Chapter 95 Inflammatory Myopathies
The inflammatory myopathies are a heterogeneous group of disorders characterized pathologically by inflammation in skeletal muscle, with resulting muscle fiber damage and subsequent clinical weakness. There are two major categories of inflammatory myopathy – idiopathic and infectious (Box 95-1). Although several types of muscular dystrophy may be associated with inflammation (e.g., facioscapulohumeral dystrophy and some of the limb-girdle dystrophies), these disorders traditionally are excluded from classification of inflammatory myopathy because they result from a fundamental genetic defect. The classic idiopathic inflammatory myopathies are usually considered autoimmune diseases, although a genetic predisposition may exist for many of these diseases based on inherited human leukocyte antigen haplotypes.
Box 95-1 The Inflammatory Myopathies
As a group, the inflammatory myopathies are the most common acquired myopathies of childhood. The infectious myositides are the most prevalent of these conditions worldwide, with acute viral myositis clearly the most common variety in North America and Europe. In contrast, the idiopathic varieties are relatively uncommon, with a combined incidence (including adult cases) of approximately 1 in 100,000 [Dalakas, 1991]. There are three major forms of idiopathic inflammatory myopathy – dermatomyositis, polymyositis, and inclusion body myositis – with dermatomyositis by far the most common form in childhood [Pachman, 1994]. Polymyositis is relatively uncommon in children and usually is seen as an overlap syndrome with other connective tissue disorders. Inclusion body myositis is exclusively a disease of adulthood and therefore is not discussed here.
Idiopathic Inflammatory Myopathies
Although clinicians frequently conceptualize the idiopathic inflammatory myopathies as a single entity (e.g., polymyositis is dermatomyositis without the rash), the disorders are clinically, histologically, and pathogenetically distinct. Banker and Victor [1966] first reported the unique vascular changes of juvenile dermatomyositis and highlighted the involvement of organs other than muscle and skin. Subsequent immunologic and pathologic observations confirmed distinct pathophysiologies for each of these disorders and recent observations have solidified this view. Although there have been occasional reports of siblings or parent/child cases of both dermatomyositis and polymyositis, most cases are sporadic.
Dermatomyositis
Clinical Features
Juvenile dermatomyositis can present at any age, including infancy, although most cases occur between ages 5 and 14 years. Females are affected more commonly than males. Onset of weakness is typically over weeks to a few months, although fatigue, muscle aches and stiffness, decreased activity, and fever can precede the onset of weakness by months. More acute, fulminate onset (over days) or an indolent, slowly progressive course (sometimes lasting a year) also has been described [Kissel et al., 1991; Pachman, 1994, 1995; Tymms and Webb, 1985]. Weakness typically affects the neck flexors and shoulder and pelvic girdle muscles first, so that the child may have difficulty getting up from the floor, riding a bicycle, running, climbing steps, or raising the arms above the head. Distal weakness is usually present, although not as severe as in proximal muscles. Complete sparing of distal strength should always lead the clinician to suspect another diagnosis, particularly one of the dystrophies. Dysphagia occurs in approximately one-third of patients, and rarely individuals also will have chewing difficulties, dysarthria, and speech delay because of oropharyngeal involvement.
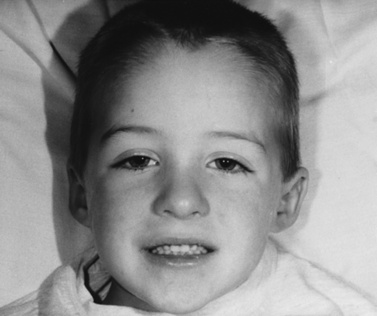
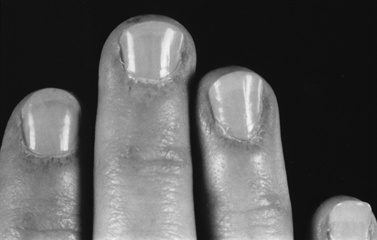
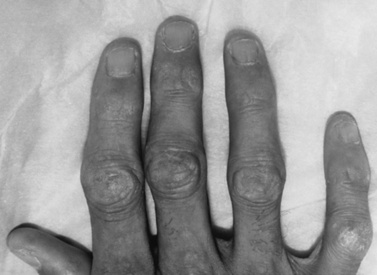
The rash frequently precedes the muscle symptoms and often provides the most important clue to diagnosis [Bowyer et al., 1986; Sontheimer, 1999]. Skin changes typically involve the fingers and periorbital regions first, although any area may be involved. A purplish discoloration of the eyelids (heliotrope rash) with periorbital edema and extension on to the cheeks and forehead is typical (Figure 95-1). On the hands, the periungual regions become erythematous and scaly, with dilated capillary loops in the nailbeds (Figure 95-2). Papular, erythematous, scaly lesions over the knuckles (Gottron’s sign) are characteristic, with similar lesions occurring on the elbows, knees, and malleoli. A more macular, erythematous rash may appear on the face, neck, and anterior chest (V sign) or on the shoulders and upper back (shawl sign). Subcutaneous calcifications, which do not occur in adult cases, are present in 30–70 percent of childhood cases. The calcinosis tends to develop over pressure points (e.g., buttocks, knees, elbows) but can occur anywhere and develops more commonly when the diagnosis has been delayed or the treatment inadequate [Bowyer et al., 1983; Pachman, 1995]. The lesions typically appear as painful, hard nodules that erupt through the skin in severe cases. Occasional patients have the characteristic rash but never develop weakness, a condition sometimes referred to as dermatomyositis sine myositis or amyopathic dermatomyositis [Euwer and Sontheimer, 1993; Stonecipher, 1993].
Associated manifestations
Although involvement of organs other than muscle and skin has become less common with the advent of effective immunosuppressive therapies, some children will develop clinical manifestations, in addition to weakness and rash, on the basis of the underlying vasculopathy. About 50 percent will have electrocardiographic abnormalities, including conduction defects and dysrhythmias, and pericarditis, myocarditis, and congestive heart failure can occur [Askari, 1984; Haupt and Hutchins, 1982; Tymms and Webb, 1985]. Pulmonary function tests may demonstrate restrictive defects and reduced diffusion capacity, even in patients with no pulmonary symptoms [Pachman, 1994]. Approximately 10 percent of patients, particularly those with antibodies to histidyl transfer ribonucleic acid synthetase (Jo-1), develop frank interstitial lung disease or bronchiolitis obliterans and organizing pneumonia [Love et al., 1991]. Vasculopathic involvement of the gastrointestinal tract can result in malabsorption, mucosal ulceration, perforation, and hemorrhage. Dysphagia and delayed gastric emptying, caused by inflammatory involvement of the alimentary skeletal and smooth muscles, are more common.
Arthralgias are common, but true arthritis occurs mainly in children with an “overlap” connective tissue disorder (see below). Ophthalmic involvement on the basis of vascular changes in the retina and conjunctiva can occur [Pachman, 1995]. Renal compromise is rare and results from acute tubular necrosis in the setting of myoglobinuria after fulminant muscle necrosis [Rose et al., 1996]. In contrast to adult dermatomyositis patients, children have demonstrated no definitive increased incidence of malignancy [Sigugeirsson et al., 1992; Fardet et al., 2009].
Laboratory Features
The laboratory evaluation of patients with inflammatory myopathy should include muscle enzyme analysis, electrodiagnostic studies, and muscle biopsy. Muscle imaging studies can indicate areas of inflammation in affected muscles but are seldom useful in making a diagnosis [Hernandez et al., 1993].
Blood tests
Creatine kinase is the most reliable, sensitive, and specific marker for muscle destruction. The creatine kinase level is elevated in about 70 percent of patients sometime during the course of the disease, with levels up to 50 times normal [Amato et al., 1996; Dalakas, 1994a; Tymms and Webb, 1985]. However, it is important to note that the creatine kinase level can be normal throughout the course of the disease and does not correlate with weakness. Rarely, serum aldolase can be elevated when the serum creatine kinase is normal; serum myoglobin, lactate dehydrogenase, aspartate aminotransferase, and alanine aminotransferase can also be elevated but provide no additional information to the creatine kinase. Erythrocyte sedimentation rate is normal or mildly elevated and does not correlate with severity. Antinuclear antibodies occur in 25–50 percent of patients, most commonly in those with overlap syndromes.
Although myositis-specific antibodies associated with specific human leukocyte antigen haplotypes can be demonstrated in a minority of patients, they usually are identified in adults. There have been isolated reports of these antibodies in childhood dermatomyositis, although their role in the pathogenesis of the inflammatory myopathies, in both adult and childhood cases, is unclear [Love et al., 1991; Plotz et al., 1995; Gunawardena et al., 2009]. Although these antibodies provide some information about prognosis [Joffe et al., 1993; Miller, 1993], their value in diagnosing and managing patients is limited, and assaying for them routinely in children with suspected inflammatory myopathy is not indicated. The only exception is the Jo-1 antibody, which is commonly present in patients with interstitial lung disease. In these Jo-1-positive patients, the interstitial lung disease often is more refractory to treatment, and may require the initiation of therapy with prednisone and a second-line immunosuppressive agent (see “Treatment” later). The presence of interstitial lung disease is also a contraindication to the use of methotrexate, which itself can cause interstitial pulmonary fibrosis.
Muscle biopsy
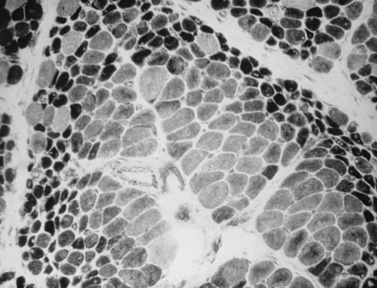
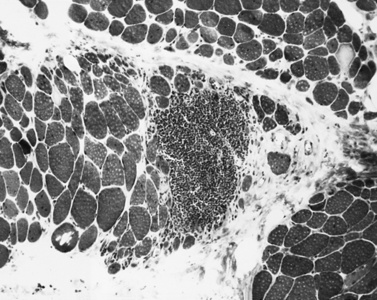
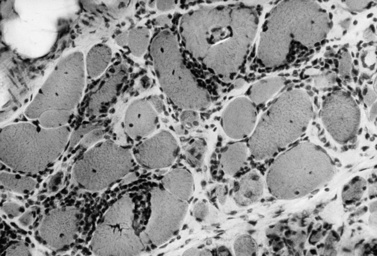
The classic pathologic features associated with dermatomyositis include a microvasculopathy with capillary loss, perivascular and perimysial inflammation, perifascicular atrophy, and scattered areas of fibers with central areas of myofibriallar loss (Figure 95-3). The most characteristic of these changes is perifascicular atrophy, which is present in approximately 50 percent of cases and is relatively specific for dermatomyositis. The inflammatory infiltrates are predominantly perimysial and perivascular, and are composed primarily of macrophages, B cells, and CD4+ cells (Figure 95-4 and see below) [Arahata and Engel, 1984]. One of the earliest demonstrable histological abnormalities on light microscopy is deposition of C5b-9 complement membrane attack complex (MAC) on small blood vessels (as well as immunoglobulin M and other complement components, including C3 and C9) [Emslie-Smith and Engel, 1990; Kissel et al., 1986]. These immunohistochemical findings can be helpful in making a diagnosis, especially when other histologic findings are absent or equivocal. Electron microscopy typically reveals small intramuscular arterioles and capillaries with endothelial hyperplasia, microvacuoles, and cytoplasmic inclusions, but is seldom needed to make a diagnosis.
Pathogenesis
More recent evidence, however, has raised many questions about the interpretation of all of these observations and suggested other possible pathogenic mechanisms. The vascular MAC and immunoglobulin deposits, for example, could be due to either immune complex deposition, or complement activation by the classical antibody-mediated or alternative pathways [Greenberg and Amato, 2004], occurring as a secondary phenomenon after the microvasculature has been damaged by some other mechanism (e.g., interferon or other cytokine-related toxicity). The fact that some patients have developed dermatomyositis with hereditary complement deficiencies argues against a primary destruction of capillaries by complement and MAC.
Recent evidence has also suggested that factors other than ischemia may be responsible for the production of perifascicular atrophy [Greenberg and Amato, 2004]. In animal models of ischemic myopathy, for example, the pathological changes are located centrally within fascicles and the perifascicular regions are preferentially spared. In addition, perifascicular atrophy is not evident in ischemic muscle damaged by vasculitis. Gene expression studies on muscle biopsies from dermatomyositis patients do not reveal upregulation of genes normally associated with ischemia, suggesting that some other process may be operative in producing these changes [Greenberg et al., 2005].
Finally, although the inflammatory infiltrate in dermatomyositis is composed primarily of macrophages, B cells, and CD4+ cells in the perivascular and perimysial regions, the CD4+ cells are not T-helper cells, but rather predominantly plasmacytoid dendritic cells [Greenberg et al., 2005], which are the body’s primary producers of type 1 interferons. Gene expression and proteomic studies on muscle biopsies and in serum suggest that increased expression of type 1 interferons in patients with dermatomyositis might be directly toxic to small vessels and muscle fibers. Furthermore, capillaries and muscle fibers in dermatomyositis over-express type 1 interferon inducible proteins, particularly in the perifascicular regions, and these changes can be seen even before the development of perifascicular atrophy [Greenberg et al., 2005; Salajegheh et al., 2009]. These findings in muscle biopsies are also supported by observations suggesting that increased interferon-α and its inducible genes are seen in serum of patients with dermatomyositis and that the levels correlate with disease activity [Walsh et al., 2007; Niewold et al., 2009]. These observations, taken together, suggest that type 1 interferon over-expression, rather than a microvasculopathy, may be the pivotal event in producing the muscle damage in dermatomyositis, although what might trigger this over-expression is currently unknown.
Treatment
Although corticosteroids are generally regarded as the most effective therapy for dermatomyositis, there is disagreement surrounding the best route of administration, dosing regimen, duration of therapy, and parameters to monitor during therapy [Amato and Barohn, 2009]. There have been no controlled trials to examine these issues, although retrospective studies have demonstrated that prednisone reduces morbidity and improves strength and function [Adams and Plotz, 1995; Chwalinska-Sadowska and Madykowa, 1990; Dalakas, 1994a; Joffe et al., 1993].
Corticosteroids
Prednisone is the usual first line of treatment for dermatomyositis. Short courses of intravenous methylprednisolone (Solu-Medrol; 20–30 mg/kg/day or every other day for 3–5 days) may be the best way to initiate therapy in severely affected individuals, especially because this therapy may prevent calcinosis [Laxer et al., 1987]. A more traditional regimen involves oral therapy (1–2 mg/kg/day), given as a single morning dose. After 3–6 weeks of daily prednisone, the dosage usually can be switched directly to an alternate-day regimen at the same dose, although occasional patients will require persistent daily therapy. The high dose is maintained while strength is carefully monitored. Although the creatine kinase level can sometimes be useful in monitoring response, it should never be the primary determinant of therapeutic decision-making.
Patients should be evaluated at least monthly during this phase of treatment. When the child’s strength has returned to normal or improvement has reached a plateau (usually within 4–6 months), the prednisone can be tapered at a rate no faster than 5 mg every 2–4 weeks. If an exacerbation occurs during the tapering, the prednisone dose is increased back to its original level and again given daily. When strength has recovered, the prednisone taper can be resumed. Although occasional patients will require daily dosing, or even split daily doses to maintain adequate disease control, alternate-day dosing is preferred because it is associated with fewer side effects. Most patients (approximately 80 percent) improve with therapy, with approximately two-thirds having a complete response [Amato et al., 1996; Pachman, 1995].
As most patients require therapy for 1–2 years to achieve remission, limiting the side effects of prednisone is an important part of management [Boumpas et al., 1993]. Patients should be started on a low-sodium, low-carbohydrate, high-protein, low-calorie diet to limit weight gain and reduce the risk of hypertension. Calcium supplementation (1000–1500 mg/day) and vitamin D reduce the risk of osteoporosis, as does physical therapy with axial exercise, as tolerated by the patient’s weakness. Baseline determination of bone density through dual-energy x-ray absorptiometry scanning is indispensable in diagnosing and following osteoporosis in this population. Therapy with bisphosphonates should be considered in children who develop definite osteoporosis, or in those expected to be on very long-term therapy [Bianchi et al., 2000]. Exercise also reduces the risk of steroid myopathy [Escalante et al., 1993]. Blood pressure, serum glucose and potassium levels, and ocular status (for cataracts and glaucoma) should be assessed periodically. Hip pain in a child on prednisone must be investigated aggressively because avascular necrosis is an uncommon but devastating development in these individuals. Growth retardation and delayed puberty can occur with prolonged therapy.
Although the rash usually responds along with the muscle response, occasional patients require separate treatment for their skin changes. Hydroxychloroquine, chloroquine, topical steroids, topical tacrolimus, and sunscreen have been used to treat the rash when weakness is mild or not apparent [Euwer and Sontheimer, 1993; Sontheimer, 1999; Stonecipher, 1993]. Treatment of calcinosis is more difficult. Colchicine, probenecid, warfarin, and phosphate buffers have been used with only limited success [Pachman, 1994]. Surgery may be indicated, but the lesions may recur or worsen despite surgery.
Other agents
Patients who respond poorly to steroids, relapse repeatedly as prednisone is tapered, or develop intolerable side effects are candidates for alternative therapy (Table 95-1). Methotrexate is probably the drug of choice in children, although there are no prospective, controlled studies to guide its use. In retrospective series, the majority of children refractory to prednisone improved with methotrexate therapy [Cagnoli et al., 1991; Joffe et al., 1993; Miller et al., 1992b]. In a recent series of 49 children with juvenile dermatomyositis initially treated aggressively with both pulsed corticosteroids and methotrexate, a prolonged, medication-free remission was attained in 28 of 49 within a median of 38 months from the time of diagnosis [Kim et al., 2009]. Methotrexate must be used with caution because of side effects, including alopecia, stomatitis, oncogenicity, infection, interstitial lung disease, and bone marrow, renal, and liver toxicity. Pulmonary function tests, blood counts, and liver function tests must be monitored. Azathioprine, mycophenolate mofetil, cyclophosphamide, and cyclosporine have also been used in refractory patients (see Table 95-1) [Amato and Barohn, 2009; Amato and Griggs, 2003a].
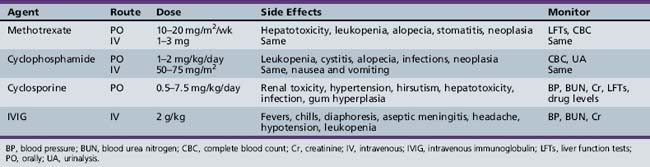
On the basis of a double-blind, placebo-controlled study suggesting benefits in 15 adult dermatomyositis patients, intravenous immunoglobulin has become an important component of the treatment of dermatomyositis [Dalakas et al., 1993]. Although originally used only for refractory cases, it is finding increasing use as both a steroid-sparing agent and a primary agent because of its low incidence of long-term side effects. Treatment is usually initiated at a dose of 2 g/kg given slowly over 2–5 days, with repeat infusions at variable intervals (usually every 2–6 weeks) for at least 3 months [Thornton and Griggs, 1994]. Flulike symptoms (i.e., headaches, myalgias, fever, chills, and nausea) may occur in as many as 50 percent of patients during the infusions but are usually mild. Rash, aseptic meningitis, transient leukopenia, renal failure, and stroke have been described rarely. A controlled trial of plasmapheresis and leukopheresis demonstrated no improvement with either modality in dermatomyositis [Miller et al., 1992a].
Rituximab is a monoclonal antibody directed against CD20 and thus depletes B cells. A small open-label study of dermatomyositis patients suggested that rituximab could be an effective therapy [Levine, 2005], and a large multicenter, double-blind, placebo-controlled trial in both adult and juvenile dermatomyositis should be completed shortly. Currently, this therapy probably should be reserved for only the most refractory cases. The dose is 750 mg/m2 (up to 1 g), given once and then repeated in 2 weeks. The course of rituximab then is to be repeated every 6–18 months.
Polymyositis
Polymyositis, although common in adults, is relatively rare in children as an isolated entity [Amato and Barohn, 2009; Amato and Griggs, 2003b; Dalakas and Hohlfeld, 2003; Hoogendijk et al., 2004; Van der Meulen et al., 2003]. In one epidemiologic study, juvenile dermatomyositis was 10–20 times more common than polymyositis [Hanissian et al., 1982]. When polymyositis does occur in children, it is often in the setting of an “overlap” condition with features of another connective tissue disorder [Pachman, 1994]. Due to the absence of the rash, the diagnosis is often delayed compared with dermatomyositis.
Clinical Features
As in dermatomyositis, patients typically present after weeks to months of neck flexor and symmetric, proximal greater than distal, arm and leg weakness. Muscle pain and tenderness are common but not always present. Dysphagia occurs in approximately one-third of patients secondary to oropharyngeal and esophageal involvement. Mild facial weakness occasionally may be demonstrated. Sensation and tendon reflexes are usually preserved. As in adults, other organs, most notably the heart and lungs, can be involved, although the incidence appears to be lower in children than in adults. Congestive heart failure or conduction abnormalities and interstitial lung disease are found in rare patients, the latter more commonly in patients with serum anti-Jo-1 antibodies [Love et al., 1991]. Polyarthritis has been reported in as many as 45 percent of polymyositis patients at the time of diagnosis.
Overlap syndromes
In children, polymyositis occurs as part of an overlap syndrome, where the myositis is associated with another connective tissue disorder, more frequently than as an isolated entity. The common overlap syndromes associated with polymyositis (and often dermatomyositis) include scleroderma, mixed connective tissue disease, Sjögren’s syndrome, systemic lupus erythematosus, polyarteritis nodosa, and rheumatoid arthritis. Involvement of the skin, joints, kidneys, eyes, and salivary glands is identical to that seen for each disorder in isolation (Figure 95-5), and the serologic abnormalities are also similar. Weakness in these patients is not always due to myositis because disuse and type 2 fiber atrophy from chronic steroid treatment of the underlying condition can also result in muscle weakness. In these individuals, it is crucial to document the nature of the muscle involvement with serologic and electromyographic studies and often muscle biopsy. The prognosis is often related to the underlying disorder, and treatment may have to be modified based on the associated condition.
Laboratory Features
Laboratory evaluation in polymyositis is similar to that in dermatomyositis. Unlike dermatomyositis, the serum creatine kinase is essentially always elevated in polymyositis. The serum creatine kinase level can be helpful in monitoring response to therapy but only in conjunction with the physical examination. Positive antinuclear antibodies are present in approximately 30 percent. Muscle-specific antibodies have been reported to be more common in polymyositis than dermatomyositis [Love et al., 1991; Plotz et al., 1995; Targoff, 1994]. Close analysis of these reports, however, indicates that the muscle histopathology in many of the antibody-positive cases does not demonstrate the typical pathologic features of polymyositis (e.g., there is usually no invasion of non-necrotic muscle fibers by CD8+ T lymphocytes) [Greenberg et al., 2002; Miller et al., 2002; Mozaffar and Pestronk, 2000]. In some cases, the biopsies are more suggestive of a microangiopathic disorder similar to that seen in dermatomyositis. Electromyography is similar to that in dermatomyositis, with a myopathic picture of increased spontaneous activity, small polyphasic motor units, and early recruitment.
Muscle biopsy
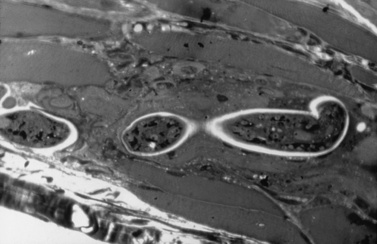
The pathologic findings of polymyositis are distinct from dermatomyositis and reflect basic differences in the underlying pathophysiology [Amato and Griggs, 2003b; Dalakas and Hohlfeld, 2003; Van der Meulen et al., 2003]. Biopsies are characterized by variability in fiber size, with scattered necrotic and regenerating fibers, and endomysial inflammation, with invasion of non-necrotic muscle fibers by CD8+ cytotoxic/suppressor T cells and macrophages (Figure 95-6) [Arahata and Engel, 1984; Engel and Arahata, 1984]. The invaded (and sometimes noninvaded) fibers express major histocompatibility complex class 1 antigen, which is not present on normal fibers [Emslie-Smith et al., 1989]. The vascular changes of dermatomyositis are not seen. Although B lymphocytes are not common, oligoclonal plasma cells have been identified in the endomysium in polymyositis [Greenberg et al., 2005b; Bradshaw et al., 2007]. Unlike in dermatomyositis, in which there are many plasmacytoid dendritic cells that produce interferons, polymyositis muscle biopsies have many endomysial myeloid dendritic cells that probably function as antigen-presenting cells [Greenberg et al., 2007].
Pathogenesis
Polymyositis results from a human leukocyte antigen-restricted, antigen-specific, cell-mediated immune response against muscle fibers. The T-cell receptors of the muscle-invading T cells have an oligoclonal pattern of gene rearrangement, suggesting that the immune response is antigen-specific [Mantegazza et al., 1993]. The antigen against which the immune response is generated and the trigger for the autoimmune attack are not known. Although speculation has centered around a viral infection triggering the immune response, viral antigens and genomes have not been identified in the muscle fibers of patients with polymyositis [Leff et al., 1992; Plotz et al., 1995]. Gene expression studies demonstrate increased expression of immunoglobulin genes [Greenberg et al., 2002], although the significance of these findings is unclear. Gene expression studies in muscle and blood in patients with polymyositis demonstrate upregulation of the type I interferon genes, though not as high as is seen in dermatomyositis [Greenberg et al., 2002]. However, as is the case in dermatomyositis, the upregulation of these genes seems to correlate with disease activity in polymyositis [Walsh et al., 2007].
Treatment
The treatment of children with polymyositis does not differ in any substantive way from that of dermatomyositis. Corticosteroids are the mainstay of treatment, with secondary agents used as indicated by the response [Amato and Barohn, 2009]. The role of intravenous immunoglobulin in polymyositis is less clear than in dermatomyositis, and should probably be reserved for refractory cases. Treatment of patients with an overlap syndrome is occasionally dictated more by the degree of joint, kidney, or skin involvement than by the muscle disease. Although the majority of patients with polymyositis respond to therapy, the response is often less robust and less sustained than in dermatomyositis. Fewer patients (approximately 20 percent) attain a complete remission, so that more long-term therapy, along with its associated side effects from medications, is frequently required. Unfortunately, no prospective studies have examined in detail the long-term prognosis of these patients.
Congenital Inflammatory Myopathy
Over the past three decades, there have been scattered reports of infants with “congenital inflammatory myopathy” or “infantile myositis” [Kinoshita et al., 1980, 1986; Nagai et al., 1992; Roddy et al., 1986; Shevell et al., 1990; Thompson, 1982]. The disorder is characterized by perinatal hypotonia and generalized weakness with elevated serum creatine kinase levels, myopathic electromyography, and striking inflammation on muscle biopsy. In one case of well-documented congenital myositis [Roddy et al., 1986], the child was later diagnosed as having Prader–Willi syndrome. Most of the other patients did not improve with corticosteroid therapy, and it is likely that many of these cases had a form of muscular dystrophy with secondary inflammation. Inflammation on biopsy is not specific for a primary inflammatory myopathy and can be demonstrated in Duchenne, facioscapulohumeral, dysferlin-related, sarcoglycan-related, calpain-related (see later), and Fukuyama-type muscular dystrophies, in Walker–Warburg syndrome, and in the Occidental type of congenital dystrophy with hypomyelination and laminin alpha-2 deficiency [Olney and Miller, 1983; Pegoraro et al., 1996].
Other Idiopathic Inflammatory Myopathies
Several other extremely rare inflammatory myositides have been described in children. Eosinophilic myositis has been described in several children with peripheral eosinophilia and focal eosinophilic infiltration in isolated muscles [Agrawal and Giesen, 1981; Kaufman et al., 1993; Nagar and Bar-Ziv, 1993]. However, several recent reports suggest that many of these cases may actually be calpainopathies (i.e., limb-girdle muscular dystrophy type 2B [Krahn et al., 2006; Amato, 2008]. Thus, diagnoses of polymyositis or eosinophilic myositis should be made with extreme caution in children, and then only after the various dystrophies have been carefully considered.
Focal nodular myositis, as the name implies, is also a focal disorder associated with mononuclear infiltration in individual muscles, causing the subacute onset of focal pain, swelling, and weakness [Flaisler et al., 1993]. Some patients go on to develop generalized polymyositis. Improvement has been reported after surgical excision of the mass but can also occur spontaneously. A myositis with noncaseating granulomas can occur in sarcoid myopathy, a condition that may respond partially to prednisone [Banker, 1994].
Inflammatory Myopathy Associated with Infections
Influenza Myositis
Influenza A, B, and, rarely, C are common upper respiratory tract pathogens. Acute infection usually is associated with myalgias when fever and other constitutional symptoms are present. The myalgias presumably are caused by the systemic release of cytokines, although a true myositis can develop [Hays and Gamboa, 1994].
Clinical Features
As respiratory symptoms subside, pain, swelling, and muscle tenderness herald the onset of myositis [Hays and Gamboa, 1994; Mejlszenkier et al., 1973; Ruff and Secrist, 1982]. The pain can be so severe as to interfere with the child’s ability to walk or perform routine activities, and muscle swelling and induration may be present. Weakness can be profound and myoglobinuria has been reported [Christianson and San Joaquin, 1990]. The symptoms are self-limited, lasting less than 1 week, although rare patients have recurrent symptoms associated with different influenza types [Ruff and Secrist, 1982].
Laboratory Features
The creatine kinase level is usually elevated, often markedly so, whereas it is typically normal in uncomplicated influenza infection. Electromyography reveals features of an active necrotizing myopathy. Muscle biopsy is rarely indicated but indicates scattered necrotic and regenerating fibers, with occasional interstitial inflammatory cells. Influenza virus has been isolated only occasionally on culture of the muscle specimen [Hays and Gamboa, 1994].
Pathogenesis
The mechanism of muscle injury in these patients is unclear. Although muscle damage may result from direct attack of the virus on muscle, electron microscopy has not revealed viral particles in the muscle biopsies, and viral cultures rarely are positive for influenza [Farrell et al., 1980]. An immune-mediated attack on muscle triggered by the virus is another possible mechanism for muscle injury.
Other Viral Myositides
Acute viral myositis is not specific for influenza infection, and similar syndromes can complicate infections with coxsackievirus, parainfluenza, mumps, measles, adenovirus, herpes simplex, cytomegalovirus, hepatitis B, Epstein–Barr virus, respiratory syncytial virus, echovirus, and, possibly, arboviruses [Hays and Gamboa, 1994]. Like influenza, the myositis associated with these viruses is usually self-limited and requires only supportive therapy. Acute and convalescent viral titers (3 or 4 weeks after infection), as well as blood, stool, urine, and throat cultures, can be useful in identifying the responsible virus but seldom add to the management of the muscle symptoms.
An inflammatory myopathy has also been described with infection by human immunodeficiency virus (HIV). This disorder usually develops in patients with acquired immune deficiency syndrome but also can occur in early HIV infection. It has been reported exclusively in adults, although HIV-infected children also are at risk. The presentation is similar to that of polymyositis, with subacute progressive, symmetric proximal weakness, an elevated creatine kinase level, and myopathic electromyography. The disorder must be distinguished from zidovudine (AZT) myotoxicity, HIV wasting syndrome, and other disorders that can complicate HIV infection [Dalakas, 1994b; Illa et al., 1991].
Trichinosis
Clinical Features
Between 2 and 12 days after ingestion, the larval form of the nematode disseminates through the bloodstream and invades the muscles [Davis et al., 1976]. Patients develop fever, headache, abdominal pain, diarrhea, generalized myalgias, and weakness during the acute systemic reaction. Periorbital edema, ptosis, subconjunctival hemorrhage, and an erythematous urticarial or petechial rash also are often present. Myalgias and weakness peak in the third week of the infection but can last for months. Severe disease can be complicated by myocarditis, pneumonitis, and central nervous system infection.
Laboratory Features
Most patients have a marked eosinophilic leukocytosis and elevated serum creatine kinase level. Serum antibodies against T. spiralis can be demonstrated 3–4 weeks after infection. Electromyography demonstrates the typical features of an inflammatory myopathy. In the early stage of infection, muscle biopsy reveals infiltration of the muscle by eosinophils and polymorphonuclear leukocytes. With more chronic infection, mononuclear inflammatory cells are more common [Gross and Ochoa, 1979]. Larvae, cysts with focal calcification, fibrosis, and granulomas may be observed months after the initial infection (Figure 95-7).
Toxoplasmosis
Toxoplasmosis is caused by the protozoon Toxoplasma gondii. The common mode of infection is through the ingestion of food, usually undercooked meat, contaminated with oocysts or cysts containing bradyzoites of the organism. The cysts mature and enter the bloodstream and lymphatics to invade other organs. Exposure to the organism is common worldwide, although symptomatic infection usually occurs in immunocompromised patients. Skeletal muscle involvement can occur in isolation or as part of more generalized infection [Banker, 1994].
Clinical and Laboratory Features
In most patients, symptoms are mild and nonspecific; they include fever, malaise, fatigue, and myalgias. Patients with more severe disease have lymphadenopathy, hepatosplenomegaly, uveitis, pneumonia, myocarditis, rash, or meningoencephalopathy. Weakness develops in those with significant muscle involvement [Gherardi et al., 1992; Pollock, 1979]. The serum creatine kinase level is elevated, and the typical myopathic picture is evident on electromyography. Muscle biopsy may reveal cysts containing the organism, along with inflammation and giant cells. The diagnosis is confirmed through serologic studies indicating immunoglobulin M reactivity to the organism.
Cysticercosis
Cysticercosis is caused by the tapeworm Taenia solium, another organism usually transmitted through eating undercooked meat. Although central nervous system infection usually dominates the clinical picture, with focal neurologic deficits, seizures, and encephalopathy, skeletal muscles also can be involved. Weakness, myalgias, muscle tenderness, and muscle pseudohypertrophy are accompanied by an elevated creatine kinase level and eosinophilia [Banker, 1994; Sawney et al., 1976]. The biopsy is characterized by fibrosis, inflammation with eosinophils, giant cells, and encysted organisms. Praziquantel (50 mg/kg/day in three divided doses for 2 weeks) is effective against central nervous system infection, but its efficacy in muscle is less clear. Albendazole (15 mg/kg/day in divided doses for 8 days) also has been used. Niclosamide and paromomycin are useful for treating the adult worms, and corticosteroids suppress the inflammatory reaction against degenerating parasites.
Bacterial Infections
Pyomyositis refers to the multifocal abscesses associated with bacterial infection of the muscle [Taksande et al., 2009]. Although bacterial infections are more common in developing countries, they are being seen with increasing frequency in developed countries as a result of HIV infection and intravenous drug abuse [Antony and Kerodle, 1996; Hsueh et al., 1996; Rodgers et al., 1993].
Muscle pain, tenderness, and fever are the initial symptoms, most commonly affecting the quadriceps, glutei, and deltoids. Most patients have a neutrophilic pleocytosis, elevated erythrocyte sedimentation rate, and normal or elevated serum creatine kinase level. Blood cultures are usually negative early in the infection until the patient becomes septic. Ultrasound, computed tomography, and magnetic resonance imaging of muscle can be useful in localizing abscesses for diagnostic needle aspiration. The most common organisms are Staphylococcus aureus, streptococci, Escherichia coli, Yersinia organisms, and Legionella organisms [Akman et al., 1996; Collazos et al., 1996; O’Neill et al., 1996]. Pyomyositis usually arises as an extension of infection from adjacent tissues or via hematologic spread of the organisms. Early in the illness, the infection may respond to appropriate antibiotics, although more severe infections require incision and drainage.
Fungal Myositides
Fungal infection of the muscles is very uncommon and usually occurs in an immunosuppressed adult patient. Candidiasis is the most common fungal myositis and almost always is associated with disseminated disease [Arena et al., 1981; Jarowski et al., 1978]. Diffuse muscle pain, tenderness, weakness, fever, and a papular erythematous rash are evident but may be obscured by other systemic involvement. Muscle biopsy demonstrates infiltration of the muscle by hyphal and yeast forms of the organism, inflammation, and hemorrhagic necrosis. Myositis also has been reported as complicating cryptococcal infection, sporotrichosis, actinomycosis, and histoplasmosis [Halverson et al., 1985; Heffner, 1993; Wrzolek et al., 1990].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Adams E.M., Plotz P.H. The treatment of myositis. How to approach resistant disease. Rheum Clin North Am. 1995;21:179.
Agrawal B.L., Giesen P.C. Eosinophilic myositis: An unusual cause of pseudotumor and eosinophilia. JAMA. 1981;246:70.
Akman I., Ostrov B., Varma B.K., et al. Pyomyositis: Report of three patients and review of the literature. Clin Pediatr (Bologna). 1996;35:397.
Amato A.A. Adults with eosinophilic myositis and calpain-3 mutations. Neurology. 2008;26(70):730.
Amato A.A., Barohn R.J. Evaluation and treatment of inflammatory myopathies. J Neurol Neurosurg Psychiatry. 2009;80:1060.
Amato A.A., Griggs R.C. Inflammatory myopathies. Curr Opin Neurol. 2003;16:569.
Amato A.A., Griggs R.C. Unicorns, dragons, polymyositis and other mythical beasts. Neurology. 2003;61:316.
Amato A.A., Gronseth G.S., Jackson C.E., et al. Inclusion body myositis: Clinical and pathological boundaries. Ann Neurol. 1996;40:581.
Antony S.J., Kerodle D.S. Nontropical pyomyositis in patients with AIDS. J Natl Med Assoc. 1996;88:865.
Arahata K., Engel A.G. Monoclonal antibody analysis of mononuclear cells in myopathies. I. Quantitative of subsets according to diagnosis and sites of accumulation and demonstration and counts of muscle fibers invaded by T cells. Ann Neurol. 1984;16:193.
Arena A.P., Perlin M., Brahman H., et al. Fever, rash, and myalgias of disseminated candidiasis during antifungal therapy. Arch Intern Med. 1981;141:1233.
Askari A.D. Inflammatory disorders of muscle: Cardiac abnormalities. Clin Rheum Dis. 1984;10:131.
Banker B.Q. Other inflammatory myopathies. In Engel A.G., Franzini-Armstrong C., editors: Myology, ed 2, New York: McGraw-Hill, 1994.
Banker B.Q., Victor M. Dermatomyositis (systemic angiopathy) of childhood. Medicine (Baltimore). 1966;45:261.
Bianchi M.L., Cimaz R., Bardare M., et al. Efficacy and safety of alendronate for the treatment of ostiopososis in diffuse connective tissue disesaes in children. Arthritis Rheum. 2000;43:1960.
Boumpas D.T., Chrousos G.P., Wilder R.L., et al. Glucocorticoid therapy for immune-mediated diseases: Basic and clinical correlates. Ann Intern Med. 1993;119:1198.
Bowyer S.L., Blane C.E., Sullivan D.B. Childhood dermatomyositis: Factors predicting functional outcome and development of dystrophic calcification. J Pediatr. 1983;103:882.
Bowyer S.L., Clark R.A.F., Ragsdale C.G., et al. Juvenile dermatomyositis: Histologic findings and pathogenic hypothesis for the associated skin changes. J Rheumatol. 1986;13:753.
Bradshaw E.M., Orihuela A., McArdel S.L., et al. A local antigen-driven humoral response is present in the inflammatory myopathies. J Immunol. 2007;178:547-1546.
Cagnoli M., Marchesoni A., Tosi S. Combined steroid, methotrexate, and chlorambucil therapy for steroid resistant dermatomyositis. Clin Exp Rheumatol. 1991;9:658.
Christianson J.C., San Joaquin V.H. Influenza-associated rhabdomyolysis in a child. Pediatr Infect Dis J. 1990;9:60.
Chwalinska-Sadowska H., Madykowa H. Polymyositis-dermatomyositis: 25 year follow-up of 50 patients – Disease course, treatment, prognostic factors. Mater Med Pol. 1990;22:213.
Collazos J., Fernandez A., Martinez E., et al. Pneumococcal pyomyositis. Case report, review of the literature, and comparison with classic pyomyositis caused by other bacteria. Arch Intern Med. 1996;156:1470.
Dalakas M.C. How to diagnose and treat the inflammatory myopathies. Semin Neurol. 1994;14:137.
Dalakas M.C. Polymyositis, dermatomyositis, and inclusion body myositis. N Engl J Med. 1991;325:1487.
Dalakas M.C. Retrovirus-related muscle disease. In Engel A.G., Franzini-Armstrong C., editors: Myology, ed 2, New York: McGraw-Hill, 1994.
Dalakas M.C., Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971.
Dalakas M.C., Illa I., Dambrosia J.M., et al. A controlled trial of high dose intravenous immunoglobulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993.
Davis M.J., Cilo M., Platitakis A., et al. Trichinosis. Severe myopathic involvement with recovery. Neurology. 1976;26:37.
Emslie-Smith A.M., Arahata K., Engel A.G. Major histocompatibility complex 1 antigen expression, immunolocalization of interferon subtypes, and T cell–mediated cytotoxicity in myopathies. Hum Pathol. 1989;20:224.
Emslie-Smith A.M., Engel A.G. Microvascular changes in early and advanced dermatomyositis: A quantitative study. Ann Neurol. 1990;27:343.
Engel A.G., Arahata K. Monoclonal antibody analysis of mononuclear cells in myopathies. II: Phenotypes of autoinvasive cells in polymyositis and inclusion body myositis. Ann Neurol. 1984;16:209.
Escalante A., Miller L., Beardmore T.D. Resistive exercise in the rehabilitation of polymyositis/dermatomyositis. J Rheumatol. 1993;20:1340.
Euwer R.L., Sontheimer R.D. Amyopathic dermatomyositis: A review. J Invest Dermatol. 1993;100:124S.
Fardet L., Dupuy A., Gain M., et al. Factors associated with underlying malignancy in a retrospective cohort of 121 patients with dermatomyositis. Medicine. 2009;88(2):91-97.
Farrell M.K., Partin J.C., Bove K.E., et al. Epidemic influenza myopathy in Cincinnati in 1977. J Pediatr. 1980;96:545.
Flaisler F., Blin D., Asencio G., et al. Focal myositis: A localized form of polymyositis? J Rheumatol. 1993;20:1414.
Gherardi R., Baidrimont M., Lionnet F., et al. Skeletal muscle toxoplasmosis in patients with acquired immunodeficiency syndrome. Ann Neurol. 1992;32:535.
Greenberg S.A., Amato A.A. Uncertainties in the pathogenesis of DM. Curr Opin Neurol. 2004;13:356.
Greenberg S.A., Bradshaw E.M., Pinkus G.S., et al. Interferon α/β-mediated innate mechanisms in dermatomyositis. Ann Neurol. 2005;57:664.
Greenberg S.A., Bradshaw E.M., Pinkus J.L., et al. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology. 2005;65:1782.
Greenberg S.A., Pinkus G.S., Amato A.A., et al. Myeloid Dendritic Cells in Inclusion-Body Myositis and Polymyositis. Muscle Nerve. 2007;35:17.
Greenberg S.A., Sanoudou D., Haslett J.N., et al. Molecular profiles of the inflammatory myopathies. Neurology. 2002;59:1170.
Gross B., Ochoa J. Trichinosis: Clinical report and histochemistry of muscle. Muscle Nerve. 1979;2:394.
Gunawardena H., Betteridge Z.E., McHugh N.J. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford). 2009;48(6):607.
Halverson P.B., Lahiri S., Wojno W.C., et al. Sporotrichal arthritis presenting as granulomatous myositis. Arthritis Rheum. 1985;28:1425.
Hanissian A.S., Masi A.T., Pitner S.E., et al. Polymyositis and dermatomyositis in children: An epidemiologic and clinical comparative analysis. J Rheumatol. 1982;9:390.
Haupt H.M., Hutchins G.M. The heart and conduction system in polymyositis-dermatomyositis. Am J Cardiol. 1982;50:998.
Hays A.P., Gamboa E.T. Acute viral myositis. In Engel A.G., Franzini-Armstrong C., editors: Myology, ed 2, New York: McGraw-Hill, 1994.
Heffner R.R. Inflammatory myopathies. A review. J Neuropathol Exp Neurol. 1993;52:339.
Hernandez R.J., Sullivan D.B., Chenevert T.L., et al. MR imaging in children with dermatomyositis: Findings and correlations with clinical and laboratory findings. Am J Roentgenol. 1993;161:359.
Hoogendijk J.E., Amato A.A., Lecky B.R., et al. Workshop report. 119th ENMC international workshop: Trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis. Naarden, The Netherlands, October 10–12, 2003. Neuromuscul Disord. 2004;14:337.
Hsueh P.R., Hsiue T.R., Hsieh W.C. Pyomyositis in intravenous drug abusers: Report of a unique case and review or the literature. Clin Infect Dis. 1996;22:858.
Illa I., Nath A., Dalakas M.C. Immunocytochemical and virological characteristics of HIV-associated inflammatory myopathies: Similarities with seronegative polymyositis. Ann Neurol. 1991;29:474.
Jarowski C.I., Fialk M.A., Murray H.W., et al. Fever, rash, and muscle tenderness. A distinctive clinical presentation of disseminated candidiasis. Arch Intern Med. 1978;138:544.
Joffe M.M., Love L.A., Leff R.L. Drug therapy of idiopathic inflammatory myopathies: Predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med. 1993;94:379.
Kaufman L.D., Kephart G.M., Seidman R.J., et al. The spectrum of eosinophilic myositis: Clinical and immunopathologic studies of three patients, and a review of the literature. Arthritis Rheum. 1993;36:1014.
Kim S., El-Hallak M., Dedeoglu F., et al. Complete and sustained remission of juvenile dermatomyositis resulting from aggressive treatment. Arthritis Rheum. 2009;60:1825.
Kinoshita M., Iwasaki Y., Wada F., et al. A case of congenital polymyositis – A possible pathogenesis of “Fukuyama type congenital muscular dystrophy”. Clin Neurol. 1980;20:911.
Kinoshita M., Nishina M., Koya N. Ten years follow-up study of steroid therapy for congenital encephalomyopathy. Brain Dev. 1986;8:281.
Kissel J.T., Halterman R.K., Rammohan K.W., et al. The relationship of complement-mediated microvasculopathy to the histologic features and clinical duration of disease in dermatomyositis. Arch Neurol. 1991;48:26.
Kissel J.T., Mendell J.R., Rammohan K.W. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med. 1986;314:331.
Krahn M., Lopez De Munain A., Streichenberger N., et al. CAPN3 mutations in patients with idiopathic eosinophilic myositis. Ann Neurol. 2006;59:905.
Laxer R.M., Stein L.D., Petty R.E. Intravenous pulse methylprednisolone treatment of juvenile dermatomyositis. Arthritis Rheum. 1987;30:328.
Leff R.L., Love L.A., Miller F.W., et al. Viruses in idiopathic inflammatory myopathies. Absence of viral candidate genomes in muscle. Lancet. 1992;339:1192.
Levine T.D. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601.
Love L.A., Leff R.L., Fraser D.D., et al. A new approach to the classification of idiopathic inflammatory myopathy: Myositis-specific autoantibodies define useful homogeneous patient groups. Medicine. 1991;70:360.
Mantegazza R., Andreetta F., Bernasconi P., et al. Analysis of T cell receptor of muscle-infiltrating T lymphocytes in polymyositis: Restricted V alpha/beta rearrangements may indicate antigen-driven selection. J Clin Invest. 1993;91:2880.
Mejlszenkier J.D., Safran A.P., Healy J.J., et al. The myositis of influenza. Arch Neurol. 1973;29:441.
Miller F.W. Myositis-specific antibodies. Touchstones for understanding the inflammatory myopathies. JAMA. 1993;270:1846.
Miller F.W., Leitman S.F., Cronin M.E., et al. Controlled trial of plasma exchange and leukopheresis in polymyositis and dermatomyositis. N Engl J Med. 1992;326:1380.
Miller L.C., Sisson B.A., Tucker L.B., et al. Methotrexate treatment of recalcitrant childhood dermatomyositis. Arthritis Rheum. 1992;35:1143.
Miller T., Al-Lozi M.T., Lopate G., et al. Myopathy with antibodies to the signal recognition particle: Clinical and pathological features. J Neurol Neurosurg Psychiatry. 2002;73:420.
Mozaffar T., Pestronk A. Myopathy with anti-Jo-1 antibodies: Pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry. 2000;68:472.
Nagai T., Hasgawa T., Saito M., et al. Infantile polymyositis: A case report. Brain Dev. 1992;14:167.
Nagar H., Bar-Ziv Y. Focal eosinophilic myositis: Unusual cause of a tumour on the chest wall. Eur J Surg. 1993;159:187.
Niewold T.B., Kariuki S.N., Morgan G.A., et al. Elevated serum interferon-alpha activity in juvenile dermatomyositis: Associations with disease activity at diagnosis and after thirty-six months of therapy. Arthritis Rheum. 2009;60(6):1815.
O’Neill D.S., Baquis G., Moral L. Infectious myositis. A tropical disease steals out of its zone. Postgrad Med. 1996;100:193.
Olney R.K., Miller R.G. Inflammatory infiltration in Fukayama type congenital muscular dystrophy. Muscle Nerve. 1983;6:75.
Pachman L.M. Inflammatory myopathy in children. Rheum Dis Clin North Am. 1994;20:919.
Pachman L.M. Juvenile dermatomyositis. Pathophysiology and disease expression. Pediatr Rheumatol. 1995;42:1071.
Pegoraro E., Mancias P., Swerdlow S.H., et al. Congenital muscular dystrophy with primary laminin 2 (merosin) deficiency presenting as inflammatory myopathy. Ann Neurol. 1996;40:782.
Plotz P.H., Rider L.G., Targoff I.N., et al. Myositis: Immunologic contributions to understanding cause, pathogenesis, and therapy. Ann Intern Med. 1995;122:715.
Pollock J.L. Toxoplasmosis appearing to be dermatomyositis. Arch Dermatol. 1979;115:736.
Roddy S.M., Ashwal S., Peckham N., et al. Infantile myositis: A case diagnosed in the neonatal period. Pediatr Neurol. 1986;2:241.
Rodgers W.B., Yodlowski M.L., Mintzer C.M. Pyomyositis in patients who have the human immunodeficiency virus. Case report and review of the literature. J Bone Joint Surg. 1993;75:588.
Rose M.R., Kissel J.T., Bickley L.S., et al. Sustained myoglobinuria: The presenting manifestation of dermatomyositis. Neurology. 1996;47:119.
Ruff R.L., Secrist D. Viral studies in benign acute childhood myositis. Arch Neurol. 1982;39:261.
Salajegheh M., Kong S.W., Pinkus J.L., et al. Interferon-Stimulated Gene 15 (ISG15) conjugates proteins in dermatomyositis muscle with perifascicular atrophy. Ann Neurol. 2009. (in-press)
Sawney B.B., Chopra J.S., Banerji A.K., et al. Pseudohypertrophic myopathy in cysticercosis. Neurology. 1976;26:270.
Shevell M., Rosenblatt B., Silver K., et al. Congenital inflammatory myopathy. Neurology. 1990;40:1111.
Sigugeirsson B., Lindelöf B., Edhag, et al. Risk of cancer in patient with dermatomyositis or polymyositis. N Engl J Med. 1992;326:363.
Sontheimer R.D. Cutaneous features of classic dermatomyositis and amyopathic dermatomyositis. Curr Opin Rheumatol. 1999;11:475.
Stonecipher M.R. Cutaneous changes of dermatomyositis in patients with normal muscle enzymes: Dermatomyositis sine myositis? J Am Acad Dermatol. 1993;28:951.
Taksande A., Vilhekar K., Gupta S. Primary pyomyositis in a child. Int J Infect Dis. 2009;13:e149.
Thompson C.E. Infantile myositis. Dev Med Child Neurol. 1982;24:307.
Thornton C.A., Griggs R.C. Plasma exchange and intravenous immunoglobulin treatment of neuromuscular disease. Ann Neurol. 1994;35:260.
Tymms K.E., Webb J. Dermatomyositis and other connective tissue diseases: A review of 105 cases. J Rheumatol. 1985;12:1140.
Van der Meulen M.F.G., Bronner M., Hoogendijk J.E., et al. Polymyositis: A diagnostic entity reconsidered. Neurology. 2003;61:316.
Walsh R.J., Kong S.W., Yao Y., et al. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007;56:3784.
Wrzolek M.A., Sher J.H., Kozlowski P.B., et al. Skeletal muscle pathology in AIDS: An autopsy study. Muscle Nerve. 1990;13:508.
