[level-membership-for-basic-science-category]
Chapter 1 Basic Principles and Pharmacodynamics
Drug Nomenclature
Trade, Brand, or Proprietary Name
 Clinical Connection: Drugs can have many different names. For example, a prototypical calcium channel blocker of the dihydropyridine class has the chemical name 3,5-dimethyl 2,6-dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate, the generic name nifedipine, and is available in the United States under several trade names including Adalat, Nifedical, and Procardia. Although marketing emphasizes trade names, the use of generic drug names is encouraged in practice to reduce prescribing errors and offers the opportunity for substitutions if appropriate.
Clinical Connection: Drugs can have many different names. For example, a prototypical calcium channel blocker of the dihydropyridine class has the chemical name 3,5-dimethyl 2,6-dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate, the generic name nifedipine, and is available in the United States under several trade names including Adalat, Nifedical, and Procardia. Although marketing emphasizes trade names, the use of generic drug names is encouraged in practice to reduce prescribing errors and offers the opportunity for substitutions if appropriate.Drug-Receptor Interactions
Although some notable exceptions exist, a fundamental principle of pharmacology is that drugs must interact with a molecular target to exert an effect. Drug interaction with molecular targets is the initiating event in a multistep process that ultimately alters tissue function. For the purposes of current discussion, the target will be referred to as a receptor. An in-depth discussion of molecular targets and a description of these processes will be presented later in this chapter (see the discussion of molecular mechanisms of drug action). Let us first consider the relationship between drug binding to its target receptors and the ultimate response of the tissue.
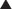
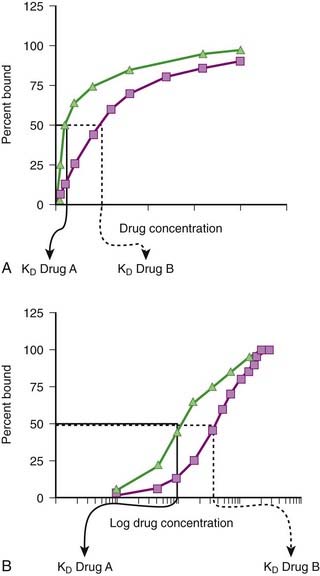
Law of Mass Action Applied to Drugs
Although the amount of drug receptor-complex formed is proportional to the concentrations of drug and receptor, this relationship is not linear but is in fact parabolic (Figure 1-1, A). Accordingly, this relationship is most often diagrammed on a semilogarithmic graph to linearize the relationship and encompass the large range of concentrations typical of the drug-receptor relationship (Figure 1-1, B).
Factors Affecting Drug-Target Interactions
Drug Binding



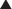
It is important to recognize that, in most cases, binding of drug to target molecules involves weaker bonds. Accordingly, the drug-receptor complex is not static, but rather there is continuous association and dissociation of the drug with the receptor as long as drug is present. A measure of the relative ease with which the association and dissociation reactions occur is the equilibrium dissociation constant (KD). Each drug-receptor combination will have a characteristic KD value. Drugs with high affinity for a given receptor display a small value for KD, and vice versa. In Figure 1-1, A and B, Drug A has a higher affinity for the receptor than Drug B. KD also represents the concentration of drug needed to bind 50% of the total receptor population. These concepts are important in the study of basic pharmacologic data regarding different compounds with affinity for the same receptor. In general, drugs with lower KD values will require lower concentrations to achieve sufficient receptor occupancy to exert an effect.
Selectivity of Drug Responses
 The cell will respond only to the spectrum of drugs that exhibit affinity for the receptors expressed by the cell.
The cell will respond only to the spectrum of drugs that exhibit affinity for the receptors expressed by the cell.

 Clinical Connection: β-Adrenergic receptor antagonists are effective drugs for a number of cardiovascular disorders. Some β-adrenergic receptor antagonists are selective for β1-adrenergic receptors to limit the potential for bronchoconstriction caused by blocking β2-adrenergic receptors. However, even β1-selective antagonists must be used cautiously in asthmatic patients, particularly at higher doses, to avoid further impairment of airway function in these patients.
Clinical Connection: β-Adrenergic receptor antagonists are effective drugs for a number of cardiovascular disorders. Some β-adrenergic receptor antagonists are selective for β1-adrenergic receptors to limit the potential for bronchoconstriction caused by blocking β2-adrenergic receptors. However, even β1-selective antagonists must be used cautiously in asthmatic patients, particularly at higher doses, to avoid further impairment of airway function in these patients.







Quantifying Drug-Target Interactions: Dose-Response Relationships
Graded Dose-Response Curves
 Measure an effect that is continuous such that, in theory, any value is possible in a given range (0% through 100%).
Measure an effect that is continuous such that, in theory, any value is possible in a given range (0% through 100%). Have a sigmoidal shape similar to the drug receptor occupancy curves shown in Figure 1-2, because the biologic response to a drug is determined by the interaction of a drug with a receptor or molecular target.
Have a sigmoidal shape similar to the drug receptor occupancy curves shown in Figure 1-2, because the biologic response to a drug is determined by the interaction of a drug with a receptor or molecular target.



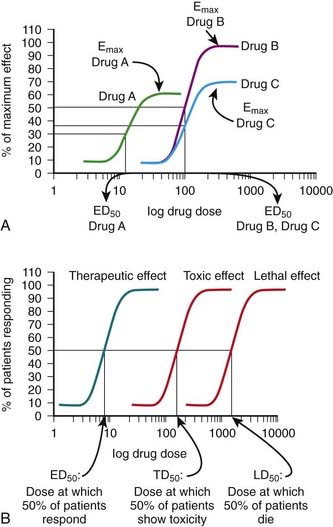
The ED50 and Emax are useful parameters to assess drugs. In Figure 1-2, A, Drug A is more potent than Drug B or Drug C, whereas Drugs B and C have equal potency. Potency is sometimes used incorrectly as a measure of therapeutic effectiveness. In fact, in most cases potency is secondary to Emax in drug selection. However, in situations in which the absorption of drug is very poor, such that only small quantities of the drug reach the target, potency can be a critical consideration. Drugs with higher Emax values have higher pharmacologic efficacy.
In Figure 1-2, A, Drug B has the greatest efficacy, followed by Drug C, whereas Drug A, despite being the most potent, has the least efficacy. Drug C is equipotent with Drug B but has less efficacy. Thus, potency and efficacy can vary independently. It is important not to confuse the pharmacologic usage of efficacy with the more general usage. Pharmacologic efficacy is a measure of the strength of effect produced by the maximum dose of drug. By definition, antagonists do not activate their receptors after binding and therefore have an intrinsic activity and efficacy of 0. Nevertheless, an antagonist may be very clinically “efficacious” or beneficial because it blocks activation of the receptor by endogenous agonist.
Quantal Dose-Response Curves
Quantal dose-response curves do the following:

 Describe the relationship between drug dosage and the frequency with which a biologic effect occurs. For example, in individuals administered an anticonvulsant medication, the percentage of individuals not experiencing a convulsive episode at any given dose is plotted in cumulative fashion (Figure 1-2, B).
Describe the relationship between drug dosage and the frequency with which a biologic effect occurs. For example, in individuals administered an anticonvulsant medication, the percentage of individuals not experiencing a convulsive episode at any given dose is plotted in cumulative fashion (Figure 1-2, B). Represent a cumulative frequency distribution for a given response.
Represent a cumulative frequency distribution for a given response.
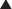 Provide an ED50 value that reflects the dose of drug that produces a response in 50% of the population (also called the median effective dose).
Provide an ED50 value that reflects the dose of drug that produces a response in 50% of the population (also called the median effective dose).
 Can be plotted for therapeutic, toxic, and lethal effects to obtain:
Can be plotted for therapeutic, toxic, and lethal effects to obtain:
Antagonism as a Mechanism of Drug Action
Physiologic (Functional) Antagonists



Pharmacokinetic Antagonists



Pharmacologic Antagonists
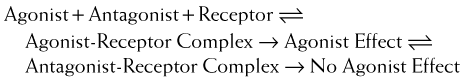
 Competitive reversible antagonism (also called competitive surmountable antagonism)
Competitive reversible antagonism (also called competitive surmountable antagonism)
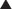 The antagonist competes directly for the target receptor with the agonist molecule. These interactions will follow the law of mass action and the same general principles described earlier, with the added facet that now two drugs are competing for receptor occupancy.
The antagonist competes directly for the target receptor with the agonist molecule. These interactions will follow the law of mass action and the same general principles described earlier, with the added facet that now two drugs are competing for receptor occupancy.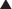
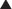
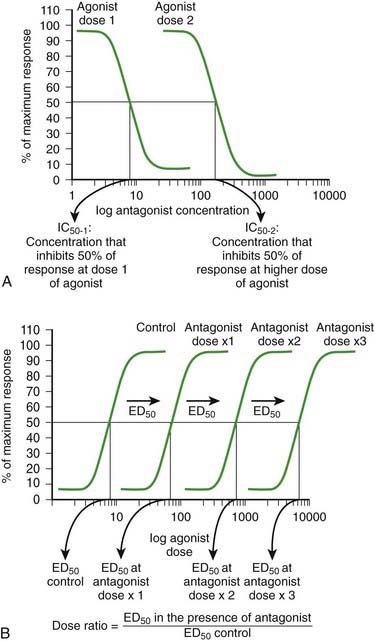
 As the concentration of antagonist increases, the number of antagonist-receptor complexes increases and the number of agonist-receptor complexes decreases. Therefore the agonist effect decreases. Figure 1-3, A shows a graded dose-response curve for increasing concentrations of antagonist in the presence of a fixed concentration of agonist. The concentration of antagonist that reduces the agonist response to 50% of maximum is the IC50, one index for quantifying antagonist effectiveness. Note that IC50 values vary with agonist starting concentration.
As the concentration of antagonist increases, the number of antagonist-receptor complexes increases and the number of agonist-receptor complexes decreases. Therefore the agonist effect decreases. Figure 1-3, A shows a graded dose-response curve for increasing concentrations of antagonist in the presence of a fixed concentration of agonist. The concentration of antagonist that reduces the agonist response to 50% of maximum is the IC50, one index for quantifying antagonist effectiveness. Note that IC50 values vary with agonist starting concentration.
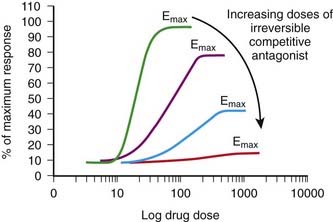
 The agonist-antagonist relationship can also be depicted on agonist dose-response curves. Figure 1-3, B illustrates graded agonist dose-response curves in the absence (control) and presence of increasing doses of antagonist.
The agonist-antagonist relationship can also be depicted on agonist dose-response curves. Figure 1-3, B illustrates graded agonist dose-response curves in the absence (control) and presence of increasing doses of antagonist.
 Increasing the concentration of antagonist produces a greater rightward displacement of the agonist dose-response curve, and the ED50 value for the agonist increases progressively. However, a maximal effect can always be reached by increasing the dose of agonist. The rightward parallel displacement of the agonist dose-response curves but with preserved Emax is characteristic of competitive reversible antagonism (see Figure 1-3).
Increasing the concentration of antagonist produces a greater rightward displacement of the agonist dose-response curve, and the ED50 value for the agonist increases progressively. However, a maximal effect can always be reached by increasing the dose of agonist. The rightward parallel displacement of the agonist dose-response curves but with preserved Emax is characteristic of competitive reversible antagonism (see Figure 1-3).
 Competitive irreversible antagonism (also called competitive insurmountable antagonism) (in older literature these are also labeled as noncompetitive antagonists)
Competitive irreversible antagonism (also called competitive insurmountable antagonism) (in older literature these are also labeled as noncompetitive antagonists)
 The antagonist competes directly with the agonist for receptor binding as described previously. However, the binding forces between the antagonist and receptor are so strong that the antagonist-receptor complex is virtually irreversible.
The antagonist competes directly with the agonist for receptor binding as described previously. However, the binding forces between the antagonist and receptor are so strong that the antagonist-receptor complex is virtually irreversible.
 Noncompetitive antagonism (also called allotropic or allosteric antagonism)
Noncompetitive antagonism (also called allotropic or allosteric antagonism)
 Noncompetitive antagonists do not directly compete with the agonist for binding at the same binding site but nevertheless impair the ability of an agonist to bind to or activate the receptor, and thus they prevent a response.
Noncompetitive antagonists do not directly compete with the agonist for binding at the same binding site but nevertheless impair the ability of an agonist to bind to or activate the receptor, and thus they prevent a response.

Molecular Mechanisms Mediating Drug Action




Receptor Coupling and Transduction Mechanisms
Extracellular Transduction Mechanisms
 Extracellular enzymes. Drugs acting via this mechanism alter the activity of extracellular enzymes involved in the synthesis or degradation of endogenous signaling molecules. These drugs affect the levels of endogenous compounds that then alter cellular function by acting on their receptors. Examples of clinical utility for this mechanism include:
Extracellular enzymes. Drugs acting via this mechanism alter the activity of extracellular enzymes involved in the synthesis or degradation of endogenous signaling molecules. These drugs affect the levels of endogenous compounds that then alter cellular function by acting on their receptors. Examples of clinical utility for this mechanism include:
 Angiotensin-converting enzyme (ACE) inhibitors, which prevent the formation of angiotensin II and are used in the treatment of hypertension
Angiotensin-converting enzyme (ACE) inhibitors, which prevent the formation of angiotensin II and are used in the treatment of hypertension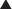

Transmembrane Transduction Mechanisms
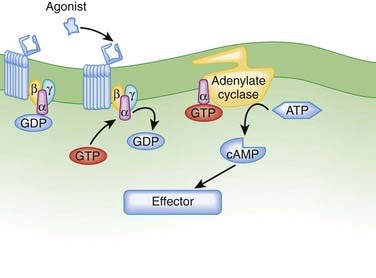
 G protein–coupled receptors (GPCRs). GPCRs, also called seven transmembrane pass receptors, are a large class of receptors that mediate the majority of endogenous transmitter and hormone driven responses (Figure 1-5).
G protein–coupled receptors (GPCRs). GPCRs, also called seven transmembrane pass receptors, are a large class of receptors that mediate the majority of endogenous transmitter and hormone driven responses (Figure 1-5).
 Ligand activation of the receptor causes the GPCR to interact with G proteins.
Ligand activation of the receptor causes the GPCR to interact with G proteins.
 Receptor-coupled enzymes. Receptor-coupled enzymes bypass the G protein coupling mechanism and link directly to cellular communication cascades. The receptor is directly coupled in some way to kinase enzymatic activity within the cell. Ligand binding stimulates the kinase enzymatic activity, which then initiates and amplifies intracellular signals and feedback responses by changing the phosphorylation status of cellular proteins. As shown in Figure 1-6, these mechanisms can be grouped into four general types that include receptors:
Receptor-coupled enzymes. Receptor-coupled enzymes bypass the G protein coupling mechanism and link directly to cellular communication cascades. The receptor is directly coupled in some way to kinase enzymatic activity within the cell. Ligand binding stimulates the kinase enzymatic activity, which then initiates and amplifies intracellular signals and feedback responses by changing the phosphorylation status of cellular proteins. As shown in Figure 1-6, these mechanisms can be grouped into four general types that include receptors:
 With guanylate cyclase activity that generate a second messenger, cyclic guanosine monophosphate (cGMP)
With guanylate cyclase activity that generate a second messenger, cyclic guanosine monophosphate (cGMP)
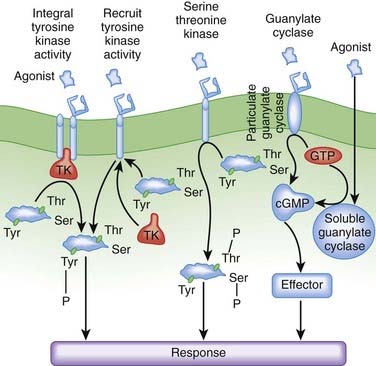
The receptor-coupled enzymes phosphorylate intracellular proteins at tyrosine (Tyr), serine (Ser), or threonine (Thr) residues to change protein function. Alternatively, cGMP generated by guanylate cyclase activates downstream enzymes (effector in Figure 1-6) that change the phosphorylation status of proteins to alter their function. Receptors with TK activity and guanylate cyclase activity are currently the most clinically useful. Examples of these two receptor-linked enzymes include the following:
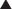 Receptors with integral TK activity
Receptors with integral TK activity



 Transmembrane ion channels. Transmembrane ion channels allow the passage of ions from one side of a membrane to another. Channels can exist in the open, closed, or inactive state, which represent different conformations of the channel protein. As shown in Figure 1-7, drugs may affect the function of these channels by directly opening or closing the channel (ligand gated channels), by influencing the voltage-dependent characteristics of the channels (voltage gated channels) and the amount of time the channel spends in a given state, or by generating second messengers that subsequently open or close the channel (second messenger gated). Common examples of functions governed by ion channels include the following:
Transmembrane ion channels. Transmembrane ion channels allow the passage of ions from one side of a membrane to another. Channels can exist in the open, closed, or inactive state, which represent different conformations of the channel protein. As shown in Figure 1-7, drugs may affect the function of these channels by directly opening or closing the channel (ligand gated channels), by influencing the voltage-dependent characteristics of the channels (voltage gated channels) and the amount of time the channel spends in a given state, or by generating second messengers that subsequently open or close the channel (second messenger gated). Common examples of functions governed by ion channels include the following:
 Neurotransmission
Neurotransmission
There are several subtypes of ion channels, based on the ways that drugs or endogenous substances regulate the channels (see Figure 1-7).
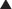 Ligand gated ion channels or receptor-operated channels. These ion channels possess a receptor for an endogenous ligand to which the drug can bind. They are composed of a multimeric protein complex that constitutes both the receptor and the ion channel. Agonist activation opens the channel, and antagonists close or inactivate the channel. Examples include the following:
Ligand gated ion channels or receptor-operated channels. These ion channels possess a receptor for an endogenous ligand to which the drug can bind. They are composed of a multimeric protein complex that constitutes both the receptor and the ion channel. Agonist activation opens the channel, and antagonists close or inactivate the channel. Examples include the following:
 Second messenger gated ion channels. These ion channels respond directly or indirectly to second messenger molecules (see second messenger section, later, for details). Drug binding to receptor elaborates a second messenger that in turn affects channel function. Examples of clinical utility include the following:
Second messenger gated ion channels. These ion channels respond directly or indirectly to second messenger molecules (see second messenger section, later, for details). Drug binding to receptor elaborates a second messenger that in turn affects channel function. Examples of clinical utility include the following:




Intracellular Transduction Mechanisms
Intracellular Receptors
Lipophilic drugs passively cross the cell membrane and thus do not require cell membrane receptors. As shown in Figure 1-8, one target for these drugs is an intracellular receptor that activates transcriptional pathways. In this mechanism, the agonist receptor complex diffuses to DNA, where it binds to DNA binding elements. Via this mechanism drugs act directly or through recruitment of coactivators or co-repressors, which increase or decrease transcription of RNA to ultimately change protein expression. This process is referred to as ligand gated transcriptional regulation. In many cases these drugs effect long-term changes by affecting gene transcription. Receptors using this coupling mechanism include:





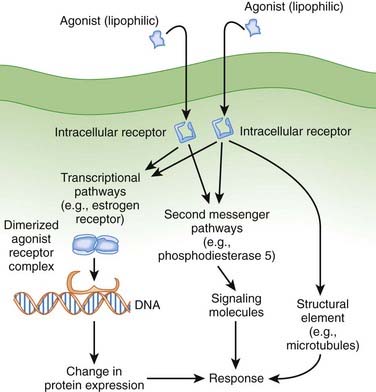
Examples of ligand gated transcription regulation with clinical utility include:
Intracellular Enzymes
Some drugs directly target intracellular enzymes, such as phosphodiesterase (PDE), that control second messenger pathways (see Figure 1-8) and thereby alter the concentrations of intracellular signaling molecules, which then effects a cellular response. As greater understanding of intracellular signaling is achieved, it is likely that more drugs using this mode of action will be developed. Often there are multiple levels of intracellular signaling molecules downstream from the enzyme being targeted. A common example of the utility of this approach is PDE5 inhibition, to prevent the breakdown of cGMP, which results in increased vasodilation. This approach is useful in the treatment of erectile dysfunction because of the ability to somewhat selectively target blood vessels in the penis.
Structural Mechanisms
Drugs can also target structural components of cells (e.g., the cytoskeleton or microtubules) to affect their function (see Figure 1-8). Examples of clinical utility include the following:


Second Messenger Systems
After formation of the drug-receptor complex and activation of a coupling mechanism (e.g., G proteins), the drug signal is transmitted to the final effector system of the cell. In many cases the transduction or coupling mechanism is linked to the final effector system via an intermediate cell signaling (second messenger) system. Drugs may also target enzymes or other processes regulating the concentrations of intracellular second messengers. This represents an important mode of drug action. In addition, it opens the possibility for synergistic or antagonist interactions among drugs that act at different sites in the same pathway. These interactions may enhance therapeutic effects or lead to adverse effects. The field of cell signaling is extremely dynamic, with new signaling molecules or new functions for established molecules discovered on a seemingly daily basis. Therefore, it is not possible to discuss the intricacies of all second messenger systems linked to clinically relevant drug actions. Nevertheless, several pathways serve as good illustrations of the involvement of cell signaling mechanisms as mediators of drug responses and as targets for future drug development. Figure 1-9 illustrates three of the best understood second messenger systems.
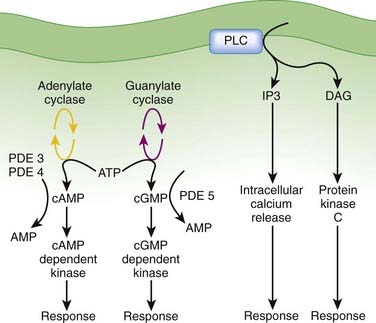
Cyclic Adenosine Monophosphate Pathway



 Agonists activating adenylate cyclase may elicit differential responses in different target tissues.
Agonists activating adenylate cyclase may elicit differential responses in different target tissues. cAMP signaling can proceed through:
cAMP signaling can proceed through:

Cyclic Guanosine Monophosphate

Phospholipase C, Inositol 1,4,5 Trisphosphate (IP3), Diacylglycerol (DAG)


 DAG activates the protein kinase C (PKC) family of enzymes.
DAG activates the protein kinase C (PKC) family of enzymes.
Activation may be isoform and tissue specific.
 The IP3-DAG-PKC pathway is very widespread and linked to many functions. This signaling mechanism is coupled to receptors for some of the major homeostatic pathways, including α-adrenergic receptors, serotonin receptors, angiotensin receptors, acetylcholine receptors, and prostaglandins, to name but a few. The widespread nature of this pathway makes it very important but also very difficult to manipulate to achieve selective therapeutic actions with minimal side effects. Nevertheless, this is an area of ongoing research, and isoform-specific inhibitors of PKC are in development for clinical applications. Enzastaurin, a selective PKC-β isoform inhibitor, is in clinical trials as an antineoplastic agent.
The IP3-DAG-PKC pathway is very widespread and linked to many functions. This signaling mechanism is coupled to receptors for some of the major homeostatic pathways, including α-adrenergic receptors, serotonin receptors, angiotensin receptors, acetylcholine receptors, and prostaglandins, to name but a few. The widespread nature of this pathway makes it very important but also very difficult to manipulate to achieve selective therapeutic actions with minimal side effects. Nevertheless, this is an area of ongoing research, and isoform-specific inhibitors of PKC are in development for clinical applications. Enzastaurin, a selective PKC-β isoform inhibitor, is in clinical trials as an antineoplastic agent. Clinical Connection: Knowledge of coupling and second messenger systems can help in understanding drug interactions. Nitrates used in the treatment of coronary ischemia stimulate guanylate cyclase to produce cGMP. PDE5 inhibitors used in the treatment of erectile dysfunction inhibit the breakdown of cGMP. Concurrent use of these two drugs can lead to excessive levels of cGMP, which in turn cause excessive vasodilation and potentially dangerous reductions in blood pressure. Consequently, patients are warned to not use PDE5 inhibitors if they are on nitrate therapy for angina.
Clinical Connection: Knowledge of coupling and second messenger systems can help in understanding drug interactions. Nitrates used in the treatment of coronary ischemia stimulate guanylate cyclase to produce cGMP. PDE5 inhibitors used in the treatment of erectile dysfunction inhibit the breakdown of cGMP. Concurrent use of these two drugs can lead to excessive levels of cGMP, which in turn cause excessive vasodilation and potentially dangerous reductions in blood pressure. Consequently, patients are warned to not use PDE5 inhibitors if they are on nitrate therapy for angina.Amplification of Drug Responses
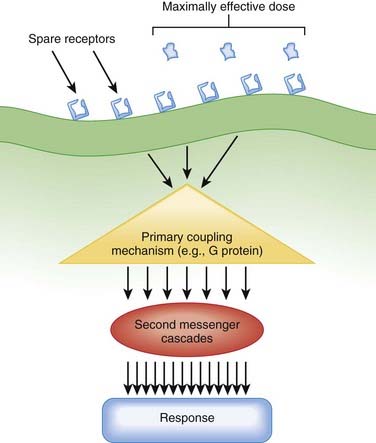
Amplification is an important component of pharmacologic responses. A great deal of amplification occurs in pharmacologic pathways, such that only a minute quantity of drug (often in the picomolar or femtomolar range) is capable of eliciting biologic responses. In general, only minute concentrations of neurotransmitters, hormones, or exogenously administered drugs need reach the molecular target to initiate a biologic response. This exquisite sensitivity of tissues to drugs results in large part from amplification of the original signal provided by the drug molecule. Amplification can occur at several points in the drug-receptor coupling and signaling systems (Figure 1-10).
 Receptor
Receptor


Factors Modifying Drug Responses
 Changes in the amount of drug at the intended molecular target
Changes in the amount of drug at the intended molecular target
 Polymorphisms in drug absorption, distribution, or metabolism are major causes of interindividual variability.
Polymorphisms in drug absorption, distribution, or metabolism are major causes of interindividual variability.
 Changes in the drug receptor itself
Changes in the drug receptor itself
 Different patients express differing numbers or composition of receptors (receptor polymorphisms), leading to differences in affinity or efficacy of the drugs they bind (see Chapter 6, on pharmacogenomics).
Different patients express differing numbers or composition of receptors (receptor polymorphisms), leading to differences in affinity or efficacy of the drugs they bind (see Chapter 6, on pharmacogenomics).
 Changes in coupling or signaling mechanisms
Changes in coupling or signaling mechanisms
Homologous desensitization or tolerance is specific to one receptor type or drug class.
Heterologous desensitization or tolerance affects many receptor types or drugs.
 Clinical Connection: Nitrates are used extensively in the treatment of coronary ischemia. Continuous administration of nitrates is known to produce a reduction in drug response. In some patients, this can occur in as little as 24 hours. Accordingly, the drug regimen for patients taking nitrates includes a daily nitrate-free period or drug holiday of approximately 8 hours each day. Such intermittent administration prevents the development of nitrate tolerance.
Clinical Connection: Nitrates are used extensively in the treatment of coronary ischemia. Continuous administration of nitrates is known to produce a reduction in drug response. In some patients, this can occur in as little as 24 hours. Accordingly, the drug regimen for patients taking nitrates includes a daily nitrate-free period or drug holiday of approximately 8 hours each day. Such intermittent administration prevents the development of nitrate tolerance.[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 1 Basic Principles and Pharmacodynamics
Drug Nomenclature
Trade, Brand, or Proprietary Name
Clinical Connection: Drugs can have many different names. For example, a prototypical calcium channel blocker of the dihydropyridine class has the chemical name 3,5-dimethyl 2,6-dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate, the generic name nifedipine, and is available in the United States under several trade names including Adalat, Nifedical, and Procardia. Although marketing emphasizes trade names, the use of generic drug names is encouraged in practice to reduce prescribing errors and offers the opportunity for substitutions if appropriate.Drug-Receptor Interactions
Although some notable exceptions exist, a fundamental principle of pharmacology is that drugs must interact with a molecular target to exert an effect. Drug interaction with molecular targets is the initiating event in a multistep process that ultimately alters tissue function. For the purposes of current discussion, the target will be referred to as a receptor. An in-depth discussion of molecular targets and a description of these processes will be presented later in this chapter (see the discussion of molecular mechanisms of drug action). Let us first consider the relationship between drug binding to its target receptors and the ultimate response of the tissue.
Law of Mass Action Applied to Drugs
Although the amount of drug receptor-complex formed is proportional to the concentrations of drug and receptor, this relationship is not linear but is in fact parabolic (Figure 1-1, A). Accordingly, this relationship is most often diagrammed on a semilogarithmic graph to linearize the relationship and encompass the large range of concentrations typical of the drug-receptor relationship (Figure 1-1, B).
Factors Affecting Drug-Target Interactions
Drug Binding
It is important to recognize that, in most cases, binding of drug to target molecules involves weaker bonds. Accordingly, the drug-receptor complex is not static, but rather there is continuous association and dissociation of the drug with the receptor as long as drug is present. A measure of the relative ease with which the association and dissociation reactions occur is the equilibrium dissociation constant (KD). Each drug-receptor combination will have a characteristic KD value. Drugs with high affinity for a given receptor display a small value for KD, and vice versa. In Figure 1-1, A and B, Drug A has a higher affinity for the receptor than Drug B. KD also represents the concentration of drug needed to bind 50% of the total receptor population. These concepts are important in the study of basic pharmacologic data regarding different compounds with affinity for the same receptor. In general, drugs with lower KD values will require lower concentrations to achieve sufficient receptor occupancy to exert an effect.
Selectivity of Drug Responses
The cell will respond only to the spectrum of drugs that exhibit affinity for the receptors expressed by the cell. Clinical Connection: β-Adrenergic receptor antagonists are effective drugs for a number of cardiovascular disorders. Some β-adrenergic receptor antagonists are selective for β1-adrenergic receptors to limit the potential for bronchoconstriction caused by blocking β2-adrenergic receptors. However, even β1-selective antagonists must be used cautiously in asthmatic patients, particularly at higher doses, to avoid further impairment of airway function in these patients.Quantifying Drug-Target Interactions: Dose-Response Relationships
Graded Dose-Response Curves
Measure an effect that is continuous such that, in theory, any value is possible in a given range (0% through 100%). Have a sigmoidal shape similar to the drug receptor occupancy curves shown in Figure 1-2, because the biologic response to a drug is determined by the interaction of a drug with a receptor or molecular target.The ED50 and Emax are useful parameters to assess drugs. In Figure 1-2, A, Drug A is more potent than Drug B or Drug C, whereas Drugs B and C have equal potency. Potency is sometimes used incorrectly as a measure of therapeutic effectiveness. In fact, in most cases potency is secondary to Emax in drug selection. However, in situations in which the absorption of drug is very poor, such that only small quantities of the drug reach the target, potency can be a critical consideration. Drugs with higher Emax values have higher pharmacologic efficacy.
In Figure 1-2, A, Drug B has the greatest efficacy, followed by Drug C, whereas Drug A, despite being the most potent, has the least efficacy. Drug C is equipotent with Drug B but has less efficacy. Thus, potency and efficacy can vary independently. It is important not to confuse the pharmacologic usage of efficacy with the more general usage. Pharmacologic efficacy is a measure of the strength of effect produced by the maximum dose of drug. By definition, antagonists do not activate their receptors after binding and therefore have an intrinsic activity and efficacy of 0. Nevertheless, an antagonist may be very clinically “efficacious” or beneficial because it blocks activation of the receptor by endogenous agonist.
Quantal Dose-Response Curves
Quantal dose-response curves do the following:
Describe the relationship between drug dosage and the frequency with which a biologic effect occurs. For example, in individuals administered an anticonvulsant medication, the percentage of individuals not experiencing a convulsive episode at any given dose is plotted in cumulative fashion (Figure 1-2, B). Represent a cumulative frequency distribution for a given response.
Provide an ED50 value that reflects the dose of drug that produces a response in 50% of the population (also called the median effective dose). Can be plotted for therapeutic, toxic, and lethal effects to obtain:
