record review of the Rochester Epidemiology Project, the average annual incidence rate has been estimated to be 5.3 new cases per 100 000 person-years (Bower et al., 1997). The prevalence, after age adjustment to the US population, has been estimated to be 1.39 per 100 000 (Golbe et al., 1988). In a review of computerized records of 15 general practices in and around London, Schrag and colleagues (1999) found an age-adjusted prevalence for PSP of 6.4 per 100 000. In other studies carried out in the United Kingdom, the prevalence of PSP ranged from 1 to 6.5 per 100 000 (Nath et al., 2001). Similar to the European studies, a prevalence of 5.82 per 100 000 has been reported in Yonago, Japan (Kawashima et al., 2004).
Like PD, PSP occurs more often in men, but its onset at a mean age of 63 years is about 10 years later than the typical onset of PD. Although no well-designed epidemiologic studies have been performed in PSP, one case-control study found that PSP patients were more likely to live in areas of relatively sparse population (Davis et al., 1988). Another study by the same investigators failed to identify any risk factor, except for low likelihood of completing at least 12 years of education, that would differentiate patients with PSP from a matched control population (Golbe et al., 1996).
Neurodiagnostic studies
Electrophysiologic studies have been helpful in documenting other abnormalities, such as sleep difficulties and seizures. Polysomnographic evaluation of 10 patients with moderate to severe PSP revealed marked sleep abnormalities; all had significant periods (2–6 hours) of insomnia (Aldrich et al., 1989). Sleep problems were correlated with worsening dementia. Another study showed marked reduction in percentage of REM sleep (Montplaisir et al., 1997). The same study also showed frontal electroencephalogram (EEG) slowing in patients with PSP. In a review of 62 patients seen over a 9-year period, Nygaard and colleagues (1989) noted seizures in 7 patients and suggested a higher-than-expected frequency of seizures in this population. This has not been our observation, but the relatively high frequency of seizures reported by Nygaard and colleagues (1989) might have been secondary to cortical infarcts. Abnormalities in motor and frontal sensory evoked potentials have been found in 8 of 13 patients with the clinical diagnosis of PSP (Abbruzzese et al., 1991).
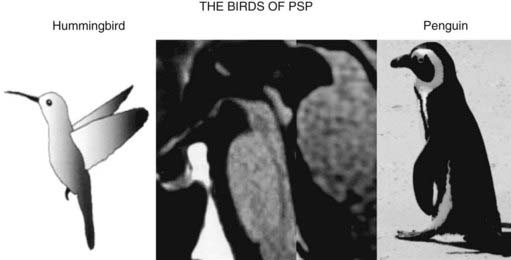
The typical findings on computed tomography or MRI scans of patients with PSP include generalized and brainstem, particularly midbrain, atrophy (Stern et al., 1989; Soliveri et al., 1999). Measuring the anteroposterior diameter of the suprapontine midbrain, Warmuth-Metz and colleagues (2001) found that in contrast to PD patients (mean 18.5 mm), PSP patients had a significantly lower diameter (13.4 mm) on axial T2-weighted MRI, and as a result, the authors concluded that this finding reliably differentiates between PD and PSP and recommended that this evaluation “should be incorporated into the diagnostic criteria for PSP.” In another study, utilizing midsagittal MRI, the average midbrain area of patients with PSP was 56 mm2, which was significantly smaller than that of patients with PD (103 mm2) or MSA-P (97.2 mm2), and this parameter, particularly the ratio of the area of the midbrain to the area of the pons, was found to reliably differentiate among the three disorders (Oba et al., 2005). On the midsagittal view of the MRI, as a result of atrophy of the rostral midbrain tegmentum, the most rostral midbrain, the midbrain tegmentum, the pontine base, and the cerebellum appear to correspond to the bill, head, body, and wing, respectively, of a hummingbird (Kato et al., 2003) or penguin (Oba et al., 2005; Sitburana and Ondo, 2009) (Fig. 9.3). The “hummingbird sign” was demonstrated in all 8 MRI scans of PSP patients but not in any of the 12 scans of PD patients or 10 scans of normal controls (Kato et al., 2003). The “morning glory sign,” a peculiar MRI finding of midbrain atrophy with concavity of the lateral margin of the midbrain tegmentum, resembling the lateral margin of the morning glory flower, is observed in PSP patients with supranuclear gaze palsy (Adachi et al., 2004). Using diffusion-weighted MRI (DWI-MRI), Seppi and colleagues (2003) were able to differentiate between PSP and PD with 90% sensitivity and 100% specificity, but this technique could not differentiate between PSP and MSA. Using diffusion tensor imaging and voxel-based morphometry in PSP, Padovani et al. (2006) provided evidence of both gray and white matter degeneration even in early stages of PSP. Using voxel-based morphometry in 15 patients with clinically proven PSP and 14 with CBD, distinct patterns of atrophy were observed that differentiated between the two disorders with 93% accuracy (Boxer et al., 2006). CBD patients had marked asymmetric (L > R) pattern of atrophy involving premotor cortex, superior parietal lobules, and striatum, whereas PSP was characterized by atrophy of the midbrain, pons, thalamus, and striatum.
Stroke risk factors and a multi-infarct state on computed tomography or MRI have been noted in patients with PSP with a higher frequency than in those with PD (Dubinsky and Jankovic, 1987). One etiology for a subgroup of PSP might be small vessel disease producing subcortical ischemia with reduction of regional cerebral blood flow, cerebral hypometabolism, and a multi-infarct state. MRIs in patients with PSP, MSA, and other parkinsonian syndromes have been associated with putaminal hypointensity on T2-weighted MRI, but this finding is less consistently noted in PSP than in the other parkinsonism-plus syndromes (Drayer et al., 1989; Stern et al., 1989). In one study, PSP could be differentiated from MSA by the presence of marked atrophy and hyperintensity of the midbrain as well as atrophy of the frontal and temporal lobes (Schrag et al., 2000).The “eye of the tiger” sign on brain MRI, typically associated with neurodegeneration with brain iron accumulation type 1 (NBIA1), formerly Hallervorden–Spatz disease (see later), has been also reported in PSP (Davie et al., 1997). PSP is associated with dorsal midbrain atrophy, and as a result of degeneration of superior colliculi, the floor of the third ventricle is flattened on sagittal MRI images (Savoiardo et al., 1994).
Positron emission tomography (PET) scanning has revealed decreased metabolic activity in the caudate, putamen, and prefrontal cortex (Foster et al., 1988; Goffinet et al., 1989; Blin et al., 1990), but the earliest sign of PSP appears to be decreased glucose metabolism in the midbrain (Mishina et al., 2004). Uptake of 18F-dopa is usually reduced in PSP but may be normal in early stages (Bhatt et al., 1991). This suggests that the parkinsonian findings in early PSP are related more to postsynaptic receptor changes than to a loss of presynaptic dopamine terminals. In another study, 18F-dopa uptake was markedly reduced in the caudate as well as in the anterior and posterior putamen of PSP patients (Brooks et al., 1990a). In contrast, the uptake was reduced only in the posterior putamen in PD patients. Similarly, dopamine transporter, imaged by [11C]-WIN PET, showed a relatively uniform reduction involving the entire striatum, whereas patients with PD had involvement chiefly of the posterior putamen (Ilgin et al., 1999). F-dopa and F-deoxyglucose PET were abnormal in 5 (33%) individuals among 15 subjects at risk of familial PSP even though they did not (yet) exhibit any symptoms (Piccini et al., 2001). Using 11C-raclopride as a D2 ligand, Brooks and colleagues (1992) showed a 24% reduction in D2 density in the caudate and a 9% reduction in the putamen of patients with PSP. A discriminant analysis of striatal 18F-dopa PET studies indicates that this technique can reliably differentiate between PD and PSP, but it is less accurate in differentiating between PD and MSA (Burn et al., 1994). By using [11C]diprenorphine, significantly reduced opioidergic binding has been demonstrated in both caudate and putamen, whereas the binding is essentially normal in PD and reduced in the putamen but not the caudate in SND (Burn et al., 1995). The cortical muscarinic acetylcholine receptors, as measured by PET and [11C]N-methyl-4-piperidyl benzilate, were found to be normal in a series of patients with PSP (Asahina et al., 1998). Using [123I]β-CIT SPECT, Pirker and colleagues (2000) showed marked reduction of striatal binding in PD, PSP, MSA, and CBD, but the pattern of abnormality (reduction in overall binding and asymmetry) did not allow a differentiation between the various disorders. Using the receptor ligand [18F]altanserin, PET scans in 8 patients with PSP showed upregulation of 5-HT2A receptors in the substantia nigra and to a lesser degree in the striatum, as compared to 13 controls, and these changes significantly correlated with the UPDRS-III and PSP-Rating Scale (Stamelou et al., 2009).
In addition to imaging studies, analysis of the CSF may also be helpful in evaluation of patients with PSP. One study, for example, found a low ratio of light (33 kDa) to heavy (55 kDa) tau protein in the CSF of patients with PSP, which differentiated (without any overlap) this group from other tauopathies such as AD, CBD, and FTD and from synucleinopathies such as PD and DLB (Borroni et al., 2008). This finding had an 87% sensitivity and 86% specificity and was validated against MRI voxel-based morphometry of brainstem gray matter.
Neuropathology, neurochemistry, and pathogenesis
The motor, neurobehavioral, and neuro-ophthalmic findings seen in patients with PSP reflect marked neuronal degeneration in the basal nucleus of Meynert, pallidum, subthalamic nucleus, superior colliculi, mesencephalic tegmentum, substantia nigra pars compacta (SNc) and SNr, locus coeruleus, red nucleus, reticular formation, vestibular nuclei, cerebellum, and spinal cord (Steele et al., 1964; Steele, 1972; Zweig et al., 1987; Juncos et al., 1991; Jellinger and Bancher, 1993; Hardman et al., 1997a, 1997b; Kanazawa et al., 2009). In addition, in contrast to PD, both the globus pallidus interna (GPi) and externa (GPe) are markedly affected in PSP, and this may contribute to thalamic inhibition and some of the parkinsonian features (Hardman and Halliday, 1999a, 1999b). Along with the degeneration of the SNc and the pedunculopontine tegmental nucleus, the glutamatergic caudal intralaminar thalamic nuclei are involved in both PSP and PD (Henderson et al., 2000). In addition there is a widespread loss of A10 dopamine neurons, clearly more severe than in PD (Murphy et al., 2008). Atrophy of the superior cerebellar peduncle has been found to be a frequent finding in the brains of patients with PSP, and this correlates with the duration of the disease (Tsuboi et al., 2003b). In one pathologic study, marked atrophy of the GPi differentiated PSP from PD and DLB (Cordato et al., 2000). Cerebellar degeneration and tau-positive inclusion bodies have been demonstrated in the Purkinje cells of patients with PSP, particularly when associated clinically with ataxia (Kanazawa et al., 2009). Although bladder dysfunction has not been considered a prominent feature in patients with PSP, the finding of neuronal loss in Onuf’s nucleus in patients with PSP suggests that bladder function should be carefully evaluated in patients with PSP (Scaravilli et al., 2000).
On the basis of a workshop sponsored by the National Institutes of Health, the following neuropathologic criteria were proposed: (1) high-density neurofibrillary tangles (NFTs) and neuropil threads in the basal ganglia and brainstem and (2) tau-positive astrocytes or their processes in areas of involvement (Hauw et al., 1994; Litvan et al., 1996a). Using rather crude pathologic criteria, De Bruin and Lees (1994) demonstrated that the clinical manifestations of PSP can vary considerably. They reviewed 90 cases reported in the literature between 1951 and 1992 that met the following two criteria: subcortical neurofibrillary degeneration and exclusion of other recognized nosologic entities. There were 51 men and 34 women (in 5 cases, the gender was not specified), with an average age at onset of 62 years and mean age at death of 67 years. The most common symptoms were unsteady gait (70.7%), stiffness (67.4%), slurred speech (67.4%), falls (60.6%), dysphagia (57.3%), and blurring of vision (21%). Vertical gaze palsy was the most common sign but was noted in only 68.5% of all cases, followed by bradykinesia in 67.4%, dysarthria in 67.4%, rigidity in 58.4%, axial dystonia in 48.3%, segmental dystonia in 20.2%, and tremor in 16.8%.
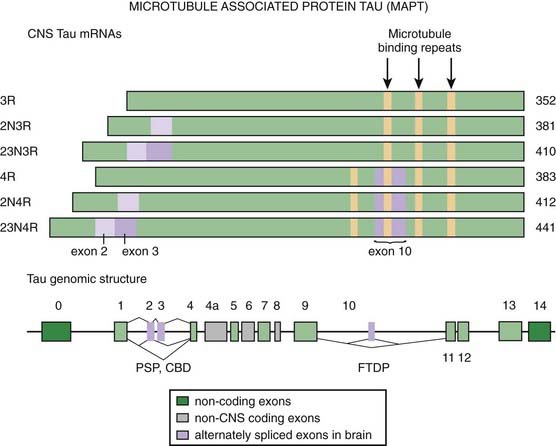
Microscopic examination reveals NFTs, granulovacuolar degeneration, gliosis, and rare Lewy bodies (Steele et al., 1964; Steele, 1972; De Bruin and Lees, 1994). One pathologic study showed no evidence of increased Lewy bodies in PSP (Tsuboi et al., 2001). The NFTs in PSP differ from those seen in AD and other neurodegenerative disorders in that PSP tangles consist of 15 nm straight tubules rather than 20–24 nm wide paired helical filaments (PHF) (Joachim et al., 1987; Jellinger and Bancher, 1993; Dickson, 1997). PHFs are composed of the microtubule-associated protein tau (MAPT) in a hyperphosphorylated state (Goedert et al., 1996). In contrast to the other neurodegenerative diseases with tau pathology, such as AD, Pick disease, CBD, and the parkinsonism–dementia complex of Guam, which are characterized by flame-shaped NFTs, the NFTs in PSP are predominantly of the globose type. The tau-containing astrocytic inclusions (“tufted astrocytes”) are more common in the basal ganglia and the brainstem of PSP, whereas they are more common in the cortex of brains of CBD. Tau inclusions in PSP oligodendrocytes are described as “coiled bodies.” Tsuboi and colleagues (2003a) showed that APOE ε4 is a risk factor for Alzheimer-type pathology in PSP. Some brains of patients with Guam parkinsonism–dementia complex also contain deposits of the TDP-43 protein (TAR-DNA binding protein 43), which binds ubiquitin, suggesting that a common pathogenic mechanism through conformational changes in this protein links this disease with FTDP, ALS, and related neurodegenerative diseases (Hasegawa et al., 2007).
The clinical and pathologic overlap between PSP, CBD, and AD provides evidence that these disorders are closely related, although the absence of amyloid deposits in PSP and CBD suggests a clear difference between the two disorders and AD (Feany and Dickson, 1996; Litvan et al., 1999; Rossor et al., 1999; Boeve et al., 2003a; Wakabayashi and Takahashi, 2004; Galpern and Lang, 2006). Among 180 cases of clinically diagnosed PSP that came to autopsy, only 137 (36%) met the pathologic criteria for PSP (Josephs and Dickson, 2003). The other diagnoses included CBD, MSA, DLBD, and Creutzfeldt–Jakob disease (CJD). The following features were seen more frequently in non-PSP cases than in PSP cases: tremor, psychosis, early dementia, asymmetric findings, absence of H1 haplotype, and presence APOE ε4. The various disorders can be differentiated by a careful histologic examination. For example, the tau in NFTs of AD shows marked ubiquitin immunoreactivity, whereas the NFT-tau from PSP does not (Flament et al., 1991; Shin et al., 1991). While the abnormal tau in AD consists chiefly of 55, 64, and 68 kDa forms, the PSP tau consists only of 64 and 68 kDa forms (Conrad et al., 1997) (Fig. 9.4). Cdk5, a kinase that is physiologically involved in the phosphorylation of tau protein, is overexpressed in PSP brains (Borghi et al., 2002). PSP and CBD also overlap with FTD in clinical, pathologic, biochemical, and genetic aspects (Boeve et al., 2003a). Tuft-shaped astrocytes seem to be more indicative of PSP than CBD, with prominent tuft-shaped astrocytes in the precentral gyrus and premotor cortex, caudate, putamen and globus pallidus, red nucleus, and superior colliculus in PSP brains (Hattori et al., 2003). Another pathologic evidence of overlap between PSP and CBD is the abundance of pathologic tau in the white matter in both disorders (Zhukareva et al., 2006). The histopathologic features of PSP also overlap closely with those of postencephalitic parkinsonism (Litvan et al., 1998c) and the parkinsonism–dementia complex of Guam, but the pallidum and the subthalamic nucleus are usually spared in the former, and the cortex seems to be more involved in the latter. Rarely, cases with clinical presentation nearly identical to that of PSP have been reported to have the pathologic picture of pallidonigroluysial atrophy (PNLA) (Kosaka et al., 1981; Ahmed et al., 2008). Of 400 cases of PSP, 8 (2%) were found to have pathologic features of PSP and PNLA is now considered a rare variant of PSP, with the latter usually presenting at a younger age, with gait and handwriting abnormalities in early stages, and having slower progression (Ahmed et al., 2008). In one study, 54% of pathologically proven cases of PSP had coexistent AD and PD, providing evidence for overlap between these neurodegenerative disorders (Gearing et al., 1994). Calbindin-D28k immunoreactivity, normally found in the medium-sized neurons and neuropil of the striatal matrix, GP, and SNr, is reduced in the GP of patients with PSP (and in striatum and SNr of patients with SND) (Ito et al., 1992). This finding suggests that calcium cytotoxicity might play a role in the marked neuronal degeneration that is found in these structures.
PSP and CBD also share pathologic tau doublet (64 and 69 kDa) as well as the predominance of 4R tau isoforms with argyrophilic grain disease (AGD). This sporadic neurodegenerative disease, which accounts for about 5% of all cases of dementia, is manifested by late-onset episodic memory loss and progressive dementia along with personality changes, irritability, agitation, delusions, and apathy. Neither clinical nor neuroimaging features can reliably differentiate between AGD and AD, and the diagnosis of AGD is based on autopsy findings. Pathologically, aggregated tau proteins are found in limbic structures in the shape of distinct argyrophilic grains and coiled bodies (Tolnay et al., 2002; Liang et al., 2005; Ferrer et al., 2008). Because of overlap in pathologic features with not only PSP and CBD, but also AD and other neurodegenerative diseases, questions have been raised whether AGD represents a distinct entity. Argyrophilic grains are present in 4–9% of adult brains and the incidence increases with age. Besides aging, oxidative stress appears to play a critical role in the development of the pathologic changes, including activation of tau kinases, which facilitate tau hyperphosphorylation in some neurons. The abnormal tau apparently binds to p62 and, after ubiquitation, aggregates, forming grains and tangles. Thrombin, which accumulates in the grains, has been thought to facilitate tau truncation, which further increases oxidative stress and contributes to the toxicity.
Molecular misreading of the ubiquitin-B (UBB) gene results in a dinucleotide deletion in ubiquitin-B mRNA, which in turn leads to accumulation of the mutant protein ubiquitin-B+1 in AD, Pick disease, FTD, PSP, and AGD, but not in synucleinopathies (PD or MSA) (Fischer et al., 2003) (Fig. 9.1). This finding provides evidence that the ubiquitin-proteasome system is impaired in these tauopathies and that ubiquitin-B+1 protein serves as a marker for these diseases. Unique haplotype in 17q21, found in 16% of Spanish as well as American PSP patients but not in any of the controls, provides further evidence that PSP is a form of a tauopathy (Pastor et al., 2004). Better understanding of the molecular pathways that are altered in various neurodegenerative disorders could be helpful in differential diagnosis based on postmortem examination of brain tissue. Using microarray technology in substantia nigra (SN) samples from six patients with PD, two patients with PSP, one patient with FTDP, and five controls, Hauser and colleagues (2005) found 142 genes that were differentially expressed in PD cases and controls, 96 in the combination of PSP-FTDP, and 12 genes that were common to all three disorders.
The marked reduction in striatal D2 receptors in PSP, demonstrated by PET studies, has also been documented in postmortem studies (Ruberg et al., 1993). In contrast to the D2 receptors, the striatal D1 receptors are spared (Pierot et al., 1988; Pascual et al., 1992). Biochemical studies show that in addition to degeneration of the nigrostriatal dopaminergic system, the cholinergic and GABAergic systems seem to be particularly affected. Cholinergic neurons have been found to degenerate in the Edinger–Westphal nucleus, the rostral interstitial nucleus of Cajal (possibly contributing to the extensor nuchal rigidity), the medial longitudinal fasciculus (contributing to vertical gaze palsy), the superior colliculus, and the pedunculopontine nucleus (Zweig et al., 1987; Juncos et al., 1991). Autoradiographic study of 18 brains of patients with pathologically confirmed PSP showed marked reduction in M2 and M4 receptors in the posterior striatal cholinergic interneurons and a reduction of M4 receptors on medium spiny projection neurons, confirming marked cholinergic dysfunction in PSP compared to Lewy body dementia (n = 45), Alzheimer disease (n = 39), and normal controls (n = 50) (Warren et al., 2007). Using the technique of an in-situ hybridization of GAD67 messenger RNA, Levy and colleagues (1995) demonstrated 50–60% reduction in the number of neurons expressing GAD67 messenger RNA in the caudate nucleus, ventral striatum, and both segments of the GP in three brains of patients with PSP. They suggest that the marked destruction of the basal ganglia output nuclei might explain the poor response to dopaminergic therapy in this disorder.
The most striking neurochemical abnormality found in PSP brains is a marked reduction in striatal dopamine, dopamine receptor density, choline acetyltransferase activity, and loss of nicotinic, rather than muscarinic, cholinergic receptors in the basal forebrain (Young, 1985; Pierot et al., 1988; Ruberg et al., 1993). Normal dopamine levels in the nucleus accumbens suggest that the mesolimbic system is relatively spared. Because of the relative sparing of the mesocorticolimbic dopaminergic system in contrast to the severe degeneration of the mesostriatal system, some investigators have suggested that the primary site of pathology in PSP is the striatum and that the changes observed in the SN are simply a result of a retrograde degeneration (Ruberg et al., 1993). The cholinergic neurons are, however, also markedly affected and may be primarily involved in PSP. The cholinergic innervation of the thalamus is particularly affected in PSP, and this finding helps to differentiate PSP from PD (Shinotoh et al., 1999). Also, reduction in acetylcholine vesicular transporter has been found to differentiate PSP from other types of neurodegenerative disorders (Suzuki et al., 2002). Suzuki and colleagues (2002) were able to correlate reductions in acetylcholine vesicular transporter and choline acetyltransferase activity in the striatum of postmortem brains of patients with PSP, but choline acetyltransferase was also significantly reduced in the inferior frontal cortex. Glutamate has been found to be increased in the striatum, pallidum, nucleus accumbens, and occipital and temporal cortex. In contrast to PD, glutathione was found to be increased in the SN of PSP patients (Perry et al., 1988). The observation that multiple neurotransmitters, particularly dopamine and acetylcholine, are affected in PSP suggests that PSP is not a primary neurotransmitter disease but a disorder in which multiple subpopulations of neurons degenerate for yet unknown reasons.
The cause of PSP is unknown, but oxidative damage, mitochondrial dysfunction, and abnormal protein (e.g., tau) processing have received the most attention (Albers and Augood, 2001). Decreased rates of adenosine triphosphate production were found in a preliminary study of muscle mitochondria function in patients with PSP, suggesting impaired mitochondrial respiratory chain activity in PSP (Di Monte et al., 1994). Further support for mitochondrial defect in PSP was later provided by additional studies. Using cybrid lines expressing mitochondrial genes, Swerdlow and colleagues (2000) found a 12.4% decrease in complex I activity (P < 0.005) in cybrid (cytoplasmic hybrid) cells but no change in complex IV activity. Cybrid cells also had significantly increased levels of several antioxidant enzymes. This study suggests a mtDNA-encoded electron transport chain enzyme defect in PSP. Further evidence for a defect in mitochondrial oxidative metabolism is the finding of significantly reduced levels of phosphocreatine and Mg2+ and increased levels of adenosine diphosphate and inorganic phosphate using phosphorus magnetic resonance spectroscopy of the brain and calf muscle in five PSP patients (Martinelli et al., 2000). Using proton magnetic resonance spectroscopic imaging, Tedeschi and colleagues (1997) found a reduced N-acetylaspartate/creatine-phosphocreatine ratio in the brainstem, centrum semiovale, and frontal and precentral cortex and N-acetylaspartate/choline in the lentiform nucleus in patients with PSP.
Research into mechanisms and treatment of PSP has been hampered by paucity of suitable animal models. Rats that are exposed systemically and chronically to annonacin, a lipophilic mitochondrial complex I inhibitor extracted from tropical fruit plants, have been shown to produce neurodegeneration resembling PSP, providing further evidence of mitochondrial dysfunction in PSP (Champy et al., 2004). Another potential animal model of PSP is a transgenic mouse (TgT34) which overexpresses wild-type human tau (T34 tau isoform) driven by the astrocyte-specific glial fibrillary acidic protein promoter (Forman et al., 2005). These transgenic mice accumulate tau in astrocytes, similar to what occurs in PSP, leading to focal neuronal degeneration. Another potential animal model of PSP is the JNPL3 mouse in which the human four repeat tau with the P301L mutation is expressed resulting in a severe phenotype within 5–6 months but without motor defects (Lewis et al., 2000).
Recent studies have drawn attention to abnormal phosphorylation of tau proteins as an important mechanism of neurodegeneration in PSP. Tau exon 10 + 16 mutation in the tau gene (MAPT, IVS10, C-U, +16) was found in a case of young-onset (age 40 years) of PSP phenotype with neuropathologic features of FTD (Morris et al., 2003). Tau is phosphorylated by serine, threonine, and tyrosine kinases, and this phosphorylation might lead to abnormal aggregation. There is growing support for the notion of altered regulation of tau gene expression in PSP (Rademakers et al., 2005). The relationship between abnormalities in tau and PSP is described in detail in the section on tauopathies.
Treatment
Although in the early stages, mild improvement in parkinsonian symptoms may be noted with levodopa or dopamine agonists (e.g., pergolide, pramipexole), most PSP patients fail to reach and maintain any meaningful improvement with these drugs (Jankovic, 1983; Litvan and Chase, 1993; Nieforth and Golbe, 1993; Jankovic, 1994; Weiner et al., 1999). The most likely reason is that in PSP, there is a marked loss of the postsynaptic receptors, particularly the D2 receptors, secondary to the loss of the postsynaptic striatal neurons (Pierot et al., 1988). Idazoxan, an experimental, potent and selective α-2 presynaptic inhibitor that increases norepinephrine (NE) transmission, was shown in a double-blind crossover study to improve motor function in nine PSP patients (Ghika et al., 1991). Physostigmine has been shown to have variable clinical effects on cognitive deficits (Blin et al., 1995). Furthermore, PSP patients have been found to be unusually sensitive to cholinergic blockade with anticholinergic drugs (Litvan et al., 1994); therefore, these drugs should be avoided in patients with PSP. On the other hand, the cholinergic drug donepezil has not been found to be beneficial in a placebo-controlled trial of 21 patients with PSP, and it might actually worsen motor function (Litvan et al., 2001b). Other drugs, including methysergide and amitriptyline, although anecdotally reported to be beneficial, have been generally disappointing (Newman, 1985). Besides amitriptyline and serotonin uptake inhibitors, AVP-923 (Zenvia), a combination of dextromethorphan and quinidine developed by Avanir, has been used to treat involuntary emotional expression disorder (IEED), or pseudobulbar affect, also known as emotional lability or incontinence (Panitch et al., 2006; Rosen and Cummings, 2007). Zolpidem, a GABA agonist and a short-acting hypnotic drug, was found to improve moderately voluntary saccadic eye movements and motor function in a small (n = 10) group of patients with PSP as compared to placebo (Daniele et al., 1999). Blepharospasm, with or without eyelid freezing, and other forms of focal dystonias, can be effectively treated with botulinum toxin injections (Jankovic and Brin, 1991; Jankovic, 2004) (Video 9.7). Electroconvulsive therapy, while helpful in some patients with PD, markedly exacerbated motor and mental symptoms in one patient with PSP (Hauser and Trehan, 1994). Cricopharyngeal myotomy is almost never performed, but severe dysphagia in advanced stages of the disease often necessitates the placement a feeding gastrostomy. There is no evidence, however, that tube feeding prevents aspiration (Finucane et al., 1999). 
Only symptomatic therapy has been used thus far, albeit with disappointing results, but it is hoped that better understanding of the pathogenesis of neuronal degeneration in PSP will lead to more effective, hypothesis-driven therapeutic interventions. It is possible, for example, that since cross-linking of tau protein by transglutaminase stabilized tau filaments into NFTs, inhibitors of transglutaminase may prevent the formation of NFTs and may have a neuroprotective effect on the disease (Zemaitaitis et al., 2000). For example, drugs such as lithium and valproic acid that inhibit abnormal phosphorylation of the tau protein by blocking the enzyme GSK-3β (also known as tau protein kinase I) might possibly exert neuroprotective effects (Chen et al., 1999). Lithium has been found to induce phosphorylation of the serine 9 residue of the glycogen synthase kinase (GSK-3β), inhibiting tau phosphorylation on the PHF-1 epitope and, therefore, this GSK-3β inhibitor may have neuroprotective properties in various tauopathies including PSP. Besides GSK-3β, lithium also inhibits inositol monophosphatase and the proteasome, protects cultured neurons against amyloid beta toxicity, prevents NFT accumulation, and causes downregulation of tau proteins by reducing tau mRNA (Rametti et al., 2008). By limiting tau phosphorylation, valproate would be expected to prevent the disturbed microtubule function, disrupted intracellular protein trafficking, formation of NFTs, and neuronal death. Valproate might also prevent elevations of intracellular calcium, increase levels of the antiapoptotic protein Bcl-2, act as a histone deacetylase inhibitor (which might interfere with apoptosis), and inhibit GSK-3β (Phiel et al., 2001; Qing et al., 2008). Other kinase inhibitors, such as noscovitine and olomoucine, might also play a role as potential therapeutic strategies in PSP.
Because of some evidence of mitochondrial complex I dysfunction in PSP (Di Monte et al., 1994), mitochondrial enhancers, such as coenzyme Q10 (CoQ10), have been studied in PSP. One randomized, placebo-controlled, phase II trial involving 21 patients with PSP showed slight but significant improvements in the scores of the total PSP Rating Scale and Frontal Assessment Battery in the group treated with a nanoparticular emulsion of CoQ10 (lipophilic emulsion, nanoQuinon, 5 mg/kg) compared to placebo (Stamelou et al., 2008). This was accompanied by a significant reduction in adenosine diphosphate (ADP) on magnetic resonance spectroscopy (MRS) in the occipital lobe and right basal ganglia. These encouraging findings from the initial study must be confirmed by a larger, phase III, clinical trial. Another drug, methylene blue (Urolene Blue), an old drug used for the treatment of urinary tract infections, thought to prevent formation of tangles in Alzheimer disease at doses of 60 mg three times a day of methylthioninium chloride (Rember, made by TauRx Therapeutics), may be potentially helpful in PSP. It is not clear whether the drug acts as a kinase inhibitor, but it has been shown to increase mitochondrial complex IV and to have a variety of antioxidant properties (Atamna et al., 2008). Another drug that is currently investigated in PSP is davunetide, an analog of vasoactive intestinal peptide which enhances the synthesis of the activity-dependent neuroprotective protein, of which Davunetide is a fragment (also known as NAP), administered as a nasal spray. This neurotrophic factor has been shown to stimulate neurite elongation and synapse formation, prevent toxicity from amyloid beta peptides, and limit tau hyperphosphorylation (Shiryaev et al., 2009).
Multiple system atrophy
Clinical features and natural history
Historically, the first case that James Parkinson described in his 1817 “Essay on the Shaking Palsy” had associated autonomic features and might have been the first case of MSA. First coined by Graham and Oppenheimer in 1969 (Graham and Oppenheimer, 1969), the term multiple system atrophy describes a syndrome characterized clinically by parkinsonism, dysautonomia, and other features previously reported as Shy–Drager syndrome (SDS), SND, and sporadic olivopontocerebellar atrophy (OPCA). The term Shy–Drager syndrome is still occasionally used in the literature, particularly by some American clinicians, as a tribute to Dr Shy, a neurologist from University of Pennsylvania and Columbia University, and Dr Drager, a urologist at Baylor College of Medicine, who first drew attention to this disorder in 1960 (Shy and Drager, 1960). In their initial report, Shy and Drager described two men who presented with symptoms of orthostatic syncope, impotence, and bladder dysfunction. They later developed parkinsonian features, including gait disturbance, mild tremor, dysarthria, constipation, and bowel and bladder incontinence. In addition to the combination of parkinsonism and autonomic failure, patients with SDS also frequently manifest cerebellar (60%) and pyramidal signs (50%). Other features described by Shy and Drager as part of the “full syndrome,” such as rectal incontinence, fasciculations, and iris atrophy, are seen rarely. The term striatonigral degeneration was introduced in the 1960s by Adams, Van Bogaert, and Van de Eecken (Aotsuka and Paulson, 1993). In an attempt to characterize SND, Gouider-Khouja and colleagues (1995) used the following clinical criteria: (1) onset after 40 years; (2) disease duration less than 10 years; (3) parkinsonian syndrome poorly responsive or unresponsive to levodopa; (4) autonomic failure; and (5) absence of family history, dementia, apraxia, supranuclear ophthalmoplegia, and “detectable focal lesions on neuroimaging study.” Dejerine and Thomas (1900) introduced the term olivopontocerebellar atrophy to describe a group of heterogeneous disorders characterized clinically by the combination of progressive parkinsonism and cerebellar ataxia and pathologically by neuronal loss in the ventral pons, inferior olives, and cerebellar cortex (Berciano, 1992). OPCA may be inherited, usually in an autosomal dominant pattern (Currier and Subramony, 1993; Rosenberg, 1995; Berciano et al., 2006), but only the sporadic OPCAs are included in the classification of MSA (Berciano, 1992; Gilman and Quinn, 1996; Wenning et al., 2004a). Although considered a sporadic disease, families with a phenotype suggestive of autosomal recessive MSA have been described (Hara et al., 2007). It is possible, however, that these familial cases represent either some forms, yet to be identified, of spinocerellar ataxia (SCA) or familial OPCA. A rating scale, the Unified Multiple System Atrophy Rating Scale (UMSARS), that assesses all important symptoms and signs of MSA has been developed and validated against related rating scales, such as the UPDRS and the International Cooperative Ataxia Rating Scale (Wenning et al., 2004b). The UMSRARS Motor Examination score seems to be the best outcome measure for future therapeutic trials (May et al., 2007).
The discovery by Papp and colleagues (1989) that the pathologic hallmark shared by all three disorders is the presence of filamentous α-synuclein-containing glial cytoplasmic inclusions (GCI) led to the recognition that these disorders are manifestations of the same pathologic process. MSA has therefore been redefined as a sporadic, progressive, adult-onset disorder characterized clinically by autonomic dysfunction (MSA-A), parkinsonism (MSA-P), and cerebellar ataxia (MSA-C) in any combination (American Academy of Neurology, 1996; Consensus Committee of the AAS and AAN, 1996; Gilman et al., 1998; Gilman, 2002; Osaki et al., 2002; Watanabe et al., 2002; Gilman et al., 2008). A second consensus statement in the diagnosis of MSA simplified the prior criteria (Consensus Committee of the AAS and AAN, 1996) and proposed the following diagnostic criteria (Gilman et al., 2008): “Definite MSA requires neuropathologic demonstration of CNS α-synuclein-positive glial cytoplasmic inclusions with neurodegenerative changes in striatonigral or olivopontocerebellar structures. Probable MSA requires a sporadic, progressive adult-onset disorder including rigorously defined autonomic failure and poorly levodopa-responsive parkinsonism or cerebellar ataxia. Possible MSA requires a sporadic, progressive adult-onset disease including parkinsonism or cerebellar ataxia and at least one feature suggesting autonomic dysfunction plus one other feature that may be a clinical or a neuroimaging abnormality.” Features that would argue against the diagnosis of MSA include: age at onset >75 years, the presence of typical PD rest tremor, neuropathy, sporadic hallucinations, dementia, white matter lesions in MRI suggestive of multiple sclerosis, and a family history of ataxia or parkinsonism.
Until the mid-1990s, the literature still used the terms SND and OPCA, and we continue to use the term OPCA for disorders that do not fit the nosology of MSA-C, such as some sporadic or autosomal dominant ataxias and other disorders such as certain hereditary, metabolic, or degenerative disorders with pathologic features of OPCA (Berciano et al., 2006).
Fearnley and Lees (1990) reviewed 10 patients, ranging in age from 47 to 50 years, with autopsy-proven SND (MSA-P). Five of these patients were misdiagnosed as having PD, largely because of good response to levodopa. Features that were helpful in differentiating SND from other parkinsonian disorders included early-onset falling, severe dysarthria and dysphonia, excessive snoring and sleep apnea, respiratory stridor, hyperreflexia, and extensor plantar responses. Cerebellar or pyramidal tract signs were present in two patients each, while autonomic symptoms were present in seven. Duration of illness ranged from 3 to 8 years, and no difference in survival was seen in levodopa responders compared to non-levodopa responders. In another series, tremor was found in only 6% of SND patients and in 71% of PD patients; the predominant features were rigidity and hypokinesia, present at onset in 84% of all SND (MSA-P) patients (Van Leeuwen and Perquin, 1988). Besides lack of tremor, the symmetrical onset of SND (MSA-P) is sometimes helpful in differentiating SND from PD, although 6 of 10 patients described by Fearnley and Lees (1990) had asymmetrical onset. In a study comparing 16 patients with pathologically proven MSA of the SND (MSA-P) variety with PD and PSP, a set of clinical criteria reliably differentiated MSA from PD but not from PSP (Colosimo et al., 1995). In addition to cerebellar and pyramidal signs, early instability with falls, and relative preservation of cognition, the following features were more typically present in MSA than in PD: autonomic dysfunction (69% versus 5%), absence of rest tremor (87% versus 40%), rapid progression (mean disease duration 7.1 years versus 13.6 years), and poor or unsustained response to levodopa (31% versus 0%). In contrast to other reports, only 43.7% of the MSA patients in this series had a symmetric onset. As in PD, PSP, and other subcortical neurodegenerative disorders, the cognitive deficit in SND consists chiefly of mild impairment of memory and executive functions, which has been attributed to “inefficient planning of memory processes” and “frontal lobe-like syndrome related to a dysfunction of the supervisory attentional system” (Pillon et al., 1995b). While pseudobulbar affect associated with emotional incontinence is typically seen in patients with PSP, pathologic laughter and crying has been also described in autopsy-proven cases of MSA, particularly the MSA-C type (Parvizi et al., 2007). Cognitive impairment, particularly associated with prefrontal dysfunction, is more severe in patients with MSA-P than those with MSA-C (Kawai et al., 2008).
MSA appears to be more common in men than in women, with symptoms first beginning in the sixth decade; death usually occurs 7–8 years after the initial symptoms and approximately 4 years after the onset of neurologic impairment (McLeod and Tuck, 1987a, 1987b). In a review of 188 pathologically proven cases of MSA, 28% patients had involvement of all four systems (parkinsonism, cerebellar dysfunction, corticospinal signs, and dysautonomia); 18% had the combination of parkinsonism, pyramidal, and autonomic findings; 11% had parkinsonian, cerebellar, and autonomic findings; another 11% had parkinsonism and dysautonomia; 10% had only parkinsonism; and parkinsonism was absent in 11% of all cases (Quinn, 1994). The clinical features and natural history of MSA were analyzed in 100 cases with probable MSA, of which 14 were confirmed at autopsy (Wenning et al., 1994b). The population consisted of 67 men and 33 women, with a median age at onset of 53 years (range: 33–76). Autonomic symptoms were present at onset in only 41% of the patients, but 97% developed autonomic dysfunction during the course of the disease. Whereas impotence was the most frequent autonomic symptom in males, urinary incontinence predominated in women. Some evidence of orthostatic hypotension was present in 68% of patients, but severe orthostatic hypotension was noted in only 15%. In contrast to PD and other parkinsonian disorders in which the latency to onset of orthostatic hypotension is usually several years, patients with MSA usually develop symptomatic orthostatic hypotension within the first year after onset of symptoms (Wenning et al., 1999), and urinary dysfunction may occur even earlier (Sakakibara et al., 2000a, 2000b). Parkinsonism was the predominant motor disorder in SND, while gait ataxia was the usual presentation of the OPCA type of MSA. Tremor was present in only 29% and was typical “pill-rolling” in only 9%. Although 29% of all patients had initial good or excellent response to levodopa, this benefit was usually short-lived; only 13% maintained a good response to levodopa. Facial dystonia (often asymmetrical) was a typical levodopa-induced complication in patients with MSA. In another study of 16 autopsy-proven cases of MSA, Litvan and colleagues (1997d) identified early severe autonomic failure, absence of cognitive impairment, early cerebellar symptoms, and early gait problems as the best predictors of the diagnosis of MSA. In a study designed to validate the clinical criteria for MSA, Litvan and colleagues (1998a) found that the accuracy was best when at least six of the following eight features were present: sporadic adult onset, dysautonomia, parkinsonism, pyramidal signs, cerebellar signs, no levodopa response, no cognitive dysfunction, and no downward gaze palsy. Wenning and colleagues (1997) examined the clinical features of 203 pathologically proven cases of MSA reported in 108 publications. The male : female ratio was 1.3 : 1, dysautonomia was present in 74%, parkinsonism in 87%, cerebellar ataxia in 54%, and pyramidal signs in 49%. The progression and prognosis were analyzed in 230 Japanese patients with MSA; the median time from onset to aid-requiring walking, confinement to wheelchair, bedridden state, and death were 3, 5, 8, and 9 years, respectively (Watanabe et al., 2002). MSA-P patients had more rapid deterioration than MSA-C patients. When patients present with parkinsonism alone, without other evidence of MSA, their MSA might be difficult to differentiate from PD during the first 6 years (Albanese et al., 1995). In one study, the following features were found to be the best predictors of MSA: dysautonomia, poor response to levodopa, speech or bulbar dysfunction, falls, and absence of dementia and of levodopa-induced confusion (Wenning et al., 2000). Early development of autonomic dysfunction has been found to be an independent predictor of poor prognosis (Tada et al., 2007). It should be noted, however, that pure autonomic failure (PAF) might herald the onset not only of MSA, but also of PD and DLB (Kaufmann et al., 2004; Mabuchi et al., 2005). In PAF, orthostatic hypotension and sudomotor dysfunction followed by constipation are the typical initial symptoms, whereas in MSA, the initial presentation usually consists of urinary problems, followed by sudomotor dysfunction or orthostatic hypotension, with subsequent progression to respiratory dysfunction (Mabuchi et al., 2005; Benarroch, 2007)). A study of 115 patients with MSA showed that autonomic dysfunction, motor impairment, and depression were most closely related to poor outcome in measures of health-related quality of life (Schrag et al., 2006a).
Prior to reclassification of sporadic OPCA as MSA-C, there were many attempts to characterize the different forms of OPCA. Approximately one-quarter of patients with sporadic OPCA, particularly those with older-onset ataxia, develop parkinsonian features and evolve into MSA-C (Gilman et al., 2000). Berciano (1992) reviewed 133 (68 familial and 65 sporadic) pathologically proven cases of OPCA. While there was a nearly 2 : 1 male preponderance in familial OPCA, no gender difference was found in the sporadic form. Age at onset was more variable in this disorder than in the other parkinsonism-plus syndromes, ranging from infancy to 66 years. Cerebellar ataxia was the presenting symptom in 73% of all patients; 8.2% began with parkinsonian symptoms, and the remainder presented with nonspecific symptoms. Dementia, gaze impairment, dysarthria, dysphagia, incontinence, and upper and lower motor neuron signs usually become apparent within a few years after onset. In one large Japanese family with OPCA, the oculomotor abnormalities consisted of limitation of upgaze and convergence, horizontal gaze nystagmus, relative sparing of pupil reactivity, and loss of vestibulo-ocular responses (Shimizu et al., 1990). Autopsy of one patient in this series revealed degeneration of the oculomotor nucleus with sparing of the Edinger–Westphal nucleus. Neuropsychological evaluation in patients with clinically diagnosed OPCA revealed emotionality, anxiety, and a tendency toward depression without cognitive decline (Brent et al., 1990). Other studies, however, noted some degree of dementia in up to 80% of patients (Berciano, 1992).
While this review focuses on the sporadic forms of OPCA, it is worth pointing out that the classification of familial cerebellar ataxias has been markedly facilitated by the discoveries of specific mutations associated with the different phenotypes. Of the autosomal dominant cerebellar ataxias with known genetic defects, SCA1, SCA2, SCA3 (Machado–Joseph disease) (Kawaguchi et al., 1994; Lu et al., 2004), SCA6, SCA12, and SCA21, and dentatorubral-pallidoluysian atrophy (DRPLA) (Komure et al., 1995; Warner et al., 1995) are associated with extrapyramidal features, including parkinsonism (Rosenberg, 1995). Young-onset, levodopa-responsive parkinsonism may be the presentation of SCA2 and may precede the onset of ataxia by 25 years (Furtado et al., 2002; Lu et al., 2002; Payami et al., 2003; Furtado et al., 2004; Lu et al., 2004). Interruption of the SCA2 CAG/CAA repeat expansion has been found to be associated with autosomal dominant parkinsonism (Charles et al., 2007).
Although rarely pathologic features of sporadic MSA are found in the autosomal dominant form of SCA (Gilman et al., 1996b), the typical sporadic MSA is genetically distinct from the inherited forms of SCA and OPCA (Bandmann et al., 1997). These disorders should be differentiated from cortical cerebellar atrophy and SCA, in which cerebellar signs are unaccompanied by autonomic features (Bürk et al., 1996; Dürr et al., 1996; Gilman and Quinn, 1996; Hammans, 1996; Osaki et al., 2002). Pathologically, there might be some similarities between the central disorders, including the presence of GCIs in rare cases of SCA, but the spinal cord is usually more atrophied in SCA than in MSA. Some features of MSA overlap with the syndrome of fragile X-associated tremor/ataxia syndrome caused by permutations of the fragile X mental retardation 1 gene (FMR1), and fragile X-associated tremor/ataxia syndrome may be a rare cause of MSA (Biancalana et al., 2005).
Several clinical studies have addressed the differentiation between MSA-P and parkinsonism and a collection of “red flags” has been generated and recently validated as having high diagnostic specificity (Kollensperger et al., 2008; Stefanova et al., 2009) (Table 9.2). The red flags were grouped into the following six categories: (1) early instability, (2) rapid progression, (3) abnormal postures (includes Pisa syndrome, disproportionate anterocollis, and/or contractures of hands or feet), (4) bulbar dysfunction (includes severe dysphonia, dysarthria, and/or dysphagia), (5) respiratory dysfunction (includes diurnal or nocturnal inspiratory stridor and/or inspiratory sighs), and (6) emotional incontinence (includes inappropriate crying and/or laughing). They proposed that a combination of two out of these six red flag categories were used as additional criteria for the diagnosis of probable MSA-P. Movement disorders other than parkinsonism that are seen in patients with MSA include dystonia, stimulus-sensitive cortical myoclonus, hemiballism, and chorea, unrelated to dopaminergic therapy (Chen et al., 1992; Steiger et al., 1992; Salazar et al., 2000). Dystonia is relatively rare in MSA patients (Rivest et al., 1990), but in one study, 46% of patients with MSA were found to have dystonia, particularly if anterocollis is considered a form of cervical dystonia (Boesch et al., 2002). Some investigators, however, believe that the neck flexion, often associated with anterior sagittal shift, is due to disproportionally increased tone in the anterior neck muscles leading to secondary fibrotic and myopathic changes (van de Warrenburg et al., 2007). Although dystonia is frequently considered to be a cause of the MSA-associated anterocollis, the mechanism of progressive neck flexion, so characteristic of MSA, particularly MSA-P, may be multifactorial. In some cases, the neck flexion has been attributed to neck extensor weakness as part of “dropped head” syndrome associated with axial myopathy (Suarez and Kelly, 1992; Oerlemans and de Visser, 1998; Askmark et al., 2001), motor neuron disease (Gourie-Devi et al., 2003), or other causes. Neck flexion, however, is not unique to MSA and can be also seen in patients with otherwise typical PD (Djaldetti et al., 1999). In some cases of PD, more frequently than in MSA, the axial postural abnormality may lead to severe flexion of the trunk, the so-called bent spine syndrome, or camptocormia (Umapathi et al., 2002; Azher et al., 2005). Another abnormal posture frequently encountered in MSA is the “Pisa syndrome” manifested by leaning of the body to one side reminiscent of the leaning tower of Pisa (Ashour et al., 2006) (Video 9.8). Besides anterocollis, another form of dystonia that is relatively frequently encountered in patients with MSA is facial and oromandibular dystonia associated with levodopa therapy. As was noted earlier in the chapter, inspiratory stridor may be a variant of laryngeal dystonia (Merlo et al., 2002). In addition to action myoclonus (Video 9.8), focal reflex myoclonus, induced by pinprick, may be seen in MSA patients (Clouston et al., 1996; Salazar et al., 2000). This form of myoclonus has a longer latency than that seen in patients with CBD (see later). In 9 of 11 patients with MSA and myoclonic tremulous movements, jerk-locked averaging technique showed premyoclonic potential, suggesting that the jerk-like movements represent a form of cortical myoclonus (Okuma et al., 2005). 
| 2 out of the following 6 needed for diagnosis of probable MSA-P: |
From Kollensperger M, et al. (European MSA Study Group). Red flags for multiple system atrophy. Mov Disord 2008;23:1093–9.
Although dysautonomia is a cardinal feature of MSA, this neurodegenerative disorder should be differentiated from PAF, which has no central component (Mathias, 1997). Autonomic dysfunction is essential for the diagnosis of MSA, and in many cases of MSA, autonomic failure, particularly impotence, precedes other neurologic symptoms or signs by several years. In contrast to PD associated with dysautonomia, in which there is predominantly peripheral (ganglionic and postganglionic) involvement, including myocardial (Goldstein et al., 2002), sympathetic denervation, the peripheral autonomic system appears to be spared in MSA and the primary lesion is preganglionic (Lipp et al., 2009). In fact, some persistence of central autonomic tone might be responsible for the frequently observed supine hypertension in MSA (Parikh et al., 2002). MSA patients also have more autonomic symptoms at baseline and more progression to global anhidrosis than patients with PD (Lipp et al., 2009). The “cold hands” sign, manifested by cold, dusky, violaceous appearance of the hands, is another characteristic feature of MSA (Klein et al., 1997). Some patients have the “cold feet” sign (Video 9.9). Liquid meal, consisting chiefly of glucose and milk, markedly reduces blood pressure in patients with MSA but not in those with PD (Thomaides et al., 1993) (Video 9.9). Respiratory disturbance, including severe obstructive sleep apnea, and vocal cord paralysis with stridor may be found in more advanced stages of the disease (Munschauer et al., 1990), but respiratory insufficiency may be the presenting symptom of MSA (Glass et al., 2006). Inspiratory stridor due to the paradoxical movement of the vocal cords (also known as Gerhardt syndrome) has been described in MSA (Eissler et al., 2001). The observation that stridor improves with botulinum toxin injections into the adductor laryngeal muscles suggests that this symptom of MSA could be due to focal laryngeal dystonia (Merlo et al., 2002). The occurrence of nocturnal or daytime stridor carries a poor prognosis, particularly when it is associated with central hypoventilation (Silber and Levine, 2000). Occasionally, however, vocal cord abductor paralysis can be seen even in the initial stages of the disease, and it has been associated with nocturnal sudden death (Isozaki et al., 1996). Early diagnosis can be made by laryngoscopy during sleep. Hypoxemia with increased alveolar-arterial oxygen gradient, associated with laryngopharyngeal movements, however, has been demonstrated in MSA patients even during wakefulness (Shimohata et al., 2007). The presence of impaired hypoxic ventilatory response has helped to differentiate MSA-C from idiopathic late-onset cerebellar ataxia (Tsuda et al., 2002). 
The natural history of MSA has been the subject of several recent studies. In contrast to the approximately 1.5% annual decline in UPDRS-III noted in patients with PD (Jankovic and Kapadia, 2001), the average annual decline in MSA-P is 28.3% (Seppi et al., 2005). Since most of the studies were based on pathologically proven cases, the prognosis in these series has been worse than otherwise predicted. In one study of 59 patients with MSA that included 25 with SDS, 24 with OPCA, and 10 with SND, the survival was poorest in SDS, followed by SND and OPCA (Saito et al., 1994). On the basis of a meta-analysis of 433 reported cases of MSA, Ben-Shlomo and colleagues (1997) found survival to range from 0.5 to 24 years (mean: 6.2 years), and cerebellar features were associated with marginally better survival. In one study of 100 patients clinically diagnosed with probable MSA (14 of whom were pathologically confirmed), nearly half of all the patients were markedly disabled or wheelchair bound within 5 years after onset, and the median survival was 9.5 years (Wenning, et al., 1994b) (Fig. 9.5). This is similar to the median survival of 8.6 years for men and 7.3 years for women in a group of 22 patients with pathologically proven MSA followed prospectively (Schrag et al., 2008). In another study, the investigators analyzed 35 pathologically confirmed cases of MSA and confirmed a direct correlation between severity of disease and nigrostriatal cell loss (Wenning et al., 1995). Although marked degeneration in the olivopontocerebellar system, particularly the cerebellar vermis, was noted in 88% of the brains, the cerebellar pathology did not correlate with the presence of cerebellar signs. Although some authors have suggested that the earlier and the more severe the involvement of the autonomic nervous system, the poorer the prognosis (Saito et al., 1994), this has not been confirmed by other studies (Ben-Shlomo et al., 1997). MSA patients usually die from aspiration, sleep apnea, or cardiac arrhythmia. Survival data and clinically relevant milestones – namely: frequent falling, cognitive disability, unintelligible speech, severe dysphagia, dependence on wheelchair for mobility, the use of urinary catheters, and placement in residential care – were determined based on a retrospective chart review of pathologically confirmed cases of PSP (n =110) and MSA (n = 42) (O’Sullivan et al., 2008). Patients with PSP had an older age of onset (P < 0.001) and reached their first clinical milestone earlier than patients with MSA (P < 0.001). Patients with PSP generally had a less favorable prognosis than those with MSA, and rarely autonomic failure in MSA was associated with shorter survival.
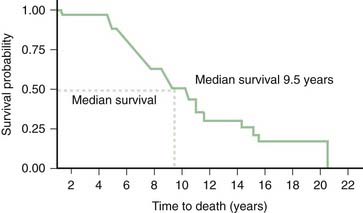
Epidemiology
The average annual incidence rate for MSA has been estimated to be 3 new cases per 100 000 person-years. (Bower et al., 1997), and the age-adjusted prevalence has been estimated at 4.4 per 100 000 (Schrag et al., 1999). Some studies have shown that, similar to PD, smoking is significantly less frequent in patients with MSA, and farming is an independent risk factor for MSA (Vanacore et al., 2005).
Neurodiagnostic studies
In addition to tests of autonomic function, patterns of plasma levels of catecholamines and their metabolites may be helpful in differentiating the various forms of autonomic failures (Cohen et al., 1987; Goldstein et al., 1989; Polinsky, 1993; Mathias, 1995; American Academy of Neurology, 1996; Parikh et al., 2002). These studies are designed primarily to localize the site of autonomic impairment and include investigation of neurogenic bladder (Fowler, 1996), sphincter electromyography (EMG) (Stocchi et al., 1997), and other investigations designed to test the integrity of the autonomic nervous system. Neurogenic sphincter electromyography, however, can be also seen in other disorders, including PD, PSP, Huntington disease, and a variety of common urologic problems (Colosimo et al., 1999; Giladi et al., 2000; Vodusek, 2001). Although anorectal dysfunction does not differentiate MSA from PD, external anal sphincter electromyography denervation is a very sensitive measure of anorectal dysfunction (Tison et al., 2000), and this abnormality occurs much earlier in MSA than in PD (Stocchi et al., 2000). Patients with SDS show a deficit in the central component of the baroreflex, whereas peripheral sympathetic pathways are affected primarily in the syndrome of PAF. While the peripheral sympathetic neurons are spared in MSA, there is some evidence that they lack the normal preganglionic activation (Parikh et al., 2002). Furthermore, the relatively well-preserved sympathetic tone is probably responsible for the supine hypertension that is seen in most patients with MSA. In neurogenic orthostatic hypotension, the rise in NE levels is minimal or absent despite a marked drop in blood pressure after either head-up tilt or an upright position. Clonidine has been found to increase growth hormone (GH) in normal controls, patients with PAF, and patients with PD but not patients with MSA (Thomaides et al., 1992; Kimber et al., 1997). Whether this clonidine-GH test can reliably differentiate between MSA and PD, as Kimber and colleagues (1997) suggested, awaits further clinical-pathologic studies. In a study of 69 MSA patients, 35 PD patients, and 90 healthy controls, the GH response to arginine was found to be significantly lower in MSA patients as compared to PD or healthy controls, a finding with over 90% sensitivity and specificity (Pellecchia et al., 2006).
While somatosensory, visual, and auditory evoked responses are often abnormal, motor evoked potentials are usually normal (Abele et al., 2000). In one study, 73% of patients with SDS had abnormal brainstem auditory evoked responses (Sinatra et al., 1988). In a study comparing polysomnograms of seven patients with SDS with those of seven control patients, significant obstructive sleep apnea without oxygen desaturation was seen in four of the five nontracheotomized SDS patients; three of these patients later died suddenly during sleep (Munschauer et al., 1990). In a more recent study, Plazzi and colleagues (1997) demonstrated that 90% of MSA patients experience some form of REM sleep behavioral disorder (RBD). A strong relationship between RBD and subsequent MSA has been demonstrated by a number of other studies.
Using PET scan technology to measure 6-[18F]fluoro-dopamine-derived radioactivity in myocardium, Goldstein and colleagues found normal rates of cardiac spillover of NE and normal production of levodopa, dihydroxyphenyl-glycol, and dihydroxyphenylacetic acid suggestive of intact cardiac sympathetic terminals in patients with SDS (Goldstein et al., 1997). This is in contrast to absent radioactivity in patients with PAF, indicating loss of postganglionic sympathetic terminals in this peripheral autonomic disorder. The value of this test in diagnosing MSA has been questioned (Mathias, 1997), however, since both sympathetic and parasympathetic failure have been associated with MSA. Neurochemical changes seen in MSA are similar to those present in PAF, and some suggest that MSA represents a central progression from PAF (McLeod and Tuck, 1987a, 1987b). Pharmacologically, these two conditions may be distinguished by supine and standing plasma NE levels. In PAF, both standing and supine NE levels are low, while in MSA, only the standing value is diminished. Besides decreased NE, acetylcholine and cerebrospinal fluid (CSF) acetylcholinesterase (AChE) levels are also reduced (Polinsky et al., 1989). This is consistent with the notion that postganglionic sympathetic neurons are intact in MSA but their function is markedly impaired in PAF.
Another imaging study used [11C]DTBZ PET and [123I]iodobenzovesamicol single-photon emission computed tomography (SPECT), and showed that RBD in patients with MSA correlates with nigrostriatal dopaminergic deficit. Furthermore, obstructive sleep apnea in MSA is related to a thalamic cholinergic deficit, possibly owing to decreased pontine cholinergic projections (Gilman et al., 2003a). The same group used PET with [11C]PMP and that subcortical AChE activity was significantly more decreased in MSA-P and PSP than in PD. The authors suggested that this reflects greater impairment in the pontine cholinergic group (PPN) and may account for the greater gait disturbances in the early stages of these two disorders compared to PD (Gilman et al., 2010). Carbon-11-labelled 2-[2-(2-dimethylaminothiazol-5-yl)ethenyl]-6-[2-(fluoro)ethoxy] benzoxazole PET, believed to image α-synuclein, in 8 patients with MSA showed high distribution volumes in the subcortical white matter, putamen and posterior cingulate cortex, globus pallidus, primary motor and anterior cingulate cortex, and substantia nigra compared to the normal controls (Kikuchi et al., 2010). Since these areas are also rich in glial cytoplasmic inclusions, the carbon-11-labelled 2-[2-(2-dimethylaminothiazol-5-yl)ethenyl]-6-[2-(fluoro)ethoxy] benzoxazole PET may be a potential surrogate marker for progression of synucleinopathies.
Neuroimaging techniques, including MRI, transcranial sonography, functional imaging (PET and SPECT), and imaging cardiac sympathetic innervation, can be very helpful in differentiating MSA from other parkinsonian disorders and neuroimaging criteria have been proposed (Brooks et al., 2009). Neuroimaging specifically designed to assess putaminal integrity may prove helpful in differentiating MSA from PD and in predicting levodopa response (Drayer et al., 1989; Stern et al., 1989). MRI in patients with SDS sometimes reveals areas of decreased signal bilaterally in the posterolateral putamen on T2-weighted images (Pastakia et al., 1986). Although increased levels of iron may contribute to this hypointensity, reactive microgliosis and astrogliosis may also play an important role. In addition to striatal (putaminal) hypointensity on T2-weighted MRI scans, a characteristic finding in patients with MSA, particularly MSA-P, is the slit-hyperintensity in the lateral margin of the putamen (Savoiardo et al., 1994; Sitburana and Ondo, 2009). This abnormality was found in 17 of 28 (61%) of patients with clinically diagnosed MSA (Konagaya et al., 1994). Although not all MSA patients with this slit-hyperintensity had parkinsonism, none of the 25 patients with clinically diagnosed PD demonstrated this finding on MRI. Linearization of putaminal margin was found in 88.8% of patients with MSA-P, but only in 8.3% of patients with MSA-C, 7.9% of patients with PD, and 7.4% of healthy subjects (Ito et al., 2007). Although hyperintense putaminal rim is quite specific for MSA, putaminal hypointensity was not found to be a useful discriminator in one study (Schrag et al., 1998). The combination of increased signal on T2 or FLAIR in the lateral putamen (indicating iron deposition) and the slit-hyperintensity at the posterolateral border of the putamen on T2-weighted images provides a sensitivity for MSA of 97% (von Lewinski et al., 2007). Using DWI-MRI, Schocke and colleagues (2002) found that regional apparent diffusion coefficients values are increased in the putamen of patients with MSA, and this evidence of striatal degeneration apparently reliably differentiates MSA from PD. DWI imaging showing abnormal signal in the middle cerebellar peduncle is apparently relatively specific for MSA-C and is not seen in PSP or PD (Nicoletti et al., 2006; Paviour et al., 2007). Ghaemi and colleagues (2002) found that both multitracer PET and three-dimensional MRI-based volumetry are very sensitive measures and that they reliably differentiate MSA from PD by demonstrating decreased putaminal volume, glucose metabolism, and postsynaptic D2 receptor density in patients with MSA. These techniques, when applied to the imaging of the midbrain, did contribute additional gain in diagnostic accuracy. In one study, however, the typical “hot cross bun” sign in the pons was present on MRI in 63% of patients (Watanabe et al., 2002). Other studies have highlighted the “hot cross bun” sign as a characteristic feature of MSA (Sitburana and Ondo, 2009). This study also drew attention to the involvement of the cortex in MSA. T2*-weighted gradient echo was more sensitive in demonstrating hypointense putaminal changes than T2-weighted fast spin echo MRI (Kraft et al., 2002) (Fig. 9.6). A diagnostic algorithm based on brain MRI has been proposed (Bhattacharya et al., 2002).
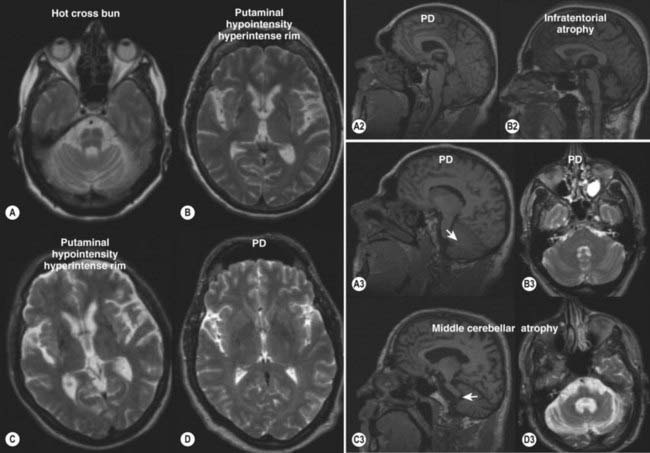
Figure 9.6 Radiologic features of MSA versus PD.
From Brooks DJ, Seppi K; for the Neuroimaging Working Group on MSA. Proposed neuroimaging criteria for the diagnosis of multiple system atrophy. Mov Disord 2009;24:949–964, with permission.
Using proton magnetic resonance spectroscopy, Davie and colleagues (1995) showed a significant reduction in the N-acetylaspartate (NAA)/creatine ratio from the lentiform nucleus in six of seven patients with MSA and in only one of nine patients with PD. Similar abnormalities were found in some patients with OPCA and most likely reflect regional neuronal loss. Further studies are needed to determine whether this technique can reliably differentiate between MSA and PD. Brain parenchyma sonography has been used in one study to differentiate PD from atypical parkinsonism, mostly MSA (Walter et al., 2003). The investigators found that 24 of 25 (96%) patients with PD exhibited hyperechogenicity, whereas only 2 of 23 (9%) patients with atypical parkinsonism showed a similar pattern. They concluded that brain parenchyma sonography might be highly specific in differentiating between PD and atypical parkinsonism. Computed tomography and MRI scans in patients with OPCA (MSA-C) typically show pancerebellar and brainstem atrophy, enlarged fourth ventricle and cerebellopontine angle cisterns, and demyelination of transverse pontine fibers on T2-weighted MRI images (Berciano, 1992). Although the MRI findings in MSA are highly specific, they have low sensitivity (Schrag et al., 2000). In a cross-sectional study of 15 MSA-P patients and 17 PD patients matched for age and disease duration, there were lower activity ratios of striatal to frontal uptake on iodobenzamide (IBZM)-SPECT; and on DWI, there were similar differences in the regional apparent diffusion coefficients (rADC). The findings from DWI were more accurate when compared to IBZM-SPECT based on the higher specificity, predictive accuracy, and positive predictive values of both the methods. Further studies using DWI validating these changes with disease progression are needed (Seppi et al., 2004).
PET scanning has revealed decreased striatal and frontal lobe metabolism (De Volder et al., 1989; Brooks et al., 1990b; Eidelberg et al., 1993) and a reduction in D2 receptor density in the striatum (Brooks et al., 1992; Antonini et al., 1997). Using [18F]fluorodeoxyglucose, [18F]fluorodopa, and [11C]raclopride (RACLO) PET scans, Antonini and colleagues (1997) showed that the combination of [18F]fluorodeoxyglucose and [11C]raclopride scans reliably differentiated between MSA and PD, but [18F]fluorodopa PET scans could not distinguish between the different forms of parkinsonism. In a study of 167 patients [18F]fluorodeoxyglucose PET was found to have high specificity and sensitivity in distinguishing between parkinsonian disorders (Tang et al., 2010). In a study of three patients with SDS, the two with more advanced stages of the disease showed reduced 18F-6-fluorodopa uptake, indicating nigrostriatal dysfunction (Bhatt et al., 1990). Gilman and colleagues (1999) found significantly reduced specific binding to the type 2 vesicular monoamine transporter using PET and [11C]dihydrotetrabenazine as a type 2 vesicular monoamine transporter ligand in striatal monoaminergic presynaptic terminal of patients with MSA. PET scans in patients with MSA-C show a reduced metabolic rate in the brainstem and cerebellum (Gilman, 2002), and these changes can be detected even before the onset of extrapyramidal features (Gilman et al., 1996a). A study of 10 patients with the sporadic OPCA form of MSA showed that the 18F-fluorodopa uptake was reduced by a mean of 21% in the putamen, and 11C-diprenorphine uptake was reduced by a mean of 22% (Rinne et al., 1995). Although the authors suggest that these findings support “subclinical” nigrostriatal dysfunction, all of the patients were clinically impaired, and some were disabled by severe ataxia and autonomic dysfunction. The reduced 11C-diprenorphine uptake suggests that in addition to involvement of the nigrostriatal projection, some MSA patients have a loss of intrinsic striatal neurons that contain presynaptic and postsynaptic mu, kappa, and delta opioid receptors.
Besides neuroimaging, transcranial ultrasonography has been reported to provide high diagnostic yield in differentiating PD from atypical parkinsonian disorders. Sonographic studies in 102 patients with PD, 34 with MSA, and 21 with PSP found marked unilateral or bilateral hyperechogenicity in 89% of 88 patients with PD, 25% of 32 patients with MSA-P, and 39% of 18 patients with PSP (Behnke et al., 2005).
Neuropathology, neurochemistry, and pathogenesis
The spectrum of pathologic changes in MSA includes cell loss, gliosis, and demyelination in the striatum (caudate and putamen), SN, locus coeruleus, inferior olives, pontine nuclei, dorsal vagal nuclei, Purkinje cells of the cerebellum, intermediolateral cell columns, and Onuf’s nucleus of the caudal spinal cord. Involvement of at least three of these areas, including the putamen and SN, is required for the pathologic diagnosis of MSA (Quinn, 1994; Ito et al., 1996). In one pathologic study of 100 MSA cases (46 men and 54 women), 34% were categorized as SND, 17% were categorized as OPCA (MSA-C), and the remainder (49%) had a mixed pathology (Ozawa et al., 2004). On the basis of the distribution of GCIs and correlation of the clinicopathologic changes, the study showed that GCIs might be contributing to neuronal damage in the MSA-C type more than the in MSA-P type, suggesting the possibility of different mechanisms of cell death in the subtypes of MSA. The brunt of the pathology in MSA brains is in the dorsolateral portion of the striatum and ventrolateral portion of the globus pallidus and SN, and the degree of response to levodopa seems to correlate inversely with the severity of the striatal efferent involvement (Ito et al., 1996). The putamen is the most prominently affected, with neuronal cell loss and deposition of iron, producing brownish pigmentation (O’Brien et al., 1990). Cholinergic neurons in the pedunculopontine nucleus and laterodorsal tegmentum and noradrenergic neurons in locus coeruleus were found to be markedly depleted in the brains of patients with MSA, whereas the serotonergic rostral raphe neurons are well preserved (Benarroch et al., 2002). Lewy bodies or NFTs are not common. Using calcineurin immunostaining, Goto and colleagues (1996) found marked neuronal loss particularly in the caudal and lateral portion of the putamen, with corresponding degeneration of GPi, GPe, and ventrolateral portion of the SNr. The same group (Goto et al., 1990b) noted selective degeneration of the met-enkephalin-containing neurons in the putamen and GPe, with relative preservation of the caudate nucleus. Previous studies have noted low levels of dopamine and increased activity of dopamine β-hydroxylase, the NE-synthesizing enzyme, in the midbrain. Vasomotor impairment in SND has been attributed to selective loss of tyrosine hydroxylase-immunoreactive neurons in the A1 and A2 regions of the medulla oblongata (Malessa et al., 1990). Loss of arginine-vasopressin synthesizing neurons in the hypothalamic suprachiasmatic nucleus has been demonstrated in the brains of patients with MSA (Benarroch et al., 2006). Calbindin-D28k immunoreactivity in the striatal projection system is markedly decreased in the brains of patients with SND (Ito et al., 1992) and in Purkinje cells of the cerebellum in patients with MSA (Wüllner et al., 2000). In the early stages of MSA-P, the medium spiny neurons staining for calbindin (localized to the matrix), but not those staining for calcineurin (localized striasomes), appear to be depleted first (Sato et al., 2007). Reduced calcium-binding capacity in these neurons might affect the bcl-2 family of proteins and lead to apoptosis of selected neuronal regions not only in PD, but also in other neurodegenerative diseases, such as MSA. The small myelinated fibers innervating the vocal cord are lost in nearly all patients with MSA, but when the large myelinated fibers of the recurrent laryngeal nerve become affected, as is seen in some patients with SND, vocal cord paralysis becomes evident and may be life-threatening (Hayashi et al., 1997).
The fundamental pathologic changes in OPCA (MSA-C), whether familial or sporadic, are a loss of Purkinje cells in the cerebellar cortex, particularly in the vermis, and degeneration of the olivopontine nuclei (Wenning et al., 1996). In addition to cerebellar atrophy, SN degeneration and depigmentation, neuronal loss in other brainstem nuclei, and demyelination of corticospinal tracts and posterior columns are seen (Koeppen et al., 1986; Matsuo et al., 1998). One clinical-pathologic study found a strong correlation between the frequency of neuronal cytoplasmic inclusions exclusively found in the pontine nucleus and the severity of olivopontocerebellar degeneration. The immunohistochemical and ultrastructural features of the neuronal inclusions were identical to those of the glial inclusions (Yokoyama et al., 2001). Detailed morphometric and biochemical studies of OPCA brains correlated reductions in aspartic and glutamic acid with Purkinje cell loss in the cerebellar cortex and with neuronal cell loss in the inferior olives (aspartic acid) (Bebin et al., 1990). In addition, quisquilate receptors appear to be decreased, while quinolinic acid metabolism is increased (Makoweic et al., 1990; Kish et al., 1991). The increased quinolinic acid phosphoribosyl-transferase activity in OPCA has been interpreted as a compensatory mechanism designed to protect quinolinic acid-sensitive granule cells (Kish et al., 1991). In addition, low glutamate dehydrogenase activity has been found in most, but not all, studies; however, this defect probably is not disease-specific (Berciano, 1992; Kostic et al., 1989). In a study of 14 brains of patients with OPCA, Kish and colleagues (1992) found a 53% reduction in dopamine in the putamen, 35% in the caudate, and 31% in the nucleus accumbens. Only two patients had severe neuronal loss in the SN and a corresponding dopamine depletion in the striatum. Mitochondrial deoxyribonucleic acid abnormalities have been postulated to be important in the pathogenesis of OPCA in some patients (Truong et al., 1990). A mitochondrial DNA G11778 mutation has been identified in a family with levodopa-responsive parkinsonism and multisystem degeneration (Simon et al., 1999).
Degeneration of the catecholaminergic neurons in the intermediate reticular formation of the rostral ventrolateral medulla seems to correlate well with autonomic failure in patients with MSA (Benarroch et al., 1998, 2000). The medial spiny neurons that give rise to both the direct pathway from the striatum to the GPi and the indirect pathway from the striatum to the GPe are affected. Gliosis, however, seems to be much more prevalent in GPe than in GPi. Upregulation of D1 receptors, determined by dopamine-stimulated adenylyl cyclase, has been demonstrated in brains of patients with PD as compared to PSP and MSA (n = 10 each); it is actually reduced in MSA (Tong et al., 2004). While the median CSF concentration of the neurotransmitter metabolites 5-hydroxyindolacetic acid and 3-methoxy-4-hydroxyphenylethyleneglycol was reduced significantly (49–70%) in MSA compared to PD, several brain-specific proteins (tau, neuron-specific enolase, myelin basic protein) were elevated (130–230%) in MSA compared with those in PD (Abdo et al., 2004). Although a combination of CSF tau and 3-methoxy-4-hydroxyphenylethyleneglycol significantly discriminated PD from MSA, it is too early to recommend routine use of CSF in differentiating these two disorders.
The discovery in 1989 by Papp and Lantos (Papp et al., 1989) of the characteristic histologic marker, the GCIs, led to improved characterization of MSA as a clinical-pathologic entity. These inclusions are particularly concentrated in the oligodendrogliocytes and have been found in all autopsied brains of patients with SDS, SND, and sporadic OPCA. This shared pathologic feature strongly argues in support of the notion that these three disorders should be regarded as variants of the same disease entity, namely, MSA (Kato and Nakamura, 1990; Arima et al., 1992; Murayama et al., 1992; Papp and Lantos, 1992; Lantos and Papp, 1994; Quinn, 1994; Wenning et al., 1994a). GCIs, argyrophilic, perinuclear inclusions with a diameter varying from 4 µm to 20 µm, are found particularly in the oligodendrogliocytes in the supplementary motor cortex, anterior central gyrus, putamen, pallidum, basal pons, and medullary reticular formation. They are composed of 20–30 nm straight tubules, and they contain ubiquitin, tau protein, α- and β-tubulin, MAP-5, αB-crystallin, and α-synuclein. Several studies have documented the presence of α-synuclein in the GCI (Spillantini et al., 1998; Tu et al., 1998; Wakabayashi et al., 1998), but the significance of this finding is uncertain (Mezey et al., 1998). Although very characteristic of MSA, GCIs have been rarely found in other disorders, such as CBD, PSP, and autosomal dominant spinocerebellar ataxia (Gilman et al., 1996b). The characteristics and distribution of the inclusions, however, may be slightly different from the typical GCIs that are seen in MSA. Originally, α-synuclein was identified as the precursor protein for the non-Aβ component (NAC) of Alzheimer disease of amyloid plaques and these plaques have been found to contain fragments of the α-synuclein protein (NAC precursor protein, or NACP). NACP/α-synuclein accumulates not only in Lewy bodies, but also as small granules in neurons and as diffuse deposits in neuronal process in PD brains (Shoji et al., 2000). In addition, NACP accumulates in cortical astrocytes. The distribution and cell type of NACP accumulation are similar in DLB, but in MSA, NACP accumulates chiefly in the oligodendroglia and olivary neurons. α-Synuclein is selectively and extensively phosphorylated at serine 129, especially by casein kinase 1 and 2, in various synucleinopathies, including MSA (Fujiwara et al., 2002). Recently, p39 immunoreactive GCIs have been reported as the hallmark of GCI in MSA (Honjyo et al., 2001). A cdk5 (cyclin-dependent kinase) activator in oligodendrocytes, p39 induces formation of GCIs in the oligodendrocytes. Midkine, a neurotrophic factor, has also been identified in the GCI, indicating a rescue of neurons via oligode (Burn and Jaros, 2001). Because of increasing evidence of dysregulation of myelin basic protein and p25α, also called tubulin polymerization promoting protein, in MSA, there is an emerging notion that MSA represents an oligodendrogliopathy (Wenning et al., 2008). Only 10% of the MSA cases are found to have Lewy bodies (Ozawa et al., 2004). A significant correlation has been found between the frequency of GCIs, severity of neuronal cell loss, and disease duration (Ozawa et al., 2004).
The autonomic failure in MSA has been largely attributed to the depletion of sympathetic preganglionic neurons in the spinal intermediolateral cell column (IML) and its afferent medullary catecholaminergic and serotonergic neurons. In 12 MSA patients who had died within 3.5 years after disease onset, 4 died suddenly and 8 died as a result of established causes (Tada et al., 2009). The investigators found that the spinal IML and medullary catecholaminergic and serotonergic systems were involved even in the early stages of MSA, and their degeneration may be responsible for sudden death in patients with MSA.
Using antibodies against certain components of myelin basic protein, several investigators found evidence of extensive myelin degeneration in MSA, thus supporting the notion of widespread oligodendroglial dysfunction in MSA (Castellani, 1998; Matsuo et al., 1998). Diffuse degeneration of the white matter, particularly involving the central tegmental tract associated with vacuolation, has been an increasingly recognized pathologic feature of MSA (Armstrong et al., 2007). Other studies found evidence of apoptosis in the glia, but not neurons, of MSA brains (Probst-Cousin et al., 1998). Furthermore, microglial activation involving translocation of NF-kappaB/Rel A to the nucleus was found to be particularly prominent in affected brain regions (Schwarz et al., 1998).
In addition to the typical findings in the brain, there is a marked loss of neurons in the lateral horns of the spinal cord, but these pathologic changes correlate poorly with dysautonomia (Gray et al., 1988). Substance P-like immunoreactivity was markedly decreased in laminae I + II of fourth thoracic and third lumbar spinal cord segments in 10 of 11 SDS patients, and all had a decrease in small and large myelinated fibers in the fourth thoracic ventral roots (Tomokane et al., 1991).
The cause of MSA is unknown, and genetic factors probably do not play an important role. Recent findings link MSA to PD and related neurodegenerative disorders as “α-synucleinopathies” (Jaros and Burn, 2000; Galvin et al., 2001). To test whether variations in the gene coding for α-synuclein (SNCA) are associated with increased risk in MSA, a candidate single nucleotide polymorphism (SNP) association study was performed on 384 most-associated SNPs in a genome-wide association study of PD in 413 MSA cases and 3974 control subjects. The 10 most significant SNPs were then replicated in an additional 108 MSA cases and 537 controls (Scholz et al., 2009). The study found that SNPs at SNCA were associated with increased risk for the development of MSA at an odds ratio of 6.2 (P = 5.5 × 10−12).
While many studies have investigated the putative role of environmental toxins in the pathogenesis of idiopathic PD, the possible role of such toxins in MSA has received little attention. We reported on 10 patients whose clinical features were consistent with MSA and in whom toxins were suspected to play an etiologic role (Hanna et al., 1999). One patient with pathologically confirmed MSA was exposed to high concentrations of various toxins, including formaldehyde, malathion, and diazinon. The other MSA patients had a history of heavy exposure to various agents, such as n-hexane, benzene, methyl-isobutyl-ketone, and pesticides. The pathologic case revealed extensive advanced glial changes, including GCIs, which were seen particularly in the deep cerebellar white matter, brainstem, cortex (superior frontal, insula, and hippocampus), and putamen. Additionally, there was notable neuronal cell loss with depigmentation of the SN and locus coeruleus. Although a cause-and-effect relationship cannot be proven, these cases suggest that environmental toxins could play a role in the pathogenesis of some cases of MSA. The inverse relationship between PD and smoking has been also found with MSA but not with PSP (Vanacore et al., 2000).
Although there is no experimental model of MSA, intraperitoneal injection of 3-acetylpyridine in rats produces neurochemical and histologic changes that are consistent with OPCA (Deutch et al., 1989). In addition to causing degeneration of the nigrostriatal dopaminergic pathway, this neurotoxin causes degeneration of the climbing fibers, which normally originate in the inferior olive and terminate in the cerebellum. Another possible animal model of MSA is the “double lesion” rat model in which an initial 6-OHDA substantia nigra lesion is followed by a quinolinic acid induced lesion in the striatum (Kollensperger et al., 2007). Transgenic mice overexpressing human wild-type α-synuclein in oligodendrocytes exhibit many of the features of MSA (Yazawa et al., 2005).
Treatment
About two-thirds of patients with MSA respond to levodopa. Parkinsonian symptoms accompanying SDS are difficult to treat, because dopaminergic drugs frequently exacerbate the already prominent symptoms of orthostatic hypotension. The addition of liberal salt, fludrocortisone, and elastic stockings can improve standing blood pressures (Mathias and Kimber, 1998). However, parkinsonian patients have difficulty putting on elastic stockings, such as the Jobst stockings. In addition, physical maneuvers such as leg-crossing and squatting can alleviate orthostatic lightheadedness (Van Lieshout et al., 1992). In a double-blind, placebo-controlled study of 97 patients with various causes of autonomic failure, including 18 patients with SDS and 22 patients with PD, midodrine, a peripheral α-adrenergic agonist, has been found to be effective in the treatment of orthostatic hypotension (Jankovic et al., 1993a, 1993b). The safety and efficacy of midodrine were later confirmed by a larger controlled study involving a total of 171 patients with orthostatic hypotension (Low et al., 1997). Wright and colleagues (1998) showed dose-dependent increases in standing systolic blood pressure with midodrine in patients with MSA. In patients with neurally mediated recurrent syncope, midodrine reduced frequency of syncope from 67% (8 of 12) to 17% (2 of 12) when compared to placebo (Kaufmann et al., 2002). The most frequent side effects associated with the drug included piloerection, scalp pruritus, urinary retention, and supine hypertension. The effects of subcutaneous injections of octreotide, a somatostatin analog, were tested in a group of nine patients with MSA (Bordet et al., 1995). The drug improved orthostatic hypotension, and it allowed patients to maintain an upright posture for a longer period of time as compared to placebo. Because fludrocortisone and midodrine, particularly when combined with liberal salt intake, increase the risk of supine hypertension, patients should be instructed to place their beds in the reverse Trendelenburg position. The use of nighttime nitroglyceride or clonidine patches has been suggested for the treatment of supine hypertension, but these measures are not always successful. Although this modest improvement was attributed to a release of NE by octreotide during maintenance of erect posture, no increase in plasma NE levels could be demonstrated.
Other agents that are used to increase standing blood pressure include indomethacin, ibuprofen, pseudoephedrine and other sympathomimetics, caffeine and dihydroergotamine, yohimbine, and NE precursors, such as 3,4-dihydroxyphenyl serine, also known as L-threo DOPS or droxidopa (McLeod and Tuck, 1987a, 1987b; Polinsky, 1993; Senard et al., 1993; Freeman et al., 1999). Droxidopa appears to increase NE in the brains of normal and NE-depleted animals, suggesting that droxidopa acts as an NE precursor. The drug may also act in the peripheral nervous system as evidenced by increased heart rate and elevated blood pressure. In 32 patients (26 MSA, 6 PAF) with symptomatic orthostatic hypotension, droxidopa (up to 300 mg twice daily) reduced the fall in systolic blood pressure during orthostatic challenge (by a mean of 22 ± 28 mmHg) and 78% of the patients were considered clinically improved. There were no reports of supine hypertension (Mathias et al., 2001). However, carbidopa blunted the effects of droxidopa and thus the drug may have limited value in patients with PD or MSA who also take carbidopa/levodopa (Kaufmann et al., 2003). Phase III studies are currently under way to determine the safety and efficacy profile of droxidopa.
Bladder problems, particularly urinary retention and incontinence, are relatively common and often troublesome manifestations of MSA (Scientific Committee of the First International Consultation on Incontinence, 2000). Increased urinary frequency due to overactive bladder is less common and often improves with 5 mg of antimuscarinic oxybutynin (Ditropan) three to four times per day and 2 mg of tolterodine (Detrol) three times per day. The latter drug may be better tolerated because it has eight times less affinity for the salivary gland, thus causing much lower frequency of dry mouth. Use of 0.4 mg of the α-blocker tamsulosin (Flomax) twice a day may be effective if the urinary frequency is associated with benign prostatic hypertrophy; this condition must be excluded prior to the use of antimuscarinic agents. Prazosin and moxisylate are specific antagonists of bladder α-adrenergic receptors. In a controlled study in 49 patients, there was improvement of symptoms in 47.6% in the prazosin group and 53.6% in the moxisylate group. Orthostatic hypotension was seen in about 23% in the prazosin group and 11% in moxisylate group. More than 35% of patients had reduction in residual volume, and there was improvement in urinary urgency, frequency, and incontinence. The dosage used was 1 mg of prazosin and 10 mg of moxisylate three times a day in an oral form (Sakakibara et al., 2000a, 2000b). The combination of intravesical prostaglandin E2 and oral bethanechol chloride has been found to be of limited usefulness in treating urinary retention (Hindley et al., 2004). Sildenafil citrate (Viagra) has been found to be safe and effective in the treatment of erectile dysfunction associated with PD, but it may unmask orthostatic hypotension in patients with MSA (Zesiewicz et al., 2000; Hussain et al., 2001; Farooq et al., 2008). Constipation represents the most frequent gastrointestinal manifestations of MSA. Psyllium (Metamucil) was found to increase stool frequency and weight but did not alter colonic transit or anorectal function. Other effective treatments for constipation include polyethylene glycol (Miralax), bisacodyl (Dulcolax), magnesium sulfate, and macrogol 3350 (Movicol). Mosapride citrate, a novel 5-HT4 agonist and partial 5-HT3 antagonist, has been found to ameliorate constipation in parkinsonian patients (Liu et al., 2005). Other recently introduced drugs for constipation include lubiprostone (Amitiza), which locally activates intestinal ClC-2 chloride channels and increases intestinal fluid secretion without altering serum electrolyte levels, and tegaserod maleate (Zelnorm), a novel selective serotonin receptor type-4 (5-HT4) partial agonist that stimulates upper gastrointestinal motility (Sullivan et al., 2006).
Despite the marked involvement of the striatum, many patients with MSA do improve with levodopa, at least initially (Fearnley and Lees, 1990). Although patients with MSA often respond to dopaminergic therapy (levodopa or apomorphine), in contrast to PD, the MSA patients may experience dyskinesias without concomitant improvement in motor functioning (Hughes et al., 1992b). The levodopa-induced dyskinesias that are seen in MSA patients seem to be more dystonic, often involving the face, rather than the choreic or stereotypic movements that are characteristically seen in patients with PD. Furthermore, MSA patients do not seem to notice a recurrence of parkinsonian symptoms until several days after levodopa withdrawal. In one study, only four of eight patients with MSA had a moderate response to levodopa; in contrast, all eight control patients with PD had a consistently good response to levodopa (Parati et al., 1993). Ataxia, present in patients with the OPCA type of MSA, does not respond to pharmacologic therapy and has to be treated by physical means, such as a cane or a walker. In a study of 20 patients with MSA, Iranzo and colleagues (2000) found vocal cord abduction dysfunction in 14 (70%); and in 3 of 3 patients, continuous positive airway pressure completely eliminated laryngeal stridor, obstructive apnea, and hemoglobin desaturation. In another study, continuous positive airway pressure effectively ameliorated nocturnal stridor in 13 patients with MSA (Iranzo et al., 2004). Tracheostomy or other airway restoration techniques must be sometimes performed in patients who have vocal cord abductor paralysis (Isozaki et al., 1996). Stridor has been reported to improve with botulinum toxin injections into the adductor laryngeal muscles (Merlo et al., 2002).
Corticobasal degeneration
Clinical features and natural history
In 1968, Rebeiz and colleagues (1968) reported three patients of Irish descent with parkinsonism, myoclonus, supranuclear palsy, and apraxia who were found at autopsy to have “corticodentatonigral degeneration with neuronal achromasia.” The full spectrum of clinical manifestations of this complex neurodegenerative disorder was not fully recognized until quite recently. Since cerebellar deficit is not a feature of the disorder, the term corticobasal ganglionic degeneration has been used to describe the predominant involvement of the cortex and the basal ganglia; the term corticobasal degeneration is also used, particularly in the European literature (Mahapatra et al., 2004). Since the initial description of CBD was based on clinical-pathologic characteristics, some have suggested the term corticobasal syndrome to draw attention to the marked clinical heterogeneity and to emphasize that CBD pathology may be encountered in PSP, FTD, speech apraxia, primary progressive aphasia, and the posterior cortical atrophy syndrome (Wadia and Lang, 2007). The most striking features of CBD include marked asymmetry of involvement; focal rigidity and dystonia with or without contractures, and hand, limb, gait, and speech apraxia (Video 9.10), although a symmetric form of CBD has been described (Hassan et al., 2010). This series of five pathologically proven patients, however, had many atypical features such as absence of limb dystonia, myoclonus, apraxia, or alien hand and 40% had positive family history, suggesting the possibility of a disorder other than CBD, despite its pathologic overlap, or the concept of CBD as a distinct clinical-pathologic entity needs to be revisited. In addition, some patients manifest coarse rest and action tremor, and cortical-type focal myoclonus (Video 9.8); and parkinsonism, and in some cases cognitive decline may precede these classic features (Bergeron et al., 1998). Other features include cortical sensory deficit, language and speech alterations (Özsancak et al., 2000), frontal lobe symptomatology, depression, apathy, irritability, and agitation (Litvan, et al., 1998b). 
The asymmetric onset differentiates CBD from most other neurodegenerative disorders, and some patients who have been categorized as having “asymmetric cortical degenerative syndrome” (Caselli, 2000) might have CBD. In a study of 14 patients with pathologically confirmed CBD, Wenning and colleagues (1998) found that asymmetric hand clumsiness was the most common presenting symptom, noted at onset in 50% of the patients. At the time of the first neurologic visit, about 3 years after onset, the following signs were present: unilateral limb rigidity (79%), bradykinesia (71%), ideomotor apraxia (64%), postural imbalance (45%), unilateral limb dystonia (43%), and cortical dementia (36%). The mean age at onset was 63 ± 7.7 years; the mean duration of symptoms from onset to death was 7.9 ± 0.7 years (range: 2.5–12.5). Patients with early bradykinesia, frontal syndrome, and two of the following three – tremor, rigidity, and bradykinesia – had a poor prognosis. These clinical features are similar to those described earlier by Rinne and colleagues (1994b), who reviewed 36 patients (20 females and 16 males), with a mean age at onset of 60.9 ± 9.7 years (range: 40–76). In the patients reported by Riley and colleagues (1990), the mean age at onset was 60 years (range: 51–71), and men were more commonly affected than women (3 : 2). Two patients died 7 and 10 years after disease onset. In a series of 147 cases collected from eight centers, the following features were most common: parkinsonism (100%), higher cortical dysfunction (93%), dyspraxia (82%), gait disorder (80%), dystonia (71%), tremor (55%), myoclonus (55%), alien limb (42%), cortical sensory loss (33%), and dementia (25%) (Kompoliti et al., 1998). In one clinical-pathologic study, the presence of varying combinations of early frontal-lobe type behavioral symptoms, nonfluent language disturbance, orobuccal apraxia, and utilization behavior were predictive of CBD rather than Alzheimer pathology (Shelley et al., 2009).
The typical features of CBD can be categorized into movement disorders (akinesia, rigidity, postural instability, limb dystonia, cortical myoclonus, and postural/intention tremor) and cortical signs, such as cortical sensory loss, apraxias (ideational and ideomotor) (Video 9.10), and the alien limb phenomenon (Video 9.11) (Riley et al., 1990; Rinne et al., 1994b; Fitzgerald et al., 2007; Brainin et al., 2008). The alien hand syndrome occurs not only in CBD but also in other disorders including strokes involving the genu or anterior rostrum of the corpus callosum and the contralateral frontomedial cortical and subcortical region (Brainin et al., 2008). Ideomotor apraxia, possibly secondary to involvement of the supplementary motor area and characterized by not knowing “how to do it” (as opposed to not knowing “what to do” in ideational apraxia), is the most typical form of apraxia (Leiguarda et al., 1994; Leiguarda and Marsden, 2000; Zadikoff and Lang, 2005; Wheaton and Hallett, 2007). This apraxia may improve with tactile stimulation, such as the use of the appropriate tool (Graham et al., 1999). Using the De Renzi ideomotor apraxia test, Soliveri and colleagues (2005) compared limb apraxia in patients with CBD (n = 24) and PSP (n = 25). They found that “awkwardness errors,” conceptually appropriate but clumsily executed actions because of fine finger motility, were the most common apraxic errors in patients with CBD, followed by spatial errors, characterized by incorrect orientation or trajectory of the arm, hand, or digits in space or in relation to the body. Sequence errors, incorrect sequences of actions or inappropriate repetition of movements, were least impaired in CBD. The order of impairment was reversed in patients with PSP. Overall, apraxia, particularly ideational or limb-kinetic apraxia, was more frequent and more severe in patients with CBD than in those with PSP, in whom ideomotor apraxia appears more common. Limb contractures, often preceded by the alien hand phenomenon, are more common in this condition than in the other parkinsonism-plus syndromes (Doody and Jankovic, 1992; Leiguarda et al., 1994; Leiguarda and Marsden, 2000). This anterior or motor alien hand syndrome must be differentiated from sensory or posterior syndrome associated with a lesion in the thalamus, splenium of corpus callosum, and temporal-occipital lobe (Hakan et al., 1998). In autopsy-proven cases of CBD, the following were found to be the best predictors of the diagnosis of CBD: limb dystonia, ideomotor apraxia, myoclonus, and asymmetric akinetic-rigid syndrome with late onset of gait or balance disturbance (Litvan et al., 1997a; Wenning et al., 1998). In some cases of CBD, the alien hand phenomenon is associated with spontaneous arm levitation and either tactile avoidance or tactile pursuit or both, each in the opposite limb (Fitzgerald et al., 2007). Arm levitation has been also described in PSP, another feature overlapping these two disorders (Barclay et al., 1999). In our study of 66 patients diagnosed clinically with CBD, 39 (59%) had dystonia (Vanek and Jankovic, 2001). 
Neurologic examination often reveals asymmetrical apraxia, oculomotility disturbance particularly manifested by impaired convergence and vertical and horizontal gaze palsy, bulbar impairment, focal myoclonus, mirror movements, hyperreflexia, Babinski sign, but no ataxia. In contrast to PSP, the vertical saccades are only slightly impaired in CBD and usually involve only upward gaze; furthermore, there is a marked increase in horizontal saccade latency in CBD, which correlates well with an “apraxia score” (Vidailhet et al., 1994). Focal myoclonus, usually involving one arm, present at rest and exacerbated by voluntary movement or in response to sensory stimulation, resembles typical cortical myoclonus but differs in several features. In contrast to the typical reflex cortical myoclonus, which is characterized by long latency (50 ms in the hand), enlarged somatosensory evoked responses (SEP), and cortical discharge preceding the movement, the reflex myoclonus associated with CBD is usually not associated with enlarged SEP and has a shorter latency from stimulus to jerk (40 ms) (Thompson et al., 1994). This suggests that the characteristic short-latency reflex myoclonus in CBD represents enhancement of a direct sensory-cortical pathway, whereas the more typical reflex cortical myoclonus involves abnormal sensorimotor cortical relays (Strafella et al., 1997). Using transcranial magnetic stimulation, Valls-Solé and colleagues (2001) found evidence of enhanced excitability, or reduced inhibition, in the motor area of the hemisphere contralateral to the alien hand sign in patients with CBD. [18F]fluorodeoxyglucose PET scanning in patients with CBD and upper limb apraxia showed marked hypometabolism in the superior parietal lobule and supplementary motor area (Peigneux et al., 2001). In another study of a 70-year-old right-handed man who had alien hand syndrome-related right parietal infarction, functional MRI identified selective activation of contralateral primary motor cortex (M1), presumably released from conscious control by intentional planning systems (Assal et al., 2007).
Although considered by some as “the most common presentation” of CBD (Grimes et al., 1999), in our experience, dementia is a late feature of CBD, and semantic memory is usually well preserved (Graham et al., 2003a, 2003b; Murray et al., 2007). This has been also suggested by a clinical-pathologic study of 15 patients with CBD followed longitudinally. Despite the persistence of apraxia, impaired executive functioning, and worsening language performance, memory remained relatively preserved (Murray et al., 2007). CBD-type syndrome, however, may be the main clinical manifestation of AD (Chand et al., 2006) and some patients with clinical presentation consistent with FTD have been found to have CBD at autopsy (Mathuranath et al., 2000b). The full spectrum of clinical features typically seen in CBD can be also present in patients with documented Pick disease, but the latter disorder is usually dominated by cognitive, behavioral, and language disturbances, such as progressive nonfluent aphasia or primary progressive aphasia (PPA) and semantic dementia (Kertesz et al., 1994; Bond et al., 1997; Litvan et al., 1997b; Hodges, 2001; Rossor, 2001; Graham et al., 2003b; Kertesz and Munoz, 2003; McMonagle et al., 2006; Rohrer et al., 2008). There are three PPA syndromes: (1) progressive nonfluent aphasia (PNFA), characterized by effortful speech with agrammatism and speech apraxia; (2) semantic PPA or semantic dementia (SemD), which involves fluent speech with loss of word and object meaning; and (3) logopenic progressive aphasia (LPA), characterized by word-finding pauses, moderate anomia, and impaired repetition of sentences (Mendez, 2010). In a clinicopathologic study of 18 patients followed in the Lille Memory Clinic over a 15-year period, patients with anarthria had a tauopathy, the agrammatics had a ubiquitin-positive, TDP-43 proteinopathy, the jargon and LPA patients had Alzheimer disease, those with typical SemD had a ubiquitin-positive TDP proteinopathy, and those with atypical SemD had either corticobasal degeneration or argyrophilic grain disease (Deramecourt et al., 2010).
Maurice Ravel, the well-known French composer, was thought to have PPA, which later evolved to right hand apraxia and other features of CBD (Amaducci et al., 2002). Another patient with PPA, a scientist with an interest in visual arts, became fascinated with Ravel’s Bolero and during the evolution of her own PPA, transformed the musical elements of the piece into visual form (’transmodal art”) (Seeley et al., 2008). Despite severe degeneration in the left inferior frontal-insular, temporal and striatal regions, this new creativity was associated with increased gray matter volume and hyperperfusion in her right posterior neocortex on various neuroimaging studies. This form of synesthesia has been described in a variety of disorders, present to a variable degree in up to 4% of normal individuals (Simner, 2007), but also has been found in patients with thalamic lesions (Ro et al., 2007) and new or increased creativity has been reported in patients with temporal lobe lesion, PD, FTD, and other neurodegenerative disorders (Pollak et al., 2007; Griffiths, 2008; Mesulam et al., 2008).
Neuropsychological testing in patients with CBD typically shows deficits in frontal-striatal-parietal cognitive domains, including attention/concentration, executive functions, verbal fluency, praxis, language, and visuospatial functioning (Pillon et al., 1995a). Patients also typically exhibit impaired graphesthesia and may present with visuospatial dysfunction (Tang-Wai et al., 2003). This profile depends on which hemisphere is primarily affected. Usually presenting as word-finding disturbances (anomia), this language disorder then evolves into impairment of the grammatical structure (syntax) and comprehension (semantics) (Mesulam, 2003). Some patients with PPA present as the “foreign accent syndrome” (Luzzi et al., 2008). In one study of 10 patients with PPA who were followed prospectively until they became nonfluent or mute, Kertesz and Munoz (2003) found that at autopsy, all had evidence of FTD: CBD in 4, Pick body dementia in 3, and tau and synuclein negative ubiquinated inclusions of the motor neuron disease in 3. Although no mutations were found in the tau gene in 25 patients with PPA, there was a significant overrepresentation of the tau H1/H1 genotype, also found in PSP and CBD (Mesulam, 2003; Sobrido et al., 2003). Imaging studies have shown that PPA is often associated with atrophy in the left frontotemporal region, and other areas such as the fusiform and precentral gyri and intraparietal sulcus are activated, possibly as a compensatory neuronal strategy (Sonty et al., 2003). These and other studies provide evidence that PPA is related to dissociation for grammatical and working memory aspects of sentence processing within the left frontal cortex (Grossman, 2002). In one study of 55 patients, CBD was divided into “motor onset” (n = 19) and “cognitive onset” (n = 36) and it was found that language was more impaired in the latter, but there was no correlation between side of atrophy or motor impairment and the severity of language dysfunction (McMonagle et al., 2006). Tau-positive pathology was present in 85% of the 19 brains and the pathologic diagnosis of CBD was confirmed in 58%.
PPA is sometimes confused with the syndrome of slowly progressive anarthria that is seen in the late anterior opercular syndrome (see later), but in the latter syndrome, there is no associated language or cognitive deficit. The majority of patients with CBD have been found to have aphasia (e.g., anomic, Broca’s, and transcortical motor aphasia) (Frattali et al., 2000) with phonologic (e.g., spelling) impairment even without clinically observable aphasia (Graham et al., 2003b). Another disorder that progresses rapidly to a nonambulatory state and muteness is motor neuron disease-inclusion body dementia (MND-ID) (Josephs et al., 2003; Kleiner-Fisman et al., 2004). This entity, a subtype of FTD, which is confirmed only pathologically, usually begins in the patient’s mid-forties and shares some features of CBD. Typically, the patients have early dysphagia suggestive of bulbar palsy but without fasciculations. Pathologically, the brains show severe caudate atrophy, with intracytoplasmic inclusions that stain with antibodies against heavy and light subunits of neurofilaments and against ubiquitin. Families with clinical features of Pick disease and the pathologic picture of CBD have been described (Brown et al., 1996). In contrast, apraxia and parkinsonism, if present, are usually late findings in Pick disease, whereas personality changes, aggressive behavior, disinhibition, cognitive deficits, elements of Klüver–Bucy syndrome, and other features of FTD are common (Cherrier and Mendez, 1999; Nasreddine et al., 1999). CBD is probably most frequently confused with PSP, chiefly because of the overlapping oculomotor findings. The CBD patients, however, have much more marked asymmetry in their motor deficits, less severe ophthalmoparesis, and more prominent apraxia and myoclonus (see Table 9.1). The neuropsychological studies show a pattern of deficits that is different from that seen in PSP or AD. When 21 patients with CBD were compared with a group of patients with AD, the CBD patients performed significantly better than the AD patients on tests of immediate and delayed recall of verbal material, whereas the AD patients (with or without extrapyramidal symptoms) performed better on tests of praxis, finger-tapping speed, and motor programming (Massman et al., 1996). The CBD and AD groups all displayed prominent deficits on tests of sustained attention/mental control and verbal fluency and exhibited mild deficits on confrontation naming. The CBD patients endorsed significantly more depressive symptoms on the Geriatric Depression Scale. A similar neuropsychological pattern was demonstrated in another study of 15 patients with CBD (Pillon et al., 1995a). The spectrum of neuropsychological deficits in CBD is broadening (Bergeron et al., 1997).
In addition to parkinsonian, aphasia, neuropsychological, and oculomotor deficits, patients with CBD may also present with progressive spasticity (Hasselblatt et al., 2007).
Neurodiagnostic studies
Computed tomography scans were abnormal in 14 of the 15 patients in one series; 8 had asymmetrical parietal lobe atrophy corresponding to the most affected side, and 6 had bilateral parietal atrophy (Riley et al., 1990). Asymmetric frontoparietal atrophy helps to differentiate CBD from PSP (Soliveri et al., 1999; Sitburana and Ondo, 2009). Another radiographic abnormality that is occasionally encountered in CBD is the “eye of the tiger” sign on brain MRI, characteristically seen in NBIA1 (formerly Hallervorden–Spatz disease) (Molinuevo et al., 1999). One patient with typical CBD clinically was found to have basal ganglia calcification, similar to Fahr disease, on MRI (Manyam et al., 2001; Brodaty et al., 2002; Warren et al., 2002; Oliveira et al., 2004). In a clinical-radiologic study of 8 patients with CBD compared to 36 controls, Yamauchi and colleagues (1998) found atrophy of the corpus callosum, especially the middle portion, which correlated with cognitive impairment and cerebral cortical metabolism measured by 18F-fludeoxyglucose PET. Hyperintensity in the subcortical white matter in the rolandic region on FLAIR images with asymmetric atrophy in the cerebral peduncle, and atrophy in the midbrain tegmentum also have been described as typical MRI findings in patients with CBD (Koyama et al., 2007). Marked asymmetry of the motor pathways (corticospinal and transcallosal fibers) in CBD can be also demonstrated by diffusion tensor tractography using MR diffusion tensor imaging (DTI) (Boelmans et al., 2009). Despite these reported abnormalities, no specific neuroimaging picture of CBD has emerged, and there is no correlation between antemortem MRI and pathologically confirmed CBD (Josephs et al., 2004). PET scans show reduced [18F]fluorodopa uptake in the caudate and putamen and markedly asymmetrical cortical hypometabolism, especially in the superior temporal and inferior parietal lobe (Eidelberg et al., 1991; Sawle et al., 1991; Blin et al., 1992). In one study, [123I]β-CIT SPECT showed marked asymmetry in reduced striatal binding in patients with CBD, similar to PD, but this did not allow reliable differentiation between PD, PSP, MSA, and CBD (Pirker et al., 2000). In two CBD patients with myoclonus, SEP showed a reduced N20 amplitude but without giant SEP (Brunt et al., 1995). Although the other neurophysiologic studies were consistent with cortical reflex myoclonus, the unusual absence of SEP may be explained either by cortical parietal atrophy or by pathologic hyperexcitability of the motor cortex due to a loss of inhibitory input from the sensory cortex (Lu et al., 1998).
Neuropathology, neurochemistry, and pathogenesis
Despite marked asymmetry in clinical findings, autopsy studies of CBD brains show predominantly bilateral atrophy of the precentral gyrus without significant asymmetry of neuropathologic changes (Cordato et al., 2001). Pathologic features in this disease include neuronal degeneration in the precentral and postcentral cortical areas, degeneration of the basal ganglia, including the SN, and the presence of achromatic neural inclusions seen not only in the cortex, but also in the thalamus, subthalamic nucleus, red nucleus, and SN (Gibb et al., 1990; Lippa et al., 1991; Lowe et al., 1992; Kumar et al., 2002). These ballooned neurons, characterized by perikaryal swelling, dispersion of Nissl substance, eccentrically located nucleus, cytoplasm vacuolation, and achromasia, show strong diffuse cytoplasmic immunoreactivity with anti-αB crystallin, a protein that is homologous with the small cell stress proteins. They also show weak, diffuse immunoreactivity with anti-ubiquitin (not present in swollen neurons in infarcted brain). While ballooned neurons are not specific for CBD and have been found in Pick disease, PSP, AD, FTD, and argyrophilic grain disease, as well as Creutzfeldt–Jakob disease, tau-containing distal astrocytic processes producing “astrocytic plaques” have been suggested by Feany and Dickson (1996) to be a distinctive pathologic feature of CBD. These cortical plaques, which are amyloid- and microglia-negative, represent clusters of miliary-like tau-positive structures within the distal processes of the astrocytes. In one review of clinical-pathologic studies, 83% of the patients with the clinical syndrome of CBD (CBS) had evidence of tauopathy in autopsied brains (Wadia and Lang, 2007). Abnormal phosphorylation of tau is not specific for CBD; it can be seen in a variety of neurodegenerative disorders. In the CBD brain, however, tau accumulates as two 64 and 68 kDa polypeptides that are not recognized by antibodies specific to the adult tau sequences encoded by exons 3 and 10 of the tau gene (Bergeron et al., 1998; Kumar et al., 2002). In contrast to the 80 nm periodicity of twisted filaments in AD, the periodicity is about 290 nm in CBD. Tau immunostains usually show granular neuronal deposits, neuropil threads, and glial inclusions. In addition, NFTs and Pick bodies, spherical cortical intraneuronal inclusions, are usually present in the cortical areas but, in contrast to Pick disease, not in the hippocampus. Another characteristic finding of CBD is the presence of corticobasal inclusions, which consist of round, fibrillary or homogeneous basophilic tau-positive inclusions that displace cellular pigment into the periphery, similar to globose neurofibrillary tangles seen in PSP (Kertesz et al., 2009). Neuronal inclusions in CBD are found predominantly in cortical pyramidal and nonpyramidal neurons and may have a distinctive perinuclear, coiled filamentous appearance. In addition to the different distribution, the Pick bodies of Pick disease and the Pick-like bodies of CBD have distinct staining characteristics. While Pick bodies are strongly argentophilic with Bodian and Bielschowsky stains but negative with the Gallyas stain, the Pick-like bodies of CBD stain with Gallyas stain but are not strongly argentophilic. Furthermore, typical Pick bodies usually do not stain with the anti-tau antibody 12E8, which detects phosphorylation at SER 262/356, while the CBD inclusions are recognized by the 12E8 antibody. Astrocytic plaques, different from thorn-shaped astrocytes that are typically seen in PSP, are also typically present in the brains of patients with CBD. In addition, coiled bodies, ubiquitin-negative, tau-immunoreactive inclusions and oligodendroglial inclusions that consist of filaments coiled around a nucleus and extend into the proximal part of the cell process are also typically present in CBD. These inclusions are found not only in CBD but also in PSP and Pick disease; they are particularly numerous in CBD and PSP.
The apparent overlap in clinical and pathologic features between CBD, PSP, and Pick disease needs clarification from further pathologic studies (Kosaka et al., 1991; Woods and McKee, 1992; Lang et al., 1994; Mori et al., 1994; Jendroska et al., 1995; Schneider et al., 1997; Boeve et al., 1999; Cordato et al., 2001; Boeve et al., 2003a; Wadia and Lang, 2007). In one study of 13 patients with clinically diagnosed CBD, pathologic examination found evidence of CBD in 7 patients; AD in 2 patients; and PSP, Pick disease, CJD, and nonspecific neurodegenerative disorder in 1 patient each (Boeve et al., 1999). This indicates marked pathologic heterogeneity and argues for a need to examine the brain before the diagnosis of CBD can be confirmed. Asymmetric parietofrontal cortical degeneration was the most consistent pathologic abnormality in this autopsy series. One of the most important studies addressing the clinical and pathologic overlap between CBD and PSP was based on 35 cases from the Queen Square Brain Bank in which there were 21 clinically and 19 pathologically diagnosed cases of corticobasal syndrome (CBS) or degeneration (CBD) (Ling et al., 2010). Of 19 pathologically confirmed CBD cases, only 5 had been diagnosed correctly in life (sensitivity = 26.3%). All had a unilateral presentation, clumsy useless limb, limb apraxia, and myoclonus, 4 had cortical sensory impairment and focal limb dystonia, and 3 had an alien limb. Eight cases of CBD had been clinically diagnosed as PSP, all of whom had vertical supranuclear palsy, and 7 had falls within the first 2 years. Of 21 cases with CBS, only 5 had CBD (positive predictive value of 23.8%); 6 others had PSP pathology, 5 had AD, and the remaining five had other non-tau pathologies. Forty-two percent of CBD cases presented clinically with a PSP phenotype and 29% of CBS cases had underlying PSP pathology. The authors suggested the CBD–Richardson syndrome for the overlap cases and concluded that CBD “is a discrete clinico-pathological entity but with a broader clinical spectrum than was originally proposed.”
Pathologic features of CBD have been reported in one family with tauopathy in which all three siblings and their grandmother exhibited parkinsonism with levodopa response in one member of the family (Uchihara and Nakayama, 2006). Since frontal lobe dysfunction was also present this family may have had FTDP rather than CBD. The clinical features of CBD, including PPA, have been described in two cases with LRRK2 G2019S mutation (Chen-Plotkin et al., 2008).
A relationship between CBD, Pick disease, and PSP is suggested by the presence of ballooned neurons and nigral basophilic inclusions, which are usually present in all three disorders. Although these disorders are clinically and pathologically similar, there are some distinguishing pathologic features. While “parietal Pick disease” has been reported, the vast majority of pathologically documented Pick cases exhibit degenerative changes predominantly in the frontotemporal distribution with “knife-edge” atrophy of the gyri. In addition, ballooned neurons and Pick bodies, strongly argentophilic and homogeneously ubiquitinated intraneuronal inclusions, are typically present in Pick disease. However, neither of the two histologic hallmarks is absolutely required for the neuropathologic diagnosis of Pick disease (Growdon and Primavera, 2000). Kertesz and colleagues (1994) proposed the concept of “Pick complex” for certain focal cortical degenerations such as PPA (with or without amyotrophic lateral sclerosis, or ALS), frontal lobe dementia, and CBD. He and his colleagues (Kertesz et al., 2000b) later suggested that there is a clinical and pathologic overlap between CBD, FTD, and PPA (Williams and Lees, 2009) (Fig. 9.7).
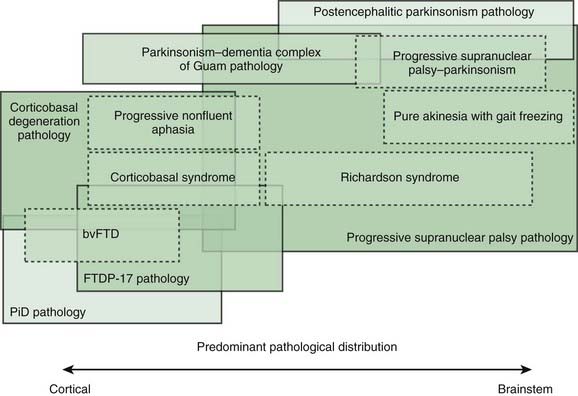
Dopamine concentration was reduced in the CBD brains throughout the striatum and SN when compared with age-matched controls (Riley et al., 1990).
Treatment
To date, no effective treatment has been found, although myoclonus may improve with clonazepam, and painful rigidity and dystonia may improve with botulinum toxin injections (Jankovic and Brin, 1991). Dopaminergic drugs are rarely, if ever, effective. Although levodopa rarely provides any improvement in patients with CBD (Kompoliti et al., 1998), levodopa-induced dyskinesia has been reported in rare autopsy-proven cases (Frucht et al., 2000). Rehabilitation has been reported to improve ideomotor apraxia following stroke, but it is not known whether similar strategies improve apraxia associated with CBD (Hanna-Pladdy et al., 2003).