[level-membership-for-neurology-category]
Chapter 59 Antiepileptic Drug Therapy in Children
Rational management of antiepileptic drug therapy in children requires an understanding of pharmacokinetics, pharmacodynamics, and toxicology of these agents. Pharmacokinetics is the study of drug absorption, distribution, metabolism, and elimination: that is, what the body does to a drug. Pharmacodynamics is the study of a drug’s biochemical and physiologic effects: that is, what the drug does to the body. Children, much more than adults, have widely varying abilities to absorb, distribute, metabolize, and eliminate drugs [Rane and Wilson, 1976]. Evidence also exists that antiepileptic drug pharmacodynamics in children differ from those in adults [Henriksen, 1988]. Application of basic pharmacokinetic and pharmacodynamic principles to antiepileptic drug therapy facilitates the attainment and maintenance of targeted serum concentrations, clinical response, control of drug interactions, and optimization of clinical response.
Pharmacokinetic Principles
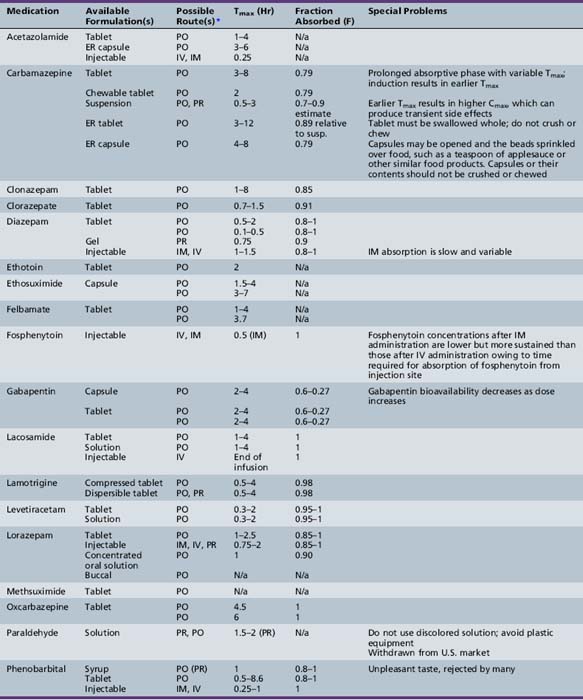
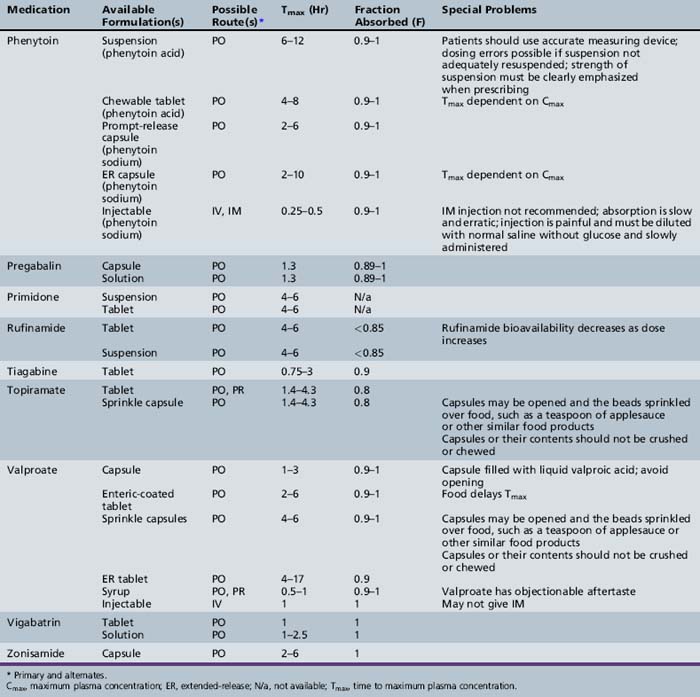
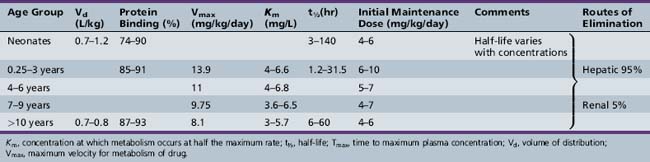
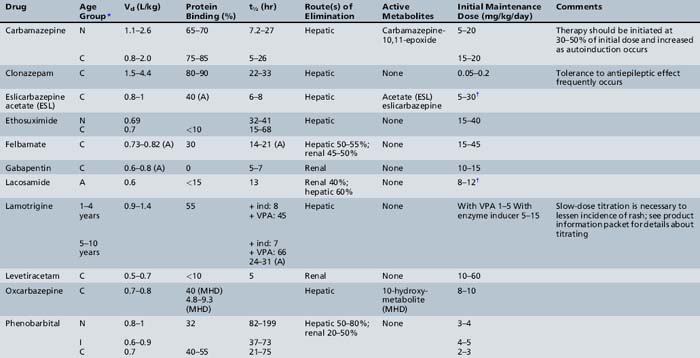
This chapter will review general pharmacokinetic principles and discuss the clinically relevant features of specific antiepileptic drugs. For quick reference, pharmacokinetic information for the antiepileptic drugs is summarized in tables, with absorption characteristics provided in Table 59-1, and volume of distribution, protein binding, half-life, and metabolites presented in Table 59-2 and Table 59-3.
Absorption
Absorption is a complex phenomenon controlled by the physicochemical properties of the drug, the formulation, and the physiologic conditions at the sites of absorption: stomach, small intestine, colon, rectum, buccal and nasal mucosa, and muscle. Most drugs are given orally as solid formulations, which must first disintegrate into small particles that are then dissolved in the gastrointestinal fluid. Only after a drug is dissolved is it available for absorption. Most drugs are absorbed by passive diffusion of the unionized form but some drugs use active transport mechanisms. Gastrointestinal absorption is influenced by age-dependent changes in physiologic variables, such as gastric emptying time, stomach and intestinal pH, absorptive area, and gastrointestinal transit time. Food intake may affect antiepileptic drug pharmacokinetics, especially absorption. Although there is concern that the ketogenic diet may alter dosing requirements, a recent study indicated that there are no significant changes in the concentrations of several antiepileptic drugs (valproate – free or total, topiramate, phenobarbital, lamotrigine, clonazepam) during the ketogenic diet [Dahlin et al., 2006].
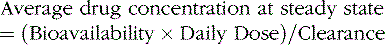
Descriptors of absorption, such as Cmax and Tmax, and the bioavailability, may vary considerably among different formulations of the same drug. The relative rates of absorption, and thus Tmax and Cmax, adhere to the following order of most rapid to slowest, and highest peak concentration to lowest: solutions > suspensions > capsules > tablets > extended-release formulations [Garnett and Cloyd, 1993]. Formulation-related differences in absorption characteristics are greatly influenced by the composition of the inactive ingredients (e.g., filler, particle size of drug) and nature of the capsule or tablet coating).
Volume of Distribution
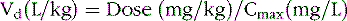
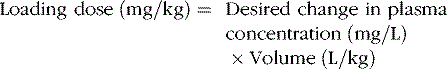
The change in plasma concentration term denotes the desired increase after administration of the loading dose. For example, if a patient has a plasma concentration of 5 mg/L and the target concentration is 20 mg/L, the desired change in concentration is 15 mg/L. For phenytoin, the Vd is approximately 0.75 L/kg, so an IV dose of 11.25 mg/kg is required. When loading doses are administered intramuscularly, orally, or rectally, resultant maximum plasma concentrations are usually less than after IV loading, owing to slower and incomplete absorption and the elimination of some drug during the absorption phase. Table 59-4 presents loading dose information for six major antiepileptic drugs.
Protein Binding
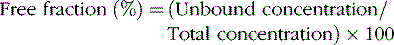
In general, only the unbound drug is able to cross the blood–brain barrier and is in equilibrium with brain and spinal fluid (Figure 59-1; see also Table 59-3). Consequently, clinical response and toxicity correlate better with the unbound drug concentration than with the total drug concentration. For drugs that have limited protein binding (less than 50 percent) or a constant ratio of unbound to total concentration, measurement of total plasma concentrations is a reliable indicator of response. However, if a drug is highly protein-bound (greater than 85 percent), measurements of total concentrations may be an unreliable guide to clinical outcomes. The fraction bound may change because of illness, age, or other drugs that compete for binding sites. In these circumstances, it may be necessary to measure the free concentration to understand the clinical effect better. Low-extraction drugs are those that have only a small portion of the drug metabolized by each pass through the liver. Most of the antiepileptic drugs are low-extraction drugs and alterations in protein binding do not affect the steady-state concentration of the unbound drug. Thus, adjustments in dose generally are not necessary when protein binding is altered because the unbound concentrations remain unchanged. Erroneously increasing the dose when total levels are low, but unbound levels have not changed, will likely result in toxicity.
Metabolism and Elimination
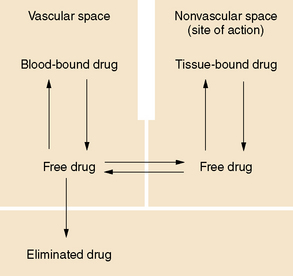
Clearance is the volume of blood from which drug is removed (cleared) per unit of time. It is identical conceptually and mathematically to the more familiar physiologic parameter of creatinine clearance. For antiepileptic drugs that are eliminated by the liver, clearance is determined by the free concentration of the drug and the intrinsic ability of hepatic enzymes to metabolize the unbound drug. Although, for most drugs, clearance is proportional to dose, some drugs exhibit nonlinear pharmacokinetics in which clearance changes with concentration (Figure 59-2).
The following points are important to remember:
 Clearance and absorption determine the amount of drug a patient requires to maintain a targeted steady-state plasma concentration.
Clearance and absorption determine the amount of drug a patient requires to maintain a targeted steady-state plasma concentration.



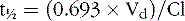
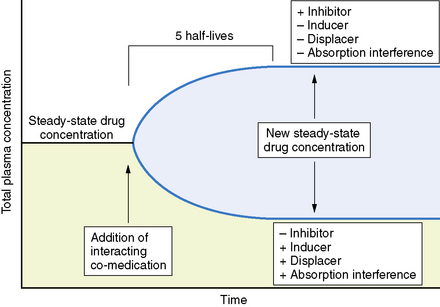
Thus, t½ is not a direct measure of Cl, but is useful in calculating the time to reach steady state or the time required for drug elimination. After initiation or modification of therapy or after any change in clearance, 5 half-lives must elapse for a new steady-state plasma concentration to be reached (see Figure 59-2). The time required to attain steady state can be explained by using the definition of t½. The plasma concentration in the body declines 50 percent in 1 t½; therefore, the percentage of drug remaining in the body, as indicated by the plasma concentration after 2, 3, 4, and 5 half-lives, is 75 percent, 87.5 percent, 93.75 percent, and 96.875 percent, respectively. The actual amount of drug remaining in the body and the additional amount of drug removed in subsequent half-lives are so small (after 5 half-lives) that, for practical purposes, it can be ignored. However, the rate of decline is most rapid in the first two half-lives, and the risk of seizures is greatest during this period.
Biotransformation (metabolism) of medications is accomplished through biochemical reactions. These reactions create compounds that are more polar (more water-soluble) and are eliminated from the body, usually through the kidneys. Phase I reactions (e.g., using the cytochrome P-450 system) involve oxidation and reduction. Phase II reactions (non-cytochrome P-450-mediated) couple the drug or its metabolite with endogenous substrates by conjugation or synthesis. The endogenous substrate may be glucuronic acid, glutathione, glucose, sulfate, or acetate, making the drug more polar and hence water-soluble [Capparelli, 1994]. Antiepileptic drugs may undergo metabolism by phase I or II reactions, or a combination of both. As persons mature, the pathways may change and the metabolite profile will be altered.
In the neonate, phase I reactions, such as hydroxylation and, to a lesser extent, dealkylation, are significantly depressed [Anderson and Lynn, 2009]. In older children, however, phase I activity generally exceeds that in adolescents or adults. As a general rule, the rate of drug metabolism reaches a peak at 0.5–2 years of age and then declines in an exponential manner, reaching adult values in early adolescence. As an example, the clearance of valproic acid, in which metabolism includes both phase I and phase II reactions, is fastest in young children and then declines to reach adult values at the age of approximately 12–15 years. Also, its metabolism shifts from phase II to phase I reactions.
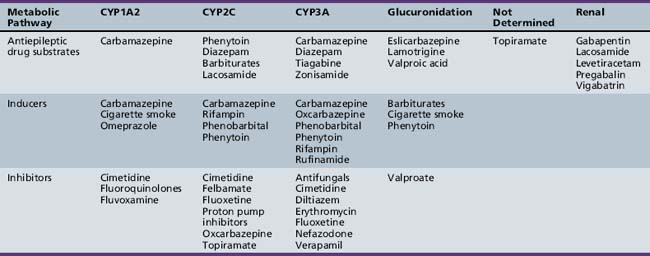
Active metabolites formed after biotransformation of the parent compound can contribute significantly to desired and toxic effects of the parent drug. Metabolites exist in both bound and unbound states. Only the unbound metabolite is available for elimination or biotransformation to an inactive metabolite. As with unbound parent drug, the unbound metabolite concentration in the plasma most closely correlates with the concentration of metabolite at the site of action. The presence of active metabolites complicates interpretation of plasma drug concentrations and clinical response. As with the parent drug, the relationship between unbound and total metabolite concentrations can be disrupted by displacement from serum proteins or changes in metabolism. In fact, parent or metabolite concentrations may be altered independently by drug interaction or physiologic change in metabolism. Table 59-5 displays the cytochrome P-450 isoform families involved in antiepileptic drug metabolism, along with known inducers and inhibitors.
Renal elimination is accomplished by glomerular filtration and/or renal tubular secretion. Glomerular filtration rate and renal tubular function in newborns are less efficient than in adults [Linday, 1994]. After 1 year of age, glomerular filtration rate, corrected for body surface area, is the same as for older children and young adults. Renal tubular function reaches adult values, normalized to body surface areas, within several months. Decreases in clearance can be better correlated with sexual maturity than with chronologic age. Making correlations with physiologic or sexual maturation and developmental age, rather than chronologic age, may be helpful in accounting for the variability observed in clearance of drugs that are renally eliminated [Finkelstein, 1994].
Specific Antiepileptic Drugs
Benzodiazepines
The benzodiazepines rapidly cross the blood–brain barrier. Brain concentrations reach equilibrium with plasma concentrations within seconds after IV administration. But the highly lipid-soluble nature of diazepam and lorazepam causes them to redistribute to other body tissues, resulting in a rapid decrease in brain concentrations and a very high Vd. This redistribution is responsible for the short duration of action after a single dose [Greenblatt et al., 1989; Leppik et al., 1983]. The benzodiazepines are metabolized by the liver and have linear elimination kinetics. Some of the metabolites exhibit antiepileptic activity and also may contribute to toxicity. Because the active metabolites have very long half-lives, repeated administration of these drugs, culminating in large total doses, may lead to long periods of sedation, usually most problematic for diazepam and chlorazepate.
Carbamates
Carbamazepine
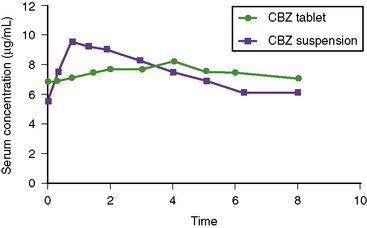
Carbamazepine is a very water-insoluble, neutral compound with a slow dissolution rate in gastrointestinal fluid. The relative bioavailability is similar for all four formulations (i.e., suspension, chewable tablet, extended-release, and regular tablet), ranging from 75 to 90 percent, although marked intrapatient and interpatient variability has been observed [Faigle and Feldmann, 1975; Maas et al., 1987]. In children on maintenance therapy, the rate of absorption is much faster for the suspension than for tablet formulations, resulting in a significantly earlier Tmax and higher Cmax (Figure 59-3). This property occasionally produces transient concentration-dependent central nervous system side effects. Administering the same daily dose in smaller amounts and giving it more frequently may correct this problem. Sustained-release preparations may permit twice-a-day dosing, eliminating the need for administration of medications during school hours. This improves medication compliance and decreases central nervous system side effects because peak concentrations will be lower. Studies conducted by the U.S. Food and Drug Administration have demonstrated that carbamazepine tablets are significantly less well absorbed after being exposed to high humidity [Meyer et al., 1992; Wang et al., 1993]. These findings emphasize the importance of storing all antiepileptic drugs at room temperature in a dry location.
The Vd in children for carbamazepine based on oral preparations ranges from 0.8 to 2.0 L/kg. Both carbamazepine and its active metabolite, carbamazepine-10,11-epoxide, are bound to several plasma proteins [Eichelbaum et al., 1975; Rawlins et al., 1975]. Approximately 65–85 percent of carbamazepine and 30–60 percent of carbamazepine-10,11-epoxide are bound to albumin. Both also bind to the reactant protein α1-acid glycoprotein. Binding to α1-acid glycoprotein increases after physiologic stress, such as from burn injuries or inflammatory diseases (e.g., juvenile rheumatoid arthritis). Increased binding to α1-acid glycoprotein can increase total carbamazepine and carbamazepine-10,11-epoxide concentrations without necessarily increasing the unbound drug concentrations. In such situations, patients may have total concentrations above the desired range but have no side effects. Adjusting dosage is not necessary; plasma concentrations return to baseline as α1-acid glycoprotein declines.
Carbamazepine is eliminated primarily by phase I cytochrome P-450-mediated metabolism (see Table 59-5), forming an active metabolite (10,11-epoxide). Carbamazepine displays dose-dependent elimination pharmacokinetics, in which dosage increases produce a less than proportionate increase in steady-state total concentration (see Figure 59-2). Carbamazepine metabolism undergoes autoinduction, in which clearance increases over time after exposure to the drug. Within 30 days after beginning therapy, carbamazepine clearance may increase by as much as 300 percent. Further increases in maintenance doses may result in further induction [Kudriakova et al., 1992]. In some children, induction causes plasma carbamazepine concentrations to remain unchanged or even decline, despite increasing doses up to 40–50 mg/kg per day [Curatolo et al., 1988]. Carbamazepine and carbamazepine-10,11-epoxide metabolism also can be induced by phenytoin or phenobarbital, resulting in as much as a 100 percent increase in clearance [Kerr and Levy, 1989]. Initial carbamazepine t½ ranges from 10 to 20 hours, but the t½ decreases because of autoinduction to 4–12 hours. Consequently, many children require three or four doses a day to maintain targeted plasma concentrations. Sustained-release preparations help overcome this problem.
Oxcarbazepine
Oxcarbazepine is rapidly absorbed, with a Tmax of less than 1 hour after a single oral dose. Absorption is nearly 100 percent. It is a prodrug that is converted extensively to 10,11-dihydro-10-hydroxy-carbamazepine (the monohydroxy derivative), which is the active compound. Less than 2 percent of the area under the concentration–time curve is attributed to the parent compound, and approximately 70 percent to the monohydroxy derivative. Only the monohydroxy derivative is measured for therapeutic monitoring [Theisohn and Heimann, 1982]. Co-administration of oxcarbazepine and food leads to an increase in the bioavailability and maximum plasma concentration; however, Tmax remains unchanged. The slight changes in the absorption parameters with food administration do not appear to be clinically significant [Degen et al., 1994].
The major advantage of oxcarbazepine is that it is not metabolized to the active epoxide metabolite, which is believed to contribute to the side-effect profile of carbamazepine. Both the parent drug and its metabolite are lipophilic, which facilitates their movement across the blood–brain barrier. The monohydroxy derivative has a volume of distribution of 0.7–0.8 L/kg [Feldmann et al., 1977; Theisohn and Heimann, 1982], and both oxcarbazepine and the monohydoxy derivative bind to plasma proteins at approximately 60 percent and 40 percent, respectively [Klitgaard and Kristensen, 1986; Patsalos et al., 1990].
Approximately 96 percent of an oral dose is eliminated in the urine, with less than 1 percent of oxcarbazepine being excreted unchanged. The greater part of the dose is conjugated and excreted as the glucuronide (9 percent oxcarbazepine glucuronide and 49 percent monohydroxylated glucuronide) [Lloyd et al., 1994; Schutz et al., 1986]. Unlike carbamazepine, oxcarbazepine does not appear to induce its own metabolism [Larkin et al., 1991].
Eslicarbazepine
Eslicarbazepine acetate is a voltage-gated channel blocker that was approved in Europe in 2009. Approval in the US is still pending. Its chemical structure is similar to both carbamazepine and oxcarbazepine [Benes et al., 1999]. It differs from carbamazepine in that it does not produce an epoxide metabolite or exhibit autoinduction. Eslicarbazepine acetate is a prodrug that undergoes first-pass hydrolytic metabolism to cleave the acetate group to form the active entity – eslicarbazepine, which is the S(+) enantiomer of licarbazepine (also known as the S(+)10-monohydroxy metabolite of oxcarbazepine) [Almeida et al., 2008b]. It appears to exhibit linear pharmacokinetics and is metabolized to a glucuronidated metabolite via several uridine diphosphate glucuronosyltransferases [Loureiro and Fernandes-Lopes, 2008]. One study in 29 children and adolescents (ages 2–17 years) shows dose-proportional pharmacokinetics [Almeida et al., 2008a]. Both a suspension and scored tablets of strengths 200 mg, 400 mg, 600 mg, and 800 mg were available in the study. The daily doses used in the study ranged from 5 to 30 mg/kg/day. The Tmax was between 0.5 and 3 hours. Eslicarbazepine acetate was cleared faster in younger children as compared to adolescents, although Cmax was similar among all age groups.
Felbamate
Approximately 95 percent of a dose of felbamate is absorbed in adults after oral administration. Absorption of the drug is fairly rapid, with Tmax values ranging from 1 to 4 hours [Wilensky et al., 1985]. The Vd of felbamate is approximately 0.8 L/kg [Devinsky et al., 1994; Wilensky et al., 1985]. Distribution of felbamate is fairly uniform throughout the entire body. It binds mainly to albumin (22–25 percent), and the amount of binding is dependent on albumin concentration [Perhach and Shumaker, 1995].
About 90 percent of felbamate is eliminated in the urine. Approximately 40–50 percent of the absorbed dose is eliminated as the unconjugated parent compound, and up to 58 percent is found as metabolites and conjugates [Shumaker et al., 1990]. Felbamate is cleared at a faster rate in children. Population pharmacokinetic analysis of data from 139 pediatric patients gives clearance values approximately 40 percent higher in younger children (2–12 years of age) than those in older children and adults (13–65 years).
Gamma-Aminobutyric Acid Analogues
Gabapentin
The absolute bioavailability after oral administration of gabapentin is 60 percent [Vollmer et al., 1986]. Absorption is linear up to daily doses of 1800 mg, after which bioavailability begins to decline, decreasing to 35 percent at daily doses of 4800 mg [Richens, 1993]. Gabapentin uses the l-amino acid transporter system in the gut to enter the body. This system is saturable and the dependence of gabepentin on it may explain the decrease in bioavailability with increasing doses above 1800 mg [Stewart et al., 1993].
Gabapentin has a Vd of approximately 0.6–0.8 L/kg [Richens, 1993] and does not bind to plasma proteins [Vollmer et al., 1986]. To enter the brain, gabapentin uses the l-amino acid transporter system, which is also saturable [Luer et al., 1999].
Gabapentin is eliminated renally. Clearance in children aged 1 month to 12 years was well correlated with creatinine clearance. Clearance was higher in children younger than 5 years of age, necessitating a 30 percent higher dose to attain equivalent concentrations as observed in children age 5 and older and in adults [Haig et al., 2001; Ouellet et al., 2001]. It does not have any significant pharmacokinetic interactions.
Pregabalin
Pregabalin is rapidly absorbed. Its bioavailability is greater than 90 percent. Unlike gabapentin, pregabalin demonstrates no changes in absorption as dose increases. Pregabalin does not bind to plasma proteins and is not metabolized in humans, with 98 percent of the dose recovered unchanged in urine [Brockbrader et al., 2000]. Like gabapentin, it does not appear to interact with other antiepileptic drugs. There were no changes in trough steady-state concentrations of concomitant antiepileptic drugs (phenytoin, lamotrigine, or valproic acid) when pregabalin was co-administered. Likewise, there were no changes in pregabalin concentrations after administration of carbamazepine, phenytoin, lamotrigine, or valproic acid [Brodie et al., 2005]. It should be noted that there did appear to be a decrease in pregabalin exposure when it was given with phenytoin; however, this was thought to be due to a possible food effect with pregabalin. Because pregabalin is eliminated mostly via the kidneys, doses may need to be adjusted in children with decreased renal function.
Hydantoins
Phenytoin
Phenytoin is a poorly water-soluble compound and is often formulated as a sodium salt. It is available as a suspension, chewable tablet, capsule, and a parenteral formulation. The nonlinear, saturation pharmacokinetics of phenytoin can make even small differences in dose clinically significant. The chewable tablet and the suspension contain phenytoin acid, whereas the capsules and the IV formulation contain phenytoin sodium, which is equivalent to only 92 percent of the acid (see Table 59-1). Thus, two 50-mg chewable tablets deliver 8 percent more phenytoin than a 100-mg phenytoin sodium capsule. The suspension is available in a 125-mg/5-mL strength. In the past, the reliability of the suspension has been questioned owing to its tendency to settle. It has been demonstrated that some agitation is sufficient to permit delivery of the specified amount of drug [Sarkar et al., 1989]. Unexpected fluctuations in plasma phenytoin concentrations associated with the use of suspensions likely have been the result of pharmacokinetics rather than formulation problems. Intramuscular injection of the parenteral solution (with a pH of 11) is contraindicated because it results in precipitation of drug that is then slowly and erratically absorbed [Kostenbauder et al., 1975]. It is also painful and causes tissue necrosis [Serrano and Wilder, 1974]. Phenytoin capsules are available in both prompt-release and extended-release formulations. Patient and family education about the differences in phenytoin dosage forms is important to maintain optimal therapy.
Bioavailability, Tmax, and Cmax are altered when large amounts of phenytoin are given as a single oral loading dose, presumably because of the formation of a multicapsule concretion. In a study involving six healthy adults, absorption was reduced by 10–15 percent and Tmax increased from 18 to 32 hours, when phenytoin sodium capsules were given in single dose of 10 mg/kg or more [Jung et al., 1980]. Dividing the total loading dose into 5-mg/kg doses given every 2–3 hours overcomes this problem. The most rapid way to attain therapeutic plasma concentrations after oral loading is to use prompt-release phenytoin sodium capsules or suspension [Goff et al., 1984].
Some reports have suggested that phenytoin is poorly absorbed in newborns because of irregular gastric emptying time, relatively high gastric pH (greater than 4), or decreased absorptive surface [Morrow and Richens, 1989; Morselli, 1983; Painter et al., 1978]. Other evidence indicates that formulation-related differences in absorption, nonlinear pharmacokinetics, and age-dependent changes in elimination account for much of the variability in plasma concentrations [Dodson, 1984].
Phenytoin Vd in children and adolescents is approximately 0.7 ± 0.1 L/kg [Koren et al., 1984]. The relatively small variability in phenytoin Vd permits precise calculation of loading doses. For example, a 15-mg/kg loading dose given intravenously will result in a postinfusion mean concentration of 20 mg/L, with a range of 15–25 mg/L. Phenytoin is also highly protein-bound to albumin.
Phenytoin undergoes extensive hepatic metabolism. A small fraction, 5 percent or less, is excreted unchanged [Battino et al., 1995]. It is metabolized by a saturable cytochrome P-450 enzymatic pathway. Phenytoin follows Michaelis–Menten or saturable elimination kinetics (see Figure 59-2). As the plasma concentration rises, phenytoin clearance decreases, owing to saturation of enzymatic metabolism. Therefore, increases in dose result in disproportionately greater increases in steady-state plasma concentration.
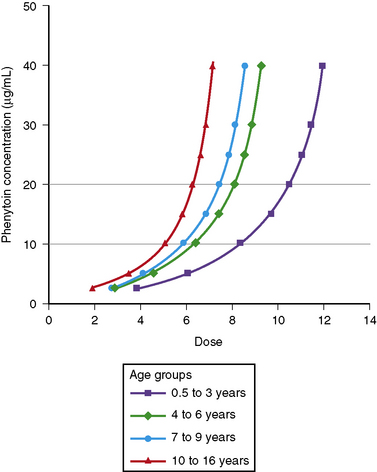
Phenytoin metabolism in neonates is reduced compared with that in older children. Within days to weeks after birth, the rate of metabolism accelerates, reaching a maximum in the 0.5- to 3-year age range. Children in this age group require the highest mg/kg daily doses (Figure 59-4). Thereafter, metabolism and dosage requirements decline gradually, reaching adult values during adolescence. Figure 59-5 illustrates that even minor alterations in dosage produce disproportionately large changes in plasma concentration. Therefore, small adjustments in daily dose are indicated as the plasma concentration approaches or exceeds Km, which usually falls in the range of 5–10 mg/L. Use of the scored 50-mg chewable tablet, the 30-mg phenytoin sodium capsule, or the suspension facilitates making such adjustments in dose.
Fosphenytoin
Fosphenytoin is a prodrug of phenytoin that is rapidly hydrolyzed by tissue and red blood cell alkaline phosphatases to phenytoin. Due to the rapid conversion to phenytoin, the pharmacokinetic information for phenytoin and not fosphenytoin is what is clinically relevent. Fosphenytoin is water-soluble and is formulated as a nontoxic, parenteral solution. The pharmacokinetics of phenytoin derived from fosphenytoin are identical to phenytoin given directly [Leppik et al., 1989]. Fosphenytoin is packaged as milligram phenytoin equivalents and is well tolerated when given intramuscularly.
Fosphenytoin absorption, not its conversion to phenytoin, appears to be the rate-limiting step in achieving therapeutic phenytoin levels. The mean half-life of fosphenytoin is 8 minutes after IV administration [Donn et al., 1987; Gerber et al., 1988; Leppik et al., 1989] and 33 minutes after intramuscular injection [Leppik et al., 1990]. Age does not affect the phosphatase enzyme responsible for the conversion of fosphenytoin to phenytoin. Fosphenytoin is more avidly bound to albumin, compared with phenytoin, resulting in brief increases of free phenytoin concentrations after infusion [Hussey et al., 1990; Jamerson et al., 1990]. The observed decrease in total phenytoin concentrations returns to those expected when the conversion from fosphenytoin to phenytoin is completed; therefore, monitoring of plasma concentrations should be done about 1 hour after IV administration of fosphenytoin. Unlike phenytoin, fosphenytoin can be given intramuscularly and avoids serious tissue necrosis from extravasation of IV phenytoin. Its major uses are treatment of status epilepticus and maintenance therapy when oral administration is not possible.
Lacosamide
Lacosamide (known as harkoside in earlier studies) was approved for use in the United States during 2009. It is completely and rapidly absorbed with a bioavailability of 100 percent [Hovinga, 2003]. Lacosamide appears to follow linear pharmacokinetics and its Cmax is between 1 and 4 hours after an oral dose [Hovinga, 2003]. There does not seem to be a food effect on lacosamide pharmacokinetics [Kellinghaus, 2009]. Protein binding has been shown to be less than 15 percent. Lacosamide is metabolized to an inactive desmethyl metabolite, which is then cleared along with the parent drug via the kidney. Lacosamide is not approved for use in young children at this time; however, there is at least one clinical trial that is investigating the safety and pharmacokinetics in children with partial seizures between the ages of 2 and 17 years. Doses being studied are 8–12 mg/kg/day.
Lamotrigine
Lamotrigine has a bioavailability approaching 100 percent. Its Tmax occurs 2–3 hours after administration of a single oral dose. With doses in the range of 15–240 mg, Tmax and area under the concentration–time curve are directly proportional to dose [Yuen and Peck, 1987]. The Vd of lamotrigine is approximately 1.5 L/kg in children [Chen et al., 1999]. It is distributed uniformly throughout the body, and approximately 55 percent of lamotrigine is bound to plasma proteins [Garnett, 1997].
Lamotrigine is extensively metabolized to an inactive glucuronide by an inducible uridine diphosphate (UDP) glucuronyltransferase reaction [Lamictal, 2004]. Clearance is greater in younger children [Battino et al., 2001]. Infants (younger than 2 months of age) metabolize lamotrigine more slowly, and metabolic rates increase during the first year of life [Mikati et al., 2002]. Estrogen-containing oral contraceptives may cause dramatic cyclical fluctuations of blood levels (decreasing during the estrogen phase and rebounding in the placebo week), and levels decrease dramatically during pregnancy [Pennell et al., 2008]. There may be an impact of age on lamotrigine concentrations [Reimers et al., 2007]. However, data for weight were not available in a majority of the patients and the effect of age could be due to the fact that older children weigh more than younger children [Reimers et al., 2007].
Levetiracetam
Levetiracetam is extremely water-soluble and has an oral bioavailability of close to 100 percent. Its Cmax is within 2 hours of dose administration. When the drug is administered with food, the rate of absorption is decreased, but the extent of absorption is unchanged. Therefore, levetiracetam can be administered without regard to timing of meals. Levetiracetam also exhibits linear pharmacokinetics after single doses ranging from 250 up to 5000 mg [Patsalos, 2000]. The Vd for levetiracetam is 0.5–0.7 L/kg, and protein binding is less than10 percent. Two-thirds of a dose is eliminated unchanged and the rest is eliminated as an inactive metabolite in the urine [Keppra package insert, 2004]. The clearance is 30–40 percent higher in children than in adults [Chhun et al., 2009; Glauser et al., 2007; May et al., 2003; Pellock et al., 2001]. Results are now available from studies in children as young as 2 months old and they confirm that the half-life of levetiracetam in younger children is approximately 5–6 hours, shorter than that seen in adults [Glauser et al., 2007]. An IV formulation and a sustained-release formulation are now on the market. It may be possible to give levetiracetam rapidly. Patients (4–32 years of age) who temporarily lost the ability to take medications by mouth were given an IV loading dose of levetiracetam via rapid infusion (given within 6 minutes). Levetiracetam concentrations ranged from 14 to 189 μg/mL and no significant changes in blood pressure or significant abnormalities in the electrocardiogram were experienced [Wheless et al., 2009]. There were also no local infusion site reactions noted; however, the formulation was diluted 1:1 with normal saline or dextrose 5 percent in water (D5W).
Drug–drug interaction information in children indicates that the concentrations of carbamazepine, valproic acid, topiramate, or lamotrigine are similar between study groups who receive levetiracetam or placebo [Otoul et al., 2007].
Rufinamide
Rufinamide is slowly absorbed due to the fact that it is lipophilic and dissolves slowly in aqueous solutions. The bioavailability is nonlinear and decreases at higher doses, with doses of 1600 mg producing less than proportional concentrations. Steady-state doses of 200, 400, and 800 mg produced morning trough concentrations of 2.81, 5.23, and 11.0 μmol/L, whereas 1600-mg doses resulted in a mean trough concentration of 19.6 μmol/L (4.7 mg/L) [Elger et al., 2010; Limite, 2005; NoAuthors, 2005]. Dissolution is the rate-limiting step in absorption; therefore, changes in gastrointestinal environment, such as with the presence of food, could have an effect on the absorption of rufinamide. It is suggested that rufinamide be administered with food. Rufinamide exposure is increased by 31–34 percent and the Cmax by 36–56 percent when the drug is given after high-fat meals [Perucca et al., 2008]. Rufinamide is metabolized to inactive metabolites via the carboxylesterase pathway. Although rufinamide does not appear to be metabolized via the cytochrome P-450, the carboxylesterase enzymes can be induced by inducers of the CYP450 enzymes. Rufinamide also appears to be a weak inhibitor of CYP2E1 and a weak inducer of CYP3A4.
Pharmacokinetic data in children are beginning to appear in the literature. The majority of the data concerning rufinamide is on file with the manufacturer; however, data on these trials have been summarized in Perucca et al. [2008]. A dosing regimen for children has been simulated from several phase II and III clinical studies involving patients with partial or generalized seizures and one study with Lennox–Gastaut syndrome patients. The simulation included 119 children (aged less than 12 years) and 99 adolescents (12–17 years) [Marchand et al., 2010], and was used in supporting material presented to the European Agency for the Evaluation of Medicinal Products. These studies involved patient populations and the presence of inducing and inhibiting polytherapy. Children who weigh less than 30 kg had greater variability in simulated rufinamide exposure measures than those children who were heavier than 30 kg. In addition, the co-administration of valproate resulted in higher rufinamide plasma concentrations. This study proposed a maximum daily dose of 600 mg for children who also receive valproic acid, and a maximum daily dose of 1000 mg for those children who do not also receive valproic acid. Indeed, valproic acid increased rufinamide steady-state plasma concentrations from 54.9 percent in male children and 70.1 percent in female children in a population pharmacokinetic study that is on file at Eisai [Perucca et al., 2008]. The increase in concentrations when valproic acid was a co-medication was approximately 3 times higher in children than in adults. (Adult concentrations were 15 percent higher.) Population studies involving children have also identified increases in rufinamide’s apparent clearance values due to the presence of inducing co-medications. Known antiepileptic drug inducers, such as carbamazepine, phenobarbital, phenytoin, or primidone, as well as vigabatrin, decreased rufinamide steady-state plasma concentrations in children from 23 to 46 percent [Perucca et al., 2008].
Tiagabine
Oral bioavailability of tiagabine is greater than 90 percent, and the bioavailability of tablets and that of the oral solution appear to be similar [Mengel et al., 1991a]. The absorption rate for tiagabine is reduced up to 65 percent when the drug is administered with food, although the extent of absorption is not affected [Mengel et al., 1991b]. Tiagabine has a Vd of 0.74–0.85 L/kg [Gustavson et al., 1997], and protein binding is approximately 96 percent [Gustavson et al., 1997; Ostergaard et al., 1995]. Tiagabine has a half-life of 5–9 hours [Gustavson and Mengel, 1995]. Tiagabine undergoes extensive metabolism by means of the cytochrome P-450 3A isoform, but no active metabolites have been identified. As observed with other drugs, clearances in children are faster than in adults; adjusting both Vd and clearance values according to body size gives values similar to those seen in adults [Gustavson et al., 1997].
Topiramate
Oral tablets of topiramate have a relative bioavailability of approximately 80 percent that of an oral solution. It is rapidly absorbed, with a Tmax of approximately 2 hours [Easterling et al., 1988]. Overall absorption is not affected by co-administration with food [Doose et al., 1992]. The protein binding of topiramate is less than 15 percent [Ben-Menachem, 1995a], and its Vd is 0.6–0.8 L/kg [Johannessen, 1997]. Elimination is mainly through the kidney, although a portion of the dose is removed by the liver [Ben-Menachem, 1995a]. Preliminary observations in pediatric patients indicate that the mean oral topiramate clearance is 44–55 percent higher in children, and they may need a higher dose than adults to achieve a similar plasma concentration [Adin et al., 2004; Glauser, 1997].
Valproic Acid
Valproic acid appears to be completely absorbed from all the available products (except for the extended-release formulation), but the rate of absorption varies significantly with dosage form (see Table 59-1). Its Tmax is 0.5–1 hour after ingestion for the syrup, 0.5–2 hours after the capsule, 1–6 hours for the enteric-coated tablet, and 3–6 hours for the sprinkle capsule [Cloyd et al., 1983, 1985, 1992]. Absorption after ingestion of the enteric-coated tablet is delayed for several hours; during this lag phase, plasma concentrations may continue to decline. Food further delays, but does not decrease, valproic acid absorption from the enteric-coated tablet [Fischer et al., 1988]. The extended-release tablet has an absolute bioavailability of 90 percent [Dutta et al., 2004]. The extended-release tablets and the enteric-coated tablets are not interchangeable. The extended-release tablets need to be prescribed at a dose that is 8–20 percent higher than the enteric-coated dose to attain similar plasma concentrations [Dutta and Zhang, 2004]. An IV formulation is also available and has been studied in children. The IV formulation should not be given intramuscularly due to toxic muscle necrosis [Gallo et al., 1997].
Valproic acid distribution volumes vary more than those of carbamazepine, phenobarbital, or phenytoin, and range from 0.12 to 0.49 L/kg. When loading doses are calculated, a volume of 0.2 L/kg is recommended. Valproic acid protein binding to albumin is saturable. At concentrations below 40–50 mg/L, the bound fraction averages 90–95 percent. As total concentration rises to the upper boundary of the therapeutic range, the percentage bound falls to 80–85 percent. Saturable protein binding results in a nonlinear relationship between total valproic acid concentration and dose. Unbound valproic acid concentrations, however, remain linear with dose [Cloyd et al., 1993; Riva et al., 1983]. The percentage of unbound valproic acid can be unpredictable and unusually high after rapid infusion in children who are critically ill [Birnbaum et al., 2003].
The elimination half-life of valproic acid is short. In the first few weeks after birth, half-life ranges from 17 to 40 hours, but rapidly declines from 3 to 20 hours in infants and young children. The full antiepileptic effect of valproic acid is obtained days to weeks after steady state is reached, and this effect continues for some time after the drug is discontinued. The lingering effect may be partly the result of its inhibitory effect on the enzymes responsible for the degradation of GABA, or may be due to the accumulation of 2-ene- valproic acid. This prolonged duration has prompted some investigators to propose single daily dosing [Stefan et al., 1984]; however, with other proposed mechanisms of action, seizure control is related to plasma valproic acid concentrations. Under these circumstances, dosing at least every half-life is indicated to minimize fluctuations in plasma concentrations. When valproic acid is given as monotherapy, the half-life may be long enough to maintain therapeutic concentrations for up to 24 hours in some children. Such children may be able to take valproic acid once a day [Cloyd et al., 1985].
Vigabatrin
Vigabatrin is a 50:50 racemic mixture of R(−) and S(+) enantiomers, with the S(+) enantiomer responsible for the drug’s pharmacologic activities and toxic effects [Haegele et al., 1983; Rey et al., 1990]. Bioavailability is approximately 75 percent in adults when the drug is given orally and is similar for both the tablet and the solution [Hoke et al., 1991]. Administration of vigabatrin with food does not significantly affect vigabatrin pharmacokinetics [Frisk-Holmberg et al., 1989]. Vigabatrin has been studied in children [Rey et al., 1989, 1990], and bioavailability appears to be somewhat lower in children than in adults [Rey et al., 1992]. Vigabatrin does not bind to plasma proteins, and it has an apparent Vd of 0.8 L/kg [Ben-Menachem, 1995b]. Clearance or the dose to concentration ratio of vigabatrin appears to be greater in children than in adults, indicating that a higher dose is needed in children to achieve a particular concentration when compared to adults [Armijo et al., 1997].
Most of a vigabatrin dose appears unchanged in the urine. Studies in infants and children show that the half-life of the inactive R(–) enantiomer, but not the half-life of the active S(+) enantiomer, increases with linear age. Vigabatrin exerts its pharmacologic effect by irreversible inhibition of GABA transaminase, causing increases in synaptic GABA concentrations by inhibiting its breakdown. Because the half-life of GABA transaminase is much longer than that of vigabatrin itself, the pharmacologic effect is determined by the half-life of the enzyme [Ben-Menachem, 1995b; Jung et al., 1977].
Zonisamide
Absolute bioavailability of zonisamide has not been assessed owing to the lack of an IV formulation. The drug reportedly is nearly completely absorbed, as indicated by early studies in which radioactive drug was measured in the urine and bile of rats [Matsumoto et al., 1983]. Human data indicate that, after multiple doses, 65 percent of zonisamide is recovered in urine (62 percent) or feces (3 percent). Food does not affect bioavailability [Zonegran Product Information, 2004]. Zonisamide is only 40 percent bound to plasma proteins, and its apparent Vd is approximately 1.45 L/kg [Zonegran Product Information, 2004]. Zonisamide is highly bound to erythrocytes, resulting in whole-blood concentrations that are higher than plasma concentrations. Binding to erythrocytes causes nonlinearity in plasma concentrations after a dose of greater than 400 mg [Matsumoto et al., 1989]. Zonisamide is eliminated by renal and hepatic routes. The metabolism of zonisamide is mediated in part by cytochrome P-450 3A4 [Zonegran Product Information, 2004]. Zonisamide–carbamazepine interaction was identified in a study including 12 children (5–16 years of age); trough concentrations decreased by 37 percent and carbamazepine-10,11-epoxide by 13 percent when carbamazepine was added to zonisamide monotherapy. The mean trough concentrations observed in 72 children undergoing zonisamide monotherapy were 27.0 ± 9.4 mg/L for a dose of 11.1 ± 2.5 mg/kg/day [Miura, 2004].
Generic Formulations
Absorption may differ among similar formulations (such as tablets) from various manufacturers (e.g., carbamazepine) [Kauko and Tammisto, 1974; Pynnonen et al., 1978; Sillanpaa, 1981]. Significant changes in serum phenytoin concentrations have been observed when the same maintenance doses of medication are taken from different manufacturers [Burkhardt et al., 2004]. Eight adult patients (ages 34 ± 49) had mean total phenytoin concentration of 17.7 ± 5.3 mg/L while on branded drug, which decreased to 12.5 ± 2.7 mg/L with generic and then increased to 17.8 ± 3.9 mg/L after the branded drug was re-introduced. Brand and generic phenytoin do not yield equivalent concentrations in some patients and substitution should not be permitted without prescriber and patient notification. For drugs with nonlinear pharmacokinetics such as phenytoin, such increases can lead to clinical toxicity or seizures [Mikati et al., 1992]. Generic medications can create significant cost savings for patients; however, the risks associated with possible changing drug concentrations need to be considered against the monetary advantages. Substitution of drugs made by differing manufacturers (brand to generic, generic to brand, or generic to generic) may lead to problems, especially when drugs with narrow therapeutic ranges or nonlinear pharmacokinetics are used. Therefore, counseling patients to keep taking medication prepared by the same manufacturer, whenever possible, usually is advisable [Nuwer et al., 1990]. Problems usually arise within 2 weeks of formulation change, so patients and parents should be particularly vigilant during this period. Most of the data available concerning generic substitution of antiepileptic drugs consists of surveys, retrospective database collection, or case studies, and are not prospectively designed to compare brand against generic medications directly.
Pharmacodynamics
Dose-Response or Concentration-Response Concept
Substantial evidence exists that both the number of patients experiencing seizure control increases and the degree of seizure control for a given patient improves with higher antiepileptic drug concentrations [Porter, 1989; Schmidt and Haenel, 1984]. The minimum effective concentration defines the concentration at which the desired pharmacologic response is first observed in some patients. The intensity of response increases in direct proportion to drug concentration, until a response is maximized. This result is the maximum pharmacologic effect for that drug. With some drugs, such as phenytoin, further increases in concentration may reduce response (e.g., phenytoin-induced exacerbation of seizures) [Troupin and Ojemann, 1975]. It is not known whether other antiepileptic drugs share this property but there are anecdotal reports of seizures increasing as antiepilpetic drugs are stepped up.
The incidence and severity of side effects also increase with drug concentration. The concentration at which side effects appear is usually, but not always, greater than the concentrations needed to achieve seizure control. The difference between concentrations that produce desired responses and those that produce toxic responses defines the therapeutic range. Optimal ranges, representing population averages, for commonly used antiepileptic drugs are listed in Table 59-6.
Table 59-6 Optimal Target Ranges for Commonly Prescribed Antiepileptic Drugs
| Drug | Target Drug Level | |
|---|---|---|
| Total (mg/L) | Free (mg/L) | |
| Carbamazepine | 4–12 | 0.5–3.6 |
| Eslicarbazepine | N/A | N/A |
| Ethosuximide | 40–100 | |
| Felbamate | 40–100 | |
| Gabapentin | 4–16 | |
| Lacosamide | 0–15* | |
| Lamotrigine | 2–20* | 1–9* |
| Levetiracetam | 5–45* | |
| MHD (oxcarbazepine active metabolite) | 10–35 | |
| Phenobarbital | 15–40 | 6–20 |
| Phenytoin | 10–20 | 1–2 |
| Primidone | 5–12 (15–40 PB) | |
| Pregabalin | 0–10* | |
| Rufinamide | 0–60* | |
| Tiagabine | 0–0.235* | |
| Topiramate | 2–25* | |
| Valproic acid | 50–150 | 6–20 |
| Vigabatrin | 20–160* | |
| Zonisamide | 7–40* | |
PB, phenobarbital.
* Range is not defined, and values are concentrations found in clinical trials.
Tolerance
Tolerance is a phenomenon in which pharmacologic or toxicologic effect diminishes with chronic use, even at the same or increasing plasma concentrations. Different mechanisms for the development of tolerance have been recognized [Froscher and Engels, 1986; Koella, 1986]. One mechanism is the result of biochemical adaptation (e.g., upregulation or downregulation of receptor sites). For example, the effectiveness of benzodiazepine therapy diminishes over time, necessitating even larger doses. Animal studies have demonstrated that chronic exposure to benzodiazepines causes a downregulation of benzodiazepine receptors, which reduces their anticonvulsant effect.
Another mechanism of tolerance is behavioral. The patient becomes progressively insensitive to drug effect without any apparent biochemical change [Siegel, 1986]. For example, many patients initiated on phenobarbital or primidone exhibit mild neurotoxicity even at low plasma concentrations but later, at the same concentration, have no symptoms [Leppik et al., 1984].
Physiologic Factors Affecting Drug Disposition in Children
General Considerations
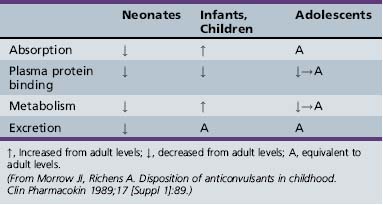
Antiepileptic drug pharmacokinetics and dosage requirements are influenced by the changing physiology of children as they age. Table 59-7 summarizes the effect of various developmental stages on absorption, protein binding, metabolism, and excretion. The physiologic changes associated with maturation produce marked alterations in antiepileptic drug pharmacokinetics. The variability with which children reach developmental milestones, along with genetic and environmental factors, causes antiepileptic drug pharmacokinetics to vary substantially, even in children of the same age. As a result of the differences between children and adults, children require more frequent and larger doses relative to body size to attain targeted plasma concentrations. Although important age-related, quantitative differences in antiepileptic drug pharmacokinetics are recognized, the pattern of drug-specific pharmacokinetics, such as saturation of absorption, protein binding, metabolism, and enzyme induction, is similar for children and for adults.
A number of physiologic and pathophysiologic processes can alter antiepileptic drug pharmacokinetics. Gastrointestinal disorders, particularly those that increase transit time, can alter the endothelial lining and decrease absorption. Antiepileptic drugs that are slowly absorbed, such as carbamazepine and phenytoin, are most likely to be affected. Physiologic stress, such as that associated with myocardial infarction, burns, traumatic injury, chronic inflammation, and surgery, increases α1-acid glycoprotein, resulting in greater binding with carbamazepine and carbamazepine-10,11-epoxide [MacKichan, 1992]. Febrile illnesses may produce increases in phenytoin clearance that persist for several weeks, resulting in lowering of plasma concentrations by as much as 50 percent [Leppik et al., 1986].
Renal failure can affect the excretion of antiepileptic drugs for which the kidney is the primary route of elimination. Both clinical response and plasma antiepileptic drug concentrations should be carefully assessed in patients with renal disease. Severe liver disease can alter the metabolism of antiepileptic drugs, resulting in accumulation of the parent drug [Kutt, 1983]. Antiepileptic drug metabolism appears to be well preserved in mild to moderate liver disease. Clinical response may vary, depending on the particular antiepileptic drug.
Plasma antiepileptic drug concentrations can fluctuate over a 24-hour cycle. A day–night cycle, in which both valproic acid and carbamazepine concentrations decrease at night, exists [Lockard and Levy, 1990; Pisani et al., 1990]. The mechanism for this alteration is presumed to be a circadian rhythm in clearance. Absorption of enteric-coated valproic acid tablets may be reduced at night [Cloyd, 1991]. In either case, considering the effect of night-time plasma antiepileptic drug concentrations when designing dosing strategies for patients with poorly controlled nocturnal seizures may be helpful.
Neonates
Gastric emptying time is irregular, absorption area is reduced, and biliary function is underdeveloped for several months or more after birth. These factors can contribute to erratic drug absorption [Morselli, 1983]. In addition, gastric pH is elevated, which may reduce the absorption of certain drugs, such as clonazepam [Meyer and Straughn, 1993]. In contrast with the oral route, rectal absorption is reliable and efficient. Protein binding is reduced because of lower plasma albumin concentrations, whereas greater extracellular water and less adipose tissue can either increase or decrease Vd. Neonatal drug metabolism and renal excretion are reduced. Reactions mediated by hepatic cytochrome P-450 enzymes reach and then exceed adult values within a few weeks after birth. By contrast, renal elimination of drugs and active metabolites may be lower than in adults until the age of 6 months.
Antiepileptic Drug Interactions
Interactions involving antiepileptic drugs may alter pharmacokinetics (see Figure 59-5). The clinical significance of pharmacokinetic interactions is determined by several factors, including concentration of the interacting drugs, the patient’s seizure control, and presence of toxicity.
Absorption
Concomitant therapy may influence absorption of antiepileptic drugs. For example, continuous tube feedings interfere with phenytoin absorption [Maynard et al., 1987]. Calcium- and magnesium-containing antacids appear to complex with phenytoin, decreasing its rate and extent of absorption. They should not be administered concurrently [Cacek, 1986; Fischer et al., 1988]. Gastric pH affects the rate and extent of benzodiazepine absorption; thus, use of antacids and histamine H2 blockers may decrease the plasma concentration of these drugs [Meyer and Straughn, 1993].
Protein Binding
Valproic acid and phenytoin compete for the same binding sites. Concurrent administration may decrease valproic acid and phenytoin binding. When this occurs, unbound concentrations briefly rise, potentially leading to clinical toxicity. Total phenytoin and valproic acid concentrations decrease; however, unbound concentrations rapidly re-equilibrate to the original concentration. In general, phenytoin and valproic acid dosing does not have to be adjusted in these circumstances. [Scheyer and Cramer, 1990]. Aspirin and other highly protein-bound drugs can have a similar effect, and measuring unbound concentrations in these circumstances can be very helpful in guiding treatement.
Metabolism
Drugs interact in many ways, such as induction and inhibition of liver enzymes. Information on how drugs are metabolized may provide insight into the potential for a drug interaction. For example, if a drug is known to be an inducer or an inhibitor of a particular P-450 isoenzyme, it may be possible to predict if a medication will affect the plasma concentrations of a second drug that is metabolized by that P-450 particular isoenzyme when the drugs are given concomitantly. This approach may be particularly useful in predicting the interaction between the many new antiepileptic drugs being developed and other possible concomitant medications (see Table 59-5).
Mechanism of Action of Antiepileptic Drugs
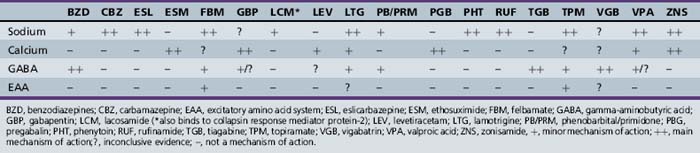
Decrease of excitatory responses may be effected by mimicking antagonists to excitatory amino acid receptors, inhibiting excitatory neurotransmitter release into the synapse, or inhibiting an enzyme needed in the synthesis of the excitatory neurotransmitter. By contrast, drugs can be used to increase inhibitory activity at the synaptic level, such as increasing GABA release into the synapse, increasing synthesis of GABA, or using an antiepileptic drug that has a GABA agonist effect. Activity at sodium or calcium channels can result in bursts of action potentials (e.g., an excitatory response) or in a release of neurotransmitter. The latter activity can be inhibitory or excitatory, depending on the neurotransmitter involved (e.g., glutamate, an excitatory neurotransmitter, versus GABA, an inhibitory neurotransmitter). The newer antiepileptic drugs appear to have multiple and/or more unique mechanisms for seizure control. Use of drugs with different mechanisms of action may lead to control of additional types of seizures. Moreover, drugs that have multiple mechanisms of action also can be advantageous because they increase the possibility of controlling seizures in patients with multiple seizure types with one or at least fewer drugs. This practice can result in fewer side effects and lower drug costs. Table 59-8 presents the mechanisms of action proposed for both the older and the newer antiepileptic drugs.
Dosage Formulations and Routes of Administration
Antiepileptic drugs are most commonly and conveniently given orally. In many circumstances, however, use of an alternative route of administration is necessary. Oral administration is not possible, for example, before and during surgery, with acute severe trauma, an inability to swallow when consciousness is impaired, or gastrointestinal illnesses. Interruption of oral administration occurs in children during frequent bouts of vomiting and diarrhea. In acute emergencies, especially within medical facilities, some antiepileptic drugs can be given by intravenous or intramuscular administration. Unfortunately, parenteral formulations are not available for most antiepileptic drugs because of their poor solubility in water. This creates a significant dilemma for those persons on maintenance antiepileptic therapy when oral administration cannot be continued. The rectal route of administration has been extremely useful for those situations, but water-insoluble antiepileptic drugs are generally not well absorbed rectally [Graves and Kriel, 1987].
Intravenous and Intramuscular Administration
Intravenous administration is a good alternative to oral therapy when patients are hospitalized. Phenytoin is very insoluble in water and thus is formulated with ethanol and propylene glycol, and adjusted to a pH of around 11. This formulation should not be given intramuscularly. Fosphenytoin, a phenytoin prodrug, is not formulated in propylene glycol and is maintained at close to physiological pH. It can be given intramuscularly, and the drug can be infused more rapidly than is possible with phenytoin, with fewer adverse effects [Leppik et al., 1989]. Other antiepileptic drugs that are available for intravenous administration include valproic acid, diazepam, lorazepam, lacosamide, levetiracetam, and phenobarbital. Rapid infusion (less than 15 minutes) of some antiepileptic drugs has been shown to be safe, making them more suitable for loading doses, as well as for bridging therapy.
Rectal Administration
When administered as a solution, diazepam is rapidly absorbed rectally, reaching the Tmax within 5–20 minutes in children [Lombroso, 1989]. By contrast, rectal administration of lorazepam is relatively slow, with a Tmax of 1–2 hours [Graves et al., 1987]. Table 59-9 provides a comprehensive list of antiepileptic drugs available for rectal administration and recommendations for use. Only one rectal formulation is available commercially (Diastat). The bioavailability of carbamazepine oral suspension diluted with an equal amount of water given rectally is similar to that of the oral tablet [Graves and Kriel, 1987]. The bioavailability of topiramate tablets crushed in water and given rectally is similar to that of oral tablets [Conway et al., 2003]. The availability of lamotrigine tablets crushed in water and given rectally is about half that of oral tablets [Birnbaum et al., 2000, 2001].
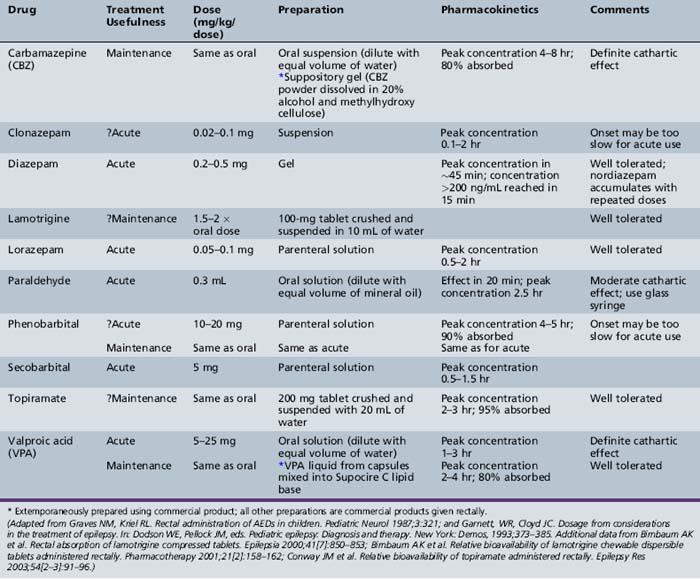
Some drugs cannot be administered rectally owing to factors such as poor absorption or poor solubility in aqueous solutions. The relative rectal bioavailability of gabapentin, oxcarbazepine, and phenytoin is so low that the current formulations are not considered to be suitable for administration by this route [Clemens et al., 2007; Fuerst et al., 1988; Kriel et al., 1997; Kvan and Johannessen, 1975; Moolenaar et al., 1981]. The dependence of gabapentin on an active transport system, and the much-reduced surface area of the rectum compared with the small intestine, may be responsible for its lack of absorption from the rectum.
Intranasal Administration
Intranasal administration is a potentially attractive alternative route when intravenous administration is impossible or impractical. Nasal access is readily available, even when a patient is having a seizure; requires little, if any, caregiver training; and, under the right circumstances, allows rapid drug absorption. Although the absorptive area of the nasal cavity is approximately the same as that of the rectum, the volume of solution that can be instilled is 0.5 mL or less [Romeo et al., 1998]. Several studies comparing intranasal midazolam with intravenous diazepam have been carried out in children. In summary, both drugs had similar onset of effect and seizure control, although intranasal midazolam has been used for preoperative sedation. Several disadvantages are associated with intranasal midazolam administration:
The rapid elimination half-life may explain the reports of seizure breakthrough within a few hours after intranasal delivery of midazolam [Scheepers et al., 2000]. Absorption of diazepam has been demonstrated after intranasal delivery [Lindhardt et al., 2001]. There are currently no commercial intranasal formulations available.
Other Routes of Administration
Lorazepam is absorbed after sublingual administration and can be useful in treatment of serial and prolonged seizures [Yager and Seshia, 1988]. Delivery of antiepileptic agents directly into the cerebral subarachnoid or intraventricular space is being investigated. Spinal intrathecal administration of baclofen for control of spasticity is currently used for persons with spinal cord disease and cerebral palsy, and can be administered by a programmable continuous pump surgically placed in the abdomen [Albright et al., 1993; Penn et al., 1989].
Monitoring of Patients on Antiepileptic Drug Therapy
Clinical Monitoring of Efficacy
Statistical interpretation of the change in seizure frequency based on modified sequential analysis has been proposed [Leppik et al., 1989]. In addition, achievement of steady state with the new drug regimen is necessary to determine if a medication has been given an adequate trial at any specific dose. The time to achieve a steady state varies considerably from drug to drug, depending on the drug’s half-life. In every case, 5 half-lives must elapse before a steady state is reached.
Interpretation of clinical response in patients in whom polypharmacy is necessary is especially difficult. Clinical improvement or deterioration after the addition of a second antiepileptic drug could be due to numerous factors. Such factors include the additive or synergistic effect of the two medications, the effect of the second drug alone, and the effect of the second drug on the metabolism and/or displacement of the initial drug. It is important to make clinical conclusions only after a steady state is reached with the new drug regimen. If improvement is obtained after the addition of the second medication, the clinician should attempt to taper and discontinue the first medication, to determine whether the clinical improvement is an effect of synergistic therapy or that of the second medication itself. Finally, it is necessary to remember that withdrawal of co-medication might have an effect on the disposition of the remaining drug(s), which could influence clinical response (see Figure 59-5).
Monitoring of Drug Concentrations
The ability to measure blood levels of antiepileptic drugs has been a major advance in the understanding of drug metabolism and disposition, and has led to more rational drug therapy. The influence of periodic monitoring of blood levels may reduce the number of therapeutic failures by 50 percent [Kutt and Penry, 1974]. As expected, drug concentrations generally are related to the dose administered; however, unexpected results may occur, especially in cases of patient noncompliance or with drugs that follow nonlinear kinetics.
Interpretation of “Optimal Therapeutic Ranges”
Numerous factors should be considered in the interpretation of drug concentrations. Medications that have rapid absorption and clearance have wide fluctuation between dosing intervals. For example, the liquid formulation of valproic acid, especially when given to a child also receiving enzyme-inducing co-medications, is rapidly absorbed and eliminated; therefore, a twofold fluctuation during dosing intervals is possible. Recording the time of the blood sample in relation to the last dose is needed to interpret laboratory results properly. Unexpectedly low concentrations are most frequently the result of poor compliance; however, they might be seen after the addition of an enzyme-inducing co-medication. Very high drug concentrations are seen with administration of high doses, in persons with genetically low clearance or some disease states, or with use of enzyme-inhibiting co-medications such as valproic acid [Perucca et al., 2001].
The terms optimal drug concentration and target range are preferred over therapeutic range, for several reasons. First, these newer terms express the concept that the optimal dose and drug concentration need to be determined for each individual patient. The ideal or optimal dose and drug concentration would be those associated with complete control of seizures with no adverse effects. Clinical effects and toxicity of antiepileptic drugs correlate better with drug concentrations than with total daily dosage [Glauser and Pippenger, 2000]. Published “optimal drug concentrations” are attempts to report ranges of concentrations at which many patients have improved seizure control without significant adverse effects. These ranges generally have not been determined by controlled studies in large populations. Obviously, more clinical experience correlating clinical response with drug concentrations is available for the older antiepileptic drugs. Although measurement of drug concentrations has contributed greatly to the development of rational drug management, routine monitoring may not be beneficial. In a randomized controlled trial of the impact of drug monitoring, no benefit with regard to improved seizure control or reduced adverse effects was observed in the study group in which levels were routinely sampled [Jannuzzi et al., 2000]. A retrospective study of antiepileptic drug monitoring in a pediatric emergency room setting failed to find a correlation between drug concentrations and clinical decisions [Kozer et al., 2003].
When to Obtain Drug Concentrations
Although enthusiasm for routine monitoring of drug concentrations has moderated, this assistance from the laboratory is helpful in clinical management when used appropriately. Laboratory specimens collected for measurement of drug concentrations should be obtained after the drug is at steady state. The time after dose of when the sample was drawn should be recorded in order to interpret where the concentration would be on a pharmacokinetic profile. Recommendations with regard to specific clinical situations in which determination of drug concentration is useful are listed in Box 59-1 [Glauser and Pippenger, 2000]. It is useful to measure the antiepileptic drug concentration after the patient has reached steady state dosing and optimal clinical outcome has been obtained. This level could be considered the “optimal drug concentration” or the “target level” for the individual patient. Then a level should be obtained when a breakthrough of the seizure pattern occurs (i.e. a single seizure in a person usually well controlled, or a doubling of the usual seizure rate). If the level is lower than the “target value,” one needs to consider noncompliance or alteration in pharmacokinetics. Noncompliance is by far the most common cause of deviation from the “target value.” In these cases, the dose should not be changed, but steps should be taken to increase compliance (pill box, education, and so on). Illnesses affecting absorption or clearance can also be detected by comparing the current value with the “target level.” Also, levels should be measured after changes in co-medication once a new steady state has been reached to determine if any changes in the levels have occurred. If a patient experiences an increase in seizures but the current concentration is at the “target,” it is possible that the seizure threshold has changed or was underestimated, and changes in dose may be needed and a new “target level” should be established.







What to Measure
Total drug concentrations generally are measured; however, it also is possible to determine levels of free or unbound drug, as well as of metabolites of the administered drug. Free drug concentrations correlate best with clinical effect and toxicity; however, they are more expensive and difficult to obtain. In most cases, the ratio of free to bound drug is relatively constant for a particular patient; therefore, total drug concentrations usually are adequate. In certain instances, however, particularly with critically ill patients under intensive care, determination of free drug levels, especially for phenytoin and valproic acid, is essential. In such patients, many drugs are typically administered, increasing the likelihood that antiepileptic drugs will be displaced from protein-binding sites. The percentage of unbound valproic acid increases with higher drug concentrations and with co-medication [Cloyd et al., 1992], or when valproic acid is rapidly administered [Birnbaum et al., 2003]. When the bound fraction is doubled, the valproic acid free fraction may be eight times higher [Fenichel, 1986].
Occasionally, measurement of antiepileptic drug metabolites is useful. With several antiepileptic drugs, metabolites are clinically active and contribute to both response and toxicity. Phenobarbital is an active metabolite present during primidone therapy. Carbamazepine-10,11-epoxide is a derivative of carbamazepine and contributes to toxicity [Schoeman et al., 1984]. Clinical monitoring of oxcarbazepine and eslicarbazepine acetate treatment is evaluated by measuring their primary metabolites.
Laboratory Tests for Idiosyncratic Reactions
In an effort to identify patients in whom serious or potentially life-threatening adverse effects may develop, obtaining complete blood counts and chemistry profiles at routine intervals has become common practice. The cost for this routine monitoring can exceed the costs of medication [Hart and Easton, 1982]. Moreover, it is very debatable whether routine laboratory monitoring can actually identify patients at risk for serious reactions or can identify significant adverse events better than clinical monitoring. Identification of life-threatening reactions is rarely made by routine laboratory screening of asymptomatic children [Camfield et al., 1986]. Fulminant or irreversible hepatic failure during valproic acid therapy is not reliably predicted by laboratory monitoring [Willmore et al., 1991]. Many reports, therefore, indicate that routine laboratory screening of asymptomatic patients is of little value [Camfield et al., 1989; Pellock and Willmore, 1991]. However, complete blood counts and chemistry profiles before, and then after, initiation of antiepileptic drug therapy are reasonable [Harden, 2000]. It is much more effective to caution patients and parents to obtain these tests immediately whenever there are signs of any possible reaction, such as unusual bleeding or bruising, significant loss of appetite, jaundice, and so on. Testing for inborn errors of metabolism may be useful for identifying young children at risk for hepatic dysfunction from valproic acid.
Adverse Drug Reactions to Antiepileptic Drugs
Adverse drug reactions are defined by the World Health Organization as “any response to a drug that is noxious or unintended, and which occurs at doses used in man for prophylaxis, diagnosis, or therapy” [Venulet and ten Ham, 1996]. The most frequent adverse drug reactions to antiepileptic drugs are dose-related, mild, and reversible. Less frequently observed but potentially more serious are the idiosyncratic adverse reactions, which generally are not related to dosage but may be due to individual peculiarities of drug metabolism, i.e., lack of pathways to process toxic metabolites. Idiosyncratic reactions are almost never seen with antiepileptic drugs that are not metabolized.
Central Nervous System Adverse Reactions
The central nervous system adverse reaction profile for children is similar to that for adults. Most of the older antiepileptic drugs – phenytoin, phenobarbital, primidone, carbamazepine, and valproic acid – and their active metabolites have similar side-effect profiles, comprising of cognitive impairment, sedation, dizziness, diplopia, ataxia, headaches, and effects on somnolence. The effects of antiepileptic drugs on cognitive function generally are concentration- (and dose-) dependent. Drug therapy is known to contribute to cognitive deficits, especially treatment with multiple drugs [Thompson and Trimble, 1982]. Removal of antiepileptic drugs frequently results in improved cognitive function and motor skills [Duncan et al., 1990]. It is important to recognize that reaction times, disordered attention, and impulsivity, even in untreated children with epilepsy, differ from those in control subjects. The differences in attention and reaction time, however, are small between children with mild as opposed to severe seizure disorders [Mitchell et al., 1992].
Side effects frequently occur in children who are given phenobarbital. Phenobarbital has been associated with a significant depression of cognitive function when it is used for treatment of febrile seizures in children. In a randomized, placebo-controlled, prospective study, children had lower mean intelligence quotient (IQ) scores both during and 6 months after stopping therapy with phenobarbital [Farwell et al., 1990]. Clinical tolerance does develop to some degree; however, an unacceptably high frequency of adverse effects from phenobarbital commonly is observed in children [Farwell et al., 1990; Wolf and Forsythe, 1978]. The adverse effects are reminiscent of complaints present in attention deficit disorders and include overactivity, aggressiveness, inattention, and irritability. The behavioral disorders are observed in 20–50 percent of children receiving phenobarbital for febrile seizures, and result in discontinuation of therapy in 20–30 percent of children [Herranz et al., 1984].
The presence of pre-existing behavior problems, especially hyperactivity, is strongly predictive of adverse reactions during phenobarbital therapy. In a study of children receiving phenobarbital for febrile seizures, 80 percent of those with abnormal behavior before drug therapy reported aggravation of pre-existing hyperactive behavior with phenobarbital, versus only 20 percent for children with normal preseizure behavior [Wolf and Forsythe, 1978]. In some cases, these adverse effects may resolve with continuation or with a lower dose of phenobarbital; however, discontinuation of therapy or alternative medication should be considered.
The newer antiepileptic drugs – gabapentin, felbamate, topiramate, lamotrigine, tiagabine, oxcarbazepine, levetiracetam, vigabatrin, and zonisamide – are thought to have fewer and less severe adverse effects; however, their adverse central nervous system effects are similar to those of the older antiepileptic drugs. Although gabapentin has a good safety profile and seems well tolerated in adults, significant behavioral side effects are observed in some children. In a nonblinded, uncontrolled series, adverse behavioral effects were reported in 47 percent of children given gabapentin for add-on therapy; however, only 12 percent of children were withdrawn from gabapentin because of these problems [Khurana et al., 1996]. In other reports, aggression, hyperactivity, temper tantrums, and increased oppositional behavior developed in 12 children taking the drug [Lee et al., 1996; Tallian et al., 1996; Wolf and Forsythe, 1978]. Most, but not all, of these children had pre-existing cognitive dysfunction, such as mental retardation, autistic features, or behavioral difficulties. The adverse cognitive changes resolved with discontinuation of gabapentin; if administration of the drug was resumed, the adverse cognitive changes tended to recur. In one 6-year-old female, the drug was clinically effective in controlling seizures as monotherapy and was well tolerated for 6 months before the aggressiveness developed [Tallian et al., 1996].
In limited pediatric trials, 36 percent of children taking tiagabine have experienced treatment-emergent adverse effects, most commonly somnolence [Gustavson et al., 1997]. In approximately 15 percent of children, decreased functioning in school, aggression, and other cognitive changes occurred within the first 4 months of initiation of topiramate therapy [Gerber et al., 2000]. A previous history of behavioral changes and concurrent lamotrigine therapy were associated factors. Central nervous system-related adverse effects from topiramate tend to decrease over time [Ritter et al., 2000]. Of greater concern are reports of the development of major depression, schizophrenic-like reactions, and organic psychoses in adults taking vigabatrin [Ferrie et al., 1996; Ring et al., 1993]. A reversible acute psychosis has been reported in a child taking vigabatrin [Canovas Martinez et al., 1995]. Development of hyperkinesia, somnolence, and insomnia in children taking vigabatrin has been reported by a number of investigators [Aicardi et al., 1996; Dalla Bernardina et al., 1995; Dulac et al., 1991]. Generally, these effects have not led to discontinuation of the drug. Meador et al. recently reported that, when compared with other antiepileptic drugs, valproic acid can significantly lower, in a dose-related fashion, the IQ of children of 3 years of age who have been exposed to valproic acid in utero [Meador et al., 2009].
Gastrointestinal Effects
Weight Gain
A survey in children and adolescents taking valproic acid found the most common side effect to be weight gain; however, for many patients, the weight gain was “beneficial.” The incidence of unwanted weight gain was 26 percent in adults and 15 percent in children and adolescents. An increase in weight was observed more than twice as frequently in females [Clark et al., 1980; Wirrell, 2003]. Weight increase was independent of dose [Covanis et al., 1982]. Weight gain also was reported in children during therapy with carbamazepine [Herranz et al., 1988; Hogan et al., 2000] and vigabatrin [Dalla Bernardina et al., 1995; Dulac et al., 1991]. Weight gain has been reported in adults taking gabapentin [DeToledo et al., 1997].
Weight Loss
Sustained weight loss was reported in children on maintenance topiramate therapy [Reiter et al., 2004]. Seventy-five percent of children and adults during clinical research trials reported a loss of appetite and decrease in body weight while taking felbamate [Bergen et al., 1995]. Zonisamide also is associated with weight loss in children [Kothare et al., 2006].
Gastric Irritation
Signs of gastric irritation, with heartburn and indigestion, are seen with use of some antiepileptic drugs. Gastric irritation during valproic acid therapy can be reduced with administration of lower doses or by taking medication with food. Use of enteric-coated preparations of valproic acid will enable most patients who did not tolerate valproic acid because of gastrointestinal symptoms (nausea/vomiting, eructation, heartburn) to resume valproic acid therapy [Clark et al., 1980; Wilder et al., 1983]. Aggressive medical intervention with antacids and H2 receptor antagonists (other than cimetidine) has been helpful for some children with clinical signs of gastritis, allowing valproic acid therapy to continue [Marks et al., 1988].
Approximately 20 percent of patients receiving ethosuximide will demonstrate dose-related adverse effects, primarily gastric distress, vomiting, hiccups, and anorexia [Sherwin, 1983]. Gastrointestinal disturbances also are encountered in 14 percent of patients taking carbamazepine, with symptoms of nausea, vomiting, anorexia, and constipation [Herranz et al., 1988]. Nausea and vomiting commonly are observed with felbamate [Dodson, 1984; Theodore et al., 1991].
Gingival Hyperplasia
Gingival hyperplasia occurs in 10–20 percent of persons treated with phenytoin and is more frequent in young persons. It is observed more frequently in persons taking higher dosages, often appears 2–3 months after beginning therapy, and reaches its maximum severity in 12–18 months [Babcock, 1965; Little et al., 1975]. Gingival hyperplasia generally resolves within 2–5 months after discontinuation or a reduction in medication [Little et al., 1975]. A vigorous program of good oral hygiene started before and continued during phenytoin therapy can be effective in minimizing gingival enlargement [Modeer and Dahllof, 1987; Pihlstrom et al., 1980]. Drugs other than antiepileptic drugs, including nifedipine and cyclosporine, have been associated with the development of gingival hyperplasia [Butler et al., 1987].
Increased Seizures
Some patients, especially those with generalized or absence seizures, may experience an increase in seizure activity when carbamazepine or phenytoin is added [Horn et al., 1986; Perucca et al., 1998; Snead and Hosey, 1985]. Vigabatrin also has been reported to increase seizure activity and even lead to status epilepticus [de Krom et al., 1995]. Aggravation of seizures occurred in 3 percent of children with generalized seizures, most often in those with nonprogressive myoclonic epilepsy, and was extremely unusual in children with partial epilepsies and infantile spasms [Lortie et al., 1997]. Oxcarbazepine has been reported to increase myoclonic jerks and absence seizures [Gelisse et al., 2004]. Phenytoin and carbamazepine may increase seizures associated with the Lennox–Gastaut syndrome, and gabapentin and pregabalin can increase or induce myoclonic seizures.
Osteomalacia
Osteomalacia (rickets) may occur in patients taking antiepileptic drugs. In a survey of institutionalized patients, 52 of 144 patients with a developmental cognitive disability ranging in age from 3 to 26 years, who were on antiepileptic drug therapy for at least 2 years, had elevations of serum alkaline phosphatase levels greater than 2 standard deviations above normal. Most patients were taking phenytoin or phenobarbital [Hunt et al., 1986]. The cause of osteomalacia during antiepileptic drug therapy appears to be multifactorial [Farhat et al., 2002]. Many antiepileptic drugs presumably enhance hepatic conversion of 25-hydroxyvitamin D to biologically inactive metabolites. Clinically significant rickets seldom develops in ambulatory children on antiepileptic drug therapy, or in institutionalized children who are not taking medication. On the other hand, clinically overt rickets was observed in 10 percent of institutionalized children on phenobarbital and/or phenytoin. In these patients, sunlight was more important for the maintenance of serum 25-hydroxyvitamin D than were supplements of vitamin D [Morijiri and Sato, 1981].
Current evidence indicates that antiepileptic drugs may decrease bone mineral density in adults. In adults, correlation with 25-hydroxyvitamin D levels was poor [Farhat et al., 2002]. In children, no correlation was observed between duration of antiepileptic drug therapy and 25-hydroxyvitamin D levels. Bone mineral density measurements tended to be lower in patients on older antiepileptic drugs than in patients on newer antiepileptic drugs, but were lower in all patients on any antiepileptic drug than in the control population. It was previously thought that older antiepileptic drugs resulted in induction of vitamin D metabolism, resulting in decreased bone mineral density; however, the lack of correlation between vitamin D levels and bone mineral density suggests that other mechanisms are operative.
Some authors recommend routine bone mineral density monitoring for patients on chronic antiepileptic drug therapy [Farhat et al., 2002]. Consensus is lacking on treatment of patients who have low bone mineral density secondary to antiepileptic drug therapy. Other factors may contribute to decreased bone mineral density, such as lack of exercise and sunlight.
Tremor and Movement Disorders
A benign essential type of tremor has been observed with use of valproic acid, beginning within 1 month of initiating therapy. It is dose-dependent, occurring in some patients with blood valproic acid levels greater than 40 mg/mL. Reduction of tremor may occur with reduction of dose [Karas et al., 1982]. Tremor also has been seen with use of lamotrigine and gabapentin [Schmidt and Kramer, 1994]. Oculogyric crises and choreoathetotic movements have developed in several adults while taking gabapentin [Buetefisch et al., 1996; Reeves et al., 1996].
Other Effects
Hair changes have been associated with antiepileptic drug treatment. Hair changes (thinning or wavy hair) are seen in 12 percent of children during valproic acid therapy [Clark et al., 1980]. Additional dose-related adverse effects of phenytoin include hirsutism, coarse facial features, and acne, especially in children and teenagers. Pancreatitis has been associated with valproic acid therapy [Coulter and Allen, 1980; Wyllie et al., 1984]. Visual field changes, which may be irreversible, may occur with vigabatrin treatment [Krauss et al., 1998]. Topiramate has been reported to cause acute myopia and secondary angle closure glaucoma. Patients present with decreased visual acuity and ocular pain. In a majority of cases, immediate discontinuation of topiramate results in a resolution of symptoms [Sankar et al., 2001; Thambi et al., 2002]. Hyponatremia occasionally is encountered in patients taking carbamazepine and oxcarbazepine [Borusiak et al., 1998; Holtmann et al., 2002; Koivikko and Valikangas, 1983]. Hyperthermia and oligohydrosis have been reported in several children taking topiramate and zonisamide. Clinical signs included fever, decreased sweating, and exercise intolerance, which resolved in a majority of cases when topiramate or zonisamide was discontinued [Arcas et al., 2001; Knudsen et al., 2003]. Topiramate and zonisamide may cause nephrolithiasis. The risk is increased with topiramate or zonisamide treatment and the ketogenic diet [Kossoff et al., 2002].
Anticonvulsant Hypersensitivity Syndrome in Children
Clinical Features
The hypersensitivity syndrome is characterized by rash, fever, and malaise, generally occurring within the first several months of therapy. Certain types of rash may be particularly alarming, such as those of Stevens–Johnson syndrome or toxic epidermal necrolysis. Often hepatic involvement is part of the syndrome, and the liver is the most frequently involved internal organ. Affected children may present with hepatomegaly and elevations in serum transaminase levels. The involvement may progress to hepatic necrosis. Other systems may be involved, including kidney, lungs, bone marrow, and the lymphatic system [Handfield-Jones et al., 1993].
Pathogenesis
Although the mechanism of the hypersensitivity syndrome is not completely understood, current evidence implicates toxic metabolites [Glauser, 2000; Verrotti et al., 2002]. The aromatic AEDs (phenobarbital, phenytoin > carbamazepine > oxcarbazepine), lamotrigine, and felbamate have been more frequently implicated in causing this syndrome. Presumably, accumulation of toxic metabolites, such as reactive aromatic epoxide intermediates, precipitates the syndrome [Glauser, 2000].
Prevention
Routine laboratory monitoring (i.e., with complete blood counts or liver function tests) will not prospectively identify which patients are at high risk for the development of an acute hypersensitivity reaction. Nevertheless, certain factors place some patients at high risk. Clinical profiles that identify patients at high risk for hypersensitivity reactions differ for specific antiepileptic drugs (valproic acid, felbamate, and lamotrigine) (Box 59-2). When one of these drugs is being considered for a patient at high risk, one should proceed with caution. Alternative therapy, if possible, should be considered. Additional laboratory tests to screen for inborn errors of metabolism may be indicated when use of valproic acid is under consideration in children younger than 2 years of age. Families need to be advised that the child is in a high-risk category and counseled on the early recognition of the syndrome.
















(From Glauser TA. Idiosyncratic reactions: New methods of identifying high-risk patients. Epilepsia 2000;41 [Suppl 8]:S16.)
Although the identification of a high-risk clinical profile is the most inexpensive and practical way available to reduce the risk of hypersensitivity syndrome, various biomarkers also have been identified [Glauser, 2000]. These biomarkers may indicate that some patients have genetic susceptibility to the accumulation of toxic metabolites due to deficient detoxification pathways. The biomarkers that have been investigated include deficient epoxide hydrolase activity and deficiencies in free radical-scavenging enzyme activity. Studies in patients of Chinese ancestry indicate a strong association between the risk of developing toxic epidermal necrolysis and Stevens-Johnson Syndrome and the presence of HLA-B* 1502. Clinicians may want to avoid use of carbamazepine in this population.
Managing Adverse Effects
Idiosyncratic reactions pose another problem. Mild, asymptomatic elevations of liver enzymes do not mandate discontinuation of therapy if not in excess of 2–3 times normal values. On the other hand, Stevens–Johnson syndrome and toxic epidermal necrolysis, clinically symptomatic hepatotoxicity, pancreatitis, and most rashes are indications for immediate cessation of the responsible medication. If the rash was mild, however, and especially if the patient had an otherwise favorable clinical response to the drug, a cautious rechallenge might be considered. Successful resumption of therapy has been reported in a number of patients, especially with valproic acid after that drug was discontinued, when a more gradual dose escalation was used [Schlumberger et al., 1994; Tavernor et al., 1995].
Selection of Drugs for Initiation of Antiepileptic Drug Therapy
The selection of a specific drug for the treatment of epilepsy is based primarily on the criteria of therapeutic efficacy expected for the patient and seizure type. Certain epileptic syndromes require specific therapy. For most patients and seizure types, however, efficacy differences are minimal, and the selection of the initial drug for antiepileptic therapy is guided equally by a consideration of potential adverse effects. Although highly effective, phenytoin is rarely a drug of first choice in children because of a higher incidence of dysmorphic effects. In children, especially those with pre-existing tendencies for attention-deficit disorders, an unacceptable incidence of behavioral problems, as well as of cognitive dysfunction, has been noted with barbiturate therapy. The principles to be used during initiation and selection of antiepileptic drug therapy in children have been summarized [Crumrine, 2002; Fenichel, 1986].
Discontinuation of Antiepileptic Drug Therapy
The decision to discontinue antiepileptic drug therapy often is as challenging as the decision to initiate and continue long-term drug therapy. Most commonly, the question arises after a patient has done well and has become seizure-free for some substantial period. At other times, the question arises because of chronic noncompliance, or because no therapy has had a measurable effect on seizure control. Many factors are considered in making these decisions, including the length of the seizure-free interval, drug history, presence of adverse effects, and the neurologic and epilepsy syndrome diagnosis of the patient. Numerous retrospective and prospective investigations are available. Ultimately, the decision to withdraw or to continue antiepileptic drug therapy is that of the patient and family, and their perception of acceptable risk of recurrence often differs from that of the treating physician [Gordon et al., 1996].
Benefits of Drug Discontinuation
The potential benefits of drug discontinuation are numerous. Most obvious is the immediate reduction in cost to the family and third-party payer with elimination of drug therapy and usually the reduction in associated expenses incurred by laboratory tests and physician and clinic charges. In addition, in many patients, the adverse, sometimes previously unrecognized effects of antiepileptic drug therapy are reversed. Patients who had been on phenobarbital, phenytoin, valproic acid, and topiramate had subtle improvement in various psychometric tasks, such as memory, vigilance attention, and visual motor performance [Gallassi et al., 1992; Lee et al., 2003], and in psychomotor speed [Aldenkamp et al., 1993], when therapy was discontinued.
Risks of Drug Discontinuation
The obvious risk of stopping antiepileptic drug therapy is the recurrence of seizures. In most cases, seizure control can be regained with resumption of the previous drug therapy. Response to therapy after a relapse may be rapid and satisfactory [Arts et al., 1988]. Chadwick and colleagues observed that approximately 90 percent of patients who experience seizures after antiepileptic drug discontinuation have a 2-year remission after therapy is resumed [Chadwick et al., 1996]. Thus, control was usually but not always regained despite increasing doses of a previously successful drug regimen.
A guideline has been published to assist the physician and the family considering discontinuation of antiepileptic drug therapy. The conclusions were based on a MEDLINE search [Camfield and Camfield, 2008].
Children who were seizure-free for 2 or more years while on antiepileptic drugs, who had a single type of partial or generalized seizure, who had a normal IQ and normal findings on neurologic examination, and a normalized electroencephalogram (EEG) on therapy had a 69 percent chance of discontinuing antiepileptic drug therapy without seizure recurrence. A similar conclusion was reached by another meta-analysis, in which 75 percent of “relatively unselected” patients were seizure-free 1 year after discontinuation, and 71 percent 2 years after discontinuation of antiepileptic drug therapy [Berg and Shinnar, 1994]. Children with remote symptomatic seizures and those with seizure onset in adolescence (rather than earlier) were at greatest risk of recurrence.
Although relapse was observed in every study, in one prospective series, relapse rates were slightly higher in children who had epileptiform activity on the last EEG before antiepileptic drug discontinuation [Andersson et al., 1997]. The authors concluded that the persistence of 3-Hz spike-wave activity during therapy also was an unfavorable prognostic sign. Children more frequently had recurrences of seizures when antiepileptic drugs were stopped after 1 year versus 3 years of therapy. In another randomized, prospective study of 6 versus 12 months of antiepileptic drug therapy in children who became seizure-free on antiepileptic drug therapy, no difference between relapse rates was observed [Peters et al., 1998]. That report again presented the predictive variables for relapse, which were partial epilepsy, a remote symptomatic etiology, an older age at onset (12 years and older), and an epileptiform EEG during therapy.
Families should discuss withdrawing antiepileptic drugs with their neurologist once children have become free of seizures for 1 year or longer while on antiepileptic drug therapy. Children whose EEGs have become normal have a somewhat better chance of successful withdrawal of therapy. However, there are critical life transitions when the risk of discontinuing therapy outweighs continuation. These are when children have been seiure-free and are eligible to begin driving, and when they are leaving home and transitioning to college or independent living. Finally, for some patients, withdrawal of antiepileptic drugs is rarely successful – for example, those with abnormalities on neurologic examination or with certain seizure syndromes such as juvenile myoclonic epilepsy, Lennox–Gastaut syndrome, or infantile spasms [Peters et al., 1998; Shinnar et al., 1994].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Adin J., Gomez M.C., Blanco Y., et al. Topiramate serum concentration-to-dose ratio: influence of age and concomitant antiepileptic drugs and monitoring implications. Ther Drug Monit. 2004;26(3):251-257.
Aicardi J., Mumford J.P., Dumas C., et al. Vigabatrin as initial therapy for infantile spasms: a European retrospective survey. Sabril IS Investigator and Peer Review Groups. Epilepsia. 1996;37(7):638-642.
Albright A.L., Barron W.B., Fasick M.P., et al. Continuous intrathecal baclofen infusion for spasticity of cerebral origin. JAMA. 1993;270(20):2475-2477.
Aldenkamp A.P., Alpherts W.C., Blennow G., et al. Withdrawal of antiepileptic medication in children – effects on cognitive function: The Multicenter Holmfrid Study. Neurology. 1993;43(1):41-50.
Almeida L., Minciu I., Nunes T., et al. Pharmacokinetics, efficacy, and tolerability of eslicarbazepine acetate in children and adolescents with epilepsy. J Clin Pharmacol. 2008;48(8):966-977.
Almeida L., Potgieter J.H., Maia J., et al. Pharmacokinetics of eslicarbazepine acetate in patients with moderate hepatic impairment. Eur J Clin Pharmacol. 2008;64(3):267-273.
Anderson G.D., Lynn A.M. Optimizing pediatric dosing: a developmental pharmacologic approach. Pharmacotherapy. 2009;29(6):680-690.
Andersson T., Braathen G., Persson A., et al. A comparison between one and three years of treatment in uncomplicated childhood epilepsy: a prospective study. II. The EEG as predictor of outcome after withdrawal of treatment. Epilepsia. 1997;38(2):225-232.
Arcas J., Ferrer T., Roche M.C., et al. Hypohidrosis related to the administration of topiramate to children. Epilepsia. 2001;42(10):1363-1365.
Armijo J.A., Cuadrado A., Bravo J., et al. Vigabatrin serum concentration to dosage ratio: influence of age and associated antiepileptic drugs. Ther Drug Monit. 1997;19(5):491-498.
Arts W.F., Visser L.H., Loonen M.C., et al. Follow-up of 146 children with epilepsy after withdrawal of antiepileptic therapy. Epilepsia. 1988;29(3):244-250.
Babcock J.R. Incidence of gingival hyperplasia associated with Dilantin therapy in a hospital population. J Am Dent Assoc. 1965;71(6):1447-1450.
Battino D., Croci D., Granata T., et al. Single-dose pharmacokinetics of lamotrigine in children: influence of age and antiepileptic comedication. Ther Drug Monit. 2001;23(3):217-222.
Battino D., Estienne M., Avanzini G. Clinical pharmacokinetics of antiepileptic drugs in paediatric patients. Part II. Phenytoin, carbamazepine, sulthiame, lamotrigine, vigabatrin, oxcarbazepine and felbamate. Clin Pharmacokinet. 1995;29(5):341-369.
Ben-Menachem E. Potential antiepileptic drugs: Topiramate. In: Levy R.H., Mattson R.H., Meldrum B.S., editors. Antiepileptic Drugs. New York: Raven Press; 1995:1063.
Ben-Menachem E. Vigabatrin: Chemistry, absorption, distribution, and elimination. In: Levy R.H., Mattson R.H., Meldrum B.S., editors. Antiepileptic Drugs. New York: Raven Press; 1995:915.
Benes J., Parada A., Figueiredo A.A., et al. Anticonvulsant and sodium channel-blocking properties of novel 10,11-dihydro-5H-dibenz[b,f]azepine-5-carboxamide derivatives. J Med Chem. 1999;42(14):2582-2587.
Berg A.T., Shinnar S. Relapse following discontinuation of antiepileptic drugs: a meta-analysis. Neurology. 1994;44(4):601-608.
Bergen D.C., Ristanovic R.K., Waicosky K., et al. Weight loss in patients taking felbamate. Clin Neuropharmacol. 1995;18(1):23-27.
Birnbaum A.K., Kriel R.L., Burkhardt R.T., et al. Rectal absorption of lamotrigine compressed tablets. Epilepsia. 2000;41(7):850-853.
Birnbaum A.K., Kriel R.L., Im Y., et al. Relative bioavailability of lamotrigine chewable dispersible tablets administered rectally. Pharmacotherapy. 2001;21(2):158-162.
Birnbaum A.K., Kriel R.L., Norberg S.K., et al. Rapid infusion of sodium valproate in acutely ill children. Pediatr Neurol. 2003;28(4):300-303.
Borusiak P., Korn-Merker E., Holert N., et al. Hyponatremia induced by oxcarbazepine in children. Epilepsy Res. 1998;30(3):241-246.
Brockbrader H., Hunt T., Strand J., et al. Pregabalin pharmacokinetics and safety in healthy volunteers: Results from two phase I studies. Neurology. 2000;54(Suppl 3):421.
Brodie M.J., Wilson E.A., Wesche D.L., et al. Pregabalin drug interaction studies: lack of effect on the pharmacokinetics of carbamazepine, phenytoin, lamotrigine, and valproate in patients with partial epilepsy. Epilepsia. 2005;46(9):1407-1413.
Buetefisch C.M., Gutierrez A., Gutmann L. Choreoathetotic movements: A possible side effect of gabapentin. Neurology. 1996;46(3):851-852.
Burkhardt R.T., Leppik I.E., Blesi K., et al. Lower phenytoin serum levels in persons switched from brand to generic phenytoin. Neurology. 2004;63(8):1494-1496.
Burstein A.H., Modica R., Hatton M., et al. Pharmacokinetics and pharmacodynamics of midazolam after intranasal administration. J Clin Pharmacol. 1997;37(8):711-718.
Butler R.T., Kalkwarf K.L., Kaldahl W.B. Drug-induced gingival hyperplasia: phenytoin, cyclosporine, and nifedipine. J Am Dent Assoc. 1987;114(1):56-60.
Cacek A.T. Review of alterations in oral phenytoin bioavailability associated with formulation, antacids, and food. Ther Drug Monit. 1986;8(2):166-171.
Camfield C., Camfield P., Smith E., et al. Asymptomatic children with epilepsy: little benefit from screening for anticonvulsant-induced liver, blood, or renal damage. Neurology. 1986;36(6):838-841.
Camfield P., Camfield C. When is it safe to discontinue AED treatment? Epilepsia. 2008;49(Suppl 9):25-28.
Camfield P., Camfield C., Dooley J., et al. Routine screening of blood and urine for severe reactions to anticonvulsant drugs in asymptomatic patients is of doubtful value. CMAJ. 1989;140(11):1303-1305.
Canovas Martinez A., Ordovas Baines J.P., Beltran Marques M., et al. Vigabatrin-associated reversible acute psychosis in a child. Ann Pharmacother. 1995;29(11):1115-1117.
Capparelli E.V. Pharmacokinetic considerations in the adolescent: non-cytochrome P450 metabolic pathways. J Adolesc Health. 1994;15(8):641-647.
Chadwick D., Taylor J., Johnson T. Outcomes after seizure recurrence in people with well-controlled epilepsy and the factors that influence it. The MRC Antiepileptic Drug Withdrawal Group. Epilepsia. 1996;37(11):1043-1050.
Chen C., Casale E.J., Duncan B., et al. Pharmacokinetics of lamotrigine in children in the absence of other antiepileptic drugs. Pharmacotherapy. 1999;19(4):437-441.
Chhun S., Jullien V., Rey E., et al. Population pharmacokinetics of levetiracetam and dosing recommendation in children with epilepsy. Epilepsia. 2009;50(5):1150-1157.
Clark J.E., Covanis A., Gupta A.K., et al. Unwanted effects of sodium valproate in children and adolescents. In: Parsonage M.J., Caldwell A.D.S., editors. The place of sodium valproate in the treatment of epilepsy. London: Academic Press; 1980:30.
Clemens P.L., Cloyd J.C., Kriel R.L., et al. Relative bioavailability, metabolism and tolerability of rectally administered oxcarbazepine suspension. Clin Drug Investig. 2007;27(4):243-250.
Cloyd J.C. Pharmacokinetic pitfalls of present antiepileptic medications. Epilepsia. 1991;32(Suppl 5):S53.
Cloyd J.C., Fischer J.H., Kriel R.L., et al. Valproic acid pharmacokinetics in children. IV. Effects of age and antiepileptic drugs on protein binding and intrinsic clearance. Clin Pharmacol Ther. 1993;53(1):22-29.
Cloyd J.C., Kriel R.L., Fischer J.H. Valproic acid pharmacokinetics in children. II. Discontinuation of concomitant antiepileptic drug therapy. Neurology. 1985;35(11):1623-1627.
Cloyd J.C., Kriel R.L., Fischer J.H., et al. Pharmacokinetics of valproic acid in children: I. Multiple antiepileptic drug therapy. Neurology. 1983;33(2):185-191.
Cloyd J.C., Kriel R.L., Jones-Saete C.M., et al. Comparison of sprinkle versus syrup formulations of valproate for bioavailability, tolerance, and preference. J Pediatr. 1992;120(4 Pt 1):634-638.
Conway J.M., Birnbaum A.K., Kriel R.L., et al. Relative bioavailability of topiramate administered rectally. Epilepsy Res. 2003;54(2–3):91-96.
Coulter D.L., Allen R.J. Pancreatitis associated with valproic acid therapy for epilepsy. Ann Neurol. 1980;7(1):92.
Covanis A., Gupta A.K., Jeavons P.M. Sodium valproate: monotherapy and polytherapy. Epilepsia. 1982;23(6):693-720.
Crumrine P.K. Antiepileptic drug selection in pediatric epilepsy. J Child Neurol. 2002;17(Suppl 2):2S2. -2S8
Curatolo P., Bruni O., Cusmai R. Use of erythromycin to inhibit carbamazepine metabolism in children with partial complex seizures. Brain Dev. 1988;10(3):206.
Dahlin M.G., Beck O.M., Amark P.E. Plasma levels of antiepileptic drugs in children on the ketogenic diet. Pediatr Neurol. 2006;35(1):6-10.
Dalla Bernardina B., Fontana E., Vigevano F., et al. Efficacy and tolerability of vigabatrin in children with refractory partial seizures: a single-blind dose-increasing study. Epilepsia. 1995;36(7):687-691.
de Krom M.C., Verduin N., Visser E., et al. Status epilepticus during vigabatrin treatment: a report of three cases. Seizure. 1995;4(2):159-162.
Degen P.H., Flesch G., Cardot J.M., et al. The influence of food on the disposition of the antiepileptic oxcarbazepine and its major metabolites in healthy volunteers. Biopharm Drug Dispos. 1994;15(6):519-526.
DeToledo J.C., Toledo C., DeCerce J., et al. Changes in body weight with chronic, high-dose gabapentin therapy. Ther Drug Monit. 1997;19(4):394-396.
Devinsky O., Vazquez B., Luciano D. New antiepileptic drugs for children: felbamate, gabapentin, lamotrigine, and vigabatrin. J Child Neurol. 1994;9(Suppl 1):S33-S45.
Dodson W.E. Antiepileptic drug utilization in pediatric patients. Epilepsia. 1984;25(Suppl 2):S132-S139.
Donn K.H., Drissel D.A., Quon C.Y., et al. Systemic availability and pharmacokinetics of phenytoin after intramuscular ACC-9653, a phenytoin prodrug. Epilepsia. 1987;28:S87.
Doose D.R., Gisclon L.G., Stellar S.M., et al. The effect of food on the bioavailability of topiramate from 100- and 400-mg tablets in healthy male subjects. Epilepsia. 1992;33(Suppl 3):105.
Dulac O., Chiron C., Luna D., et al. Vigabatrin in childhood epilepsy. J Child Neurol. 1991(Suppl 2):S30-S37.
Duncan J.S., Shorvon S.D., Trimble M.R. Effects of removal of phenytoin, carbamazepine, and valproate on cognitive function. Epilepsia. 1990;31(5):584-591.
Dutta S., Reed R.C., Cavanaugh J.H. Absolute bioavailability and absorption characteristics of divalproex sodium extended-release tablets in healthy volunteers. J Clin Pharmacol. 2004;44(7):737-742.
Dutta S., Zhang Y. Bioavailability of divalproex extended-release formulation relative to the divalproex delayed-release formulation. Biopharm Drug Dispos. 2004;25(8):345-352.
Easterling D.E., Zakzewshi T., Moyer M.D., et al. Plasma pharmacokinetics of topiramate, a new anticonvulsant in humans. Epilepsia. 1988;29:662.
Eichelbaum M., Ekbom K., Bertilsson L., et al. Plasma kinetics of carbamazepine and its epoxide metabolite in man after single and multiple doses. Eur J Clin Pharmacol. 1975;8(5):337-341.
Elger C.E., Stefan H., Mann A., et al. A 24-week multicenter, randomized, double-blind, parallel-group, dose-ranging study of rufinamide in adults and adolescents with inadequately controlled partial seizures. Epilepsy Res. 2010;88(2–3):255-263.
Faigle J.W., Feldmann K.F. Pharmacokinetic data of carbamazepine and its major metabolites in man. In: Schneider H., Janz D., Gardner-Thorpe C., et al, editors. Clinical pharmacology of antiepileptic drugs. Berlin: Springer-Verlag; 1975:159.
Farhat G., Yamout B., Mikati M.A., et al. Effect of antiepileptic drugs on bone density in ambulatory patients. Neurology. 2002;58(9):1348-1353.
Farwell J.R., Lee Y.J., Hirtz D.G., et al. Phenobarbital for febrile seizures–effects on intelligence and on seizure recurrence. N Engl J Med. 1990;322(6):364-369.
Feldmann K.F., Brechbuhler S., Faigle J.S., et al. Pharmacokinetics and metabolism of GP 47 680, a compound related to CBZ in animals and man. In: Meinardi H., RA J., editors. Advances in epileptology. Amsterdam/Lisse: Sets and Zeitlinger; 1977:290.
Fenichel G.M. Anticonvulsant therapy in children. Int Pediatr. 1986;1:231.
Ferrie C.D., Robinson R.O., Panayiotopoulos C.P. Psychotic and severe behavioural reactions with vigabatrin: a review. Acta Neurol Scand. 1996;93(1):1-8.
Finkelstein J.W. The effect of developmental changes in adolescence on drug disposition. J Adolesc Health. 1994;15(8):612-618.
Fischer J.H., Barr A.N., Paloucek F.P., et al. Effect of food on the serum concentration profile of enteric-coated valproic acid. Neurology. 1988;38(8):1319-1322.
Frisk-Holmberg M., Kerth P., Meyer P. Effect of food on the absorption of vigabatrin. Br J Clin Pharmacol. 1989;27(Suppl 1):23S-25S.
Froscher W., Engels H.G. Tolerance to the anticonvulsant effect of clonazepam: Tolerance to beneficial adverse effects of antiepileptic drugs. New York: Raven Press; 1986.
Fuerst R.H., Graves N.M., Kriel R.L., et al. Absorption and safety of rectally administered phenytoin. Eur J Drug Metab Pharmacokinet. 1988;13(4):257-260.
Gallassi R., Morreale A., Di Sarro R., et al. Cognitive effects of antiepileptic drug discontinuation. Epilepsia. 1992;33(Suppl 6):S41-S44.
Gallo B.V., Slater J.D., Toledo C., et al. Pharmacokinetics and muscle histopathology of intramuscular valproate. Epilepsy Res. 1997;28(1):11-15.
Garnett W.R. Lamotrigine: pharmacokinetics. J Child Neurol. 1997;12(Suppl 1):S10-S15.
Garnett W.R., Cloyd J.C. Dosage form considerations in the treament of pediatric epilepsy. In: Dotson W.E., Pellock J.M., editors. Pediatric epilepsy: Diagnosis and therapy. New York: Demos; 1993:241.
Gelisse P., Genton P., Kuate C., et al. Worsening of seizures by oxcarbazepine in juvenile idiopathic generalized epilepsies. Epilepsia. 2004;45(10):1282-1286.
Gerber N., Mays D.C., Donn K.H., et al. Safety, tolerance and pharmacokinetics of intravenous doses of the phosphate ester of 3-hydroxymethyl-5,5-diphenylhydantoin: a new prodrug of phenytoin. J Clin Pharmacol. 1988;28(11):1023-1032.
Gerber P.E., Hamiwka L., Connolly M.B., et al. Factors associated with behavioral and cognitive abnormalities in children receiving topiramate. Pediatr Neurol. 2000;22(3):200-203.
Glauser T.A. Preliminary observations on topiramate in pediatric epilepsies. Epilepsia. 1997;38(Suppl 1):S37-S41.
Glauser T.A. Idiosyncratic reactions: new methods of identifying high-risk patients. Epilepsia. 2000;41(Suppl 8):S16-S29.
Glauser T.A., Mitchell W.G., Weinstock A., et al. Pharmacokinetics of levetiracetam in infants and young children with epilepsy. Epilepsia. 2007;48(6):1117-1122.
Glauser T.A., Pippenger C.E. Controversies in blood-level monitoring: reexamining its role in the treatment of epilepsy. Epilepsia. 2000;41(Suppl 8):S6-S15.
Goff D.A., Spunt A.L., Jung D., et al. Absorption characteristics of three phenytoin sodium products after administration of oral loading doses. Clin Pharm. 1984;3(6):634-638.
Gordon K., MacSween J., Dooley J., et al. Families are content to discontinue antiepileptic drugs at different risks than their physicians. Epilepsia. 1996;37(6):557-562.
Graves N.M., Kriel R.L. Rectal administration of antiepileptic drugs in children. Pediatr Neurol. 1987;3(6):321-326.
Graves N.M., Kriel R.L., Jones-Saete C. Bioavailability of rectally administered lorazepam. Clin Neuropharmacol. 1987;10(6):555-559.
Greenblatt D.J., Ehrenberg B.L., Gunderman J., et al. Kinetic and dynamic study of intravenous lorazepam: comparison with intravenous diazepam. J Pharmacol Exp Ther. 1989;250(1):134-140.
Gustavson L.E., Boellner S.W., Granneman G.R., et al. A single-dose study to define tiagabine pharmacokinetics in pediatric patients with complex partial seizures. Neurology. 1997;48(4):1032-1037.
Gustavson L.E., Mengel H.B. Pharmacokinetics of tiagabine, a gamma-aminobutyric acid-uptake inhibitor, in healthy subjects after single and multiple doses. Epilepsia. 1995;36(6):605-611.
Haegele K.D., Schoun J., Alken R.G., et al. Determination of the R(−)- and S(+)-enantiomers of gamma-vinyl-gamma-aminobutyric acid in human body fluids by gas chromatography–mass spectrometry. J Chromatogr. 1983;274:103-110.
Haig G.M., Bockbrader H.N., Wesche D.L., et al. Single-dose gabapentin pharmacokinetics and safety in healthy infants and children. J Clin Pharmacol. 2001;41(5):507-514.
Handfield-Jones S.E., Jenkins R.E., Whittaker S.J., et al. The anticonvulsant hypersensitivity syndrome. Br J Dermatol. 1993;129(2):175-177.
Harden C.L. Therapeutic safety monitoring: what to look for and when to look for it. Epilepsia. 2000;41(Suppl 8):S37-S44.
Hart R.G., Easton J.D. Carbamazepine and hematological monitoring. Ann Neurol. 1982;11(3):309-312.
Henriksen O. Specific problems of children with epilepsy. Epilepsia. 1988;29(Suppl 3):S6-S9.
Herranz J.L., Armijo J.A., Arteaga R. Effectiveness and toxicity of phenobarbital, primidone, and sodium valproate in the prevention of febrile convulsions, controlled by plasma levels. Epilepsia. 1984;25(1):89-95.
Herranz J.L., Armijo J.A., Arteaga R. Clinical side effects of phenobarbital, primidone, phenytoin, carbamazepine, and valproate during monotherapy in children. Epilepsia. 1988;29(6):794-804.
Hogan R.E., Bertrand M.E., Deaton R.L., et al. Total percentage body weight changes during add-on therapy with tiagabine, carbamazepine and phenytoin. Epilepsy Res. 2000;41(1):23-28.
Hoke K.D., Chi E.M., Antony K.K., et al. Bioequivalence and relative bioavailability of vigabatrin. Epilepsia. 1991;32(Suppl 3):7.
Holtmann M., Krause M., Opp J., et al. Oxcarbazepine-induced hyponatremia and the regulation of serum sodium after replacing carbamazepine with oxcarbazepine in children. Neuropediatrics. 2002;33(6):298-300.
Horn C.S., Ater S.B., Hurst D.L. Carbamazepine-exacerbated epilepsy in children and adolescents. Pediatr Neurol. 1986;2(6):340-345.
Hovinga C.A. SPM-927 (Schwarz Pharma). IDrugs. 2003;6(5):479-485.
Hunt P.A., Wu-Chen M.L., Handal N.J., et al. Bone disease induced by anticonvulsant therapy and treatment with calcitriol (1,25-dihydroxyvitamin D3). Am J Dis Child. 1986;140(7):715-718.
Hussey E.K., Dukes G.E., Messenheimer J.A., et al. Evaluation of the pharmacokinetic interaction between diazepam and ACC-9653 (a phenytoin prodrug) in healthy male volunteers. Pharm Res. 1990;7(11):1172-1176.
Jamerson B.D., Donn K.H., Dukes G.E., et al. Absolute bioavailability of phenytoin after 3-phosphoryloxymethyl phenytoin disodium (ACC-9653) administration to humans. Epilepsia. 1990;31(5):592-597.
Jannuzzi G., Cian P., Fattore C., et al. A multicenter randomized controlled trial on the clinical impact of therapeutic drug monitoring in patients with newly diagnosed epilepsy. The Italian TDM Study Group in Epilepsy. Epilepsia. 2000;41(2):222-230.
Johannessen S.I. Pharmacokinetics and interaction profile of topiramate: Review and comparison with other new antipileptic drugs. Epilepsia. 1997;38(Suppl 1):S18.
Jung D., Powell J.R., Walson P., et al. Effect of dose on phenytoin absorption. Clin Pharmacol Ther. 1980;28(4):479-485.
Jung M.J., Lippert B., Metcalf B.W., et al. gamma-Vinyl GABA (4-amino-hex-5-enoic acid), a new selective irreversible inhibitor of GABA-T: effects on brain GABA metabolism in mice. J Neurochem. 1977;29(5):797-802.
Karas B.J., Wilder B.J., Hammond E.J., et al. Valproate tremors. Neurology. 1982;32(4):428-432.
Kauko K., Tammisto P. Comparison of two generically equivalent carbamazepine preparations. Ann Clin Res. 1974;6(0 Suppl 11):21-25.
Kellinghaus C. Lacosamide as treatment for partial epilepsy: mechanisms of action, pharmacology, effects, and safety. Ther Clin Risk Manag. 2009;5:757-766.
Keppra. UCB Pharma I., editor. Levetiracetam; product information. Smyrna, GA, 2004.
Kerr B.M., Levy R.H. Carbamazepine-carbamazepine epoxide. In: Levy R.H., Mattson R., Meldrum B., editors. Antiepileptic drugs. New York: Raven Press; 1989:505.
Khurana D.S., Riviello J., Helmers S., et al. Efficacy of gabapentin therapy in children with refractory partial seizures. J Pediatr. 1996;128(6):829-833.
Klitgaard N.A., Kristensen O. Use of saliva for monitoring oxcarbazepine therapy in epileptic patients. Eur J Clin Pharmacol. 1986;31(1):91-94.
Knudsen J.F., Thambi L.R., Kapcala L.P., et al. Oligohydrosis and fever in pediatric patients treated with zonisamide. Pediatr Neurol. 2003;28(3):184-189.
Koella W.P. Tolerance: Its various forms and their nature. In: Frey H.H., Froscher W., Koella W.P., editors. Tolerance to beneficial and adverse effects of antiepileptic drugs. New York: Raven Press; 1986:1.
Koivikko M.J., Valikangas S.L. Hyponatraemia during carbamazepine therapy in children. Neuropediatrics. 1983;14(2):93-96.
Koren G., Brand N., Halkin H., et al. Kinetics of intravenous phenytoin in children. Pediatr Pharmacol (New York). 1984;4(1):31-38.
Kossoff E.H., Pyzik P.L., Furth S.L., et al. Kidney stones, carbonic anhydrase inhibitors, and the ketogenic diet. Epilepsia. 2002;43(10):1168-1171.
Kostenbauder H.B., Rapp R.P., McGovren J.P., et al. Bioavailability and single-dose pharmacokinetics of intramuscular phenytoin. Clin Pharmacol Ther. 1975;18(4):449-456.
Kothare S.V., Kaleyias J., Mostofi N., et al. Efficacy and safety of zonisamide monotherapy in a cohort of children with epilepsy. Pediatr Neurol. 2006;34(5):351-354.
Kozer E., Scolnik D., Agamata W.M., et al. Utility of antiepileptic drug monitoring in the pediatric emergency department. Ther Drug Monit. 2003;25(1):17-21.
Krauss G.L., Johnson M.A., Miller N.R. Vigabatrin-associated retinal cone system dysfunction: electroretinogram and ophthalmologic findings. Neurology. 1998;50(3):614-618.
Kriel R.L., Birnbaum A.K., Cloyd J.C., et al. Failure of absorption of gabapentin after rectal administration. Epilepsia. 1997;38(11):1242-1244.
Kudriakova T.B., Sirota L.A., Rozova G.I., et al. Autoinduction and steady-state pharmacokinetics of carbamazepine and its major metabolites. Br J Clin Pharmacol. 1992;33(6):611-615.
Kutt H. Effects of acute and chronic diseases on the disposition of antiepileptic drugs. In: Morselli P.L., Pippenger C.E., Penry J.K., editors. Antiepileptic drug therapy in pediatrics. New York: Raven Press; 1983:293.
Kutt H., Penry J.K. Usefulness of blood levels of antiepileptic drugs. Arch Neurol. 1974;31(5):283-288.
Kvan L., Johannessen S. Lack of efficacy of phenytoin suppositories in antiepileptic treatment. Acta Neurol Scand. 1975;54:103.
Lamictal. GlaxoSmithKline, editor. Lamotrigine: Product information. Research Triangle Park, NC, 2004.
Larkin J.G., McKee P.J., Forrest G., et al. Lack of enzyme induction with oxcarbazepine (600 mg daily) in healthy subjects. Br J Clin Pharmacol. 1991;31(1):65-71.
Lee D.O., Steingard R.J., Cesena M., et al. Behavioral side effects of gabapentin in children. Epilepsia. 1996;37(1):87-90.
Lee S., Sziklas V., Andermann F., et al. The effects of adjunctive topiramate on cognitive function in patients with epilepsy. Epilepsia. 2003;44(3):339-347.
Leppik I.E., Boucher B.A., Wilder B.J., et al. Pharmacokinetics and safety of a phenytoin prodrug given i.v. or i.m. in patients. Neurology. 1990;40(3 Pt 1):456-460.
Leppik I.E., Boucher R., Wilder B.J., et al. Phenytoin prodrug: preclinical and clinical studies. Epilepsia. 1989;30(Suppl 2):S22-S26.
Leppik I.E., Cloyd J.C., Miller K. Development of tolerance to the side effects of primidone. Ther Drug Monit. 1984;6(2):189-191.
Leppik I.E., Derivan A.T., Homan R.W., et al. Double-blind study of lorazepam and diazepam in status epilepticus. JAMA. 1983;249(11):1452-1454.
Leppik I.E., Fisher J., Kriel R., et al. Altered phenytoin clearance with febrile illness. Neurology. 1986;36(10):1367-1370.
Limite A.I. Rufinamide: CGP 33101, E 2080, RUF 331, Xilep. Drugs R D. 2005;6(4):249-252.
Linday L.A. Developmental changes in renal tubular function. J Adolesc Health. 1994;15(8):648-653.
Lindhardt K., Gizurarson S., Stefansson S.B., et al. Electroencephalographic effects and serum concentrations after intranasal and intravenous administration of diazepam to healthy volunteers. Br J Clin Pharmacol. 2001;52(5):521-527.
Little T.M., Girgis S.S., Masotti R.E. Diphenylhydantoin-induced gingival hyperplasia: its response to changes in drug dosage. Dev Med Child Neurol. 1975;17(4):421-424.
Lloyd P., Flesch G., Dieterle W. Clinical pharmacology and pharmacokinetics of oxcarbazepine. Epilepsia. 1994;35(Suppl 3):S10-S13.
Lockard J.S., Levy R.H. Valproate and paroxysmal cyclicity compared to other anticonvulsants in the monkey model. New York: Raven Press; 1990.
Lombroso C.T. Intermittent home treatment of status and clusters of seizures. Epilepsia. 1989;30(Suppl 2):S11-S14.
Lortie A., Chiron C., Dumas C., et al. Optimizing the indication of vigabatrin in children with refractory epilepsy. J Child Neurol. 1997;12(4):253-259.
Loureiro and Fernandes-lopes. Hepatic UDP-glucuronosyltransferases responsible for eslicarbazepine glucuronidation. Proceedings of the Annual Meeting of the Portuguese Pharmacological Society 2008. 2008. Lisbon, Portugal
Luer M.S., Hamani C., Dujovny M., et al. Saturable transport of gabapentin at the blood-brain barrier. Neurol Res. 1999;21(6):559-562.
Maas B., Garnett W.R., Pellock J.M., et al. A comparative bioavailability study of carbamazepine tablets and a chewable tablet formulation. Ther Drug Monit. 1987;9(1):28-33.
MacKichan J.J. Influence of protein binding and use of unbound (free) drug concentrations. In: Evans W.E., Schentag J.J., Jusko W.J., editors. Applied pharmacokinetics: Principles of therapeutic drug monitoring. ed 3. Vancouver, WA: Applied Therapeutics; 1992:S1-S48.
Marchand M., Fuseau E., Critchley D.J. Supporting the recommended paediatric dosing regimen for rufinamide in Lennox-Gastaut syndrome using clinical trial simulation. J Pharmacokinet Pharmacodyn. 2010;37(1):99-118.
Marks W.A., Morris M.P., Bodensteiner J.B., et al. Gastritis with valproate therapy. Arch Neurol. 1988;45(8):903-905.
Matsumoto K., Miyazaki H., Fujii T., et al. Absorption, distribution and excretion of 3-(sulfamoyl[14C]methyl)-1,2-benziosoxazole (AD-810) in Rats, Dogs and Monkeys and of AD-810 in Men. Arzneimittelforschung. 1983;33(7):961-968.
Matsumoto K., Miyazaki H., Fujii T., et al. Binding of sulfonamides to erythrocytes and their components. Chem Pharm Bull (Tokyo). 1989;37(7):1913-1915.
May T.W., Rambeck B., Jurgens U. Serum concentrations of Levetiracetam in epileptic patients: the influence of dose and co-medication. Ther Drug Monit. 2003;25(6):690-699.
Maynard G.A., Jones K.M., Guidry J.R. Phenytoin absorption from tube feedings. Arch Intern Med. 1987;147(10):1821.
Meador K.J., Baker G.A., Browning N., et al. Cognitive function at 3 years of age after fetal exposure to antiepileptic drugs. N Engl J Med. 2009;360(16):1597-1605.
Mengel H., Gustavson L.E., Sorensen H.J., et al. Bioavailability and tolerability of a tiagabine HC1 tablet formulation versus capsules and oral solution in normal subjects. Epilepsia. 1991;32(Suppl 3):6.
Mengel H., Gustavson L.E., Sorensen H.J., et al. Effect of food on the bioavailability of tiagabine HC1. Epilepsia. 1991;32(Suppl 3):6.
Meyer M.C., Straughn A.B. Biopharmaceutical factors in seizure control and drug toxicity. Am J Hosp Pharm. 1993;50(12 Suppl 5):S17-S22.
Meyer M.C., Straughn A.B., Jarvi E.J., et al. The bioinequivalence of carbamazepine tablets with a history of clinical failures. Pharm Res. 1992;9(12):1612-1616.
Mikati M., Bassett N., Schachter S. Double-blind randomized study comparing brand-name and generic phenytoin monotherapy. Epilepsia. 1992;33(2):359-365.
Mikati M.A., Fayad M., Koleilat M., et al. Efficacy, tolerability, and kinetics of lamotrigine in infants. J Pediatr. 2002;141(1):31-35.
Mitchell W.G., Zhou Y., Chavez J.M., et al. Reaction time, attention, and impulsivity in epilepsy. Pediatr Neurol. 1992;8(1):19-24.
Miura H. Zonisamide monotherapy with once-daily dosing in children with cryptogenic localization-related epilepsies: clinical effects and pharmacokinetic studies. Seizure. 2004;13S:S17-S23.
Modeer T., Dahllof G. Development of phenytoin-induced gingival overgrowth in non-institutionalized epileptic children subjected to different plaque control programs. Acta Odontol Scand. 1987;45(2):81-85.
Moolenaar F., Jelsma B.H., Wisser J., et al. Manipulation of rectal absorption rate of phenytoin in man. Pharm Weekbl. 1981;3:175.
Morijiri Y., Sato T. Factors causing rickets in institutionalised handicapped children on anticonvulsant therapy. Arch Dis Child. 1981;56(6):446-449.
Morrow J.I., Richens A. Disposition of anticonvulsants in childhood. Clin Pharmacokinet. 1989;17(Suppl 1):89-104.
Morselli P.L. Development of physiological variables important for drug kinetics. New York: Raven Press; 1983.
NoAuthors. Rufinamide: CGP 33101, E 2080, RUF 331, Xilep. Drugs R D. 2005;6(4):249-252.
Nuwer M.R., Browne T.R., Dodson W.E., et al. Generic substitutions for antiepileptic drugs. Neurology. 1990;40(11):1647-1651.
Ostergaard L.H., Gram L., Mogens D. Potential antiepileptic drugs: Tiagabine. In: Levy R.H., Mattson R.H., Meldrum B.S., editors. Antiepileptic drugs. New York: Raven Press; 1995:1058.
Otoul C., De Smedt H., Stockis A. Lack of pharmacokinetic interaction of levetiracetam on carbamazepine, valproic acid, topiramate, and lamotrigine in children with epilepsy. Epilepsia. 2007;48(11):2111-2115.
Ouellet D., Bockbrader H.N., Wesche D.L., et al. Population pharmacokinetics of gabapentin in infants and children. Epilepsy Res. 2001;47(3):229-241.
Painter M.J., Pippenger C., MacDonald H., et al. Phenobarbital and diphenylhydantoin levels in neonates with seizures. J Pediatr. 1978;92(2):315-319.
Patsalos P.N. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther. 2000;85(2):77-85.
Patsalos P.N., Elyas A.A., Zakrzewska J.M. Protein binding of oxcarbazepine and its primary active metabolite, 10-hydroxycarbazepine, in patients with trigeminal neuralgia. Eur J Clin Pharmacol. 1990;39(4):413-415.
Pellock J.M., Glauser T.A., Bebin E.M., et al. Pharmacokinetic study of levetiracetam in children. Epilepsia. 2001;42(12):1574-1579.
Pellock J.M., Willmore L.J. A rational guide to routine blood monitoring in patients receiving antiepileptic drugs. Neurology. 1991;41(7):961-964.
Penn R.D., Savoy S.M., Corcos D., et al. Intrathecal baclofen for severe spinal spasticity. N Engl J Med. 1989;320(23):1517-1521.
Pennell P.B., Peng L., Newport D.J., et al. Lamotrigine in pregnancy: clearance, therapeutic drug monitoring, and seizure frequency. Neurology. 2008;70(22 Pt 2):2130-2136.
Perhach J.L., Shumaker R.C. Felbamate: Absorption, distribution, and excretion. In: Levy R.H., Mattson R.H., Meldrum B.S., editors. Antiepileptic drugs. New York: Raven Press; 1995:810.
Perucca E., Cloyd J., Critchley D., et al. Rufinamide: clinical pharmacokinetics and concentration-response relationships in patients with epilepsy. Epilepsia. 2008;49(7):1123-1141.
Perucca E., Dulac O., Shorvon S., et al. Harnessing the clinical potential of antiepileptic drug therapy: dosage optimisation. CNS Drugs. 2001;15(8):609-621.
Perucca E., Gram L., Avanzini G., et al. Antiepileptic drugs as a cause of worsening seizures. Epilepsia. 1998;39(1):5-17.
Peters A.C., Brouwer O.F., Geerts A.T., et al. Randomized prospective study of early discontinuation of antiepileptic drugs in children with epilepsy. Neurology. 1998;50(3):724-730.
Pihlstrom B.L., Carlson J.F., Smith Q.T., et al. Prevention of phenytoin associated gingival enlargement – a 15-month longitudinal study. J Periodontol. 1980;51(6):311-317.
Pisani F., Oteri G., Caputo M., et al. Diurnal fluctuations in plasma drug levels at steady-state: Studies with valproic acid and carbamazepine. New York: Raven Press; 1990.
Porter R.J. General principles – how to use antiepileptic drugs. In: Levy R.H., Mattson R., Meldrum B., et al, editors. Antiepileptic drugs. New York: Raven Press, 1989.
Pynnonen S., Mantyla R., Iisalo E. Bioavailability of four different pharmaceutical preparations of carbamazepine. Acta Pharmacol Toxicol (Copenh). 1978;43(4):306-310.
Rane A., Wilson J.T. Clinical pharmacokinetics in infants and children. Clin Pharmacokinet. 1976;1(1):2-24.
Rawlins M.D., Collste P., Bertilsson L., et al. Distribution and elimination kinetics of carbamazepine in man. Eur J Clin Pharmacol. 1975;8(2):91-96.
Reeves A.L., So E.L., Sharbrough F.W., et al. Movement disorders associated with the use of gabapentin. Epilepsia. 1996;37(10):988-990.
Reimers A., Skogvoll E., Sund J.K., et al. Lamotrigine in children and adolescents: the impact of age on its serum concentrations and on the extent of drug interactions. Eur J Clin Pharmacol. 2007;63(7):687-692.
Reiter E., Feucht M., Hauser E., et al. Changes in body mass index during long-term topiramate therapy in paediatric epilepsy patients – a retrospective analysis. Seizure. 2004;13(7):491-493.
Rey E., Pons G., Olive G. Vigabatrin. Clinical pharmacokinetics. Clin Pharmacokinet. 1992;23(4):267-278.
Rey E., Pons G., Olive G., et al. Pharmacokinetics of vigabatrin (GVC) in children with measurement of R(−) and S(+) enantiomers. Proceedings of the 18th International Epilepsy Congress. 1989. New Delhi, Oct 17–22
Rey E., Pons G., Richard M.O., et al. Pharmacokinetics of the individual enantiomers of vigabatrin (gamma-vinyl GABA) in epileptic children. Br J Clin Pharmacol. 1990;30(2):253-257.
Richens A. Clinical pharmacokinetics of gabapentin. In: Chadwick D., editor. New trends in epilepsy management: the role of gabapentin. London: Royal Society of Medicine Services; 1993:41.
Ring H.A., Crellin R., Kirker S., et al. Vigabatrin and depression. J Neurol Neurosurg Psychiatry. 1993;56(8):925-928.
Ritter F., Glauser T.A., Elterman R.D., et al. Effectiveness, tolerability, and safety of topiramate in children with partial-onset seizures. Topiramate YP Study Group. Epilepsia. 2000;41(Suppl 1):S82-S85.
Riva R., Albani F., Franzoni E., et al. Valproic acid free fraction in epileptic children under chronic monotherapy. Ther Drug Monit. 1983;5(2):197-200.
Romeo V.D., deMeireles J., Sileno A.P., et al. Effects of physicochemical properties and other factors on systemic nasal drug delivery. Adv Drug Deliv Rev. 1998;29(1–2):89-116.
Sankar P.S., Pasquale L.R., Grosskreutz C.L. Uveal effusion and secondary angle-closure glaucoma associated with topiramate use. Arch Ophthalmol. 2001;119(8):1210-1211.
Sarkar M.A., Garnett W.R., Karnes H.T. The effects of storage and shaking on the settling properties of phenytoin suspension. Neurology. 1989;39(2 Pt 1):207-209.
Scheepers M., Scheepers B., Clarke M., et al. Is intranasal midazolam an effective rescue medication in adolescents and adults with severe epilepsy? Seizure. 2000;9(6):417-422.
Scheyer R.D., Cramer J.A. Pharmacokinetics of antiepileptic drugs. Semin Neurol. 1990;10(4):414-421.
Schlumberger E., Chavez F., Palacios L., et al. Lamotrigine in treatment of 120 children with epilepsy. Epilepsia. 1994;35(2):359-367.
Schmidt D., Haenel F. Therapeutic plasma levels of phenytoin, phenobarbital, and carbamazepine: individual variation in relation to seizure frequency and type. Neurology. 1984;34(9):1252-1255.
Schmidt D., Kramer G. The new anticonvulsant drugs. Implications for avoidance of adverse effects. Drug Saf. 1994;11(6):422-431.
Schoeman J.F., Elyas A.A., Brett E.M., et al. Correlation between plasma carbamazepine-10,11-epoxide concentration and drug side-effects in children with epilepsy. Dev Med Child Neurol. 1984;26(6):756-764.
Schutz H., Feldmann K.F., Faigle J.W., et al. The metabolism of 14C-oxcarbazepine in man. Xenobiotica. 1986;16(8):769-778.
Serrano E.E., Wilder B.J. Intramuscular administration of diphenylhydantoin. Histologic follow-up studies. Arch Neurol. 1974;31(4):276-278.
Sherwin A.L. How to use ethosuximide. New York: Raven Press; 1983.
Shinnar S., Berg A.T., Moshe S.L., et al. Discontinuing antiepileptic drugs in children with epilepsy: a prospective study. Ann Neurol. 1994;35(5):534-545.
Shumaker R.C., Fantel C., Kelton E., et al. Evaluation of the elimination of (14C) felbamate in healthy men. Epilepsia. 1990;31:642.
Siegel S. Environmental modulation of tolerance: Evidence from benzodiazepine research: Tolerance to beneficial and adverse effects of antiepileptic drugs. New York: Raven Press; 1986.
Sillanpaa M. Carbamazepine. Pharmacology and clinical uses. Acta Neurol Scand Suppl. 1981;88:1-202.
Snead O.C.3rd, Hosey L.C. Exacerbation of seizures in children by carbamazepine. N Engl J Med. 1985;313(15):916-921.
Stefan H., Burr W., Fichsel H., et al. Intensive follow-up monitoring in patients with once daily evening administration of sodium valproate. Epilepsia. 1984;25(2):152-160.
Stewart B.H., Kugler A.R., Thompson P.R., et al. A saturable transport mechanism in the intestinal absorption of gabapentin is the underlying cause of the lack of proportionality between increasing dose and drug levels in plasma. Pharm Res. 1993;10(2):276-281.
Tallian K.B., Nahata M.C., Lo W., et al. Gabapentin associated with aggressive behavior in pediatric patients with seizures. Epilepsia. 1996;37(5):501-502.
Tavernor S.J., Wong I.C., Newton R., et al. Rechallenge with lamotrigine after initial rash. Seizure. 1995;4(1):67-71.
Thambi L., Kapcala L.P., Chambers W., et al. Topiramate-associated secondary angle-closure glaucoma: a case series. Arch Ophthalmol. 2002;120(8):1108.
Theisohn M., Heimann G. Disposition of the antiepileptic oxcarbazepine and its metabolites in healthy volunteers. Eur J Clin Pharmacol. 1982;22(6):545-551.
Theodore W.H., Raubertas R.F., Porter R.J., et al. Felbamate: a clinical trial for complex partial seizures. Epilepsia. 1991;32(3):392-397.
Thompson P.J., Trimble M.R. Anticonvulsant drugs and cognitive functions. Epilepsia. 1982;23(5):531-544.
Troupin A.S., Ojemann L.M. Paradoxical intoxication – a complication of anticonvulsant administration. Epilepsia. 1975;16(5):753-758.
Venulet J., ten Ham M. Methods for monitoring and documenting adverse drug reactions. Int J Clin Pharmacol Ther. 1996;34(3):112-129.
Verrotti A., Trotta D., Salladini C., et al. Anticonvulsant hypersensitivity syndrome in children: incidence, prevention and management. CNS Drugs. 2002;16(3):197-205.
Vollmer K.O., von Hodenberg A., Kolle E.U. Pharmacokinetics and metabolism of gabapentin in rat, dog and man. Arzneimittelforschung. 1986;36(5):830-839.
Wang J.T., Shiu G.K., Ong-Chen T., et al. Effects of humidity and temperature on in vitro dissolution of carbamazepine tablets. J Pharm Sci. 1993;82(10):1002-1005.
Wheless J.W., Clarke D., Hovinga C.A., et al. Rapid infusion of a loading dose of intravenous levetiracetam with minimal dilution: a safety study. J Child Neurol. 2009;24(8):946-951.
Wilder B.J., Karas B.J., Penry J.K., et al. Gastrointestinal tolerance of divalproex sodium. Neurology. 1983;33(6):808-811.
Wilensky A.J., Friel P.N., Ojemann L.M., et al. Pharmacokinetics of W-554 (ADD 03055) in epileptic patients. Epilepsia. 1985;26(6):602-606.
Willmore L.J., Triggs W.J., Pellock J.M. Valproate toxicity: risk-screening strategies. J Child Neurol. 1991;6(1):3-6.
Wirrell E.C. Valproic acid-associated weight gain in older children and teens with epilepsy. Pediatr Neurol. 2003;28(2):126-129.
Wolf S.M., Forsythe A. Behavior disturbance, phenobarbital, and febrile seizures. Pediatrics. 1978;61(5):728-731.
Wyllie E., Wyllie R., Cruse R.P., et al. Pancreatitis associated with valproic acid therapy. Am J Dis Child. 1984;138(10):912-914.
Yager J.Y., Seshia S.S. Sublingual lorazepam in childhood serial seizures. Am J Dis Child. 1988;142(9):931-932.
Yuen W.C., Peck A.W. Lamotrigine pharmacokinetics: Oral and IV infusion in man. Epilepsia. 1987;28:528.
ZonegranProductInformation. Zonegran. Zonisamide: Product information. I. Eisai. 2004. Teaneck, NJ
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 59 Antiepileptic Drug Therapy in Children
Rational management of antiepileptic drug therapy in children requires an understanding of pharmacokinetics, pharmacodynamics, and toxicology of these agents. Pharmacokinetics is the study of drug absorption, distribution, metabolism, and elimination: that is, what the body does to a drug. Pharmacodynamics is the study of a drug’s biochemical and physiologic effects: that is, what the drug does to the body. Children, much more than adults, have widely varying abilities to absorb, distribute, metabolize, and eliminate drugs [Rane and Wilson, 1976]. Evidence also exists that antiepileptic drug pharmacodynamics in children differ from those in adults [Henriksen, 1988]. Application of basic pharmacokinetic and pharmacodynamic principles to antiepileptic drug therapy facilitates the attainment and maintenance of targeted serum concentrations, clinical response, control of drug interactions, and optimization of clinical response.
Pharmacokinetic Principles
This chapter will review general pharmacokinetic principles and discuss the clinically relevant features of specific antiepileptic drugs. For quick reference, pharmacokinetic information for the antiepileptic drugs is summarized in tables, with absorption characteristics provided in Table 59-1, and volume of distribution, protein binding, half-life, and metabolites presented in Table 59-2 and Table 59-3.
Absorption
Absorption is a complex phenomenon controlled by the physicochemical properties of the drug, the formulation, and the physiologic conditions at the sites of absorption: stomach, small intestine, colon, rectum, buccal and nasal mucosa, and muscle. Most drugs are given orally as solid formulations, which must first disintegrate into small particles that are then dissolved in the gastrointestinal fluid. Only after a drug is dissolved is it available for absorption. Most drugs are absorbed by passive diffusion of the unionized form but some drugs use active transport mechanisms. Gastrointestinal absorption is influenced by age-dependent changes in physiologic variables, such as gastric emptying time, stomach and intestinal pH, absorptive area, and gastrointestinal transit time. Food intake may affect antiepileptic drug pharmacokinetics, especially absorption. Although there is concern that the ketogenic diet may alter dosing requirements, a recent study indicated that there are no significant changes in the concentrations of several antiepileptic drugs (valproate – free or total, topiramate, phenobarbital, lamotrigine, clonazepam) during the ketogenic diet [Dahlin et al., 2006].
Descriptors of absorption, such as Cmax and Tmax, and the bioavailability, may vary considerably among different formulations of the same drug. The relative rates of absorption, and thus Tmax and Cmax, adhere to the following order of most rapid to slowest, and highest peak concentration to lowest: solutions > suspensions > capsules > tablets > extended-release formulations [Garnett and Cloyd, 1993]. Formulation-related differences in absorption characteristics are greatly influenced by the composition of the inactive ingredients (e.g., filler, particle size of drug) and nature of the capsule or tablet coating).
Volume of Distribution
The change in plasma concentration term denotes the desired increase after administration of the loading dose. For example, if a patient has a plasma concentration of 5 mg/L and the target concentration is 20 mg/L, the desired change in concentration is 15 mg/L. For phenytoin, the Vd is approximately 0.75 L/kg, so an IV dose of 11.25 mg/kg is required. When loading doses are administered intramuscularly, orally, or rectally, resultant maximum plasma concentrations are usually less than after IV loading, owing to slower and incomplete absorption and the elimination of some drug during the absorption phase. Table 59-4 presents loading dose information for six major antiepileptic drugs.
Protein Binding
In general, only the unbound drug is able to cross the blood–brain barrier and is in equilibrium with brain and spinal fluid (Figure 59-1; see also Table 59-3). Consequently, clinical response and toxicity correlate better with the unbound drug concentration than with the total drug concentration. For drugs that have limited protein binding (less than 50 percent) or a constant ratio of unbound to total concentration, measurement of total plasma concentrations is a reliable indicator of response. However, if a drug is highly protein-bound (greater than 85 percent), measurements of total concentrations may be an unreliable guide to clinical outcomes. The fraction bound may change because of illness, age, or other drugs that compete for binding sites. In these circumstances, it may be necessary to measure the free concentration to understand the clinical effect better. Low-extraction drugs are those that have only a small portion of the drug metabolized by each pass through the liver. Most of the antiepileptic drugs are low-extraction drugs and alterations in protein binding do not affect the steady-state concentration of the unbound drug. Thus, adjustments in dose generally are not necessary when protein binding is altered because the unbound concentrations remain unchanged. Erroneously increasing the dose when total levels are low, but unbound levels have not changed, will likely result in toxicity.
Metabolism and Elimination
Clearance is the volume of blood from which drug is removed (cleared) per unit of time. It is identical conceptually and mathematically to the more familiar physiologic parameter of creatinine clearance. For antiepileptic drugs that are eliminated by the liver, clearance is determined by the free concentration of the drug and the intrinsic ability of hepatic enzymes to metabolize the unbound drug. Although, for most drugs, clearance is proportional to dose, some drugs exhibit nonlinear pharmacokinetics in which clearance changes with concentration (Figure 59-2).
The following points are important to remember:
Clearance and absorption determine the amount of drug a patient requires to maintain a targeted steady-state plasma concentration.
Thus, t½ is not a direct measure of Cl, but is useful in calculating the time to reach steady state or the time required for drug elimination. After initiation or modification of therapy or after any change in clearance, 5 half-lives must elapse for a new steady-state plasma concentration to be reached (see Figure 59-2). The time required to attain steady state can be explained by using the definition of t½. The plasma concentration in the body declines 50 percent in 1 t½; therefore, the percentage of drug remaining in the body, as indicated by the plasma concentration after 2, 3, 4, and 5 half-lives, is 75 percent, 87.5 percent, 93.75 percent, and 96.875 percent, respectively. The actual amount of drug remaining in the body and the additional amount of drug removed in subsequent half-lives are so small (after 5 half-lives) that, for practical purposes, it can be ignored. However, the rate of decline is most rapid in the first two half-lives, and the risk of seizures is greatest during this period.
Biotransformation (metabolism) of medications is accomplished through biochemical reactions. These reactions create compounds that are more polar (more water-soluble) and are eliminated from the body, usually through the kidneys. Phase I reactions (e.g., using the cytochrome P-450 system) involve oxidation and reduction. Phase II reactions (non-cytochrome P-450-mediated) couple the drug or its metabolite with endogenous substrates by conjugation or synthesis. The endogenous substrate may be glucuronic acid, glutathione, glucose, sulfate, or acetate, making the drug more polar and hence water-soluble [Capparelli, 1994]. Antiepileptic drugs may undergo metabolism by phase I or II reactions, or a combination of both. As persons mature, the pathways may change and the metabolite profile will be altered.
In the neonate, phase I reactions, such as hydroxylation and, to a lesser extent, dealkylation, are significantly depressed [Anderson and Lynn, 2009]. In older children, however, phase I activity generally exceeds that in adolescents or adults. As a general rule, the rate of drug metabolism reaches a peak at 0.5–2 years of age and then declines in an exponential manner, reaching adult values in early adolescence. As an example, the clearance of valproic acid, in which metabolism includes both phase I and phase II reactions, is fastest in young children and then declines to reach adult values at the age of approximately 12–15 years. Also, its metabolism shifts from phase II to phase I reactions.
Active metabolites formed after biotransformation of the parent compound can contribute significantly to desired and toxic effects of the parent drug. Metabolites exist in both bound and unbound states. Only the unbound metabolite is available for elimination or biotransformation to an inactive metabolite. As with unbound parent drug, the unbound metabolite concentration in the plasma most closely correlates with the concentration of metabolite at the site of action. The presence of active metabolites complicates interpretation of plasma drug concentrations and clinical response. As with the parent drug, the relationship between unbound and total metabolite concentrations can be disrupted by displacement from serum proteins or changes in metabolism. In fact, parent or metabolite concentrations may be altered independently by drug interaction or physiologic change in metabolism. Table 59-5 displays the cytochrome P-450 isoform families involved in antiepileptic drug metabolism, along with known inducers and inhibitors.
Renal elimination is accomplished by glomerular filtration and/or renal tubular secretion. Glomerular filtration rate and renal tubular function in newborns are less efficient than in adults [Linday, 1994]. After 1 year of age, glomerular filtration rate, corrected for body surface area, is the same as for older children and young adults. Renal tubular function reaches adult values, normalized to body surface areas, within several months. Decreases in clearance can be better correlated with sexual maturity than with chronologic age. Making correlations with physiologic or sexual maturation and developmental age, rather than chronologic age, may be helpful in accounting for the variability observed in clearance of drugs that are renally eliminated [Finkelstein, 1994].
Specific Antiepileptic Drugs
Benzodiazepines
The benzodiazepines rapidly cross the blood–brain barrier. Brain concentrations reach equilibrium with plasma concentrations within seconds after IV administration. But the highly lipid-soluble nature of diazepam and lorazepam causes them to redistribute to other body tissues, resulting in a rapid decrease in brain concentrations and a very high Vd. This redistribution is responsible for the short duration of action after a single dose [Greenblatt et al., 1989; Leppik et al., 1983]. The benzodiazepines are metabolized by the liver and have linear elimination kinetics. Some of the metabolites exhibit antiepileptic activity and also may contribute to toxicity. Because the active metabolites have very long half-lives, repeated administration of these drugs, culminating in large total doses, may lead to long periods of sedation, usually most problematic for diazepam and chlorazepate.
Carbamates
Carbamazepine
Carbamazepine is a very water-insoluble, neutral compound with a slow dissolution rate in gastrointestinal fluid. The relative bioavailability is similar for all four formulations (i.e., suspension, chewable tablet, extended-release, and regular tablet), ranging from 75 to 90 percent, although marked intrapatient and interpatient variability has been observed [Faigle and Feldmann, 1975; Maas et al., 1987]. In children on maintenance therapy, the rate of absorption is much faster for the suspension than for tablet formulations, resulting in a significantly earlier Tmax and higher Cmax (Figure 59-3). This property occasionally produces transient concentration-dependent central nervous system side effects. Administering the same daily dose in smaller amounts and giving it more frequently may correct this problem. Sustained-release preparations may permit twice-a-day dosing, eliminating the need for administration of medications during school hours. This improves medication compliance and decreases central nervous system side effects because peak concentrations will be lower. Studies conducted by the U.S. Food and Drug Administration have demonstrated that carbamazepine tablets are significantly less well absorbed after being exposed to high humidity [Meyer et al., 1992; Wang et al., 1993]. These findings emphasize the importance of storing all antiepileptic drugs at room temperature in a dry location.
The Vd in children for carbamazepine based on oral preparations ranges from 0.8 to 2.0 L/kg. Both carbamazepine and its active metabolite, carbamazepine-10,11-epoxide, are bound to several plasma proteins [Eichelbaum et al., 1975; Rawlins et al., 1975]. Approximately 65–85 percent of carbamazepine and 30–60 percent of carbamazepine-10,11-epoxide are bound to albumin. Both also bind to the reactant protein α1-acid glycoprotein. Binding to α1-acid glycoprotein increases after physiologic stress, such as from burn injuries or inflammatory diseases (e.g., juvenile rheumatoid arthritis). Increased binding to α1-acid glycoprotein can increase total carbamazepine and carbamazepine-10,11-epoxide concentrations without necessarily increasing the unbound drug concentrations. In such situations, patients may have total concentrations above the desired range but have no side effects. Adjusting dosage is not necessary; plasma concentrations return to baseline as α1-acid glycoprotein declines.
Carbamazepine is eliminated primarily by phase I cytochrome P-450-mediated metabolism (see Table 59-5), forming an active metabolite (10,11-epoxide). Carbamazepine displays dose-dependent elimination pharmacokinetics, in which dosage increases produce a less than proportionate increase in steady-state total concentration (see Figure 59-2). Carbamazepine metabolism undergoes autoinduction, in which clearance increases over time after exposure to the drug. Within 30 days after beginning therapy, carbamazepine clearance may increase by as much as 300 percent. Further increases in maintenance doses may result in further induction [Kudriakova et al., 1992]. In some children, induction causes plasma carbamazepine concentrations to remain unchanged or even decline, despite increasing doses up to 40–50 mg/kg per day [Curatolo et al., 1988]. Carbamazepine and carbamazepine-10,11-epoxide metabolism also can be induced by phenytoin or phenobarbital, resulting in as much as a 100 percent increase in clearance [Kerr and Levy, 1989]. Initial carbamazepine t½ ranges from 10 to 20 hours, but the t½ decreases because of autoinduction to 4–12 hours. Consequently, many children require three or four doses a day to maintain targeted plasma concentrations. Sustained-release preparations help overcome this problem.
Oxcarbazepine
Oxcarbazepine is rapidly absorbed, with a Tmax of less than 1 hour after a single oral dose. Absorption is nearly 100 percent. It is a prodrug that is converted extensively to 10,11-dihydro-10-hydroxy-carbamazepine (the monohydroxy derivative), which is the active compound. Less than 2 percent of the area under the concentration–time curve is attributed to the parent compound, and approximately 70 percent to the monohydroxy derivative. Only the monohydroxy derivative is measured for therapeutic monitoring [Theisohn and Heimann, 1982]. Co-administration of oxcarbazepine and food leads to an increase in the bioavailability and maximum plasma concentration; however, Tmax remains unchanged. The slight changes in the absorption parameters with food administration do not appear to be clinically significant [Degen et al., 1994].
The major advantage of oxcarbazepine is that it is not metabolized to the active epoxide metabolite, which is believed to contribute to the side-effect profile of carbamazepine. Both the parent drug and its metabolite are lipophilic, which facilitates their movement across the blood–brain barrier. The monohydroxy derivative has a volume of distribution of 0.7–0.8 L/kg [Feldmann et al., 1977; Theisohn and Heimann, 1982], and both oxcarbazepine and the monohydoxy derivative bind to plasma proteins at approximately 60 percent and 40 percent, respectively [Klitgaard and Kristensen, 1986; Patsalos et al., 1990].
Approximately 96 percent of an oral dose is eliminated in the urine, with less than 1 percent of oxcarbazepine being excreted unchanged. The greater part of the dose is conjugated and excreted as the glucuronide (9 percent oxcarbazepine glucuronide and 49 percent monohydroxylated glucuronide) [Lloyd et al., 1994; Schutz et al., 1986]. Unlike carbamazepine, oxcarbazepine does not appear to induce its own metabolism [Larkin et al., 1991].
Eslicarbazepine
Eslicarbazepine acetate is a voltage-gated channel blocker that was approved in Europe in 2009. Approval in the US is still pending. Its chemical structure is similar to both carbamazepine and oxcarbazepine [Benes et al., 1999]. It differs from carbamazepine in that it does not produce an epoxide metabolite or exhibit autoinduction. Eslicarbazepine acetate is a prodrug that undergoes first-pass hydrolytic metabolism to cleave the acetate group to form the active entity – eslicarbazepine, which is the S(+) enantiomer of licarbazepine (also known as the S(+)10-monohydroxy metabolite of oxcarbazepine) [Almeida et al., 2008b]. It appears to exhibit linear pharmacokinetics and is metabolized to a glucuronidated metabolite via several uridine diphosphate glucuronosyltransferases [Loureiro and Fernandes-Lopes, 2008]. One study in 29 children and adolescents (ages 2–17 years) shows dose-proportional pharmacokinetics [Almeida et al., 2008a]. Both a suspension and scored tablets of strengths 200 mg, 400 mg, 600 mg, and 800 mg were available in the study. The daily doses used in the study ranged from 5 to 30 mg/kg/day. The Tmax was between 0.5 and 3 hours. Eslicarbazepine acetate was cleared faster in younger children as compared to adolescents, although Cmax was similar among all age groups.
Felbamate
Approximately 95 percent of a dose of felbamate is absorbed in adults after oral administration. Absorption of the drug is fairly rapid, with Tmax values ranging from 1 to 4 hours [Wilensky et al., 1985]. The Vd of felbamate is approximately 0.8 L/kg [Devinsky et al., 1994; Wilensky et al., 1985]. Distribution of felbamate is fairly uniform throughout the entire body. It binds mainly to albumin (22–25 percent), and the amount of binding is dependent on albumin concentration [Perhach and Shumaker, 1995].
About 90 percent of felbamate is eliminated in the urine. Approximately 40–50 percent of the absorbed dose is eliminated as the unconjugated parent compound, and up to 58 percent is found as metabolites and conjugates [Shumaker et al., 1990]. Felbamate is cleared at a faster rate in children. Population pharmacokinetic analysis of data from 139 pediatric patients gives clearance values approximately 40 percent higher in younger children (2–12 years of age) than those in older children and adults (13–65 years).
Gamma-Aminobutyric Acid Analogues
Gabapentin
The absolute bioavailability after oral administration of gabapentin is 60 percent [Vollmer et al., 1986]. Absorption is linear up to daily doses of 1800 mg, after which bioavailability begins to decline, decreasing to 35 percent at daily doses of 4800 mg [Richens, 1993]. Gabapentin uses the l-amino acid transporter system in the gut to enter the body. This system is saturable and the dependence of gabepentin on it may explain the decrease in bioavailability with increasing doses above 1800 mg [Stewart et al., 1993].
Gabapentin has a Vd of approximately 0.6–0.8 L/kg [Richens, 1993] and does not bind to plasma proteins [Vollmer et al., 1986]. To enter the brain, gabapentin uses the l-amino acid transporter system, which is also saturable [Luer et al., 1999].
Gabapentin is eliminated renally. Clearance in children aged 1 month to 12 years was well correlated with creatinine clearance. Clearance was higher in children younger than 5 years of age, necessitating a 30 percent higher dose to attain equivalent concentrations as observed in children age 5 and older and in adults [Haig et al., 2001; Ouellet et al., 2001]. It does not have any significant pharmacokinetic interactions.
Pregabalin
Pregabalin is rapidly absorbed. Its bioavailability is greater than 90 percent. Unlike gabapentin, pregabalin demonstrates no changes in absorption as dose increases. Pregabalin does not bind to plasma proteins and is not metabolized in humans, with 98 percent of the dose recovered unchanged in urine [Brockbrader et al., 2000]. Like gabapentin, it does not appear to interact with other antiepileptic drugs. There were no changes in trough steady-state concentrations of concomitant antiepileptic drugs (phenytoin, lamotrigine, or valproic acid) when pregabalin was co-administered. Likewise, there were no changes in pregabalin concentrations after administration of carbamazepine, phenytoin, lamotrigine, or valproic acid [Brodie et al., 2005]. It should be noted that there did appear to be a decrease in pregabalin exposure when it was given with phenytoin; however, this was thought to be due to a possible food effect with pregabalin. Because pregabalin is eliminated mostly via the kidneys, doses may need to be adjusted in children with decreased renal function.
Hydantoins
Phenytoin
Phenytoin is a poorly water-soluble compound and is often formulated as a sodium salt. It is available as a suspension, chewable tablet, capsule, and a parenteral formulation. The nonlinear, saturation pharmacokinetics of phenytoin can make even small differences in dose clinically significant. The chewable tablet and the suspension contain phenytoin acid, whereas the capsules and the IV formulation contain phenytoin sodium, which is equivalent to only 92 percent of the acid (see Table 59-1). Thus, two 50-mg chewable tablets deliver 8 percent more phenytoin than a 100-mg phenytoin sodium capsule. The suspension is available in a 125-mg/5-mL strength. In the past, the reliability of the suspension has been questioned owing to its tendency to settle. It has been demonstrated that some agitation is sufficient to permit delivery of the specified amount of drug [Sarkar et al., 1989]. Unexpected fluctuations in plasma phenytoin concentrations associated with the use of suspensions likely have been the result of pharmacokinetics rather than formulation problems. Intramuscular injection of the parenteral solution (with a pH of 11) is contraindicated because it results in precipitation of drug that is then slowly and erratically absorbed [Kostenbauder et al., 1975]. It is also painful and causes tissue necrosis [Serrano and Wilder, 1974]. Phenytoin capsules are available in both prompt-release and extended-release formulations. Patient and family education about the differences in phenytoin dosage forms is important to maintain optimal therapy.
Bioavailability, Tmax, and Cmax are altered when large amounts of phenytoin are given as a single oral loading dose, presumably because of the formation of a multicapsule concretion. In a study involving six healthy adults, absorption was reduced by 10–15 percent and Tmax increased from 18 to 32 hours, when phenytoin sodium capsules were given in single dose of 10 mg/kg or more [Jung et al., 1980]. Dividing the total loading dose into 5-mg/kg doses given every 2–3 hours overcomes this problem. The most rapid way to attain therapeutic plasma concentrations after oral loading is to use prompt-release phenytoin sodium capsules or suspension [Goff et al., 1984].
Some reports have suggested that phenytoin is poorly absorbed in newborns because of irregular gastric emptying time, relatively high gastric pH (greater than 4), or decreased absorptive surface [Morrow and Richens, 1989; Morselli, 1983; Painter et al., 1978]. Other evidence indicates that formulation-related differences in absorption, nonlinear pharmacokinetics, and age-dependent changes in elimination account for much of the variability in plasma concentrations [Dodson, 1984].
Phenytoin Vd in children and adolescents is approximately 0.7 ± 0.1 L/kg [Koren et al., 1984]. The relatively small variability in phenytoin Vd permits precise calculation of loading doses. For example, a 15-mg/kg loading dose given intravenously will result in a postinfusion mean concentration of 20 mg/L, with a range of 15–25 mg/L. Phenytoin is also highly protein-bound to albumin.
Phenytoin undergoes extensive hepatic metabolism. A small fraction, 5 percent or less, is excreted unchanged [Battino et al., 1995]. It is metabolized by a saturable cytochrome P-450 enzymatic pathway. Phenytoin follows Michaelis–Menten or saturable elimination kinetics (see Figure 59-2). As the plasma concentration rises, phenytoin clearance decreases, owing to saturation of enzymatic metabolism. Therefore, increases in dose result in disproportionately greater increases in steady-state plasma concentration.
Phenytoin metabolism in neonates is reduced compared with that in older children. Within days to weeks after birth, the rate of metabolism accelerates, reaching a maximum in the 0.5- to 3-year age range. Children in this age group require the highest mg/kg daily doses (Figure 59-4). Thereafter, metabolism and dosage requirements decline gradually, reaching adult values during adolescence. Figure 59-5 illustrates that even minor alterations in dosage produce disproportionately large changes in plasma concentration. Therefore, small adjustments in daily dose are indicated as the plasma concentration approaches or exceeds Km, which usually falls in the range of 5–10 mg/L. Use of the scored 50-mg chewable tablet, the 30-mg phenytoin sodium capsule, or the suspension facilitates making such adjustments in dose.
Fosphenytoin
Fosphenytoin is a prodrug of phenytoin that is rapidly hydrolyzed by tissue and red blood cell alkaline phosphatases to phenytoin. Due to the rapid conversion to phenytoin, the pharmacokinetic information for phenytoin and not fosphenytoin is what is clinically relevent. Fosphenytoin is water-soluble and is formulated as a nontoxic, parenteral solution. The pharmacokinetics of phenytoin derived from fosphenytoin are identical to phenytoin given directly [Leppik et al., 1989]. Fosphenytoin is packaged as milligram phenytoin equivalents and is well tolerated when given intramuscularly.
Fosphenytoin absorption, not its conversion to phenytoin, appears to be the rate-limiting step in achieving therapeutic phenytoin levels. The mean half-life of fosphenytoin is 8 minutes after IV administration [Donn et al., 1987; Gerber et al., 1988; Leppik et al., 1989] and 33 minutes after intramuscular injection [Leppik et al., 1990]. Age does not affect the phosphatase enzyme responsible for the conversion of fosphenytoin to phenytoin. Fosphenytoin is more avidly bound to albumin, compared with phenytoin, resulting in brief increases of free phenytoin concentrations after infusion [Hussey et al., 1990; Jamerson et al., 1990]. The observed decrease in total phenytoin concentrations returns to those expected when the conversion from fosphenytoin to phenytoin is completed; therefore, monitoring of plasma concentrations should be done about 1 hour after IV administration of fosphenytoin. Unlike phenytoin, fosphenytoin can be given intramuscularly and avoids serious tissue necrosis from extravasation of IV phenytoin. Its major uses are treatment of status epilepticus and maintenance therapy when oral administration is not possible.
Lacosamide
Lacosamide (known as harkoside in earlier studies) was approved for use in the United States during 2009. It is completely and rapidly absorbed with a bioavailability of 100 percent [Hovinga, 2003]. Lacosamide appears to follow linear pharmacokinetics and its Cmax is between 1 and 4 hours after an oral dose [Hovinga, 2003]. There does not seem to be a food effect on lacosamide pharmacokinetics [Kellinghaus, 2009]. Protein binding has been shown to be less than 15 percent. Lacosamide is metabolized to an inactive desmethyl metabolite, which is then cleared along with the parent drug via the kidney. Lacosamide is not approved for use in young children at this time; however, there is at least one clinical trial that is investigating the safety and pharmacokinetics in children with partial seizures between the ages of 2 and 17 years. Doses being studied are 8–12 mg/kg/day.
Lamotrigine
Lamotrigine has a bioavailability approaching 100 percent. Its Tmax occurs 2–3 hours after administration of a single oral dose. With doses in the range of 15–240 mg, Tmax and area under the concentration–time curve are directly proportional to dose [Yuen and Peck, 1987]. The Vd of lamotrigine is approximately 1.5 L/kg in children [Chen et al., 1999]. It is distributed uniformly throughout the body, and approximately 55 percent of lamotrigine is bound to plasma proteins [Garnett, 1997].
Lamotrigine is extensively metabolized to an inactive glucuronide by an inducible uridine diphosphate (UDP) glucuronyltransferase reaction [Lamictal, 2004]. Clearance is greater in younger children [Battino et al., 2001]. Infants (younger than 2 months of age) metabolize lamotrigine more slowly, and metabolic rates increase during the first year of life [Mikati et al., 2002]. Estrogen-containing oral contraceptives may cause dramatic cyclical fluctuations of blood levels (decreasing during the estrogen phase and rebounding in the placebo week), and levels decrease dramatically during pregnancy [Pennell et al., 2008]. There may be an impact of age on lamotrigine concentrations [Reimers et al., 2007]. However, data for weight were not available in a majority of the patients and the effect of age could be due to the fact that older children weigh more than younger children [Reimers et al., 2007].
Levetiracetam
Levetiracetam is extremely water-soluble and has an oral bioavailability of close to 100 percent. Its Cmax is within 2 hours of dose administration. When the drug is administered with food, the rate of absorption is decreased, but the extent of absorption is unchanged. Therefore, levetiracetam can be administered without regard to timing of meals. Levetiracetam also exhibits linear pharmacokinetics after single doses ranging from 250 up to 5000 mg [Patsalos, 2000]. The Vd for levetiracetam is 0.5–0.7 L/kg, and protein binding is less than10 percent. Two-thirds of a dose is eliminated unchanged and the rest is eliminated as an inactive metabolite in the urine [Keppra package insert, 2004]. The clearance is 30–40 percent higher in children than in adults [Chhun et al., 2009; Glauser et al., 2007; May et al., 2003; Pellock et al., 2001]. Results are now available from studies in children as young as 2 months old and they confirm that the half-life of levetiracetam in younger children is approximately 5–6 hours, shorter than that seen in adults [Glauser et al., 2007]. An IV formulation and a sustained-release formulation are now on the market. It may be possible to give levetiracetam rapidly. Patients (4–32 years of age) who temporarily lost the ability to take medications by mouth were given an IV loading dose of levetiracetam via rapid infusion (given within 6 minutes). Levetiracetam concentrations ranged from 14 to 189 μg/mL and no significant changes in blood pressure or significant abnormalities in the electrocardiogram were experienced [Wheless et al., 2009]. There were also no local infusion site reactions noted; however, the formulation was diluted 1:1 with normal saline or dextrose 5 percent in water (D5W).
Drug–drug interaction information in children indicates that the concentrations of carbamazepine, valproic acid, topiramate, or lamotrigine are similar between study groups who receive levetiracetam or placebo [Otoul et al., 2007].
Rufinamide
Rufinamide is slowly absorbed due to the fact that it is lipophilic and dissolves slowly in aqueous solutions. The bioavailability is nonlinear and decreases at higher doses, with doses of 1600 mg producing less than proportional concentrations. Steady-state doses of 200, 400, and 800 mg produced morning trough concentrations of 2.81, 5.23, and 11.0 μmol/L, whereas 1600-mg doses resulted in a mean trough concentration of 19.6 μmol/L (4.7 mg/L) [Elger et al., 2010; Limite, 2005; NoAuthors, 2005]. Dissolution is the rate-limiting step in absorption; therefore, changes in gastrointestinal environment, such as with the presence of food, could have an effect on the absorption of rufinamide. It is suggested that rufinamide be administered with food. Rufinamide exposure is increased by 31–34 percent and the Cmax by 36–56 percent when the drug is given after high-fat meals [Perucca et al., 2008]. Rufinamide is metabolized to inactive metabolites via the carboxylesterase pathway. Although rufinamide does not appear to be metabolized via the cytochrome P-450, the carboxylesterase enzymes can be induced by inducers of the CYP450 enzymes. Rufinamide also appears to be a weak inhibitor of CYP2E1 and a weak inducer of CYP3A4.
Pharmacokinetic data in children are beginning to appear in the literature. The majority of the data concerning rufinamide is on file with the manufacturer; however, data on these trials have been summarized in Perucca et al. [2008]. A dosing regimen for children has been simulated from several phase II and III clinical studies involving patients with partial or generalized seizures and one study with Lennox–Gastaut syndrome patients. The simulation included 119 children (aged less than 12 years) and 99 adolescents (12–17 years) [Marchand et al., 2010], and was used in supporting material presented to the European Agency for the Evaluation of Medicinal Products. These studies involved patient populations and the presence of inducing and inhibiting polytherapy. Children who weigh less than 30 kg had greater variability in simulated rufinamide exposure measures than those children who were heavier than 30 kg. In addition, the co-administration of valproate resulted in higher rufinamide plasma concentrations. This study proposed a maximum daily dose of 600 mg for children who also receive valproic acid, and a maximum daily dose of 1000 mg for those children who do not also receive valproic acid. Indeed, valproic acid increased rufinamide steady-state plasma concentrations from 54.9 percent in male children and 70.1 percent in female children in a population pharmacokinetic study that is on file at Eisai [Perucca et al., 2008]. The increase in concentrations when valproic acid was a co-medication was approximately 3 times higher in children than in adults. (Adult concentrations were 15 percent higher.) Population studies involving children have also identified increases in rufinamide’s apparent clearance values due to the presence of inducing co-medications. Known antiepileptic drug inducers, such as carbamazepine, phenobarbital, phenytoin, or primidone, as well as vigabatrin, decreased rufinamide steady-state plasma concentrations in children from 23 to 46 percent [Perucca et al., 2008].
Tiagabine
Oral bioavailability of tiagabine is greater than 90 percent, and the bioavailability of tablets and that of the oral solution appear to be similar [Mengel et al., 1991a]. The absorption rate for tiagabine is reduced up to 65 percent when the drug is administered with food, although the extent of absorption is not affected [Mengel et al., 1991b]. Tiagabine has a Vd of 0.74–0.85 L/kg [Gustavson et al., 1997], and protein binding is approximately 96 percent [Gustavson et al., 1997; Ostergaard et al., 1995]. Tiagabine has a half-life of 5–9 hours [Gustavson and Mengel, 1995]. Tiagabine undergoes extensive metabolism by means of the cytochrome P-450 3A isoform, but no active metabolites have been identified. As observed with other drugs, clearances in children are faster than in adults; adjusting both Vd and clearance values according to body size gives values similar to those seen in adults [Gustavson et al., 1997].
Topiramate
Oral tablets of topiramate have a relative bioavailability of approximately 80 percent that of an oral solution. It is rapidly absorbed, with a Tmax of approximately 2 hours [Easterling et al., 1988]. Overall absorption is not affected by co-administration with food [Doose et al., 1992]. The protein binding of topiramate is less than 15 percent [Ben-Menachem, 1995a], and its Vd is 0.6–0.8 L/kg [Johannessen, 1997]. Elimination is mainly through the kidney, although a portion of the dose is removed by the liver [Ben-Menachem, 1995a]. Preliminary observations in pediatric patients indicate that the mean oral topiramate clearance is 44–55 percent higher in children, and they may need a higher dose than adults to achieve a similar plasma concentration [Adin et al., 2004; Glauser, 1997].
Valproic Acid
Valproic acid appears to be completely absorbed from all the available products (except for the extended-release formulation), but the rate of absorption varies significantly with dosage form (see Table 59-1). Its Tmax is 0.5–1 hour after ingestion for the syrup, 0.5–2 hours after the capsule, 1–6 hours for the enteric-coated tablet, and 3–6 hours for the sprinkle capsule [Cloyd et al., 1983, 1985, 1992]. Absorption after ingestion of the enteric-coated tablet is delayed for several hours; during this lag phase, plasma concentrations may continue to decline. Food further delays, but does not decrease, valproic acid absorption from the enteric-coated tablet [Fischer et al., 1988]. The extended-release tablet has an absolute bioavailability of 90 percent [Dutta et al., 2004]. The extended-release tablets and the enteric-coated tablets are not interchangeable. The extended-release tablets need to be prescribed at a dose that is 8–20 percent higher than the enteric-coated dose to attain similar plasma concentrations [Dutta and Zhang, 2004]. An IV formulation is also available and has been studied in children. The IV formulation should not be given intramuscularly due to toxic muscle necrosis [Gallo et al., 1997].
Valproic acid distribution volumes vary more than those of carbamazepine, phenobarbital, or phenytoin, and range from 0.12 to 0.49 L/kg. When loading doses are calculated, a volume of 0.2 L/kg is recommended. Valproic acid protein binding to albumin is saturable. At concentrations below 40–50 mg/L, the bound fraction averages 90–95 percent. As total concentration rises to the upper boundary of the therapeutic range, the percentage bound falls to 80–85 percent. Saturable protein binding results in a nonlinear relationship between total valproic acid concentration and dose. Unbound valproic acid concentrations, however, remain linear with dose [Cloyd et al., 1993; Riva et al., 1983]. The percentage of unbound valproic acid can be unpredictable and unusually high after rapid infusion in children who are critically ill [Birnbaum et al., 2003].
The elimination half-life of valproic acid is short. In the first few weeks after birth, half-life ranges from 17 to 40 hours, but rapidly declines from 3 to 20 hours in infants and young children. The full antiepileptic effect of valproic acid is obtained days to weeks after steady state is reached, and this effect continues for some time after the drug is discontinued. The lingering effect may be partly the result of its inhibitory effect on the enzymes responsible for the degradation of GABA, or may be due to the accumulation of 2-ene- valproic acid. This prolonged duration has prompted some investigators to propose single daily dosing [Stefan et al., 1984]; however, with other proposed mechanisms of action, seizure control is related to plasma valproic acid concentrations. Under these circumstances, dosing at least every half-life is indicated to minimize fluctuations in plasma concentrations. When valproic acid is given as monotherapy, the half-life may be long enough to maintain therapeutic concentrations for up to 24 hours in some children. Such children may be able to take valproic acid once a day [Cloyd et al., 1985].
Vigabatrin
Vigabatrin is a 50:50 racemic mixture of R(−) and S(+) enantiomers, with the S(+) enantiomer responsible for the drug’s pharmacologic activities and toxic effects [Haegele et al., 1983; Rey et al., 1990]. Bioavailability is approximately 75 percent in adults when the drug is given orally and is similar for both the tablet and the solution [Hoke et al., 1991]. Administration of vigabatrin with food does not significantly affect vigabatrin pharmacokinetics [Frisk-Holmberg et al., 1989]. Vigabatrin has been studied in children [Rey et al., 1989, 1990], and bioavailability appears to be somewhat lower in children than in adults [Rey et al., 1992]. Vigabatrin does not bind to plasma proteins, and it has an apparent Vd of 0.8 L/kg [Ben-Menachem, 1995b]. Clearance or the dose to concentration ratio of vigabatrin appears to be greater in children than in adults, indicating that a higher dose is needed in children to achieve a particular concentration when compared to adults [Armijo et al., 1997].
Most of a vigabatrin dose appears unchanged in the urine. Studies in infants and children show that the half-life of the inactive R(–) enantiomer, but not the half-life of the active S(+) enantiomer, increases with linear age. Vigabatrin exerts its pharmacologic effect by irreversible inhibition of GABA transaminase, causing increases in synaptic GABA concentrations by inhibiting its breakdown. Because the half-life of GABA transaminase is much longer than that of vigabatrin itself, the pharmacologic effect is determined by the half-life of the enzyme [Ben-Menachem, 1995b; Jung et al., 1977].
