Sarcomas of Soft Tissue*
Lee J. Helman and Robert G. Maki
• In the United States, there are 11,280 new cases annually; fewer than 1% of cancer diagnoses.
• No specific etiologic agent is identifiable in the majority of cases.
• Occasional cases are related to previous radiation, chemical exposure, alkylating chemotherapeutic agents, or chronic lymphedema.
• Genetic conditions related to soft-tissue sarcoma include neurofibromatosis, tuberous sclerosis, basal cell nevus syndrome, Gardner syndrome, and Li-Fraumeni syndrome.
Diagnosis and Evaluation of Extent of Disease
• Core-needle biopsy (large lesions) or excisional biopsy (small lesions).
• Pathological review of histologic subtype, grade, and assessment of margins (excisional biopsies).
• Magnetic resonance imaging or computed tomography (CT) of primary site.
• Chest x-ray for low-grade tumors and high-grade T1 lesions, chest CT for high-grade T2 tumors.
• High-grade histology, deep location, and T2 tumor size are independent adverse prognostic factors for distant metastasis and survival.
• Presentation with recurrent disease and positive surgical margin (gross or microscopic) are independent adverse prognostic factors for local recurrence.
• Individual patient prognosis may be estimated by using a nomogram or newer techniques.
• The American Joint Committee on Cancer (International Union Against Cancer) system uses criteria that include grade, size, and location relative to the investing muscular fascia, nodal status, and distant metastases. Other means to characterize risk of recurrence of soft-tissue sarcomas include nomograms and Bayesian belief networks.
• Surgical resection with an adequate margin of normal tissue; for extremity lesions, a limb-sparing approach is possible in more than 90% of patients and offers survival comparable to amputation without the associated morbidity.
• For most patients, local control is improved with preoperative or postoperative radiotherapy, with size and primary site also impacting its use.
• The role of chemotherapy for high-risk patients remains controversial, but chemotherapy is used at several major centers for high-risk patients, especially for extremity tumors with known chemotherapy sensitivity, preoperatively when possible.
• In addition to surgery, 3 years of adjuvant imatinib is the standard of care for high-risk gastrointestinal stromal tumor (GIST).
• Local recurrence rates vary depending on the anatomic primary site and the adequacy of local therapy; for extremity lesions, approximately 20% of patients experience locally recurrent disease.
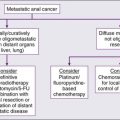
• Systemic cytotoxic chemotherapy with selective use of surgery is the mainstay of therapy for patients with metastatic disease.
• Kinase-directed therapy is clinically beneficial for patients with GIST; pazopanib was shown active in patients with other types of sarcoma who failed to respond to other systemic therapies, but has comparatively limited benefit.
• For the small subset of patients who experience isolated (solitary) lung metastases, 20% to 50% 3-year survival rates have been reported with metastasis resection.
Introduction
Soft-tissue sarcomas (STSs) comprise a group of relatively rare, anatomically and histologically diverse neoplasms. Most STSs share a common embryologic origin, arising primarily from tissues derived from the mesodermal or ectodermal germ layers, distinct from carcinomas, which arise from the endodermal germ layer, although some sarcomas such as angiosarcomas also have an endodermal origin. Although the somatic soft tissues account for as much as 75% of total body weight, neoplasms of the soft tissues are comparatively rare, accounting for less than 1% of adult malignancies and 15% of pediatric malignancies. The relative rarity of these tumors, coupled with the histologic diversity of tumors, has led to studies that include diverse STS subtypes in a single study group, making it difficult to develop specific therapies for specific tumor types, especially in adult STSs. The annual incidence of STSs in the United States is about 11,280 new cases, comparable to the incidence of testicular cancer.1 However, an estimated 3900 patients die annually of STS—a rate nearly tenfold greater than is seen with testicular cancer—emphasizing the comparatively high overall mortality rate that is seen with this type of tumor.
Etiology and Epidemiology
Environmental Factors
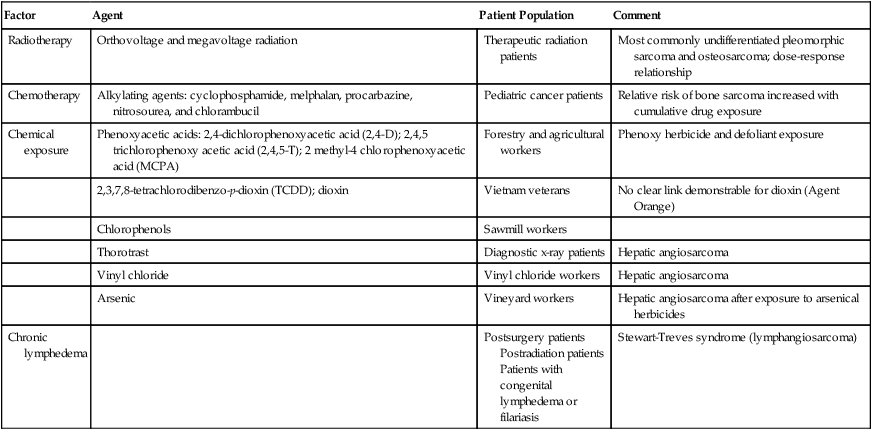
No specific etiologic agent may be identified in the majority of patients with STS. There are a number of recognized associations between environmental factors and the subsequent development of sarcoma; these are summarized in Table 93-1. The development of sarcoma has been reported after the use of ionizing radiation for the treatment of lymphoma2; solid tumors of the head and neck,3 breast,4,5 gynecologic organs, and skin; and benign conditions including endometriosis, tuberculous arthritis,6 and benign thymic enlargement. The vast majority of radiation-associated sarcomas are high-grade (87%), and the predominant histologies include undifferentiated pleomorphic sarcoma (formerly termed malignant fibrous histiocytoma), angiosarcoma, malignant peripheral nerve sheath tumor, osteosarcoma and others; translocation-associated sarcomas are rare, in comparison, and teleologically it is difficult to understand how random DNA damage leads to a specific chromosomal translocation.7–10
Table 93-1
Soft-Tissue Sarcoma: Predisposing Environmental Factors
| Factor | Agent | Patient Population | Comment |
| Radiotherapy | Orthovoltage and megavoltage radiation | Therapeutic radiation patients | Most commonly undifferentiated pleomorphic sarcoma and osteosarcoma; dose-response relationship |
| Chemotherapy | Alkylating agents: cyclophosphamide, melphalan, procarbazine, nitrosourea, and chlorambucil | Pediatric cancer patients | Relative risk of bone sarcoma increased with cumulative drug exposure |
| Chemical exposure | Phenoxyacetic acids: 2,4-dichlorophenoxyacetic acid (2,4-D); 2,4,5 trichlorophenoxy acetic acid (2,4,5-T); 2 methyl-4 chlorophenoxyacetic acid (MCPA) | Forestry and agricultural workers | Phenoxy herbicide and defoliant exposure |
| 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD); dioxin | Vietnam veterans | No clear link demonstrable for dioxin (Agent Orange) | |
| Chlorophenols | Sawmill workers | ||
| Thorotrast | Diagnostic x-ray patients | Hepatic angiosarcoma | |
| Vinyl chloride | Vinyl chloride workers | Hepatic angiosarcoma | |
| Arsenic | Vineyard workers | Hepatic angiosarcoma after exposure to arsenical herbicides | |
| Chronic lymphedema | Postsurgery patients Postradiation patients Patients with congenital lymphedema or filariasis |
Stewart-Treves syndrome (lymphangiosarcoma) |
By criteria that were described initially by Cahan and colleagues,11 radiation-induced sarcomas appear no sooner than 4 years after the therapeutic radiation12,13 and often appear decades later. More recently, however, a shorter 2-year period after diagnosis has been used to define radiation-related disease, and the median time from radiation to development of a secondary sarcoma is 8 to 10 years. Contrary to early theories, recent studies have suggested that both orthovoltage and megavoltage treatments are sarcomagenic9,14 at doses from 8.8 to 70 Gy. In a carefully conducted case-control analysis, the Late Effects Study Group found 64 cases of osteosarcoma in 9170 patients who had survived more than 2 years after the diagnosis of a variety of cancers. A dose-response relationship was found between the radiation dose and the subsequent development of osteosarcoma, with a relative risk ranging from 0.6-fold in patients who received less than 10 Gy to 38.3-fold in those who received more than 60 Gy.15 A complete understanding of the underlying biology of radiation-induced malignancy remains elusive. Host-related factors (especially young age) and treatment (especially its intensity) both seem to play a role in a complex relationship.16 Underlying genetic susceptibility is also important and is governed by factors such as deletion or mutation of tumor suppressor and DNA repair genes. This scenario likely means that subgroups of the populations have a greater risk than was previously anticipated, whereas others are at lower risk than was believed. The identification of osteosarcoma as a common second malignancy following radiation therapy for retinoblastoma is an example of a high-risk scenario. It is probable that the greater intensity of multimodality treatments for cancer that have been introduced in recent years with the goal of improving cancer control and survival will result in an increase in the rate of radiation-induced malignancies. For example, the use of radiation after lumpectomy for ductal carcinoma in situ may result in unnecessary second cancers, which are avoidable in over 95% of patients with the use of mastectomy and reconstruction. The most common postradiation cancer appears to be sarcoma, especially considering its baseline incidence rates.16
Sarcomas have also been weakly associated with exposure to various chemical agents. A number of conflicting reports have emerged that suggest a relationship between occupational exposure to phenoxyacetic acids (found in some herbicides) and chlorophenols (found in some wood preservatives). Studies from Sweden demonstrated a link between phenoxy herbicide exposure in forestry workers and the subsequent development of sarcoma.17–20 However, additional investigations in the United States, New Zealand, and Finland have not confirmed this relationship.23–23 Similarly, there has been no demonstrable increase in the risk of sarcoma in Vietnam veterans exposed to Agent Orange (dioxin or TCDD [2,3,7,8-tetrachlorodibenzo-p-dioxin]).24 Hepatic angiosarcomas have been associated with exposure to a number of compounds, including Thorotrast (a colloidal suspension of thorium dioxide that was formerly used as an intravenous contrast agent in radiologic imaging procedures),27–27 vinyl chloride,30–30 and arsenic.31,32
Recent studies have suggested a relationship between exposure to alkylating chemotherapeutic agents and the subsequent development of sarcomas. Osteosarcomas have been reported after cyclophosphamide treatment for pediatric acute lymphoblastic leukemia.33,34 In the recent report from the Late Effects Study Group, prior chemotherapy, particularly with melphalan, procarbazine, nitrosoureas, or chlorambucil, was found to be an independent risk factor for the development of sarcoma.15 The relative risk of sarcoma increased with cumulative drug exposure.
Chronic lymphedema may be a factor in the development of angiosarcoma. These neoplasms have been noted to arise in the chronically lymphedematous arms of women who were treated for breast cancer with radical mastectomy (Stewart-Treves syndrome).35,36 Lower-extremity angiosarcomas have also been observed in patients with congenital lymphedema or filariasis complicated by chronic lymphedema.37
A recent history of trauma is often elicited from sarcoma patients, particularly those with extremity sarcoma. Usually, the interval between the traumatic event and the diagnosis of sarcoma is short, making a causal relationship unlikely. Some reports have suggested, however, that chronic inflammatory processes may be a risk factor for sarcoma. Shrapnel, bullets, intramuscular iron injections, and foreign-body implants have been implicated.38
Genetic Predisposition
Germline mutations can play an important role in the development of STSs (Table 93-2). These genetic changes are identified with or similar to the genetic changes that are seen in corresponding sporadic sarcomas (Table 93-3). Mechanistically, the proteins encoded by the altered genes are involved in maintenance of the genome through DNA repair and the cell cycle.
Table 93-2
Germline Mutations Associated with Soft-Tissue Sarcoma
| Syndrome | Inheritance Pattern | Locus | Gene | Associated Soft-Tissue Sarcomas |
| Familial gastrointestinal stromal tumor syndrome | AD | 4q12 | KIT | Gastrointestinal stromal tumor |
| Familial desmoid fibromatosis (Gardner syndrome) | AD | 5q21 | APC | Desmoid fibromatosis |
| Li-Fraumeni syndrome | AD | 17p13,22q11 | TP53, CHK2 | Multiple types |
| Neurofibromatosis type I (von Recklinghausen disease) | AD | 17q11 | NF1 | Malignant peripheral nerve sheath tumors |
| Retinoblastoma | AD | 13q14 | RB1 | Multiple types |
| Rhabdoid predisposition syndrome | AD | 22q11 | SNF5/INII | Malignant rhabdoid tumors |
| Werner syndrome | AR | 8p11–12 | WRN | Multiple types |
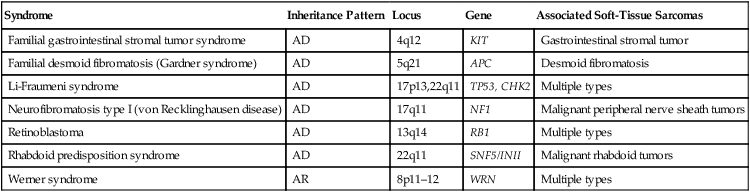
Table 93-3
Selected Molecular Alterations Identified in Sarcoma
| Tumor Type | Characteristic Cytogenetic Events | Molecular Events | Frequency (%) | Diagnostic Utility? |
| Alveolar soft part sarcoma | t(X;17)(p 11;q25) | ASPL-TFE3 fusion | >90 | Yes |
| Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12) | EWSR1-NR4A3 fusion | >75 | Yes |
| Clear cell sarcoma | t(12;22)(q13;q12) | EWSR1-ATF1 fusion | >75 | Yes |
| Desmoplastic small round cell tumor | t(11;22)(q13;q12) | EWSR1-WT1 fusion | >75 | Yes |
| Dermatofibrosarcoma protuberans | Ring form of chromosomes 17 and 22 | COLIA1-PDGFB fusion | >75 | Yes |
| t(17;22)(q21;q13) | COLIA1-PDGFB fusion | 10 | Yes | |
| Ewing sarcoma/peripheral primitive neuroectodermal tumor | t(11;12)(q24;q12) | EWSR1-FLII fusion | >80 | Yes |
| t(21;22)(q12;q12) | EWSR1-ERG fusion | 5-10 | Yes | |
| t(2;22)(q33;q12) | EWSR1-FEV fusion | <5 | Yes | |
| Fibrosarcoma, infantile | t(12;15)(q13;q26) | ETV6-NTRK3 fusion | >75 | Yes |
| Trisomies 8,11,17 and 20 | >75 | Yes | ||
| Gastrointestinal stromal tumor | Monosomies 14 and 22 | >75 | Yes | |
| Deletion of 1p | KIT or PDGFRA mutation | >25 >90 |
No Yes |
|
| Inflammatory myofibroblastic tumor | 2p23 rearrangement | ALK fusion genes | 50 | Yes |
| Leiomyosarcoma | Deletion of 1p | >50 | No | |
| Liposarcoma | ||||
| Well-differentiated | Ring form involving chromosome 12q | >75 | Yes | |
| Myxoid/round cell | t(12;16)(q13;p11) | FUS-DDIT3 fusion | >75 | Yes |
| Pleomorphic | Complex | EWSR1-DDIT3 fusion | <5 | Yes |
| Undifferentiated pleomorphic sarcoma | Complex | >90 | No | |
| Malignant peripheral nerve sheath tumor | Complex; NF1 loss | >90 | No | |
| Rhabdoid tumor | Deletion of 22q | INI1 inactivation | >90 | Yes |
| Rhabdomyosarcoma | ||||
| Alveolar | t(2;13)(q35;q14) | PAX3-FOXO1 fusion | >75 | Yes |
| t(1;13)(p26;q14), double minutes | PAX7-FOXO1 fusion | 10-20 | Yes | |
| Embryonal | Trisomies 2q, 8, and 20 | >75 | Yes | |
| Loss of heterozygosity at 11p15 | >75 | Yes | ||
| Synovial sarcoma | ||||
| Monophasic | t(X;18)(p11;q11) | SS18-SSX1 or SS18- | >90 | Yes |
| Biphasic | t(X;18)(p11;q11) | SSX2 fusion SS18-SSX1 fusion |
>90 | Yes |
The epidemiological relationship between the development of STS and inherited syndromes associated with a predisposition to neoplasia (e.g., neurofibromatosis and Li-Fraumeni syndrome) has been appreciated for decades.39,40 For example, patients with neurofibromatosis have a 7% to 10% lifetime risk of developing a malignant peripheral nerve sheath tumor (MPNST).41 A sudden increase in the size of any neurofibroma suggests malignant transformation.42,43 The mechanisms underlying the transformation from a benign neurofibroma to MPNST are not well understood.
However, loss-of-function mutations in the NF1 gene, which are found in patients with neurofibromatosis, result in activation of one of the ras signaling pathways, a well-known mechanism that has been identified in a variety of cancers.44 It has been observed that secondary MPNSTs (arising from a prior neurofibroma) have deletions of 17p (particularly 17p12–17p13.1) and mutations at the region of the TP53 tumor suppressor gene.47–47 Thus it is postulated that an initial alteration in the NF1 gene contributes to the formation of a benign neurofibroma through activation of the ras pathway and that secondary mutations in the TP53 gene allow the transformation into MPNST. Because mutations in TP53 lead to an inability to control DNA damage via the cell cycle, they also enable rapid accumulation of other mutations, which encode mutant proteins that undoubtedly play an important role in sarcomagenesis.
Li-Fraumeni syndrome was identified when relatives of pediatric STS patients were noted to have an increased frequency of diverse and often multiple primary cancers.39,48 The neoplasms that were noted in relatives included some STSs, premenopausal breast cancers, brain tumors, adrenocortical carcinomas, leukemias, and occasional germ cell tumors.49 Follow-up of Li-Fraumeni kindreds over 2 decades has revealed that the majority of individuals have cancer at young ages, with 79% of those affected younger than age 45 years at the time of diagnosis of malignancy.50 The observed cancer distribution in families is believed to fit a rare autosomal dominant mode of genetic transmission with high penetrance.51 Recent molecular genetic studies have identified germline TP53 mutations in the majority of patients with Li-Fraumeni syndrome.52 Germline mutations in CHK2, another component of the cell-cycle checkpoint machinery encoded by a gene located on 22q11, are responsible for another subgroup of patients with Li-Fraumeni syndrome, and other Li-Fraumeni–like patients’ kindreds without TP53 alteration have been described.53
Pediatric patients with familial retinoblastoma have a 13q chromosomal deletion54 and an increased incidence of osteosarcoma and other neoplasms, including STS.15,55,56 The retinoblastoma (Rb1) protein is expressed ubiquitously in normal cells and is a well-known tumor suppressor, involved in maintaining the integrity of the genome through control of the cell cycle. Interestingly, not only is the Rb1 gene that is mutated in osteosarcomas associated with retinoblastoma, but abnormality or absence of the Rb1 gene product has also been observed in multiple other malignancies, including sporadic osteosarcomas, breast cancer,57 small cell lung cancer,58 and STSs.59
Familial adenomatous polyposis, a subset of which includes Gardner syndrome with its desmoid tumors, is caused by germline mutations in the APC gene.62–62 Patients with Gardner syndrome experience desmoid fibromatosis at a younger age than do patients with sporadic desmoids.63 With the use of prophylactic colectomy, progressive mesenteric desmoid tumor is now a significant cause of death of patients who have familial adenomatous polyposis,64 as are second neoplasms such as ampullary and duodenal cancers. The APC gene is involved in the Wnt cell-signaling pathway. One of the normal functions of the APC protein is to bind β-catenin.65 Thus loss-of-function mutations of APC result in the activation of transcription of oncogenes by β-catenin. Consistent with the importance of this pathway in desmoid tumors, APC loss and CTNNB1 (encoding β-catenin) mutations are also identified in sporadic desmoid fibromatosis.68–68
Rhabdoid predisposition syndrome is due to inactivating germline mutations in the INI1 gene.69 Patients with this syndrome experience one or more extrarenal and/or renal rhabdoid tumors. INI1 is a member of the SWI/SNF protein complex, which controls gene expression globally through its ability to alter chromatin structure.70 Thus loss of INI1 gene expression results in other changes in gene expression, specifically activation of oncogenes. Rhabdoid tumors have loss-of-function mutations in both copies of the INI1 gene.71
Werner syndrome is a rare genetic instability syndrome caused by mutations in the WRN gene.72,73 Affected patients age prematurely and are at greatly increased risk for a variety of cancers, including STSs. The WRN gene encodes a protein involved in DNA repair74 and loss of WRN protein function leads to genetic instability, accumulation of genetic mutations, and, ultimately, predisposition to rapid aging and cancer.
Germline mutations in the KIT oncogene are found in patients with familial gastrointestinal stromal tumor (GIST) syndrome.75,76 Activating KIT or PDGFRA mutations are also identified in over 90% of sporadic gastrointestinal stromal tumors (GISTs).77 Patients with the familial syndrome experience, to varying degrees, skin hyperpigmentation, urticaria pigmentosa, and cutaneous mast cell disease in addition to one or more GISTs.78 Activating KIT mutations have been shown to lead to ligand-independent activation of the KIT receptor tyrosine kinase pathway, which results in dysregulated cell growth, and are thought to be the first step in the pathogenesis of GISTs.75 Interestingly, the identification of the important role of KIT in the pathogenesis of GISTs has led to treatment with imatinib,79 the oral multitargeted receptor tyrosine kinase inhibitor (see “Prognostic Factors as Therapeutic Targets” and “Gastrointestinal Stromal Tumors” below).
Chromosomal Rearrangements
A large number of sarcomas have been found to have consistent chromosomal abnormalities (see Table 93-3).80 These chromosomal rearrangements are important diagnostically, may be important prognostically (see “Potential Molecular Prognostic Factors” below), and have shed light on the pathogenesis of sarcomas. Some of the specific translocations, such as those seen in dermatofibrosarcoma protuberans or tenosynovial giant cell tumor provide targets for pharmacologic therapy (see the section on treatment of GISTs). Benign soft-tissue neoplasms also harbor chromosomal rearrangements.
Chromosomal translocations are the most common cytogenetic abnormality in soft-tissue neoplasms and are likely responsible for the initiation of tumorigenesis in most cases.80 Deletions and trisomies have also been reported and are thought to represent secondary changes involved in tumor progression. Deletions tend to represent loss of tumor suppressor genes, whereas trisomies suggest the presence of an oncogene. Although we know much about the principal tumorigenic events in many sarcomas, defining the secondary changes has been much more problematic and is an area of intense study with the advent of large-scale tumor DNA and RNA sequencing and related genomic techniques.
Cloning and molecular analysis of the various genetic aberrations that characterize different sarcomas have revealed the different pathogenetic mechanisms that underlie these tumors. Translocations typically create chimeric transcription factors or growth factors that result in deregulation of transcription or growth control. A typical example of a chimeric transcription factor is the PAX3-FOXO1 fusion protein, which has been shown to activate a complex myogenic transcriptional program when the protein is expressed in a fibroblast cell line.81 Infantile fibrosarcoma is characterized by a translocation involving chromosomes 12 and 15 that encodes a chimeric ETV6-NTRK3 constitutively activated growth factor receptor, which remarkably is found in other cancers such as secretory breast cancer, a subset of acute myelogenous leukemia, and a form of salivary gland carcinoma.82 Other oncogenic proteins appear to act by a mechanism that remodels chromatin structure, which is known to have a profound influence on gene expression (e.g., INI1 mutations in rhabdoid tumors).71 Specific chromosomal rearrangements are very useful in the diagnosis of STSs. Beyond the obvious benefit of providing further objective proof of a diagnosis in morphologically typical cases, the detection of chromosomal aberrations may facilitate the diagnosis of lesions that are difficult to characterize by standard histopathological, ultrastructural, and immunohistochemical techniques.83,84 For example, the presence of the translocation t(X;18)(p11;q11) has been used to confirm the diagnosis of synovial sarcoma in poorly differentiated cases that were diagnostically very challenging.87–87 Similarly, the finding of the characteristic translocation t(11;22)(q24;q12) in a small round blue cell tumor supports the diagnosis of Ewing sarcoma/primitive neuroectodermal tumor (PNET).88 By the same token, a morphologic sarcoma subtype without an expected translocation may suggest a variant translocation, as has been observed in Ewing sarcoma–like small round blue cell tumors that lack the EWSR1-FLI1 translocation but rather contain CIC-DUX4 or a pericentric X chromosome inversion.91–91
Translocations may be identified by a variety of genetic analyses, spanning older techniques such as cytogenetic analysis to fluorescence in situ hybridization, reverse transcriptase polymerase chain reaction, and RNA sequencing. A detailed description of these techniques is beyond the scope of this chapter, but each technique has its advantages and disadvantages. Cytogenetic analysis requires fresh (living) tissue, because the cells need to be cultured before karyotypic analysis, and it largely has been supplanted by newer techniques. Fluorescence in situ hybridization does not require fresh or frozen tissue and frequently may be used with paraffin-embedded material; it is a standard diagnostic tool in many pathology laboratories. It is noted that evidence of a translocation by fluorescence in situ hybridization is not absolutely diagnostic of a sarcoma subtype, because one gene, such as EWSR1, may be involved in many different types of sarcomas, and such data must be incorporated with other data regarding the tumor sample. Reverse transcriptase polymerase chain reaction is a more sensitive technique that may be performed on fresh, frozen, or paraffin-embedded tissues. The major drawback of reverse transcriptase polymerase chain reaction is the relatively high false-positive rate, which results from its sensitivity. Meticulous care is required to prevent problems from contamination. Sequencing of the RNA complement of the genome allows for detection of mutations, translocations, and, to some degree, gene amplification, and is expected to be used more frequently over time.92
Many sarcomas are characterized by several different translocations, most of which are mutually exclusive (see Table 93-3). For instance, alveolar rhabdomyosarcoma is characterized by a translocation involving chromosomes 2 and 13, which results in fusion of the PAX3 and FOXO1 genes, or by a translocation involving chromosomes 1 and 13, which results in fusion of the PAX7 and FOXO1 genes.93–96 Sarcomas within a subtype may also have differences in the specific exons that are involved in each of these different translocations. It has been proposed that this heterogeneity may result in differences in prognosis (see “Potential Molecular Prognostic Factors” below). For example, Ewing sarcoma/PNET and synovial sarcoma possess genetic variations that have been suggested to have prognostic significance, further underscored by the Ewing-like sarcomas that lack EWSR1 fusions but contain other translocations.89,91,97–102 Future research will hopefully establish whether cytogenetic and molecular factors may be used as a basis for therapeutic decisions and the prediction and evaluation of response to treatment.
The identification of genetic alterations with high specificity for different sarcomas will also identify specific therapeutic targets. This has already resulted in the successful treatment of a variety of sarcomas, for example, GISTs, dermatofibrosarcoma protuberans (DFSP), and tenosynovial giant cell tumor/pigmented villonodular synovitis (TGCT/PVNS), and to a lesser degree, angiosarcoma, desmoid tumor, and Ewing sarcoma.79,103–105 Approximately 90% of GISTs harbor activating mutations in the KIT oncogene, which result in ligand-independent activation of the KIT receptor tyrosine kinase pathway.77 Imatinib, a small molecule drug that is administered orally and inhibits the KIT receptor tyrosine kinase, is highly efficacious in the treatment of GIST (see section of treatment of GISTs). DFSP is characterized by translocations involving the COL1A1 and PDGFB genes, which result in activation of the platelet-derived growth factor-β (PDGF-β) pathway in what is believe to be an autocrine fashion. Imatinib is active against the PDGF-β pathway and has been shown to be effective in the treatment of a small number of DFSPs. In a similar way, TGCT/PVNS with its t(1;2) involving COL6A3-CSF1 is responsive to imatinib by virtue of its inhibition of the CSF1 receptor.106
It is noteworthy to mention two common benign soft-tissue tumors—leiomyomas and lipomas—that have a high frequency of chromosomal rearrangements of chromosome 12q that involves the high-mobility protein group gene HMGIC/HMGA2.107,108 These translocations are not seen in the corresponding leiomyosarcomas or liposarcomas, indicating that in these tumors, the benign form is not a precursor of the malignant counterpart. Other benign soft-tissue neoplasms contain specific translocations, such as nodular fasciitis [t(17;22)(p13;q13) MYH9-USP6] or angiomatoid fibrous histiocytoma [t(12;22)(q13;q12) EWSR1-ATF1], underscoring the idea that translocations can occur in tumors anywhere in the spectrum of connective tissue tumors from benign to malignant.109,110
Sarcomas with Complex Karyotypes
A second major subset of STSs is characterized by aneuploidy and the lack of specific fusion genes. This group of sarcomas includes such tumors as leiomyosarcomas, MPNST, and fibrosarcomas (see Tables 93-3 and 93-4). These tumors tend to occur in older patients and appear to have a relatively high frequency of mutations in the p53 and retinoblastoma (Rb) signaling pathways.111,112 These tumors are characterized by chromosomal gains and losses that presumably target tumor suppressor genes (in the event of losses) and oncogenes (in the event of gains). A special case appears to be well-differentiated/dedifferentiated liposarcoma, with amplifications of cell-cycle regulatory genes CDK4 and HDM2 and others on chromosome 12q. The cooperation of these genes to create a unique type of aneuploid sarcoma remains a current challenge in the field. Supporting the clinical data from syndromic sarcomas, alterations disrupting chromosomal mechanics and DNA repair appear to be involved in sarcomagenesis.
Table 93-4
Sarcomas with Complex Karyotypes
| Type of Sarcoma | Resembles |
| Fibrosarcoma (other than congenital) | Fibrous tissue |
| Leiomyosarcoma | Smooth muscle |
| Undifferentiated pleomorphic sarcoma | Poorly differentiated |
| Osteosarcoma | Bone |
| Chondrosarcoma (types other than extraskeletal myxoid) | Cartilage |
| Liposarcoma (types other than myxoid) | Fat |
| Embryonal rhabdomyosarcoma | Skeletal muscle |
| Malignant peripheral nerve sheath tumor* | Nerve sheath |
| Angiosarcoma | Endothelium |
Pathology
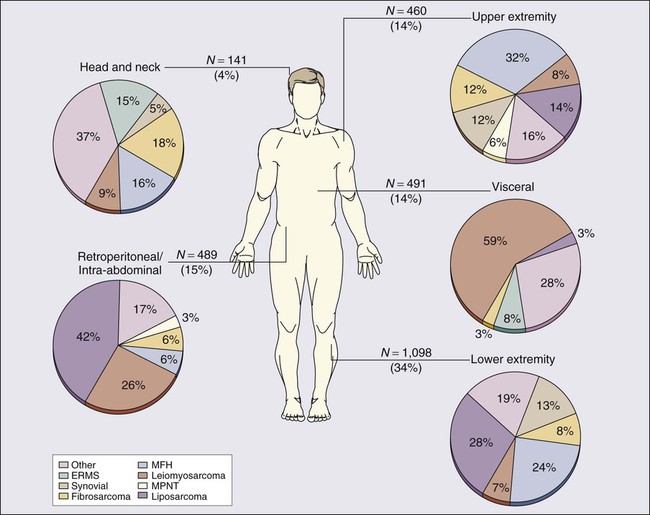
Prospectively collected databases of STS patients, some of which contain data on more than 10,000 patients, demonstrate that STSs are described in essentially all anatomic sites. Figure 93-1 outlines the anatomic sites and site-specific histologic subtypes of 4207 sarcomas treated at a single referral institution. Approximately half of all STSs occur in the extremities (lower: 34%; upper: 14%), where the most common histopathological subtypes are well-differentiated, myxoid, and round cell liposarcoma (28%) and undifferentiated pleomorphic sarcoma (UPS, formerly termed malignant fibrous histiocytoma) (24%). Retroperitoneal sarcomas make up 15% of all STSs, well-differentiated or dedifferentiated liposarcoma being the predominant histologic subtype (42%). Visceral sarcomas make up an additional 14%, whereas the head and neck sarcomas make up approximately 4%.
Classification
An alternative way to index STSs is by their differentiation. Tumors may be grouped broadly into adipocytic tumors, fibroblastic/myofibroblastic tumors, so-called fibrohistiocytic tumors, smooth muscle tumors, pericytic (perivascular) tumors, PNETs, skeletal muscle tumors, vascular tumors, osseous tumors, and tumors of uncertain differentiation (Table 93-5).113 Classification is based on clinical, histologic, ultrastructural, immunohistochemical, and genetic features. Electron microscopic evidence of cellular substructures, neurofibrils, microfilaments, actin-myosin complexes, and dense bodies, has historically been used to define the tissue of origin.114 However, the widespread availability of commercial antibodies for immunohistochemical analysis has all but eliminated the need for electron microscopic examination. Immunohistochemical staining for proteins that are characteristic of smooth muscle (smooth muscle actin and desmin), skeletal muscle (muscle-specific actin, desmin, and myogenin), blood vessels (factor VIII, CD34, and CD31), and epithelial tissue (epithelial membrane antigen and cytokeratins) often facilitates reliable classification, with genetic proof of a sarcoma translocation considered to be iron-clad evidence of a specific sarcoma subtype.115,116 However, because the identical translocation may be found in more than one type of cancer, such as the ASPL-TFE3 translocation of both alveolar soft part sarcoma and papillary renal cancer, translocation data cannot be used in isolation.

The tissue of origin classification scheme is the basis for the 2013 World Health Organization classification system for sarcomas.113,117 The World Health Organization classification system is reproducible for most sarcomas. As the degree of histologic differentiation declines, however, the determination of the tissue of origin becomes increasingly difficult. In particular, despite advanced immunohistochemical and molecular analyses, determining the tissue of origin for some soft-tissue tumors may be difficult, occasionally arbitrary, and sometimes impossible. This leads to significant disparities in diagnoses among pathologists. Discrepancies between the original histologic diagnosis and the subsequent diagnosis by an expert reviewer have been noted in as many as 25% of cases, although these data predate modern immunohistochemical and genetic analyses.118,119 Review of suspicious tissue specimens at an expert center is therefore wise, because the degree of expertise in correctly diagnosing some of the 50 or more types of rare and unusual sarcoma is directly related to the number of sarcomas that a pathologist has seen, and, increasingly, the specific molecular diagnostic tools needed to identify rare sarcoma subtypes.
It is important to classify STSs as precisely as possible because of major differences in their clinical behavior and in their susceptibility to different therapies. For example, a few STSs, including epithelioid sarcoma, clear cell sarcoma, angiosarcoma, and rhabdomyosarcoma have a greater risk of regional lymph node metastasis.120,121 In one single-institution study, the overall rate of nodal metastasis at the time of sarcoma presentation was under 3%; however, the rate was much higher for angiosarcoma (13%), embryonal rhabdomyosarcoma (14%), and epithelioid sarcoma (17%).120,121
Patterns of distant metastases also differ for subtypes of sarcoma. For example, myxoid/round cell liposarcoma tends to metastasize to soft-tissue sites, including the retroperitoneum as well as to the spine and pelvis and other bone marrow sites,122,123 and patients with myxoid/round cell liposarcoma often present with metastatic disease. If a myxoid/round cell liposarcoma is identified in the abdomen, the thighs should be examined for an occult primary tumor. The majority of what had been suspected to be primary myxoid liposarcomas of the abdomen and retroperitoneum are actually misdiagnosed well differentiated / dedifferentiated liposarcomas that have myxoid features (and usually metastasize far less often than myxoid/round cell liposarcoma).124
Histologic Grading
Histologic classification alone does not always provide enough information to predict the clinical behavior of STSs. For many sarcomas, histologic grading provides additional information that can aid in predicting biological behavior and planning treatment. The spectrum of grades varies among specific histologic subtypes (Fig. 93-2). For example, liposarcomas exhibit wide variations in grade, whereas Ewing sarcomas/PNETs are always considered high-grade. In careful comparative multivariate analyses, histologic grade has been the most important prognostic factor in assessing the risk for distant metastasis and tumor-related mortality.127–127 Several grading systems have been proposed, but there is no consensus regarding the specific morphologic criteria that should be employed in the grading of STSs. The two most important criteria appear to be the mitotic index and the extent of tumor necrosis.
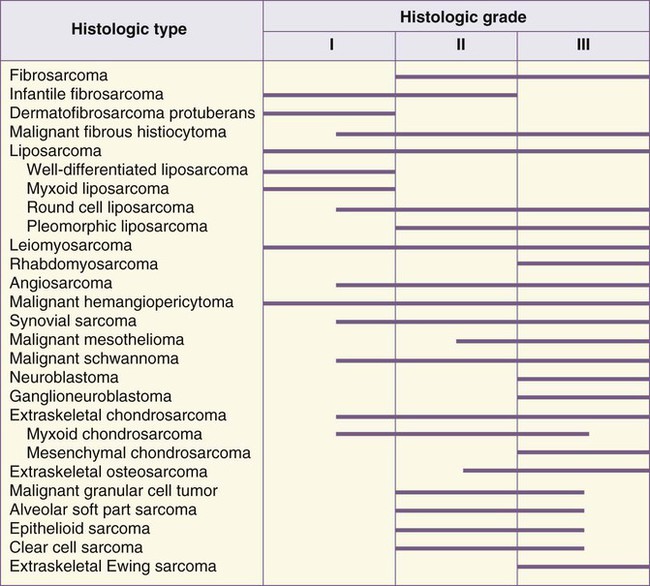
Two of the most commonly used grading systems are the U.S. National Cancer Institute (NCI) system developed by Costa128 and the FNCLCC system (Federation Nationale des Centres de Lutte Contre le Cancer) developed by the French Federation of Cancer Centers Sarcoma Group.129 The NCI system is based on the tumor’s histologic subtype and amount of tumor necrosis, but cellularity, nuclear pleomorphism, and mitotic index are considered for certain subtypes. The FNCLCC system uses a score generated by evaluation of three parameters: tumor differentiation, mitotic rate, and amount of tumor necrosis. The prognostic values of these two grading systems were retrospectively compared in a population of 410 adult patients with nonmetastatic STS.130 Significant discrepancies were observed in one-third of cases. An increased number of grade III tumors, a reduced number of grade II tumors, and better correlation with overall and metastasis-free survival were observed in favor of the FNCLCC system.130 Thus in the absence of other comparative data, the FNCLCC system appears to be the best presently available grading system. The FNCLCC has thus been incorporated into the American Joint Committee on Cancer (AJCC) version 7 STS staging system.131
Clinical Presentation and Diagnosis
The majority of patients with STS present with a painless mass, although pain is noted at presentation in up to one-third of cases132; synovial sarcoma in particular may be most associated with a painful mass, among the sarcomas. Delay in diagnosis of sarcomas is common, the most common incorrect diagnosis for extremity and trunk lesions being lipoma or hematoma.
Biopsy
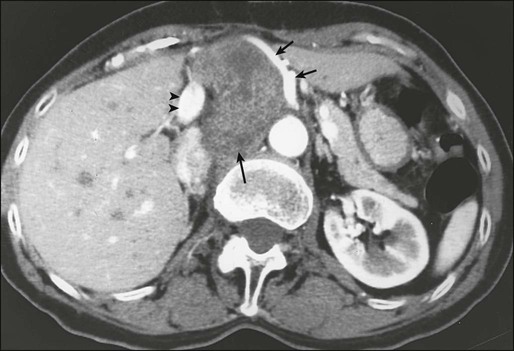
Biopsy of the primary tumor is essential for most patients seen with soft-tissue masses. In general, any soft-tissue mass in an adult that is asymptomatic or enlarging, is larger than 5 cm, or persists beyond 4 to 6 weeks should be biopsied. The preferred biopsy approach is generally the least-invasive technique required to allow a definitive histologic diagnosis and assessment of grade. In most centers, core-needle biopsy provides satisfactory tissue for diagnosis135–135 and has been demonstrated to result in substantial cost savings compared with open biopsy; core-needle biopsy (usually with multiple passes) also yields adequate material for molecular testing, with fine-needle aspiration considered inadequate by most practitioners.135 Direct palpation may be used to guide needle biopsy of most superficial lesions, but less-accessible sarcomas often require an image-guided biopsy to safely sample the most heterogeneous component of the mass. Needle tract tumor recurrences after closed biopsy are rare, but have been reported,136 leading some surgeons to advocate tattooing the biopsy site for subsequent excision or for inclusion in radiotherapy treatment volumes (Fig. 93-3). Because of the frequent difficulty in accurately diagnosing these lesions even when adequate tissue is available, fine-needle aspiration is not recommended for initial diagnosis. The major utility of fine-needle aspiration in most centers is in the diagnosis of suspected recurrent sarcoma.
Imaging
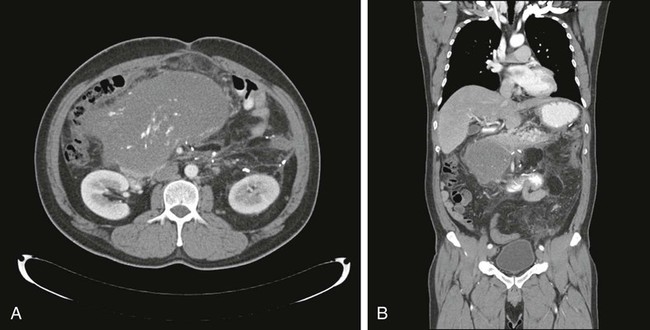
For soft-tissue masses of the extremities, magnetic resonance imaging (MRI) has been regarded as the imaging modality of choice (Fig. 93-4). This is because MRI enhances the contrast between tumor and muscle and between tumor and adjacent blood vessels and provides multiplanar definition of the lesion.137,138 Despite the fact that a study by the Radiation Diagnostic Oncology Group comparing MRI and computed tomography (CT) in patients with malignant bone (N = 183) and soft-tissue (N = 133) tumors showed no specific advantage of MRI over CT from a diagnostic standpoint,139 the majority of musculoskeletal radiologists and almost all surgical oncologists prefer MRI for soft-tissue tumors. For pelvic lesions, the multiplanar capability of MRI may provide superior single-modality imaging, although similar techniques are now commonly available with CT scans (Fig. 93-5). The multiplanar capability is also helpful for visualization of disease in noncoplanar ways when conformal radiotherapy technique is being employed and is an especially helpful adjunct in using image fusion techniques or to visualize peritumoral edema that may harbor sarcoma cells. In the retroperitoneum and abdomen, CT usually provides satisfactory anatomic definition of the lesion (Fig. 93-6) and is a very useful modality for imaging metastatic disease in the lungs and abdomen, given the averaging of signal that occurs with normal patient respiration and peristalsis during the much lengthier MRI. Occasionally, MRI with gradient sequence imaging may better delineate the relationship of the tumor to midline vascular structures, particularly the inferior vena cava and aorta. Older and more invasive studies such as angiography or cavography are almost never used for the evaluation of STSs.
The utility of 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) with CT registration images (PET-CT) in the evaluation and treatment of STS has been a subject of numerous studies and has been reviewed in detail elsewhere. The technique uses radiolabeled glucose analogs,20 which are taken up at increased rates by malignant tumors. Pilot studies of PET in STS suggest that by evaluating tumor metabolic activity, PET scans may allow for noninvasive assessment of tumor grade.140 PET may be helpful in the assessment of locally recurrent STS141 and in the evaluation of response to therapy.142,143 The role and cost-effectiveness of PET in the staging of STS remain undefined; further prospective studies will be required to fully define the role for FDG-PET in the diagnosis, evaluation, and treatment of STS. For example, for the vast majority of patients with metastatic GIST, PET-CT appears to add very little to the assessment of response to tyrosine kinase inhibitors, given the excellent image quality of contrast-enhanced CT scan.144
Staging
The relative rarity of STSs, the anatomic heterogeneity of these lesions, and the presence of more than 50 recognized histologic subtypes of variable grade have made it difficult to establish a functional system that can accurately stage all forms of this family of neoplasms. The seventh edition of the AJCC and the International Union Against Cancer (UICC) is the most widely used staging system for STSs.131 This staging system was first published in 1977, and incorporates histologic grade into the conventional TNM system (Table 93-6). The 2010 seventh edition’s classification system functionally eliminated tumor depth in staging, and classifies node-positive disease without metastases (N1M0) as stage III as compared with stage IV in the sixth edition of 2002. In addition, a unique staging system for GIST has been generated.131 For the detailed background to these changes, the reader is referred to a more complete discussion (SB Edge et al., eds: AJCC Staging Manual, 7th ed.).
Table 93-6
American Joint Committee on Cancer Staging System, Seventh Edition
| Stage | Grade | Tumor | Nodes | Metastasis |
| IA | G1, GX | T1a, T1b | N0 | M0 |
| IB | G1, GX | T2a, T2b | N0 | M0 |
| IIA | G2, G3 | T1a, T1b | N0 | M0 |
| IIB | G2 | T2a, T2b | N0 | M0 |
| III | G3 | T2a, T2b | N0 | M0 |
| Any | Any | N1 | M0 | |
| IV | Any G | Any T | Any N | M1 |
| G: PRIMARY TUMOR GRADE | ||||

From AJCC Cancer Staging Manual, 7th ed. New York, Springer Verlag, 2010.
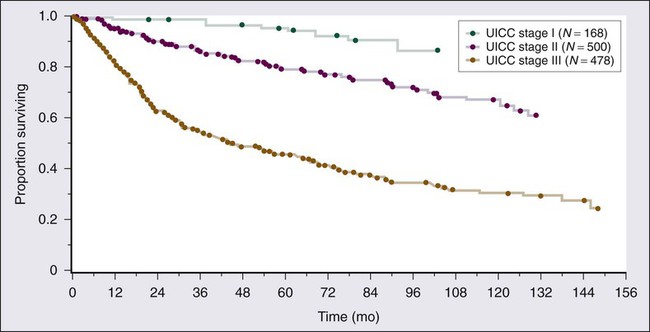
Three distinct histologic grades (G1 to G3) are recognized, using the French (FNCLCC) grading system, in the interest of consistency between institutions. Histologic grade and tumor size are the primary determinants of clinical stage (see Table 93-6). Tumor size is further substaged as “a” (a superficial tumor that arises outside the investing fascia) or “b” (a deep tumor that arises beneath the fascia or invades the fascia), although it does not impact stage in AJCC version 7. Figure 93-7 outlines stage-specific survival plots based on previous versions of the AJCC criteria.
Nomograms and newer techniques such as Bayesian belief networks may better help specify risk for discussion with patients.145,146 These have been generated for both specific anatomic sites as well as for specific sarcoma histologies. It is difficult to envision a unique staging system for each of the more than 50 STS histologies, which argues that a multidimensional risk assessment as may be performed with nomograms should supersede traditional staging systems in the interest of accuracy, although this comes at the cost of simplicity. As the first example of this technique being used in STSs, Kattan and colleagues constructed and validated a nomogram to predict the probability of 12-year sarcoma-specific death based on a prospective series of patients.145 This tool is useful for individual patient counseling, follow-up scheduling, and clinical trial eligibility assessment and is further facilitated by being also available for personal computers and handheld devices. Nomograms have since been developed for specific anatomic sites such as the retroperitoneum, or for specific sarcoma subtypes such as liposarcoma.147–150 These useful tools may be found online at www.nomograms.org.
Prognostic Factors
Conventional Clinicopathological Factors
The initial study of prognostic factors in extremity sarcoma from Memorial Sloan-Kettering Cancer Center evaluated clinicopathological prognostic factors in a series of 423 patients with localized extremity STS seen from 1968 to 1978.142 This analysis was among the first to discriminate between specific clinical end points, and clearly established the clinical profile of what is now accepted as the high-risk patient with extremity STS: the patient with a large (in this case >5 cm), high-grade, deep lesion. The adverse prognostic significance of a high tumor grade, deep tumor location, and tumor size larger than 5 cm was also noted in the recent report of the French Federation of Cancer Centers study of 546 patients with sarcomas of the extremities, head and neck, trunk wall, retroperitoneum, and pelvis.126
A follow-up report evaluated clinicopathological prognostic factors that had been documented prospectively in a population of 1041 patients with extremity STS.125 The end points for the multivariate analyses were local recurrence, distant recurrence (metastasis), and disease-specific survival. Results of the regression analyses for each of these end points are summarized in Table 93-7 as an example of the key prognostic factors that are ascertained in such studies. These results, by using prospectively acquired data, confirm the initial observations made at that institution by using an independent data set.151 In addition, the previously unappreciated prognostic significance of specific histologic subtypes and the increased risk for adverse outcome associated with a microscopically positive surgical margin or locally recurrent disease were noted. Unlike other solid tumors, the adverse prognostic factors for local recurrence of a STS are somewhat different from those that predict distant metastasis and tumor-related mortality (Table 93-7).125 Therefore staging systems that are designed to stratify patients according to risk of distant metastasis and tumor-related mortality by using these prognostic factors (such as the AJCC/UICC system) do not stratify patients according to risk of local recurrence.
Table 93-7
Multivariate Analysis of Prognostic Factors in Patients with Extremity Soft-Tissue Sarcoma
| End Point | Adverse Prognostic Factor* | Relative Risk |
| Local recurrence | Age >50 years | 1.6 |
| Local recurrence at presentation | 2.0 | |
| Microscopically positive margin | 1.8 | |
| Fibrosarcoma | 2.5 | |
| Malignant peripheral nerve tumor | 1.8 | |
| Distant recurrence | Size 5-10 cm | 1.9 |
| Size >10 cm | 1.5 | |
| High-grade | 4.3 | |
| Deep location | 2.5 | |
| Local recurrence | 1.5 | |
| Leiomyosarcoma | 1.7 | |
| Other nonliposarcoma histology | 1.6 | |
| Disease-specific survival | Size >10 cm | 2.1 |
| Deep location | 2.8 | |
| Local recurrence at presentation | 1.5 | |
| Leiomyosarcoma | 1.9 | |
| Malignant peripheral nerve sheath tumor | 1.9 | |
| Microscopically positive margin | 1.7 | |
| Lower-extremity site | 1.6 |
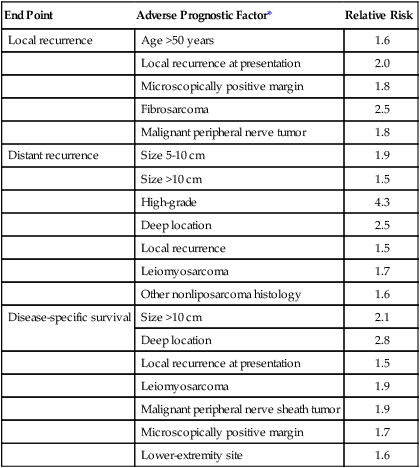
*Adverse prognostic factors identified are independent by Cox regression analysis.
Modified from Pisters PWT, Leung DHY, Woodruff JM, et al. Analysis of prognostic factors in 1041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679.
It should be emphasized that the prognostic factors that have been identified are derived primarily from studies of patients with localized extremity sarcomas. Despite the fact that extremity sarcomas make up the majority of sarcomas, these results do not necessarily apply to the populations of STS patients from other sites. For example, the AJCC staging system for STSs of retroperitoneum or viscera is nonsensical with respect to primary tumor depth (indicated as superficial or deep), because all tumors from this anatomic site are deep tumors, and staging systems specific for each clinical scenario may be more appropriate means to estimate clinical outcomes. Separate reviews of prognostic factors for sarcomas of the retroperitoneum,152,153 head and neck,154–157 gastrointestinal tract, colon and rectum,160 uterus,161 synovial sarcomas,162,163 UPS,166–166 and Ewing sarcoma167–170 also have been reported.
Potential Molecular Prognostic Factors
As clinicopathological factors affecting patient outcomes were being identified, examination of the tissue itself for factors predicting outcome became another topic of interest, and continues to this day in the form of more detailed molecular profiling of RNA expression, comparative genomic hybridization, tumor DNA or RNA sequencing, and analysis of DNA modifications such as methylation patterns in specific tumors. Some of the first parameters that were evaluated for prognostic significance included p53,171 HDM2 (the human version of the murine p53 interacting protein MDM2),171 Ki-67,171 altered expression of the retinoblastoma gene product (pRb)59,172 in high-grade sarcomas, and the importance of the specific type of SS18-SSX fusion transcripts in synovial sarcoma99 or EWSR1-FLI1 fusion transcripts in Ewing sarcoma.97 Notably, whereas many individual immunohistochemical markers have been examined as a prognostic factor in outcome, data are generally conflicting. It is well recognized that there is variability of immunohistochemistry with both technique, which may vary between laboratories, and assessment; what may be 2+ staining to one investigator is 3+ or 4+ staining to another.
As one of the first examples of analyzing multiple immunohistochemical markers with respect to outcome, Heslin and colleagues evaluated the potential prognostic significance of pRb, p53, HDM2, and Ki-67 by immunohistochemical techniques in a population of 121 patients with primary, high-grade extremity sarcomas, and compared these factors with conventional clinicopathological prognostic factors (median follow-up: 64 months).171 Clinicopathological and molecular factors that were found to be statistically significant adverse prognostic factors in both univariate and multivariate analyses for the separate end points of distant metastasis and tumor-related mortality included tumor size greater than 5 cm, microscopically positive surgical margin, and a Ki-67 score greater than 20 (>20% nuclear staining). Overexpression of p53 or HDM2 or deletion of pRb did not correlate with an increased risk of distant metastasis or tumor-related mortality. Similarly, data with respect to specific SS18-SSX translocation type (i.e., SSX1 or SSX2) and outcome have yielded conflicting results,99,173 and the specific EWSR1-FLI1 translocation does not appear to affect overall outcomes for patients.97,98,174
With the increasing use of genomic analyses such as RNA expression arrays, comparative genomic hybridization or tumor RNA sequencing, it is possible that expression or mutation-specific stratification may be possible within specific histologies, in a manner analogous to current use of this technique to stratify breast cancer.175 Additionally, these profiles have recently been applied to determining metastatic potential of primary tumors, and this approach could clearly be applicable to STSs, in which the presence or absence of metastases remains the most important prognostic factor.176 Phosphoprotein profiling could become useful for patient risk stratification, as has been examined in stage III rhabdomyosarcoma to predict outcome.177 This approach also holds future promise for both prognostic and therapeutic approaches, although concerns regarding phosphoprotein lability and tissue handling are concerns in the proper conduct of such studies. Although specific cellular and molecular parameters have been identified as having independent prognostic significance, there is currently no consensus on how specific molecular prognostic factors should be used in clinical practice. Validated markers may affect outcome prognostication in the future, but for the time being remain investigational.
Prognostic Factors as Therapeutic Targets
The prognosis of GISTs is often poor when treated by surgery alone,178 and these tumors do not respond to conventional systemic chemotherapy. Arguably the most exciting discovery in GIST research is the finding of the activity of kinase-directed therapy, which is discussed later in the chapter. The protooncogene KIT is the cellular homolog of the oncogene v-Kit (identified in a feline sarcoma virus). KIT (also termed CD117) encodes a transmembrane tyrosine kinase receptor that is structurally similar to platelet-derived growth factor and provides selective targets of key aberrations in the molecular signaling implicated in the pathogenesis of GISTs and other tumors (e.g., DFSP). An interesting additional feature of KIT expression in GIST is that different types and locations of mutations in KIT are independent risk factors in predicting disease-free survival independent of treatment with kinase receptor inhibitors.179 KIT expression was uniformly evident in a study of GIST cases not treated with imatinib.77,179 Of interest, KIT was highly phosphorylated in all cases, even in those samples that lacked demonstrable KIT mutations.77 Mutations are found in specific regions of the KIT gene (most commonly exon 9 and exon 11), and are associated with differing prognosis (see the following discussion). In the era before imatinib, the subset of patients with exon 11 KIT mutations resulting in single amino acid substitutions (i.e., missense codon mutations) fared much better than patients with deletion/insertion mutations of exon 11 (5-year recurrence-free survival rate of 89% ± 11% vs. 37% ± 10%, respectively). A potential explanation for this finding is that exon 11 missense mutations are detected in lower-grade, favorable outcome GISTs.179 Against this explanation is the fact that the metastatic GISTs with KIT exon 9 mutations appear to have a lower risk of relapse compared with exon 11 KIT mutation or PDGFRA mutant GISTs. Although it is conceivable that this type of mutation is a surrogate for the behavior of a GIST, it is also plausible that this type of mutation represents the initial pathogenetic mechanism, making it a true prognostic marker and target. It is apparent that the mutation in KIT is context dependent; KIT mutation and ETS transcription factor family member ETV1 overexpression appear to synergize to drive GIST development.180
Molecular Therapeutic Targets in Sarcomas: Gastrointestinal Stromal Tumor
We have just now begun to see the implications and success of targeting genetic alterations in sarcomas that drive oncogenesis. GISTs were previously thought to be gastrointestinal leiomyosarcomas that were particularly resistant to cytotoxic chemotherapy. It was subsequently demonstrated that these tumors were derived from interstitial cells of Cajal or their precursors and were frequently characterized by point mutations in the KIT receptor tyrosine kinase and were clearly distinct from leiomyosarcomas.181,182 Subsequently, the tumors were treated with imatinib, which targets the KIT kinase, with dramatic results.183 KIT mutations were observed in exon 11 (juxtamembrane domain, seen in 71% of tumors), exon 9 (the extracellular region, 13%), exon 13 (first lobe of the split-kinase domain, 4%), and exon 17 (phosphotransferase domain, 4%). The subset of patients with exon 11 mutations resulting in single-point amino acid substitutions (i.e., missense codon mutations) fared much better than did patients with deletion/insertion mutations of exon 11 (5-year recurrence-free survival rate of 89% ± 11% vs. 37% ± 10%, respectively).179 In addition, tumors that lack KIT mutations appear to have mutations in platelet-derived growth factor (PDGF)-α receptor gene PDGFRA, and those that have no mutation in KIT or PDGFRA are driven by loss of succinate dehydrogenase expression, mutation in BRAF, or other genetic alterations, all changes that are mutually exclusive of KIT mutations.184 Finally, the type of mutation that is identified appears not only to predict response to imatinib, but also to suggest whether other kinase inhibitors, such as sunitinib, may have beneficial effects.185 Although to date the most common adult STS demonstrate few consistent mutations in kinase genes that are amenable to target inhibition, new options for treatment likely will be derived from more careful genetic analysis of specific sarcoma subtypes by using newer sequencing techniques, comparative genomic hybridization, analysis of tumor microRNA and DNA methylation patterns.92
Treatment of Localized Primary Soft-Tissue Sarcoma
Surgery
Limb-Sparing Surgery Versus Amputation
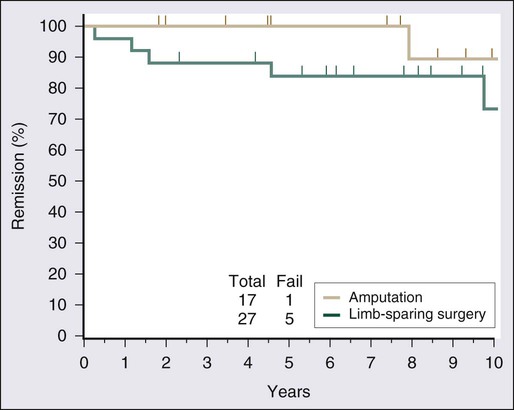
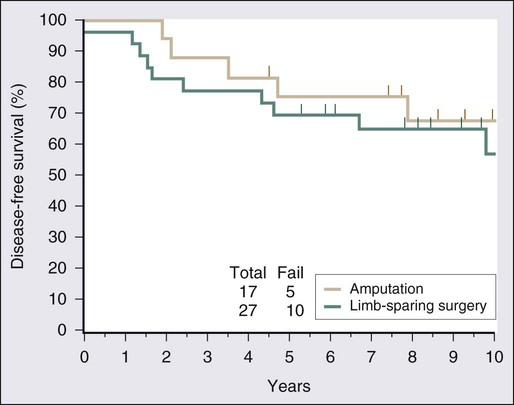
Surgical resection remains the cornerstone of therapy for localized STS, and the prototypical situations concern the management of lesions arising in the extremity, the most common anatomic site. Since the early 1980s, there has been a marked decline in the rate of amputation as the primary therapy for extremity STS. With the widespread application of multimodality treatment strategies, fewer than 10% of patients undergo amputation.187,188 The current use of limb-sparing multimodality treatment approaches for patients with extremity sarcoma is largely based on a randomized prospective study from the U.S. NCI in which patients with extremity sarcomas that were amenable to limb-sparing surgery were randomly assigned to receive amputation or limb-sparing surgery with postoperative radiotherapy.189,190 Both arms of this trial included postoperative chemotherapy with doxorubicin, cyclophosphamide, and methotrexate. With more than 9 years of follow-up evaluation, 5 (19%) of 27 patients randomly assigned to receive limb-sparing surgery and postoperative radiation with chemotherapy had local recurrences, compared with 1 (6%) of 17 patients in the amputation plus chemotherapy arm (P = 0.22; Fig. 93-8).190 The disease-free survival rate was 63% for limb-sparing surgery versus 71% for amputation (P = 0.52; Fig. 93-9), and the overall survival rate was 70% for limb-sparing surgery versus 71% for amputation (P = 0.97). This study established that for patients for whom limb-sparing surgery is an option, a multimodality approach using limb-sparing surgery combined with postoperative radiotherapy yields disease-related survival rates comparable to those for amputation while simultaneously preserving a functional extremity.
Currently, at least 90% of patients with localized extremity sarcomas can undergo limb-sparing procedures.187,191 Most surgeons consider definite major vascular, bony, or nerve involvement to be relative indications for amputation. Complex en bloc bone, vascular, and nerve resections with interposition grafting may be undertaken, but the associated morbidity is high. Therefore, for a few patients with critical involvement of major bony or neurovascular structures, amputation remains the only surgical option but offers the prospect of prompt rehabilitation with excellent local control and survival.190 An approach approved in Europe for selected patients with locally advanced tumors is limb perfusion, in which a tourniquet is placed proximal to the tumor on a limb, the blood flushed out, and then a perfusate of tumor necrosis factor (TNF) and chemotherapy administered through a recirculating pump. This technique has allowed for limb salvage even for patients with otherwise unresectable disease, with chronic edema the most common long-term complication.194–194 However, TNF is unavailable in the United States, either for patient care or clinical trials.
Completeness of Resection
Satisfactory local resection involves resection of the primary tumor with a margin of normal tissue around the lesion. The width of the margin should differ depending on whether or not adjuvant radiotherapy is used. It is clear that dissection along the tumor pseudocapsule (enucleation) is associated with local recurrence rates ranging between 33% and 63%.197–197 Wide local excision with a margin of normal tissue around the lesion is associated with local recurrence rates in the range of 10% to 31%, as was noted in the control arms (surgery alone) of the randomized trials evaluating postoperative radiotherapy.198,199
In contrast to malignant melanoma, a cancer in which there are randomized data to address adequate margin size, no comparable data are available to define what constitutes a satisfactory gross resection margin for a sarcoma, because the anatomic situation with each STS is unique. In general, every effort should be made to achieve a wide margin (2 cm is a frequently cited arbitrary choice) around the tumor mass, except in the immediate vicinity of functionally important neurovascular structures, where, in the absence of frank neoplastic involvement, dissection is performed in the immediate perineural or perivascular tissue planes. The 2-cm choice is unnecessary if radiotherapy is also used, because substantial modification of the surgical approach with much closer margins of resection (e.g., 1 to 2 mm) is made possible, and even large lesions may be managed conservatively in that setting. Technical details of the surgical approach to extremity sarcomas are beyond the scope of this chapter, but are comprehensively reviewed in surgical atlases.200 At the same time, it is also important to bear in mind that involved (i.e., positive) resection margins remain an adverse finding even when adjuvant radiotherapy is used, notwithstanding the amelioration of risk that radiation treatment provides. Data from Memorial Sloan-Kettering Cancer Center, Princess Margaret Hospital, and Massachusetts General Hospital suggest an additional absolute reduction of local control of approximately 10% to 15% for patients with positive margins compared with those with microscopically negative surgical margins.200–203 As a result, radiation therapy should never be considered adequate therapy after incomplete surgical resection.
In considering the existing outcome data, it is important to bear in mind that these data consider the rubric “positive margins” in a uniform way, although in reality, this is unlikely to be the case. In fact, positive resection margins have different causes. One is oncologically inadequate surgery in which positive resection margins might have been avoidable in another surgeon’s hands. When this is the case, microscopically positive surgical margins may be considered a technical failure. Alternatively, positive resection margins may arise in anatomically adverse presentations in which locally advanced disease challenges the goals of conservative resection from the outset. In another study from the Princess Margaret Hospital, Gerrand and colleagues evaluated the type of microscopically positive margin as a prognostic factor and defined four groups in this setting.204 Patients with low-grade liposarcomas and microscopically positive surgical margins (group 1) have a low risk of local failure, as do those in whom a positive margin is anticipated before surgery to preserve critical structures and to whom radiotherapy is given to sterilize the minimal residual disease (group 2). However, two categories of patients with positive margins are associated with a higher risk of local recurrence: (a) patients who are seen after unplanned excision who have a positive margin on subsequent reexcision (group 3) and (b) patients with unanticipated positive margins occurring during primary sarcoma resection (group 4). For group 3, an “unplanned excision” is defined as an excisional biopsy or resection that is carried out without adequate preoperative staging or consideration of the need to remove normal tissue around tumor, an adverse feature reported by the same authors previously.205 These data appear to support the premise that, provided that adjuvant radiotherapy is administered, a very small amount of residual disease resulting from a “planned” positive margin at the site of a critical anatomic structure (group 2, local recurrence rate of 3.6% and 95% confidence interval [CI], 0% to 10.4%) is not associated with the same deleterious risk that occurs with a positive margin that follows major contamination because of a “shell out” intralesional surgery (group 3, local recurrence rate of 32%; 95% CI, 11% to 53%) or inadvertent contamination of the wound (group 4, local recurrence rate of 38%; 95% CI, 14% to 61%).204 These data seem particularly relevant to anatomic sites where achievement of adequate resection margins is a perennial problem, such as the head and neck, as evidenced by recent results from a prospective series where the outcome approaches that of extremity and body wall sarcomas.206 We caution, however, that such results are probably not attainable without a defined management protocol and joint multidisciplinary assessment before treatment is undertaken, because there exist issues that merit discussion at the individual case level (e.g., the complex relationship between tissues to be resected and reconstructed and the radiotherapy volumes and doses, all of which can influence each other in the decision algorithm).
Lymph Node Dissection
Given the low (less than 3%) prevalence of lymph node metastasis in adults with sarcomas,117,118 there is no role for routine regional lymph node dissection. Patients with angiosarcoma, embryonal rhabdomyosarcoma, and epithelioid histologies have an increased incidence of lymph node metastasis and should be carefully examined for adenopathy by physical examination, imaging, and sentinel lymph node sampling. Therapeutic lymph node dissection (curative) results in a 34% actuarial survival rate120; therefore the rare patients (who more commonly have evidence of other metastatic disease) with regional nodal involvement who have no evidence of extranodal disease should undergo therapeutic lymphadenectomy. Patients with adverse features at the time of dissection (i.e., extracapsular extension beyond the lymph nodes into perinodal fat or positive or doubtful margins on the neurovascular bundle) or in whom treatment into the next grossly uninvolved lymph node echelon is not feasible with surgery should also be considered for additional adjuvant nodal irradiation. The principles underlying this approach have been outlined.207
Surgery Alone
Although the majority of patients with extremity STS should be treated with preoperative or postoperative radiotherapy, recent reports suggest that concomitant radiotherapy might not be required for selected patients with completely resected, small, primary STSs (Table 93-8).208–211 Rydholm and colleagues reported their experience with 70 patients with subcutaneous or intramuscular extremity sarcomas treated with wide surgical resection and microscopic assessment of surgical margins.210 Negative histologic margins were obtained for 32 of 40 subcutaneous and 24 of 30 intramuscular tumors. The 56 patients with microscopically negative margins received no postoperative radiotherapy, yet only 4 (7%) experienced local recurrence. A study from Brigham and Women’s Hospital reported similar results for a selected group of 74 patients with primary extremity STS treated by surgery without radiotherapy.211 The 10-year actuarial local control rate was 93% ± 4%. The absolute gross margin was a significant predictor of local recurrence; patients with a close gross margin of less than 1 cm had a 10-year local control rate of 87% ± 6% compared with 100% for patients with a closest gross margin of 1 cm or greater (P = 0.04). The generally favorable local control rates with surgery alone that these and other investigators200,212 reported in these series of highly selected patients are comparable to local recurrence rates observed for more heterogeneous patient populations treated with conventional multimodality therapy incorporating preoperative or postoperative radiotherapy (Table 93-9).68,198,213–219 These data support the hypothesis that selected patients with small, primary STSs may be treated with surgical resection alone without preoperative or postoperative radiotherapy.
Table 93-8
Selected Results of Surgery Alone for Patients with Soft-Tissue Sarcoma
| First Author | Institution | No. of Patients | Selection Criteria | Adjuvant Radiation (No.) | Local Recurrence (%) |
| Geer209 | MSKCC | 174 | T1 size, primary tumor | 117 | 10 |
| Rydholm210 | Lund, Sweden | 56 | G/M margin negative | 0 | 7 |
| Baldini211 | BWH | 74 | T1 size, G/M margin negative | 0 | 7 |
| Karakousis208 | RPCI | 116 | 2-cm G margin | 0 | 10 |
| Fabrizio582 | Mayo | 34 | Not stated | 0 | 15 |

Table 93-9
Selected Studies* Examining Local Control with Surgery and Radiotherapy for Localized Soft-Tissue Sarcoma
| Radiotherapy Approach | First Author | Radiation Dose (GY) | Study Design | No. of Patients | Local Failure (%) | Subset |
| Preoperative EBRT | Suit213 | 50-56 | Retrospective | 89 | 17 | |
| Barkley214 | 50 | Retrospective | 110 | 10 | ||
| Brant215 | 50.4 | Retrospective | 58 | 9 | ||
| O’Sullivan221 | 50 | RCT | 94 | 7 | ||
| Brachytherapy | Pisters203 | 42-45 | RCT | 119 | 9 | (high-grade) |
| 45 | 23 | (low-grade) | ||||
| Postoperative EBRT | Lindberg217 | 60-75 | Retrospective | 300 | 22 | |
| Karakousis218 | 45-60 | Retrospective | 53 | 14 | ||
| Suit213 | 60-68 | Retrospective | 131 | 12 | ||
| Yang198 | 45 + 18 | RCT | 91 | 0 | (high-grade) | |
| 50 | 5 | (low-grade) | ||||
| O’Sullivan221 | RCT | 96 | 7 |
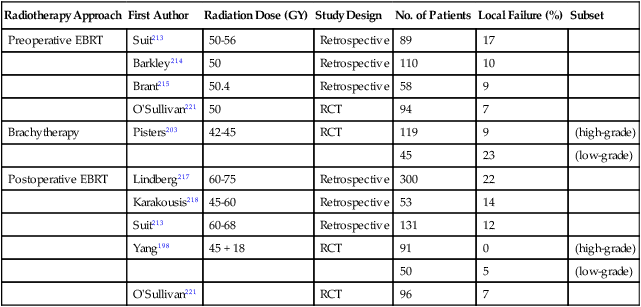
EBRT, External beam radiotherapy; RCT, randomized controlled trial.
*Randomized controlled trials and selected nonrandomized retrospective series.
It is difficult to define the precise selection criteria that should be used to identify patients with primary sarcoma who can safely undergo treatment by surgery without radiotherapy. Most investigators have limited this approach to patients with carefully selected T1 tumors that may be resected with clear margins (see Table 93-8). In contrast, Karakousis and colleagues did not consider absolute tumor size but instead used surgical resection alone for all patients in whom a minimum intracompartmental margin of 2 cm could be maintained circumferentially, irrespective of tumor size.208 Karakousis and colleagues updated the results for high-grade STS of the policy of limiting the use of postoperative radiation treatment for tumors resected with positive or “narrow” (<2 cm) resection margins.220 This approach has yielded useful data because the consistent application of this treatment approach resulted in a local recurrence rate of 19% with “wide” margin surgery alone compared with 24% after “narrow” margin surgery and adjuvant radiotherapy.220 Although the results provide some clarity about the 2 cm or greater margin benchmark, it would be useful to also have similar data from other groups for a variety of margin widths to draw conclusions about when it is safe to withhold radiotherapy. Moreover, the authors acknowledge the potential to treat a greater proportion of cases with radiotherapy and lower the 19% local recurrence rate in some of those “favorable cases” that are currently treated with surgery alone by widening the indication for adjuvant radiotherapy in a proportion of these patients.220 This view is consistent with contemporary observations such as the local recurrence rate of 7% in patients who received combined modality treatment such as those in the recent Canadian randomized trial (see “Preoperative or Postoperative Radiotherapy” below).221
Factors other than anatomic location, tumor size, and the feasibility of achieving an R0 resection (macroscopically and microscopically complete) should be considered in selecting patients for treatment by surgery alone. For example, the issue of whether the patient has had a prior “unplanned” excision (see earlier) is important. At the Princess Margaret Hospital, a significantly higher rate of local recurrence was apparent in patients who were treated after unplanned excision on the outside than in patients who received their treatment at their institution (22% vs. 7%, P = 0.03).205 It is important to remember that unplanned excision is very common in the community setting, where small soft-tissue lesions are often excised without image guidance under the presumption that they are benign. Therefore, although it is reasonable to attempt a reexcision if it is considered feasible, patients who have undergone unplanned excision should also be strongly considered for adjuvant radiation.
Preoperative or Postoperative Radiotherapy
Conservative (limb-sparing) surgery and radiotherapy have been combined to optimize local control for patients with localized STS. Radiotherapy may be administered preoperatively,213–215,222,223 postoperatively,217,224,225 or by interstitial techniques (brachytherapy).68,216,226–230
Local Control
Data from two randomized controlled trials (RCTs)73,198 have confirmed earlier retrospective reports suggesting that surgery combined with radiotherapy results in superior local control compared with surgery alone.214,217,219 Yang and colleagues from the NCI recently reported on a RCT of postoperative external beam radiotherapy (EBRT).198 In this trial, 141 patients with localized extremity STSs amenable to limb-sparing resection were randomly assigned to receive postoperative EBRT or no radiotherapy. All patients with high-grade lesions received postoperative chemotherapy. In the subset of 91 patients with high-grade lesions, no local recurrences have been noted in the 44 patients who received postoperative radiotherapy (with chemotherapy) versus nine local recurrences (19%) in the 47 patients who received postoperative chemotherapy alone (P < 0.001). In the 50 patients with low-grade sarcomas, 1 (4%) of 26 patients who received adjuvant radiotherapy has had a local recurrence versus 8 (33%) of 24 patients treated by surgical resection alone (P = 0.016). However, no improvement in survival was noted with adjuvant radiotherapy in the entire cohort of patients or in any subgroup.
The second RCT of postoperative radiotherapy was conducted at Memorial Sloan-Kettering Cancer Center, where investigators studied adjuvant brachytherapy for patients with extremity and superficial trunk STSs.203 One hundred sixty-four patients with extremity or superficial trunk STSs were randomly assigned to receive adjuvant brachytherapy (42 to 45 Gy with an iridium-192 implant) or no postoperative radiotherapy after complete resection of their sarcomas. Random assignment took place in the operating room after gross total resection, thereby limiting the potential bias that might influence the extent of surgical resection in a comparative trial. Sixty-eight of 119 patients with high-grade tumors also received chemotherapy. With a median follow-up of 76 months, 5-year actuarial local control rates were significantly better in the group treated with adjuvant brachytherapy (82%) than in those who received surgery alone (69%). Subset analysis demonstrated that the local control advantage of brachytherapy was confined to patients with high-grade lesions, for whom the 5-year local control rate was 89% (vs. 66% in the surgery-only group). Patients with low-grade STSs did not appear to experience the same local control benefit with adjuvant brachytherapy.203,230 As was noted in the NCI RCT,184 the improvement in local control did not translate into any detectable survival difference between the brachytherapy and no-brachytherapy arms of the trial.
Local failure rates with combined-modality regimens incorporating surgery and radiotherapy are generally less than 15% (see Table 93-9). Despite theoretical advantages that may favor preoperative radiation, brachytherapy, or postoperative radiation, there does not appear to be a major difference in local control rates among these radiation techniques, although at present, data comparing the approaches are sparse.
Relationship Between Local Control and Survival
Whether local control affects overall survival for patients with STS remains unclear and highly controversial.231–235 Only an adequately powered prospective randomized trial can assess the precise nature of any relationship between local control and overall survival. Three RCTs have evaluated local control and survival in the context of defining treatment approaches for STS. In a randomized trial of amputation versus conservative surgery plus radiation from the NCI, local recurrence rates were 19% in the limb-sparing arm versus 6% in the amputation arm (P = 0.022).189,190 Despite this, overall survival rates were equivalent at 70% for limb-sparing surgery and 71% for amputation (P = 0.97). In the randomized trials of postoperative radiotherapy,198,203 the improvement in local control that was noted in patients who were treated with surgery plus radiotherapy did not translate into any detectable survival advantage. None of the currently available data from prospective RCTs support the hypothesis that better local control enhances survival in patients with sarcoma. Methodologically, the available trials are problematic for this issue because the outcome of interest (i.e., a difference in survival consequent on a differential in local control) would require a prohibitively large sample size. Thus, it is most improbable for these trials to be capable of demonstrating an effect with their modest sample sizes that were intended for evaluation of different outcomes. Indeed, it is most unlikely if even a meta-analysis of the trials could demonstrate this. Furthermore, data from nonrandomized studies support the concept that there is little, if any, relationship between local control and survival. In a recent series from Sweden, the outcome of patients who were treated with an inadequate excision was compared with that of patients who had an adequate operation.234 Local recurrence was 3.5 times more common after inadequate excision, but there was no difference in the incidence or timing of distant metastases. The power of the RCTs that have been reported so far to detect a difference in survival is relatively small, and a large number of patients may be required to demonstrate that prevention of local recurrence affects survival.232 Stotter and colleagues have argued that local recurrence is a time-dependent variable and should be considered as such in multivariate studies.231 Analysis in this fashion of the data from a nonrandomized study demonstrates a statistically significant relationship between local control and survival. Other retrospective analyses have yielded similar conclusions.236,237
For a more detailed description of the methodologic problems associated with time-dependent variables and the use of surrogate end points that emerge after the initial sarcoma treatment, the reader is referred elsewhere.238 In this context, it is clearly important to distinguish between the well-defined adverse prognostic impact of subsequent local recurrence on survival125,235,239 and the unproved positive effect of improved local control (i.e., prevention of local recurrence with improved local therapy) on survival. The former phenomenon might be a manifestation of more aggressive tumor biology; that is, biologically more aggressive lesions might recur locally and metastasize more frequently.
Treatment Sequencing: Preoperative Versus Postoperative Treatment
Of further interest, an improvement in overall survival (a crude rate of 85% vs. 72% in favor of postoperative radiotherapy, P = 0.048) emerged initially from the Canadian SR2 RCT, but dissipated after longer follow up (Fig. 93-10).221,240 Thus in terms of overall survival, it appears reasonable to treat patients with postoperative EBRT, as local control rates are comparable to preoperative techniques, but major wound complication rates are significantly lower. On the other hand, given the lower risk of late effects such as fibrosis with preoperative radiation, consideration of longer-term end points should be a consideration in any patient in whom radiation therapy is to be considered.241 Also, in anatomic sites where wound complications are rarely seen (e.g., the upper extremity), the rationale for wound complication avoidance as a reason to favor the use of postoperative radiotherapy is not as sound. In particular, the obvious advantage to preoperative radiotherapy in the proximal arm and shoulder is apparent where avoidance of large volume and higher dose irradiation that may treat the lung or brachial plexus may be achieved. These principles are also reasonable in the head and neck based on data from Princess Margaret Hospital.206
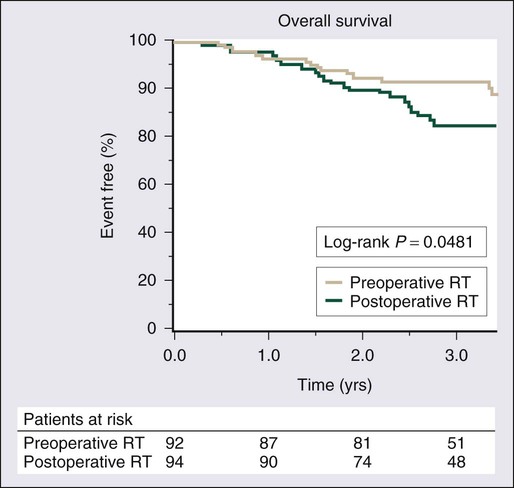
In contrast to EBRT, which requires 5 to 7 weeks of treatment, with brachytherapy, the patient’s entire local treatment (surgery plus radiation) may be completed in the 2 weeks following primary tumor resection. Brachytherapy has potential cost advantages242 and has implications in terms of overall patient convenience. Accordingly, if the necessary expertise is available, brachytherapy provides an excellent, cost-effective alternative for patients with high-grade lesions, although such expertise is increasingly uncommon. Brachytherapy should not be used for patients with low-grade sarcomas.230
The only randomized study of preoperative versus postoperative EBRT for patients with localized extremity STS was conducted by the National Cancer Institute of Canada Clinical Trials Group/Canadian Sarcoma Group. In this trial, 190 patients with extremity STS were randomly assigned to preoperative versus postoperative EBRT.221 The radiotherapy parameters for this protocol required a field margin of 5 cm around the gross tumor volume for the initial phase of treatment (i.e., treatment to 50 Gy in 25 fractions), and this generally included any peritumoral edema that was seen on MRI, irrespective of the grade or size of the tumor. Subsequently, a reduced-volume field was treated to a total combined dose of 66 Gy in all postoperative cases and in those preoperative patients in whom the resection margins were involved.
The results of this trial are complex because the primary end point that powered the trial, and hence its sample size, was the cumulative incidence of acute wound complications 120 days after protocol surgery in both arms of the study. Nevertheless, the local control rates after 3.3 years of median follow-up are identical in both arms of the study (7%). As might be expected, local wound complications were more common in the preoperative radiation cohort, whereas increased late complications such as radiation field fibrosis were more common in the postoperative group, who were treated with a larger treatment field with a higher total dose of EBRT.243
It is clear that although field size and radiation dose may be minimized with preoperative radiotherapy,244 major wound complications after preoperative radiotherapy and surgery have been reported to be in the 20% to 35% range.245,246 In the Canadian Sarcoma Group RCT, wound complications were defined as secondary wound surgery, hospital admission for wound care, deep packing, or prolonged dressings within 120 days after tumor resection. By these criteria, preoperative radiation had a significantly higher rate (35% vs.17%, P = 0.01) of wound complications than did postoperative EBRT. Of note, the risk was confined to the lower extremity.221
Taken in isolation, the wound complication fact alone may be expected to cause some groups to continue to favor postoperative radiotherapy. The Canadian RCT demonstrates that late tissue outcomes strongly favor the preoperative approach, with equivalent longer-term local tumor control.247 The rates of grade 2 or greater fibrosis and edema were significantly higher in the postoperative arm compared with preoperative radiotherapy, and were independently associated with the larger irradiation volumes and doses used in postoperative radiotherapy.247 Short-term functional outcome in the SR2 trial has also been reported and continues to be collected prospectively.248 Two validated instruments—the Toronto Extremity Salvage Score (TESS) and the Short Form-36 quality-of-life instrument (SF-36)—were applied, as was the observer-based Musculoskeletal Tumor Society (MSTS).248 Patients who were treated with postoperative radiotherapy had better function with higher MSTS Rating Scale, TESS, and SF-36 bodily pain scores at 6 weeks after surgery than those who were treated with preoperative-radiation, but there were no differences at later time points up to 1 year. Thus the timing of radiotherapy has minimal impact on the function of STS patients in the first year after surgery, but thereafter, significant factors likely come into play. These include the apparently deteriorating late tissue sequelae caused by larger doses and volumes. Of interest, patients who experience wound complications appear to continue to suffer some impaired function. Further follow-up is required to assess the ongoing evolution of these competing risks.
Conformal Radiotherapy and Intensity-Modulated Radiotherapy
STS presents in virtually any anatomic site, and the capacity for unusual presentation is nearly limitless. This can result in circumstances in which conventionally delivered radiotherapy is impossible because of the magnitude of the volume to be treated, uncertainty in defining the target for radiotherapy, or, more usually, because of the proximity of normal tissues to the intended target volume. Although some presentations are extremely problematic (e.g., uncertain targets because of organ mobility or imprecise anatomic issues resulting from poor definition of tumor location related to imaging limitations or inadequate surgical and/or pathological description), others may be addressed by novel methods of radiotherapy delivery. The most commonly available advance over standard EBRT is intensity-modulated radiotherapy, an advanced form of 3D conformal radiotherapy in which radiation beams are not only shaped at their perimeters, but also include variable intensity across the profiles of the beams. This permits the creation of greatly improved conformation of dose to targets of irregular shape while generating high-dose gradients between tumor and normal tissues. A full discussion of the potential uses of intensity-modulated radiotherapy in STS is beyond the scope of this chapter but is discussed in detail elsewhere.249 It may be administered preoperatively, postoperatively, or as a sole modality with specific indications. Some applications highlight its use in lesions adjacent to the spine or critical anatomic structures of the head and neck and in the retroperitoneum to permit liver avoidance (as well as to permit spinal cord, kidney, and intestinal dose limitation), especially in lesions involving the right upper abdominal quadrant. Avoidance of late toxicity to anatomic structures such as weight-bearing bone that are at risk for fracture after treatment of extremity sarcomas seems also to be feasible.250 Although these approaches are promising, their precise contribution and role need to be evaluated.251
Conventional Radiotherapy Without Surgery
Radiotherapy alone has been employed as primary therapy for patients with locally advanced, inoperable STS and patients who present with stage IV disease. Efforts to use radiation as the primary treatment have demonstrated that high doses (more than 65 Gy) are required to achieve local control rates between 30% and 60%,252,253 and that there appears to be an inverse relationship between tumor size and local control rates. In a series of 35 patients treated with high-dose (>65 Gy) primary radiotherapy for tumor sizes less than 5 cm, 5 to 10 cm, and larger than 10 cm, the local control rates were 88%, 53%, and 33%, respectively.254 In general, local control rates with radiation alone are inferior to those after surgery; therefore, primary radiation should be reserved for patients who are medically unfit for surgery, have technically unresectable tumors, or refuse surgery as initial therapy. However, some caution is necessary in interpreting such results in a scientifically valid manner. Encumbered with such adverse selection factors, the outcome of radiotherapy would never be comparable to that of surgery. Moreover, surgery has the added advantage over radiotherapy alone because its use for locally advanced cases is ordinarily also combined with adjuvant radiotherapy.
Particle-Based Radiotherapy (Emphasizing Proton Beam Therapy)
The use of radiation employing particles (electrons, neutrons, protons, and even portions of whole atoms such as carbon ions) provides another means to control the radiation field. Proton therapy allows for more accurate dose administration (lower “exit dose” of the proton beam) compared with photon based radiotherapy with more accurate discrimination between normal tissue and tumor compared with conformal photon-radiation therapy or intensity-modulated radiotherapy.255
Neutrons have been used in a number of pilot studies, primarily in patients with locally advanced disease, with 60% to 70% local control rates,258–258 but are more a curiosity now given the increasing use of proton beam radiation clinically. As an example, one study reviewed the results of 220 patients with locally advanced sarcomas who were treated with neutron radiotherapy. Ninety-four patients with gross residual disease after resection were treated with neutron therapy alone; among these patients, 27% had major morbidity, 26% had 5-year survival, and 56% had local control. One hundred four patients with microscopically positive margins of resection received a neutron boost dose; they had 7% morbidity, 65% 5-year survival, and 78% local control rates. These results suggest that for patients with gross residual disease, neutron beam radiation may provide improved local control compared with conventional external beam treatment, but these data should continue to be interpreted with caution when one considers the late tissue sequelae that appear to result from neutron beam radiation use. Comparative studies are needed to define the precise role of particle beam-based therapy in the treatment of STS. Apart from unresectable disease, it seems unclear where the benefit of proton and other particle-based therapy (e.g., carbon ion) might accrue when one considers the exceptionally favorable results of conventional photon-based treatment combined with surgery and the more adverse normal tissue tolerance to particle-based therapy.
As one clear situation in which the greatest possible targeting is desired, proton beam radiation has proved useful in rhabdomyosarcoma in parameningeal locations, wherein children’s developing brains may be less affected by the penumbra of the radiation field,259 Proton beam radiation also has found use in unresectable sarcomas.260,261 There are no comparative data examining this newest approach for sarcoma radiation therapy and this still relatively scarce resource seems best directed to clinical situations in which the most care with nearby structures (e.g., brain, spinal cord) must be taken. Chondrosarcoma of the skull base or chordoma of the clivus are good examples of situations in which novel technique may be most effective relative to other options.262,263
Adjuvant Chemotherapy
The statistical technique of meta-analysis may overcome the problem of inadequate power of small RCTs, and meta-analyses based on individual patient data may minimize other potential biases (e.g., exclusion of unpublished trials, variable follow-up, post randomization exclusions, and differing definition of end points) that are inherent in analyses that are limited to published results. In 1997, the Sarcoma Meta-Analysis Collaboration (SMAC) published an individual patient data meta-analysis of outcomes for 1568 STS patients included in 14 RCTs that completed accrual by December 1992.264,265 Significant improvements were found in local and distant relapse-free intervals and recurrence-free survival for all patients, and these improvements did translate into a significant overall survival benefit in the prospectively defined subgroup—almost 60% of the patients—with extremity sarcomas, but not in all patients (Table 93-10).
Table 93-10
Doxorubicin-Based and Ifosfamide-Based Chemotherapy for Localized Soft-Tissue Sarcoma: 2008 Sarcoma Meta-Analysis of Adjuvant Clinical Trials Results
| RELATIVE RISKS AND 95% CONFIDENCE INTERVALS FOR LOCAL RECURRENCE, DISTANT RECURRENCE, OVERALL RECURRENCE, AND SURVIVAL | ||||||||
| Local Recurrence | Distant Recurrence | Overall Recurrence | Survival | |||||
| Treatment | RR | 95% CI | RR | 95% CI | RR | 95% CI | RR | 95% CI |
| Doxorubicin | 0.75 | 0.56-1.01 | 0.69 | 0.56-0.86 | 0.69 | 0.56-0.86 | 0.84 | 0.68-1.03 |
| Doxorubicin with ifosfamide | 0.66 | 0.39-1.12 | 0.61 | 0.41-0.92 | 0.61 | 0.41-0.92 | 0.56 | 0.36-0.85 |
| Combined | 0.73 | 0.56-0.94 | 0.67 | 0.56-0.82 | 0.67 | 0.56-0.82 | 0.77 | 0.64-0.93 |
| RELATIVE RISK REDUCTIONS AND 95% CONFIDENCE INTERVALS FOR LOCAL RECURRENCE, DISTANT RECURRENCE, OVERALL RECURRENCE, AND SURVIVAL | ||||||||
| Local Recurrence | Distant Recurrence | Overall Recurrence | Survival | |||||
| Treatment | ARR | 95% CI | ARR | 95% CI | ARR | 95% CI | ARR | 95% CI |
| Doxorubicin | 3% | 1%-7% | 9% | 4%–14% | 9% | 4%-14% | 5% | 6%-21% |
| Doxorubicin with ifosfamide | 5% | 1%-12% | 10% | 1%–19% | 12% | 3%-21% | 11% | 3%-19% |
| Combined | 4% | 0%-7% | 9% | 5%–14% | 10% | 5%-15% | 6% | 2%-11% |
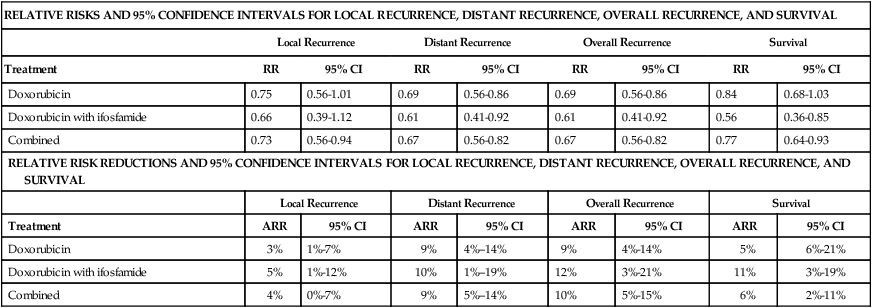
Adapted from Pervaiz N, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573.
These data were updated in a 2008 meta-analysis that also included newer studies that contain ifosfamide as part of their backbone, but did not include data from the largest trial of adjuvant chemotherapy, which was a negative study for improved overall survival.265 For the 18 studies and 1953 patients included in the meta-analysis, the odds ratios (ORs) for local recurrence was 0.73 (95% CI, 0.56 to 0.94; P = 0.02) in favor of chemotherapy. For distant and overall recurrence the OR was 0.67 (95% CI; 0.56 to 0.82; P = 0.0001) in favor of chemotherapy. In terms of survival, doxorubicin alone had an OR of 0.84 (95% CI; 0.68 to 1.03; P = 0.09), which is not statistically significant. However, the OR for doxorubicin combined with ifosfamide was 0.56 (95% CI; 0.36 to 0.85; P = 0.01) in favor of chemotherapy. With the understanding that publication and other biases may affect interpretation of meta-analyses, these are the strongest data supporting the use of adjuvant chemotherapy for patients with primary extremity or trunk STS, without reference to histology.
Full reports have been published on other phase II and RCTs (Table 93-11) examining anthracycline- and ifosfamide-based regimens supported by growth factors. In the Italian Cooperative Group study,267 accrual was terminated based on an early stopping rule, after half the planned number of patients had been recruited. The median recurrence-free survival and overall survival (Table 93-11) were superior for the chemotherapy group, but a high cumulative incidence of late distant relapses was noted (2-year: 28% vs. 45%, P = 0.08;4-year: 44% vs. 45%, P = 0.94 for chemotherapy vs. control groups, respectively). The 4-year overall survival rate remained better for the chemotherapy group (69% vs. 50%, P = 0.04). At the last published analysis, after a median follow-up of 90 months (range: 56 months to 119 months), the intention-to-treat analysis continued to demonstrate a trend towards improved overall survival (P = 0.07).268 The 5-year overall survival estimates, a reasonable end point for the survival analysis of adjuvant treatment in STSs, were 66% and 46% for the treatment and the control groups, respectively (P = 0.04).268
Table 93-11
Largest Randomized Controlled Trials with Ifosfamide Adjuvant Chemotherapy Versus Observation in Soft-Tissue Sarcoma
| Study | Chemotherapy Regimen | No. of Patients | Disease Sites | Follow-up | Survival | |
| RFS | OS | |||||
| Italian Cooperative*268 | Epirubicin + Ifosfamide + filgrastim (×5) | 53 | Limb (grade III >5 cm) | 59 months (median) | 48 months | 75 months |
| Control | 51 | 16 months | 46 months | |||
| P = 0.04 | P = 0.03 | |||||
| Australian Cooperative†269 | Ifosfamide + doxorubicin + dacarbazine (IFADIC) + filgrastim (×6) | 31 | Limb 47, Trunk 12 (grades II/III) | 41 months (mean) | 77% | NR |
| Control | 28 | 57% | NR | |||
| EORTC (Woll et al., ASCO abstract)271 | Ifosfamide + doxorubicin + sargramostim Control |
175 176 |
Trojani grades II to III STS at any site | NR | P = 0.1 52% (5-years) 52% (P = NS) |
P = 0.4 64% (5-years) 69% (P = NS) |
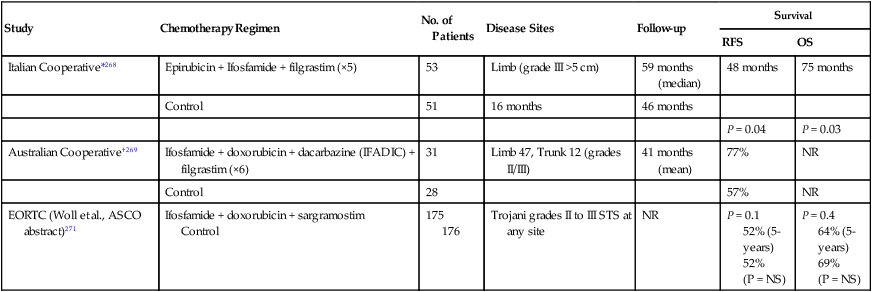
*Median recurrence-free plus overall survival.
†(1) Comparison percent recurrence-free plus overall survival after mean observation period of 41 (range: 8 to 84) months. (2) Overall survival plotted but actuarial results not reported at defined follow-up time(s).
The authors of a second study, a prospective randomized feasibility trial, concluded that a regimen of 6 cycles of ifosfamide, doxorubicin, and dacarbazine given concurrently with postoperative hyperfractionated radiotherapy (during cycles 3 and 4 when doxorubicin was omitted from the regimen) was manageable and tolerable.269 It did not translate into significant benefits in recurrence-free survival (P = 0.1 vs. control arm), time to local failure (P = 0.09), or overall survival (P = 0.4), but the small number of patients (31 vs. 28) precludes meaningful conclusions regarding benefit. A third small RCT, from another Italian center, examined 41 patients who received either an intensive epirubicin + ifosfamide combination (n = 19) versus single-agent epirubicin (n = 16), in comparison with local therapy alone (n = 43); the study was closed early for poor accrual and was accordingly underpowered for efficacy end points.270
In the largest randomized study of adjuvant chemotherapy to date, Woll and colleagues randomly assigned 351 patients, from 1995 to 2003, 1 : 1 between observation alone or 5 cycles of chemotherapy with doxorubicin (75 mg/m2 per cycle) + ifosfamide (5 g/m2 over 24 hours per cycle), every 21 days, with lenograstim (3 μg/kg SC daily for 14 days/cycle). In the preliminary analysis of data, 5-year recurrence-free survival rate was 52% in both arms, and 5-year overall survival rate was 69% on the observation arm versus 64% on the chemotherapy arm.271
Can blanket recommendations be made regarding the use of adjuvant chemotherapy in adult STS? In an editorial272 that accompanied the report of the Italian Cooperative Group Trial that still addresses many issues faced today,267 Bramwell concluded that a specific standard of care was not yet clear and that the situation would take some time to change. Guidelines from the United States and Canada support the use of chemotherapy in selected patients.273 European guidelines recommend adjuvant chemotherapy only after shared decision making and careful selection of patients.274 With the current state of knowledge, chemotherapy is a reasonable standard of care for those patients willing to accept significant toxicity for what may be little if any improvement in overall survival. Observation after primary tumor control is a reasonable option for many people, most so in those sarcomas already known to be relatively chemotherapy resistant. Those people who are most likely to benefit from adjuvant chemotherapy are those who can tolerate ifosfamide chemotherapy; the meta-analysis of 2008 indicated that the benefit from chemotherapy appeared to be more an issue of delivery of ifosfamide rather than doxorubicin.265
Neoadjuvant Chemotherapy
• Destruction of the primary tumor may reduce the risk of contamination at surgery and permit closer margins with less tissue loss and functional disability but improved local control, which has been observed consistently in randomized clinical trials.
• Extensive delays in initiating chemotherapy resulting from complex surgery and/or radiotherapy are avoided; for rapidly growing tumors, this earlier elimination of micrometastases may improve survival, although this has not been proved.
• Treating a patient with an intact tumor allows the medical oncologist to assess the effects of preoperative therapy and thus judge whether the chosen chemotherapeutic regimen has activity against the specific tumor in the specific patient being treated. The importance of this opportunity to determine whether an empirically chosen regimen will or will not be beneficial may be useful when dealing with marginally effective chemotherapy. However, the lack of data supporting regimens other than anthracycline + ifosfamide in the adjuvant setting presently limits the usefulness of such information.
Numerous neoadjuvant treatment approaches and preoperative drug regimens have been explored. Intraarterial administration of drugs such as doxorubicin and cisplatin has been evaluated, in some cases in conjunction with radiotherapy, isolated limb perfusion, or hyperthermia. The intraarterial route delivers drugs more directly to the tumor but is more complex, expensive, and prone to complications than is the intravenous (IV) route. In the one small RCT on route of administration, there were no differences in rates of local control or overall failure between neoadjuvant chemotherapy given intraarterially or intravenously.275,276 In a series of nonrandomized studies reflecting the evolution of neoadjuvant treatment at their center over a 20-year period, the University of California, Los Angeles (UCLA) group treated a total of 498 patients with neoadjuvant chemotherapy.277 As one example of its studies, the combination of chemotherapy and 28 Gy of radiotherapy before surgery was noted by investigators to provide the best local control with the lowest complication rate. On the basis of results of the RCT described earlier, intraarterial doxorubicin was replaced by IV doxorubicin, and cisplatin and ifosfamide were added in a follow-up protocol.277 In the whole group of 498 patients, the overall local recurrence rates were 11% at 5 years and 15% at 10 years, and corresponding overall survival rates were 71% and 66%. The local recurrence rate was lower and overall survival rate higher for patients who had no residual tumor (38%) or greater than 95% necrosis (14%) after neoadjuvant chemotherapy, compared with those who had less than 95% necrosis. In a multivariate analysis, pathological necrosis was an independent predictor of local recurrence and overall survival. The percentage of patients with 95% or greater necrosis increased to 48% with the addition of ifosfamide, compared with 13% for patients in all other protocols combined.
Pisters and colleagues reviewed the long-term results of neoadjuvant chemotherapy given at the University of Texas MD Anderson Cancer Center for stage IIIB extremity sarcomas between 1986 and 1990.278 All patients received doxorubicin-based regimens; at that time, ifosfamide was rarely used (three patients). In 75 patients, the overall clinical objective response rate (complete response plus partial response) was 27%. At a median follow-up of 85 months, the 5-year actuarial local recurrence-free survival, overall recurrence-free survival, and overall survival rates were 83%, 52%, and 59%, respectively. In contrast with the UCLA group’s results277 for pathological response, there were no differences in any outcomes between responding and nonresponding patients, as defined at that time.
In a separate analysis at MD Anderson, 65 patients (42 extremity sarcomas and 23 retroperitoneal sarcomas) were treated at the same center between 1991 and 1996 with doxorubicin- or ifosfamide-based neoadjuvant chemotherapy; 34% achieved a radiographic partial response and 9% a minor response.279 Patients having partial response had higher rates of negative-margin resections, local recurrence-free survival, and overall survival than did nonresponders. Postoperative morbidity was also evaluated in a larger cohort of 105 patients (71 extremity and 34 retroperitoneal STS) treated at MD Anderson during the same period, of whom 50 received ifosfamide as well as doxorubicin.280 The authors found no evidence that preoperative chemotherapy increased surgical complications (e.g., wound infections and other wound problems), length of hospital stay, rate of readmission, or rate of reoperation. Similar outcomes have been observed in other reported series.281
The European Organization for the Research and Treatment of Cancer (EORTC) has performed the only RCT assessing preoperative chemotherapy for STS (3 cycles of doxorubicin and ifosfamide plus filgrastim) versus no preoperative chemotherapy (control).282 A total of 150 patients with high-risk STS (≥8 cm any grade, or grades II/III tumors <8 cm, or grades II/III local recurrence tumors/inadequate surgery) were randomly assigned, of whom 134 were considered eligible for outcome assessment. Radiotherapy was indicated for tumors that were excised with close or microscopically positive margins and was given in 46% of patients in the chemotherapy arm and 54% of patients in the control arms. Limb salvage was possible in 89% of patients, and chemotherapy did not affect postoperative wound healing. Grade 4 toxicities were rare, although there was one death as a consequence of neutropenic fever. At a median follow-up of 7.3 years, the 5-year recurrence-free survival and overall survival rates were 56% versus 52% (P = 0.36) and 65% versus 64% (P = 0.22) for the chemotherapy and control arms, respectively. Although originally planned as a phase III trial with adequate numbers to detect a 15% difference in 5-year overall survival, the study was closed after completion of the phase II section because of slow accrual. Most groups have concluded that for large high-grade tumors, preoperative chemotherapy is feasible, does not increase postoperative morbidity, increases the rate of operability, and may enhance local control. The beneficial effects on distant metastases and overall survival, if any, were marginal.
Combined Preoperative Chemotherapy and Radiotherapy
With the advances that have been made with combined-modality treatment of other solid tumors, there has been interest in combined-modality preoperative treatment (concurrent or sequential chemotherapy and radiation) for patients with localized STSs. Concurrent doxorubicin-based chemoradiation has been employed extensively by Eilber and colleagues at UCLA.283,284 This treatment protocol involved intraarterial doxorubicin with a hypofractionation schedule to be coincident with intraarterial chemotherapy (35 Gy of EBRT delivered in 10 daily fractions, which was reduced to 17.5 Gy in 5 daily fractions to minimize local toxicity). A subsequent prospective randomized trial from the same group that remains unpublished compared preoperative intraarterial doxorubicin with IV doxorubicin, both followed by 28 Gy of radiation delivered over 8 days followed by surgical resection. No differences in local recurrence or survival were noted.
The combination of regional chemotherapy and concurrent radiotherapy that was originally pioneered by Eilber and colleagues has been modified and used by other groups.287–287 Investigators from the University of Illinois treated 55 patients with a 10-day preoperative regimen of intraarterial doxorubicin (10 mg/m2 per day) with concomitant radiotherapy (25 Gy: 2.5 Gy per fraction in 10 fractions).287 With a mean follow-up of 94 months, local control was 85%. Complications related to the therapy occurred in 26% of patients and required further operative management in 7% of patients. Temple and colleagues treated a group of 42 patients with a similar regimen of 60 to 90 mg of doxorubicin that was infused intraarterially or intravenously over a 3-day period followed by sequential radiotherapy (30 Gy: 3 Gy per fraction in 10 fractions).286 Resection of the residual posttreatment mass was performed 4 to 6 weeks later. At a median follow-up of 6 years, local control was achieved in 39 of 40 patients, although two patients were excluded from this analysis because clear margins were not obtained at the time of surgery. Intraarterial infusion-related complications occurred in 4 (11%) of 35 patients. Objective radiographic and pathological response rates were not reported; therefore, the efficacy of concurrent chemoradiation therapy in achieving cytoreduction to an extent sufficient to convert lesions that are resectable by amputation only to lesions that are amenable to a limb-sparing approach remains anecdotal. Moreover, whether preoperative chemoradiation approaches offer local control advantages over conventional treatment approaches that combine surgery with preoperative or postoperative radiotherapy is also not apparent. In fact, with current local control rates with surgery and radiation alone exceeding 90%, it is difficult to appreciate how this outcome would be furthered with preoperative chemotherapy unless the strategy involved a radiotherapy dose reduction to ameliorate normal tissue toxicity.
Alternative chemotherapy-radiation sequencing has been used by investigators from Massachusetts General Hospital, who have reported mature data from a sequential chemoradiation strategy in the treatment of patients with large (>8 cm), localized, high-grade extremity STSs.288 The Massachusetts General Hospital protocol involves interdigitating courses of chemotherapy and radiotherapy: three courses of doxorubicin, ifosfamide, mesna, and dacarbazine and two 22-Gy courses of radiation (11 fractions each) for a total preoperative radiation dose that is lower than usual (44 Gy). This was followed by surgical resection with microscopic assessment of surgical margins. An additional 16-Gy (8 fractions) boost dose was delivered for microscopically positive surgical margins. The strategy, therefore, simultaneously addresses the dual problems of local control and metastatic risk. The outcomes of 48 patients who were treated with this regimen between June 1989 and March 1999 were compared with those of a matched series of historic controls (treated between January 1988 and March 1997).288 The 5-year actuarial local control, distant metastasis-free survival, and overall survival rates for the sequential chemotherapy-radiation group are 92%, 75%, and 87%, respectively. For the matched historic controls, these rates are 86%, 47%, and 58%, respectively. The protocol was toxic, with 29% experiencing confluent moist skin desquamation, 25% requiring hospitalization at some time for febrile neutropenia. Wound-healing complications evaluated by using recently described criteria221 were apparent in 29% of cases and confined to the lower extremities, as was also observed in the Canadian Sarcoma Group treatment sequencing RCT.221 One patient died from late marrow dysfunction attributed to chemotherapy. Although the results are encouraging from a tumor standpoint (Fig. 93-11), they require prospective comparative studies for confirmation, especially because of the local and systemic toxicity associated with this approach.289 Although a Radiation Therapy Oncology Group phase II study yielded similar outcomes compared with the data of DeLaney and colleagues,288,290 they, too, are characterized by concerning features of significant toxicity, including marrow dysplasia and treatment-related deaths.
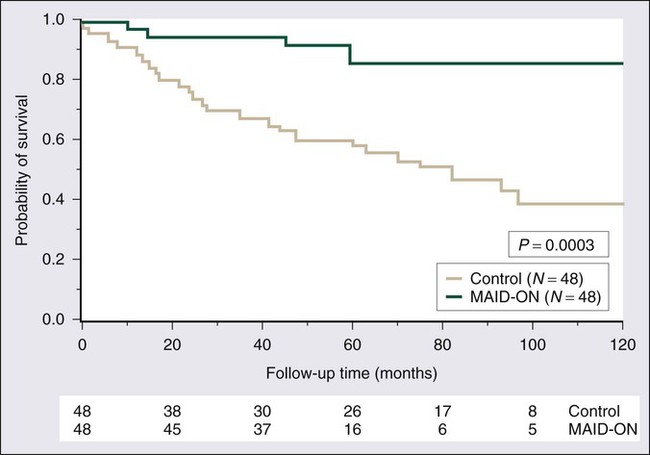
Hyperthermic Isolated Limb Perfusion and Whole-Body Hyperthermia with Chemotherapy
Hyperthermic isolated limb perfusion and whole-body hyperthermia are two investigational techniques that continue to receive considerable attention, particularly in Europe. Hyperthermic isolated limb perfusion (with TNF-α, interferon-α, and melphalan) has been used as an neoadjuvant therapy to render tumors resectable and as a primary therapy to avoid amputation for nonresectable extremity STS.291,292 Overall response rates in early series, which recruited 55, 35, and 41 patients, respectively, ranged from 72% to 91%, and limb salvage surgery was possible in 84% to 91% of cases. Small numbers of patients experienced subsequent local recurrence, and some of these patients required amputations. Not surprisingly, many patients ultimately died of distant metastases.
Various techniques of regional or whole-body hyperthermia have been combined with a variety of chemotherapy regimens.293–296 A group from Munich evaluated preoperative chemotherapy (4 cycles of doxorubicin, ifosfamide, and etoposide) combined with regional hyperthermia (RHT) followed by surgery and adjuvant treatment (same chemotherapy ± radiation). Median follow-up times were 58 months for the RHT-91 protocol (59 patients) and 30 months for the RHT-95 protocol.296 All patients had grades II/III tumors 5 cm or larger with extracompartmental extension. Clinical response rates were 42% and 33% for the two protocols, with respective local progression-free survival rates of 58% and 57%. Corresponding overall survival rates were 42% and 48%, respectively. The possible utility of hyperthermia as part of local tumor control was further evaluated in a phase III RCT (EORTC 62961/European Society for Hyperthermic Oncology RHT-95). The assumption was made that preoperative chemotherapy with doxorubicin, ifosfamide, and etoposide is effective for patients with high-risk localized disease in this setting, and patients were randomized to receive preoperative etoposide, ifosfamide, and doxorubicin (EIA, 4 cycles) plus RHT (2 fractions) versus EIA chemotherapy alone. As a result, no difference was expected in terms of overall survival; improved local control was confirmed in those patients treated with hyperthermia.297