Chapter 15 Neuroinflammation
Introduction
Perinatal Brain Damage
The consequences of perinatal injury include a spectrum of disorders, such as mental retardation, cerebral palsy, epilepsy, vision and hearing loss, learning difficulties, and school failure. Periventricular white matter damage, including periventricular leukomalacia, is most frequently observed in human preterm neonates [Volpe, 2009]. Full-term neonates with perinatal encephalopathy generally develop gray matter damage that most frequently affects the neocortex, basal ganglia, and hippocampus [Volpe, 2001]. Cerebrovascular occlusion leading to perinatal stroke may be arterial or venous, but excludes global injuries due to hypoxic-ischemic injury. Neurodevelopmental disability, including cerebral palsy, epilepsy, and behavior disorders, as well as impaired vision and language, is common after perinatal stroke [Kirton and deVeber, 2009].
The pathophysiology of perinatal brain damage (see Chapters 17 and 18) has proven to be multifactorial, with sensitizing factors occurring in utero that make the brain more vulnerable to secondary insults occurring around birth, such as hypoxia-ischemia (HI), and excess release of glutamate leading to excitotoxicity [Dammann et al., 2002; Mesples et al., 2005b; Nelson and Chang, 2008; Degos et al., 2008]. Systemic inflammation linked to chorioamnionitis has been recognized as a key sensitizing factor, while CNS inflammatory responses have been shown to play a key modulatory role in the amplitude of brain damage and subsequent adverse neurological outcome.
Systemic Inflammation and Perinatal Brain Damage
Epidemiological studies have shown a strong association between fetal infection/inflammation (chorioamnionitis) and brain damage in the newborn and/or neurological handicap in survivors [Dammann and Leviton, 2007]. Experimental studies have confirmed a sensitizing effect of systemic inflammation on perinatal brain lesions induced by hypoxic-ischemic or excitotoxic insults [Dommergues et al., 2000; Eklind et al., 2005]. In addition, some experimental data also suggest that perinatal exposure to infectious/inflammatory factors can alter, in a more or less subtle manner, the programs of brain development that will result in lasting neurological deficits. The relationship between this latter observation and human diseases remains to be fully demonstrated, although clinical evidence is supportive of this hypothesis [Volpe, 2009].
Sensitizing Effect
As mentioned above, perinatal brain damage may be caused by a combination of several insults. In this so-called multiple hit hypothesis, systemic infection/inflammation linked to chorioamnionitis can act as predisposing factors, making the brain more susceptible to a second stress (sensitization process). Indeed, systemic injection of low doses of lipopolysaccharide (LPS) to developing rats makes the newborn brain significantly more susceptible to hypoxic-ischemic insult [Eklind et al., 2005]. Similarly, systemic injection of interleukin-1-beta (IL-1β) or LPS to newborn mice or rats makes the brain much more sensitive to an excitotoxic insult [Dommergues et al., 2000]. The mechanisms by which sensitization is working are not yet fully understood, but could include changes in gene transcription and modifications of glutamate receptor activity.
Disruption of Brain Programming
Systemic infection/inflammatory factors can also alter brain development by themselves, even if they do not induce major clastic lesions. Accordingly, injection of Escherichia coli to pregnant rabbits induces diffuse white matter cell death [Debillon et al., 2000], and injection of Ureaplasma parvum, a pathogen frequently observed in chorioamnionitis, to pregnant mice induces myelin defects and loss of interneurons in the offspring [Normann et al., 2009]. Similarly, injection of LPS to pregnant rats induces transient central inflammation and myelination defects in the offspring [Rousset et al., 2006]. Of major concern, exposure of newborn mice to low doses of systemic IL-1β induces a moderate and transient inflammatory response during the neonatal period, that may be sufficient to disrupt oligodendrocyte maturation, myelin formation, and axonal development [Favrais and Gressens, personal communication]. These white matter abnormalities are moderate during the developmental period but persist until adulthood. They lead to permanent deficiencies in cognitive testing without detectable effects on motor function. The relevance of these abnormal behaviors to human pathology remains to be confirmed. The underlying molecular mechanisms include alterations of the transcription of genes implicated in oligodendrogenesis, myelin formation and axonal maturation.
Blood–Brain Barrier
At birth, the blood–brain barrier (BBB) in the neonate is substantially more mature than is commonly thought. The tight junctions are present early in embryonic development [Kniesel et al., 1996], restricting entrance of proteins into the brain in a controllable fashion, and by birth the BBB is functional with no fenestrations [Engelhardt, 2003]. The presence of the barrier substantially affects leukocyte passage but does not guarantee minimal leukocyte transmigration. The use of direct inflammatory challenge, such as intrastriatal injections of IL-1β or tumor necrosis factor alpha (TNFα) in rats of different ages, does not show a linear decline of leukocyte transmigration with age [Anthony et al., 1997], but rather that the newborn CNS is more resistant to inflammatory stress than the juvenile brain. The reported magnitude of BBB disturbance following HI or focal stroke in neonatal rodents varies and depends on the aspect of barrier studied [Svedin et al., 2007; Faustino et al., 2009]. Degradation of the extracellular matrix plays a role in neonatal ischemic injury. Excessive activation of matrix metalloproteinase-9 (MMP-9) early after HI is deleterious to the immature brain, as demonstrated by smaller injury size in MMP-9 knockout mice [Svedin et al., 2007] and following pharmacological inhibition of this protease [Leonardo et al., 2008].
Crosstalk between the Periphery and the CNS
The precise molecular mechanisms by which circulating mediators of inflammation have a deleterious effect on perinatal brain lesions remain a matter for debate [Hagberg and Mallard, 2005]. Circulating cytokines do not seem to cross the intact BBB easily, although this issue is contested. Different alternative pathways have been proposed to link serum cytokines with brain damage [Malaeb and Dammann, 2009]. Several major crosstalk mechanisms are outlined in Figure 15-1. Circulating cytokines could initially alter the permeability of the BBB to inflammatory mediators and cells. They could also act directly on parts of the brain lacking the BBB, such as the circumventricular organs, meninges, and choroid plexus, or, as demonstrated in the adult brain, indirectly through activation of the vagal nerve. Cytokine effects also could be mediated by cyclo-oxygenases (Cox) located in the BBB. In particular, cytokines could activate the inducible isoform Cox-2 to enhance the local production of prostaglandin E2 (PGE2), which could have deleterious effects on the developing brain. This latter mechanism has been demonstrated in a mouse model of perinatal excitotoxic brain damage [Favrais et al., 2007]. Some of these deleterious effects could involve an autocrine/paracrine loop, leading to excess production of inflammatory cytokines by brain cells.
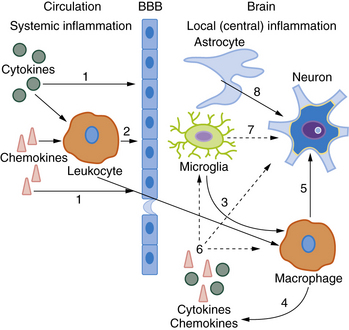
Glial Cells
Macrophages are seen in abundance following neonatal HI [McRae et al., 1995; Ivacko et al., 1996] and focal stroke [Dingman et al., 2006; Denker et al., 2007], producing inflammatory cytokines, high levels of nitric oxide, MMPs, and complement molecules. The early postinjury macrophage population is predominantly comprised of resident microglia rather than invading monocytes [Denker et al., 2007]. The notion that microglia contribute to, rather than limit, acute ischemic injury in the immature brain comes from findings that reduction in injury is associated with diminished microglial activation/monocyte infiltration [Arvin et al., 2002; Dommergues et al., 2003]. At the same time, several studies have shown that anti-inflammatory drugs thought to protect adult brain by reducing macrophage accumulation after stroke protect neonatal brain without directly affecting inflammatory mechanisms associated with microglial activation [Tikka et al., 2001; Fox et al., 2005; van den Tweel et al., 2005; Dingman, et al. 2006]. Distinct steps of microglial maturation and differentiation (such as expression of class II histocompatibility complex [MHC], cathepsin, and other molecules), and the propensity of neurons to undergo apoptosis in the developing brain, may account for this age-dependence of the microglial response. Complement activation – C3 and C1q deposition in particular – is deleterious after HI, whereas sensitizing of neonatal rodents with cobra venom factor or deletion of the C1q gene confers protection [Cowell et al., 2003; Ten et al., 2005]. The mechanisms of protection are not completely understood but may include decreased C3 deposition and reduction in neutrophil activation [Ten et al., 2005].
The relative contribution of proinflammatory mechanisms in astrocytes, as opposed to other roles of these cells in ischemic injury, is not well understood but astrocytes express MHC and can upregulate inducible nitric oxide synthase (iNOS) and increase cytokine production. Mast cells have been shown to play an injurious role in neonatal HI [Jin et al., 2007] and focal stroke [Biran et al., 2008]. The injurious effects of these cells are thought to depend on TGF-β and IL-9 [Mesples et al., 2005a]. Less is known about the contribution of the T and B cell infiltration that is seen at later time points after injury.
Individual Inflammatory and Cell Signaling Molecules
Cytokines and Chemokines
Clinical data on the pathophysiological role of inflammatory cytokines in perinatal brain damage, including in term babies, continue to emerge [Grether and Nelson, 1997; Foster-Barber and Ferriero, 2002; Bartha et al., 2004].
Overall, it appears that IL-1 potentiates ischemic brain injury. Following HI or transient middle cerebral artery occlusion (MCAO) in neonatal rats, brain IL-1β expression is increased rapidly [Hagberg et al., 1996; Denker et al., 2007] following a major rapid systemic increase of IL-1β protein [Denker et al., 2007]. Brain IL-1β levels can be further amplified by concomitant infection or manipulations within the oxidant pathways [Doverhag et al., 2008; Girard et al., 2008]. The pro-inflammatory shift in balance between IL-1β and IL-1ra following HI when combined with infection (modeled by endotoxin or LPS exposure) can play a role in the initiation of perinatal brain damage [Girard et al., 2008]. IL-1β-induced local inflammatory reaction and chemokine expression, with the consequent leukocyte attraction and BBB disruption, is age-dependent [Anthony et al., 1997]. Studies that use genetic deletion of IL-1β or IL-1α alone, or in combination (IL-1αβ knockout), however, show no protection after HI injury [Hedtjarn et al., 2005], indicating the presence of multiple or desynchronized pro- and anti-injurious effects of IL-1, which are all abrogated in knockout mice. IL-18, a pro-inflammatory cytokine from the IL-1 family, also contributes to HI injury [Hedtjarn et al., 2002].
TNFα can exhibit pleiotropic functions in the ischemic adult brain. It may lead to apoptosis by reacting with Fas-associated death domain (FADD) and caspase-8, whereas signaling through TNFR2 can lead to anti-inflammatory and anti-apoptotic functions, depending on the cell origin of TNFα production and the type of receptor involved. In adult, TNFα-induced sensitization prior to cerebral ischemia is shown to be associated with a preconditioning response, whereas TNFα sensitization post stroke is detrimental [Hallenbeck, 2002]. In neonatal brain injury the pathophysiological role of TNFα may also be complex. A key role of TNFα in injury in an excitotoxic model combined with prior inflammatory challenge (IL-1β) has been recently demonstrated [Aden et al., 2009]. A TNFα blocker, etanercept, did not affect brain damage when given before injury, but substantially ameliorated injury when given after the combined inflammatory and excitotoxic insult, suggesting that TNFα may act by producing an imbalance between pro- and anti-inflammatory cytokines in injured brain [Aden et al., 2009].
Type 2 T helper (Th2) cytokines play multiple roles in neonatal brain injury. The IL-9/IL-9 receptor pathway, which is most active in the newborn brain, has a direct anti-apoptotic action in the newborn neocortex [Fontaine et al., 2008], and contributes to HI and excitotoxic injury in the developing brain, presumably by activating mast cells [Dommergues et al., 2000; Patkai et al., 2001]. IL-10, a Th2 cytokine that is synthesized in the CNS and can act on hematopoietic and nonhematopoietic cells, can reverse injury caused by IL-1β, TNFα, and IL-6. In a white matter lesion model in P5 rats, protection is seen when IL-10 is administered post insult, whereas treatment prior to or at the time of insult is ineffective [Mesples et al., 2003].
Chemokines play a key role in crosstalk between peripheral and local responses. The pathophysiological role of the CC-class chemokine, monocyte chemoattractant protein 1 (MCP-1), in neonatal brain injury is evident from protection by functional inactivation of MCP-1 post insult [Galasso et al., 2000] or in mice with depleted IL-1 converting enzyme [Xu et al., 2001]. The role of the CXC-(Cys-X-Cys chemokine) family chemokines after neonatal brain injury is less understood but may be crucial for BBB modulation after inflammatory challenge of injured immature brain [Anthony et al., 1998]. Information on SDF-1 (CXCL12), which has been suggested to play a role in homing stem cells to regions of ischemic injury and repair in the adult, is rather scarce in the neonate but the timing for SDF-1-mediated chemotaxis and recruitment of reparative cells in the neonate may be narrow [Miller and Tran, 2005].
Intracellular Reactive Oxidant Metabolism
Oxidative stress and inflammation are tightly linked. Once activated, the inflammatory cells generate reactive oxygen species (ROS), which, in turn, trigger an inflammatory response. Superoxide anion, which is generated via Cox, xanthine oxidase, and NADPH (nicotinamide adenine dinucleotide phosphate) oxidase and is utilized via superoxide dismutase (SOD) in the cytosol and mitochondria, plays an important role in ischemic injury. Copper Zinc (CuZn) SOD overexpression exacerbates brain injury after HI by increased superoxide utilization and hydrogen peroxide (H2O2) accumulation in injured developing brain [Fullerton et al., 1998; Sheldon et al., 2004]. Enhanced activity of anti-oxidative metabolism is protective [Sheldon et al., 2008], supporting the notion of the pro-injurious role of oxidative stress in neonatal ischemic brain injury.
The role of NADPH oxidase in neonatal brain injury is now better understood. Genetic deletion of gp91-phox (the catalytic subunit of nicotinamide adenine dinucleotide phosphate) increases the extent of brain injury in two neonatal models, the HI model in P9 mice and an excitotoxic model in P5 mice [Doverhag et al., 2008], rather than decreasing injury, as seen in adult stroke. More severe HI injury parallels reduced NADPH oxidase activity and enhanced local neuroinflammation.
NO generation can have opposing roles in the process of ischemic injury, depending on the NOS isoform and cell type. iNOS inhibition is neuroprotective in neonatal HI models [Peeters-Scholte et al., 2002; Dingman et al., 2006], but inhibition is not necessarily associated with reduced proliferation of microglial cells.
Intracellular Inflammatory Signaling Pathways
The transcription factor nuclear factor-κβ (NF-κβ) is involved in the regulation of inflammation and neuronal death after stroke. It is normally located in the cytoplasm as a heterodimer composed of p65 and p50 subunits, bound to the endogenous inhibitor protein Iκβ (Nfκβ subunit). Dissociation of this complex by phosphorylation of Iκβ allows it to translocate to the nucleus, bind to functional κβ sites, and induce an array of genes involved in inflammation. NF-κβ plays a dual role in HI [Nijboer et al., 2008a, b]. NF-κβ inhibition during the early peak of activation prevents upregulation and accumulation of p53 in the nucleus and caspase-3 activation, and results in major neuroprotection weeks after injury [Nijboer et al., 2008a], whereas prolongation of inhibition abolishes the effect on neuronal apoptosis and even exacerbates injury [Nijboer et al., 2008a]. Taken together, these results demonstrate the crucial role of timing in the effects of potentially neuroprotective strategies.
Data on the contribution of mitogen-activated protein kinases (MAPK) to neonatal ischemic injury continue to accumulate. The extracellular signal-regulated kinase (ERK1/2) activation is generally neuroprotective in brain injury, whereas MAPK p38 activation is deleterious after HI [Hee Han et al., 2002]. The contribution of p38 to injury may depend on the model used, however [Fox et al., 2005]. Targeted deletion of the brain-specific c-Jun N-terminal kinase (JNK) isoform has been shown to reduce the overall JNK activity and protect mice against HI [Pirianov et al., 2007].
Anti-Inflammatory Strategies and Neuroprotection
The progressive elucidation of the pathophysiological mechanisms by which inflammation can harm the developing brain allows the design of new neuroprotective strategies specifically targeting mediators of neuroinflammation. Different strategies have been tested in relevant animal models; some of them, especially those for which clinically relevant drugs are available [Wolfberg et al., 2007], are summarized in Table 15-1.
Table 15-1 Potential Targets and Drugs for Protection Against Neuroinflammation
| Target | Examples of Drugs with Potential Clinical Use |
|---|---|
| Inflammation | Glucocorticoids, hypothermia |
| BBB integrity | ? |
| MMP-9 | ? |
| Cox | Nonsteroidal anti-inflammatory drugs (Cox-2 inhibitors) |
| Microglia | Minocycline, melatonin, cannabinoids, hypothermia |
| Mast cells | Cromolyn, histamine receptor blockers |
| Cytokines | Etanercept (soluble TNFα receptor), IL-1 receptor antagonist, IL-10, tianeptine |
| Chemokines | Broad-spectrum chemokine inhibitors |
| ROS | N-acetyl-cysteine, melatonin, hypothermia |
| iNOS | Aminoguanidine, tilarginine acetate |
| NF-κβ | ? |
| JNK | JIP peptide |
| Mitochondria | Caspase antagonists, minocycline |
| Multiple known targets | Hypothermia, erythropoietin, topiramate |
BBB, blood–brain barrier; IL, interleukin; iNOS, inducible nitric oxide synthase; JIP,jnk interacting protein; JNK, Jun N-terminal kinase; MMP, matrix metalloproteinase; NF, nuclear factor; ROS, reactive oxygen species; TNF, tumor necrosis factor.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Adén U., Favrais G., Plaisant F., et al. Systemic inflammation sensitizes the neonatal brain to excitotoxicity through a pro-/anti-inflammatory imbalance: Key role of tnfalpha pathway and protection by etanercept. Brain Behav Immun. 2009. Epub ahead of print
Anthony D., Dempster R., Fearn S., et al. CXC chemokines generate age-related increases in neutrophil-mediated brain inflammation and blood-brain barrier breakdown. Curr Biol. 1998;8(16):923-926.
Anthony D.C., Bolton S.J., Fearn S., et al. Age-related effects of interleukin-1 beta on polymorphonuclear neutrophil-dependent increases in blood-brain barrier permeability in rats. Brain. 1997;120(Pt 3):435-444.
Arvin K.L., Han B.H., Du Y., et al. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol. 2002;52(1):54-61.
Bartha A.I., Foster-Barber A., Miller S.P., et al. Neonatal encephalopathy: association of cytokines with MR spectroscopy and outcome. Pediatr Res. 2004;56(6):960-966.
Biran V., Cochois V., Karroubi A., et al. Stroke induces histamine accumulation and mast cell degranulation in the neonatal rat brain. Brain Pathol. 2008;18(1):1-9.
Cowell R.M., Plane J.M., Silverstein F.S. Complement activation contributes to hypoxic-ischemic brain injury in neonatal rats. J Neurosci. 2003;23(28):9459-9468.
Dammann O., Kuban K.C., Leviton A. Perinatal infection, fetal inflammatory response, white matter damage, and cognitive limitations in children born preterm. Ment Retard Dev Disabil Res Rev. 2002;8(1):46-50.
Dammann O., Leviton A. Perinatal brain damage causation. Dev Neurosci. 2007;29(4–5):280-288.
Debillon T., Gras-Leguen C., Vérielle V., et al. Intrauterine infection induces programmed cell death in rabbit periventricular white matter. Pediatr Res. 2000;47(6):736-742.
Degos V., Loron G., Mantz J., et al. Neuroprotective strategies for the neonatal brain. Anesth Analg. 2008;106(6):1670-1680.
Denker S., Ji S., Lee S.Y., et al. Macrophages are Comprised of Resident Brain Microglia not Infiltrating Peripheral Monocytes Acutely after Neonatal Stroke. J Neurochem. 2007;100(4):893-904.
Dingman A., Lee S.Y., Derugin N., et al. Aminoguanidine inhibits caspase-3 and calpain activation without affecting microglial activation following neonatal transient ischemia. J Neurochem. 2006;96(5):1467-1479.
Dommergues M.A., Patkai J., Renauld J.C., et al. Proinflammatory cytokines and interleukin-9 exacerbate excitotoxic lesions of the newborn murine neopallium. Ann Neurol. 2000;47(1):54-63.
Dommergues M.A., Plaisant F., Verney C., et al. Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience. 2003;121(3):619-628.
Doverhag C., Keller M., Karlsson A., et al. Pharmacological and genetic inhibition of NADPH oxidase does not reduce brain damage in different models of perinatal brain injury in newborn mice. Neurobiol Dis. 2008;31(1):133-144.
Eklind S., Mallard C., Arvidsson P., et al. Lipopolysaccharide induces both a primary and a secondary phase of sensitization in the developing rat brain. Pediatr Res. 2005;58(1):112-116.
Engelhardt B. Development of the blood-brain barrier. Cell Tissue Res. 2003;314(1):119-129.
Faustino J., Liu B., Lee S., et al. Blockade of endogenous cytokine-induced neutrophil chemoattractant protein 1 exacerbates injury after neonatal stroke. San Diego: Stroke meeting; 2009.
Favrais G., Schwendimann L., Gressens P., et al. Cyclooxygenase-2 mediates the sensitizing effects of systemic IL-1-beta on excitotoxic brain lesions in newborn mice. Neurobiol Dis. 2007;25(3):496-505.
Fontaine R.H., Cases O., Lelièvre V., et al. IL-9/IL-9 receptor signaling selectively protects cortical neurons against developmental apoptosis. Cell Death Differ. 2008;15(10):1542-1552.
Foster-Barber A., Ferriero D.M. Neonatal encephalopathy in the term infant: neuroimaging and inflammatory cytokines. Ment Retard Dev Disabil Res Rev. 2002;8(1):20-24.
Fox C., Dingman A., Derugin N., et al. Minocycline confers early but transient protection in the immature brain following focal cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2005;25(9):1138-1149.
Fullerton H.J., Ditelberg J.S., Chen S.F., et al. Copper/zinc superoxide dismutase transgenic brain accumulates hydrogen peroxide after perinatal hypoxia ischemia. Ann Neurol. 1998;44(3):357-364.
Galasso J.M., Miller M.J., Cowell R.M., et al. Acute excitotoxic injury induces expression of monocyte chemoattractant protein-1 and its receptor, CCR2, in neonatal rat brain. Exp Neurol. 2000;165(2):295-305.
Girard S., Kadhim H., Larouche A., et al. Pro-inflammatory disequilibrium of the IL-1 beta/IL-1ra ratio in an experimental model of perinatal brain damages induced by lipopolysaccharide and hypoxia-ischemia. Cytokine. 2008;43(1):54-62.
Grether J.K., Nelson K.B. Maternal infection and cerebral palsy in infants of normal birth weight. JAMA. 1997;278(3):207-211.
Hagberg H., Gilland E., Bona E., et al. Enhanced expression of interleukin (IL)-1 and IL-6 messenger RNA and bioactive protein after hypoxia-ischemia in neonatal rats. Pediatr Res. 1996;40(4):603-609.
Hagberg H., Mallard C. Effect of inflammation on central nervous system development and vulnerability. Curr Opin Neurol. 2005;18(2):117-123.
Hallenbeck J.M. The many faces of tumor necrosis factor in stroke. Nat Med. 2002;8(12):1363-1368.
Hedtjärn M., Leverin A.L., Eriksson K., et al. Interleukin-18 involvement in hypoxic-ischemic brain injury. J Neurosci. 2002;22(14):5910-5919.
Hedtjärn M., Mallard C., Iwakura Y., et al. Combined deficiency of IL-1beta18, but not IL-1alphabeta, reduces susceptibility to hypoxia-ischemia in the immature brain. Dev Neurosci. 2005;27(2–4):143-148.
Hee Han B., Choi J., Holtzman D.M. Evidence that p38 mitogen-activated protein kinase contributes to neonatal hypoxic-ischemic brain injury. Dev Neurosci. 2002;24(5):405-410.
Ivacko J.A., Sun R., Silverstein F.S. Hypoxic-ischemic brain injury induces an acute microglial reaction in perinatal rats. Pediatr Res. 1996;39(1):39-47.
Jin Y., Silverman A.J., Vannucci S.J. Mast cell stabilization limits hypoxic-ischemic brain damage in the immature rat. Dev Neurosci. 2007;29(4–5):373-384.
Kirton A., deVeber G. Advances in perinatal ischemic stroke. Pediatr Neurol. 2009;40(3):205-214.
Kniesel U., Risau W., Wolburg H. Development of blood-brain barrier tight junctions in the rat cortex. Brain Res Dev Brain Res. 1996;96(1–2):229-240.
Leonardo C.C., Eakin A.K., Ajmo J.M., et al. Delayed administration of a matrix metalloproteinase inhibitor limits progressive brain injury after hypoxia-ischemia in the neonatal rat. J Neuroinflammation. 2008;11:5-34.
Malaeb S., Dammann O. Fetal inflammatory response and brain injury in the preterm newborn. J Child Neurol. 2009;24(9):119-126.
McRae A., Gilland E., Bona E., et al. Microglia activation after neonatal hypoxic-ischemia. Brain Res Dev Brain Res. 1995;84(2):245-252.
Mesples B., Plaisant F., Gressens P. Effects of interleukin-10 on neonatal excitotoxic brain lesions in mice. Brain Res Dev Brain Res. 2003;141(1–2):25-32.
Mesplès B., Fontaine R.H., Lelièvre V., et al. IL-9/IL-9 receptor signaling selectively protects cortical neurons against developmental apoptosis. Neurobiol Dis. 2005;18:193-205.
Mesplès B., Plaisant F., Fontaine R.H., et al. Pathophysiology of neonatal brain lesions: lessons from animal models of excitotoxicity. Acta Paediatr. 2005;94(2):185-190.
Miller R.J., Tran P.B. Chemokinetics. Neuron. 2005;47(5):621-623.
Nelson K.B., Chang T. Is cerebral palsy preventable? Curr Opin Neurol. 2008;21(2):129-135.
Nijboer C.H., Heijnen C.J., Groenendaal F., et al. Strong neuroprotection by inhibition of NF-kappab after neonatal hypoxia-ischemia involves apoptotic mechanisms but is independent of cytokines. Stroke. 2008;39(7):2129-2137.
Nijboer C.H., Heijnen C.J., Groenendaal F., et al. A dual role of the NF-kappab pathway in neonatal hypoxic-ischemic brain damage. Stroke. 2008;39(9):2578-2586.
Normann E., Lacaze-Masmonteil T., Eaton F., et al. A novel mouse model of Ureaplasma-induced perinatal inflammation: effects on lung and brain injury. Pediatr Res. 2009;65(4):430-436.
Patkai J., Mesples B., Dommergues M.A., et al. Deleterious effects of IL-9-activated mast cells and neuroprotection by antihistamine drugs in the developing mouse brain. Pediatr Res. 2001;50(2):222-230.
Peeters-Scholte C., Koster J., Veldhuis W., et al. Neuroprotection by selective nitric oxide synthase inhibition at 24 hours after perinatal hypoxia-ischemia. Stroke. 2002;33(9):2304-2310.
Pirianov G., Brywe K.G., Mallard C., et al. Deletion of the c-Jun N-terminal kinase 3 gene protects neonatal mice against cerebral hypoxic-ischaemic injury. J Cereb Blood Flow Metab. 2007;27(5):1022-1032.
Rousset C.I., Chalon S., Cantagrel S., et al. Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr Res. 2006;59(3):428-433.
Sheldon R.A., Christen S., Ferriero D.M. Genetic and pharmacologic manipulation of oxidative stress after neonatal hypoxia-ischemia. Int J Dev Neurosci. 2008;26(1):87-92.
Sheldon R.A., Jiang X., Francisco C., et al. Manipulation of antioxidant pathways in neonatal murine brain. Pediatr Res. 2004;56(4):656-662.
Svedin P., Hagberg H., Savman K., et al. Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia-ischemia. J Neurosci. 2007;27(7):1511-1518.
Ten V.S., Sosunov S.A., Mazer S.P., et al. C1q-deficiency is neuroprotective against hypoxic-ischemic brain injury in neonatal mice. Stroke. 2005;36(10):2244-2250.
Tikka T., Fiebich B.L., Goldsteins G., et al. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21(8):2580-2588.
van den Tweel E.R., van Bel F., Kavelaars A., et al. Long-term neuroprotection with 2-iminobiotin, an inhibitor of neuronal and inducible nitric oxide synthase, after cerebral hypoxia-ischemia in neonatal rats. J Cereb Blood Flow Metab. 2005;25(1):67-74.
Volpe J.J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50(5):553-562.
Volpe J.J. The encephalopathy of prematurity–brain injury and impaired brain development inextricably intertwined. Semin Pediatr Neurol. 2009;16(4):167-178.
Wolfberg A.J., Dammann O., Gressens P. Anti-inflammatory and immunomodulatory strategies to protect the perinatal brain. Semin Fetal Neonatal Med. 2007;12(4):296-302.
Xu H., Barks J.D., Schielke G.P., et al. Attenuation of hypoxia-ischemia-induced monocyte chemoattractant protein-1 expression in brain of neonatal mice deficient in interleukin-1 converting enzyme. Brain Res Mol Brain Res. 2001;90(1):57-67.