Chapter 23 Psychiatry
Selective Serotonin Reuptake Inhibitors (SSRIs)


MOA (Mechanism of Action)
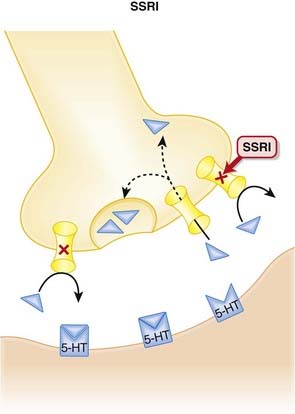
 The monoamine hypothesis suggests that depression is caused by a deficiency of synaptic neurotransmitters such as serotonin (5-HT), norepinephrine, and dopamine. Serotonin, in particular, is associated with mood.
The monoamine hypothesis suggests that depression is caused by a deficiency of synaptic neurotransmitters such as serotonin (5-HT), norepinephrine, and dopamine. Serotonin, in particular, is associated with mood.





Pharmacokinetics






Contraindications
 SSRIs and monoamine oxidase inhibitors (MAOIs): SSRIs increase serotonin concentrations in the synapse, whereas MAOIs inhibit the breakdown of serotonin. Concomitant use can therefore lead to excessive serotonin (see details in Side Effects). When switching from an SSRI to an MAOI, or vice versa, allow for a washout period of at least 1 to 2 weeks.
SSRIs and monoamine oxidase inhibitors (MAOIs): SSRIs increase serotonin concentrations in the synapse, whereas MAOIs inhibit the breakdown of serotonin. Concomitant use can therefore lead to excessive serotonin (see details in Side Effects). When switching from an SSRI to an MAOI, or vice versa, allow for a washout period of at least 1 to 2 weeks.

Side Effects
Serious


Non-Serious
 Sexual dysfunction may be both mechanical, as serotonin inhibits functions such as erections, ejaculation, lubrication, and orgasm, and central, as serotonin has an inhibitory effect on dopamine, a neurotransmitter believed to play an important role in arousal. Note: sexual dysfunction can also accompany depression.
Sexual dysfunction may be both mechanical, as serotonin inhibits functions such as erections, ejaculation, lubrication, and orgasm, and central, as serotonin has an inhibitory effect on dopamine, a neurotransmitter believed to play an important role in arousal. Note: sexual dysfunction can also accompany depression.

Important Notes
 As with other antidepressants, when an SSRI is being discontinued, the dose should be tapered gradually in order to avoid discontinuation symptoms, including dizziness, nausea, headache, and others. The incidence and severity appears to vary between SSRI, and longer half-life agents such as fluoxetine appear to be less likely to induce a discontinuation syndrome.
As with other antidepressants, when an SSRI is being discontinued, the dose should be tapered gradually in order to avoid discontinuation symptoms, including dizziness, nausea, headache, and others. The incidence and severity appears to vary between SSRI, and longer half-life agents such as fluoxetine appear to be less likely to induce a discontinuation syndrome.


Advanced
Drug Interactions
 The SSRIs inhibit multiple CYP450 isozymes. Fluoxetine and paroxetine inhibit the CYP2D6 isoenzyme, and this can lead to clinically important drug interactions with drugs such as tricyclic antidepressants (TCAs), carbamazepine, or vinblastine. There is also a very serious interaction with thioridazine (see Contraindications).
The SSRIs inhibit multiple CYP450 isozymes. Fluoxetine and paroxetine inhibit the CYP2D6 isoenzyme, and this can lead to clinically important drug interactions with drugs such as tricyclic antidepressants (TCAs), carbamazepine, or vinblastine. There is also a very serious interaction with thioridazine (see Contraindications).
Evidence
Depression

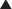


Premenstrual Syndrome
 A 2007 Cochrane review (31 trials, N = 844 participants) found that SSRIs were highly effective in the treatment of premenstrual symptoms, both physical and behavioral, compared with placebo. There were 2.5 times as many withdrawals because of adverse events among SSRI-treated subjects as among placebo-treated subjects.
A 2007 Cochrane review (31 trials, N = 844 participants) found that SSRIs were highly effective in the treatment of premenstrual symptoms, both physical and behavioral, compared with placebo. There were 2.5 times as many withdrawals because of adverse events among SSRI-treated subjects as among placebo-treated subjects.
FYI
 The SSRIs were the first class of antidepressants that were discovered using “rational drug design.” The strategy was based on the observation that TCAs inhibited noradrenaline (NA) or 5-HT reuptake to various extents. Scientists then discovered some nontricyclic compounds that were also reuptake inhibitors, acting on either NA or 5-HT to varying degrees. This led to the approval of the first such agent, zimeldine, which was withdrawn from the market after a few years.
The SSRIs were the first class of antidepressants that were discovered using “rational drug design.” The strategy was based on the observation that TCAs inhibited noradrenaline (NA) or 5-HT reuptake to various extents. Scientists then discovered some nontricyclic compounds that were also reuptake inhibitors, acting on either NA or 5-HT to varying degrees. This led to the approval of the first such agent, zimeldine, which was withdrawn from the market after a few years.Tricyclic Antidepressants (TCAs)
Description
TCAs are a class of antidepressants with a common chemical (tricyclic) structure and mode of action.


MOA (Mechanism of Action)
 The monoamine hypothesis suggests that depression is caused by a deficiency of synaptic neurotransmitters such as serotonin (5-HT), NA, and dopamine. Serotonin, in particular, is associated with mood.
The monoamine hypothesis suggests that depression is caused by a deficiency of synaptic neurotransmitters such as serotonin (5-HT), NA, and dopamine. Serotonin, in particular, is associated with mood.










Contraindications
 Avoid concomitant use of TCAs and MAOIs: TCAs increase serotonin concentrations in the synapse, whereas MAOIs inhibit the breakdown of serotonin. Concomitant use can therefore lead to excessive serotonin. When switching from a TCA to an MAOI, or vice versa, allow for a washout period of at least 2 weeks.
Avoid concomitant use of TCAs and MAOIs: TCAs increase serotonin concentrations in the synapse, whereas MAOIs inhibit the breakdown of serotonin. Concomitant use can therefore lead to excessive serotonin. When switching from a TCA to an MAOI, or vice versa, allow for a washout period of at least 2 weeks.



Important Notes
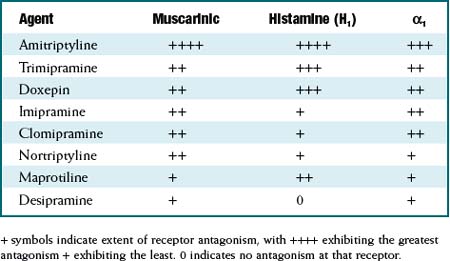
 The TCAs are grouped as a class based on their chemical structure. Although as a class they are considered to be 5-HT or noradrenaline reuptake inhibitors, the degree of reuptake inhibition differs markedly among agents. See Table 23-1.
The TCAs are grouped as a class based on their chemical structure. Although as a class they are considered to be 5-HT or noradrenaline reuptake inhibitors, the degree of reuptake inhibition differs markedly among agents. See Table 23-1. Similarly, the affinities for blockade of receptors that mediate the side effects experienced by patients also differ markedly among agents. See Table 23-2.
Similarly, the affinities for blockade of receptors that mediate the side effects experienced by patients also differ markedly among agents. See Table 23-2.

TABLE 23-1 Extent of Serotonin and Noradrenaline Reuptake Inhibition among TCAs
| Agent | Serotonin (5HT) | Noradrenaline(NA) |
|---|---|---|
| Clomipramine | +++ | + |
| Amitriptyline | ++ | ± |
| Imipramine | + | + |
| Trimipramine | 0 | + |
| Doxepin | + | ++ |
| Nortriptyline | ± | ++ |
| Desipramine | 0 | +++ |
+ symbols indicate extent of reuptake inhibition, with +++ exhibiting the greatest reuptake inhibition + exhibiting the least. 0 indicates no inhibition of the reuptake transporter for that neurotransmitter.

Evidence


FYI
 TCAs have been around for 50 years. Imipramine was the first TCA synthesized, and it was based on the tricyclic structure of the antipsychotic chlorpromazine. A study in 1958 found imipramine lacked efficacy in psychosis, but, surprisingly, a subgroup of patients with depression improved on imipramine. TCAs became the drugs of choice for treating depression for the next 30 years.
TCAs have been around for 50 years. Imipramine was the first TCA synthesized, and it was based on the tricyclic structure of the antipsychotic chlorpromazine. A study in 1958 found imipramine lacked efficacy in psychosis, but, surprisingly, a subgroup of patients with depression improved on imipramine. TCAs became the drugs of choice for treating depression for the next 30 years.
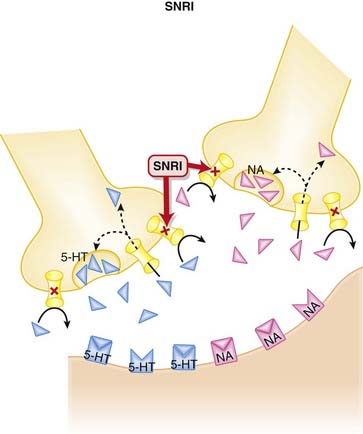
Serotonin Noradrenaline Reuptake Inhibitors (SNRIs)


MOA (Mechanism of Action)
 The monoamine hypothesis suggests that depression is caused by a deficiency of synaptic neurotransmitters such as serotonin (5-HT), NA, and dopamine. Serotonin, in particular, is associated with mood.
The monoamine hypothesis suggests that depression is caused by a deficiency of synaptic neurotransmitters such as serotonin (5-HT), NA, and dopamine. Serotonin, in particular, is associated with mood.






Pharmacokinetics
 Compared with the SSRIs, the SNRIs have shorter elimination half-lives. Venlafaxine is available in an extended-release(ER) dosage form.
Compared with the SSRIs, the SNRIs have shorter elimination half-lives. Venlafaxine is available in an extended-release(ER) dosage form.







Side Effects
 Gastrointestinal (GI) distress, attributed to inhibition of serotonin reuptake, appears to be most common with venlafaxine. Stimulation of serotonin receptors in the brain likely mediates nausea. Serotonin receptors are also found in the gut, and serotonin appears to have an effect on GI motility that becomes intolerable in some patients, leading to cramping and diarrhea.
Gastrointestinal (GI) distress, attributed to inhibition of serotonin reuptake, appears to be most common with venlafaxine. Stimulation of serotonin receptors in the brain likely mediates nausea. Serotonin receptors are also found in the gut, and serotonin appears to have an effect on GI motility that becomes intolerable in some patients, leading to cramping and diarrhea.






Important Notes
 When an SNRI is being discontinued, the dose should be tapered gradually in order to avoid discontinuation symptoms, including aggression, agitation, convulsions, dysphoric mood, electric shock sensations, and others. These symptoms have been particularly evident with venlafaxine.
When an SNRI is being discontinued, the dose should be tapered gradually in order to avoid discontinuation symptoms, including aggression, agitation, convulsions, dysphoric mood, electric shock sensations, and others. These symptoms have been particularly evident with venlafaxine. The theory behind the development of the SNRIs was to try and increase the levels of two neurotransmitters (serotonin, noradrenaline) simultaneously, while avoiding many of the bothersome side effects of the TCAs, which also act as reuptake inhibitors of these two neurotransmitters. There is evidence to suggest that dual reuptake inhibition is more efficacious than selective inhibition (see Evidence section).
The theory behind the development of the SNRIs was to try and increase the levels of two neurotransmitters (serotonin, noradrenaline) simultaneously, while avoiding many of the bothersome side effects of the TCAs, which also act as reuptake inhibitors of these two neurotransmitters. There is evidence to suggest that dual reuptake inhibition is more efficacious than selective inhibition (see Evidence section).



Evidence
Venlafaxine versus Fluoxetine for Depression
 A 2005 Cochrane review (10 trials, N = 1831 participants) compared fluoxetine with venlafaxine. The authors found venlafaxine to be significantly better than fluoxetine (an SSRI) for improving depression rating scores. Some side effects were more common with venlafaxine than with fluoxetine, including dry mouth, dizziness, sweating, and nausea.
A 2005 Cochrane review (10 trials, N = 1831 participants) compared fluoxetine with venlafaxine. The authors found venlafaxine to be significantly better than fluoxetine (an SSRI) for improving depression rating scores. Some side effects were more common with venlafaxine than with fluoxetine, including dry mouth, dizziness, sweating, and nausea.Venlafaxine for Generalized Anxiety Disorder
 A 2003 Cochrane review (8 trials, N = 2058 participants) examined the efficacy of various antidepressants for generalized anxiety disorder (GAD). Based on two trials, the authors found venlafaxine to be statistically better than placebo for treatment response, assessed by Clinical Global Impression (CGI) scores.
A 2003 Cochrane review (8 trials, N = 2058 participants) examined the efficacy of various antidepressants for generalized anxiety disorder (GAD). Based on two trials, the authors found venlafaxine to be statistically better than placebo for treatment response, assessed by Clinical Global Impression (CGI) scores.Milnacipran for Depression
 A 2009 Cochrane review (16 trials, N = 2277 participants) compared milnacipran with other antidepressants for depression. Milnacipran was associated with fewer withdrawals because of adverse events (a measure of tolerability) compared with the TCAs (odds ratio [OR] 0.55), and weak evidence suggested fewer adverse events of sleepiness or drowsiness, dry mouth, or constipation versus these agents.
A 2009 Cochrane review (16 trials, N = 2277 participants) compared milnacipran with other antidepressants for depression. Milnacipran was associated with fewer withdrawals because of adverse events (a measure of tolerability) compared with the TCAs (odds ratio [OR] 0.55), and weak evidence suggested fewer adverse events of sleepiness or drowsiness, dry mouth, or constipation versus these agents.

Noradrenergic and Specific Serotonergic Antidepressants (NaSSAs)

MOA (Mechanism of Action)
 NaSSAs have a dual mechanism of action:
NaSSAs have a dual mechanism of action:
 Inhibition of α2 autoreceptors and heteroreceptors
Inhibition of α2 autoreceptors and heteroreceptors
• An autoreceptor is a receptor that when bound by ligand reduces release of that ligand into the synapse. The α2 receptor is a classic example of an autoreceptor, as when it is bound by noradrenaline (NA) it inhibits NA release.
• A heteroreceptor is like an autoreceptor, although when bound it can mediate the release of other neurotransmitters in addition to its own ligand.
• Thus inhibition of these autoreceptors and heteroreceptors prevents the negative feedback of NA on 5-HT and NA neurotransmission (Figure 23-4). Thus neurotransmission is sustained.

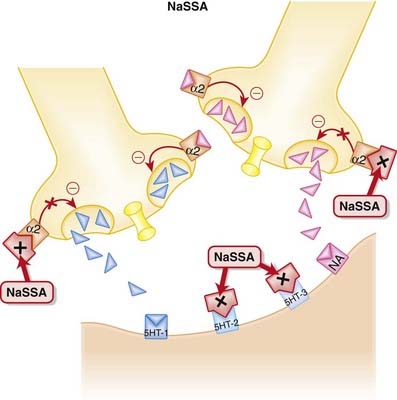

Side Effects



Important Notes







Monoamine Oxidase Inhibitors (MAOIs)
Description
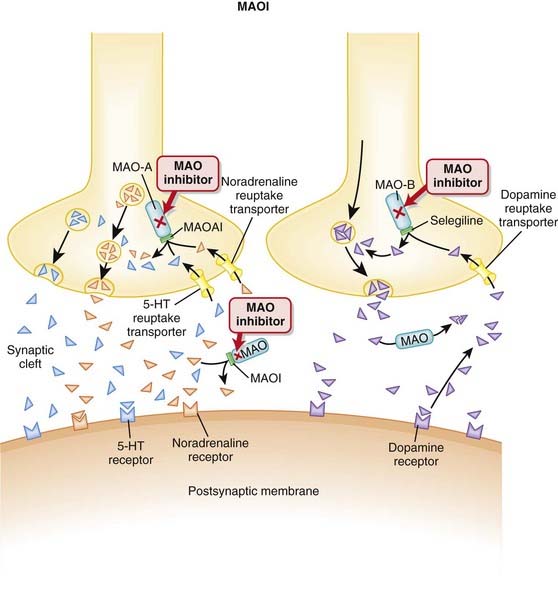
MAOIs are a heterogeneous group of agents that inhibit the monoamine oxidase (MAO) enzyme.





MOA (Mechanism of Action)
 MAO degrades catecholamines, serotonin, and other endogenous amines in the CNS as well as in the periphery.
MAO degrades catecholamines, serotonin, and other endogenous amines in the CNS as well as in the periphery.












Contraindications
 Drug interaction: with sympathomimetics: nonselective MAOIs may potentiate the hypertensive effects of sympathomimetics, leading to a hypertensive crisis that can be fatal. Methylphenidate, dopamine, epinephrine, norepinephrine, and similar agents (methyldopa, l-dopa, l-tryptophan, l-tyrosine, phenylalanine) should be avoided.
Drug interaction: with sympathomimetics: nonselective MAOIs may potentiate the hypertensive effects of sympathomimetics, leading to a hypertensive crisis that can be fatal. Methylphenidate, dopamine, epinephrine, norepinephrine, and similar agents (methyldopa, l-dopa, l-tryptophan, l-tyrosine, phenylalanine) should be avoided.
Side Effects
 Sleep disturbances include insomnia and reduction in rapid eye movement (REM) sleep. The insomnia is likely a central stimulatory effect from the increased monoamines, although a mechanism has not been established. Moclobemide, a reversible and selective MAO-A inhibitor, may cause fewer problems with sleep.
Sleep disturbances include insomnia and reduction in rapid eye movement (REM) sleep. The insomnia is likely a central stimulatory effect from the increased monoamines, although a mechanism has not been established. Moclobemide, a reversible and selective MAO-A inhibitor, may cause fewer problems with sleep.



Important Notes
 MAO-A in the gut breaks down tyramine, a chemical that stimulates the release of norepinephrine. Tyramine is typically found in aged foods such as cheese, wine, beer, yogurt, and yeast. Ingestion of these foods leads to increased tyramine, and because its breakdown is inhibited, there is increased norepinephrine release, leading to hypertensive crisis.
MAO-A in the gut breaks down tyramine, a chemical that stimulates the release of norepinephrine. Tyramine is typically found in aged foods such as cheese, wine, beer, yogurt, and yeast. Ingestion of these foods leads to increased tyramine, and because its breakdown is inhibited, there is increased norepinephrine release, leading to hypertensive crisis.




Evidence
MAO-B Inhibitors versus Dopaminergics for Early Parkinson’s Disease
 A 2009 Cochrane review (two trials, N = 593 patients) compared MAO-B inhibitors with a dopamine agonist or levodopa in patients with early Parkinson’s. No difference in deaths was found between selegiline and either agent, but patients treated with selegiline were more likely to need add-on therapy during follow-up than patients receiving levodopa (OR 12.02) or a dopamine agonist (OR 2.00). Motor fluctuations were reduced by MAO-B inhibitors versus levodopa (OR 0.55) but not dopamine agonists. Withdrawals because of adverse events were less common with MAO-B inhibitors compared with dopamine agonists (OR 0.11).
A 2009 Cochrane review (two trials, N = 593 patients) compared MAO-B inhibitors with a dopamine agonist or levodopa in patients with early Parkinson’s. No difference in deaths was found between selegiline and either agent, but patients treated with selegiline were more likely to need add-on therapy during follow-up than patients receiving levodopa (OR 12.02) or a dopamine agonist (OR 2.00). Motor fluctuations were reduced by MAO-B inhibitors versus levodopa (OR 0.55) but not dopamine agonists. Withdrawals because of adverse events were less common with MAO-B inhibitors compared with dopamine agonists (OR 0.11).

Noradrenaline and Dopamine Reuptake Inhibitors (NDRIs)

MOA (Mechanism of Action)





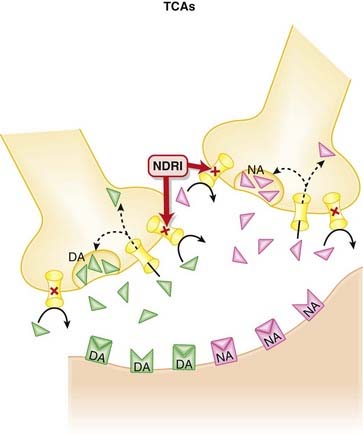



Contraindications







Important Notes





Evidence
Smoking Cessation: Antidepressants versus Placebo
 A 2007 Cochrane review (53 trials) compared antidepressants to placebo or alternative pharmacotherapies for smoking cessation or relapse prevention. The review included 31 RCTs of bupropion versus placebo or no treatment for smoking cessation, finding that bupropion doubled the odds of cessation across these studies (OR 1.94). In four trials, nortriptyline also improved the odds of quitting smoking (OR 2.34).
A 2007 Cochrane review (53 trials) compared antidepressants to placebo or alternative pharmacotherapies for smoking cessation or relapse prevention. The review included 31 RCTs of bupropion versus placebo or no treatment for smoking cessation, finding that bupropion doubled the odds of cessation across these studies (OR 1.94). In four trials, nortriptyline also improved the odds of quitting smoking (OR 2.34).Smoking Cessation: versus Varenicline (Partial Nicotine Agonist)
 A 2007 Cochrane review (7 trials, N = 7267 participants) assessed the efficacy and tolerability of varenicline versus other interventions and placebo for smoking cessation. The authors included three double-blind randomized controlled trials that compared bupropion with varenicline and when the data from these studies were combined, found that more varenicline subjects were abstinent or continuously abstinent from smoking at 12 months compared with bupropion (relative risk [RR] 1.52).
A 2007 Cochrane review (7 trials, N = 7267 participants) assessed the efficacy and tolerability of varenicline versus other interventions and placebo for smoking cessation. The authors included three double-blind randomized controlled trials that compared bupropion with varenicline and when the data from these studies were combined, found that more varenicline subjects were abstinent or continuously abstinent from smoking at 12 months compared with bupropion (relative risk [RR] 1.52).
Lithium

MOA (Mechanism of Action)
 Decades after the discovery of lithium’s utility in bipolar disorder, the mechanism behind its efficacy is still poorly understood.
Decades after the discovery of lithium’s utility in bipolar disorder, the mechanism behind its efficacy is still poorly understood.
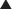


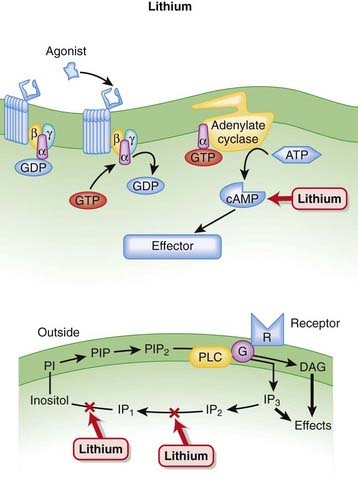
Pharmacokinetics




Contraindications
 In the presence of the following conditions, lithium should be used only with extreme caution, when other treatments have failed:
In the presence of the following conditions, lithium should be used only with extreme caution, when other treatments have failed:
Side Effects






Important Notes
 Lithium is unique among all psychotropic agents in that it lacks any sedative, euphoriant, or depressive effects in normal individuals who do not suffer from psychiatric illness.
Lithium is unique among all psychotropic agents in that it lacks any sedative, euphoriant, or depressive effects in normal individuals who do not suffer from psychiatric illness.Advanced
Drug Interactions
 A drug interaction between lithium and diuretics has a unique mechanism. In response to decreased volume secondary to diuretic use, the kidneys will try to retain Na+ in an effort to retain water. When the kidney reabsorbs Na+, it will also reabsorb Li+, as it has a hard time differentiating between these two monovalent cations.
A drug interaction between lithium and diuretics has a unique mechanism. In response to decreased volume secondary to diuretic use, the kidneys will try to retain Na+ in an effort to retain water. When the kidney reabsorbs Na+, it will also reabsorb Li+, as it has a hard time differentiating between these two monovalent cations.
Evidence
Bipolar Disorder
 A 2007 Health Technology Assessment (United Kingdom) review (45 trials) compared various mood stabilizers in preventing relapse in bipolar disorder. The authors found that lithium, valproate, lamotrigine, and olanzapine were more efficacious than placebo as maintenance therapy for relapse prevention. Lithium and olanzapine were effective for preventing manic relapses, but not for depressive relapses.
A 2007 Health Technology Assessment (United Kingdom) review (45 trials) compared various mood stabilizers in preventing relapse in bipolar disorder. The authors found that lithium, valproate, lamotrigine, and olanzapine were more efficacious than placebo as maintenance therapy for relapse prevention. Lithium and olanzapine were effective for preventing manic relapses, but not for depressive relapses.Lithium versus Placebo for Maintenance Treatment in Mood Disorders
 A 2001 Cochrane review (nine trials, N = 825 participants) compared lithium to placebo for treatment of mood disorder. Lithium was most beneficial at preventing relapse when used in bipolar disorder (OR 0.29). No significant benefit for relapse prevention was found in unipolar disorder. Because of low event rates, no conclusions could be drawn about the impact of lithium on suicide.
A 2001 Cochrane review (nine trials, N = 825 participants) compared lithium to placebo for treatment of mood disorder. Lithium was most beneficial at preventing relapse when used in bipolar disorder (OR 0.29). No significant benefit for relapse prevention was found in unipolar disorder. Because of low event rates, no conclusions could be drawn about the impact of lithium on suicide.FYI
 The discovery of lithium, one of the most important drugs in psychiatry, is an example of serendipity and scientific acumen. In the late 1940s an Australian scientist was administering lithium salt to guinea pigs to increase the solubility of urates. After noting that lithium made the animals lethargic, he decided to give lithium carbonate to agitated or manic psychiatric patients. Lithium appeared to have a positive effect on mania, paving the way for its use for this indication.
The discovery of lithium, one of the most important drugs in psychiatry, is an example of serendipity and scientific acumen. In the late 1940s an Australian scientist was administering lithium salt to guinea pigs to increase the solubility of urates. After noting that lithium made the animals lethargic, he decided to give lithium carbonate to agitated or manic psychiatric patients. Lithium appeared to have a positive effect on mania, paving the way for its use for this indication.First-Generation Antipsychotics






MOA (Mechanism of Action)
 Dopamine is believed to play a key role in schizophrenia and thought disorders. Patients with schizophrenia experience hallucinations (visual, auditory, and tactile experiences in the absence of stimulation—for example, seeing something that is not there) and delusions (beliefs that are not true).
Dopamine is believed to play a key role in schizophrenia and thought disorders. Patients with schizophrenia experience hallucinations (visual, auditory, and tactile experiences in the absence of stimulation—for example, seeing something that is not there) and delusions (beliefs that are not true).



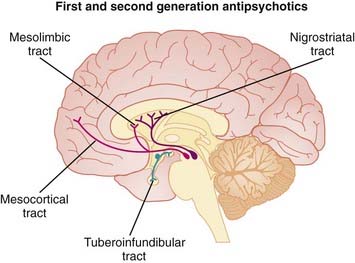
Pharmacokinetics
 Most antipsychotics are highly lipophilic (and therefore easily enter the brain) and accumulate in well-perfused tissues such as brain and lung. Because of their lipophilic nature, most antipsychotics have the potential to cross the placenta and to enter breast milk.
Most antipsychotics are highly lipophilic (and therefore easily enter the brain) and accumulate in well-perfused tissues such as brain and lung. Because of their lipophilic nature, most antipsychotics have the potential to cross the placenta and to enter breast milk.


 Several first-generation antipsychotics are available in parenteral formulations, both standard intravenous and intramuscular formulations to achieve faster onset, and depot formulations to achieve prolonged effects (Table 23-3).
Several first-generation antipsychotics are available in parenteral formulations, both standard intravenous and intramuscular formulations to achieve faster onset, and depot formulations to achieve prolonged effects (Table 23-3).TABLE 23-3 Onset and Duration of Action of Depot and Intramuscular Formulations of First- Generation Antipsychotics
| Onset | Duration | |
|---|---|---|
| Depot | ||
| Haloperidol decanoate | 3 weeks | |
| Fluphenazine decanoate | 3-4 weeks | |
| Pipotiazine palmitate | 3-6 weeks | |
| Intramuscular (IM) | ||
| Haloperidol IM | 30-45 minutes | |
| Chlorpromazine IM | 15-30 minutes | |
| Fluphenazine IM | <1 hour | |






Side Effects
Common
 Anticholinergic effects include dry mouth, constipation, difficulty urinating, and loss of accommodation.
Anticholinergic effects include dry mouth, constipation, difficulty urinating, and loss of accommodation.







Important Notes
 There is increasing concern about the metabolic side effects associated with long-term use of antipsychotics. The risk of some of these side effects may be lower with first-generation antipsychotics compared with second generation antipsychotics (see Evidence section).
There is increasing concern about the metabolic side effects associated with long-term use of antipsychotics. The risk of some of these side effects may be lower with first-generation antipsychotics compared with second generation antipsychotics (see Evidence section).


Evidence
Risk of Diabetes in First- versus Second-Generation Antipsychotics
 A 2008 systematic review (11 studies) compared diabetes risk among various antipsychotics. The majority of studies in this review were cross-sectional or retrospective cohort studies, which somewhat limits confidence in the analysis. The authors found an increased risk of diabetes in patients taking second-generation versus first-generation antipsychotics (RR 1.32). Data were insufficient to include aripiprazole, ziprasidone, and amisulpride in this analysis. Relative risks ranged from a low of 1.16 with risperidone to a high of 1.39 with clozapine. Differences in risk were statistically significant for all second-generation agents except risperidone.
A 2008 systematic review (11 studies) compared diabetes risk among various antipsychotics. The majority of studies in this review were cross-sectional or retrospective cohort studies, which somewhat limits confidence in the analysis. The authors found an increased risk of diabetes in patients taking second-generation versus first-generation antipsychotics (RR 1.32). Data were insufficient to include aripiprazole, ziprasidone, and amisulpride in this analysis. Relative risks ranged from a low of 1.16 with risperidone to a high of 1.39 with clozapine. Differences in risk were statistically significant for all second-generation agents except risperidone.


Second-Generation Antipsychotics


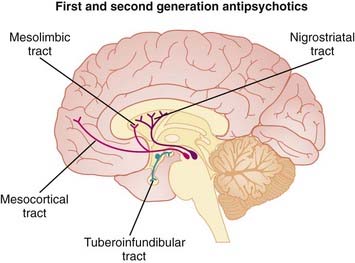
MOA (Mechanism of Action) (Figure 23-9)
 Dopamine is believed to play a key role in schizophrenia and thought disorders. Patients with schizophrenia experience hallucinations (visual, auditory, and tactile experiences in the absence of stimulation—for example, seeing something that is not there) and delusions (beliefs that are not true).
Dopamine is believed to play a key role in schizophrenia and thought disorders. Patients with schizophrenia experience hallucinations (visual, auditory, and tactile experiences in the absence of stimulation—for example, seeing something that is not there) and delusions (beliefs that are not true).






Pharmacokinetics






Contraindications







Side Effects
 The side effect profile of atypical antipsychotics varies widely owing to their heterogeneous receptor binding profiles.
The side effect profile of atypical antipsychotics varies widely owing to their heterogeneous receptor binding profiles.Serious
All Agents
 Increased mortality in elderly patients: These agents are associated with increased risk of death in elderly patients with dementia, from a variety of causes, largely cardiovascular or infectious. The mechanism is still unclear at this time.
Increased mortality in elderly patients: These agents are associated with increased risk of death in elderly patients with dementia, from a variety of causes, largely cardiovascular or infectious. The mechanism is still unclear at this time. Endocrine:
Endocrine:

Clozapine
 Agranulocytosis can be fatal and necessitates weekly or biweekly blood monitoring of all patients taking clozapine. The risk is about 1% in the first 3 months and then decreases to about 0.01% after 1 year. Although agranulocytosis is a risk with all antipsychotics, it is much more common with clozapine compared to other agents.
Agranulocytosis can be fatal and necessitates weekly or biweekly blood monitoring of all patients taking clozapine. The risk is about 1% in the first 3 months and then decreases to about 0.01% after 1 year. Although agranulocytosis is a risk with all antipsychotics, it is much more common with clozapine compared to other agents.






Important Notes
 Clozapine is itself considered to be atypical among atypical antipsychotics, with a unique side effect profile and a greater degree of success in treatment-resistant patients. Thus many consider clozapine to be in a class of its own.
Clozapine is itself considered to be atypical among atypical antipsychotics, with a unique side effect profile and a greater degree of success in treatment-resistant patients. Thus many consider clozapine to be in a class of its own.



Advanced
Drug Interactions
 Clozapine is metabolized by CYP1A2 and CYP3A4, and its metabolism is affected by notable inducers or inhibitors of these isozymes, such as the following:
Clozapine is metabolized by CYP1A2 and CYP3A4, and its metabolism is affected by notable inducers or inhibitors of these isozymes, such as the following:



Evidence
Schizophrenia


Psychosis and Aggression Associated with Alzheimer’s Disease
 A 2006 Cochrane review (10 trials) found that although risperidone and olanzapine were useful in reducing aggression and risperidone was useful for psychosis, these agents also had serious harmful effects such as a higher incidence of stroke as well as EPS and other adverse events. There were insufficient data to assess cognitive function.
A 2006 Cochrane review (10 trials) found that although risperidone and olanzapine were useful in reducing aggression and risperidone was useful for psychosis, these agents also had serious harmful effects such as a higher incidence of stroke as well as EPS and other adverse events. There were insufficient data to assess cognitive function.
FYI
 Clozapine is widely recognized as being the atypical antipsychotic with the greatest efficacy; however, its 50-year history has been tumultuous. From its early trials in the 1960s, clozapine was noted as being “atypical” because of its lack of disabling neurologic side effects. However, the lack of EPS was interpreted as an indication that it lacked antipsychotic efficacy as well, as at that time the two effects were considered to be connected.
Clozapine is widely recognized as being the atypical antipsychotic with the greatest efficacy; however, its 50-year history has been tumultuous. From its early trials in the 1960s, clozapine was noted as being “atypical” because of its lack of disabling neurologic side effects. However, the lack of EPS was interpreted as an indication that it lacked antipsychotic efficacy as well, as at that time the two effects were considered to be connected.

Benzodiazepines





MOA (Mechanism of Action)
 γ-Aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the CNS. GABA binds to three different types of receptors: GABAA, GABAB, and GABAC.
γ-Aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the CNS. GABA binds to three different types of receptors: GABAA, GABAB, and GABAC.
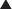




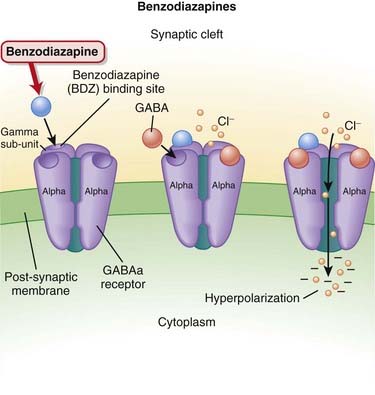
Pharmacokinetics

 The elimination half-lives of BZDs play a key role in determining how they are used (see Important Notes for further discussion).
The elimination half-lives of BZDs play a key role in determining how they are used (see Important Notes for further discussion).

TABLE 23-4 Elimination Half-Lives of Various Benzodiazepines
| Half-life Duration | Half-Life (Hours) |
|---|---|
| Short Half-Life Agents | |
| Triazolam | 2-3 |
| Intermediate Half-Life Agents | |
| Alprazolam | 12-15 |
| Lorazepam | 10-20 |
| Oxazepam | 10-20 |
| Temazepam | 10-40 |
| Chlordiazepoxide* | 15-40 |
| Long Half-Life Agents | |
| Diazepam* | 20-80 |
| Flurazepam* | 40-100 |






















Evidence
Generalized Anxiety Disorder
 A 2007 systematic review examined three commonly used BZDs—diazepam (12 trials), lorazepam (7 trials), and alprazolam (4 trials)—versus placebo. All included studies were DBRCTs. The authors chose withdrawal from study as their primary outcome measure, and fewer patients treated with BZD withdrew for any reason (RR 0.78) or for lack of efficacy (RR 0.29). However, more BZD-treated patients withdrew because of an adverse event compared with placebo (RR 1.54).
A 2007 systematic review examined three commonly used BZDs—diazepam (12 trials), lorazepam (7 trials), and alprazolam (4 trials)—versus placebo. All included studies were DBRCTs. The authors chose withdrawal from study as their primary outcome measure, and fewer patients treated with BZD withdrew for any reason (RR 0.78) or for lack of efficacy (RR 0.29). However, more BZD-treated patients withdrew because of an adverse event compared with placebo (RR 1.54).Older Adults with Insomnia
 A 2005 systematic review examined the risks and benefits of sedative use for insomnia in adults 60 years of age or older. The review included 20 DBRCTs, covering sleep quality, sleep time, awakenings, or latency of sleep onset. BZDs (830 participants) and non-BZDs (1099 participants) were the main interventions used, versus placebo (468 participants). Based on four studies, the authors calculated a number needed to treat (NNT) of 13 for improvement in sleep quality with sedatives. However, the number needed to harm (NNH) was six versus placebo. No difference in sleep quality was detected across three studies that compared BZDs with atypical BZDs. Sedatives increased sleep time by 25 minutes overall. Cognitive adverse effects were significantly more common with sedatives than with placebo (OR 4.78).
A 2005 systematic review examined the risks and benefits of sedative use for insomnia in adults 60 years of age or older. The review included 20 DBRCTs, covering sleep quality, sleep time, awakenings, or latency of sleep onset. BZDs (830 participants) and non-BZDs (1099 participants) were the main interventions used, versus placebo (468 participants). Based on four studies, the authors calculated a number needed to treat (NNT) of 13 for improvement in sleep quality with sedatives. However, the number needed to harm (NNH) was six versus placebo. No difference in sleep quality was detected across three studies that compared BZDs with atypical BZDs. Sedatives increased sleep time by 25 minutes overall. Cognitive adverse effects were significantly more common with sedatives than with placebo (OR 4.78).
Alcohol Withdrawal
 A 2005 Cochrane review found that BZDs were significantly better for alcohol withdrawal seizures compared with placebo and had similar efficacy to other drugs, or anticonvulsants specifically. BZDs elicited similar changes in alcohol withdrawal scores compared with other drugs. The findings of the review are complicated by the heterogeneity among trials.
A 2005 Cochrane review found that BZDs were significantly better for alcohol withdrawal seizures compared with placebo and had similar efficacy to other drugs, or anticonvulsants specifically. BZDs elicited similar changes in alcohol withdrawal scores compared with other drugs. The findings of the review are complicated by the heterogeneity among trials.


Central Nervous System Stimulants


MOA (Mechanism of Action)
 Methylphenidate and amphetamine work by increasing neurotransmitter levels in the synapse, specifically, levels of dopamine (DA) and noradrenaline (NA). They accomplish this through a variety of mechanisms.
Methylphenidate and amphetamine work by increasing neurotransmitter levels in the synapse, specifically, levels of dopamine (DA) and noradrenaline (NA). They accomplish this through a variety of mechanisms.










Contraindications



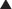




Evidence
Safety of Methylphenidate in Adults
 A 2009 systematic review (26 trials, 811 participants) examined the safety of methylphenidate versus placebo for a variety of disorders, including ADHD, in adults. The trials were of relatively short duration (1 to 12 weeks) and therefore provide no information about chronic use. No serious (life-threatening or irreversible) adverse effects were reported in any studies. Dry mouth, anorexia, changes in mood, “jitteriness,” depression or sadness, weight loss, and vertigo were the most common adverse events that occurred more frequently with methylphenidate than placebo. The five studies reporting cardiovascular changes found slight increases in blood pressure and pulse rate.
A 2009 systematic review (26 trials, 811 participants) examined the safety of methylphenidate versus placebo for a variety of disorders, including ADHD, in adults. The trials were of relatively short duration (1 to 12 weeks) and therefore provide no information about chronic use. No serious (life-threatening or irreversible) adverse effects were reported in any studies. Dry mouth, anorexia, changes in mood, “jitteriness,” depression or sadness, weight loss, and vertigo were the most common adverse events that occurred more frequently with methylphenidate than placebo. The five studies reporting cardiovascular changes found slight increases in blood pressure and pulse rate.