Structures Supporting Cancer Clinical Trials
Jeffrey S. Abrams, Margaret Mooney, James A. Zwiebel, Michaele C. Christian and James H. Doroshow
• Cancer clinical trials provide the evidence on which sound oncology practice is based.
• Providing a greater efficiency in implementing clinical trials and achieving enrollment rapidly has been a major goal of the National Cancer Institute, cancer centers, the biopharmaceutical industry, and patient advocacy groups.
• Physicians in private practice have many opportunities to participate in clinical trials sponsored by the National Cancer Institute and/or the biopharmaceutical industry.
• Physicians and patients have online access to all U.S. sponsored clinical trials at clinicaltrials.gov and have access to additional information about cancer trials at cancer.gov.
• Successful participation in clinical trials includes the following clinical components:
 The attitude and commitment of the physician
The attitude and commitment of the physician
 Sufficient preparation and infrastructure
Sufficient preparation and infrastructure
 Trained staff, including at least some of the following:
Trained staff, including at least some of the following:



• Affiliation with an institution or network that provides scientific and administrative support, such as the following:



 Biopharmaceutical industry network
Biopharmaceutical industry network

 Access to an authorized Institutional Review Board
Access to an authorized Institutional Review Board
 Access to adequate laboratory facilities to process protocol-required specimens
Access to adequate laboratory facilities to process protocol-required specimens
 Adherence to good clinical practices
Adherence to good clinical practices
 Accurate and timely data reporting
Accurate and timely data reporting
 Proper maintenance of primary source documentation
Proper maintenance of primary source documentation
 Adequate preparation for on-site audits
Adequate preparation for on-site audits

• Many organizations now provide access to clinical trials and/or provide the necessary training and certification; relevant Web sites are included in this chapter.
National Cancer Institute–Sponsored Clinical Trials Activities
The NCI supports the development of more than 100 agents for cancer treatment and prevention (many in collaboration with pharmaceutical and biotechnology companies) and has an extensive clinical trials system that encompasses treatment, prevention, and cancer control studies. More than 800 trials are active at any given time, and several hundred new trials open each year. In the treatment area, the NCI has programs for early therapeutics development (primarily phases I and II trials), including many sites with grants and contracts to complete these early trials. The NCI also supports a large program of Clinical Trials Cooperative Groups that conduct later-phase trials, predominantly pilot studies and phase III trials. Clinical Trials Cooperative Groups funded by the NCI provide a standing mechanism for performing large-scale multicenter treatment, cancer control, diagnostic, and prevention trials. Phase III trials are conducted nationally, and a member of any NCI-supported Cooperative Group is able to participate in trials conducted by any of the Groups, because they all share a common menu of trials on the Cancer Trials Support Unit Web site (www.ctsu.org). Over the years, this system has supported an experienced cadre of clinical researchers, biostatisticians, and research support staff who can respond to new clinical discoveries by organizing definitive phase III clinical trials. Because of their size and complexity, these trials require extensive infrastructure support to manage the necessary regulatory and data-reporting tasks. Although these Cooperative Group trials have provided a significant proportion of the evidence on which oncology practice is based, public advocacy for more rapid progress, increasing fiscal pressures on medical practice, and the accelerated pace of drug discovery caused the NCI to review and restructure aspects of its clinical trials program beginning in 1998. The major goals of these restructuring efforts were to increase access to NCI trials for patients and their physicians; eliminate barriers to participation in clinical trials; improve the coordination and cooperation among the functionally diverse elements of the NCI clinical trials program, including relationships with industry and the U.S. Food and Drug Administration (FDA); improve the prioritization process for developing clinical trials leading to increased scientific quality; enhance the standardization of tools for clinical trial design and data capture; increase operational efficiency so that trials could be executed in a more timely manner; and most recently, restructure the Cooperative Groups into a tightly integrated network by reducing the number of Groups and changing the review criteria for funding to emphasize collaboration and cooperation.
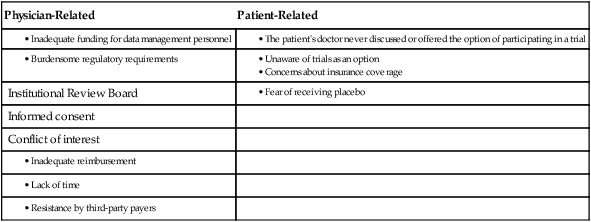
Through surveys among physicians and the public, it was found that the obstacles to accrual in adult oncology were multifactorial (Table 20-1).3–3 With these barriers in mind, the NCI undertook a series of comprehensive analyses of how it conducts and funds clinical trials by involving a wide range of stakeholders that included Cooperative Group and Cancer Center leaders, patient advocates, representatives from the FDA and the pharmaceutical industry, and government staff. The detailed reports of these reviews are available online: the 2005 review (http://integratedtrials.nci.gov/ict/ctwg_report_June2005.pdf from 2005), the 2008 review (http://ccct.cancer.gov/files/OEWG-Report.pdf), and most recently, the 2010 Institute of Medicine (IOM) Report (http://books.nap.edu/openbook.php?record_id=12879). Several important projects were launched based on the recommendations, all of which were aimed at modernizing NCI’s clinical trials regulatory and data collection systems, opening trial access to more patients and investigators, and simplifying the role of local Institutional Review Boards (IRBs) in multiinstitutional clinical trials. New opportunities were created for community physicians to participate in a broad array of clinical trials, and new tools were created to enable this participation. Two major initiatives, the Cancer Trials Support Unit (CTSU) and the Central Institutional Review Board (CIRB), were uniformly supported by all stakeholders and have now become integral parts of NCI’s clinical trials system; they are described next.
Cancer Trials Support Unit
The NCI CTSU is designed to facilitate one-stop online access to a broad menu of phase III trials, large phase II trials, and selected international collaborative trials by a national network of NCI investigators. Network investigators can access the CTSU menu of treatment trials from a public Web site (www.ctsu.org). The scientific leadership for each study remains within the organization that developed the trial, but patient enrollment can come from any network physician across the country, and all patient enrollment is handled through CTSU’s central registration system, the Oncology Patient Enrollment Network. By providing more physicians and their patients with the opportunity to choose from a broader menu of trials, the CTSU promotes faster accrual to individual trials, allows increased access and additional treatment options to more patients nationwide, and renders trials involving uncommon cancers more feasible. Although the clinical trials menu and centralized patient enrollment are the most visible aspects of the CTSU, another major function of the CTSU is its centralized regulatory database. For all physicians in the network, the CTSU maintains important demographic information about their sites or practices, including clinical trial group affiliation and academic/practice affiliation(s), Office for Human Research Protection assurance numbers for their sites, IRB approvals for specific protocols, and conflict of interest forms for investigators. This process enables physicians, nurses, and clinical research associates to register once annually instead of having to register for each clinical trial group or trial in which they participate. Centralizing regulatory data has reduced the workload for investigators in the field, consolidated duplicative work, and allowed clinical trial group staff to partially offload this activity to the CTSU and focus instead on protocol development and analysis.
Central Institutional Review Board
In NCI-sponsored multicenter trials, the identical protocol is carried out at many sites, often including as many as 100 different practices; each site requires its own local IRB (LIRB) to conduct an initial full-board review and subsequent annual reviews, adverse event reviews, and amendment reviews. These multiple IRB reviews create a largely redundant, time-consuming workload at these sites, compounding the ever-mounting pressures on the nation’s IRB system, which have been well documented.4 To provide an idea of the scope of the duplicative effort that occurs, consider that NCI has more than 8000 registered investigators at more than 1500 sites. On average, there are 160 ongoing phase III trials and 30 new trials entering the NCI system annually, resulting in approximately 16,000 IRB reviews (3000 initial reviews) conducted each year.4 In addition, investigators often mention that the amount of time, paperwork, and funding required for them to obtain IRB approval is a serious barrier to opening trials. These factors provided the impetus for the NCI to develop a new, centralized approach to human subjects protection for its large phase III trials program.
The benefits to research participants include study review by persons who represent a broad range of oncology expertise. The benefits to LIRBs include the ability to carry out local review without convening the entire Board. Continuing reviews, amendments, and nonlocal adverse events are handled by the CIRB as well. The benefits to investigators include time and effort saved because they can download an already completed IRB application for each study, as well as eliminating the need to submit amendments, continuing reviews, and nonlocal adverse events to their IRB. An economic analysis demonstrated that local sites have a substantial cost savings as members of the CIRB.5 In addition, subjects are enrolled in trials more quickly because the full LIRB does not need to meet. In 2011, the NCI launched a pilot program to change the model for the NCI CIRBs to that of a full IRB (i.e., single IRB of record) for participating institutions. Thus in the future the NCI CIRB will function in a fashion similar to commercial IRBs.
NCI National Clinical Trials Network
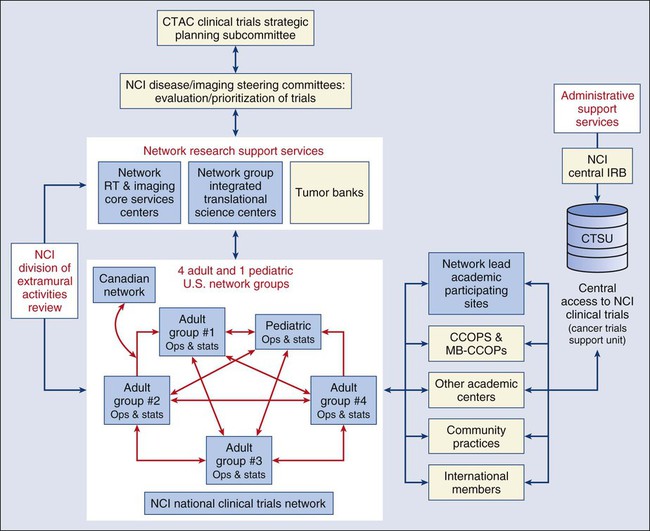
The 2010 IOM report (http://iom.edu/Reports/2010/A-National-Cancer-Clinical-Trials-System-for-the-21st-Century-Reinvigorating-the-NCI-Cooperative.aspx) noted that, despite its impressive record of accomplishment, the current NCI-supported trials system had become less efficient, had difficulty prioritizing studies, and was underfunded for the number of trials it conducts. The IOM report recommended that the existing adult Cooperative Groups be consolidated into a smaller number of Groups, each with greater capabilities and appropriate incentives to promote a more integrated system overall. Consolidation should promote efficiency by encouraging a structural makeover of clinical trial group operations centers, statistics, and data management centers.6 It should also facilitate prioritization in clinical research by focusing trials more strictly than previously on questions unlikely to be addressed by the private sector. As a result of the IOM review, as well as input received by the NCI from stakeholders across the oncology community, the NCI developed a comprehensive plan to transform the previous NCI-sponsored Clinical Trials Cooperative Group Program that funded multiple separate organizations conducting cancer treatment trials into a new, consolidated, and integrated program referred to as the NCI National Clinical Trials Network (NCTN).
In the NCTN Program, Network Groups are expected to collaborate with each other and with NCI to achieve the overall goal of the Program. Member institutions/sites of Network Groups will be able to enroll patients in all adult phase III trials, as well as select early phase trials and trials in adolescents and young adults, irrespective of the specific Network Group that is leading the trial. Network Groups will also provide trial operations, data management, and statistical support for approved, multi-center phase II and phase III trials originating outside the Network Group. The NCTN Program, including each of its six key components, is illustrated in Figure 20-1. Although the reconfiguration of the former Cooperative Group program previously described will not officially be in place until 2014, several of the Cooperative Groups have already publicly announced their consolidation plans (http://www.cancer.gov/ncicancerbulletin/032211/page9) and are working together to develop new clinical trials.
Other National Cancer Institute–Sponsored Structures Supporting Clinical Trials
Community Clinical Oncology Program
For practices or groups or networks of practices that already have research experience and can commit to enrolling significant numbers of patients in clinical trials, NCI grant funding through the Community Clinical Oncology Program (CCOP) mechanism offers many opportunities. CCOPs must document the ability to enroll 50 patients per year in cancer treatment studies plus 50 to 75 patients in prevention studies and/or trials focused on symptom management and cancer control. The grants provide important up-front funding to enable sites to hire critical staff from the start to support the substantial patient accrual that is required. In addition, special minority-based CCOPs are funded to provide additional support for groups or networks that serve predominantly minority populations. Minority-based CCOPs strive to increase cancer prevention and control activities in minority and underserved communities in addition to increasing access to clinical trials for minority patients. The CCOP program, funded by the NCI’s Division of Cancer Prevention, has been in existence since 1983. In 2010, 47 CCOPs and 16 minority-based CCOPs received funding for participation in NCI-approved clinical trials. Overall, the program comprises 3375 participating physicians and 395 participating hospitals. CCOP sites accrued more than 12,015 patients/participants to cancer treatment, prevention, and control trials in 2009. Details on these programs can be found at the NCI Division of Cancer Prevention’s Web site (http://dcp.cancer.gov/programs-resources/programs/ccop).
Phases I and II Early Therapeutics Clinical Trials Networks
• The integration of NCI’s funded tumor biology and translational science programs (e.g., the NCI-designated Cancer Centers and the Specialized Programs of Research Excellence ) with clinical investigators managing the ET-CTN. This integration will require the formation of interdisciplinary teams for investigational agent development. These project teams will bring together the necessary expertise to both formulate key questions and execute clinical trials in the development of specific investigational agents.
• Addition of laboratories with validated assays that will carry out the molecular characterization of patient tumors both before and after treatment.
• The establishment of a centralized regulatory, data management, and pharmacologic core to support the ET-CTN, making multisite studies easier to perform even in the earliest stages of drug development.
• The extension of the NCI’s CIRB to these early phase multicenter trials by establishment of a new review board with the requisite expertise to focus on early phase clinical trials.
National Community Cancer Centers Program
The NCI Community Cancer Centers Program (NCCCP) was launched as a pilot program in 2007.7 The ultimate aim of the NCCCP was to expand cancer research further into the community and enhance the delivery of the latest, most advanced cancer care to a greater number of Americans in the communities in which they live. To achieve the program’s stated aims, NCI established a public-private partnership with a selected group of participating community hospitals and health systems. After 3 years, most sites were able to achieve specific accomplishments, such as:
• Enhancing research capacity in essential areas, particularly in trial capacity and infrastructure (e.g., more phase II trials and biospecimen collection);
• Improving quality of care for key indicators of breast and colon cancer treatment;
• Leveraging a high level of matching funds for implementing NCCCP activities; and
• Establishing stronger relationships with critical cancer physicians.
Biopharmaceutical Industry–Sponsored Cancer Clinical Trials
Expectations of Clinical Research Sites
Staffing is foremost among the critical components for an effective research practice listed in Box 20-1. The number and precise composition of the necessary staff depend on the number of patients who are enrolled in clinical trials and the nature of the practice. In some settings in which the number of patients in studies is small, one good research nurse can perform many of the required functions. At more active sites, research nurses, clinical research associates, and research pharmacists perform separate functions. For budgeting purposes, for example, it is often estimated that one full-time clinical research associate can handle 25 new patients and up to 50 patients in follow-up in a year. Some of the structures that support clinical trials participation, such as the CCOP program, provide substantial up-front funding to support salaries for the necessary staff in return for a commitment to substantial accrual. Many others, including the CTSU and the Cooperative Groups, provide funding only on a “per-case” basis, and thus start-up costs must be borne by the site.
Educational and Training Tools
As has been described previously, participation in clinical trials adds many complexities to the care of cancer patients and can require that physicians, nurses, and other office staff acquire new and different skills. Fortunately, because of the widespread interest in clinical trials in the cancer community, many resources exist for gaining information about clinical trials and for acquiring the necessary skills. Professional societies are a good source for educational programs and materials, and some, such as the Oncology Nursing Society (http://www.oncc.org/), the Society of Clinical Research Associates (http://www.socra.org/), and the American Society of Health-System Pharmacists (http://www.ashp.org/), actually offer certification programs that can serve as important career development incentives to office staff. The American Society of Clinical Oncology (http://www.asco.org) also has a very useful Web site with links to a variety of sites that provide information about available clinical trials and detailed information about chemotherapy agents for physicians and nurses. NCI-sponsored Cooperative Groups provide regular educational activities for physicians, statisticians, nurses, and data managers who participate in their trials. Furthermore, the biopharmaceutical industry sponsors a wide range of educational activities that are conducted by both academic institutions and professional societies (e.g., the American Society of Clinical Oncology and the American Association of Cancer Research) about the clinical trials process in general and about the responsibilities of the individual clinical investigator.
The NCI’s home page (http://www.cancer.gov/) provides a gateway to the many Web sites at the NCI and provides links to many other useful sites (Box 20-2). It contains extensive information about cancer in general and cancer statistics, as well as detailed information about clinical trials. Useful information on insurance coverage for patients in clinical trials, including the coverage offered by specific insurance carriers, can also be obtained from www.cancer.gov. Subsequent to the U.S. Food and Drug Administration Amendment Act, U.S. federal law now mandates registration of phases II-IV clinical trials with results reporting in many cases. This comprehensive listing of all trials conducted in the United States, whether they are federally or privately sponsored, can be found at www.clinicaltrials.gov.