Chapter 87 Normal Muscle
Embryology and Development
Primitive cells, called premyoblasts, are the precursors of muscle in the embryonic paraxial mesoderm. The paraxial mesoderm coalesces to form segmented epithelial spheres on either side of the notochord, referred to as somites. Craniofacial muscles arise rostral of the first somite from paraxial and prechordal mesoderm [Lu et al., 2002]. Discrete condensations of mesenchymal cells, called dermatomyotomes and located dorsomedial to the notochord, give rise to the axial muscles, and overlying skin and condensations from the lateral somite give rise to the limb muscles (Figure 87-1) [McLennon, 1994; Buckingham et al., 2003]. Connective tissue and tendons arise from the somatopleural mesoderm, somites, and neural crest. Sclerotome, which is ventral in location, forms the skeleton.
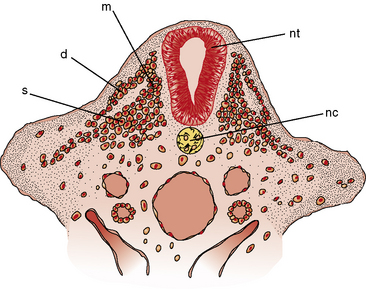
Premyoblasts express the paired box transcription factors, Pax-3 and Pax-7. Bone morphogenetic protein 4 (BMP4), released from adjacent neural tube and lateral plate mesoderm, inhibits gene expression of myogenic transcription factors, MyoD and Myf-5. Wnts (Wnt 1, 3, 7a, and 1) and sonic hedgehog signals from adjacent notochord, neural tube, and surface ectoderm activate Myf-5 and MyoD (members of a family of transcription factors called myogenic regulatory factors) in premyoblasts [Pownall et al., 2002]. These committed cells, myoblasts, migrate to the myotome. The myoblasts divide rapidly within the first several weeks of pregnancy, after a quantal cell cycle under the influence of fetal growth factors. The primordial cells stream ventrally and penetrate between the ectoderm and somatopleura. As the cells migrate, they split and recombine, such that individual muscles are formed from several adjacent myotomes. In the limb bud, the cells coalesce into dorsal and ventral masses.
In a coordinated fashion, the MyoD family of growth factors form heterodimeric DNA complexes with other basic helix–loop helix transcription factors to regulate an array of gene expression [Kassar-Duchossoy et al., 2004]. Collectively, the transcription factors govern the assignment to skeletal muscle lineage, migration of progenitor cells from the hypoaxial domain of the dermatomyotome to the limb, condensation of premuscle masses, and formation of primary and secondary myotubes [Cossu and Biressi, 2005].
The maturation of premyoblasts to myoblasts begins with the cessation of DNA synthesis. The postmitotic myoblasts elongate and begin attachment and fusion with other myoblasts, end to end, to form a long and slender primary myotube (Figure 87-2). The process is facilitated by the appearance of several fetal adhesion molecules on the surface of the myoblast [Schnorrer and Dickson, 2004]. Secondary and tertiary myotubes are formed from side-to-side fusion of myoblasts to existing myotubes and require innervation for the process. Satellite cells provide nuclei to the polar ends of the elongating myotube. Individual muscles begin to form after the initial myotubes appear and the ingrowth of innervation. In the absence of innervation, maturation beyond primary myotubes does not occur, and the muscle develops abnormally.
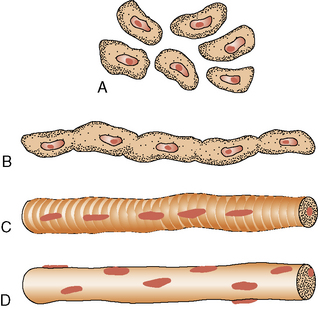
Fig. 87-2 Development of the muscle fiber.
A, Premyoblasts. B, Myoblast fusion. C, Myotube. D, Muscle fiber.
At about 4 weeks’ gestation, the contractile proteins appear and polymerize to form myofilaments, which are produced predominantly in the polar region of the myotube. By the fifth week, the myofilaments aggregate to form the myofibrils, with simultaneous formation of characteristic striations. A microscopic cross-section of the muscle fiber at this stage reveals a tubular structure, with the contractile proteins located around the periphery of the fiber and the center containing the nuclei. The basic shape of an anatomic muscle is apparent by 7 weeks. Movement begins simultaneously with innervation at 8 weeks. In the latter stages of early fetal development, neuron sprouting is intense. Initially, individual mammalian muscle fibers are multiply innervated. Between 16 and 25 weeks’ gestation and in association with secondary myotube formation, all but one synapse is eliminated. There is constant denervation and reinnervation as neuromuscular interaction forms physiologic innervations of the motor units. Multiple motor neurons compete for innervation. Competition weakens some synapses and strengthens others, so that, ultimately, a single input prevails. This process of synaptic elimination is stimulated by local factors produced by muscle [Wyatt and Balice-Gordon, 2003]. Several growth factors and receptors appear on the cell surface to facilitate integration of nerve terminals to the muscle cell. The fetal (g) form of the acetylcholine receptor is present until 31 weeks’ gestation, after which only the adult type (e) is found. Rhythmic movements that are spinally mediated start shortly after innervation. Most of the fetal membrane proteins that promote growth and differentiation, including the major histocompatibility gene products [Kaparti et al., 1988], are not present on normal mature muscle.
Distinctive muscle fiber types (see next section) develop after innervation. Primitive fibers, typed as IIc, are undifferentiated and are believed to be precursors of the mature fibers: types I, IIa, and IIb [Landon, 1982]. The myosin in type IIc fibers is immunologically distinct from that of other fiber types [Thornell et al., 1984]. Type I fibers appear at about 18 weeks and are smaller than type II fibers until after birth, when they become somewhat larger. Most muscle fibers in the 20- to 26-week-old fetus are type IIc fibers. Type IIa and IIb fibers appear during the last month of gestation. Only a small percentage of type IIc fibers persist after birth. Differentiation into distinct fiber types is determined by neural influences and causes the biochemical and physiologic diversity of mature fibers. Calcineurin, a Ca2+-calmodulin-regulated phosphatase, plays a critical role in the differentiation of physiologic properties in the muscle fiber [Matlin et al., 2001]. Continuous firing of motoneurons causes an increase in intracellular calcium, which activates calcineurin. Activated calcineurin binds and dephosphorylates two kinds of transcription factors: nuclear factor of activated T cell (NFAT) and myocyte enhancer factor 2 (MEF2), resulting in significant activation of the slow myosin heavy chain 2 gene (slow MyHC2) promoter. In contrast, in fast fibers, high-amplitude calcium sparks induced by infrequent phasic firing of the motor nerves are insufficient to keep activation of calcineurin. When calcineurin is inactivated, phosphorylated NFAT cannot enter the nucleus and the slow fiber-specific program is downregulated, resulting in the predominant transcription of genes encoding fast fiber-specific proteins [Jiang et al., 2004].
Without innervation, muscle fibers atrophy and undergo cell death. Similarly, spinal motor neurons become nonviable without innervating muscle, so that muscle and neuronal development are ultimately interdependent [McLennon,1994].
Satellite cells are mononuclear muscle cells that lie beneath the basement membrane of mature muscle fibers. They were first described by Alexander Mauro in 1960. Satellite cells are abundant in fetal muscle, compose 1–5 percent of the total nuclei of mature muscle, and diminish with aging. The typical satellite cell during embryonic development migrates from dorsal somite to dermatomyotome. Transcription factor Pax-7 is a key protein involved in myogenic determination of satellite cells. Transcription factor Pax-3 is needed for the migration of these cells from the dorsal somite and Pax-7 is required for the transition to forming mature muscle cells. The dependency on Pax-7 is felt to rest on the stage of maturation of the animal. Pax-7 are not essential for maturation of satellite cells in adults [Lepper et al., 2009]. The satellite cells express M-cadherin, c-met, foxk-1, and CD 34 antigens [Wernig et al., 2004; Beauchamp et al., 2000]. Muscle injury activates satellite cell proliferation and maturation into myofibers. This is marked by expression of muscle regulatory factors proteins Myf-5 and MyoD [Martin et al., 2006].
While most of the satellite cells are derived from the primordial somite, they can also be derived from hematopoietic stem cells and vascular progenitor cells (endothelium, pericyte, and mesangioblast) [Shi and Garry, 2006]. They are the primary stem cell for regeneration of injured muscle and are capable of forming not only individual muscle fibers, but also complete muscle fascicles [Alameddine et al., 1989; Anderson, 2000; Schultz, 1985].
Muscle is quite malleable and is under a number of neuronal, hormonal, and usage influences that determine muscle function and contractile properties [Pette, 2001]. This plasticity of muscle results in changes in fiber type distribution, contractile proteins, calcium uptake of the sarcoplasmic reticulum, and altered energy metabolism.
Anatomy and Structure
Morphology
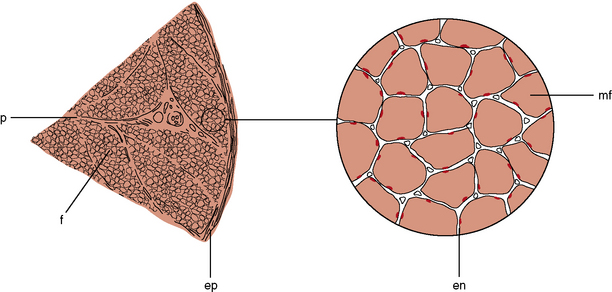
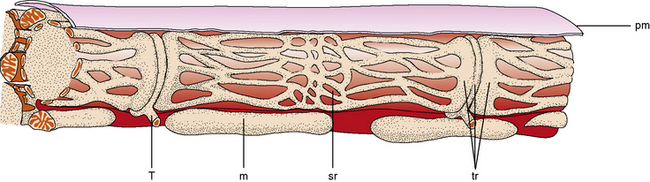
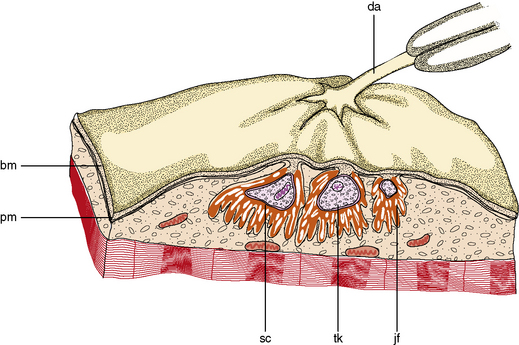
The muscle as an organ comprises all the individual anatomic muscles. Each anatomic muscle has an origin and an insertion on the skeleton, and bridges one or more bony articulations. Contraction of the anatomic muscle thus causes movement across a joint. The anatomic muscle is enclosed within a thick sheet of connective tissue called the epimysium. Separating the muscle into individual fascicles is the perimysium, which is contiguous with the epimysium. Within the perimysium are the nutrient blood vessels, intramuscular nerves, and muscle spindles (Figure 87-3). The confluence of the perimysial and epimysial connective tissue forms the tendons at either end of the muscle belly. The muscle fascicle, which is bounded by perimysial connective tissue, is a wedge-shaped structure comprising several hundred individual muscle fibers. Surrounding each muscle fiber is a network of fine connective tissue, called the endomysium. The terminal axons and a rich capillary network reside within the endomysium. Within the muscle fiber are large groups of myofibrils, which contain myofilaments. Myofilaments are made of contractile proteins.
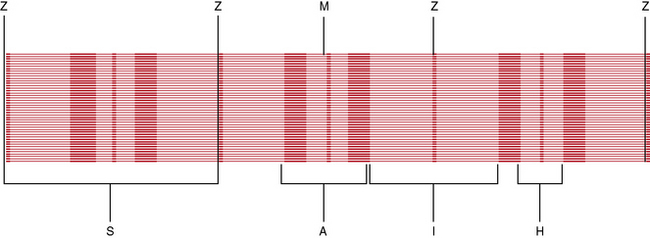
Sarcomere
The most striking feature of skeletal muscle on microscopic examination is the characteristic striations (“striated” muscle), which are especially prominent under polarized light. The striations are caused by the difference in the refractive index of the contractile proteins that are in phase with each other in the myofibrils. A repeating unit is called a sarcomere; it comprises interdigitating myofilaments and is bounded by the Z line (Figure 87-4). The sarcomere has a length of about 2.5–3.0 μm and a diameter of 1.0 μm. The Z disk anchors the thin filaments of actin that extend into each adjacent sarcomere and are located in the I band. The M line bisects the sarcomere and also divides the A band, which is formed by an array of thick filaments composed of myosin. The area within the A band in which the thin and thick filaments do not overlap is called the H band. A refers to anisotropic and I refers to isotropic in connection with the refractile indexes under polarized light.
Contractile and Sarcomeric Proteins
The two major proteins involved in muscle contraction are actin and myosin, which jointly interact with adenosine triphosphate to convert chemical energy to mechanical work. The actin system is complex. The actin molecule has four major domains surrounding a cleft containing adenosine triphosphate or adenosine diphosphate, and is tightly bound to a divalent cation [Pollard, 1993]. There are at least 48 classes of actin-binding proteins. The thin filaments are composed of two chains of about 400 polymerized actin molecules arranged in a double helix (Figure 87-5). The actin molecule has a molecular weight of about 42,000 daltons and is rich in 3-methyl histidine, a unique amino acid found exclusively in muscle. The actin molecule is the same in both fast and slow skeletal muscle fibers. Alpha-actinin is a major component of the Z disk and plays a role in the binding or cross-linking of actin to other cytoskeletal structures [Critchley, 1993]. Capping proteins anchor the barbed end of actin to the Z line [Cooper et al., 2010]. Mutation in the α-actin gene has been associated with congenital myopathy with actin filament aggregate myopathies and nemaline myopathy [Schroder et al., 2004; Nowak et al., 1999], hypertrophic and dilated cardiomyopathies [Marston and Hodgkinson, 2001]. The role of actin structure in its interaction with myosin and in its effect on polymerization of actin molecules is thought to cause the phenotypic variability in clinical presentation. The mechanochemical myosin molecule is a complex protein with a molecular weight of about 500,000 daltons. The myosin molecule consists of at least six polypeptides with two heavy chains and four light chains. The contractile properties of a muscle fiber are related to myosin. Myosin heavy chains are different for type I, IIa, and IIb fibers. The two heavy chains form a double helix that extends along the tail of the molecule. The light chains are of the following three types: two alkali-dissociated and one phosphorylated. The light chains are different for type I and II fibers, but are the same for IIa and IIb fibers. The structure of myosin is hexameric, composed of two heavy, two alkali-dissociated, and two phosphorylated chains. There are nine isoforms of the myosin heavy chain: I, IIA, IIB, IID/X, IIA, α, neonatal, embryonic, and extraocular. The various isoforms of the heavy and light chains create a spectrum of isomyosins that are specific for fast-twitch skeletal muscle, slow-twitch skeletal muscle, cardiac muscle, smooth muscle, brain, and platelets [Whalen, 1985]. Embryonic and neonatal muscle contains distinct sets of isomyosins. Skeletal muscle isomyosins are determined by neural influences (see above). “Pure” muscle fibers contain a single myosin heavy chain isoform. “Hybrid” fibers contain more than one myosin isoform, as in muscle fibers transitioning from one fiber type to another during reinnervation. The maximal force, velocity, and power produced by muscle fibers are determined to a large extent by the properties of myosin isoforms [Lutz and Lieber, 2002]. Mutations in myosin gene MYH7 cause hyaline body myopathy with or without cardiomyopathy and familial cardiomyoneuromyopathy. Respiratory failure occurs with mutations in exons 37 and 38 of the MYH7 gene [Goebel and Laing, 2009; Selcen et al., 2002].
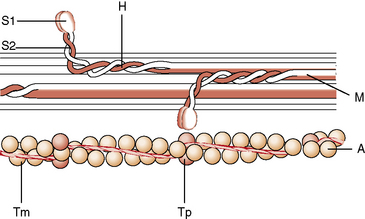
At one end, the myosin molecule evaginates to form globular heads. The myosin head is called the S2 region and is attached to the heavy chains. The S2 region is flexible at either end [Huxley, 1982]. One of each type of light chain is associated with the globular heads. The LC1 (light chain 1), or P light chain, somehow modulates the contractile response by specific phosphatase and kinase, and exists as P1 and P2 isoforms [Westwood et al., 1984]. The cross-links between actin and myosin occur at the globular heads during excitation. Thus, the myosin molecule is a bipolar structure, with the heavy chains of the tail ordered along the backbone of the thick filament and the heads projected from the side. The heads have a repeating helical arrangement that forms about three cross-bridges per repeating unit [Harrington and Rodgers, 1984]. C proteins exist as several isoforms and can be seen as cross-stripes on the A band on electron microscopy [Fishman, 1993]. The function of C proteins is unknown but is probably regulatory of cross-bridge movements.
Actin and myosin have an inherent affinity, and troponin and tropomyosin are regulatory proteins that enable the actin–myosin interaction to be controlled by the calcium ion [Grabarek et al., 1990]. Tropomyosin is another helical structure, comprising two long filaments that reside within the groove of the actin chains [Smillie, 1993]. One tropomysin molecule binds one troponin complex and positions tropinin molecules regularly along the actin filament with a periodicity of 385 A. The troponin complex holds tropomyosin in an “off” or “on” state of contraction position on the actin helix, depending on the level of calcium [Lehman et al., 2001]. There are at least two isoforms of tropomyosin. Fast-twitch skeletal muscle contains both α and β isoforms; slow-twitch muscle contains only β-tropomyosin [Heeley et al., 1985]. Because of the close association with actin, tropomyosin probably also has a structural role.
The troponin molecules are located periodically along tropomyosin and comprise three subunits. Troponin T anchors troponin to tropomyosin. Troponin C binds calcium (Ca2+), which induces a conformational change of the protein and results in activation of actomyosin adenosine triphosphatase, which causes contraction [Gergeley et al., 1993]. Troponin I inhibits actomyosin adenosine triphosphatase. The contraction of striated muscle is regulated by the troponin (Tn) complex, which acts as a Ca2+ sensor. Ca2+ binding to the regulatory sites of troponin C (TnC) strengthens the interaction of TnC with troponin I (TnI), and weakens the inhibitory activity of TnI for actin and the affinity of troponin T (TnT) for tropomyosin (Tm). Tm then moves toward the groove of the helical actin filament, switching on the myosin active site, which leads to muscle contraction. Tn binds to actin-Tm in the absence of Ca2+, and the changes in interactions among these thin filament components in the presence of Ca2+ promote strong binding of myosin to actin [Gomes et al., 2002, Brown and Cohen, 2005]. Mutations of sarcomeric genes for troponin TI and tropomyosin are some of several causes of nemaline myopathy [Wallgren-Pettersson, 2002] and cardiomyopathies [Gomes et al., 2004].
Titin, the largest known protein of about 3–3.7 megadaltons, constitutes about 10 percent of myofibrillary proteins and is important for maintaining tension during stretch and recentering the sarcomere after relaxation [Tskhovrebova and Trinick, 2003]. Opposing molecules of titin span the sarcomere. The NH2 termini of the titin molecules attach to the Z disk, attaching myosin to the Z disk. The COOH ends extend and overlap in the M band, interconnecting myosin and actin, and providing elasticity to the sarcomere. It is encoded by a single gene (TTN0), which is located on the long arm of chromosome 2. The protein spans 3–4 megadaltons and consists of repeating immunoglobulin and fibronectin 111 domains. The first 200 amino acids of the titin molecule reside at the periphery of the Z disk and mark its boundary. Titin interacts with several proteins, including telethonin, ankyrin, filamin, nebulin, and α-actinin. Titin plays a major role in myofibrillogenesis, and over- or under-expression of titin leads to disruption of assembly of the Z disk and sarcomere. It has a major function as a “molecular blueprint” that specifies the precise assembly of structural regulatory and contractile proteins of sarcomere, and it gives the sarcomere its distinct biomechanical properties and integrity during stretch, relaxation, and contraction [Kontrogianni-Konstantopoulos et al., 2009]. Mutations in the titin gene cause familial dilated cardiomyopathy [Geruli et al., 2002].
Nebulin is another large protein of the skeletal muscle sarcomere and measures 500–800 kDa. The COOH terminus is attached to the Z disk, while the NH2 terminus projects into the I band and is closely associated with actin. The NH2 terminal binds to the actin-capping protein, and the COOH terminal binds to titin and myopalladin. Nebulin is encoded by the NEB gene, which is located on the short arm of chromosome 2 [Donner et al., 2004]. Nebulin regulates the length of thin filaments in sarcomeres. Mice with null mutations of the NEB gene have variable length and generate less force. The role of the NEB gene is felt to be in stabilizing thin filaments at a determined length and is critical for generation of optimal force. Like titin, nebulin interacts with several structural and functional proteins [Kontrogianni-Konstantopoulos et al., 2009]. Mutations of nebulin causes nemaline myopathy [Lekhotari et al., 2006] and distal myopathy [Wallgren-Pettersson et al., 2007].
Obscurin is the third and most recently discovered member of a family of giant proteins involved in maintaining the structure of the sarcomere. It measures 720–900 kDa and is encoded by the OBSCN gene, located on the short arm of chromosome 1. Immunohistochemical techniques indicate that obscurin is localized at the M line and Z disk in skeletal and cardiac muscle. Obscurin binds and interacts with numerous muscle proteins, including titin, ankyrin, calmodulin, and myomesin. Obscurin plays an important role in myofibrillogenesis by allowing lateral alignment and fusion of myofibrils into larger bundles. It also has an important part in assembly and stabilization of A bands and M bands in the sarcomere. Mutations of the obscurin gene produce hypertrophic cardiomyopathy by causing loss of binding with titin [Arimura et al., 2007].
Sarcotubular System
A major constituent of the muscle fiber is the sarcotubular system (Figure 87-6). The sarcolemma is the plasma membrane of the muscle cell and is surrounded by the basement membrane and endomysial connective tissue. The sarcolemma is an excitable membrane and shares many properties with the membrane of the neuron. The T tubules are contiguous with the sarcolemma and extend into the interior of the muscle fiber as a tubular system in communication with the sarcolemma. Depolarization of the sarcolemma is propagated throughout the interior of the muscle fiber through this system [Peachey, 1985]. The T tubules project into the interior of the muscle fibers in the area of the junction of the I band and A band, where they come in immediate contact with a second tubular system within the sarcoplasm called the sarcoplasmic reticulum. The T tubule, bounded on either side by the sarcoplasmic reticulum, is a triad. The sarcoplasmic reticulum forms a fine plexus around the myofibrils. Excitation of the sarcolemma and T tubules causes release of calcium from the sarcoplasmic reticulum and initiation of contraction by the myofilaments.
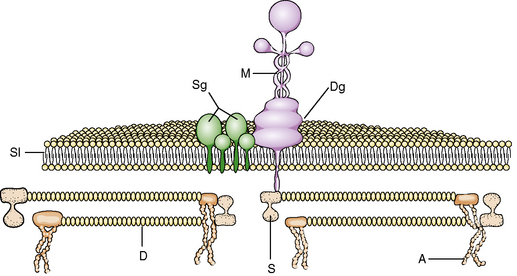
Several important proteins are associated with the junction of the T tubules and the sarcoplasmic reticulum. The voltage sensors of the T-tubule calcium channels are regulated by dihydropyridine receptors. Ryanodine receptors mediate the calcium release of the sarcoplasmic reticulum during muscle activation. Calcium adenosine triphosphatase pumps calcium back into the sarcoplasmic reticulum during relaxation. Calsequestrin is a calcium-binding protein that increases the capacity of calcium in the sarcoplasmic reticulum [Franzini-Armstrong, 1999]. Some patients with myasthenia gravis, especially associated with thymoma, have anti-skeletal muscle antibodies to ryanodine receptor antigens, as well as to titin [Skeie et al., 2003; Suzuki et al., 2009]. Autosomal-dominant and autosomal-recessive mutations of the ryanodine receptor gene (RYR) cause central core disease [Ferreiro et al., 2002; Jungbluth et al., 2005], myopathy with type 1 fiber dominance [Morrison, 2008], and central core disease with rods [Scacheri et al., 2000].
Cytoskeletal Proteins
The structural integrity of the muscle is maintained against the physical forces of contraction exerted on the sarcolemma membrane by an intricate system of cytoskeletal proteins (Figure 87-7). These proteins, through a complex pattern of arrangement, anchor the internal structure of the muscle to the basement membrane. Advances in techniques of molecular biology have led to identification and characterization of several of these proteins (Table 87-1).
| Protein | Location | Disease |
|---|---|---|
| PLASMA MEMBRANE | ||
| Dystrophin | Xp21 DMD | BMD, DMD |
| α-Sarcoglycan | 17q12–q21 | LGMD-2D |
| β-Sarcoglycan | 4q12 | LGMD-2E |
| γ-Sarcoglycan | 13q12 | LGMD-2C |
| δ-Sarcoglycan | 5q33–34 | LGMD-2F |
| Dysferlin | 2p13 | LGMD-2D, Miyoshi’s myopathy |
| Caveolin-3 | 3p25 | LGMD-1C |
| EXTRACELLULAR MATRIX | ||
| α2-Laminin | 6q2 | Merosin-deficient CMD |
| Collagen VI | 21q22 | Ullrich’s CMD, Bethlem’s myopathy |
| SARCOPLASMIC | ||
| Calpain-3 | 15q15.1 | LGMD-2A |
| TRIM32 | 9q31–34 | LGMD-2H |
| Myotubularin | Xq28 | Myotubular myopathy |
| Desmin | 11q21–23 | Desmin-related myopathy |
| Plectin | 8q24–qter | Epidermolysis bullosa simplex with late-onset muscular dystrophy |
| GLYCOSYLATION-RELATED | ||
| Fukutin | 9q31–33 | Fukuyama CMD |
| FKRP | 19q1 | LGMD-D2, CMD-1C |
| POMGnT1 | 1p32–34 | Muscle-eye-brain CMD (O-mannose β-1,2-N-acetylglucosaminyl transferase) |
| POMT1 | 9q34 | Walker–Warburg CMD (O-mannosyltransferase-1) |
| SARCOMERIC PROTEINS | ||
| Titin | 2q | LGMD-2J |
| Myotilin | 5q22–34 | LGMD-1A |
| Telethonin | 17q11–12 | LGMD-2G |
| Actin | 1q42 | Nemaline myopathy |
| Tropomyosin-3 | 1q21–23 | Nemaline myopathy |
| Tropomyosin-2 | 9p13 | Nemaline myopathy |
| Nebulin | 2q21–22 | Nemaline myopathy |
| Slow troponin T | 19q13 | Nemaline myopathy |
| Ryanodine receptor (RYR1) | 19q13.1 | Central core disease, central core disease with rods |
| Myosin heavy chain (MyH 7) | 14q12 | Myopathy with hyaline bodies, cardiomyopathy, cardiomyoneuropathy |
| NUCLEAR PROTEINS | ||
| Emerin | Xq28 | EDMD |
| Lamin A/C | 1q11–21 | LGMD-1B |
BMD, Becker muscular dystrophy; CMD, congenital muscular dystrophy; DMD, Duchenne muscular dystrophy; EDMD, Emery–Dreifuss muscular dystrophy; LGMD, limb girdle muscular dystrophy.
Recent updates from http://www.neuromuscular.wustl.edu and http://www.Genetests.com http://neuromuscular_wustl.edu.
(Adapted from Vainzof M, Zatz M: Protein defects in neuromuscular diseases, Braz J Med Biol Res 36:543–555, 2003.)
Dystrophin
The identification and characterization of dystrophin led to an understanding of the role of cytoskeletal proteins in providing strength and stability to the muscle membrane. Dystrophin is a large protein molecule of about 427 kilodaltons and is composed of 3685 amino acids. It constitutes 0.01 percent of total muscle protein and 5 percent of the sarcolemmal cytoskeletal proteins [Hoffman et al., 1987]. The protein has the shape of a rod, is about 150 nm long, and is arranged as an antiparallel homodimer. It is abundant at the myotendinous junction [Samitt and Bonilla, 1990] and at the postsynaptic membrane of the neuromuscular junction [Byers et al., 1991]. Immunocytochemical studies have localized dystrophin to the cytoplasmic surface of the sarcolemma [Bies et al., 1992]. Dystrophin and two other structural proteins, spectrin and vinculin, are located at the sites of attachment of sarcomeres to the cytoplasmic membrane overlying both the I bands and M lines [Porter et al., 1992]. These findings demonstrate that dystrophin forms an integral part of a muscle’s cytoskeleton and links the contractile apparatus to the sarcolemma. There is no difference in the expression of dystrophin in fast- and slow-twitch muscle fibers or in intrafusal muscle fibers [Zubrzycka-Gaarn et al., 1988].
Dystrophin has a binding site for the filamentous form of actin at the 5′ end or domain. The central rod domain contains a number of repeats; it demonstrates homology with spectrin and gives the molecule a flexible rod-shaped structure [Pons et al., 1990]. The rod domain is highly conserved in vertebrates, and antibodies against the human dystrophin rod domain cross-react with amphibian and other vertebrate species [Sherratt et al., 1992]. The third domain is rich in cysteine [Suzuki et al., 1992], and the fourth domain, the carboxy terminus, binds with the dystrophin–glycoprotein complex [Ervasti and Campbell, 1991]. The dystrophin–glycoprotein complex consists of dystroglycans (α and β), the sarcoglycans (α,β,γ,δ,ε), sarcospan, the syntrophin, and dystrobrevin [Crawford et al., 2000]. The peripheral members of the complex include neuronal nitric oxide synthase, caveolin, laminin, and merosin [Watkins et al., 2000]. Even though the RNA levels of these proteins are normal in patients with dystrophin deficiency, the protein expression is reduced. The deficiency of these proteins contributes further to the pathologic effects of dystrophin deficiency [Chen et al., 2000].
The gene coding for the protein dystrophin is located on the short arm of the X chromosome near the region Xp21. The dystrophin gene is the largest gene identified so far, covering more than 2.5 megabases (Mb), and contains at least 79 exons [Gutmann and Fischbeck, 1989]. The large size of the gene is responsible for the high rate of mutation in this gene. A 14-kilobase dystrophin messenger RNA is expressed in skeletal, cardiac, and smooth muscle cells. Smaller amounts are expressed in the brain. Isoforms of dystrophin, which are smaller in size, are expressed in nearly all tissues examined. Deficiency of the brain isoform of dystrophin, Dp 140 and Dp 71, is associated with cognitive handicaps. Dystrophin in the brain is important in maintaining synaptic plasticity and participates in cellular signaling pathways involved in modeling synapses [Blake and Kroger, 2000]. Dystrophin isoform Dp 260 is expressed in retina alone [Pillers et al., 1999].
The most important function of dystrophin is to provide mechanical support and structural integrity to the sarcolemma [Lapidos et al., 2004]. Dystrophin is part of the linkage system from the actin cytoskeleton out through the sarcolemmal membrane and basal lamina to the extracellular matrix. Dystrophin plays an important role in maintenance of calcium homeostasis in muscle fibers [Blake et al., 2002]. Muscle fibers deficient in dystrophin show an increase in intracellular calcium and are derived from leaks in the cell membrane and sarcoplasmic reticulum [Carlson, 1998]. Hypercontracted muscle fibers seen on histological examination are the earliest signs of increase in intracellular calcium. The increase in intracellular calcium is believed to have a role in muscle fiber necrosis and apoptosis [Sandri and Carbano, 1999]. Dystrophin within the dystrophin–glycoprotein complex participates in this intracellular signaling. It plays a role in regulation of calcium-dependent kinases in the cell by interacting with calmodulin [Anderson et al., 1996]. Alteration in calcium signaling in dystrophin-deficient muscles plays a major role in altering the normal maturation process of regenerating and developing muscle fibers [Chen et al., 2000].
Through its linkage to neuronal nitric oxide synthase, dystrophin has a role in regulation of blood flow through muscle during exercise. Abnormalities in modulation of neuronal nitric oxide contribute to muscle fiber damage in dystrophinopathies [Sander et al., 2000]. Deletions or abnormalities of the dystrophin gene cause a deficiency or absence of dystrophin, resulting in the X-linked Duchenne’s and Becker’s muscular dystrophies.
Dystrophin–Glycoprotein Complex
Biochemical investigation of dystrophin led to the discovery of a large oligomeric complex of glycoprotein localized in the sarcolemma. This dystrophin–glycoprotein complex binds the muscle cytoskeleton to a component of the extracellular matrix, laminin [Campbell and Kahl, 1989; Ervasti et al., 1990]. The dystrophin–glycoprotein complex consists of cytoskeletal, transmembrane, and extracellular components, and is composed of two subgroups of protein complexes: the dystroglycans and sarcoglycans.
Dystroglycan consists of a transmembrane and an extracellular component encoded by a single messenger RNA. The precursor protein is processed by an unknown protease to α- and β-dystroglycan molecules. The size of dystroglycan molecule is expressed in various tissues and its molecular weight varies, based upon differential glycosylation in those tissues. In muscle α-dystroglycan weighs 43 kDa, and in brain it weighs 120 kDa. The synthesized protein undergoes glycosylation as it passes from endoplasmic reticulum to the sarcolemmal membrane. The α-dystroglycan is a heavily glycosylated protein located on the extracellular side of the sarcolemmal membrane. The glycosylation is mediated by six glycosyl transferases: protein-O-mannosyl transferase 1 (POMT1), protein-O-mannosyl transferase 2 (POMT2), protein-O-mannose 1,2-N-acetylglucosaminyltransferase 1 (POMGnT1), fukutin (FKTN), fukutin-related protein (FKRP), and LARGE. Mutations in these enzymes cause myopathies, and brain and eye malformations [Mercuri et al., 2009].
Alpha-dystroglycan plays an active role in basement membrane assembly. It binds to β-dystroglycan (transmembrane component) with its carboxy-terminal region on the intracellular side. Alpha-dystroglycan binds to several proteins – laminins, agrin, and perlecan – to anchor into the extracellular matrix. Beta-dystroglycan binds to the WW domain and EF hands of the dystrophin molecule. The interactions between beta-dystroglycan, caveolin and dystrophin regulates the insertion of the dystrophin molecule into the sarcolemma. Rapsyn, an essential protein for formation of the neuromuscular junction, binds to β-dystroglycan. The dystroglycan complex plays a pivotal role in linking the cytoskeleton to the extracellular matrix. Dystroglycans with dystrophin are important in the formation of the neuromuscular junction. Binding of agrin to α-dystroglycan is a critical step in the stabilization of acetylcholine receptor clustering at the neuromuscular junction. The agrin-mediated signaling is mediated by muscle-specific tyrosine kinase (MuSK), which is part of the protein complex at the neuromuscular junction. Antibodies directed against MuSK result in myasthenia gravis (the acetylcholine receptor antibody-negative form). The dystroglycan complex also plays an important part in normal migration of neurons in the cerebral cortex [Martin and Freeze, 2003; Michele and Campbell, 2003]. The assembly and integration of these proteins occur in the presence of dystrophin; in its absence, these proteins may not be degraded, properly assembled, or integrated into the sarcolemma [Ibraghimov-Beskrovnaya et al., 1992].
No primary mutations in dystroglycans have been identified in human disease. Defective glycosylation of α-dystroglycans causes congenital muscular dystrophies with brain involvement: Fukuyama’s congenital muscular dystrophy, muscle-eye-brain disease, Walker–Warburg syndrome, and limb girdle muscular dystrophy type 2I [Mercuri et al., 2009].
Sarcoglycans
The sarcoglycan complex consists of α-sarcoglycan (adhalin), β-sarcoglycan, γ-sarcoglycan, and δ-sarcoglycan [Bonnemann et al., 1995]. The sarcoglycan genes α, β, and γ are located on 17q12–21, 4q12, and 13q12 chromosomes, respectively. Expression of α- and γ-sarcoglycan is limited to skeletal and cardiac muscles [Yamamoto et al., 1994]. The sarcoglycans form a tetrameric complex in the Golgi apparatus, and then transition to the sarcolemma as a completely assembled structure. The sarcoglycans consist of a single transmembrane domain, a small intracellular domain, and a large extracellular domain, and their molecular weight ranges from 35 to 50 kilodaltons. The sarcoglycan complex, which is composed of α-, β-, γ-, and δ-sarcoglycan, is part of the dystrophin-associated glycoprotein complex; it acts as a link between the extracellular matrix and the cytoskeleton, confers structural stability to the sarcolemma, and protects muscle fibers from mechanical stress during muscle contraction. Alpha-sarcoglycan is present only in cardiac and skeletal muscles, but the other sarcoglycans are found in smooth muscle cells as well. Gamma-sarcoglycan bind to dystrophin, and δ-sarcoglycan can bind to β-dystroglycan. The sarcoglycans interact with filamin, a protein that is important for actin reorganization and which is involved in signal transduction associated with cell migration.
Mutations in any of the sarcoglycan genes cause destabilization of the complex, produce a decrease in the amount of all sarcoglycan proteins, and cause the different forms of limb girdle muscular dystrophy [Strauband Campbell, 1997]. The diseases associated with sarcoglycan gene mutations (α-sarcoglycan [SGCA], limb girdle muscular dystrophy type 2D; β-sarcoglycan [SGCB], limb girdle muscular dystropy type 2E; γ-sarcoglycan [SGSG], limb girdle muscular dystrophy type 2C; δ-sarcoglycan [SGCD], limb girdle muscular dystrophy type 2F) are rare disorders in the general population but represent a sizable proportion of all muscular dystrophies with normal dystrophin (about 10–20 percent of cases). Limb girdle muscular dystrophy type 2D is the most common sarcoglycanopathy, followed by types 2C and 2E; the rarest form is type 2F [Boito et al., 2003; Manzur and Muntoni, 2009].
The measurement of α-sarcoglycan is a useful screening test to look for sarcoglycan gene mutations in patients with muscular dystrophy [Duggan et al., 1997]. In sarcoglycan knockout mice, α-sarcoglycan has been successfully injected and expressed in tibialis anterior muscle of mice with adeno-associated virus 1 as a vector [Rodino- Klapac et al., 2008]. A phase 1 human trial for limb girdle muscular dystrophy type 2D, with gene transfer of human sarcoglycan with adeno-associated virus, is in progress (http:/www.clinicaltrials.gov).
Utrophin
Initially described as dystrophin-related protein or dystrophin-like protein, utrophin is an autosomally encoded protein. It is smaller in size than dystrophin, and it has been localized to the sarcolemmal postsynaptic membrane at the neuromuscular junction and myotendinous junction in the mature muscle fiber [Khurana et al., 1991]. At the neuromuscular junction, it is preferentially concentrated at the acetylcholine receptor-rich crests. Utrophin is present in all tissues tested so far. In the brain, it is localized at the inner plasma face of astrocytic foot processes at the abluminal aspect of the blood–brain barrier [Khurana et al., 1992]. Utrophin has a similar amino acid sequence as the carboxy terminus of dystrophin, and it copurifies with α-sarcoglycan. The gene coding for utrophin is localized to the long arm of human chromosome 6, at 6q24. The transcription of utrophin occurs preferentially at the nuclei close to the neuromuscular junctions (subsynaptic nuclei). The transcription of the utrophin gene is regulated by growth factors released from neurite, such as heregulin, a neurite-associated growth factor that is a member of the neuregulin family of growth factors. In muscles, growth factors increase the transcription of acetylcholine receptor, sodium channels, and utrophin [Fishbach and Rosen, 1997]. In dystrophin-deficient states, utrophin content is increased and localizes to sarcolemma as well. A number of approaches to upregulate the expression of utrophin in muscles are being explored as treatment for Duchenne’s muscular dystrophy. Miura and colleagues have shown an increase in utrophin expression in mdx mouse skeletal muscle, using a peroxisome proliferator activated receptor beta/delta, GW501516. The treated mice showed an increase in utrophin, expression of β-dystroglycan in sarcolemma, and an increase in sarcolemmal strength [Miura et al., 2009].
Dysferlin
Dysferlin is a 237-kilodalton protein composed of 2080 amino acids. It has a single transmembrane segment at the carboxy terminus. The protein is anchored at the sarcomere by the carboxy terminus, and the rest of the protein is intracytoplasmic. It is distributed at the sarcolemma, similar to dystrophin and the dystrophin–glycoprotein complex. However, the protein is not associated with dystrophin and the dystrophin–glycoprotein complex. The dysferlin protein is coded by a gene located on chromosome 2p13. The gene is relatively large and spans 150 kilobases. It is composed of 55 exons and is expressed in skeletal and cardiac muscles and placenta. A shorter transcript of 3.5 kilobases is expressed in all regions of the brain. Proteins with homology to dysferlin are seen in other tissues. Fer-1 is present in testes and is required for maturation of spermatozoa. Otoferlin, with 55 percent homology to dysferlin, is expressed in the inner hair cells. Myoferlin, with 68 percent homology, is present in the lungs, placenta, heart, and cytoplasm and nuclear membranes of skeletal muscles [Bansal et al., 2003].
The ferlin family of cytoplasmic proteins has motifs that are homologous to so-called C2 domains. The C2 domains are typically found in proteins that function in signal transduction or in membrane trafficking, such as protein kinase C and synaptotagmins [Anderson et al., 1999]. These domains interact with multiple targets, including calcium, phospholipids, and other proteins, to mediate signaling events for membrane fusions of vesicles to form plugs over the sarcolemmal membrane defects [Glover and Brown, 2007; De Luna et al., 2004; Rizo and Sudhof, 1998]. The clinical disorders with deficiency, such as distal myopathy of Miyoshi and limb girdle muscular dystrophy type 2B, are due to defects in the processes of membrane trafficking or fusion [Bajaoui et al., 1995; Liu et al., 1998; Chiu et al., 2009]. Myopathies can occur with overexpression of dysferilin protein [Glover et al., 2010].
Caveolin
Caveolae are flask-shaped plasma membrane invaginations that participate in membrane trafficking, sorting, transport, and signal transduction. They are abundant in fibroblasts, adipocytes, endothelial cells, and smooth and striated muscle cells. Caveolin, a family of 21- to 24-kilodalton proteins, is a major constituent of the caveolae. Caveolins form the protein scaffolding upon which the specific proteins and lipids interacting with it are concentrated. Caveolin-3 is expressed in muscles. The human caveolin-3 gene maps to chromosome 3p25, and it codes for a protein of 150 amino acids. The caveolin protein is localized to the sarcolemmal membrane on immunocytochemical studies [Betz et al., 2001].
Mutations in the caveolin-3 gene produce autosomal-dominant limb girdle muscular dystrophy type 1C, hyperCKemia, hereditary rippling muscle disease, familial hypertrophic cardiomyopathy, and long QT syndrome, and have been associated with sudden infant death syndrome [Gazzerro et al., 2009; Woodman et al., 2004]. The mutant gene produces marked reduction in the caveolin-3 protein, consistent with the dominantly inherited disorder [Cagliani et al., 2003].
Merosin (Laminin-α2)
Laminin-α2 is a major component of laminin in the basal lamina of postnatal muscle. It is also expressed in cardiac muscle, pancreas, lung, kidney, adrenal gland, skin, testis, meninges, choroid plexus, and brain. Expression of the laminin-α2 gene occurs in cells of mesenchymal origin. The gene for laminin-α2 is localized to chromosome 6q22–23 [Vuolteenaho et al., 1994]. The mature myofiber laminin is a heterodimer composed of laminin-α2, β1, and γ1. The laminin interacts with sarcolemma through the dystrophin–glycoprotein and vinculin–integrin complexes. It interacts with basal lamina and extracellular matrix through perlecan, nidogen, and a series of other proteins. Developing and embryonic myofibers express different isoforms of laminin. The laminin isoform expression changes to the α2 isoform from the α1 isoform at the time of birth. During myogenesis, laminin-α2 helps maintain the survival and stability of myotubes [Gullberg et al., 1999]. Mutations of laminin-α2 in children manifest as congenital muscular dystrophy, complete absence of muscle merosin, and abnormalities of cerebral deep white matter [Jones et al., 2001; Philpot et al., 1995]. Prenatal diagnosis can be made by immunostaining the chorionic villus sample for merosin [Yamamoto et al., 2004].
Intermediate Filaments
Intermediate filaments are cytoskeletal proteins measuring between 10 and 12 nm in thickness [Omary et al., 2004]. Their size is intermediate between actin and the thicker myofilaments, such as myosin. Intermediate filaments are seen in cells derived from all three embryonic lineages. Desmin, vimentin, nestin, and lamin are expressed in muscle.
Desmin is the major intermediate filament in skeletal muscle. It is also expressed in cardiac and certain smooth muscles. Desmin is located at the Z band beneath the sarcolemma and is denser at the myotendinous junction [Tidball, 1992]. Desmin is involved with the alignment of sarcomeres with one another and the plasma membrane by forming a a three-dimensional scaffold around the Z disks. It plays a role in the functional and spatial relationship between the nucleus and plasma membrane, and it is presumed to regulate myogenesis because it is more strongly expressed in immature myofibers [Fuchs and Weber, 1994; Gallanti et al., 1992].
Desmin is a 53-kilodalton protein organized in three domains. The highly conserved central α-helical core of 310 amino acids is flanked by a nonhelical amino-terminal head and carboxy-terminal tail. The core is formed of four helical rods: A1, A2, B1, and B2. The desmin polypeptide chains are intertwined in a coiled-coil tense into a homodimer [Geisler and Weber, 1982]. The desmin filaments interact with other filaments and other proteins to form a cross-linking network, which attaches to membranes. A gene located on chromosome 2q35 encodes the desmin protein [Viegas-Pequignot et al., 1989].
Mutations of the desmin gene result in the desmin-related myopathies, also known as myofibrillar myopathies, and cardiomyopathies [Dalakas et al., 2000, Goldfarb et al., 2004; Schroder and Schoser, 2009].
Vimentin is an intermediate filament that is also expressed in immature muscle cells during myogenesis. After the postnatal period, its expression in muscle is primarily in regenerating and immature cells [Bornemann and Schmalbruch, 1993]. It is also expressed in tumors of rhabdoid origin.
Nestin is a protein that is expressed with desmin in developing muscle cells [Sjoberg et al., 1994]. Postnatally, the protein is found with desmin in regenerating muscle fibers in patients with dystrophic and inflammatory myopathies.
Plectin, a large protein (more than 500 kilodaltons) in the family of plakins, is a cytoskeletal linker protein for desmin and keratin organization; it is expressed in a wide variety of tissues, including muscle and skin. It has a high-affinity binding site for intermediate filaments at its COOH terminus. The amino terminus has an actin-binding site, and this site shares similarities with the actin-binding domains of α-actinin, dystrophin, and utrophin. Plectin provides mechanical strength to tolerate the forces of physical stress. Plectin is encoded by a gene localized to 8q24 and has 32 exons [Liu et al., 1996; Boyer et al., 2009]. Deficiency of plectin has been identified as the cause of recessive epidermolysis bullosa with congenital muscular dystrophy [Whiche, 1998].
Other important cytoskeletal proteins include tubulins and anchor proteins. Microtubules formed from tubulins and other intermediate filaments play an important role in maintaining the cytoplasmic architecture. Anchor proteins link the cytoskeletal proteins to the plasma membrane. Tropomodulin is an integral component of the cytoskeletal structure in the postsynaptic region of the neuromuscular junction [Sussman et al., 1993].
Nuclear Membrane-Related Proteins: Emerin and Lamin A/C
Emerin is a protein associated with the inner nuclear membrane. It is attached to the inner nuclear membrane by its carboxy terminus and extends into the nucleoplasm. Emerin is composed of 254 amino acids and is coded by a gene on the X chromosome. Emerin is important in the organization of nuclear membrane during cell division [Fairley et al., 1999]. Mutation of this gene causes X-linked Emery–Dreifuss muscular dystrophy [Haraguchi et al., 2004].
Lamins are major components of the nuclear lamina, which is the major structural framework of nuclei in eukaryotic cells. In humans, two forms of type A (A and C) and B (B1 and B2) are present. Lamins have the helical rod portion and nonhelical portions at the amino and carboxy termini. Lamins differ from other intermediate filaments by the presence of a nuclear localization signal close to their carboxy terminus. The lamins interact with themselves, lamin-associated proteins, and chromatin. The lamins play a role in DNA replication, chromatin organization, spatial arrangement of nuclear pores, nuclear growth, and mechanical stability of the nucleus [Loewinger and McKeon, 1988; Maniotis et al., 1997]. Mice with null mutations for lamin A gene show chromatin clumping, nuclear fragmentation, nuclear invaginations, and inclusions in myonuclei at the myotendinous junctions, indicating susceptibility to mechanical stress in nuclear membranes deficient in lamin A [Mittelbronn et al., 2008].
The nuclear lamins belong to type 5 intermediate filaments. Lamin A is expressed during and after differentiation, and lamin B is expressed thoughout development and cell differentiation, but is absent in mature cells. The LMNA gene is located on the 1q21–22 region and has 12 exons. Through alternative splicing, lamins A and C are formed. Lamin A is produced by post-translational modification of precursor prelamin A. Post-translational modification of prelamin A includes farnesylation, proteolytic cleavage of the terminal three amino acids, carboxymethylation, and excision of 15 carboxy-terminal amino acids to produce the mature lamin A. The enzyme zinc metalloproteinase is responsible for the first and second steps in proteolysis. Lamin A has 566 amino acids; lamin C, formed by alternative splicing, has 98 amino acids and shares a similarity with the carboxy-terminus amino acids of lamin A [Jacob and Garg, 2006].
Mutations in the lamin A/C gene cause autosomal-dominant Emery–Dreifuss muscular dystrophy, Charcot–Marie–Tooth disease type 2, dilated cardiomyopathy with conduction defects, Dunnigan-type familial partial lipodystrophy, mandibuloacral dysplasia type B, and restrictive dermopathy [Benedetti and Merlini, 2004; van der Kooi et al., 2002].
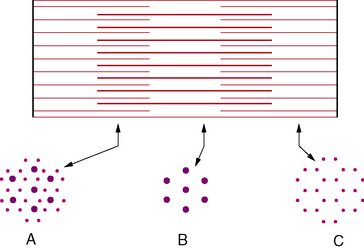
Muscle Fiber Types
Clinical use commonly divides human muscle fibers into types I and II [Dubowitz and Brook, 1973]. Type I fibers have slow-twitch characteristics and are rich in mitochondria and substrates for aerobic (oxidative) metabolism. They are slightly smaller than type II fibers and have a less well-developed sarcoplasmic reticulum and T-tubule systems. Type II fibers have fast-twitch characteristics and are rich in glycogen and other substrates and enzymes needed for anaerobic (glycolytic) metabolism. Type II fibers can be subdivided into type IIa and IIb fibers. Type IIb fibers are the prototype for the type II fibers, whereas type IIa fibers are intermediate, with both oxidative and glycolytic properties. Type I fibers are generally found in muscles needed for sustained tonic contraction, such as the antigravity muscles of the limbs and trunk. Type I fibers have a more luxurious capillary blood supply. Type II fibers are usually found in muscles concerned with rapid, forceful movement. Table 87-2 summarizes the properties of muscle fiber types. Division of muscle fibers into three basic types is probably an oversimplification. There is a continuum of physiologic properties dependent on the innervation of the motor neuron, which contributes to the plasticity of muscle that enables it to adapt to a variety of demands [Buchthal and Schmalbruch, 1980].
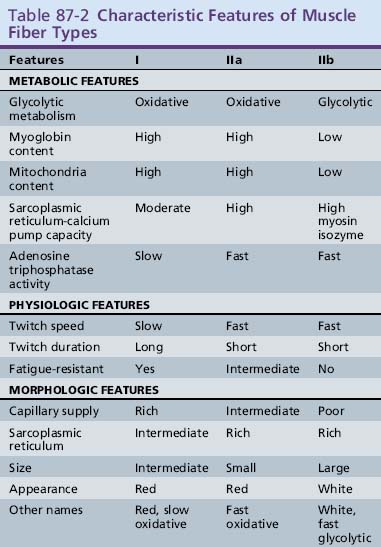
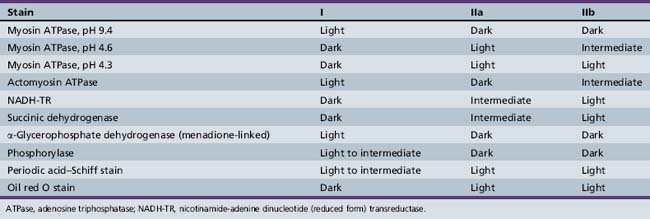
The biochemical characteristics of the fiber types can be visualized using histochemical reactions. Type I fibers manifest a strong reaction when stained for the mitochondrial enzymes, succinic dehydrogenase and nicotinamide-adenine dinucleotide (reduced form) transreductase. Nicotinamide-adenine dinucleotide (reduced form) transreductase is also found in abundance in sarcoplasmic reticulum. Myosin adenosine triphosphatase activity can also be used to differentiate fiber types because of the relative abundance of myosin adenosine triphosphatase in type II fibers. Preincubation at various pH levels provides different staining characteristics to various muscle fibers. Type II fibers have an alkali-stable, acid-labile adenosine triphosphatase, whereas type I fibers have the opposite characteristic. Table 87-3 summarizes the histologic and histochemical properties of the muscle fiber types.
All muscle fibers of a motor unit are of a single type. Most of the physiologic, metabolic, and morphologic properties of the muscle fiber depend on innervation from the anterior horn cell. Small α motor neurons innervate type I fibers, and large α motor neurons innervate type II fibers. Reinnervation by the axons of another anterior horn cell alters the muscle fiber properties so that they are identical to the fibers of the donor motor unit [Buller et al., 1960]. The clinical significance of this phenomenon is fiber-type grouping that occurs with reinnervation and can be detected by electromyography and muscle histochemistry. The fibers of a motor unit are normally distributed randomly over several millimeters in the muscle. When a muscle fiber loses its innervation, reinnervation from adjacent, intact axons changes its fiber type. The fiber type can also be altered by changing the characteristics of its innervation [Edgerton et al., 1985], especially its frequency and the quantity of impulses. Prolonged tonic stimulation to a type II fiber can induce type I histochemical characteristics in it [Pette and Vrborá, 1985]. The plasticity of muscle, which alters its characteristics to match changing patterns of innervation, makes the muscle system the most adaptable organ.
Function
Motor Unit
Contraction of a muscle fiber is initiated by depolarization of its anterior horn cell, which is the final common pathway for all cortical and spinal influences that affect movement. Depolarization of the anterior horn cell membrane is propagated along its axon in the peripheral nerve. At the nerve terminal, axon twigs run parallel within the muscle fascicle to the muscle fibers and innervate the fibers at the neuromuscular junction. Each anterior horn cell innervates about 50–200 muscle fibers, depending on the muscle. The anterior horn cell, its peripheral axon, and the innervated muscle fibers compose the motor unit and can be considered a functional unit of the muscle. The muscle fibers within the motor unit are not contiguous but are scattered fairly randomly. In the rat, the motor unit is dispersed over 8–76 percent of the cross-sectional area of the muscle, with some clustering of a few fibers in the motor unit territory [Bodine-Fowler et al., 1990]. When the anterior horn cell discharges, all the muscle fibers innervated by that anterior horn cell contract almost simultaneously. The finer and more coordinated the movement of a muscle, the fewer the fibers in the motor unit. Eye muscles and some hand muscles may have 30–50 fibers in the motor unit, whereas some back muscles have hundreds. In general, the larger the muscle, the larger the size of the motor units [McComas, 1991]. For example, there are about 100 fibers per motor unit in the intrinsic muscles of the hand, 200 in the extensor digitorum brevis, and 900 in the larger muscles.
Neuromuscular Transmission
The neuromuscular junction is formed by specialized portions of the nerve and muscle plasma membrane, which are separated by the synaptic cleft (Figure 87-8). The area on the muscle is the end plate and is located near the center of the muscle at the end-plate zone. The synaptic knob is the terminal dilatation of the nerve axon and contains synaptic vesicles of acetylcholine. The motor neuron action potential is propagated down the motor nerve by saltatory conduction. Na+ channels at the nodes of Ranvier produce current. Within the muscle, the motor nerve fiber branches into terminal fibers that individually innervate muscle fibers within the motor unit. The nerve terminals are unmyelinated, and the membrane excitability is maintained by both K+ and Na+. Antibodies to the K+ antigen cause acquired neuromyotonia, such as Isaac’s syndrome [Newsome-Davis et al., 2003; Tan et al., 2008].
Depolarization of the nerve terminus opens voltage-gated calcium channels at the synaptic knob. As calcium enters the nerve ending, exocytosis of the synaptic vesicles occurs, releasing acetylcholine. Antibodies directed against the voltage-gated calcium channels cause Lambert–Eaton syndrome by reducing the quantal release of acetylcholine [Newsome-Davis, 2004]. Acetylcholine is synthesized from acetyl coenzyme A and choline, and exists in a reserve compartment that is not readily available for release and in a mobilization compartment of vesicles. Each synaptic vesicle contains a quantum of about 10,000 acetylcholine molecules. The vesicles are attached to the active zone of the presynaptic membrane. Fifty to 300 vesicles fuse with a nerve terminus depolarization. The process of vesicle formation, fusion, and release is a complex system involving many proteins [Ruff, 2003].
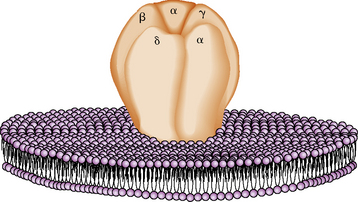
Acetylcholine combines with specific acetylcholine receptors located on the postsynaptic membrane of the muscle sarcolemma. There are about 15,000/μm2 receptors in the junctional fold concentrated at the tops of the secondary folds. The acetylcholine receptor is a transmembrane glycoprotein with a molecular weight of 250,000 daltons and is composed of four subunits: α, β, δ, and γ [Momoi and Lennon, 1982]. The muscle acetylcholine receptor monomer is constructed with two α subunits and one of each of the other subunits in order: α1, γ, δ, β1 [Lindstrom, 2003]. The acetylcholine receptor monomer contains two acetylcholine-binding sites and the cation-specific channel, whose opening is regulated. The receptor is barrel-shaped, traverses the sarcolemma, and projects above its surface. The subunits are arranged around the central channel-like staves (Figure 87-9) [Kistler et al., 1982]. The acetylcholine receptors are anchored to dystrophin-related protein complexes and connected to the cytoskeleton by dystrophoglycans and sarcoglycan protein complexes. The structure and function of the acetylcholine receptor are remarkably similar in all species studied. Congenital myasthenic syndromes are caused by genetic mutations of the receptor and other synapse structures, whereas myasthenia gravis is caused by antibodies to receptor proteins.
The binding of two acetylcholine molecules with the α subunits of the receptor causes a conformational change in the M2 portion of α-helical structures of each of the five subunits. This change results in a rotational realignment of the helices to open the center of the receptor for a few milliseconds and thereby alter the sodium and potassium permeability by opening ion channels. Increased ion conductance across the sarcolemma causes a localized depolarization of the muscle end plate. A single quantum of acetylcholine interacting with the end plate causes a miniature end-plate potential insufficient to engender a propagated membrane discharge. When depolarization of the terminal axon occurs, 50–300 acetylcholine quanta are released. Sufficient interactions between acetylcholine molecules and receptors cause the summation of potentials to reach the threshold potential for the end plate. Thus, an end-plate potential is achieved, and the depolarization is propagated along the sarcolemma and T tubules. Depolarization of the end plate is a statistical phenomenon; given sufficient numbers of acetylcholine molecules and receptors, an interaction will probably occur, followed by depolarization [Katz and Miledi, 1972]. Interference with the release of acetylcholine or a reduction in the number of receptors lessens the likelihood of successful neuromuscular transmission.
Excitation–Contraction Coupling
The major function of muscle is generating movement or force by contraction. This function is accomplished through a series of events called excitation–contraction coupling, which is the linkage of the electrical and mechanical properties of muscle. Depolarization of the T tubules at the triad is sensed by dihydropyridine (a calcium-channel blocker) receptors, which are associated with charged molecules within the tubular membrane. Movement of these charged molecules, which can be inhibited by dantrolene [Hui, 1983], causes a conformational change in the “feet,” which are structures that bridge the membranes of the T tubules and sarcoplasmic reticulum [Peachey, 1985]. Calsequestrin is a major protein of the sarcoplasmic reticulum that binds calcium and is located close to the sarcoplasmic reticulum calcium efflux channels in register with the “feet” processes [Stadhouders, 1990]. Interaction of these membrane structures at the triad causes the release of calcium from the sarcoplasmic reticulum into the sarcoplasm. During tetany, calcium concentrations decrease in the sarcoplasmic reticulum and increase in the sarcoplasm [Somlyo et al., 1981]. This release of calcium is critical for the mechanics of muscle contractions. Calcium-binding proteins in the sarcoplasm, including calmodulin [Kennelly et al., 1989] and parvalbumin [Heizmann et al., 1990], are essential for regulation of the contractile proteins.
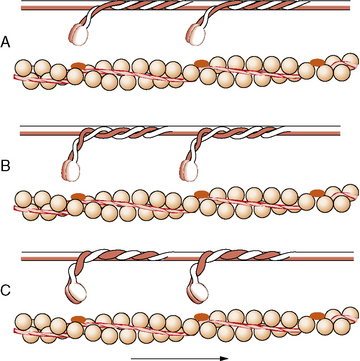
When it is released by the sarcoplasmic reticulum, calcium binds with calmodulin; the complex acts as a second messenger that activates the myosin light chain troponin C kinase [Kennelly et al., 1989]. This reaction causes an unmasking of a myosin-binding site by displacing troponin I and tropomyosin. The myosin head swings away from the backbone of the thick filament at the hinge area of the S2 region [Horowits et al., 1989]. The myosin head then attaches to actin at an angle, and the hydrolysis of adenosine triphosphate causes the myosin head to swivel to a more acute angle, moving the chains toward one another (Figure 87-10). Repetition of this event causes shortening of the sarcomere [Vale and Milligan, 2000]. Each attachment, swivel, and reattachment shortens the muscle about 1 percent, or about 14 nm [Ford et al., 1977]. Myosin hydrolyzes adenosine triphosphate slowly, unless combined with actin. Each stroke of the myosin–actin cross-bridge attachment consumes one molecule of adenosine triphosphate: adenosine triphosphate binding to myosin, dissociation of actin and myosin, adenosine triphosphate hydrolysis, reassociation of actin and myosin, and release of hydrolysis products. The rate of stroke cycling and therefore the contraction velocity are dependent on the myosin adenosine triphosphatase isoform [Pollack, 1996]. The exact mechanism of the phase transition that brings about muscle contraction is still not fully known [Brooks, 2003]. The entire process constitutes excitation–contraction coupling.
The tension generated by a muscle is directly proportional to the number of cross-links between the myosin heads of thick filaments and the actin of the thin filaments, as determined by length–tension relationships [Pollack and Sugi, 1984]. On cross-section, each myosin heavy chain is surrounded by six actin light chains. This geometric array is found in the area of the sarcomere where there are overlapping myofilaments, as in the A band (Figure 87-11). The more foreshortened the sarcomere, the more cross-links between the filaments. However, only the thin filaments occur in the I band. Each muscle has a length at which it generates maximum force and at which the greatest interaction between actin and myosin occurs [Huxley, 1996; Lieber, 1986].
Evidence suggests that contraction of a muscle occurs by heavy and light myofilaments sliding toward one another. When a muscle contracts, the distance between the Z lines decreases at the expense of the I and H bands. The A band remains unchanged (Figure 87-12). During stretch, the I band increases in length as the actin filament arrays slide in the direction opposite to the myosin filament arrays; thus, the distance between Z lines increases. This sliding is produced by the formation and breakage of the cross-links between actin and myosin.
Neuromuscular transmission is terminated by calcium sequestering within the synaptic knob. Muscle contraction is terminated when the T tubules repolarize; this causes reaccumulation of calcium within the sarcoplasmic reticulum, using a pump that depends on adenosine triphosphate [Peachey, 1985]. Unlike the nerve and muscle plasma membrane, the contraction process has no refractory period. As long as calcium is free in the sarcoplasm and adequate energy stores exist, the muscle will stay contracted. Muscle can stay contracted without excitation of the membrane system, and energy is required for the reuptake of calcium into the sarcoplasmic reticulum. Failure of calcium reuptake from the sarcoplasm when energy stores are depleted inhibits relaxation of the muscle. This mechanism causes rigor mortis and the cramping during exercise found in energy-deficiency disorders, such as McArdle’s disease.
Neural Control of Muscle Contraction
The sarcomere is the contracting unit of muscle. Whole-muscle contraction force is proportional to the number of sarcomeres contracting in parallel, whereas whole-muscle contraction velocity is proportional to the number of sarcomeres acting in series [Lieber, 1986]. This interaction generally applies to the muscle fiber and to the architecture of muscle. The more fibers in parallel, the more force is generated by contraction. The longer the muscle fiber, the more sarcomeres are in series and the greater the contraction velocity.
Motor Unit System
The motor unit consists of a single lower motor neuron and the muscle fibers it innervates. The motor unit is the final common pathway of the motor system [Miles, 1994]. The following two control mechanisms are used by the nervous system in generating muscle contraction force:
There is a rank order of recruitment within a motor neuron pool, so that increasingly larger motor units are recruited for increasing demands of force. Smaller motor units are recruited initially [Henneman, 1985]. Similarly, the rate of discharge of motor units is proportional to the degree of force generated. Current information indicates that the firing rates of motor neurons are not controlled individually, but that the nervous system controls the firing rate of motor neuron pools to modulate force generation. This mechanism has been termed common drive [De Luca, 1985]. In general, the recruitment of additional motor units is more important in generating force and power than modulating firing rates [Brooks, 2003].
Motor unit recruitment is the process by which different motor units are activated to produce a specific degree and type of muscle force [Petajan, 1991]. In the graded generation of force, the lower force threshold, slow-twitch motor units are recruited first. Increasing force output raises the firing rate of these first-recruited motor neuron pools in fine gradations. Further force generation requires recruitment of large motor neurons with higher force threshold, fast-twitch motor units. This recruitment causes a 5- to 10-fold increase in the graded force. Normally, a motor unit discharges up to rates of 5–15 Hz before an additional neuron is recruited. The firing rate of the fastest-firing motor unit divided by the number of recruited neurons is normally less than 5 (e.g., three neurons firing at a rate of 15 Hz). When this recruitment ratio is greater than 10 (e.g., two neurons firing at a rate of 20), it suggests a loss of motor units. At maximal exertion, there is fusion of contractions of the slow-twitch motor units. Because of the short twitch duration of the large motor units, they may not achieve contraction fusion with voluntary effort [De Luca et al., 1982]. During normal activity, slow-twitch motor units discharge more frequently and are active more often than fast-twitch motor units [Hennig and Lomo, 1985].
Gamma Efferent System
The γ efferent system plays a major role in controlling muscle contraction via its effects on the muscle spindle. The muscle spindle is a specialized sensory organ located within the perimysial connective tissue (Figure 87-13). It is an encapsulated structure containing 2–10 small muscle fibers (intrafusal fibers) and special sensory nerve endings. The intrafusal fibers comprise nuclear bag fibers, nuclear chain fibers, and an intermediate type of fiber. Each has distinct ultrastructural and histochemical characteristics [Barker et al., 1972]. The intrafusal fibers of the spindle are arranged in parallel with the extrafusal fibers of the muscle.
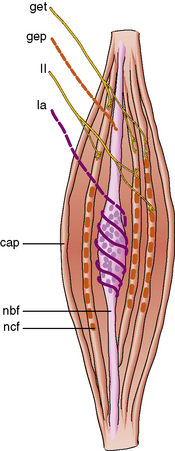
The sensory endings are of two types [Scalzi and Price, 1972]. The primary endings (annulospiral) are supplied by large, myelinated, rapidly conducting Ia afferent fibers, and end in a spiral or cagelike arrangement around nuclear bag fibers. These endings are sensitive to both stretch of the spindle and the velocity of stretch. Secondary endings of group II sensory nerves terminate in a variety of endings, including cylindrical, flat, and annulospiral or ring-shaped endings, mostly on nuclear chain fibers [Schroder et al., 1989]. Secondary endings are sensitive to the stretch of the spindle.
The fusimotor fibers compose as much as 30 percent of the total number of ventral root fibers. Most are γ efferents, although a few are β efferents [Barker et al., 1972]. Multiterminal plate endings (i.e., flower-spray) are located on nuclear bag fibers from myelinated γ efferents and probably cause dynamic innervation. Trail endings, however, primarily innervate nuclear chain fibers and arise from smaller myelinated and unmyelinated fibers. Trail fibers supply static innervation.
The afferent fibers of primary endings synapse directly on the anterior horn cells, which supply the extrafusal fibers of the same muscle. The secondary ending afferents probably synapse on motor neurons of other muscles [Scalzi and Price, 1972]. Stretch of a muscle causes distortion of the primary endings and generates a signal to the motor neuron to cause contraction. This signal is the mechanism of the deep tendon reflex. If the muscle were shortened, spindle discharge would decrease, causing muscle relaxation; therefore, spindles serve as a feedback mechanism to maintain muscle length and tone.
The γ efferent system operates in concert with the α motor neuron to effect movement [van der Meulen et al., 1972]. Stimulation of the γ motor neuron causes stretching of the nuclear bag fibers, distortion of the primary endings, and reflex stimulation of the α motor neuron, as discussed previously. The γ motor neurons are regulated by several descending tracts, whose main purpose is to maintain the sensitivity to stretch throughout muscle contraction so that different muscle groups can adjust to the needs of postural control.
The other major sensory innervation of muscle is the Golgi tendon organ [Stuart et al., 1972], which is a netlike structure located around the tendon fascicles supplied by fast-conducting myelinated afferent fibers. The output causes stimulation of inhibitory internuncial neurons, which, in turn, synapse on α motor neurons. The sensory organ is arranged in series with the muscle and thus is more sensitive to changes in muscle force than to changes in length. Excessive stretch produces an inhibitory influence on the α motor neurons.
Muscle Metabolism
At rest, high-energy phosphate compounds are produced from the metabolism of available fats and glucose [Hultman, 1995]. Increased lipid supply inhibits glycolysis and carbohydrate use. Excess glucose can be metabolized directly from the circulation or stored as glycogen until needed. With maximal exertion, phosphocreatine and muscle glycogen are the fuels for energy production. Above 60 percent maximal exertion, type II fibers are preferentially recruited, and carbohydrates are the preferred fuel after a normal meal. Fat utilization is increased with starvation or a high fat intake before exercise. At exercise intensities below 60 percent, fat is the major fuel corresponding to the recruitment of type I fibers.
There is a constant relationship among force, phosphate concentrations, and pH [Weiner et al., 1990]. As energy is expended, phosphocreatine concentrations decrease, phosphate increases, and pH decreases. The adenosine triphosphate concentration remains stable [Kushmerick, 1995]. Thus, fatigue is relative to the degree of acidosis and free phosphate concentrations. A lactate–proton co-transport system is important for reducing the accumulation of lactate and H+ within the muscle [Juel, 1997]. Recovery from fatigue parallels the normalization of these metabolic parameters [Boska et al., 1990].
Lipids can be used, giving a relatively higher yield of adenosine triphosphate per mole of lipid consumed. Mobilizing lipids for energy consumption is a slower process than mobilizing carbohydrates. Activities that require endurance or prolonged effort are associated with higher lipid metabolism. Training increases the vascular perfusion of muscle and oxygen delivery, and increases energy production by aerobic metabolism. Training also increases the capacity of the electron transfer system, oxidative phosphorylation, and the lactate–proton co-transport system [Juel, 1997]. Training increases lactate clearance in the muscle by increasing muscle oxidation of lactate and increasing the lactate transporter protein, monocarboxylate transporter [Dubouchaud et al., 2000]. In general, muscle metabolism is quite plastic and adapts relatively rapidly to altered functional requirements.
Muscle has metabolic functions besides energy production. Muscle protein anabolism and catabolism are controlled by many factors in the inverse relationship. The major factors that promote protein synthesis and muscle growth are exercise, amino acid availability, and insulin [Kimball et al., 2002; Wolfe, 2002]. In the adult male, muscle represents about 40 percent of body weight. It is a vast storehouse for protein, amino acids, fat, and glycogen. Muscle metabolism is intimately involved with metabolism of other tissues, especially the liver. Most lactate in the body is produced from resting muscle. The cell-to-cell lactate shuttle hypothesis suggests that lactate is an intermediate metabolite rather than an end product. It is produced by many tissues, especially muscle, and is used as an energy source in the heart and other highly oxidative tissues and as a precursor of glucose in liver gluconeogenesis (Cori’s cycle). Similarly, during fasting or other stresses, muscle releases amino acids, lactate, and other precursors for use by the liver during gluconeogenesis. There is clinical evidence that children with chronic hypoglycemia, as is found in the glycogen storage diseases, have secondary muscle degradation from the chronic muscle catabolism that provides the amino acids necessary for gluconeogenesis [Slonim et al., 1983].
Clinical Laboratory Tests
Modern investigation of muscle disease includes clinical biochemistry and immunology, electromyography, muscle biopsy pathology, molecular genetics, imaging of muscle, and other modalities [Younger and Gordon, 1996].
Clinical Biochemistry
Several muscle sarcoplasmic enzymes are used in the clinical evaluation of muscle disease. These enzymes are released from the muscle fiber when it is damaged. Of these enzymes, aldolase and creatine kinase are used most commonly, especially the latter. Creatine kinase exists as several isozymes, including M and B. BB is found almost exclusively in brain tissue, whereas MM is found almost exclusively in muscle and heart tissue. MB is found in several tissues. Determination of creatine kinase isozymes is helpful in differentiating brain from muscle disease, but not muscle from heart disease [Vainzof et al., 1985]. Creatine kinase is not found in the liver or erythrocytes.
The normal range of creatine kinase enzyme activity in the blood varies with age, gender, and daily activities. In an inactive male, however, this range may rise to 2–3 times that of the upper limits. After vigorous extended activity, such as running a marathon or a similar effort, plasma creatine kinase activity may approach 10 times that of normal [Brown et al., 1982]. Elevation of plasma creatine kinase also occurs after trauma, seizures [Wyllie et al., 1985], electroconvulsive therapy [Taylor et al., 1981], and injections. Increased activities of these enzymes are suggestive, but not diagnostic, of myopathic disease. Creatine kinase can also be mildly elevated in neuropathic diseases. Other enzymes released include transaminases, lactate dehydrogenase, carbonic anhydrase III, and pyruvate kinase. None is as useful as creatine kinase. Serum myoglobin also correlates with muscle breakdown [Norregaard-Hansen and Hein-Sorensen, 1982]. Other biochemical methods that have been used in investigating muscle disease include measuring urinary excretion of creatine and 3-methyl histidine. Their clinical application has not been commonly accepted for various reasons.
Electromyography
Electromyography is the recording of electrical activity by a needle electrode of action potentials of muscle fibers firing singly or in groups near the electrode [Daube, 1991].
The second part of clinical electromyography is recording the spontaneous resting properties of the muscle. Normally, muscle is electrically silent during rest. End-plate noise can be recorded if the needle is placed near the end-plate zone. End-plate noise is generated by miniature end-plate potentials and single fiber discharges produced by needle irritation [Shahani and Young, 1985]. Spontaneous activities, such as fibrillations, positive sharp waves, fasciculations, or others, usually indicate underlying disease, most commonly neuropathic disorders. Spontaneous repetitive discharges are found in various neuropathic and myopathic disorders, such as the myotonias.
The third part of electromyography is recording motor unit potentials to evaluate the electrical properties of muscle during contraction. The motor unit potential usually has two or three phases and is 5–15 milliseconds in duration. The voltage usually does not exceed 5 mV. With a primary muscle disease, a frequent finding is brief, small-amplitude, polyphasic potentials. With chronic neuropathic disease, a common finding is prolonged, large-amplitude, polyphasic potentials. With graded contraction, an increase in the firing rate of individual motor units occurs. Further effort causes an increase in the number of recruited motor units and is proportional to the degree of muscle contractions. Thus, motor units are recruited in response to the degree of effort, and there is a recruitment order of motor units that is relatively fixed [Henneman et al., 1974]. Single-fiber electromyography is a technique for studying the characteristics of neuromuscular transmission and the localized organization of muscle fibers within a motor unit [Sanders et al., 1996]. A special needle electrode is introduced into the muscle at multiple sites. The number of single muscle fiber action potentials belonging to one motor unit recorded in a given area gives a quantitative measure of the fiber density. Recording from two fibers of the same motor unit enables one to measure “jitter,” or the mean consecutive difference in time between the firing of the two fibers, which is usually between 10 and 55 milliseconds. An increase in jitter is commonly observed with defects of neurotransmission. Single-fiber electromyography also detects blocking when the second fiber fails to discharge, which is the basis for the decremental response during repetitive stimulation in patients with myasthenia gravis. These subjects are discussed in other chapters.
Muscle Biopsy
The method of muscle biopsy has been reviewed in detail elsewhere [Dubowitz and Brook, 1973]. It can be performed by either an open biopsy or a needle biopsy technique [Heckmatt et al., 1984]. Either method is relatively simple and can be performed on an outpatient basis. The muscle chosen for biopsy should be one affected clinically, but not severely so.
Histochemistry often reveals the most significant information. Adenosine triphosphatase reactions are used to determine fiber typing. The random distribution of the fiber type on the adenosine triphosphatase reaction is a major feature of normal muscle. Normally, there are about equal numbers of type I, IIa, and IIb fibers, although up to 50 percent of a single type can be found in some muscles [Dubowitz and Brook, 1973]. Oxidative stains and the periodic acid–Schiff stain also reflect fiber typing (see Table 87-3). The oxidative reactions that include succinic dehydrogenase and nicotinamine-adenine dinucleotide transreductase are helpful in identifying mitochondria and the sarcotubular system. Many other histochemical reactions facilitate the study of specific diseases, such as the myophosphorylase reaction that aids in diagnosis of McArdle’s disease. Figure 87-14 illustrates the major histologic and histochemical features of muscle.
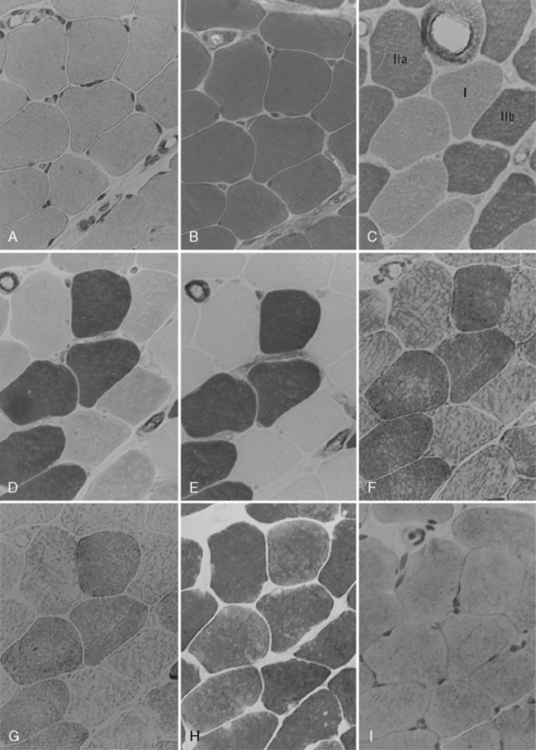
Immunohistochemistry is becoming increasingly important for the study of normal and diseased muscle. The absence of dystrophin on immunohistochemical staining of muscle tissue is diagnostic of Duchenne’s and related muscular dystrophies. Certain congenital myopathies can be classified based on immunohistochemical staining of other cytoskeletal proteins [Duggan et al., 1997; Philpot et al., 1995; Wallgren-Pettersson et al., 1995].
Electron microscopy is rarely needed for accurate diagnosis, but it is often necessary in disorders of the mitochondria and to determine the ultrastructure of central cores, nemaline rods, and other inclusions in congenital myopathy [Younger and Gordon, 1996].
Muscle Imaging
Ultrasonography, computed tomography, and magnetic resonance imaging are used in the diagnosis and management of patients with neuromuscular disease [Clague et al., 1995]. Ultrasonography is a cost-effective method for localization of diseased muscle for biopsy purposes. It is useful for imaging dystrophic muscle [Heckmatt and Dubowitz, 1988]. Magnetic resonance imaging is particularly useful for the diagnosis of inflammatory muscle disease. Inflammation is easily identified and can be distinguished from other causes of weakness [Stonecipher et al., 1994].
Molecular Genetics
Techniques for mapping and sequencing the human genome have brought about an entirely new approach to the diagnosis of human neuromuscular disease [Nawrotzki et al., 1996]. High deletion frequencies, unstable tandem repeat sequences, genomic duplications, and triplet repeat expansions are common abnormalities that enable a molecular genetic diagnosis to be made for such diseases as Duchenne’s and Becker’s muscular dystrophy, some forms of limb girdle dystrophy, myotonic muscular dystrophy, congenital muscular dystrophy, and many others (see Table 87-1). Often, the defective gene product, such as dystrophin in the X-linked dystrophies, can be measured or visualized in muscle samples for diagnostic purposes.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Alameddine H.S., Dehaupas M., Fardeau M. Regeneration of skeletal muscle fibers from autologous satellite cells multiplied in vitro: An experimental model for testing cultured cell myogenicity. Muscle Nerve. 1989;12:544.
Anderson J.E. A role for nitric oxide in muscle repair: Nitric oxide-medicated activation of muscle satellite cells. Mol Biol Cell. 2000;11:1859.
Anderson J.T., Rogers R.P., Jarrett H.W. Ca2+-calmodulin binds to the carboxyl-terminal domain of dystrophin. J Biol Chem. 1996;271:6605.
Anderson L.V., Davison K., Moss J.A., et al. Dysferlin is a plasma membrane protein and is expressed early in human development. Hum Mol Genet. 1999;8:855.
Arimura T., Matsumoto Y., Okazaki O., et al. Structural analysis of obscurin gene in hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2007;362:281-287.
Bajaoui K., Hirabayashi K., Hentati F., et al. Linkage of Miyoshi myopathy (distal autosomal recessive muscular dystrophy) locus to chromosome 2p12-14. Neurology. 1995;45:768.
Bansal D., Miyake K., Vogel S., et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423(6936):168.
Barker D., Harker D., Stacey M.J., et al. Fusimotor innervation. In: Banker B.Q., Przybylski R.J., van der Meulen J.P., et al, editors. Research in muscle development and the spindle. Amsterdam: Excerpta Medica, 1972.
Beauchamp J.R., Heslop L., Yu D.S., et al. Expression of CD34 and Myf5 defines majority of quiescent adult skeletal satellite cells. J Cell Biol. 2000;151:1221-1234.
Benedetti S., Merlini L. Laminopathies: From the heart of the cell to the clinics. Curr Opin Neurol. 2004;17:553.
Betz R.C., Schoser B.G., Kasper D., et al. Mutations in CAV3 cause mechanical hyperirritability of skeletal muscle in rippling muscle disease. Nat Genet. 2001;28:218.
Bies R.D., Caskey C.T., Fenwick R. An intact cysteine-rich domain is required for dystrophin function. J Clin Invest. 1992;90(2):666.
Blake D.J., Kroger S. The neurobiology of Duchenne muscular dystrophy: Learning lessons from muscle? Trends Neurosci. 2000;23:92.
Blake D.J., Weir A., Newey S., et al. Function and genetics of dystrophin and dystrophin related proteins in muscles. Physiol Rev. 2002;82:291-329.
Bodine-Fowler S., Garfinkel A., Roy R.R., et al. Spatial distribution of muscle fibers within the territory of a motor unit. Muscle Nerve. 1990;13:1133.
Boito C., Fanin M., Siciliano G., et al. Novel sarcoglycan gene mutations in a large cohort of Italian patients. J Med Genet. 2003;40(5):67.
Bonnemann C.G., Modi R., Noguchi S., et al. Beta-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat Genet. 1995;11:266.
Bornemann A., Schmalbruch H. Anti-vimentin staining in muscle pathology. Neuropathol Appl Neurobiol. 1993;19:414.
Boska M.D., Moussavi R.S., Carson P.J., et al. The metabolic basis of recovery after fatiguing exercise of human muscle. Neurology. 1990;40:240.
Boyer J., Bernstein M.A., Boudreau-Lariveire C. Plakins in striated muscle. Muscle Nerve. 2009:299-308.
Brooks S.V. Current topics in skeletal muscle physiology. Adv Physiol Educ. 2003;27:171.
Brown J.H., Cohen C. Regulation of muscle contraction by tropomyosin and tropoinin: How structure illuminates function. Adv Protein Chem. 2005;71:121-159.
Brown L.A., McClune J.M., Wang H.C. Creatine kinase activity following strenuous exercise. JAMA. 1982;248:2971.
Buchthal F., Schmalbruch H. Motor unit of mammalian muscle. Physiol Rev. 1980;60:90.
Buckingham M., Bajard L., Chang T., et al. The formation of skeletal muscle: From somite to limb. J Anat. 2003;202:59.
Buller A.J., Eccles J.C., Eccles R.M. Interactions between motor neurons and muscles in respect of the characteristic speeds of their responses. J Physiol. 1960;150:417.
Byers T.J., Kunkel L.M., Watkins S.C. The subcellular distribution of dystrophin in mouse skeletal, cardiac, and smooth muscle. J Cell Biol. 1991;115:411.
Cagliani R., Bresolin N., Prelle A., et al. A CAV3 microdeletion differentially affects skeletal muscle and myocardium. Neurology. 2003;61(11):1513.
Campbell K.P., Kahl S.D. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989;338:259.
Carlson D.G. The dystrophinopathies: an alternative to the structural hypothesis. Neurobiol Dis. 1998;5:3-15.
Chen Y.W., Zhao P., Borup R., et al. Expression profiling in the muscular dystrophies: Identification of novel aspects of molecular pathophysiology. J Cell Biol. 2000;151(6):1321.
Chiu Y.H., Hornsey M.A., Klinge L., et al. Attenuated muscle regeneration is a key factor in dysferilin deficient muscular dystrophy. Hum Mol Genet. 2009;18:1976-1989.
Clague J.E., Roberts N., Gibson H., et al. Muscle imaging in health and disease. Source Neuromusc Disord. 1995;5:171.
Cooper J.A., Kim T., Sept D. The interaction of capping protein with the barbed end of the actin filament. J Mol Biol. 2010 Dec 17;404(5):794-802.
Cossu G., Biressi S.,. Satellite cells, myoblasts and other myogenic progenitors: possible origin, phenotypic features and role in muscle regeneration. Semin Cell Dev Biol. 2005;16:623-631.Vale R. Guidebook to the cytoskeletal and motor proteins. New York: Oxford University Press; 1993.
Crawford G.E., Gaulkner J.A., Crosbie R.H., et al. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J Cell Biol. 2000;150:1399.
Critchley D.R. Alpha-actinins. In: Kreis T., Vale R., editors. Guidebook to the cytoskeletal and motor proteins. New York: Oxford University Press, 1993.
Dalakas M.C., Park K.Y., Semino-Mora C., Lee H.S., et al. Desmin myopathy, a skeletal myopathy caused by mutations in desmin gene. N Engl J Med. 2010 Mar 16;342(11):770-780.
Daube J.R. Needle examination in clinical electromyography. Muscle Nerve. 1991;14:685.
De Luca C. Control properties of motor units. J Exp Biol. 1985;115:125.
De Luca C.J., LeFever R.S., McCue M.P., et al. Behavior of human motor units in different muscles during linearly varying contractions. J Physiol. 1982;329:158.
De Luna N., Gallardo E., Illa I. In vivo and in vitro dysferlin expression in human muscle satellite cells. J Neuropathol Exp Neurol. 2004;63(10):1104.
Donner K., Sandbacks M., Lehtokari V.L., et al. Complete genomic structure of the human nebulin gene and identification of alternatively spliced transcripts. Eur J Hum Genet. 2004;12:744-751.
Dubouchaud H., Butterfield G.E., Wolfel E.E., et al. Endurance training, expression, and physiology of LDH, MCT1 and MCT2 in human skeletal muscle. Am J Physiol Endocrinol Metab. 2000;278:E571.
Dubowitz V., Brook M.H. Muscle biopsy: A modern approach. Philadelphia: WB Saunders; 1973.
Duggan D.J., Gorospe J.R., Fanin M., et al. Mutations in the sarcoglycan genes in patients with myopathy. N Engl J Med. 1997;336:618.
Edgerton V.R., Martin T.P., Bodine S.C., et al. How flexible is the neural control of muscle properties? J Exp Biol. 1985;115:393.
Ervasti J.M., Campbell K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121.
Ervasti J.M., Ohlendieck K., Kahl S.D., et al. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315.
Fairley E.A., Kendrick-Jones J., Ellis J.A. The Emery-Dreifuss muscular dystrophy phenotype arises from aberrant targeting and binding of emerin at the inner nuclear membrane. J Cell Sci. 1999;112:2571.
Ferreiro A., Monnier N., Romero N.B., et al. A recessive form of central core disease, transiently presenting a multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann Neurol. 2002;51:750.
Fishbach G.D., Rosen K.M. ARIA: Neuromuscular junction neuregulin. Annu Rev Neurosci. 1997;20:429.
Fishman D.A. C-proteins. In: Kreis T., Vale R., editors. Guidebook to the cytoskeletal and motor proteins. New York: Oxford University Press, 1993.
Ford L.E., Huxley A.F., Simmons E.M. Tension responses to sudden length change in stimulated frog muscle fibers near slack length. J Physiol. 1977;269:441.
Franzini-Armstrong C. The sarcoplasmic reticulum and the control of muscle contraction. FASEB J. 1999;12:S266.
Fuchs E., Weber K. Intermediate filaments: Structure, dynamics, function, and disease. Annu Rev Biochem. 1994;63:345.
Gallanti A., Prelle A., Moggio M., et al. Desmin and vimentin as markers of regeneration in muscle diseases. Acta Neuropathol (Berl). 1992;85:88.
Gazzerro E., Sotglia F., Bruno C., et al. Caveolinopathies: From the biology of caveolin-3 to human diseases. Eur J Hum Genet. 2009:1-11.
Geisler N., Weber K. The amino acid sequence of chicken muscle desmin provides a common structural model for intermediate filament proteins. EMBO J. 1982;1:1649.
Gergeley J., Garbarek Z., Tao T. Troponins. In: Kreis T., Vale R., editors. Guidebook to the cytoskeletal and motor proteins. New York: Oxford University Press, 1993.
Geruli B., Gramlich M., Atherton J., et al. Mutations of TTN, encoding the giant muscle filament titin, causes familial dilated cardiomyopathy. Nat Genet. 2002;30:201-204.
Glover L., Brown R.L. Dysferilin in membrane traffiking and patch repair. Traffic. 2007;8:785-794.
Glover L., Newton K., Krishnan G., et al. Dysferlin overexpression in skeletal muscle produces a progressive myopathy. Ann Neurol. 2010;67:384-393.
Goebel H.N., Laing N.G. Actinopathies and Myosinopathies. Brain Pathol. 2009;19:516-522.
Goldfarb L.G., Vicart P., Goebel H.H., et al. Desmin myopathy. Brain. 2004;127:723.
Gomes A.V., Barnes J.A., Harada K., et al. Role of troponin T in disease. Mol Cell Biochem. 2004;263(1):115.
Gomes A.V., Potter J.D., Szczesna-Cordary D. The role of troponins in muscle contraction. IUBMB Life. 2002;54:323.
Grabarek Z., Tan R.Y., Wang T., et al. Inhibition of mutant troponin C activity by an intradomain disulphide bond. Nature. 1990;345:132.
Gullberg D., Tiger C.F., Velling T. Laminins during muscle development and in muscular dystrophies. Cell Mol Life Sci. 1999;56:442-460.
Gutmann D.H., Fischbeck K.H. Molecular biology of Duchenne and Becker’s muscular dystrophy: Clinical applications. Ann Neurol. 1989;26:189.
Haraguchi T., Holaska J., Yamane M., et al. Emerin binding to Btf, a death-promoting transcriptional repressor, is disrupted by a missense mutation that causes Emery-Dreifuss muscular dystrophy. Eur J Biochem. 2004;271(5):1035.
Harrington W.F., Rodgers M.E. Myosin. Annu Rev Biochem. 1984;53:35.
Heckmatt J., Dubowitz V. Real time ultrasound imaging of muscle. Muscle Nerve. 1988;11:56.
Heckmatt J.Z., Moosa A., Hutson C., et al. Diagnostic needle biopsy: A practical and reliable alternative to open biopsy. Arch Dis Child. 1984;59:528.
Heeley D.H., Dhoot G.K., Perry S.V. Factors determining the subunit composition of tropomyosin in mammalian skeletal muscle. Biochem J. 1985;226:461.
Heizmann C., Rohrenbech J., Kamphuis W. Parvalbumin: Molecular and functional aspects. Adv Exp Med Biol. 1990;269:57.
Henneman E. The size-principle: A deterministic output emerges from a set of probabilistic connections. J Exp Biol. 1985;115:105.
Henneman E., Clamann J.P., Gillies J.O., et al. Rank order of motoneurones within a pool: Law of combination. J Neurophysiol. 1974;37:1388.
Hennig R., Lomo T. Firing patterns of motor units in normal rats. Nature. 1985;314:164.
Hoffman E.P., Brown R.H., Kunkel L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919.
Horowits R., Maruyama K., Podolsky R.J. Elastic behavior of connecting filaments during thick filament movement in activated skeletal muscle. J Cell Biol. 1989;109:2169.
Hui C.S. Pharmacological studies of charge movement in frog skeletal muscle. J Physiol. 1983;337:509.
Hultman E. Fuel selection of muscle fibre. Proc Nutr Soc. 1995;54:107.
Huxley H.E. A personal view of muscle and motility mechanisms. Annu Rev Physiol. 1996;58:1.
Huxley H.E. The mechanism of force production in muscle. In: Schotland D.L., editor. Disorders of the motor unit. New York: John Wiley & Sons, 1982.
Ibraghimov-Beskrovnaya O., Ervasti J.M., Leveille C.J., et al. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696.
Jacob K.N., Garg A. Laminopathies: Multisystem dystrophy syndromes. Mol Genet Metab. 2006;87:289-302.
Jiang H., Jordan T., Li J., et al. Innervation-dependent and fiber type-specific transcriptional regulation of the slow myosin heavy chain 2 promoter in avian skeletal muscle fibers. Dev Dyn. 2004;231:292-302.
Jones K., Morgan G., Johnston H., et al. The expanding phenotype of laminin alpha 2 (merosin) abnormalities: Case series and review. J Med Genet. 2001;38:649.
Juel C. Lactate-proton cotransport in skeletal muscle. Physiol Rev. 1997;77:321.
Jungbluth H., Zhou H., Hartley L., et al. Minicore myopathy with ophthalmoplegia caused by mutations in the ryanodine receptor type 1 gene. Neurology. 2005;65:1930-1935.
Kaparti G., Pouliot Y., Carpenter S. Expression of immunoreactive major histocompatibility complex products in human skeletal muscles. Ann Neurol. 1988;23:64.
Kassar-Duchossoy L., Gayarud-Morel B., Gomes D., et al. Mrf4 determines muscle identitiy inMyf5: MyoD double–mutant mice. Nature. 2004;431:466.
Katz B., Miledi R. The statistical nature of the acetylcholine potential and its molecular components. J Physiol. 1972;224:665.
Kennelly P.J., Starovasnik M., Krebs E.G. Activation of rabbit skeletal light chain kinase by calmodulin. A mechanistic overview. Adv Exp Med Biol. 1989;255:155.
Khurana T.S., Watkins S.C., Chafey P., et al. Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul Disord. 1991;1:185.
Khurana T.S., Watkins S.C., Kunkel L.M. The subcellular distribution of chromosome-6 encoded dystrophin related protein in the brain. J Cell Biol. 1992;119:357.
Kimball S.R., Farrell P.A., Jefferson L.S. Invited review: Role of insulin in translational control of protein synthesis in skeletal muscle by amino acids or exercise. J Appl Physiol. 2002;93:1168.
Kistler J., Stroud R.M., Klymkowsky M.W., et al. Structure and function of an acetylcholine receptor. Biophys J. 1982;37:371.
Kontrogianni-Konstantpoulos A., Ackermann A., Bowman A., et al. Muscle giants: Molecular scaffolds in sarcomerogenesis. Physiol Rev. 2009;89:1217-1267.
Kushmerick M.J. Skeletal muscle: A paradigm for testing principles of bioenergetics. J Bioenerg Biomembr. 1995;27:555.
Landon D.N. Skeletal muscle: Normal morphology, development, and innervation. In: Mastaglia F.L., Walton J., editors. Skeletal muscle pathology. New York: Churchill Livingstone, 1982.
Lapidos K.A., Kakkar R., McNally E.M., et al. The dystrophin glycoprotein complex: Signaling strength and integrity for the sarcolemma. Circ Res. 2004;94(8):1023.
Lepper C., Conway S.J., Fan C.-M., et al. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirement. Nature. 2009 Jul 30;460(7255):627-631.
Lehman W., Rosol M., Tobacman L., et al. Troponin organization on relaxed and activated thin filaments revealed by electron microscopy and three dimensional reconstruction. J Mol Biol. 2001;307:739-744.
Lekhotari V.L., Pelin K., Sandbacka M., et al. Identification of 45 novel mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Hum Mutat. 2006;27:946-956.
Lieber R.C. Skeletal muscle adaptability. I. Review of basic properties. Dev Med Child Neurol. 1986;28:390.
Lindstrom J.M. Nicotinic acetylcholine receptors of muscles and nerves. Ann N Y Acad Med. 2003;998:41.
Liu C.G., Maercker C., Castanon M.J., et al. Human plectin: Organization of the gene, sequence analysis, and chromosome localization (8q24). Proc Natl Acad Sci USA. 1996;93:4278.
Liu J., Aoki M., Illa I., et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20:31.
Loewinger L., McKeon F. Mutations in the nuclear lamin proteins resulting in their aberrant assembly in the cytoplasm. EMBO J. 1988;7:2301.
Lu J., Bassel-Duby R., Hawkins A., et al. Control of facial muscle development by MyoR and capsulin. Science. 2002;298:2378.
Lutz G.J., Lieber R. Studies of myosin isoforms in muscle cells: Single cell mechanics and gene transfer. Clin Orthop Relat Res. 2002;1(Suppl 403):S51.
Maniotis A.J., Chen C.S., Ingber D.E. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci USA. 1997;94:849.
Manzur A.Y., Muntoni F. Daignsosis and new treatments in muscular dystrophies. J Neurol Neurosurg Psychiatry. 2009;80:706-714.
Marston S., Hodgkinson J. Cardiac and skletal myopathies: Can genotype explain phenotype? J Muscle Res Cell Motil. 2001;2:1-4.
Martin C.M., Russell J.L., Ferdous A., et al. Molecular signatures define myogenic stem cell populations. Stem Cell Rev. 2006;2:37-42.
Martin P.T., Freeze H. Glycobiology of neuromuscular disorders. Glycobiology. 2003;13(8):67R.
Matlin C.A., Delday M.I., Siclair D.D., et al. Impact of manipulations of myogenesis in utero on performance of adult skeletal muscle. Reproduction. 2001;122:359.
McComas A.J. Motor unit estimation: Methods, results and present studies. Muscle Nerve. 1991;14:585.
McLennon I.S. Neurogenic and myogenic regulation of skeletal muscle formation: A critical re-evaluation. Prog Neurobiol. 1994;44:119.
Mercuri E., Messina S., Bruno C., et al. Congenital muscular dystrophies with defective glycosylation of dystroglycan: A population study. Neurology. 2009;72:1802-1809.
Michele D.E., Campbell K.P. Dystrophin-glycoprotein complex post-translational processing and dystroglycan function. J Biol Chem. 2003;278:15457.
Miles T.S. The control of human motor units. Clin Exp Pharmacol Physiol. 1994;21:511.
Mittelbronn M., Sullivan T., Stewart C.L., et al. Myonuclear degeneration in LMNA null mice. Brain Pathol. 2008;18:338-343.
Miura P., Chakkalakal J.V., Boudreault L., et al. Pharmacological activation of PPARB/D stimulates utophin A expression in skeletal muscle fibers and restores sarcolemmal integrity in mature mdx mice. 2009:1-30. HMG Advance Access published september 10 2009
Momoi M.Y., Lennon V.A. Purification and biochemical characterization of nicotinic acetylcholine receptors of human muscle. J Biol Chem. 1982;257:12757.
Morrison L. Unraveling RYR1 mutations and muscle biopsies. Neurology. 2008;70:99-100.
Nawrotzki R., Blake D.J., Davies K.E. The genetic basis of neuromuscular disorders. Trends Genet. 1996;12:294.
Newsome-Davis J. Lambert-Eaton syndrome. Rev Neurol (Paris). 2004;160:177.
Newsome-Davis J., Buckley C., Clover L., et al. Autoimmune disorders of the neuronal potassium channels. Ann N Y Acad Sci. 2003;998:2001.
Norregaard-Hansen K., Hein-Sorensen O. Significance of serum myoglobin in neuromuscular diseases and carrier detection of Duchenne muscular dystrophy. Acta Neurol Scand. 1982;66:259.
Nowak K.J., Wattanasarichaigoon D., Goebel H.H., et al. Mutations in the skeletal muscle alpha-actin gene in patients with actin and nemalin myopathy. Nat Genet. 1999;23:208.
Omary M.B., Coulombe P.A., McLean W.H.I. Intermediate filament proteins and their associated diseases. N Engl J Med. 2004;351:2087.
Peachey L.D. Excitation-contraction coupling: The link between the surface and the interior of a muscle cell. J Exp Biol. 1985;115:91.
Petajan J.H. Motor unit recruitment. Muscle Nerve. 1991;14:489.
Pette D. Historical perspectives: Plasticity of mammalian skeletal muscle. J Appl Physiol. 2001;90:1119.
Pette D., Vrborá G. Neural control of phenotypic expression in mammalian muscle fibers. Muscle Nerve. 1985;8:676.
Philpot J., Sewry C., Pennock J., et al. Clinical phenotype in congenital muscular dystrophy: Correlation with expression of merosin in skeletal muscle. Neuromuscul Disord. 1995;5:301.
Pillers D.A.H., Fitzgerald K.M., Duncan N.M., et al. Duchenne/Becker muscular dystrophy: Correlation of phenotype by electroretinography with sites of dystrophin mutations. Hum Genet. 1999;105:2.
Pollack G.H. Phase transitions and the molecular mechanism of contraction. Biophys Chem. 1996;59:315.
Pollack G.H., Sugi H. Contractile mechanisms in muscle. New York: Plenum Press; 1984.
Pollard T.D. Actin and actin binding proteins. In: Kreis T., Vale R., editors. Guidebook to the cytoskeletal and motor proteins. New York: Oxford University Press, 1993.
Pons F., Augier N., Heilig R., et al. Isolated dystrophin molecules as seen by electron microscopy. Proc Natl Acad Sci USA. 1990;87:7851.
Porter G.A., Dmytrenko G.M., Winkelmann J.C., et al. Dystrophin colocalizes with beta-spectrin in distinct subsarcolemmal domains in mammalian skeletal muscle. J Cell Biol. 1992;117:997.
Pownall M.E., Gustafsson M.K., Emerson C.P.Jr. Myogenic regulatory factors and specific progenitors in vertebrate embryos. Annu Rev Cell Dev Biol. 2002;18:747.
Rizo J., Sudhof T.C. C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem. 1998;273:15879.
Rodino-Klapac L.R., Lee J.S., Mulligan R.C., et al. Lack of toxicity of alpha sarcoglycan overexpression supports clinical gene transfer trial in LGMD2D. Neurology. 2008;71:240-247.
Ruff R.L. Neurophysiology of the neuromuscular junction. Ann N Y Acad Sci. 2003;998:1.
Samitt C.E., Bonilla E. Immunocytochemical study of dystrophin at the myotendinous junction. Muscle Nerve. 1990;13:493.
Sander M., Chavoshan B., Harris S.A., et al. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2000;97:13818.
Sanders D.B., Stalberg E.V., Nandedkar S.D. Analysis of the electromyographic interference pattern. J Clin Neurophysiol. 1996;13:385.
Sandri M., Carbano U. Apoptosis of skeletal muscles during development and disease. Int J Biochem Cell Biol. 1999;31:1373-1390.
Scacheri P.C., Hoffman E.P., Fratkini J.D., et al. A novel ryanodine receptor gene mutation causing both cores and rods in congenital myopathy. Neurology. 2000;55:1689.
Scalzi H.A., Price H.M. Electron microscopic observations of the sensory region of the mammalian muscle spindle. In: Banker B.Q., Przybylski R.J., van der Meulen J.P., et al, editors. Research in muscle development and the spindle. Amsterdam: Excerpta Medica, 1972.
Schnorrer F., Dickson B.J. Muscle building: Mechanisms of myotube guidance and attachment site selection. Dev Cell. 2004;7:9.
Schroder J.M., Bodden H., Hamacher A., et al. Scanning electron microscopy of teased intrafusal muscle fibers from rat muscle spindle. Muscle Nerve. 1989;12:221.
Schroder J.M., Durling H.J., Laing N.G. Actin myopathy with nemaline bodies, intranuclear rods nd heterozygous mutations on ACTA 1. Acta Neuropathol (Berl). 2004;108:250-256.
Schroder R., Schoser B. Myofibrillar myopathies: A clinical and myopathological guide. Brain Pathol. 2009;19:483-492.
Schultz E. Satellite cells in normal, regenerating and dystrophic muscle. Adv Exp Med Biol. 1985;182:73.
Selcen D., Krueger B.R., Engel A.G. Familial cardioneuromyopathy with hyaline masses and nemline rods: a novel phenotype. Ann Neurol. 2002;51:224-234.
Shahani B.T., Young R.R. Clinical electromyography. In: Baker A.B., Joyut R.J., editors. Clinical Neurology. Philadelphia: Harper & Row, 1985.
Sherratt T.G., Vulliamy T., Strong P.N. Evolutionary conservation of the dystrophin central rod domain. Biochem J. 1992;287:755.
Sjoberg G., Jiang W.-Q., Ringertz N.R., et al. Colocalization of nestin and vimentin/desmin in skeletal muscle cells demonstrated by three-dimensional fluorescence digital imaging microscopy. Exp Cell Res. 1994;214:447.
Skeie G.O., Romi F., Aarli J.A., et al. Pathogenesis of myositis and myasthenia associated with titin and ryanodine receptor antibodies. Ann N Y Acad Sci. 2003;998:343.
Slonim A.E., Coleman R.A., McElligot M.A., et al. Improvement of muscle function in acid maltase deficiency by high protein therapy. Neurology. 1983;33:34.
Smillie L.B. Tropomyosins. In: Kreis T., Vale R., editors. Guidebook to the cytoskeletal and motor proteins. New York: Oxford University Press, 1993.
Somlyo A.V., Gonzalez-Serratos H., Shuman H., et al. Calcium release and ionic changes in the sarcoplasmic reticulum of tetanized muscle: An electron probe study. J Cell Biol. 1981;90:577.
Stadhouders A.M. The structural basis of excitation-contraction coupling in muscular contraction. Acta Anaesthesiol Belg. 1990;41:65.
Stonecipher M.R., Jorizzo J.L., Monu J., et al. Dermatomyositis with normal muscle enzyme concentrations. A single-blind study of the diagnostic value of magnetic resonance imaging and ultrasound. Arch Dermatol. 1994;130:1294.
Straub V., Campbell K.P. Muscular dystrophies and the dystrophin-glycoprotein complex. Curr Opin Neurol. 1997;10:168.
Stuart D.G., Mosher C.G., Gerlach R.L. Properties and central connections of Golgi tendon organs with special reference to locomotion. In: Banker B.Q., Przybylski R.J., van der Meulen J.P., et al, editors. Research in muscle development and the spindle. Amsterdam: Excerpta Medica, 1972.
Sussman M.A., Bilak M., Kedes L., et al. Tropomodulin is highly concentrated at the postsynaptic domain of human and rat neuromuscular junctions. Exp Cell. 1993;209:388.
Suzuki A., Yoshida M., Yamamoto H., et al. Glycoprotein-binding site of dystrophin is confined to the cysteine-rich domain and the first half of the carboxy-terminal domain. FEBS Lett. 1992;308:154.
Suzuki S., Utsugisawa K., Yoshikawa H., et al. Autoimmune targets in heart and skeletal muscle sin myathenia gravis. Arch Neurol. 2009;66:1334-1338.
Tan K.M., Lennon V.A., Klein C.J., et al. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology. 2008;70:1883-1890.
Taylor R.J., Von Witt R.J., Fry A.H. Serum creatine phosphokinase activity in psychiatric patients receiving electroconvulsive therapy. J Clin Psychiatry. 1981;42:103.
Thornell L.E., Billeter R., Butler-Browne G.S., et al. Development of fiber types in human fetal muscle. J Neurol Sci. 1984;66:107.
Tidball J.C. Desmin at myotendinous junctions. Exp Cell Res. 1992;199:341.
Tskhovrebova L., Trinick J. Titin: Properties and family relationships. Nat Rev Mol Cell Biol. 2003;4:679.
Vale R.D., Milligan R.A. The way things move: Looking under the hood of molecular motor proteins. Science. 2000;288:88.
Vainzof M., Zatz M., Otto P.A., et al. Serum CK-MB activity in progressive muscular dystrophy: is it of nososlogic value. Am J Med Genet. 1985 Sep;22(1):81-87.
van der Kooi A.J., Bonne G., Eymard B., et al. Lamin A/C mutations with lipodystrophy, cardiac abnormalities, and muscular dystrophy. Neurology. 2002;59:620.
van der Meulen J.P., Hellekson C.J., Hinnas R.W. The central control of the muscle spindle. In: Banker B.Q., Przybylski R.J., van der Meulen J.P., et al, editors. Research in muscle development and the spindle. Amsterdam: Excerpta Medica, 1972.
Viegas-Pequignot E., Li Z.L., Dutrillaux B., et al. Assignment of human desmin gene to band 2q35 by nonradioactive in site hybridization. Hum Genet. 1989;83:33.
Vuolteenaho R., Nissinen M., Sainio K., et al. Human laminin M chain (merosin): Complete primary structure, chromosomal assignment, and expression of the M and A chain in human fetal tissues. J Cell Biol. 1994;124:381.
Wallgren-Pettersson C. Nemaline and myotubular myopathies. Semin Pediatr Neurol. 2002;9:132.
Wallgren-Pettersson C., Clarke A., Samson F., et al. The myotubular myopathies: Differential diagnosis of the X linked recessive, autosomal dominant, and autosomal recessive forms and present state of DNA studies. J Med Genet. 1995;32:673.
Wallgren-Pettersson C., Lekhotari V.L., Kalimo H., et al. Distal myopathy caused by missense mutations in the nebulin gene. Brain. 2007;130:1465-1476.
Watkins S.C., Cullen M.J., Hoffman E.P., et al. Plasma membrane cytoskeleton of muscle: A fine structural analysis. Microsc Res Tech. 2000;48:131.
Weiner M.W., Moussavi R.S., Baker A.J., et al. Constant relationship between force phosphate concentration, and pH in muscles with differential fatigability. Neurology. 1990;40:1888.
Wernig A., Bone M., Irintchev A., et al. Mcadherin is a reliable markerof quiescent satellite cellsin mouse skeletal muscle. Basic Appl Myol. 2004;14:161-168.
Westwood S.A., Hudlicka O., Perry S.V. The effect of contractile activity on the phosphorylation of the P light chain of myosin of rabbit skeletal muscle. Biochem J. 1984;218:841.
Whalen R.G. Myosin isoenzymes as molecular markers for muscle physiology. J Exp Biol. 1985;115:43.
Whiche G. Role of plectin in cytoskeletal organization and dynamics. J Cell Sci. 1998;111:247.
Wolfe R.R. Regulation of muscle protein by amino acids. J Nutr. 2002;132:3219S.
Woodman S.E., Sotgia F., Galbiati F., et al. Cavelinopathies : Mutations in caveoline cause four distinct autosomal dominant muscle diseases. Neurology. 2004;62:538-543.
Wyatt R.M., Balice-Gordon R.J. Activity-dependent elimination of neuromuscular synapses. J Neurocytol. 2003;32:777.
Wyllie E., Leuders H., Pippenger C., et al. Postictal serum creatine kinase in the diagnosis of seizure disorders. Arch Neurol. 1985;42:123.
Shi X., Garry D.J. Muscle stem cells in development, regeneration and disease. Genes and Development. 2006;20:1692-1708.
Yamamoto H., Mizuno Y., Hayashi K., et al. Expression of dystrophin-associated protein 35DAG (A4) and 50DAG (A2) is confined to striated muscles. J Biochem (Tokyo). 1994;115:162.
Yamamoto L., Gollop T., Naccache N., et al. Protein and DNA analysis for the prenatal diagnosis of [alpha]2-laminin-deficient congenital muscular dystrophy. Diagn Mol Pathol. 2004;13(3):167.
Younger D.S., Gordon P.H. Diagnosis in neuromuscular diseases. Neurol Clin. 1996;14:135.
Zubrzycka-Gaarn E.E, Bulman D.E., Karpati G., et al. The Duchenne muscular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature. 1988 Jun 2;333(6172):466-469.
