Prostate Cancer
William G. Nelson, H. Ballentine Carter, Theodore L. DeWeese, Emmanuel S. Antonarakis and Mario A. Eisenberger
• Prostate cancer is the most commonly diagnosed life-threatening cancer in men (241,740 cases and 28,170 deaths in 2012).
• Small prostate cancers are present in 29% of men between ages 30 and 40 and 64% of men between ages 60 and 70.
• The lifetime risk of a prostate cancer diagnosis is 1 in 6, and the risk of dying from prostate cancer is 1 in 35.
• Age, family history, diet and lifestyle, and ethnicity are risk factors for prostate cancer development.
• Germline mutations in RNASEL and MSR1, encoding proteins that function in host responses to infection, appear responsible for some cases of hereditary prostate cancer.
• An inflammatory lesion, termed proliferative inflammatory atrophy (PIA), is an early precursor to prostate cancer.
• Somatic inactivation of GSTP1, encoding a carcinogen-detoxification enzyme, may initiate prostatic carcinogenesis by increasing the vulnerability of prostate cells to damage mediated by oxidant and electrophilic carcinogens.
• Gene fusions, involving TMPRSS2 and ETS family transcription factor genes, may contribute to the androgen dependence of prostate cancers.
• Defects in the functions of NKX3.1, PTEN, and CDKN1B are common in prostate cancer cells.
Screening, Diagnosis, and Staging
• Prostate cancer screening using specific antigen (PSA) testing reduces the risk of prostate cancer death but may also lead to overdiagnosis of non-life-threatening disease.
• Transrectal ultrasound (TRUS)-guided core needle biopsies are used to diagnose prostate cancer.
• Stage, histologic grade (Gleason score), and serum PSA levels are prognostic factors.
• Management options include observational strategies (watchful waiting and active surveillance), anatomic radical prostatectomy (with or without robot-assisted laparoscopic techniques), external beam radiation therapy, and brachytherapy.
• A progressive rise in the serum PSA after treatment indicates prostate cancer recurrence.
• Depending on the approach used, side effects associated with treatment of localized prostate cancer can include urinary, bowel, and sexual dysfunction.
• Salvage therapy for prostate cancer recurrences after initial treatment include external radiation after surgery, or include surgery, brachytherapy, or cryosurgery after external beam radiation.
• Adjuvant androgen suppression can improve survival for some men with prostate cancer treated with external beam radiation therapy.
• Adjuvant external radiation improves survival for some men treated with radical prostatectomy.
• Androgen suppression, most often accomplished via the use of luteinizing hormone–releasing hormone (LHRH) analogs or antagonists, with or without antiandrogens, is the most commonly used treatment.
• Side effects can include loss of libido, hot flashes, gynecomastia, loss of lean muscle mass and bone density, and the development of metabolic syndrome.
• Docetaxel and cabazitaxel chemotherapy improves the survival of men with progressive androgen-independent prostate cancer.
• Second-line treatments targeting the androgen-signaling pathway, including abiraterone acetate and enzalutamide, prolong survival of men previously treated with androgen suppression and taxane chemotherapy.
• Bisphosphonates and denosumab antagonize loss of bone density accompanying androgen deprivation, and reduce skeletal complications associated with metastatic prostate cancer progression.
• Sipuleucel-T, a dendritic cell vaccine, has shown a survival benefit in men with advanced prostate cancer. Other immunotherapies are under development in clinical trials.
Introduction
In 2012, an estimated 241,740 prostate cancer diagnoses will be made in the United States, accompanied by an estimated 28,170 prostate cancer deaths.1 Beginning around 1994-1996, with widespread use of serum prostate-specific antigen (PSA) testing and digital rectal examination (DRE) for prostate cancer screening, and with increased treatment of clinically localized prostate cancer with surgery or radiation therapy, age-adjusted prostate cancer death rates have fallen steadily. Although this trend might indicate a beneficial impact of prostate cancer screening and/or early prostate cancer treatment on prostate cancer mortality, mass screening of the general population for prostate cancer remains controversial.2 One challenge for prostate cancer screening is the prevalence of the disease in the United States: autopsy series have revealed small prostate cancers in as many as 29% of men between ages 30 and 40 and 64% of men between ages 60 and 70.3 Obviously, not all of these men are at risk for symptomatic or life-threatening prostate cancer progression. In fact, many such men, if diagnosed with prostate cancer, may be at greater risk for treatment-associated morbidity.
Currently, for U.S. men, the lifetime risk of a diagnosis of prostate cancer is about 1 in 6, whereas the lifetime risk of death from prostate cancer is on the order of 1 in 35.1 Over the past two decades, treatment approaches for men with prostate cancer have changed dramatically, with improvement in established prostate cancer treatments and the introduction of new prostate cancer treatment approaches. Now, men diagnosed with prostate cancer often face a bewildering array of treatment choices. Clearly, the physicians that care for these men must weigh the risks of prostate cancer progression against the potential for side effects from treatment, in the context of other health risks and life choices, to best use the current collection of treatments for the greatest benefit.
Prostate Anatomy and Function
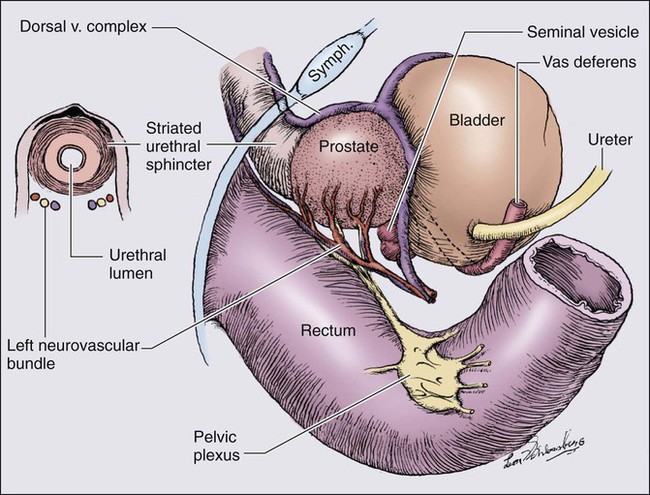
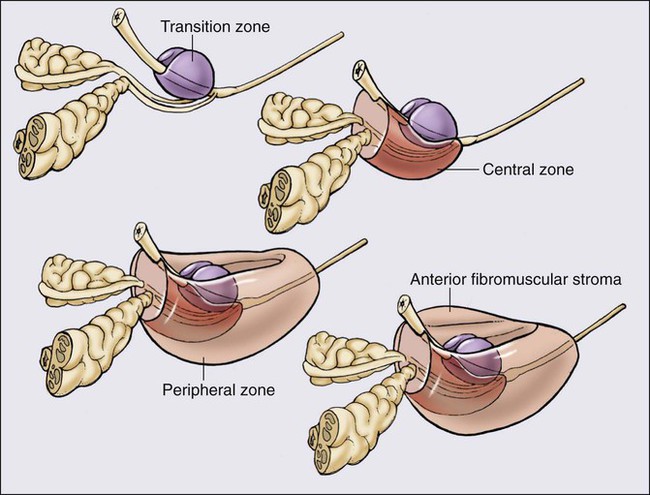
The prostate is a male sex accessory gland that surrounds the urethra and contributes secretions to the ejaculate (Figure 84-1). Located in the pelvis, the prostate sits adjacent to the bladder and rectum, is surrounded incompletely by a thin capsule composed of collagen, elastin, and smooth muscle, and at the apex of the gland, forms part of the urethral sphincter apparatus.4 Nerves to the corpora cavernosa of the penis, needed for penile erection, travel through fascia along the posterolateral surface of the prostate. These nerves can be recognized as a neurovascular bundle by urologists and preserved during radical prostatectomy to minimize sexual dysfunction postoperatively.5,6 The prostate parenchyma has been divided into three zones that can be seen by transrectal ultrasonography, and recognized readily by surgical pathologists examining radical prostatectomy specimens: a central zone, surrounding the ejaculatory ducts and accounting for some 25% of the prostate; a transition zone, near the prostatic urethra with 10% of prostate tissue normally; and a peripheral zone, with the bulk of prostate tissue encompassing the posterolateral region of the prostate (Figure 84-2).7,8
The prostate requires androgenic hormones, and an intact androgen receptor, for normal growth and development. In the prostate, the major circulating androgenic hormone, testosterone, produced by Leydig cells in the testes upon stimulation by luteinizing hormone (LH), is converted by 5α-reductase (nicotinamide-adenine dinucleotide phosphate-dependent δ4-3-ketosteroid 5α-oxidoreductase) to 5α-dihydrotestosterone (DHT).9 DHT, a more potent androgen than testosterone, binds to intracellular androgen receptors, alters androgen receptor conformation to promote dissociation from chaperone proteins, triggers androgen receptor dimerization and transport into the cell nucleus, and activates the expression of selected target genes.10 Stereotypically, androgen receptor target genes are characterized by the presence of androgen response element (ARE) DNA sequences within the transcriptional regulatory region, permitting direct binding and trans-activation by the androgen receptor.11 For genes like KLK3 (encoding PSA), which are activated by the androgen receptor selectively in prostate cells, and not in cells of other tissues, the transcriptional regulatory region also contains additional DNA sequences (prostate-specific enhancer or PSE) conferring prostate-specific expression.12
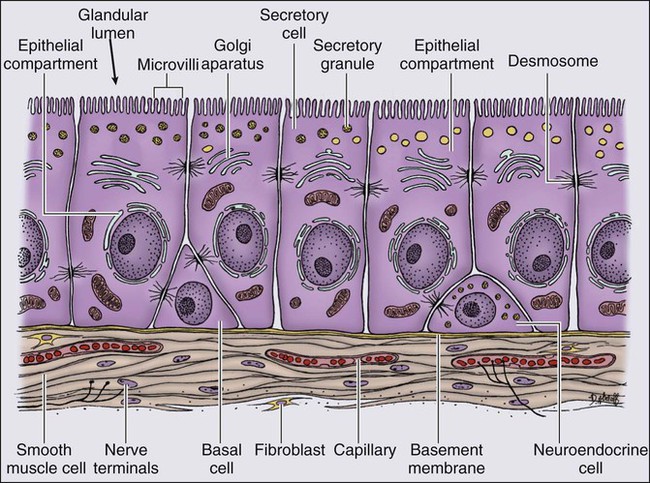
The normal prostate epithelium is composed of basal epithelial cells, characterized by the expression of cytokeratins K5 and K14, and p63, columnar secretory epithelial cells, which express the androgen receptor, PSA, cytokeratins K8 and K18, prostate-specific membrane antigen (PSMA), and prostate-specific acid phosphatase (PAP); and rare neuroendocrine cells, that secrete chromogranin A, neuron-specific enolase, and synaptophysin (Figure 84-3).13 The basal epithelial cell compartment likely contains pluripotent prostatic stem cells, capable of self-renewal proliferation and of differentiation. In contrast, columnar secretory cells, specialized to produce secretions for the ejaculate, are terminally differentiated, particularly under the influence of androgenic hormones. The prostate epithelium is in turn supported by a stroma containing fibroblasts, smooth muscle cells, nerves, and blood vessels. Stromal cells, which also express the androgen receptor, secrete polypeptide growth factors, such as keratinocyte growth factor (KGF), that contribute to the regulation of epithelial homeostasis via a paracrine signaling mechanism.14,15 Abnormal stromal–epithelial interactions, with disordered regulation of epithelial cell proliferation and differentiation, may contribute to the pathogenesis of both prostate cancer and BPH.16
Prostate cancer cells, and PIN cells, arise from the prostatic epithelium. Even though transformed, such cells typically retain many of the phenotypic attributes characteristic of differentiated columnar secretory cells, including the expression of androgen receptor, PSA, PSMA, and PAP. Prostate cancers reminiscent of basal epithelial cells are exceptionally rare; prostate cancers with features of neuroendocrine cells are somewhat more common.17 However, unlike normal columnar epithelial cells, neoplastic prostate epithelial cells are capable of proliferation. This has led to the concept that the target cell for neoplastic transformation in the prostate may be an “intermediate” cell, in transit from a basal epithelial stem cell to a differentiated columnar secretory epithelial cell, with properties of both stem cells and differentiated cells.18,19 Another feature of neoplastic prostate epithelial cells, as compared with normal basal or columnar secretory cells, is that the neoplastic cells appear to use androgen receptor signaling not only for differentiation, but also for proliferation, as most prostate cancer cells tend, at least initially, to display some dependence on androgens for maintenance of growth and survival. Somatic fusions between an androgen-regulated gene, TMPRSS2, at chromosome 21q22, and genes encoding members of the ETS family of transcription factors, commonly found in prostate cancers, may provide a mechanistic explanation by which androgen signaling can promote prostate cancer cell growth.20,21 Ultimately, in life-threatening prostate cancer, prostate cancer cells escape from the prostate gland, proliferate in lymph nodes, in bones, and in other organs and become less and less dependent on androgenic hormones.
Genetics and Epidemiology
Genetic Predisposition to Prostate Cancer
Familial clusters of prostate cancer have been recognized since at least 1956, when Morganti et al. reported that men with prostate cancer were more likely to have relatives with prostate cancer than men without a prostate cancer diagnosis.22 In a study conducted more than three decades later, when detailed family histories were collected from men with prostate cancer and their spouses, the men with prostate cancer were more likely to have a brother or father with prostate cancer.23 Twin studies, comparing the tendency for concordant prostate cancer development between monozygotic twins, sharing all of their genes, and dizygotic twins, sharing half of their genes, have also hinted at a significant contribution of hereditary to prostate cancer: in a study of 44,788 pairs of twins in Sweden, Denmark, and Finland,24 42% of the prostate cancer cases (with a 95% confidence interval of 29% to 50%) were attributed to heredity. In principle, familial clustering of prostate cancer cases could be a result of inherited susceptibility genes, shared exposure to carcinogenic stresses, or to some sort of detection or diagnosis bias (e.g., the brother of a man diagnosed with prostate cancer may be more likely to pursue screening for prostate cancer). To discover the genetic contributions to prostate cancer in spite of these potential sources of bias, both linkage analyses of genetic loci in high-risk prostate cancer families and genomewide association studies (GWAS) of prostate cancer susceptibility in large populations have been conducted. Each approach has generated a number of candidate genes or gene regions (as many as 30 or more), with several in common, confirming the contribution of heredity to prostate cancer risk but underscoring the complexity of inherited prostate cancer susceptibility.25
Among the growing number of prostate cancer susceptibility genes discovered by mapping studies are RNASEL and MSR1.26,27 RNASEL encodes a latent endoribonuclease component of an interferon-inducible 2′,5′-oligoadenylate-dependent RNA decay pathway that functions to degrade viral and cellular RNA upon viral infection.28 MSR1 encodes subunits of a trimeric class A macrophage scavenger receptor capable of binding bacterial lipopolysaccharide and lipoteichoic acid, and oxidized high and low density serum lipoproteins (oxidized HDL and LDL).29 For both RNASEL and MSR1, not only have mutations been linked to prostate cancer susceptibility in families but variant alleles have been predicted to account for as many as 13% and 3% of sporadic prostate cancer cases, respectively.27,30,31 The identification of RNASEL and MSR1 as candidate prostate cancer susceptibility genes has intensified interest in the possibility that infection and/or inflammation might contribute to the pathogenesis of human prostate cancer. In mice, targeted disruption of RnaseL leads to diminished interferon-α activity and increased susceptibility to viral infection,32 whereas targeted disruption of Msr-A leads to increased vulnerability to infection with Listeria monocytogenes, Staphylococcus aureus, Escherichia coli, and Herpes simplex virus type 1.29,33–35 Further genetic support for this etiologic mechanism has come from analyses of common variants of other genes encoding participants in host inflammatory responses, including TLR4 and other members of toll-like receptor signaling pathways, MIC-1, IL1-RN, and COX-2, have also been associated with prostate cancer risk.36–40
An increased risk for prostate cancer has long been known for men in breast cancer families carrying BRCA2 mutations, characterized by disease with an aggressive natural history arising before age 55 years.41 Nonetheless, a role for BRCA2 genotyping in general prostate cancer practice has not been established. However, as evidence has accumulated that several additional germline DNA sequence variants may be associated with prostate cancer, the possibility that genetic testing might be used to aid in prostate cancer screening, detection, diagnosis, or risk stratification has emerged. In one analysis, five such sequence variants, three single nucleotide polymorphisms (SNPs) at 8q24 and one each at 17q12 and 17q24.3, were found to have a marked association with prostate cancer, especially for men with a family history of the disease, showing a 9.46-fold increased risk (with 95% confidence interval of 3.62 to 24.72) for a prostate cancer diagnosis.42
Epidemiology of Prostate Cancer
Accumulated epidemiological evidence implicates the environment as the major contributor to the development of most prostate cancers. Prostate cancer incidence and mortality display wide geographic variation, with high rates of prostate cancer incidence and mortality in the United States and Western Europe, and low prostate cancer risk more characteristic of Asia.43 African Americans in the United States have very high prostate cancer risk.44 The geographic variation in prostate cancer incidence and mortality can best be explained by lifestyle influences, because Asian immigrants to North America typically adopt a higher prostate cancer risk.47–47
The key aspect of lifestyle in the United States most likely responsible for high prostate cancer incidence and mortality is the diet, generally rich in animal fats and meats and poor in fruits and vegetables. In the Health Professions Follow-up Study, a prospective cohort study involving 51,529 men, total fat intake, animal fat intake, and consumption of red meats were associated with increased risks of prostate cancer development.48 Red meat consumption was similarly correlated with prostate cancer risks in the Physicians Health Study49 and in a large cohort study in Hawaii.50 The cooking of red meats at high temperatures, or on charcoal grills, is known to lead to the formation of both heterocyclic aromatic amine and polycyclic aromatic hydrocarbon carcinogens.51,52 Ingestion of 2-amino-1-methyl-6-phenylimidazopyridine (PhIP), one of the heterocyclic amine carcinogens that appear in “well-done” red meats, leads to prostate cancer in rats.53 Consumption of dairy products also appears to increase prostate cancer risk, an effect that may be more attributable to calcium intake than to dietary fat or protein.54 In contrast, adequate consumption of vegetables and antioxidant micronutrients is accompanied by reduced prostate cancer risk. Consumption of tomatoes, which contain lycopene, and of cruciferous vegetables, which contain sulforaphane, may protect against prostate cancer development.55,56 Antioxidant micronutrients, such as vitamin E and selenium, may reduce prostate cancer risk only when correcting dietary deficiencies: a large trial (SELECT) of supplementation with vitamin E and selenium to prevent prostate cancer failed to show a benefit.57
Prostate Inflammation and Prostate Cancer
Chronic or recurrent inflammation is known to play a causative role in the development of many human cancers, including cancers of the liver, esophagus, stomach, large intestine, and bladder. Inflammatory changes have been recognized in prostate tissues for many years, leading to speculation that inflammation might contribute in some way to prostate cancer development.58 However, over the past few years, evidence has accumulated in support of a more critical role for prostatic inflammation in the pathogenesis of prostate cancer. Inflammatory changes are present in almost all radical prostatectomy specimens from men with prostate cancer. Because inflammation in the prostate is not usually associated with symptoms, the prevalence of prostate inflammation is not known, and the association with prostate cancer has been difficult to test.59,60 A syndrome of irritative voiding symptoms and pelvic pain, perhaps attributable to inflammation near the prostatic urethra, is reported by some 9% or more of men between 40 and 79 years of age, with as many as 50% of such men suffering more than one episode by age 80 years.61 Most episodes of symptomatic prostatitis are not clearly attributable to specific infectious agents. Even so, sexually transmitted infections do appear to increase prostate cancer risk.62,63 Nonetheless, if prostate infection and inflammation lead to prostate cancer, the mechanism does not appear likely to involve direct transformation of prostate epithelial cells by microbial DNA. Instead, the production of microbicidal oxidants by inflammatory cells, such as superoxide, nitric oxide, and peroxynitrite, may promote prostate cancer development by triggering cell and genome damage.64,65 Increased production of oxidants by inflammatory cells in the prostate may be why decreased prostate cancer risk has been associated with intake of a variety of antioxidants or of nonsteroidal antiinflammatory drugs, and why RNASEL and MSR1, two of the prostate cancer susceptibility genes identified thus far, encode proteins that function in host responses to infections.
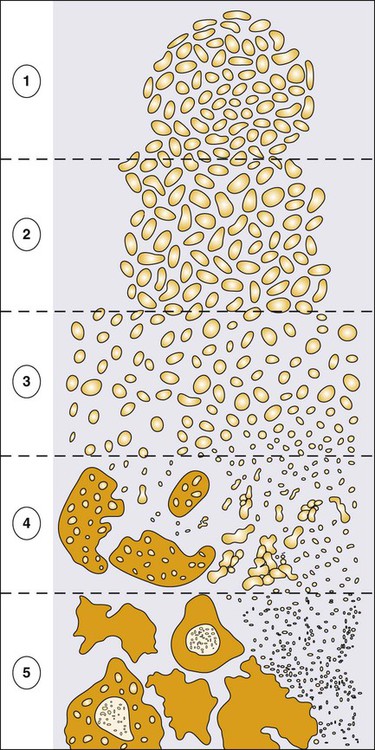
Despite these provocative hints, the contribution of prostate inflammation to prostatic carcinogenesis has been difficult to assess. However, in 1999, De Marzo et al. provided the most compelling linkage of prostate inflammation to prostate cancer by proposing that a prostate lesion, termed proliferative inflammatory atrophy (PIA), might be a precursor to PIN and to prostate cancer (Figure 84-4).66 Areas of the prostate, containing epithelial cells that do not fully differentiate into columnar secretory cells, have long been recognized as focal atrophy lesions by prostate pathologists.58,67 The term PIA has been used to describe those focal atrophy lesions that contain proliferating epithelial cells, are associated with chronic inflammation, and are often located adjacent to PIN lesions and/or prostate cancers.68 The epithelial cells in PIA lesions typically express high levels of stress-response polypeptides such as GSTP1, GSTA1, and cyclooxygenase 2 (COX-2). Loss of GSTP1 expression in rare PIA lesions, attributable to de novo GSTP1 CpG island hypermethylation, may be what leads to the development of PIN and prostate cancer.69
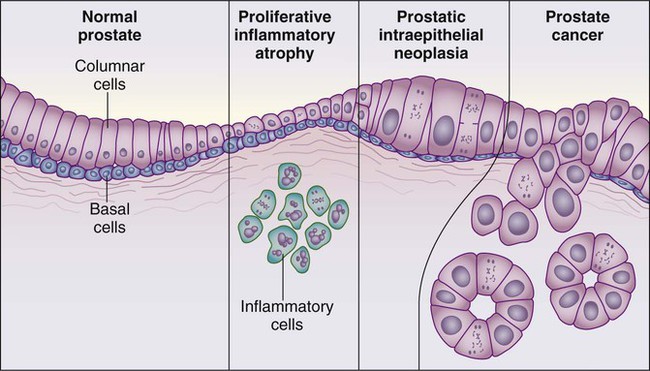
Etiological and Biological Characteristics
Somatic Genome Alterations in Prostate Cancer Cells
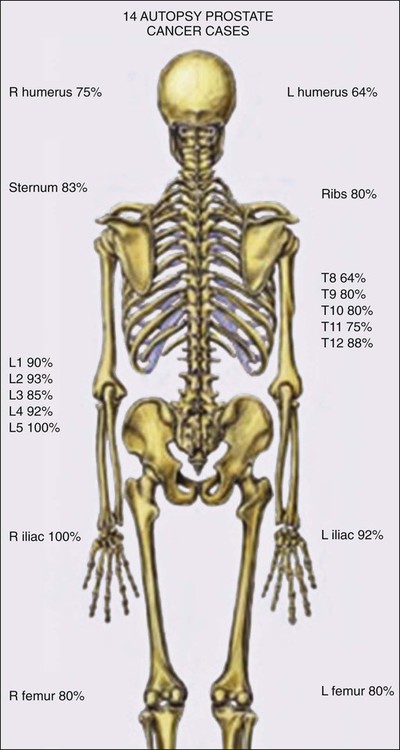
Prostate cancer cells typically contain a plethora of somatic genome alterations, including gene mutations, gene deletions, gene amplifications, chromosomal rearrangements, and changes in DNA methylation (Figure 84-5). In the United States, prostate cancer diagnoses are typically made in men between 60 and 70 years of age, whereas small prostate cancers have been detected at autopsy in nearly 30% of men between 30 and 40 years of age.3 Thus, the somatic genome changes present in prostate cancers have often accumulated over many decades. The acquisition of somatic genome changes in the prostate may be influenced by lifestyle as well: although small prostate cancers have been detected at autopsy in men from geographic regions with low prostate cancer mortality, these small prostate cancers are usually only present in much older men.72–72 Also, in the United States, prostates removed at radical prostatectomy for prostate cancer usually contain more than one prostate cancer lesion (Figure 84-6).
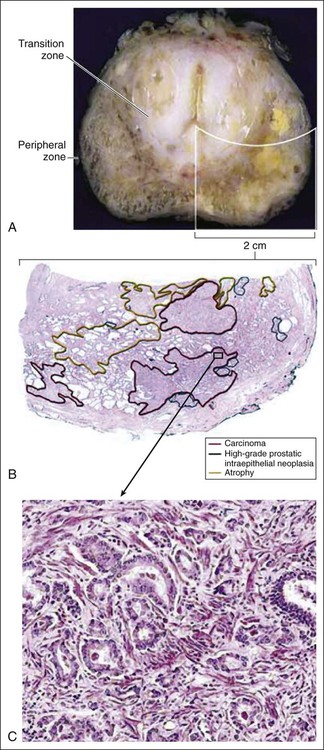
Over the years, several techniques have been used to catalog genome accidents in prostate cancer cells, including karyotyping, fluorescence in situ hybridization (FISH), comparative genome hybridization, loss of heterozygosity analyses, and genomewide microarray and/or sequencing approaches. In one such study, each prostate cancer case exhibited a mean of 3866 base mutations (range 3192 to 5865), 20 nonsilent coding sequence mutations (range 13 to 43), and 108 rearrangements (range 43 to 213).73 In another, DNA hypermethylation was found at 5408 regions of the genome, with 73% of the regions near genes (5′, 3′, or intron–exon junctions), and 27% of the regions at conserved intergenic sites.74 Often, these analyses reveal different chromosomal abnormalities in different cancer cases, in different cancer lesions in the same cancer case, and in different areas within the same cancer lesion. The propensity to develop such a heterogeneous collection of somatic genome lesions over so many years, and in a manner so sensitive to environment and lifestyle, suggests strongly that prostate cancers likely arise as a consequence of either chronic or recurrent exposure to genome-damaging stresses, defective protection against genome damage, or some combination of both processes. The resultant genomic instability may be the reason some prostate cancers progress to threaten life.75,76
One characteristic somatic genome alteration, a rearrangement, drives the production of fusion transcripts between an androgen-regulated gene, TMPRSS2, at chromosome 21q22, and members of the ETS family of transcription factors (Figure 84-7).20 Fusion partners for TMPRSS2 include ERG (also at chromosome 21q22), ETV1 (at chromosome 7p21), and ETV4 (at chromosome 17q21).20,77,78 The gene rearrangements may result from a mishap during the androgen receptor transcriptional trans-activation of TMPRSS2 in which tangling or untangling of DNA by TOP2B triggers DNA double-strand breaks that recombine with ETS family partner genes via nonhomologous end-joining (Figure 84-8).79,80 The appearance of the resultant fusion transcripts provides a plausible mechanism for the dependence of prostate cancer cells on androgenic hormones for growth and survival, as the expression of ETS family transcription factors can be stimulated by androgen action. TMPRSS2–ERG fusions have been detected in ~60% of prostate cancers and in >20% of PIN lesions.81 ERG is highly expressed by many prostate cancers, and not at all by others, though neither ERG expression nor the presence of ERG fusion transcripts appears to have great prognostic significance.81–84 Nonetheless, the presence of TMPRSS2-ERG fusion transcripts does seem to define a molecular subset of prostate cancer. Rarer somatic alterations, including SPOP mutations (6% to 15%) and SPINK1 overexpression (~10%), are restricted to cases devoid of TMPRSS2-ETS family rearrangement rearrangements.85,86

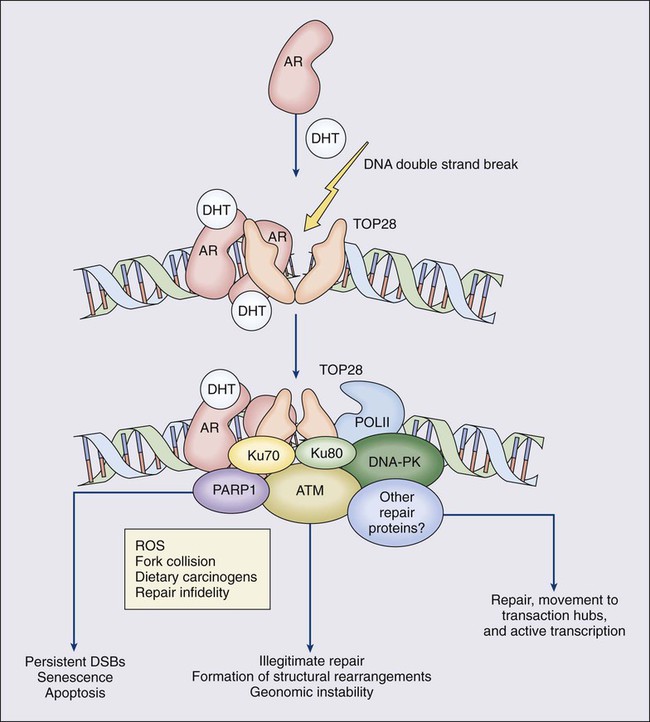
Hypermethylation of CpG island sequences encompassing the regulatory region of GSTP1, encoding the π–class glutathione S-transferase (GST) is the most common somatic genome change yet reported for prostate cancer.87,88 GSTs catalyze the detoxification of carcinogens, and of other reactive chemical species, via conjugation with the intracellular scavenger glutathione. In mice, targeted disruption of π-class GST genes leads to increased skin tumors after treatment with the carcinogen 7,12-dimethylbenzanthracene (DMBA).90 Similarly, human prostate cancer cells devoid of GSTP1 appear especially vulnerable to genome damage mediated by exposure to N-OH-PhIP, the charred meat carcinogen that causes prostate cancer when fed to rats, and by exposure to oxidant stresses.91 In the normal prostate epithelium, GSTP1 is present at high levels in basal cells, and in lower levels in columnar secretory cells, though the enzyme can be induced in columnar epithelial cells subjected to genome-damaging stresses. In contrast, the enzyme is almost never present in prostate cancer cells. In nearly all cases, the absence of GSTP1 expression in prostate cancer cells can be attributed to hypermethylation of GSTP1 CpG island sequences, a somatic genome change that prevents GSTP1 transcription. Absence of GSTP1 expression and GSTP1 CpG island hypermethylation may also be characteristic of cells comprising PIN lesions, thought to be precursors to prostate cancer.92 The mechanism by which hypermethylated GSTP1 CpG island alleles arise during prostatic carcinogenesis remains to be elucidated. Nonetheless, prostate cells carrying inactivated GSTP1 genes appear to enjoy some sort of selective growth advantage early during the development of prostate cancer.
NKX3.1 encodes a prostate-specific homeobox gene essential for normal prostate development that may be a target for somatic loss on chromosome 8p21.93 NKX3.1 has been shown to bind DNA and to repress PSA expression via interactions with ETS transcription factors.94,95 Mice carrying one or two disrupted Nkx3.1 alleles manifest prostatic epithelial hyperplasia and dysplasia.96,97 In men, loss of 8p21 DNA sequences occurs early during prostatic carcinogenesis, with 63% of PIN lesions and >90% of prostate cancers, showing loss of heterozygosity at polymorphic 8p21 marker sequences in one report.98 However, although mapping studies have indicated that NKX3.1 lies within a common region of deletion, encompassing 2 megabases at 8p21, molecular pathology analyses have not yet established NKX3.1 as a somatic target for inactivation during prostatic carcinogenesis because somatic NKX3.1 mutations have not been identified. Nonetheless, loss of NKX3.1 expression does appear to accompany prostate cancer progression.
PTEN, a tumor suppressor gene encoding a phosphatase active against both proteins and lipid substrates, appears to be a common target for somatic alteration during prostate cancer progression.99–106 PTEN is an inhibitor of the phosphatidylinositol 3′-kinase/protein kinase B (PI3K/Akt) signaling pathway needed for cell cycle progression and cell survival. Although PTEN is expressed by normal prostate epithelial cells, and by cells present in PIN lesions, the expression of PTEN is often diminished in prostate cancers, with many prostate cancers containing collections of neoplastic cells with no PTEN.107 PTEN defects have been found in a wide variety of cancers and cancer cell lines.101 For prostate cancer, a number of somatic PTEN alterations have been reported, including homozygous deletions, loss of heterozygosity, mutations, and probable CpG island hypermethylation. However, despite common losses of 10q sequences near PTEN in prostate cancers, somatic mutations at the remaining PTEN alleles are not as frequent. In a study of prostate cancer metastases recovered at autopsy, somatic PTEN alterations were even more common than in primary prostate cancers, and a significant heterogeneity in PTEN defects in different metastatic deposits from the same patient was also evident.106 Haploinsufficiency for PTEN may contribute to the phenotype of transformed cells in the prostate. Pten+/– mice display prostatic hyperplasia and dysplasia, and crosses of Pten+/– mice with Nkx3.1+/– mice have revealed that Pten+/–Nkx3.1+/– mice and Pten+/–Nkx3.1–/– mice develop lesions reminiscent of human PIN.110–110
Defective regulation of p27, a cyclin-dependent kinase inhibitor encoded by CDKN1B, may also accompany prostatic carcinogenesis.111,112 In PIN cells and prostate cancer cells, p27 levels are almost always diminished, though the mechanism(s) for the reduction in p27 levels appear complex: somatic loss of DNA CDKN1B sequences at 12p12-13 have been reported for only 23% of localized prostate cancers, 30% of prostate cancer lymph node metastases, and 47% of distant prostate cancer metastases.113 In place of CDKN1B gene alterations, p27 polypeptide levels may be lowered indirectly by inadequate PTEN repression of the PI3K/Akt signaling pathway.116–116 In this way, low p27 levels may be as much a result of loss of PTEN function as of CDKN1B alterations. The critical contribution of PTEN to epithelial growth regulation in the prostate is evident in mice, where disruption of Cdkn1b alleles leads to prostatic hyperplasia, and Pten+/–Cdkn1b–/– mice develop prostate cancer by 3 months of age.111,117
Metastatic prostate cancer is almost always treated with androgen deprivation, antiandrogens, or a combination of androgen deprivation and antiandrogens.118,119 However, despite such treatment, androgen-independent prostate cancer cells eventually emerge and progress to threaten life. Curiously, in these cells, androgen receptor expression and androgen receptor signaling remain intact despite the absence of androgens.120,121 Somatic alterations of AR have been reported for many prostate cancers, especially for androgen-independent prostate cancers. AR amplification, accompanied by high-level expression of androgen receptors, may promote the growth of androgen-independent prostate cancer cells by increasing the sensitivity of the cells to low androgen levels.122 AR mutations, encoding androgen receptors with altered ligand specificity have also been detected; for some of the mutant androgen receptors, even antiandrogens can act as agonist ligands.125–125 When 44 mutant androgen receptors from prostate cancers were evaluated for transcriptional regulatory capabilities, 16% of the receptors had lost transcriptional activation activity, 45% of the receptors had gained some transcriptional regulatory ability, 32% of the receptors maintained some partial transcriptional modulatory activity, and the remaining 7% behaved like wild-type receptors.126 In addition to somatic AR gene changes, androgen-independent prostate cancer cells with wild-type androgen receptors may activate androgen receptor signaling even in the absence of androgens, via posttranslational modifications of the androgen receptor and/or androgen receptor coactivators in response to other growth factor signaling pathways.120,127–130
Changes in Gene Expression in Prostate Cancers
Alterations in gene expression in prostate cancers have been catalogued using cDNA microarray technologies.131–142 Among the many genes exhibiting over- or underexpression in prostate cancers, the products of at least two genes appear consistently increased, and the product of a third gene appears to become elevated during androgen-independent progression. Hepsin, located at 19q11-13.2, encodes a transmembrane serine protease, expressed at high levels in many normal tissues.143 Hepsin may contribute to prostate cancer progression: forced overexpression of hepsin in mouse prostates leads to disorganization of the epithelial basement membrane and increased metastasis.144 α-Methylacyl-CoA racemase (AMACR), a mitochondrial and peroxisomal enzyme that acts on pristanoyl-CoA and C27-bile acyl-CoA substrates to catalyze the conversion of R– to S-stereoisomers in order to permit metabolism by β-oxidation, has been reported to be overexpressed in almost all prostate cancers.145,146 Germline AMACR mutations lead to adult-onset neuropathy.147 Immunohistochemistry studies, which have revealed that AMACR is occasionally present in normal prostate cells, increased in PIN cells, and further elevated in prostate cancer cells, have prompted the use of antibodies against AMACR as tools for prostate cancer diagnosis by surgical pathologists.146,148 The polycomb protein enhancer of zeste homolog 2 (EZH2), a transcriptional regulatory protein, is elevated in metastatic androgen-independent prostate cancer.149 The mechanism by which EZH2 contributes to prostate cancer progression has not been established. However, elevated EZH2 expression in primary prostate cancers portends a poor prognosis.149
Telomere Shortening During Prostatic Carcinogenesis
Telomeres, containing repeat DNA sequences at the termini of chromosomes, protect against loss of chromosome sequences during genome replication. DNA ends tend to shorten each generation as a consequence of bidirectional DNA synthesis (the “end-replication” problem); the telomere repeat sequences serve as templates for the enzyme telomerase, which can extend the chromosome termini and maintain chromosome integrity through cell division.150 Growth dysregulation accompanying the development of most human cancers tends to lead to cell proliferation in the absence of telomerase, and to shortened chromosome telomeres.151 Critically shortened telomere sequences may promote genome instability by increasing illegitimate DNA recombination.152,153 Mice carrying disrupted genes needed for a functioning telomerase show increased numbers of cancers, especially when crossed to mice with defective p53 genes.154 In the prostate, short telomere repeat sequences appear characteristic of cells both in PIN lesions and in prostate cancer.157–157 At some point, most cancer cells activate the expression of telomerase, providing some maintenance of chromosome termini. Telomerase expression has been detected in prostate cancers, but not at high levels in normal prostate tissues or in BPH.155
Prostate Cancer Screening, Early Detection, and Prevention
Clinical Evaluation
Digital Rectal Examination
In men with early-stage prostate cancers, physical findings, if present, are usually limited to an abnormal DRE, used for both screening and staging. Palpable areas of induration, or asymmetric firmness of the gland, suggest the presence of prostate cancer, but these findings can also be caused by prostate inflammation (especially granulomatous prostatitis), by benign prostatic hyperplasia (BPH), and by prostatic stones. DRE has only fair reproducibility in the hands of experienced examiners.158 When used alone for detection of prostate cancer, DRE misses from 23% to 45% of the cancers that are subsequently detected by prostate biopsies done for serum PSA elevations or for transrectal ultrasound (TRUS) abnormalities.161–161 In addition, prostate cancers detected by DRE are at an advanced pathological stage in more than 50% of men.162,163
The positive predictive value of DRE (the fraction of men who have prostate cancer if the DRE is abnormal) ranged from 4% to 11% in men with PSA levels from 0.0 to 2.9 ng/mL, and from 33% to 83% in men with PSA levels of 3.0 to 9.9 ng/mL or more.164 When DRE and PSA are used in prostate cancer screening, detection rates are higher with PSA than with DRE and highest with both tests together.165 Furthermore, a DRE abnormality tends to be associated with the presence of high-grade cancer.166 Thus DRE and PSA are generally considered to be complementary tests and a prostate biopsy is usually recommended for men with an abnormality on DRE that is suspicious for prostate cancer regardless of the PSA level.
Serum PSA
PSA is a member of the human kallikrein gene family of serine proteases encoded by KLK3 located on chromosome 19.167 A component of the ejaculate, PSA is produced by columnar secretory cells in the prostate. PSA expression is regulated by androgens, becoming detectable in serum at puberty accompanying increases in luteinizing hormone and testosterone. In the absence of prostate cancer, serum PSA levels increase with age and prostate volume and are generally higher in African Americans. Cross-sectional population data suggest that the serum PSA increases 4% per milliliter of prostate volume, and that 30% and 5% of the variance in PSA can be accounted for by prostate volume and age, respectively.168
Serum PSA elevations likely occur as a result of disruption of the normal prostate architecture, permitting PSA to diffuse into the prostate parenchyma and gain access to the circulation. This can occur in the setting of both benign and malignant prostate diseases (prostatitis, BPH, and prostate cancer) and as a result of prostate manipulation (prostate massage and prostate biopsy).169 Although the presence of some type of prostate disease is the most important determinant driving elevation of the serum PSA, an increased serum PSA is not specific for prostate cancer. Furthermore, not all men with prostate disease have elevated serum PSA levels.
Treatments targeting the prostate gland (for BPH or for prostate cancer) can lower serum PSA by decreasing the number of prostatic epithelial cells capable of producing PSA, and by decreasing the amount of PSA produced by each cell. Modulation of sex steroid hormone levels for treatment of BPH or prostate cancer, radiation therapy for prostate cancer, and surgical ablation of prostate tissue for BPH or prostate cancer can all lead to decreases in serum PSA. 5α-Reductase inhibitors, like finasteride and dutasteride, lower PSA levels by 50% after 12 months of treatment.170 Thus for men treated with these agents for 12 months or more, the serum PSA level should be doubled to estimate the “true” PSA value. Interpretation of serum PSA values should always take into account the presence of prostate disease, previous diagnostic procedures, and prostate-targeted treatments.
Serum PSA and Prostate Cancer Detection
The serum PSA value, along with the findings at DRE, correlate directly with the risk of prostate cancer at biopsy (Table 84-1). Furthermore, the PSA level at a young age anticipates the future risk of being diagnosed with prostate cancer decades later (Figure 84-9). Gann et al. first showed that the risk of a prostate cancer diagnosis, including the diagnosis of life-threatening disease, incrementally and directly with PSA over the decade after a baseline measurement, even at low PSA levels (below 4.0 ng/mL); a finding that has now been confirmed by many others.173–173 These observations may allow a more targeted approach to prostate cancer screening using a baseline PSA value to direct screening intensity.174,175
Table 84-1
Positive Predictive Value of DRE and PSA in a Multicenter Screening Trial
| DRE | PSA | PPV (%) |
| Abnormal | Any | 21.4 |
| Any | >4 | 31.5 |
| 4–10 | 26.1 | |
| >10 | 52.9 | |
| Normal | >4 | 24.4 |
| Abnormal | <4 | 10.0 |
| 4–10 | 40.8 | |
| >10 | 69.1 |
DRE, Digital rectal examination; PPV, positive predictive value; PSA, prostate-specific antigen.
Data from Catalona WJ, Richie JP, Ahmann FR, et al. Comparison of digital rectal examination and serum prostate specific antigen in the early detection of prostate cancer: results of a multicenter clinical trial of 6,630 men. J Urol 1994;151:1283.
In the early years of PSA testing, most clinicians used PSA as a dichotomous test (i.e., the PSA was “elevated” or not) with elevations triggering a prostate biopsy. However, data from the placebo arm of the Prostate Cancer Prevention Trial (PCPT176), which accrued men with a serum PSA value <3.0 ng/mL, has suggested that there is no PSA cut-off level with both high sensitivity and high specificity for prostate cancer, and that virtually no level is low enough to exclude the presence of prostate cancer. Instead, serum PSA tests may be more appropriately thought of as measures of a continuum of prostate cancer risk, with the risk of cancer (and of high-grade cancer) rising directly with the PSA level, an observation first made by Gann et al. using a cohort population study design.171 In recent years, more emphasis has been placed on integrating several variables associated with the risk of prostate cancer (e.g., age, family history, race, DRE findings, prior biopsy results) to construct risk calculators for estimating the need for a prostate biopsy.177,178 The future will likely see further integration of genome and other molecular biomarker tools in prostate cancer screening algorithms.
PSA “Density”
The major source of serum PSA in men without prostate cancer is the transition zone (TZ) epithelium, not the epithelium of the peripheral zone.179 Because benign prostate enlargement is a growth of the transition zone tissue, and because serum PSA levels are largely a reflection of transition zone volume in men with benign prostate enlargement, adjusting the serum PSA for either prostate volume (PSA density) or more specifically, the transition zone volume (PSA-TZ), has been shown to improve the distinction between those with prostate cancer and those with benign prostate enlargement.180–183 PSA density is also directly associated with the presence of high-grade cancer on prostate biopsy, and is therefore useful for identifying those men less likely to have high-grade cancer most appropriate for active surveillance.184
PSA “Velocity”
Carter et al., using frozen serum samples from an aging study cohort, demonstrated that a PSA “velocity” (PSAV; the rate of change of serum PSA in ng/mL per year upon repeated testing) of 0.75 ng/mL per year or greater had a specificity of 90% for distinguishing men with prostate cancer in the setting of benign prostatic enlargement, and a specificity of 100% for distinguishing men with prostate cancer in the absence of benign prostatic enlargement when PSA levels were in the range of 4.0 to 10.0 ng/mL (Figure 84-9).185
The PSAV tends to be higher in men with high-grade and high-stage prostate cancer as compared to men with lower grade and stage disease.186,187 Furthermore, men with a PSAV above 2.0 ng/mL per year in the year before a diagnosis of prostate cancer appear to be at an increased risk of prostate cancer death after surgical intervention when compared with men with a PSA velocity of 2.0 ng/mL per year or less.188 For PSAV measured 10 to 15 years before prostate cancer diagnosis (when most men had PSA levels below 4.0 ng/mL), a PSAV less than 0.35 ng/mL per year has been associated with a prostate cancer–specific survival 25 years later of 92%, compared with 54% with a PSAV greater than 0.35 ng/mL per year.189 There is also a direct relationship between the number of times the PSAV exceeds a given threshold (e.g., 0.4 ng/mL per year), a concept known as “risk count,” and the risk of high-grade cancer.190,191 Thus a continuously increasing serum PSA should raise the suspicion of high-grade prostate cancer if there is no other explanation for the rise.
Molecular Forms of PSA
PSA in the bloodstream circulates in both bound and unbound forms. Most of the detectable PSA in the serum (65% to 90%) is bound to α1-antichymotrypsin (“complexed” PSA or cPSA, whereas the rest (10% to 35%) remains unbound (“free” PSA or fPSA).167 The serum PSA tests in common use for prostate cancer detection and monitoring effectively measure the sum of fPSA and cPSA, providing a determination of the “total” serum PSA (tPSA). Assays that can distinguish fPSA and cPSA have been developed and approved by the U.S. FDA specifically for use in the early detection of prostate cancer. In general, men with prostate cancer have a greater fraction of serum tPSA bound to α1-antichymotrypsin (cPSA), and a commensurately lower fraction of tPSA that is unbound (fPSA), than men without prostate cancer. This difference is thought to be due to the differential expression of PSA isoforms by cells in the transition zone (the zone of origin for BPH) tissue as compared with peripheral zone (the zone where most prostate cancers arise) tissue.
The %fPSA appears most useful in distinguishing men with and without prostate cancer in the setting of tPSA levels between 2 and 10 ng/mL.192 An fPSA of 25% and a PSA density of 0.078 were shown to have comparable specificity for prostate cancer diagnosis at biopsy (at a sensitivity of 95%), but %fPSA does not require a TRUS for determination.193 Most urologists use %fPSA determinations for decisions about the need for a repeat biopsy in a man with a persistently elevated serum PSA and previous negative prostate biopsies, where the possibility of a missed prostate cancer may be a concern. New assays for cPSA exhibit comparable specificity and sensitivity for prostate cancer diagnosis as %fPSA, with the potential advantage of using one assay for cPSA versus two assays to determine the %fPSA (measuring both tPSA and the %fPSA).
Benign PSA (BPSA), a degraded isoform of fPSA, is preferentially found in nodular tissue involved in benign prostatic enlargement and has been shown to be elevated among men with benign prostatic hyperplasia.194 A second isoform, proPSA, is an inactive precursor of PSA that contains differing leader sequences of amino acids. When compared to men without prostate cancer, the tissues and serum of prostate cancer patients have an increased proportion of proPSA that is fPSA, a difference that has been used to improve the discrimination of men with and without prostate cancer.195 A Beckman Coulter “Prostate Health Index” (PHI) uses proPSA, fPSA, and tPSA in a formula (proPSA/fPSA) × (tPSA)1/2 that was shown to improve prostate cancer detection over total and %fPSA.196 Additionally, there is some evidence that proPSA is associated with a more aggressive prostate cancer phenotype.197
Urine Biomarkers of Prostate Cancer
PCA-3, a noncoding prostate-specific RNA, was found to be overexpressed in prostate cancer tissue compared with benign tissue.198,199 Urine assays for PCA3 have been developed that appear to improve the specificity of prostate cancer detection in response to an abnormal serum PSA,200 When compared with %fPSA, a PCA3 score (the ratio of PCA3 RNA to PSA mRNA × 1000) has been shown to be a superior predictor of prostate cancer on a repeat biopsy when an initial biopsy was negative.201 Although PCA3 may have a higher specificity when used for prostate cancer detection, urine testing for PCA3 exhibits a lower sensitivity when compared with serum PSA testing.
A number of other urinary molecular biomarkers have been evaluated for prostate cancer detection, including hypermethylated GSTP1 genes, AMACR proteins, and TMPRSS2-ERG fusion mRNA transcripts. In a multicenter evaluation of urinary DNA methylation markers, including GSTP1, the negative predictive value of the urine test for prostate cancer diagnosis was 87% for men with a PSA of 4.1 to 10.0 ng/mL.202 Although such DNA methylation urine tests might spare men unnecessary repeat biopsies, the addition of TMPRSS2-ERG mRNA urine testing (expected to be present in half of men with prostate cancer) to PCA3 RNA urine testing may provide a positive predictive value as high as 95%, steering men who harbor prostate cancer despite a previous negative biopsy toward a definitive diagnosis.199,203 The discovery and development of new molecular biomarkers is progressing very quickly; in the future, such biomarkers (or biomarker panels) will need to be incorporated into algorithms along with standard measures of risk (e.g., age, family history, race) to selectively identify men who should undergo further evaluation for the presence of prostate cancer.204
TRUS-Guided Prostate Biopsy
TRUS is not an accurate method for localizing early prostate cancer and is not recommended for use in prostate cancer screening. The primary role of TRUS in prostate cancer detection and diagnosis is to ensure accurate sampling of prostate tissue by prostate biopsies in men suspected of harboring cancer based on serum PSA levels and DRE.205 This is best accomplished by targeting peripheral zone lesions that appear hypoechoic by TRUS for biopsy, along with performing systematic sampling biopsies of areas without hypoechoic lesions in the prostate periphery.
TRUS-guided prostate biopsies are performed routinely with an 18-guage needle fired from a spring-loaded gun through a port mounted on the TRUS probe. Most commonly, in preparation for a biopsy procedure, men are administered a fluoroquinolone antibiotic and given a cleansing enema. Injection of a local anesthetic around the periphery of the prostate is used by most urologists to reduce discomfort associated with prostate biopsy. Major complications, such as bleeding and/or infection requiring hospitalization, are rare, although hematuria and hematospermia are common sequelae of the procedure. More recently, a rise in fluoroquinolone-resistant E. coli strains have been implicated in higher rates of hospitalization for postbiopsy sepsis,206 leading to more liberal use of targeted antibiotic prophylaxis, based on rectal swab cultures done prior to a biopsy.207
The optimal biopsy technique, including the number and placement of biopsies for tissue procurement that will minimize the chance of missing a relevant cancer remains controversial. Nonetheless, the best evidence available suggests that biopsies placed more laterally within the peripheral zone of the prostate may be important to exclude prostate cancer in men with elevated serum PSA values and a nonsuspicious DRE. Magnetic resonance imaging (MRI) may have a role in improving the specificity of prostate cancer detection and in identification of sites within the prostate for directing prostate biopsies.208 At this point, it is not clear whether this use of MRI will improve health outcomes or reduce (vs. increase) the costs of care.
Screening for Prostate Cancer
Opportunistic PSA-based screening has been widespread in the United States since the early 1990s. It has been estimated that 45% to 70% of the 30% decline in prostate cancer mortality that occurred in the 1990s could be attributed to the stage migration and earlier treatment of prostate cancer associated with PSA testing.209 Ecological studies of prostate cancer mortality comparing countries with different rates of PSA uptake, suggest that the detection of more aggressive disease with PSA testing could explain mortality differences.210
Two randomized trials of prostate cancer screening reported different results in 2009.211,212 In the Prostate, Lung, Colon, and Ovary (PLCO) trial, no difference in mortality was found between the screening and control groups, whereas in the European Randomized Study of Screening for Prostate Cancer (ERSPC), there was a 21% reduction in prostate cancer deaths in men randomized to screening at a median follow-up of 9 years. However, the absolute reduction in deaths for men screened was only 0.71 per 1000 men, with 48 men diagnosed with prostate cancer and treated to prevent one death, raising a concern about overdiagnosis and overtreatment even despite the mortality reduction. Updated reports from both trials have shown few changes with longer follow-up.213,214 With these conflicting data, as might be expected, champions of prostate cancer screening dismiss the results of the PLCO trial because of high contamination rates in the control arm, high rates of prescreening in the population prior to initiation of the trial, and low rates of follow-up for positive screening tests. Nevertheless, the findings of the PLCO trial do hint that in the setting of ongoing widespread screening as is seen in the United States, further intensification of screening would not likely improve health outcomes.215
The worry about overdiagnosis and overtreatment of prostate cancer in the United States has led many to reconsider the value of serum PSA testing for prostate cancer screening. A draft recommendation on prostate cancer screening from the U.S. Preventive Services Task Force (USPSTF) was released in the fall of 2011 recommending against prostate cancer screening for men of all ages, regardless of race or family history.216 These USPSTF guidelines may prompt an increase in shared decision making between patients and physicians prior to pursuing prostate cancer screening,217 a more nuanced and targeted approach to screening for those who desire to pursue it,175 and an individualized approach to prostate cancer treatment that includes active surveillance if a diagnosis of prostate cancer is made rather than the rush to curative intervention for all.218
Prostate Cancer Prevention
The high lifetime risks of prostate cancer development, the morbidities associated with treatment of established prostate cancer, and the inability to eradicate life-threatening metastatic prostate cancer offer compelling reasons for prostate cancer prevention. In addition, epidemiological data, indicating a dominant role for lifestyle factors in prostate cancer development, suggest that prostate cancer risk modification may be feasible, if only through lifestyle modification. Also, because prostatic carcinogenesis takes many decades, there may be a broad window of opportunity to change lifestyle in an effort to retard prostate cancer development. Clearly, although the specific lifestyle factors fostering prostate cancer development have not been conclusively identified, it is likely that consumption of a diet rich in fruits, vegetables, and antioxidant micronutrients, and poor in saturated fats and “well-done” red meats, may significantly reduce risks of prostate cancer development, and of the development of other diseases characteristic of life in the developed world. As the etiology of prostate cancer is even better understood, new opportunities for prostate cancer prevention may also arise. For example, if prostate inflammation contributes to prostate cancer development, antiinflammatory drugs might be considered candidate prostate cancer prevention drugs.219 For drugs to be developed and tested for prostate cancer prevention, randomized clinical trials, capable of assessing both drug safety and drug efficacy, will be required.220 Ideally, such trials can be targeted at men with a high risk for prostate cancer development.
Thus far, two classes of agents, 5α-reductase inhibitors and antioxidant micronutrients, have been subjected to large randomized clinical trials; neither class of agents has shown a convincing prostate cancer prevention benefit. In the Prostate Cancer Prevention Trial (PCPT), the propensity for the 5α-reductase inhibitor finasteride to reduce the prevalence of prostate cancer in healthy men age 55 years and older when given for 7 years was tested.221 For the trial, men (n = 18,882) with a PSA of 3.0 ng/mL or less and a normal DRE were randomized to treatment with finasteride (5 mg/d) or to placebo.221 While on study, men with a PSA elevation or an abnormal rectal examination were subjected to prostate biopsy; in addition, at the end of the treatment period, a prostate biopsy was planned for all of the men in the trial. Prostate cancer was detected in 18.4% of the men treated with finasteride versus 24.4% of men receiving placebo (P < 0.001).221 However, high-grade prostate cancers appeared more commonly associated with finasteride treatment than with placebo (6.4% vs. 5.1%).221 Similarly, in the REDUCE trial, men (n = 6729) with an elevated serum PSA (2.5 to 10.0 ng/mL) and a recent negative prostate biopsy were randomized to receive the 5α-reductase inhibitor dutasteride (0.5 mg/d) or placebo.222 For this trial, men underwent prostate biopsies after 2 and 4 years of treatment. With a result reminiscent of PCPT, overall prostate cancer detection was reduced from 25.1% to 19.9% for men receiving dutasteride, but high-grade prostate cancer, which was similar in the first 2 years of the trial for men treated with dutasteride versus placebo, was increased in dutasteride-treated men in the last 2 years of the trial from <0.1% to 0.5%. The findings of PCPT and REDUCE may mean that 5α-reductase inhibitors prevent or treat low-grade cancers better than high-grade cancers. These mixed results, a reduction in overall prostate cancer prevalence but an increase in high-grade prostate cancers, make prescribing 5α-reductase inhibitors to healthy men for the purpose of preventing prostate cancer very problematic, an opinion shared by the FDA, which has not approved either drug for such an indication.223
Epidemiological studies have provided compelling evidence that intake of selenium and of vitamin E might diminish prostate cancer risks, especially in the setting of inadequate dietary consumption.224–230 As a result, a prospective, randomized, placebo-controlled clinical trial of selenium and vitamin E (SELECT; n = 35,533) was undertaken to test the ability of the antioxidant micronutrients to prevent prostate cancer.231 Selenium (200 µg selenomethionine), α–tocopherol (400 mg), the combination of selenium and α–tocopherol, or neither, were given to men randomized to four different treatment groups using a 2 × 2 factorial design, for 7 to 12 years.231 The men studied were age 55 years or older (50 years or older for African Americans) with an unremarkable DRE and a serum PSA of 4.0 ng/mL or less.231 Unfortunately, the trial failed to show any reduction in prostate cancer and showed a slight increase in prostate cancer incidence among men taking α–tocopherol.57,232 When this result is considered in the context of the findings from epidemiology studies and previous smaller clinical trials, the data tend to suggest that men with low blood levels of the antioxidant micronutrients, who tend to be at the highest risk for prostate cancer development, might be the only men with any chance for benefit from supplementation.225,226,230,233 For this reason, correction of antioxidant micronutrient deficiencies might ultimately prove more generally safe and effective than widespread supplementation (and its risk of oversupplementation), which may carry a threat of harm with little benefit.
Pathology and Pathways of Spread
Histopathology of Prostate Cancer
Microscopic analysis of prostate tissue by a surgical pathologist is needed for the diagnosis of prostate cancer, for determining prostate cancer stage after prostatectomy, and for histologic grading, via the assignment of a Gleason score, to predict the behavior of prostate cancer. The majority of prostate cancers are adenocarcinomas, though other types of cancers can appear. Most often, prostate cancer diagnoses are made using core-needle biopsy specimens, which sample small amounts of prostate tissue. For many prostate cancer cases, needle biopsies contain only small numbers of prostate cancer cells among more plentiful noncancerous glands. Also, several prostate conditions, including acute, chronic, or granulomatous prostate inflammation, epithelial atrophy, and PIN, exhibit histologic features that mimic some of those present in prostate cancers.234 Thus, prostate cancers can be difficult to recognize in needle biopsy specimens, and difficult to distinguish from other prostate abnormalities. Experienced prostate pathologists use a combination of architectural, cytologic, and ancillary findings to make a diagnosis of prostate cancer on needle biopsy.236–236 In addition, because normal prostate glands, but not glands present in prostate cancers, contain basal epithelial cells, immunohistochemical staining for basal epithelial cell markers, such as cytokeratins K5 and K14 and the nuclear marker p63, can be used to help distinguish benign from malignant glands in prostate tissue samples. Immunohistochemical staining for AMACR, a prostate cancer biomarker discovered through cDNA microarray transcriptome profiling, can aid in prostate cancer diagnosis.146,148,237 Neither of these immunohistochemistry reagents perfectly distinguishes prostate cancer: the absence of basal epithelial cell markers is not always diagnostic of prostate cancer, and AMACR expression is absent in some prostate cancers and present in PIN.234 For all of these reasons, second-opinion interpretations of prostate biopsy findings, especially when foci of atypical glands suspicious for cancer are identified, are often helpful.
High-grade PIN (HGPIN), a lesion characterized by the proliferation of malignant-appearing prostate epithelial cells within the confines of otherwise normal glandular structures, is identified in about 5% of men subjected to prostate biopsies.238,239 The evidence that HGPIN is a likely precursor to prostate cancer includes the findings that (1) HGPIN is more commonly present in prostates that also contain prostate cancer, (2) HGPIN and prostate cancer both tend to arise in the peripheral zone of the prostate and are often directly contiguous, and (iii) HGPIN and prostate cancer express similar biomarkers and share many somatic genome abnormalities.240,241 The notion that HGPIN lesions might be prostate cancer precursors has stimulated interest in possibly treating men with HGPIN to prevent prostate cancer.241 Unfortunately, the natural history of individual HGPIN lesions is not known. Furthermore, HGPIN lesions, which can only be recognized by sampling prostate biopsies, are not easily monitored. These limitations have hindered the use of HGPIN as a response surrogate for cancer prevention drug development. Because HGPIN is not currently treated, the major significance of the finding of HGPIN in a prostate biopsy in the apparent absence of prostate cancer is that prostate cancer may have been missed by the prostate sampling strategy used for the biopsy procedure. On repeat biopsy of men with HGPIN on initial biopsy, the cancer detection rate was 20% among men who underwent standard extended biopsy sampling (14 cores or less), and 31% among men who underwent a “saturation” biopsy sampling (20 biopsy cores or more).242 The National Comprehensive Cancer Network (NCCN) recommends a repeat biopsy within 1 year for men with HGPIN if the lesion was multifocal (2 cores or more involved) and/or the initial biopsy was not performed using an extended approach (more than 6 cores).243
Of interest, unlike prostate cancer, prostate inflammation, or BPH, HGPIN lesions are not thought to perturb prostate architecture enough to elevate the serum PSA. Recently, increasing attention has been afforded the notion that PIA (proliferative inflammatory atrophy) lesions might be precursors to PIN and/or prostate cancer.244 Like PIN, PIA lesions tend to arise in the peripheral zone of the prostate, where prostate cancers arise, and some PIA cells acquire somatic genome alterations reminiscent of prostate cancer cells.66,245 At present, men with PIA lesions are not subjected to any kind of treatment, and presence of PIA on an initial prostate biopsy is not thought to predict the detection of prostate cancer on repeat biopsy. The major significance of PIA to the diagnosis of prostate cancer by prostate biopsy may be propensity for such lesions to occasionally exhibit features that mimic prostate cancer.234
The most frequently used approach to histologic grading of prostate cancer is the application of Gleason scoring.246 The Gleason grade refers to architectural prostate cancer patterns, numbered 1 (well differentiated) to 5 (poorly differentiated). Because prostate cancers are often heterogeneous, Gleason scoring (sometimes referred to as the “combined” Gleason grade) is accomplished by adding the Gleason grade of the most abundant pattern to the Gleason grade of the second most abundant pattern (e.g., a Gleason score of 4 + 3 = 7). The Gleason score, when applied by an expert pathologist, is one of the best tools available for predicting cancer-specific outcomes; the higher the Gleason score the greater the risk of cancer progression with or without treatment.247 Of note, because Gleason scoring applies pattern grades to the architecture of cancer within the prostate, metastatic prostate cancers detected by biopsies of metastatic deposits are not assigned a Gleason score. Gleason grading practices have evolved over time. As part of a 2005 International Society of Urological Pathology (ISUP) Consensus Conference, a revised approach was formulated and adopted, subtly changing the definitions of Gleason patterns 3 and 4 and improving the interrater reproducibility of Gleason grading.248 With the newer ISUP grading strategy, a Gleason score of 3 + 3 = 6 can be expected to have a very low risk of prostate cancer recurrence after primary therapy.
Life-Threatening Prostate Cancer Progression
Prostate cancer progression has long been understood to involve metastases to bones, which are often painful, and to lymph nodes. The predilection of the disease for bones may reflect a hospitable microenvironment capable of providing growth factor support in a collaboratively malignant milieu (Figure 84-10).249 In turn, prostate cancer cells perturb bone homeostasis in such a way as to increase osteoblast activity, which can be readily discerned by bone scanning. This activity forms the basis for bone scan imaging, such as with 99mTc-methylene diphosphonate (MDP) and 18F-sodium fluoride (NaF), in which osteoblastic activity can be visualized throughout the entire skeleton.250 Reportedly, NaF positron emission tomography/computed tomography (NaF PET/CT), in which osteoblastic activity is coregistered with bone radiography, offers the highest sensitivity and specificity for bone metastasis.251
More recent autopsy studies have provided newer insights into the phenotype(s) of lethal prostate cancer. In addition to expected bone and lymph node metastases, men dying of prostate cancer tend to exhibit common metastases to liver (64%), the dura (43%), and elsewhere.252 The most troubling feature of lethal prostate cancer may be its heterogeneity. In different metastases from the same case, the expression of androgen receptor, and its target PSA, appears markedly variable, present in the majority of cells in some lesions but mostly absent for others.253 This likely reflects ongoing genome and epigenome instability. Genomewide assessment of somatic genetic and epigenetic defects in lethal prostate cancers at autopsy show that although disseminated metastatic disease emerges from a single clone, new defects continue to appear in individual metastases.254 Of interest, DNA hypermethylation changes tend to be maintained in all metastatic lesions in most cases, at least as much as any of the genetic alterations, whereas DNA hypomethylation, leading to activation of repressed embryonic genes, continues to evolve variably lesion to lesion.255 Of course, the greatest worry about ongoing genomic, epigenomic, and phenotypic heterogeneity in metastatic prostate cancer is the likelihood that treatment resistance will almost certainly emerge, regardless of the agent used.
Clinical Manifestations/Patient Evaluation/Staging
Evaluation of the Extent of Prostate Cancer
The extent of prostate cancer is correlated with clinical tumor stage (Table 84-2), Gleason score (the sum of two Gleason grades; Figure 84-11), and serum PSA level.256 Nomograms, incorporating clinical stage (estimated using DRE findings), the serum PSA level, and the Gleason score, have been shown to be capable of predicting both pathological prostate cancer extent determined on radical prostatectomy specimens and the long-term outcome following primary tumor treatment.259–259 The most commonly used risk stratification scheme today is that of D’Amico et al., who suggested that men with prostate cancer can be stratified into low risk (stages T1c to 2a, and serum PSA less than 10 ng/mL, and Gleason score of 6 or less), intermediate risk (stage T2b, or serum PSA between 10 and 20 ng/mL, or Gleason score of 7), and high risk (stage T2c, or serum PSA greater than 20 ng/mL, or Gleason score of 8 or greater) groups. The fraction of men free of prostate cancer 10 years after radical prostatectomy is significantly different for the risk categories: 83% of men with low-risk prostate cancer, 46% of men with intermediate-risk prostate cancer, and 29% of men with high-risk prostate cancer.259
Table 84-2
TNM Staging for Prostate Cancer (from http://www.cancerstaging.org)
| PRIMARY TUMOR (T) | |
| CLINICAL | |
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Clinically inapparent tumor neither palpable nor visible by imaging |
| T1a | Tumor incidental histologic finding in 5% or less of tissue resected |
| T1b | Tumor incidental histologic finding in more than 5% of tissue resected |
| T1c | Tumor identified by needle biopsy (e.g., because of elevated PSA) |
| T2 | Tumor confined within prostate1 |
| T2a | Tumor involves one-half of one lobe or less |
| T2b | Tumor involves more than one-half of one lobe but not both lobes |
| T2c | Tumor involves both lobes |
| T3 | Tumor extends through the prostate capsule2 |
| T3a | Extracapsular extension (unilateral or bilateral) |
| T3b | Tumor invades seminal vesicle(s) |
| T4 | Tumor is fixed or invades adjacent structures other than seminal vesicles, such as external sphincter, rectum, bladder, levator muscles, and/or pelvic wall |
| PATHOLOGICAL (PT)3 | |
| pT2 | Organ confined |
| pT2a | Unilateral, one-half of one side or less |
| pT2b | Unilateral, involving more than one-half of side but not both sides |
| pT2c | Bilateral knee |
| pT3 | Extraprostatic extension |
| pT3a | Extraprostatic extension or microscopic invasion of bladder neck4 |
| pT3b | Seminal vesicle invasion |
| REGIONAL LYMPH NODES (N) | |
| CLINICAL | |
| NX | Regional lymph nodes were not assessed |
| N0 | No regional lymph node metastasis |
| N1 | Metastases in regional lymph node(s) |
| PATHOLOGIC | |
| pNX | Regional nodes not sampled |
| pN0 | No positive regional nodes |
| pN1 | Metastases in regional node(s) |
| DISTANT METASTASIS (M)5 | |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
| M1a | Nonregional lymph node(s) |
| M1b | Bone(s) |
| M1c | Other site(s) with or without bone disease |
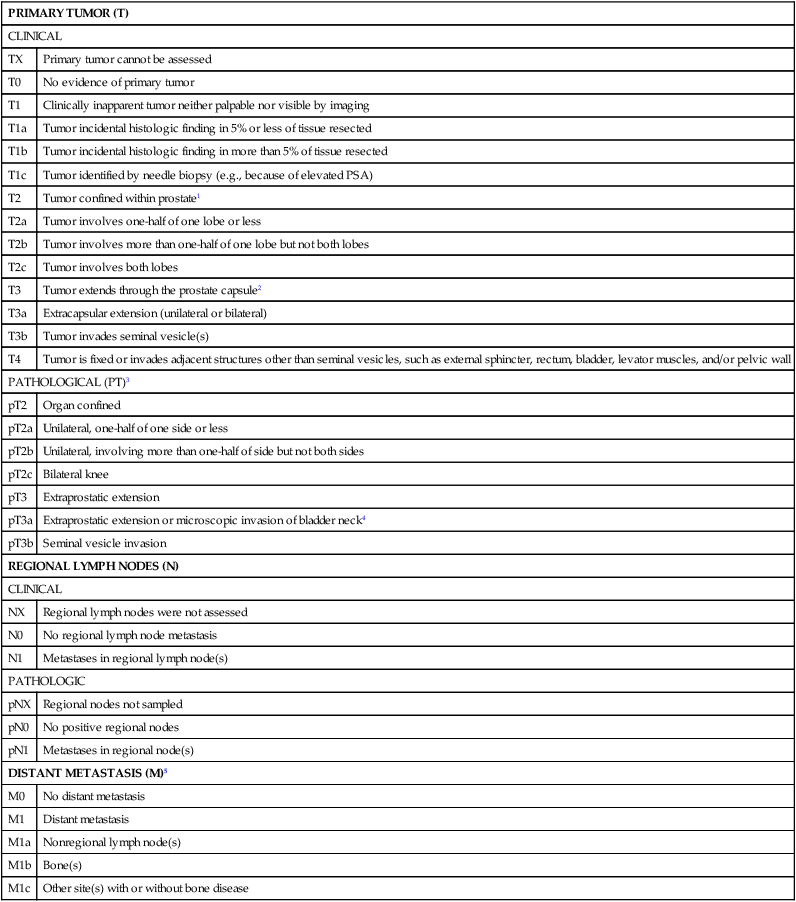
1Tumor found in one or both lobes by needle biopsy, but not palpable or reliably visible by imaging, is classified as T1c.
2Invasion into the prostatic apex or into (but not beyond) the prostatic capsule is classified not as T3 but as T2.
3There is no pathological T1 classification.
4Positive surgical margin should be indicated by an R1 descriptor (residual microscopic disease).
5When more than one site of metastasis is present, the most advanced category is used. pM1c is most advanced.
A number of pretreatment tools are available for prediction of the pathological extent of tumors and the probability of biochemical recurrence.260,261 When used before initiating prostate cancer treatment, risk stratification of men with prostate cancer aids in counseling such men about the expected outcome of prostate cancer treatment, providing estimates of the chance that local treatments might be curative.
Radiographic Imaging
Although CT scanning is used routinely by radiation oncologists for prostate cancer treatment planning, no imaging technique available today has been proven to add additional useful information when used to evaluate the extent of prostate cancer in men with low- and intermediate-risk disease.262 TRUS and MRI give the most accurate definition of prostatic architecture and anatomy, but current imaging technologies do not provide very precise assessments of cancer extent within the prostate or the presence of microscopic foci of prostate cancer that have escaped the confines of the prostate gland. Radionuclide bone scans detect metastatic prostate cancer in less than 1% of men with a serum PSA value less than or equal to 20 ng/mL and are not recommended for the initial evaluation of men with low- or intermediate-risk prostate cancer.263 PET has not yet been found to be useful in the evaluation of men with prostate cancer and has no place in the prostate cancer staging.264 New imaging technologies, including three-dimensional color Doppler, contrast-enhanced color Doppler, magnetic resonance spectroscopy, and high-resolution MRI with magnetic nanoparticles, have great potential for improving the assessment of local and distant prostate cancer extent.262,265,266 Cross-sectional imaging of the pelvis, by CT scan or MRI, for the purpose of detecting lymph node metastases, and radionuclide bone scans for the detection of bony metastases, should be reserved for men with high-risk prostate cancer.
111In-capromab pendetide, a radioimmunoconjugate featuring a monoclonal antibody to an intracellular domain of prostate-specific membrane antigen was approved by the FDA for use in the evaluation of men with clinically localized prostate cancer but is rarely used today for assessment of prostate cancer extent, even for men with high-risk prostate cancer, in large part because of low scan sensitivity and frequent difficulties in scan interpretation. There may be a role for 111In-capromab pendetide imaging with single photon emission computed tomography used in coregistration with computed tomography (SPECT/CT) for prognostic staging.267 New nuclear medicine agents capable of detecting PSMA appear promising in early development.250 Perhaps, one of these tools may overcome the inadequacies of 111In-capromab.
Serum Biomarker Assays and “Molecular” Classification for Prognosis
The serum PSA level at the time of a prostate cancer diagnosis is correlated with both intermediate and long-term outcomes including tumor volume, stage, grade, and freedom from relapse after treatment.268 However, the serum PSA cannot be used alone to predict disease extent for an individual patient. Instead, PSA values are more typically used as part of multivariable models for predicting the extent of disease and the probability that a cancer will behave in an aggressive manner.269,270 The NCCN recommends the use of the D’Amico classification scheme using serum PSA, clinical stage, and biopsy Gleason score to stratify men into low-, intermediate-, and high-risk groups for selecting management strategies and for counseling patients about prognosis.271 Other PSA forms used in the detection and diagnosis of prostate cancer, such as %fPSA, proPSA, and so forth, are not part of commonly employed risk stratification and prognosis tools.
“Molecular” classification of tumor cells for prognosis refers to the detection of circulating prostate cancer cells and/or cell fragments, either indirectly, by identifying mRNA species, like those encoding PSA or PSMA, characteristically expressed by epithelial cells from the prostate,272 or directly, by recovering “circulating” prostate cancer cells by a variety of isolation methods.275–275 To detect prostate lineage mRNAs or prostate cancer DNAs, polymerase chain reaction (PCR) approaches, capable of astonishing sensitivity, are typically used. Circulating tumor cells (CTCs) appear early in the course of prostate cancer, though simple enumeration of the cells does not provide clear prognostic information for those with localized cancers.276,277 Further, the presence of CTCs at diagnosis does not accurately predict the development of metastatic disease posttreatment.
Primary Therapy
Selection of Treatment Approach
Men thought to have localized prostate cancer face a number of management options, including observational strategies (watchful waiting, active surveillance), radical prostatectomy, interstitial brachytherapy, and external beam radiation therapy. Furthermore, cryotherapy, high-intensity focused ultrasound (HIFU), and focal therapy are also being presented to some patients as treatment alternatives despite limited long-term data to support their use. A number of issues should be considered by patients and physicians before deciding on a management strategy. These include the specific side effect profile of different treatments, the possibility that certain comorbid conditions might increase the severity of side effects or the risk of complications, the characteristics of the cancer and the risk of progression without treatment, patient comorbidities that might limit the need for treatment, and individual patient preferences.278
The NCCN guidelines for the management of clinically localized prostate cancer reflect the needed consideration of both life expectancy and the risk profile of the individual patient’s cancer in making management recommendations (http://www.nccn.org; see Table 84-3).271
Table 84-3
Management Options for Localized Prostate Cancer by Risk Profile and Life Expectancy
| Risk Profile | Criteria | Management Option |
| Very low | Stage T1c, and Gleason score ≤6, and PSA <10 ng/mL, and <3 biopsy cores with cancer, ≤50% involvement of any core with cancer, PSA density <0.15 ng/mL/cm3 | Active surveillance if life expectancy <20 years |
| Low | Stage T1c or T2a, and Gleason score ≤6, and PSA <10 ng/mL | Active surveillance if life expectancy <10 years; radiation (external beam or brachytherapy) or surgery if life expectancy ≥10 years |
| Intermediate | Stage T2b or T2c, or Gleason score 7, or PSA 10–20 ng/mL | Active surveillance or external radiation with/without brachytherapy or surgery if life expectancy <10 years Surgery or external radiation with/without ADT with/without brachytherapy if life expectancy ≥10 years |
| High | Stage T3a, or Gleason score 8–10, or PSA >20 ng/mL | External radiation + ADT or surgery |
ADT, androgen deprivation therapy.
Adapted from Carter HB. Management of low (favourable)-risk prostate cancer. BJU Int 2011;108:1684-1695.
Observational Strategies
Watchful waiting was a common strategy in the pre-PSA era when most cancers were detected at an incurable stage and the morbidity of primary treatments was high. The primary intent of watchful waiting was to avoid treatment until symptoms emerged and non-curative palliative treatment might be necessary.279 In contrast, active surveillance, a more modern approach, involves the selective delayed curative treatment of men found to have disease progression while being carefully monitored. Active surveillance implies a much more diligent monitoring regimen than watchful waiting.
The Scandinavian Prostate Cancer Group Study 4 (SPCG-4), a trial conducted before the PSA screening had been adopted, randomized men (n = 695) with localized prostate cancer to surgery or watchful waiting (Figure 84-12).280 Although these men would not have been considered optimal candidates for active surveillance by current criteria, as most of the men had palpable disease by DRE, surgery was accompanied by a 40% reduction in prostate cancer deaths for men with 15 men needing treatment to prevent a single prostate cancer death. Of note, the benefits of surgery were restricted to men under the age of 65 years. The Prostate cancer Intervention Versus Observation Trial (PIVOT), a study conducted in the setting of common PSA screening, randomized men (n = 731) with a mean age of 67 years to surgery or watchful waiting.281 The distribution of prostate cancer cases among the study subjects included 43% low-risk, 36% intermediate-risk, and 20% high-risk disease. These data, which have been presented but not yet published, suggest that through 12 years of follow-up there has been no difference in cancer-specific survival attributable to treatment. Thus older men with low-risk, screen-detected, prostate cancers may not derive great benefits from radical prostatectomy.
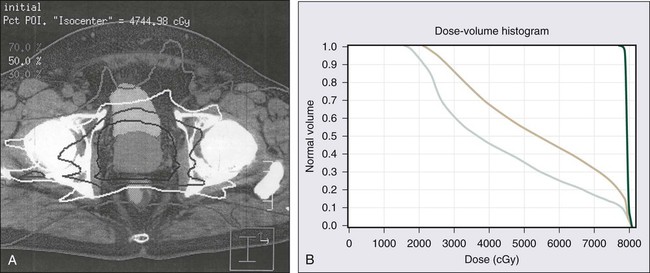
Active surveillance seeks to individualize prostate cancer care primarily among men thought to harbor low-grade prostate cancers who would otherwise be fit for curative treatment. The intent is to intervene early with curative therapy if the disease should progress,279 while sparing men with more indolent prostate cancer the side effects of aggressive primary therapy.282,283 This management approach, which relies on diligent monitoring for signs of disease progression, has been called a variety of names, including “expectant management with curative intent” and others, but active surveillance is the terminology most often used today.270,278,284
The rationale for active surveillance has been derived from medical evidence reflecting a variety of reported studies, including competing risk analyses, surgical series, nonrandomized cohort studies, and randomized trials. Taken together, the findings indicate that using a time horizon of 10 to 15 years, less than 3% of men diagnosed with well-differentiated tumors (Gleason score 6 or below) classified as low-risk (based on a serum PSA <10.0 ng/mL and tumor stage of T2a or less) will die of prostate cancer whether treated or not.281,285–288 Such data clearly call into question the need for treating men with low-risk disease and a life expectancy of less than 10 to 15 years. Unfortunately, curative intervention for favorable-risk prostate cancer appears to be undertaken as commonly as curative intervention for higher-risk disease.291–291 This is of concern because at least half of newly diagnosed men with prostate cancer have favorable risk disease, and 80% to 90% of these men undergo some form of treatment even when the age at diagnosis is above 75 years. Overtreatment of nonthreatening prostate cancer probably reflects the combination of a fear of harm from cancer, of litigation if aggressive treatment is not recommended, and of misaligned incentives that favor treatment in spite of evidence that nonintervention may be the most rational option.
Despite these biases driving overtreatment of favorable-risk prostate cancer, there has been a growing interest in active surveillance approaches as evidenced by the number of academic centers reporting outcomes from single-arm cohort studies.184,218,270,278,284 As of yet, there is no standard approach to selection of men for active surveillance, nor are there established guidelines for monitoring regimens, or for criteria to be used as triggers for curative intervention, for men managed by active surveillance. The “ideal” candidate may be a man with favorable-risk prostate cancer (very low-risk to low-risk), a life expectancy less than 15 years, and a personal desire to avoid the side effects of prostate cancer treatments. Typical monitoring strategies tend to involve DRE and serum PSA determinations at 3- to 6-month intervals, with repeated prostate biopsies at intervals of 1 to 4 years. The most common intervention triggers are a rising serum PSA and an increase in Gleason score of cancer seen on repeat biopsy, a finding that may result from undersampling by the initial biopsy or from “dedifferentiation” of a low-grade cancer. When managed in this way, approximately 25% to 50% of men will undergo curative intervention within 5 to 10 years of surveillance. Of the men proceeding to curative treatment, 25% will do so because of personal preference and without any trigger, 30% to 40% will do so because of a higher Gleason score, and 30% to 40% will do so because of an inexorably rising serum PSA.270,284 In one active surveillance series for men with prostate cancer (30% intermediate-risk disease and 70% with favorable-risk disease), the 10-year prostate cancer actuarial survival was 97%.292 An ongoing trial, the Prostate Testing for Cancer and Treatment (ProtecT) trial, which started in the United Kingdom in 2001, has randomized men with prostate cancer, ages 50 to 69 years to conformal radiotherapy, radical prostatectomy, or active surveillance.293 Results from this trial may further inform the difficult decision that men with localized prostate cancer face in choosing between active surveillance and curative intervention.
Radical Prostatectomy
Radical prostatectomy is most often recommended for treatment of men with clinically localized prostate cancer who have a life expectancy of at least 10 years or more (Table 84-3). Although there are not specific or universally accepted age limits for radical prostatectomy, increasing age is associated with a lower likelihood of receiving surgical treatment for prostate cancer. For example, among men with low-risk or intermediate-risk disease, 3 in 4 at ages 56 to 65 years, 2 in 5 at ages 66 to 75, and 1 to 2 in 10 at age >75 years undergo surgical intervention for prostate cancer.294
Radical prostatectomy was not commonly recommended for localized prostate cancer prior to the 1980s because of blood loss, and associated complications of incontinence and erectile dysfunction. But by 1990, surgery was the most commonly chosen option for management of prostate cancer.295 The increasing rates of radical prostatectomy relative to other management options was in large part due to the description of an anatomic approach to radical retropubic prostatectomy (RRP) that dramatically decreased operative morbidity, to the widespread use of PSA testing that led to an earlier diagnosis of disease, and to the ease of detection of prostate cancer with TRUS-directed prostate biopsies.6
Radical prostatectomy can be performed under regional or general anesthesia using a perineal, retropubic, or laparoscopic approach. Radical perineal prostatectomy is not a commonly performed procedure today; rather, the most common approach to removal of the prostate for treatment of prostate cancer is robot-assisted laparoscopic radical prostatectomy (RALRP). With RALP, the robotic system (a console connected to robotic arms) translates the movement of the surgeon’s fingers into movement of robotic arms attached to the laparoscopic instruments used to perform the surgery. Claims of advantages of RALP over other approaches have been widely touted by surgeons, hospitals, and the device manufacturer, resulting in increased usage of surgery overall, especially in older men.296 However, there is no evidence for improved disease-free or functional outcomes with RALP compared to RRP.299–299 Instead, both disease-free and quality-of-life outcomes after radical prostatectomy depend more on the experience of the surgeon than on the surgical approach used.300 With any surgical approach, prior pelvic surgery or radiation therapy tends to be associated with an increased risk of surgical complications.
Hemorrhage and injury to surrounding structures (blood vessels, obturator nerve, ureter, and rectum) are the most common intraoperative complications of radical prostatectomy. In the immediate postoperative period, complications can include deep venous thrombosis and pulmonary emboli, urine anastomotic leak, and postoperative bleeding. The operative mortality rate (death within 30 to 60 days) after radical prostatectomy is from 0.4% to 1.6%, depending on age and comorbidity.301 Following surgery, most men spend 1 to 2 nights in the hospital and have an indwelling urinary catheter for 1 to 2 weeks to allow healing of the vesicourethral anastomosis. After catheter removal, the most common long-term complications of surgery are urinary incontinence as a result of intrinsic sphincter deficiency, and erectile dysfunction from injury to the cavernous nerves innervating the corporal bodies of the penis, both of which negatively impact quality of life.
Urinary Function after Radical Prostatectomy
Urinary incontinence rates after radical prostatectomy vary greatly in different reports: from 5% to as high as 50%, with as many as 5% to 10% of men undergoing another procedure for incontinence after radical prostatectomy.297 Some of the variation in reported incontinence rates may be attributable to differences in definitions of incontinence (stress incontinence vs. more severe difficulties with urinary control), in the time after surgery when urinary continence was assessed (urinary control can improve over as long as a year following surgery), in whether incontinence was reported by treating surgeons in case series or by patients in survey questionnaires, and in the age of the patients undergoing surgery (older age is associated with higher incontinence rates). In the best case series, as many as 95% of men are completely dry 2 years after radical prostatectomy, and as many as 98% of men report no significant urinary problems.302,303 Using validated questionnaires to assess urinary incontinence and bother, Sanda et al.304 found urinary incontinence (any pad use) two years after treatment in 20% of men postsurgery, with a moderate-to-severe urinary problem in 7% (Table 84-4). Most men after radical prostatectomy have some degree of stress incontinence for a period of time that usually resolves within the first postoperative year, after which incontinence is not likely to improve further.
Table 84-4
Percentage of Men’s Quality-of-Life Concerns 24 Months after Treatment of Localized Prostate Cancer
| Quality-of-Life Domain | Prostatectomy | External Beam Radiation Therapy | Brachytherapy | |
| Urinary irritation or obstruction | Dysuria | <1 | 1 | 5 |
| Hematuria | 0 | <1 | 1 | |
| Weak stream | 4 | 10 | 11 | |
| Frequency | 10 | 14 | 20 | |
| Urinary incontinence | Leaking >1 time per day | 14 | 7 | 10 |
| Frequent dribbling | 5 | 2 | 3 | |
| Any pad use | 20 | 5 | 8 | |
| Leaking problem | 8 | 5 | 6 | |
| OVERALL URINARY PROBLEM | 7 | 11 | 16 | |
| Bowel function | Urgency | 2 | 16 | 9 |
| Frequency | <1 | 10 | 7 | |
| Fecal incontinence | <1 | 2 | 5 | |
| Bloody stools | <1 | 5 | 3 | |
| Rectal pain | 2 | 4 | 4 | |
| OVERALL BOWEL PROBLEM | 1 | 11 | 8 | |
| Sexual function | Poor erections | 58 | 60 | 48 |
| Difficulty with orgasm | 42 | 50 | 38 | |
| Erections not firm | 64 | 66 | 54 | |
| Erections not reliable | 51 | 51 | 40 | |
| Poor sexual function | 53 | 58 | 43 | |
| OVERALL SEXUALITY PROBLEM | 43 | 37 | 29 | |
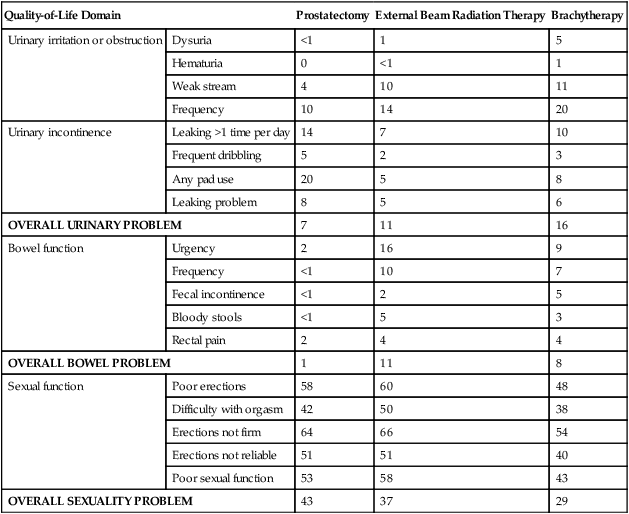
Adapted from Sanda MG et al. Quality of life and satisfaction with outcome among prostate-cancer survivors. N Engl J Med 2008;358:1250-1261.
Surgical technique has significant consequences for urinary control following radical prostatectomy. Both the striated urinary sphincter musculature and smooth muscle surrounding the urethra can be injured during surgery.305 Postoperative strictures at the site of the vesicourethral anastomosis can also affect return of urinary control.306 Avoidance of strictures (scars) by performance of a careful, tension-free, vesicourethral anastomosis has led to improved urinary control rates by expert surgeons.307,308 Men with persistent or severe urinary incontinence after radical prostatectomy can be treated with periurethral injections of bulking agents (silicone) that increase resistance at the bladder outlet, a pelvic sling placed through the perineum that repositions the urethra, and an artificial urinary sphincter—a three-piece device made up of a compressing urethral cuff, a fluid-filled reservoir placed in the abdomen that transmits pressure to the urethral cuff, and an activating pump placed in the scrotum that transfers fluid from the cuff to the reservoir allowing the cuff to open and the bladder to empty.
Erectile Function after Radical Prostatectomy
In 1982, Walsh and Donker described the anatomy of the nerves traversing the lateral surface of the prostate en route to the corpora cavernosa of the penis, discerning the proximity of the nerves to vascular structures (the neurovascular bundles) visible at the time of radical prostatectomy.5 This revelation led Walsh et al. to propose a modification of the radical prostatectomy procedure to preserve the neurovascular bundles in an effort to maintain erectile function postoperatively.6 Wide adoption of this modification led to improvements in erectile function following radical prostatectomy, and this approach is routinely used today when preservation of these nerves will not compromise removal of all cancer.
As is the case with urinary continence after radical prostatectomy, reports of return of erectile function vary greatly from 31% to 86% with study differences likely due to definitions of potency used, the methods for assessing return of sexual function, the cohort studied, and timing of assessment after surgery.309 Return of erections after surgery inversely correlates with age, and is directly associated with the quality of preoperative erections, the frequency of sexual activity preoperatively, and the quality and extent of nerve preservation. In a study using validated questionnaires to assess sexual function, sexual dysfunction was found to remain a big problem one year after treatment in 26% of men who underwent radical prostatectomy, with distress related to sexual dysfunction described by 44% of sex partners postoperatively.304 Most of the improvement in erections occurs within the first 2 years after surgery.310
Common approaches to managing erectile dysfunction after surgery include PDE-5 inhibitors, vacuum erection devices, vasoactive agents placed intraurethrally or injected directly, and placement of a penile prosthesis. Current thinking is that injury to the nerves (even when preserved at surgery) supplying corporal tissues results in injury to the corporal tissues that may be ameliorated by increased penile blood flow. Thus most sexual medicine experts encourage men to aggressively pursue attempts to achieve erections as soon as possible after surgery. If spontaneous erections do not occur with stimulation and PDE-5 inhibitors, men are encouraged to pursue other options described above, the greatest success achieved being with penile injection therapy.311
Control of Prostate Cancer by Radical Prostatectomy
Because PSA is a prostate-specific biomarker, successful removal of all prostate tissue after radical prostatectomy should result in an undetectable serum PSA. A detectable PSA after surgery, and/or a rising PSA, is an indication of residual prostate cancer that is referred to as a biochemical recurrence. Most series define a biochemical recurrence after surgery as a serum PSA that is above 0.2 ng/mL or rising. Biochemical recurrence rates vary directly with the risk assignment before surgery (low-risk, intermediate-risk, high-risk; see Table 84-3), age, the extent and grade of the cancer found on final pathological assessment. In modern surgical series, for men with localized prostate cancer, biochemical recurrence-free survival at 5, 10, and 15 years is reported at 84%, 74%, and 66%, respectively.312
Biochemical recurrence is not a surrogate for cancer-specific mortality because most men with a biochemical recurrence as the only evidence of residual disease do not die of prostate cancer. A study of the natural history of biochemical recurrence in a cohort of men (n = 450) who underwent surgery for presumed localized prostate cancer, had rising serum PSA, and received no treatment until the development of overt metastases, at a median follow-up of 8 years, revealed that only 30% of the men had developed metastatic disease.313 For these men, the median time from surgery to biochemical recurrence was 3 years and the median metastasis-free survival was 10 years. The Gleason score and PSA doubling time were independent predictors of metastasis-free survival, with no evidence of metastases in 94% of men with prostate cancer and a Gleason score of 6 or less versus 19% of those with Gleason scores of 8 to 10, and no evidence of metastases in 72% of men with recurrent prostate cancer and a PSA doubling time of >15 months versus 7% of those with a PSA doubling time of 3 to 9 at 10 years, respectively.
Cancer-specific survival after radical prostatectomy was recently evaluated in a multiinstitutional study of men (n = 11,521) with prostate cancer diagnosed in the PSA era, revealing a cumulative incidence of prostate cancer deaths at 15 years of only 7%.287 Predictors of prognosis include the Gleason grade and the presence of seminal vesicle invasion, with prostate cancer mortality rates of 0.2% to 1.2%, 4.2% to 6.5%, 6.6% to 11%, and 26% to 37% for Gleason scores of 6 or less, 3+4, 4+3, and 8 to 10, respectively, and 0.8% to 1.5%, 2.9% to 10%, 15% to 27%, and 22% to 30% for organ-confined cancer, cancer with extraprostatic extension, cancer with seminal vesicle invasion, and cancer accompanied by lymph node metastasis, respectively.287 Because these results were from a single-arm study without a comparison group, it is not possible to know to what extent the favorable surgical results for screen-detected cancers were a result of surgical intervention for a lethal phenotype versus the intrinsic indolent biological behavior of many such cancers. Hopefully, results from PIVOT, comparing surgery with watchful waiting, and from ProtecT, comparing conformal radiotherapy, prostatectomy, and active surveillance will provide more definitive answers to this question.
Radiation Therapy
Radiation therapy has been used in the management of prostate cancer for nearly a century. Following Roentgen’s discovery of the x-ray in 1895,314 and the isolation of radium by Pierre and Marie Curie in 1898,315 several pioneering physicians began treating prostate disorders, including prostate cancer, with radiation. In 1910, Paschkis and Tittinger inserted radium into the prostatic urethra with a cystoscope in what may be the use of radiation for prostate cancer. Not long after, Hugh Hampton Young from Johns Hopkins reported the relatively large experience of treating prostate cancer patients with urethral and rectal radium “applicators.”316 These and other early studies revealed that even when radiation was applied in this crude manner, it could improve symptoms and kill prostate cancer. However, treatment was technically difficult for the physician and uncomfortable for the patient. In 1928, the first report on the use of externally delivered low-energy kilovoltage radiation for prostate cancer was proffered by Barringer.317 The associated dosimetry was not well worked out and, thus men were treated until their skin turned red. These types of low-energy radiation machines were used until cobalt machines became available and provided the first opportunity to treat more deeply seated tumors in the body. The first reported series of prostate cancer patients treated with 60Co therapy was by George et al. in 1965 and featured men with unresectable disease.318 During this time (beginning in the late 1950s), the megavoltage linear accelerator was being developed at Stanford University.319 Using this new technology, the pioneering work of Bagshaw, Kaplan, Del Regato, and others, ushered in the modern era of radiation therapy for prostate cancer by offering the possibility of cure with radiation therapy in this disease.320,321
External Beam Radiotherapy for Localized Prostate Cancer using 3D-Conformal and Intensity-Modulated Approaches
For more than four decades, external beam radiotherapy has been widely used for the definitive management of clinically localized prostate cancer. More recently, technical advances have permitted the safe delivery of 80 Gy to the prostate, while minimizing radiation exposure of nearby normal tissues. Historically, a four-field pelvic box with shielding of the posterior wall of the rectum and anal canal was used to treat the prostate, seminal vesicles, and proximal lymphatic drainage to a dose of 45 to 50 Gy in 1.8- to 2.0-Gy fractions, with a boost to the prostate to 65 to 70 Gy. Even at these doses, external beam radiation therapy was fairly effective: although overall survival numbers were generally higher for men treated with radical prostatectomy (often younger and healthier men), cause-specific survival rates were not significantly different.322 Although the four-field box radiation therapy technique is of historic significance only, it is worth noting that side effects (rectal bleeding, dysuria, bowel dysfunction, etc.) associated with this technique were higher than those seen with contemporary technologies and delivery.
To improve on these outcomes, new technical innovations, such as CT-based simulation/treatment planning, the introduction of multileaf collimators in modern linear accelerators, and advances in treatment planning software have allowed for increased precision and accuracy in prostate cancer radiotherapy. For three-dimensional conformal radiation therapy (3D-CRT), three-dimensional reconstructions of pelvic CT images are generated to create more accurate target volumes (e.g., prostate and seminal vesicles) for treatments, enabling better identification of critical structures (e.g., bladder and rectum) to be avoided. An iterative process is then used to design beam arrangements that will deliver a prescribed dose to the regions of interest and minimize dose to a given volume of an adjacent organ. In this way, the incident radiation volume “conforms” to target volume. The treatment target volumes and normal organs can then be visualized in three dimensions, creating a “beam’s-eye view,” a portrayal of the target area as if looking straight down the path of the radiation beam. During treatment delivery, computerized multileaf collimators then shape each individual beam to conform to the shape of the target in the beam’s-eye view. Intensity-modulated radiation therapy (IMRT) builds on the advantages of 3D-CRT by using treatment planning software capable of even further optimizing dose distribution for prostate cancer treatment, by modifying not just the orientation and shape of the incident beams but also the intensity across the designated treatment volume (Figure 84-13).
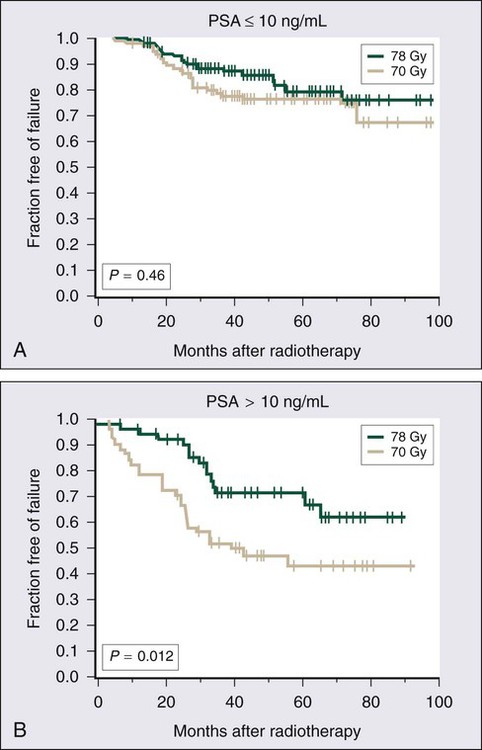
Complications of 3D-CRT and IMRT
Although the use of 3D-CRT was intended to minimize the effects of high-dose radiation on normal tissues, increased late complications, such as rectal and urinary toxicity, have been noted with escalating radiation doses used in 3D-CRT.323,324 Rectal toxicity encompasses urgency, frequency, pain, fecal incontinence, or bleeding. In a Fox Chase Cancer Center case series, the 5-year incidence of grade 3 or 4 rectal toxicity at a dose of 75 to 76 Gy was 8%, which upon better shielding of the anterior rectal wall, could be reduced to 2%. IMRT seems to be accompanied by less rectal complications. In one case series, men with clinically localized prostate cancer treated with IMRT to a total dose >81 Gy at Memorial Sloan-Kettering Cancer Center exhibited a reduction in late rectal toxicity when compared with 3D-CRT.325 With a median follow-up of 24 months, grade 2 or higher rectal bleeding was seen in 4% of men receiving IMRT, with grade 3 rectal toxicity in only 0.5% (and no grade 4 rectal toxicity) at 3 years. Urinary toxicity, including urethral strictures, tends to be similar for both 3D-CRT and IMRT, with as many as 15% of men suffering late grade 2 urinary complications. Because this may be the result of high-dose radiation to the urethra, decreasing urethral doses for men with prostate cancer limited to the peripheral zone of the prostate may be a means of reducing late urinary toxicity, but better diagnostic imaging will be necessary to identify men with such cancers. Overall, for external beam radiation therapy, 9% to 11% of men treated can expect to have some level of rectal or urinary distress 1 year after treatment (Table 84-4).304
Data on the incidence of erectile dysfunction after external beam radiotherapy has been widely variable. Sexual function has many facets that are difficult to evaluate or quantify. A commonly used qualitative definition of sexual potency is the ability to achieve spontaneous erections sufficient for intercourse. Although potency rates after radical prostatectomy have increased with use of nerve-sparing techniques, the mechanism of radiation-related erectile dysfunction appears unrelated to the neurovascular bundles. Zelefsky et al. specifically addressed this topic by performing duplex ultrasound studies before and after prostaglandin injection to stimulate erections in men with radiation-related erectile dysfunction.326 A diminished peak penile blood flow rate (<25 mL/min) was evident in 63% of such men, with abnormal distensibility of the corpora cavernosa in 32%. Thus, the primary mechanism of radiation therapy-associated impotence may be vascular damage rather than nerve damage. Fisch et al. demonstrated a correlation between incidence of erectile dysfunction postradiotherapy and radiation dose to the vascular penile bulb.327 Men receiving >70 Gy to >70% of the bulb of the penis are at greatest risk of experiencing radiation therapy–associated erectile dysfunction. A steady decline in potency rates over time is characteristic for men treated with external beam radiation therapy. In a large series (n = 434) of men treated with radiation therapy at Stanford University, 86% of the men remained potent at 15 months after treatment, but only 50% were potent 6 years later, and only 30% maintained erectile function for the remainder of their lives.328 Sildenafil administration resulted in improvement of erectile function in 74% of men with radiation therapy–associated erectile dysfunction in a study at Memorial Sloan-Kettering Cancer Center.329 Men who do not respond to sildenafil may respond to intracavernosal prostaglandin injections.
Cancer Control by External Beam Radiation Therapy
With the ready availability of serum PSA testing, outcome comparisons between primary prostate cancer treatments tend to focus on PSA as a marker of prostate cancer recurrence (“PSA relapse”–free survival). Of course, if successful in controlling cancer, surgical removal of the prostate eliminates PSA production by both nonneoplastic and neoplastic cells, whereas radiation therapy may not abolish PSA production by residual prostate epithelial cells even as cancer cells are killed. Nonetheless, a rising serum PSA after radiotherapy for prostate cancer is correlated with the appearance of progressive or metastatic prostate cancer upon further follow-up.330 Furthermore, the rate of serum PSA rise may help distinguish between local versus distant treatment failure. Men with slow rates of serum PSA increases are more likely to have local prostate cancer recurrences, whereas men with a rapid serum PSA rise appear more likely to have distant prostate cancer metastates.331 In 2006, the American Society for Therapeutic Radiology and Oncology (ASTRO) issued a consensus statement establishing the definition of recurrence following radiation therapy as a serum PSA level that is 2.0 ng/mL over the nadir value.332 This definition is an update to the 1997 ASTRO criteria for biochemical recurrence (three consecutive rises of the serum PSA determined at least 3 months apart333) and is now preferred for evaluating and comparing radiation treatment strategies. Before these criteria were formulated, treatment outcomes in different studies and case series were difficult to compare because of the lack of uniformity in defining treatment failure.
External beam radiation therapy using 3D-CRT and IMRT approaches achieves effective prostate cancer control.324,334–336 In a large cohort of men (n = 1100) treated with 3D-CRT at Memorial Sloan-Kettering Cancer Center, the PSA relapse-free survivals for men with low-risk prostate cancer treated to radiation doses of 64.8 to 70.2 Gy versus 81 Gy, were 77% and 98%, respectively.336 Men with intermediate-risk and high-risk prostate cancers also showed significant improvement. In a randomized trial of radiation dose escalation, higher-dose treatment was associated with improvement in prostate cancer control rates for men with prostate cancer, especially men with a pretreatment PSA >10 ng/mL (Figure 84-14).334,337 In this study, men (n = 305) with stage T1–T3 prostate cancer received either radiation therapy to a total dose of 70 Gy using conventional radiotherapeutic techniques, or to 78 Gy using a 3D-CRT approach. Results revealed a freedom-from-PSA relapse of 64% and 70% at 6 years for the 70-Gy and 78-Gy groups, respectively (P = 0.03). Men with a pretreatment serum PSA >10 ng/mL were found to have the most significant benefit from radiation dose escalation, with freedom-from-PSA relapse rates of 62% for the 78-Gy arm and 43% for the 70-Gy arm (P = 0.01). Overall survival was not significantly different between the two radiation doses, but a trend toward an improved freedom-from-distant metastasis was evident in men treated on the 78-Gy arm, 98% versus 88% at 6 years (P = 0.056).
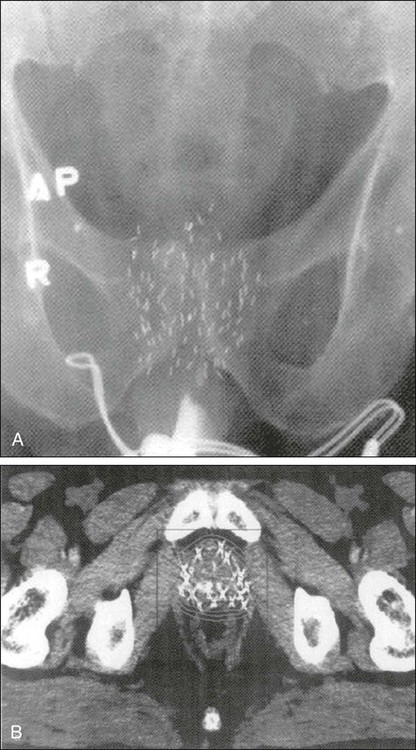
Brachytherapy
Prostate brachytherapy involves the implantation of radioactive sources into the prostate using image guidance (Figure 84-15). In principle, brachytherapy offers an attractive means for radiation dose escalation and conformality in the treatment of clinically localized prostate cancer. However, the most attractive feature for many men with prostate cancer is that this option may be most convenient, allowing a rapid return to normal lifestyle and activity. A typical brachytherapy procedure is performed in 2 hours, often under spinal anesthesia, and does not require an overnight hospital stay. The most common sources in use today are 125I (Iodine) and 103Pd (Palladium) (Table 84-5). Thus far, there have been no compelling data to support the superiority of one isotope or the other. Theoretically, the higher initial dose rate emitted by 103Pd might be advantageous for the treatment of cancers, like prostate cancer, with a relatively low αβ ratio (more radioresistant); however, retrospective data for prostate cancer have been inconclusive.338 To date, no long-term prospectively randomized data exist directly comparing 125I with 103Pd. Most experience with prostate brachytherapy has been with permanent low dose-rate (LDR) implants. Several centers have collected experience with temporary high dose-rate (HDR) implants for prostate cancer brachytherapy.341–341 Although this treatment approach is not widely used, the available results appear comparable with permanent brachytherapy for clinically localized prostate cancer.
Table 84-5
Physical Differences Between 125I and 103Pd Radioactive Seeds
| 125I | 103Pd | |
| Year introduced | 1965 | 1986 |
| Photon energy (keV) | 28 | 21 |
| Half-life (d) | 59.4 | 17 |
| Initial dose rate (for monotherapy) | 7 cGy/h | 18–20 cGy/h |
| RBE | 1.4 | 1.9 |
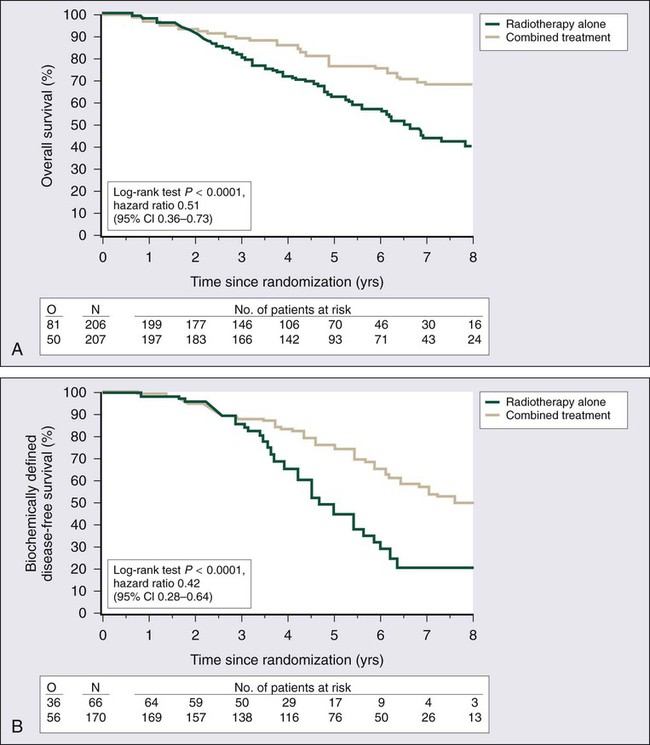
Brachytherapy may exhibit its greatest benefits in the treatment of men with low-risk, clinically localized, prostate cancer, though in the absence of a prospective randomized trial, this recommendation has been based on retrospective studies with varying follow-up time. One of the largest of these studies reported outcomes for more than 2500 men with prostate cancer treated at 11 different institutions with permanent interstitial brachytherapy.342 The median follow-up for this group of men with stage T1 or T2 prostate cancer was 63 months and all were treated with either 125I or 103Pd without use of hormonal therapy. Men with low-risk, intermediate-risk, and high-risk prostate cancer had 8-year actuarial PSA relapse-free survivals of 82%, 70%, and 48%, respectively, using ASTRO criteria. The dosimetric quality of the implant was critical to outcome: for men in which 90% of the prostate (D90) received ≥130 Gy, the 8-year PSA relapse-free survival was 93%, whereas for men in which the prostate D90 was <130 Gy, the 8-year PSA relapse-free survival was 76%. There were few, if any, differences attributable to the use of 125I versus 103Pd implants.
The addition of supplemental external beam radiotherapy to brachytherapy remains somewhat controversial. Davis et al. examined the radial distance of extraprostatic extension of prostate cancer and found it to be almost always 5 mm or less, which would be within a typical brachytherapy dose distribution.343 Thus generous periprostatic margins in brachytherapy planning should obviate the need for supplemental external beam radiotherapy. Proponents of combinations of brachytherapy and external beam radiation therapy emphasize both the advantage of higher delivered doses and the ability to smooth out cold spots inherent with brachytherapy, the so-called spackle effect. Sylvester et al. have recently reported a retrospective review of 232 men with clinically localized prostate cancer treated with either 125I or 103Pd brachytherapy and external beam radiation therapy administered before the implant.344 At a median follow-up of 9.4 years, the biochemical relapse-free survival for the entire study group was 74%, with biochemical relapse-free survival for low-risk prostate cancer of 85.8%, for intermediate risk disease of 80.3%, and for the high-risk group of 67.8%. These results compare favorably with all other surgical and/or radiation series with respect to long-term durable prostate cancer control.
Relative contraindications to the use of prostate brachytherapy are large prostate size, pre-implant obstructive urinary symptoms, history of prior TURP, and the presence of perineural prostate cancer invasion on prostate biopsy. Large prostate size has been perceived to be associated with a higher risk of urinary morbidity postimplant and with unsuitability for implant because of pubic arch interference. Men with a prostate volume of greater than 50 mL have been either counseled against brachytherapy or placed on androgen deprivation therapy (ADT) in an attempt to reduce gland size. Nonetheless, the implantation of large prostates with radioactive seeds has been described with acceptable morbidity.345,346 In one case series, postimplant dosimetry quality was found to be independent of prostate size or use of ADT.346 The correlation of preimplantation obstructive urinary symptoms and postimplantation urinary obstruction has not been fully resolved. Terk et al. reported that a high International Prostate Symptom Score (I-PSS), a measure of obstructive urinary symptoms, predicted postimplant urinary retention.347 With the use of α–blockers before and after implant procedures, others have noted no association between preimplant I-PSS and urinary obstruction.348 A prospective study examining preimplant urinary flow rate and postvoid residual, in addition to I-PSS, showed no association of obstructive urinary symptoms with postimplant urinary retention or long-term urinary function.349 TURP is thought to be a relative contraindication to prostate brachytherapy as it has been associated with unacceptably high rates of urinary incontinence. This could possibly be attributable to seed placement approaches that result in a high central dose to the TURP defect. However, by using a peripheral source loading approach to limit dose to the TURP defect to 110% of the prescription dose, the incidence of urinary incontinence may be reduced.350
A phenomenon peculiar to prostate brachytherapy that deserves mention is the so-called PSA spike. With a time of onset between 12 and 30 months postimplantation, approximately one-third of men with prostate cancer treated with brachytherapy will experience a transient increase in serum PSA.351 This spike may be due to radiation-associated prostatitis that compromises prostate architecture, permitting more PSA to appear in the serum. Notably, such PSA spikes portend no worse long-term outcome.
Toxicity of Brachytherapy
The short- and long-term sequelae of brachytherapy for prostate cancer differ from those of external beam radiotherapy and radical prostatectomy. Kleinberg et al. described the morbidity outcomes of the early Memorial Sloan-Kettering Cancer Center experience with permanent transperineal brachytherapy, reporting that the most common side effects were nocturia and dysuria (80% and 48%, respectively, 2 months after implantation).352 By 12 months following implantation, these figures had declined (to 45% and 20%). Urinary retention is seen in 3% to 14% of men and usually lasts 1 week or less. The most bothersome late complications of prostate cancer brachytherapy are urethral stricture and urinary incontinence. Ragde et al. reported a 5.1% incidence of urinary incontinence in men treated with prostate cancer brachytherapy and followed for 7 years; each of the men with incontinence had a history of TURP.353 Urethral stricture appeared in 14.4% of the men. Others have also seen an increased incidence of urinary incontinence in the setting of a history of TURP.354 In a cohort study of Medicare beneficiaries (n = 2124) who were treated with brachytherapy, urinary incontinence was noted in 6.6% and bladder outlet obstruction requiring intervention was found in 8.3%.355 In all, as many as 16% of men treated with brachytherapy for prostate cancer can expect to suffer distressful urinary symptoms 2 years after treatment (Table 84-4).304 As is the case for external beam radiation therapy, rectal complications after brachytherapy tend to emerge within 3 years of implant placement, with rectal injury in 5% to 10% of men treated.356,355,357,304
As popularity of prostate brachytherapy for clinically localized prostate cancer grew in the 1990s, a commonly cited advantage of the treatment modality was a lower incidence of treatment-associated erectile dysfunction compared to external beam radiotherapy or radical prostatectomy. Undoubtedly, this selling point tipped the scales in favor of brachytherapy for many men faced with selecting a treatment for early-stage prostate cancer. For instance, Stock et al. reported a 2-year potency rate of 94% after implant, whereas Wallner et al. reported a 3-year potency rate of 86%.358,359 Studies with longer follow-up, however, have shown a continued decrease in sexual potency over time. With more long-term follow-up in other case series, only 57% of men retained potency at 5 years.356 Even Stock et al. subsequently reported a 6-year potency rate of 59% in their case series.360 Notably, their study found that 70% of men with normal erectile function before implant retained potency at 6 years, whereas men with “erectile function sufficient for intercourse” but suboptimal erections had only a 34% 6-year potency rate. Using postimplant dosimetry studies, Merrick et al. demonstrated that dose to the penile bulb correlated with postimplant erectile dysfunction. In the majority of men who retained potency, the dose delivered to 50% of the penile bulb was less than 50 Gy.361 Potentially, this knowledge may result in improved morbidity outcomes with technical attention to this dose threshold. Erectile dysfunction is not the sole complication of prostate brachytherapy with the potential to affect sexual quality of life, however, with reports of hematospermia in 28%, orgasmalgia in 15%, and alteration in the intensity of orgasm in 38%.362 These side effects tend to be transient in most men.
Proton Beam Radiotherapy
Though only available at a few centers worldwide, there has been an interest in proton beam therapy for prostate cancer. The unique physical properties of protons make them ideal for the treatment of disease in proximity to critical strictures. Specifically, protons deposit the majority of their energy at the very end of their linear tracks, a phenomenon termed the “Bragg peak.” The dose falls off very rapidly at depths beyond the Bragg peak. This is particularly useful in the treatment of prostate cancer, to minimize rectal and bladder dose. Investigators from Loma Linda University reported their experience treating men (n = 1255) with T1–T3 prostate cancer.363 Men were treated with protons alone to 74 cobalt Gray equivalents (CGE) or with photons to 45 Gy followed by proton boost to 75 CGE. The median follow-up was 62 months and the 8-year actuarial biochemical disease-free survival rate was 73%. In a recent prospective, randomized trial of 70.2 GyE versus 79.2 GyE (combination of photons plus protons) for men (n = 393) with stage T1b-T2b prostate cancer and a serum PSA <15 ng/mL, at a median follow-up of 5.5 years, 61.4% of the men treated with 70.2 GyE versus 80.4% of men treated with 79.2 GyE were free of biochemical treatment failure, supporting the concept that higher doses of radiation results in a statistically significant reduction in the risk of recurrence of localized prostate cancer.364
Adjuvant Endocrine Therapy
ADT has been found to improve treatment outcomes in randomized trials of men with high-risk prostate cancer treated with external beam radiation therapy but not as convincingly for men treated with radical prostatectomy (Figure 84-16).367–367 D’Amico et al. reported results of a large retrospective study (n = 1586) of men treated with 3D-CRT plus or minus ADT for low-risk, intermediate-risk, and high-risk prostate cancer.368 In this study, the median radiation dose was 70.2 Gy and ADT was used for 2 months before radiation therapy, during treatment, and for 2 months after treatment was completed. With a median follow-up of 51 months, the 5-year PSA relapse-free survival for men with low-risk prostate cancer was 92% with the addition of ADT versus 84% without. Men with intermediate- and high-risk prostate cancer also fared significantly better when given ADT. Radiation Therapy Oncology Group (RTOG) Trial 94-08, which completed accrual in 2001 and was reported in 2012, was designed to ascertain whether men (n = 1917) with stage T1b–T2 prostate cancer and a serum PSA of 20 ng/mL or less benefit from the addition of “complete androgen blockade” given for 4 months before and concomitantly with external beam radiation therapy.369 Results of this trial revealed that the addition of androgen suppression to radiation significantly improved disease-specific and overall survival, reducing overall deaths from 8% to 4% at 10 years.
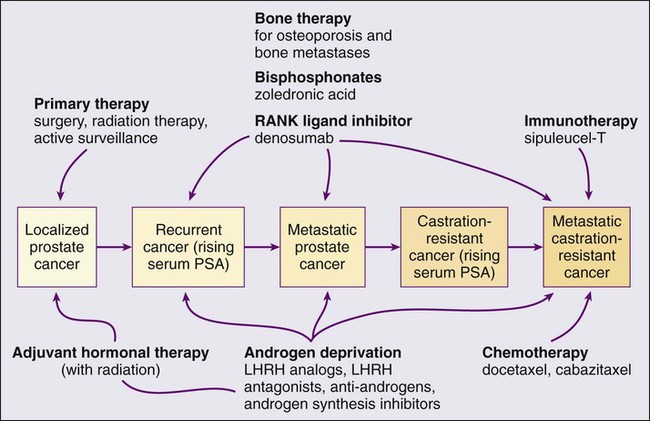
ADT has also been used with prostate cancer brachytherapy, both to reduce the size of the prostate gland and to improve outcomes. Most prostate glands exhibit some decrease in volume after 3 months of ADT, with an average 30% to 40% reduction, and little further volume decreases.370 About 10% of prostate glands will show no volume reduction at all in response to androgen deprivation. Decreasing the size of the prostate may reduce pubic arch interference in selected men. Blank et al. reported that men treated with ADT tended to have smaller prostates that required fewer seed implants.371 However, at this point there are no data to suggest that smaller prostate volumes correlate with reduced acute or late morbidity from brachytherapy. Furthermore, although prospective randomized trials have demonstrated improved survival in those men with locally advanced prostate cancer treated with external beam radiotherapy and ADT, it is not clear that these results can be extrapolated to men treated with brachytherapy. A retrospective matched-pair analysis of men (n = 60) with prostate cancer treated at Memorial Sloan-Kettering Cancer Center showed no benefit for the addition of ADT to brachytherapy for men with low-, intermediate-, or high-risk prostate cancers.372
Postprostatectomy Adjuvant Radiation Therapy
Pathological features that portend a higher risk of local recurrence are common after radical prostatectomy. A positive surgical margin is associated with an approximately 50% risk of prostate cancer recurrence.373–377 Other features associated with recurrence are extracapsular extension, seminal vesicle invasion, and Gleason score of 7 or greater. Several retrospective series have now demonstrated improved PSA relapse-free survival with the addition of adjuvant radiation therapy following radical prostatectomy in men with these risk factors.380–380 In one study, men (n = 149) with pathological stage T3N0 prostate cancer and an undetectable postoperative serum PSA, adjuvant radiation therapy was given to a median dose of 64.8 Gy to some men (n = 52), whereas the remainder (n = 97) underwent no further treatment.378 In a matched-pair analysis, the 5-year freedom-from-PSA relapse rate was 89% in the adjuvant radiation therapy group versus 55% for treatment with surgery alone (P < 0.01).
Three prospective randomized trials have been completed that tested the benefits of adjuvant radiation therapy versus observation following radical prostatectomy in men with prostate cancer and poor pathological features. The European Organization for Research and Treatment of Cancer (EORTC) has published clinical trial results revealing an improvement in biochemical relapse-free survival and locoregional progression-free survival for the men with pT3 tumors or pT2/T3 prostate cancer and positive surgical margins treated with adjuvant radiation therapy.366 In a second study targeting the same patient population conducted by the Southwest Oncology Group (SWOG), men with prostate cancer who received adjuvant radiation not only enjoyed better biochemical and local control, but also appeared likely to have better metastasis-free survival and overall survival; these improvements were not yet statistically significant even at a median follow-up of 10 years.381 Nonetheless, men treated with adjuvant radiation therapy were less likely to need hormonal therapy at 5 years (10% vs. 21%, P < 0.001). Avoidance of ADT is likely of significant clinical benefit, reducing or delaying significant morbidity, including hot flashes, diminished bone density, sexual dysfunction, cognitive dysfunction, and overall reduced quality of life. Finally, a third randomized trial comparing adjuvant radiation and observation after prostatectomy, which enrolled only men with pT3 disease regardless of surgical margin status, was reported by the German Cancer Study Group.382 As with the other two studies, men treated with adjuvant radiation experienced a superior biochemical relapse-free survival with median follow-up of only 3.3 years. Thus, overall, data available today provide a convincing case for adjuvant radiation treatment after prostatectomy. However, it is clear that not all men benefit from such treatment. What is needed for the future is a more precise way to stratify men for adjuvant radiation therapy: one such group may be men with positive surgical margins after radical prostatectomy.
Salvage Radiotherapy after Radical Prostatectomy
Salvage radiotherapy refers to the use of radiation therapy postprostatectomy in the setting of recognized prostate cancer recurrence. As many as 27% to 53% of men who undergo radical prostatectomy for prostate cancer will have a detectable PSA within 10 years of surgery.383 Subsequently, approximately 25% of men who undergo radical prostatectomy will be treated with salvage radiation therapy for recurrent prostate cancer.384 In the setting of persistent or rising serum PSA following radical prostatectomy, it is important to rule out distant metastatic prostate cancer with bone scan, chest radiography, and CT scan of the abdomen and pelvis, prior to the initiation of salvage radiation therapy. Partin et al. correlated the rate of serum PSA rise with likelihood of local versus distant relapse after surgery.385 A serum PSA rise of 0.75 ng/mL/year was associated with local recurrence. In 1999, an ASTRO consensus panel concluded that treatment of men with local prostate cancer recurrence after radical prostatectomy and a pre–radiation therapy serum PSA <1.5 ng/mL was more likely to be successful.386 In addition, doses above 64 Gy were recommended in the salvage setting. Several studies have demonstrated a Gleason score greater than 7 to be associated with a low likelihood of successful salvage after prostate cancer recurrence postprostatectomy.387,388 Caddedu et al. reported no men free of PSA relapse treated with salvage radiation therapy after prostatectomy for prostate cancers with Gleason scores of 8 or above.388 Similarly, Song et al. found only 2 of 14 men with Gleason score of 8 or above to be prostate cancer free at the time of analysis.387 These results indicate that men with recurrent prostate cancer and a Gleason score of 8 or greater are unlikely to benefit from salvage radiation therapy because of the likelihood of microscopic systemic prostate cancer metastases.
However, the largest multiinstitutional retrospective series to date (n = 501 men) has provided a clearer view of the likelihood of benefit for various subgroups of men with local prostate cancer recurrence after prostatectomy.389 In this study, predictors of a poor response to salvage radiation included Gleason scores of 8 to 10, preradiation PSA level of greater than 2 ng/mL, PSA doubling time after prostatectomy of 10 months or less, negative surgical margins and seminal vesicle invasion. Nonetheless, a significant fraction of men with one or more of these negative prognostic features still experienced a durable response to salvage radiation, particularly if the radiation was given prior to a PSA level of 2 ng/mL. Even for the group of men with the worst combination of prognostic features, a Gleason score of 8 to 10 and a preradiation PSA of ≥2 ng/mL, salvage radiation produced a progression-free survival of 12% at 4 years. In another large retrospective study, Trock et al. reported that men treated with salvage radiotherapy resulted in a 3-fold improvement in prostate cancer–specific survival compared with men who received no salvage treatment (HR = 0.32, 95% CI of 0.19 to 0.54; P < 0.001).390 Importantly, this benefit was limited to men with a prostate-specific antigen doubling time of less than 6 months and remained after adjustment for pathological stage and other established prognostic factors. Although these findings hint at marked advantage to salvage radiation therapy after prostatectomy, the studies are retrospective and the results may be confounded by selection bias. Prospective studies are needed to definitively determine whether patients with one or more poor prognostic features derive benefit from salvage radiation. However, given the available data and considering the limited morbidity of this treatment, especially with IMRT, it is reasonable to consider salvage radiation for postprostatectomy patients with PSA recurrence regardless of prognostic factors.
The role of ADT concomitant with radiation therapy given as an adjuvant to surgery as salvage for prostate cancer recurrence following surgery has not been established. In the RTOG 85-31 Trial, a subgroup of men with pathological T3N0 prostate cancer who underwent radical prostatectomy received adjuvant radiation therapy to a dose of 60 to 65 Gy and then were randomized to immediate androgen deprivation or androgen deprivation initiated at the time of PSA relapse.391 At 5 years, 65% of men treated with immediate androgen deprivation versus 42% of men treated with delayed androgen deprivation were free of PSA relapse. There are no prospective trials examining the addition of ADT to salvage radiation therapy for local prostate cancer recurrence after prostatectomy. Taylor et al. reported a benefit to the addition of ADT to salvage radiation therapy in a retrospective case series.392 In this series, adjuvant ADT was given to men who received salvage radiation therapy for a median duration of 24 months. At 5 years, 81% of men receiving ADT (vs. 54% not treated with androgen deprivation) were free of PSA relapse. In contrast, Song et al. reported identical median disease-free survivals of 26 months for men treated with or without concurrent ADT along with salvage radiation therapy for prostate cancer recurrence.387 The 1999 ASTRO Consensus Panel concluded that there was insufficient evidence to support routine use of ADT with postprostatectomy radiation therapy.386
In general, men receiving radiation therapy postprostatectomy experience little in the way of additional morbidity. The incidence of urinary incontinence does not seem to be increased and erectile function does not seem to be worsened in men treated with adjuvant radiation therapy after prostatectomy.393,394 Bastasch et al. reported that 100% of men who were potent after nerve-sparing radical prostatectomy remained potent after adjuvant IMRT.395 Despite these promising reports, side effects remain possible. Katz et al. noted that 19% of men experienced grade 2 or 3 genitourinary toxicity (hematuria or urethral stricture), and 12% of men experienced grade 2 bowel toxicity (with no grade 3 or higher toxicity noted), following salvage radiation with 3D conformal techniques.396