[level-membership-for-surgery-category]
Chapter 41 Primary sclerosing cholangitis
Overview
Primary sclerosing cholangitis (PSC) is a chronic, cholestatic liver disease of unknown etiology characterized by diffuse inflammatory destruction of intrahepatic and/or extrahepatic bile ducts that results in bile stasis, hepatic fibrosis, cirrhosis, and end-stage liver disease. Despite the increased awareness of PSC, the exact etiology and an effective medical therapy have yet to be identified. The disease is associated with specific human leukocyte antigen (HLA) haplotypes and has a close association with inflammatory bowel disease (IBD), which suggests that PSC is an immune-mediated disorder; but unlike other typical autoimmune diseases, there is a 2:1 male predominance. Life expectancy is reduced in PSC patients, because disease progression often results in complications of liver failure and cholangiocarcinoma (see Chapters 50A, 50B, and 50C). Among eligible candidates, surgical therapy has moved largely to orthotopic liver transplantation (OLT), as it remains the only effective, life-extending treatment for advanced-stage PSC (see Chapter 97A).
Epidemiology and Demographics
PSC mainly affects males during the fourth decade of life (Angulo & Lindor, 1999; Chapman, 2003). In the United States, population-based studies reported an age-adjusted incidence for PSC of 1.25 per 100,000 men and 0.54 per 100,000 women per year (Bambha et al, 2003). In the same study, the calculated prevalence of PSC was 20.9 and 6.3 per 100,000 men and women, respectively (Bambha et al, 2003). The worldwide prevalence and geographic distribution of PSC are undefined, as most epidemiologic data come from Northern Europe and the upper Midwest in the United States. Of interest is the strong association of PSC with IBD. Approximately 75% to 80% of Northern European patients with PSC suffer from IBD, with chronic ulcerative colitis (CUC) being most common, seen in about 90% (Chapman et al, 1980; Olsson et al, 1991; Wiesner et al, 1989). Conversely, only approximately 5.5% of patients with CUC have PSC (Olsson et al, 1991).
Smoking is the only environmental factor known to influence PSC susceptibility, and it is associated with a reduced risk of PSC. In a study from The Netherlands, the incidence of current smoking was 19% in patients with PSC compared with 38% in control subjects (Van Erpecum et al, 1996). Similarly, a U.S. study showed that 4.9% of patients with PSC were reported to smoke compared with 26.1% of a control population matched for age and gender. This reported difference was not attributable to the prevention of CUC (Loftus et al, 1996b). Nevertheless, smoking and alcohol consumption should be discouraged in PSC patients, because both are associated with the development of cholangiocarcinoma in this population (Bergquist et al, 1998; Chalasani et al, 2000).
Adenocarcinoma of the bile ducts appears in 8% to 15% of patients with PSC, rendering it a premalignant condition (Chalasani et al, 2000; De Groen et al, 1999; Foutch et al, 1990; see Chapters 50A, 50B, and 50C). In a recent study, 161 PSC patients without evidence of cholangiocarcinoma upon entry were followed over a median of 11.5 years. During that period, 59 patients (36.6%) died, 50 patients (31.1%) underwent liver transplantation, and 11 patients (6.8%) had cholangiocarcinoma develop (Burak et al, 2004). No association was found between the duration of PSC and the incidence of cholangiocarcinoma, and multivariate analysis revealed that variceal bleeding was the only significant risk factor for the diagnosis of cancer (risk ratio, 24.2; 95% confidence interval, 3.3% to 67%) (Burak et al, 2004).
Clinical Presentation
PSC can affect any age group, but it is more common among men in their fourth decade of life (Wiesner & LaRusso, 1980; MacCarty et al, 1983). PSC can also occur in childhood, and children with PSC frequently have liver disease with features similar to autoimmune hepatitis (AIH) (El-Shabrawi et al, 1987; Wilschanski et al, 1995). In fact, the overlap between PSC and AIH in children can be as high as 35% (Feldstein et al, 2003). In contrast, overlap of PSC and AIH affects only about 5% of adults (Kaya et al, 2000; Van Buuren et al, 2000).
The clinical presentation of PSC varies depending on the disease stage at the time of diagnosis. PSC can be diagnosed in asymptomatic individuals who come to medical attention because of abnormal liver function tests. Approximately 15% to 40% of patients are asymptomatic at the time of diagnosis (Talwalkar & Lindor, 2005). There are no pathognomonic signs or symptoms when the disease becomes clinically apparent. The symptomatic patient may be seen initially with signs, symptoms, and complications of cholestatic liver disease or hepatic failure. In a recent study, the most common first symptoms were abdominal pain (20%), pruritus (10%), diarrhea (8%), jaundice (6%), fatigue (6%), and fever (4%) (Kaplan et al, 2007). Symptoms of bacterial cholangitis are uncommon, unless dominant biliary strictures or biliary stones are present.
Physical examination may reveal jaundice, hepatomegaly, splenomegaly, and excoriations. Ascites and peripheral edema are observed with the development of biliary cirrhosis and portal hypertension (see Chapter 70A). These findings are less common today, because PSC patients are usually evaluated before the development of end-stage liver disease. A commonly encountered clinical scenario is a patient with CUC who presents with a cholestatic pattern of liver enzymes. Further medical evaluation results in the concurrent diagnosis of PSC. As with other chronic liver diseases, health-related quality of life is significantly impaired among PSC patients compared with healthy individuals (Younossi et al, 2001).
Diagnostic Criteria
The diagnosis of PSC is made based on clinical presentation, biochemical profile, and characteristic cholangiographic appearance of the bile ducts. Before the 1960s, PSC was a diagnosis of exclusion following abdominal exploration and biopsy of a suspected bile duct neoplasm (Schwartz & Dale, 1958). In the 1960s, diagnosing PSC became more frequent, when its association with IBD was recognized (Thorpe et al, 1967). With the advent of endoscopic retrograde cholangiopancreatography (ERCP) and percutaneous transhepatic cholangiography (PTC) in the 1970s, the characteristic “beaded” or “pruned tree” cholangiographic appearance obviated the need for exploratory surgery to diagnose PSC and resulted in its increased recognition (Wiesner & LaRusso, 1980; see Chapter 18).
The first formal diagnostic criteria included 1) absence of previous operative trauma to the biliary system; 2) sclerosis and stenosis involving all or most of the extrahepatic bile ducts; 3) exclusion of malignant disease involving the biliary tree, such as cholangiocarcinoma; and 4) absence of calculi in the gallbladder and common bile duct (Holubitsky & McKenzie, 1964). Following the introduction of ERCP, the criteria were expanded to include a subset of PSC cases recognized to have intrahepatic bile duct involvement alone (MacCarty et al, 1983). In addition, serial ERCP examinations of PSC patients demonstrated the subsequent development of cholangiocarcinoma (Chapman et al, 1980). As such, malignancy arising in the setting of sclerosing cholangitis was eliminated as an exclusionary criterion. Imaging techniques (i.e., ERCP) have also shown biliary tract calculi in many patients with established PSC, suggesting the removal of calculi as an exclusionary criterion (Kaw et al, 1995).
The diagnosis of PSC currently relies on 1) characteristic cholangiographic abnormalities of the biliary tree; 2) compatible clinical and biochemical findings, typically of ductal cholestasis with elevated serum alkaline phosphatase level for at least 6 months duration; and 3) exclusion of other causes of secondary sclerosing cholangitis (SSC). The clinical and cholangiographic features of SSC can mimic PSC, but SSC originates from known pathologic processes. Several well-described causes of SSC are listed in Table 41.1 (Abdalian & Heathcote, 2006). Currently, the most common presentation is an asymptomatic patient with persistently elevated levels of alkaline phosphatase first noted on routine serum biochemical screening. In the proper clinical context, a characteristic cholangiographic appearance of the bile ducts is sufficient for making the diagnosis.
AIDS, acquired immunodeficiency syndrome
Liver biopsy is not always necessary for diagnosing PSC, nor does it contribute new information that affects the clinical management of patients. In a study of 79 patients with PSC who had an already established diagnosis by cholangiography, liver biopsy did not affect management (Burak et al, 2003). Thus the role of liver biopsy in PSC is to 1) exclude other causes of liver disease, 2) diagnose small-duct PSC (discussed later), and 3) define the disease stage for determining prognosis and assessing efficacy of treatment prior to entering in therapeutic trials. Small-duct PSC comprises approximately 5% of histologically confirmed PSC cases (Angulo et al, 2002) and has significantly better long-term prognosis compared with classic PSC. However, some patients with small-duct disease can progress to classic PSC (Angulo et al, 2002). Patients with small-duct PSC usually have IBD and are seen intially with a cholestatic pattern of liver enzymes but normal cholangiography. Nevertheless, liver biopsy reveals classic histologic features of PSC. In the majority of patients, the history, serum biochemical profile, and cholangiography distinguish PSC from other causes of chronic cholestatic liver disease. The differential diagnosis of PSC is presented in Table 41.2.
Table 41.2 Differential Diagnosis of Primary Sclerosing Cholangitis
Biochemical and Serologic Abnormalities
Biochemical cholestasis of at least 6 months’ duration gives reason to suspect PSC. Alkaline phosphatase remains the most commonly elevated liver enzyme, often showing a threefold to fourfold increase. Nevertheless, a normal alkaline phosphatase level does not exclude the diagnosis of PSC, because normal levels have been reported in patients with cholangiographically proven disease (Balasubramanian et al, 1988). Therefore a normal alkaline phosphatase level should not dissuade further investigation if the clinical history (e.g., presence of IBD) and other evidence suggest liver disease.
Aminotransferase levels are often modestly elevated in patients with PSC, except in children, in whom these levels can be markedly increased. On average, adults with PSC have aminotransferase levels less than three times the upper limit of normal (Lee & Kaplan, 1995). PSC patients with highly elevated aminotransferases may show concomitant serologic evidence and histologic features of AIH (Czaja, 1998).
Serum bilirubin levels are normal in 60% of patients at diagnosis (Talwalkar & Lindor, 2005), but bilirubin levels will markedly rise as PSC progresses. An abrupt, sustained rise of bilirubin may herald a dominant biliary stricture, a bile duct stone, or the development of cholangiocarcinoma; therefore it should prompt additional investigation. Serum copper and ceruloplasmin levels and hepatic and urinary copper values are often abnormal. Hepatic copper levels can be elevated to the degree seen in Wilson disease and primary biliary cholangitis (PBC) (LaRusso et al, 1984); the copper increase is a reflection of prolonged cholestasis.
No autoantibody is pathognomonic for diagnosing PSC. The prevalence of antineutrophil cytoplasmic antibodies (ANCAs), anticardiolipin antibodies, and antinuclear antibodies (ANAs) in patients with PSC is 84%, 66%, and 53%, respectively (Angulo et al, 2000). Antimitochondrial antibodies (AMAs) and anti–smooth muscle antibodies are rare in patients with PSC. Approximately 25% of patients have hypergammaglobulinemia, and immunoglobulin M (IgM) levels are the most commonly elevated component (Wiesner & LaRusso, 1980). Autoantibody testing is helpful to identify those PSC patients with concurrent AIH, but antibody titers are not important in following PSC activity.
Imaging Studies
Visualization of the biliary tree is essential for establishing the diagnosis of PSC, and ERCP is the gold standard imaging technique (see Chapter 18). Typical cholangiographic findings of PSC include multifocal stricturing and beading throughout the biliary tree characteristic of alternating fibrosis and ectasia of the bile ducts (Fig. 41.1). Strictures of the bile ducts are the hallmark of the disease. They are often diffusely distributed and annular with intervening segments of dilated ducts. The classic cholangiographic findings involve both the intrahepatic and extrahepatic biliary tree. Strictures can vary in length and confluency, ranging from a bandlike appearance 1 to 2 mm in length to dense, annular strictures several centimeters in length. Approximately 25% of patients have biliary outpouchings that resemble diverticula. Similarly, 30% to 40% of patients may have mural irregularities that produce a shaggy appearance, varying from a fine brush border to frank nodularity (MacCarty et al, 1983).
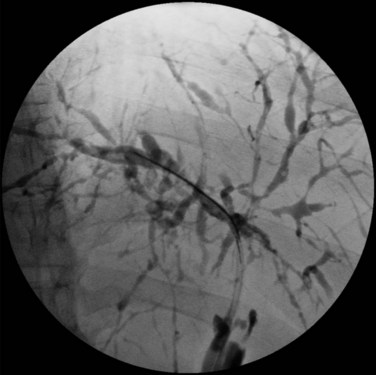
In a study of 86 patients with PSC, cholangiographic findings revealed involvement of the intrahepatic and extrahepatic ducts in 80 and 85 patients, respectively, when there was adequate visualization of the intrahepatic biliary tree. Involvement of only the intrahepatic and proximal extrahepatic ducts was seen in 20% of patients, and a very small number of patients had only small-duct PSC with a normal cholangiogram (MacCarty et al, 1983). Gallbladder and cystic duct involvement is less severe but is present in as many as 15% of patients (Brandt et al, 1988). ERCP should be considered in all patients suspected of having PSC. In addition to establishing the diagnosis, ERCP defines the extent and distribution of disease, identifies benign dominant strictures for endoscopic dilation or stenting, and allows brushings for cytology studies to screen for cholangiocarcinoma.
As many as 10% of cholangiocarcinomas have radiologic features that mimic PSC. Cholangiographic features that may suggest malignant degeneration of established PSC are markedly dilated biliary ducts or ductal segments, presence of a polypoid mass of 1 cm or greater in diameter, and progressive stricture formation. Nevertheless, these cholangiographic features are not specific for cholangiocarcinoma and can be seen in the absence of malignancy (MacCarty et al, 1985).
Despite the high diagnostic and therapeutic capabilities of ERCP, the procedure is invasive and is associated with clinical complications. The Mayo Clinic experience has shown that among PSC patients undergoing ERCP, the estimated rate of procedural complications requiring hospitalization is more than 10%. PSC patients undergoing ERCP are expected to have longer procedural time and a higher incidence of cholangitis compared with non-PSC patients (see Chapter 43), but the risk of pancreatitis, perforation, and bleeding are similar (Bangarulingam et al, 2009).
Magnetic resonance cholangiography (MRC) is a noninvasive substitute for ERCP (Fig. 41.2; see Chapter 17). Recent studies have estimated the sensitivity and specificity of MRC for diagnosing PSC to be between 80% to 82% and 87% to 98%, respectively (Talwalkar et al, 2004; Berstad et al, 2006). These studies concluded that MRC has diagnostic accuracy comparable to ERCP and results in cost savings when used as the initial diagnostic strategy. In another study, MRC was shown to be better than ERCP in identifying peripheral intrahepatic bile duct strictures (Vitellas et al, 2002).
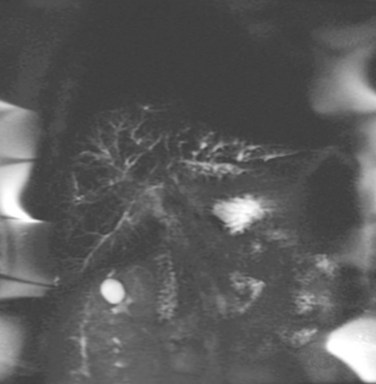
In a retrospective, nonblinded study of PSC patients with known biliary tract carcinoma, MRC findings of a well-defined mass exhibiting abnormal signal intensity were classified as definite cholangiocarcinoma in six or seven patients (Vitellas et al, 2002). MRC will likely replace diagnostic ERCP, but at present it is less sensitive and does not allow for biliary biopsy and cytology or therapeutic intervention.
PTC is useful when access to the biliary tree by ERCP is not technically feasible. Abdominal ultrasonography (US) and computed tomography (CT) are valuable in affirming ductal dilation and evaluating complications of PSC such as biliary stones, cirrhosis, and bile duct malignancy (see Chapters 13 and 16). US is often able to show large duct dilation and mural thickening of the common bile duct in patients with confirmed abnormalities on ERCP. It can further be used to detect gallstones or biliary tract calculi, which are important in the evaluation of bacterial cholangitis, worsening cholestasis, or jaundice.
Morphologic features of ductal abnormalities and cirrhosis can be seen on CT. Atrophy of the left lateral segment and hypertrophy of the caudate lobe may differentiate cirrhosis associated with PSC from that seen in other types of cirrhosis (Caldwell et al, 2001). CT can also complement cholangiography in the evaluation of malignancy with its ability to detect peripheral intrahepatic cholangiocarcinoma and metastatic spread within the liver parenchyma and the abdomen (Campbell et al, 1998, 2001). However, perihilar lymphadenopathy is very common in PSC with or without cholangiocarcinoma, and this finding cannot be taken as direct evidence of malignancy or metastasis.
Pathology
PSC can affect the entire biliary system. On rare occasions, it may be limited to the small intrahepatic ducts and be detectable only on histology. Small-duct PSC is thought to account for 5% of histologically confirmed PSC cases, although its true prevalence remains unknown (Angulo et al, 2002).
Liver biopsy findings alone are rarely sufficient to establish the diagnosis of PSC, which is characterized by damage to, atrophy of, and ultimately loss of bile ducts, thus sharing histologic features with other hepatobiliary diseases (Scheuer, 1998). Liver biopsy specimens from patients show portal tract inflammation and sclerosis (Fig. 41.3). Afflicted bile ducts are surrounded by a cuff of lightly inflamed sheets of fibrous tissue and edema, causing the fibrotic layers to separate and form the characteristic onion skin appearance. This classic histologic finding is nearly pathognomonic, but it is seen in fewer than 10% of PSC patients (Ludwig et al, 1981). Affected bile ducts will eventually atrophy and be replaced by rounded scars.
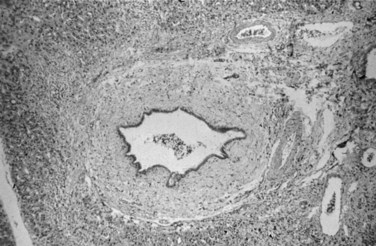
On occasion, similar fibroobliterative cholangitis can be seen in 1) PBC, 2) mechanical obstruction of larger bile ducts, 3) ductopenic rejection following liver transplantation, and 4) after intraarterial infusion of 5-fluorouracil. Involvement of the large intrahepatic and extrahepatic ducts distinguishes PSC from PBC. Inflammation in early-stage PSC is generally milder than that of PBC, but this distinction is difficult to make (Scheuer, 1998). Granulomas, thought to be classically associated with PBC, may be seen in about 4% of biopsies from PSC patients, making the distinction between PSC and PBC even more challenging (Ludwig et al, 1995).
Distinguishing between PSC-related histopathologic features and changes as a result of simple obstruction of distal bile ducts makes the liver biopsy interpretation difficult. Canalicular cholestasis is nonspecific and can occur as a result of any cause of biliary obstruction. As PSC progresses, however, the histopathologic changes of chronic cholestasis spill into the hepatic parenchyma. The most commonly used liver histology grading system proposed by Ludwig is based on these parenchymal changes. Disease was designated as stage 1, portal; stage 2, periportal; stage 3, septal; and stage 4, cirrhotic (Table 41.3; Ludwig et al, 1986). Histologic changes in the same liver can be markedly varied from segment to segment at any given point in time. Whereas histologic staging was formerly used as an independent predictor of the disease’s natural history, the revised PSC survival model does not take into account the histologic stage (Kim et al, 2000).
| Portal stage (stage I) | Portal edema, inflammation, ductal proliferation; abnormalities do not extend beyond the limiting plate |
| Periportal stage (stage II) | Periportal fibrosis and inflammation with or without ductal proliferation; piecemeal necrosis may be present |
| Septal stage (stage III) | Septal fibrosis or bridging necrosis can be identified |
| Cirrhotic stage (stage IV) | Biliary cirrhosis evident |
Etiopathogenesis
The strong association of PSC with IBD has drawn much attention to the potential role of the inflamed colon. The hypothesis suggests that inflammation of the colon may increase permeability to various intraluminal products—toxins, bacteria, inflammatory mediators—that ultimately lead to liver disease. Bacteria or their toxic metabolic products have been considered but have not been conclusively shown to have a pathogenetic role in PSC development (Eade & Brooke, 1969; Palmer et al, 1980). However, investigators have shown that the absence of low-grade, chronic portal vein bacteremia in patients with CUC could potentially elicit the immune response that is responsible for PSC (Warren et al, 1966). Similarly, histologic evidence of bacteremia manifested as portal phlebitis is mild or absent in most PSC patients (Ludwig et al, 1981). Additional information showed that colonic bacteria might metabolize primary bile acids into toxic bile salts that are then absorbed through the ulcerated colon into the portal circulation, but no evidence of bile acid metabolism abnormalities in patients with PSC or CUC has been found (Siegel et al, 1977).
Animal models of PSC have reported that bacterial chemotactic peptides can lead to portal inflammation and histologic changes of PSC (Hobson et al, 1988: Lichtman et al, 1990). Suppression of the inflammatory response was reported in an animal model following inhibition of tumor necrosis factor (TNF). Nevertheless, in a clinical trial of PSC patients with pentoxifylline, a TNF inhibitor, no beneficial effect on symptoms or liver tests was seen (Bharucha et al, 2000). Clinical observations also cast doubt upon the role of the inflamed colonic mucosa in the development of PSC. First, PSC develops in approximately 25% of patients without evidence of IBD. Invasive screening of asymptomatic patients with PSC reveals that many have no endoscopic or histologic evidence of IBD. Second, lack of association between the severity of colonic disease and the likelihood of development and severity of PSC strengthens the skepticism that CUC may not directly cause PSC. Third, failure of proctocolectomy to modify the natural history of PSC argues against a direct causative role of CUC in PSC (Cangemi et al, 1989; Steckman et al, 1984). Thus, the putative contribution of increased colonic permeability in CUC resulting in inflammation of bile ducts remains unclear.
Genetic factors seem to play a role in the development of PSC. First, there have been reports of familial PSC among siblings (Quigley et al, 1983; Jorge et al, 1987). Second, various HLA associations with PSC have been reported, and HLA B8, DR3, DR2, and haplotype A1, B8, and DR3 have been found more frequently in patients with PSC compared with controls (Chapman et al, 1983; Donaldson et al, 1991; Schrumpf et al, 1982; Wiencke et al, 2007). PSC associated with DR4 is a more aggressive disease, although this correlation has not been universally found (Mehal et al, 1994; Olerup et al, 1995). Additional associations with DRB3*0101, DRB1*0301, DQA1*0501, DQB1*0201 and DRB1*1301, and DQA1*0103 and DQB1*0603 have been reported (Donaldson & Norris, 2002). Polymorphisms in the TNF-α receptor have been described as a possible genetic link to PSC. G to A substitution at position −308 in the TNF-α gene has been associated with susceptibility to PSC (Mitchell et al, 2001). A functional variant of stromelysin, matrix metalloproteinase 3, may also influence PSC susceptibility and disease progression (Satsangi et al, 2001). Moreover, variations in the MICA gene (major histocompatibility complex class I–related MIC gene family) have a role in PSC predisposition. Independent of other HLA haplotypes, the MICA 002 allele appears to significantly reduce the risk of PSC, and the MICA 008 allele increases the risk of developing PSC (Norris et al, 2001). Finally, the CCR5-δ32 mutation, characterized by a 32-base pair deletion in the CCR5 gene of T cells, has been associated with susceptibility to PSC development and severity (Eri et al, 2004).
Immune-mediated damage to cholangiocytes seems to be the most likely mechanism leading to PSC. Theoretically, certain HLA molecules and haplotypes may contribute by eliciting an immune response against antigenic epitopes present on biliary epithelia. Enhanced expression of MHC class II antigens (i.e., HLA DR) on cholangiocytes in early-stage PSC has drawn suspicion regarding its role in altered immunity in disease pathogenesis. Aberrant expression of HLA DR is also apparent in PBC and extrahepatic biliary obstruction, suggesting that expression of this antigen is an epiphenomenon rather than an implicit cause of PSC (Broome et al, 1990; Chapman et al, 1988; Van Milligen De Wit et al, 1995).
Decreased hepatic clearance of circulating immune complexes, generalized complement activation, and sharing of a specific epitope between human colonic and biliary epithelial cells have also been reported (Bodenheimer et al, 1983; Minuk et al, 1985; Das et al, 1990; Mandal et al, 1994), yet none of these observations have proven pathogenic associations with PSC. Enhanced interactions of intracellular adhesion molecule 1 (ICAM1) present on cholangiocytes with its cognate ligand on T lymphocytes, leukocyte function–associated antigen 1 (ITGAL), may play a role in PSC development. Genetic polymorphisms of ICAM1 have been implicated in susceptibility to PSC. Homozygote status of the E469E allele for ICAM1 has been associated with protection against PSC (Yang et al, 2004). Both enhanced expression on proliferating cholangiocytes and increased serum levels of ICAM1 have been reported in PSC patients (Van Milligen De Wit et al, 1995; Adams et al, 1991; Bloom et al, 1995).
Associated Diseases
PSC is strongly associated with IBD, most commonly CUC. PSC patients may also have biochemical, serologic, and histologic features of AIH, and a variety of other diseases have been reported to associate with PSC. Given the lack of large studies, it is unclear whether these weak associations are true or simply coincidental. An abridged list of associated diseases is presented in Table 41.4.
Table 41.4 Diseases Associated with Primary Sclerosing Cholangitis
Inflammatory Bowel Diseases
The close association of PSC with IBD, particularly CUC, is widely recognized. IBD is seen in approximately 70% to 80% of patients with PSC, and CUC accounts for 85% to 90% of those patients; Crohn disease is responsible for the rest (Fausa et al, 1991; Loftus et al, 1997). Patients with PSC and Crohn disease may have milder liver disease than patients with PSC and CUC, and PSC has not been seen in Crohn disease involving only the small intestine (Rasmussen et al, 1997). Typically, the diagnosis of IBD is established 8 to 10 years before the liver disease is evident, although there is no clear temporal association, and cases of IBD occurring years after diagnosis of PSC have also been reported (Chapman et al, 1980; Loftus et al, 1996a, 1998). No direct correlation has been found between the severity of bowel disease and the severity of liver disease. Furthermore, therapy of the IBD does not alter the course of the liver disease. For instance, proctocolectomy, the most aggressive treatment for CUC, has no effect on PSC natural history (Cangemi et al, 1989). Colitis is usually milder in patients with both CUC and PSC in comparison to patients with CUC alone.
PSC may play a role in the development of colorectal dysplasia in the setting of CUC, as patients with CUC and PSC show a higher risk of colonic dysplasia compared with patients who have only CUC (Broome et al, 1995a, 1995b). In a study from Sweden, the absolute cumulative risk of developing colorectal dysplasia/carcinoma in patients with PSC and CUC was 9%, 31%, and 50% after 10, 20, and 25 years of disease duration, respectively. In patients with CUC alone, the parallel risk was 2%, 5%, and 10%, respectively (Broome et al, 1995b). Subsequent studies also confirmed the observation of Broom and others, where predicted risk of colorectal carcinoma with PSC and concomitant IBD after 10 years of disease duration was up to 25% (Kornfeld et al, 1997; Claessen et al, 2009). Other investigators have reported that patients with both PSC and CUC were five times more likely to develop colonic dysplasia, based on mucosal biopsy, compared with patients with CUC alone (Brentnall et al, 1996). Whether this finding reflects the fact that PSC patients have milder pancolonic disease that remained undetected for a longer period of time is uncertain. Interestingly, a retrospective case-control study found a similar prevalence of PSC in patients with both CUC and colorectal carcinoma compared with those who had CUC but no neoplasia, that is, no carcinoma or dysplasia (Nuako et al, 1998).
Colonic dysplasia has also been reported in PSC patients without IBD (Broome et al, 1995a, 1995b). However, the risk of colorectal carcinoma in patients with PSC at 10 and 20 years of disease duration was much lower (2% and 2%) than in patients with PSC and concomitant IBD (14% and 31%). PSC patients with CUC continue to have increased risk of colorectal dysplasia and neoplasia, even after liver transplantation (Bleday et al, 1993). The increased neoplastic potential is especially critical in PSC patients following liver transplant, because of the life-long immunosuppression. Annual colonoscopy with surveillance biopsies is recommended in patients with PSC and CUC, because screening and early detection improves survival (Narumi et al, 1995; Loftus et al, 1998). Annual colonoscopy is also suggested for PSC patients without IBD.
Autoimmune Hepatitis
An overlap syndrome between PSC and AIH has been described (Gohlke et al, 1996; Luketic et al, 1997). These patients typically fulfill definite criteria for both, having elevated serum alkaline phosphatase and aminotransferases and increased IgG and antinuclear and/or anti–smooth muscle antibody titers. Liver biopsy reveals moderate to severe interface hepatitis with or without biliary damage, and aminotransferase levels are elevated beyond what would be expected for classic PSC, typically less than three times normal. IBD may also be present in patients with concurrent PSC and AIH but at a lower frequency than in PSC alone. In patients with overlap syndrome, AIH may respond to immunosuppressive therapy. Patients who are seen with AIH but do not respond entirely to immunosuppressant therapy and subsequently develop a cholestatic biochemical profile should be suspected of having concomitant PSC.
Natural History
PSC is an insidious and progressive disease that frequently leads to end-stage liver disease and liver failure. Patients with early-stage PSC are generally asymptomatic. Advanced-stage PSC is marked by complications of chronic cholestasis and end-stage liver disease. Prior appendectomy may delay the onset of PSC but does not affect the prevalence or severity of the disease (Florin et al, 2004). In the absence of liver transplantation, the median survival from the time of diagnosis is about 12 years. Even asymptomatic patients have decreased survival compared to age-matched controls (Wiesner et al, 1989). In a study of 45 PSC patients who were asymptomatic at the time of diagnosis, 76% had progression of PSC, and 31% had liver failure develop with a mean follow-up of 6.25 years (Porayko et al, 1990). Early diagnosis of PSC is associated with longer survival because of presumed lead-time bias (Broome et al, 1996; Okolicsanyi et al, 1996).
The progressive pattern and severe complications of PSC warrant close medical management and intervening treatment. Prognostic models for PSC have been developed to predict survival and identify the ideal timing for liver transplantation. Cox multivariable regression analysis has been widely used to define the variables for these models. The revised Mayo PSC natural history model uses six independent, reproducible parameters to estimate the survival of PSC patients. These variables include age, bilirubin, albumin, aspartate aminotransferase, and history of variceal bleeding (Kim et al, 2000). Recently the Model for End-Stage Liver Disease (MELD) has been widely used to ensure organ allocation priority to end-stage liver disease patients with the highest mortality risk (Kamath et al, 2001).
Small-duct PSC, although rare, has a favorable long-term prognosis. In a multiinstitutional and multinational study, the 83 small-duct PSC patients were matched by age, gender, year of diagnosis, and institution to two patients with large-duct PSC. Approximately 23% patients with small-duct PSC progressed to large-duct PSC over a median of 7.4 years. Patients with small-duct PSC did not develop cholangiocarcinoma, unless the disease progressed to large-duct PSC. The same study showed patients with small-duct PSC had a statistically longer transplantation-free survival: 13 years compared with 10 years. Two patients with small-duct PSC who underwent liver transplantation did have a recurrence of small-duct PSC in the graft after 9 years (Bjornsson et al, 2008). In children with PSC who have progressive disease, the median survival despite medical therapy, and in the absence of liver transplantation, was reported to be 12.7 years (Feldstein et al, 2003).
Specific Complications
Cholelithiasis and Choledocholithiasis (See Chapters 30 and 35)
Although PSC is a disease affecting mainly young men, it is surprising that 25% to 30% will have calculi either in the gallbladder or biliary tree. In a study of 121 patients with PSC, 26% had gallstones, half of which were pigment stones, although the spectrum of pathology in the gallbladder is not limited to stones (Brandt et al, 1988). The same study showed that PSC directly involved the gallbladder in 15% of patients. The gallbladder can also suffer an unusual form of acalculous cholecystitis characterized by a diffuse lymphoplasmacytic infiltrate (Jessurun et al, 1998). Finally, in patients with PSC, gallbladder polyps require special attention. In a study of 102 patients with PSC, 14 patients (13.7%) had an intraluminal gallbladder mass, and eight of these lesions (57%) were adenocarcinomas (see Chapter 49). Recent case series showed that adenocarcinomas of the gallbladder can occur in polyps less than 1 cm (Karlsen et al, 2008), thus in PSC patients with gallbladder polyps, cholecystectomy is recommended (Buckels et al, 2002).
Radiologic studies have shown that intrahepatic calculi are present in approximately 8% of PSC patients (Dodd et al, 1997). Biliary calculi can serve as a nidus for the development of bacterial cholangitis in PSC patients, although this is uncommon in the absence of dominant biliary strictures or prior bile duct surgery. Evidence of bacterial cholangitis requires endoscopic evaluation to remove possible bile duct calculi and/or to dilate biliary strictures. Bacterial cholangitis commonly occurs in patients with PSC following ERCP, thus prophylactic coverage with antibiotics prior to (i.e., a single intravenous dose) and following ERCP (i.e., oral ciprofloxacin for 10 days) is recommended for PSC patients.
Dominant Biliary Strictures
Dominant strictures occur in 20% to 45% of PSC patients and present with progression of jaundice, pruritus, bacterial cholangitis, and right upper quadrant pain. ERCP is necessary to evaluate the bile ducts and allow therapeutic dilation with or without biliary stenting. In a prospective study, 12 symptomatic PSC patients with major ductal strictures were treated with repeated balloon dilation. Eight patients showed sustained improvement following an average of three treatment sessions to obtain satisfactory dilation of the affected bile ducts (Wagner et al, 1996). A retrospective study of 25 PSC patients with symptomatic dominant strictures showed that endoscopic stenting was technically successful in 21 patients (84%) and was associated with significant improvement of serum liver tests. Moreover, 12 (57%) of 21 patients with PSC remained asymptomatic with stable liver biochemistries, and four patients (19%) had clinical and biochemical relapse over a median follow-up of 29 months. All four patients with relapse responded favorably to additional endoscopic therapy (Van Milligen De Wit et al, 1996). The same investigators reported that in 16 patients with symptomatic PSC, short-term biliary stent placement (median, 9 days) was effective, with 13 patients (81%) remaining symptom free and without biochemical evidence of cholestasis after a median follow-up of 19 months (Van Milligen De Wit et al, 1997).
Despite these findings, it is unclear whether dominant biliary strictures are directly responsible for the cholestasis of PSC patients. In a retrospective study of 125 patients with PSC, 56 (45%) had dominant strictures, defined by stenosis of the common bile duct less than 1.5 mm in diameter and/or stenosis of the right or left hepatic duct less than 1.0 mm. Of interest, in PSC patients with (n = 56) or without (n = 69) dominant strictures up to 12 months after cholangiography, alkaline phosphatase and bilirubin levels were not significantly different (Bjornsson et al, 2004).
Cholangiocarcinoma
The most ominous complication of PSC is the development of cholangiocarcinoma (see Chapters 50A, 50B, and 50C), which is estimated to occur in 8% to 15% of patients during the course of the disease (Chapman, 2003; Claessen et al, 2009). In a study of 161 patients with PSC, approximately 7% had cholangiocarcinoma develop over a mean follow-up of 11.5 years (Burak et al, 2004). Among PSC patients, the estimated annual incidence of cholangiocarcinoma is approximately 1.5% (Gores, 2003). The cumulative risk of biliary tract malignancy is approximately 11.2% at 10 years from diagnosis of PSC (Kornfeld et al, 1997). Moreover, at the time of liver transplantation, approximately 10% of explanted livers have histopathologic evidence of unsuspected cholangiocarcinoma.
Cholangiocarcinoma is usually diagnosed at an advanced stage because of its insidious nature. Many of the signs and symptoms associated with cholangiocarcinoma are typical of PSC itself, making early detection of cholangiocarcinoma difficult. Suspicion for cholangiocarcinoma should be raised when a patient with PSC is seen with rapidly progressive jaundice, weight loss, or abdominal discomfort. In a study comparing PSC patients who developed cholangiocarcinoma with those who did not, the investigators found no clinical or biochemical features that could herald the onset of biliary malignancy in the year prior to diagnosis (Bergquist et al, 1998). In patients with PSC, risk factors for developing cholangiocarcinoma include age, liver histologic stage, concurrent CUC (Rosen & Nagorney, 1991), smoking (Bergquist et al, 1998), and history of variceal bleeding (Burak et al, 2004).
In PSC patients, dysplasia of the biliary epithelium is likely the greatest predisposing factor of cholangiocarcinoma. In a study from the United Kingdom, biliary dysplasia was detected in approximately 20% of liver biopsies from 26 patients with PSC and cholangiocarcinoma, but not in a single case of 60 PSC patients who were free of biliary malignancy for a follow-up period of 2 years (Fleming et al, 2001). Moreover, recognition of biliary dysplasia by three independent pathologists showed moderate reproducibility, further suggesting dysplasia as a feasible indicator of present or developing biliary malignancy (Fleming et al, 2001). Likewise, colonic dysplasia seems to be a risk factor for cholangiocarcinoma, with risk increased significantly in patients with PSC and CUC (Broome et al, 1995b); therefore detection of dysplasia in either the biliary tree or colon requires vigilant surveillance for cholangiocarcinoma and may warrant consideration for liver transplantation under certain circumstances.
Early detection of cholangiocarcinoma, and thus promise for cure, has been hampered by the low sensitivity and specificity of standard diagnostic approaches and techniques. Cholangiocarcinoma of the large bile ducts is not easily detectable by cross-sectional imaging. In this case, ERCP is perhaps the best approach to evaluate for bile duct cancer. To this end, cholangiographic features suggestive of cholangiocarcinoma have been described, but the accurate distinction between benign and malignant biliary disease is often impossible. In fact, about 10% of malignant-appearing biliary strictures are benign (Hadjis et al, 1985). Biliary brush cytology specimens and fine-needle biopsies obtained at ERCP are at best 30% to 40% sensitive for securing the diagnosis of cholangiocarcinoma. A novel test for early detection of cholangiocarcinoma is fluorescence in situ hybridization (FISH) on bile duct cytology specimens. In a study on detection of malignant biliary strictures, the sensitivity of FISH (34%) was better than that of standard cytology (15%, P < .01), but the specificity of FISH and cytology was similar, at 91% and 98%, respectively (P = .06) (Kipp et al, 2004). A recent study of 498 patients undergoing ERCP for pancreatobiliary stricture demonstrated that polysomy of FISH had a significantly higher sensitivity (42.9%) than routine cytology (20.1%) with identical specificity (99.6%) (Fritcher et al, 2009).
Carbohydrate antigen 19-9 (CA19-9) has been used with questionable accuracy to aid the diagnosis of cholangiocarcinoma. In patients with PSC, a cutoff serum level of 129 U/mL is estimated to have a diagnostic sensitivity of 89% and specificity of 98% for diagnosing cholangiocarcinoma (Levy et al, 2005). Similar elevations of CA19-9 can be seen in patients with pancreatic malignancies, bacterial cholangitis, and in active smokers. In PSC patients, periodic CA19-9 testing is recommended, and a sustained rise should raise suspicion for possible development of cholangiocarcinoma. Others have shown that a carcinoembryonic antigen (CEA) level greater than 5 ng/mL was 100% specific but only 38% sensitive for detection of cholangiocarcinoma (Nichols et al, 1993). Another study using an index of CA19-9 and CEA showed an 86% diagnostic accuracy for detecting cholangiocarcinoma. Although most of these patients had radiologically occult disease, and several went on to liver transplantation, the great majority still died from cholangiocarcinoma (Ramage et al, 1995). At present, we have no reliable means to detect cholangiocarcinoma early, at a curable stage, although work is underway to find dependable molecular markers for early cholangiocarcinoma detection.
Endoscopic ultrasound (EUS; see Chapter 14) is better than CT and magnetic resonance imaging (MRI) to evaluate regional lymph nodes for possible cholangiocarcinoma metastasis, including the option to obtain a biopsy of questionable lesions (Gores, 2000). Positron emission tomography (PET) is a promising tool for detection of cholangiocarcinoma in PSC (see Chapter 15), and it can detect small cholangiocarcinomas arising in the setting of PSC with 100% accuracy compared with control groups (Keiding et al, 1998). A blinded study in Sweden examined 24 pretransplant PSC patients without evidence of malignancy on CT, MRI, and US, and PET correctly identified three of three patients with explanted liver pathology showing cholangiocarcinoma. But PET failed to detect a high-grade hilar ductal dysplasia and was falsely positive in a patient with benign epithelioid granulomas (Prytz et al, 2006), therefore, further investigations are necessary to define the role of PET in the diagnosis and routine surveillance of cholangiocarcinoma.
Nonspecific Primary Sclerosing Cholangitis Complications
Many patients with PSC complain of fatigue and pruritus. The etiology of fatigue is unclear in chronic cholestatic liver diseases, including PSC, and this can affect patients’ quality of life. In PSC patients, the pruritus can be debilitating; although its mechanism is not well defined, endogenous opioids and retention of other unknown factors usually excreted in the bile may play some role in its development (Bergasa & Jones, 1995; Spivey et al, 1994; Jones & Bergasa, 1990).
Patients with PSC may also suffer from steatorrhea and subsequent deficiencies of fat-soluble vitamins. In a pretransplant group of PSC patients, deficiencies of vitamins A, D, and E were present in 82%, 57%, and 43% of patients, respectively (Jorgensen et al, 1995). These deficiencies can be treated with simple vitamin supplementation. In patients with PSC and steatorrhea, consideration should be given to exclude celiac sprue or chronic pancreatitis, because either entity can coexist with PSC, and both conditions are treatable causes of fat malabsorption (Hay et al, 1988). Metabolic bone disease is also very common in PSC. Osteopenic bone disease can be severe in advanced-stage PSC, with 50% of patients having a bone mineral density below the fracture threshold (Hay et al, 1991). Although the majority of PSC patients are males, bone biopsies revealed findings consistent with osteoporosis rather than osteomalacia.
Patients with PSC have complications of portal hypertension, just as patients with other causes of liver cirrhosis do (see Chapter 70A, Chapter 70B, Chapter 74, Chapter 75A, Chapter 75B, Chapter 75C ). In a study of 283 patients with PSC, 102 patients (36%) had esophageal varices, including 57 (56%) of moderate to large size. A recent study of PSC patients reported that platelet count, albumin level, and advanced histologic disease were independent predictors of esophageal varices (Zein et al, 2004), which are usually managed with endoscopic banding. If these measures are ineffective, a shunting procedure (TIPS) can be performed as a bridge to liver transplantation. Patients with advanced-stage PSC develop ascites, spontaneous bacterial peritonitis, and encephalopathy. Treatment of such complications is similar to that of other causes of end-stage liver disease, with liver transplantation considered the optimal management (see Chapter 70A, Chapter 70B , 74, 75, and 97A).
Treatment
Therapy of Specific Primary Sclerosing Cholangitis Complications
Cholelithiasis and Choledocholithiasis
Symptomatic gallbladder disease in early-stage PSC patients should be treated with cholecystectomy. In an asymptomatic patient with PSC, presence of an intraluminal gallbladder mass requires cholecystectomy. In a study of PSC patients who underwent cholecystectomy, 13.7% of patients were found to have a gallbladder mass, and 57% were adenocarcinomas (Buckels et al, 2002). Cholecystectomy is not normally advocated for polyps less than 1 cm, but it may be justified in PSC patients; adenocarcinomas have been reported in recent case series of polyps less than 1 cm (Karlsen et al, 2008).
Following the diagnosis of choledocholithiasis, the therapeutic intervention of choice is ERCP (see Chapter 27). During this procedure, endoscopic sphincterectomy is performed with removal of biliary stones and dilation of possible strictures. Temporary biliary stent placement is typically recommended but depends on the clinical circumstances.
Dominant Biliary Stricture and Recurrent Bacterial Cholangitis
Dominant biliary stricture should be evaluated by ERCP or percutaneous transhepatic cholangiography (PTC) to ensure comprehensive imaging of the biliary tree and to permit biliary brushings and biopsies of affected areas to exclude cholangiocarcinoma. The main concern is to scrutinize whether the observed biliary stricture represents cholangiocarcinoma or a benign lesion. Dominant stricture is defined as a common duct diameter less than 1.5 mm and a hepatic duct less than 1 mm (Stiehl et al, 2002).
Therapy of Nonspecific Complications
Pruritus
Patients with advanced-stage PSC frequently complain of intense pruritus. This distressing symptom can improve using various medical therapies (Angulo & Lindor, 1999). Cholestyramine is a nonabsorbable bile acid–binding resin that decreases the intestinal absorption of bile acids and alleviates pruritus. Phenobarbital has been used in conjunction with cholestyramine to treat PSC patients with nocturnal pruritus. Ursodeoxycholic acid (ursodiol)—a hydrophilic bile acid that likely replaces hydrophobic, toxic bile acids from the bile pool—may also improve pruritus. Antihistamines, such as hydroxyzine and diphenhydramine, can be used as supplements to cholestyramine or ursodeoxycholic acid, particularly for nocturnal pruritus, because of their sedative properties. Rifampin may also improve pruritus, although its potential side effects, specifically drug-induced hepatitis, make it a second-line agent. Opiate antagonists like naloxone, nalmefene, and naltrexone are also used to alleviate pruritus (Bergasa, 2004).
Steatorrhea, Fat-Soluble Vitamin Deficiency, and Hepatic Osteodystrophy
Prolonged cholestasis causes decreased intestinal bile acid concentration. Therefore, patients with advanced-stage PSC may develop fat malabsorption and steatorrhea, although PSC patients who develop steatorrhea should first be evaluated for other coexisting causes of steatorrhea, including celiac sprue and pancreatic insufficiency (Hay et al, 1988). Because of intraluminal bile acid deficiency, steatorrhea may improve from dietary changes, such as lowering daily fat intake and substituting medium-chain triglycerides for long-chain ones.
Therapy of Hepatobiliary Disease
A double-blind, randomized, controlled study involving 105 PSC patients and standard dose UDCA (13 to 15 mg/kg body weight/day) failed to demonstrate clinical benefit in the treatment group compared with controls over a median follow-up of 2.2 years. Subsequently, pilot studies demonstrated promise with high-dose UDCA in PSC. A small, double-blind, placebo-controlled study from the United Kingdom reported that PSC patients (n = 13) who received high-dose (20 mg/kg body weight/day) UDCA had significant improvement in liver biochemistries, cholangiographic appearance, and reduction of liver fibrosis compared with patients taking placebo (n = 13) (Mitchell et al, 2001). In a similar U.S. study, 30 PSC patients were treated with high-dose (25 to 30 mg/kg body weight/day) UDCA, with reported improvement of the Mayo PSC risk score at the end of therapy (Harnois et al, 2001). Unfortunately, two separate large, randomized, double-blind, placebo-controlled trials indexing a total of 219 and 150 patients, respectively, with extended follow-up over 5 years, showed that long-term, high-dose UDCA (17 to 30 mg/kg body weight/day) improved liver tests but did not decrease clinical end points of cirrhosis, varices, cholangiocarcinoma, liver transplantation, or death (Olsson et al, 2005; Lindor et al, 2009). In fact, Lindor and others demonstrated that adverse events were more likely to occur in the high-dose UDCA group than in the placebo group.
Although UDCA plays a limited role in slowing the progression of liver disease, it has been shown to affect the frequency of colonic neoplasia or cancer in patients with PSC and CUC. In a cross-sectional study of 59 patients with both PSC and CUC who were undergoing colonoscopic surveillance, UDCA use was associated with decreased prevalence of colonic dysplasia (Tung et al, 2001). In another randomized, placebo-controlled trial of 52 patients with concurrent PSC and CUC, use of UDCA resulted in a relative risk of 0.26 for developing colorectal dysplasia or cancer (Pardi et al, 2003). Additional, prospective, randomized controlled studies are needed to verify the proposed chemopreventive effect of UDCA in patients with PSC and CUC.
Surgical Therapy
Reconstructive Biliary Surgery
In 1988, Cameron and colleagues reported the Johns Hopkins experience of reconstructive biliary surgery in 31 PSC patients who underwent this procedure for persistent jaundice or recurrent bacterial cholangitis (Cameron et al, 1988). This surgical approach included excision of the hepatic duct bifurcation and extrahepatic biliary tree, dilation of the intrahepatic ducts, insertion of Silastic transhepatic biliary stents, and bilateral hepaticojejunostomies. Five of 31 patients had cirrhosis. Of the 26 patients without cirrhosis, the 1-, 3-, and 5-year survival rates were 92%, 87%, and 71%, respectively. The mean bilirubin in the noncirrhotic group improved from 9.9 mg/dL to 4.3 mg/dL after 5 years. In contrast, two of the five patients with cirrhosis died after surgery, and only one survived the after the first postoperative year. The authors concluded that primary biliary reconstruction should be considered in noncirrhotic patients with severe stricturing at or near the hepatic duct bifurcation or in the distal biliary tree. Patients with cirrhosis should be referred for liver transplantation.
Over the past decade, biliary reconstructive surgery for PSC has become less common, because endoscopic techniques for bile duct dilation (see Chapter 27) and stenting, as well as outcomes with liver transplantation, have improved (see Chapter 97A). In addition, hesitation arose after several centers reported increased difficulty performing liver transplantation in patients who had previous biliary surgery. Despite this, biliary-enteric anastomosis may still be indicated in noncirrhotic patients with primarily extrahepatic disease. To compare efficacy of these different modes of therapy, Ahrendt and colleagues (1998) retrospectively reviewed 146 patients with PSC seen at the Johns Hopkins Hospital from 1980 to 1994. Of these, 50 patients underwent resection of the extrahepatic bile ducts and long-term transhepatic stenting (replaced every 2.2 months on average), 54 patients were managed nonoperatively with endoscopic balloon dilation with or without percutaneous stenting, 28 patients were treated medically, and 21 patients received liver transplantation (Ahrendt et al, 1998). Of the 50 patients managed with resection of the extrahepatic biliary tree, three patients (6%) died prior to hospital discharge, two of whom had cirrhosis. Of 21 patients, four (19%) died after liver transplant; there were no postprocedural deaths in the endoscopically managed group. In noncirrhotic patients managed with resection, bilirubin levels fell significantly from 8.2 mg/dL preoperatively to just over 3 mg/dL after 5 years. By comparison, those patients managed nonoperatively with endoscopic or percutaneous procedures had a slight lowering of their serum bilirubin levels at 1 and 3 years but returned to pretreatment levels by 5 years. Most significant, however, was the improvement in 5-year and transplant-free survival in patients managed with resection (85% and 78%, respectively) versus those managed with endoscopic dilation and/or percutaneous stenting (59% and 46%, respectively). Part of the difference in survival was due to the development of cholangiocarcinoma in three of the endoscopically managed patients. As the majority of cholangiocarcinomas occur in the perihilar region, this may explain why none of the patients who underwent resection died from this complication.
Myburgh (1994) reported on 24 patients with biliary enteric bypass without the need for transanastomotic stenting. With the Hepp-Couinaud technique, a side-to-side anastomosis 2.5 to 3.5 cm wide was performed between a Roux-en-Y loop of jejunum and the right and left hepatic ducts at their confluence. Without applying stents, the need of stent replacement, and thus the increased risk for bacterial cholangitis, was eliminated. Among the 16 noncirrhotic patients who underwent bypass, survival was 100%, and 11 remained free of jaundice with a median follow-up of 6.5 years; however, patients with cirrhosis derived minimal benefit (Myburgh, 1994).
It appears that biliary reconstructive surgery may be an effective palliative measure for selected noncirrhotic patients with primarily extrahepatic PSC. However, the association of biliary-enteric drainage surgical procedures with development of cholangiocarcinoma is a significant concern (Tocchi et al, 2001). In our practice, we refer PSC patients with dominant biliary strictures for endoscopic dilation, given the high degree of success attained by this procedure and its relative ease and low morbidity/mortality compared with biliary surgery. We recommend biliary reconstructive surgery for those few PSC patients who have persistent cholestasis but no cirrhosis; the biliary obstruction is not amenable to endoscopic treatment, and they are not candidates for liver transplantation.
Orthotopic Liver Transplantation
Orthotopic liver transplantation (OLT; see Chapter 97A) remains the most effective treatment for PSC. At the Mayo Clinic, the 1- and 5-year patient survival rates for PSC are 92% and 86%, respectively. These rates compare favorably with results of OLT for other chronic liver diseases. Wiesner and colleagues (1996) reported outcomes for patients who underwent OLT for PSC and identified risk factors that adversely affected outcome. These factors can be divided into those that influence OLT outcome and those that are specific for PSC. The former include residence in the intensive care unit or being on life support prior to transplantation, age greater than 65 years, poor nutritional status, Child-Pugh class C, and renal failure requiring dialysis prior to or after transplantation. These factors are also predictive of increased blood loss, prolonged ICU stay, and major postoperative complications. Risk factors specific for PSC are disease severity, previous biliary or shunt surgery, concurrent bile duct cancer, and presence of IBD.
Controversy surrounds the impact of prior biliary surgery on subsequent OLT for PSC. There is little doubt that prior biliary surgery increases the technical difficulty of OLT, but it is unknown whether this event affects survival. In a series of 26 patients, Farges and colleagues (1995) reported increased operative time, increased blood loss, and severe complications—including death—in 12 OLT patients who had undergone previous right upper quadrant abdominal operations. In a combined series of 216 patients from the University of Pittsburgh and Mayo Clinic, prior biliary tract and/or portal hypertensive surgery was associated with increased mortality the first 5 years after OLT, but this did not reach statistical significance (Abu-Elmagd et al, 1993). At the University of California at San Francisco, increased operative time and blood loss were reported, but not increased mortality, in PSC patients with history of prior colectomy or biliary surgery other than simple cholecystectomy (Narumi et al, 1995). Ahrendt and others (1998) reported the Johns Hopkins experience with 21 PSC patients who had undergone OLT; they found significantly increased operative time and blood loss in patients with previous biliary tract operations, as well as increased operative mortality, but this finding did not reach statistical significance. These reports suggest that prior biliary tract surgery increases the technical difficulty of OLT for PSC with a trend toward increased mortality.
After OLT, complications occur that are unique to PSC patients. Increased rates of biliary strictures have been noted in patients transplanted for PSC. This increase may not represent disease recurrence, as other factors may also cause biliary stricturing, such as ischemia related to chronic rejection or possible chronic low-grade bacterial cholangitis resulting from the Roux-en-Y anastomosis, which is performed much more frequently in patients with PSC. However, a University of Pittsburgh study found a significantly greater incidence of biliary strictures in allografts of patients transplanted for PSC versus patients who had OLT and choledochojejunostomy anastomosis for other end-stage liver disease (Sheng et al, 1993). Because cholangiographic and other clinical and biochemical criteria for recurrent disease have not been widely accepted, there is no consensus regarding incidence of recurrent PSC in liver allografts. Careful analysis at our institution concluded that 20% of patients transplanted for PSC developed recurrent disease based on characteristic cholangiographic and histologic features (Graziadei et al, 1999). PSC may recur earlier at a higher ratio after living-donor liver transplantation, particularly when the graft is from a biologically related living donor (Tamura et al, 2007). Proposed risk factors for recurrent PSC include IBD, prolonged ischemia time, number of cellular rejection episodes, prior biliary surgery, cytomegalovirus infection, and lymphotoxic cross match (Gautam et al, 2006).
Acute and chronic ductopenic rejection can be severe and steroid resistant, often resulting in graft loss. In a study of 100 consecutive PSC patients transplanted at Baylor University, it was reported that chronic rejection and disease recurrence occurred in 13% and 16%, respectively. These events adversely affected both graft and patient survival, markedly so in those patients with chronic rejection (Jeyarajah et al, 1998). Five-year graft survival rates were 33% and 65%, respectively, for patients with chronic rejection and disease recurrence compared with 76% for patients free of chronic rejection or recurrence. The authors suggested that chronic rejection and disease recurrence after transplantation for PSC are distinct entities, as evidenced by the difference in outcome, and that they should be managed accordingly.
Because many patients with PSC have CUC, concern has arisen over the possibility that OLT and attendant lifelong immunosuppression may increase the risk of colorectal carcinoma. In a study of 108 patients with PSC and concomitant IBD who underwent OLT, Loftus and others reported a fourfold, but not statistically significant, increase in colon carcinoma in the group that did not have a prior colectomy compared with the expected colon cancer in a group with comparable (pre-OLT) duration of IBD. This finding, however, did not affect patient survival (Loftus et al, 1998). Goss and colleagues (1997) also reported no difference in PSC patient survival in groups with or without a prior colectomy. Given the lack of impact on survival, we do not recommend prophylactic proctocolectomy in PSC patients with IBD who undergo OLT. Nonetheless, the risk of colonic neoplasia in these transplanted PSC patients warrants annual surveillance colonoscopy with biopsies and colectomy, even when only low-grade dysplasia is detected.
PSC patients with cholangiocarcinoma undergoing OLT merit additional discussion (see Chapter 97E). Survival after OLT for patients with PSC and cholangiocarcinoma is dependent upon how and when the cancer is detected. Generally speaking, in PSC patients in whom cholangiocarcinoma was known preoperatively, OLT has been ineffective; indeed, recurrence of cholangiocarcinoma occurred almost uniformly. However, incidental cholangiocarcinomas, defined as tumors less than 1 cm in diameter discovered only at the time of pathologic sectioning of the explanted liver, portend a better prognosis. In a UCLA study, 8% of patients transplanted for PSC had an incidental cholangiocarcinoma with 5-year survival of 83%, which is not significantly different from the survival of patients undergoing OLT without incidental cholangiocarcinoma (Goss et al, 1997).
Resection of Cholangiocarcinoma
In the absence of PSC, surgical resection has been the most effective and only potentially curative treatment for cholangiocarcinoma. In patients with PSC and cholangiocarcinoma, resection is discouraged: cholangiocarcinoma is often multifocal, underlying parenchymal disease may preclude resection, and reported recurrent disease with death occurs in more than 90% of patients. This is because PSC is considered a premalignant disease. Historically, cholangiocarcinoma has been regarded as a contraindication for liver transplantation given high tumor recurrence in the transplanted organ, and disease-free and overall survival are no different from those of resection. However, selected liver transplant centers using radiation therapy, chemotherapy, and abdominal exploration prior to transplantation have shown promising outcomes for patients with hilar cholangiocarcinoma, with 5-year survival of 80% (Heimbach, 2008).
Proctocolectomy
The presence of PSC in patients with CUC affects the management of CUC. Much has already been said regarding the increased risk of dysplasia and colon carcinoma in patients with PSC. We currently recommend removal of the colon only for indications pertinent to IBD. However, the decision to perform a Brooke ileostomy or an ileal pouch–anal anastomosis (IPAA) is greatly influenced by the presence of PSC. In a retrospective analysis of patients with PSC and CUC treated with either Brooke ileostomy or IPAA, 8 (26%) of 31 patients with the ileostomy developed peristomal varices and subsequent bleeding, but none of the 40 patients who underwent IPAA developed perianastomotic varices or perineal bleeding (Kartheuser et al, 1996). The cumulative risk of pouchitis at 10 years after IPAA was 61% in patients with PSC compared with 36% in patients with CUC alone (Penna et al, 1996). Therefore in PSC patients who need proctocolectomy, we recommend IPAA and not Brooke ileostomy, because treating pouchitis is simpler than managing bleeding peristomal varices.
Abdalian R, Heathcote EJ. Sclerosing cholangitis: a focus on secondary causes. Hepatology. 2006;44:1063-1074.
Abu-Elmagd KM, et al. Efficacy of hepatic transplantation in patients with primary sclerosing cholangitis. Surg Gynecol Obstet. 1993;177:335-344.
Adams DH, et al. Increased expression of intercellular adhesion molecule 1 on bile ducts in primary biliary cirrhosis and primary sclerosing cholangitis. Hepatology. 1991;14:426-431.
Ahrendt SA, et al. Primary sclerosing cholangitis: resect, dilate, or transplant? Ann Surg. 1998;227:412-423.
Angulo P, Lindor KD. Primary sclerosing cholangitis. Hepatology. 1999;30:325-332.
Angulo P, Maor-Kendler Y, Lindor KD. Small-duct primary sclerosing cholangitis: a long-term follow-up study. Hepatology. 2002;35:1494-1500.
Angulo P, et al. Serum autoantibodies in patients with primary sclerosing cholangitis. J Hepatol. 2000;32:182-187.
Balasubramanian K, Wiesner RH, LaRusso NF. Primary sclerosing cholangitis with normal serum alkaline phosphatase activity. Gastroenterology. 1988;95:1395-1398.
Bambha K, et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology. 2003;125:1364-1369.
Bangarulingam SY, et al. Complications of endoscopic retrograde cholangiopancreatography in primary sclerosing cholangitis. Am J Gastroenterol. 2009;104:855-860.
Bergasa NV. Pruritus in chronic liver disease: mechanisms and treatment. Curr Gastroenterol Rep. 2004;6:10-16.
Bergasa NV, Jones EA. The pruritus of cholestasis: potential pathogenic and therapeutic implications of opioids. Gastroenterology. 1995;108:1582-1588.
Bergquist A, et al. Risk factors and clinical presentation of hepatobiliary carcinoma in patients with primary sclerosing cholangitis: a case-control study. Hepatology. 1998;27:311-316.
Berstad AE, et al. Diagnostic accuracy of magnetic resonance and endoscopic retrograde cholangiography in primary sclerosing cholangitis. Clin Gastroenterol Hepatol. 2006;4:514-520.
Bharucha AE, et al. A pilot study of pentoxifylline for the treatment of primary sclerosing cholangitis. Am J Gastroenterol. 2000;95:2338-2342.
Bjornsson E, et al. Dominant strictures in patients with primary sclerosing cholangitis. Am J Gastroenterol. 2004;99:502-508.
Bjornsson E, et al. The natural history of small-duct primary sclerosing cholangitis. Gastroenterology. 2008;134:975-980.
Bleday R, et al. Increased risk of early colorectal neoplasms after hepatic transplant in patients with inflammatory bowel disease. Dis Colon Rectum. 1993;36:908-912.
Bloom S, Fleming K, Chapman R. Adhesion molecule expression in primary sclerosing cholangitis and primary biliary cirrhosis. Gut. 1995;36:604-609.
Bodenheimer HCJr, et al. Elevated circulating immune complexes in primary sclerosing cholangitis. Hepatology. 1983;3:150-154.
Brandt DJ, et al. Gallbladder disease in patients with primary sclerosing cholangitis. Am J Roentgenol. 1988;150:571-574.
Brentnall TA, et al. Risk and natural history of colonic neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis. Gastroenterology. 1996;110:331-338.
Broome U, et al. Distribution of HLA-DR, HLA-DP, and HLA-DQ antigens in liver tissue from patients with primary sclerosing cholangitis. Scand J Gastroenterol. 1990;25:54-58.
Broome U, et al. Subclinical time span of inflammatory bowel disease in patients with primary sclerosing cholangitis. Dis Colon Rectum. 1995;38:1301-1305.
Broome U, et al. Primary sclerosing cholangitis and ulcerative colitis: evidence for increased neoplastic potential. Hepatology. 1995;22:1404-1408.
Broome U, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut. 1996;38:610-615.
Buckels DC, et al. In primary sclerosing cholangitis, gallbladder polyps are frequently malignant. Am J Gastroenterol. 2002;97:1138-1142.
Burak K, et al. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am J Gastroenterol. 2004;99:523-526.
Burak KW, et al. Is there a role for liver biopsy in primary sclerosing cholangitis? Am J Gastroenterol. 2003;98:1155-1158.
Caldwell SH, et al. Imaging and clinical characteristics of focal atrophy of segments 2 and 3 in primary sclerosing cholangitis. J Gastroenterol Hepatol. 2001;16:220-224.
Cameron JL, et al. Resection of hepatic duct bifurcation and transhepatic stenting for sclerosing cholangitis. Ann Surg. 1988;207:614-620.
Campbell WL, et al. Biliary tract carcinoma complicating primary sclerosing cholangitis: evaluation with CT, cholangiography, US, and MR imaging. Radiology. 1998;207:41-50.
Campbell WL, et al. Using CT and cholangiography to diagnose biliary tract carcinoma complicating primary sclerosing cholangitis. Am J Roentgenol. 2001;177:1095-1100.
Cangemi JR, et al. Effect of proctocolectomy for chronic ulcerative colitis on the natural history of primary sclerosing cholangitis. Gastroenterology. 1989;96:790-794.
Chalasani N, et al. Cholangiocarcinoma in patients with primary sclerosing cholangitis: a multicenter case-control study. Hepatology. 2000;31:7-11.
Chapman RW. The management of primary sclerosing cholangitis. Curr Gastroenterol Rep. 2003;5:9-17.
Chapman RW, et al. Association of primary sclerosing cholangitis with HLA-B8. Gut. 1983;24:38-41.
Chapman RW, et al. Expression of HLA-DR antigens on bile duct epithelium in primary sclerosing cholangitis. Gut. 1988;29:422-427.
Chapman RWG, et al. Primary sclerosing cholangitis: a review of its clinical features, cholangiography, and hepatic histology. Gut. 1980;21:870-877.
Claessen MM, et al. High lifetime risk of cancer in primary sclerosing cholangitis. J Hepatol. 2009;50:158-164.
Czaja AJ. Frequency and nature of the variant syndromes of autoimmune liver disease. Hepatology. 1998;28:360-365.
Das KM, Vecchi M, Sakamakis S. A shared and unique epitope(s) on human colon, skin and biliary epithelium detected by monoclonal antibody. Gastroenterology. 1990;98:464-469.
De Groen PC, et al. Biliary tract cancers. N Engl J Med. 1999;341:1368-1378.
Dodd GDIII, et al. Bile duct calculi in patients with primary sclerosing cholangitis. Radiology. 1997;203:443-447.
Donaldson PT, Norris S. Evaluation of the role of MHC class II alleles, haplotypes and selected amino acid sequences in primary sclerosing cholangitis. Autoimmunity. 2002;35:555-564.
Donaldson PT, et al. Dual association of HLA DR2 and DR3 with primary sclerosing cholangitis. Hepatology. 1991;13:129-133.
Eade MN, Brooke BN. Portal bacteremia in cases of ulcerative colitis submitted to colectomy. Lancet. 1969;1:1008-1009.
El-Shabrawi M, et al. Primary sclerosing cholangitis in childhood. Gastroenterology. 1987;92:1226-1235.
Eri R, et al. CCR5-Delta32 mutation is strongly associated with primary sclerosing cholangitis. Genes Immun. 2004;5:444-450.
Farges O, et al. Primary sclerosing cholangitis: liver transplantation or biliary surgery? Surgery. 1995;117:146-155.
Fausa O, Schrumpf E, Elgjo K. Relationship of inflammatory bowel disease and primary sclerosing cholangitis. Semin Liver Dis. 1991;11:31-39.
Feldstein AE, et al. Primary sclerosing cholangitis in children: a long-term follow-up study. Hepatology. 2003;38:210-217.
Fleming KA, et al. Biliary dysplasia as a marker of cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol. 2001;34:360-365.
Florin TH, Pandeya N, Radford-Smith GL. Epidemiology of appendicectomy in primary sclerosing cholangitis and ulcerative colitis: its influence on the clinical behaviour of these diseases. Gut. 2004;53:973-979.
Foutch PG, et al. Endoscopic retrograde wire-guided brush cytology for diagnosis of patients with malignant obstruction of the bile duct. Am J Gastroenterol. 1990;85:791-795.
Fritcher EG, et al. A multivariable model using advanced cytologic methods for the evaluation of indeterminate pancreatobiliary strictures. Gastroenterology. 2009;136:2180-2186.
Gautam M, Cheruvattath R, Balan V. Recurrence of autoimmune liver disease after liver transplantation: a systematic review. Liver Transpl. 2006;12:1813-1824.
Gohlke F, et al. Evidence for an overlap syndrome of autoimmune hepatitis and primary sclerosing cholangitis. J Hepatol. 1996;24:699-705.
Gores GJ. Early detection and treatment of cholangiocarcinoma. Liver Transpl. 2000;6:S30-S34.
Gores GJ. Cholangiocarcinoma: current concepts and insights. Gastroenterology. 2003;125:1536-1538.
Goss JA, et al. Orthotopic liver transplantation for primary sclerosing cholangitis: a 12-year single-center experience. Ann Surg. 1997;225:472-481.
Graziadei IW, et al. Long-term results of patients undergoing liver transplantation for primary sclerosing cholangitis. Hepatology. 1999;30:1121-1127.
Hadjis NS, Collier NA, Blumgart LH. Malignant masquerade at the hilum of the liver. Br J Surg. 1985;72:659-661.
Harnois DM, et al. High-dose ursodeoxycholic acid as a therapy for patients with primary sclerosing cholangitis. Am J Gastroenterol. 2001;96:1558-1562.
Hay JE, et al. Primary sclerosing cholangitis and celiac disease: a novel association. Ann Intern Med. 1988;109:713-717.
Hay JE, et al. The metabolic bone disease of primary sclerosing cholangitis. Hepatology. 1991;14:257-261.
Heimbach JK. Successful liver transplantation for hilar cholangiocarcinoma. Curr Opin Gastroenterol. 2008;24:384-388.
Hobson CH, et al. Enterohepatic circulation of bacterial chemotactic peptide in rats with experimental colitis. Gastroenterology. 1988;94:1006-1013.
Holubitsky IB, McKenzie AD. Primary sclerosing cholangitis of the extrahepatic bile ducts. Can J Surg. 1964;7:277-283.
Jessurun J, Bolio-Solis A, Manivel JC. Diffuse lymphoplasmacytic acalculous cholecystitis: a distinctive form of chronic cholecystitis associated with primary sclerosing cholangitis. Hum Pathol. 1998;29:512-517.
Jeyarajah DR, et al. Recurrent primary sclerosing cholangitis after orthotopic liver transplantation: is chronic rejection part of the disease process? Transplantation. 1998;66:1300-1306.
Jones EA, Bergasa NV. The pruritus of cholestasis: from bile acids to opiate agonists. Hepatology. 1990;11:884-887.
Jorge AD, Esley C, Ahumada J. Family incidence of primary sclerosing cholangitis associated with immunological diseases. Endoscopy. 1987;19:114-117.
Jorgensen RA, et al. Serum lipid and fat-soluble vitamin levels in primary sclerosing cholangitis. J Clin Gastroenterol. 1995;20:215-219.
Kamath PS, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33:464-470.
Kaplan GG, et al. The burden of large and small duct primary sclerosing cholangitis in adults and children: a population-based analysis. Am J Gastroenterol. 2007;102:1042-1049.
Karlsen TH, Schrumpf E, Boberg KM. Gallbladder polyps in primary sclerosing cholangitis: not so benign. Curr Opin Gastroenterol. 2008;24:395-399.
Kartheuser AH, et al. Comparison of surgical treatment of ulcerative colitis associated with primary sclerosing cholangitis: ileal pouch–anal anastomosis versus Brooke ileostomy. Mayo Clin Proc. 1996;71:748-756.
Kaw M, et al. Biliary tract calculi in primary sclerosing cholangitis. Am J Gastroenterol. 1995;90:72-75.
Kaya M, Angulo P, Lindor KD. Overlap of autoimmune hepatitis and primary sclerosing cholangitis: an evaluation of a modified scoring system. J Hepatol. 2000;33:537-542.
Keiding S, et al. Detection of cholangiocarcinoma in primary sclerosing cholangitis by positron emission tomography. Hepatology. 1998;28:700-706.
Kim WR, et al. A revised natural history model for primary sclerosing cholangitis. Mayo Clin Proc. 2000;75:688-694.
Kipp BR, et al. A comparison of routine cytology and fluorescence in situ hybridization for the detection of malignant bile duct strictures. Am J Gastroenterol. 2004;99:1675-1681.
Kornfeld D, Ekbom A, Ihre T. Survival and risk of cholangiocarcinoma in patients with primary sclerosing cholangitis: a population-based study. Scand J Gastroenterol. 1997;32:1042-1045.
LaRusso NF, et al. Primary sclerosing cholangitis. N Engl J Med. 1984;310:899-903.
Lee Y-M, Kaplan MM. Primary sclerosing cholangitis. N Engl J Med. 1995;332:924-933.
Levy C, et al. The value of serum CA 19-9 in predicting cholangiocarcinomas in patients with primary sclerosing cholangitis. Dig Dis Sci. 2005;50:1734-1740.
Lichtman SN, et al. Hepatic inflammation in rats with experimental small intestinal bacterial overgrowth. Gastroenterology. 1990;98:414-423.
Lindor KD, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology. 2009;50:808-814.
Loftus EVJr, Sandborn WJ, Lindor KD. Interactions between chronic liver disease and inflammatory bowel disease. Inflamm Bowel Dis. 1997;3:288-302.
Loftus EVJr, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis. Gastroenterology. 1996;110:432-440.
Loftus EVJr, et al. Primary sclerosing cholangitis is associated with nonsmoking: a case-control study. Gastroenterology. 1996;110:1496-1502.
Loftus EVJr, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis following orthotopic liver transplantation. Hepatology. 1998;27:685-690.
Ludwig J, Colina F, Poterucha JJ. Granulomas in primary sclerosing cholangitis. Liver. 1995;15:307-312.
Ludwig J, et al. Primary sclerosing cholangitis. Contemp Issues Surg Pathol. 1986;8:193-213.
Ludwig J, et al. Morphologic features of chronic hepatitis associated with primary sclerosing cholangitis or chronic ulcerative colitis. Hepatology. 1981;1:632-640.
Luketic VA, et al. An atypical presentation for primary sclerosing cholangitis. Dig Dis Sci. 1997;42:2009-2016.
MacCarty RL, et al. Primary sclerosing cholangitis: findings on cholangiography and pancreatography. Radiology. 1983;149:39-44.
MacCarty RL, et al. Cholangiocarcinoma complicating primary sclerosing cholangitis: cholangiographic appearances. Radiology. 1985;156:43-46.
Mandal A, et al. Autoantibodies in sclerosing cholangitis against a shared peptide in biliary and colon epithelium. Gastroenterology. 1994;106:185-192.
Mehal WZ, et al. HLA DR4 is a marker for disease progression in primary sclerosing cholangitis. Gastroenterology. 1994;106:160-167.
Minuk GY, et al. Abnormal clearance of immune complexes from the circulation of patients with primary sclerosing cholangitis. Gastroenterology. 1985;88:166-170.
Mitchell SA, et al. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology. 2001;121:900-907.
Myburgh JA. Surgical biliary drainage in primary sclerosing cholangitis. Arch Surg. 1994;129:1057-1062.
Narumi S, et al. Liver transplantation for sclerosing cholangitis. Hepatology. 1995;22:451-457.
Nichols JC, et al. Diagnostic role of serum CA 19-9 for cholangiocarcinoma in patients with primary sclerosing cholangitis. Mayo Clin Proc. 1993;68:874-879.
Norris S, et al. Mapping MHC-encoded susceptibility and resistance in primary sclerosing cholangitis: the role of MICA polymorphism. Gastroenterology. 2001;120:1475-1482.
Nuako KW, et al. Primary sclerosing cholangitis and colorectal carcinoma in patients with chronic ulcerative colitis: a case-control study. Cancer. 1998;82:822-826.
Okolicsanyi L, et al. Primary sclerosing cholangitis: clinical presentation, natural history and prognostic variables: an Italian multicentre study. Eur J Gastroenterol Hepatol. 1996;8:685-691.
Olerup O, et al. HLA-DR and HLA-DQ are not markers for rapid disease progression in primary sclerosing cholangitis. Gastroenterology. 1995;108:870-878.
Olsson R, et al. Prevalence of primary sclerosing cholangitis in patients with ulcerative colitis. Gastroenterology. 1991;100:1319-1323.
Olsson R, et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology. 2005;129:1464-1472.
Palmer KR, Duerden BJ, Holdworth CD. Bacteriological and endotoxin studies in cases of ulcerative colitis submitted to surgery. Gut. 1980;21:851-854.
Pardi DS, et al. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003;124:889-893.
Penna C, et al. Pouchitis after ileal pouch–anal anastomosis for ulcerative colitis occurs with increased frequency in patients with associated primary sclerosing cholangitis. Gut. 1996;38:234-239.
Porayko MK, et al. Patients with asymptomatic primary sclerosing cholangitis frequently have progressive disease. Gastroenterology. 1990;98:1594-1602.
Prytz H, et al. Dynamic FDG-PET is useful for detection of cholangiocarcinoma in patients with PSC listed for liver transplantation. Hepatology. 2006;44:1572-1580.
Quigley EMM, et al. Familial occurrence of primary sclerosing cholangitis and ulcerative colitis. Gastroenterology. 1983;85:1160-1165.
Ramage JK, et al. Serum tumor markers for the diagnosis of cholangiocarcinoma in primary sclerosing cholangitis. Gastroenterology. 1995;108:865-869.
Rasmussen HH, et al. Hepatobiliary dysfunction and primary sclerosing cholangitis in patients with Crohn’s disease. Scand J Gastroenterol. 1997;32:604-610.
Rosen CB, Nagorney DM. Cholangiocarcinoma complicating primary sclerosing cholangitis. Semin Liver Dis. 1991;11:26-30.
Satsangi J, et al. A functional polymorphism of the stromelysin gene (MMP-3) influences susceptibility to primary sclerosing cholangitis. Gastroenterology. 2001;121:124-130.
Scheuer PJ. Ludwig symposium on biliary disorders—part II. Pathologic features and evolution of primary biliary cirrhosis and primary sclerosing cholangitis. Mayo Clin Proc. 1998;73:179-183.
Schrumpf E, et al. HLA antigens and immunoregulatory T cells in ulcerative colitis associated with hepatobiliary disease. Scand J Gastroenterol. 1982;17:187-191.
Schwartz SI, Dale WA. Primary sclerosing cholangitis: review and report of six cases. Arch Surg. 1958;77:439-451.
Sheng R, et al. Biliary strictures in hepatic transplants: prevalence and types in patients with primary sclerosing cholangitis vs those with other liver diseases. Am J Roentgenol. 1993;161:297-300.
Siegel JH, Barnes S, Morris JS. Bile acids in liver disease associated with inflammatory bowel disease. Digestion. 1977;15:469-481.
Spivey JR, et al. Methionine-enkephalin concentrations correlate with stage of disease but not pruritus in patients with primary biliary cirrhosis. Am J Gastroenterol. 1994;89:2028-2032.
Steckman M, Drossman DA, Lesesne HR. Hepatobiliary disease that precedes ulcerative colitis. J Clin Gastroenterol. 1984;6:425-428.
Stiehl A, et al. Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: outcome after endoscopic treatment. J Hepatol. 2002;36:151-156.
Talwalkar JA, Lindor KD. Primary sclerosing cholangitis. Inflamm Bowel Dis. 2005;11:62-72.
Talwalkar JA, et al. Cost-minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology. 2004;40:39-45.
Tamura S, et al. Recurrence of primary sclerosing cholangitis after living donor liver transplantation. Liver Int. 2007;27:86-94.
Thorpe MEC, Scheuer PJ, Sherlock S. Primary sclerosing cholangitis, the biliary tree, and ulcerative colitis. Gut. 1967;8:435-448.
Tocchi A, et al. Late development of bile duct cancer in patients who had biliary-enteric drainage for benign disease: a follow-up study of more than 1,000 patients. Ann Surg. 2001;234:210-214.
Tung BY, et al. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med. 2001;134:89-95.
Van Buuren HR, et al. High prevalence of autoimmune hepatitis among patients with primary sclerosing cholangitis. J Hepatol. 2000;33:543-548.
Van Erpecum KJ, et al. Risk of primary sclerosing cholangitis is associated with nonsmoking behavior. Gastroenterology. 1996;110:1503-1506.
Van Milligen De Wit AW, et al. Lack of complications following short-term stent therapy for extrahepatic bile duct strictures in primary sclerosing cholangitis. Gastrointest Endosc. 1997;46:344-347.
Van Milligen De Wit AWM, Van Deventer SJH, Tytgat GNJ. Immunogenetic aspects of primary sclerosing cholangitis: implications for therapeutic strategies. Am J Gastroenterol. 1995;90:893-900.
Van Milligen De Wit AWM, et al. Endoscopic stent therapy for dominant extrahepatic bile duct strictures in primary sclerosing cholangitis. Gastrointest Endosc. 1996;44:293-299.
Vitellas KM, et al. MR cholangiopancreatography in patients with primary sclerosing cholangitis: interobserver variability and comparison with endoscopic retrograde cholangiopancreatography. Am J Roentgenol. 2002;179:399-407.
Wagner S, et al. Endoscopic management of biliary tract strictures in primary sclerosing cholangitis. Endoscopy. 1996;28:546-551.
Warren KW, Athanassiades S, Monge JI. Primary sclerosing cholangitis: a study of 42 cases. Am J Surg. 1966;111:23-38.
Wiencke K, et al. Primary sclerosing cholangitis is associated with extended HLA-DR3 and HLA-DR6 haplotypes. Tissue Antigens. 2007;69:161-169.
Wiesner RH, LaRusso NF. Clinicopathologic features of the syndrome of primary sclerosing cholangitis. Gastroenterology. 1980;79:200-206.
Wiesner RH, et al. Primary sclerosing cholangitis: natural history, prognostic factors, and survival analysis. Hepatology. 1989;10:430-436.
Wiesner RH, et al. Liver transplantation for primary sclerosing cholangitis: impact of risk factors on outcome. Liver Transplant Surg. 1996;2:99-108.
Wilschanski M, et al. Primary sclerosing cholangitis in 32 children: clinical, laboratory, and radiographic features, with survival analysis. Hepatology. 1995;22:1415-1422.
Yang X, et al. Susceptibility to primary sclerosing cholangitis is associated with polymorphisms of intercellular adhesion molecule-1. J Hepatol. 2004;40:375-379.
Younossi ZM, et al. Health-related quality of life in chronic liver disease: the impact of type and severity of disease. Am J Gastroenterol. 2001;96:2199-2205.
Zein CO, Lindor KD, Angulo P. Prevalence and predictors of esophageal varices in patients with primary sclerosing cholangitis. Hepatology. 2004;39:204-210.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 41 Primary sclerosing cholangitis
Overview
Primary sclerosing cholangitis (PSC) is a chronic, cholestatic liver disease of unknown etiology characterized by diffuse inflammatory destruction of intrahepatic and/or extrahepatic bile ducts that results in bile stasis, hepatic fibrosis, cirrhosis, and end-stage liver disease. Despite the increased awareness of PSC, the exact etiology and an effective medical therapy have yet to be identified. The disease is associated with specific human leukocyte antigen (HLA) haplotypes and has a close association with inflammatory bowel disease (IBD), which suggests that PSC is an immune-mediated disorder; but unlike other typical autoimmune diseases, there is a 2:1 male predominance. Life expectancy is reduced in PSC patients, because disease progression often results in complications of liver failure and cholangiocarcinoma (see Chapters 50A, 50B, and 50C). Among eligible candidates, surgical therapy has moved largely to orthotopic liver transplantation (OLT), as it remains the only effective, life-extending treatment for advanced-stage PSC (see Chapter 97A).
Epidemiology and Demographics
PSC mainly affects males during the fourth decade of life (Angulo & Lindor, 1999; Chapman, 2003). In the United States, population-based studies reported an age-adjusted incidence for PSC of 1.25 per 100,000 men and 0.54 per 100,000 women per year (Bambha et al, 2003). In the same study, the calculated prevalence of PSC was 20.9 and 6.3 per 100,000 men and women, respectively (Bambha et al, 2003). The worldwide prevalence and geographic distribution of PSC are undefined, as most epidemiologic data come from Northern Europe and the upper Midwest in the United States. Of interest is the strong association of PSC with IBD. Approximately 75% to 80% of Northern European patients with PSC suffer from IBD, with chronic ulcerative colitis (CUC) being most common, seen in about 90% (Chapman et al, 1980; Olsson et al, 1991; Wiesner et al, 1989). Conversely, only approximately 5.5% of patients with CUC have PSC (Olsson et al, 1991).
Smoking is the only environmental factor known to influence PSC susceptibility, and it is associated with a reduced risk of PSC. In a study from The Netherlands, the incidence of current smoking was 19% in patients with PSC compared with 38% in control subjects (Van Erpecum et al, 1996). Similarly, a U.S. study showed that 4.9% of patients with PSC were reported to smoke compared with 26.1% of a control population matched for age and gender. This reported difference was not attributable to the prevention of CUC (Loftus et al, 1996b). Nevertheless, smoking and alcohol consumption should be discouraged in PSC patients, because both are associated with the development of cholangiocarcinoma in this population (Bergquist et al, 1998; Chalasani et al, 2000).
Adenocarcinoma of the bile ducts appears in 8% to 15% of patients with PSC, rendering it a premalignant condition (Chalasani et al, 2000; De Groen et al, 1999; Foutch et al, 1990; see Chapters 50A, 50B, and 50C). In a recent study, 161 PSC patients without evidence of cholangiocarcinoma upon entry were followed over a median of 11.5 years. During that period, 59 patients (36.6%) died, 50 patients (31.1%) underwent liver transplantation, and 11 patients (6.8%) had cholangiocarcinoma develop (Burak et al, 2004). No association was found between the duration of PSC and the incidence of cholangiocarcinoma, and multivariate analysis revealed that variceal bleeding was the only significant risk factor for the diagnosis of cancer (risk ratio, 24.2; 95% confidence interval, 3.3% to 67%) (Burak et al, 2004).
Clinical Presentation
PSC can affect any age group, but it is more common among men in their fourth decade of life (Wiesner & LaRusso, 1980; MacCarty et al, 1983). PSC can also occur in childhood, and children with PSC frequently have liver disease with features similar to autoimmune hepatitis (AIH) (El-Shabrawi et al, 1987; Wilschanski et al, 1995). In fact, the overlap between PSC and AIH in children can be as high as 35% (Feldstein et al, 2003). In contrast, overlap of PSC and AIH affects only about 5% of adults (Kaya et al, 2000; Van Buuren et al, 2000).
The clinical presentation of PSC varies depending on the disease stage at the time of diagnosis. PSC can be diagnosed in asymptomatic individuals who come to medical attention because of abnormal liver function tests. Approximately 15% to 40% of patients are asymptomatic at the time of diagnosis (Talwalkar & Lindor, 2005). There are no pathognomonic signs or symptoms when the disease becomes clinically apparent. The symptomatic patient may be seen initially with signs, symptoms, and complications of cholestatic liver disease or hepatic failure. In a recent study, the most common first symptoms were abdominal pain (20%), pruritus (10%), diarrhea (8%), jaundice (6%), fatigue (6%), and fever (4%) (Kaplan et al, 2007). Symptoms of bacterial cholangitis are uncommon, unless dominant biliary strictures or biliary stones are present.
Physical examination may reveal jaundice, hepatomegaly, splenomegaly, and excoriations. Ascites and peripheral edema are observed with the development of biliary cirrhosis and portal hypertension (see Chapter 70A). These findings are less common today, because PSC patients are usually evaluated before the development of end-stage liver disease. A commonly encountered clinical scenario is a patient with CUC who presents with a cholestatic pattern of liver enzymes. Further medical evaluation results in the concurrent diagnosis of PSC. As with other chronic liver diseases, health-related quality of life is significantly impaired among PSC patients compared with healthy individuals (Younossi et al, 2001).
Diagnostic Criteria
The diagnosis of PSC is made based on clinical presentation, biochemical profile, and characteristic cholangiographic appearance of the bile ducts. Before the 1960s, PSC was a diagnosis of exclusion following abdominal exploration and biopsy of a suspected bile duct neoplasm (Schwartz & Dale, 1958). In the 1960s, diagnosing PSC became more frequent, when its association with IBD was recognized (Thorpe et al, 1967). With the advent of endoscopic retrograde cholangiopancreatography (ERCP) and percutaneous transhepatic cholangiography (PTC) in the 1970s, the characteristic “beaded” or “pruned tree” cholangiographic appearance obviated the need for exploratory surgery to diagnose PSC and resulted in its increased recognition (Wiesner & LaRusso, 1980; see Chapter 18).
The first formal diagnostic criteria included 1) absence of previous operative trauma to the biliary system; 2) sclerosis and stenosis involving all or most of the extrahepatic bile ducts; 3) exclusion of malignant disease involving the biliary tree, such as cholangiocarcinoma; and 4) absence of calculi in the gallbladder and common bile duct (Holubitsky & McKenzie, 1964). Following the introduction of ERCP, the criteria were expanded to include a subset of PSC cases recognized to have intrahepatic bile duct involvement alone (MacCarty et al, 1983). In addition, serial ERCP examinations of PSC patients demonstrated the subsequent development of cholangiocarcinoma (Chapman et al, 1980). As such, malignancy arising in the setting of sclerosing cholangitis was eliminated as an exclusionary criterion. Imaging techniques (i.e., ERCP) have also shown biliary tract calculi in many patients with established PSC, suggesting the removal of calculi as an exclusionary criterion (Kaw et al, 1995).
The diagnosis of PSC currently relies on 1) characteristic cholangiographic abnormalities of the biliary tree; 2) compatible clinical and biochemical findings, typically of ductal cholestasis with elevated serum alkaline phosphatase level for at least 6 months duration; and 3) exclusion of other causes of secondary sclerosing cholangitis (SSC). The clinical and cholangiographic features of SSC can mimic PSC, but SSC originates from known pathologic processes. Several well-described causes of SSC are listed in Table 41.1 (Abdalian & Heathcote, 2006). Currently, the most common presentation is an asymptomatic patient with persistently elevated levels of alkaline phosphatase first noted on routine serum biochemical screening. In the proper clinical context, a characteristic cholangiographic appearance of the bile ducts is sufficient for making the diagnosis.
AIDS, acquired immunodeficiency syndrome
Liver biopsy is not always necessary for diagnosing PSC, nor does it contribute new information that affects the clinical management of patients. In a study of 79 patients with PSC who had an already established diagnosis by cholangiography, liver biopsy did not affect management (Burak et al, 2003). Thus the role of liver biopsy in PSC is to 1) exclude other causes of liver disease, 2) diagnose small-duct PSC (discussed later), and 3) define the disease stage for determining prognosis and assessing efficacy of treatment prior to entering in therapeutic trials. Small-duct PSC comprises approximately 5% of histologically confirmed PSC cases (Angulo et al, 2002) and has significantly better long-term prognosis compared with classic PSC. However, some patients with small-duct disease can progress to classic PSC (Angulo et al, 2002). Patients with small-duct PSC usually have IBD and are seen intially with a cholestatic pattern of liver enzymes but normal cholangiography. Nevertheless, liver biopsy reveals classic histologic features of PSC. In the majority of patients, the history, serum biochemical profile, and cholangiography distinguish PSC from other causes of chronic cholestatic liver disease. The differential diagnosis of PSC is presented in Table 41.2.
Table 41.2 Differential Diagnosis of Primary Sclerosing Cholangitis
Biochemical and Serologic Abnormalities
Biochemical cholestasis of at least 6 months’ duration gives reason to suspect PSC. Alkaline phosphatase remains the most commonly elevated liver enzyme, often showing a threefold to fourfold increase. Nevertheless, a normal alkaline phosphatase level does not exclude the diagnosis of PSC, because normal levels have been reported in patients with cholangiographically proven disease (Balasubramanian et al, 1988). Therefore a normal alkaline phosphatase level should not dissuade further investigation if the clinical history (e.g., presence of IBD) and other evidence suggest liver disease.
Aminotransferase levels are often modestly elevated in patients with PSC, except in children, in whom these levels can be markedly increased. On average, adults with PSC have aminotransferase levels less than three times the upper limit of normal (Lee & Kaplan, 1995). PSC patients with highly elevated aminotransferases may show concomitant serologic evidence and histologic features of AIH (Czaja, 1998).
Serum bilirubin levels are normal in 60% of patients at diagnosis (Talwalkar & Lindor, 2005), but bilirubin levels will markedly rise as PSC progresses. An abrupt, sustained rise of bilirubin may herald a dominant biliary stricture, a bile duct stone, or the development of cholangiocarcinoma; therefore it should prompt additional investigation. Serum copper and ceruloplasmin levels and hepatic and urinary copper values are often abnormal. Hepatic copper levels can be elevated to the degree seen in Wilson disease and primary biliary cholangitis (PBC) (LaRusso et al, 1984); the copper increase is a reflection of prolonged cholestasis.
No autoantibody is pathognomonic for diagnosing PSC. The prevalence of antineutrophil cytoplasmic antibodies (ANCAs), anticardiolipin antibodies, and antinuclear antibodies (ANAs) in patients with PSC is 84%, 66%, and 53%, respectively (Angulo et al, 2000). Antimitochondrial antibodies (AMAs) and anti–smooth muscle antibodies are rare in patients with PSC. Approximately 25% of patients have hypergammaglobulinemia, and immunoglobulin M (IgM) levels are the most commonly elevated component (Wiesner & LaRusso, 1980). Autoantibody testing is helpful to identify those PSC patients with concurrent AIH, but antibody titers are not important in following PSC activity.
Imaging Studies
Visualization of the biliary tree is essential for establishing the diagnosis of PSC, and ERCP is the gold standard imaging technique (see Chapter 18). Typical cholangiographic findings of PSC include multifocal stricturing and beading throughout the biliary tree characteristic of alternating fibrosis and ectasia of the bile ducts (Fig. 41.1). Strictures of the bile ducts are the hallmark of the disease. They are often diffusely distributed and annular with intervening segments of dilated ducts. The classic cholangiographic findings involve both the intrahepatic and extrahepatic biliary tree. Strictures can vary in length and confluency, ranging from a bandlike appearance 1 to 2 mm in length to dense, annular strictures several centimeters in length. Approximately 25% of patients have biliary outpouchings that resemble diverticula. Similarly, 30% to 40% of patients may have mural irregularities that produce a shaggy appearance, varying from a fine brush border to frank nodularity (MacCarty et al, 1983).
In a study of 86 patients with PSC, cholangiographic findings revealed involvement of the intrahepatic and extrahepatic ducts in 80 and 85 patients, respectively, when there was adequate visualization of the intrahepatic biliary tree. Involvement of only the intrahepatic and proximal extrahepatic ducts was seen in 20% of patients, and a very small number of patients had only small-duct PSC with a normal cholangiogram (MacCarty et al, 1983). Gallbladder and cystic duct involvement is less severe but is present in as many as 15% of patients (Brandt et al, 1988). ERCP should be considered in all patients suspected of having PSC. In addition to establishing the diagnosis, ERCP defines the extent and distribution of disease, identifies benign dominant strictures for endoscopic dilation or stenting, and allows brushings for cytology studies to screen for cholangiocarcinoma.
As many as 10% of cholangiocarcinomas have radiologic features that mimic PSC. Cholangiographic features that may suggest malignant degeneration of established PSC are markedly dilated biliary ducts or ductal segments, presence of a polypoid mass of 1 cm or greater in diameter, and progressive stricture formation. Nevertheless, these cholangiographic features are not specific for cholangiocarcinoma and can be seen in the absence of malignancy (MacCarty et al, 1985).
Despite the high diagnostic and therapeutic capabilities of ERCP, the procedure is invasive and is associated with clinical complications. The Mayo Clinic experience has shown that among PSC patients undergoing ERCP, the estimated rate of procedural complications requiring hospitalization is more than 10%. PSC patients undergoing ERCP are expected to have longer procedural time and a higher incidence of cholangitis compared with non-PSC patients (see Chapter 43), but the risk of pancreatitis, perforation, and bleeding are similar (Bangarulingam et al, 2009).
Magnetic resonance cholangiography (MRC) is a noninvasive substitute for ERCP (Fig. 41.2; see Chapter 17). Recent studies have estimated the sensitivity and specificity of MRC for diagnosing PSC to be between 80% to 82% and 87% to 98%, respectively (Talwalkar et al, 2004; Berstad et al, 2006). These studies concluded that MRC has diagnostic accuracy comparable to ERCP and results in cost savings when used as the initial diagnostic strategy. In another study, MRC was shown to be better than ERCP in identifying peripheral intrahepatic bile duct strictures (Vitellas et al, 2002).
In a retrospective, nonblinded study of PSC patients with known biliary tract carcinoma, MRC findings of a well-defined mass exhibiting abnormal signal intensity were classified as definite cholangiocarcinoma in six or seven patients (Vitellas et al, 2002). MRC will likely replace diagnostic ERCP, but at present it is less sensitive and does not allow for biliary biopsy and cytology or therapeutic intervention.
PTC is useful when access to the biliary tree by ERCP is not technically feasible. Abdominal ultrasonography (US) and computed tomography (CT) are valuable in affirming ductal dilation and evaluating complications of PSC such as biliary stones, cirrhosis, and bile duct malignancy (see Chapters 13 and 16). US is often able to show large duct dilation and mural thickening of the common bile duct in patients with confirmed abnormalities on ERCP. It can further be used to detect gallstones or biliary tract calculi, which are important in the evaluation of bacterial cholangitis, worsening cholestasis, or jaundice.
Morphologic features of ductal abnormalities and cirrhosis can be seen on CT. Atrophy of the left lateral segment and hypertrophy of the caudate lobe may differentiate cirrhosis associated with PSC from that seen in other types of cirrhosis (Caldwell et al, 2001). CT can also complement cholangiography in the evaluation of malignancy with its ability to detect peripheral intrahepatic cholangiocarcinoma and metastatic spread within the liver parenchyma and the abdomen (Campbell et al, 1998, 2001). However, perihilar lymphadenopathy is very common in PSC with or without cholangiocarcinoma, and this finding cannot be taken as direct evidence of malignancy or metastasis.
Pathology
PSC can affect the entire biliary system. On rare occasions, it may be limited to the small intrahepatic ducts and be detectable only on histology. Small-duct PSC is thought to account for 5% of histologically confirmed PSC cases, although its true prevalence remains unknown (Angulo et al, 2002).
Liver biopsy findings alone are rarely sufficient to establish the diagnosis of PSC, which is characterized by damage to, atrophy of, and ultimately loss of bile ducts, thus sharing histologic features with other hepatobiliary diseases (Scheuer, 1998). Liver biopsy specimens from patients show portal tract inflammation and sclerosis (Fig. 41.3). Afflicted bile ducts are surrounded by a cuff of lightly inflamed sheets of fibrous tissue and edema, causing the fibrotic layers to separate and form the characteristic onion skin appearance. This classic histologic finding is nearly pathognomonic, but it is seen in fewer than 10% of PSC patients (Ludwig et al, 1981). Affected bile ducts will eventually atrophy and be replaced by rounded scars.
On occasion, similar fibroobliterative cholangitis can be seen in 1) PBC, 2) mechanical obstruction of larger bile ducts, 3) ductopenic rejection following liver transplantation, and 4) after intraarterial infusion of 5-fluorouracil. Involvement of the large intrahepatic and extrahepatic ducts distinguishes PSC from PBC. Inflammation in early-stage PSC is generally milder than that of PBC, but this distinction is difficult to make (Scheuer, 1998). Granulomas, thought to be classically associated with PBC, may be seen in about 4% of biopsies from PSC patients, making the distinction between PSC and PBC even more challenging (Ludwig et al, 1995).
Distinguishing between PSC-related histopathologic features and changes as a result of simple obstruction of distal bile ducts makes the liver biopsy interpretation difficult. Canalicular cholestasis is nonspecific and can occur as a result of any cause of biliary obstruction. As PSC progresses, however, the histopathologic changes of chronic cholestasis spill into the hepatic parenchyma. The most commonly used liver histology grading system proposed by Ludwig is based on these parenchymal changes. Disease was designated as stage 1, portal; stage 2, periportal; stage 3, septal; and stage 4, cirrhotic (Table 41.3; Ludwig et al, 1986). Histologic changes in the same liver can be markedly varied from segment to segment at any given point in time. Whereas histologic staging was formerly used as an independent predictor of the disease’s natural history, the revised PSC survival model does not take into account the histologic stage (Kim et al, 2000).
| Portal stage (stage I) | Portal edema, inflammation, ductal proliferation; abnormalities do not extend beyond the limiting plate |
| Periportal stage (stage II) | Periportal fibrosis and inflammation with or without ductal proliferation; piecemeal necrosis may be present |
| Septal stage (stage III) | Septal fibrosis or bridging necrosis can be identified |
| Cirrhotic stage (stage IV) | Biliary cirrhosis evident |
Etiopathogenesis
The strong association of PSC with IBD has drawn much attention to the potential role of the inflamed colon. The hypothesis suggests that inflammation of the colon may increase permeability to various intraluminal products—toxins, bacteria, inflammatory mediators—that ultimately lead to liver disease. Bacteria or their toxic metabolic products have been considered but have not been conclusively shown to have a pathogenetic role in PSC development (Eade & Brooke, 1969; Palmer et al, 1980). However, investigators have shown that the absence of low-grade, chronic portal vein bacteremia in patients with CUC could potentially elicit the immune response that is responsible for PSC (Warren et al, 1966). Similarly, histologic evidence of bacteremia manifested as portal phlebitis is mild or absent in most PSC patients (Ludwig et al, 1981). Additional information showed that colonic bacteria might metabolize primary bile acids into toxic bile salts that are then absorbed through the ulcerated colon into the portal circulation, but no evidence of bile acid metabolism abnormalities in patients with PSC or CUC has been found (Siegel et al, 1977).
Animal models of PSC have reported that bacterial chemotactic peptides can lead to portal inflammation and histologic changes of PSC (Hobson et al, 1988: Lichtman et al, 1990). Suppression of the inflammatory response was reported in an animal model following inhibition of tumor necrosis factor (TNF). Nevertheless, in a clinical trial of PSC patients with pentoxifylline, a TNF inhibitor, no beneficial effect on symptoms or liver tests was seen (Bharucha et al, 2000). Clinical observations also cast doubt upon the role of the inflamed colonic mucosa in the development of PSC. First, PSC develops in approximately 25% of patients without evidence of IBD. Invasive screening of asymptomatic patients with PSC reveals that many have no endoscopic or histologic evidence of IBD. Second, lack of association between the severity of colonic disease and the likelihood of development and severity of PSC strengthens the skepticism that CUC may not directly cause PSC. Third, failure of proctocolectomy to modify the natural history of PSC argues against a direct causative role of CUC in PSC (Cangemi et al, 1989; Steckman et al, 1984). Thus, the putative contribution of increased colonic permeability in CUC resulting in inflammation of bile ducts remains unclear.
Genetic factors seem to play a role in the development of PSC. First, there have been reports of familial PSC among siblings (Quigley et al, 1983; Jorge et al, 1987). Second, various HLA associations with PSC have been reported, and HLA B8, DR3, DR2, and haplotype A1, B8, and DR3 have been found more frequently in patients with PSC compared with controls (Chapman et al, 1983; Donaldson et al, 1991; Schrumpf et al, 1982; Wiencke et al, 2007). PSC associated with DR4 is a more aggressive disease, although this correlation has not been universally found (Mehal et al, 1994; Olerup et al, 1995). Additional associations with DRB3*0101, DRB1*0301, DQA1*0501, DQB1*0201 and DRB1*1301, and DQA1*0103 and DQB1*0603 have been reported (Donaldson & Norris, 2002). Polymorphisms in the TNF-α receptor have been described as a possible genetic link to PSC. G to A substitution at position −308 in the TNF-α gene has been associated with susceptibility to PSC (Mitchell et al, 2001). A functional variant of stromelysin, matrix metalloproteinase 3, may also influence PSC susceptibility and disease progression (Satsangi et al, 2001). Moreover, variations in the MICA gene (major histocompatibility complex class I–related MIC gene family) have a role in PSC predisposition. Independent of other HLA haplotypes, the MICA 002 allele appears to significantly reduce the risk of PSC, and the MICA 008 allele increases the risk of developing PSC (Norris et al, 2001). Finally, the CCR5-δ32 mutation, characterized by a 32-base pair deletion in the CCR5 gene of T cells, has been associated with susceptibility to PSC development and severity (Eri et al, 2004).
Immune-mediated damage to cholangiocytes seems to be the most likely mechanism leading to PSC. Theoretically, certain HLA molecules and haplotypes may contribute by eliciting an immune response against antigenic epitopes present on biliary epithelia. Enhanced expression of MHC class II antigens (i.e., HLA DR) on cholangiocytes in early-stage PSC has drawn suspicion regarding its role in altered immunity in disease pathogenesis. Aberrant expression of HLA DR is also apparent in PBC and extrahepatic biliary obstruction, suggesting that expression of this antigen is an epiphenomenon rather than an implicit cause of PSC (Broome et al, 1990; Chapman et al, 1988; Van Milligen De Wit et al, 1995).
Decreased hepatic clearance of circulating immune complexes, generalized complement activation, and sharing of a specific epitope between human colonic and biliary epithelial cells have also been reported (Bodenheimer et al, 1983; Minuk et al, 1985; Das et al, 1990; Mandal et al, 1994), yet none of these observations have proven pathogenic associations with PSC. Enhanced interactions of intracellular adhesion molecule 1 (ICAM1) present on cholangiocytes with its cognate ligand on T lymphocytes, leukocyte function–associated antigen 1 (ITGAL), may play a role in PSC development. Genetic polymorphisms of ICAM1 have been implicated in susceptibility to PSC. Homozygote status of the E469E allele for ICAM1 has been associated with protection against PSC (Yang et al, 2004). Both enhanced expression on proliferating cholangiocytes and increased serum levels of ICAM1 have been reported in PSC patients (Van Milligen De Wit et al, 1995; Adams et al, 1991; Bloom et al, 1995).
Associated Diseases
PSC is strongly associated with IBD, most commonly CUC. PSC patients may also have biochemical, serologic, and histologic features of AIH, and a variety of other diseases have been reported to associate with PSC. Given the lack of large studies, it is unclear whether these weak associations are true or simply coincidental. An abridged list of associated diseases is presented in Table 41.4.
Table 41.4 Diseases Associated with Primary Sclerosing Cholangitis
Inflammatory Bowel Diseases
The close association of PSC with IBD, particularly CUC, is widely recognized. IBD is seen in approximately 70% to 80% of patients with PSC, and CUC accounts for 85% to 90% of those patients; Crohn disease is responsible for the rest (Fausa et al, 1991; Loftus et al, 1997). Patients with PSC and Crohn disease may have milder liver disease than patients with PSC and CUC, and PSC has not been seen in Crohn disease involving only the small intestine (Rasmussen et al, 1997). Typically, the diagnosis of IBD is established 8 to 10 years before the liver disease is evident, although there is no clear temporal association, and cases of IBD occurring years after diagnosis of PSC have also been reported (Chapman et al, 1980; Loftus et al, 1996a, 1998). No direct correlation has been found between the severity of bowel disease and the severity of liver disease. Furthermore, therapy of the IBD does not alter the course of the liver disease. For instance, proctocolectomy, the most aggressive treatment for CUC, has no effect on PSC natural history (Cangemi et al, 1989). Colitis is usually milder in patients with both CUC and PSC in comparison to patients with CUC alone.
PSC may play a role in the development of colorectal dysplasia in the setting of CUC, as patients with CUC and PSC show a higher risk of colonic dysplasia compared with patients who have only CUC (Broome et al, 1995a, 1995b). In a study from Sweden, the absolute cumulative risk of developing colorectal dysplasia/carcinoma in patients with PSC and CUC was 9%, 31%, and 50% after 10, 20, and 25 years of disease duration, respectively. In patients with CUC alone, the parallel risk was 2%, 5%, and 10%, respectively (Broome et al, 1995b). Subsequent studies also confirmed the observation of Broom and others, where predicted risk of colorectal carcinoma with PSC and concomitant IBD after 10 years of disease duration was up to 25% (Kornfeld et al, 1997; Claessen et al, 2009). Other investigators have reported that patients with both PSC and CUC were five times more likely to develop colonic dysplasia, based on mucosal biopsy, compared with patients with CUC alone (Brentnall et al, 1996
[/not-level-membership-for-surgery-category]
