Stem Cells, Cell Differentiation, and Cancer
Piero Dalerba, Michael F. Clarke, Irving L. Weissman and Maximilian Diehn
• Many tumors originate in organs and tissues that undergo a continuous process of cell turnover, which is sustained by a minority population of stem cells (e.g., the colon, breast, lung, prostate, brain, and bone marrow).
• Stem cells have four fundamental properties: the ability to give rise to new stem cells with intact and unlimited growth potential (self-renewal), the ability to give rise to a progeny of specialized cells (differentiation), the ability to migrate into new tissue locations and establish tissue growth (migration and tissue repair), and the ability to balance the previous three properties according to a genetic program that places constraints on their numerical expansion (homeostatic control).
• In many tissues, stem cells are the only long-lived cells. This observation suggests that early transforming events, either genetic mutations or epigenetic modifications, are likely to accumulate in stem cells.
• In addition to oncogenes that control cell survival and proliferation, there is a class of oncogenes that regulate self-renewal. In some cancers, tumor growth might be sustained by progenitor cells, which do not naturally self-renew but have aberrantly acquired this ability during disease progression.
• Experimental data suggest that, in many leukemias and solid tumors (e.g., breast, colon, head and neck, pancreas, bladder, and prostate carcinomas), only a specific, phenotypically distinct subset of cancer cells is able to form tumors when transplanted in mice.
• Currently, most antitumor drugs are evaluated on the basis of their capacity to rapidly reduce tumor size. However, because self-renewing cancer cells are often minority populations, treatments that simply reduce tumor size may not effectively target these cells.
• To result in cure, therapies must eradicate self-renewing cancer cells. The ability to identify these cells should allow the identification of new diagnostic markers and therapeutic targets.
Introduction
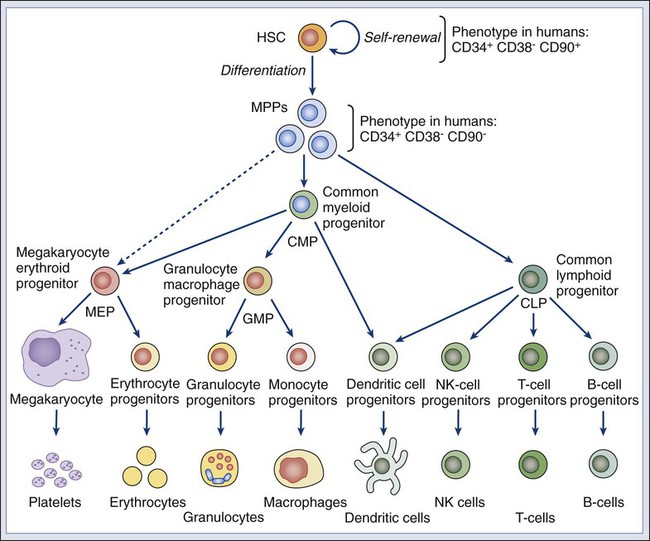
Many tumors originate from tissues that naturally undergo a continuous process of cell turnover. In such tissues, cell maturation is arranged according to a hierarchical system in which a minority population of stem cells is able to perpetuate itself through a process called “self-renewal” while also giving rise to several stages of intermediate progenitors and, eventually, to terminally differentiated cells (Fig. 7-1). As opposed to stem cells, intermediate progenitors and terminally differentiated cells have a limited expansion potential and are unable to self-renew.1,2 Because stem cells are frequently found in small numbers, they must be isolated and tested prospectively to define their molecular and biochemical properties. Although most tissues are likely to regenerate from tissue-specific stem cells, stem cells have been identified in only a few of them. The stem cells that give rise to the lymphohematopoietic system, called hematopoietic stem cells (HSCs), are among the best characterized and have been isolated from mice and humans.3–6 HSCs have important applications in cancer therapy, where bone marrow transplantation is used to regenerate the hematopoietic system after myeloablative treatments.6 The ability to isolate HSCs with high degrees of purity enables the execution of tumor-free autologous bone-marrow transplants in cancer patients.7,8
Understanding the biology of the normal tissues from which tumors originate, and especially of their stem cell populations, can provide important insights into cancer biology. Several aspects of stem cell biology are relevant to cancer.9,10 First, many cancer cells share with normal stem cells the capacity to self-renew, and emerging evidence suggests that similar molecular pathways underpin this property in the two cell types.11–16 Second, because stem cells are long-lived and often represent the only long-lived cells of normal adult tissues, they also represent the most likely target population where transforming events, either genetic mutations or epigenetic modifications, progressively accumulate, at least in the initial phases of the disease.2 Third, increasing evidence suggests that many tumor tissues, akin to normal ones, contain a minority “cancer stem cell” (CSC) population with indefinite proliferative potential that drives tumor growth and metastasis.17–20 Finally, many physiological properties of stem cells are mirrored in cancer cells and help define their biology. For example, because stem cells are long-lived and are necessary to maintain tissue homeostasis over the lifetime of an organism, they are frequently endowed with high levels of enzymatic systems that protect them from environmental hazards. These very same enzymatic systems endow specific subsets of cancer cells with resistance to antitumor agents and might help explain treatment failure.
Properties of Normal Stem Cells
HSCs are the most studied and best understood type of somatic stem cells and serve as a model for stem cells in other tissues.21,22 Hematopoiesis is a tightly regulated process in which a small pool of HSCs gives rise to an increasingly diversified population of oligo-lineage intermediates, which in turn form the full repertoire of mature elements of the lymphohematopoietic system (e.g., erythrocytes, platelets, granulocytes, macrophages, and lymphocytes; Fig. 7-1).23,24 These mature cell types perform a diverse set of functions, ensuring tissue oxygenation, blood coagulation, and immunity. HSCs have four fundamental properties: self-renewal, differentiation, migration, and homeostatic control. First, HSCs need to self-renew to maintain the stem cell pool. Self-renewal is not synonymous with proliferation. Self-renewal is a cell division in which one or both of the daughter cells remain undifferentiated and maintain the identity of a stem cell, endowed with unlimited growth potential. Second, HSCs must undergo differentiation: they must generate a progeny of cells that expand and progressively specialize in order to replenish the various populations of mature cells that are damaged, aged, or otherwise lost. Third, HSCs are able to enter the circulation and migrate from one blood-forming site to another (e.g., from the marrow of one bone to that of another) to replace HSCs that have been lost and to maintain a constant output of blood cells.25 Finally, HSCs must balance self-renewal, differentiation, migration, and tissue repair according to a strict genetic program, which ensures homeostatic control of their numbers and regulates their expansion in response to various types of stress, such as bleeding or infection.28–28
In mice, HSCs represent less than 0.05% of bone marrow cells and are admixed with a heterogeneous pool of other immature progenitors. This pool includes a specific subset of immature multipotent progenitors (MPPs), which are able to differentiate into the full variety of hematopoietic lineages but lack self-renewal capacity.29 These populations form a hierarchy in which HSCs give rise to MPPs, which in turn give rise to oligo-lineage progenitors and the various mature cell types of the blood and lymphoid tissues (Fig. 7-1).6,24 As HSCs mature to become MPPs, they increase their proliferation rate but lose the ability to self-renew. Only HSCs can reconstitute hematopoiesis in the long term, for the lifetime of the animal, whereas MPPs can reconstitute hematopoiesis for a short period, usually less than 16 weeks.30
A similar hierarchy is also mirrored in human blood tissues, in which HSCs and MPPs are characterized by a CD34+CD38− phenotype and by the lack of mature blood cell lineage markers (Lin−) but can be separated on the basis of the differential expression of CD90 in HSCs (Fig. 7-1).31 More generally, the hierarchical organization of hematopoietic tissues can be successfully applied to model the cellular composition and developmental dynamics of many, if not most, adult mammalian tissues, such as the brain, mammary gland, and intestinal epithelium.32–35
Genetic Regulation of Self-Renewal
The long-term survival of a tissue, either normal or neoplastic, is dependent on its capacity to self-renew, whereas its overall size is determined by the balance between the rates of cell proliferation and cell death across its various components.36 In normal tissues, stem cell numbers are under tight genetic regulation, resulting in the long-term maintenance of a constant tissue size.28–28 In contrast, tumor tissues have escaped this homeostatic regulation. Within a tumor, the number of cells with the ability to self-renew is constantly expanding, resulting in continuous tissue growth. Thus it is not surprising that many known oncogenes are able to expand stem cell numbers, either by protecting them from apoptosis or by inhibiting their differentiation. For example, enforced expression of Bcl2 results in an expansion of HSCs,37 whereas enforced expression of c-myb and c-myc prevents HSC differentiation along the erythroid lineage.38,39
Because the size of both normal and cancer tissues is dependent of the number of cells able to self-renew, it is also not surprising that a specific subset of oncogenes and/or tumor-suppressor genes might directly activate and/or disable self-renewal pathways.9,40 Indeed, the basic molecular machinery that ensures the unlimited growth capacity of most cancer cells (e.g., the telomerase enzymatic complex) is fundamental to ensure self-renewal of most normal stem cells.13–13 The capacity to modulate self-renewal, however, is not a necessary attribute of all oncogenes and/or tumor-suppressor genes; enforced expression of Bcl2, for instance, although able to expand the number of self-renewing HSCs, is unable per se to endow self-renewal properties on more mature cell types, such as MPPs.37
Among cancer genes with direct control over self-renewal functions, the best example is probably the Bmi1 oncogene. Bmi1 is a member of the Polycomb gene family, a group of transcriptional repressors involved in the epigenetic regulation of developmental and differentiation processes. In mice, Bmi1 can cooperate with c-myc to induce lymphoma.41 In mice genetically deficient for Bmi1 (Bmi1−/−), the number of HSCs is markedly reduced at birth, and their transplantation in lethally irradiated animals is able to reconstitute hematopoiesis, but only in a transient and self-limiting fashion, indicating a cell autonomous defect of self-renewal.14 Bmi1 exerts similar roles across many types of adult stem cells, such as neural and mammary stem cells.42,43 Lack of Bmi1 expression is associated with upregulation of tumor suppressors, such as the cyclin-dependent kinase inhibitors encoded by the alternative transcripts of the Cdkn2a locus (i.e. the p16Ink4a and p19Arf proteins), and with overexpression of transcription factors that promote differentiation, such as selected members of the Hox gene family.14 This association suggests that, in stem cells, the function of Bmi1 is to prevent the expression of a cascade of genes whose coordinated activity can lead to loss of self-renewal capacity. Remarkably, in a mouse model of leukemia initiated in Bmi1−/− mice, leukemic cells are initially able to expand and cause the death of their primary hosts but are unable to sustain long-term growth when transplanted in secondary syngeneic animals, where they eventually arrest, differentiate, and undergo apoptosis.15 Infection of Bmi1−/− leukemic cells with a retrovirus encoding for Bmi1 completely rescues this defect, allowing leukemic cells to grow indefinitely and be serially passaged in mice.15 Similar observations can also be obtained in human breast cancer, where downregulation of BMI1 by means of enforced expression of microRNA-200c associates with reduced expansion and tumorigenicity of cancer cells.16 Once more, this defect can be rescued by enforced expression of a modified version of the BMI1 messenger RNA, which is insensitive to the inhibitory action of microRNA-200c.16 Interestingly, human BMI1 appears to induce telomerase activity.44 Again, in many human model systems, cancer cells lacking telomerase, although capable of substantial short-term proliferation and numerical expansion, progressively exhaust their growth capacity and irreversibly arrest.13 These studies reinforce the concept that, in order to endow cancer cells with unlimited growth potential—a property frequently referred to as “immortality”36—constitutive activation of proliferation pathways is not sufficient. It is also necessary to ensure activation of self-renewal pathways and/or inactivation of pathways that prevent self-renewal.
The Wnt pathway was first implicated in a mouse model of breast cancer, in which aberrant expression of Wnt1, caused by insertion of the mouse mammary tumor virus (MMTV) close to the Wnt1 gene, resulted in mammary tumors.45,46 Wnt1 belongs to a large family of secreted proteins that bind to receptors of both the Frizzled and low-density lipoprotein receptor–related protein (Lrp) families, resulting in activation of β-catenin.47 In the absence of Wnt stimulation, β-catenin is degraded by the adenomatous polyposis coli (Apc), glycogen synthase kinase–3β (Gsk3β), and Axin protein complex.48 Wnt/β-catenin signaling plays a pivotal role in the self-renewal of normal stem cells.49 Activation of β-catenin signaling by Wnt proteins allows expansion of stem/progenitor cells, both in vitro and in vivo, and across different tissues, including the bone marrow, skin, mammary gland, and small intestinal epithelium.50–53 Constitutive activation of the β-catenin pathway is oncogenic54,55 and is almost invariably observed in colon cancer, most frequently by inactivating mutations of human APC.56 Current evidence suggests that β-catenin signaling is key for cancer cells to maintain a stem/progenitor cell phenotype.19,57 In colon cancer cells, inhibition of the β-catenin pathway induces expression of p21cip1/waf1, a cell-cycle inhibitor, followed by proliferation arrest and acquisition of a differentiated phenotype.57 Enforced expression of c-myc, an oncogene whose transcription is activated by β-catenin, inhibits p21cip1/waf1 expression and allows cancer cells to continue proliferating in the absence of β-catenin signaling, revealing yet another role for a classic oncogene, c-myc, in the regulation of cell differentiation.57
The Notch family of receptors, first identified as regulators of wing patterning in the Drosophila fruit fly, control development and differentiation in many tissues and across many animal species.58 In vitro stimulation of HSCs with selected Notch ligands (i.e., Jagged-1 and Delta) transiently increases the activity of stem/progenitor cells, both in vitro and in vivo, suggesting that Notch activation promotes expansion of either HSCs or MPPs.59,60 Constitutive activation of the Notch pathway is endowed with powerful oncogenic effects in both mouse and human cells. The mouse oncogene int-3 encodes a constitutively active, truncated variant of the Notch-4 receptor.61 In humans, aberrant activation of NOTCH1, either by point-mutation or chromosomal translocation, is a common occurrence in T-cell acute lymphoblastic leukemia (T-ALL).62,63 Importantly, inhibition of NOTCH1 signaling can induce apoptosis in T-ALL cell lines and is now being explored as a promising therapeutic strategy.64
The Shh pathway provides yet another example of a pathway with key roles in both tissue development, stem-cell homeostasis, and oncogenesis. Like the Wnt factors, Shh is a secreted molecule and a powerful morphogen.65 Shh acts as a ligand for receptors encoded by members of the Patched gene family65 and activates signaling circuitries that regulate self-renewal in selected types of stem cells, such as bladder stem cells.66 Germline mutations in the SHH gene cause aberrations of embryo development in Drosophila and holoprosencephaly in humans.40 With relation to human cancer, germline mutations in Patched-1 (PTCH1) cause Gorlin or basal cell nevus syndrome (BCNS), whereas sporadic mutations in PTCH1 are observed in the majority of sporadic basal cell carcinomas (BCC) and a substantial fraction of medulloblastomas.40 Most importantly, pharmacological inhibition of the SHH pathway has recently shown powerful antitumor activity in clinical trials, against both human BCCs and medulloblastomas harboring PTCH1 mutations, and is rapidly changing treatment guidelines for these diseases.67–70
Target Cells for Malignant Transformation
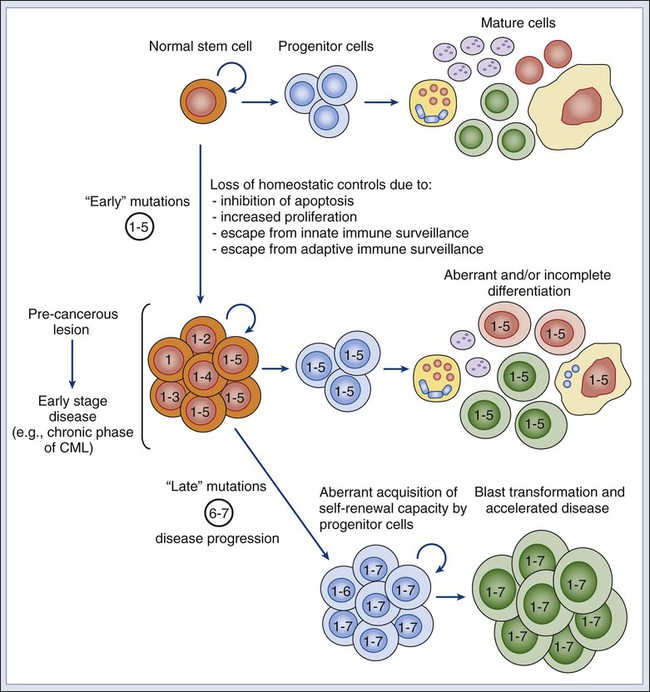
If oncogenic mutations often target signaling pathways that regulate self-renewal, then are stem cells the targets of neoplastic transformation? Several lines of evidence suggest that stem cells might be involved in the initial phases of cancer development. First, the fact that multiple and sequential mutations are necessary for a cell to become cancerous71,72 suggests that, in many cases, mutations might need, at first, to accumulate in a stem cell. Because mature cells often have a limited life span, it is unlikely that the full repertoire of mutations necessary to achieve malignant transformation might occur during their relatively short life. Second, many tumors arise in tissues that are known to contain stem cell populations, which intrinsically are endowed with the ability to self-renew. Because cancer cells must undergo self-renewal to achieve unlimited growth, stem cells might be more easily poised to undergo early malignant transformation steps compared with more mature cells, which naturally lack this fundamental property and would need to aberrantly activate self-renewal pathways to become malignant. However, even if the first set of initiating events is likely to target a stem cell population, it is possible that, as part of disease progression, the aberrant progeny of mutated stem cells might acquire additional changes and eventually gain the capacity to self-renew. In this scenario, the malignant clone would acquire a second population of self-renewing cells, whose phenotypic identity might resemble that of a progenitor rather than a stem cell (Fig. 7-2).2
Experimental evidence in support of a stem cell origin of cancer, at least during early initiation phases, is provided by elegant experiments in a mouse model of glioblastoma, caused by conditional deletion of the Neurofibromatosis-1 (Nf1), Trp53, and Pten tumor-suppressor genes.73 In this model, combined deletion of the three genes in brain tissues leads to rapid development of high-grade gliomas with 100% penetrance. This effect, however, can be observed only when the deletion is specifically targeted to cell compartments that include stem cell populations, either by conditional knockout of the tumor-suppressor genes in cells expressing the neural stem/progenitor marker Nestin or by somatic ablation of the tumor-suppressor genes in the mouse brain’s subventricular zone, where neural stem cells reside, which is achieved by stereotactic injection of an adenovirus encoding the Cre recombinase in mice genetically engineered to carry Nf1, Trp53, and Pten alleles between loxP sites. Most remarkably, somatic ablation of the same tumor suppressors in other areas of the brain, such as the striatum, is unable to generate any tumor. Moreover, tumors generated from adenovirus injection in the subventricular zone are able to disseminate throughout the brain parenchyma, following natural migration pathways of neural stem cells and exhibiting phenotypic hallmarks of multilineage differentiation along the astrocytic, neuronal, and oligodendrocytic lineages.
Although it is likely that a first set of “early” transforming mutations might accumulate in the stem cell compartment, it is also possible that more mature, downstream progenitors that originated as the progeny of mutated stem cells might acquire a second set of “late” mutations, which might constitute the ultimate transforming event giving rise to cancer.74 In this scenario, mutated stem cells might represent a “reservoir” population of precancerous cells, whereas fully transformed progenitors might sustain the growth of the full-blown neoplastic mass (Fig. 7-2).75 This concept is supported by studies on normal hematopoietic development and hematologic malignancies, where the developmental hierarchy of the different cellular compartments is best understood, and where phenotypic differences between HSCs and MPPs allow a careful dissection of their relative contributions. For example, in mice, simultaneous deletion of the Trp53 gene and the Cdkn2a locus (encoding the p16Ink4a and p19Arf tumor suppressor proteins) leads to abnormal and selective acquisition of self-renewal properties by MPPs, which remain capable of producing the various differentiated cell types, but also acquire self-renewal and become capable of supporting the production of blood tissues upon serial transplantation, in essence behaving as stem cells.76 This observation indicates that genetic mutations commonly observed in human cancer might be responsible for a progressive and pathological “unleashing” of self-renewal properties in cells that are usually not endowed with them.
Similar observations have also been made for different forms of human leukemia, such as acute myelogenous leukemia (AML) and chronic myelogenous leukemia (CML). One of the most frequent mutations in AML is the t(8;21) translocation, which results in the expression of a chimeric AML1/ETO transcript.77 Marrow samples from patients with t(8;21) AML treated at Hiroshima Hospital, when collected at clinical remission, contained HSCs (CD34+CD90+CD38−Lin−) that had up to 90% incidence of the chimeric AML1/ETO transcript.78 When these mutated HSCs were analyzed in vitro for their differentiation capacity, they gave rise to normal myeloerythroid progenies, showing that the mutation, alone and by itself, was insufficient to confer a fully transformed phenotype. Remarkably, marrow samples from the same patients, when collected at relapse, contained high numbers of cells with an MPP phenotype (CD34+CD90−CD38−Lin−) that gave rise to leukemic “blast” colonies in vitro.78 This observation is common to many forms of AML79 and is associated with the expansion of a pathological population of MPPs that have abnormally gained the capacity to self-renew.80 Taken together, these observations suggest that, in human AML, a first set of mutations (e.g., AML1/ETO) might initially accumulate in stem cells and that subsequent mutations in either the stem cells or their progeny might lead to overt leukemia.2
A similar scenario is observed in patients with CML, where leukemic cells are characterized by the t(9;22) BCR/ABL chromosomal translocation. In the initial “chronic phase” of human CML, the leukemic population undergoes a multilineage differentiation process, similar to that sustained by normal HSCs.81,82 At this initial stage, β-catenin signaling is active in HSCs but not in their descendant progenitor cells. However, when CML progresses to “myeloid blast crisis,” patients experience an AML-like disease. In this second stage, the leukemic clone acquires a second, phenotypically distinct, self-renewing population whose surface marker profile corresponds to that of a granulocyte-macrophage progenitor cell (GMP) and that is able to transplant the disease to immunodeficient mice.83 This second population is characterized by de novo aberrant activation of the β-catenin pathway, often as a result of a pathological mis-splicing of the GSK3β inhibitory enzyme.84 Thus disease progression in human CML appears to result from activation of self-renewal pathways in a descendant progenitor cell population or, more likely, failure to shut down these pathways during differentiation processes. Once again, these studies suggest that, in human CML, early mutational events (e.g., BCR/ABL) accumulate in HSCs during the chronic phase of the disease, whereas during a blast crisis a second set of mutations leads to the rise of a second self-renewing population, aberrantly originated from a progenitor cell, not an HSC (Fig. 7-2).
Evidence for Cancer Stem Cells
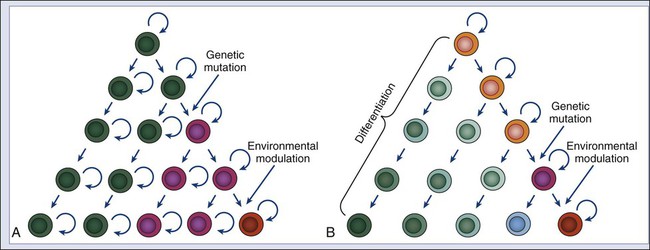
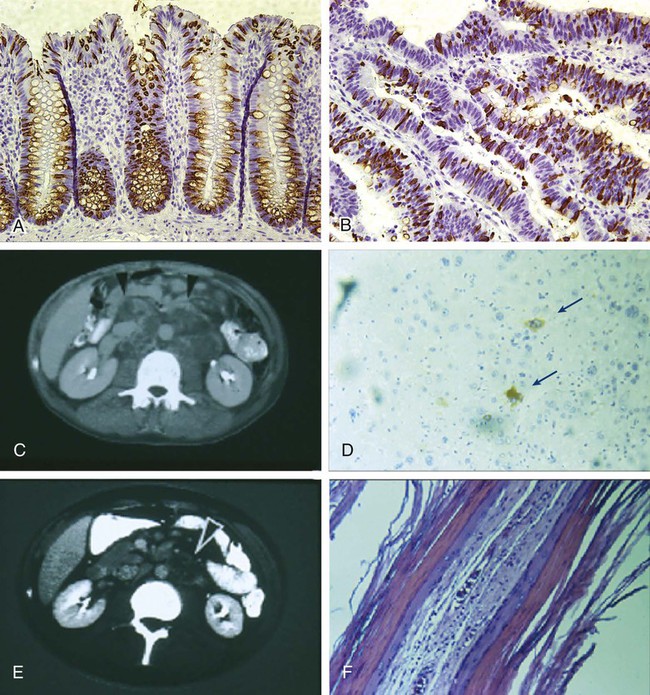
It has long been known that tumor tissues contain heterogeneous populations of cancer cells that are characterized by different phenotypes.85 This diversity can arise in part from accumulation of sequential and/or divergent mutations caused by genetic instability and in part by the variable action of environmental factors (Fig. 7-3, A). In addition, however, a tumor can also be viewed as an aberrant organ, sustained in its growth by a “pathological” stem cell population, which originates from genetic mutations and/or epigenetic modifications that lead to increased cell proliferation and tissue expansion. It is also possible that, in this aberrant organ, the “pathological” stem cell population could undergo a multilineage differentiation process reminiscent of the one observed in normal tissues, giving rise to multiple, phenotypically diverse populations of cancer cells that, although genetically identical, might differ in their epigenetic status and their ability to self-renew (Fig. 7-3, B).9,10 Several lines of evidence indicate that multilineage differentiation does occur in cancer tissues and contributes substantially to cellular heterogeneity,86 in addition to genetic instability and environmental factors, which undoubtedly contribute to the observed variability.87 Indeed, it is well documented that the cell composition of cancer tissues frequently mirrors that of their normal counterparts in terms of differentiated cell types.86 A classic example is the frequent presence of abundant mucus-producing goblet-like cells in human colon carcinomas (Fig. 7-4, A and B). Perhaps one of the most striking examples of abnormal differentiation in cancer is observed in some germ cell tumors, such as teratomas and teratocarcinomas, where mature organs such as teeth, skin, and hair can be found simultaneously admixed within the tumor mass. Remarkably, within many of these tumors, only a minority of cancer cells express immature cell markers such as α-fetoprotein (Fig. 7-4, D). The terminally differentiated cancer cells that form the teeth and hair in the tumors are most likely unable to proliferate and to contribute substantially to long-term tumor growth and metastasis. On the contrary, the minority population of immature cancer cells expressing α-fetoprotein is likely to be at the source of long-term tumor growth, as well as to represent the progenitor population from which the other more mature populations arise. Preliminary data on teratoma formation by human embryonic stem cells lend support to this working model.88
If a tumor is viewed as an abnormal organ, then the principles of stem cell biology can be applied to model its physiology and clinical behavior.9,10 Historically, this concept was first explored using clonogenic assays, where single-cell suspensions of cancer cells were tested in limiting dilution for their capacity to form colonies, either in vitro or in vivo.89 Indeed, in vivo clonogenic assays had been previously used to provide the first experimental evidence of HSCs, because they showed that only a minority of normal bone marrow cells, when transplanted in irradiated mice, was able to engraft and give rise to blood-forming colonies in the spleen.90 When applied to tumors, clonogenic assays showed that only a minority of cancer cells, ranging from 1 : 100 to 1 : 10,000, was able to form colonies.89 These results were reproduced across many tumor types and suggested the existence of minority populations of stem-like cancer cells, but investigators were unable to formally prove it, because they could not rule out stochastic effects in cell survival and engraftment.91
To prove that tumor tissues contain a specific subset of cancer cells that is preferentially responsible for sustaining tumor growth, it is necessary to isolate different populations of cancer cells in parallel and compare their properties side by side in a prospective assay—for example, a transplantation assay in which cancer cells are tested for their tumorigenic capacity.92 This type of experiment was first performed in human AML, where it was shown that, in many cases, the “tumor-initiating” capacity was enriched in a phenotypic subset of the leukemic clone. In these studies, the subset of leukemic cells that displayed the highest capacity of establishing human AML in immunodeficient mice was the population characterized by the CD34+CD38− phenotype.80,93,94 As previously discussed, the human CD34+CD38− population corresponds to a very immature subset of hematopoietic cells that contains HSCs (i.e., CD90+CD34+CD38−) and MPPs (i.e., CD90−CD34+CD38−),31 both of which are involved in the natural history of these leukemic disorders.78,80 Although characterized by the highest frequency of tumorigenic cells, the CD34+CD38− population is not the only self-renewing population observed in AML.95 As observed for CML during blast crisis, a second tumorigenic population, characterized by a more mature, granulocyte-macrophage progenitor–like phenotype (CD34+CD38+CD110−CD45RA+), can also be frequently detected in AML. This second population is hierarchically related to the first (i.e., it originates as the progeny of CD34+CD38− cells, but not vice versa) and is characterized by a much lower frequency of tumorigenic cells.80
Recently, tumorigenic and non-tumorigenic subsets of cancer cells have also been isolated from epithelial solid tumors, such as breast and colon carcinomas.17,18 As in the case of leukemias, tumorigenic cancer cells could be distinguished from non-tumorigenic ones based on their differential expression of surface markers. In breast cancer, the tumorigenic cell population was characterized as CD44+CD24−/low,17 whereas in colon cancer, it was characterized as EpCAMhighCD44+CD166+.18 In both experimental systems, as few as 100 to 200 cells from the tumorigenic population were able to consistently form tumors when injected into immunodeficient mice, whereas as many as 104 cells from other cell populations within the same tumors repeatedly failed to do so. In both cases, cancer tissues originated from the tumorigenic subset contained the full repertoire of cell populations observed in the parent tissues, including the tumorigenic subset itself and other non-tumorigenic populations. Importantly, these cancer tissues could be serially passaged in mice, each time with similar results. Lineage-tracing experiments performed in colon cancer showed that monoclonal cancer tissues, originated by injection of a single tumorigenic cancer cell bearing the EpCAMhighCD44+ phenotype and infected with a reporter lentivirus encoding for the enhanced green fluorescence protein (EGFP), reproduced the full cellular diversity of the parent tissues, including the presence of EGFP+EpCAMlowCD44− non-tumorigenic cancer cells, and a variety of multiple EGFP+ differentiated cell types, including MUC2+ goblet cells.86 These experiments provided formal proof that: (1) non-tumorigenic populations originate as the progeny of tumorigenic ones and (2) tumorigenic cells can undergo multilineage differentiation processes akin to those that allow normal stem cells to sustain the generation of complex and diverse tissues. Similar findings have been recently obtained using different types of mouse experimental cancer models.96,97
Taken together, these data indicate the presence of a cellular hierarchy within many cancer tissues, in which only a fraction of the cells have the ability to engraft in mice. These cells can expand indefinitely and differentiate into multiple lineages of specialized cell types, thus showing two hallmark properties of stem cells: self-renewal and differentiation. Based on the fulfillment of these two properties, the tumorigenic subset of cancer cells is often called the CSC subset. It is important to remember, however, that this definition is operational and was introduced to identify a subgroup of cancer cells characterized by unique functional properties but which, as observed in leukemia, might not always necessarily correspond to the stem cell population of their respective normal tissues. In colon carcinomas, for instance, CSCs display a surface marker phenotype (EpCAMhighCD44+CD166+) that is characteristic of cells found at the bottom of normal colon crypts and that defines a cell compartment probably encompassing both stem and multipotent progenitor subsets.86,98–100 Because of our limited understanding of differentiation hierarchies in both normal breast and normal colon epithelia, it is currently impossible to dissect with precision whether, in breast and colon cancer, CSC activity is found in stem cells, in downstream multipotent progenitors, or in both, and this question remains the subject of active investigations.
CSCs have also been identified in many other human tumors, including glioblastomas and several types of epithelial cancer, such as head and neck, pancreas, prostate, and bladder carcinomas.101–105 Interestingly, CD44 can be used for the isolation of CSCs across many epithelial carcinomas. This finding is in agreement with the notion that CD44 is often expressed heterogeneously within normal epithelia, usually in a gradient, with the highest levels being observed at the most basal layers, where normal stem/progenitor cells are usually thought to reside. Importantly, histologic analysis of well-differentiated head and neck carcinomas showed that, in these tumors, CD44 expression is often inversely correlated to expression of mature cells markers, such as involucrin, and positively associated with expression of BMI1, a key element of the self-renewal machinery in many types of stem cells.102 Box 7-1 discusses alternative models for cancer cell heterogeneity.
Clinical Implications of CSCs
The existence of CSCs has profound implications for the development of clinical oncology. One implication is concerned with the prediction of patient outcomes at the time of diagnosis. If tumor tissues vary in CSC content, then tumors containing a higher fraction of CSCs might be characterized by a more aggressive biology and worse clinical outcomes. Indeed, a number of studies have shown that tumors whose bulk gene expression profiles are similar to those of purified CSCs are associated with poor outcomes, likely reflecting a higher CSC content and a more immature differentiation state. This association was first shown for breast cancer, where a 186-gene signature, called the “invasiveness gene signature” (IGS), was derived by comparing the gene-expression profile of breast CSCs with that of normal breast epithelial cells.106 The IGS was used to stratify patients with breast cancer who had early-stage disease in different groups, based on the similarity of their whole tumor’s gene expression profile with the IGS. Remarkably, high similarity to the IGS was associated with reduced overall and metastasis-free survival. Similar studies have identified CSC signatures in human AML by comparing the gene-expression profile of different cell subsets within the leukemic population.95,107 Once again, high similarity to AML-CSC signatures was associated with adverse outcomes, independent of other clinical and pathological parameters. Conceptually similar findings have been made in a variety of other cancer types, including head and neck, bladder, and colon cancer.86,103,108–110 Taken together, these studies document the prognostic significance of CSCs in the natural history of human tumors and underscore their potential relevance for the future clinical management of patients with cancer.
The existence of CSCs also has important implications for the clinical evaluation of antitumor therapies. Historically, most antitumor agents have been developed on the basis of their ability to reduce tumor size. Tumor shrinkage is dependent on the elimination of large numbers of cancer cells, but not necessarily of the CSCs, which often constitute a minority of the cancer tissue. This observation implies that CSCs might be spared even when tumors appear to be responding. Indeed, in diseases such as metastatic lung or breast cancer, although standard treatments frequently shrink tumors, tumors rapidly recur, and only limited impact on patient survival is usually observed.111,112 Because anticancer therapies are often screened on the basis of their ability to shrink tumors rapidly, agents that selectively target CSCs could be overlooked. Indeed, in the initial phases of the screen, CSC-specific agents might cause only a modest reduction in tumor growth. However, if complete elimination of CSCs could be achieved, these treatments could permanently eradicate tumor tissues. Perhaps the best clinical evidence for this concept comes from patients with teratocarcinoma. Platinum-based chemotherapy is curative in most of these patients.113 Many patients, however, are left with residual masses (see Fig. 7-4, E), which usually undergo surgical resection. In most cases, histologic analysis of residual tumor tissues reveals that cancer cells expressing α-fetoprotein have been eliminated, leaving only differentiated cancer cells in a mature teratoma (see Fig. 7-4, F). Metastases rarely develop in patients with mature teratomas, and long-term cure is frequently achieved, suggesting that selective elimination of immature cancer cell populations can be sufficient to permanently abolish tumor growth. Similar observations have also been obtained in experimental models of mouse glioblastoma.114
The lack of strong associations between tumor shrinkage and tumor eradication led to the hypothesis that CSCs may be intrinsically resistant to chemotherapy and radiotherapy. This hypothesis is supported by the observation that normal adult stem cells, such as HSCs, are frequently found in a G0 quiescent state, which reduces their sensitivity to antiproliferative agents, and express high levels of members of the ABC transporter family, which pump many drugs out of the cell and increase resistance to cytotoxic agents.115,116 If the same is true for CSCs, then these cells may be significantly more resistant to cytotoxic agents than their non-tumorigenic progeny. In support of this concept, for instance, is the observation that the antitumor drug cytosine arabinoside (Ara-C), although capable of killing the majority of AML blast cells, selectively spares the CD34+CD38−CD123+ subset of leukemic CSCs.116 Similarly, it has been shown that CSCs can be resistant to both chemotherapy and radiotherapy in many types of solid tumors, and several mechanisms mediating this resistance have been uncovered. For example, glioblastoma CSCs have been shown to be radioresistant compared with their non-tumorigenic counterparts.117 This resistance is mediated, in part, by enhanced activation of DNA repair pathways and rapid repair of radiation-induced DNA damage. Similar observations have also been made for breast CSCs, in this case as the result of enhanced free radical scavenging, which prevents radiation-induced DNA damage.118,119 CSCs have also been shown to be preferentially resistant to chemotherapy in a variety of solid tumors. For example, colon CSCs are preferentially resistant to antitumor agents such as irinotecan and cyclophosphamide. In the case of the latter, resistance appears mediated by overexpression of enzymes of the aldehyde dehydrogenase family, which can oxidize and inactivate the cytotoxic metabolite of cyclophosphamide, 4-HC/aldophosphamide.120
Overall, preclinical studies suggest that CSCs might hold the key to understanding many aspects of cancer resistance to antitumor therapies in human patients. However, to formally address this question, CSCs must be enumerated before and after therapy, thus requiring repeated in vivo tumor sampling. This type of analysis can be performed in the case of patients undergoing neoadjuvant treatment regimens, because they receive chemotherapy and/or radiotherapy immediately before surgery. Remarkably, neoadjuvant treatment of breast cancer with standard chemotherapy has been shown to cause an increase in the percentage content of CSCs in vivo.121 Intriguingly, neoadjuvant treatment of HER2+ tumors with lapatinib, an HER2 inhibitor, was found not to increase the fraction of CSCs within tumors, suggesting that, in HER2+ tumors, therapies targeting HER2 might directly target the CSC subset.121 These studies confirm that CSCs can respond differentially to different drugs in human patients and encourage efforts to overcome CSC-specific resistance mechanisms and to develop CSC-targeted therapies. Moreover, given the variety of resistance mechanisms displayed by CSCs, both tumor-specific and patient-specific, characterization of the dominant resistance mechanisms in a tumor’s CSCs may become integral to the future development of personalized therapies.
Another promising target is CD47, a cell surface receptor that is overexpressed by CSCs in many tumor types.122 In normal physiology, CD47 mediates a “don’t eat me” signal that prevents normal cells, such as healthy red blood cells, from being eliminated by macrophages.123 CD47 thus acts as an inhibitor of the normal physiological pathways that mediate the removal of aged and/or damaged cells by macrophage scavenging, a process also known as “programmed cell removal.”124,125 CD47 is upregulated by HSCs when they enter the bloodstream to recirculate, thus protecting them from phagocytosis.126 Targeted inhibition of CD47 leads to eradication of CSCs, and even entire tumors, in many in vivo experimental models, including leukemias, lymphomas, and various forms of solid cancer.122,127–129 These observations suggest that, as part of neoplastic transformation, cancer cells commonly disable pathways that are meant to ensure homeostatic control of cell numbers by means of active cell elimination. These pathways include “suicide” or “programmed cell death” pathways (i.e., elimination by apoptosis) and “programmed cell removal” pathways (i.e., elimination by phagocytosis).124,125 Although several molecular mechanisms are known to mediate the escape of cancer cells from apoptosis (e.g., Bcl2 overexpression and p53 inactivation), only one is currently known to inhibit programmed cell removal: overexpression of CD47. Future investigations will shed new light on the molecular regulation of CD47 and reveal other molecules involved in programmed cell removal, such as those mediating positive “eat me” (i.e., opsonization) signals.130 These investigations will provide new molecular targets for therapeutic interventions.
Future Implications of CSCs
The identification of CSCs across many tumor types is key to the future development of clinical oncology, especially for the discovery of new prognostic biomarkers and the design of novel antitumor agents. The study of CSCs, however, is technically challenging. CSCs often represent a minority of the cancer cell populations and need to be purified from primary tissues in order to evaluate their functional properties in the context of their original tissue microenvironment. Future studies will need to devise new experimental platforms to address this challenge, such as microfluidic platforms that are able to perform precise measurements on small biopsy samples with single-cell resolution.86
The ability to identify and quantify CSCs should improve our ability to evaluate the curative potential of both traditional and investigational therapies. Although cancer cell lines are useful experimental tools, especially suited for the in vitro dissection of individual biochemical pathways, they have often proven unreliable when used to model the clinical efficacy of antitumor drugs.131 Human xenografts established in immunodeficient mice recapitulate more accurately the morphology and phenotypic diversity of the patients’ original tumors, including their three-dimensional structure and the presence of both tumorigenic and non-tumorigenic cell populations.18 Xenograft models may help evaluate the differential effect of antitumor drugs on different cancer cell populations. New antitumor agents could therefore be tested for their ability to eliminate CSCs in vivo, across multiple xenografts, and those with the greatest activity could be selected for evaluation in clinical trials, with the hope that they will pave the way to a new generation of more effective cancer therapies.