Pathology, Biomarkers, and Molecular Diagnostics
Wilbur A. Franklin, Dara L. Aisner, Miriam D. Post, Paul A. Bunn and Marileila Varella Garcia
• Biomarker development has profoundly affected basic understanding of carcinogenesis and expanded means of intervention in human cancers. Biomarkers have been applied with variable success in three broad areas that correspond to phases of tumor development and progression: (1) early detection, (2) diagnosis, and (3) prediction of clinical outcome (prognosis) and response to targeted treatment.
• For early detection, biomarkers go through five stages of development: (1) identification of promising directions; (2) validation of a clinical assay; (3) demonstration that the biomarker can detect disease before it is clinically relevant; (4) evaluation of the biomarker during prospective screening; and (5) quantification of the effect of screening on reducing the burden of disease in a population.
• Early detection using quite different approaches has changed the natural history of cervical, colorectal, and lung cancer. Progress depends on improving our understanding of the biology of site-specific carcinogenesis and using intervention strategies informed by basic biology.
• Cancer diagnosis still relies on an evaluation of tumor tissue histology, a complex process associated with interindividual variation; however, new approaches to automated quantitative histologic evaluation and immunohistochemical analyses are under active development.
• New molecular diagnostic approaches that utilize assessment of quantitative messenger RNA signatures, fluorescent-in-situ hybridization, quantitation of microRNAs, and genetic sequencing can now be accomplished with accurate use of formalin-fixed paraffin-embedded tissues.
• Molecular testing can provide guidance in prioritizing therapies for patients most likely to benefit and can identify patients who have been proven not to benefit from a targeted therapy.
• Molecular testing can also provide clinicians with a rationale to guide specific patients toward specific clinical trials.
• As new molecular techniques have been applied to cancer diagnosis and treatment, the necessity of developing standardized processing approaches for solid tumor specimens has been appreciated and has helped to change tissue collection and specimen-handling routines.
Since the time of Virchow more than 150 years ago, diagnosis and treatment of cancer have depended on the appearance of tumor cells under the microscope and the diverse and often subtle morphologic differences that distinguish benign from malignant cells. Careful study of cellular features and later of molecular features of both hematologic and solid tumors has led to the idea that cancer results from a series of genetic and phenotypic changes in progenitor cells that occur over a period of many years. In epithelial tumors, the final histologic changes that define transformation of premalignant lesions into malignancy are the invasion of stromal tissues, the elicitation of a stromal response, including angiogenesis, and metastasis to lymph nodes and distant sites.1 The clinical implications of this multistep process are best illustrated in the cervix and colon, where early intervention has resulted in reduced mortality from cancer. Once invasive carcinoma has occurred, testing becomes more urgent and intervention more aggressive. This chapter reviews concepts of solid tumor carcinogenesis and biomarker development for early detection and distinguishes early detection testing from biomarker applications in diagnosis, prognosis, and treatment of established solid tumors. Hematologic biomarkers are described elsewhere in this volume. Finally, current best practices in molecular testing are discussed.
Early Detection
Remarkable strides have recently been made in reducing morbidity and mortality through early intervention in several different kinds of solid tumors. The triggers for intervention have until now been largely based on cytologic evaluation of exfoliated cells or advances in imaging technology. To date, few molecular markers have been sufficiently sensitive, specific, or accessible to be useful in population-based studies for early detection or prevention studies. Reasons for this lack of usefulness are numerous. Sensitive biomarkers for risk assessment and early detection must be inexpensive, noninvasive, and easily applied to be useful for population-based screening. However, such biomarkers are rarely sufficiently specific for diagnosis, prognosis, or response to treatment. A formal process to guide the development of early detection biomarkers has been defined by Pepe et al.2 In this construct, biomarkers must pass through five phases of development that correspond to the strength of evidence that the biomarker will be a useful population screening tool. These phases of development are analogous to the structure for therapeutic drug development3 that has been used for many years. The phases defined by Pepe and colleagues are listed in Table 17-1 and are the framework for the discussion in this chapter of biomarker development in individual organs.
Table 17-1
Phases of Biomarker Development by Pepe and Colleagues
| Early Detection Research Network | ||
| Phase | Category | Descriptor |
| 1 | Preclinical/exploratory | Promising directions identified |
| 2 | Clinical assay and validation | Clinical assay detects established disease |
| 3 | Retrospective/longitudinal | Biomarker detects disease early, before it becomes clinical, and a screen positive rule is defined |
| 4 | Prospective screening | Extent and characteristics of disease detected by the test and the false referral rate are identified |
| 5 | Cancer control | Impact of screening on reducing the burden of disease on the population is quantified |

The Cervix and a Validated Biomarker
Invasive cervical carcinoma is one of the few tumors that results from well-defined premalignant lesions that can be identified and effectively treated, reducing morbidity and mortality from a tumor that is the third most common in women worldwide.4 More than 60 years ago, it was recognized that cellular abnormalities in cervical epithelium detectable in exfoliated cells could predict the presence of carcinoma.5 By 1988, premalignant cervical cytology had been codified as the Bethesda System for reporting cervical/vaginal cytologic diagnoses.6 Population-based light microscopic screening for atypical cells in cervical smears with surgical removal of detected abnormal epithelium in Great Britain is estimated to have reduced the prevalence of invasive squamous carcinoma by 80% after 1988.7 Conventional cervical cytology is thus one of the most effective early detection biomarkers in use today.
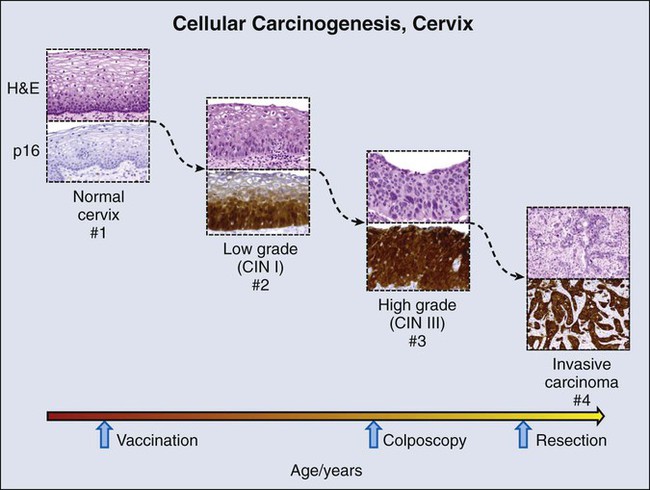
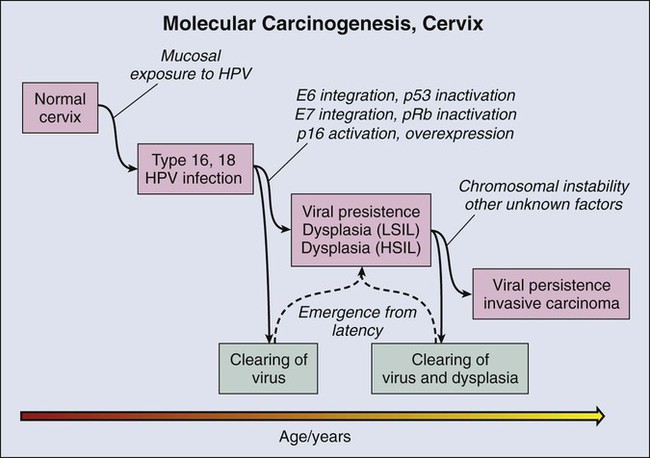
Delineation of molecular carcinogenesis in the cervix (Figs. 17-1 and 17-2) has provided alternative methods for detection, risk assessment, and diagnosis. Nearly all cervical carcinomas and their precursor lesions are caused by human papillomavirus (HPV).8 HPV is a DNA tumor virus that in 1934 was shown by Peyton Rous to produce squamous tumors in rabbits.11–11 It was not until 1974 that zur Hausen recognized the association between HPV and cervical cancer.14–14 Persistent infection with high-risk strains of HPV, most commonly type 16 and 18,15 is an essential step in cervical carcinogenesis (reviewed in reference 16).16 The virus encodes three proteins that stimulate cell proliferation and survival: E5, E6, and E7 (reviewed in reference 17).17 The viral proteins E6 and E7 interfere with p53 and Rb, respectively,18,19 resulting in resistance to apoptosis and chromosomal instability. The HPV genome may be integrated into the host cell genome, accounting for long-term persistence of the virus. Integration of E6 and E7 genes20,21 is crucial to progression of the infected cell to a malignant state, whereas other viral genes may be lost.
Detection of HPV DNA in clinical samples can be accomplished by a number of methods, with the dominant techniques based on polymerase chain reaction (PCR) or hybridization with signal amplification (reviewed in reference 22).22 In the United States, the predominant assay is the Food and Drug Administration (FDA)-approved Hybrid Capture 2 method (Qiagen, Germantown, Md.). In this technique, viral DNA hybridizes with an HPV RNA probe cocktail that detects multiple high-risk HPV types. The RNA : DNA hybrid is captured by hybrid-specific antibody that coats the surface of a microplate well. The hybrids are detected by a second hybrid-specific antibody conjugated to alkaline phosphatase. Each hybrid may bind to multiple antibodies, amplifying signal. Antibody-conjugated alkaline phosphatase cleaves a chemiluminescent substrate, emitting a light signal that is quantified with use of a luminometer. Light emitted above a standardized threshold intensity indicates the presence of virus. The test covers 11 HPV types in addition to HPV 16 and 18 and has a reported analytic sensitivity of 1.09 picograms/mL. A reengineered version of this hybrid capture test for low resource settings, careHPV, covers 14 HPV types, requires no running water, and has a turnaround time of 2.5 hours. The test has been successfully used in rural China with a clinical sensitivity of 90% for the detection of cervical intraepithelial neoplasia types 2 and 3.23
Understanding of the molecular oncogenesis of cervical squamous carcinoma has provided a standardized biomarker of risk. More than 95% of HPV infections clear up without further consequences. However, persistent infection for months to years with HPV type 16 or 18 carries a 40% risk of squamous intraepithelial lesions (SILs),24 and untreated high-grade SILs have been estimated to result in invasive carcinoma in approximately 30% of persons over 5 to 10 years.24,25 Persistent HPV infection thus indicates high risk for eventual invasive carcinoma and represents an additional biomarker that could be useful in the prevention of invasive carcinoma.
Testing for HPV DNA has been found to be more sensitive than cytologic screening for detection of high-grade SILs26,27 but somewhat less specific. In view of the number of infections that are self-limited, relying on a single HPV test may result in greater numbers of negative colposcopy examinations. To address this problem, co-testing strategies have been evaluated whereby both cytology and HPV testing are performed on initial examination.28 Negative results have a high negative predictive value, and the rescreening interval can be extended from 1 to 3 years, reducing overall screening expense. Persons with positive results of a viral DNA test but negative cytologic findings have been rescreened at 1 year and examined with colposcopy if positive results are found. Whether this strategy or other strategies for cervical cancer risk management will be universally adopted remains to be determined.22,29 What is clear is that HPV testing provides a means for improving sensitivity for detection of early cervical lesions, further reducing mortality from invasive cervical carcinoma while potentially reducing screening expense.
Finally, HPV is not only a biomarker for risk assessment and colposcopic intervention but is an immunogen that has been successfully incorporated into bivalent (types 16 and 18, Cervarix) and quadrivalent (types 6, 11, 16, and 18, Gardasil) vaccines to prevent infection and carcinogenesis (reviewed in references 30 and 31).30,31 An in-depth discussion of HPV vaccines for tumor prevention is beyond the scope of this chapter. However, the many facets of HPV expression in cervical carcinoma and the emerging role of HPV in other tumors, including vulvar and vaginal condylomata, certain skin cancers, and head and neck carcinoma, illustrate how a valuable biomarker that is integral to oncogenesis can be used to improve the accuracy of estimating tumor risk and the specificity of treatment. Unfortunately, such clearly defined and easily accessible biomarkers are rare. Rather, the more usual case is described in the next sections for colon and lung, where potential biomarkers are defined but have not found a role in early detection, diagnosis, prevention, or treatment.
Colon and Multistep Carcinogenesis with Hereditary Components
Like cervical carcinoma, colonic adenocarcinoma can be effectively prevented by extirpation of premalignant epithelium, which in the colon is represented by polyps. Several recent trials have indicated that removal of polyps by colonoscopy34–34 reduces the frequency of colorectal carcinoma (CRC), and consensus opinion is that endoscopic polypectomy has a significant impact on the incidence of CRC. Recent data have also suggested that colonoscopic screening reduces CRC mortality by more than 50%34 and that the less extensive sigmoidoscopy reduces mortality from distal CRC by a nearly equal percentage (50%).35 The latter sigmoidoscopic study was a large, randomized, population-based screening trial. Colonoscopic screening for colon polyps beginning at age 50 years has become a standard of care.
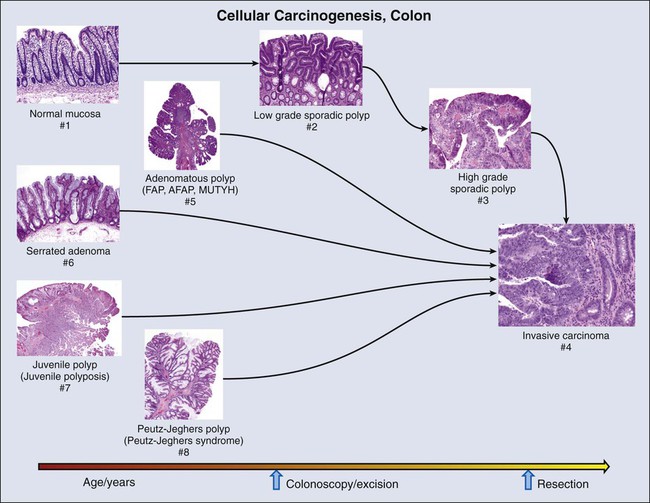
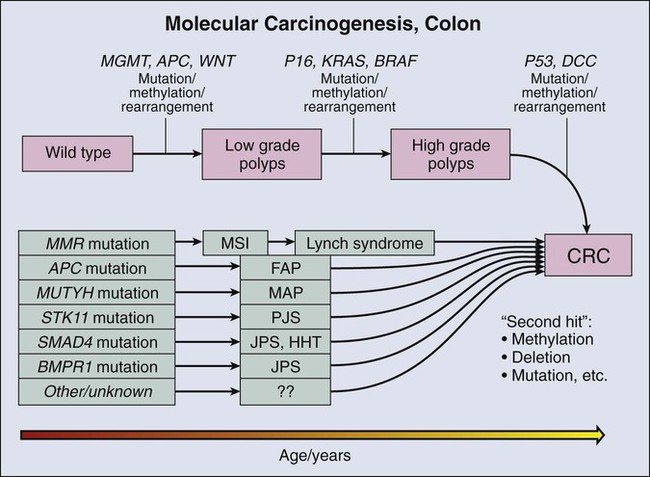
Molecular biomarkers to date have not played a direct role in reduction of morbidity and mortality in persons with CRC. However, molecular studies have facilitated understanding of colon carcinogenesis (Figs. 17-3 and 17-4) and provided a rationale for colonoscopic surveillance and polypectomy. At the mid twentieth century, an association between benign polyps was suspected, but at that time no direct evidence was available to show that polyps undergo transition to CRC.36 Suspicions were finally confirmed in the 1980s, when the clonal nature of both sporadic polyps and CRC was documented.37 Activating mutations in the KRAS oncogene and inactivating mutations in several tumor suppressor genes were described.38 The same mutations were demonstrated in both polyps and carcinoma, and the number of mutations was found to increase with the histologic grade of the adenoma. This finding suggested a progression to invasive carcinoma through the accumulation of genetic and epigenetic changes. These changes provide a proliferative and survival advantage to mutant cells that continue to proliferate and mutate until a malignant phenotype is reached. The combined data from these studies led to the proposal of a multistep model of colon carcinogenesis39 that continues as the standard model.40
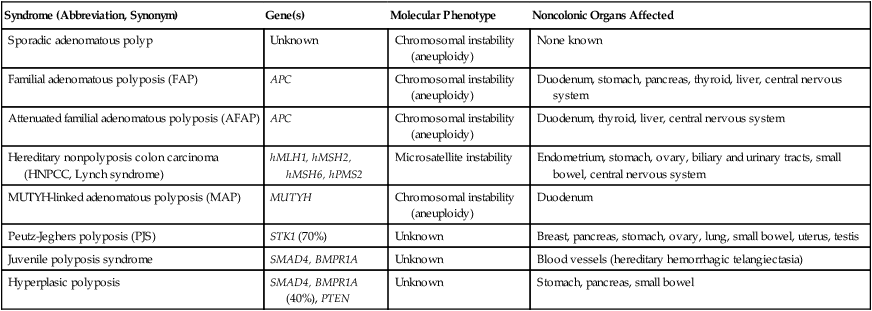
Several forms of hereditary colon cancer are recognized (reviewed in reference 41)41 and are listed in Table 17-2.
Table 17-2
| Syndrome (Abbreviation, Synonym) | Gene(s) | Molecular Phenotype | Noncolonic Organs Affected |
| Sporadic adenomatous polyp | Unknown | Chromosomal instability (aneuploidy) | None known |
| Familial adenomatous polyposis (FAP) | APC | Chromosomal instability (aneuploidy) | Duodenum, stomach, pancreas, thyroid, liver, central nervous system |
| Attenuated familial adenomatous polyposis (AFAP) | APC | Chromosomal instability (aneuploidy) | Duodenum, thyroid, liver, central nervous system |
| Hereditary nonpolyposis colon carcinoma (HNPCC, Lynch syndrome) | hMLH1, hMSH2, hMSH6, hPMS2 | Microsatellite instability | Endometrium, stomach, ovary, biliary and urinary tracts, small bowel, central nervous system |
| MUTYH-linked adenomatous polyposis (MAP) | MUTYH | Chromosomal instability (aneuploidy) | Duodenum |
| Peutz-Jeghers polyposis (PJS) | STK1 (70%) | Unknown | Breast, pancreas, stomach, ovary, lung, small bowel, uterus, testis |
| Juvenile polyposis syndrome | SMAD4, BMPR1A | Unknown | Blood vessels (hereditary hemorrhagic telangiectasia) |
| Hyperplasic polyposis | SMAD4, BMPR1A (40%), PTEN | Unknown | Stomach, pancreas, small bowel |
Familial Adenomatous Polyposis
The first hereditary colon carcinoma to be described, familial adenomatous polyposis (FAP), is characterized in its classic form by the occurrence of hundreds to thousands of polyps in the colon of affected persons with the formation of invasive carcinomas at an early age. Polyps may also sometimes be found in the upper gastrointestinal tract. As long ago as 1882, siblings were described with numerous and recurring polyps,42 and by the mid twentieth century, FAP was established as a hereditary autosomal-dominant disorder.43 A gene mutation associated with FAP was identified on chromosome 5q by linkage analysis44,45 and positional cloning studies.46–49 The gene, adenomatous polyposis coli (APC), is now known to encode a tumor suppressor that modulates wnt signaling through binding and stabilization of a complex of proteins that includes GSK3b, β−catenin, and axin family members. Mutations in APC truncate the protein at its carboxyl terminus and disrupt the protein signaling complex, which results in deregulation of cell cycle control, cell migration, differentiation, and apoptosis (reviewed in reference 50).50 In addition to being responsible for FAP, APC mutation is present in approximately 60% of sporadic colon adenocarcinomas51,52 and an approximately equal number of sporadic polyps,51,53 suggesting that APC mutation is an early event in sporadic carcinogenesis, as well as in hereditary disease. An attenuated form of FAP with smaller numbers of tumors (<100) has been described.54,55 This condition is referred to as attenuated FAP54 and may be driven by the mutations in APC in the 3′ and 5′ ends rather the central portion of the gene that is affected in classical FAP.55
Hereditary Nonpolyposis Colon Cancer
The most common form of hereditary colon cancer is hereditary nonpolyposis colon cancer (HNPCC). HNPCC is a familial syndrome that differs from FAP in that it manifests a smaller numbers of polyps compared with FAP. The earliest description of this disorder can again be traced back to the early twentieth century,56 but the hereditary character and syndromic features of the disease were firmly established by the diligent kindred studies of Lynch and Lynch,57 after whom the syndrome is named (Lynch syndrome). In brief, the syndrome consists of CRC that occurs in close relatives, often younger than 50 years,58,59 without adenomatous polyposis. HNPCC is also associated with an increased frequency of tumors in the endometrium, stomach, ovary, hepatobiliary tract, upper urinary tract, small bowel, and central nervous system.41 In contrast to FAP polyps, which are frequently found on the left side of the colon, persons with HNPCC frequently have flat lesions on the right side of the colon with a distinct histologic appearance referred to as “serrated adenoma” (Fig. 17-3).
The molecular genetic underpinnings of HNPCC were deciphered beginning with the linkage to microsatellite markers on chromosome 2p.60 The genetic basis of the disease was established when it was found that patients with the syndrome harbored mutations in the mismatch repair (MMR) genes hMLH1,61,62 hMSH2,63,64 hMSH6,65 and hPMS2,66,67 with mutations in hMLH1 and hMSH2 accounting for more than 90% of HNPCC cases. The germline mutations are heterozygous, and colon carcinogenesis results from silencing of the nonmutant allele by methylation68,69 or by acquired allelic loss.
The phenotypic consequence of gene silencing is loss of MMR protein and the resulting inability to repair DNA replication errors, ultimately leading to accumulation of mutations contributing to carcinogenesis. At a clinical level, protein loss can be the demonstrated by immunochemistry and optimally requires four separate stains. The inability of affected cells to repair DNA replication errors results in insertions or deletions in short tandem repeats (microsatellites) that exist throughout the genome and is referred to as “microsatellite instability” (MSI).70,71 Currently, MSI is documented by testing of a standard set of five microsatellite markers recommended by a consensus panel.72 Results are scored on the basis of the number of unstable loci and are classified as MSI-High (MSI-H), MSI-Low (MSI-L), or MSI-Stable (MSI-S). More than 90% of HNPCC tumors are MSI-H, but approximately 15% of all sporadic tumors are also MSI-H,73 and thus the test is not specific for hereditary disease.
MUTYH-Associated Polyposis
A third hereditary variant of colon carcinoma is MUTYH-associated polyposis (MAP), which is the newest polyposis syndrome (first described in 2002).74 MAP is a disorder similar to attenuated FAP and is characterized by adenomatous polyps that may number in the hundreds but not in the thousands, usually ranging from 10 to 1000.77–77 Polyps may also occur in extra colonic sites, usually the duodenum. Adenomas tend to occur in the proximal colon.76 MUTYH is a glycosylase and part of the base excision repair pathway. Oxidative damage to DNA frequently results in the modification of guanine (G) to 8-oxoG. 8-oxoG can pair with adenine (A) during DNA replication, a mismatch that is recognized by MUTYH, which removes the mismatched A and restores the correct base, cytosine (C). The damaged G (8-oxoG) is then replaced with an intact G. Mutation of MUTYH at specific hot spots hinders recognition of the 8-oxoG : A mispairing, resulting in G : C to A : T transversion at sites of oxidative damage. The mutational hot spots are found in exon 7, resulting in amino acid substitution Y179C, and in exon 13, resulting in substitution G496D. Germline mutation at both hot spots (biallelic mutation) occurs in 0.3% of patients with CRC. Biallelic mutation confers 28-fold relative risk for CRC compared with control subjects. Monoallelic mutation is more common with a frequency of approximately 2% in CRC but also occurs in approximately the same frequency in control populations. Most studies suggest that monoallelic mutation confers little or no increased risk of CRC.75,76 Among control patients without polyposis, biallelic mutation is rare.76
Hamartomatous Polyposes
The fourth hereditary form of colon cancer is the hamartomatous polyposes, a category that includes Peutz-Jeghers syndrome (PJS) and juvenile polyposis syndrome. First described in the 1890s,78,79 PJS was named in 1954 by Bruwer and colleagues83 in recognition of the work of Peutz80 and Jeghers.81,82 PJS is an autosomal-dominant condition characterized by multiple histologically distinct polyps (Fig. 17-3) that occur in the large and small bowel in association with melanin pigmentation of the lips, mouth, nose, anus, and extremities.84 The condition is associated with a high risk for cancer in not only the large and small intestine but also in the esophagus, stomach, pancreas, breast, uterus, ovary, and lung.87–87 Gene mapping has identified a locus on chromosome 19.3 p1388,89 that encodes a serine-threonine kinase, STK11 (LKB1), a gene that is mutated in more than 90% of affected individuals.90–94 A variety of mutations including deletions, truncations, and missense point mutations may occur over nearly the full coding region of the gene.86 STK11 protein is multifunctional, playing a role in cell cycle arrest, wnt signaling, and the tuberous sclerosis complex pathway, with effects on mammalian target of rapamycin signaling.84 Mutation, often associated with loss of heterozygosity,95,96 results in loss of function, and the gene is thus considered to be a tumor suppressor gene. The overall frequency of PJS is estimated at 0.5 to 2.0 per 100,000 live births.97
Juvenile Polyposis
Juvenile polyps are characteristically solitary peculated lesions that contain tortuous, often cystically dilated crypts, stromal granulation tissue and inflammation, and denudation of the surface epithelium.98 These lesions frequently occur in children, and there is no evidence that solitary juvenile polyps pose an increased of risk for carcinoma.100–100 However, multiple juvenile polyps may occur in young persons and their family members,103–103 and the presence of >5 juvenile polyps or a single juvenile polyp in a person with a family member with juvenile polyposis now defines the condition, termed “juvenile polyposis.” This form of polyposis carries a lifetime risk of colon carcinoma of 39% and a relative risk of 34.104 Fifty percent to 60% of affected individuals carry germline mutations in SMAD4 or BMPR1A.105–107 Nearly all patients with SMAD4 mutations also have hereditary hemorrhagic telangiectasia108 (overlap syndrome). Both SMAD4 and BMPR1A are involved in the transforming growth factor–β (TGF-β)/bone morphogenetic protein pathway.
Other Polyposes
Finally, a variety of polyposis syndromes with potential genetic linkage have been described and are best exemplified by hyperplastic polyposis syndrome (serrated polyposis syndrome).109,110 This condition consists of the occurrence of multiple sessile polyps variously referred to as hyperplastic polyps or serrated adenomas, frequently on the right side of the colon and numbering from 5 to >100.111 Although familial associations of the syndrome112 and linkage to high risk of colon carcinoma113 have been reported, the genetic basis is complex and associated with mutations in several driver genes, including BRAF and KRAS.110 No single genetic mechanism has been linked to the syndrome, and the true frequency is not established. To date, the number of reported cases is small.
Biomarkers for Colon Cancer Screening
Genetic tests
Although the number of highly penetrant mutations that predispose to colon carcinoma is large, the proportion of colon carcinomas that can be accounted for by these mutations is approximately 5%41 and has been overestimated in the past.73 Markers of hereditary predisposition and molecular progression in sporadic tumors that have been delineated to date have not been easily converted into an early detection test suitable for broad population-based applications for several reasons. First, the types of accessible specimens that can be used for tests for early detection consist primarily of blood and fecal DNA. Although DNA from invasive tumors can be detected in the blood by PCR-based mutation testing, it is not as useful at earlier polyp stages. In addition, collection and processing of fecal specimens for a biomarker that is expected to be present at a low level and heavily contaminated by nonneoplastic cells is a logistical challenge. Second, some of the genetic abnormalities identified in colon carcinogenesis do not readily lend themselves to screening assays. Mutations in APC, for example, consist mainly of truncations and deletions that may occur over a wide region of the gene, and thus designing a sensitive clinical assay has been difficult. Finally, the wide availability of colonoscopy services that combine polyp detection and excision reduces the cost and improves the attractiveness of direct and systematic colonoscopic population-based surveillance.
Other Screening Biomarkers
Biomarkers that have been most successful for colon cancer screening are fecal tests such as the guaiac fecal occult blood test (FOBT), the fecal immunochemical test, and the stool DNA (sDNA) test. The greatest experience has been with the FOBT. This test is based on a color change whereby α-guaiaconic acid extracted from the wood of tropical Guaiacum tree is rapidly oxidized by hydrogen peroxide to a blue-colored quinone in the presence of heme derived from blood. A wide variability in results occurs114 as a consequence of the level of compliance with collection methods, the type or brand of kit used, the frequency of testing, user interpretation, and other factors. Despite a lack of standardization, FOBT screening with follow-up colonoscopy is reported to reduce both incidence115 and mortality116,117 from colon carcinoma. The former is attributed to polyp removal. The fecal immunochemical test is a second test for occult blood and is reported to be more sensitive and specific than the guaiac assays. It has been shown to have a cancer detection rate similar to colonoscopy but has a lower sensitivity for polyps.118
Occult blood tests measure host response (hypervascularity and bleeding) to a tumor. The sDNA test, however, aims to measure a direct product of tumor cells. A recent version of the sDNA test measures multiple markers in stool using an array-based platform119 and for this reason has been referred to as “next generation,” although this technology does not use deep sequencing methods and should not be confused with “next generation sequencing.” The biomarkers that are incorporated into the platform include four methylation probes, as well as mutated KRAS and hemoglobin. Patterns of gene methylation have been identified in colon tumors that have been characterized as CpG island methylation high and CpG island methylation low (reviewed in reference 120).120 When applied to a preliminary cohort of 678 subjects with polyps or colorectal carcinoma, the combination test including methylation markers, KRAS mutation, and occult blood had a sensitivity of 85% for carcinoma and 54% for polyps.
Despite the availability of these fecal-based tests, the target in early colon cancer intervention appears to be shifting from detection of invasive carcinoma to prevention by detection and thorough clearing of adenomatous polyps from the colon. Defining the biology of colon carcinogenesis and confirming that polyps are the precursor lesions along with improvements in colonoscopic methods has led to reduction in mortality from colon cancer. Colonoscopy enables direct identification of potential precursor lesions and at the same time provides a means for ablating them, which is strikingly evident in the new sigmoidoscopy findings,35 indicating that although sigmoidoscopy reduces mortality from visible distal colon tumors, mortality from proximal lesions is unaffected. It seems likely that colonoscopy will be the main vehicle for early detection and intervention in colon cancer for the foreseeable future.
Lung Carcinogenesis and a Known Carcinogen
For several decades, lung carcinoma has had the highest mortality of all tumors, accounting for more than twice as many deaths as the next most lethal tumor.121,122 Initial large-scale efforts during the 1970s and 1980s to reduce mortality by early detection using chest radiographs and/or sputum cytology were not successful. Chemoprevention using β-carotene and/or alpha tocopherol dietary supplementation also failed.123–126 Recently, however, early detection efforts have begun to yield some success in reducing mortality even in this problematic tumor. Low-dose computed tomography (LDCT) has a higher clinical sensitivity for detecting smaller tumors than does a chest radiograph. Early studies in smokers documented a 2.7% prevalence of malignant disease detected by LDCT versus 0.7% in the same patients screened by chest radiograph.127 In a multicenter study of more than 30,000 smokers, LDCT detected lung cancer in 1.3% at baseline and an additional 0.3% at annual follow-up examinations for an overall total of detection rate of approximately 1.5%.128 Although this study was not randomized and had no unscreened control arm, it showed a survival advantage in comparison with historical control subjects. Recently, a large randomized trial of CT versus radiography conducted annually in three rounds found that LDCT detected carcinoma in 2.4% of the screened high-risk population and resulted in a 20% reduction in lung cancer mortality that was not observed in the chest radiograph control group.129 The weight of evidence supporting improved long-term mortality rates in smokers has prompted the publication of multisociety consensus guidelines recommending annual LDCT screening for smokers and former smokers ages 55 to 74 years with access to appropriate follow-up.130
Premalignant Lesions in the Lung
Unlike mortality reductions that have been achieved in the colon and cervix, where preinvasive and therefore nonmalignant lesions are targeted, the benefits of LDCT screening for lung cancer have been achieved through early detection of invasive tumors. A strategy to identify in situ premalignant lesions could potentially reduce lung cancer mortality to a much lower rate. Unfortunately, what are thought to be premalignant lesions in the lung are difficult to detect and may occur at unpredictable locations anywhere in the central and alveolar airways (Figs. 17-5 and 17-6), exemplifying the challenges confronting prevention efforts in some organ systems.
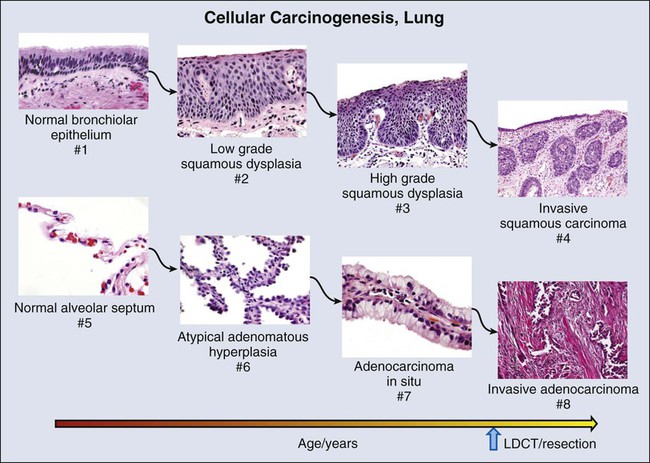
Squamous Dysplasia
The predominant premalignant lesion of the central airways is squamous dysplasia131 (also referred to as “atypia”). Squamous dysplasia was documented more than 50 years ago in association with carcinoma,132 as well as in smokers without concurrent carcinoma.133 Central squamous lesions are flat, inconspicuous, and only rarely identified by white light fiberoptic bronchoscopy. They are somewhat better visualized by laser-induced fluorescence emission bronchoscopy.134 This method relies on a loss of autofluorescence, and signal from dysplasias may be mimicked by fluorescence loss caused by mucosal hemorrhage and other reactive changes. Thus, although sensitivity of laser-induced fluorescence emission bronchoscopy is high, specificity is low.
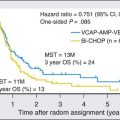
Prospective studies on the power of dysplasia to prospectively predict future central airway malignancy are few and face the forbidding challenges of inaccessibility of the bronchi and the prolonged follow-up that is required to achieve significant results in a sufficiently powered cohort. Sputum studies indicate that atypia of cells exfoliated from the central airways is associated with elevated risk for incidental carcinoma. The hazard ratio increases with the proximity of the sputum collection date to lung cancer diagnosis, but sputum atypia even 3 years prior to lung cancer diagnosis is associated with increased risk of lung carcinoma.135 Confirmation of sputum analyses in biopsy studies to identify sources of dysplastic epithelium will require a large and disciplined multicenter trial involving bronchial mapping and revisiting dysplastic sites to evaluate the long-term disposition of discovered lesions.
The corollary question of whether ablation of premalignant central airway lesions could reduce lung cancer mortality remains an open one. A recent chemoprevention study136 using Iloprost, a prostacyclin agonist, has suggested that reversal of premalignant dysplasia morphology may be possible, but the study was a small one, and the end point was purely histologic. The long-term effect on mortality of agents that may alter the premalignant morphology of central airway epithelium remains unknown.
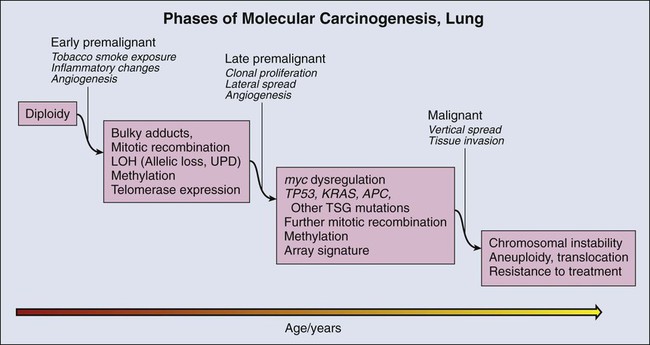
Molecular Changes during Lung Carcinogenesis
As in other organs, most notably the colon, molecular changes that accompany the morphologic changes of dysplasia have been investigated in considerable detail. These studies are informed by the well-established role of tobacco smoke as a carcinogen in lung cancer. The dominant carcinogenic compounds found in cigarette smoke are the polyaromatic hydrocarbons and nicotine-derived nitrosamine ketone.137 These compounds are metabolized to highly reactive intermediates that are inactivated largely by mitochondrial glutathione transferases. However, repair is imperfect, and a small proportion of the intermediates react directly with DNA, forming bulky adducts. Unless removed by the DNA repair system, adducts can create errors of replication. Over time, molecular alterations accumulate, providing a selective advantage to mutant cells for growth and survival.
Components of this pathway have been associated with increased risk for future lung cancer. Case control studies indicate that current smokers with tumors have higher levels of adducts in blood or tissue but that nonsmokers and former smokers do not have these higher levels.138 Bulky adducts appear to reflect ongoing smoking-related DNA damage rather than biological changes specifically associated with malignant transformation. Clinically applicable adduct levels predictive of lung cancer have not been established.
Glutathione transferase and DNA repair enzyme mutations and polymorphisms that affect DNA repair activity could represent a risk factor for lung cancer because efficient deactivation of reactive carcinogens and repair of DNA damage caused by bulky adducts are important in protecting potential precursor cells from critical mutational changes. However, single-nucleotide polymorphism (SNP) association studies in many carcinogen metabolizing and DNA repair enzymes have demonstrated only weak and inconsistent associations and have not been sufficiently informative to guide intervention in potentially affected persons.139–143
Tumor Suppressor Gene Methylation
DNA damage in lung premalignancy engenders a number of genetic and epigenetic changes that provide a cellular survival or proliferative advantage to affected bronchial epithelial cells. Perhaps the earliest molecular events may be epigenetic and include DNA methylation, histone modification, and alterations in micro (mi)RNA expression (reviewed in references 144 and 145).144,145 The greatest experience regarding the use of epigenetic changes in the context of early detection of lung cancer has been obtained in regard to methylation. Methylation refers to the conversion of cytosine to 5′ methylcytosine by DNA methyltransferase. In lung carcinogenesis, methylation preferentially occurs in CpG-rich regions (CpG islands) that are present in normally unmethylated promoter regions of critical genes (hypermethylation) and may silence gene expression. In lung tumors, interest has focused on genes such as p16,146 which are silenced in tumors but expressed in normal control tissue and are considered to be tumor suppressor genes (TSGs). The development of a simple and sensitive assay based on the conversion of methyl cytosine to uracil in the presence of metabisulfite followed by PCR with primers specific for the methylated bases, termed methylation-specific PCR,147 has greatly facilitated the assessment of methylation status as a risk marker for lung carcinoma.
Methylation of key TSG promoter regions in cells exfoliated from bronchial surfaces into the sputum is nearly universally associated with prevalent lung carcinomas148 and frequently occurs in sputa of smokers without a tumor as well. TSG methylation is not present in sputum of people who have never smoked. This finding initially led to optimism that methylation could be used to screen selected populations at risk for lung carcinoma. However, the predictive power of single gene methylation or methylation of a small number of genes is limited by the ubiquity of methylation in smokers and the consequent high rate of false-positive results. This problem has led to the strategy of testing promoter regions in large numbers of genes to estimate overall methylation burden.149 Identification of panels of genes to optimize the predictive power of methylation testing is a process that is still under way. In receiver-operator curve analyses, a seven-gene methylation profile selected from a 31-gene panel achieved a predictive accuracy of 71% to 77%. However, with a sensitivity set at 75%, the false-positive rate was 75%.150 Although TSG promoter methylation may prove problematic as a general screening tool, it may be useful in specific niche populations or to supplement other methods of screening such as LDCT, and ongoing studies aim to further evaluate the usefulness of this approach.
Aneuploidy
A second molecular consequence of tobacco exposure is chromosomal instability, which is detected as aneuploidy by fluorescence in situ hybridization (FISH) in bronchial epithelial cells in situ and in sputum. Aneuploidy is nearly universal in lung carcinoma. It is also frequently observed in high-grade dysplastic lesions in the central airways.151 Aneuploidy in sputum has been associated with prevalent carcinoma152,153 and has recently been shown to have a sensitivity of 76% within a detection window of 18 months before the appearance of invasive carcinoma.154 However, FISH performed on sputum is labor intensive and difficult to perform both in the specimen preparation and slide interpretation. FISH requires a high level of expertise at every step, which has led to the development of automated methods of FISH screening. However, to date, these automated methods have not been broadly applied. It seems likely that FISH may most successfully be applied in restricted settings in conjunction with other technologies.
High-Throughput Technologies
High-throughput methodologies have been suggested as possible early detection tools. Recently, genome-wide association studies have identified an SNP variant at 15q15.2 that is significantly associated with cancer risk, with an overall odds ratio of 1.15.155 The low odds ratio suggests that the SNP variant itself will not represent a useful predictor. However, this region of the gene still needs to be explored for more powerful biomarkers.
Gene expression microarrays have identified consistent changes in the transcriptome of epithelium brushed from nonmalignant bronchial surfaces that distinguish smokers from nonsmokers.156 An additional set of changes are reported to identify subjects with cancer from smokers without cancer at an accuracy of 83%.157 Finally, large-scale genomic sequencing studies have identified differences in the “genomic landscape” between smokers and nonsmokers.158 It is expected that manageable numbers of biomarkers and clinically applicable testing platforms will emerge from these exploratory studies that will improve risk assessment and management of high-risk smokers, as well as patients with already invasive carcinomas.
Other Organ Sites
Premalignant cellular and molecular changes are described in several other tumors, including breast, prostate, bladder, and esophagus. A detailed review of these changes may be found in appropriate chapters of this text (see Chapters 74, 83, 84, and 91). However, identification of these changes has not led to mortality reductions.
Prognosis and Prediction
Protein Biomarkers for Prediction of Outcome
IHC Markers
IHC staining for estrogen receptors (ERs) and progesterone receptors (PRs) was one of the earliest applications to find a place in patient management. Until the early 1980s, antibodies to these proteins were not widely available. However, with the development of monoclonal antibody technology, antibodies that could identify hormone receptors were created159 and found to reliably label receptors even in FFPE sections of breast carcinomas.160,161 Unexpected results of this testing were that these receptors are typically found in the cell nucleus and that the intratumoral and intertumoral range of expression is large. Because of this finding, semiquantitative scoring that takes into account intensity of staining and percentage of cells stained has been used to establish expression levels that define positive and negative results.162,163 These scoring systems continue to prove their prognostic value to the present time.164
A second IHC test that has been successfully applied to the management of breast carcinoma is the test for HER2/neu (ERBB2) expression. HER2 is a tyrosine kinase receptor (TKR) and a member of the epidermal growth factor receptor (EGFR) family of signaling molecules. Its role in human carcinogenesis was initially discovered through DNA screening, where it was found to be amplified in 30% of the tested breast cancers.165 RNA and protein were shown to be overexpressed in tumors that are amplified, as well as in a subset of nonamplified tumors.166 HER2 testing became crucial when it was found that HER2 protein overexpression was a determining factor for in vitro and xenograft responsiveness to anti-HER2 monoclonal antibody treatment.167,168 In early clinical trials, anti-HER2 IHC was used as the method for determination of HER2 status and determined eligibility for treatment with the anti-HER2 antibody drug, Herceptin.169 A standardized approach to HER2 IHC testing was established several years later when expert panels agreed upon criteria for determining HER2 status (Fig. 17-7).170
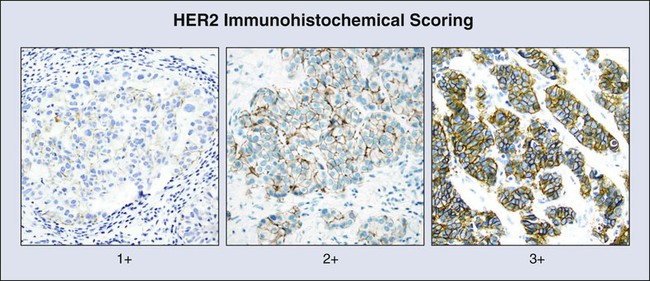
Recently, tests for multiple IHC markers have been combined into panels that include ER, PR, HER2, and the proliferation marker Ki-67,171 as well as several other markers thought to be of prognostic importance.172 These panels have been found to provide prognostic information that is comparable with that of multiplex mRNA expression assays discussed later and have the added advantage of accessibility in local laboratories and ready correlation with histologic features.
A substantial subset (~15%) of breast tumors do not express ER, PR, or HER2 and are now referred to as triple-negative tumors.173 These lesions are notable for their aggressive histology and poor long-term outcome. This category overlaps but is not identical to the basaloid-like breast cancer category defined by microarray-based expression profiling174 and includes nonhereditary tumors that frequently express perturbations of BRCA1 pathway gene expression. The triple-negative category thus has increased the importance of accuracy in both positive and negative IHC testing and reemphasizes the need for local validation and controls for accurate IHC procedures (see the Best Practices section in this chapter).
Finally, EGFR is expressed at a high level in most non–small cell lung carcinomas, including both adenocarcinoma and squamous carcinoma, and expression correlates with gene amplification and copy number.175,176 When a scoring system was applied in a recent phase 3 trial testing the effectiveness of the anti-EGFR chimeric (mouse/human) monoclonal antibody, Cetuximab, in advanced cases of non–small cell carcinoma, only patients with the highest levels of EGFR expression (scores >200) experienced a survival benefit.177 This result suggests that EGFR expression levels determined that IHC may be a reasonable selection criterion for anti-EGFR antibody treatment. Properly scored EGFR IHC expression may prove to be a valuable predictor of response to anti-EGFR monoclonal antibody treatment.
Measurement of Gene Expression for Prognosis and Prediction of Treatment Response
The analysis of gene expression in cancer was accelerated greatly during the 1990s with the introduction of microarray technology that allowed simultaneous measurement of expression of large numbers of genes.178 The underlying technology of microarrays involves the binding of gene probes to specific addresses on a solid matrix. A fluorescence-labeled complementary DNA library is then hybridized to the solid matrix, and the level of expression at each address is measured by fluorescence microscopic scanning of the array. Many variations on this basic concept have resulted in high-density arrays that can be used to evaluate expression levels of virtually all genes in a single tissue sample. Large-scale gene expression profiling has been used to cluster tumors according similarities in gene expression patterns and to identify new subsets of lung and breast tumors,174,179–182 with prognostic implications. However, this method creates unwieldy amounts of data, and to produce a clinically relevant result, it has been necessary to filter expression data to identify profiles that can be used for clinical decision making.
This refinement has been most thoroughly applied in breast cancer, where dominant competing platforms—Mammaprint (Agendia, Amsterdam, The Netherlands) and Oncotype DX (Genomic Health, Redwood City, Calif.)—have been created primarily for predicting likelihood of benefit from chemotherapy in early-stage breast cancer (reviewed in reference 183).183 To create these platforms, RNA markers are tested in tissue samples and the most powerful and efficient combinations of markers are assembled into a panel to provide an estimate of the likelihood of benefit from therapy. Available platforms have varying tissue requirements, with some platforms needing unfixed frozen tissue and others effective with FFPE tissues (see the Best Practices section). Preanalytical processing is thus a major consideration in the selection of a microarray platform. The use of this kind of gene expression testing is growing, but the role these tests will ultimately play in the dawning era of high-throughput molecular profiling and specifically targeted treatment remains to be determined.
DNA Sequencing as a Biomarker Predicting Response to Treatment
The large number of mutations found in tumors of all types during the past few decades has prompted comprehensive sequencing and gene expression analysis of virtually all solid tumor types through The Cancer Genome Atlas (TCGA).184 A number of genetic aberrations discovered by the TCGA and earlier efforts have been exploited for treatment of patients with cancer, with the primary focus of interest being the tyrosine kinase signaling pathway. TKRs (Fig. 17-8) are transmembrane immunoglobulin-like molecules185 containing an extracellular ligand-binding region, a transmembrane region, and an intracellular tyrosine kinase region that contains tyrosine residues whose phosphorylation regulates signal transduction. Specific mutations in tyrosine kinase domains enhance phosphorylation and stimulate downstream intracellular signaling. The physiological result is accelerated cell proliferation, extended cell survival, and increased angiogenesis and cellular migration, properties that are hallmarks of carcinogenesis.1
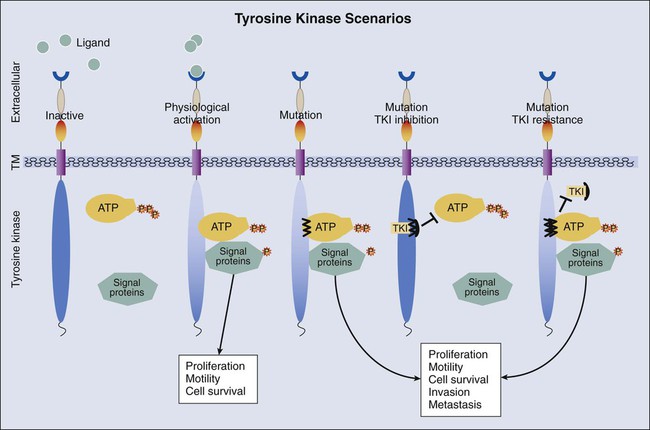
A number of small-molecule tyrosine kinase inhibitors (TKIs; Table 17-3) specific for these receptors have been designed to block phosphorylation and suppress tumor growth and spread. Also, the extracellular domains of these molecules have proven to be antigenic, and therefore the extracellular ligand binding domain is also a target for therapeutic antibody development. Several of these drugs have successfully transferred to the clinic, and the number of such drugs available for cancer treatment is likely to continue to expand, creating an ongoing demand for more comprehensive molecular testing of malignancies of virtually all types.
Table 17-3
Treatable Molecular Lesions in Solid Tumors
| Tumor(s) | Gene(s)/Protein | Molecular Lesion(s) | Drug |
| Breast | ER | Overexpression | Fulvestrant/tamoxifen |
| Breast | ER/PGR | Overexpression | Exemestane/letrozole |
| Breast/stomach | HER2/NEU | Amplification/overexpression | Everolimus/lapatinib/pertuzumab/ trastuzumab |
| Colon | KRAS | Absence of mutation | Cetuximab/panitumumab |
| Colon | KRAS | Activating mutations | MEK inhibitors (AZD6244, others) |
| Colon/liver/melanoma/NSCLC | CTNNB1 | Activating mutation (wnt pathway) | Porcupine inhibitors (LGK974, others) |
| Colon/NSCLC | NRAS | Activating mutations | MEK inhibitors (AZD6244, others) |
| Colon/NSCLC | PIK3CA | Activating mutations | PIK3CA inhibitor (BKM120, others) |
| Colon/NSCLC | EGFR | Overexpression/increased gene copy number | Cetuximab/panitumumab |
| GIST | C-KIT/PDGFRα | Activating mutations | Imatinib/midostaurin (PKC412) |
| Lymphoma | CD20 antigen | Express/overexpression | Rituximab/tositumomab/ofatumumab/ ibritumomab-tiuxetan (radiolabeled) |
| Lymphoma | CD25 antigen | Express/overexpression | Denileukin-diftitox (antibody-toxin conjugate) |
| Lymphoma | CD30 antigen | Express/overexpression | Brentuximab-vedotin (antibody-toxin conjugate) |
| Melanoma | KIT | Activating mutations | Imatinib |
| Melanoma/colon/lung | BRAF | Activating mutations | Vemurafenib, dabrafenib |
| NSCLC | ALK | Gene rearrangement (multiple fusion partners) | Crizotinib |
| NSCLC | EGFR | Activating mutation (small deletion/point mutation) | Erlotinib/gefitinib |
| NSCLC | RET | Translocation | Vandetanib/cabozantinib/sunitinib |
| NSCLC | ROS | Gene rearrangement (multiple fusion partners) | Crizotinib/sunitinib |
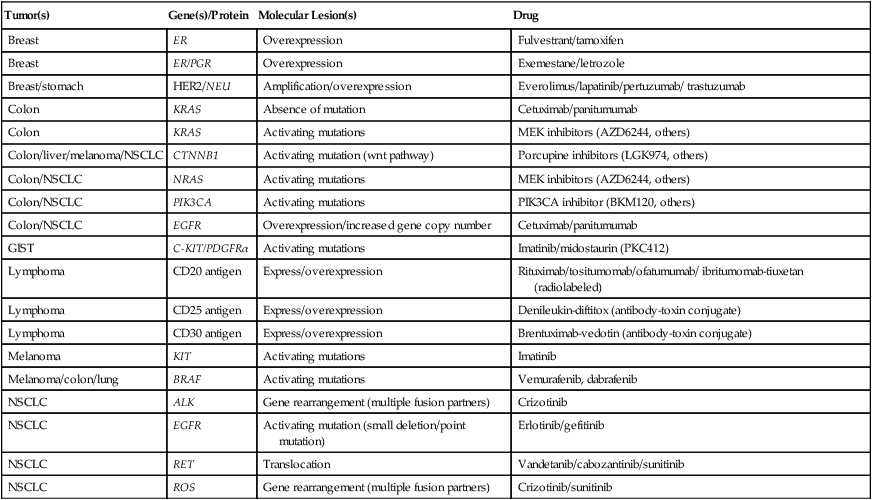
GIST, Gastrointestinal stromal tumor; NSCLC, non–small cell lung cancer/carcinoma.
Gastrointestinal Stromal Tumors
GISTs were, until recently, a rare and ill-defined neoplasm of somewhat mysterious origin. In the early 1990s these tumors were defined as spindle cell malignancies that exhibited no evidence of cell of origin.186 The status of these tumors began to change as it was recognized in the mid 1990s that GIST tumors express CD34 but not desmin187 and thus displayed a distinct immunophenotype. A watershed discovery was reported by Hirota et al.188 in 1998. These investigators described nearly uniform expression of KIT (CD117) in GIST tumors and suggested that they may arise from the immunophenotypically similar paraganglionic interstitial cells of Cajal. Most importantly, they identified deletions and point mutations in the juxtamembrane portion of the KIT TKR gene. These mutations were shown to activate KIT signaling, and KIT was thus a potential therapeutic target. KIT is a member of the TKR signaling family and is encoded at chromosome 4q11-q12. In GIST tumors, mutations in the KIT gene (reviewed in references 189, 190, and 191)191–191 are located predominantly in the juxtamembrane domain (exon 11), with two thirds of tumors harboring mutations in this region of the gene. Exon 11 mutations allow adenosine triphosphate to phosphorylate receptor in the absence of ligand (stem cell factor). Phosphorylation of the receptor triggers downstream signaling of RAF, MEK, MAPK, PIK3CA, and STAT3, transforming mutated cells to malignant status. Approximately 10% of mutations are found in exon 9, which encodes the extracellular domain of the molecule. These mutations induce a change in receptor conformation mimicking that seen in the presence of ligand and resulting in receptor phosphorylation. Exon 9 mutations are commonly seen in large and small intestinal tumors but rarely in gastric neoplasms. Finally, mutations in exons 13 (tyrosine kinase domain) and 17 (activation loop) are occasionally found in GIST tumors. The effects of exons 13 and 17 mutations on the receptor are less well established, but the net result of all these mutations is receptor activation and oncogenic transformation of mutant cells. KIT mutations occur in approximately three fourths of GIST tumors.191–191
Tumors that do not contain a KIT mutation may contain a mutation in the closely homologous tyrosine kinase receptor gene, platelet-derived growth factor receptor (PDGFR), alpha polypeptide (PDGFRA).192,193 PDGFR protein has two isoforms, α and β, and it is the α form that is mutated. Approximately 10% of GIST tumors contain mutations in PDGFRA.191–191 The mutated loci in PDGFRA are distributed similarly to KIT, but frequencies differ. The most frequent mutation site (exon 18) encodes a portion of the tyrosine kinase domain (TK2) and occurs in 6% of all GISTs. Juxtamembrane mutations in PDGFRA are relatively infrequent, occurring in <1% of GISTs.
Introduction of TKIs that block signaling by mutant receptor has had a profound effect on the outlook for this tumor. Prior to 2000, the outlook for patients with an advanced GIST was bleak. Most patients experienced a recurrence after surgical resection, and responses to chemotherapy were poor. Only 10% of patients were disease free after extended follow-up.194,195 A dramatic reversal of this situation occurred when it was realized that GIST is exquisitely sensitive to tyrosine kinase blockade. The drug successfully used to treat GIST was originally designed to block signaling by the BCR-ABL fusion protein in chronic myelogenous leukemia. Structural similarities between the intended target of the drug and KIT led to the discovery that the TKI now known as imatinib strongly inhibits mutated KIT.196 Subsequent clinical trials demonstrated high response rates and prolonged progression-free survival (PFS),199–199 leading to FDA approval of the drug as discussed elsewhere in this volume. KIT testing is now used routinely to identify persons likely to respond to the drug.
PDGFRA mutations are not equivalent to those in KIT. More than half of PDGFRA mutations encode a D842V amino acid substitution that is resistant to imatinib, whereas other mutations in PDGFRA are sensitive.200 Alternative drugs that show activity against the D842V resistance mutation such as midostaurin (PKC412)201 are under investigation.
Colon Carcinoma
KRAS as a Prognostic Marker in CRC
KRAS mutation occurs in approximately 40% of colon carcinomas. The independent effect of KRAS mutation on outcome in colon cancer is still unclear and remains controversial.202 In a series of 3,439 patients with CRC, it was found that of the 12 possible mutations on codons 12 and 13, only the glycine to valine mutation on codon 12 (8.6%) had a statistically significant impact on outcome.203 Smaller series have shown similar results, but retrospective data from other large randomized studies have failed to consistently demonstrate a meaningful effect of KRAS mutation on outcome in CRC. In a study of a large number of untreated patients, there was no difference in PFS (7.3 weeks) between mutant and nonmutant tumors.204
KRAS as a Predictor of Treatment Response in CRC
In 2007, a study published in the New England Journal of Medicine205 reported that administration of the monoclonal antibody, Cetuximab, to patients with advanced CRC expressing immunohistochemically detectable EGFR resulted in improved survival compared with untreated control subjects. Both treatment and control arms had been treated with a fluoropyrimidine, irinotecan, and oxaliplatin or had contraindications to treatment with these drugs. The difference in survival, although not large, suggested that this specific treatment could be useful in managing CRC and prompted a closer look at responders versus nonresponders. Several subsequent studies indicated that KRAS mutation status dictates response to anti-EGFR monoclonal antibody treatment in persons with CRC. In the randomized studies summarized in Table 17-4,204,206–208 wild-type tumors treated with anti-EGFR antibody (Cetuximab or Panitumumab) have a PFS advantage of several months and a significantly reduced hazard ratio in comparison with mutated tumors in first-line and third-line settings.
Table 17-4
KRAS Mutation as a Predictive Biomarker in Clinical Trials of Metastatic Colorectal Carcinoma
| Study | Treatment | No. Patients | KRAS Mutant* | Hazard Ratio | KRAS Wild Type* | Hazard Ratio | Reference |
| Amado | Panitumumab vs. BSC† (third line) | 427 | 7.4 wk | 0.99 | 12.3 wk | 0.45 | Amado et al., 2008204 |
| Karapetis | Cetuximab vs. BSC† (third line) | 572 | 1.8 mo | 0.99 | 3.7 mo | 0.40 | Karapetis et al., 2008206 |
| Van Cutsem (CRYSTAL) | FOLFIRI ± Cetuximab (first line) | 540 | 7.6 mo | 1.07 | 9.9 mo | 0.68 | Van Cutsem et al., 2009207 |
| Bokemeyer (OPUS) | FOLFOX ± C (first line) | 233 | 5.5 mo | 1.83 | 7.7 mo | 0.57 | Bokemeyer et al., 2009208 |
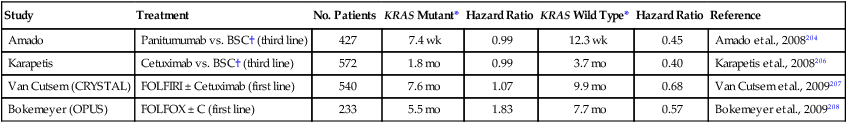
It has recently been suggested that not all mutations are equally predictive of poor response to monoclonal anti-EGFR therapy. Patients with codon 13 mutation (p.G13D) appear to respond to such treatment with a significantly longer overall survival and reduced hazard ratio in comparison with patients with other KRAS mutations.209
BRAF Mutation and Prognosis
A second mutation in colon carcinoma of clinical importance is BRAF. BRAF mutations were first identified in colon cancer through a survey of BRAF mutations in all human tumors.210 The reported frequency of BRAF mutation in clinical samples ranges from approximately 5% to 15%.211–214 BRAF mutation is more frequent in tumors on the right side of the colon.217–217 MSI-H occurs in approximately half of BRAF-mutated tumors.214,217,218 BRAF mutation is associated with aggressive tumor behavior and shortened survival, but only in tumors that are MSI stable214,217,219,220; mutation in MSI-H tumors carries no survival disadvantage. MSI in BRAF-positive tumors is attributed to methylation of MMR rather than mutation and appears to be unrelated to HNPCC and germline mutation of MMR genes.221,222 BRAF mutation in colon tumors confers resistance to anti-EGFR monoclonal antibody inhibitors,213,223–225 and BRAF mutated tumors respond poorly to BRAF inhibitors. Resistance to BRAF inhibitors is thought to be due to activation of EGFR in response to BRAF inhibition.226 It has been suggested that BRAF-positive tumors may therefore respond to dual inhibition of both EGFR and BRAF.
Other Mutations
Additional mutations of predictive importance are NRAS and PIK3CA, which occur at frequencies of <5%211,213,227 and 10% to 15%,213,225,227 respectively, in colon carcinoma. PIK3CA mutations overlap those found in other genes, but NRAS mutations are nonoverlapping with mutations in other genes, including KRAS and BRAF. Mutations in either PIK3CA or NRAS are associated with poor response to EGFR monoclonal antibody therapy.213,225 Finally, initial results of comprehensive molecular analysis by the TCGA have been reported and identify large numbers of mutations that are likely to become targets of specific molecular inhibitors.227 Comprehensive molecular testing is expected to contribute greatly to colon carcinoma management in the near future.
Lung Cancer—A Heterogeneous Tumor
For many years, molecular characterization of lung cancer was impeded by the extreme heterogeneity of this disease as reflected in the most recent revision of the anatomic classification of lung carcinoma.228 Since the 1920s, lung tumors have been divided into small cell (“oat cell”) lung carcinoma (SCLC) and non–small cell lung carcinoma (NSCLC) that differ not only morphologically but express different classes of cell surface molecules, possess different mutational profiles, and exhibit different biological behavior. Small cell carcinomas express a number of markers that are also found on neural cells and in current classifications are categorized as one of several different types of “neuroendocrine” carcinomas. The vast majority of NSCLC does not express neuroendocrine markers but instead expresses large quantities of Erb-B cell surface TKR. SCLC is highly genetically unstable and appears to be driven predominantly by seven transmembrane receptors. In NSCLC, the level of genetic instability is more variable, and the TKRs are major tumor cell drivers. Finally, SCLCs are uniformly aggressive and are particularly sensitive to mitotic inhibitors.
EGFR
The original isolation of EGF from mouse salivary gland in 1962,229 the demonstration of its binding to cell membrane receptor,230 and sequencing of the receptor231 established the role of intercellular signaling in epithelial cell growth and maintenance. By the 1990s, overexpression of EGFR had been demonstrated in squamous carcinoma. Monoclonal antibodies to EGFR that were effective in localizing EGFR in situ became available in the late 1990s and documented the broad expression of EGFR in adenocarcinoma.175 EGFR expression in NSCLC was found to be closely linked to gene copy number.176
Further studies of EGFR were driven by of the discovery of a subset of patients who were mostly nonsmokers with adenocarcinoma who responded dramatically to EGFR TKIs (Fig. 17-9). Simultaneously, two groups reported that mutations in the EGFR tyrosine kinase domain were present in a subset of adenocarcinomas and that their presence correlated with sensitivity to TKI.232,233 The mutations most commonly consisted of either single base substitution in exon 21 (c.2573G>C, p.L858R) or in-frame deletion within exon 19 ranging from 9 to 18 base pairs in the region coding for the adenosine triphosphate binding pocket of the tyrosine kinase domain. These mutations resulted in strong constitutive activation of the encoded EGFR protein.
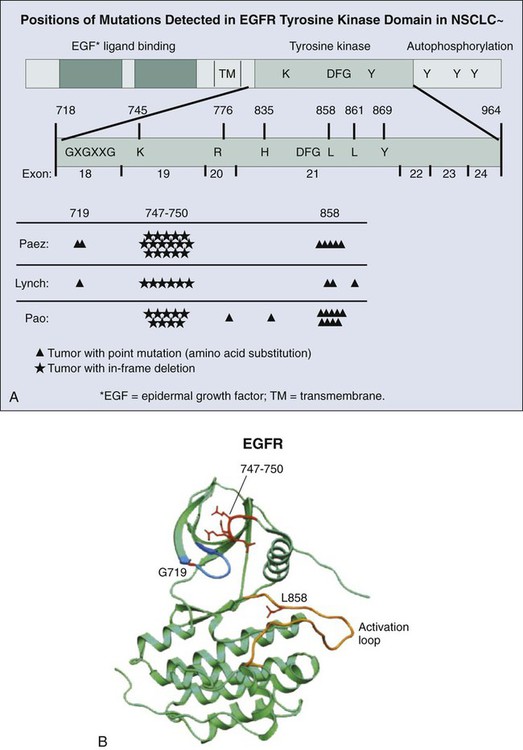
Activating mutations of EGFR result in tumor susceptibility to TKI therapy.234 However, responses to therapy are temporary, and hopes of a long-lasting therapy are generally unrealized because of resistance to targeted therapy that inevitably arises in treated patients. Two common mechanisms of resistance to EGFR TKI are MET amplification237–237 and a point mutation in exon 20 of EGFR (c.2369 C>T, p.T790M),238 the effect of which is depicted in Figure 17-8. As a result of the point mutation in EGFR exon 20, a bulky methionine side chain is thought to sterically hinder binding of TKIs. Tumors cells harboring this mutation may exist in low numbers in untreated tumors and emerge through selective pressure exerted by targeted therapy. In addition, insertions in exon 20 of EGFR are associated with primary resistance to TKI therapy,239 which also is thought to be due to steric hindrance of TKIs.
KRAS
KRAS mutations are more common in lung cancer than are EGFR mutations and, unlike EGFR mutations, they occur predominantly in smokers.240,241 Co-existence of KRAS and EGFR mutation is uncommon but has been reported.242 Because of its position “downstream” of EGFR signaling, the hypothesis that an activating mutation in KRAS would render a tumor nonsusceptible to TKI therapy in lung tumors has been extensively investigated with conflicting results.243
ALK and Other Large-Scale Rearrangements
Point mutations and small deletions are not the only molecular mechanisms driving lung cancer. Overexpression of oncogenes may also be driven by gene activation through chromosomal rearrangements. Rearrangements that fuse gene promoters to the tyrosine kinase domain of growth factors, growth factor receptors, or transcription factors may result in inappropriate signaling from the fusion products. Examples of activation by gene fusion in lung cancer involve ALK, ROS1, and RET. In 2007, Soda et al.244 discovered a transcript derived from a patient with NSCLC that could transform T3T fibroblasts. Further investigation demonstrated the transcript to be fusion of a gene called echinoderm microtubule-associated protein-like 4 (EML4) with the anaplastic lymphoma kinase (ALK) gene (Fig. 17-10), which is also rearranged in large cell lymphomas. This rearrangement has been detected in 4% to 7% of pulmonary adenocarcinomas, with an increased proportion seen when cohorts are selected based on clinicopathological features including lack of smoking history.245–248 The driver of tumorigenesis in this rearrangement is thought to be ALK, which is not typically expressed in adult lung tissue. Juxtaposition of a 5′ partner with a promoter results in high-level expression of the kinase domain of ALK and leads to inappropriate signaling by this molecule.244,249
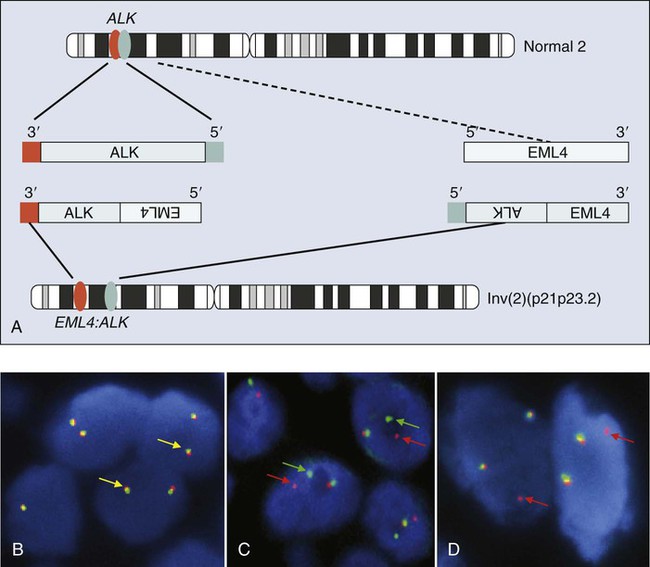
Less than 5 years after its initial discovery, more than 10,000 lung cancers have been tested for the presence of this rearrangement. More than 13 molecular variants of EML4–ALK fusion have been detected,246,250 and three other rare activating gene partners have been identified, including TRK-fused gene (TFG),251 KIF5B,252 and KLC1.253 A novel targeted therapy agent, crizotinib, has been found to be effective against tumors driven by this molecular marker254 and received approval for treatment of these specific tumors by the U.S. Food and Drug Administration. Numerous other ALK inhibitors are in initial clinical trials.
Summary
These three examples, EGFR, KRAS, and ALK, underscore the diversity of molecular alterations in NSCLC, and with ongoing extensive efforts, “driver mutations” have been identified in a significant proportion of lung cancers. Molecular testing can provide guidance for prioritizing therapies to patients who are likely to receive the greatest benefit and, conversely, can identify patients proven not to benefit from a targeted therapy. Finally, molecular testing can provide clinicians with a rationale to guide certain patients toward clinical trials of targeted therapeutics. These selected examples of targets are by no means the only ones of interest in NSCLC, in which a growing number of molecularly targeted agents are under investigation in association with specific molecular alterations. These alterations include mutations in BRAF, HER2, PIK3CA, and rearrangements in RET, among others.255
Melanoma
Several targetable mutations have been found in melanoma in recent years involving the BRAF, NRAS, and KIT genes (see Table 17-3). Of these, the most common is BRAF.256
BRAF
BRAF protein is a serine-threonine kinase that is encoded on chromosome 7q34. It is an important signal transduction molecule that mediates signals from RAS to MEK, ultimately promoting cell proliferation and mobility. Activating missense mutations in BRAF were first identified in a screening of more than 500 cells lines from multiple organ sites.210 Mutations were found in 60% of melanoma lines and tumors at lower frequencies in carcinomas of the colon (15%), lung (3%), breast (3%), and ovary (4%), as well as in glioma (11%). BRAF mutation is now thought to occur in approximately half of primary and metastatic melanoma clinical samples,257–260 with some variation according to melanoma subtype. Activating BRAF mutations largely occur in exon 15, which encodes the catalytic domain of BRAF. By far the most frequent single base change (c.1799T>A) results in an amino acid change, V600E.210,260 This mutation activates continuous intracellular signaling, resulting in cell proliferation, increased tumor cell survival, and enhanced mobility. Two compounds selectively block the kinase activity of mutated BRAF, vemurafenib and dabrafenib. In clinical trials, both compounds have produced high response rates and improved PFS or overall survival in tumors with BRAF mutations.261–264 These studies reinforce the need for accurate molecular characterization of melanoma to allow for selection of effective treatment.
KIT
A second mutation target in melanoma is KIT, reviewed in references 256 and 265 (previously discussed in regard to GIST). KIT mutations differentially affect subsets of melanoma. Mutation frequencies in sun-damaged cutaneous, acral, and mucosal melanomas range from 2% to 20%, but KIT mutations are absent from cutaneous tumors arising in non–sun-damaged skin.256,266 In addition, a different spectrum of mutations affects melanoma compared with GIST.268–268 Whereas KIT mutations in GIST are often short deletions or insertions in exon 11, the vast majority of mutations in melanoma reported to date are point mutations. Mutations in exon 9, which constitute more than 10% of GIST mutations, have not been found in melanoma. Mutations in exon 13, which encode the kinase domain of KIT, are more common in melanoma than in GIST. Finally, KIT gene amplification is frequent in melanoma268–268 but is not described in GIST.189
Therapeutic trials testing the effectiveness of KIT inhibitors on KIT-mutated tumors have had limited success. In a recent phase 2 trial enrolling 51 patients with metastatic melanoma who had genetic alterations of KIT, durable complete responses to imatinib have been reported in two patients with mutations in exon 11 and partial durable responses have been reported in four others for an overall response rate of 16%.268 Another study has reported similar findings (23% overall partial response rate) in tumors with exon 11 and 13 mutations.269 Imatinib blockade of KIT may be a life-extending alternative for patients with BRAF wild-type/KIT-mutated metastatic tumors of specific subtype. This new clinically relevant development will add urgency to the implementation of multiplex testing at many institutions that are equipped to perform molecular testing.
NRAS
NRAS, which is encoded on chromosome 1, is a member of the RAS family of signaling molecules and was originally identified as an oncogene in neuroblastoma cells.270,271 Mutation in melanoma was reported shortly after discovery of the gene.272 NRAS is altered in approximately 20% to 30% of melanomas of all types256,260 except for uveal melanoma. Mutations are restricted mainly to exon 3, codon 61 (>80%), with a smaller proportion occurring at exon 2, codon 13.260 These mutations are thought to lock NRAS in its activated state. However, NRAS mutation by itself may not be sufficient for malignant transformation because both sporadic and congenital nevi of several types have been shown to harbor NRAS mutation without evidence of malignant transformation.273,274 To date, no drugs are known to directly and effectively target NRAS signaling. However, downstream signal transduction has been the subject of ongoing preclinical investigation.256,265
GNAQ/GNA11/BAP1
Mutations in GNAQ, GNA11, and BAP1 are confined largely to uveal melanomas, blue nevi, and meningeal melanoma. The GNAQ and GNA11 genes encode membrane-bound guanosine-5-triphosphatases that regulate signaling through seven transmembrane receptors. Activating mutations in either GNAQ or GNA11 are reported in more than 80% of uveal melanomas and blue nevi275,276 at codon Q209 and R183 hot spots. BRCA1-associated protein 1 (BAP1) is encoded on chromosome 3p21.1 and is mutated in more than 80% of metastatic uveal melanomas.277 BAP1 mutation has also recently been described in a familial syndrome with development of clinically and histologically characteristic tumors with some features of melanoma. This mutation also occurs in sporadic tumors with similar features.278 BAP1 mutations cause protein truncation and therefore inactivation of the protein. For this reason, BAP1 is thought to represent a tumor suppressor gene. BAP1 mutations are of limited prognostic value and are not yet therapeutic targets but illustrate the highly specific relationships that may exist between morphology and molecular pathology.
Current Technologies and Best Practices for Solid Tumors
Preanalytical Processing
Tissue Collection and Processing
Tissues obtained for solid-tumor molecular testing may come from surgical resections or biopsies (fine-needle aspirates and cores). Most often, the tissue needed for molecular testing is derived from diagnostic material, unless special arrangements are made to specifically collect tissues for molecular studies. Issues regarding specific specimen types are discussed here (also see Chapter 25).
Biopsies
Improved imaging and tissue sampling techniques in recent years, exemplified by advances in lung sampling methods,279,280 have resulted in increasing reliance on small biopsies for diagnosis and monitoring of treatment. The primary objective of these tissue acquisition methods has been to obtain tissue for unequivocal diagnosis, which can require multiple immunohistochemical stains in addition to conventionally stained sections. Meeting this objective can leave little remnant tissue behind for ancillary studies. When tumor is of an advanced stage and surgery is not appropriate, remnant tissue from these diagnostic biopsies may be the only tissue available for molecular testing. Because morbidity from biopsy procedures has declined and the value of information derived from molecular testing has become more critical, performing biopsies solely for molecular analysis has gained increasing acceptance by clinicians and patients alike. The major limitation of biopsy specimens for molecular testing is small tumor cell yield. Also, testing of DNA extracted from paraffin sections must take into account shearing of DNA caused by sectioning. Despite these challenges, isolation of tumor DNA from small specimens of many types including tissue cores, cell block preparations, smears, and imprinted slides has been well documented.281–284 Individual laboratories must validate testing protocols for each specimen type.
Enriching for Tumor Cells
Because nonmalignant cells invariably accompany (“contaminate”) all clinical samples, specimens must be enriched for tumors to a level that is consistent with the analytical sensitivity of the platform that will be used for testing. As a rule of thumb, a minimum of 100 viable tumor cells in a 10-µm section is required for a single PCR-based assay. The quality of the biopsy specimen may be more important than quantity for critical molecular studies.285,286
Choosing Methods of Analysis
A large number of testing methods are now available for molecular analysis, and choosing an appropriate platform can be challenging. For mutation determinations, several testing platforms are available. These platforms include Sanger sequencing, which remains the gold standard for mutation detection, high-resolution melting curve analysis, allele-specific PCR, amplification refractory system (ARMS), often coupled with a bifunctional self-probing primer (Scorpions), pyrosequencing, and allele-specific tests that may involve placement of a lock on mutant DNA and digestion of wild type. In addition, multiplex tests can detect multiple mutations, and Next Generation (“NextGen”) sequencing permits mutational testing for expanded panels, or potentially, the entire genome or exome. Each of these platforms must be independently evaluated and validated in the specific setting of the testing laboratory. Among the parameters that must be assessed are (1) amount and quality of starting material (DNA), (2) assay sensitivity, (3) specificity, (4) coverage of genome, (5) turnaround time, (6) simplicity, (7) start-up costs, (8) cost per test, and (9) clinical validation. The approach to laboratory validation of any individual method is highly complex and requires appropriate evaluation and documentation of a host of performance characteristics, including accuracy, reproducibility, robustness, interfering substances, analytic sensitivity, analytic specificity, and limit of detection.287 Several methods currently used for the identification of mutations in clinical samples are briefly discussed in the following sections.
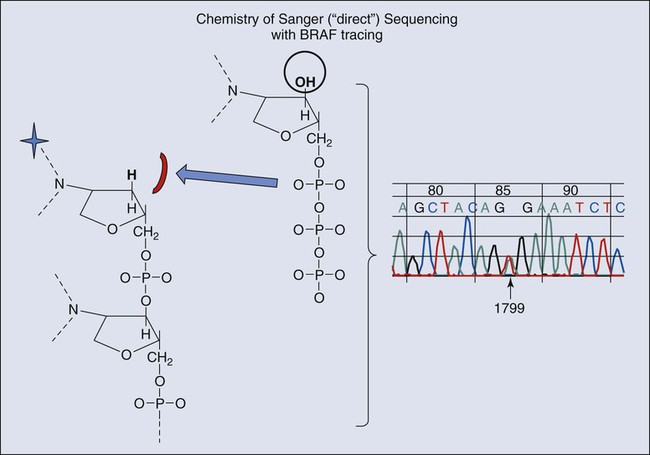
Sanger Sequencing
Sanger sequencing has been the gold standard for many years and, like many other assays, is based on the dideoxy-chemistry illustrated in Figure 17-11. The sequencing reaction is read as a color code that distinguishes oligonucleotides of variable length with four specific color labels. A ladder of nucleotides is created that can be identified by their electrophoretic mobility and color of terminal fluorescent nucleotide. The identification of abnormal (mutant) peaks in chromatograms of this ladder can be facilitated by computer programs that not only create the chromatogram but compare them with reference sequences (RefSeq) and identify abnormalities that represent mutations and polymorphisms.
High-Resolution Melting Analysis
High-resolution melting analysis (Fig. 17-12) measures differences in melting-point temperatures between matched and mismatched DNA doublets. Mismatching causes a reduction in melting-point temperatures that can be quantified using software that is readily available in quantitative PCR instrumentation. The advantage of melting-point analysis is that it is sensitive and requires relatively small amounts of DNA (<5 ng). It also has excellent coverage, detecting any mutational abnormality that may be present in the amplified segment. However, any perturbation of DNA structure, including untargeted SNPs and adducts, may produce aberrant melting-point curves, and nonspecific background for this method is relatively high. The specificity of this reaction is therefore low without confirmation by a second technology, typically direct sequencing, which can negate the rapid turnaround and relative low cost of the assay. Many investigators regard this test as a screening assay that requires confirmation of abnormal results by a second assay.
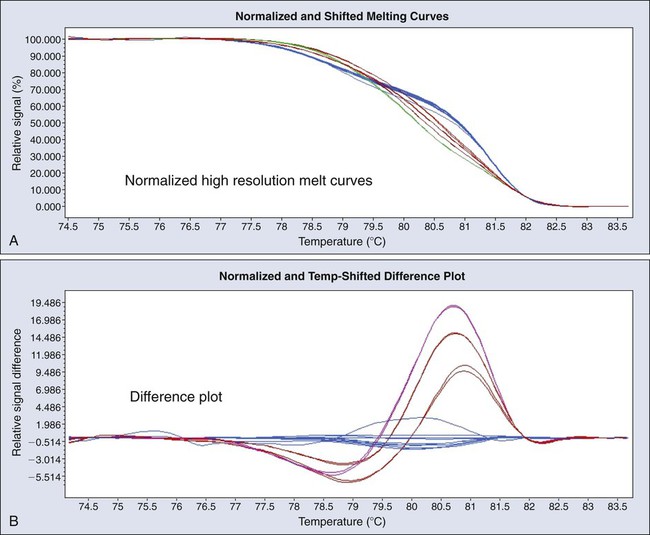
Multiplex Assays
MASSarray (Sequenom, San Diego, Calif.) and SNaPshot (ABI, New York, NY) are based on similar chemistry but use different methods to measure the outcome of sequencing reactions. For MASSarray, a mass biased probe is used, which is detected by mass spectrometry.288 For SNaPshot, a fluorescent probe is used, and standard sequencing capillary gel electrophoresis is used to detect mutant or wild type peaks. PCR reactions are performed with multiplex primers, and panels of primer sets are constructed to detect mutations at multiple sites. The chemistry is based on single base extension from a probe complimentary to the region of interest. Primers are positioned immediately adjacent to the mutated base, and addition of a dideoxynucleotide at the mutation site results in the formation of a labeled oligonucleotide. Probe sets can be designed at different lengths for specific alleles so that they can be multiplexed into panels of up to eight or nine analytes per panel. Amplified probe sets can be measured by the appropriate instrumentation, either mass spectroscopy (MASSarray) or capillary gel electrophoresis (SNaPshot, Fig. 17-13). The amplification incorporated into this technology results in a high level of sensitivity so that only 10% concentration of tumor cells is required for mutation detection.
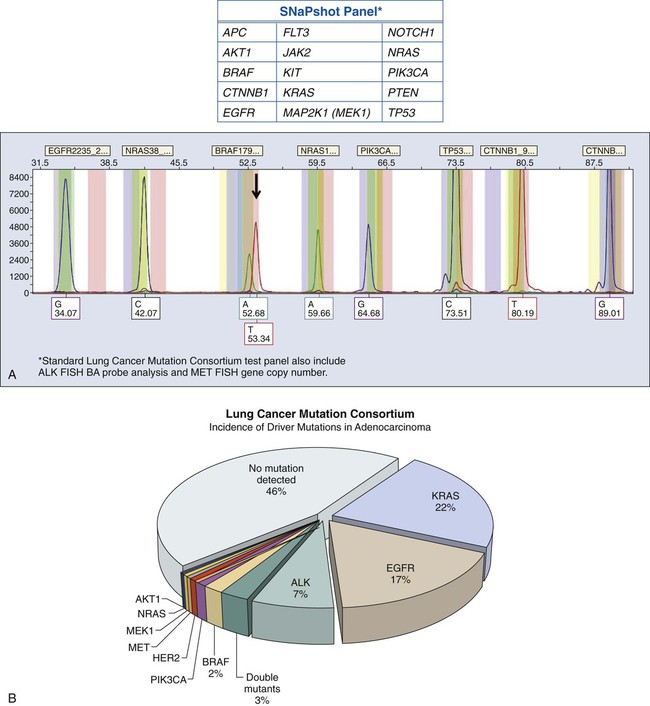
NextGen Sequencing
Advances in sequencing technology are making possible the complete sequencing of the cancer genome158,289,290 through such programs as TCGA, described elsewhere in this volume. It is now nominally possible to sequence the entire genome of a single tumor for individual patients at an accessible charge. Demand for genome-based selection of targeted anticancer agents is expected to rapidly increase. There is thus a developing need for flexible deep sequencing platforms to provide high-density sequencing data with broad coverage in a clinical setting. Two such platforms are currently available, Ion Torrent and MiSeq. Ion Torrent exploits the release of a hydrogen ion by DNA polymerase during sequencing (see Fig. 17-11) and measures changes in pH using an integrated semiconductor array for nonoptical sequencing.291 The alternative MiSeq platform is a “sequence by synthesis” approach that utilizes serial additions of fluorescent-labeled nucleotides to immobilized products across a two-dimensional array that allows for parallel sequencing of thousands of nucleotide strands simultaneously. Published performance characteristics of MiSeq compare favorably with Ion Torrent.292 Both platforms must be thoroughly validated for each target mutation before introduction into a clinical setting. These systems are being rapidly introduced in kit format and are expanding sequencing capabilities of clinical laboratories from single gene or small multiplex sequencing to large-scale parallel sequencing of hundreds of genes simultaneously. It is expected that in the near future, complete sequence coverage of all relevant genomic treatment targets will be available to treating physicians at a local clinical laboratories.
Assays for Gene Rearrangements
Large-scale genomic rearrangements are not easily detected by conventional gene sequencing, and specialized methods have been used for decades to demonstrate innumerable complex abnormalities for which nomenclature is now standardized293 and uniformly applied in searchable databases (http://cgap.nci.nih.gov/Chromosomes/Mitelman). Large-scale genomic changes could potentially be identified by molecular methods such as array CGH, SNP arrays, or NextGen sequencing. However, at the present time, array CGH and SNP arrays test only copy number,294 and NextGen sequencing is not yet sufficiently refined to routinely detect rearrangements in a clinical setting. Only FISH is validated for application to clinical samples to identify complex rearrangements. FISH has the advantages of maintaining histologic architecture, accuracy on small sample sizes, and a wide availability in clinical laboratories. The large number of FISH tests currently in active use cannot be fully addressed here because of space limitations. However, lung cancer provides an example of the relevance of discovery and clinical testing for large-scale gene rearrangements to targeted therapy, as discussed in the next section.
FISH Assays and ALK Rearrangement
Copy number changes and chromosomal rearrangement are ubiquitous in lung carcinoma, but to date, only a small number of these changes are recurrent. One exception is the recently discovered ALK rearrangement. In these rearrangements, ALK is the oncogenic driver, switched on by the rearrangement. The EML4-ALK fusion is generated by a paracentric chromosomal inversion in the short arm of chromosome 2, between bands p21 and p23.2 (see Fig. 17-10). These two genes originally are separated by approximately 12.8 MB. The other described ALK fusions in lung cancer, TFG-ALK,251 KIF5B–ALK,252 KLC1-ALK,253 and PTPN3-ALK,295 are caused by chromosomal translocations t(2;10)(p23.2; p11.22), t(2;3)(p23.2;q12.1), t(2;9)(p23.2;q31.3), and t(2;14)(p23.2;q32.1), respectively.
Numerous technical platforms are available for molecular diagnosis of ALK rearrangements in lung cancer, but the most commonly used in the clinical setting is FISH. The standard FISH assay uses a dual-color break-apart (BA) probe set encompassing the 3′ and the 5′ sequences immediately adjacent to the breakpoint area in ALK (intron 19), labeled in orange and green fluorophores, respectively.296 The chromogenic in situ hybridization BA assay for ALK rearrangements is also commercially available. Despite interesting results and a high level of concordance with FISH,297,298 the ALK chromogenic in situ hybridization assay has not yet been comprehensively explored. The ALK BA FISH probe was used in the initial clinical studies with crizotinib254,299 and is approved by the FDA as a companion diagnostic test. The ALK BA FISH assay is DNA based and is thus robust and resistant to technical artifacts. It has multiple other advantages, such as being highly suitable for FFPE tissue sections, and detects all variants of EML4–ALK, as well as any of the reported or uncharacterized partners. On the other hand, the FISH platform requires specialized resources (e.g., a fluorescence microscope and laboratory space protected from light), along with highly trained personnel.
Recently, two other receptor kinase genes were identified as activated in lung cancers by fusions, ROS1251 and RET.300 Integrated molecular and histopathology-based screening systems have been successful in detecting a large number of fusion partners in lung cancer for these two genes, and preclinical studies have confirmed their tumorigenic potential.252 Clinical trials of targeted agents for ROS1 are currently under way, and rapid expansion of this field is expected.
RNA Assays
This situation could change with the development of specific targeted agents that require an RNA companion diagnostic and by improvement in sensitivity, specificity, and preanalytical requirements of RNA-based testing platforms. As previously mentioned, predictive RNA testing has been most widely tested in the Mammaprint and OncoDx platforms. Mammaprint was originally based on expression microarray testing of a 70-gene panel that required high-quality RNA. Formalin fixation and paraffin embedding produce chemical changes301 in RNA, and RNA may degrade and shear in storage.302 An antidote is to shorten the template to 50 to 100 bases and to use random primers rather than reverse transcribing from the polyA tail of mRNA. Mammaprint has adapted to the need for FFPE testing by incorporating these steps in a new multiplex PCR based-technology.303 OncoDx has used an RT-PCR–based approach to measure a multigene panel.304 Both of these platforms require careful normalization with consistently expressed housekeeping genes to ensure quality of results. Finally, a promising new platform that uses florescent nano bar codes305 linked to sequence-specific probes is being tested with promising early results. A major advantage of this system is that there is no requirement for PCR.