Chapter 42 Channelopathies
Epilepsy Syndromes
Dravet’s Syndrome (Severe Myoclonic Epilepsy of Infancy, Severe Myoclonic Epilepsy of Infancy – Borderline)
Clinical Features
Dravet’s syndrome was described first by Charlotte Dravet in 1978 and then in the publication Advances in Epileptology in 1982 [Dravet et al., 1982]. Dravet described a group of children using the name “severe myoclonic epilepsy of infancy” (SMEI). Over time, it became recognized that some children have an incomplete form of the disease, leading to the terminology of “severe myoclonic epilepsy of infancy – borderline.”
Development is universally normal in the first year of life. As seizure types become more varied and more frequent, there is often developmental regression or cessation of developmental progress. Severe mental retardation is present in many children with Dravet’s syndrome, but the degree of cognitive impairment is associated with seizure control in many patients [Wolff et al., 2006]. Behavioral issues seem to become more of a concern after age 2. Hyperactivity and autistic traits can be present and very prominent. As children enter into adolescence, hyperkinetic behavior tends to improve and is replaced with overall slowed behavior. Ataxia may also become prominent.
Not all children with Dravet’s syndrome present with what are now considered the classical features, as described above. Some children may not have all of the varied seizure types and have little developmental regression. Myoclonic seizures need not be present for a diagnosis to be made. These seemingly less affected children continue to be exquisitely sensitive to seizure exacerbation due to elevated body temperature, as well as to anticonvulsants that are sodium channel blockers (e.g., carbamazepine, phenytoin, fosphenytoin, oxcarbazepine, lamotrigine, and zonisamide). In some cases these features may suggest the diagnosis. Recently, this syndrome was recognized in a large percentage of children (11 of 14) presenting with seizures and encephalopathy after receiving vaccines (vaccine encephalopathy) [Berkovic et al., 2006]. Logically, it would seem that, for many children, their first fever likely occurs with the first or second set of immunizations. A child was reported to have “hemiconvulsion-hemiplegia syndrome” after a prolonged episode of hemiconvulsion, and subsequently was identified to have the genetic mutation associated with Dravet’s syndrome [Sakakibara et al., 2009].
Genetics/Pathophysiology
Mutations in a sodium channel, SCN1A, were initially identified in 7 of 7 children with severe myoclonic infantile epilepsy [Claes et al., 2001]. Approximately 80 percent of children with a clinical diagnosis of Dravet’s syndrome have a mutation in this gene. This channel was initially implicated in generalized epilepsy with febrile seizures plus (GEFS+; see below). The sensitivity to body temperature in both of these syndromes led investigators to evaluate for mutations in the SMEI population. The majority of the children with Dravet’s syndrome have a de novo mutation, although some of the families have a higher than expected history of febrile seizures. The phenotype of patients can be predicted by mutation in most cases, as the majority of patients have a truncation mutation or a mutation that affects the function of the channel pore. Patients with less severe phenotypes often have point mutations that do not result in as severe an effect on the function of the sodium channel, although the correlation of specific SCN1A mutation to phenotype is not a tight one.
Recent discoveries related to the cell type-specific localization of SCN1A added to our understanding of how loss of function of a sodium channel, logically a cause of hypoexcitablity of individual neurons, could lead to network hyperexcitabilty and, consequently, seizures. This seeming contradiction can be explained by the finding that the loss of SCN1A function leads to selective loss of sodium channel function in inhibitory interneurons [Yu et al., 2006], causing inhibitory dysfunction and secondary hyperexcitability.
Clinical Laboratory Tests
Magnetic resonance imaging (MRI) in patients with Dravet’s syndrome is usually without any focal abnormalities. In one study that evaluated 58 children with Dravet’s syndrome, 60 percent had SCN1a mutations and 22 percent had abnormal MRI findings, the majority with cortical atrophy and others with cerebellar atrophy, white matter hyperintensity, mesial temporal sclerosis, and focal cortical dysplasia. Abnormal findings were more likely in patients without a genetic mutation [Striano et al., 2007]. Other studies have suggested that mild, diffuse atrophy and ventriculomegaly may develop over time.
Treatment
Seizure control is the primary treatment goal in this disorder. Medications that are known to block the sodium channel often will exacerbate seizures and should be avoided [Guerrini et al., 1998]. Prior to clinical diagnosis, a worsening of seizures while being treated with one of these medications should raise suspicion of Dravet’s syndrome. Topiramate, valproic acid, benzodiazepines, and levetiracetam have been helpful. Nonpharmacologic treatments, such as vagal nerve stimulation or the ketogenic diet [Caraballo and Fejeman, 2006], have been useful in some patients. Combination therapy with stiripentol, clobazam, and either depakote or topiramate has been reported to be more effective than other combinations of medication. In an initial report by Chiron et al., 15 of 21 patients responded to stiripentol [Chiron et al., 2000]. Acetazolamide has not been shown to be beneficial. A recent report with the calcium channel blocker, verapamil, has suggested that this may be helpful, but more research is required [Iannetti et al., 2009].
Avoidance of hot temperatures, both environmental and elevated body temperature, has been used by many families to reduce seizures. Antipyretics, such as acetaminophen, have been helpful, although there is a recent report of four children with transient liver abnormalities that may be associated with use of this medication [Nicolai et al., 2008]. Helmets may be indicated in some patients. Due to the severity of the cognitive impairment, appropriate support must be initiated for the family [Nolan et al., 2008]. Medications for behavioral issues may also be necessary (see Chapter 49).
Generalized Epilepsy with Febrile Seizures Plus (GEFS+)
Clinical Features
This is a familial epilepsy syndrome characterized by febrile seizures in childhood in several generations of family members, often with continuation of febrile seizures into adulthood. Some seizures related to fever may be prolonged. Some family members may also have generalized epilepsies, such as absence epilepsy, myoclonic astatic epilepsy, or, rarely, Dravet’s syndrome. Seizure types include generalized tonic clonic, myoclonic, absence, and atonic seizures. There is variable penetrance of seizures in these familial cohorts. Phenotype also varies among family members. Many family members may have resolution of seizures by age 12. The majority of these patients have normal development and intelligence. There has also been a report of temporal lobe epilepsy with mesial temporal sclerosis associated with the SCN1A mutation [Mantegazza et al., 2005].
Genetics/Pathophysiology
SCN1B was a mutation first reported in a large family with this syndrome [Wallace et al., 1998]. Mutations in other sodium channels – SCN1A [Escayg et al., 2000a] and SCN2A [Sugawara et al., 2001] – have been found subsequently. The majority of these mutations have been point mutations. In patients with SCN1A mutations, a difference in phenotype from GEFS+ and Dravet’s syndrome can often be predicted, given the location of the mutation (distance from the pore), as well as alteration in transcription of the gene. Nonsense and truncation mutations are more likely to be associated with Dravet’s syndrome. Sodium channel mutations do not account for all of the mutations in GEFS+; there also have been reports of mutations identified in GABAA receptor subunit genes,GABRG2 and GABRD (gamma 2 and delta subunits) [Harkin et al., 2002]. GABAA receptors are ligand-gated chloride channels that provide the majority of inhibition in brain beyond the neonatal period, and mutations resulting in GABAA receptor dysfunction result in increased central nervous system excitability that has been associated with a number of genetic epilepsies.
Benign Familial Neonatal Seizures
Clinical Features
Benign familial neonatal seizures are an autosomal-dominant epilepsy presenting with seizures in the first or second week of life, most commonly starting on day of life 2 or 3, resolving within weeks to months. Most seizures have stopped at 4–5 months of life. Seizures are usually multifocal clonic seizures or focal seizures. The feature suggesting this entity is the presence of similar seizures in parents and first-degree relatives, occurring at the same age. Development is characteristically normal during this time period, as well as after seizures stop. Fifteen percent of children will develop epilepsy later in life, usually in childhood or as a young adult. There are some children who progress to medically refractory epilepsy with encephalopathy [Steinlein et al., 2007].
Genetics/Pathophysiology
Mutations in potassium channels KCNQ2 [Singh et al., 1998; Biervert et al., 1998] and KCNQ3 [Charlier et al., 1998] (found on chromosomes 20 and 8 respectively) have been reported in families with benign neonatal seizures. These mutations also have been reported in some families with benign rolandic epilepsy [Hahn and Neubauer, 2009]. Recently, mutations in these genes also have been reported in a small percentage of patients with idiopathic generalized epilepsy, suggesting it may play some role in the etiology of these epilepsies. Mutations can cause alteration in function or complete loss of function of the potassium channel. Approximately 50 percent of mutations lead to shortening of expressed protein [Heron et al., 2007]. The age specificity of the seizures in this disorder is thought to emanate from brain developmental changes during the neonatal period. GABA, which acts as an inhibitory neurotransmitter later in life, can be excitatory in the early neonatal period due to developmental changes in the chloride gradient that result in opening of GABAA receptor chloride channels, producing membrane depolarization in early development rather than membrane hyperpolarization, as it does in mature neurons. In contrast, opening of potassium channels is hyperpolarizing throughout development, and due to the paucity of GABAergic inhibition, potassium channel-mediated inhibition is uniquely critical in the newborn. This may explain why impairment or absence of potassium channel inhibition results in seizures specifically at this time and why only a small fraction of patients with KCNQ2/3 mutations have seizures later in life.
Developmental Delay, Epilepsy and Neonatal Diabetes (DEND)
This is a rare syndrome, presenting with neonatal diabetes, developmental delay, seizures and mild dysmorphic features, which has been associated with a mutation in the KCNJ11 gene that encodes for a subunit of the adenosine triphosphate (ATP)-sensitive potassium channel. This channel is found on pancreatic islet cells, as well as neurons, and neonates with this disorder usually present with diabetes and subsequently develop seizures and global developmental delay [Gloyn et al., 2004]. Dysmorphic features, including downturned mouth, bilateral ptosis, prominent metopic suture, and contractures, have also been described [Gloyn et al., 2004]. There have been reports of infantile spasms in some of these children [Bahi-Buisson et al., 2007], as well as others with tonic-clonic and myoclonic seizures. Seizures have been very refractory to traditional antiepileptic medications. In contrast, patients are very responsive to treatment with sulfonylurea medications such as glibenclamide, leading to improvement in diabetes as well as developmental outcomes and seizures.
Other “Idiopathic” Epilepsies
Autosomal-Dominant Nocturnal Frontal Lobe Epilepsy
Autosomal-dominant nocturnal frontal lobe epilepsy is a familial epilepsy characterized by frontal lobe seizures that typically occur at night and usually present as arousal from sleep with bizarre hypermotor behaviors, such as spinning, thrashing and rocking. Seizures can occur several times per night. Nicotinic receptor mutations have been found in many of these familial cohorts [Steinlein et al., 1995], although there are several families for which no gene mutation has been identified. These ligand-gated receptors allow sodium and potassium to cross the cell membrane. Many patients are responsive to carbamazepine and phenytoin.
Benign Familial Infantile–Neonatal Seizures
Benign familial infantile–neonatal seizures is an epilepsy syndrome that has been described as being similar to benign neonatal seizures but occurs at a slightly older age. Mutations have been found in a sodium channel, SCN2A1, in some cohorts [Herlenius et al., 2007].
Childhood Absence Epilepsy
Childhood absence epilepsy has been linked to mutations in GABA receptors (GABRA1 and GABRG2) [Baulac et al., 2001; Wallace et al., 2001] and chloride channels (CLCN2). Mutations have also been described in a calcium channel, CACNA1H, but this mutation may represent an ethnic variant present in Chinese Han patients, as these findings were not present in a large European cohort [Chen et al., 2003]. The families with CLCN2 also had members with generalized tonic-clonic seizures on awakening and juvenile myoclonic epilepsy [Baykan et al., 2004].
Juvenile Myoclonic Epilepsy
Juvenile myoclonic epilepsy is a seizure disorder that usually presents in adolescents with myoclonic seizures that are more likely to occur in the early morning after awakening, as well as generalized tonic-clonic seizures that also tend to occur in the morning hours. Several gene mutations have been found in these patients, although the majority of patients have yet to have an underlying etiology determined. It appears that, similar to childhood absence epilepsy, this is likely a polygenic disorder. Channels that have been identified include GABA receptors (GABRA1 and GABRD) [Cossette et al., 2002], calcium channels (CACNB4) [Escayg et al., 2000b], and chloride channels (CLCN2) [Baykan et al., 2004]. In addition, a gene that is not a direct channel gene but enhances calcium influx into the cell and can stimulate programmed cell death (EFHC1) [Suzuki et al., 2004] has also been identified as being involved in this epilepsy syndrome.
Familial Pain Syndromes
Clinical Features
Inherited erythromelalgia, primary erythermalgia
Inherited erythromelalgia (IEM), or primary erythermalgia, is a pain syndrome characterized by episodes of redness and swelling of the hands and feet, associated with burning pain. These episodes can be triggered by mild warmth or exercise. Many patients prefer to avoid or are unable to wear socks and shoes due to heat inducing an episode. Some patients also report involvement of the ears, nose, and other parts of their face, as well as the upper legs. Erythema can become constant, and edema may be associated [Drenth and Waxman, 2007]. Families have reported symptoms starting in the first year of life. Age of onset can vary from childhood to adulthood, and can be familial or sporadic [Drenth et al., 2008; Han et al., 2009]. About 15 percent of cases are familial, and in these cases onset is often in the first decade of life.
Paroxysmal extreme pain disorder
Paroxysmal extreme pain disorder (PEPD), formerly called familial rectal pain, is characterized by severe pain, which most commonly occurs in the perirectal region but can also involve genitals, limbs, and face, especially the periorbital region. Pain is associated with flushing. Stimulation of the region by bowel movements, contact in the perianal region, eating, or sudden changes in temperature can induce pain episodes. Areas of pain and redness can spread to other parts of the body, including orbits and face. Cardiac abnormalities, occurring at the time of the episode, have been reported in some of these cases. In addition, harlequin skin changes and pupillary abnormalities have been seen. Tonic episodes that are nonepileptic in nature can occur with the pain episodes and are secondary to the intense severity of the pain. Episodes can last seconds to minutes, and rarely 1–2 hours. Onset is usually sudden and very commonly the paroxyms are provoked. Limb attacks can be associated with weakness lasting up to 24 hours after the pain has resolved. Constipation is a common problem due to the episodes being induced by passing stool. Symptoms have been reported as early as at the time of delivery and have been suspected to occur in utero [Fertleman et al., 2007].
Genetics/Pathophysiology
Mutations in SCN9A, a sodium channel, have been associated with IEM and PEPD. Mutations are thought to lead to hyperexcitability of the sodium channel. Mutations in IEM allow the channel to be activated by smaller than normal depolarizations and the channel remains open longer, once activated. Familial cohorts with IEM have demonstrated mutations in this channel [Drenth et al., 2005], although a study looking at a more heterogeneous population with IEM only found 1 of 15 patients with a mutation [Drenth et al., 2005], suggesting that other factors might also be involved in this disease. Mutations in PEPD lead to prolonged action potentials and repetitive neuronal firing when stimulated [Jarecki et al., 2008]. Mutations in this same channel that lead to loss of function are associated with congenital indifference to pain [Goldberg et al., 2007; Nilsen et al., 2009]. It remains unclear why different mutations lead to different phenotypes and different pain syndromes.
Treatment
Treatment for IEM, including use of sodium channel blockers, has not been very effective, although there have been reports of some relief with lidocaine, mexiletine [Choi et al., 2009], and carbamazepine [Fischer et al., 2009]. Response to medications may vary with different mutations. Carbamazepine has been helpful in treating PEPD, but topiramate and gabapentin have not.
Migraine and Ataxia Syndromes
Familial Hemiplegic Migraines
Clinical Features
Familial hemiplegic migraine often presents in the first or second decade of life with severe headache, often unilateral, and is associated with unilateral weakness that can last up to 24 hours, or rarely several days. Coma has been reported in a small number of patients. Some patients can have a less impressive clinical presentation with unilateral paresthesias and hemianopsia. Rarely, seizures can occur during hemiplegia. In some patients, ataxia and dysarthria can be seen between attacks, or for a short duration while recovering from an attack. Family members also have been reported with benign paroxysmal torticollis of infancy [Giffin et al., 2002], although this is rare and it is unclear whether this is or is not related to the gene mutation.
Headaches can at times have features of basilar migraine, including vertigo, visual symptoms, tinnitus, dysarthria, and ataxia. Some patients have been reported to have progressive ataxia later in life [Terwindt et al., 1998]. In addition, cognitive impairment has been noted in affected patients [Marchioni et al., 1995].
Genetics/Pathophysiology
Many patients with familial hemiplegic migraine have a calcium channel mutation (CACNA1A) [Stam et al., 2008]; if this is present, patients seem to be more likely to have ataxia and coma, and to be more prone to delayed cerebral edema after minor head injury [Terwindt et al., 1998; Kors et al., 2001; Stam et al., 2009]. Mutations in CACNA1A also have been reported in patients with alternating hemiplegia of childhood, which phenotypically has some overlap with hemiplegic migraine [de Vries et al., 2008], as well as in patients with episodic ataxia type 2 (see below) and spinocerebellar ataxia type 6. Mutations in SCN1A and in ATP1A2 also have been found in some patients with this clinical syndrome [De Fusco et al., 2003; Dichgans et al., 2005a]. Interestingly, febrile seizures also have been reported in patients with mutations in either SCN1A or ATP1A2 [de Vries et al., 2009; De Fusco et al., 2003].
Clinical Laboratory Tests
MRI findings are not pathognomonic, but cerebellar atrophy [Terwindt et al., 1998], particularly in the superior cerebellar vermis, has occasionally been reported. MR spectroscopy of this region demonstrated metabolic abnormalities consistent with neuron loss [Dichgans et al., 2005b]. EEG during events can demonstrate slowing in the affected hemisphere, and mild asymmetries with minimal unilateral slowing have been noted on EEGs performed between episodes [Marchioni et al., 1995].
Episodic Ataxia
Clinical Features
This disorder is characterized by intermittent periods of ataxia, (also see Chapter 67). There are two commonly described disorders: episodic ataxia type 1 and type 2. They differ slightly from one another in clinical presentation, allowing clinical separation.
Genetics/Pathophysiology
Both disorders are inherited in an autosomal-dominant fashion, although penetrance is not always complete. Ion channel abnormalities also have been found in both disorders. Type 1 has been associated with point mutations in KCNA1, located on chromosome 12 [Browne et al., 1994]. This is a potassium channel that has no intervening introns. Episodic ataxia type 2 has been linked to mutations in CACNA1A (a calcium channel). Mutations that interfere with splicing or lead to a premature stop have been linked to the episodic ataxia type 2 phenotype [van den Maagdenberg et al., 2002]. This appears to lead to loss of function of the calcium channel. Familial hemiplegic migraine (see above) and spinocerebellar ataxia type 6 (see below) also have been reported to have mutations in this gene.
Spinocerebellar Ataxia
Clinical Features
Several progressive ataxias have been described and reported as due to a variety of etiologies (see Chapter 67). Channelopathies are responsible for one subtype, now called spinocerebellar ataxia type 6. This presents as a slowly progressive cerebellar degeneration with ataxia, dysmetria, and other cerebellar signs as a prominent clinical feature. Spasticity and cranial neuropathies are not prominent. There can be some overlap with episodic ataxia type 2, with episodes of truncal ataxia lasting for several hours to days and often precipitated by stress or exertion. There also may be some features associated with basilar migraine or familial hemiplegic migraine.
Genetics/Pathophysiology
Spinocerebellar ataxia type 6 has been reported to be associated with triplet repeats in the CACNA1A gene [Zhuchenko et al., 1997]. Unlike the gene changes associated with other triplet repeat disorders, this one seems relatively stable and is a smaller expansion than that typically seen in association with an abnormal phenotype. It is unclear if symptoms are related to channel dysfunction or the cytotoxic effects of the repeat, as is seen in other diseases. The overlap between these phenotypes suggests that there is a pathological role in the abnormal function of the calcium channel. Mutations in this gene also have been reported in cohorts with familial hemiplegic migraine – which usually have point mutations. Cohorts with episodic ataxia type 2 also have been reported to have mutations in this gene that often lead to splicing errors or premature stops.
Muscle Channelopathies
Contraction in skeletal muscle occurs by generating and propagating action potentials. The release of intracellular calcium stores triggers mechanical contraction. This process is dependent on proper functioning of ion channels. Mutations in muscle ion channel genes have been identified, which cause a variety of diseases collectively called muscle channelopathies. Voltage-gated ion channels share similar structural features. The ion-conducting pores are selective for a specific ion. Mutations in muscle channel genes cause changes in channel function that alter membrane excitability and cause neuromuscular symptoms. Understanding the pathophysiological mechanisms involved in these diseases is an on-going focus of many studies. Clinical symptoms and their underlying neurophysiology are being linked with abnormal muscle membrane function. The overall incidence of muscle channelopathies is estimated at 1 in 100,000 [Meola et al., 2009].
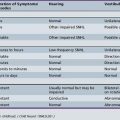
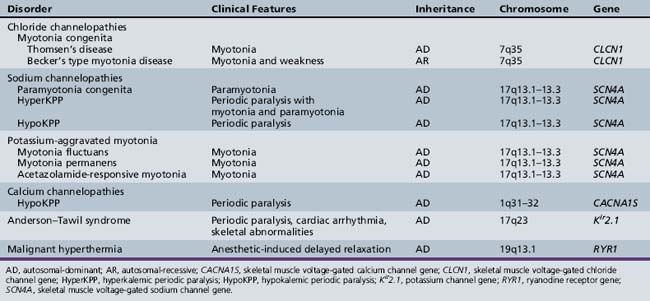
Skeletal muscle disorders associated with ion channelopathies produce symptoms of myotonia, weakness, or both [Hudson et al., 1995]. Table 42-1 summarizes the various disorders (see also Chapter 96).
Myotonia Congenita
Clinical Features
Patients with Becker’s or autosomal-recessive myotonia congenita may have transient muscle weakness on initiation of movement after a period of rest or quiescence. Muscle strength normalizes after several muscle contractions. Both forms of myotonia congenita are more severe in men than women [Platt and Griggs, 2009].
Genetics
The point mutation for the skeletal muscle chloride channel is located on chromosome 7q35 [Koch et al., 1992]. Both recessive and dominant forms are due to mutations in the voltage-gated chloride channel gene (CLCN1).
Pathophysiology
In normal skeletal muscle, a nerve stimulus causes depolarization of the sarcolemma, propagating an action potential that results in muscle contraction, followed by relaxation. Increased excitability of the muscle fibers in myotonia results in a single nerve stimulus causing repetitive action potentials. Skeletal muscle has a high chloride conductance, which accounts for up to 85 percent of resting membrane conductance [Bryant and Morales-Aguilera, 1971]. In myotonia congenita, reduced sarcolemmal chloride conductance causes enhanced excitability of the muscle cells, resulting in myotonic discharges [Lipicky et al., 1971].
Sodium Channel Disorders
Paramyotonia Congenita (Eulenberg’s Disease)
Paramyotonia congenita is a syndrome consisting of episodic paralysis and myotonia that is triggered by cold and exercise. As opposed to the warm-up phenomenon seen in patients with myotonia congenita, paradoxical myotonia that quickly worsens with exercise can be demonstrated. Facial muscles (particularly eyelids), pharyngeal muscles, and hand muscles are particularly involved. The legs are more mildly affected [Miller et al., 2004]. Myotonia may be present for a brief time, but weakness may persist for hours. Muscle hypertrophy is present in about 30 percent of patients [Matthews et al., 2008]. Paramyotonia congenita is present from infancy and symptoms manifest within the first decade of life.
Genetics
Paramyotonia congenita is caused by a mutation of the SCN4A gene, with exons 22 and 24 being active areas of interest [Matthews et al., 2008].
Pathophysiology
Episodes of weakness experienced by individuals with paramyotonia congenita are caused by intermittent loss of fiber excitability. This is due to persistent depolarization of the membrane resting potential, as sodium channels do not inactivate completely, resulting in a persistent influx of sodium current [Cannon, 2006].
Clinical laboratory tests
Muscle biopsy is rarely necessary, but shows an absence of type 2B fibers. Clinical electrophysiology may be useful as the compound muscle action potential (CMAP) will show a gradual and prolonged decrement after exercise. With repeated exercise testing and muscle cooling, the decrement is exacerbated [Fournier et al., 2006].
Hyperkalemic Periodic Paralysis
Hyperkalemic periodic paralysis shares features with paramyotonia congenita. The disorder can occur at any age, but may appear in infancy. The frequency of attacks increases during adolescence, and decreases after the fourth decade [Miller et al., 2004]. Mild myotonia of the eyelids and finger extensors may be noted on examination between attacks. Myotonia may be cold-induced but is not exacerbated by exercise [Subramony et al., 1986], which distinguishes it from paramyotonia congenita. Moderate exercise may trigger attacks of flaccid muscle weakness, which may be brief or last for several hours. Attacks also may be precipitated or made worse by eating foods containing high potassium, exposure to cold, rest after moderate exercise, glucocorticoids, or emotional stress [Lehmann-Horn et al., 2004]. Permanent weakness may develop in older individuals [Bradley et al., 1990]. Before confirming a diagnosis of hyperkalemic periodic paralysis, other causes of potassium elevation should be excluded. Adrenal insufficiency, rhabdomyolysis, hypoaldosteronism, or medications such as angiotensin-converting enzyme inhibitors should be ruled out.
Genetics
Between 60 and 70 percent of patients have a mutation in the SCN4A gene on chromosome 17, which encodes skeletal muscle voltage-gated sodium channels [Fontaine et al., 1990]. Genetic testing should focus on the common sites of mutations (T704M and M1592V) in the SCN4A gene. It has also been confirmed that hyperkalemic periodic paralysis and paramyotonia congenita are allelic disorders [Ptacek et al., 1991]. These are inherited in autosomal-dominant fashion.
Pathophysiology
Abnormal inactivation of voltage-gated sodium channels causes membrane depolarization. The abnormal sodium channel gating creates a gain of function, resulting in increased sodium current that depolarizes affected muscle and leads to weakness [Venance et al., 2006].
Clinical Laboratory Tests
Serum potassium elevation by 1.5–3 mmol/L during an attack and electrocardiogram changes (peaked T waves) confirm the diagnosis. Myotonia is detectable on EMG in approximately 50 percent of patients [Lehmann-Horn et al., 2004] and supports the diagnosis. Muscle biopsy in patients with periodic paralysis may exhibit vacuoles and tubular aggregates.
Potassium-Aggravated Myotonias
There are several other myotonic disorders, all associated with allelic point mutations in the gene encoding SCN4A. All are characterized by stiffness following ingestion of potassium or strenuous exercise. All share the absence of sensitivity to cold. Myotonia fluctuans is a mild form of myotonia precipitated by rest and resolved with continued exercise. The onset of myotonia is delayed for 10 or more minutes after exercise. Pain sometimes accompanies the myotonia, but muscle weakness is not a feature [Heine et al., 1993]. Acetazolamide-responsive myotonia (atypical myotonia congenita) is a painful myotonia precipitated by exercise and is insensitive to cold [Ptacek et al., 1994a]. Myotonia permanens is a very severe disorder, with EMG showing continuous myotonic activity. As a result, generalized muscle hypertrophy is noted, as well as serum creatine kinase elevation.
Calcium Channel Disorders
Genetics
Mutations in the CACNA1S gene account for about 70 percent of cases of hypokalemic periodic paralysis [Ptacek et al., 1994b]. CACNA1S codes for the dihydropyridine receptor. The gene maps to chromosome 1q31–32. About 12 percent of patients have a mutation in the SCN4A gene [Jurkat-Rott et al., 2000]. There may be further gene heterogeneity to account for other patients who do not test positive for either of these mutations.
Pathophysiology
In hypokalemic periodic paralysis, the mechanism for depolarization-induced attacks of weakness is not well characterized. A loss of function leading to reduced current density and slower activation is a result of calcium channel mutations. There are also sodium channel mutations that enhance inactivation, producing a net loss of function defect as well [Venance et al., 2006].
Andersen–Tawil Syndrome
Andersen–Tawil syndrome is characterized by periodic paralysis, cardiac ventricular ectopy, and skeletal abnormalities [Tawil et al., 1994]. It was first described in 1971 [Andersen et al., 1971]. Clinical symptoms of intermittent weakness begin in the first or second decade. Episodes can be triggered by prolonged rest or rest following exertion. Proximal weakness may develop and be permanent. Potassium may be elevated, normal, or reduced during attacks, and levels do not correlate well with the frequency or severity of weakness. ECG abnormalities include prolonged QT interval, prominent U waves, premature ventricular contractions, ventricular bigeminy, and polymorphic ventricular tachycardia. Patients with ventricular ectopy may be asymptomatic, or may present with palpitations, syncope, or cardiac arrest [Tristani-Firouzi et al., 2002]. The physical phenotype includes small mandible, hypertelorism, low-set ears, clinodactyly, syndactyly, and a broad nasal bridge [Donaldson et al., 2004]. The disorder is autosomal-dominant and maps to chromosome 17q23, which includes the KCNJ2 gene that encodes the kir2.1 potassium channel receptor.
Malignant Hyperthermia
Genetics
Normal contraction of skeletal muscle depends on interaction between the ryanodine receptor and the dihydropyridine receptor. The ryanodine receptor encodes the skeletal muscle sarcoplasmic reticulum calcium release gene. The receptor maps to chromosome 19. The dihydropyridine receptor encodes the voltage-dependent calcium channel. Mutations in the RYR1 gene account for 50–70 percent of cases of malignant hyperthermia [McCarthy et al., 2000]. Mutations also have been identified in the CACNA2D1 and CACNA1S calcium channel genes [Jurkat-Rott et al., 2002].
Pathophysiology
Hypersensitive channels caused by mutations in the RYR1 gene allow efflux of calcium from the sarcoplasmic reticulum into the muscle cell. Sustained muscle contraction and rigidity result, which also causes an elevation in body temperature. This excessive muscle contraction results in depletion of ATP and increased glycogenolysis, which produces lactic acidosis and a metabolic acidosis. Oxygen consumption is escalated and results in hypoxemia and hypercapnia with excess carbon dioxide production. Patients may develop rhabdomyolysis, hyperkalemia, and complications, including renal failure or cardiac dysrhythmia and arrest [Platt and Griggs, 2009].
Clinical Laboratory Tests
Confirmation of malignant hyperthermia is sought by contracture testing. This procedure relies on the in vitro response of freshly biopsied muscle to various concentrations of caffeine and halothane, which raise the intracellular calcium content. Test sensitivity is almost 100 percent and specificity 87–90 percent [Allen et al., 1998].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Allen G.C., Larach M.G., Kunselman A.R. The sensitivity and specificity of the caffeine-halothane contracture test: a report from the North American Malignant Hyperthermia Registry. The North American Malignant Hyperthermia Registry of MHAUS. Anesthesiology. 1998;88:579-588.
Andersen E.D., Krasilnikoff P.A., Overvad H. Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies. A new syndrome. Acta Paediatr Scand. 1971;60:559-564.
Bahi-Buisson N., Eisermann M., Nivot S., et al. Infantile spasms as an epileptic feature of DEND syndrome associated with an activating mutation in the potassium adenosine triphosphate(ATP) channel, Kir6.2. J Child Neurol. 2007;22(9):1147-1150.
Baulac S., Huberfeld G., Gourfinkel-An I., et al. First genetic evidence of GABAa receptor dysfunction in epilepsy: a mutation in the gamma-2-subunit gene. Nat Genet. 2001;28:46-48.
Baykan B., Madia F., Bebek N., et al. Autosomal recessive idiopathic epilepsy in an inbred family from Turkey: identification of a putative locus on Chromosome 9q32–33. Epilepsia. 2004;45:479-487.
Berkovic S., Harbin L., McMahon J., et al. De-novo mutations of the sodium channel geen SCN1A in alleged vaccine encephalopathy. Lancet Neurol. 2006;5:488-492.
Biervert C., Schroeder B., Kubisch C., et al. A potassium channel mutaionin neonatal human epilepsy. Science. 1998;279:403-406.
Bradley W.G., Taylor R., Rice D.R., et al. Progressive myopathy in hyperkalemic periodic paralysis. Arch Neurol. 1990;47(9):1013-1017.
Browne D.L., Gancher S.T., Nutt J.E., et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA 1. Nat Genet. 1994;8(2):136-140.
Bryant S.H., Morales-Aguilera A. Chloride conductance in normal and myotonic muscle fibres and the action of monocarboxylic aromatic acids. J Physiol. 1971;219:367-383.
Cannon S.C. Pathomechanisms in channelopathies of skeletal muscle and brain. Annu Rev Neurosci. 2006;29:387-415.
Caraballo R., Fejerman N. Dravet syndrome: a study of 53 patients. Epilepsy Res. 2006;70(Suppl 1):s231-s238.
Charlier C., Singh N., Ryan S., et al. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53-55.
Chen Y., Llu J., Pan H., et al. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol. 2003;54:239-243.
Chiron C., Marchand M., Tran A., et al. Stiripental in severe myoclonic epilepsy in infancy: a randomized placebo-controlled syndrome –dedicated trial. Lancet. 2000;356:1638-1642.
Choi J., Dib-Hajj S., Han C., et al. Mexiletine-responsive erythrolelalgia due to a new Nav1.7 mutations showing use dependent current fall-off. Exp Neurol. 2009;216(3):383-389.
Claes L., Del-Favero J., Ceulemans B., et al. De Novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy or infancy. Am J Hum Genet. 2001;68:1327-1332.
Cossette P., Liu L., Bridebois K., et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184-189.
De Fusco M., Marconi R., Silvestr L., et al. Haplo insuffiency of ATP1A2 encoding the Na/K puma a2 subunit associated with familial hemiplegic migraine type 2. Nat Genet. 2003;33:192-196.
de Vries B., Stam A., Beker F., et al. CACNA1A mutation linking hemiplegic migraine and alternating hemiplegia of childhood. Cephalgia. 2008;28:887-891.
de Vries B., Stam A., Kirkpatrick M., et al. Familial hemiplegic migraine is associated with febrile seizures in an FHM2 family with a novel de novov ATP1A2 mutation. Epilepsia. 2009;50(11):2503-2504.
Dichgans M., Freilinger T., Eckstein G., et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;336:371-377.
Dichgans M., Herzog J., Freilinger T., et al. H-MRS alterations in the cerebellum of patients with familial hemiplegic migraine type 1. Neurology. 2005;64:608-613.
Donaldson M.R., Yoon G., Fu Y.H., et al. Andersen-Tawil syndrome: a model of clinical variability, pleiotropy, and genetic heterogeneity. Ann Med. 2004;36(Suppl 1):92-97.
Dravet C., Roger J., Bureau M., et al. Myoclonic epilepsies in childhood. In: Akimoto H., Kazamamtsuri H., Seino, et al. Advances in Epileptology, XIIIth Epilepsy International Symposium. NewYork: Raven Press; 1982:135-140.
Drenth J., Morsche R., Guillet G., et al. SCN9A mutations define primary erythermalgia as a neuropath disorder of voltage gated sodium channels. J Invest Dermatol. 2005;124:1333-1338.
Drenth J., Morsche R., Manssur S., et al. Primary erythermalgia as a Sodium channelopathy. Arch Dermatol. 2008;144(3):320-324.
Drenth J., Waxman S. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest. 2007;117(12):3603-3609.
Escayg A., Macdonald B., Meisler M., et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24:343-345.
Escayg A., De Waard M., Lee D., et al. Coding and non coding variation of the human calcium-channel beta(4)-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. 2000;66:1531-1539.
Fertleman C., Ferrie C., Aicardi J., et al. Paroxysmal extreme pain disorder (previously familial rectal pain syndrome). Neurology. 2007;69:586-595.
Fischer T., Gilmore E., Estacion M., et al. A novel NAv1.7 Mutation Producing Carbamazepine-Responsive Erythromelalgia. Ann Neurol. 2009;65:733-741.
Fontaine B., Khurana T.S., Hoffman E.P., et al. Hyperkalemic periodic paralysis and the adult muscle sodium channel alpha-subunit gene. Science. 1990;250:1000-1002.
Fournier E., Viala K., Gervais H., et al. Cold extends electromyography distinction between ion channel mutations causing myotonia. Ann Neurol. 2006;60:356-365.
Giffin N., Benton S., Goadsby P. Benign paroxysmal torticollis of infancy: four new cases and a linkage to CACNA1A mutation. Dev Med Child Neurol. 2002;44:490-493.
Gloyn A., Pearson E., Antcliff J., et al. Activating mutations in the gene encoding the ATP-sensitive potassium channel subunit Kir6.2 and permanent neonatal diabetes. NEJM. 2004;350(18):1838-1849.
Goldberg Y., MacFarlane J., MacDonald M., et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet. 2007;71:311-319.
Guerrini R., Darvet C., Genton P., et al. Lamotrigine and seizures aggravation in severe myoclonic epilepsy. Epilepsia. 1998;39(5):508-512.
Hahn A., Neubauer B. Sodium and potassium channel dysfunctions in rare and common idiopathic epilepsy syndromes. Brain Dev. 2009;31(7):515-520.
Han C., Dib-Hajj S., Lin Z., et al. Early and late-onset inherited erythromelalgia: genotype-phenotype correlation. Brain. 2009;132:1711-1722.
Harkin L., Bowser D., Dibbons L., et al. Truncation of the GABA-A-receptor gamma-2-subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2002;70:530-536.
Heine R., Pika U., Lehmann-Horn F. A novel SCH4A mutation causing myotonia aggravated by cold and potassium. Hum Mol Genet. 1993;2:1349-1353.
Herlenius E., Heron S., Grinton B., et al. SCN2A mutations and benign familial neonatal infantile seizures: The phenotypic spectrum. Epilepsia. 2007;48(6):1138-1142.
Heron S.E., Cox K., Griston B.E., et al. Deletions or duplications in KCNQ2 can cause benign familial neonatal seizures. J Med Genet. 2007;44(12):791-796.
Hudson A.J., Ebers G.C., Bulman D.E. The skeletal muscle sodium and chloride channel diseases. Brain. 1995;118:547-563.
Iannetti P., Parisi P., Spalice A., et al. Addition of verapamil in the treatment of severe myoclonic epilepsy in infancy. Epilepsy Res. 2009;85:89-95.
Jarecki B., Sheets P., Jackson J., et al. Paroxysmal extreme pain disorder mutations within the D3/S4–S5 linker of Nav1.7 cause moderate destabilization of fast inactivation. J Physiol. 2008;586(17):1437-1453.
Jurkat-Rott K., Lerche H., Lehmann-Horn F. Skeletal muscle channelopathies. J Neurol. 2002;249(11):1493-1502.
Jurkat-Rott K., Mitrovic N., Hang C., et al. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci USA. 2000;97:9549-9554.
Kors E., Terwindt G., Vermeulen F., et al. Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann Neurol. 2001;49:753-760.
Koch M.C., Steinmeyer K., Lorenz C., et al. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992;257:797-800.
Lehmann-Horn F., Rudel R., Jurkat-Rott K. Non-dystrophic myotonias and periodic paralyses. In: Engel A.G., Franzini-Armstrong C., editors. Myology: Basic and Clinical. ed 3. NewYork: McGraw-Hill; 2004:1257-1300.
Links T.R., Zwarts M.J., Oosterhuis H.J.G.H. Improvement of muscle strength in familial hypokalemic periodic paralysis with acetazolamide. J Neurol Neurosurg Psychiatry. 1988;51:1142-1145.
Lipicky R.J., Bryant S.H., Salmon J.H. Cable parameters, sodium, potassium, chloride, and water content, and potassium efflux in isolated external intercostal muscle of normal volunteers and patients with myotonia congenita. J Clin Invest. 1971;50:2091-2103.
Mantegazza M., Gambardell A., Rusconni R., et al. Identification of an Nav1.1 sodium channel (SCN1A) loss-of-funtion mutation associated with familial simple febrile seizures. Proc Natl Acad Sci USA. 2005;102:18177-18182.
Marchioni E., Galimberti C., Soragna D., et al. Familial hemiplegic migraine versus migraine with prolonged aura. Neurology. 1995;45:33-37.
Matthews E., Tan S.V., Fialho D., et al. What causes paramyotonia in the United Kingdom? Common and new SCN4A mutations revealed. Neurology. 2008;70:50-53.
McCarthy T.V., Quane K.A., Lynch P.J. Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum Mutat. 2000;15:410-417.
Meola G., Hanna M.G., Fontaine B. Diagnosis and new treatment in muscle channelopathies. J Neurol Neurosurg Psychiatry. 2009;80:360-365.
Miller T.M., as da Silva M.R., Miller H.A., et al. Correlating phenotype and genotype in the periodic paralyses. Neurology. 2004;63:1647-1655.
Nicolai J., Gunning B., Leroy P., et al. Acute hepatic injury in four children with Dravet syndrome: valproic acid, topiramate or acetaminophen. Seizure. 2008;17(1):92-97.
Nilsen K., Nicholas A., Woods C., et al. Two Novel SCN9A mutations causing insensitivity to pain. Pain. 2009;143:155-158.
Nolan K., Camfield C., Camfield P. Coping with a child with Dravet syndrome: insights from the families. J Child Neurol. 2008;23(6):690-694.
Platt D., Griggs R. Skeletal Muscle Channelopathies: New insights into the periodic paralyses and nondystrophic myotonias. Curr Opin Neurol. 2009;22(5):524-531.
Ptacek L.J., Tyler F., Trimmer J.S., et al. Analysis in a large hyperkalemic periodic paralysis pedigree supports tight linkage to a sodium channel locus. Am J Hum Genet. 1991;49:378-382.
Ptacek L.J., Tawil R., Griggs R.C., et al. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell. 1994;77:863-868.
Ptacek L.J., Tawil R., Griggs R.C., et al. Sodium channel mutations in acetazolamide responsive myotonia congenita, paramyotonia congenita, and hyperkalemic periodic paralysis. Neurology. 1994;44:1500-1503.
Sakakibara T., Nakagawa E., Saito Y., et al. Hemiconvulsion-Hemiplegia syndrome in a patient with severe myoclonic epilepsy in infancy. Epilepsia. 2009;50(9):2158-2162.
Singh N., Charlier C., Stauffer D., et al. A novel potassium channel gene KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25-29.
Stam A., Luijckx G.-J., Poll-The B., et al. Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218L mutation. J Neurol Neurosurg Psychiatry. 2009;80:1125-1129.
Stam A., Vanmolkot K., Kremer H., et al. CACNA1A R1347Q: a frequent recurrent mutation in hemiplegic migraine. Clin Genet. 2008;74:481-485.
Steinlein O., Conrad C., Weidner B. Benign familial neonatal convulsions: always benign? Epilepsy Res. 2007;73:245-249.
Steinlein O., Mulley J., Propping P., et al. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11:201-203.
Striano P., Mancardi M., Biancheri R., et al. Brain MRI findings in severe myoclonic epilepsy in infancy and genotype-phenotype correlations. Epilepsia. 2007;48(6):1092-1096.
Subramony S.H., Wee A.S., Mishra S.K. Lack of cold sensitivity in hyperkalemic periodic paralysis. Muscle Nerve. 1986;9:700-703.
Sugawara T., Tsurubuchi Y., Agarwaka K., et al. A missense mutation of the Na Channel alpha II subunit gene Nav1.2 in a patient with febrile and afebrile seizure causes channel dysfunction. Proc Natl Acad Sci USA. 2001;98:6384-6389.
Suzuki T., Delgado-Escueta A., Aguan K., et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet. 2004;36:842-849.
Tawil R., Ptacek L.J., Pavlakis S.G., et al. Andersen’s syndrome: potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann Neurol. 1994;35:326-330.
Terwindt G., Ophoff R., Haan J., et al. Variable clinical expression of mutations in the P/Q type calcium channel gene in familial hemiplegic migraine. Neurology. 1998;50:1105-1110.
Tristani-Firouzi M., Jensen J.L., Donaldson M.R., et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest. 2002;110:381-388.
Venance S.L., Cannon S.C., Fialho D., et al. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. 2006;129(1):8-17.
van den Maagdenberg A., Kors E., Brunt E., et al. Episodic ataxia type 2 Three novel truncation mutations and one novel missense mutation in the CACNA1A gene. J Neurol. 2002;249:1515-1519.
Wallace R., Marini C., Petrou S., et al. Mutant GABAa receptor gamma-2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49-52.
Wallace R., Wang D., Singh R., et al. Febrile seizures and generlaized epilepsy associated with a mutation in the Na+ channel beta-1 subunit gene CN1B. Nat Genet. 1998;19:366-370.
Wolff M., Casse-Perrot C., Dravet C. Severe myoclonic epilepsy of infants (dravet syndrome):natural history and neuropsychological findings. Epilepsia. 2006;47(Suppl 2):45-48.
Yu F.H., Mantegazza M., Westenbroek R.E., et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142-1149.
Zhuchenko O., Bailey J., Bonnen P., et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutatmine expansions in the alpha(1A)-voltage-dependent calcium channel. Nat Genet. 1997;15:62-69.