Chapter 21 Ataxia
Pathophysiology and clinical syndromes
Ataxia is the type of clumsiness produced by dysfunction of the cerebellum or cerebellar pathways. The pathophysiology of the signs and symptoms has been detailed in the earlier chapter on motor control (Chapter 2). The core symptoms are difficulty with balance and gait, clumsiness of the hands, and dysarthria. The differential diagnosis is very long and includes all types of neurologic pathologic processes. While most patients presenting with ataxia will have a sporadic disorder, recently there has been increased attention on the genetic ataxias because of rapid advances in research.
Sporadic ataxia
Table 21.1 lists the principal categories. A series of 112 patients with sporadic ataxia with the following criteria were studied: (1) progressive ataxia; (2) onset after 20 years; (3) informative and negative family history (no similar disorders in first- and second-degree relatives; parents older than 50 years); and (4) no established symptomatic cause (Abele et al., 2002). Thirty-two patients (29%) met the clinical criteria of possible (7%) or probable (22%) multiple system atrophy (MSA). With genetic testing, Friedreich ataxia was found in five patients (4%), the spinocerebellar ataxia (SCA) 2 mutation in one (1%), the SCA3 mutation in two (2%) and the SCA6 mutation in seven (6%). The disease remained unexplained in 65 patients (58%). Antigliadin antibodies were present in 14 patients, 10 patients with unexplained ataxia (15%) and 4 patients with an established diagnosis (9%); this interesting aspect will be discussed further below.
Degenerative ataxia
MSA is likely the most common disorder, certainly in adults (Bhidayasiri and Ling, 2008; Gilman et al., 2008; Wenning et al., 2008). In addition to ataxia, patients have parkinsonism and autonomic dysfunction (including impotence). The disorder has also been called olivopontocerebellar atrophy when the emphasis is on ataxia, striatonigral degeneration when the emphasis is on bradykinesia and rigidity, and Shy–Drager syndrome when the emphasis is on autonomic dysfunction. Early falls are a prominent feature. Variable clinical features include pyramidal signs, tremor, dysarthria, dystonia, and mild dementia. There is typically a poor response to levodopa, but clearly at times there is some response, and this can be confusing. Responses are never dramatic and typically unsustained. The pathologic hallmark of the disorder, in addition to neuronal cell loss, is the glial cytoplasmic inclusion (GCI) (Yoshida, 2007). MSA is described in detail in Chapter 9.
Another degenerative cause is progressive myoclonic epilepsy since ataxia is typically a part of this syndrome. Often myoclonus and ataxia become difficult to separate. These syndromes are described in Chapter 20.
Strokes
Hemisensory loss and hemiataxia is typically due to a thalamic lesion.
Isolated gait ataxia can be seen with a lesion of the pontomedullary junction.
Toxic/metabolic
The childhood hyperammonemias are a cause of intermittent ataxia.
Celiac disease or sprue is an interesting cause of ataxia and possibly myoclonus as well (Hadjivassiliou et al., 1998). Celiac disease itself is a gluten-sensitive enteropathy with malabsorption. The gastrointestinal disorder can be reversed with a gluten-free diet, but the cerebellar degeneration does not necessarily get better. Curiously, it is now clear that up to 40% of patients with sporadic ataxia have antigliadin antibodies, but no sign of celiac disease (Pellecchia et al., 1999; Burk et al., 2001; Bushara et al., 2001; Hadjivassiliou et al., 2003, 2008c; Lin et al., 2010). This has been disputed, but the power of that study may have been too low (Abele et al., 2003). Antigliadin antibodies have also been seen in a similar percentage of patients with genetic ataxias (Bushara et al., 2001). This has also been disputed (Hadjivassiliou et al., 2003), but the percent abnormal may depend on the numbers of the specific SCA types. It is not clear what this means. In some of these patients, there are abnormalities of the white matter and prominent headache; at least these patients have some symptomatic response to a gluten-free diet (Hadjivassiliou et al., 2001). Antibodies to gangliosides were found in 64% of patients with mixed ataxias, suggesting that the increase in antigliadin antibodies may not be specific (Shill et al., 2003). In gluten-associated ataxia, there can also be antibodies directed to tissue transglutaminase, either type 2 (Hadjivassiliou et al., 2006) or, likely more specifically, type 6 (Hadjivassiliou et al., 2008a). Antigliadin antibodies in patients with ataxia bind to the neural antigen synapsin I (Alaedini et al., 2007). There may well be an increase in autoimmunity in sporadic cerebellar ataxia (Hadjivassiliou et al., 2008b).
There is open-label evidence that a gluten-free diet may benefit patients with antigliadin antibodies (Hadjivassiliou et al., 2008c). Intravenous immunoglobulin (IVIg) has improved the ataxia in three patients with overt celiac disease and ataxia (Souayah et al., 2008). IVIg therapy has appeared to help two patients, as well as two other patients with anti-GAD antibodies (see below for this entity), suggesting a role for immunotherapy (Nanri et al., 2009).
Paraneoplastic
The paraneoplastic causes are very important to keep in mind (Anderson et al., 1988; Bolla and Palmer, 1997). The clinical syndrome is often a rapidly progressive one over a relatively short period of time and then a plateau without further change. What appears to be happening is a rapid destruction of Purkinje cells. Even if the cancer is found and successfully treated, the disorder may not improve because the cells are irreversibly damaged. Nevertheless, the best treatment is certainly treatment of the cancer.
There are three types of anti-Purkinje cell antibodies. Anti-Yo (PCA-1) is seen with tumors of breast, ovary, and adnexa. Atypical anticytoplasmic antibody (anti-Tr or PCA-Tr) is seen with Hodgkin disease, and tumors of the lung and colon. PCA-2 has been identified mostly with lung tumors; 3 of 10 patients had ataxia (Vernino and Lennon, 2000).
There are three antineuronal antibodies. Anti-Hu (ANNA-1) can be seen in possible conjunction with encephalomyelitis (Lucchinetti et al., 1998). It is associated with small-cell lung tumor, tumors of breast and prostate, and neuroblastoma. Anti-Ri (ANNA-2) is found with tumors of breast and ovary. Atypical anti-Hu is seen with tumors of the lung and colon, adenocarcinoma, and lymphoma.
Anti-CV2 (CRMP) antibody is associated with a syndrome of ataxia and optic neuritis (de la Sayette et al., 1998). It has been seen with small-cell lung carcinoma. The CV2 antigen is expressed by oligodendrocytes. Interestingly, this is one syndrome where improvement has been seen with removal of the tumor.
Antibodies directed to a serum protein, Ma1, have been found in patients with testicular and other tumors (Dalmau et al., 1999; Gultekin et al., 2000). The antibodies are anti-Ma and anti-Ta (Ta is Ma2). These patients may also have limbic encephalitis. Ma1 is a phosphoprotein highly limited to brain and testis.
Antibodies directed to amphiphysin are rarely associated with a cerebellar syndrome (Saiz et al., 1999). This is a marker for small-cell lung carcinoma. Antibodies to Zic4 are associated with cerebellar degeneration and small-cell lung cancer (Sabater et al., 2008).
Antibodies against a glutamate receptor can be seen with cancer, and this causes a pure cerebellar syndrome (Sillevis Smitt et al., 2000). Patients with Hodgkin disease can develop paraneoplastic cerebellar ataxia because of the generation of autoantibodies against mGluR1, and this is mediated both by functional and degenerative effects (Coesmans et al., 2003).
Other new antibodies are being identified (Jarius et al., 2010). Tests that are currently commercially available include Hu, Ma, Ta, Yo, Ri, amphiphysin, Zic4 and CV2.
In a series of 50 patients with paraneoplastic cerebellar degeneration out of 137 with any neurologic syndrome, 19 had anti-Yo, 16 anti-Hu, 7 anti-Tr, 6 anti-Ri, and 2 anti-mGluR1 (Shams’ili et al., 2003). While 100% of patients with anti-Yo, anti-Tr, and anti-mGluR1 antibodies had ataxia, 86% of anti-Ri and only 18% of anti-Hu patients had paraneoplastic cerebellar degeneration. In 42 patients (84%), a tumor was detected; the most common were gynecological and breast cancer (anti-Yo and anti-Ri), lung cancer (anti-Hu), and Hodgkin lymphoma (anti-Tr and anti-mGluR1). All patients received antitumor therapy and 7 had some neurologic improvement. The functional outcome was best in the anti-Ri patients, with 3 out of 6 improving neurologically; 5 were able to walk at the time of last follow-up or death. Survival was worse with anti-Yo and anti-Hu compared with anti-Tr and anti-Ri.
Autoimmune
Ataxia can be associated with anti-GAD antibodies (Saiz et al., 1997; Abele et al., 1999; Honnorat et al., 2001). There can be a pure ataxia syndrome and one with an associated peripheral neuropathy. In one series of 14 patients, 13 were women and 11 had late-onset diabetes (Honnorat et al., 2001). Anti-GAD antibodies are better known for association with stiff-person syndrome, but the relationship is not clear. In one case with antibodies in the cerebrospinal fluid, the antibody blocked GABAergic transmission in the rat cerebellum (Mitoma et al., 2000). As with stiff-person syndrome, patients can exhibit other forms of autoimmunity. IVIg therapy may be useful (Nanri et al., 2009).
Demyelinating
Ataxia is common in multiple sclerosis. A patient with leukoencephalopathy with neuroaxonal spheroids (LENAS), a rare disease of cerebral and cerebellar white matter, had a 14-year course of progressive neurologic decline consistent with a clinical diagnosis of probable MSA, with prominent cerebellar dysfunction and dysautonomia (Moro-De-Casillas et al., 2004).
Genetic ataxia
One of the most active areas in movement disorders and genetics is in determining the genes for numerous types of hereditary ataxias. Additionally, we are beginning to understand some of the mechanisms of neurodegeneration. On the other hand, specific therapies are still in the future. Many of the genes can be tested commercially. This is helpful, but it is important to remember that genetic testing can have significant consequences, both emotionally and socially – and not only for the individual tested, but also for the family. Hence, testing should be done with care and clear informed consent (Tan and Ashizawa, 2001).
Moseley et al. (1998) determined the incidence of spinocerebellar ataxia (SCA) types 1, 2, 3, 6, and 7 and Friedreich ataxia (FA) among a large panel of ataxia families in the United States. They collected DNA samples and clinical data from patients representing 361 families with adult-onset ataxia of unknown etiology. Patients with a clinical diagnosis of FA were specifically excluded. Among the 178 dominant kindreds, they found SCA1 expansion at a frequency of 5.6%, SCA2 expansion at a frequency of 15.2%, SCA3 expansion at a frequency of 20.8%, SCA6 expansion at a frequency of 15.2%, and SCA7 expansion at a frequency of 4.5%. Among patients with apparently recessive or negative family histories of ataxia, 6.8% and 4.4% tested positive for a CAG expansion at one of the dominant loci, and 11.4% and 5.2% of patients with apparently recessive or sporadic forms of ataxia had FA expansions. Among the FA patients, the repeat sizes for one or both FA alleles were relatively small, with sizes for the smaller allele ranging from 90 to 600 GAA repeats. The clinical presentation for these patients was atypical for FA including adult onset of disease, retained tendon reflexes, normal plantar response, and intact or partially intact sensation. The incidence of the SCAs has also been explored in other countries, such as Australia (Storey et al., 2000), Taiwan (Soong et al., 2001), and Thailand (Sura et al., 2009). The pattern does differ somewhat in different countries (Schols et al., 2004). The epidemiologic patterns are updated in the supplementary material in Wardle et al. (2009).
Looking specifically at patients with onset at age 18 or later (“late onset”), a study in the southeast Wales population showed the most frequent defined diagnoses to be SCA6, dentatorubral-pallidoluysian atrophy (DRPLA), and SCA8 (Wardle et al., 2009).
A very detailed compendium of the genetic ataxic disorders can be found at: http://www.neuromuscular.wustl.edu/ataxia/aindex.html.
Here, the principal disorders will be reviewed. There are several good reviews (Di Donato, 1998; Subramony et al., 1999; Evidente et al., 2000; Klockgether, 2000; Stevanin et al., 2000; Devos et al., 2001; Di Donato et al., 2001; Schols et al., 2004; Klockgether, 2008; Manto and Marmolino, 2009; Durr, 2010).
Dominant ataxia
Table 21.3 gives the identified SCAs and their known genetic disorder. Many of them are due to expanded trinucleotide repeats. SCA3 is identical to Machado–Joseph disease and is often referred to as SCA3/MJD. DRPLA is often included in such lists. It shares the mutation disorder of CAG expansion and may have prominent ataxia as part of its manifestation. Some details about the proteins are known and these are listed in Table 21.4.
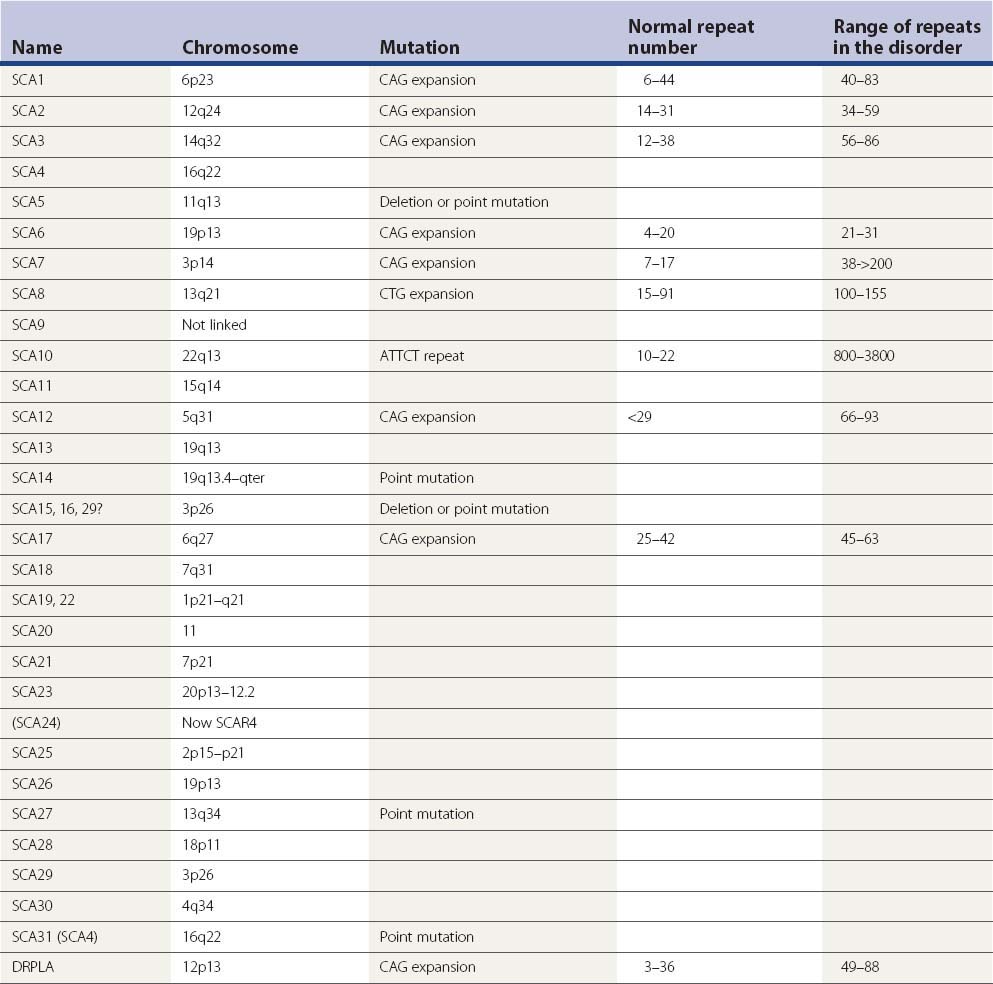
| Name | Protein |
|---|---|
| SCA1 | Ataxin-1 |
| SCA2 | Ataxin-2 |
| SCA3 (Machado–Joseph disease) | Ataxin-3 |
| SCA4 (with sensory axonal neuropathy) | |
| SCA5 (Lincoln ataxia) | Beta III spectrin (SPTBN2) |
| SCA6 | α1a component of the voltage-dependent calcium channel: CACNL1A4 |
| SCA7 | Ataxin-7 |
| SCA8 | (mutation is in noncoding region) |
| SCA9 | |
| SCA10 | Ataxin-10 |
| SCA11 | Tau tubulin kinase 2 (TTBK2) |
| SCA12 | Protein phosphatase 2A, regulatory subunit B (PPP2R2B) |
| SCA13 | KCNC3 |
| SCA14 | Protein kinase Cγ (PRKCG) |
| SCA15, 16, 29? | Inositol 1,4,5-triphosphate receptor, type 1 (ITPR1) |
| SCA17 (also called HDL4) | TATA binding protein (TBP) |
| SCA18 (with sensorimotor neuropathy) | |
| SCA19 | |
| SCA20 | |
| SCA21 | |
| SCA22 | |
| SCA23 | |
| (SCA24) Now SCAR4 | |
| SCA25 (with sensory neuropathy) | |
| SCA26 | |
| SCA27 | Fibroblast growth factor 14 (FGF14) |
| SCA28 | ATPase family gene 3-like 2 (AFG3L2) |
| SCA29 (congenital, non-progressive) | |
| SCA30 | |
| SCA31 (Japanese form of SCA4) | Pleckstrin homology domain-containing protein, family G, member 4 (PLEKHG4, puratrophin-1) |
| DRPLA | Atrophin-1 or DRPLA protein |
Generally, the cerebellar syndrome is similar in the different disorders. Patients experience gradual onset of balance and gait difficulty, dysarthria and clumsiness of the hands. There may be visual symptoms such as blurry vision or diplopia. Age of onset is highly variable. Sometimes clinical features can help differentiate the different disorders. SCA12, for example, may be somewhat unique in that it may present with an action tremor that may look like essential tremor (O’Hearn et al., 2001). Other manifestations may help predict the genotype, but virtually any constellation of signs and symptoms can occur with any phenotype. Some guidelines are noted below.
Lining up the ADCAs and the SCAs gives a start (Table 21.5). Subramony has suggested some phenotypic clues (Subramony et al., 1999), and these are updated in Table 21.6.
Table 21.5 Relationship between ADCAs and SCAs
| ADCA type | SCA type |
|---|---|
| I (ataxia plus) | 1, 2, 3, 4, 8, 9, 12, 17, 27, 28, DRPLA |
| II (with pigmentary maculopathy) | 7 |
| III (pure ataxia) | 5, 6, 11, 14, 15, 16, 22, 26, 30, 31 |
| Ataxia and epilepsy | 10 |
| Early onset with learning disability | 13 |
| Age at onset | Young adult: SCA1, 2, 3, 21 |
| Older adult: SCA6 | |
| Childhood onset: SCA2, 7, 13, 25, 27, DRPLA | |
| Prominent anticipation | SCA7, DRPLA |
| Upper motor neuron signs | SCA1, 3, 7,12 |
| Some in SCA6, 8 | |
| Rare in SCA2 | |
| Slow saccades | Early, prominent: SCA2, 7 |
| Late: SCA1, 3, 28 | |
| Rare: SCA6 | |
| Extrapyramidal signs | Early chorea: DRPLA |
| Akinetic-rigid, Parkinson: SCA2, 3, 12, 21 | |
| Generalized areflexia | SCA2, 4, 19, 21, 22 |
| Late: SCA3 | |
| Rare: SCA1 | |
| Visual loss | SCA7 |
| Dementia | Prominent: SCA17, DRPLA |
| Early: SCA2, 7 | |
| Otherwise: rare | |
| Myoclonus | DRPLA, SCA2, 14, 19 |
| Tremor | SCA2, 8, 12, 15, 16, 19, 27 |
| Seizures | SCA10 |
The pathophysiology of the triplet repeat ataxias has been extensively studied (Koeppen, 2005; Paulson, 2007; Soong and Paulson, 2007; Zoghbi and Orr, 2009). It appears that the mutated protein is toxic to the cell. For example, in a mouse model of SCA1, the gene was made conditional; when the gene was turned off, the animals improved (Zu et al., 2004). Similarly, treatment of a mouse model of SCA1 with RNA interference (RNAi) can improve the disorder. Recombinant adeno-associated virus vectors expressing short hairpin RNAs were injected into the cerebellum with marked benefit (Xia et al., 2004).
There is no treatment for the SCAs. Lithium improved neurologic function and hippocampal dendritic arborization in a mouse model of SCA1 (Watase et al., 2007). Mode of action of lithium in this circumstance is not clear. Human studies have been initiated.
Then there are the autosomal dominant episodic ataxias (Table 21.7) (Evidente et al., 2000; Kullmann et al., 2001; Jen, 2008; Tomlinson et al., 2009).
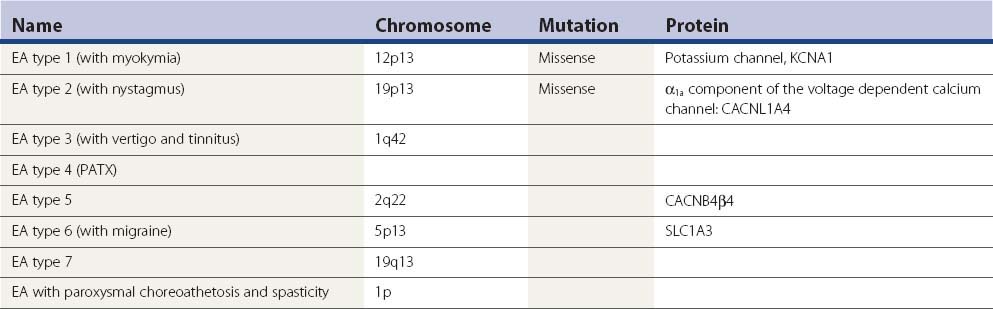
EA-2 has intermittent attacks of ataxia, dysarthria, nausea, vertigo, diplopia, and oscillopsia lasting minutes to days. There may be interictal nystagmus or mild ataxia. Episodes are provoked by stress and exercise, but not startle. About half of the patients also have migraine. The attacks may respond to acetazolamide. The potassium channel blocker 4-aminopyridine can also be effective (Strupp et al., 2004). The gene for EA-2 is the same as the gene for SCA6, but the nature of the mutation differs. A different missense or truncation mutation in this gene causes familial hemiplegic migraine. There can be phenotypic overlaps between all three conditions. Two patients have been described that progressed to a late life dystonia (Spacey et al., 2005). Such patients may also have weakness; a disorder at the neuromuscular junction has been demonstrated with single fiber EMG (Jen et al., 2001; Maselli et al., 2003).
EA-3, called periodic vestibulocerebellar ataxia, is an autosomal dominant disorder characterized by defective smooth pursuit, gaze-evoked nystagmus, ataxia, and vertigo (Damji et al., 1996).
EA-4 is characterized by vestibular ataxia, vertigo, tinnitus, and interictal myokymia; attacks are diminished by acetazolamide (Steckley et al., 2001).
Episodic ataxia with paroxysmal choreoathetosis and spasticity has age of onset of 2–15 years (Auburger et al., 1996). There are attacks of ataxia, with involuntary movements and dystonia of extremities, paresthesias, and headache. Episodes last about 20 minutes and occur twice per day to twice per year. Precipitating factors include alcohol, fatigue, emotional stress, and physical exercise. In some there is spastic paraplegia which may persist between attacks. Treatment is with acetazolamide.
EA-5 has been identified in one family and is due to a mutation in a calcium channel gene (Escayg et al., 2000). In other families with a mutation in this gene, there is epilepsy.
Recessive ataxia
There are a large number of recessive ataxias. The most common of these are discussed here, but even many of these are rare. The ones discussed in this chapter are summarized in Table 21.8. Others can be found in the genetics table in Chapter 1 (Table 1.5). The full list of designated autosomal recessive ataxias (SCAR) and ataxias with spasticity (SPAX) can be found there. The terminology is rather complex and some disorders have multiple names from appearing in multiple lists. Another abbreviation for autosomal recessive ataxia is ARCA, and below it will be noted that ARCA1 is SCAR8 and ARCA2 is SCAR9.
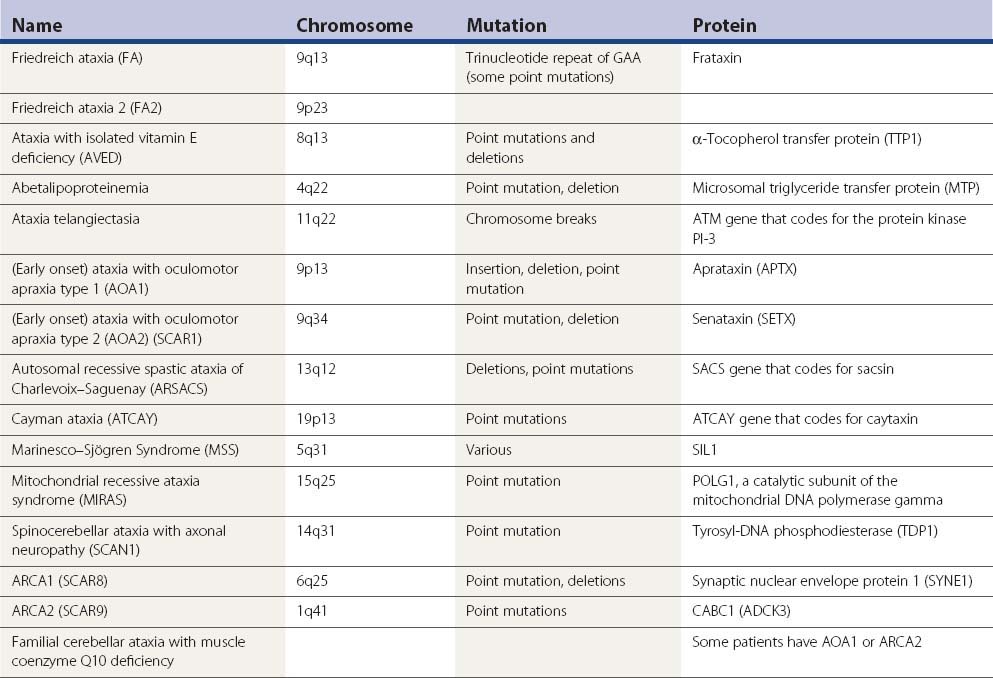
Friedreich ataxia (FA)
The major autosomal recessive ataxia to consider is FA, the most common cause of hereditary ataxia with a prevalence of about 1 in 50 000 persons (Bradley et al., 2000; Evidente et al., 2000; Pandolfo, 2008; Schulz et al., 2009a). Age of onset is generally before 20 years. The clinical features, in addition to ataxia, are dysarthria, sensory loss, and corticospinal tract signs with absent reflexes. There is an axon loss peripheral neuropathy. There may be skeletal abnormalities such as kyphoscoliosis, cardiomyopathy, and diabetes. The cardiomyopathy is hypertrophic with possible associated muscular subaortic stenosis or hypokinetic-dilated left ventricle. Abnormal ECGs are common.
The genetic abnormality is usually an expanded trinucleotide repeat of GAA in a gene on chromosome 9q coding for the protein called frataxin, and a commercial genetic test is available. Normal repeat length is 6–28, and patients have 66–1700. Six percent of the time there is a point mutation. Frataxin is a mitochondrial protein encoded by nuclear DNA. It is believed that frataxin is involved in iron transport, and in FA there is excessive iron accumulation in mitochondria. Using functional MRI, increased iron has been detected in patients in the dentate nucleus (Waldvogel et al., 1999). The excess iron may lead to toxic free radical damage. There are significant reductions in the activities of complex I, complex II/III, and aconitase in FA heart muscle (Bradley et al., 2000).
For treatment, there has been considerable enthusiasm for idebenone, a molecule structurally related to coenzyme Q10. Early studies showed that it could reduce heart size and improve cardiac function, but without clear effect on the ataxia (Rustin et al., 1999; Lerman-Sagie et al., 2001; Hausse et al., 2002; Rustin et al., 2002; Buyse et al., 2003, Mariotti et al., 2003). There are also negative studies on cardiac function (Schols et al., 2001). Some studies, in children and using higher doses, have begun to show beneficial effects on the ataxia. In an open-label trial of 9 patients, age range 11–19 years, treated with idebenone at 5 mg/kg/day, there was a significant reduction in ataxia scores after 3 months of treatment (Artuch et al., 2002). In a subsequent long-term study with 5–20 mg/kg/day by this same group over a 3–5-year period, 10 children in the age range of 8–18 years showed stable neurologic function (Pineda et al., 2008). Adults of age 18–46 over the same period showed neurologic deterioration.
In another study, 48 FA patients, aged 9–17 years, were enrolled in a 6-month, randomized, double-blind, placebo-controlled study (Di Prospero et al., 2007). The patients received placebo or one of three doses of idebenone (approximately 5 mg/kg, 15 mg/kg, and 45 mg/kg). Whereas an overall analysis did not show a significant neurologic improvement, there were indications of a dose-dependent response. A secondary analysis, excluding patients who required wheelchair assistance, showed a significant improvement in the ICARS (International Cooperative Ataxia Rating Scale). The authors suggested that higher doses may be necessary to have a beneficial effect on neurologic function. Further trials are ongoing (Schulz et al., 2009b), but it is not clear that the drug will really have an impact on quality of life (Brandsema et al., 2010).
There are also some attempts at treatment using coenzyme Q10. Cardiac muscle 31P-MRS has been demonstrated to improve with combination therapy with coenzyme Q10 (CoQ10) and vitamin E (Lodi et al., 2001). Some of these patients were followed for 4 years, and a few of them had suggestive benefit with less progression than predicted (Hart et al., 2005).
There are other approaches being considered in treatment of FA also (Mancuso et al., 2010). Because of the mitochondrial iron accumulation, iron chelation might be useful. A 6-month trial of deferiprone was conducted in nine adolescents (Boddaert et al., 2007). By MRI, there was reduction of iron in the dentate nucleus, and the youngest patients may have had some neurologic improvement. Histone deacetylase (HDAC) inhibitors may increase frataxin expression and this has been tested in a mouse model (Rai et al., 2008).
Now that the gene can be identified, the phenotype has been widened in recent years. (1) Late-onset FA with age of onset more than 25 characterized by a more benign course; (2) FA with retained reflexes (FARR); and (3) the Acadian or Louisiana form with slower progression (Evidente et al., 2000). FA can even present with peripheral neuropathy alone, without ataxia (Panas et al., 2002).
A second gene was identified whose mutation can lead to classic FA. This has been localized to chromosome 9p (Christodoulou et al., 2001).
Ataxia with isolated vitamin E deficiency (AVED)
This is a rare, but important, disorder that has a phenotype similar to FA (Di Donato et al., 2010). Additionally, there can be ophthalmoplegia and retinitis pigmentosa. This disorder is due to a defect in the TTP1 gene coding for α-tocopherol transfer protein. This protein incorporates α-tocopherol into lipoproteins secreted by the liver. As in the conditions when vitamin E is deficient because of malabsorption, this disorder can be treated with vitamin E (Gabsi et al., 2001). It should not be missed!
Abetalipoproteinemia
Abetalipoproteinemia is a rare autosomal recessive deficiency of apoB-containing lipoproteins caused by a microsomal triglyceride transfer protein (MTP) deficiency. Coming on in teenage years or early adulthood, the syndrome is due to vitamin E deficiency. The main characteristics are ataxia and polyneuropathy, as well as acanthocytosis, celiac syndrome, and retinal degeneration (Triantafillidis et al., 1998). It should be treated with high doses of vitamin E.
Ataxia telangectasia (AT)
This disorder is due to a mutation in the ATM gene that codes for a protein kinase that plays an important role in cell cycle control, apoptosis, and DNA double-strand break repair. This abnormality leads to increased incidence of malignancy as well as neurodegeneration. The mutations are scattered over the gene. The disorder begins early in childhood, often age 1–2 years (Woods and Taylor, 1992; Di Donato et al., 2001). There is ataxia, truncal more than appendicular, and dysarthria. Oculomotor apraxia – difficulty in initiating saccades – occurs early. Many other features are associated such as dystonia, masked facies, choreoathetosis, myoclonus, tremor, long tract signs and, eventually, peripheral neuropathy and cognitive decline. There are the well-known oculocutaneous telangectasias as well as immunodeficiencies with reduced immunoglobulins and T-cell deficiencies, recurrent sinopulmonary infections, and malignancies, particularly leukemia or lymphoma. Alpha-fetoprotein levels are elevated and chromosome breaks can be identified.
Early-onset ataxia with ocular motor apraxia and hypoalbuminemia (EAOH or AOA1 and AOA2)
Two disorders recognized in Japan, early-onset ataxia with hypoalbuminemia and ataxia with ocular motor apraxia are now considered the same clinical entity because of the identification of a common mutation in the aprataxin gene (APTX). In six patients from four families, cerebellar ataxia and peripheral neuropathy were noted in all, ocular motor apraxia was observed in five patients, and choreiform movements of the limbs and mental deterioration were observed in five patients (Shimazaki et al., 2002). All patients had hypoalbuminemia and hypercholesterolemia; brain MRI or CT showed marked cerebellar atrophy. Nerve biopsy revealed depletion of large myelinated fibers in three of the five patients examined. Many other patients have been described (D’Arrigo et al., 2008). This disorder is also common in Portugal, but rare in Germany (Habeck et al., 2004). Genetic testing is available. Ataxia with oculomotor apraxia type 2 (AOA2) has also been identified with a similar clinical picture but with mutations in the senataxin (SETX) gene (Criscuolo et al., 2006). In a series of 19 patients, other features noted were a frequent polyneuropathy and elevation of the serum alpha-fetoprotein (Tazir et al., 2009). The SETX gene is also mutated in ALS4, a motor neuron disorder with early onset, and some patients may share this phenotype (Schols et al., 2008).
Autosomal recessive spastic ataxia of Charlevoix–Saguenay (ARSACS)
This disorder of ataxia and pyramidal signs with high prevalence in northeastern Quebec is due to a mutation in the gene, SACS, that codes for a protein sacsin (Engert et al., 2000). Even in Quebec, the clinical syndrome is genetically heterogeneous (Thiffault et al., 2006).
Although this entity has been thought to be geographically distinct, a family in Tunisia was identified with the disorder (Mrissa et al., 2000), and more cases with variable phenotype are also being recognized (Takiyama, 2007). Commercial genetic testing is available for this disorder.
Cayman ataxia
Cayman ataxia is a recessive congenital ataxia restricted to one area of Grand Cayman Island that appears to be due to one of two mutations (Bomar et al., 2003). The gene ATCAY or Atcay encodes a neuron-restricted protein called caytaxin. Caytaxin contains a CRAL-TRIO motif common to proteins that bind small lipophilic molecules. Mutations in caytaxin are also responsible for the jittery mouse. Mutations in another protein containing a CRAL-TRIO domain, alpha-tocopherol transfer protein (TTPA), cause a vitamin E-responsive ataxia.
Marinesco–Sjögren syndrome (MSS)
This syndrome is characterized by ataxia, cataracts from infancy, learning disability, myopathy, and short stature. Mutations in the gene for SIL1, a protein in the endoplasmic reticulum, cause the disorder (Senderek et al., 2005). SIL1 appears to function as a nucleotide exchange factor for the heat-shock protein 70 (HSP70) (Anttonen et al., 2005).
Mitochondrial recessive ataxia syndrome (MIRAS)
Most commonly mutations in POLG1, a catalytic subunit of the mitochondrial DNA polymerase gamma, produce progressive weakness of the eye muscles, dysphagia, and some somatic weakness (Cagnoli et al., 2008). Particularly in Finland, mutations can also cause ataxia, perhaps with sensory axonal peripheral neuropathy (Hakonen et al., 2005). An alternate phenotype is SANDO (sensory ataxia, neuropathy, dysarthria, and ophthalmoparesis). A wide spectrum of neurologic disease can be produced by mutations in POLG1 (Milone and Massie, 2010; Tzoulis et al., 2010).
Spinocerebellar ataxia with axonal neuropathy (SCAN1)
This disorder appears due to a mutation in tyrosyl-DNA phosphodiesterase (TDP1), a protein in the DNA repair pathway (Takashima et al., 2002; Hirano et al., 2007).
Autosomal recessive cerebellar ataxia type 1 (ARCA1)
This syndrome is characterized by middle-age onset, slow progression and moderate disability, significant dysarthria, mild oculomotor abnormalities, occasional brisk reflexes in the lower extremities, normal nerve conduction studies, and diffuse cerebellar atrophy on imaging (Dupre et al., 2007). SYNE1 mutations are causative (Gros-Louis et al., 2007).
Autosomal recessive cerebellar ataxia type 2 (ARCA2)
These patients present with progressive ataxia characterized by cerebellar atrophy and seizures, and studies suggestive of coenzyme Q10 deficiency including mildly elevated lactate levels (Lagier-Tourenne et al., 2008; Mollet et al., 2008). Mutations in the same gene were found by two groups, one that called it the CABC1 gene (Mollet et al., 2008) and the other referring to it as the ADCK3 gene (Lagier-Tourenne et al., 2008).
Familial cerebellar ataxia with muscle coenzyme Q10 deficiency
Six patients with muscle CoQ10 deficiency (26–35% of normal) presented with cerebellar ataxia, pyramidal signs, and seizures (Musumeci et al., 2001). All six patients responded to CoQ10 supplementation; strength increased, ataxia improved, and seizures became less frequent. In a study of muscle biopsies in 135 patients with undefined cerebellar ataxia, 13 were found to have deficient CoQ10 (Lamperti et al., 2003). The mutation in one family was in the aprataxin gene (Quinzii et al., 2005). This diagnosis is a potentially important cause of familial ataxia because it is at least partially treatable (Artuch et al., 2006). The disorder appears to be recessive, the gene defect is not identified in all cases (Montero et al., 2007), but some of these patients certainly have ARCA2.
Frequency of the autosomal recessive ataxias
In a review of 102 patients with autosomal recessive (or sporadic) ataxia, a diagnosis was made in 57 of them; 36 were affected with FA, seven with AOA2, four with AT, three with AOA1, three with MSS, two with ARSACS, one with AVED, and one with ARCA2 (Anheim et al., 2010). Genetic testing is currently available for FA, AVED, MSS, AOA1, AOA2 and MIRAS.
X-linked ataxia
Fragile X-associated tremor/ataxia syndrome (FXTAS)
Attention has been drawn to the fact that the fragile X premutation has been associated with tremor with appearance similar to essential tremor (Leehey et al., 2003; Amiri et al., 2008). Ataxia and executive dysfunction are also prominent in this disorder (Hagerman and Hagerman, 2004). Women can be affected as well as men (Hagerman et al., 2004). FXTAS has a similar clinical appearance to MSA, but in a review of 77 patients with an MSA diagnosis, only one person was identified with FXTAS (Biancalana et al., 2005); in another review of 426 patients, only 4 had the premutation (Kamm et al., 2005). Continuing studies show this disorder to be rare (Reis et al., 2008). The neuropathology of this syndrome is characterized by intranuclear inclusions (Greco et al., 2006). An increased signal in the middle cerebellar peduncle can sometimes be seen by MRI.
Diagnostic plan
In the evaluation of a patient with ataxia, Table 21.9 outlines a reasonable approach. After a good history and physical examination, laboratory tests should be done to follow up clinical suspicions. A nice algorithm is presented by Schulz et al., (2009a). A detailed algorithm for recessive or sporadic ataxia is presented by Anheim et al. (2010).
Recovery from cerebellar injury; therapy
Weakness, deconditioning, and spasticity are often seen in conjunction with ataxia and contribute substantially to its morbidity. All other factors being equal, stronger, more athletic individuals tend to tolerate moderate ataxia better. Specific physical treatments for ataxia with the application of weights might be helpful. The use of added mass to treat certain tremors has a sound mechanical basis. Because the addition of mass lowers the resonant frequency of a limb, it reduces its response to high-frequency oscillating driving signals. Indeed, Hewer et al. (1972) found better results in patients who had a tremor frequency greater than 7 Hz. In general, the more severe the tremor in terms of amplitude, the more weight was needed to obtain improvement. With regard to ataxia, up to a point, additional mass also improved ataxia of arm movement and of gait. However, beyond this, increased weight was associated with poorer performance. The optimal weight value varied by individual and was not clearly related to the severity of ataxia. Within the limits of fatigue tolerance, which unfortunately may pose a significant restriction, weight therapy remains a reasonable treatment option for some patients. It is also possible to use devices with incorporated viscous damping (Aisen et al., 1993).
Pharmacologic therapy has been reviewed (Ogawa, 2004), and there has been an evidence-based review (Trujillo-Martin et al., 2009). Unfortunately, there are not any excellent treatments and the evidence itself is generally limited.
Several agents have been reported to show some ataxia-ameliorating effects. Botez et al. (1996) found low levels of the dopamine metabolite homovanillic acid in the cerebrospinal fluid of patients with FA and olivopontocerebellar atrophy. In a double-blind trial, amantadine produced significant improvements in both movement time and reaction time in patients (Botez et al., 1996). Unfortunately, this was not associated with functional improvement. Research conducted largely in Japan has emphasized the potential utility of thyrotropin releasing hormone (TRH) and many analogs in cerebellar ataxia (Sobue et al., 1983), but these results are generally not dramatic and have not been widely reproduced.
The dense and widespread distribution of serotonergic terminals throughout the cerebellum and spinocerebellar tracts suggests that serotonin plays a major role in regulating cerebellar functions. While some studies have shown benefit in ataxic patients from oral administration of L-5-hydroxytryptophan (L-5-HTP) (Trouillas et al., 1995), there have also been a number of negative results (Wessel et al., 1995). Moreover, L-5-HTP has been associated with a somewhat high rate of unpleasant gastrointestinal side effects, chiefly nausea and diarrhea, even when administered with a peripheral decarboxylase blocker. In addition, L-5-HTP has been associated with a syndrome resembling eosinophilia–myalgia although the possible role of contaminants in the preparation has not been fully determined (Michelson et al., 1994). Some attention has shifted toward trials of alternative serotonin agonists. Buspirone is a selective serotonin 1A receptor agonist which has been found by Lou et al. (1995) in an open-label study and Trouillas et al. (1997) in both an open and in a randomized double-blind study to produce some small benefit. There has been a double-blind study at NINDS with negative results (Massaquoi et al., unpublished). Ondansetron, a 5-HT3 antagonist, may help some patients (Mandelcorn et al., 2004).
There have been reports of a small but persistent improvement in ataxia, and possibly arrest of symptom progression in patients treated with physostigmine, either orally or via a transdermal patch (Aschoff et al., 1996), but double-blind studies have failed to demonstrate significant benefit (Wessel et al., 1997). In anecdotal reports (Helveston et al., 1996a, 1996b; Hurd et al., 1996), the antioxidant N-acetylcysteine has been reported to improve ataxia along with a number of other problems in different ataxias, but controlled trials have not been done. There is a double-blind crossover trial of branched-chain amino acid therapy which has suggested some improvement over a 4-week period (Mori et al., 2002). The proposed explanation of efficacy is that this therapy improved glutamatergic transmission in the cerebellum. A small trial of D-cycloserine was successful (Ogawa et al., 2003), and one patient improved significantly on piracetam (Vural et al., 2003). In an open trial, 10 patients were improved with gabapentin (Gazulla et al., 2004).
Three patients with different types of ataxia have responded favorably to varenicline (Zesiewicz and Sullivan, 2008; Zesiewicz et al., 2009). Varenicline is a highly selective partial agonist at α4β2 nicotinic acetylcholine receptors and a full agonist at α7 nicotinic receptors. There are acetylcholine receptors in the cerebellum, and stimulation of α4β2 nicotinic acetylcholine receptors can improve alcohol-induced ataxia in an animal model. However, the mechanism of action is not at all certain for any symptomatic effect should this prove useful.
Riluzole has shown some efficacy in a randomized, double-blind, placebo-controlled trial of 40 patients presenting with cerebellar ataxias of different etiologies. Patients were treated with riluzole (100 mg/day) or placebo for 8 weeks. The number of patients with an improvement in ataxia was significantly higher in the riluzole group (Ristori et al., 2010).
Surgical ablation or high-frequency electrical stimulation of the ventral intermediate nucleus of the thalamus (VIM) can be effective in reducing cerebellar tremor (Narabayashi, 1992; Nguyen and Degos, 1993); however, these procedures do not significantly lessen ataxia.
Abele M., Burk K., Schols L., et al. The aetiology of sporadic adult-onset ataxia. Brain. 2002;125(Pt 5):961-968.
Abele M., Schols L., Schwartz S., Klockgether T. Prevalence of antigliadin antibodies in ataxia patients. Neurology. 2003;60(10):1674-1675.
Abele M., Weller M., Mescheriakov S., et al. Cerebellar ataxia with glutamic acid decarboxylase autoantibodies. Neurology. 1999;52(4):857-859.
Aisen M.L., Arnold A., Baiges I., et al. The effect of mechanical damping loads on disabling action tremor. Neurology. 1993;43(7):1346-1350.
Alaedini A., Okamoto H., Briani C., et al. Immune cross-reactivity in celiac disease: anti-gliadin antibodies bind to neuronal synapsin I. J Immunol. 2007;178(10):6590-6595.
Amiri K., Hagerman R.J., Hagerman P.J. Fragile X-associated tremor/ataxia syndrome: an aging face of the fragile X gene. Arch Neurol. 2008;65(1):19-25.
Anderson N.E., Rosenblum M.K., Posner J.B. Paraneoplastic cerebellar degeneration: clinical-immunological correlations. Ann Neurol. 1988;24(4):559-567.
Anheim M., Fleury M., Monga B., et al. Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics. 2010;11(1):1-12.
Anttonen A.K., Mahjneh I., Hamalainen R.H., et al. The gene disrupted in Marinesco-Sjogren syndrome encodes SIL1, an HSPA5 cochaperone. Nat Genet. 2005;37(12):1309-1311.
Artuch R., Aracil A., Mas A., et al. Friedreich’s ataxia: idebenone treatment in early stage patients. Neuropediatrics. 2002;33(4):190-193.
Artuch R., Brea-Calvo G., Briones P., et al. Cerebellar ataxia with coenzyme Q10 deficiency: diagnosis and follow-up after coenzyme Q10 supplementation. J Neurol Sci. 2006;246(1–2):153-158.
Aschoff J.C., Kailer N.A., Walter K. [Physostigmine in treatment of cerebellar ataxia]. Nervenarzt. 1996;67(4):311-318.
Auburger G., Ratzlaff T., Lunkes A., et al. A gene for autosomal dominant paroxysmal choreoathetosis/spasticity (CSE) maps to the vicinity of a potassium channel gene cluster on chromosome 1p, probably within 2 cM between D1S443 and D1S197. Genomics. 1996;31(1):90-94.
Bhidayasiri R., Ling H. Multiple system atrophy. Neurologist. 2008;14(4):224-237.
Biancalana V., Toft M., Le Ber I., et al. FMR1 premutations associated with fragile X-associated tremor/ataxia syndrome in multiple system atrophy. Arch Neurol. 2005;62(6):962-966.
Boddaert N., L.e. Quan Sang K.H., Rotig A., et al. Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood. 2007;110(1):401-408.
Bolla L., Palmer R.M. Paraneoplastic cerebellar degeneration. Case report and literature review. Arch Intern Med. 1997;157(11):1258-1262.
Bomar J.M., Benke P.J., Slattery E.L., et al. Mutations in a novel gene encoding a CRAL-TRIO domain cause human Cayman ataxia and ataxia/dystonia in the jittery mouse. Nat Genet. 2003;35(3):264-269.
Botez M.I., Botez-Marquard T., Elie R., et al. Amantadine hydrochloride treatment in heredodegenerative ataxias: a double blind study. J Neurol Neurosurg Psychiatry. 1996;61(3):259-264.
Bradley J.L., Blake J.C., Chamberlain S., et al. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum Mol Genet. 2000;9(2):275-282.
Brandsema J.F., Stephens D., Hartley J., Yoon G. Intermediate-dose idebenone and quality of life in Friedreich ataxia. Pediatr Neurol. 2010;42(5):338-342.
Burk K., Bosch S., Muller C.A., et al. Sporadic cerebellar ataxia associated with gluten sensitivity. Brain. 2001;124(Pt 5):1013-1019.
Bushara K.O., Goebel S.U., Shill H., et al. Gluten sensitivity in sporadic and hereditary cerebellar ataxia. Ann Neurol. 2001;49(4):540-543.
Buyse G., Mertens L., Di Salvo G., et al. Idebenone treatment in Friedreich’s ataxia: Neurological, cardiac, and biochemical monitoring. Neurology. 2003;60(10):1679-1681.
Cagnoli C., Brussino A., Di Gregorio E., et al. Mutations in the POLG1 gene are not a relevant cause of cerebellar ataxia in Italy. J Neurol. 2008;255(7):1079-1080.
Christodoulou K., Deymeer F., Serdaroglu P., et al. Mapping of the second Friedreich’s ataxia (FRDA2) locus to chromosome 9p23-p11: evidence for further locus heterogeneity. Neurogenetics. 2001;3(3):127-132.
Coesmans M., Smitt P.A., Linden D.J., et al. Mechanisms underlying cerebellar motor deficits due to mGluR1-autoantibodies. Ann Neurol. 2003;53(3):325-336.
Criscuolo C., Chessa L., Di Giandomenico S., et al. Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology. 2006;66(8):1207-1210.
D’Arrigo S., Riva D., Bulgheroni S., et al. Ataxia with oculomotor apraxia type 1 (AOA1): clinical and neuropsychological features in 2 new patients and differential diagnosis. J Child Neurol. 2008;23(8):895-900.
Dalmau J., Gultekin S.H., Voltz R., et al. Ma1, a novel neuron- and testis-specific protein, is recognized by the serum of patients with paraneoplastic neurological disorders. Brain. 1999;122(Pt 1):27-39.
Damji K.F., Allingham R.R., Pollock S.C., et al. Periodic vestibulocerebellar ataxia, an autosomal dominant ataxia with defective smooth pursuit, is genetically distinct from other autosomal dominant ataxias. Arch Neurol. 1996;53(4):338-344.
de la Sayette V., Bertran F., Honnorat J., et al. Paraneoplastic cerebellar syndrome and optic neuritis with anti-CV2 antibodies: clinical response to excision of the primary tumor. Arch Neurol. 1998;55(3):405-408.
Devos D., Schraen-Maschke S., Vuillaume I., et al. Clinical features and genetic analysis of a new form of spinocerebellar ataxia. Neurology. 2001;56(2):234-238.
Di Donato S. The complex clinical and genetic classification of inherited ataxias. I. Dominant ataxias. Ital J Neurol Sci. 1998;19(6):335-343.
Di Donato I., Bianchi S., Federico A. Ataxia with vitamin E deficiency: update of molecular diagnosis. Neurol Sci. 2010;31(4):511-515.
Di Donato S., Gellera C., Mariotti C. The complex clinical and genetic classification of inherited ataxias. II. Autosomal recessive ataxias. Neurol Sci. 2001;22(3):219-228.
Di Prospero N.A., Baker A., Jeffries N., Fischbeck K.H. Neurological effects of high-dose idebenone in patients with Friedreich’s ataxia: a randomised, placebo-controlled trial. Lancet Neurol. 2007;6(10):878-886.
Dupre N., Gros-Louis F., Chrestian N., et al. Clinical and genetic study of autosomal recessive cerebellar ataxia type 1. Ann Neurol. 2007;62(1):93-98.
Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9(9):885-894.
Engert J.C., Berube P., Mercier J., et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet. 2000;24(2):120-125.
Escayg A., De Waard M., Lee D.D., et al. Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. 2000;66(5):1531-1539.
Evidente V.G., Gwinn-Hardy K.A., Caviness J.N., Gilman S. Hereditary ataxias. Mayo Clin Proc. 2000;75(5):475-490.
Gabsi S., Gouider-Khouja N., Belal S., et al. Effect of vitamin E supplementation in patients with ataxia with vitamin E deficiency. Eur J Neurol. 2001;8(5):477-481.
Gazulla J., Errea J.M., Benavente I., Tordesillas C.J. Treatment of ataxia in cortical cerebellar atrophy with the GABAergic drug gabapentin. A preliminary study. Eur Neurol. 2004;52(1):7-11.
Gilman S., Wenning G.K., Low P.A., et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670-676.
Greco C.M., Berman R.F., Martin R.M., et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain. 2006;129(Pt 1):243-255.
Gros-Louis F., Dupre N., Dion P., et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet. 2007;39(1):80-85.
Gultekin S.H., Rosenfeld M.R., Voltz R., et al. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain. 2000;123(Pt 7):1481-1494.
Habeck M., Zuhlke C., Bentele K.H., et al. Aprataxin mutations are a rare cause of early onset ataxia in Germany. J Neurol. 2004;251(5):591-594.
Hadjivassiliou M., Aeschlimann P., Strigun A., et al. Autoantibodies in gluten ataxia recognize a novel neuronal transglutaminase. Ann Neurol. 2008;64(3):332-343.
Hadjivassiliou M., Boscolo S., Tongiorgi E., et al. Cerebellar ataxia as a possible organ-specific autoimmune disease. Mov Disord. 2008;23(10):1370-1377.
Hadjivassiliou M., Grunewald R.A., Chattopadhyay A.K., et al. Clinical, radiological, neurophysiological, and neuropathological characteristics of gluten ataxia. Lancet. 1998;352(9140):1582-1585.
Hadjivassiliou M., Grunewald R.A., Lawden M., et al. Headache and CNS white matter abnormalities associated with gluten sensitivity. Neurology. 2001;56(3):385-388.
Hadjivassiliou M., Grunewald R., Sharrack B., et al. Gluten ataxia in perspective: epidemiology, genetic susceptibility and clinical characteristics. Brain. 2003;126(Pt 3):685-691.
Hadjivassiliou M., Maki M., Sanders D.S., et al. Autoantibody targeting of brain and intestinal transglutaminase in gluten ataxia. Neurology. 2006;66(3):373-377.
Hadjivassiliou M., Sanders D.S., Woodroofe N., et al. Gluten ataxia. Cerebellum. 2008;7(3):494-498.
Hagerman P.J., Hagerman R.J. The fragile-X premutation: a maturing perspective. Am J Hum Genet. 2004;74(5):805-816.
Hagerman R.J., Leavitt B.R., Farzin F., et al. Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am J Hum Genet. 2004;74(5):1051-1056.
Hakonen A.H., Heiskanen S., Juvonen V., et al. Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am J Hum Genet. 2005;77(3):430-441.
Hart P.E., Lodi R., Rajagopalan B., et al. Antioxidant treatment of patients with Friedreich ataxia: four-year follow-up. Arch Neurol. 2005;62(4):621-626.
Hausse A.O., Aggoun Y., Bonnet D., et al. Idebenone and reduced cardiac hypertrophy in Friedreich’s ataxia. Heart. 2002;87(4):346-349.
Helveston W., Cibula J.E., Hurd R., et al. Abnormalities of antioxidant metabolism in a case of Friedreich’s disease. Clin Neuropharmacol. 1996;19(3):271-275.
Helveston W., Hurd R., Uthman B., Wilder B.J. Abnormalities of glutathione peroxidase and glutathione reductase in four patients with Friedreich’s disease [letter]. Mov Disord. 1996;11(1):106-107.
Hewer R.L., Cooper R., Morgan M.H. An investigation into the value of treating intention tremor by weighting the affected limb. Brain. 1972;95:579-590.
Hirano R., Interthal H., Huang C., et al. Spinocerebellar ataxia with axonal neuropathy: consequence of a Tdp1 recessive neomorphic mutation? EMBO J. 2007;26(22):4732-4743.
Honnorat J., Saiz A., Giometto B., et al. Cerebellar ataxia with anti-glutamic acid decarboxylase antibodies: study of 14 patients. Arch Neurol. 2001;58(2):225-230.
Hurd R.W., Wilder B.J., Helveston W.R., Uthman B.M. Treatment of four siblings with progressive myoclonus epilepsy of the Unverricht-Lundborg type with N-acetylcysteine. Neurology. 1996;47(5):1264-1268.
Jarius S., Wandinger K.P., Horn S., et al. A new Purkinje cell antibody (anti-Ca) associated with subacute cerebellar ataxia: immunological characterization. J Neuroinflammation. 2010;7:21.
Jen J.C. Hereditary episodic ataxias. Ann NY Acad Sci. 2008;1142:250-253.
Jen J., Wan J., Graves M., et al. Loss-of-function EA2 mutations are associated with impaired neuromuscular transmission. Neurology. 2001;57(10):1843-1848.
Kamm C., Healy D.G., Quinn N.P., et al. The fragile X tremor ataxia syndrome in the differential diagnosis of multiple system atrophy: data from the EMSA Study Group. Brain. 2005;128:1855-1860.
Klockgether T. Recent advances in degenerative ataxias. Curr Opin Neurol. 2000;13(4):451-455.
Klockgether T. The clinical diagnosis of autosomal dominant spinocerebellar ataxias. Cerebellum. 2008;7(2):101-105.
Koeppen A.H. The pathogenesis of spinocerebellar ataxia. Cerebellum. 2005;4(1):62-73.
Kullmann D.M., Rea R., Spauschus A., Jouvenceau A. The inherited episodic ataxias: how well do we understand the disease mechanisms? Neuroscientist. 2001;7(1):80-88.
Lagier-Tourenne C., Tazir M., Lopez L.C., et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet. 2008;82(3):661-672.
Lamperti C., Naini A., Hirano M., et al. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology. 2003;60(7):1206-1208.
Leehey M.A., Munhoz R.P., Lang A.E., et al. The fragile X premutation presenting as essential tremor. Arch Neurol. 2003;60(1):117-121.
Lerman-Sagie T., Rustin P., Lev D., et al. Dramatic improvement in mitochondrial cardiomyopathy following treatment with idebenone. J Inherit Metab Dis. 2001;24(1):28-34.
Liu C.S., Soong B.W., Lee Y.C., et al. Gluten sensitivity: associated sporadic cerebellar ataxia in Taiwan. Acta Neurol Taiwan. 2010;19(4):263-269.
Lo Y.L. Clinical and immunological spectrum of the Miller Fisher syndrome. Muscle Nerve. 2007;36(5):615-627.
Lodi R., Hart P.E., Rajagopalan B., et al. Antioxidant treatment improves in vivo cardiac and skeletal muscle bioenergetics in patients with Friedreich’s ataxia. Ann Neurol. 2001;49(5):590-596.
Lou J.-S., Goldfarb L., McShane L., et al. Use of buspirone for treatment of cerebellar ataxia. An open-label study. Arch Neurol. 1995;52:982-988.
Lucchinetti C.F., Kimmel D.W., Lennon V.A. Paraneoplastic and oncologic profiles of patients seropositive for type 1 antineuronal nuclear autoantibodies. Neurology. 1998;50(3):652-657.
Mancuso M., Orsucci D., Choub A., Siciliano G. Current and emerging treatment options in the management of Friedreich ataxia. Neuropsychiatr Dis Treat. 2010;6:491-499.
Mandelcorn J., Cullen N.K., Bayley M.T. A preliminary study of the efficacy of ondansetron in the treatment of ataxia, poor balance and incoordination from brain injury. Brain Inj. 2004;18(10):1025-1039.
Manto M., Marmolino D. Cerebellar ataxias. Curr Opin Neurol. 2009;22(4):419-429.
Mariotti C., Solari A., Torta D., et al. Idebenone treatment in Friedreich patients: One-year-long randomized placebo-controlled trial. Neurology. 2003;60(10):1676-1679.
Maselli R.A., Wan J., Dunne V., et al. Presynaptic failure of neuromuscular transmission and synaptic remodeling in EA2. Neurology. 2003;61(12):1743-1748.
Michelson D., Page S.W., Casey R., et al. An eosinophilia-myalgia syndrome related disorder associated with exposure to L-5-hydroxytryptophan. J Rheumatol. 1994;21(12):2261-2265.
Milone M., Massie R. Polymerase gamma 1 mutations: clinical correlations. Neurologist. 2010;16(2):84-91.
Mitoma H., Song S., Ishida K., et al. Presynaptic impairment of cerebellar inhibitory synapses by an autoantibody to glutamate decarboxylase. J Neurol Sci. 2000;175(1):40-44.
Mollet J., Delahodde A., Serre V., et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am J Hum Genet. 2008;82(3):623-630.
Montero R., Pineda M., Aracil A., et al. Clinical, biochemical and molecular aspects of cerebellar ataxia and Coenzyme Q10 deficiency. Cerebellum. 2007;6(2):118-122.
Mori M., Adachi Y., Mori N., et al. Double-blind crossover study of branched-chain amino acid therapy in patients with spinocerebellar degeneration. J Neurol Sci. 2002;195(2):149-152.
Moro-De-Casillas M.L., Cohen M.L., Riley D.E. Leucoencephalopathy with neuroaxonal spheroids (LENAS) presenting as the cerebellar subtype of multiple system atrophy. J Neurol Neurosurg Psychiatry. 2004;75(7):1070-1072.
Moseley M.L., Benzow K.A., Schut L.J., et al. Incidence of dominant spinocerebellar and Friedreich triplet repeats among 361 ataxia families. Neurology. 1998;51(6):1666-1671.
Mrissa N., Belal S., Hamida C.B., et al. Linkage to chromosome 13q11–12 of an autosomal recessive cerebellar ataxia in a Tunisian family. Neurology. 2000;54(7):1408-1414.
Musumeci O., Naini A., Slonim A.E., et al. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology. 2001;56(7):849-855.
Nanri K., Okita M., Takeguchi M., et al. Intravenous immunoglobulin therapy for autoantibody-positive cerebellar ataxia. Intern Med. 2009;48(10):783-790.
Narabayashi H. Analysis of intention tremor. Clin Neurol Neurosurg. 1992;94(Suppl):S130-S132.
Nguyen J.P., Degos J.D. Thalamic stimulation and proximal tremor. A specific target in the nucleus ventrointermedius thalami. Arch Neurol. 1993;50(5):498-500.
Ogawa M. Pharmacological treatments of cerebellar ataxia. Cerebellum. 2004;3(2):107-111.
Ogawa M., Shigeto H., Yamamoto T., et al. D-Cycloserine for the treatment of ataxia in spinocerebellar degeneration. J Neurol Sci. 2003;210(1–2):53-56.
O’Hearn E., Holmes S.E., Calvert P.C., et al. SCA-12: Tremor with cerebellar and cortical atrophy is associated with a CAG repeat expansion. Neurology. 2001;56(3):299-303.
Panas M., Kalfakis N., Karadima G., et al. Friedreich’s ataxia mimicking hereditary motor and sensory neuropathy. J Neurol. 2002;249(11):1583-1586.
Pandolfo M. Friedreich ataxia. Arch Neurol. 2008;65(10):1296-1303.
Paulson H.L. Dominantly inherited ataxias: lessons learned from Machado-Joseph disease/spinocerebellar ataxia type 3. Semin Neurol. 2007;27(2):133-142.
Pellecchia M.T., Scala R., Filla A., et al. Idiopathic cerebellar ataxia associated with celiac disease: lack of distinctive neurological features [see comments]. J Neurol Neurosurg Psychiatry. 1999;66(1):32-35.
Pineda M., Arpa J., Montero R., et al. Idebenone treatment in paediatric and adult patients with Friedreich ataxia: Long-term follow-up. Eur J Paediatr Neurol. 2008;12(6):470-475.
Quinzii C.M., Kattah A.G., Naini A., et al. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology. 2005;64(3):539-541.
Rai M., Soragni E., Jenssen K., et al. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS ONE. 2008;3(4):e1958.
Reis A.H., Ferreira A.C., Gomes K.B., et al. Frequency of FMR1 premutation in individuals with ataxia and/or tremor and/or parkinsonism. Genet Mol Res. 2008;7(1):74-84.
Ristori G., Romano S., Visconti A., et al. Riluzole in cerebellar ataxia: a randomized, double-blind, placebo-controlled pilot trial. Neurology. 2010;74(10):839-845.
Rustin P., Rotig A., Munnich A., Sidi D. Heart hypertrophy and function are improved by idebenone in Friedreich’s ataxia. Free Radic Res. 2002;36(4):467-469.
Rustin P., von Kleist-Retzow J.C., Chantrel-Groussard K., et al. Effect of idebenone on cardiomyopathy in Friedreich’s ataxia: a preliminary study. Lancet. 1999;354(9177):477-479.
Sabater L., Bataller L., Suárez-Calvet M., et al. ZIC antibodies in paraneoplastic cerebellar degeneration and small cell lung cancer. J Neuroimmunol. 2008:201-202. 163–165
Saiz A., Arpa J., Sagasta A., et al. Autoantibodies to glutamic acid decarboxylase in three patients with cerebellar ataxia, late-onset insulin-dependent diabetes mellitus, and polyendocrine autoimmunity. Neurology. 1997;49(4):1026-1030.
Saiz A., Dalmau J., Butler M.H., et al. Anti-amphiphysin I antibodies in patients with paraneoplastic neurological disorders associated with small cell lung carcinoma. J Neurol Neurosurg Psychiatry. 1999;66(2):214-217.
Schols L., Arning L., Schule R., et al. “Pseudodominant inheritance” of ataxia with ocular apraxia type 2 (AOA2). J Neurol. 2008;255(4):495-501.
Schols L., Bauer P., Schmidt T., et al. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3(5):291-304.
Schols L., Vorgerd M., Schillings M., et al. Idebenone in patients with Friedreich ataxia. Neurosci Lett. 2001;306(3):169-172.
Schulz J.B., Boesch S., Burk K., et al. Diagnosis and treatment of Friedreich ataxia: a European perspective. Nat Rev Neurol. 2009;5(4):222-234.
Schulz J.B., Di Prospero N.A., Fischbeck K. Clinical experience with high-dose idebenone in Friedreich ataxia. J Neurol. 2009;256(Suppl 1):42-45.
Senderek J., Krieger M., Stendel C., et al. Mutations in SIL1 cause Marinesco-Sjogren syndrome, a cerebellar ataxia with cataract and myopathy. Nat Genet. 2005;37(12):1312-1314.
Shams’ili S., Grefkens J., de Leeuw B., et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain. 2003;126(Pt 6):1409-1418.
Shill H.A., Alaedini A., Latov N., Hallett M. Anti-ganglioside antibodies in idiopathic and hereditary cerebellar degeneration. Neurology. 2003;60(10):1672-1673.
Shimazaki H., Takiyama Y., Sakoe K., et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia: the aprataxin gene mutations. Neurology. 2002;59(4):590-595.
Sillevis Smitt P., Kinoshita A., De Leeuw B., et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med. 2000;342(1):21-27.
Sobue I., Takayanagi T., Nakanishi T., et al. Controlled trial of thyrotropin releasing hormone tartrate in ataxia of spinocerebellar degenerations. J Neurol Sci. 1983;61(2):235-248.
Soong B.W., Lu Y.C., Choo K.B., Lee H.Y. Frequency analysis of autosomal dominant cerebellar ataxias in Taiwanese patients and clinical and molecular characterization of spinocerebellar ataxia type 6. Arch Neurol. 2001;58(7):1105-1109.
Soong B.W., Paulson H.L. Spinocerebellar ataxias: an update. Curr Opin Neurol. 2007;20(4):438-446.
Souayah N., Chin R.L., Brannagan T.H., et al. Effect of intravenous immunoglobulin on cerebellar ataxia and neuropathic pain associated with celiac disease. Eur J Neurol. 2008;15(12):1300-1303.
Spacey S.D., Materek L.A., Szczygielski B.I., Bird T.D. Two novel CACNA1A gene mutations associated with episodic ataxia type 2 and interictal dystonia. Arch Neurol. 2005;62(2):314-316.
Steckley J.L., Ebers G.C., Cader M.Z., McLachlan R.S. An autosomal dominant disorder with episodic ataxia, vertigo, and tinnitus. Neurology. 2001;57(8):1499-1502.
Stevanin G., Durr A., Brice A. Clinical and molecular advances in autosomal dominant cerebellar ataxias: from genotype to phenotype and physiopathology. Eur J Hum Genet. 2000;8(1):4-18.
Storey E., du Sart D., Shaw J.H., et al. Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients with spinocerebellar ataxia. Am J Med Genet. 2000;95(4):351-357.
Strupp M., Kalla R., Dichgans M., et al. Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology. 2004;62(9):1623-1625.
Subramony S.H., Vig P.J., McDaniel D.O. Dominantly inherited ataxias. Semin Neurol. 1999;19(4):419-425.
Sura T., Eu-Ahsunthornwattana J., Youngcharoen S., et al. Frequencies of spinocerebellar ataxia subtypes in Thailand: window to the population history? J Hum Genet. 2009;54(5):284-288.
Takashima H., Boerkoel C.F., John J., et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet. 2002;32(2):267-272.
Takiyama Y. Sacsinopathies: sacsin-related ataxia. Cerebellum. 2007;28:1-7. Feb
Tan E., Ashizawa T. Genetic testing in spinocerebellar ataxias: defining a clinical role. Arch Neurol. 2001;58(2):191-195.
Tazir M., Ali-Pacha L., M’Zahem A., et al. Ataxia with oculomotor apraxia type 2: a clinical and genetic study of 19 patients. J Neurol Sci. 2009;278(1–2):77-81.
Thiffault I., Rioux M.F., Tetreault M., et al. A new autosomal recessive spastic ataxia associated with frequent white matter changes maps to 2q33–34. Brain. 2006;129(Pt 9):2332-2340.
Tomlinson S.E., Hanna M.G., Kullmann D.M., et al. Clinical neurophysiology of the episodic ataxias: insights into ion channel dysfunction in vivo. Clin Neurophysiol. 2009;120(10):1768-1776.
Triantafillidis J.K., Kottaras G., Sgourous S., et al. A-beta-lipoproteinemia: clinical and laboratory features, therapeutic manipulations, and follow-up study of three members of a Greek family. J Clin Gastroenterol. 1998;26(3):207-211.
Trouillas P., Serratrice G., Laplane D., et al. Levorotatory form of 5-hydroxytryptophan in Friedreich’s ataxia. Results of a double-blind drug-placebo cooperative study. Arch Neurol. 1995;52:456-460.
Trouillas P., Xie J., Adeleine P., et al. Buspirone, a 5-hydroxytryptamine1A agonist, is active in cerebellar ataxia. Results of a double-blind drug placebo study in patients with cerebellar cortical atrophy. Arch Neurol. 1997;54(6):749-752.
Trujillo-Martin M.M., Serrano-Aguilar P., Monton-Alvarez F., Carrillo-Fumero R. Effectiveness and safety of treatments for degenerative ataxias: a systematic review. Mov Disord. 2009;24(8):1111-1124.
Tzoulis C., Neckelmann G., Mork S.J., et al. Localized cerebral energy failure in DNA polymerase gamma-associated encephalopathy syndromes. Brain. 2010;133(Pt 5):1428-1437.
Vernino S., Lennon V.A. New Purkinje cell antibody (PCA-2): marker of lung cancer-related neurological autoimmunity. Ann Neurol. 2000;47(3):297-305.
Vural M., Ozekmekci S., Apaydin H., Altinel A. High-dose piracetam is effective on cerebellar ataxia in patient with cerebellar cortical atrophy. Mov Disord. 2003;18(4):457-459.
Waldvogel D., van Gelderen P., Hallett M. Increased iron in the dentate nucleus of patients with Friedrich’s ataxia. Ann Neurol. 1999;46(1):123-125.
Wardle M., Majounie E., Muzaimi M.B., et al. The genetic aetiology of late-onset chronic progressive cerebellar ataxia. A population-based study. J Neurol. 2009;256(3):343-348.
Watase K., Gatchel J.R., Sun Y., et al. Lithium therapy improves neurological function and hippocampal dendritic arborization in a spinocerebellar ataxia type 1 mouse model. PLoS Med. 2007;4(5):e182.
Wenning G.K., Stefanova N., Jellinger K.A., et al. Multiple system atrophy: a primary oligodendrogliopathy. Ann Neurol. 2008;64(3):239-246.
Wessel K., Hermsdorfer J., Deger K., et al. Double-blind crossover study with levorotatory form of hydroxytryptophan in patients with degenerative cerebellar diseases. Arch Neurol. 1995;52:451-455.
Wessel K., Langenberger K., Nitschke M.F., Kompf D. Double-blind crossover study with physostigmine in patients with degenerative cerebellar diseases. Arch Neurol. 1997;54(4):397-400.
Woods C.G., Taylor A.M. Ataxia telangiectasia in the British Isles: the clinical and laboratory features of 70 affected individuals. Q J Med. 1992;82(298):169-179.
Xia H., Mao Q., Eliason S.L., et al. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med. 2004;10(8):816-820.
Yoshida M. Multiple system atrophy: alpha-synuclein and neuronal degeneration. Neuropathology. 2007;27(5):484-493.
Zesiewicz T.A., Sullivan K.L. Treatment of ataxia and imbalance with varenicline (chantix): report of 2 patients with spinocerebellar ataxia (types 3 and 14). Clin Neuropharmacol. 2008;31(6):363-365.
Zesiewicz T.A., Sullivan K.L., Freeman A., Juncos J.L. Treatment of imbalance with varenicline Chantix(R): report of a patient with fragile X tremor/ataxia syndrome. Acta Neurol Scand. 2009;119(2):135-138.
Zoghbi H.Y., Orr H.T. Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J Biol Chem. 2009;284(12):7425-7429.
Zu T., Duvick L.A., Kaytor M.D., et al. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci. 2004;24(40):8853-8861.