Chapter 89 Peripheral Neuropathies
Anatomy
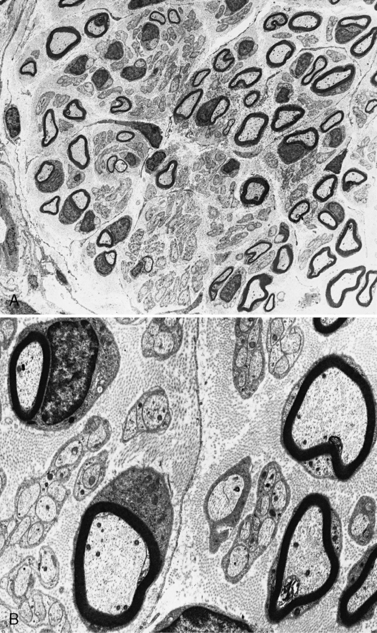
The peripheral nervous system consists of cranial nerves III through XII, the spinal roots, the nerve plexuses, the peripheral nerves, and the autonomic ganglia. (The autonomic nervous system is addressed separately and is not discussed in this chapter.) Neuronal cell bodies subserving the nerves in the peripheral nervous system reside in the brainstem; anterior horn cells of the spinal cord; intermediolateral cell column, where the autonomic system originates; and the dorsal root ganglia for afferent sensory function. Peripheral nerves innervate all skeletal muscles via large myelinated nerve fibers. Sensory input from skin, joints, and muscles is transmitted via a combination of unmyelinated and myelinated nerve fibers from the periphery to the central nervous system (CNS). The outer connective tissue sheath of a peripheral nerve, the epineurium, encases bundles of nerves in fascicles. Each fascicle has a sheath termed the perineurium. Myelinated and unmyelinated nerve fibers surrounded by collagenous fibers, the endoneurium, are present in fascicles (Figure 89-1). Peripheral nerves are very vascular, with arteries and arterioles in the epineurium, arterioles in the perineurium, and primarily capillaries in the endoneurium. The venous system is similarly represented.
Facial Nerve Paralysis (Bell’s Palsy)
Cranial nerve VII is a complex nerve that has motor, sensory, and autonomic components (Figure 89-2). The nonmotor functions of the facial nerve are mediated by parasympathetic afferent fibers, which innervate lacrimal and salivary glands; efferent fibers, which subserve taste; and other fibers that mediate the auditory reflex. Motor functions are subserved by somatic afferent fibers innervating muscle fibers of facial movement.
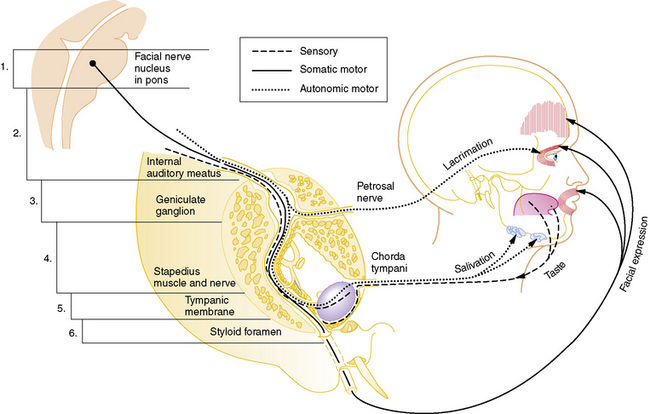
Fig. 89-2 Schematic pathway of the facial nerve (cranial nerve VII).
(Modified from Haymaker W: Bing’s local diagnosis in neurological diseases, 15th ed. St. Louis, 1969, Mosby; Rasmussem AT: The principal nervous pathways, New York, 1951, Macmillan; Swaiman KF, Wright FS: Pediatric neuromuscular diseases, St. Louis, 1979, Mosby.)
Clinical Features
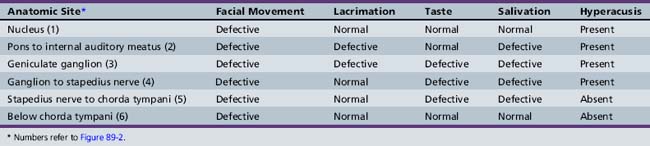
The incidence of Bell’s palsy is 2.7 per 100,000 below the age of 10 years, and 10.1 per 100,000 during the second decade of life [Katusik et al., 1986]. Female and male incidence is equal. Ear pain near the mastoid process is the first manifestation of impending facial nerve involvement half of the time. Unilateral inability to close the eyelid and maintain normal facial movement is the initial indication of motor involvement. Facial weakness develops rapidly over several hours to 3 days, resulting in paresis to complete paralysis. Bell’s palsy commonly follows an upper respiratory tract infection, indicating possible postinfectious demyelination. Drinking and eating are impaired because of the inability to close the mouth on the involved side. Depending on the site of facial nerve involvement (see Figure 89-2 and Table 89-1), lacrimation may be decreased and taste distorted.
The differential diagnosis of facial paralysis includes trauma, acute and chronic infections of the inner ear, herpes simplex infection, herpes zoster, Mycoplasma pneumoniae infection [Klar et al., 1985], Epstein–Barr virus infection [Andersson and Sterner, 1985], and Lyme disease (Borrelia spp. infection) (Box 89-1). Hypertension may be responsible for an acute facial paresis resembling Bell’s palsy [Lloyd et al., 1966]. Melkersson–Rosenthal and Möbius’ syndromes have facial paralysis. Guillain–Barré syndrome, motor neuron disorders, myasthenia gravis, various myopathies, leprosy, and Tangier disease may cause facial paresis. The Melkersson–Rosenthal syndrome is marked by orofacial edema, facial nerve palsy, fissured tongue, and epithelial noncaseating granulomas with perivascular infiltrates [Cousin et al., 1998; Kaminagukura and Jorge, 2009].
Box 89-1 Facial Weakness in Childhood













































Bilateral, congenital facial paralysis, usually in conjunction with ophthalmoplegia involving cranial nerve VI, presents in the newborn as Möbius’ syndrome. The condition is the result of aplasia or hypoplasia of cranial nerves and nuclei VI and VII [Jaradeh et al., 1996]. Muscles innervated by other cranial nerves may be involved. Arthrogryposis may be present. Mental retardation may be diagnosed later. Möbius’ syndrome may be sporadic or inherited as a dominant condition linked to chromosomes 13q12.2–13 or 3q [Kremer et al., 1996; Nishikawa et al., 1997]. Differential diagnosis includes traumatic delivery with skull fracture and injury to the nerve in the facial canal, sacral pressure on the facial nerve during delivery, or pressure over the parotid area from forceps application with nerve compression. Möbius’ syndrome may be related in some instances to maternal ingestion of ergotamine in early pregnancy [Smets et al., 2004].
Simple asymmetry of facial expression when infants and children are crying is common. A rare syndrome, the cardiofacial syndrome, referred to as Cayler’s syndrome, is due to congenital hypoplasia of the depressor anguli oris causing the asymmetric crying facies syndrome. Unilateral partial lower facial weakness is noted in infants when smiling or crying. The syndrome is associated with congenital heart disease [Caksen et al., 1996; Cayler et al., 1971; Nelson and Eng, 1972; Punal et al., 2001]. Anomalies of the head and neck, heart, skeleton, genitourinary tract, and CNS accompany the syndrome [Lin et al., 1997]. Cardiofacial syndrome links to chromosome 22q11.2. Variations include one infant without cardiac involvement and another with hypoparathyroidism [Giannotti and Mingarelli, 1994; Lindsay et al., 1995; Stewart and Smith, 1997]. Frameshift mutation of the EYA1 gene has been described [Shimasaki et al., 2004]. Microduplication of 22q11.2 is associated with velocardiofacial syndrome, DiGeorge’s syndrome, conotruncal anomaly face syndrome, and in some but not all children with Caylor’s cardiofacial syndrome [Cotter et al., 2005]. The majority of children with asymmetrical crying facies are normal.
Laboratory Findings
Uncomplicated facial palsy that resolves relatively quickly does not need detailed evaluation. Palsy that persists or seems atypical requires study. Evaluation for infection is appropriate, but peripheral leukocyte and differential counts and erythrocyte sedimentation rate are usually normal. Cerebrospinal fluid (CSF) studies may show pleocytosis and blood–brain barrier alterations in Lyme disease (Borrelia spp. infection) [Roberg et al., 1991]. Imaging of the cranium is performed to exclude skull fracture, osteomyelitis, mastoiditis, increased intracranial pressure, calcification, and osteopetrosis. Magnetic resonance imaging (MRI) often reveals gadolinium enhancement of the facial nerve in typical cases of Bell’s palsy. This has been used in adults to predict the outcome of acute facial nerve palsy during the first few days after onset of symptoms [Kress et al., 2004]. The time to recovery from onset may increase from 6 to 14 weeks if enhancement on postcontrast images is present [Yetiser et al., 2003]. MRI can be used in children to find rare tumors invading the facial nerve [Koerbel et al., 2003]. Small lesions inside the temporal bone and at the cerebellopontine angle can be detected using gadolinium-enhanced MRI [Becelli et al., 2003]. Electrodiagnostic techniques may be useful in assessing the severity of facial nerve involvement and predicting the outcome. These studies include determination of nerve conduction velocity [Langworth and Taverner, 1963], detection of the level or threshold of response to stimulation [Campbell et al., 1962; Devi et al., 1978], nerve conduction latencies, and electrical stimulation of the tongue [Peiris and Miles, 1965]. Serial studies may be helpful [Fisch, 1984]. Nerve degeneration can be detected electrically by 72 hours after onset of paralysis.
Because Bell’s palsy may be a component of a more widespread disease affecting other cranial and peripheral nerves, obtaining a complete electromyographic evaluation and extremity nerve conduction velocities is sometimes helpful [Bueri et al., 1984].
Treatment and Prognosis
The prognosis for recovery in children is good [Singhi and Jain, 2003]. Most children do not need drug therapy [Salman and MacGregor, 2001]. A number of drugs have been used, particularly steroids. Surgical decompression has been recommended in some patients when progressive paralysis and thus nerve degeneration occur [Jenkins et al., 1985]. Prednisone is the most widely used drug; it is given in high dosage for 1 week, followed by slow withdrawal of the drug in the second week. It should be started within 1 week of disease onset. The degree to which steroid therapy can alter the natural history of Bell’s palsy is unknown, although it appears to be more helpful in completely paralyzed than paretic individuals [Wolf et al., 1978]. Most children recover completely without treatment. Recovery usually begins within 2–4 weeks, reaching its maximum within 6–12 months. In meta-analyses comparing steroids to antiviral treatment, usually acyclovir, there is no added advantage in adding antiviral treatment to steroid treatment [Quant et al., 2009; Sullivan et al., 2007]. Wetting solutions to maintain corneal moisture and an eye patch for protection may be needed when the child cannot close the eye.
Brachial Plexus
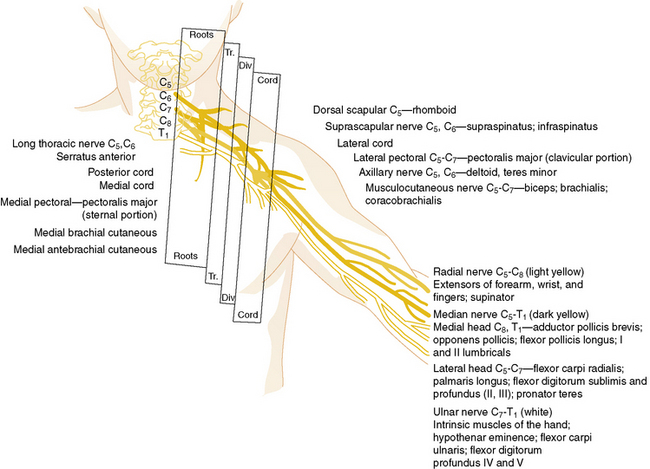
Nerve roots from the fifth cervical through the first thoracic nerves form the three primary trunks of the brachial plexus. Once formed, they divide promptly into anterior and posterior divisions (Figure 89-3). The posterior divisions join to form the posterior cord, which gives rise to the upper and lower subscapular, thoracodorsal, axillary, and radial nerves. The anterior divisions of the fifth, sixth, and seventh nerves form the lateral cord, and the anterior divisions of the eighth cervical and first thoracic nerves form the medial cord. The lateral cord subsequently gives rise to the musculocutaneous nerve and a branch to the coracobrachialis. The medial cord gives rise to the ulnar, medial antebrachial cutaneous, and medial brachial cutaneous nerves. Additional branches from the lateral and medial cords unite to form the median nerve.
Injury
Birth-related brachial plexus injuries occur in 0.5–1 in 1000 live births. Although traditionally thought to be associated with excessive traction to the affected extremity during breech delivery or traction to the head and neck during vertex delivery [Adler and Patterson, 1967], brachial plexus injuries can occur in the absence of fetal trauma [Ouzounian et al., 1997]. One study showed that half of cases of Erb’s palsy occur in normal-sized infants without trauma at delivery [Graham et al., 1997]. Brachial plexus injury is observed often in infants with shoulder dystocia [Greenwald et al., 1984; Rossi et al., 1982; Tada et al., 1984], although other studies question this assumption [Ecker et al., 1997; Graham et al., 1997]. Birth-related brachial plexus injury is associated with clavicle fracture in 10 percent of cases and humerus fracture in 10 percent.
The most common brachial plexus injury is Erb’s palsy, which composes 80–90 percent of newborn brachial plexus injuries. Damage of the fifth and sixth cervical nerves or the upper trunk of the brachial plexus will result in paralysis of the upper arm. The infant with this condition typically lies with the humerus adducted and internally rotated with the elbow extended, forearm pronated, and wrist flexed. The deltoid, biceps, brachialis, supinator, and extensors of the wrist and finger muscles are paralyzed (Figure 89-4). The biceps and radioperiosteal reflexes are absent. Unless there has been avulsion of nerve roots, recovery occurs in part if not completely. It is important to protect the arm and brachial plexus from further injury by pinning the forearm in a sleeve sling in front of the chest. Two to three weeks later, passive range of motion is appropriate to prevent contracture of the shoulder. Although quick recovery may be observed, complete recovery may take many months. Good return of hand and arm function by 6 months is a good prognostic sign [DiTaranto et al., 2004].
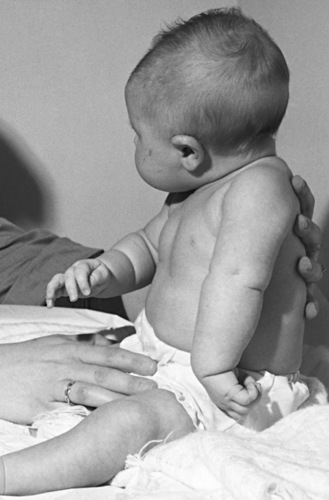
Fig. 89-4 Erb’s palsy caused by birth injury.
(Courtesy of Division of Pediatric Neurology, University of Minnesota Medical School.)
Newborn brachial plexus injury can cause phrenic nerve palsy in a small percentage of infants with diaphragmatic paralysis and significant respiratory sequelae plus failure to thrive [Bowerson et al., 2010].
Surgery for newborn brachial nerve lesions is becoming more popular, especially when electrical physiologic and neuroimaging data indicate primary injury to the plexus and no nerve root evulsion. Primary nerve reconstruction, followed by appropriate tendon transfers, provides return of function in select patients [Tonkin et al., 1996; Piatt, 2004]. Surgical intervention may benefit 20–25 percent of all patients, including those with conducting neuroma of the upper trunk [Chen et al., 2008].
Injury to the lower trunk of the plexus, involving the eighth cervical nerve and first thoracic nerve, is known as Klumpke paralysis. This condition is rare, composing less than 1 percent of newborn brachial plexus injuries [al-Qattan et al., 1995]. Weakness of the forearm extensors, flexors of the wrist and fingers, and intrinsic muscles of the hand occurs. Horner’s syndrome is often present as a result of involvement of the sympathetic fibers accompanying the first thoracic nerve. The elbow is typically flexed, the forearm is supinated, the wrist is extended, and a clawlike deformity of the hand with hyperextension of the wrist and fingers is present. The triceps reflex may be diminished, and the grasp reflex is usually absent. As with Erb’s palsy, protection of the plexus, initially followed by passive range of motion therapy – in this instance, to the wrist and fingers – is appropriate during recovery.
The most severe newborn brachial plexus injury occurs when all three trunks of the brachial plexus are involved. Total brachial plexus involvement is present 10 percent of the time in newborn injuries. The arm hangs limply from the shoulder, with no muscle movement. This is associated with tendon areflexia and decreased sensation over the arm and hand. The outlook for recovery after complete brachial plexus injury is poorer than with isolated upper or lower trunk injury because of the larger distribution of involvement and the usually greater severity of nerve injury. In this circumstance, nerve roots have often been avulsed or nerves in the plexus divided by severe stretching. In up to 10–20 percent of newborn brachial plexus injuries, bilateral involvement is present. Rare instances of brachial neuritis have been reported in children and may be associated with diphtheria-pertussis-tetanus vaccinations [Martin and Weintraub, 1973].
Inherited Conditions
Recurrent inherited brachial plexus neuritis, referred to as hereditary neuralgic amyotrophy, is carried on chromosome 17q25.3, where mutations in SEPT9, encoding the septin-9 protein, have been identified [Hannibal et al., 2009; Ueda et al., 2010]. It is a dominantly inherited condition typically diagnosed in childhood [Guillozet and Mercer, 1973; Wiederholt, 1974]. This recurrent syndrome presents with pain and is followed by brachial plexus-innervated muscle weakness, but often includes facial paresis. The long thoracic nerve is involved, with scapular winging present. Lumbar plexus involvement may be noted. Tendon reflexes are reduced. Residual weakness may develop after exacerbations. Autonomic nerves are affected [Dunn et al., 1978]. Short stature, hypotelorism, small face and palpebral fissures, prominent epicanthal folds, and syndactyly are part of the syndrome [Laccone et al., 2008]. Axonal loss is demonstrated by electrical physiologic testing. The condition is believed to be an immune complex disease that causes damage to blood vessel walls. In most instances, recovery occurs spontaneously, and there is no specific treatment [Rubin, 2001].
Hereditary neuropathy with liability to pressure palsies may present in children. This autosomal-dominantly inherited condition, due to a deletion involving thePMP22gene, carried on chromosome 17p11.2–p12, usually causes transient paralyses in the territory of individual peripheral nerve trunks but may cause recurrent brachial neuropathy. The condition is painless, demyelination is corroborated electrophysiologically, and recovery is common [Chance, 2006; Windebank et al., 1995].
Inflammatory Neuropathies
Acute Inflammatory Demyelinating Polyradiculoneuropathy (Guillain–Barré Syndrome)
Acute inflammatory demyelinating polyradiculoneuropathy is commonly known as Guillain–Barré syndrome (see Chapter 90). It is an inflammatory, demyelinating disorder of spinal nerve roots and peripheral nerves of acute to subacute onset, associated with a T-cell-mediated immune response [Hughes et al., 1999; Vucic et al., 2009]. An antecedent, presumably viral infection, triggers inflammation and demyelination [Arnason and Soliven, 1993; Hartung et al., 1995a]. Cytomegalovirus, Epstein–Barr virus, Mycoplasma pneumoniae, vaccinia virus, and Campylobacter jejuni bacterial diarrhea are recognized causes of prodromal illness preceding acute inflammatory demyelinating polyradiculoneuropathy [Hartung et al., 1995b; Kuwabara, 2004]. An antecedent infectious disease is recognized in 65–74 percent of cases 3 days to 6 weeks before the onset of symptoms caused by demyelination [Beghi et al., 1985; Hartung et al., 1995b; Korinthenberg et al., 2007], and 8 percent of cases are preceded by vaccination. The resultant neuropathy is predominantly motor [Guillain et al., 1916], and is typically accompanied by mild sensory loss and sensory ataxia. Severe sensory deficit is rare [Dawson et al., 1988]. Autonomic involvement occurs in children and may be quite severe for some [Hodson et al., 1984].
Guillain–Barré syndrome can now be divided into at least four subtypes. The most important in the Western world is acute inflammatory demyelinating polyradiculoneuropathy (AIDP) [Vucic et al., 2009]. The other subtypes include acute motor axonal neuropathy (AMAN) and acute motor and sensory axonal neuropathy (AMSAN); these are more prevalent in Asia and Central and South America. In Colombia, AMAN accounted for 30 percent of cases [Ortiz-Corredor et al., 2007]. The fourth and rarest subtype, representing 1 percent of cases, is the Miller Fisher syndrome of ataxia, areflexia, and ophthalmoplegia.
Pathology
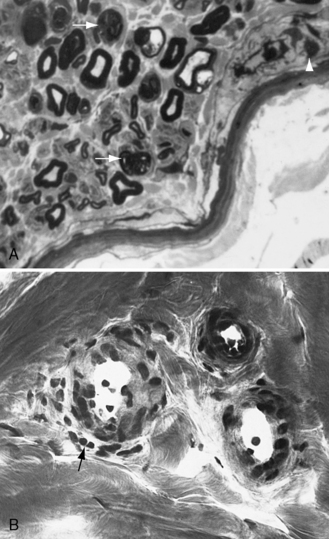
Demyelination and mononuclear cell infiltration of peripheral nerve may be found on nerve biopsy in AIDP. Varying degrees of edema, endoneurial lymphocytic cell infiltration, and segmental demyelination occur (Figure 89-5). Macrophage invasion of Schwann cells can be seen ultrastructurally [Brechenmacher et al., 1987]. Spinal roots and proximal and distal nerves display focal and perivenular lymphocytes and macrophages [Hall et al., 1992]. Complement fixing antibodies to peripheral nerve myelin may be demonstrated in some cases [Koski et al., 1987]. Children with previous diarrhea are more likely to have autoantibodies [Nishimoto et al., 2008]. Axonal degeneration is present in AMAN and AMSAN, with antibodies to gangliosides on the axolemma that target macrophages to invade the axon at the node of Ranvier, producing axonal degeneration and subsequent muscle weakness [Hughes and Cornblath, 2005; Uncini and Yuki, 2009]. In AMAN, antibodies against GM1, GM1b, GD1a, and GalNAc-G1a have been found in children [Nishimoto et al., 2008]. Antibodies to GalNaccGd1a are found in AMSAN and to GQ1b in Miller Fisher syndrome [Hughes and Cornblath, 2005]. CSF protein is often elevated after the first week, usually accompanied by a lack of inflammatory cellular response – often with fewer than 10 lymphocytes/mm3.
Clinical Characteristics
The overall incidence of AIDP is 1–2 per 100,000 each year; in the population younger than 18 years, the incidence is 0.8 per 100,000 each year [Beghi et al., 1985]. The incidence rate for males is 1.5 times that for females [Koobatian et al., 1992]. Muscle pain may be the initial manifestation of the disease in children, involving the anterior and posterior thighs, buttocks, and lower back [Ropper and Shahani, 1984]. Pain and paresthesias after infection are quickly followed by symmetric, progressive weakness of extremity and facial muscles. Bladder dysfunction, mental alterations, headache, ataxia, and meningismus may be accompanying features. Tendon reflexes are diminished or lost. The disease may progress for days to several weeks (median of 7 days) before plateauing [Korinthenberg et al., 2007], and then improves over a period of months. Weakness may proceed rapidly with complete paralysis, including bulbar paralysis, developing within 24 hours of onset. Ventilatory support may be necessary, even early in the course of the illness. Eighty percent of patients make an excellent recovery.
Unusual childhood variants of Guillain–Barré syndrome include:
MRI often reveals gadolinium enhancement of the spinal nerve roots. Electrophysiologic evaluation usually provides evidence of peripheral nerve demyelination, often accompanied by changes indicative of axonal involvement. Distal motor latencies are prolonged to more than 150 percent of normal. Nerve conduction velocities are reduced to below 70 percent of normal. Compound motor action potentials are significantly reduced, with prolonged terminal dispersions, these being the most common electrophysiologic findings in children [Bradshaw and Jones, 1992]. Children younger than 10 years of age have greater slowing of motor nerve conduction velocities than children older than 10. More profound slowing corresponds to high antibody levels against peripheral nerve myelin during the first week of illness [Rudnicki et al., 1992]. In cases where the pathologic change is one of mainly axonal degeneration, nerve conduction studies reveal normal or mildly slow conduction with diminished amplitude of compound muscle action potentials.
The youngest reported patients are neonates. A 4-month-old child presented with advancing hypotonia [Carroll et al., 1977]. Several other children younger than 6 months of age have been reported. The rate of progression, degree of impairment, and time before improvement vary greatly; however, recovery does not continue after 2 years from onset [Ropper, 1986]. Poor prognosis is related to the need for ventilation, and failure to improve after 3 weeks of reaching peak deficit [Winer et al., 1985]. The mortality rate approaches 5 percent in adults but should be negligible in children; 10–15 percent of adults remain permanently disabled [Arnason, 1984].
Management
Plasmapheresis may be effective if begun within 7 days of illness onset [French Cooperative Group, 1987; Greenwood et al., 1984; Guillain-Barré Syndrome Study Group, 1985; Osterman et al., 1984; Smith and Hughes, 1992]. If patients lose the ability to walk unaided, have significant reduction in respiratory capacity, or have bulbar insufficiency, plasmapheresis or intravenous immunoglobulin therapy is indicated. Although most plasmapheresis studies have been performed on adults, a study on children demonstrated shortened recovery time in 5 of 6 children aged 5–15 years [Lamont et al., 1991]. A retrospective study showed faster recovery of independent ambulation in 9 children who received plasmapheresis, compared with 14 who did not receive plasmapheresis [Epstein and Sladky, 1990]. Intravenous gamma globulin is more convenient in small children and shortens the course of illness [Kleyweg et al., 1988; Sladky, 2004; Tasdemir et al., 2006]. In adults, intravenous immunoglobulin given during the first week is at least as effective as plasmapheresis and may be of greater efficacy [van der Meché et al., 1992]. There is no documented benefit to using both therapies [Bril et al., 1996; Hughes and Plasma Exchange/Sandoglobin Guillain–Barré Syndrome Trial Group, 1996].
Chronic Inflammatory Demyelinating Polyradiculoneuropathy
Chronic inflammatory demyelinating polyradiculopathy (CIDP) was the cause of polyneuropathy in 8.8–11.6 percent of children in two series of childhood polyneuropathy [Ouvrier and McLeod, 1988; Sladky et al., 1986]. A low frequency of antecedent events is described. Subacute onset of weakness over 2 months with predominantly lower-extremity motor system involvement, accompanied by sensory loss and diminished tendon reflexes, is typical [Nevo et al., 1996; Simmons et al., 1997]. Pain and cranial neuropathies are uncommon. Progressive weakness for 2 months is required for the diagnosis of CIDP, although 1 in 5 affected children reaches the nadir by this time [Simmons et al., 1997]. For some children with CIDP, a monophasic disease progresses beyond 2 months, and for others a relapsing-remitting course is present [Markowitz et al., 2008]. A subacute steroid-responsive inflammatory polyneuropathy, which progresses for up to 2 months, has also been recognized [Rodriguez-Casero et al., 2005].
Investigations
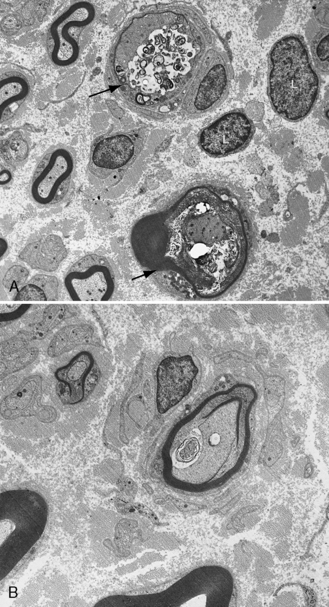
Cerebrospinal protein is elevated, with a normal cell count. Slowed conduction velocities, prolonged distal latencies, and temporal dispersion are present electrophysiologically. MRI often reveals gadolinium enhancement of the spinal nerve roots. Sural nerve biopsy, if performed, shows demyelination, remyelination, and usually a T-cell lymphocyte and macrophage inflammatory infiltrate. Onion bulb formation indicates chronicity of the condition (Figure 89-6). Circulating CD4+ and CD25+ T-regulatory lymphocytes are reduced in CIDP patients, and there is increased frequency of genotype GA13–16 of the SH2D2A gene, encoding a T-cell-specific adapter protein [Notturno et al., 2008]. These findings seemingly alter immune responses, predisposing affected individuals to chronic illness.
Management
Childhood CIDP has been shown to improve with therapy [Colan et al., 1980; Gorson et al., 1997; Nevo et al., 1996; Sladky et al., 1986; Uncini et al., 1991], which most often begins with intravenous immunoglobulin; however, most children respond to corticosteroids, with slow tapering to prevent relapses [Rabie and Nevo, 2009]. In children resistant to oral prednisone or intravenous methylprednisolone and intravenous immunoglobulin, cyclosporine may be of benefit. Two infants and one child responded to treatment without adverse effects. A small group of patients require intense immunosuppression and some require regular intravenous immunoglobulin infusions for a prolonged period.
Hereditary Neuropathies
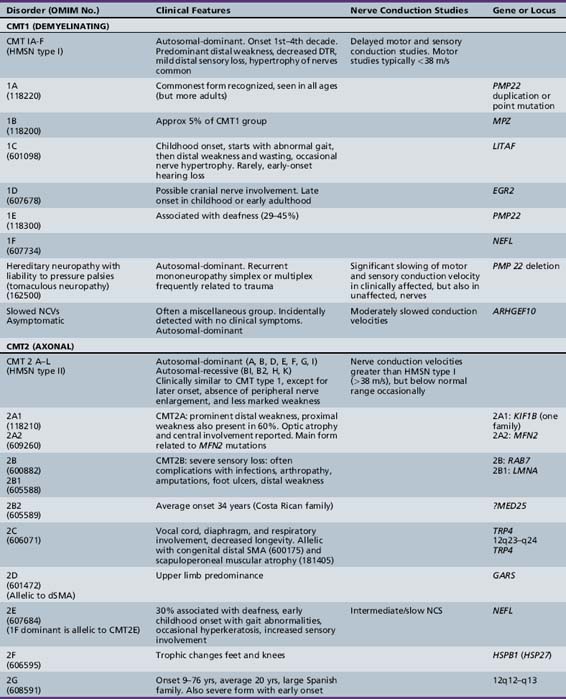
Approximately 70 percent of chronic neuropathies seen in children are hereditary. In Dyck’s classification of these conditions [Dyck, 1984], the broad categories of hereditary motor (HMN), hereditary sensory and autonomic (HSAN), and hereditary motor and sensory neuropathies (HMSN) are subdivided on the basis of clinical, genetic, and (rarely) biochemical characteristics. The main basis for inclusion in one of these three groups is whether the peripheral motor, peripheral sensory, or both nerve groups are mainly affected. The hereditary peripheral motor neuropathies are called the spinal muscular atrophies in other classifications and are described in Chapter 88. The original Dyck classification of the hereditary motor and sensory neuropathies did not allow for the inclusion of autosomal and X-linked recessive forms of HMSN types I and II. Nor could it have included the striking recent advances in the understanding of the molecular biology of these disorders.
The classification of Charcot–Marie–Tooth disease and related disorders has become complex and confused, and is likely to remain so because of the ever-expanding list of gene mutations that are continuing to be discovered. A current summary of the hereditary polyneuropathies is presented in Table 89-2.
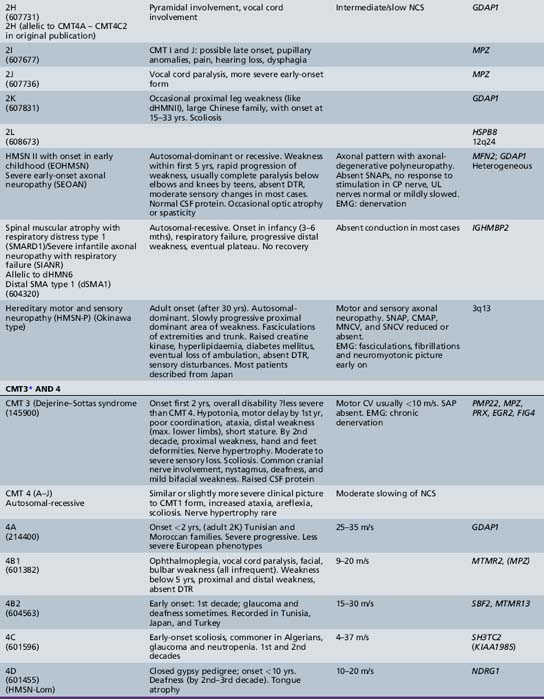
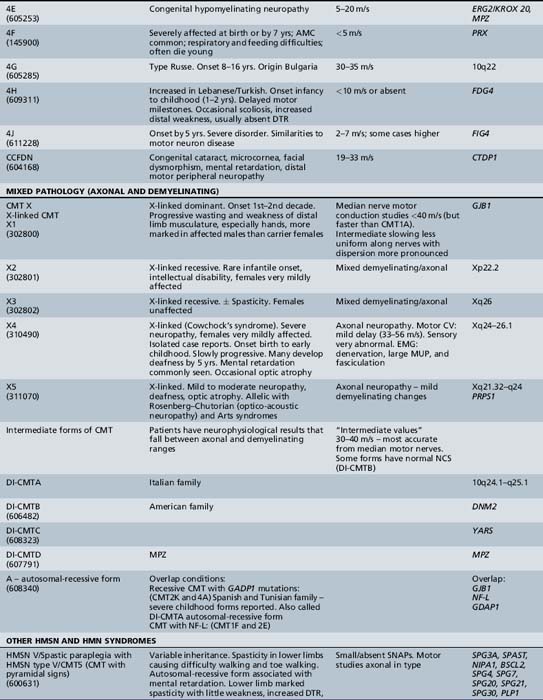
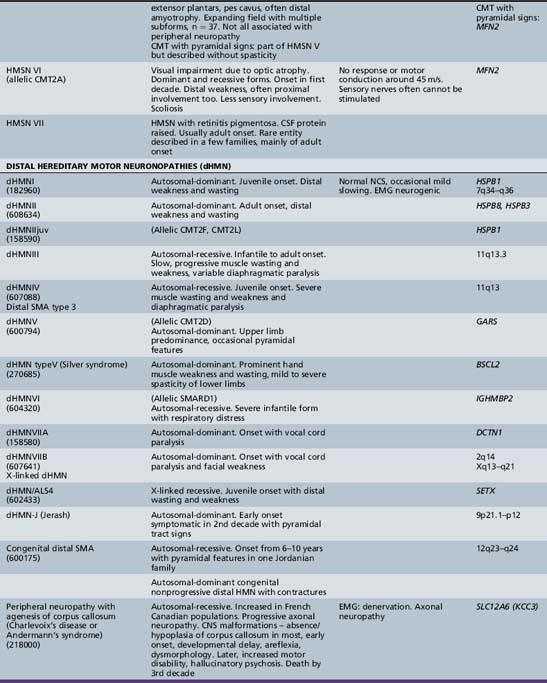
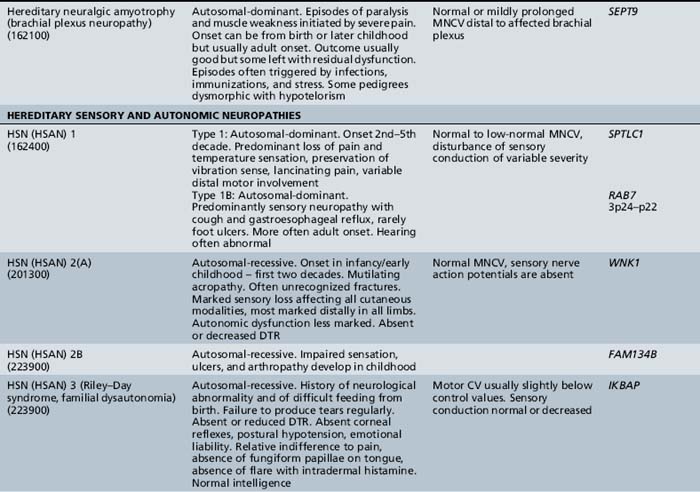

Hereditary Motor and Sensory Neuropathy (Charcot–Marie–Tooth Disease)
HMSN (CMT disease) type I is a heterogeneous disorder causing a progressive motor and sensory demyelinating neuropathy. It is a dominantly inherited disorder with onset in the first or second decade of life. In certain families, the defective gene maps to chromosome 1 [Chance et al., 1990; Lebo et al., 1990], where it has been shown to cause mutations of myelin protein zero (P0), the most abundant protein in peripheral myelin [Hayasaka et al., 1993]. Much more commonly, the defective gene links to the short arm of chromosome 17 in the region 17p11.2 [Middleton-Price et al., 1990; Raeymakers et al., 1989; Vance et al., 1989]. In most type I families, affected members have a 1.5 million basepair tandem DNA duplication at that site [Lupski et al., 1991, Hallam et al., 1992; Raeymakers et al., 1991]. This region contains the gene for the myelin protein PMP22, increased amounts of which may lead to excessive or faulty production of myelin. Rare cases of HMSN type I have been due to point mutations of the PMP22 gene. The disease carried on chromosome 17 is now designated CMT type IA, and the condition carried on chromosome 1 is called CMT type IB. Although cases of HMSN types IA and IB caused by point mutations tend to be more severely affected than cases caused by the duplication on chromosome 17, the frequent exceptions to the rule make it difficult to separate these subtypes on clinical or neurophysiologic grounds. Cases of autosomal-dominant HMSN type I, in which no abnormalities of the PMP22 or MPZ genes can be demonstrated, are designated CMT types IC to 1F, the causative genes for which are shown in Table 89-2.
Pathology
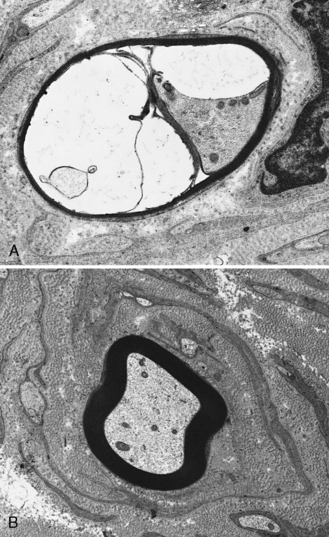
The peripheral nerves in CMT type I are often easily palpated because they are hypertrophied as a result of excessive Schwann cell and fibroblast activity. This creates redundant wrappings of Schwann cell investments around nerve fibers accompanied by excessive collagen deposition, known as onion bulbs [Thomas et al., 1974]. Sural nerve biopsies disclose a loss of myelinated fibers, especially the large myelinated fibers. With advancing age, there is progressive reduction in the myelinated fiber density, an increased number of fibers undergoing demyelination, and increased frequency of onion bulb formations [Low et al., 1978]. In this setting, onion bulb formations are the result of repeated episodes of demyelination and remyelination (Figure 89-7). Some loss of axons is eventually observed, indicating that, although demyelination is predominant, some axonal degeneration is part of the disease process and is responsible for the progression of the weakness and wasting [Nordborg et al., 1984].
Clinical Characteristics
The disease is typically recognized during the first decade of life or in early adolescence, but it may be evident even at birth. A large variation in the age of onset and severity of disease among individuals in the same family has sometimes been observed [Birouk et al., 1997; Hagberg and Lyon, 1981]. Early onset, although uncommon, usually results in greater disability later in life than that seen in individuals with later onset, who are often able to ambulate and work until old age.
Clinical Neurophysiology
Motor and sensory conduction velocity is within the “demyelinating” range and thus is less than 60 percent of normal values in infants; in patients older than 3 years, it is less than 38 m/sec in all peripheral nerves. Conduction velocity changes little with increasing age, but there is a progressive reduction in the amplitude of the evoked muscle action potential, indicating axonal loss [Roy et al., 1989].
Genetics
HMSN type I is dominantly inherited. In addition, autosomal-recessive [Gabreels-Festen et al., 1992; Harding and Thomas, 1980] and X-linked forms of a similar disorder exist [Hahn et al., 1990; Rozear et al., 1987]. Autosomal-recessive demyelinating forms, which are now designated CMT type 4, tend to have an earlier onset and a higher incidence of ataxia, complete areflexia, and kyphoscoliosis than dominant types. Several recessive varieties have been cloned or linked to specific chromosomal regions, e.g., in inbred Tunisian (chromosome 8q13–21) [Ben Othmane et al., 1993a], Bulgarian (chromosome 8q24) [Kalaydjieva et al., 1996], and Algerian (chromosome 5q23–33) [Kessali et al., 1997] families. The genes for these conditions are listed in Table 89-2. Another form with florid myelin sheath changes, known as hereditary motor and sensory neuropathy with focally folded myelin or congenital dysmyelinating neuropathy, has been linked to chromosome 11q23. Even this latter condition, however, is heterogeneous. CMT type 4B1 is due to mutations of the myotubularin gene; CMT type 4B2, which is characteristically associated with glaucoma, is due to a mutation of the SBF2 gene; and CMT type 4B3 (also known as CMTIVH) is caused by FGD4 mutations. Similar histopathological changes are sometimes seen with MPZ mutations.
Hereditary Motor and Sensory Neuropathy Type II
Hereditary motor and sensory neuropathy (Charcot–Marie–Tooth disease) type II (CMT2) is heterogeneous and is usually dominantly inherited. Autosomal-recessive and X-linked forms of the disorder are known and will be discussed further [Hahn et al., 1990; Harding and Thomas, 1980; Rozear et al., 1987]. In fact, CMT2 has now been linked to at least 13 loci and 10 genes (see Table 89-2). Whilst CMT2A was initially linked to a missense mutation in the kinesin family member 1B-ss (KIF1B) gene in one pedigree, no such mutations have been identified in other affected kindreds. Mutations in the gene encoding a mitochondrial fusion protein, mitofusin 2 (MFN2), are now thought to be responsible for about 25 percent of CMT2A cases [Zuchner et al., 2004; Verhoeven et al., 2006]) (see below).
CMT2B is caused by missense mutations in RAB 7, which encodes the small guanosine triphosphatase late endosomal protein, which may have a role in axonal transport [Houlden et al., 2004]. It may be confused with hereditary sensory and autonomic neuropathy type I because of the presence of moderate sensory loss, with complicating ulcers in up to 50 percent of cases [Verhoeven et al., 2003]. Other subtypes of autosomal-dominant CMT2 are listed in Table 89-2.
The pathology is marked by axonal degeneration. Nerve hypertrophy and palpable nerve enlargement are not present. The onion bulb formation typical of type I disease is rarely seen. In one variant, focally enlarged myelinated axons with aggregations of neurofilaments have been described [Goebel et al., 1986]. In addition to the focal accumulation of intra-axonal neurofilaments in the myelinated fibers, similar structures have been seen without axonal enlargement in the nonmyelinated axons. Similar structures have been noted in fibroblasts and endothelial cells in muscle and skin. Whether these intra-axonal filaments describe a different disease process is unknown. Nerve conduction velocities are low-normal to slightly reduced.
A rather severe form of HMSN type II was designated hereditary motor and sensory neuropathy of neuronal type with onset in early childhood (EOHMSNA). Two large series of such patients were reported by [Ouvrier et al., 1981] and by [Gabreels-Festen et al., 1991]. The disorder accounts for up to 5 percent of childhood chronic neuropathies. The features of the disorder include sporadic occurrence or evidence of autosomal-recessive or, less commonly, dominant inheritance. In addition, onset of weakness within the first 5 years of life, rapid progression of weakness to almost complete paralysis below the elbows and knees by the second decade, moderate sensory changes in most cases, clinical electrophysiologic studies consistent with an axonal degenerative polyneuropathy, and histologic studies indicating neuronal (axonal) atrophy and degeneration of peripheral motor and sensory neurons are noted. CSF is usually normal. The condition advances rather rapidly, so that most patients are nonambulatory by the mid- to late teens. Approximately 50 percent of such Australian cases studied have been shown to be due to autosomal-dominant (CMT 2A) or recessive (compound heterozygous or homozygous) mutations of the mitofusin (MFN2) gene. Mitofusin2, a large transmembrane protein, promotes membrane fusion and is involved in the maintenance of the morphology of axonal mitochondria. Families with MFN2 mutations have been reported in which some members had variable upper motor neuron signs or optic atrophy in addition to peripheral neuropathy. MFN2 mutations thus have also been implicated in the causation of CMT disease associated with pyramidal tract features (HMSN type V) and with cases of CMT disease complicated by optic atrophy (HMSN type VI). A similar EOHMSNA phenotype due to mutations of the ganglioside-derived differentiation-associated protein 1 (GDAP1) gene has been seen, mainly in North African and Mediterranean countries.
Congenital and Early Infantile Axonal Types
Some infants are born with arthrogryposis secondary to an axonal neuropathy. This condition is often a nonprogressive disorder, presumably secondary to an intrauterine insult. It is not clear whether these disorders are hereditary. Other cases have been described in which infants a few weeks old have presented with talipes and, later, rapidly progressive respiratory distress, leading to death or permanent ventilatory support within the first year of life [Appleton et al., 1994; Vanasse and Michaud, 1992]. In some of these cases, clinical electrophysiologic and histopathologic examinations have been consistent with a form of spinal muscular atrophy affecting the diaphragm and distal muscles preferentially (SMARD) [Grohmann et al., 2003]. In others, investigations have confirmed the presence of an axonal neuropathy [Wilmshurst et al., 2000]. Mutations of the gene encoding the immunoglobulin mu binding protein (IGHMBP2) cause both of these disorders.
Hereditary Motor and Sensory Neuropathy Type III (Dejerine–Sottas Disease) and Hypomyelinating Neuropathies
The most extreme form of hypomyelinating neuropathy, in which the patient is severely affected at birth by a condition resembling congenital spinal muscular atrophy, is sometimes termed congenital amyelinating neuropathy [Charnas et al., 1988; Karch and Urich, 1975; Kasman et al., 1976; Palix and Coignet, 1978; Seitz et al., 1986]. In this group, arthrogryposis is common, presumably because of lack of limb movements in utero. Due to respiratory problems and swallowing difficulties, these infants usually die when very young. Nerve conduction velocity is either unrecordable or extremely low. Peripheral nerve histopathologic examination reveals no (amyelination) or very little myelin and virtually absent onion bulbs. Such cases probably represent examples of the extreme end of the spectrum of clinical and pathologic severity resulting from myelin protein mutations. Cases of central and peripheral hypomyelination in association with juvenile-onset cataracts were due to mutations of DRCTNNB1A, which encodes hyccin, a membrane protein [Ugur and Tolin, 2008]. SOX10 mutations can also cause central and peripheral hypomyelination in combination with Waardenburg’s syndrome or with Hirschsprung’s disease. Less extreme but severe infantile hypomyelinating cases merge into the Dejerine–Sottas syndrome (HMSN type III). In the majority of cases, hypotonia and delay in walking to beyond the second year are observed, but most HMSN type III toddlers eventually do walk, although coordination is never fully normal. Ataxia is present in all patients and is probably due to a proprioceptive deficit. Distal weakness, more marked in the lower limbs, is present early. Proximal weakness and deformity of the hands and feet are not common in early childhood. Both become increasingly more obvious with advancing age, so that by the second decade proximal weakness is the rule. Deep tendon reflexes are usually absent. Clinical enlargement of nerves, with the median and other peripheral nerves feeling thicker than a pencil, is present in most HMSN type III patients by the end of the first decade or earlier. By the time adequate testing can be performed, moderate to severe sensory loss is present in most patients. By comparison, such marked sensory loss is rarely seen in CMT type I. Scoliosis often occurs early and tends to progress. Nerve conduction velocities are typically lower than 10 m/sec, probably because of loss of saltatory conduction. Prognosis is variable [Guzzetta et al., 1982]. Some patients clearly deteriorate over the first two decades [Tyson et al., 1997], but most patients we have been able to follow have shown little change over many years, except for the progression of limb deformities and scoliosis [Ouvrier et al., 1987].
Many, perhaps the majority of, cases of congenital hypomyelinating neuropathy and Dejerine–Sottas syndrome are the result of myelin protein mutations. Both (dominant) heterozygous PMP22 and myelin P0, as well as homozygous myelin P0 mutations, have been reported to cause the Dejerine–Sottas phenotype [Tyson et al., 1997; Valentijn et al., 1995; Warner et al., 1996]. In addition, homozygous inheritance of the chromosome 17p11.2–12 duplication (in which the patient has four copies of the PMP22 gene), compound heterozygous states involving a deletion and a point mutation of the PMP22 gene [Gonnaud et al., 1997], and a case of apparent homozygosity for HMSN type II [Sghirlanzoni et al., 1992], caused by a myelin P0 mutation [Taroni et al., 1996], have all been associated with the Dejerine–Sottas phenotype. Periaxin, EGR2, and FIG4 gene mutations are also rarely causative. The complexity of the genetics of the Dejerine–Sottas syndrome makes it important to study such cases and their families, wherever possible, by detailed molecular genetic analysis before genetic counseling is given. In addition to the preceding studies, two remarkable cases of recovery from a congenital hypomyelinating neuropathy were reported by [Ghamdi et al., 1997] and [Levy et al., 1997].
Hereditary Motor and Sensory Neuropathy Types IV to VII
The term hereditary motor and sensory neuropathy type IV was applied by Dyck to Refsum’s disease, but is not widely used. Dyck designated patients with spastic paraplegia, who had abnormalities in quantitative testing of sensation, sensory nerve conduction, and histometric evaluation of sural nerve, as well as a pattern of autosomal-dominant inheritance, as having HMSN type [Harding and Thomas 1984] reported 25 cases of peroneal muscular atrophy with pyramidal features. In contrast to [Dyck et al., 1975], these authors believed that peroneal muscular atrophy with pyramidal features is a separate disorder from hereditary spastic paraplegia but that it also demonstrates heterogeneity. The combination of hereditary motor and sensory neuropathy with optic atrophy was designated hereditary motor and sensory neuropathy type VI by [Dyck et al., 1975]. First described by Vizioli, this rare condition may be transmitted by autosomal-dominant or recessive inheritance [Ippel et al., 1995]. The combination of a neuropathy with familial retinitis pigmentosa, first reported by [Massion-Verniory et al., 1946] from Belgium, was designated HMSN type VII by [Dyck et al., 1975]. In the original family, a 34-year-old man rather abruptly developed visual and gait difficulties that worsened over the next 9 years. Two of his sisters had pigmentary retinopathy without evidence of neuropathy. CSF protein was elevated. A nerve biopsy was unhelpful. Cases of this combination are rare and, to the authors’ knowledge, have not been reported in childhood.
X-Linked Forms
X-linked dominant forms (x-linked dominant charcot–marie–tooth disease (CMTX1, HMSNX1)
Clinical Manifestations
The clinical features of X-linked dominant CMT (CMTX1) are broadly similar to those of CMT type IA, but the disease is more severe in CMTX males and less severe in females than in CMT type Ia. Gait difficulties are usually the first manifestation. Wasting of leg muscles and, subsequently, of the intrinsic muscles of the hands is more marked in CMTX1 males than females and more obvious than in males with CMT type Ia. The patellar reflexes are retained in about 50 percent of affected females but in only 10 percent of males. The ankle jerks are absent in most cases [Nicholson and Nash, 1993]. Sensory loss is present in more than 75 percent of affected adult men and women. Clinical hypertrophy of nerves is not a feature. Despite the presence of the condition, few adults with the disorder are sufficiently disabled for disease to interfere with work until age 50 or 60. Affected men often require canes to stabilize their gait when elderly, but they are not often wheelchair-bound.
Clinical Neurophysiology
Motor nerve conduction velocities in CMTX1 males tend to be lower than normal but faster than those in typical HMSN type I cases. [Nicholson and Nash 1993] found a mean median motor nerve velocity of 31 m/sec in a group of 20 X-linked HMSN males from two kindreds, compared with a mean median motor nerve conduction velocity of 45 m/sec in 32 affected females.
Genetics
The gene, GJB1, for the gap junction protein (connexin 32) is localized to the DXYS1 region at Xq13.1 on the proximal long arm of the X chromosome near the centromere [Bergoffen et al., 1993a; Fischbeck et al., 1986]. Direct sequencing of the GJB1 gene has revealed numerous mutations in CMTX1 individuals [Bergoffen et al., 1993b]. There is remarkable variability in the GJB1 mutations, with mostly different mutations in tested CMTX1 patients [Scherer et al., 1994]. Connexin 32 is normally expressed in myelinated peripheral nerve and appears to be localized mainly at the nodes of Ranvier and Schmidt–Lanterman incisures. It is postulated that connexin 32 may form intracellular gap junctions that connect the folds of Schwann cell cytoplasm, allowing transfer of ions, nutrients, and other small molecules around and across the compact myelin to the innermost myelin layers, perhaps also providing indirect sustenance to the axon as well [Bergoffen et al., 1993b].
X-linked recessive forms
Four different X-linked recessive forms of CMTX (CMTX2–5) have also been reported. Female carriers are usually asymptomatic. The specific characteristics of these disorders are still poorly defined, except for CMTX5, in which a combination of deafness and progressive optic atrophy complicates the progressive polyneuropathy. This condition is also known as Arts syndrome, optico-acoustic neuropathy, or the Rosenberg–Chutorian syndrome, and is due to loss of function mutations in PRPS1 (phosphoribosylpyrophosphate synthetase 1), which result in reduced de novo purine synthesis. Treatment with S-adenosylmethionine (SAM) appears to be of benefit [Kim et al., 2007].
Distal Hereditary Motor Neuropathies
Traditionally, the distal hereditary neuropathies (dHMN) have been grouped with the spinal muscular atrophies. They are defined clinically by the presence of predominantly distal weakness with absent sensory changes. Motor nerve conduction velocities are normal or only mildly slow. Sensory conduction is normal. Sural nerve biopsies are also normal, but electromyography and biopsy studies of distal muscles show evidence of denervation. With the advances of molecular biology [Irobi et al., 2006], it has become clear that there is sometimes overlap, with sensory as well as motor neuron involvement, both clinically evident and apparent on laboratory investigation. A good example of this overlap is seen in the disorders caused by mutations of the gene encoding the immunoglobulin mu binding protein (IGHMBP2), in which respiratory failure in infancy is accompanied by either a severe sensorimotor polyneuropathy (SIANR) or, more commonly, spinal muscular atrophy (SMARD or dHMNVI). The types of dHMN are listed in Table 89-2.
Hereditary Sensory and Autonomic Neuropathies
Type I
Hereditary sensory and autonomic neuropathy type I is a dominantly inherited disorder, usually beginning in the second or third decade of life [Dyck, 1984]. The condition is mapped to 9q22.1–22.3 [Nicholson et al., 1996]. The disorder is caused by mutations of the SPTLC1 gene encoding serine palmitoyl transferase [Dawkins et al., 2001]. In some instances, genetic analysis has shown that a similar disease may be inherited as an autosomal-recessive condition rather than as the typical dominantly inherited condition with variable expression [Kondo et al., 1974]. Characteristically, the patient experiences lightning-like pains and develops perforating ulcers of the feet.
Cutaneous sensation and tendon reflexes are lost in the lower extremities [Berginer et al., 1984]. Pes cavus, hammer toes, and peroneal muscular weakness develop. Variability of disease expression is so great that some affected relatives may be asymptomatic, but 100 percent penetrance is evident by the age of 30 years, as reported in a large kindred by [Nicholson et al., 1996]. Spontaneous amputations of the distal limbs have occurred. Pathologic studies from such individuals have demonstrated nerve degeneration and bone necrosis [Teot et al., 1985]. As individuals advance into adulthood, the loss of all peripheral sensory modalities, except those of the face, has been reported [Nance and Kirby, 1985]. Hereditary deafness is an associated feature [Fitzpatrick et al., 1976; Nicholson et al., 1996]. Increased immunoglobulin A levels were found in members of three unrelated families [Whitaker et al., 1974]. In a separate family, both immunoglobulin A and immunoglobulin G levels rose as the disease progressed [Iwabuchi et al., 1976]. Sural nerve biopsy reveals marked loss of myelinated nerve fibers, especially large myelinated fibers, and loss of unmyelinated nerve fibers [Danon and Carpenter, 1985]. Treatment is limited and is generally preventive. Protection of the feet from injury by wearing appropriate shoes and avoiding jumping and falling is important. Meticulous attention to proper toenail trimming and treatment of plantar ulcers are other important measures. If conservative efforts fail, amputation of affected extremities is necessary [Gwathmey and House, 1984].
Type II
Hereditary sensory and autonomic neuropathy type II occurs as a congenital sensory neuropathy that is recessively inherited [Ohta et al., 1973]. The disorder is due to mutations of the HSN2 gene [Lafreniere et al., 2004]. In addition to loss of pain and temperature sensation, affected children have great difficulty recognizing objects by touch. Recurrent infections are common in the digits, and fractures may occur. As the disease progresses, mutilation of fingers and toes occurs. Motor nerve conduction is normal but sensory action potentials are absent. Sural nerve biopsies reveal a marked decrease in myelinated fibers of all calibers, as well as of unmyelinated fibers. There are possibly several subtypes of this disorder, one of which is thought to be nonprogressive [Ferrière et al., 1992].
Type III
Hereditary sensory and autonomic neuropathy type III is otherwise known as familial dysautonomia or the Riley–Day syndrome (see Chapter 98).
Type IV
Hereditary sensory and autonomic neuropathy type IV is a congenital recessively inherited sensory neuropathy, usually associated with mental retardation [Dyck, 1984]. Anhidrosis is a feature of the condition [Gillespie and Perucca, 1960; Pinsky and DiGeorge, 1966; Swanson, 1963; Vassalla et al., 1968]. [Rosemberg et al., 1994] summarized the findings in 32 cases: 16 sporadic and 16 from seven kindreds. Of the 16 sporadic cases, parental consanguinity was present in 4. The condition is similar to HSAN type II, except for the sweating deficit and possibly the preservation of fungiform papillae on the tongue. Oral lacerations and self-mutilation are particularly prominent, leading to infection and scarring of the tongue, lips, and gums, and sometimes to osteomyelitis of the jaw. The condition differs from HSAN type III, in that overflow tears are preserved in type IV and excessive sweating is a feature of dysautonomia. The anhidrosis leads to episodes of apparently unexplained fevers and hyperthermia, often associated with febrile convulsions, particularly in infancy.
The incidence and degree of mental retardation are apparently higher than in HSAN type II, although normal intellect has been reported. While often attributed to episodes of hyperthermia, mental disability occurs in the absence of overt episodes of the latter. When measured, IQs have varied from 41 to 92. Microcephaly is sometimes described [Rosemberg et al., 1994]. Decreased CSF levels of substance P were found in four patients with HSAN type IV. This may be a secondary effect of the loss of primary sensory neurons, which might, in turn, be the result of a prenatal deficiency of nerve growth factor [Iwanaga et al., 1996]. The long-term prognosis is poorly defined but, in the authors’ experience, rather unsatisfactory. Chronic infections, particularly of the bones and joints, cause multiple hospitalizations and deformities. Death before the age of 4 years occurs in approximately 20 percent of cases [Rosemberg et al., 1994], usually secondary to hyperthermia or sepsis.
This disorder is of autosomal-recessive inheritance. [Indo et al., 1996] reported four unrelated patients with consanguineous parents who had deletion, splice, and missense mutations of NTRK1, a tyrosine kinase receptor for nerve growth factor. Mouse models lacking NTRK1 show diminished responses to painful stimuli and develop skin ulcerations, missing digits, and corneal opacities. They also demonstrate deficits in cholinergic basal forebrain projections to the hippocampus and cortex [Smeyne et al., 1994]. The striking similarity of these mouse models to human HSAN type IV suggests that a deficit in NTRK1 is responsible for this disorder, which also explains the cognitive difficulties.
Type V
Individuals with HSAN type V do not perceive pain or temperature normally, but other sensory modalities are relatively well preserved [Dyck et al., 1983; Low et al., 1978]. Strength and deep tendon reflexes are normal, as are conventional nerve conduction studies. Nerve biopsy discloses absence of small myelinated fibers but preservation of the large and also the unmyelinated ones. These findings explain the preservation of tendon reflexes and the normal nerve conduction studies. The disorder is due to mutations of the nerve growth factor beta gene (NGFB) [Einarsdottir et al., 2004].
Other Inherited Sensory Neuropathies
Several entities have been described that do not fit into the preceding categories. These include sensory neuropathy in association with spasticity [Cavanagh et al., 1979], skeletal dysplasia [Axelrod et al., 1983], keratitis [Donaghy et al., 1987], growth hormone deficiency [Liberfarb et al., 1993], ichthyosis and anterior chamber syndrome [Quinlivan et al., 1993], deafness, and ovarian agenesis [Linssen et al., 1994]. In addition, two different types of sensory neuropathy have been described in Navajo children [Appenzeller et al., 1976; Johnsen et al., 1993]. Most of these conditions appear to be of autosomal-recessive inheritance, but an X-linked sensory neuropathy was described by [Jestico et al., 1985].
Congenital Indifference to Pain
The term insensitivity to pain was previously applied to patients in whom analgesia is the result of abnormalities of peripheral nerves, cutaneous nerve endings or central sensory pathways, whereas indifference to pain applied to those who have normal sensory pathways but fail to appreciate the painful nature of stimuli [Manfredi et al., 1981]. [Dyck et al., 1983] have emphasized the fact that precise analysis of some early cases of indifference to pain showed abnormalities of the peripheral sensory system when sophisticated methods were used. However, a case with a normal morphometric nerve study has been reported [Landrieu et al., 1990]. More recently, by sequence analysis of the SCN9A gene in three northern Pakistani families segregating autosomal-recessive “channelopathy-associated insensitivity to pain,” [Cox et al., 2006] identified three distinct homozygous nonsense mutations. These children appeared to be of normal intelligence. All could correctly perceive sensations of touch, warm and cold temperature, proprioception, tickle, and pressure, but not painful stimuli. [Goldberg et al., 2007] identified 10 different mutations in the SCN9A gene, 9 of which were truncating mutations, in affected members of nine different families with congenital insensitivity to pain. Thus, although the terminology has again been blurred, the molecular nature of this condition has now been clarified!
Acquired Sensory Neuropathy
Acquired sensory neuropathies are rare in childhood. [Roach et al., 1985] reported self-injurious behavior in a 2-year-old child with a sensory neuropathy following organophosphate ingestion. A delayed sensory neuropathy resulting from chlorpyriphos (another organophosphate) was reported in 14- and 15-year-old siblings by [Kaplan et al., 1993]. Sensory neuropathy of acute or subacute onset has been reported after administration of excessive doses of pyridoxine [Albin et al., 1987; Schaumburg et al., 1983]. Acute sensory neuropathy is a monophasic disorder characterized by the rapid onset of generalized paresthesia, ataxia, and areflexia associated with a severe, mainly proprioceptive, sensory defect. The site of the lesion is thought to be at the dorsal root ganglion and is probably mediated by an autoimmune response. Fernandez et al. [1994] described such a case in a 9-year-old boy. Leprosy may be associated with sensory neuropathy, which presents even in childhood, but the neuropathy is usually patchy or thermally distributed. Associated clinical features, coupled with family history, usually make diagnosis easy once suspicion is aroused [Sabin and Thomas, 1984]. It is exceptional for amyloidosis to present before the age of 15 years. Diagnosis is readily apparent on sural nerve biopsy [O’Connor et al., 1984].
Management of the Childhood Polyneuropathies
Specific Treatment
It is hoped that, as the genes causing the various forms of CMT disease are isolated and their gene products identified, more specific therapies may become available. Recent trials of high-dose vitamin C have not shown any clinical benefit in adults and children with CMT type 1A, despite promising effects in a transgenic mouse model [Burns et al., 2009].
Friedreich’s Ataxia
Friedreich’s ataxia is an autosomal-recessive disease characterized by slowly progressive ataxia commencing in childhood. The ataxia affects all motor functions, and the gait disturbance eventually requires use of a wheelchair, usually in the second decade of life. Intellect is preserved. Skeletal deformities of pes cavus, hammer toes, and kyphoscoliosis are typical. Cardiomyopathy is often present. A sensory neuropathy resulting from progressive degeneration of dorsal ganglion cells, with “dying back” of their central and peripheral axons, is the principal cause of disease progression [Lamarche et al., 1984]. This process results in an alteration in sensory nerve potentials and conduction [Dunn, 1973; Oh and Halsey, 1973]. Motor nerve conduction velocities remain typically in the low-normal range at the time when sensory conduction velocities are markedly impaired. These findings are in addition to those of the spinal cord posterior column and corticospinal tract degeneration. A loss of myelinated nerve fibers, particularly those of large caliber, is seen in peripheral sensory nerves, and is responsible for the prominent impairment of vibratory and proprioceptive sensation. Axonal degeneration, with some secondary demyelination in the later stages, is the predominant finding in nerve biopsies.
When strict diagnostic criteria are applied, all cases of Friedreich’s ataxia are inherited by autosomal-recessive transmission [Barbeau, 1976]. The Friedreich’s ataxia locus was linked to chromosome 9 in typical families [Chamberlain et al., 1988]. Subsequent testing of late-onset and Acadian cases, as well as of families with early-onset ataxia with retained tendon reflexes, also indicated linkage homogeneity [Chamberlain et al., 1989; De Michele et al., 1994; Keats et al., 1990; Klockgether et al., 1993; Palau et al., 1995]. In 1996, Campuzano and colleagues identified the responsible gene, X25, in the critical region on chromosome 9q13. The gene encodes a 210-amino acid protein, frataxin, the function of which is currently unknown. Most Friedreich’s patients (98 percent) are homozygous for an unstable GAA trinucleotide in the first intron of X25, but a few (approximately 1 percent) have point mutations of the frataxin gene. It is thought that the triplet GAA expansion causes disease by suppressing frataxin gene expression, which, in some way, causes spinal cord, heart, and pancreatic degeneration. This is the first autosomal-recessive disease in which a triplet repeat expansion has been demonstrated.
Further studies have shown that virtually all patients with typical Friedreich disease, Acadian Friedreich disease, late-onset Friedreich disease, and early-onset ataxia with retained tendon reflexes carry two copies of the GAA triplet expansion. Cases with larger GAA expansions tend to have earlier onset and are more likely to show additional manifestations of the disease, such as optic atrophy, hearing loss, and possibly cardiomyopathy [Montermini et al., 1997] (see Chapter 67).
Giant Axonal Neuropathy
Giant axonal neuropathy was first described in 1972 by Berg and colleagues in a 6-year-old girl as a slowly progressive peripheral neuropathy resembling Friedreich’s ataxia. With reports of four more children, it became clear that this disorder is a generalized one of increased, abnormal cytoplasmic microfilaments involving a number of body tissues, including Schwann cells, fibroblasts, and vascular endothelium [Carpenter et al., 1974; Igisu et al., 1975; Koch et al., 1977; Ouvrier et al., 1974]. Axons are swollen with densely packed neurofilaments. The initial descriptions of children younger than 9 years of age noted the presence of unusually kinky hair (Figure 89-8). Parental consanguinity was recorded for six involved children, suggesting autosomal-recessive inheritance [Donaghy et al., 1988; Gambarelli et al., 1977; Igisu et al., 1975; Ouvier et al., 1974]. Reports of siblings with the disease further suggested recessive inheritance [Jones et al., 1979; Takebe et al., 1981]. The responsible gene, gigaxonin, located on chromosome 16q24 [Bomont et al., 2000], is a cytoskeletal protein that may play a role in neurofilament architecture. Progressive gait disturbance, kinky hair, long curly eyelashes, a prominently high forehead, and variable CNS involvement, including optic atrophy, abnormal ocular motility, mental retardation, and spasticity, are hallmarks of the condition [Dooley et al., 1981; Fois et al., 1985; Kirkham et al., 1980]. Leukoencephalopathy on brain imaging studies may be accompanied clinically by deterioration of cognitive function [Stollhoff et al., 1991].
As the disease advances, motor function declines. Not all systems are equally affected; for example, some wheelchair-bound children retain normal mental capacity [Boltshauser et al., 1977; Pfeiffer et al., 1977]. Rapid progression of the sensory motor neuropathy leads to early death, although milder variants have been recorded, including not-infrequent cases with absence of the characteristic “woolly” hair [Bruno et al., 2004]. Abnormal EEGs and visual-, auditory-, and somatosensory-evoked potentials have been recorded [Majnemer et al., 1986]. Nerve conduction studies show evidence of axonal neuropathy.
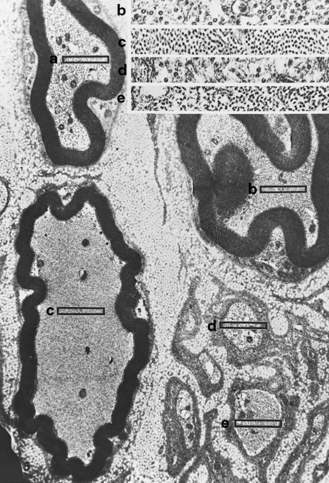
Thickened, distal axons, distended by an increased number of neurofilaments and accompanied by axonal loss, constitute the peripheral nerve pathologic findings [Asbury et al., 1972; Carpenter et al., 1974]. Other pathologic descriptions note the presence of cytoplasmic microfilaments in Schwann cells, endothelial cells, perineurial cells, endoneurial fibroblasts, and melanocytes [Bonerandi et al., 1981; Klymkowsky and Plummer, 1985; Prineas et al., 1976] (Figure 89-9). Skin biopsy may demonstrate accumulation of microfilaments in endothelial cells, melanocytes, and fibroblasts [Takebe et al., 1981]. Neurofilaments that accumulate in sural nerve have the same protein composition as normal neurofilaments [Ionasescu et al., 1983; Pena, 1982]. One newborn with hypotonia had axonal swelling in peripheral nerves and distal degeneration of long spinal cord tracts without involvement of the cerebral cortex or brainstem [Kinney et al., 1985]. Severe cerebellar degeneration and optic atrophy clinically were demonstrated at autopsy to be caused by degeneration and gliosis of the optic nerves and tracts [Thomas et al., 1987]. Ultrastructurally, the hair has characteristic longitudinal grooves [Treiber-Held et al., 1994].
Infantile Neuroaxonal Dystrophy (Seitelberger’s Disease)
Infantile neuroaxonal dystrophy is a progressive central and peripheral nervous system disorder, characterized pathologically by the presence of axonal spheroids. The disorder is usually due to mutations of the PLA2G6 gene, which encodes iPLA2-VIA a, a cytosolic Ca2+ independent phospholipid important in membrane homeostasis [Khateeb et al., 2006]. Onset of symptoms is recognized between ages 1 and 2 years. Loss of mental and motor milestones denotes the regressive nature of the condition. Seizures typically develop. Progressive motor sensory neuropathy causes loss of sensation, in particular, with subsequent limb mutilation. Hypotonia associated with diminished tendon reflexes in an infant showing regression of milestones is a clear sign of the disorder. Urinary retention may become a problem as the disease advances. Hypothalamic failure results in diabetes insipidus and hypothyroidism. The autonomic nervous system is affected, with decreased tearing and poor temperature regulation clinically apparent. One infant presented unusually early in the neonatal period with hypertonia, axonal spheroids on biopsy, and mineralization of the basal ganglia observed by imaging studies and at autopsy [Venkatesh et al., 1994]. MRI shows cerebellar atrophy and cerebellar hyperintensity on T2-weighted images [Tanabe et al., 1993], corresponding to marked loss of cerebellar neurons and astrogliosis found at autopsy. The MRI changes are characteristic early in the course of the disease and may aid in diagnosis. Children with infantile neuroaxonal dystrophy do not survive beyond their preschool years. Later-onset forms of neuroaxonal dystrophy have been reported.
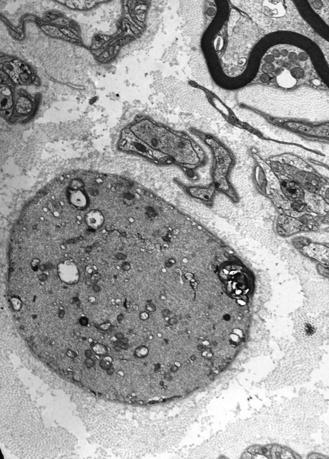
Central, peripheral, and autonomic neurons and nerve fibers contain spheroids of varying sizes with a predilection for involvement of sensory nerves [Itoh et al., 1993]. Sural nerve biopsy displays periodic acid–Schiff-positive, ballooned axons, the spheroids that contain mitochondria, glycogen-like granules, dense bodies, vesicles, and electron-lucent material [Shimono et al., 1976] (Figure 89-10). Membranotubular and granulovesicular profiles are found ultrastructurally in spheroids [Malandrini et al., 1995]. Peripheral nerve lesions mimic those found in the CNS [Duncan et al., 1970; Martin and Martin, 1972]. Biceps muscle biopsy may be useful for diagnostic purposes, revealing axonal spheroid bodies in intermuscular nerve fibers, neuromuscular junctions, and motor nerve endings [Miike et al., 1986]. Early muscle biopsy may require an intense ultrastructural investigation of presynaptic neurons at the neuromuscular junction to demonstrate dystrophic axons [Wakai et al., 1993]. Immunohistochemistry of spheroids is positive for ubiquitin and negative for beta-tubulin and beta-amyloid. Lack of reactivity to tubulin in spheroids suggests that loss of microtubules may be part of the mechanism in the formation of axonal spheroids [Moretto et al., 1993]. Activation of the ubiquitin system is an attempt on the part of the neuron to remove abnormal proteins. The morphologic findings of infantile neuroaxonal dystrophy are remarkably similar to those reported in alpha-N-acetylgalactosaminidase deficiency (Schindler’s disease) [Wolfe et al., 1995].
Krabbe’s Disease (Globoid Cell Leukodystrophy)
Galactosylceramide lipidosis, known as Krabbe’s disease or globoid cell leukodystrophy, is an autosomal-recessive neurodegenerative disorder that presents most often in infants of 3–5 months of age (see Chapter 71). Deficiency of galactosylceramide beta-galactosidase results from mutations and heterogeneous deletions in the galactocerebrosidase gene on 14q31 [Luzi et al., 1995; Rafi et al., 1995; Tatsumi et al., 1995]. The clinical profile is a highly irritable infant with spasticity, optic atrophy, intellectual delay, and polyneuropathy. The disease leads to a loss of milestones, microcephaly, severe failure to thrive, and progressive demyelinating peripheral neuropathy with delayed nerve conduction velocities. Hyperacusis is part of the irritability. CSF protein is elevated. Average age of death is 13 months. An earlier, neonatal onset is associated with failure to thrive, infantile spasms, and hemiplegia [Hagberg, 1984]. A small number of patients have later-onset forms of the condition, up to age 50 [Rafi et al., 1995], causing spasticity, motor polyneuropathy, dysarthria, and optic atrophy [Loonen et al., 1985].
The central and peripheral manifestations are due to abnormalities in the metabolism of myelin with accumulation of galactosylceramide, which causes an infiltration of macrophages that subsequently transform into globoid cells in the brain [Suzuki, 1984]. The peripheral neuropathy is characterized by uniform marked slowing of conduction velocities [Miller et al., 1985]. Hypomyelination is noted pathologically in infantile Krabbe’s disease. Segmental demyelination is more important in later-onset globoid cell leukodystrophy and can be demonstrated on sural nerve biopsy. Ultrastructurally, curvilinear lamellar cytoplasmic inclusions are present in Schwann cells and histiocytes creating the globoid cell.
Krabbe’s disease may present initially as a peripheral neuropathy. A 13-year-old with scoliosis and pes cavus displayed distal leg weakness and sensory loss. Electrophysiologic nerve study indicated demyelinating neuropathy. Four years later, spasticity developed, and the diagnosis was established [Marks et al., 1997].
Metabolic Neuropathies
Diabetes Mellitus
It has been known for a long time that up to 10 percent of children with insulin-dependent diabetes mellitus have symptoms and signs caused by peripheral neuropathy associated with diabetes [Gamstorp et al., 1966]. These bilaterally symmetric changes, especially in the lower extremities, include mild distal weakness, loss of touch and pain sensation, and decreased ankle tendon reflexes. Despite good management of diabetes, impaired nerve function occurs.
In a large prospective study of nerve conductions and autonomic nervous system function in diabetic children, slowed sensory nerve conduction velocities and impaired autonomic function were present in 25 percent of children at the time of diagnosis [Solders et al., 1997]. Follow-up measurements of nerve conduction velocities and parasympathetic function (R-R variations) 2 years later showed deterioration from baseline of sensory and motor nerve conductions and autonomic nerve function. The degree of deterioration correlated with blood sugar control. After 3 years of diabetes, all motor and sensory nerve conduction times were slowed [Hyllienmark et al., 1995]. In contrast with the electrophysiologic changes, only 4 percent of children at onset of diabetes have symptoms or signs suggestive of neuropathy, despite early nerve conduction slowing. In another approach to peripheral nerve function, vibration perception threshold was measured in diabetic boys. Compared with age-matched control subjects, impaired vibration perception was present in diabetic boys in the absence of clinical symptoms of peripheral or autonomic neuropathy [Olsen et al., 1994].
Mildly impaired autonomic nervous system function is found in 30–50 percent of children with diabetes when measured soon after diagnosis [Donaghue et al., 1996; Verrotti et al., 1995]. The percentage involved does not change over 3 years [Donaghue et al., 1996]; abnormalities are not related to age, duration of diabetes, or degree of blood sugar control. Heart rate variation during Valsalva maneuver and maximum/minimum 30:15 ratio are the most sensitive indices to detect autonomic abnormalities in children [Verrotti et al., 1995]. Further, asymptomatic diabetic children have diminished sensation of bladder filling [Barkai and Szabó, 1993]. If cardiovascular autonomic dysfunction is present in addition, loss of bladder filling sensation is even more striking. Loss of bladder filling sensation correlates with duration of diabetes and poor blood sugar control.
It is clear that careful clinical and electrophysiologic follow-up is required to monitor altered peripheral and autonomic nerve function in childhood-onset diabetes mellitus. In another study comparing diabetic and nondiabetic children, both motor and sensory peripheral nerve abnormalities were related to poor blood sugar control, as measured by hemoglobin A1c and duration of diabetes [Fiçicioglu et al., 1994]. These results emphasize the importance of metabolic control and duration of diabetes in the pathogenesis of diabetic neuropathy. The findings suggest that peripheral neuropathy is common in young, insulin-dependent diabetics. As nerve function tests are easy to conduct and are sensitive, they may help to achieve better metabolic control and prevent diabetic complications.
Uremic Neuropathy
Uremic neuropathy is rarely diagnosed in children. When recognized, it is characterized by burning sensations in the feet and a symmetric motor sensory neuropathy with progressive muscle weakness. Motor nerve and sensory nerve conduction velocities are decreased early in the course of the disease, often before clinical symptoms appear [Di Paolo et al., 1982]. In particular, the proximal 1a sensory and motor nerve conduction velocities are significantly slowed early, compared with distal values in uremic neuropathy [Panayiotopoulos and Lagos, 1980]. The H-reflex response latency can be used to detect early changes in some patients [Knoll and Dierker, 1980]. Parathyroid hormone levels have been correlated with early peripheral nerve conduction delays in some instances [Di Paolo et al., 1982] but not in others [Aiello et al., 1982]. The facial nerve is a sensitive indicator of uremic neuropathy [Mitz et al., 1984]. In vitro inhibition of tubulin 6s polymerization by uremic toxins suggests a mechanism for initiating uremic neuropathy [Braguer et al., 1986]. The plasma of uremic patients contains high concentrations of middle molecules in the 300- to 1500-dalton range, which cause toxic effects [Babb et al., 1981]. A b4-2 peak correlates with the neuropathy [Faguer et al., 1983].
The predominant change in the peripheral nerve is a demyelinating neuropathy, but some individuals experience a progressive axonal neuropathy with secondary segmental demyelination. An acute axonal neuropathy is rarely observed [Said et al., 1983]. In the more rapidly progressive neuropathies, ischemic changes related to accelerated hypertension and possibly to septicemia may be etiologically related to rapid progression [McGonigle et al., 1985]. Biotin therapy has been recommended for patients with advanced renal failure before the neuropathy becomes manifest [Yatzidis et al., 1984].
Acute Intermittent Porphyria
Acute intermittent porphyria has produced symptoms within the first decade of life, including autism [Luder et al., 2009], but it typically presents after puberty. Gene carriers are at risk of developing potentially fatal neurogenic attacks if exposed to precipitating factors [Wassif et al., 1994], including certain medications like tricyclic antidepressants, barbiturates, halothane, valproic acid, sulfonamides, estrogens, and alcohol. Axonal damage, observed on both sural nerve biopsy [Defanti et al., 1985] and autopsy evaluation, suggests that the neuropathy is primarily axonal [Yamada et al., 1984]. The neuropathy is predominantly motor, with associated abdominal pain, dysautonomia, and central nervous system involvement [Pischik and Kauppinen, 2009].
Acute, severe, colicky abdominal pain is a typical manifestation of an acute episode, accompanied by CNS and peripheral nervous system impairment. Peripherally, motor weakness is most striking, but sensory impairment may also occur. A flaccid paralysis resembling Guillain–Barré syndrome has been observed. The proximal muscles may be more involved than distal muscles when weakness occurs. Reflexes are often diminished or absent. The motor cranial nerves, particularly cranial nerves III, VII, and X, may be involved. Urinary retention or incontinence occurs. Centrally, mental changes, consisting of confusion, delirium, and hallucinations, may develop. Autonomic function involving the sympathetic and parasympathetic nervous systems may be affected. Typically, the symptoms remit with resolution of the episode, but parasympathetic dysfunction has been detected during remission and when the patient is otherwise asymptomatic [Laiwah et al., 1985]. No successful treatment has been described. Lead poisoning and Guillain–Barré syndrome must be considered in the differential diagnosis.
Vitamin Deficiency
Thiamine, or vitamin B1, is a water-soluble vitamin that serves as a coenzyme in carbohydrate metabolism. Classic thiamine deficiency in children causes peripheral neuropathy, encephalopathy, and high-output cardiac failure [Debuse, 1992]. Infantile beriberi disease has been described in infants born to mothers with thiamine deficiency. Symptoms consist of anorexia, vomiting, and lethargy occurring within the first 3 months of life, and may be associated with vasoconstriction, hypotension, and severe metabolic acidosis. The infant may be pale, puffy, and listless on examination. Cranial nerve dysfunction has been noted, with ptosis, optic atrophy, and laryngeal paralysis causing a hoarse cry. Cardiac enlargement with heart failure has also been found. A thiamine-dependent state also has been described [Mandel et al., 1984]. Treatment allows for reversal of symptoms, with or without sequelae [Román, 1994].
Vitamin E deficiency may be associated with peripheral neuropathy and ataxia [Jackson et al., 1996]. It may also occur in children with cystic fibrosis, chronic cholestasis, abetalipoproteinemia, and short bowel syndrome [Sokol, 1990].
Congenital Pernicious Anemia
Two forms of pernicious anemia in children are associated with peripheral neuropathy. One occurs as a congenital or early-onset disease in children [Heisel et al., 1984]. Congenital pernicious anemia occurs before age 5 years as a result of a vitamin B12 deficiency caused by isolated absence of gastric intrinsic factor. There is adequate gastric free acid and a normal gastric mucosa. The patient has a macrocytic anemia and a decreased serum vitamin B12 level. There is an absence of antibodies to the parietal cell and intrinsic factor. In at least one case, autoantibodies to intrinsic factor and parietal cells were present [van Loon et al., 2009]. Infants have peripheral neuropathy, muscle weakness, ataxia, fever, and macrocytic anemia. There may be a delay in acquiring motor milestones and growth failure. Treatment is with parenteral vitamin B12, which results in recovery to normal hemoglobin levels in 4–6 weeks.
Infants who are breastfed by strictly vegetarian mothers or who have undiagnosed pernicious anemia are at risk for developing a megaloblastic anemia because of a deficiency of vitamin B12. A loss of acquired milestones with seeming neurologic regression occurs [Higginbottom et al., 1978]. Cystathioninuria, glycinuria, methylmalonic aciduria, 3-hydroxy propionic aciduria, and formic aciduria may develop. Inadequate tissue availability of vitamin B12 results in increased concentrations of methylmalonic acid and homocysteine, caused by inhibition of methylmalonyl-CoA mutase and methionine synthase, respectively. It can be prevented by supplementing the vegetarian mother’s diet with vitamin B12 (cobalamin).
Abetalipoproteinemia
Abetalipoproteinemia, known as Bassen–Kornzweig syndrome, is characterized by progressive ataxic neuropathy, retinitis pigmentosa, steatorrhea, hypolipidemia, deficiency of fat-soluble vitamins, and acanthocytosis [Bassen and Kornzweig, 1950; Selimoglu et al., 2001]. The onset of symptoms in this autosomal-recessive disorder occurs between early childhood and late teen years. Apoprotein B, required for synthesis and structural integrity of chylomicrons and low-density and very low-density lipoproteins, is missing in plasma because of lack of synthesis [Salt et al., 1960]. The result is absence of lipoproteins. Intestinal fat absorption is absent from birth and results in a progressive deficiency of fat-soluble vitamins A, D, E, and K [Seckeler and Linden, 2008]. Many of the clinical problems of the syndrome are due to vitamin E deficiency, and treatment with vitamin E is recommended [Miller and Lloyd, 1982]. Sensory neuropathy exists with reduced amplitude responses [Lowry et al., 1984].
Pathology
Degeneration of posterior columns, spinal cerebellar pathways, and cerebellum, caused by vitamin E deficiency [Mino, 1993], is the major pathologic change. Loss of anterior horn cells is found in the spinal cord. Posterior column degeneration leads to abnormal somatosensory-evoked potentials [Brin et al., 1986]. Sural nerve biopsy shows decreased numbers of large myelinated fibers and clusters of regenerating fibers [Wichman et al., 1985]. The posterior fundus of the eye shows a loss of photoreceptors, loss or attenuation of pigment epithelium, and preservation of submacular pigment epithelium, with an excessive accumulation of lipofuscin [Cogan et al., 1984]. Macrophage-like pigmented cells invade the retina.
Clinical Laboratory Tests
The most significant finding is the inborn absence of the major apoprotein of low-density plasma lipoproteins [Lange and Steck, 1984], leading to an abnormal serum and red-cell lipid profile, along with spiny erythrocytes (acanthocytosis). Fat-soluble vitamin deficiency is typically present. The electroretinogram may be nonrecordable because of the pigmentary retinopathy [Judisch et al., 1984]. There are wide variations in the appearance of the acanthocytes. Erythrocyte membrane phospholipids and cholesterol are increased [Iida et al., 1984].
Management
High doses of vitamin E provide clinical amelioration and electrophysiologic improvement, with increased evoked motor unit potentials and improved nerve conduction velocities [Brin et al., 1986; Hegele and Angel, 1985; Runge et al., 1986]. In addition to large doses of oral vitamin E therapy, a low-fat diet and supplements of the other fat-soluble vitamins are recommended. If these dietary changes are started early enough, the progressive retinopathy can most likely be prevented.
Alpha-Lipoprotein Deficiency (Tangier Disease)
Tangier disease is an uncommon familial disorder of lipoprotein metabolism, notable for the absence of normal high-density lipoprotein from plasma and the accumulation of cholesterol esters in multiple organs. Extremely low plasma cholesterol levels are present. Physical examination shows patches of yellow-orange lymphoid tissue in the tonsils and pharynx, and hepatosplenomegaly. Yellow patches are present on the surface of the liver, and liver biopsy specimens contain cholesterol esters. Fiberscopic rectal examination shows orange-brown spots present throughout the rectum. Foam cells are present in the lymphoid tissue of both the pharynx and rectal mucosa, and rectal mucosal biopsy is recommended for diagnostic purposes [Tarao et al., 1984]. Widespread storage of lipid material by the reticuloendothelial system is present, and foamy histiocytes can be found easily [Dechelotte et al., 1985]. Onset is between age 2 and 67 years [Pietrini et al., 1985]. Sural nerve biopsy shows a reduction of smaller myelinated and unmyelinated fibers [Gibbels et al., 1985], and abnormalities in the paranodal regions. There is lipid deposition, redundant myelin foldings, myelin splitting and vesiculation, and small tomacula [Cai et al., 2006]. Abnormal lipid storage is found in Schwann cells of unmyelinated fibers. Conduction block is present on electrophysiologic nerve study, associated with mutation of the adenotriphosphate-binding cassette transporter-1 gene on 9q22–q31 [Théaudin et al., 2008].
Clinical Characteristics
Recurrent neuropathy occurs in children with fluctuating asymmetric sensory involvement, mostly in the lower extremities, sometimes accompanied by progressive development of weakness of both distal and proximal muscles. Complaints of numbness and tingling in the distal extremities are early symptoms, followed by signs of the neuropathy and weakness. A dissociated loss of pain and temperature sensation may occur. More severe forms of neuropathy have been described in older individuals, suggesting more than one type of neuropathic involvement [Marbini et al., 1985; Pietrini et al., 1985]. Facial diplegia may develop. Hand muscle atrophy occurs. Presentation may be confused for autoimmune-mediated neuropathy due to asymmetric neuropathy.
Biochemistry
Tangier disease is characterized by low levels of apo-A-1 and high-density lipoproteins. A higher than normal amount of pro-apo-A-1 lipoprotein is present in Tangier disease, but there is no deficiency of the converting enzyme activity to mature apo-A-1 [Bojanovski et al., 1984]. The turnover rate from pro-apo-lipoprotein to mature apo-lipoprotein may be abnormal [Edelstein et al., 1984]. In normal circumstances, high-density lipoproteins bind to monocytes at the cell surface, and the lipoprotein is internalized and subsequently resecreted without significant degradation. Conversely, high-density lipoproteins bind to Tangier monocytes more actively than usual, and when internalized, they are found in secondary lysosomes; that is, when they are resecreted, most of the high-density lipoprotein is degraded. At least one of the abnormal mechanisms in this disease is apparently the abnormal cellular metabolism of high-density lipoproteins in Tangier monocytes [Schmitz et al., 1985]. Another biochemical abnormality demonstrated in Tangier disease is a significant reduction of endoneurial sodium, potassium adenosine triphosphatase, and magnesium adenosine triphosphatase activities, demonstrated in six sural nerve biopsies from patients with the condition [Sutherland and Pollock, 1984]. All of these patients had either mononeuropathy multiplex or a progressive axonal neuropathy. The reduced level of the adenosine triphosphatase activity may relate to altered endoneurial lipid metabolism and impaired axoplasmic flow.
Metachromatic Leukodystrophy
Metachromatic leukodystrophy is a recessively inherited disease affecting children and adults. The basic disorder is a failure of the catabolism of sulfatide, the sulfate ester of galactose cerebroside. This lipid is a component of the myelin membrane and is probably also a component of neuronal membranes [McKhann, 1984]. The storage of metachromatic material in Schwann cell lysosomes and macrophages associated with segmental demyelination is well recognized in the late infantile, juvenile, and adult forms (Figure 89-11) [Luijten et al., 1978]. Given the abnormal storage of lipid in the central and peripheral nervous system, a combination of upper and lower motor neuron disease is recognized clinically. Infants may present with cranial neuropathy and multiple cranial nerve enhancement on MRI [Singh et al., 2009]. Nerve conduction velocities are slowed, and the latencies of evoked potentials are prolonged. Sensory nerve conduction velocities are generally delayed earlier and more severely than are motor nerve conduction velocities [Takakura et al., 1985].
Clinically, in the infantile form of metachromatic leukodystrophy, the infant acquires normal milestones until 1–2 years of age and then begins to show signs of the disease. An unsteady gait is an early sign, accompanied by loss of language, intellectual deterioration, and later, spasticity. Spinal fluid protein is elevated, and nerve conduction velocities are slowed [Haberlandt et al., 2009]. The juvenile form of the disease occurs after several years of age, again with gait disturbance followed by intellectual deterioration. Given the greater variability of presentation in the adult form, dementia may be the initial presentation, without other obvious neurologic deficits. The disease can be suspected when evoked potentials are delayed and peripheral nerve conduction velocities are slowed [Wulff and Trojaborg, 1985].
Bone marrow transplantation has shown some promise, at least for delaying the progression of the late infantile and juvenile forms of metachromatic leukodystrophy [Bayever et al., 1985; Krivit et al., 1990; Lipton et al., 1986]. Follow-up studies indicate arrest of CNS disease but not peripheral neuropathy.
Refsum’s Disease (Heredopathia Atactica Polyneuritiformis) and Peroxisome Biogenesis Disorders
Refsum’s disease is a recessively inherited condition caused by a deficiency in phytanic acid oxidase activity, allowing the accumulation of phytanic acid in body tissues. There is a spectrum of conditions – namely, Zellweger’s syndrome, neonatal adrenoleukodystrophy, and infantile Refsum’s disease – characterized by absence of catalase-positive peroxisomes and general impairment of peroxisomal functions. Anorexia, ataxia, ichthyosis, and sensorineural hearing loss were originally described [Refsum, 1946; Refsum et al., 1949]. Malabsorption and steatorrhea may be present early [Mandel et al., 1992]. In the classic form of the disease, clinical characteristics can be divided into three groups. Congenital abnormalities, such as skeletal deformities unrelated to the level of phytanic acid, constitute one group. Retinitis pigmentosa, which develops slowly, is another clinical group, unrelated to the plasma phytanic acid level. A third group of lesions includes the neuropathy, rash, and cardiac arrhythmias, which can deteriorate or improve according to plasma phytanic acid level [Gibberd et al., 1985]. The sensorimotor neuropathy is of the chronic hypertrophic demyelinating type with reduced nerve conduction velocities and raised CSF protein levels. Exacerbations of the neuropathy and rash can be precipitated by a low calorie intake, with mobilization of phytanic acid from adipose tissue.
Patients with Refsum’s disease lack the ability to degrade phytanic acid to pristanic acid and carbon dioxide. This defect can be analyzed in fibroblasts [Skjeldal et al., 1986]. The gene responsible for the metabolic defect codes for phytanoyl-CoA hydroxylase. Serial plasma exchanges to reduce the level of phytanic acid have been correlated with significant clinical improvement, in particular of the neuropathy and cerebellar ataxia [Hungerbeuhler et al., 1985]. When this is combined with a diet low in phytanic acid, clinical improvement, which correlates with a lowered serum phytanic acid level, can be sustained.
An infantile form of Refsum’s disease is now recognized as a result of excessive phytanic acid storage. Pigmentary retinopathy is again observed, along with hypotonia, severe hearing impairment, and severe developmental delay [Weleber et al., 1984]. Facial dysmorphism and hepatomegaly have also been described [Budden et al., 1986]. Two patients had intracranial hemorrhage secondary to a vitamin K-responsive clotting defect, and both had steatorrhea. The affected infant may have protracted diarrhea and low serum cholesterol level. There is an absence of the normal hepatic peroxisomes [Van den Branden et al., 1986] compared with the adult form of Refsum’s disease, in which peroxisomes and fibroblasts are not diminished [Beard et al., 1985]. The plasma of patients with the infantile but not the adult form of Refsum’s disease contains increased amounts of pipecolic acid and at least two abnormal bioacids [Poulos et al., 1984]. Cultured skin fibroblasts show a marked increase in the concentration of the long-chain fatty acid, hexacosanoic acid (26C) [Poulos and Sharp, 1984]. Infantile Refsum’s disease appears to share several biochemical features with the cerebrohepatorenal syndrome (Zellweger’s disease) and adrenoleukodystrophy. Both patients with infantile Refsum’s disease and patients with Zellweger’s disease have a deficiency in phytanic acid oxidase activity, and all three are considered neonatal peroxisomal disorders [Aubourg et al., 1986; Poulos et al., 1985].
Chédiak–Higashi Syndrome
Chédiak–Higashi syndrome is a rare autosomal-recessive disorder. Patients have hypopigmentation, pancytopenia, susceptibility to infection, and an increased risk of lymphoreticular malignancy. Psychomotor retardation, seizures, and muscular weakness have been described. Treatment is bone marrow transplantation, which improves hematologic and immune defects. Peripheral neuropathy with moderate axon loss persists and becomes progressive [Misra et al., 1991; Tardieu et al., 2005]. In advanced neuropathy, the pathology includes abnormal granules in Schwann cells [Lockman et al., 1967].
Spinocerebellar degeneration has been observed and is often associated with features of parkinsonism [Pettit and Berdal, 1984]. Muscle biopsy in advanced disease shows neurogenic muscular atrophy secondary to the peripheral neuropathy. In addition, acid phosphatase-positive granules indicating abnormal lysosomes are found in normal-appearing muscle fibers. These correspond ultrastructurally to autophagic vacuoles containing glycogen particles and membranous structures [Uchino et al., 1993]. These findings indicate that Chédiak–Higashi syndrome is a generalized lysosomal disorder. The defective gene has been localized to 1q42–44 [Barbosa et al., 1997], with mutations throughout the CHS1LYST gene that governs vesicle trafficking [Kaplan et al., 2008].
Toxic Neuropathies
Diphtheria
Diphtheria is an uncommon disease in the Western Hemisphere because most individuals are immunized with diphtheria-pertussis-tetanus vaccine. The disease occurs in unimmunized children and in adults who have lost immunity. An exotoxin produced by Corynebacterium diphtheriae infection in the throat produces cardiomyopathy and neuropathy. A systemic radiculoneuropathy may develop, with onset 1–16 weeks after infection and marked by sensory loss [Gaskill and Korb, 1946]. Clinically, blurred vision and swallowing difficulties mark the onset of diphtheric neuropathy. A generalized motor sensory demyelinating polyneuropathy with distal, symmetric involvement is recognized [Créange et al., 1995]. Lower cranial nerves are affected. Ventilation-dependent respiratory failure may occur, and the clinical course may be biphasic [Logina and Donaghy, 1999]. Proximal and distal muscle weakness and muscle tenderness are present. Proprioceptive modalities are altered in the lower extremities. Spinal fluid protein may be elevated, with or without pleocytosis. Recovery is usually complete within several months [Buzzi, 1982].
Neuropathy of Serum Sickness
Serum sickness is a systemic illness resulting from hypersensitivity to an injected foreign protein, such as tetanus or diphtheria antisera, producing encephalomyelitis, neuropathy, or brachial plexus neuropathies [Igbal and Arnason, 1984; McCombe and Pender, 1991]. Fever, joint swelling, abdominal pain, diarrhea, and cutaneous eruptions develop within days of receiving foreign protein. Deposition of antigen–antibody complexes is associated with disease [McCombe and Pender, 1991].
Botulism
Three forms of botulism exist, the most common being the infantile form that occurs between 1 and 38 weeks of age. A heat-labile neurotoxin is elaborated by Clostridium botulinum under anaerobic conditions. The toxin produces a presynaptic blockade by disabling synaptic acetylcholine-containing vesicle exocytosis and preventing the release of acetylcholine from cholinergic nerve endings [Glickman et al., 1976; Montal, 2010]. Human pathogenic neurotoxins types A, B, E, and F are produced by a diverse group of anaerobic spore-forming bacteria, including Clostridium botulinum groups I and II, Clostridium butyricum, and Clostridium baratii. Laboratory diagnosis of botulism is based on the detection of botulinum neurotoxin in a patient via mouse lethality assay [Lindström and Korkeala, 2006]. A newer genetic test uses a macroarray GeneDisc Cycler system, testing simultaneously for bont/A, bont/B, bont/E, and bont/F genes that encode for the respective neurotoxins A, B, E, F. The test is specific and can be used on food and clinical samples [Fach et al., 2010].
Infantile Botulism
A history of constipation in a 1- to 6-month-old infant, followed by a subacute progression over 4–5 days of bulbar and extremity weakness with feeding difficulties, ptosis, hypotonia, and respiratory embarrassment, constitutes the presentation of this condition [Pickett et al., 1976]. A spectrum of disease is apparent, from those not requiring hospitalization who have only mild feeding difficulties, mild hypotonia, and perhaps failure to thrive, to more severe involvement, including severe hypotonia, decreased gag reflex, dysphagia, ptosis, and constipation. Respiratory arrest and death are possible [Thompson et al., 1980]. Large and sluggishly reacting pupils are typically present, tendon reflexes are reduced, and the gag reflex may be absent. Ingestion of contaminated food, often honey, is the usual source of the organism.
Infants secrete botulinum toxin in their stools from the organism that grows in the intestine. Examination of the stool for the presence of the toxin is helpful. Electromyography helps to establish the diagnosis. Miniature end-plate potentials are decreased in frequency, consistent with the decreased release of acetylcholine. The evoked compound muscle action potential amplitude is reduced, with high rates of repetitive nerve stimulation (20 and 50 Hz), and an incremental response occurs because of enhanced acetylcholine release [Cornblath et al., 1983]. Muscle needle examination typically shows an increase in fibrillations and positive waves indicative of denervation. Supportive care is the mainstay of treatment, and complete recovery is anticipated. Botulism immunoglobulin (BIG) given intravenously may shorten the duration and diminish the potential complications of the disorder [Domingo et al., 2008; Thompson et al., 2005]. Gastrointestinal tract infection from Clostridium botulinum, with subsequent production of toxin and the development of clinical disease, has been described in an adult [Chia et al., 1986].
Classic Botulism
The classic form of botulism typically seen in adults, but only occasionally seen in children and adolescents, is caused by the contamination of commercial food products with botulinum toxin. Microgram amounts of toxin can produce severe paralysis and interruption of autonomic nervous system function. Inadequate cooking of foods during the canning process allows the Clostridium botulinum spores to germinate, and the toxin is produced within the closed container. The syndrome develops 12–36 hours after ingestion of the toxin-containing food. Early symptoms are blurred vision, diplopia, dizziness, dysarthria, and dysphagia, and are followed by a descending paralysis and dyspnea. Respiratory paralysis and death may develop [Cherington, 1974]. With supportive care, recovery is expected within weeks to months. Electrophysiologically, incremental response on repetitive nerve stimulation and small compound motor action potentials are usually present. Single-fiber electromyography may be necessary to demonstrate increased jitter and impulse blocking [Cruz Martínez et al., 1985].
Wound Botulism
Wound contamination with Clostridium botulinum leads to subsequent production of botulinum toxin after bacterial incubation, producing clinical botulism 4–14 days after the wound has been infected. The condition has been described in a small number of individuals, ranging from adolescents to older adults [de Jesus et al., 1973; Merson and Dowell, 1973]. The disease may be severe, producing diffuse muscle weakness and bulbar dysfunction requiring ventilatory support. Prompt recognition and treatment may be necessary to prevent respiratory failure. Botulinum antitoxins are available for treatment of wound and food-borne botulism.
Antibiotic-Induced Neuropathy
A number of antibiotic, antifungal, and antituberculous drugs have been known to cause peripheral neuropathy in a small percentage of cases. For example, chloramphenicol can cause mild, primarily sensory, peripheral neuropathy after long-term use at relatively high doses. Nitrofurantoin may produce a polyneuropathy of sudden onset on rare occasion. Isoniazid causes an axonal neuropathy responsive to pyridoxine therapy in 1–2 percent of patients. The primarily sensory neuropathy begins with paresthesias. Ethambutol, an antituberculous drug, occasionally causes a sensory neuropathy associated with optic neuritis. Linezolid, typically used to treat multidrug-resistant tuberculosis, may cause neuropathy [Anger et al., 2010; Schecter et al., 2010]. In an adult treated with linezolid for methicillin-resistant Staphylococcus aureus, a painful axonal sensorimotor neuropathy is slowly improving.
Pyridoxine-Induced Polyneuropathy
Pyridoxine taken in large doses can cause a sensory neuropathy, with paresthesias, diffuse sensory loss, sensory ataxia, and autonomic dysfunction [Albin et al., 1987; Schaumburg et al., 1983]. Weakness is now reported in rare instances [Gdynia et al., 2008; Scott et al., 2008]. Sural nerve biopsy shows marked loss of large myelinated nerve fibers [Santoro et al., 1991]. Recovery may be incomplete. A chronic neuropathy from lower-dose ingestion has similar and milder symptoms and a better prognosis [Dordain and Deffond, 1994]. There is a dose-dependent relationship to the development of neuropathy [Berger et al., 1992]. Most reports in the literature concern adults; however, a report of an 18-year-old with pyridoxine-dependent seizures describes onset of sensory neuropathy by 2 years of age as a result of 2 g/day of pyridoxine therapy for seizure control. The neuropathy did not progress from age 2 to 18 years. Electrophysiologic studies indicate a pure sensory neuronopathy [McLachlan and Brown, 1995].
Nitrous Oxide-Induced Polyneuropathy
Severe myeloneuropathy and macrocytic anemia associated with a low vitamin B12 level are reported after prolonged exposure to nitrous oxide [Jameson et al., 1999; Stacy et al., 1992]. B12 supplementation alone does not result in improvement, but the addition of methionine does arrest the progression of neuropathy. Chronic nitrous oxide exposure inhibits methionine synthetase activity, which remains suppressed after nitrous oxide exposure has ended, underscoring the need for treatment with methionine. Continuous exposure to nitrous oxide for longer than 3 hours causes neuropathy, especially in B12-deficient individuals [Louis-Ferdinand, 1994; Richardson, 2010]. Nitrous oxide can be safely used as an anesthetic in patients with HMSN, CMT disease [Isbister et al., 2008].
Chemotherapeutic Agent-Induced Neuropathy
Peripheral neuropathy may develop after the use of chemotherapeutic agents to treat neoplasms. Painful sensorimotor peripheral neuropathy may develop after high-dose cytosine arabinoside therapy, and is manifested pathologically by axonal degeneration and scattered destruction of myelin sheaths [Borgeat et al., 1986]. The neuropathy is marked by dysesthesias, muscle aching, and progressive weakness. Cytosine arabinoside neuropathic pain may respond to carbamazepine [Malapert and Degos, 1989]. In 1 percent of patients treated with high-dose cytosine arabinoside, demyelinating polyneuropathy develops, resulting in quadriparesis and need for ventilatory support [Openshaw et al., 1996]. In addition to peripheral neuropathy, brachial plexus neuropathy has been observed with high-dose cytosine arabinoside [Scherokman et al., 1985].
Adenine arabinoside has also been reported to cause a sensory axonal neuropathy with pain and tingling in the feet [Lok et al., 1984], which may involve the distal arms in addition [Kanterewicz et al., 1990].
Vincristine is often used in children to treat acute lymphoblastic leukemia and may cause acute axonal sensorimotor neuropathy [Akbayram et al., 2010; Baker and Lipson, 2010]. Less commonly, it may cause autonomic and cranial nerve neuropathy [Shapiro and Young, 1984]. Tingling sensations, absent tendon reflexes, particularly at the heel cords, distal weakness, and impaired vibration and superficial sensation develop after exposure to vincristine. Vincristine neuropathy is most profound in children with HMSN type I and is associated with incomplete recovery of neuropathic deficits. Neuropathy is generally less severe in HMSN type II, with better recovery of nerve function after drug use is discontinued [Igarashi et al., 1995], but not always [Nishikawa et al., 2008]. Vincristine should be avoided in HMSN type I [Graf et al., 1996]. Clinical recovery from vincristine neuropathy in children after discontinuation of treatment is generally good [Ramchandren et al., 2009].
Cisplatin may cause profound axonal loss in peripheral nerves, producing a sensory neuropathy. Paresthesias and numbness occur months after initiating cancer therapy with the agent. Proprioception is also impaired, with diminished vibration and joint position sensation. Occasionally, mild distal muscle weakness is noted. In select protocols treating germ-cell tumors in children and using cisplatin as one therapeutic agent, the incidence of neuropathy was very low [Göbel et al., 1989; Hartmann et al., 1988]. High-tone hearing loss was noted, which may be due to the combination of cisplatin and etoposide. Cisplatin and suramin cause axonal sensorimotor polyneuropathy, whereas tacrolimus may cause demyelinating neuropathy [Peltier and Russell, 2002]. Vitamin E may be protective against the development of cisplatin neuropathy [Pace et al., 2010].
Vaccine-Induced Polyneuropathy
The diphtheria-pertussis-tetanus vaccine may rarely produce segmental demyelination and axonal neuropathy [Reinstein et al., 1982] from the tetanus toxoid component. The influenza, or 1976 swine flu, vaccine allegedly caused Guillain–Barré syndrome, but this has been questioned by re-evaluation of the statistical evidence on which the initial allegation rested [Hurwitz et al., 1981]. Likewise, no statistically significant association exists between Guillain–Barré syndrome and the 2009 H1N1 vaccination [Centers for Disease Control and Prevention, 2010]. Hepatitis B vaccination has been associated with neuropathy in case reports from adults. Acute sensory neuropathy has been reported in a 13-year-old following bacille Calmette–Guérin (BCG) vaccination [Wilmshurst et al., 1999].
Heavy Metal Neuropathy
Slowing of nerve conduction occurs in children environmentally exposed to lead, who are otherwise asymptomatic [Schwartz et al., 1988]. Concern is raised for developmental problems, cognitive deficits, and attention-deficit hyperactivity disorder in children with chronic lead exposure.
Rare instances of arsenic poisoning, usually perpetrated purposely against an individual, have been reported, producing an axonal, primarily sensory, neuropathy. Exposure may be by overdose, producing a subacute neuropathy, or may be more chronic, causing a slowly progressing neuropathy. Environmental exposure from contaminated water is associated with skin pigmentation, palmar and plantar keratosis, gastrointestinal symptoms, liver disease, cognitive impairment, and peripheral neuropathy Mazumder et al., 1992].
Mercury poisoning was described in Minamata Bay in Japan. The poisoning resulted from industrial discharge of short-chain alkyl mercury compounds, which were picked up by microorganisms and subsequently entered the food chain, concentrated in fish and shellfish. Transplacental transfer of methylmercury occurred [Yorifuji et al., 2009].
Organic mercury poisoning in children produces acrodynia, or pink disease, consisting of generalized erythema, especially of the hands, feet, and face, with swelling of the hands and feet. Stomatitis, loss of teeth, increased perspiration, and irritability are present. Elemental mercury is absorbed from a saturated atmosphere of mercury vapor, leading to subacute exposure. Several patients with predominantly axonal motor neuropathy resulting from vapor exposure have been recorded [Ross, 1964; Swaiman and Flagler, 1971]. Chronic exposure results in sensory and motor neuropathy, encephalopathy, and autonomic dysfunction. Chronic exposure may produce a subclinical neuropathy detected electrophysiologically [Zampollo et al., 1987]. Removing the source of mercury is important, and chelation therapy may be of value. High mercury levels in children from inorganic exposure can be successfully treated [Erkek et al., 2010].
Accidental ingestion of rat poison or insecticides containing thallium causes an acute or subacute syndrome of gastrointestinal disturbance and neuropathy. A sensory and motor axonal severe peripheral neuropathy with abdominal pain, nausea, vomiting, and alopecia is typical. One transplacental case is known. If death does not ensue, recovery is usually complete [Desenclos et al., 1992; Rangel-Guerra et al., 1990]. Thallium was banned in the United States as a rodenticide in 1972.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Adler J.B., Patterson R.L. Erb’s palsy: long term results of treatment in eighty-eight cases. J Bone Joint Surg. 1967;49A:1052.
Aiello I., Serra G., Gilli P., et al. Uremic neuropathy: correlations between electroneurographic parameters and serum levels of parathyroid hormone and aluminum. Eur Neurol. 1982;21:396.
Akbayram S., Akgun C., Doğan M., et al. Use of pyridoxine and pyridostigmine in children with vincristine-induced neuropathy. Indian J Pediatr. 2010;77:681.
Albin R.L., Albers J.W., Greenberg H.S., et al. Acute sensory neuropathy-neuronopathy from pyridoxine overdose. Neurology. 1987;37:1729.
al-Qattan M.M., Clarke H.M., Curtis C.G. Klumpke’s birth palsy. Does it really exist? J Hand Surg. 1995;20:19.
Andersson J., Sterner G. A 16-month-old boy with infectious mononucleosis, parotitis and Bell’s palsy. Acta Paediatr Scand. 1985;74:629.
Anger H.A., Dworkin F., Sharma S., et al. Linezolid use for treatment of multidrug-resistant and extensively drug-resistant tuberculosis, New York City, 2000-06. J Antimicrob Chemother. 2010;65:775.
Appenzeller O., Kornfeld M., Snyder R. Acromutilating, paralyzing neuropathy with corneal ulceration in Navajo children. Arch Neurol. 1976;33:733.
Appleton R., Riordan A., Tedman B., et al. Congenital peripheral neuropathy presenting as apnoea and respiratory insufficiency. Dev Med Child Neurol. 1994;36:545.
Arnason B.G.W. Acute inflammatory demyelinating polyneuropathies. In Dyck P.J., Thomas P.K., Lambert E.H., et al, editors: Peripheral neuropathy, ed 2, Philadelphia: WB Saunders, 1984.
Arnason B.G.W., Soliven B. Acute inflammatory demyelinating polyradiculoneuropathy. In: Dyck P.J., Thomas P.K., Griffin J.W., et al, editors. Peripheral neuropathy. Philadelphia: WB Saunders, 1993.
Asbury A.K., Gary M.K., Cox S.C., et al. Giant axonal neuropathy: a unique case with segmental neurofilamentous masses. Acta Neuropathol. 1972;20:237.
Aubourg P., Scotta J., Rocchicciolo F., et al. Neonatal adrenoleukodystrophy. J Neurol Neurosurg Psychiatry. 1986;49:77.
Axelrod F.B., Pearson J., Tepperberg, et al. Congenital sensory neuropathy with skeletal dysplasia. J Pediatr. 1983;102:727.
Babb A.L., Ahmad S., Bergstrom J., et al. The middle molecule hypothesis in perspective. Am J Kidney Dis. 1981;1:46.
Baker S.K., Lipson D.M. Vincristine-induced peripheral neuropathy in a neonate with congenital acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2010;32:114.
Barbeau A. Friedreich’s ataxia 1976 – an overview. Can J Neurol Sci. 1976;3:389.
Barbosa M.D., Barratt F.J., Tchernev V.T., et al. Identification of mutations in two major mRNA isoforms of the Chediak-Higashi syndrome gene in human and mouse. Hum Mol Genet. 1997;6:1091.
Barkai L., Szabó L. Urinary bladder dysfunction in diabetic children with and without subclinical cardiovascular autonomic neuropathy. Eur J Pediatr. 1993;152:190.
Bassen F.A., Kornzweig A.L. Malformation of the erythrocytes in a case of atypical retinitis pigmentosa. Blood. 1950;5:381.
Bayever E., Ladisch S., Philippart M., et al. Bone-marrow transplantation for metachromatic leukodystrophy. Lancet. 1985;2:471.
Beard M.E., Sapirstein V., Kolodny E.H., et al. Peroxisomes in fibroblasts from skin of Refsum’s disease patients. J Histochem Cytochem. 1985;33:480.
Becelli R., Perugini M., Carboni A., et al. Diagnosis of Bell palsy with gadolinium magnetic resonance imaging. J Craniofac Surg. 2003;14:51.
Beghi E., Kurland L.T., Mulder D.W., et al. Guillain-Barré syndrome: clinicoepidemiologic features and effect of influenza vaccine. Arch Neurol. 1985;42:1053.
Ben Othmane K., Hentati F., Lennon F., et al. Linkage of a locus (CMT4A) for autosomal recessive Charcot-Marie-Tooth disease to chromosome 8q. Hum Mol Genet. 1993;10:1625.
Berg B.O., Rosenberg S., Asbury A.K. Giant axonal neuropathy. Pediatrics. 1972;49:894.
Berger A.R., Schaumburg H.H., Schroeder C., et al. Dose response, coasting, and differential fiber vulnerability in human toxic neuropathy: a prospective study of pyridoxine neurotoxicity. Neurology. 1992;42:1367.
Berginer V., Baruchin A., Ben-Yakar Y., et al. Plantar ulcers in hereditary sensory neuropathy: a plea for conservative treatment. Int J Dermatol. 1984;23:664.
Bergoffen J., Scherer S.S., Wang S., et al. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039.
Bergoffen J., Trofatter J., Pericek-Vance M.A., et al. Linkage localisation of X-linked Charcot-Marie-Tooth disease. Am J Hum Genet. 1993;52:312.
Birouk N., Gouider R., Le Guern E., et al. Charcot-Marie-Tooth disease type Ia with 17p11.2 duplication. Clinical and electrophysiological phenotype study and factors influencing disease severity in 119 cases. Brain. 1997;120:813.
Bojanovski D., Gregg R.E., Brewer H.B.Jr. Tangier disease: in vitro conversion of proapo-A-I Tangier to mature apo-A-I Tangier. J Biol Chem. 1984;259:6049.
Boltshauser E., Bischoff A., Isler W. Giant axonal neuropathy. J Neurol Sci. 1977;31:269.
Bomont P., Cavalier L., Blondeau F., et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB / kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet. 2000;26:370-374.
Bonerandi J.J., Gambarelli D., Livet M.O., et al. Melanocytic involvement in giant axonal neuropathy. J Cutan Pathol. 1981;8:313.
Borgeat A., De Muralt B., Stalder M. Peripheral neuropathy associated with high-dose ara-C therapy. Cancer. 1986;58:852.
Bowerson M., Nelson V.S., Yang L.J. Diaphragmatic paralysis associated with neonatal brachial plexus palsy. Pediatr Neurol. 2010;42:234.
Bradshaw D.Y., Jones H.R. Guillain-Barré syndrome in children: clinical course, electrodiagnosis, and prognosis. Muscle Nerve. 1992;15:500.
Braguer D., Gallice P., Monti J.P., et al. Inhibition of microtubule formation by uremic toxins: action mechanism and hypothesis about the active component. Clin Nephrol. 1986;25:212.
Brechenmacher C., Vital C., Deminiere C., et al. Guillain-Barré syndrome: an ultrastructural study of peripheral nerve in 65 patients. Clin Neuropathol. 1987;6:19.
Bril V., Ilse W.K., Pearce R., et al. Pilot trial of immunoglobulin versus plasma exchange in patients with Guillain-Barré syndrome. Neurology. 1996;46:100.
Brin M.F., Pedley T.A., Lovelace R.E., et al. Electrophysiologic features of abetalipoproteinemia: functional consequences of vitamin E deficiency. Neurology. 1986;36:669.
Bruno C., Bertini E., Federico A., et al. Clinical and molecular findings in patients with giant axonal neuropathy. Neurology. 2004;62:13-16.
Budden S.S., Kennaway N.G., Buist N.R., et al. Dysmorphic syndrome with phytanic acid oxidase deficiency, abnormal very long chain fatty acids, and pipecolic acidemia: studies in four children. J Pediatr. 1986;108:33.
Bueri J.A., Cohen L.G., Panizza M.E., et al. Peripheral nerve involvement in Bell’s palsy. Arq Neuropsiquiatr. 1984;42:341.
Burns J., Ouvrier R.A., Yiu E.M., et al. Ascorbic acid for Charcot–Marie–Tooth disease type 1A in children: a randomised, double-blind, placebo-controlled, safety and efficacy trial. Lancet Neurol. 2009;8:537-544.
Buzzi S. Diphtheria toxin treatment of human advanced cancer. Cancer Res. 1982;42:2054.
Cai Z., Blumbergs P.C., Cash K., et al. Paranodal pathology in Tangier disease with remitting-relapsing multifocal neuropathy. J Clin Neurosci. 2006;13:492.
Caksen H., Kurtoglu S., Ustünbau H.B., et al. A case of the cardiofacial syndrome (Cayler’s syndrome). Acta Paediatr Jpn. 1996;38:3256.
Campbell E.D.R., Hickey R.P., Nixon K.H., et al. Value of nerve-excitability measurements in prognosis of facial palsy. Br Med J. 1962;2:7.
Campuzano V., Montermini L., Moltò M.D., et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423.
Carpenter S., Karpati G., Andermann F., et al. Giant axonal neuropathy: a clinically and morphologically distinct neurological disease. Arch Neurol. 1974;31:312.
Carroll J.E., Jedziniak M., Guggenheim M.A. Guillain-Barré syndrome: Another cause of the “floppy infant”. Am J Dis Child. 1977;131:699.
Cavanagh N.P., Eames R.A., Galvin R.J., et al. Hereditary sensory neuropathy with spastic paraplegia. Brain. 1979;102:79.
Cayler G., Blumenfeld C.M., Anderson R.L. Further studies of patients with the cardiofacial syndrome. Chest. 1971;60:161.
Centers for Disease Control and Prevention. Preliminary results: surveillance for Guillain-Barré syndrome after receipt of influenza A (H1N1) 2009 monovalent vaccine – United States, 2009–2010. MMWR Morb Mortal Wkly Rep. 2010;59:657.
Chamberlain S., Shaw J., Rowland A., et al. Mapping of mutation causing Friedreich’s ataxia to human chromosome 9. Nature. 1988;334:248.
Chamberlain S., Shaw J., Wallis J., et al. Genetic homogeneity at the Friedreich ataxia locus on chromosome 9. Am J Hum Genet. 1989;44:518.
Chance P.F., Bird T.D., O’Connel P., et al. Genetic linkage and heterogeneity in type 1 Charcot-Marie-Tooth disease (hereditary motor and sensory neuropathy type I). Am J Hum Genet. 1990;47:915.
Chance P.F. Inherited focal, episodic neuropathies: hereditary neuropathy with liability to pressure palsies and hereditary neuralgic amyotrophy. Neuromolecular Med. 2006;8:159.
Charnas L., Trapp B., Griffin J. Congenital absence of peripheral myelin: abnormal Schwann cell development causes lethal arthrogryposis multiplex congenita. Neurology. 1988;38:966.
Chen F., Miyahara R., Matsunaga Y., et al. Schwannoma of the brachial plexus presenting as an enlarging cystic mass: report of a case. Ann Thorac Cardiovasc Surg. 2008;14:311.
Cherington M. Botulism: ten-year experience. Arch Neurol. 1974;30:432.
Chia J.K., Clark J.B., Ryan C.A., et al. Botulism in an adult associated with food-borne intestinal infection with Clostridium botulinum. Med Int. 1986;315:239.
Cogan D.G., Rodrigues M., Chu F.C., et al. Ocular abnormalities in abetalipoproteinemia: a clinicopathologic correlation. Ophthalmology. 1984;91:991.
Colan R.V., Snead O.C., Oh S.J., et al. Steroid-responsive polyneuropathy with subacute onset in childhood. J Pediatr. 1980;97:374.
Cornblath D.R., Sladky J.T., Sumner A.J. Clinical electrophysiology of infantile botulism. Muscle Nerve. 1983;6:448.
Cotter P.D., Nguyen H., Tung G., Rauen K.A. Incidence of microduplication 22q11.2 in patients referred for FISH testing for velo cardiofacial and DiGeorge syndromes. Eur J Hum Genet. 2005;13:1245.
Cox J.J., Reimann F., Nicholas A.K., et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894-898.
Créange A., Meyrignac C., Roualdes B., et al. Diphtheric neuropathy. Muscle Nerve. 1995;18:1460.
Cruz Martínez A., Anciones B., Ferred M.T., et al. Electrophysiologic study in benign human botulism type B. Muscle Nerve. 1985;8:580.
Danon M.J., Carpenter S. Hereditary sensory neuropathy: biopsy study of an autosomal dominant variety. Neurology. 1985;35:1226.
Dawkins J.L., Hulme D.J., Brahmbhatt S.B., et al. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type 1. Nat Genet. 2001;27:309-312.
Dawson D.M., Samuels M.A., Morris J. Sensory form of acute polyneuritis. Neurology. 1988;38:1728.
Debuse P.J. Shoshin beriberi in an infant of a thiamine-deficient mother. Acta Paediatr. 1992;81:723.
Dechelotte P., Kantelip B., de Laguillaumie B.V., et al. Tangier disease: a histological and ultrastructural study. Pathol Res Pract. 1985;180:424.
Defanti C.A., Sghirlanzoni A., Bottacchi E., et al. Porphyric neuropathy: a clinical, neurophysiological and morphological study. Ital J Neurol Sci. 1985;6:521.
de Jesus P.V., Slater R., Spitz L.K., et al. Neuromuscular physiology of wound botulism. Arch Neurol. 1973;29:425.
De Michele G., Filla A., Cavalcanti F., et al. Late onset Friedreich’s disease: clinical features and mapping of mutation to the FRDA locus. J Neurol Neurosurg Psychiatry. 1994;57:977.
Desenclos J.C., Wilder M.H., Coppenger G.W., et al. Thallium poisoning: an outbreak in Florida, 1988. South Med J. 1992;85:1203.
Devi S., Challenor Y., Duarte N., et al. Prognostic value of minimal excitability of facial nerve in Bell’s palsy. J Neurol Neurosurg Psychiatry. 1978;41:649.
Di Paolo B., Cappelli P., Spisni C., et al. New electrophysiological assessments for the early diagnosis of encephalopathy and peripheral neuropathy in chronic uremia. Int J Tissue React. 1982;4:301.
DiTaranto P., Campagna L., Price A.E., et al. Outcome following nonoperative treatment of brachial plexus birth injuries. J Child Neurol. 2004;19:87.
Domingo R.M., Haller J.S., Gruenthal M. Infant botulism: two recent cases and literature review. J Child Neurol. 2008;23:1336.
Donaghue K.C., Fung A.T., Fairchild J.M., et al. Prospective assessment of autonomic and peripheral nerve function in adolescents with diabetes. Diabet Med. 1996;13:65.
Donaghy M., Brett E.M., Ormerod I.E.C., et al. Giant axonal neuropathy, observations on a further patient. J Neurol Neurosurg Psychiatry. 1988;51:991.
Donaghy M., Hakin R.N., Bamford J.M., et al. Hereditary sensory neuropathy with neurotrophic keratitis. Description of an autosomal recessive disorder with a selective reduction of small myelinated nerve fibres and a discussion of the classification of the hereditary sensory neuropathies. Brain. 1987;110:563.
Dooley J.M., Oshima Y., Becker L.E., et al. Clinical progression of giant-axonal neuropathy over a twelve year period. Can J Neurol Sci. 1981;8:321.
Dordain G., Deffond D. Pyridoxine neuropathies. Review of the literature. Therapie. 1994;49:333.
Duncan C., Strub R., McGarry P., et al. Peripheral nerve biopsy as an aid to diagnosis in infantile neuroaxonal dystrophy. Neurology. 1970;20:1024.
Dunn H.G. Nerve conduction studies in children with Friedreich’s ataxia and ataxia-telangiectasia. Dev Med Child Neurol. 1973;15:324.
Dunn H.G., Daube J.R., Gomez M.R. Heredofamilial brachial plexus neuropathy (hereditary neuralgic amyotrophy with brachial predilection) in childhood. Dev Med Child Neurol. 1978;20:28.
Dyck P.J.. Neuronal atrophy and degeneration predominantly affecting peripheral sensory and autonomic neurons. Dyck P.J., Thomas P.K., Lambert E.H., et al, editors. Peripheral neuropathy, vol 2. Philadelphia: WB Saunders, 1984.
Dyck P.J., Mellinger J.F., Reagan T.J., et al. Not “indifference to pain” but varieties of hereditary sensory and autonomic neuropathy. Brain. 1983;106:373.
Dyck P.J., Thomas P.K., Lambert E.H. Peripheral neuropathy. Philadelphia: WB Saunders; 1975. vol 2
Ecker J.L., Greenberg J.A., Norwitz E.R., et al. Birth weight as a predictor of brachial plexus injury. Obstet Gynecol. 1997;89:643.
Edelstein C., Gordon J.I., Vergani C.A., et al. Comparative in vitro study of the pro-apolipoprotein A-I to apolipoprotein A-I converting activity between normal and Tangier plasma. J Clin Invest. 1984;74:1098.
Einarsdottir E., Carlsson A., Minde J., et al. A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Hum Mol Genet. 2004;15(13):799-805.
Epstein M.A., Sladky J.T. The role of plasmapheresis in childhood Guillain-Barré syndrome. Ann Neurol. 1990;28:65.
Erkek N., Senel S., Sarac A., et al. Being alive after a severe inorganic mercury intoxication. Eur J Pediatr. 2010;196:625.
Fach P., Fenicia L., Knutsson R., et al. An innovative molecular detection tool for tracking Clostridium botulinum types A, B, E, F and other botulinum neurotoxin producing Clostridia based on the genedisc cycler. Int J Food Microbiol. (Apr 8):2010.
Faguer P., Man N.K., Cueille G., et al. Improved separation and quantification of the “middle molecule”: b4-2 in uremia. Clin Chem. 1983;29:703.
Fernandez J.M., Dávalos A., Ferrer I., et al. Acute sensory neuronopathy: report of a child with remarkable clinical recovery. Neurology. 1994;44:762.
Ferrière G., Guzzetta F., Kulakowski S., et al. Nonprogressive type II hereditary sensory autonomic neuropathy: A homogeneous clinicopathologic entity. J Child Neurol. 1992;7:364.
Fiçicioglu C., Aydin A., Haktan M., et al. Peripheral neuropathy in children with insulin-dependent diabetes mellitus. Turk J Pediatr. 1994;36:97.
Fischbeck K.H., Ar-Rushdi H.N., Pericek-Vance M.A., et al. X-linked neuropathy: Gene localisation with DNA probes. Ann Neurol. 1986;20:527.
Fisch U. Progressive value of electrical tests in acute facial paralysis. Am J Otolaryngol. 1984;5:494.
Fitzpatrick D.B., Hooper R.E., Seife B. Hereditary deafness and sensory radicular neuropathy. Arch Otolaryngol. 1976;102:552.
Fois A., Balestri P., Farnetani M.A., et al. Giant axonal neuropathy: endocrinological and histological studies. Eur J Pediatr. 1985;144:274.
French Cooperative Group on Plasma Exchange in Guillain-Barré syndrome. Efficiency of plasma exchange in Guillain-Barré syndrome: role of replacement fluids. Ann Neurol. 1987;22:753.
Gabreels-Festen A.A.W.M., Gabreels F.J.M., Jennekens F.G.I., et al. Autosomal recessive form of hereditary motor and sensory neuropathy type I. Neurology. 1992;42:1755.
Gabreels-Festen A.A.W.M., Joosten E.M.G., Gabreels F., et al. Hereditary motor and sensory neuropathy of neuronal type with onset in early childhood. Brain. 1991;114:1855.
Gambarelli D., Hassoun J., Pellissier J.F., et al. Giant axonal neuropathy: involvement of peripheral nerve, myenteric plexus and extra-neuronal area. Acta Neuropath. 1977;39:261.
Gamstorp I., et al. Peripheral neuropathy in juvenile diabetes. Diabetes. 1966;15:411.
Gaskill H.S., Korb M. Occurrence of multiple neuritis in cases of cutaneous diphtheria. Arch Neurol Psychiatry. 1946;55:559.
Gdynia H.J., Müller T., Sperfeld A.D., et al. Severe sensorimotor neuropathy after intake of highest dosages of vitamin B6. Neuromuscul Disord. 2008;18:156.
Ghamdi M., Armstrong D., Miller G. Congenital hypomyelinating neuropathy: a reversible case. Pediatr Neurol. 1997;16:71.
Giannotti A., Mingarelli R. Cayler cardiofacial syndrome and del 22q11: part of the CATCH22 phenotype [letter]. Am J Med Genet. 1994;53:303.
Gibbels E., Schaefer H.E., Runne U., et al. Severe polyneuropathy in Tangier disease mimicking syringomyelia or leprosy: clinical, biochemical, electrophysiological, and morphological evaluation, including electron microscopy of nerve, muscle, and skin biopsies. J Neurol. 1985;232:283.
Gibberd F.B., Billimoria J.D., Goldman J.M., et al. Heredopathia atactica polyneuritiformis: Refum’s disease. Acta Neurol Scand. 1985;72:1.
Gillespie J.B., Perucca L.G. Congenital generalized indifference to pain (congenital analgia). Am J Dis Child. 1960;100:124.
Glickman R.M., Khorana J., Kilgore A. Botulism toxin: mechanism of presynaptic blockade. Science. 1976;193:1256.
Göbel U., Bamberg M., Haas R.J., et al. Non-testicular germ cell tumors: analysis of the therapy study MAKEI83/86 and changes in the protocol for the follow-up study. Klin Padiatr. 1989;201:247.
Goebel H.H., Vogel P., Gabriel M. Neuropathologic and morphometric studies in hereditary motor and sensory neuropathy type II with neurofilament accumulation. Ital J Neurol Sci. 1986;7:325.
Goldberg Y.P., MacFarlane J., MacDonald M.L., et al. Loss-of-function mutations in the Na(v)1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet. 2007;71:311-319.
Gorson K.C., Allam G., Simovic D., et al. Improvement following interferon-alpha 2A in chronic inflammatory demyelinating polyneuropathy. Neurology. 1997;48:777.
Graf W.D., Chance P.F., Lensch M.W., et al. Severe vincristine neuropathy in Charcot-Marie-Tooth disease type 1A. Cancer. 1996;77:1356.
Graham E.M., Forouzan I., Morgan M.A. A retrospective analysis of Erb’s palsy cases and their relation to birth weight and trauma at delivery. J Matern Fetal Med. 1997;6:1.
Greenwald A.G., Schute P.C., Shiveley J.L. Brachial plexus birth palsy: a 10-year report on the incidence and prognosis. J Pediatr Orthop. 1984;4:689.
Greenwood R.J., Newsom-Davis J., Hughes R.A., et al. Controlled trial of plasma exchange in acute inflammatory polyradiculoneuropathy. Lancet. 1984;1:877.
Grohmann K., Varon R., Stolz P., et al. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Ann Neurol. 2003;54:719.
Guillain G., Barré J.A., Strohl A. Sur un syndrome de radiculo-nevrite avec hyperalbuminose de liquide cephalo-rachidien sans réaction cellulaire: remarques sur les caractères cliniques et graphiques des reflexes tendineux. Bull Mem Soc Med Hop Paris. 1916;40:1462.
Guillozet N., Mercer R.D. Hereditary recurrent brachial neuropathy. Am J Dis Child. 1973;125:884.
Guzzetta F., Ferrière G., Lyon G. Congenital hypomyelination polyneuropathy: pathological findings compared with polyneuropathies starting later in life. Brain. 1982;105:395.
Gwathmey F.W., House J.H. Clinical manifestations of congenital insensitivity of the head and classification of syndromes. J Hand Surg. 1984;9A:863.
Haberlandt E., Scholl-Bürgi S., Neuberger J., et al. Peripheral neuropathy as the sole initial finding in three children with infantile metachromatic leukodystrophy. Eur J Paediatr Neurol. 2009;13:257.
Hagberg B. Krabbe’s disease: clinical presentation of neurological variants. Neuropediatrics. 1984;15:11.
Hagberg B., Lyon G. Pooled European series of hereditary peripheral neuropathies in infancy and childhood: a “correspondence work shop” report of the European federation of child neurology societies (EFCNS). Neuropediatrics. 1981;12:9.
Hahn A.F., Brown W.F., Koopman W.J., et al. X-linked dominant hereditary motor and sensory neuropathy. Brain. 1990;113:1511.
Hallam P.J., Harding A.E., Berciano J., et al. Duplication of part of chromosome 17 is commonly associated with hereditary motor and sensory neuropathy type I (Charcot-Marie-Tooth disease type 1). Ann Neurol. 1992;31:570.
Hall S.M., Hughes R.A.C., Atkinson P.F., et al. Motor nerve biopsy in severe Guillain-Barré syndrome. Ann Neurol. 1992;31:441.
Hannibal M.C., Ruzzo E.K., Miller L.R., et al. SEPT9 gene sequencing analysis reveals recurrent mutations in hereditary neuralgic amyotrophy. Neurology. 2009;72:1755.
Harding A.E., Thomas P.K. Peroneal muscular atrophy with pyramidal features. J Neurol Neurosurg Psychiatry. 1984;47:168.
Harding A.E., Thomas P.K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103:259.
Hartmann O., Pinkerton C.R., Philip T., et al. Very-high-dose cisplatin and etoposide in children with untreated advanced neuroblastoma. J Clin Oncol. 1988;6:44.
Hartung H.P., Pollard J.D., Harvey G.K., et al. Immunopathogenesis and treatment of the Guillain-Barré syndrome – part I. Muscle Nerve. 1995;18:137.
Hartung H.P., Pollard J.D., Harvey G.K., et al. Immunopathogenesis and treatment of the Guillain-Barré syndrome – part II. Muscle Nerve. 1995;18:154.
Hayasaka K., Takada G., Ionasescu V.V., et al. Mutation of the myelin Po gene in Charcot-Marie-Tooth neuropathy type IB. Hum Mol Genet. 1993;2:1369.
Hegele R.A., Angel A. Arrest of neuropathy and myopathy in abetalipoproteinemia with high-dose vitamin E therapy. Can Med Assoc J. 1985;132:41.
Heisel M.A., Siegel S.E., Falk R.E., et al. Congenital pernicious anemia: report of seven patients, with studies of the extended family. J Pediatr. 1984;105:564.
Herbert P.N., Forte T., Heinen R.J., et al. Tangier disease: one explanation of lipid storage. N Engl J Med. 1978;299:519.
Higginbottom M.C., Sweetman L., Nyhan W.L. A syndrome of methylmalonic aciduria, homocystinuria, megaloblastic anemia and neurologic abnormalities in a vitamin B-12 deficient breast-fed infant of a strict vegetarian. N Engl J Med. 1978;299:317.
Hodson A.K., Hurwitz B.J., Albrecht R. Dysautonomia in Guillain-Barré syndrome with dorsal root ganglioneuropathy, wallerian degeneration, and fatal myocarditis. Ann Neurol. 1984;15:88.
Houlden H., Blake J., Reilly M.M. Heriditary Sensory neuropathies. Curr Opin Neurol. 2004;17:569-577.
Hughes R.A.C., The Plasma Exchange/Sandoglobin Guillain Barré Syndrome Trial Group. Comparison of plasma exchange, intravenous immunoglobulin, and plasma exchange followed by intravenous immunoglobulin in the treatment of the Guillain Barré syndrome. Ann Neurol. 1996;3:551. (abstract)
Hungerbeuhler J.P., Meier C., Rousselle L., et al. Refsum’s disease: management by diet and plasmapheresis. Eur Neurol. 1985;24:153.
Hurwitz E.S., Schonberger L.B., Nelson D.B., et al. Guillain-Barré syndrome and the 1978-1979 influenza vaccine. N Engl J Med. 1981;304:1557.
Hyllienmark L., Brismar T., Ludvigsson J. Subclinical nerve dysfunction in children and adolescents with IDDM. Diabetologia. 1995;38:685.
Igarashi M., Thompson E.I., Rivera G.K. Vincristine neuropathy in type I and type II Charcot-Marie-Tooth disease (hereditary motor sensory neuropathy). Med Pediatr Oncol. 1995;25:113.
Igbal A., Arnason B.G. Neuropathy of serum sickness. In: Dyck P.J., Thomas P.K., Lambert E.H., et al, editors. Peripheral neuropathy. Philadelphia: WB Saunders, 1984.
Igisu H., Ohta M., Tabira T., et al. Giant axonal neuropathy: a clinical entity affecting the central as well as peripheral nervous system. Neurology (Minneapolis). 1975;25:717.
Iida H., Takashima Y., Maeda S., et al. Alterations in erythrocyte membrane lipids in abetalipoproteinemia: phospholipid and fatty acyl composition. Biochem Med. 1984;32:79.
Indo Y., Tsuruta M., Hayashida Y., et al. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nature Genet. 1996;13:485.
Ionasescu V., Searby C., Rubenstein P., et al. Giant axonal neuropathy: normal protein composition of neurofilaments. J Neurol Neurosurg Psychiatry. 1983;46:551.
Ippel E., Wittebol-Post D., Jennekens F.G.I., et al. Genetic heterogeneity of hereditary motor and sensory neuropathy type VI. J Child Neurol. 1995;10:459.
Irobi J., Dierick I., Jordanova A., et al. Unraveling the genetics of distal hereditary motor neuronopathies. Neuromolecular Med. 2006;8:131-146.
Isbister G.K., Burns J., Prior F., et al. Safety of nitrous oxide administration is patients with Charcot-Marie-Tooth disease. J Neurol Sci. 2008;268:160.
Itoh K., Negishi H., Obayashi C., et al. Infantile neuroaxonal dystrophy – immunohistochemical and ultrastructural studies on the central and peripheral nervous systems in infantile neuroaxonal dystrophy. Kobe J Med Sci. 1993;39:133.
Iwabuchi S., Yoshino Y., Goto H., et al. Analysis of serum immunoglobulins in hereditary sensory radicular neuropathy. J Neurol Sci. 1976;30:29.
Iwanaga R., Matsuishi T., Ohnishi A., et al. Serial magnetic resonance images in a patient with congenital sensory neuropathy with anhidrosis and complications resembling heat stroke. J Neurol Sci. 1996;142:79.
Jackson C.E., Amato A.A., Barohn R.J. Isolated vitamin E deficiency. Muscle Nerve. 1996;19:1161.
Jameson M., Roberts S., Anderson N.E., et al. Nitrous oxide-induced vitamin B(12) deficiency. J Clin Neurosci. 1999;6:164.
Jaradeh S., D’Cruz O., Howard J.F.Jr, et al. Mobius syndrome: electrophysiologic studies in seven cases. Muscle Nerve. 1996;19:1148.
Jenkins H.A., Herzog J.A., Coker N.J. Bell’s palsy in children: cases of progressive facial nerve degeneration. Ann Otol Rhinol Laryngol. 1985;94:331.
Jestico J.V., Urry P.A., Efphimiou J. An hereditary sensory and autonomic neuropathy transmitted as an X-linked recessive trait. J Neurol Neurosurg Psychiatry. 1985;48:1259.
Johnsen S.D., Johnson P.C., Stein S.R. Familial sensory autonomic neuropathy with arthropathy in Navajo children. Neurology. 1993;43:1120.
Jones M.Z., Nigro M.A., Barre P.S. Familial giant axonal neuropathy. J Neuropath Exp Neurol. 1979;38:324. (abstract)
Judisch G.F., Rhead W.J., Miller D.K. Abetalipoproteinemia: report of an unusual patient. Ophthalmologica. 1984;189:73.
Kalaydjieva L., Hallmayer J., Chandler D., et al. Gene mapping in Gypsies identifies a novel demyelinating neuropathy on chromosome 8q24. Nature Genet. 1996;14:214.
Kaminagakura E., Jorge J.Jr. Melkersson Rosenthal syndrome: a histopatholgic mystery and dermatologic challenge. J Cutan Pathol. 2009;38:241.
Kanterewicz E., Bruguera M., Viola C., et al. Toxic neuropathy after adenine arabinoside treatment in chronic hbsag-positive liver disease. J Clin Gastroenterol. 1990;12:90.
Kaplan J., DeDomenico K., Ward D.M. Chediak-Higashi syndrome. Curr Opin Hematol. 2008;15:22.
Kaplan J.G., Kessler J., Rosenberg N., et al. Sensory neuropathy associated with Dursban (chlorpyrifos) exposure. Neurology. 1993;43:2193.
Karch S.B., Urich H. Infantile polyneuropathy with defective myelination: an autopsy study. Dev Med Child Neurol. 1975;17:504.
Kasman M., Bernstein L., Schulman S. Chronic polyradiculoneuropathy of infancy: a report of three cases with familial incidence. Neurology. 1976;26:565.
Katusik S., Beard C.M., Wiederholt W.C., et al. Incidence, clinical features and prognosis in Bell’s palsy, Rochester, Minnesota 1968-1982. Ann Neurol. 1986;20:622.
Keats B.J.B., Ward L.J., Shaw J., et al. “Acadian” and “classical” forms of Friedreich ataxia are most probably caused by mutations at the same locus. Am J Med Genet. 1990;33:266.
Kessali M., Zemmouri R., Guilbot A., et al. A clinical, electrophysiological, neuropathologic and genetic study of two large Algerian families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease. Neurology. 1997;48:867.
Khateeb S., Flusser H., Ofir R., et al. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Amer J Hum Genet. 2006;79:942-948.
Kim H.J., Sohn K.M., Shy M.E., et al. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (cmtx5). Am J Hum Genet. 2007;81(3):552-558.
Kinney R.B., Gottfried M.R., Hodson A.K., et al. Congenital giant axonal neuropathy. Arch Pathol Lab Med. 1985;109:639.
Kirkham T.H., Guitton D., Coupland S.G. Giant axonal neuropathy: visual and oculomotor deficits. Can J Neurol Sci. 1980;7:177.
Klar A., Gross-Kieselstein E., Hurvitz H., et al. Bilateral Bell’s palsy due to Mycoplasma pneumoniae infection. Isr J Med Sci. 1985;21:692.
Kleyweg R.P., van der Meché F.G.A., Meulstee J. Treatment of Guillain-Barré syndrome with high-dose gammaglobulin. Neurology. 1988;38:1639.
Klockgether T., Chamberlain S., Wullner U., et al. Late onset Friedreich’s ataxia. Molecular genetics, clinical neurophysiology, and magnetic resonance imaging. Arch Neurol. 1993;50:803.
Klymkowsky M.W., Plummer D.J. Giant axonal neuropathy: a conditional mutation affecting cytoskeletal organization. J Cell Biol. 1985;100:245.
Knoll O., Dierker E. Detection of uremic neuropathy by reflex response latency. J Neurol Sci. 1980;47:305.
Koch T., Schultz P., Williams R., et al. Giant axonal neuropathy: a childhood disorder of microfilaments. Ann Neurol. 1977;1:438.
Koerbel A., Prevedello D.M., Tatsui C.E., et al. Posterior fossa gangliocytoma with facial nerve invasion: case report. Arq Neuropsiquiatr. 2003;61:274.
Kondo K., Horikawa Y., Japan N. Genetic heterogeneity of hereditary sensory neuropathy. Arch Neurol. 1974;30:336.
Koobatian T.J., Birkhead G.S., Schramm M.M., et al. The use of hospital discharge data for public health surveillance of Guillain-Barré syndrome. Ann Neurol. 1992;30:618.
Korinthenberg R., Schessl J., Kirschner J. Clinical presentation and course of childhood Guillain-Barré syndrome: a prospective multicentre study. Neuropediatrics. 2007;38:10.
Koski C.L., Sanders M.E., Swoveland P.T., et al. Activation of terminal components of complement in patients with Guillain-Barré syndrome and other demyelinating neuropathies. J Clin Invest. 1987;80:1492.
Kremer H., Kuyt L.P., van den Helm B., et al. Localization of a gene for Möbius syndrome to chromosome 3q by linkage analysis in a Dutch family. Hum Mol Genet. 1996;5:1367.
Kress B., Griesbeck F., Stippich C., et al. Bell palsy: quantitative analysis of MR imaging data as a method of predicting outcome. Radiology. 2004;230:504.
Krivit W., Shapiro E., Kennedy W., et al. Treatment of late infantile metachromatic leukodystrophy by bone marrow transplantation. N Engl J Med. 1990;322:28.
Kuwabara S. Guillain-Barré syndrome: epidemiology, pathophysiology and management. Drugs. 2004;64:597.
Laccone F., Hannibal M.C., Neesen J., et al. Dysmorphic syndrome of hereditary neuralgic amyotrophy associated with a SEPT9 gene mutation—a family study. Clin Genet. 2008;74:279.
Lafreniere R.G., MacDonald M.L., Dube M.P., et al. Identification of a novel gene (HSN2) causing hereditary sensory and autonomic neuropathy type II through the study of Canadian genetic isolates. Am J Hum Genet. 2004;74:1064-1073.
Laiwah A.C., Macphee G.J., Boyle P., et al. Autonomic neuropathy in acute intermittent porphyria. J Neurol Neurosurg Psychiatry. 1985;48:1025.
Lamarche J.B., Lemieux B., Lieu H.B. The neuropathology of “typical” Friedreich’s ataxia in Quebec. Can J Neurol Sci. 1984;11:592.
Lamont P.J., Johnston H.M., Berdoukas V.A. Plasmapheresis in children with Guillain-Barré syndrome. Neurology. 1991;41:1928.
Lange Y., Steck T.L. Mechanism of red blood cell acanthocytosis and echinocytosis in vivo. J Membr Biol. 1984;77:153.
Landrieu P., Said G., Allaire C. Dominantly transmitted congenital indifference to pain. Ann Neurol. 1990;27:574-578.
Langworth E.P., Taverner D. The prognosis in facial palsy. Brain. 1963;86:465.
Lebo R., Lynch E., Wiegant J., et al. Multicolor fluorescence in situ hybridization dissects demyelinating Charcot-Marie-Tooth disease gene region. J Neurol Sci. 1990;98(Suppl):106.
Levy B.K., Fenton G.A., Loaiza S., et al. Unexpected recovery in a newborn with severe hypomyelinating neuropathy. Pediatr Neurol. 1997;16:245.
Liberfarb R.M., Jackson A.H., Eavey R.D., et al. Unique hereditary sensory and autonomic neuropathy with growth hormone deficiency. J Child Neurol. 1993;8:271.
Lin A.E., Ardinger H.H., Ardinger R.H.Jr, et al. Cardiovascular malformations in Smith-Lemli-Opitz syndrome. Am J Med Genet. 1997;68:270.
Lindsay E.A., Greenberg F., Shaffer L.G., et al. Submicroscopic deletions at 22q11.2: variability of the clinical picture and delineation of a commonly deleted region. Am J Med Genet. 1995;56:191.
Lindström M., Korkeala H. Laboratory diagnostics of botulism. Clin Microbiol Rev. 2006;19:298.
Linssen W.H.J.P., Van den Bent M.J., Brunner H.G., et al. Deafness, sensory neuropathy, and ovarian dysgenesis: A new syndrome or a broader spectrum Perrault syndrome. Am J Med Genet. 1994;51:81.
Lipton M., Lockman L.A., Ramsay N.K., et al. Bone marrow transplantation in metachromatic leukodystrophy. Birth Defects. 1986;22:57.
Lloyd A.V., Jewitt D.E., Still J.D. Facial paralysis in children with hypertension. Arch Dis Child. 1966;41:292.
Lockman L.A., Kennedy W.R., White J.G. Chédiak-Higashi syndrome: electrophysiological and electron microscopic observation on the peripheral neuropathy. J Pediatr. 1967;70:942.
Logina I., Donaghy. Diphtheritic polyneuropathy: a clinical study and comparison with Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 1999;67:433.
Lok A.S., Wilson L.A., Thomas H.C. Neurotoxicity associated with adenine arabinoside monophosphate in the treatment of chronic hepatitis B virus infection. J Antimicrob Chemother. 1984;14:93.
Loonen M.C., Van Diggelen O.P., Janse H.C., et al. Late-onset globoid cell leucodystrophy (Krabbe’s disease): clinical and genetic delineation of two forms and their relation to the early-infantile form. Neuropediatrics. 1985;16:137.
Louis-Ferdinand R.T. Myelotoxic, neurotoxic and reproductive adverse effects of nitrous oxide. Adverse Drug React Toxicol Rev. 1994;13:193.
Low P.A., McLeod J.G., Prineas J.W. Hypertrophic Charcot-Marie-Tooth disease: light and electron microscope studies of the sural nerve. J Neurol Sci. 1978;35:93.
Lowry N.J., Taylor M.J., Belknapp W., et al. Electrophysiological studies in five cases of abetalipoproteinemia. Can J Neurol Sci. 1984;11:60.
Luder A.S., Mamet R., Farbstein I., et al. Awareness is the name of the game: clinical and biochemical evaluation of a case of a girl diagnosed with acute intermittent porphyria associated with autism. Cell Mol Biol. 2009;55:19.
Luijten J.A., Straks W., Blikkendaal-Lieftinck L.F., et al. Metachromatic leukodystrophy: a comparative study of the ultrastructural findings in the peripheral nervous system of three cases, one of the later infantile, one of the juvenile and one of the adult form of the disease. Neuropaediatrie. 1978;9:338.
Lupski J.R., Montes de Oca-Luna R., Slaugenhaupt S., et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219.
Luzi P., Rafi M.A., Wenger D.A. Characterization of the large deletion in the GALC gene found in patients with Krabbe’s disease. Hum Mol Genet. 1995;4:2335.
Majnemer A., Rosenblatt B., Watters G., et al. Giant axonal neuropathy: central abnormalities demonstrated by evoked potentials. Ann Neurol. 1986;19:394.
Malandrini A., Cavallaro T., Fabrizi G.M., et al. Ultrastructure and immunoreactivity of dystrophic axons indicate a different pathogenesis of Hallervorden-Spatz disease and infantile neuroaxonal dystrophy. Virchows Arch. 1995;427:415.
Malapert D., Degos J.D. Painful legs and moving toes. Neuropathy caused by cytarabine. Rev Neurol (Paris). 1989;145:869.
Mandel H., Berant M., Hazani A., et al. Thiamine-responsive anemia syndrome. N Engl J Med. 1984;311:836.
Mandel H., Meiron D., Schutgens R.B., et al. Infantile Refsum’s disease: gastrointestinal presentation of a peroxisomal disorder. J Pediatr Gastroenterol Nutr. 1992;14:83.
Manfredi M., Bini G., Cruccu G., et al. Congenital absence of pain. Arch Neurol. 1981;38:507-511.
Marbini A., Gemignani F., Ferrarini G., et al. Tangier disease: a case with sensorimotor distal polyneuropathy and lipid accumulation in striated muscle and vasa nervorum. Acta Neuropathol (Berl). 1985;67:121.
Markowitz J.A., Jeste S.S., Kang P.B. Child neurology: chronic inflammatory demyelinating polyradiculoneuropathy in children. Neurology. 2008;71:74.
Marks H.G., Scavina M.T., Kolodny E.H., et al. Krabbe’s disease presenting as a peripheral neuropathy. Muscle Nerve. 1997;20:1024.
Martin G.I., Weintraub M.I. Brachial neuritis and seventh nerve palsy: a rare hazard of DPT vaccination. Clin Pediatr. 1973;12:506.
Martin J.J., Martin L. Infantile neuroaxonal dystrophy: ultrastructural study of the peripheral nerves and of the motor end plates. Eur Neurol. 1972;8:239.
Massion-Verniory L., Dumont E., Potvin A.M. Amyotrophie neurogene avec la retinite pigmentaire. Rev Neurol (Paris). 1946;78:561.
Mazumder D.N., Das Gupta J., Chakraborty A.K., et al. Environmental pollution and chronic arsenicosis in south Calcutta. Bull World Health Organ. 1992;70:481.
McCombe P.A., Pender M.P. Lack of neurological abnormalities in Lewis rats with experimental chronic serum sickness. Clin Exp Neurol. 1991;28:139.
McGonigle R.J., Bewick M., Weston M.J., et al. Progressive, predominantly motor uraemic neuropathy. Acta Neurol Scand. 1985;71:379.
McKhann G.M. Metachromatic leukodystrophy: clinical and enzymatic parameters. Neuropediatrics. 1984;15:4.
McLachlan R.S., Brown W.F. Pyridoxine dependent epilepsy with iatrogenic sensory neuronopathy. Can J Neurol Sci. 1995;22:50.
Merson M., Dowell V.R. Epidemiologic, clinical and laboratory aspects of wound botulism. N Engl J Med. 1973;289:1005.
Middleton-Price H.R., Harding A.E., Monteiro C., et al. Linkage of hereditary motor and sensory neuropathy type I to the pericentromeric region of chromosome 17. Am J Hum Genet. 1990;46:92.
Miike T., Ohtani Y., Nishiyama S., et al. Pathology of skeletal muscle and intramuscular nerves in infantile neuroaxonal dystrophy. Acta Neuropathol. 1986;69:117.
Miller D.P.R., Lloyd J.K. Effect of large oral doses of vitamin E on the neurological sequelae of patients with abetalipoproteinemia. Ann N Y Acad Sci. 1982;393:133.
Miller R.G., Gutmann L., Lewis R.A., et al. Acquired versus familial demyelinative neuropathies in children. Muscle Nerve. 1985;8:205.
Mino M. Vitamin A and E deficiency in children, including the marginal deficiency. Nippon Rinsho. 1993;51:972.
Misra V.P., King R.H., Harding A.E., et al. Peripheral neuropathy in the Chediak-Higaski syndrome. Acta Neuropathol (Berl). 1991;81:354.
Mitz M., Di Benedetto M., Klingbeil G.E., et al. Neuropathy in end-stage renal disease secondary to primary renal disease and diabetes. Arch Phys Med Rehabil. 1984;65:235.
Montal M. Botulinum neurotoxin: a marvel of protein design. Annu Rev Biochem. 2010;79:591.
Montermini L., Richter A., Morgan K., et al. Phenotypic variability in Friedreich ataxia: role of the associated GAA triplet repeat expansion. Ann Neurol. 1997;41:675.
Moretto G., Sparaco M., Monaco S., et al. Cytoskeletal changes and ubiquitin expression in dystrophic axons of Seitelberger’s disease. Clin Neuropathol. 1993;12:34.
Nance P.W., Kirby R.L. Rehabilitation of an adult with disabilities due to congenital sensory neuropathy. Arch Phys Med Rehabil. 1985;66:123.
Nelson K.B., Eng G.D. Congenital hypoplasia of the depressor anguli oris muscle: differentiation from congenital facial palsy. J Pediatr. 1972;81:16.
Nevo Y., Pestronk A., Kornberg A.J., et al. Childhood chronic inflammatory demyelinating neuropathies: clinical course and long-term follow-up. Neurology. 1996;47:98.
Nicholson G.A., Dawkins J.L., Blair I.P., et al. The gene for hereditary sensory neuropathy type I (HSN-I) maps to chromosome 9q22.1-q22.3. Nat Genet. 1996;13:101.
Nicholson G., Nash J. Intermediate nerve conduction velocities define X-linked Charcot-Marie-Tooth neuropathy families. Neurology. 1993;43:2558.
Nishikawa M., Ichiyama T., Hayashi T., et al. Möbius-like syndrome associated with a 1;2 chromosome translocation. Clin Genet. 1997;51:122.
Nishikawa T., Kawakami K., Kumamoto T., et al. Severe neurotoxicities in a case of Charcot-Marie-Tooth disease type 2 caused by vincristine for acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2008;30:519.
Nishimoto Y., Susuki K., Yuki N. Serologic marker of acute motor axonal neuropathy in childhood. Pediatr Neurol. 2008;39:67.
Nordborg C., Conradi N., Sourander P., et al. Hereditary motor and sensory neuropathy of demyelinating and remyelinating type in children: ultrastructural and morphometric studies on sural nerve biopsy specimens from ten sporadic cases. Acta Neuropathol (Berl). 1984;64:1.
Notturno F., Pace M., De Angelis M.V., et al. Susceptibility to chronic inflammatory demyelinating polyradiculoneuropathy is associated to polymorphic GA repeat in the SH2D2A gene. J Neuroimmunol. 2008;197:124.
O’Connor C.R., Rubinow A., Brandwein S., et al. Familial amyloid polyneuropathy: a new kinship of German ancestry. Neurology. 1984;34:1096.
Oh S.J., Halsey J.H. Abnormality in nerve potentials in Friedreich’s ataxia. Neurology. 1973;23:52.
Ohta M., Ellefson R.D., Lambert E.H., et al. Hereditary sensory neuropathy, type II: clinical, electrophysiologic, histologic and biochemical studies of a Quebec kinship. Arch Neurol. 1973;29:23.
Olsen B.S., Nir M., Kjaer I., et al. Elevated vibration perception threshold in young patients with type 1 diabetes in comparison to non-diabetic children and adolescents. Diabet Med. 1994;11:888.
Openshaw H., Slatkin N.E., Stein A.S., et al. Acute polyneuropathy after high dose cytosine arabinoside in patients with leukemia. Cancer. 1996;78:1899.
Ortiz-Corredor F., Peña-Preciado M., Díaz-Ruíz J. Motor recovery after Guillain-Barré syndrome in childhood. Disabil Rehabil. 2007;29:883.
Osterman P.G., Lundemo G., Pirskanem R., et al. Beneficial effects of plasma exchange in acute inflammatory polyradiculoneuropathy. Lancet. 1984;2:1296.
Ouvrier R.A., McLeod J.G.. Chronic peripheral neuropathy in childhood: an overview. Aust Paed J. 1988(Suppl 1):80.
Ouvrier R.A., McLeod J.G., Conchin T.E. The hypertrophic forms of hereditary motor and sensory neuropathy. A study of hypertrophic Charcot-Marie-Tooth disease (HMSN Type I) and Dejerine-Sottas disease (HMSN Type III) in childhood. Brain. 1987;110:121.
Ouvrier R.A., McLeod J.G., Morgan G.J., et al. Hereditary motor and sensory neuropathy of neuronal type with onset in early childhood. J Neurol Sci. 1981;51:181.
Ouvrier R.A., Prineas J., Walsh J.C., et al. Giant axonal neuropathy: a third case. Proc Aust Assoc Neurol. 1974;11:137.
Ouzounian J.G., Korst L.M., Phelan J.P. Permanent Erb palsy: a traction-related injury? Obstet Gynecol. 1997;89:139.
Pace A., Giannarelli D., Galiè E., et al. Vitamin E neuroprotection for cisplatin neuropathy: a randomized, placebo-controlled trial. Neurology. 2010;74:762.
Palau F., De Michele G., Vilchez J.J., et al. Early-onset ataxia with cardiomyopathy and retained tendon reflexes maps to Friedreich’s ataxia locus on chromosome 9q. Ann Neurol. 1995;37:359.
Palix C., Coignet J. Un cas de polyneuropathie périphérique neo-natale par amyélinisation. Pédiatrie. 1978;33:201.
Panayiotopoulos C.P., Lagos G. Tibial nerve H-reflex and F-wave studies in patients with uremic neuropathy. Muscle Nerve. 1980;3:423.
Peiris O.A., Miles D.W. Galvanic stimulation of the tongue as a prognostic index in Bell’s palsy. Br Med J. 1965;2:1162.
Peltier A.C., Russell J.W. Recent advances in drug-induced neuropathies. Curr Opin Neurol. 2002;15:633.
Pena S.D. Giant axonal neuropathy: an inborn error of organization of intermediate filaments. Muscle Nerve. 1982;5:166.
Pettit R.E., Berdal K.G. Chédiak-Higashi syndrome: neurologic appearance. Arch Neurol. 1984;41:1001.
Pfeiffer J., Schlote W., Bischoff A.S., et al. General giant axonal neuropathy. Acta Neuropathol. 1977;40:213.
Piatt J.H. Birth injuries of the brachial plexus. Pediatr Clin North Am. 2004;51:421-440.
Pickett J., Berg B., Chaplin E., et al. Syndrome of botulism in infancy: clinical and electrophysiologic study. N Engl J Med. 1976;295:770.
Pietrini V., Rizzuto N., Vergani C., et al. Neuropathy in Tangier disease: a clinicopathologic study and a review of the literature. Acta Neurol Scand. 1985;72:495.
Pinsky L., DiGeorge A.M. Congenital familial sensory neuropathy with anhidrosis. J Pediatr. 1966;68:1.
Pischik E., Kauppinen R. Neurological manifestations of acute intermittent porphyria. Cell Mol Biol. 2009;55:72.
Poulos A., Sharp P. Plasma and skin fibroblast C26 fatty acids in infantile Refsum’s disease. Neurology. 1984;34:1606.
Poulos A., Sharp P., Fellenberg A.J., et al. Cerebro-hepato-renal (Zellweger) syndrome, adrenoleukodystrophy, and Refsum’s disease: plasma changes and skin fibroblast phytanic acid oxidase. Hum Genet. 1985;70:172.
Poulos A., Sharp P., Whiting M. Infantile Refsum’s disease (phytanic acid storage disease): a variant of Zellweger’s syndrome. Clin Genet. 1984;26:579.
Prineas J.W., Ouvrier R.A., Wright R.G., et al. Giant axonal neuropathy: a generalized disorder of cytoplasmic microfilament formation. J Neuropathol Exp Neurol. 1976;35:458.
Punal J.E., Siebert M.F., Angueira F.B., et al. Three new patients with congenital unilateral facial nerve palsy due to chromosome 22q11 deletion. J Child Neurol. 2001;16:450.
Quant E.C., Jeste S.S., Muni R.H., et al. The benefits of steroids versus steroids plus antivirals for treatment of Bell’s palsy: a meta-analysis. BMJ. 2009;339:3354.
Quinlivan R., Robb S., Hughes R.A., et al. Congenital sensory neuropathy in association with ichthyosis and anterior chamber cleavage syndrome. Neuromusc Disord. 1993;3:217.
Rabie M., Nevo Y. Childhood acute and chronic immune-mediated polyradiculoneuropathies. Eur J Paediatr Neurol. 2009;13:209.
Raeymakers P., Timmerman V., DeJonghe P., et al. Localization of the mutation in an extended family with Charcot-Marie-Tooth neuropathy (HMSNI). Am J Hum Genet. 1989;45:953.
Raeymakers P., Timmerman V., Nelis E., et al. Duplication in chromosome 17p11.2-12 in Charcot-Marie-Tooth neuropathy type 1a (CMT1a). Neuromusc Disord. 1991;1:93.
Rafi M.A., Luzi P., Chen Y.Q., et al. A large deletion together with a point mutation in the GALC gene is a common mutant allele in patients with infantile Krabbe’s disease. Hum Mol Genet. 1995;4:1285.
Ramchandren S., Leonard M., Mody R.J., et al. Peripheral neuropathy in survivors of childhood acute lymphoblastic leukemia. J Peripher Nerv Syst. 2009;14:184.
Rangel-Guerra R., Martínez H.R., Villarreal H.J. Thallium poisoning. Experience with 50 patients. Gac Med Mex. 1990;126:487.
Refsum S.. Heredopathia atactica polyneuritiformis: a familial syndrome not hitherto described: a contribution to the clinical study of the hereditary diseases of the nervous system. Acta Psychiatry Scand. 1946(Suppl 38):1.
Refsum S., Salomonsen L., Skatvedt M. Heredopathia atactica polyneuritiformis in children. J Pediatr. 1949;35:335.
Reinstein L., Pargament J.M., Goodman J.S. Peripheral neuropathy after multiple tetanus toxoid injections. Arch Phys Med Rehabil. 1982;63:332.
Richardson P.G. Peripheral neuropathy following nitrous oxide abuse. Emerg Med Australas. 2010;22:88.
Roach E.S., Abramson J.S., Lawless M.R. Self-injurious behavior in acquired sensory neuropathy. Neuropediatrics. 1985;16:159.
Roberg M., Ernerudh J., Fosberg P., et al. Acute peripheral facial palsy: CSF findings and etiology. Acta Neurol Scand. 1991;83:55.
Rodriguez-Casero M.V., Shield L.K., Kornberg A.J. Subacute inflammatory demyelinating polyneuropathy in children. Neurology. 2005;64:1786.
Román G.C. An epidemic in Cuba of optic neuropathy, sensorineural deafness, peripheral sensory neuropathy and dorsolateral myeloneuropathy. J Neurol Sci. 1994;127:11.
Ropper A.H. Severe acute Guillain-Barré syndrome. Neurology. 1986;36:429.
Ropper A.H., Shahani B.T. Pain in Guillain-Barré syndrome. Arch Neurol. 1984;41:511.
Rosemberg S., Marie S.K., Kliemann S. Congenital insensitivity to pain with anhidrosis (hereditary sensory and autonomic neuropathy type IV). Pediatric Neurol. 1994;11:50.
Ross A.T. Mercuric polyneuropathy with albumino-cytologic dissociation and eosinophilia. JAMA. 1964;188:830.
Rossi L.N., Vassella F., Mumenthaler M. Obstetrical lesions of the brachial plexus. Eur Neurol. 1982;21:1.
Roy E.P.3d, Gutmann L., et al. Longitudinal conduction studies in hereditary motor and sensory neuropathy type 1. Muscle Nerve. 1989;12:52.
Rozear M.P., Pericak-Vance M.A., Fischbeck K., et al. Hereditary motor and sensory neuropathy, X-linked: a half century follow-up. Neurology. 1987;37:1460.
Rubin D.I. Neuralgic amyotrophy: clinical features and diagnostic evaluation. Neurologist.. 2001;7(6):350-356.
Rudnicki S., Vriesendorp F., Koski C.L., et al. Electrophysiologic studies in the Guillain-Barré syndrome: effects of plasma exchange and antibody rebound. Muscle Nerve. 1992;15:57.
Runge P., Muller D.P., McAllister J., et al. Oral vitamin E supplements can prevent the retinopathy of abetalipoproteinaemia. Br J Ophthalmol. 1986;70:166.
Sabin T.D., Thomas T.R. Leprosy. In: Dyck P.J., Thomas P.K., Lambert E.H., et al, editors. Peripheral neuropathy, vol 2. Philadelphia: WB Saunders; 1984. ed 2
Said G., Boudier L., Selva J., et al. Different patterns of uremic polyneuropathy: clinicopathologic study. Neurology. 1983;33:567.
Salman M.S., MacGregor D.L. Should children with Bell’s palsy be treated with corticosteroids? A systematic review. J Child Neurol. 2001;16(8):565-568.
Salt H.B., Wolff O.H., Lloyd J.K., et al. On having no beta-lipoprotein: a syndrome comprising abetalipoproteinaemia, acanthocytosis and steatorrhoea. Lancet. 1960. IIL325
Santoro L., Ragno M., Nucciotti R., et al. Pyridoxine neuropathy. A four-year electrophysiological and clinical follow-up of a severe case. Acta Neurol. 1991;13:13.
Schaumburg H., Kaplan J., Windebank A., et al. Sensory neuropathy from pyridoxine abuse. A new megavitamin syndrome. N Engl J Med. 1983;309:445.
Schecter G.F., Scott C., True L., et al. Linezolid in the treatment of multidrug-resistant tuberculosis. Clin Infect Dis. 2010;50:49.
Scherer S.S., Deschennes S., Fischbeck K., et al. Connexin 32 is expressed by myelinating Schwann cells. St. Paul, Minnesota. Proceedings of the Peripheral Nerve Society Meeting. 1994:36. (abstract)
Scherokman B., Filling-Katz M.R., Tell D. Brachial plexus neuropathy following high-dose cytarabine in acute monoblastic leukemia. Cancer Treat Rep. 1985;69:1005.
Schmitz G., Assmann G., Robenek H., et al. Tangier disease: a disorder of intracellular membrane traffic. Proc Natl Acad Sci USA. 1985;82:6305.
Schwartz J., Landrigan P.J., Feldman R.G., et al. Threshold effect in lead-induced peripheral neuropathy. J Pediatr. 1988;112:12.
Scott K., Zeris S., Kothari M.J. Elevated B6 levels and peripheral neuropathies. Electromyogr Clin Neurophysiol. 2008;48:219.
Seckeler M.D., Linden J. Maternal abetalipoproteinemia resulting in multiple fetal anomalies. Neonatology. 2008;94:310.
Seitz R.J., Wechsler W., Mosny D.S., et al. Hypomyelination neuropathy in a female newborn presenting as arthrogryposis multiplex congenita. Neuropediatrics. 1986;17:1326.
Selimoglu M.A., Esrefoglu M., Gundogdu C., et al. Abetalipoproteinemia: a case report. Turk J Pediatr. 2001;43:243.
Sghirlanzoni A., Pareyson D., Balestrini M.R., et al. HMSN III phenotype due to homozygous expression of a dominant HMSN-II gene. Neurology. 1992;42:2201.
Shapiro W.R., Young D.F. Neurological complications of antineoplastic therapy. Acta Neurol Scand. 1984;100:125.
Shimasaki N., Watanabe K., Hara M., et al. EYA1 mutation in a newborn female presenting with cardiofacial syndrome. Pediatr Cardiol. 2004;25(4):411-413.
Shimono M., Ohta M., Asada M., et al. Infantile neuroaxonal dystrophy: ultrastructural study of peripheral nerve. Acta Neuropathol (Berl). 1976;36:71.
Simmons Z., Wald J.J., Albers J.W. Chronic inflammatory demyelinating polyradiculoneuropathy in children: I. Presentation, electrodiagnostic studies, and initial clinical course, with comparison to adults. Muscle Nerve. 1997;20:1008.
Singhi P., Jain V. Bell’s palsy in children. Semin Pediatr Neurol. 2003;10:289.
Singh R.K., Leshner R.T., Kadom N., et al. Isolated cranial nerve enhancement in metachromatic leukodystrophy. Pediatr Neurol. 2009;40:380.
Skjeldal O.H., Stokke O., Norseth J., et al. Phytanic acid oxidase activity in cultured skin fibroblasts: diagnostic usefulness and limitations. Scand J Clin Lab Invest. 1986;46:283.
Sladky J.T. Guillain-Barré syndrome in children. J Child Neurol. 2004;19:191.
Sladky J.T., Brown M.J., Berman P.H. Chronic inflammatory demyelinating polyneuropathy of infancy: a corticosteroid-responsive disorder. Ann Neurol. 1986;20:76.
Smets K., Zecic A., Willems J. Ergotamine as a possible cause of Möbius sequence: additional clinical observation. J Child Neurol. 2004;19:398.
Smeyne R.J., Klein R., Schnapp A., et al. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994;368:246.
Smith G.D., Hughes R.A.C. Plasma exchange treatment and prognosis of Guillain-Barre syndrome. Q J Med. 1992;85:751.
Sokol R.J. Vitamin E and neurologic deficits. Adv Pediatr. 1990;37:119.
Solders G., Thalme B., Aguirre-Aquino M., et al. Nerve conduction and autonomic nerve function in diabetic children. A 10-year follow-up study. Acta Paediatr. 1997;86:361.
Stacy C.B., Di Roco A., Gould R.J. Methionine in the treatment of nitrous-oxide-induced neuropathy and myeloneuropathy. J Neurol. 1992;239:401.
Stewart H.S., Smith J.C. Two patients with asymmetric crying facies, normal cardiovascular systems and deletion of chromosome 22q11. Clin Dysmorphol. 1997;6:165.
Stollhoff K., Albani M., Goebel H.H. Giant axonal neuropathy and leukodystrophy. Pediatr Neurol. 1991;7:69.
Sullivan F.M., Swan I.R., Donnan P.T., et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med. 2007;357:1598.
Sutherland W.H., Pollock M. Endoneurial adenosine triphosphatase activity in Tangier disease and other peripheral neuropathies. Muscle Nerve. 1984;7:447.
Suzuki K. Biochemical pathogenesis of genetic leukodystrophies: comparison of metachromatic leukodystrophy and globoid cell leukodystrophy (Krabbe’s disease). Neuropediatrics. 1984;15:32.
Swaiman K.F., Flagler D.G. Penicillamine therapy of the Guillain-Barré syndrome caused by mercury poisoning. Neurology. 1971;21:456.
Swanson A.G. Congenital insensitivity to pain with anhidrosis. Arch Neurol. 1963;8:299.
Tada K., Tsuyuguchi Y., Kawai H. Birth palsy: natural recovery course and combined root avulsion. J Pediatr Orthop. 1984;4:279.
Takabe Y., Koide N. Giant axonal neuropathy report of two siblings with endocrinological and histological studies. Neuropediatrics. 1981;12:392-404.
Takakura H., Nakano C., Kasagi S., et al. Multimodality evoked potentials in progression of metachromatic leukodystrophy. Brain Dev. 1985;7:424.
Takebe Y., Koide N., Takahashi G. Giant axonal neuropathy: report of two siblings with endocrinological and histological studies. Neuropediatrics. 1981;12:392.
Tanabe Y., Iai M., Ishii M., et al. The use of magnetic resonance imaging in diagnosing infantile neuroaxonal dystrophy. Neurology. 1993;43:110.
Tarao K., Iwamura K., Fujii K., et al. Japanese adult siblings with Tangier disease and statistical analysis of reported cases. Tokai J Exp Clin Med. 1984;9:379.
Tardieu M., Lacroix C., Neven B., et al. Progressive neurologic dysfunctions 20 years after allogeneic bone marrow transplantation for Chediak-Higashi syndrome. Blood. 2005;106:40.
Taroni F., Botti S., Sghirlanzoni A., et al. PMP22 and MPZ point mutations in Italian families with hereditary neuropathy with liability to pressure palsies (HNPP) and Dejerine-Sottas disease (DSD). Am J Hum Genet. 1996;59:A288. Abstract 1688
Tasdemir H.A., Dilber C., Kanber Y., Uysal S. Intravenous immunoglobulin for Guillain-Barré syndrome: how effective?. J Child Neurol. 2006;21:972.
Tatsumi N., Inui K., Sakai N., et al. Molecular defects in Krabbe’s disease. Hum Mol Genet. 1995;4:1865.
Teot L., Arnal F., Humeau C., et al. Ultrastructural aspects of nerves, bones, and vessels in hereditary sensory neuropathy. J Orthop Res. 1985;3:226.
The Guillain-Barré syndrome study group. Plasmapheresis and acute Guillain-Barré syndrome. Neurology. 1985;35:1096.
Théaudin M., Couvert P., Fournier E., et al. Lewis–Sumner syndrome and tangier disease. Arch Neurol. 2008;65:968.
Thomas C., Love S., Powell H.C., et al. Giant axonal neuropathy: correlation of clinical findings with postmortem neuropathology. Ann Neurol. 1987;22:79.
Thomas P.K., Calne D.B., Stewart G. Hereditary motor and sensory polyneuropathy (peroneal muscular atrophy). Ann Hum Genet. 1974;38:111.
Thompson J.A., Filloux F.M., Van Orman C.B., et al. Infant botulism in the age of botulism immune globulin. Neurology. 2005;64:2029.
Thompson J.A., Glasgow L.A., Warpinski J.R., et al. Infant botulism: clinical spectrum and epidemiology. Pediatrics. 1980;66:936.
Tonkin M.A., Eckersley J.R., Gschwind C.R. The surgical treatment of brachial plexus injuries. Aust N Z J Surg. 1996;66:29.
Treiber-Held S., Budjarjo-Welim H., Riemann D., et al. Giant axonal neuropathy: a generalized disorder of intermediate filaments with longitudinal groves in the hair. Neuropediatrics. 1994;25:89.
Tyson J., Ellis D., Fairbrother U., et al. Hereditary demyelinating neuropathy of infancy. A genetically complex syndrome. Brain. 1997;120:47.
Uchino M., Uyama E., Hirano T., et al. A histochemical and electron microscopic study of skeletal muscle in an adult case of Chédiak-Higashi syndrome. Acta Neuropathol (Berl). 1993;86:521.
Ueda M., Kawamura N., Tateishi T., et al. Phenotypic spectrum of hereditary neuralgic amyotrophy caused by the SEPT9 R88W mutation. J Neurol Neurosurg Psychiatry. 2010;81:94.
Uncini A., Parano E., Lange D.J., et al. Chronic inflammatory demyelinating polyneuropathy in childhood: clinical and electrophysiological features. Childs Nerv Syst. 1991;7:191.
Uncini A., Yuki N. Electrophysiologic and immunopathologic correlates in Guillain-Barré syndrome subtypes. Expert Rev Neurother. 2009;9:67.
Valentijn L.J., Ouvrier R.A., van den Bosch N.H., et al. Dejerine-Sottas neuropathy is associated with a de novo PMP22 mutation. Hum Mutat. 1995;5:76.
Vanasse M., Michaud J. Congenital axonal neuropathy. Pediatr Neurol. 1992;8:404. (abstract)
Vance J.M., Nicholson G.A., Yamaoka L.H., et al. Linkage of Charcot-Marie-Tooth neuropathy type 1a to chromosome 17. Exp Neurol. 1989;104:186.
Van den Branden C., Vamacq J., Wybo I., et al. Phytol and peroxisome proliferation. Pediatr Res. 1986;20:411.
van der Meché F.G.A., Schmitz P.I.M., Dutch Guillain-Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med. 1992;326:1123.
van Loon M., Postels D.G., Heikens G.T., et al. Severe pernicious anaemia in an 8-year-old African girl. Ann Trop Paediatr. 2009;29:231.
Vassalla F., Emrich H.M., Kraus-Ruppert R., et al. Congenital sensory neuropathy with anhidrosis. Arch Dis Child. 1968;43:124.
Venkatesh S., Coulter D.L., Kemper T.D. Neuroaxonal dystrophy at birth with hypertonicity and basal ganglia mineralization. J Child Neurol. 1994;9:74.
Verhoeven K., Claeys, Kristl G., et al. MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain. 2006;129:2093-2102.
Verhoeven K., De Jonghe P., Van de Putte T., et al. Slowed Conduction and thin myelination of peripheral nerves associated with mutant rho Guanine-nucleotide exchange factor 10. Am J Hum Genet. 2003;73:926-932.
Verrotti A., Chiarelli F., Blasetti A., et al. Autonomic neuropathy in diabetic children. J Paediatr Child Health. 1995;31:545.
Vucic S., Kiernan M.C., Cornblath D.R. Guillain-Barré syndrome: an update. J Clin Neurosci. 2009;16:733.
Wakai S., Asanuma H., Tachi N., et al. Infantile neuroaxonal dystrophy: axonal changes in biopsied muscle tissue. Pediatr Neurol. 1993;9:309.
Warner L.E., Hilz M.J., Appel S.H., et al. Clinical phenotypes of different MPZ (Po) mutations may include Charcot-Marie-Tooth type IB, Dejerine-Sottas, and congenital hypomyelination. Neuron. 1996;17:451.
Wassif W.S., Deacon A.C., Floderus Y., et al. Acute intermittent porphyria: diagnostic conundrums. Eur J Clin Chem Clin Biochem. 1994;32:915.
Weleber R.G., Tongue A.C., Kennaway N.G., et al. Ophthalmic manifestations of infantile phytanic acid storage disease. Arch Ophthalmol. 1984;102:1317.
Whitaker J.N., Falchuck Z.M., Engel W.K., et al. Hereditary sensory neuropathy: association with increased synthesis of immunoglobulin A. Arch Neurol. 1974;30:359.
Wichman A., Buchthal F., Pezeshkpour G.H., et al. Peripheral neuropathy in abetalipoproteinemia. Neurology. 1985;35:1279.
Wiederholt W.C. Hereditary brachial neuropathy: report of two families. Arch Neurol. 1974;30:252.
Wilmshurst J.M., Macleod M.J., Hughes E., et al. Acute sensory neuropathy in an adolescent girl following BCG vaccination. Eur J Paediatr Neurol. 1999;3:277.
Wilmshurst J.M., Surtees R., Cox T., Robinson R.O. Cerebellar ataxia, anterior horn cell disease, learning difficulties, and dystonia: a new syndrome. Dev Med Child Neurol. 2000;42:775.
Windebank A.J., Schenone A., Dewald G.W. Hereditary neuropathy with liability to pressure palsies and inherited brachial plexus neuropathy: two genetically distinct disorders. Mayo Clin Proc. 1995;70:743.
Winer J.B., Hughes R.A., Greenwood R.J., et al. Prognosis in Guillain-Barré syndrome. Lancet. 1985;1:1202.
Wolfe D.E., Schindler D., Desnick R.J. Neuroaxonal dystrophy in infantile alpha-N-acetylgalactosaminidase deficiency. J Neurol Sci. 1995;132:44.
Wolf S.M., Wagner J.H.Jr, Davidson S., et al. Treatment of Bell palsy with prednisone: a prospective randomized study. Neurology. 1978;28:158.
Wulff C.H., Trojaborg W. Adult metachromatic leukodystrophy: neurophysiologic findings. Neurology. 1985;35:1776.
Yamada M., Konodo M., Tanaka M., et al. An autopsy case of acute porphyria with a decrease of both uroporphyrinogen I synthetase and ferrochelatase activities. Acta Neuropathol (Berl). 1984;64:6.
Yatzidis H., Koutsicos D., Agroyannis B., et al. Biotin in the management of uremic neurologic disorders. Nephron. 1984;36:183.
Yetiser S., Kazkayas M., Altinok D., et al. Magnetic resonance imaging of the intratemporal facial nerve in idiopathic peripheral facial palsy. Clin Imaging. 2003;27(2):77-81.
Yorifuji T., Kashima S., Tsuda T., et al. What has methylmercury in umbilical cords told us? – Minamata disease. Sci Total Environ. 2009;408:272.
Zampollo A., Baruffini A., Cirla A.M., et al. Subclinical inorganic mercury neuropathy: neurophysiological investigations in 17 occupationally exposed subjects. Ital J Neurol Sci. 1987;8:249.
Züchner S., Mersiyanova I.V., Muglia M. Mutations in the mitochondrial gtpase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449-451.
