Chapter 22 The paroxysmal dyskinesias
Introduction
The overwhelming majority of individuals with hyperkinetic movement disorders have symptoms that are continuous or continual (e.g., chorea, dystonia, tardive dyskinesia), except for relief with sleep, and with some variation in intensity during periods of stress and relaxation, or other factors such as voluntary movements (e.g., action dystonia, intention myoclonus, intention tremor) or maintaining certain postures (e.g., essential tremor). The symptoms and signs of dopa-responsive dystonia sometimes have a diurnal pattern, being absent or slight in the morning hours and becoming more pronounced as the day proceeds (see Chapter 12). Some dyskinesias (Table 22.1) are characterized as occurring intermittently, such as myoclonus (Fahn et al., 1986; Hallett et al., 1987) and startle syndromes (hyperekplexia) (Andermann and Andermann, 1986) that can be triggered by a variety of stimuli. Restless legs syndrome is best described as a diurnal disorder, manifesting itself primarily (1) in the evenings with abnormal crawling sensations that are relieved by the patient moving about and (2) during the night with periodic movements in sleep (Hening et al., 1986; Walters et al., 1991). Sandifer syndrome is a tilting downwards of the head after eating a meal, occurring in boys, and associated with gastroesophageal reflux (Menkes and Ament, 1988). Movements that occur as a result of akathisia relieve the sensation of inner restlessness. These movements occur intermittently and are usually complex (see Chapter 19). Stereotypies are also complex movements, which appear largely in individuals with learning disability, autism, and schizophrenia (Fahn, 1993). They are not always present but occur frequently and almost continually, but in some patients they appear as intermittent bursts (Tan et al., 1997). The explanation as to why these patients make these movements is not known, but suggestions such as “being in touch with the environment” have been proposed. Perhaps the commonest dyskinesias that occur intermittently are tics, which are suppressible to varying degrees (Koller and Biary, 1989). Although tics and myoclonic jerks, since they commonly occur out of a normal background, could possibly be considered paroxysmal, this term is usually reserved for an entirely different set of hyperkinetic movement disorders, which is the topic of this chapter. The term paroxysmal dyskinesia has been applied to these disorders.
Table 22.1 Classic movement disorders that usually appear in bursts or with specific actions, but are not considered paroxysmal dyskinesias
The movements usually occur so frequently that they are not distinguished with a “paroxysmal” label.
The common neurologic paroxysmal disorders are epilepsy and migraine. Movement disorders that appear “out of the blue” and are transient and recurring are uncommon, and often present to the clinician as confusing diagnostic problems. Not uncommonly, the history that is provided by the patient does not convey the information that the episodes of abnormal movements occur at intermittent intervals, and the clinician might overlook the category of paroxysmal dyskinesias. Therefore, if the examination does not reveal the presence of a movement disorder, the clinician needs to consider the possibility that he or she is dealing with a paroxysmal dyskinesia, and thereby ask the appropriate questions that can lead to the proper diagnosis. To compound the problem, nonfamilial paroxysmal movement disorders are often psychogenic in etiology (Bressman et al., 1988); therefore, the clinician has the problem of determining the etiologic distinction of psychogenic versus organic.
The pathophysiology of paroxysmal dyskinesias is not understood, and “epilepsy of the basal ganglia” has been a serious consideration but difficult to prove. Some paroxysmal dyskinesias are supplementary sensorimotor seizures, including many of the hypnogenic variety (Lüders, 1996). The classification of the paroxysmal dyskinesias is still incomplete and evolving (Demirkiran and Jankovic, 1995), and treatment for many of them is often unsuccessful, but treatment for some can be highly successful. A most welcome burst of research on the genetics of the paroxysmal dyskinesias is shedding new light in the classification of these disorders (Nutt and Gancher, 1997), their diagnoses, and their mechanism of action as channelopathies (Griggs and Nutt, 1995) now that the genes that have so far been discovered for these disorders are those that code for some of the ion channels.
Definitions: transient, paroxysmal, episodic, and periodic
Some pediatric neurologists have utilized the term “transient” for a number of movement disorders in children, but the concept of paroxysmal dyskinesias is becoming more widely recognized among pediatric neurologists (Lotze and Jankovic, 2003). Excluding tic disorders, Fernandez-Alvarez (1998) reviewed the 356 movement disorder cases under the age of 18 years that had been seen in his department’s clinic and reported that 19% of them were classified as transient dyskinesias (Table 22.2). Sometimes these transient movements can be mistaken for seizures (Donat and Wright, 1990). An accurate diagnosis can be reassuring to the family because these movements are not seizures and are almost always benign. The diagnosis depends on the clinical features; diagnostic tests are normal and unnecessary.
Table 22.2 Transient movement disorders in children (clinic of Fernandez-Alvarez, 1998)
Kotagal and colleagues (2002) evaluated their cases of paroxysmal nonepileptic events. These are reviewed in Chapter 25. These were most common in adolescents and the school-age group. In the preschool group, the most common diagnoses were stereotypies, hypnic jerks, parasomnias, and Sandifer syndrome.
The term “periodic” is defined by both Dorland’s and Webster’s as recurring at regular intervals of time. Since the paroxysmal dyskinesias do not recur at regular intervals, the term “periodic” would not be appropriate. Despite this, in neurology this term is used for the condition of familial periodic paralysis (Rowland and Gordon, 2005) and familial periodic ataxia (e.g., Vighetto et al., 1988), even though muscle weakness and ataxia, respectively, in these disorders do not occur at regular intervals. Some authors (e.g., Griggs et al., 1978) had used the term paroxysmal ataxia in preference to periodic ataxia, and others use the term episodic ataxia (Zasorin et al., 1983). But the literature today uses “episodic ataxia.”
According to dictionary definitions, either “paroxysmal” or “episodic” would be an appropriate term for the dyskinesias that are under discussion here. By common usage, and with few exceptions (e.g., Margolin and Marsden, 1982), “paroxysmal” has been chosen in preference over “episodic” for the choreoathetotic and dystonic types and is used in this chapter. The term “episodic” is also applied here to the ataxias in keeping with the current trend in the literature. The term “paroxysmal” has been utilized to indicate that the symptoms occur suddenly out of a background of normal motor behavior. It does not define the frequency, severity, duration, aggravating factors, or type of dyskinesia of the attack. These features vary and are important in the current nosology and classification of the paroxysmal dyskinesias.
Historical aspects
This is a condensed review of historical highlights of the paroxysmal and episodic dyskinesias. For a more complete discussion, see the review by Fahn (1994). A review of paroxysmal dyskinesias in the Japanese literature is available in English by Hishikawa and colleagues (1973).
Earliest descriptions: reported as epilepsy
Although Gowers (1885) is often credited with the first report of movement-induced seizures, it is possible that his cases actually represented paroxysmal dyskinesia. One of his patients was a boy whose attack lasted 15 seconds, but the boy was said to be unconscious during his initial attack. Later, he remained awake during the attacks. Another patient was a girl whose attacks started at the age of 11 years and occurred when she arose suddenly after prolonged sitting. But at least one of her attacks was said to be associated with a terrified expression, flushed facies, and dilated pupils. Subsequent to Gowers, a number of reports of “movement-induced seizures” appeared in the literature. Many of these reports have been published under the designation of reflex epilepsy and tonic seizures induced by movement. But unlike most motor convulsions, there was no alteration in the state of consciousness. Moreover, some of these reports had more than tonic contraction, namely, they included sustained twisting, athetosis, and chorea. These characteristics are today referred to as paroxysmal dystonia and paroxysmal choreoathetosis, rather than convulsive seizures. Even the presence of choreoathetosis did not lead the earliest interpreters of these brief attacks to conclude they were a movement disorder; instead, they were considered to be a form of epilepsy, the cerebral site of these “seizures” being in the basal ganglia or in the subcortical region.
After the report of Gowers, the next report of movement-induced paroxysmal movements appears to be that of Spiller in 1927. Spiller described two patients with brief tonic spasms that were brought on by voluntary movement of the involved limbs and, in one of them, also by passive manipulation. Spiller called this “subcortical epilepsy.” Wilson (1930) described a 5-year-old boy who had brief attacks of unilateral torsion and tonic spasm that lasted up to 3 minutes and were precipitated by fright or excitement. There was no loss of consciousness. The attacks could be preceded by pain. Wilson considered this to be reflex tonic epilepsy and thought it also to be subcortical in origin. In more recent times, the concept that these attacks of tonic, often twisting, contractions without loss of consciousness are uncommon seizure disorders has continued (Whitty et al., 1964; Burger et al., 1972). It would appear that today these movement-induced involuntary movements would be considered paroxysmal choreoathetosis/dystonia rather than convulsive movements of the reflex epilepsy type. Differentiation of the attacks between cortical seizures and paroxysmal dyskinesias is sometimes difficult. Clouding of consciousness, if it occurs, would point to a seizure disorder.
By 1966 and 1967, when papers using the term “paroxysmal choreoathetosis” began regularly to appear (Stevens, 1966; Kertesz, 1967; Mushet and Dreifuss, 1967), particularly those cases induced by movements, there were still occasional papers referring the condition to a seizure disorder. As is discussed below in the section on paroxysmal hypnogenic dyskinesias, nocturnal epilepsy is now considered to be the leading cause.
Reported as a paroxysmal disorder of involuntary movements
In 1940, Mount and Reback (1940) introduced a new concept, that of labeling attacks of tonic spasms plus choreic and athetotic movements as a paroxysmal type of movement disorder. They described a 23-year-old man who had had both large and small “spells” since infancy. Both types were preceded by a sensory aura of tightness in parts of the body or by a feeling of tiredness. The movements involved the arms and legs and were usually a combination of sustained twisted posturing and chorea and athetosis. The small attacks lasted from 5–10 minutes. Longer attacks were considered large and also involved the neck (retrocollis), eyes (upward gaze), face (ipsilateral to the limbs if the limb involvement was unilateral), and speech. These large attacks lasted for as long as 2 hours, and the movements were considered to resemble those seen in Huntington disease. There was never a loss of consciousness or clonic convulsive movements, biting of the tongue, or loss of sphincter control. Drinking alcohol, coffee, tea, or cola would usually bring on an attack. Fatigue, smoking, and concentrating were other precipitating factors. The attacks would clear more rapidly if the patient lay down and would be aborted by sleep. The patient had an average of one large and two small attacks a day. Between attacks, the neurologic examination was normal. Phenytoin and phenobarbital had no effect, and scopolamine was the only drug that was found to reduce the frequency, severity, and duration of the attacks. The family history revealed 27 other members who had similar attacks; the pedigree showed autosomal dominant inheritance with what appears to be complete penetrance. Mount and Reback called this disorder “familial paroxysmal choreoathetosis.”
Mount and Reback’s paper became the seminal paper in the field of paroxysmal dyskinesias. Following its publication, most of the reports in the literature referenced it over the next five decades. However, the next report of a large family with similar attacks of muscle spasms did not refer to it. In 1961 Forssman (1961) described a family with autosomal dominant inheritance in which there were attacks lasting from 4 minutes to 3 hours.
The next large family was described in 1963 by Lance. Like Forssman (1961), Lance also did not relate this to nor refer to Mount and Reback’s report, nor did he mention the report by Forssman. In fact, Lance considered his patients to have a form of epilepsy. Later, Lance (1977) was to write one of the definitive papers in this field, containing a useful classification scheme in which he related his family to those of Mount and Reback (1940), Forssman (1961), and Richards and Barnett (1968).
Although there were reports of patients whose paroxysmal dyskinesias were induced by sudden movement, they were not denoted by any special terminology until 1967, when Kertesz (1967) introduced the label “paroxysmal kinesigenic choreoathetosis” (PKC). This label has developed into a most useful and widely accepted designation, since the kinesigenic feature has proven to be so characteristic. Kinesigenicity has an important place in the classification of the paroxysmal dyskinesias, and Demirkiran and Jankovic (1995) recommended that the term paroxysmal kinesigenic dyskinesia (PKD) be used instead, because the movements can be other than choreoathetotic. That suggestion is followed in this chapter. As is pointed out below, the PKD designation can be applied to some patients who do not have the dyskinesias triggered by sudden movement (or startle).
Kertesz reported 10 new cases of paroxysmal dyskinesia and reviewed the literature. Among the important features of his paper, Kertesz differentiated the kinesigenic variety (induced by sudden movement) from that described by Mount and Reback, by Forssman, and by Lance, which were aggravated not by movement but by alcohol, caffeine, and fatigue. It should be noted that Kertesz differentiated the kinesigenic type from that reported by Mount and Reback (1940) and by Lance (1963), but he failed to mention the paper by Forssman (1961).
Although phenytoin was recognized earlier as a very useful agent for PKD, carbamazepine was later found to be as useful and was introduced as a treatment by Kato and Araki (1969). This drug currently appears to be the one that is most commonly used for this disorder.
After Lance’s paper in 1963, Richards and Barnett (1968) reported the next big family with the same type of paroxysmal dyskinesia as Mount and Reback’s case and thought that Lance’s family (1963) represented a variant, since there were only tonic spasms and no movements in that family. Richards and Barnett emphasized the nonkinesigenic nature of the attacks and felt that the terms rigidity, tremor, dystonia, torsions spasm, athetosis, chorea, and hemiballism could all be used for such movements, often blending into each other. To emphasize the postural and increased tone, they added “dystonic” to the label. They recommended avoiding the term “epilepsy” until the pathophysiology is better known. Richards and Barnett coined the term “paroxysmal dystonic choreoathetosis” (PDC), which was later adopted by Lance in 1977. The terms “paroxysmal nonkinesigenic choreoathetosis” and “paroxysmal dystonia” are sometimes used instead of PDC (Bressman et al., 1988). We have adopted the term paroxysmal nonkinesigenic dyskinesia (PNKD) proposed by Demirkiran and Jankovic (1995).
The original cases that were reported as PNKD were idiopathic and usually familial. It was not long before symptomatic cases began to be reported in which the attacks of movements were reported as a PNKD: perinatal encephalopathy by (Rosen, 1964), encephalitis (Mushet and Dreifuss, 1967), and head injury (Whitty et al., 1964; Robin, 1977). However, earlier reports of symptomatic PNKD had been described as a manifestation of multiple sclerosis, but considered as a form of epilepsy (Matthews, 1958; Joynt and Green, 1962; Verheul and Tyssen, 1990). Many other etiologies have been reported since the cases in the 1950s and 1960s (see below).
One of the most enlightening papers (Lance, 1977) achieved the following:
Instead of Lance’s proposed classification (item 5 in the preceding list) based on duration of the attacks, the classification scheme that is adopted here is the one based on precipitating factors suggested by Demirkiran and Jankovic (1995).
The next historical advances were the recognition that (1) idiopathic PNKD can occur sporadically and not just in families (Bressman et al., 1988) and that (2) sporadic PNKD is often psychogenic in origin (Bressman et al., 1988; Fahn and Williams, 1988).
Paroxysmal hypnogenic dyskinesia (PHD)
Horner and Jackson (1969) described two families in which several members of the family had attacks of involuntary movement that occurred during sleep. These appear to be the first cases of paroxysmal hypnogenic dyskinesia (PHD) to be reported. Family W is of particular interest because some affected members had classic PKD, some had hypnogenic, and others had a combination. Case 3 in this family began with the hypnogenic variety at age 8. By age 11, daytime attacks also occurred, sometimes triggered by sudden movement. Gradually, the hypnogenic episodes disappeared, leaving the patient with kinesigenic dyskinesia that responded to anticonvulsants. Lugaresi and his colleagues (Lugaresi and Cirignotta, 1981; Lugaresi et al., 1986) independently rediscovered and eventually popularized the syndrome of paroxysmal hypnogenic dyskinesias.
In addition to these short-duration attacks, Lugaresi and colleagues (1986) reported long-duration hypnogenic attacks. Such long-duration attacks occur in a minority of individuals with paroxysmal hypnogenic dyskinesia. These longer attacks last from 2 to 50 minutes and do not respond to medication, including anticonvulsants, tricyclics, benzodiazepines, and antipsychotics.
There has long been considerable speculation as to whether the short-duration hypnogenic attacks could be a manifestation of epilepsy, since they respond so well to anticonvulsants. The lack of abnormal electroencephalographic (EEG) findings during the attack has been used to argue against this concept. However, there is accumulating evidence that many paroxysmal hypnogenic dyskinesias are indeed due to seizures. Tinuper and colleagues (1990) described three patients with this disorder who had EEG evidence for frontal lobe seizures as a cause of the attacks. Sellal and colleagues (1991) and Meierkord and colleagues (1992) studied a series of patients with hypnogenic dystonia and concluded that these represent seizure disorders, particularly of frontal lobe epilepsy because repeated nocturnal EEG recordings often reveal epileptic patterns of abnormalities. Seizures arising near the mesial posterior frontal supplementary sensorimotor area may be a particular culprit in inducing paroxysmal hypnogenic dyskinesias in children (Bass et al., 1995). These types of seizures tend to be brief, frequent, and with bilateral tonic posturing, gross proximal limb movements, and preserved consciousness. Dystonic and other dyskinetic features may result from spread of epileptic activity from the mesial frontal region to the basal ganglia because there are close anatomic connections between them. It appears that the short-lasting attacks of paroxysmal hypnogenic dyskinesias are most likely due to seizures, but the question remains whether patients without abnormal EEGs and more prolonged hypnogenic attacks could have something more akin to the paroxysmal dyskinesias. In a family with autosomal dominant nocturnal frontal lobe epilepsy, interictal EEGs were normal, but ictal video-EEG studies showed that the attacks were partial seizures with frontal lobe origin (Scheffer et al., 1995). Fish and Marsden (1994) have reviewed epilepsy masquerading as a movement disorder, and they concluded that most cases of hypnogenic dyskinesias are considered to be due to epilepsy, as did Lüders (1996), with cyclic alternating EEG pattern believed to be a provocative factor (Terzano et al., 1997).
The genetics of hypnogenic dyskinesias/seizures is being explored. A large autosomal dominant Australian family (Oldani et al., 1998) and a Norwegian family (Nakken et al., 1999) have been described with mutations in the nicotinic acetylcholine receptor alpha 4 subunit (CHRNA4) gene, located on chromosome 20q13.2–q13.3. A second acetylcholine receptor subunit, CHRNB2, is also associated with autosomal dominant nocturnal frontal lobe epilepsy (Phillips et al., 2001). Another family with autosomal dominant hypnogenic frontal-lobe epilepsy has been mapped to 15q24 (Phillips et al., 1998).
In addition to epilepsy mimicking hypnogenic paroxysmal dyskinesias, there is a syndrome of infantile convulsions and paroxysmal dyskinesias, referred to as the “infantile convulsions with choreoathetosis” (ICCA) syndrome (Lee et al., 1998). The gene for this disorder has been mapped to the pericentromeric region of chromosome 16 (see the discussion in the section on PKD and also the section “Molecular genetics of paroxysmal dyskinesias”). Single photon emission computed tomography (SPECT) studies revealed alterations in local cerebral perfusion in the sensorimotor cortex, the supplementary motor areas, and the pallidum (Thiriaux et al., 2002).
Transient paroxysmal dyskinesias in infancy
Snyder (1969) introduced a new type of paroxysmal dyskinesia that he called “paroxysmal torticollis in infancy.” He described 12 cases of intermittent head tilting in young infants. The age at onset was between 2 and 8 months of age, except for three cases, in which the first attacks occurred at 14, 17, and 30 months. The attacks would occur about two to three times a month and last from 10 minutes to 14 days, usually 2–3 days. The head would tilt to either side and often rotate slightly to the opposite side. There is no distress unless a parent attempts to straighten the head, upon which the baby cries. In some cases, the head tilting is associated with vomiting, pallor, and agitation for a short period. The infant is normal between attacks, which disappear after months or years, usually around age 2 or 3 years. Subsequently, a number of similar cases have been described (Gourley, 1971; Sanner and Bergstrom, 1979; Bratt and Menelaus, 1992), including familial cases (Lipson and Robertson, 1978). Sanner and Bergstrom (1979) reported a patient whose father had a similar condition in early infancy, suggesting that this disorder is hereditary.
The clinical picture of paroxysmal torticollis in infancy (Video 22.1) that has evolved is that the trunk can also be involved, with lateral curvature concave to the same side as the head tilting, and the ipsilateral leg can be flexed. Onset can be as early as the first months of life and can recur every couple of weeks until they disappear before the age of 2 years. Each attack can last a couple of hours to a couple of weeks. In between attacks, the child is normal. The main differential diagnosis is a posterior fossa tumor and Sandifer syndrome (Menkes and Ament, 1988). 
In 1988, the clinical spectrum expanded with the report by Angelini and colleagues (1988) under the name “transient paroxysmal dystonia in infancy.” They described nine patients who had onset of the paroxysmal dyskinesias between 3 and 5 months of age, except for one patient who had an onset at age 1 month. Three had a history of perinatal brain damage, six did not. The attacks consisted of opisthotonus, increased muscle tone with twisting of the limbs, and, in three, neck and trunk twisting, thereby linking this with “paroxysmal torticollis in infancy.” The attacks lasted several minutes, with a maximum of 2 hours in one patient. They would occur from several times per day (Video 22.2) to once a month. Remission occurred between the ages of 8 and 22 months, with two not yet having reached a remission. 
Dunn (1981) described an infant with head turning and posturing of the right arm lasting from 45 minutes to 18 hours. There were six attacks from age 26 months to age 40 months. The author did not mention the possible diagnosis of paroxysmal torticollis in infancy and made a diagnosis of paroxysmal dystonic choreoathetosis instead. One should consider the possibility that PNKD may occur in infancy and disappear over several months. If so, then the paroxysmal torticollis in infancy of Snyder and the paroxysmal dystonia in infancy of Angelini may represent the lowest age spectrum of PNKD and a benign form of the disorder.
Some patients with benign paroxysmal torticollis of infancy come from kindreds with familial hemiplegic migraine linked to CACNA1A mutation, and after recovering from these episodes as they reach childhood, they might have migraines (Giffin et al., 2002). In fact, a family with individual members of the kindred having one or more of the following: paroxysmal tonic upgaze, benign paroxysmal torticollis of infancy, and episodic ataxia, with familial migraine and a CACNA1A mutation (as in EA-2) has been reported (Roubertie et al., 2008). The clinical picture of benign paroxysmal torticollis of infancy has been summarized to consist of attacks usually lasting less than a week, recurring from every few days to every few months, improving by age 2 years, and ending by age 3; there is very frequently a family history of migraine (Rosman et al., 2009).
Intrauterine cocaine exposure can be associated with multiple transient dyskinesias. Beltran and Coker (1995) described four infants who tested positive for cocaine metabolite at birth with subsequent transient dystonic reactions, beginning at 3 hours to 3 months of age and persisting for several months.
The clinical syndrome of transient paroxysmal dyskinesia of infancy appears distinct from the syndrome referred to as “benign paroxysmal tonic upgaze of childhood” (Ouvrier and Billson, 1988; Deonna et al., 1990; Echenne and Rivier, 1992; Campistol et al., 1993), which is a sustained tonic conjugate upward deviation of the eyes that begins in infancy and eventually disappears in childhood. This appears to be an autosomal dominant disorder (Guerrini et al., 1998). An infant with tonic upgaze was found to have a partial tetrasomy of 15q (Joseph et al., 2005). Ataxia may be present, and there can be clumsiness and delayed walking. The ocular deviations lessen in the morning hours and disappear with sleep. Acetazolamide is not effective; however, Campistol and colleagues (1993) reported levodopa to be effective. Perhaps the tonic upgaze with diurnal fluctuations would be a better term than “paroxysmal.” Not all cases of infantile transient tonic upgaze disturbance are benign, although the mean age of offset is 2.5 years (Hayman et al., 1998). Ouvrier and Billson (2005) published a recent review of this disorder. The family reported by Roubertie and colleagues (2008) suggests that tonic upgaze and transient torticollis and EA-2 may be related. A secondary cause of paroxysmal tonic upgaze has been reported: a 1-year-old girl with a hypomyelinating leukoencephalopathy, who presented in the neonatal period with episodes of sustained paroxysmal tonic upward gaze, roving eye movements, pendular nystagmus, and severe hypotonia, with the later appearance of pyramidal and extrapyramidal signs and no development (Blumkin et al., 2007).
Another paroxysmal ocular disorder, known as paroxysmal ocular downward deviation has been described in normal and brain-damaged infants (Yokochi, 1991; Miller and Packard, 1998). The ocular displacement was accompanied by closure of the upper eyelids, and the episode lasted seconds.
The syndrome of “benign myoclonus of infancy” can be mistaken for infantile spasms, but the benign EEG and clinical course allow for a clear distinction. The movements are sudden myoclonic and shuddering episodes of the head and shoulders (Lombroso and Fejerman, 1977; Fejerman, 1984). The jerks often repeat in a series; consciousness remains intact.
Another benign paroxysmal disorder in infants is spasmus nutans. It consists of a slow (2.4 Hz) tremor of the head, usually horizontal, and often an associated pendular nystagmus (Antony et al., 1980). It tends to disappear within 6 months and must be differentiated from congenital nystagmus (Fernandez-Alvarez, 1998).
Episodic (paroxysmal) ataxias and tremor
Intermittent ataxia can be due to metabolic defects such as Hartnup disease (Baron et al., 1956), pyruvate decarboxylase deficiency (Lonsdale et al., 1969; Blass et al., 1970, 1971), and maple syrup urine disease (Dancis et al., 1967). Fever often triggers the attacks of ataxia. In one case with pyruvate decarboxylase deficiency (Blass et al., 1971), choreoathetosis tended to accompany the chorea. Paroxysmal ataxia and dysarthria have also been reported to occur in multiple sclerosis (Andermann et al., 1959; Espir et al., 1966; DeCastro and Campbell, 1967; Miley and Forster, 1974; Gorard and Gibberd, 1989), which as remarked previously, is a disorder that also can cause paroxysmal choreoathetosis/dystonia. The attacks of paroxysmal ataxia due to multiple sclerosis last seconds, and are thus much shorter than the attacks described below. They also can respond to carbamazepine. Paroxysmal ataxia has also been reported in Behçet disease (Akmandemir et al., 1995).
A second family from the same region in North Carolina also had ocular motility problems. These were abnormal smooth pursuit with normal saccades, dampened opticokinetic nystagmus, inability to suppress the vestibulo-ocular reflex, gaze-evoked nystagmus, and episodic attacks of horizontal diplopia, oscillopsia, ataxia, nausea, vertigo, and tinnitus (Damji et al., 1996). This family’s disorder was considered part of the same neurologic disorder that is referred to as periodic vestibulo-cerebellar ataxia, like Farmer and Mustian’s family. Of special clinical significance is the lack of dysarthria. Genetic studies showed that the ataxia of this family is distinct from the two types of episodic ataxias (EA-1 and EA-2) that were previously characterized genetically (see below).
Hill and Sherman (1968) described another family, in which onset was in childhood in many of the affected members and there was no development of progressive ataxia. White (1969) described another family that experienced childhood onset and a benign course. All the families showed autosomal dominant inheritance.
An important advance was the discovery by Griggs and colleagues (1978) that acetazolamide can effectively prevent attacks. These authors showed this benefit in one kindred with familial paroxysmal ataxia. The following year, Donat and Auger (1979) had similar results in another kindred. Fahn (1983, 1984) reported a woman who had paroxysmal tremor, both intention and resting, associated with ataxia and postural instability during the attack; acetazolamide eliminated the attacks. Factor and colleagues (1991) reported an infant who had three attacks of coarse tremor and an orofacial dyskinesia that resembled that seen with tardive dyskinesia. Each attack lasted several hours before spontaneously clearing. Tetrahydrobiopterin, the cofactor for the enzymes tyrosine hydroxylase and phenylalanine hydroxylase, was reduced. The child responded to levodopa.
Mayeux and Fahn (1982) reported a patient with PNKD in a background of hereditary ataxia. Onset of PNKD was at age 10; onset of ataxia was at age 19. During an attack, which could last from 10 minutes to 4 hours, there was an accompanying increase of ataxia. Initially there was an 8-month response to acetazolamide. After the drug was no longer effective, the patient’s PNKD responded to clonazepam. This patient might be a link between familial PNKD and paroxysmal ataxia.
Several other reports of acetazolamide-responsive familial paroxysmal ataxia have been reported (Aimard et al., 1983; Zasorin et al., 1983; Koller and Bahamon-Dussan, 1987). Although computed tomography (CT) has been normal, magnetic resonance imaging (MRI) studies have revealed selective atrophy in the anterior cerebellar vermis (Vighetto et al., 1988).
Families with a combination of periodic ataxia and persistent, continuous electrical activity in several muscles, reported either as myokymia (Van Dyke et al., 1975; Hanson et al., 1977; Gancher and Nutt, 1986; Brunt and Van Weerden, 1990) or as neuromyotonia (Vaamonde et al., 1991), have been described. Description of the attacks, which are brief and are sometimes preceded by sudden movement, include dyskinetic movements and sustained posturing as well as ataxia, dysarthria, and vertigo. This type of paroxysmal ataxia is now called episodic ataxia 1 (EA-1).
In 1986, Gancher and Nutt (1986) classified the hereditary episodic ataxias into three syndromes. In one group are those cases associated with persistent myokymia or neuromyotonia (now called EA-1). They described the attacks being precipitated by fatigue, excitement, stress, and physical trauma, but the family reported by Vaamonde and colleagues (1991) had attacks triggered by sudden movement, and kinesigenicity is now recognized as a feature. There is no dizziness or vertigo. The attacks last 2 minutes or less. Acetazolamide and anticonvulsants are usually ineffective. The gene for this type of paroxysmal ataxia has been located at chromosome 12p13 (Litt et al., 1994).
The second group (now known as EA-2) is featured by attacks of ataxia (with or without interictal nystagmus and with or without persistent ataxia), responding to acetazolamide or amphetamines. The attacks are precipitated by exercise, fatigue, or stress and occasionally by carbohydrate or alcohol ingestion. In addition to ataxia, the attacks are accompanied by vertigo, headache, nausea, and malaise. The attacks last for several hours or until the patient falls asleep. In recent years, additional families have been reported with these features (Bain et al., 1991; Baloh and Winder, 1991; Hawkes, 1992). The siblings reported by Bain and colleagues (1991) had persistent diplopia due to superior oblique paresis as part of the syndrome. Using [31P] nuclear magnetic resonance spectroscopy, Bain and his colleagues (1992) found the pH levels in the cerebellum to be increased in untreated subjects with acetazolamide-responsive paroxysmal ataxia; the pH dropped to normal with treatment. The gene for this type of paroxysmal ataxia has been mapped to chromosome 19p13 (von Brederlow et al., 1995; Vahedi et al., 1995).
Gancher and Nutt (1986) listed a third group, which is kinesigenic. Typical PKD can occur in some members of the family. The attacks of ataxia last minutes to hours, whereas the PKD lasts seconds. The disorder can resolve with time. Acetazolamide appears to be ineffective, but phenytoin is effective for both the kinesigenic ataxia and the PKD. In their review, Griggs and Nutt (1995) place this third type with associated PKD in the first group of paroxysmal ataxias. Genotyping has now definitively placed this as EA-1 (Nutt and Gancher, 1997).
A case was reported in which a young girl had attacks of ataxia associated with fevers and accompanied by vertical supranuclear ophthalmoplegia (Nightingale and Barton, 1991). The ataxia and eye findings can last days.
Miscellaneous paroxysmal disorders
Keane (1984) reported two patients with post-traumatic periodic, rhythmic movements of the tongue. The attacks occurred about every 20 seconds and each attack lasted 10 seconds. They consisted of three undulations per second. Eventually, the movements diminished. Other cases of episodic lingual dyskinesias were associated with epilepsy (Jabbari and Coker, 1981) and pontine ischemia (Postert et al., 1997).
A few paroxysmal dyskinesias are mentioned here, but are not discussed further. These are Sandifer syndrome (prolonged head tilting in children following eating, due to gastroesophageal reflux) (see the review by Menkes and Ament, 1988); hyperekplexia (excessive startle syndrome with complex movements) (see the review by Andermann and Andermann, 1986 and Chapter 20); stereotypy (Duchowny et al., 1988; Tan et al., 1997), and paroxysmal bursts of myoclonus and tics. Classically, stereotypy, myoclonus, and tics are each recognized as a specific class of movement disorders, and they characteristically present as paroxysmal bursts of their type of movement. As a result, their discussion should be separated from discussion of conditions labeled as “paroxysmal.” Shuddering attacks in children (Holmes and Russman, 1986) are brief bursts of rapid shivering-like movements of the head and arms, occurring up to 100 times per day; they can begin in infancy or in older children, and they resolve over time. The attacks lasts several seconds without impairment of consciousness. The frequency of shuddering movements as seen on EMG or EEG was similar to that of essential tremor (Kanazawa, 2000).
Classification of the paroxysmal dyskinesias
Because the various movement phenomena in any of the paroxysmal dyskinesias can vary from chorea/ballism to the sustained contractions of dystonia in any given patient, Demirkiran and Jankovic (1995) suggested that these disorders be labeled with the more generic names paroxysmal kinesigenic dyskinesia (PKD), paroxysmal nonkinesigenic dyskinesia (PNKD) (whether short-lasting or long-lasting), and paroxysmal exertion-induced dyskinesia (PED) regardless of the duration of the attack. This classification system is used here. But the paroxysmal dyskinesias that last seconds and might not be induced by sudden movement are also included in the PKD category because of their otherwise similar short duration of attacks and response to anticonvulsants.
The highlights of various categories of paroxysmal dyskinesias are presented in outline format in Table 22.3.
| A. Paroxysmal kinesigenic dyskinesia (PKD) |
Paroxysmal kinesigenic dyskinesia (PKD)
Clinical features

Kinast and colleagues (1980) reported as Case 4 a patient with typical brief dyskinesias occurring many times a day, preceded by paresthesias, and responding to anticonvulsants but not induced by sudden movement. Although this was technically not PKD, because sudden movements did not trigger any attacks, the clinical features otherwise resemble the attacks that are seen in PKD. As was mentioned above, this type of paroxysmal dyskinesia is placed in this category of PKD. Since the attacks of PNKD are usually so prolonged and because they do not ordinarily respond to anticonvulsants, this case does not fit into the PNKD category. See the discussion on the classification of these brief attacks as symptomatic PKD below.
Plant (1983) emphasized the focal and unilateral nature of PKD in many patients (Table 22.4).
Table 22.4 Laterality of types of paroxysmal kinesigenic dyskinesia (Plant, 1983)
| Laterality of attacks | No. |
|---|---|
| Unilateral – one side only | 25 |
| Unilateral – either side | 12 |
| Unilateral and bilateral | 11 |
| Bilateral only | 22 |
| Not stated | 3 |
| Total | 73 |
Primary paroxysmal kinesigenic dyskinesia
The etiology of most case reports of PKD has been idiopathic and predominantly hereditary, inheritance being autosomal dominant. For some unexplained reason males are more often affected than females, with a ratio of 3.75 : 1 (75 males and 20 females reported in Fahn, 1994). A large series of 150 cases were reported from a questionnaire in Japan. This gender imbalance was supported by the additional 26 cases reported by Houser and colleagues (1999), consisting of 23 men and 3 women, and another 150 case from Japan (Nagamitsu et al., 1999). Adding all these cases together brings the total to 218 men and 53 women or a ratio of 4.1 : 1. Age at onset shows a wide range, usually starting in childhood between the ages of 6 and 16 years, but can range from 6 months to 40 years (Frucht and Fahn, 1999; Li et al., 2005). Excluding the cases by Nagamitsu and colleagues (1999), the mean age at onset is 12 years; the median age is also 12 years. Familial cases might be more common among the Japanese (Kishimoto, 1957; Fukuyama and Okada, 1967; Kato and Araki, 1969) and Chinese (Jung et al., 1973). The survey reported by Nagamitsu and colleagues (1999) found 53 sporadic cases and 97 familial ones.
There is one report of PKD developing in a patient who had essential tremor (Nair et al., 1991). EEGs are generally normal, and CT scans are also normal (Goodenough et al., 1978; Kinast et al., 1980; Suber and Riley, 1980; Bortolotti and Schoenhuber, 1983; Lou, 1989) with a few exceptions, such as the case reported by Watson and Scott (1979) with suggested brainstem atrophy, and the one by Gilroy (1982) with an ill-defined unilateral hemispheric lesion. However, Hirata and colleagues (1991) demonstrated an abnormal EEG with rhythmic 5 Hz discharges over the entire scalp during episodes of PKD, raising the possibility that the PKD might have an epileptogenic basis. A patient was reported who developed PKD shortly after initiation of therapy with methylphenidate for attention-deficit hyperactivity disorder. Attacks persisted long after methylphenidate was discontinued and responded to treatment with carbamazepine (Gay and Ryan, 1994). The authors believe that the patient had a hereditary susceptibility for PKD that was triggered by the drug.
The attacks tend to diminish with age. Fortunately, PKD responds dramatically to anticonvulsants. The early literature indicates that phenytoin was the most popular, followed by phenobarbital and primidone. Carbamazepine appears to be the drug that is most commonly used. Valproate has also been effective (Suber and Riley, 1980), although Hwang and colleagues (1998) report that both carbamazepine and phenytoin were superior to valproate. Other anticonvulsants are effective also, including oxcarbazepine (Gokcay and Gokcay, 2000; Tsao, 2004; Chillag and Deroos, 2009), lamotrigine (Pereira et al., 2000; Uberall and Wenzel, 2000), levetiracetam (Chatterjee et al., 2002), and topiramate (Y.G. Huang et al., 2005). There is one report where high dosage of only phenytoin was effective (Bonakis et al., 2009). There is a report of response to levodopa (Loong and Ong, 1973), but another report of lack of effect with this drug (Garello et al., 1983). Analogously, there is one report of three patients with PKD worsening with haloperidol (Przuntek and Monninger, 1983), but Garello and colleagues (1983) reported no effect from this drug (as well as levodopa) in two brothers. The calcium channel blocker flunarizine, which is also a neuroleptic, was effective in a 7-year-old girl, who did not respond to carbamazepine or methylphenidate (Lou, 1989).
Homan and colleagues (1980) reported that children with PKD need doses of phenytoin that are similar to those used to treat epilepsy, whereas adults can respond to lower doses. These authors also describe a patient who might have had interictal chorea (the patient was described as fidgety) and suggested that this might represent a possible link to benign hereditary chorea. However, the fidgetiness that was described might not have been chorea. Perhaps these movements might be interictal myoclonus, similar to the two patients with PKD recently reported (Cochen De Cock et al., 2006). Another report of interictal movements was by Bird and colleagues (1978) in which a woman had anxiety-induced dystonia/choreoathetosis and random, adventitious small jerky movements when she was not having an attack (first reported by Perez-Borja and colleagues 1967); her daughter had delayed milestones and persistent choreoathetosis. However, it seems likely that the daughter’s choreoathetosis is not idiopathic but secondary, so it seems that paroxysmal dyskinesias and hereditary benign chorea should not be linked on the basis of a couple of these cases.
The pathophysiology of PKD is still unclear, and its relationship with epilepsy remains speculative. Because movement-induced seizures can occur (e.g., the case of Falconer et al., 1963) and because PKD responds dramatically to anticonvulsants, these are not sufficient reasons to consider PKD a form of epilepsy. The retention of consciousness and lack of postictal phenomena, as well as the presence of dystonia and choreoathetosis, should be sufficient to disqualify PKD from the epilepsies. However, Beaumanoir and colleagues (1996) described a boy with PKD and normal EEGs and consciousness, who later had a longer attack with clouding of consciousness and recording of postictal abnormalities on the EEG to support the diagnosis of reflex epilepsy. There is an emerging syndrome of infantile epilepsy, referred to as the ICCA syndrome, followed by childhood paroxysmal dyskinesias, as was mentioned earlier in the chapter (Lee et al., 1998; Thiriaux et al., 2002).
Franssen and colleagues (1983) investigated the contingent negative variation in one patient with PKD. Contingent negative variation is a slow cerebral potential that follows a warning stimulus, which prepares the subject to expect an imperative stimulus requiring a decision or motor response. The slow negative wave component of the contingent negative variation was more pronounced than that in control subjects. It returned to normal after phenytoin treatment. Mir and colleagues (2005) later demonstrated reduced intracortical inhibition, reduced early phase transcallosal inhibition, and reduced first phase of spinal reciprocal inhibition in PKD patients; treatment with carbamazepine normalized the abnormality of transcallosal inhibition.
The differential diagnosis of PKD is focal epilepsy, tetany, hyperekplexia, tics, stereotypies, and hysteria, as was noted in the misdiagnosis of the case reported by Waller (1977). The clinical features are so distinctive, particularly if triggered by sudden movement, that there is little likelihood of not diagnosing the condition correctly once one is aware of its existence. Similarly, the markedly effective response to anticonvulsants sets PKD apart from the other disorders. One case of primary PKD has been observed to be the result of a consistent ictal discharge arising focally from the supplementary sensorimotor cortex, with a concomitant discharge recorded from the ipsilateral caudate nucleus without spread to other neocortical areas (Lombroso, 1995), which suggests that some primary PKDs could be epileptic in origin. The nonkinesigenic, brief attacks of hemidystonia, often precipitated by hyperventilation, and controlled with anticonvulsants, has been considered a sign of epilepsy (Kotagal et al., 1989; Newton et al., 1992). So each case of such suspected nonkinesigenic paroxysmal dyskinesia needs to be evaluated for a convulsive disorder.
In addition to the cases of PKD described in the historical highlights earlier in the chapter, a number of other reports should be cited to make the review complete (Zacchetti et al., 1983; Boel and Casaer, 1984; Lang, 1984). One case of paroxysmal torticollis induced by sudden movement or stimulus appeared 5 years after the onset of classic spasmodic torticollis that had begun at age 20 years (Lagueny et al., 1993). After failing to respond to alcohol, tiapride, haloperidol, carbamazepine, clobazam, and valproate, the patient was treated effectively with injections of botulinum toxin.
There have been reports of two autopsies in PKD. Case 4 of Kertesz (1967) died, apparently by suicide, and a postmortem examination revealed no clear-cut abnormality in brain, just the presence of some melanin pigment in macrophages in the locus coeruleus. Stevens (1966) had earlier reported the postmortem findings of one of his patients, which were also essentially normal, showing only a slight asymmetry of the substantia nigra.
SPECT scans measuring cerebral blood flow were studied in two children with PKD, revealing increased perfusion in the contralateral basal ganglia at the onset of an attack in one (Ko et al., 2001) and in the contralateral thalamus in the other (Shirane et al., 2001). Physiologic studies indicate that there is surround inhibition in PKD (Shin et al., 2010).
Three independent laboratories have mapped autosomal dominant PKD with infantile convulsions (ICCA syndrome) to chromosome 16p11.2–q12.1 (Tomita et al., 1999; Bennett et al., 2000; Swoboda et al., 2000) and labeled EKD1 (episodic kinesigenic dyskinesia 1). A second locus on this chromosome at 16q13–q22.1 has been found in other families, referred to as EKD2 (Valente et al., 2000). This locus does not overlap with those of the other families. This family is distinct from the others by not having infantile convulsions. It has been given the designation of DYT19. Evidence for a third locus, called EKD3, has been suggested because three families did not map to the two known ones on chromosome 16 (Spacey et al., 2002).
A familial atonic form of PKD has been reported (Fukuda et al., 1999).
Secondary paroxysmal kinesigenic dyskinesia
The overwhelming majority of reported cases of PKD are idiopathic or familial. Although not reported as often, symptomatic PKD is probably more common. Table 22.5 lists the most common causes of symptomatic PKD, the most common being associated with multiple sclerosis and head injury. Pseudohypoparathyroidism has recently been added to this list (C.W. Huang et al., 2005). In one family with X-linked mutations in the thyroid hormone transporter gene MCT8, paroxysmal dyskinesias accompanied global retardation (Brockmann et al., 2005).
In symptomatic PKD, like primary PKD, attacks that last seconds are sometimes induced not by sudden movement but by hyperventilation. These also usually respond to anticonvulsants, such as carbamazepine, and are also seen in multiple sclerosis (Verheul and Tyssen, 1990; Sethi et al., 1992). Fahn (unpublished) has encountered a patient who had attacks lasting seconds, induced by hyperventilation and without being induced by sudden movement, following a mild cerebral ischemic episode, that also responded to carbamazepine. These pharmacologic responses suggest that the briefness of the attack is more important than the sudden movement to distinguish the classification of the paroxysmal dyskinesias. Sethi and colleagues (1992) reported success in treating three patients with paroxysmal dystonia (not induced by movement, but triggered by hyperventilation and lasting many seconds) with acetazolamide with or without a combination of carbamazepine.
Multiple sclerosis
Although few of the paroxysmal dyskinesias that are associated with multiple sclerosis are triggered by sudden movement, an occasional patient with multiple sclerosis will manifest typical PKD (Matthews, 1958). In fact, the presenting symptom of multiple sclerosis can be PKDs as in the case reported by Roos and colleagues (1991); these attacks were associated with a lesion in the caudate nucleus and responded to phenytoin. In three of the eight patients reported by Berger and colleagues (1984) with paroxysmal dyskinesia associated with multiple sclerosis, the attacks were induced by sudden movement; they were relieved by anticonvulsants. The patient with PKD with multiple sclerosis reported by Burguera and colleagues (1991) had a lesion in the left thalamus demonstrated by MRI. The PKD was the presenting symptom, as in other cases of demyelinating disease. Gatto and colleagues (1996) reported medullary lesions in multiple sclerosis and bilateral paroxysmal dystonia.
Head trauma
Case 3 of Whitty and colleagues (1964) was a 13-year-old boy with onset 9 months after mild head trauma. Robin (1977) reported a 33-year-old man with severe head injury who developed PKD 8 months later. In two of the three cases of post-traumatic paroxysmal dyskinesias reported by Drake and colleagues (1986), the movements were induced by sudden movement of the affected body part. Richardson and colleagues (1987) reported another post-traumatic case. These post-traumatic cases of PKD responded to anticonvulsants, similar to idiopathic PKD. Attacks of dystonia lasting several seconds and induced by tactile stimulation were reported secondary to a head injury; they disappeared within 2 months without treatment (George et al., 1990). Nijssen and Tijssen (1992) reported another case of tactile-induced dyskinesias as a result of a thalamic infarct.
Perinatal hypoxic encephalopathy
Rosen (1964) appears to have been the first to report a case of PKD associated with perinatal hypoxic encephalopathy, with the onset at age 12. This boy’s attacks were usually triggered by a combination of startle and body contact. Mushet and Dreifuss (1967) described a 9-year-old boy who developed brief attacks of athetosis and dystonia. They usually occurred when he was startled but also could occur following sudden movement. At age 6 months, he had a febrile illness, which was retrospectively thought to be encephalitis. He had considerable motor regression and was not able to walk, nor did he gain syntactical speech. His dyskinetic attacks were not suppressed by anticonvulsants but did respond to anticholinergics.
Basal ganglia calcifications
PKD has been reported to occur with basal ganglia calcifications with or without hypoparathyroidism (Arden, 1953). Subsequent cases of hypoparathyroidism were reported (Tabaee-Zadeh et al., 1972; Barabas and Tucker, 1988). The clinical syndrome resembles that of primary infantile convulsions and childhood PKD (Hattori and Yorifuji, 2000). Calciferol was effective in controlling these attacks. The case reported by Soffer and colleagues (1977) was not noted to have the attacks induced by sudden movement, but the briefness of the attack resembles that of PKD.
Hemiatrophy
Case 2 of the five cases described by Kinast and colleagues (1980) had attacks of left hemidystonia lasting 1 minute and occurring up to 50 times a day. The major precipitating factor was not sudden movement, but stress and the anticipation of movement (also reported in one patient by Franssen et al., 1983). Technically, like their Case 4 described above, this patient does not fulfill the criterion of attacks induced by sudden movement. This is another example, because of the brief duration, the frequency of the attacks, and their response to phenytoin, that otherwise resembles PKD, and again points out why such nonkinesigenic cases are placed under the PKD rubric in the classification scheme used here. Examination of this revealed left-sided hemiatrophy and hyperreflexia with a normal CT scan. Because the hemiatrophy syndrome can be associated with a delayed-onset movement disorder (Buchman et al., 1988), it seems reasonable to consider it an etiologic factor in this particular case.
Gilroy (1982) reported a 32-year-old man with an abnormal right hemisphere on CT scan who had multiple daily brief attacks of left hemidystonia present since the age of 5 that were typical of PKD. The speculation is that the PKD was secondary to pathology in the involved hemisphere. An arteriogram and cortical biopsy did not shed further light on the pathology.
Cerebral infarcts and hemorrhages
With the advent of magnetic resonance imaging, more cases of PKD have been reported as a result of cerebral infarcts with putaminal infarct (Merchut and Brumlik, 1986), thalamic infarct (Video 22.4) (Camac et al., 1990; Nijssen and Tijssen, 1992; Milandre et al., 1993), an infarct probably in the cortex (Fuh et al., 1991), and medullary hemorrhage (LeDoux, 1997). As was mentioned above, the case of Nijssen and Tijssen (1992) had attacks stimulated by touch of the affected limb. The attacks secondary to infarcts (Merchut and Brumlik, 1986; Fuh et al., 1991) or to multiple sclerosis (Sethi et al., 1992) can be painful tonic spasms. Riley (1996) reported a patient who had paroxysmal attacks of tightening of his throat muscles and elevation of his tongue to the roof of his mouth associated with a remote hemorrhage in the medulla. Moyamoya disease has been reported to cause PKD (and PNKD) (Gonzalez-Alegre et al., 2003). 
Other etiologies
PKD has also been reported to occur in a patient with progressive supranuclear palsy (Adam and Orinda, 1986), in hyperglycemia in the presence of a lenticular vascular malformation (Vincent, 1986), in subacute sclerosing panencephalitis (Ondo and Verma, 2002), and in spinal cord lesion (Cosentino et al., 1996). A case of spinal cord glioma was associated with paroxysmal kinesigenic segmental myoclonus (Marrufo et al., 2007). Another case of spinal cord compression was associated with PKD (Yulug et al., 2008). A case of PKD was reported in which it was the first symptom in a patient who developed Huntington disease (Scheidtmann et al., 1997). HIV infection has been reported to be associated with PKD and PKND (Mirsattari et al., 1999). A lesion in the medulla has been associated with PKD (Jabbari et al., 1999). Peripheral trauma to a foot was subsequently followed by paroxysmal hemidystonic episodes (Chiesa et al., 2008). An adult with PKD and cramp-fasciculation syndrome was associated with voltage-gated potassium channel-complex protein antibody encephalitis, with the patient responding to plasma exchange and intravenous immunoglobulin (Aradillas and Schwartzman, 2011).
Paroxysmal nonkinesigenic dyskinesia (PNKD)
Clinical features
As with PKD, the attacks of PNKD consist of any combination of dystonic postures, chorea, athetosis, and ballism. They can be unilateral – always on one side or on either side – or bilateral. Unilateral episodes can be followed by a bilateral one. They can affect a single region of the body or be generalized. Involvement of the neck can be a combination of torticollis and head tremor (Hughes et al., 1991). The major distinctions from PKD are the longer duration of each attack, the smaller frequency of the attacks, and a host of different aggravating factors for the attacks. The attacks last minutes to hours, sometimes longer than a day. Usually, they range from 5 minutes to 4 hours (Video 22.5). They are primed by consuming alcohol, coffee, or tea; by psychological stress or excitement; and by fatigue. There are usually no more than three attacks per day, and attacks may be months apart. The attacks can be severe enough to cause a patient to fall down. Speech is often affected, with inability to speak due to dystonia, but there is never any alteration of consciousness. The attacks can sometimes be aborted if the patient goes to sleep. As with PKD, patients very often report variable sensations at the beginning of the paroxysms. These can consist of paresthesias, a feeling of stiffness, crawling sensations, or a tense feeling. 
A form of PNKD, known as intermediate PDC, and more recently as paroxysmal exertion-induced dyskinesia, is triggered only by prolonged exercise and not the other precipitants. This was first described by Lance (1977) and subsequently reported in another family by Plant and colleagues (1984) and in a sporadic case by Nardocci and colleagues (1989). Under the classification scheme of Demirkiran and Jankovic (1995), this form, which is discussed separately, is called paroxysmal exertional dyskinesia (PED).
Primary paroxysmal nonkinesigenic dyskinesia (Mount–Reback syndrome)
The initial reports of PNKD were familial (Mount and Reback, 1940; Forssman, 1961; Lance, 1963; Weber, 1967; Richards and Barnett, 1968; Lance, 1977; Horner and Jackson, 1969; Tibbles and Barnes, 1980; Walker, 1981; Mayeux and Fahn, 1982; Przuntek and Monninger, 1983; Jacome and Risko, 1984), with hereditary transmission being autosomal dominant. In 1980 Kinast and colleagues (Case 4) and Dunn in 1981 each described a child with PNKD without a positive family history. Bressman and colleagues (1988) later described seven sporadic cases of PNKD, and Nardocci and colleagues (1989) added another one. The familial cases of idiopathic PNKD still greatly outnumber sporadic cases, according to the reports in the literature. However, the sporadic cases are much more difficult to diagnose, and they have the difficulty of the need to be differentiated from a psychogenic etiology (Bressman et al., 1988; Fahn and Williams, 1988). On the basis of the experience of Bressman and her colleagues (1988), the sporadic form may actually be more common than the familial form but is just rarely reported.
For some unexplained reason males are slightly more often affected than females, with a ratio of 1.4 : 1 (32 males and 23 females reported in the reviewed English language literature) (see Fahn, 1994). Age at onset shows a wide range, usually in childhood between the ages of 6 and 16 years, but can range from 2 months to 30 years. The mean age at onset is 12 years; the median is also 12 years. CT scans are normal (Mayeux and Fahn, 1982; Jacome and Risko, 1984).
The EEGs are generally normal, but the case of Jacome and Risko (1984) may be of interest. The patient had unilateral PNKD and had normal interictal EEGs. Photic stimulation at low frequencies induced paroxysmal lateralized epileptiform discharges from the contralateral hemisphere. From this the authors suggest that the disorder might have some epileptogenic basis.
Sleep aborted the episodes in one family that had myokymia in addition to the PNKD (Byrne et al., 1991). The presence of myokymia links this particular family to several with paroxysmal ataxia, in which myokymia is a feature (Van Dyke et al., 1975; Vaamonde et al., 1991).
A family reported by Kurlan and colleagues (1987) had some atypical features for classic PNKD. The long-duration attacks were painful dystonic spasms that were not precipitated by alcohol, caffeine, or excitement, but could follow exposure to cold or heat or result from exertional cramping. Other members of the family had only exertional cramping without PNKD. The authors suggested that exertional cramping might be a forme fruste of PNKD. It is also possible that the PNKD in this family falls into the category of the intermediate form of paroxysmal dyskinesia that was reported by Lance (1977) and Plant and colleagues (1984). Also unusual was the presence of some fixed dystonia, which had not been reported previously. Bressman and colleagues (1988) also described some sporadic cases of PNKD, in which the patients had some interictal dystonia.
Lance (1977) mentioned that autopsies performed on two patients with PNKD revealed no pathology. His Case II.4 had normal macroscopic findings. His Case IV.2 died of sudden infant death syndrome; macroscopic and microscopic findings were normal.
The attacks can diminish spontaneously with age (Lance, 1977; Kinast et al., 1980; Bressman et al., 1988). Unfortunately, most patients have persistence of their attacks, and they are difficult to treat. As a general rule, PNKD does not respond to the same type of anticonvulsants that so effectively treat PKD. An occasional patient will respond to such agents as carbamazepine, valproate, and gabapentin (Chudnow et al., 1997). Clonazepam, as introduced for PNKD by Lance (1977), appears to be the most successful agent, for both for primary PNKD and symptomatic PNKD. A number of other drugs have been tried, sometimes with success. These include antimuscarinics (Mount and Reback, 1940), chlordiazepoxide (Perez-Borja et al., 1967; Walker, 1981), acetazolamide (Mayeux and Fahn, 1982; Bressman et al., 1988), oxazepam and other benzodiazepines (Kurlan and Shoulson, 1983; Kurlan et al., 1987), sublingual lorazepam (Dooley and Brna, 2004), and L-tryptophan (Kurlan et al., 1987).
Kurlan and Shoulson (1983) treated one patient with familial PNKD on alternate-day oxazepam. He had marked benefit from diazepam but only for 4 weeks. Clonazepam and oxazepam gave relief for 2–3 weeks each. Eventually he was placed on a regimen of 40 mg oxazepam given on alternate days. The concept was that the benzodiazepine receptors became desensitized on daily doses. Alternate-day administration prevented this desensitization.
Przuntek and Monninger (1983) and Coulter and Donofrio (1980) carried out trials of the dopamine receptor antagonist haloperidol and reported benefit. In the obverse, Przuntek and Monninger (1983) found that levodopa worsened one patient.
Levetiracetam was found helpful in one family (Szczałuba et al., 2009).
Chronic stimulation of the ventral intermediate nucleus (VIM) of the thalamus was effective in reducing the frequency, duration, and intensity of attacks in one patient with PNKD (Loher et al., 2001). At least one patient with PNKD has been treated successfully with deep brain stimulation of the globus pallidus interna (Yamada et al., 2006).
In contrast to idiopathic PKD, which is so distinctive, the major difficulty in the diagnosis of sporadic PNKD is to differentiate it from a psychogenic movement disorder. The problem is that the disappearance of the movements with placebo or psychotherapy could be coincidental since the attacks disappear spontaneously. However, if the paroxysms are frequent and the attacks are prolonged, then repeated trials with placebo can be informative. If such trials consistently produce remissions, then one can be convinced that the diagnosis is a psychogenic disorder (see Chapter 25).
Three different genetic mappings have been made for PNKD. The first type, originally described by Mount and Reback (1940), is referred to genetically as familial paroxysmal dyskinesia type 1 (FPD1) and was mapped to 2q33–q35 (Fink et al., 1996; Fouad et al., 1996; Hofele et al., 1997; Matsuo et al., 1999). One of these families (Przuntek and Monninger, 1983; Hofele et al., 1997) shows a fair response to diazepam. The gene for this disorder has been identified as myofibrillogenesis regulator 1 (MR1) (Lee et al., 2004). Bruno and colleagues (2007) analyzed 14 families with PNKD and 8 had MR1 mutations. Patients with PNKD with MR1 mutations had their attack onset in youth (infancy and early childhood). Typical attacks consisted of a mixture of chorea and dystonia in the limbs, face, and trunk, and typical attack duration lasted from 10 minutes to 1 hour. These attacks resembled the phenotype presented by Mount and Reback (1940). Caffeine, alcohol, and emotional stress were prominent precipitants. Attacks had a favorable response to benzodiazepines, such as clonazepam and diazepam. Attacks in families without MR1 mutations were more variable in their age at onset, precipitants, clinical features, and response to medications. Several were induced by persistent exercise. A Serbian family with PKND and the MR1 mutation had similar clinical features, with attacks starting in the first 6 months of life (Stefanova et al., 2006). The frequency and severity of attacks showed an age-dependent incremental-decremental pattern with a peak between 13 and 15 years of age. Some of the non-MR1 affected patients with PNKD are suspected of having paroxysmal exertional dyskinesia (Bruno et al., 2007).
The second type of PNKD has additional clinical features, including perioral paresthesias, double vision, and headache during attacks, and some also have a constant spastic paraparesis; this type was mapped to chromosome 1p (Auburger et al., 1996) and has been called choreoathetosis/spasticity episodica (CSE). The locus is very close to the SLC2A1 gene, suggesting that the family might be positive for a GLUT1 mutation. The third type of PNKD has been seen in familial infantile convulsions, in which the gene has been mapped to the pericentromeric region of chromosome 16 (Szepetowski et al., 1997). This has been called infantile convulsions with paroxysmal choreoathetosis (ICCA). The gene has been identified as the sodium/glucose cotransporter (Roll et al., 2002). A fourth type is linked to chromosome 2q31 and has been designated PNKD2 and also listed with the dystonias as DYT20. A fifth type is a family with PNKD and epilepsy, found to be due to a mutation in the Ca-sensitive potassium channel gene on chromosome 10q22 (KCNMA1 gene) (Du et al., 2005). Further discussion about the genetics of the paroxysmal dyskinesias is in a later section in this chapter.
Secondary paroxysmal nonkinesigenic dyskinesia
The overwhelming majority of reported cases of PNKD are idiopathic or familial in etiology, but a number of cases of symptomatic PNKD have been reported. Table 22.6 lists the most common causes of symptomatic PNKD.
The most common cause of symptomatic PNKD, just like PKD, is multiple sclerosis (Matthews, 1958; Joynt and Green, 1962; Lance, 1963; Berger et al., 1984; Verheul and Tyssen, 1990; Sethi et al., 1992). In multiple sclerosis, the paroxysmal movements may only be ocular, lasting several minutes (MacLean and Sassin, 1973). One patient with paroxysmal hemidystonia associated with multiple sclerosis later developed psychogenic PNKD (Morgan et al., 2005). Another patient had paroxysmal laughter in addition to paroxysmal dyskinesias (Aguirregomozcorta et al., 2008).
The next two most common causes are perinatal encephalopathy (Lance, 1963; Erickson and Chun, 1987; Bressman et al., 1988) and psychogenic (Video 22.6) (Bressman et al., 1988; Fahn and Williams, 1988; Lang, 1995). A longer discourse on psychogenic PNKD is presented in Chapter 25. 
Other causes of PNKD are encephalitis (Video 22.7) (Mushet and Dreifuss, 1967; Bressman et al., 1988), cystinuria (Cavanagh et al., 1974), succinic semialdehyde dehydrogenase deficiency (improved with vigabatrin) (Leuzzi et al., 2007), hypoparathyroidism (Soffer et al., 1977; Yamamoto and Kawazawa, 1987; Dragasevic et al., 1997), pseudohypoparathyroidism (Prashantha and Pal, 2009), basal ganglia calcifications without altered serum calcium (Micheli et al., 1986), thyrotoxicosis (Fischbeck and Layzer, 1979), transient ischemic attacks (Margolin and Marsden, 1982; Bennett and Fox, 1989) including basilar ischemia causing episodic tongue and alternating limb movements (Li and Lee, 2009), infantile hemiplegia (Huffstutter and Myers, 1983), head trauma (Perlmutter and Raichle, 1984; Drake et al., 1986), hypoglycemia (Newman and Kinkel, 1984; Winer et al., 1990; Schmidt and Pillay, 1993), AIDS (Nath et al., 1987), diabetes (Haan et al., 1988), anoxia (Bressman et al., 1988), brain tumor (Bressman et al., 1988), hypoglycemia induced by an insulinoma (Shaw et al., 1996; Debruyne et al., 2009), poststreptococcal autoimmune neuropsychiatric disease (PANDAS) (Dale et al., 2002; Senbil et al., 2008), celiac disease (Hall et al., 2007), Sjögren syndrome (Alonso-Navarro et al., 2009), and the Allan–Herndon–Dudley syndrome, which is an X-linked recessive disorder due to a mutation in the monocarboxylate transporter 8 (MCT8) gene resulting in decreased transport of thyroid hormone into neurons (Fuchs et al., 2009). The patient with AIDS (Nath et al., 1987) had two attacks of dystonia, but details are lacking in regard to the duration or characteristics of the attacks. Moyamoya disease has been reported to cause PNKD (and PKD) (Gonzalez-Alegre et al., 2003). 
There is a case report of one person who developed paroxysmal hemidystonia after starting fluoxetine, which cleared on discontinuing the drug (Dominguez-Moran et al., 2001). Related to transient ischemic attacks mentioned above is the case of one patient who had paroxysmal hemidystonia precipitated by assuming an upright position after sitting or lying, associated with occlusion of the contralateral internal carotid artery (ICA) and near-total occlusion of the ipsilateral internal carotid artery (Sethi et al., 2002). The authors called this orthostatic paroxysmal dystonia. A SPECT study demonstrated decreased perfusion in the contralateral frontoparietal cortex during the typical dystonic spell. PNKD has now been reported in the antiphospholipid syndrome (Engelen and Tijssen, 2005). A series of 17 cases of secondary paroxysmal dyskinesias was reported by Blakeley and Jankovic (2002), who found that 9 patients had PNKD, 2 patients had PKD, 5 patients had mixed PKD/PNKD, and 1 patient had PHD.
Micheli and colleagues (1987) reported an interesting case of a youth with learning disability who had received dopamine receptor-blocking drugs since the age of 3. At age 16 he developed paroxysmal dystonia. The attacks could be precipitated by stress but not by movement, caffeine, cold, fatigue, or hyperventilation. The episodes lasted from 30 minutes to 3 hours. They did not respond to anticonvulsants but were abolished with trihexyphenidyl 20 mg/day. It is possible that the PNKD in this youth represented a variant of tardive dystonia (Burke et al., 1982; Kang et al., 1986).
A PET study was performed on one patient with post-traumatic paroxysmal hemidystonia (Perlmutter and Raichle, 1984). Decreased oxygen metabolism, decreased oxygen extraction, increased blood volume, and increased blood flow in the contralateral basal ganglia were found.
The syndrome commonly known as “alternating hemiplegia of childhood” typically contains periods of prolonged dystonic attacks, along with other elements of the syndrome (Bourgeois et al., 1993). The syndrome begins before 18 months of age. In addition to prolonged periods of dystonia, there are attacks of nystagmus, dyspnea, and autonomic phenomena. Episodes of quadriplegia appear either when a hemiplegia shifts from one side to the other or as an isolated manifestation. The episodes were often followed by developmental deterioration. Eventually, there is cognitive impairment and a choreoathetotic movement disorder. Sleep relieves the weakness and other paroxysmal phenomena, but they can reappear after awakening. The attacks can last from a few minutes to several days. Flunarizine and aripiprazole can be partially effective (Haffejee and Santosh, 2009). Some infants manifest paroxysmal dystonia before the classic features of alternating hemiplegia develop (Andermann et al., 1994). Magnetic resonance spectroscopy was normal in the case reported by Nezu and colleagues (1997). A review of the phenotypic data on 103 patients with this syndrome has been published (Sweney et al., 2009). Paroxysmal eye movements were the most frequent early symptom, manifesting in the first 3 months of life in 83% of patients. Paroxysmal episodes of focal dystonia or flaccid, alternating hemiplegia appeared by 6 months of age in 56% of infants. A European consortium reported that the natural history of alternating hemiplegia is highly variable and unpredictable, and the course is not always steadily progressive and degenerative (Panagiotakaki et al., 2010).
Paroxysmal exertional dyskinesia (PED)
Lance (1977) was the first to describe what he called an intermediate form of PDC (now called PNKD). Today, this family would appear to have PED. The family had attacks that were briefer than those in classic PNKD, lasting from 5 to 30 minutes, and in which the attacks were precipitated by prolonged exercise and not by cold, heat, stress, ethanol, excitement, or anxiety. The spasms affected mainly the legs. Plant and colleagues (1984) reported a second family. In both families, the inheritance pattern was that of autosomal dominant transmission. In neither family did anyone derive any benefit from barbiturates levodopa, or clonazepam. Sporadic cases are also seen (Video 22.8), such as the case reported by Nardocci and colleagues (1989) (Case 3). This patient also had interictal chorea without any family history of a similar condition. This patient was helped by clonazepam. Wali (1992) reported another sporadic case; this was an 18-year-old man in whom attacks of right hemidystonia lasting about 10 minutes were precipitated by prolonged running (about 10 minutes) or by cold. The EEG and the CT scan were normal; anticonvulsants were not helpful. Demirkiran and Jankovic (1995) mentioned seeing five patients, three being females. The largest series of sporadic cases is that by Bhatia and colleagues (1997). Familial cases appear to be autosomal dominant (Kluge et al., 1998). A large family with PED with four affected members had onset at 9–15 years of age and a male : female ratio of 3 : 1 (Munchau et al., 2000). The 26 reported cases consisted of 13 women and 13 men. The age at onset ranged from 2 to 30 years, all but six beginning in childhood. 
Some patients labeled as PKD, e.g., Cases 1 and 3 of Jung and colleagues (1973), and PNKD e.g., the family of Kurlan and colleagues (1987), have attacks that occur after prolonged exercise. It is possible that such patients have a variant of PNKD or a combination of PNKD and one of the other paroxysmal dyskinesias. However, if the attacks last only seconds and respond to anticonvulsants, they fit clinically with these features of PKD.
The family reported by Schloesser and colleagues (1996) supports the notion that PED is a variant of PNKD. The father of the proband was affected by exertional cramping, and two other men in the family had PED. Women in the family had more prolonged attacks, fitting those of PNKD.
Ictal and interictal cerebral perfusion SPECT studies have been conducted (Kluge et al., 1998). During the motor attacks, decreased perfusion of the frontal cortex and increased cerebellar perfusion were observed. The perfusion of the basal ganglia also decreased. No cortical hyperperfusion indicative of an epileptic nature was seen. The authors conclude that PED represents a paroxysmal movement disorder rather than epilepsy. However, an opposite finding in a case of PED has also been reported (Yoon et al., 2007). In this 16-year-old boy, there was increased perfusion in the somatosensory cortex.
In a patient with familial PED, different stimuli and maneuvers in triggering dystonic attacks in the arm were studied. Motor paroxysms could be provoked by muscle vibration, passive movements, transcranial magnetic stimulation, magnetic stimulation of the brachial plexus, and electrical nerve stimulation but not by sham stimulation (Meyer et al., 2001). The authors conclude that dystonic attacks are triggered by proprioceptive afferents.
Guerrini and colleagues (1999) described a family with PED and autosomal recessive rolandic epilepsy, with the gene mapped to chromosome 16p12–11.2. That family’s dystonia resembles the ICCA syndrome, and the genetic mapping overlaps with that syndrome. Perniola and colleagues (2001) described another family with autosomal dominant PED and epilepsy; fasting and stress were also precipitating factors.
In a study in which cerebrospinal fluid (CSF) monoamine metabolites were measured before and after the attack, a twofold increase in the concentration of homovanillic acid and 5-hydroxyindoleacetic acid was detected after the attack (Barnett et al., 2002).
A case of PED foot dystonia in an adult was found to be due to the earliest symptom of Parkinson disease, as established by dopamine transporter binding and SPECT, and effectively treated with dopaminergic medication (Katzenschlager et al., 2002). Two other adults (second cousins) who presented with paroxysmal exercise-induced dystonia later developed clinical features of Parkinson disease (Bruno et al., 2004b).
Autosomal dominant PED accompanied by epilepsy was found to be due to glucose transporter 1 (GLUT1) deficiency (Suls et al., 2008; Weber et al., 2008; Zorzi et al., 2008; Leen et al., 2010). The syndrome also includes mild developmental delay, reduced CSF glucose levels, hemolytic anemia with echinocytosis, and altered erythrocyte iron concentrations. The clinical spectrum of GLUT1 deficiency was evaluated by videotape in 57 patients, and a wide range of movement disorders are seen (Pons et al., 2010). These include abnormal gait (ataxic-spastic and ataxic), action limb dystonia (86%), mild chorea (75%), cerebellar tremor (70%), myoclonus (16%), and dyspraxia (21%). Nonepileptic paroxysmal events were encountered in 28% of patients, and included episodes of ataxia, weakness, parkinsonism, and nonkinesigenic dyskinesias. Alternating hemiplegia has also been reported with GLUT1 deficiency (Rotstein et al., 2009).
PEDs are difficult to treat. Those with GLUT1 deficiency can be treated with a ketogenic diet. Posteroventral pallidotomy has been reported to ameliorate attacks of PED (Bhatia et al., 1998). A pair of homozygous twins with GLUT1 deficiency had PED, migraine, absence seizures and writer’s cramp with an excellent response to a ketogenic diet (Urbizu et al., 2010).
Table 22.7 lists the major distinguishing features of nonepileptic PKD, PNKD, and PED.
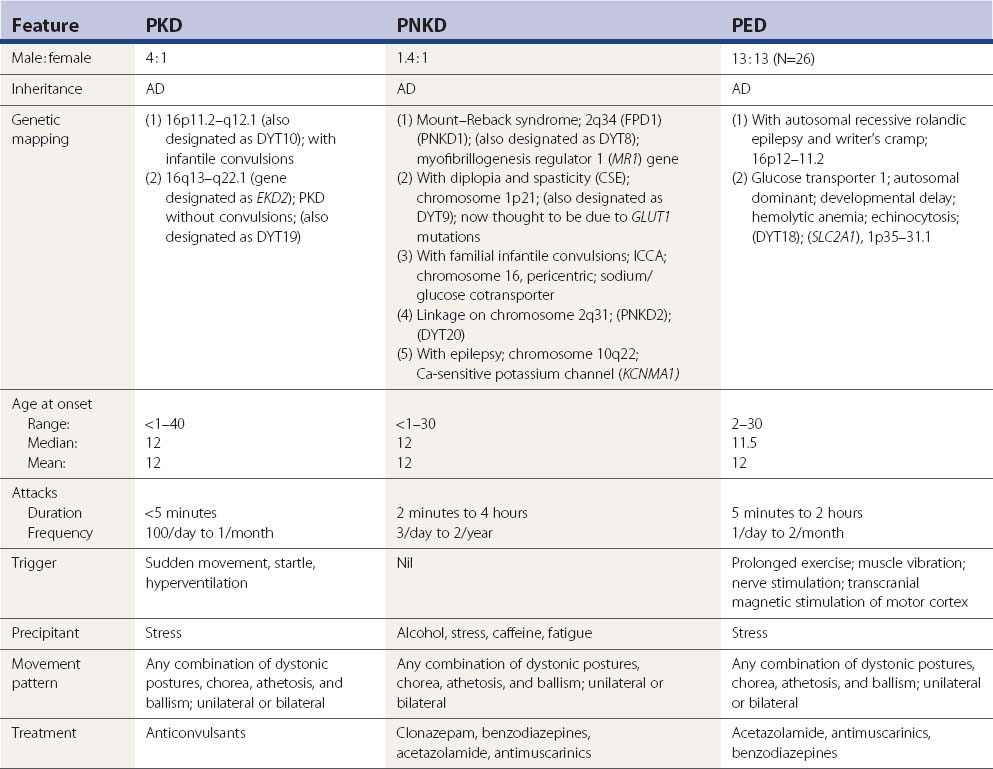
Episodic ataxias
The paroxysmal ataxias, called episodic ataxias, have been distinguished by their clinical features and by their genotypes. These characteristics are presented and summarized in Table 22.8.
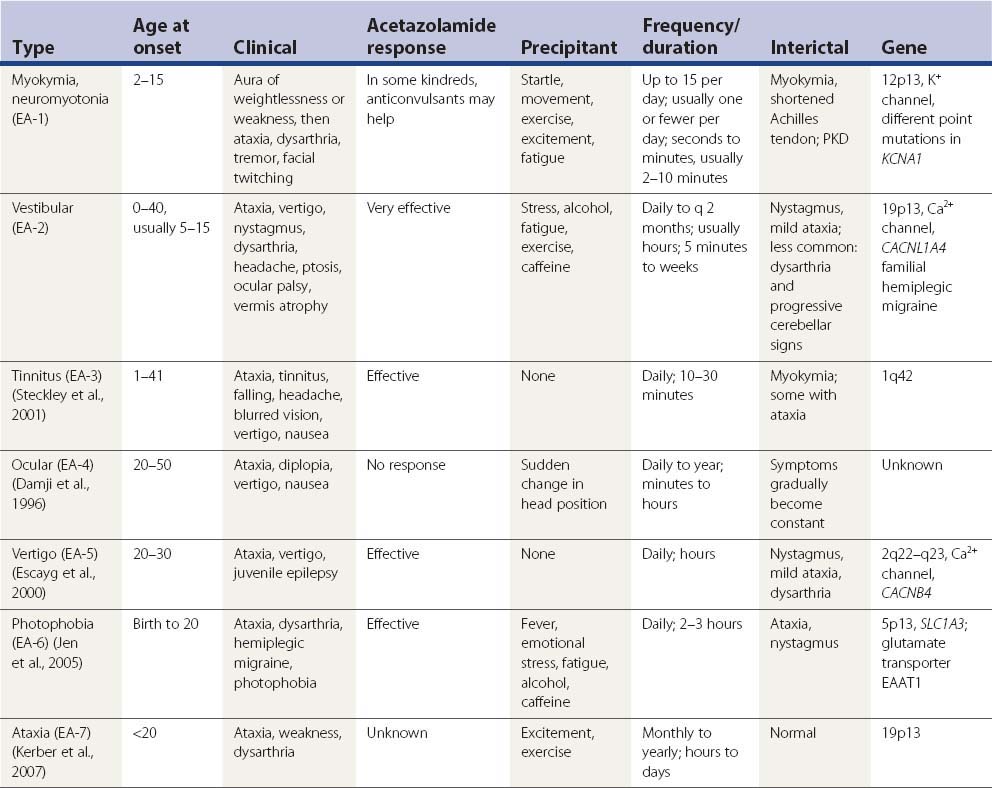
The genes for four of the hereditary episodic ataxias have been determined. In one of them, EA-1, the type associated with myokymia or neuromyotonia, has been found to be due to point mutations in the gene Kv1.1 for the voltage-gated potassium channel (KCNA1) (Browne et al., 1994; Comu et al., 1996). The gene is located on chromosome 12p13 (Litt et al., 1994). Different mutations have been found in different families (D’Adamo et al., 1998; Zerr et al., 1998; Eunson et al., 2000). The attacks are brief, lasting seconds to minutes; myokymia is present during and between attacks. The attacks can be triggered by sudden movement (Nutt and Gancher, 1997), and these can respond to anticonvulsants. Some families have episodes of myokymia without ataxia (Eunson et al., 2000). In one family, mutations in the KCNA1 gene can cause severe neuromyotonia resulting in marked skeletal deformities without episodic ataxia, and others in the family had the ataxia (Kinali et al., 2004).
Another episodic ataxia, EA-2, is the type that has a vestibular component and does not have myokymia as an associated feature. The attacks last between 15 minutes and a few days and respond to acetazolamide. The gene was mapped to chromosome 19p13 (Vahedi et al., 1995; von Brederlow et al., 1995) and the syndrome was found to be mutations in the gene for the calcium channel CACNA1A (Spacey et al., 2005). Spontaneous mutations (Yue et al., 1998) as well as familial cases have been described. There is clinical heterogeneity, with attacks varying from pure ataxia to combinations of symptoms that suggest involvement of the cerebellum, brainstem, and cortex. Oculographic findings were localizing to the vestibulocerebellum and posterior vermis. Some affected individuals exhibited a progressive ataxia syndrome that is phenotypically indistinguishable from the dominantly inherited spinocerebellar ataxia (SCA) syndromes. SCA6 has been found to be due to an expanded CAG repeat on the same gene (Zhuchenko et al., 1997). Two patients with childhood onset EA-2 developed adult-onset dystonia (Spacey et al., 2005). About one-half of the affected individuals have migraine headaches and several have episodes that are typical of basilar migraine (Baloh et al., 1997). Both familial hemiplegic migraine and EA2 are caused by mutations in the Ca2+ channel gene CACNL1A4 and can be considered as allelic channelopathies (Ophoff et al., 1996). When progressive ataxia is present, there is cerebellar atrophy on MRI scans, and not all cases have a CAG expansion (Yue et al., 1997). On the other hand, cases with the CAG expansion and cerebellar atrophy can also have EA-2 (Jodice et al., 1997). A family with the phenotype of EA-2 was found not to have a mutation of the CACNL1A gene on chromosome 19p13, indicating genetic heterogeneity for this disorder (Hirose et al., 2003).
EA-1 and EA-2 are described above. Not all families with migraine and episodic vertigo could be linked to the EA-2 markers on chromosome 19p, indicating genetic heterogeneity for this type of episodic ataxia (Baloh et al., 1996). EA-3 is an autosomal dominant episodic ataxia manifested by vestibular ataxia, vertigo, tinnitus, and interictal myokymia; attacks are diminished by acetazolamide (Steckley et al., 2001). EA-1 and EA-2 genetics were excluded. The fourth type of paroxysmal ataxia with ocular motility problems can be precipitated by sudden head movement (Damji et al., 1996). The gene for this fourth type of EA has not yet been mapped, but is genetically distinct from EA-1 and EA-2. The fifth type was seen in a French-Canadian family. The attacks began after age 20 years, but earlier in life there was epilepsy (Escayg et al., 2000). The sixth type ranges from more severe with younger onset with episodes of ataxia, seizures, migraine, and alternating hemiplegia from birth (Jen et al., 2005) to milder forms with attacks accompanied by nausea, vomiting, photophobia, phonophobia, vertigo, diplopia, and dysarthria (de Vries et al., 2009). The seventh type maps to the same region as EA-2, without any interictal symptoms, and without mutations in the potassium channel gene (Kerber et al., 2007).
Another family with episodic ataxia that does not map to the disorders labeled as EA-1 to EA-7 has been described (Damak et al., 2009). This family has not been linked to any chromosomal site yet. It appears to be autosomal dominant, with onset between 48 and 56 years of age; the most severely affected had daily attacks with slowly progressive and disabling permanent cerebellar ataxia and a poor response to acetazolamide. Sporadic cases of episodic ataxias occur. Four patients with autopsies had ataxia resembling EA-2, but had not responded to acetazolamide, nor did their molecular genetics workup fit EA-2 (Julien et al., 2001).
Molecular genetics of paroxysmal dyskinesias
A number of paroxysmal dyskinesias have been mapped to chromosome 16. PKD with infantile convulsions has been mapped to 16p11.2–q12.1 and called EKD1 and designated as DYT10 (Tomita et al., 1999; Bennett et al., 2000; Swoboda et al., 2000). PKD without convulsions has been mapped to 16q13–q22.1, with the gene referred to as EKD2 and designated as DYT19 (Valente et al., 2000). The ICCA syndrome has been reported without linkage to these two loci (Spacey et al., 2002). A family with infantile convulsions has an associated PNKD, and the abnormal gene has been mapped to chromosome 16 (Szepetowski et al., 1997). The gene in this region that appears to be responsible for ICCA is the sodium/glucose cotransporter gene (KST1) (Roll et al., 2002). It also appears to be responsible for benign familial infantile convulsions. Perhaps this disorder with its genetic mapping has been referred to by others as PKD with infantile convulsions. Guerrini (2001) proposes that disparate reports of families suggest that the same gene could be responsible for autosomal recessive rolandic epilepsy, PED, writer’s cramp (RE-PED-WC), and PNKD, with specific mutations accounting for each of these mendelian disorders. Evidence is for a major gene or a cluster of genes for epilepsy and paroxysmal dyskinesia to the pericentromeric region of chromosome 16. These families need to be tested for the KST1 mutation. Caraballo and colleagues (2001) have diagrammed where the gene loci for these different disorders have been mapped on chromosome 16 (Fig. 22.1).
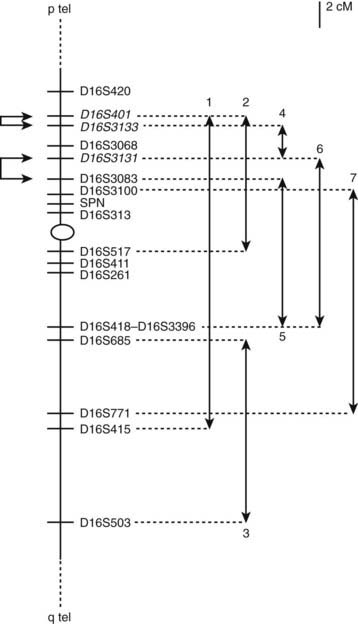
Figure 22.1 Schematic representation of the genetic map of human chromosome 16p12–q12. Critical regions are indicated: 1 = benign familial infantile convulsions (BFIC) (7 families, Carabello et al., 2001); 2 = ICCA (Szepetowski et al., 1997); 3 = PKD (Valente et al., 2000); 4 = reflex epilepsy–PED–writer’s cramp (RE-PED-WC) syndrome (Guerrini et al., 1999); 5 = PKD and BFIC (Tomita et al., 1999); 6 = PKD and BFIC (Swoboda et al., 2000); 7 = PKD (Bennett et al., 2000).
From Caraballo R, Pavek S, Lemainque A, et al. Linkage of benign familial infantile convulsions to chromosome 16p12-q12 suggests allelism to the infantile convulsions and choreoathetosis syndrome. Am J Hum Genet 2001;68(3):788–794.
Six pedigrees with PKD have been found on the Chinese mainland, and they were not linked to the two known loci and so represent another familial form of PKD (Zhou et al., 2008).
An autosomal dominant form of PED with development delay and erythrocyte changes has been found to be due to glucose transporter 1 (GLUT1) deficiency (Weber et al., 2008; Zorzi et al., 2008). There is a cation leak from the erythrocytes that alters the intracellular concentration of sodium, potassium, and calcium. This leads to hemolytic anemia and echinocytosis as detected by electron microscopy. Many more papers have reported GLUT1 deficiency causing PED (Suls et al., 2008; Schneider et al., 2009; Leen et al., 2010).
Two papers independently reported the mapping of a gene for PNKD to chromosome 2q31–q36 (Fink et al., 1996; Fouad et al., 1996), and the region was narrowed to 2q33–q35 by Fink et al. (1997). Another family was mapped by Raskind and colleagues (1998), who narrowed the region to 2q34. And Jarman and colleagues (1997) mapped the gene to a large British family. Although a cluster of sodium channel genes is near the PNKD locus, the mutated gene has now been found to be the myofibrillogenesis regulator 1 (MR1) (Lee et al., 2004). The mutations in this gene were associated with PNKD in 50 individuals from eight families; they cause changes (Ala to Val) in the N-terminal region of two MR1 isoforms. The MR1L isoform is specifically expressed in the brain and is localized to the cell membrane while the MR1S isoform is ubiquitously expressed and shows diffuse cytoplasmic and nuclear localization. The MR1 gene is homologous to the hydroxyacylglutathione hydrolase (HAGH) gene, which functions in a pathway to detoxify methylglyoxal, a compound that is present in coffee and alcoholic beverages and is produced as a by-product of oxidative stress. The finding of the MR1 gene locus suggests a mechanism whereby alcohol, coffee, and stress may act as precipitants of attacks in PNKD. However, there is genetic heterogeneity in PNKD, because there is a Canadian family without this mutation, with links to a separate locus at 2q31 instead (Spacey et al., 2006); it has been designated as PNKD2 and also DYT20, while the MR1 gene disorder is called PNKD1 and DYT8.
Auburger and colleagues (1996) studied a family with PNKD with associated spasticity; their mutated gene was mapped to a potassium channel gene cluster on chromosome 1p. The attacks are said to consist of dystonia of limbs, imbalance, dysarthria, diplopia, and sometimes headache. The attacks last about 20 minutes and can be precipitated by exercise, stress, lack of sleep, and alcohol consumption. Some affected members had persistent spastic paraparesis, which would make this family’s condition different from classic Mount–Reback syndrome. This condition has been called choreoathetosis/spasticity episodica (CSE). The locus is very close to the GLUT1 locus, suggesting that the family might be positive for a GLUT1 mutation.
A large Australian family with autosomal dominant hypnogenic frontal lobe seizures/dystonia has been found to have a missense mutation in the second transmembrane domain of the nicotinic acetylcholine receptor alpha 4 subunit (CHRNA4) gene, located on chromosome 20q13.2–q13.3 (Oldani et al., 1998). A second locus has been found on chromosome 15q24 (Phillips et al., 1998, 2001) and a third locus has been found on chromosome 1 in a large Italian family (Gambardella et al., 2000).
For a detailed review of the genetics of the paroxysmal dyskinesias, the reader is referred to the article by Weber and Lerche (2009).
Miscellany
There are probably a number of other types of paroxysmal dyskinesias that do not fit well into the classification scheme listed previously (Pourfar et al., 2005). For example, a case of truncal flexion spasms that persist repeatedly in attacks lasting several hours has been described (Brown et al., 1991). These were considered to have a spinal origin, presumably involving the propriospinal pathways. We have seen a patient with multiple brief dystonic spasms, lasting seconds, in a woman following cardiac arrhythmias. These attacks have all the clinical features of symptomatic PKD, except that they were not precipitated by sudden movement or startle. Like PKD, they responded dramatically to low dosages of carbamazepine. Perhaps the major delimiter in classifying the paroxysmal dyskinesias should not be whether they are kinesigenic or not, but the duration of the attacks. Like tics, stereotypies can occur episodically (Video 22.9). 
A syndrome in infant girls is self-stimulatory (previously called masturbatory) behavior that occurs episodically (Fleisher and Morrison, 1990; Mink and Neil, 1995; Nechay et al., 2004; Yang et al., 2005). The episodes appear between 3 and 14 months of age and consist of the child’s applying her suprapubic region to a firm edge of furniture, such as the arm of a sofa (Video 22.10). There is often stiffening of the lower extremities. The episodes last less than a minute to hours at a time. Most commonly this behavior is misdiagnosed as epilepsy or paroxysmal dystonia. 
Adam A.M., Orinda D. Focal paroxysmal kinesigenic choreoathetosis preceding the development of Steele-Richardson-Olszewski syndrome [letter]. J Neurol Neurosurg Psychiatry. 1986;49:957-968.
Aguirregomozcorta M., Ramio-Torrenta L.I., Gich J., et al. Paroxysmal dystonia and pathological laughter as a first manifestation of multiple sclerosis. Mult Scler. 2008;14(2):262-265.
Aimard G., Vighetto A., Trillet M., et al. Ataxie paroxystique familiale sensible a l’acetazolamide. Rev Neurol (Paris). 1983;139:251-257.
Akmandemir F.G., Eraksoy M., Gurvit I.H., et al. Paroxysmal dysarthria and ataxia in a patient with Behçet’s disease. J Neurol. 1995;242:344-347.
Alonso-Navarro H., Arroyo M., Parra A., Jiménez-Jiménez F.J. Paroxysmal dystonia associated to primary Sjögren’s syndrome. Mov Disord. 2009;24(5):788-790.
Andermann F., Andermann E. Excessive startle syndromes: startle disease, jumping, and startle epilepsy. Adv Neurol. 1986;43:321-338.
Andermann F., Cosgrove J.B.R., Lloyd-Smith D., et al. Paroxysmal dysarthria and ataxia in multiple sclerosis. Neurology. 1959;9:211-215.
Andermann F., Ohtahara S., Andermann E., et al. Infantile hypotonia and paroxysmal dystonia: a variant of alternating hemiplegia of childhood. Mov Disord. 1994;9:227-229.
Angelini L., Rumi V., Lamperti E., Nardocci N. Transient paroxysmal dystonia in infancy. Neuropediatrics. 1988;19:171-174.
Antony J.H., Ouvrier R.A., Wise G. Spasmus nutans, a mistken identity. Arch Neurol. 1980;37:373-375.
Aradillas E., Schwartzman R.J. Kinesigenic dyskinesia in a case of voltage-gated potassium channel-complex protein antibody encephalitis. Arch Neurol. 2011;68(4):529-532.
Arden F. Idiopathic hypoparathyroidism. Med J Aust. 1953;2:217-219.
Auburger G., Ratzlaff T., Lunkes A., et al. A gene for autosomal dominant paroxysmal choreoathetosis spasticity (CSE) maps to the vicinity of a potassium channel gene cluster on chromosome 1p, probably within 2 cM between D1S443 and D1S197. Genomics. 1996;31:90-94.
Bain P.G., Larkin G.B.R., Calver D.M., Obrien M.D. Persistent superior oblique paresis as a manifestation of familial periodic cerebellar ataxia. Br J Ophthalmol. 1991;75:619-621.
Bain P.G., O’Brien M.D., Keevil S.F., Porter D.A. Familial periodic cerebellar ataxia: A problem of cerebellar intracellular pH homeostasis. Ann Neurol. 1992;31:147-154.
Baloh R.W., Winder A. Acetazolamide-responsive vestibulocerebellar syndrome: Clinical and oculographic features. Neurology. 1991;41:429-433.
Baloh R.W., Foster C.A., Yue Q., Nelson S.F. Familial migraine with vertigo and essential tremor. Neurology. 1996;46:458-460.
Baloh R.W., Yue Q., Furman J.M., Nelson S.F. Familial episodic ataxia: Clinical heterogeneity in four families linked to chromosome 19p. Ann Neurol. 1997;41:8-16.
Barabas G., Tucker S.M. Idiopathic hypoparathyroidism and paroxysmal dystonic choreoathetosis [letter]. Ann Neurol. 1988;24:585.
Barnett M.H., Jarman P.R., Heales S.J.R., Bhatia K.P. Further case of paroxysmal exercise-induced dystonia and some insights into pathogenesis. Mov Disord. 2002;17(6):1386-1387.
Baron D.N., Dent C.E., Harris H., et al. Hereditary pellagra-like skin rash with temporary cerebellar ataxia, constant renal amino-aciduria and other bizarre biochemical features. Lancet. 1956;2:421-428.
Bass N., Wyllie E., Comair Y., et al. Supplementary sensorimotor area seizures in children and adolescents. J Pediatr. 1995;126:537-544.
Beaumanoir A., Mira L., van Lierde A. Epilepsy or paroxysmal kinesigenic choreoathetosis? Brain Dev. 1996;18:139-141.
Beltran R.S., Coker S.B. Transient dystonia of infancy, a result of intrauterine cocaine exposure? Pediatr Neurol. 1995;12:354-356.
Bennett D.A., Fox J.H. Paroxysmal dyskinesias secondary to cerebral vascular disease – reversal with aspirin. Clin Neuropharmacol. 1989;12:215-216.
Bennett L.B., Roach E.S., Bowcock A.M. A locus for paroxysmal kinesigenic dyskinesia maps to human chromosome 16. Neurology. 2000;54:125-130.
Berger J.R., Sheremata W.A., Melamed E. Paroxysmal dystonia as the initial manifestation of multiple sclerosis. Arch Neurol. 1984;41:747-750.
Bhatia K.P., Soland V.L., Bhatt M.H., et al. Paroxysmal exercise-induced dystonia: Eight new sporadic cases and a review of the literature. Mov Disord. 1997;12:1007-1012.
Bhatia K.P., Marsden C.D., Thomas D.G.T. Posteroventral pallidotomy can ameliorate attacks of paroxysmal dystonia induced by exercise. J Neurol Neurosurg Psychiatry. 1998;65:604-605.
Bird T.D., Carlson C.B., Horning M. Ten year follow-up of paroxysmal choreoathetosis: A sporadic case becomes familial. Epilepsia. 1978;19:129-132.
Blakeley J., Jankovic J. Secondary paroxysmal dyskinesias. Mov Disord. 2002;17(4):726-734.
Blass J.P., Avigan J., Uhlendorf B.W. A defect in pyruvate decarboxyalase in a child with an intermittent movement disorder. J Clin Invest. 1970;49:423-432.
Blass J.P., Kark R.A.P., Engel W.K. Clinical studies of a patient with pyruvate decarboxylase deficiency. Arch Neurol. 1971;25:449-460.
Blumkin L., Lev D., Watemberg N., Lerman-Sagie T. Hypomyelinating leukoencephalopathy with paroxysmal tonic upgaze and absence of psychomotor development. Mov Disord. 2007;22(2):226-230.
Boel M., Casaer P. Paroxysmal kinesigenic choreoathetosis. Neuropediatrics. 1984;15:215-217.
Bonakis A., Papageorgiou S.G., Potagas C., et al. A case of refractory secondary paroxysmal kinesigenic dyskinesia with high sensitivity to phenytoin monotherapy. Parkinsonism Relat Disord. 2009;15(1):68-70.
Bortolotti P., Schoenhuber R. Paroxysmal kinesigenic choreoathetosis [letter]. Arch Neurol. 1983;40:529.
Bourgeois M., Aicardi J., Goutieres F. Alternating hemiplegia of childhood. J Pediatr. 1993;122:673-679.
Bratt H.D., Menelaus M.B. Benign paroxysmal torticollis of infancy. J Bone Joint Surg [Br]. 1992;74:449-451.
Bressman S.B., Fahn S., Burke R.E. Paroxysmal non-kinesigenic dystonia. Adv Neurol. 1988;50:403-413.
Brockmann K., Dumitrescu A.M., Best T.T., et al. X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene. J Neurol. 2005;252:663-666.
Brown P., Thompson P.D., Rothwell J.C., et al. Paroxysmal axial spasms of spinal origin. Mov Disord. 1991;6:43-48.
Browne D.L., Gancher S.T., Nutt T.G., et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136-140.
Bruno M.K., Hallett M., Gwinn-Hardy K., et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: New diagnostic criteria. Neurology. 2004;63:2280-2287.
Bruno M.K., Lee H.Y., Auburger G.W., et al. Genotype-phenotype correlation of paroxysmal nonkinesigenic dyskinesia. Neurology. 2007;68(21):1782-1789.
Bruno M.K., Ravina B., Garraux G., et al. Exercise-induced dystonia as a preceding symptom of familial Parkinson’s disease. Mov Disord. 2004;19(2):228-230.
Brunt E.R.P., Van Weerden T.W. Familial paroxysmal kinesigenic ataxia and continuous myokymia. Brain. 1990;113:1361-1382.
Buchman A.S., Goetz C.G., Klawans H.L. Hemiparkinsonism with hemiatrophy. Neurology. 1988;38:527-530.
Burger L.J., Lopez R.I., Elliott F.A. Tonic seizures induced by movement. Neurology. 1972;22:656-659.
Burguera J.A., Catala J., Casanova B. Thalamic demyelination and paroxysmal dystonia in multiple sclerosis. Mov Disord. 1991;6:379-381.
Burke R.E., Fahn S., Jankovic J., et al. Tardive dystonia: Late-onset and persistent dystonia caused by antipsychotic drugs. Neurology. 1982;32:1335-1346.
Byrne E., White O., Cook M. Familial dystonic choreoathetosis with myokymia; A sleep responsive disorder. J Neurol Neurosurg Psychiatry. 1991;54:1090-1092.
Camac A., Greene P., Khandji A. Paroxysmal kinesigenic dystonic choreoathetosis associated with a thalamic infarct. Mov Disord. 1990;5:235-238.
Campistol J., Prats J.M., Garaizar C. Benign paroxysmal tonic upgaze of childhood with ataxia – a neuroophthalmological syndrome of familial origin. Dev Med Child Neurol. 1993;35:436-439.
Caraballo R., Pavek S., Lemainque A., et al. Linkage of benign familial infantile convulsions to chromosome 16p12-q12 suggests allelism to the infantile convulsions and choreoathetosis syndrome. Am J Hum Genet. 2001;68(3):788-794.
Cavanagh N.P., Bicknell J., Howard F. Cystinuria with mental retardation and paroxysmal dyskinesia in 2 brothers. Arch Dis Child. 1974;49:662-664.
Chatterjee A., Louis E.D., Frucht S. Levetiracetam in the treatment of paroxysmal kinesiogenic choreoathetosis. Mov Disord. 2002;17(3):614-615.
Chiesa V., Tamma F., Gardella E., et al. Possible post-traumatic paroxysmal kinesigenic dyskinesia. Mov Disord. 2008;23(16):2428-2430.
Chillag K.L., Deroos S.T. Oxcarbazepine use in paroxysmal kinesigenic dyskinesia: report on four patients. Pediatr Neurol. 2009;40(4):295-297.
Chudnow R.S., Mimbela R.A., Owen D.B., Roach E.S. Gabapentin for familial paroxysmal dystonic choreoathetosis. Neurology. 1997;49:1441-1442.
Cochen De Cock V., Bourdain F., Apartis E., et al. Interictal myoclonus with paroxysmal kinesigenic dyskinesia. Mov Disord. 2006;21(9):1533-1535.
Comu S., Giuliani M., Narayanan V. Episodic ataxia and myokymia syndrome: A new mutation of potassium channel gene Kv1.1. Ann Neurol. 1996;40:684-687.
Cosentino C., Torres L., Flores M., Cuba J.M. Paroxysmal kinesigenic dystonia and spinal cord lesion. Mov Disord. 1996;11:453-455.
Coulter D.L., Donofrio P. Haloperidol for nonkinesiogenic paroxysmal dyskinesia [letter]. Arch Neurol. 1980;37:325-326.
D’Adamo M.C., Liu Z.P., Adelman J.P., et al. Episodic ataxia type-1 mutations in the hKv1.1 cytoplasmic pore region alter the gating properties of the channel. EMBO J. 1998;17:1200-1207.
Dale R.C., Church A.J., Surtees R.A.H., et al. Post-streptococcal autoimmune neuropsychiatric disease presenting as paroxysmal dystonic choreoathetosis. Mov Disord. 2002;17(4):817-820.
Damak M., Riant F., Boukobza M., et al. Late onset hereditary episodic ataxia. J Neurol Neurosurg Psychiatry. 2009;80(5):566-568.
Damji K.F., Allingham R.R., Pollock S.C., et al. Periodic vestibulocerebellar ataxia, an autosomal dominant ataxia with defective smooth pursuit, is genetically distinct from other autosomal dominant ataxias. Arch Neurol. 1996;53:338-344.
Dancis J., Hutzler J., Rokkones T. Intermittent branched-chain ketonuria: Variant of maple-syrup-urine disease. N Engl J Med. 1967;276:84-89.
Debruyne F., Van Paesschen W., Van Eyken P., et al. Paroxysmal nonkinesigenic dyskinesias due to recurrent hypoglycemia caused by an insulinoma. Mov Disord. 2009;24(3):460-461.
DeCastro W., Campbell J. Periodic ataxia. JAMA. 1967;200:892-894.
Demirkiran M., Jankovic J. Paroxysmal dyskinesias: Clinical features and classification. Ann Neurol. 1995;38:571-579.
Deonna T., Roulet E., Meyer H.U. Benign paroxysmal tonic upgaze of childhood – a new syndrome. Neuropediatrics. 1990;21:213-214.
de Vries B., Mamsa H., Stam A.H., et al. Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch Neurol. 2009;66(1):97-101. Erratum in: Arch Neurol 2009;66(4):497
Dominguez-Moran J.A., Callejo J.M., Fernandez-Ruiz L.C., Martinez-Castrillo J.C. Acute paroxysmal dystonia induced by fluoxetine. Mov Disord. 2001;16:767-769.
Donat J.R., Auger R. Familial periodic ataxia. Arch Neurol. 1979;36:568-569.
Donat J.F., Wright F.S. Episodic symptoms mistaken for seizures in the neurologically impaired child. Neurology. 1990;40:156-157.
Dooley J.M., Brna P.M. Sublingual lorazepam in the treatment of familial paroxysmal nonkinesigenic dyskinesia. Pediatr Neurol. 2004;30(5):365-366.
Dragasevic N., Petkovic-Medved B., Svetel M., et al. Paroxysmal hemiballism and idiopathic hypoparathyroidism. J Neurol. 1997;244:389-390.
Drake M.E.Jr, Jackson R.D., Miller C.A. Paroxysmal choreoathetosis after head injury [letter]. J Neurol Neurosurg Psychiatry. 1986;49:837-843.
Du W., Bautista J.F., Yang H., et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37(7):733-738.
Duchowny M.S., Resnick T.J., Deray M.J., Alvarez L.A. Video EEG diagnosis of repetitive behavior in early childhood and its relationship to seizures. Pediatr Neurol. 1988;4:162-164.
Dunn D.W. Paroxysmal dystonia. Am J Dis Child. 1981;135:381-382.
Echenne B., Rivier F. Benign paroxysmal tonic upward gaze. Pediatr Neurol. 1992;8:154-155.
Engelen M., Tijssen M.A.J. Paroxysmal non-kinesigenic dyskinesia in antiphospholipid syndrome. Mov Disord. 2005;20(1):111-113.
Erickson G.R., Chun R.W. Acquired paroxysmal movement disorders. Pediatr Neurol. 1987;3:226-229.
Escayg A., De Waard M., Lee D.D., et al. Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. 2000;66(5):1531-1539.
Espir M.L.E., Watkins S.M., Smith H.V. Paroxysmal dysarthria and other transient neurological distrubances in disseminated sclerosis. J Neurol Neurosurg Psychiatry. 1966;29:323-330.
Eunson L.H., Rea R., Zuberi S.M., et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol. 2000;48:647-656.
Factor S.A., Coni R.J., Cowger M., Rosenblum E.L. Paroxysmal tremor and orofacial dyskinesia secondary to a biopterin synthesis defect. Neurology. 1991;41:930-932.
Fahn S. Paroxysmal tremor. Neurology. 1983;33(Suppl 2):131.
Fahn S. Atypical tremors, rare tremors, and unclassified tremors. Findley LJ, Capildeo R. Movement Disorders: Tremor. New York: Oxford University; 1984. pp. 431–443
Fahn S. Motor and vocal tics. In: Kurlan R., editor. Handbook of Tourette’s Syndrome and Related Tic and Behavioral Disorders. New York: Marcel Dekker; 1993:3-16.
Fahn S. The paroxysmal dyskinesias. In: Marsden C.D., Fahn S., editors. Movement Disorders 3. Oxford: Butterworth-Heinemann; 1994:310-345.
Fahn S., Marsden C.D., Van Woert M.H. Definition and clinical classification of myoclonus. Adv Neurol. 1986;43:1-5.
Fahn S., Williams D.T. Psychogenic dystonia. Adv Neurol. 1988;50:431-455.
Falconer M., Driver M., Serafetinides E. Seizures induced by movement: Report of a case relieved by operation. J Neurol Neurosurg Psychiatry. 1963;26:300-307.
Farmer T.W., Mustian V.M. Vestibulocerebellar ataxia. Arch Neurol. 1963;8:471-480.
Fejerman N. Mioclonias benignas de la infancia temprana. An Esp Pediatr. 1984;21:725-731.
Fernandez-Alvarez E. Transient movement disorders in children. J Neurol. 1998;245(1):1-5.
Fink J.K., Hedera P., Mathay J.G., Albin R.L. Paroxysmal dystonic choreoathetosis linked to chromosome 2q: Clinical analysis and proposed pathophysiology. Neurology. 1997;49:177-183.
Fink J.K., Rainier S., Wilkowski J., et al. Paroxysmal dystonic choreoathetosis: Tight linkage to chromosome 2q. Am J Hum Genet. 1996;59:140-145.
Fischbeck K.H., Layzer R.B. Paroxysmal choreoathetosis associated with thyrotoxicosis. Ann Neurol. 1979;6:453-454.
Fish D.R., Marsden C.D. Epilepsy masquerading as a movement disorder. In: Marsden C.D., Fahn S., editors. Movement Disorders 3. Oxford: Butterworth-Heinemann; 1994:346-358.
Fleisher D.R., Morrison A. Masturbation mimicking abdominal pain or seizures in young girls. J Pediatr. 1990;116:810-814.
Forssman H. Hereditary disorder charactrized by attacks of muscular contractions, induced by alcohol amongst other factors. Acta Med Scand. 1961;170:517-533.
Fouad G.T., Servidei S., Durcan S., et al. A gene for familial paroxysmal dyskinesia (FPD1) maps to chromosome 2q. Am J Hum Genet. 1996;59:135-139.
Franssen H., Fortgens C., Wattendorff A.R., van Woerkom T.C.A.M. Paroxysmal kinesigenic choreoathetosis and abnormal contingent negative variation. A case report. Arch Neurol. 1983;40:381-385.
Frucht S., Fahn S. Paroxysmal kinesigenic dyskinesia in infancy. Mov Disord. 1999;14:694-695.
Fuchs O., Pfarr N., Pohlenz J., Schmidt H. Elevated serum triiodothyronine and intellectual and motor disability with paroxysmal dyskinesia caused by a monocarboxylate transporter 8 gene mutation. Dev Med Child Neurol. 2009;51(3):240-244.
Fuh J.L., Chang D.B., Wang S.J., et al. Painful tonic spasms: an interesting phenomenon in cerebral ischemia. Acta Neurol Scand. 1991;84:534-536.
Fukuda M., Hashimoto O., Nagakubo S., Hata A. A family with an atonic variant of paroxysmal kinesigenic choreoathetosis and hypercalcitoninemia. Mov Disord. 1999;14:342-344.
Fukuyama S., Okada R. Hereditary kinesthetic reflex epilepsy. Report of five families of peculiar seizures induced by sudden movements. Adv Neurol Sci (Tokyo). 1967;11:168-197.
Gambardella A., Annesi G., DeFusco M., et al. A new locus for autosomal dominant nocturnal frontal lobe epilepsy maps to chromosome 1. Neurology. 2000;55:1467-1471.
Gancher S.T., Nutt J.G. Autosomal dominant episodic ataxia: A heterogeneous syndrome. Mov Disord. 1986;1:239-253.
Garello L., Ottonello G.A., Regesta G., Tanganelli P. Familial paroxysmal kinesigenic choreoathetosis: Report of a pharmacological trial in 2 cases. Eur Neurol. 1983;22:217-221.
Gatto E.M., Zurru M.C., Rugilo C. Medullary lesions and unusual bilateral paroxysmal dystonia in multiple sclerosis. Neurology. 1996;46:847-848.
Gay C.T., Ryan S.G. Paroxysmal kinesigenic dystonia after methylphenidate administration. J Child Neurol. 1994;9:45-46.
George M.S., Pickett J.B., Kohli H., et al. Paroxysmal dystonic reflex choreoathetosis after minor closed head injury. Lancet. 1990;336:1134-1135.
Giffin N.J., Benton S., Goadsby P.J. Benign paroxysmal torticollis of infancy: four new cases and linkage to CACNA1A mutation. Dev Med Child Neurol. 2002;44(7):490-493.
Gilroy J. Abnormal computed tomograms in paroxysmal kinesigenic choreoathetosis. Arch Neurol. 1982;39:779-780.
Gokcay A., Gokcay F. Oxcarbazepine therapy in paroxysmal kinesigenic choreoathetosis. Acta Neurol Scand. 2000;101:344-345.
Gonzalez-Alegre P., Ammache Z., Davis P.H., Rodnitzky R.L. Moyamoya-induced paroxysmal dyskinesia. Mov Disord. 2003;18(9):1051-1056.
Goodenough D.J., Fariello R.G., Annis B.L., Chun R.W. Familial and acquired paroxysmal dyskinesias. A proposed classification with delineation of clinical features. Arch Neurol. 1978;35:827-831.
Gorard D.A., Gibberd F.B. Paroxysmal dysarthria and ataxia – associated MRI abnormality. J Neurol Neurosurg Psychiatry. 1989;52:1444-1445.
Gourley I.M. Paroxysmal torticollis in infancy. Can Med Assoc J. 1971;105:504-505.
, Gowers WR, 1885. Epilepsy and Other Chronic Convulsive Diseases. Their Causes, Symptoms and Treatment. pp. 75–76. (Reprinted by New York: Dover in 1964)
Griggs R.C., Moxley R.T.III, Lafrance R.A., McQuillen J. Hereditary paroxysmal ataxia: Response to acetazolamide. Neurology. 1978;28:1259-1264.
Griggs R.C., Nutt J.G. Episodic ataxias as channelopathies. Ann Neurol. 1995;37:285-287.
Guerrini R. Idiopathic epilepsy and paroxysmal dyskinesia. Epilepsia. 2001;42:36-41.
Guerrini R., Belmonte A., Carrozzo R. Paroxysmal tonic upgaze of childhood with ataxia: a benign transient dystonia with autosomal dominant inheritance. Brain Dev. 1998;20:116-118.
Guerrini R., Bonanni P., Nardocci N., et al. Autosomal recessive rolandic epilepsy with paroxysmal exercise-induced dystonia and writer’s cramp: Delineation of the syndrome and gene mapping to chromosome 16p12-11.2. Ann Neurol. 1999;45:344-352.
Haan J., Kremer H.P.H., Padberg G. Paroxysmal chreoathetosis as presenting symptom of diabetes mellitus. J Neurol Neurosurg Psychiatry. 1988;52:133.
Haffejee S., Santosh P.J. Treatment of alternating hemiplegia of childhood with aripiprazole. Dev Med Child Neurol. 2009;51(1):74-77.
Hall D.A., Parsons J., Benke T. Paroxysmal nonkinesigenic dystonia and celiac disease. Mov Disord. 2007;22(5):708-710.
Hallett M., Marsden C.D., Fahn S. Myoclonus. Handbook of Clinical Neurology, vol. 49, Extrapyramidal Disorders. Amsterdam: Elsevier; 1987.
Hanson P.A., Martinez L.B., Cassidy R. Contractures, continuous muscle discharges, and titubation. Ann Neurol. 1977;1:120-124.
Hattori H., Yorifuji T. Infantile convulsions and paroxysmal kinesigenic choreoathetosis in a patient with idiopathic hypoparathyroidism. Brain Dev. 2000;22:449-450.
Hawkes C.H. Familial paroxysmal ataxia: report of a family. J Neurol Neurosurg Psychiatry. 1992;55:212-213.
Hayman M., Harvey A.S., Hopkins I.J., et al. Paroxysmal tonic upgaze: A reappraisal of outcome. Ann Neurol. 1998;43:514-520.
Hening W., Walters A., Kavey N., et al. Dyskinesias while awake and periodic movements in sleep in restless legs syndrome: Treatment with opioids. Neurology. 1986;36:1363-1366.
Hill W., Sherman H. Acute intermittent familial cerebellar ataxia. Arch Neurol. 1968;18:350-357.
Hirata K., Katayama S., Saito T., et al. Paroxysmal kinesigenic choreoathetosis with abnormal electroencephalogram during attacks. Epilepsia. 1991;32:492-494.
Hirose H., Arayama T., Takita J., et al. A family of episodic ataxia type 2: No evidence of genetic linkage to the CACNA1A gene. Int J Mol Med. 2003;11(2):187-189.
Hishikawa Y., Furuya E., Yamamoto J., Nan’no H. Dystonic seizures induced by movement. Arch Psychiatr Nervenkr. 1973;217:113-138.
Hofele K., Benecke R., Auburger G. Gene locus FPD1 of the dystonic Mount-Reback type of autosomal-dominant paroxysmal choreoathetosis. Neurology. 1997;49:1252-1257.
Holmes G.L., Russman B.S. Shuddering attacks: evaluation using electoencephaloraphic frequency modulation radiotelemetry and videotape monitoring. Am J Dis Child. 1986;140:72-73.
Homan R.W., Vasko M.R., Blaw M. Phenytoin plasma concentrations in paroxysmal kinesigenic choreoathetosis. Neurology. 1980;30:673-676.
Horner F.H., Jackson L.C. Familial paroxysmal choreoathetosis. Barbeau A, Brunette J-R. Progress in Neuro-Genetics. Amsterdam: Excerpta Medica Foundation; 1969. pp. 745–751
Houser M.K., Soland V.L., Bhatia K.P., et al. Paroxysmal kinesigenic choreoathetosis: a report of 26 patients. J Neurol. 1999;246:120-126.
Huang C.W., Chen Y.C., Tsai J.J. Paroxysmal dyskinesia with secondary generalization of tonic-clonic seizures in pseudohypoparathyroidism. Epilepsia. 2005;46(1):164-165.
Huang Y.G., Chen Y.C., Du F., et al. Topiramate therapy for paroxysmal kinesigenic choreoathetosis. Mov Disord. 2005;20(1):75-77.
Huffstutter W.M., Myers G.J. Paroxysmal motor dysfunction. Ala J Med Sci. 1983;20:311-313.
Hughes A.J., Lees A.J., Marsden C.D. Paroxysmal dystonic head tremor. Mov Disord. 1991;6:85-86.
Hwang W.J., Lu C.S., Tsai J.J. Clinical manifestations of 20 Taiwanese patients with paroxysmal kinesigenic dyskinesia. Acta Neurol Scand. 1998;98:340-345.
Jabbari B., Coker S.B. Paroxysmal rhythmic lingua movements and chronic epilepsy. Neurology. 1981;31:1364-1367.
Jabbari B., Khajevi K., Rao K. Medullary dystonia. Mov Disord. 1999;14:698-700.
Jacome D.E., Risko M. Photic induced-driven PLEDs in paroxysmal dystonic choreoathetosis. Clin Electroencephalogr. 1984;15:151-154.
Jarman P.R., Davis M.B., Hodgson S.V., et al. Paroxysmal dystonic choreoathetosis – genetic linkage studies in a British family. Brain. 1997;120:2125-2130.
Jen J.C., Wan J., Palos T.P., et al. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65(4):529-534.
Jodice C., Mantuano E., Veneziano L., et al. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum Mol Genet. 1997;6:1973-1978.
Joseph K., Avallone J., Difazio M. Paroxysmal tonic upgaze and partial tetrasomy of chromosome 15: A novel genetic association. J Child Neurol. 2005;20(2):165-168.
Joynt R.J., Green D. Tonic seizures as a manifestation of multiple sclerosis. Arch Neurol. 1962;6:293-299.
Julien J., Denier C., Ferrer X., et al. Sporadic late onset paroxysmal cerebellar ataxia in four unrelated patients: a new disease? J Neurol. 2001;248:209-214.
Jung S.-S., Chen K.-M., Brody J.A. Paroxysmal choreoathetosis: Report of Chinese cases. Neurology. 1973;23:749-755.
Kanazawa O. Shuddering attacks – Report of four children. Pediatr Neurol. 2000;23:421-424.
Kang U.J., Burke R.E., Fahn S. Natural history and treatment of tardive dystonia. Mov Disord. 1986;1:193-208.
Kato M., Araki S. Paroxysmal kinesigenic choreoathetosis. Report of a case relieved by carbamazepine. Arch Neurol. 1969;20:508-513.
Katzenschlager R., Costa D., Gacinovic S., Lees A.J. [I-123]-FP-CIT-SPECT in the early diagnosis of PD presenting as exercise-induced dystonia. Neurology. 2002;59(12):1974-1976.
Keane J.R. Galloping tongue: Post-traumatic, episodic, rhythmic movements. Neurology. 1984;34:251-252.
Kerber K.A., Jen J.C., Lee H., et al. A new episodic ataxia syndrome with linkage to chromosome 19q13. Arch Neurol. 2007;64(5):749-752.
Kertesz A. Paroxysmal kinesigenic choreoathetosis. An entity within the paroxysmal choreoathetosis syndrome. Description of 10 cases, including 1 autopsied. Neurology. 1967;17:680-690.
Kinali M., Jungbluth H., Eunson L.H., et al. Expanding the phenotype of potassium channelopathy: severe neuromyotonia and skeletal deformities without prominent Episodic Ataxia. Neuromuscul Disord. 2004;14(10):689-693.
Kinast M., Erenberg G., Rothner A.D. Paroxysmal choreoathetosis: report of five cases and review of the literature. Pediatrics. 1980;65:74-77.
Kishimoto K. A novel case of conditionally responsive extrapyramidal syndrome. Ann Rep Res Inst Environ Med Nagoya Univ. 1957;6:91-101.
Kluge A., Kettner B., Zschenderlein R., et al. Changes in perfusion pattern using ECD-SPECT indicate frontal lobe and cerebellar involvement in exercise-induced paroxysmal dystonia. Mov Disord. 1998;13:125-134.
Ko C.H., Kong C.K., Ngai W.T., Ma K.M. Ictal Tc-99m ECD SPECT in paroxysmal kinesigenic choreoathetosis. Pediatr Neurol. 2001;24:225-227.
Koller W., Bahamon-Dussan J. Hereditary paroxysmal cerebellopathy: Responsiveness to acetazolamide. Clin Neuropharmacol. 1987;10:65-68.
Koller W.C., Biary N.M. Volitional control of involuntary movements. Mov Disord. 1989;4:153-156.
Kotagal P., Luders H., Morris H.H., et al. Dystonic posturing in complex partial seizures of temporal lobe onset: A new lateralizing sign. Neurology. 1989;39:196-201.
Kotagal P., Costa M., Wyllie E., Wolgamuth B. Paroxysmal nonepileptic events in children and adolescents. Pediatrics. 2002;110(4):E46.
Kurlan R., Behr J., Medved L., Shoulson I. Familial paroxysmal dystonic choreoathetosis: a family study. Mov Disord. 1987;2:187-192.
Kurlan R., Shoulson I. Familial paroxysmal dystonic choreoathetosis and response to alternate-day oxazepam therapy. Ann Neurol. 1983;13:456-457.
Lagueny A., Ellie E., Burbaud P., et al. Paroxysmal stimulus-sensitive spasmodic torticollis. Mov Disord. 1993;8:241-242.
Lance J.W. Sporadic and familial varieties of tonic seizures. J Neurol Neurosurg Psychiatry. 1963;26:51-59.
Lance J.W. Familial paroxysmal dystonic choreoathetosis and its differentiation from related syndromes. Ann Neurol. 1977;2:285-293.
Lang A.E. Focal paroxysmal kinesigenic choreoathetosis [letter]. J Neurol Neurosurg Psychiatry. 1984;47:1057-1060.
Lang A.E. Psychogenic dystonia: A review of 18 cases. Can J Neurol Sci. 1995;22:136-143.
LeDoux M.S. Paroxysmal kinesigenic dystonia associated with a medullary hemorrhage. Mov Disord. 1997;12:819.
Lee H.Y., Xu Y., Huang Y., Ahn A.H., et al. The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum Mol Genet. 2004;13(24):3161-3170.
Lee W.L., Tay A., Ong H.T., et al. Association of infantile convulsions with paroxysmal dyskinesias (ICCA syndrome): confirmation of linkage to human chromosome 16p12-q12 in a Chinese family. Hum Genet. 1998;103:608-612.
Leen W.G., Klepper J., Verbeek M.M., et al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain. 2010;133(Pt 3):655-670.
Leuzzi V., Di Sabato M.L., Deodato F., et al. Vigabatrin improves paroxysmal dystonia in succinic semialdehyde dehydrogenase deficiency. Neurology. 2007;68(16):1320-1321.
Li J.Y., Lee C.H. Episodic tongue hyperkinesias and alternating limb movements associated with basilar artery ischemia. Mov Disord. 2009;24(8):1249-1251.
Li Z., Turner R.P., Smith G. Childhood paroxysmal kinesigenic dyskinesia: report of seven cases with onset at an early age. Epilepsy Behav. 2005;6:435-439.
Lipson E.H., Robertson W.C.Jr. Paroxysmal torticollis of infancy: Familial occurrence. Am J Dis Child. 1978;132:422-423.
Litt M., Kramer P., Browne D., et al. A gene for episodic ataxia/myokymia maps to chromosome 12p13. Am J Hum Genet. 1994;55:702-709.
Loher T.J., Krauss J.K., Burgunder J.M., et al. Chronic thalamic stimulation for treatment of dystonic paroxysmal nonkinesigenic dyskinesia. Neurology. 2001;56:268-270.
Lombroso C.T. Paroxysmal choreoathetosis: an epileptic or non-epileptic disorder. Ital J Neurol Sci. 1995;16:271-277.
Lombroso C.T., Fejerman N. Benign myoclonus of early infancy. Ann Neurol. 1977;1:138-143.
Lonsdale D., Faulkner W.R., Price J.W., et al. Intermittent cerebellar ataxia associated with hyperpyruvic acidemia, hyperalaninemia, and hyperalaninuria. Pediatrics. 1969;43:1025-1034.
Loong S.C., Ong Y.Y. Paroxysmal kinesigenic choreoathetosis: Report of a case relieved by L-dopa. J Neurol Neurosurg Psychiatry. 1973;36:921-924.
Lotze T., Jankovic J. Paroxysmal kinesigenic dyskinesias. Semin Pediatr Neurol. 2003;10:68-79.
Lou H.C. Flunarizine in paroxysmal choreoathetosis. Neuropediatrics. 1989;20:112.
Lüders H.O. Paroxysmal choreoathetosis. Eur Neurol. 1996;36:20-23.
Lugaresi E., Cirignotta F. Hypnogenic paroxysmal dystonia: epileptic seizure or a new syndrome? Sleep. 1981;4:129-138.
Lugaresi E., Cirignotta F., Montagna P. Nocturnal paroxysmal dystonia. J Neurol Neurosurg Psychiatry. 1986;49:375-380.
MacLean J.B., Sassin J.F. Paroxysmal vertical ocular dyskinesia. Arch Neurol. 1973;29(2):117-119.
Margolin D.L., Marsden C.D. Episodic dyskinesias and transient cerebral ischemia. Neurology. 1982;32:1379-1380.
Marrufo M., Politsky J., Mehta S., et al. Paroxysmal kinesigenic segmental myoclonus due to a spinal cord glioma. Mov Disord. 2007;22(12):1801-1803.
Matsuo H., Kamakura K., Saito M., et al. Familial paroxysmal dystonic choreoathetosis – clinical findings in a large Japanese family and genetic linkage to 2q. Arch Neurol. 1999;56:721-726.
Matthews W.B. Tonic seizures in disseminated sclerosis. Brain. 1958;81:193-206.
Mayeux R., Fahn S. Paroxysmal dystonic choreoathetosis in a patient with familial ataxia. Neurology. 1982;32:1184-1186.
Meierkord H., Fish D.R., Smith S.J.M., et al. Is nocturnal paroxysmal dystonia a form of frontal lobe epilepsy? Mov Disord. 1992;7:38-42.
Menkes J.H., Ament M.E. Neurologic disorders of gastroesophageal function. Adv Neurol. 1988;49:409-416.
Merchut M.P., Brumlik J. Painful tonic spasms caused by putaminal infarction. Stroke. 1986;17:1319-1321.
Meyer B.U., Irlbacher K., Meierkord H. Analysis of stimuli triggering attacks of paroxysmal dystonia induced by exertion. J Neurol Neurosurg Psychiatry. 2001;70:247-251.
Micheli F., Fernandez Pardal M.M., Casas Parera I., Giannaula R. Sporadic paroxysmal dystonic choreoathetosis associated with basal ganglia calcifications [letter]. Ann Neurol. 1986;20:750.
Micheli F., Fernandez Pardal M., de Arbelaiz R., et al. Paroxysmal dystonia responsive to anticholinergic drugs. Clin Neuropharmacol. 1987;10:365-369.
Milandre L., Brosset C., Gabriel B., Khalil R. Mouvements involontaires transitoires et infarctus thalamiques. (Transient dyskinesias associated with thalamic infarcts – report of five cases. Rev Neurol. 1993;149:402-406.
Miley C.E., Forster F.M. Paroxysmal signs and symptoms in multiple sclerosis. Neurology. 1974;24:458-461.
Miller V.S., Packard A.M. Paroxysmal downgaze in term newborn infants. J Child Neurol. 1998;13:294-295.
Mink J.W., Neil J.J. Masturbation mimicking paroxysmal dystonia or dyskinesia in a young girl. Mov Disord. 1995;10:518-520.
Mir P., Huang Y.Z., Gilio F., et al. Abnormal cortical and spinal inhibition in paroxysmal kinesigenic dyskinesia. Brain. 2005;128:291-299.
Mirsattari S.M., Berry M.E.R., Holden J.K., et al. Paroxysmal dyskinesias in patients with HIV infection. Neurology. 1999;52:109-114.
Morgan J.C., Hughes M., Figueroa R.E., Sethi K.D. Psychogenic paroxysmal dyskinesia following paroxysmal hemidystonia in multiple sclerosis. Neurology. 2005;65(6):E12.
Mount L.A., Reback S. Familial paroxysmal choreoathetosis. Arch Neurol Psychiatry. 1940;44:841-847.
Munchau A., Valente E.M., Shahidi G.A., et al. A new family with paroxysmal exercise induced dystonia and migraine: a clinical and genetic study. J Neurol Neurosurg Psychiatry. 2000;68:609-614.
Mushet G.R., Dreifuss F.E. Paroxysmal dyskinesia. A case responsive to benztropine mesylate. Arch Dis Child. 1967;42:654-656.
Nagamitsu S., Matsuishi T., Hashimoto K., et al. Multicenter study of paroxysmal dyskinesias in Japan – Clinical and pedigree analysis. Mov Disord. 1999;14:658-663.
Nair K.R., Bhaskaran R., Marsden C.D. Essential tremor associated with paroxysmal kinesigenic dystonia. Mov Disord. 1991;6:92-93.
Nakken K.O., Magnusson A., Steinlein O.K. Autosomal dominant nocturnal frontal lobe epilepsy: An electroclinical study of a Norwegian family with ten affected members. Epilepsia. 1999;40:88-92.
Nardocci N., Lamperti E., Rumi V., Angelini L. Typical and atypical forms of paroxysmal choreoathetosis. Dev Med Child Neurol. 1989;31:670-674.
Nath A., Jankovic J., Pettigrew L.C. Movement disorders and AIDS. Neurology. 1987;37:37-41.
Nechay A., Ross L.M., Stephenson J.B.P., O’Regan M. Gratification disorder (“infantile masturbation”): a review. Arch Dis Child. 2004;89:225-226.
Newman R.P., Kinkel W.R. Paroxysmal choreoathetosis due to hypoglycemia. Arch Neurol. 1984;41:341-342.
Newton M.R., Berkovic S.F., Austin M.C., et al. Dystonia, clinical lateralization, and regional blood flow changes in temporal lobe seizures. Neurology. 1992;42:371-377.
Nezu A., Kimura S., Ohtsuki N., et al. Alternating hemiplegia of childhood: Report of a case having a long history. Brain Dev. 1997;19:217-221.
Nightingale S., Barton M.E. Intermittent vertical supranuclear ophthalmoplegia and ataxia. Mov Disord. 1991;6:76-78.
Nijssen P.C.G., Tijssen C.C. Stimulus-sensitive paroxysmal dyskinesias associated with a thalamic infarct. Mov Disord. 1992;7:364-366.
Nutt J.G., Gancher S.T. A 10-year follow up – genotypic analysis disproves phenotypic classification. Mov Disord. 1997;12:472.
Oldani A., Zucconi M., Asselta R., et al. Autosomal dominant nocturnal frontal lobe epilepsy – a video-polysomnographic and genetic appraisal of 40 patients and delineation of the epileptic syndrome. Brain. 1998;121:205-223.
Ondo W.G., Verma A. Physiological assessment of paroxysmal dystonia secondary to subacute sclerosing panencephalitis. Mov Disord. 2002;17(1):154-157.
Ophoff R.A., Terwindt G.M., Vergouwe M.N., et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543-552.
Ouvrier R.A., Billson M.D. Benign paroxysmal tonic upgaze of childhood. J Child Neurol. 1988;3:177-180.
Ouvrier R., Billson F. Paroxysmal tonic upgaze of childhood – a review. Brain Dev. 2005;27(3):185-188.
Panagiotakaki E., Gobbi G., Neville B., et al. Evidence of a non-progressive course of alternating hemiplegia of childhood: study of a large cohort of children and adults. Brain. 2010;133(12):3598-3610.
Parker H.L. Periodic ataxia. Mayo Clin Proc. 1946;38:642-645.
Pereira A.C., Loo W.J., Bamford M., Wroe S.J. Use of lamotrigine to treat paroxysmal kinesigenic choreoathetosis. J Neurol Neurosurg Psychiatry. 2000;68:796-797.
Perez-Borja C., Tassinari A.C., Swanson A.G. Paroxysmal choreoathetosis and seizures induced by movement (reflex epilepsy). Epilepsia. 1967;8:260-270.
Perlmutter J.S., Raichle M.E. Pure hemidystonia with basal ganglion abnormalities on positron emission tomography. Ann Neurol. 1984;15:228-233.
Perniola T., Margari L., de Iaco M.G., et al. Familial paroxysmal exercise-induced dyskinesia, epilepsy, and mental retardation in a family with autosomal dominant inheritance. Mov Disord. 2001;16:724-730.
Phillips H.A., Favre I., Kirkpatrick M., et al. CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet. 2001;8:225-231.
Phillips H.A., Scheffer I.E., Crossland K.M., et al. Autosomal dominant nocturnal frontal-lobe epilepsy: genetic heterogeneity and evidence for a second locus at 15q24. Am J Hum Genet. 1998;63:1108-1116.
Plant G. Focal paroxysmal kinesigenic choreoathetosis. J Neurol Neurosurg Psychiatry. 1983;46:345-348.
Plant G.T., Williams A.C., Earl C.J., Marsden C.D. Familial paroxysmal dystonia induced by exercise. J Neurol Neurosurg Psychiatry. 1984;47:275-279.
Pons R., Collins A., Rotstein M., et al. The spectrum of movement disorders in Glut-1 deficiency. Mov Disord. 2010;25(3):275-281.
Postert T., Amoiridis G., Pohlau D., et al. Episodic undulating hyperkinesias of the tongue associated with brainstem ischemia. Mov Disord. 1997;12:619-621.
Pourfar M., Guerrini R., Parain D., Frucht S.J. Classification conundrums in paroxysmal dyskinesias: a new subtype or variations on classic themes? Mov Disord. 2005;20:1047-1051.
Prashantha D.K., Pal P.K. Pseudohypoparathyroidism manifesting with paroxysmal dyskinesias and seizures. Mov Disord. 2009;24(4):623-624.
Przuntek H., Monninger P. Therapeutic aspects of kinesigenic paroxysmal choreoathetosis and familial paroxysmal choreoathetosis of the Mount and Reback type. J Neurol. 1983;230:163-169.
Raskind W.H., Bolin T., Wolff J., et al. Further localization of a gene for paroxysmal dystonic choreoathetosis to a 5-cM region on chromosome 2q34. Hum Genet. 1998;102:93-97.
Richards R.N., Barnett H.J. Paroxysmal dystonic choreoathetosis. A family study and review of the literature. Neurology. 1968;18:461-469.
Richardson J.C., Howes J.L., Celinski M.J., Allman R.G. Kinesigenic choreoathetosis due to brain injury. Can J Neurol Sci. 1987;14:626-628.
Riley D.E. Paroxysmal kinesigenic dystonia associated with a medullary lesion. Mov Disord. 1996;11:738-740.
Robin J.J. Paroxysmal choreoathetosis following head injury. Ann Neurol. 1977;2:447-448.
Roll P., Massacrier A., Pereira S., et al. New human sodium/glucose cotransporter gene (KST1): identification, characterization, and mutation analysis in ICCA (infantile convulsions and choreoathetosis) and BFIC (benign familial infantile convulsions) families. Gene. 2002;285(1–2):141-148.
Roos R., Wintzen A.R., Vielvoye G., Polder T.W. Paroxysmal kinesiogenic choreoathetosis as presenting symptom of multiple sclerosis. J Neurol Neurosurg Psychiatry. 1991;54:657-658.
Rosen J.A. Paroxysmal choreoathetosis. Associated with perinatal hypoxic encephalopathy. Arch Neurol. 1964;11:385-387.
Rosman N.P., Douglass L.M., Sharif U.M., Paolini J. The neurology of benign paroxysmal torticollis of infancy: report of 10 new cases and review of the literature. J Child Neurol. 2009;24(2):155-160.
Rotstein M., Doran J., Yang H., et al. Glut1 deficiency and alternating hemiplegia of childhood. Neurology. 2009;73(23):2042-2044.
Roubertie A., Echenne B., Leydet J., et al. Benign paroxysmal tonic upgaze, benign paroxysmal torticollis, episodic ataxia and CACNA1A mutation in a family. J Neurol. 2008;255(10):1600-1602.
Rowland L.P., Gordon P.H. Familial periodic paralysis. 11th ed. Rowland LP. Merritt’s Neurology. Philadelphia: Lippincott Williams & Wilkins; 2005. pp. 911–915
Sanner G., Bergstrom B. Benign paroxysmal torticollis in infancy. Acta Paediatr Scand. 1979;68:219-223.
Scheffer I.E., Bhatia K.P., Lopes-Cendes I., et al. Autosomal dominant nocturnal frontal lobe epilepsy – a distinctive clinical disorder. Brain. 1995;118:61-73.
Scheidtmann K., Schwarz J., Holinski E., et al. Paroxysmal choreoathetosis a disorder related to Huntington’s disease? J Neurol. 1997;244:395-398.
Schloesser D.T., Ward T.N., Williamson P.D. Familial paroxysmal dystonic choreoathetosis revisited. Mov Disord. 1996;11:317-320.
Schmidt B.J., Pillay N. Paroxysmal dyskinesia associated with hypoglycemia. Can J Neurol Sci. 1993;20:151-153.
Schneider S.A., Paisan-Ruiz C., Garcia-Gorostiaga I., et al. GLUT1 gene mutations cause sporadic paroxysmal exercise-induced dyskinesias. Mov Disord. 2009;24(11):1684-1688.
Sellal F., Hirsch E., Maquet P., et al. Postures et mouvements anormaux paroxystiques au cours du sommeil: dystonie paroxystique hypnogenique ou epilepsie partielle? (Abnormal paroxysmal movements during sleep: hypnogenic paroxysmal dystonia or focal epilepsy?). Rev Neurol. 1991;147:121-128.
Senbil N., Yapici Z., Gürer Y.K. Paroxysmal non-kinesigenic and hypnogenic dyskinesia associated with streptococcal infection. Pediatr Int. 2008;50(2):255-256.
Sethi K.D., Hess D.C., Huffnagle V.H., Adams R.J. Acetazolamide treatment of paroxysmal dystonia in central demyelinating disease. Neurology. 1992;42:919-921.
Sethi K.D., Lee K.H., Deuskar V., Hess D.C. Orthostatic paroxysmal dystonia. Mov Disord. 2002;17(4):841-845.
Shaw C., Haas L., Miller D., Delahunt J. A case report of paroxysmal dystonic choreoathetosis due to hypoglycaemia induced by an insulinoma. J Neurol Neurosurg Psychiatry. 1996;61:194-195.
Shin H.W., Kang S.Y., Hallett M., Sohn Y.H. Extended surround inhibition in idiopathic paroxysmal kinesigenic dyskinesia. Clin Neurophysiol. 2010;121(7):1138-1141.
Shirane S., Sasaki M., Kogure D., et al. Increased ictal perfusion of the thalamus in paroxysmal kinesigenic dyskinesia. J Neurol Neurosurg Psychiatry. 2001;71:408-410.
Snyder C.H. Paroxysmal torticollis in infancy. Am J Dis Child. 1969;117:458-460.
Soffer D., Licht A., Yaar I., Abramsky O. Paroxysmal choreoathetosis as a presenting symptom in idiopathic hypoparathyroidism. J Neurol Neurosurg Psychiatry. 1977;40:692-694.
Spacey S.D., Adams P.J., Lam P.C., et al. Genetic heterogeneity in paroxysmal nonkinesigenic dyskinesia. Neurology. 2006;66(10):1588-1590.
Spacey S., Materek L.A., Szczgielski B.I., Bird T.D. Two novel CACNA1A gene mutations associated with episodic ataxia type 2 and interictal dystonia. Arch Neurol. 2005;62:314-316.
Spacey S.D., Valente E.M., Wali G.M., et al. Genetic and clinical heterogeneity in paroxysmal kinesigenic dyskinesia: Evidence for a third EKD gene. Mov Disord. 2002;17(4):717-725.
Spiller W.G. Subcortical epilepsy. Brain. 1927;50:171-187.
Steckley J.L., Ebers G.C., Cader M.Z., McLachlan R.S. An autosomal dominant disorder with episodic ataxia, vertigo, and tinnitus. Neurology. 2001;57:1499-1502.
Stefanova E., Djarmati A., Momcilovic D., et al. Clinical characteristics of paroxysmal nonkinesigenic dyskinesia in Serbian family with Myofibrillogenesis regulator 1 gene mutation. Mov Disord. 2006;21(11):2010-2015.
Stevens H. Paroxysmal choreo-athetosis. Arch Neurol. 1966;14:415-420.
Suber D.A., Riley T.L. Valproic acid and normal computerized tomographic scan in kinesigenic familial paroxysmal choreoathetosis [letter]. Arch Neurol. 1980;37:327.
Suls A., Dedeken P., Goffin K., et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain. 2008;131(Pt 7):1831-1844.
Sweney M.T., Silver K., Gerard-Blanluet M., et al. Alternating hemiplegia of childhood: early characteristics and evolution of a neurodevelopmental syndrome. Pediatrics. 2009;123(3):e534-541.
Swoboda K.J., Soong B.W., McKenna C., et al. Paroxysmal kinesigenic dyskinesia and infantile convulsions – clinical and linkage studies. Neurology. 2000;55:224-230.
Szczałuba K., Jurek M., Szczepanik E., et al. A family with paroxysmal nonkinesigenic dyskinesia: genetic and treatment issues. Pediatr Neurol. 2009;41(2):135-138.
Szepetowski P., Rochette J., Berquin P., et al. Familial infantile convulsions and paroxysmal choreoathetosis: A new neurological syndrome linked to the pericentromeric region of human chromosome 16. Am J Hum Genet. 1997;61:889-898.
Tabaee-Zadeh M.J., Frame B., Kapphahn K. Kinesiogenic choreoathetosis and idiopathic hypoparathyroidism. N Engl J Med. 1972;286:762-763.
Tan A., Salgado M., Fahn S. The characterization and outcome of stereotypic movements in nonautistic children. Mov Disord. 1997;12:47-52.
Terzano M.G., Monge-Strauss M.F., Mikol F., et al. Cyclic alternating pattern as a provocative factor in nocturnal paroxysmal dystonia. Epilepsia. 1997;38:1015-1025.
Thiriaux A., de St Martin A., Vercueil L., et al. Co-occurrence of infantile epileptic seizures and childhood paroxysmal choreoathetosis in one family: Clinical, EEG, and SPECT characterization of episodic events. Mov Disord. 2002;17(1):98-104.
Tibbles J.A., Barnes S.E. Paroxysmal dystonic choreoathetosis of Mount and Reback. Pediatrics. 1980;65:149-151.
Tinuper P., Cerullo A., Cirignotta F., et al. Nocturnal paroxysmal dystonia with short-lasting attacks: Three cases with evidence for an epileptic frontal lobe origin of seizures. Epilepsia. 1990;31:549-556.
Tomita H., Nagamitsu S., Wakui K., et al. Paroxysmal kinesigenic choreoathetosis locus maps to chromosome 16p11.2-q12.1. Am J Hum Genet. 1999;65:1688-1697.
Tsao C.Y. Effective treatment with oxcarbazepine in paroxysmal kinesigenic choreoathetosis. J Child Neurol. 2004;19:300-301.
Uberall M.A., Wenzel D. Effectiveness of lamotrigine in children with paroxysmal kinesigenic choreoathetosis. Dev Med Child Neurol. 2000;42:699-700.
Urbizu A., Cuenca-León E., Raspall-Chaure M., et al. Paroxysmal exercise-induced dyskinesia, writer’s cramp, migraine with aura and absence epilepsy in twin brothers with a novel SLC2A1 missense mutation. J Neurol Sci. 2010;295(1–2):110-113.
Vaamonde J., Artieda J., Obeso J.A. Hereditary paroxysmal ataxia with neuromyotonia. Mov Disord. 1991;6:180-182.
Vahedi K., Joutel A., van Bogaert P., et al. A gene for hereditary paroxysmal cerebellar ataxia maps to chromosome 19p. Ann Neurol. 1995;37:289-293.
Valente E.M., Spacey S.D., Wali G.M., et al. A second paroxysmal kinesigenic choreoathetosis locus (EKD2) mapping on 16q13-q22.1 indicates a family of genes which give rise to paroxysmal disorders on human chromosome 16. Brain. 2000;123:2040-2045.
Van Dyke D.H., Griggs R.C., Murphy M.J., Goldstein M.N. Hereditary myokymia and periodic ataxia. J Neurol Sci. 1975;25:109-118.
Verheul G.A.M., Tyssen C.C. Multiple sclerosis occurring with paroxysmal unilateral dystonia. Mov Disord. 1990;5:352-353.
Vighetto A., Froment J.C., Trillet M., Aimard G. Magnetic resonance imaging in familial paroxysmal ataxia. Arch Neurol. 1988;45:547-549.
Vincent F.M. Hyperglycemia-induced hemichoreoathetosis: the presenting manifestation of a vascular malformation of the lenticular nucleus. Neurosurgery. 1986;18:787-790.
von Brederlow B., Hahn A., Koopman W.J., et al. Mapping the gene for acetazolamide responsive hereditary paryoxysmal cerebellar ataxia to chromosome 19p. Hum Mol Genet. 1995;4:279-284.
Wali G.M. Paroxysmal hemidystonia induced by prolonged exercise and cold. J Neurol Neurosurg Psychiatry. 1992;55:236-237.
Walker E.S. Familial paroxysmal dystonic choreoathetosis: a neurologic disorder simulating psychiatric illness. Johns Hopkins Med J. 1981;148:108-113.
Waller D.A. Paroxysmal kinesigenic choreoathetosis or hysteria? Am J Psychiatry. 1977;134:1439-1440.
Walters A.S., Hening W.A., Chokroverty S. Review and videotape recognition of idiopathic restless legs syndrome. Mov Disord. 1991;6:105-110.
Watson R.T., Scott W.R. Paroxysmal kinesigenic choreoathetosis and brain-stem atrophy [letter]. Arch Neurol. 1979;36:522.
Weber M.B. Familial paroxysmal dystonia. J Nerv Ment Dis. 1967;145:221-226.
Weber Y.G., Lerche H. Genetics of paroxysmal dyskinesias. Curr Neurol Neurosci Rep. 2009;9(3):206-211.
Weber Y.G., Storch A., Wuttke T.V., et al. GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J Clin Invest. 2008;118(6):2157-2168.
White J.C. Familial periodic nystagmus, vertigo, and ataxia. Arch Neurol. 1969;20:276-280.
Whitty C.W.M., Lishman W.A., FitzGibbon J.P. Seizures induced by movement: a form of reflex epilepsy. Lancet. 1964;1:1403-1406.
Wilson S.A.K. The Morrison Lectures on nervous semeiology, with special reference to epilepsy. Lecture III. Symptoms indicating increase of neural function. Br Med J. 1930;2:90-94.
Winer J.B., Fish D.R., Sawyers D., Marsden C.D. A movement disorder as a presenting feature of recurrent hypglycaemia. Mov Disord. 1990;5:176-177.
Yamada K., Goto S., Soyama N., et al. Complete suppression of paroxysmal nonkinesigenic dyskinesia by globus pallidus internus pallidal stimulation. Mov Disord. 2006;21:576-580.
Yamamoto K., Kawazawa S. Basal ganglion calcification in paroxysmal dystonic choreoathetosis [letter]. Ann Neurol. 1987;22:556.
Yang M.L., Fullwood E., Goldstein J., Mink J.M. Masturbation in infancy and early childhood presenting as a movement disorder: 12 cases and a review of the literature. Pediatrics. 2005;116:1427-1432.
Yokochi K. Paroxysmal ocular downward deviation in neurologically impaired infants. Pediatr Neurol. 1991;7:426-428.
Yoon J.H., Lee P.H., Yoon S.N. Subtraction brain SPECT imaging in a patient with paroxysmal exercise-induced dystonia: role of the primary somatosensory cortex. Arch Neurol. 2007;64(11):1652-1656.
Yue Q., Jen J.C., Nelson S.F., Baloh R.W. Progressive ataxia due to a missense mutation in a calcium-channel gene. Am J Hum Genet. 1997;61:1078-1087.
Yue Q., Jen J.C., Thwe M.M., et al. De novo mutation in CACNA1A caused acetazolamide-responsive episodic ataxia. Am J Med Genet. 1998;77:298-301.
Yulug B., Bakar M., Ozer H.H., et al. Paroxysmal kinesigenic dyskinesia and cervical disc prolapse with cord compression: more than a coincidence? J Neuropsychiatry Clin Neurosci. 2008;20(2):237-239.
Zacchetti O., Sozzi G., Zampollo A. Paroxysmal kinesigenic choreoathetosis. Case report. Ital J Neurol Sci. 1983;3:345-347.
Zasorin N.L., Baloh R.W., Myers L.B. Acetazolamide-responsive episodic ataxia syndrome. Neurology. 1983;33:1212-1214.
Zerr P., Adelman J.P., Maylie J. Episodic ataxia mutations in Kv1.1 alter potassium channel function by dominant negative effects or haploinsufficiency. J Neurosci. 1998;18:2842-2848.
Zhou J., Li G., Chen C., et al. Familial pure paroxysmal kinesigenic dyskinesia in Han population from the Chinese mainland: a new subtype? Epilepsy Res. 2008;80(2–3):171-179.
Zhuchenko O., Bailey J., Bonnen P., et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15:62-69.
Zorzi G., Castellotti B., Zibordi F., et al. Paroxysmal movement disorders in GLUT1 deficiency syndrome. Neurology. 2008;71(2):146-148.