Chapter 39 Neurotransmitter-Related Disorders
For ease of classification, these disorders can be divided into five groups:
Monoaminergic Neurotransmitter Deficiency States with Hyperphenylalaninemia
Overview
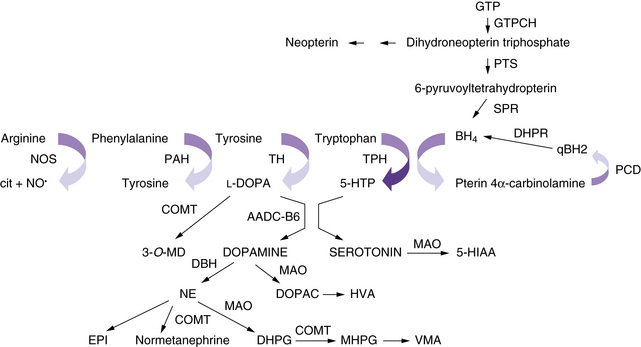
The neurotransmitter deficiency in infants in this group arises as a result of defects in BH4 metabolism (Figure 39-1). Patients are usually identified by elevated phenylalanine levels on newborn screening, as BH4 is required for phenylalanine hydroxylation in the liver. The accompanying neurotransmitter deficiency results from the lack of BH4, an obligatory co-factor required for the synthesis of catecholamines and serotonin. Although most academic biochemical genetics clinics that monitor children with phenylketonuria systematically perform the additional studies required to diagnose this group of disorders, children occasionally are not identified until they have progressive neurologic symptoms or clear evidence of developmental delay despite a phenylalanine-restricted diet. In the past, these patients were referred to as atypical phenylketonurics. In some cases, such afflicted infants are overlooked because screening is performed before an adequate interval of protein intake, resulting in a false-negative result on newborn testing.
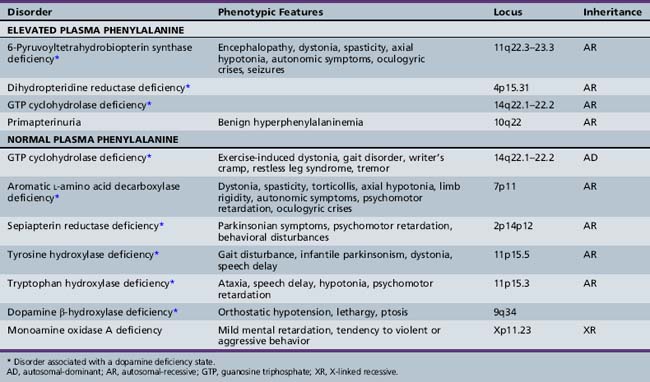
Approximately 1–3 percent of patients with hyperphenylalaninemia have an associated BH4 deficiency state. It is critical to identify such children so that BH4 and neurotransmitter precursors can be supplemented as early as possible. The two most commonly identified disorders in children presenting with hyperphenylalaninemia in the newborn period are 6-pyruvoyltetrahydropterin synthase deficiency, accounting for 60 percent of mutations in atypical phenylketonurics, and dihydropteridine reductase deficiency, affecting 30 percent of cases [Longo, 2009]. 6-Pyruvoyltetrahydropterin synthase deficiency results in inadequate BH4 synthesis; dihydropteridine reductase deficiency results in decreased regeneration of BH4 from dihydrobiopterin (see Figure 39-1). Both are autosomal-recessive disorders in which hyperphenylalaninemia results from a deficiency of BH4. Because of the involvement of BH4 in catecholamine and serotonin synthesis, such infants also have a manifest deficiency of neurotransmitter metabolites in addition to hyperphenylalaninemia. Other conditions in this category include autosomal-recessive guanosine triphosphate cyclohydrolase (GTPCH) deficiency and primapterinuria (Table 39-2).
Role of BH4 in the Central Nervous System
Because BH4 is required for the hydroxylation of aromatic amino acids, its importance in the central nervous system (CNS) becomes immediately apparent. Tyrosine and tryptophan are required for the synthesis of catecholamines and serotonin. A BH4-dependent process can be strongly suspected when plasma phenylalanine levels return to normal after BH4 supplementation. A dose of 5–10 mg/kg of BH4 is the usual recommended dose for correction of peripheral hyperphenylalaninemia, and 10–20 mg/kg per day has been recently advocated in classic phenylketonuria. Because BH4 crosses the blood–brain barrier poorly, lifelong supplementation with the neurotransmitter precursors l-DOPA and 5-hydroxytryptophan, along with carbidopa to prevent peripheral decarboxylation, is necessary in most of the disorders mentioned earlier. A much higher dose of BH4, approximately 20 mg/kg, can return BH4 levels in the cerebrospinal fluid to normal, but it remains prohibitively expensive, and no studies exist as to the possible additional benefit of such a regimen. Dosing can be monitored by assessing clinical response and periodically measuring cerebrospinal fluid levels of neurotransmitter metabolites [Hyland, 2007]. Verifying normalization of serum prolactin levels has been advocated, but sensitivity is limited, as is detection of overadministration of precursors [Concolino et al., 2008]. The greater requirement of tyrosine hydroxylase for BH4 in comparison with tryptophan hydroxylase may explain the more severe impairment in the catecholaminergic system compared with the serotonergic system.
6-Pyruvoyltetrahydropterin Synthase Deficiency
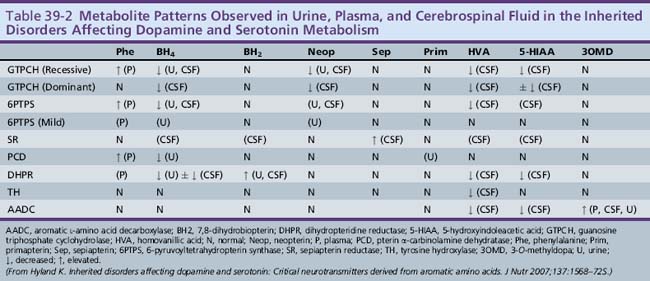
6-Pyruvoyltetrahydropterin synthase catalyzes the elimination of inorganic triphosphate from dihydroneopterin triphosphate to form 6-pyruvoyltetrahydropterin. Patients have elevated neopterin to biopterin ratios in urine and plasma. Reduced 6-pyruvoyltetrahydropterin synthase activity can be documented in red blood cells. In the classic form of the disorder, patients have reduced catecholamine and serotonin metabolites and an increased neopterin to biopterin level in cerebrospinal fluid. Patients are usually detected by newborn screening, as with phenylketonurics. They exhibit progressive signs of neurologic involvement in the first few months of life, including extrapyramidal signs, axial and truncal hypotonia, hypokinesia, feeding difficulties, choreoathetotic or dystonic limb movements, and autonomic symptoms. Many of these patients, despite early diagnosis and supplementation with BH4 and neurotransmitter precursors, continue to manifest delay in development [Dudesek et al., 2001]. A “peripheral” form of the disorder is associated with nearly one-third of known mutations with the human PTS gene [Thony and Blau, 2006], and is characterized by normal central neurotransmitter levels and less significant or transient hyperphenylalaninemia [Niederwieser et al., 1987]. Patients with the peripheral form have an excellent prognosis for normal neurologic development, provided the hyperphenylalaninemia is corrected by diet or BH4 administration.
Dihydropteridine Reductase Deficiency
Dihydropteridine reductase deficiency manifests in a variety of phenotypes, all with hyperphenylalaninemia. The clinical presentation is similar to that of central 6-pyruvoyltetrahydropterin synthase deficiency. Without folinic acid to restore methyltetrahydrofolate status in the CNS, these patients can have progressive calcification of the basal ganglia and subcortical regions, despite treatment with BH4 and neurotransmitter precursors [Woody et al., 1989]. A juvenile variant has been reported in which siblings were developmentally normal until 6 years of age, at which time they developed progressive encephalopathy, epilepsy, and pyramidal, cerebellar, and extrapyramidal features on clinical examination [Larnaout et al., 1998]. Diagnosis can be confirmed by the pattern of urine pterins and documentation of abnormal dihydropteridine reductase activity in skin fibroblasts [Milstien et al., 1980]. Results of phenylalanine loading tests are abnormal, and phenylalanine status improves or returns to normal with BH4 supplementation. Cerebrospinal fluid neurotransmitter and pterin analysis reveals reduced concentrations of homovanillic acid and 5-hydroxyindoleacetic acid, decreased or normal BH4 levels, and elevated dihydrobiopterin levels.
Pterin-4α-Carbinolamine Dehydratase Deficiency (Primapterinuria)
Pterin-4α-carbinolamine dehydratase deficiency, or primapterinuria, is a cause of mild hyperphenylalaninemia [Blau et al., 1988; Adler et al., 1992]. These infants are usually identified on newborn screening but generally have a benign course with normal development. Phenylalanine hydroxylase catalyzes the conversion of phenylalanine to tyrosine, during which BH4 is converted to the unstable carbinolamine 4α-hydroxytetrahydrobiopterin [Citron et al., 1993]. Carbinolamine dehydratase catalyzes the dehydration of carbinolamine to quinonoid dihydropterin (qBH2). The decreased rate of dehydration is responsible for the production of 7-biopterin in some mildly hyperphenylalaninemic individuals [Thony et al., 1998]. Urine studies reveal an excess of 7-substituted pterins, reduced biopterin levels, and an increased neopterin to biopterin ratio.
Monoaminergic Neurotransmitter Deficiency States without Hyperphenylalaninemia
Segawa’s Disease or Autosomal-Dominant Dopa-Responsive Dystonia
The best-described and most widely identified entity among this group of disorders is autosomal-dominant dopa-responsive dystonia caused by GTPCH deficiency, or Segawa’s disease [Segawa et al., 1976]. Identification and treatment of this disorder can be extremely rewarding because patients often benefit greatly from directed treatment of the associated dopamine deficiency state [Harwood et al., 1994]. Patients with a classic presentation of exercise-induced dystonia are not difficult to recognize. This diagnosis should also be considered in patients with spastic diplegia, especially when significant fluctuation in gait or worsening gait at the end of the day is noted, and in patients with more atypical presentations, including writer’s cramp, asymmetric limb dystonia, tremor, or restless leg-type symptoms. In patients with a classic presentation, many clinicians can make the diagnosis on a presumptive basis, after observing remission of symptoms with a trial of l-DOPA/carbidopa.
As of 2009 there are more than 85 known mutations of the GCH-1 gene, and in a large review of dopa-responsive dystonia, it was found that 60 percent are point mutations [Clot et al., 2009]. Although inheritance is autosomal-dominant, penetrance is incomplete, and variable expressivity among family members with the same mutation is well documented [Steinberger et al., 1998]. For instance, one might see spastic diplegia, writer’s cramp, restless leg syndrome, and more typical exercise-induced dystonia phenotypes among different members of the same family. The female to male ratio in sporadic cases is 4:1, and investigators have confirmed increased penetrance of GTPCH-I gene mutations in females [Ichinose et al., 1994; Furukawa et al., 1998]. Of note, a review of 47 patients with GCH-1 mutations or deletions found 40 patients manifested dystonic symptoms in the first decade of life [Clot et al., 2009].
Not surprisingly, mood and sleep disorders appear to be unusually prevalent in families with the disorder. Recent study of the neurologic and psychiatric phenotype of affected individuals within three extended families showed a higher frequency of major depressive disorder and obsessive-compulsive disorder, as well as disrupted sleep in 55 percent of patients [Van Hove et al., 2006]. These data help to support the idea that the disease carries an increased burden of attentional difficulties, anxiety, dysphoria, depression, and sleep disorders.
Aromatic l-Amino Acid Decarboxylase or Dopa-Decarboxylase Deficiency
Aromatic l-amino acid decarboxylase is a pyridoxine-dependent enzyme that decarboxylates l-DOPA and 5-hydroxytryptophan to make dopamine and serotonin, respectively. Patients with this disorder typically present in the first few months of life with dystonia or intermittent limb spasticity, axial and truncal hypotonia, oculogyric crises, autonomic symptoms, and ptosis [Hyland et al., 1992]. Neonatal symptoms, including poor suck and feeding difficulties, ptosis, lethargy, and hypothermia, are common. Neurologic signs and symptoms are clearly evident within the first few months of life in all patients described to date. These patients demonstrate multisystemic involvement with a wide array of neurologic difficulties, including problems with sleep, attention, emotional regulation, and cognitive function that extend well beyond their motor difficulties. As they get older, gross motor delays with fluctuating tone, ataxia, and expressive speech impairment are prominent features, even in the patients with the best outcomes.
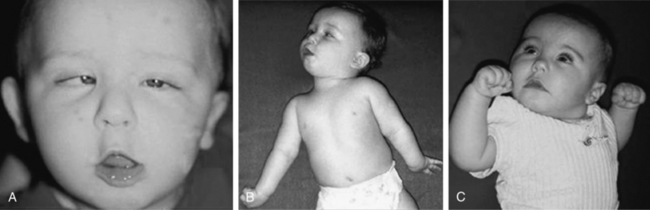
The phenomenology of the movement disorder is remarkably similar among the cases and, not surprisingly, shares a number of features in common with children with BH4 deficiency disorders, such as 6-pyruvoyltetrahydropterin synthase deficiency and dihydropteridine reductase deficiency, and the autosomal-recessive form of GTPCH deficiency. Intermittent oculogyric crises and limb dystonia, generalized athetosis, and an overall paucity of voluntary movement become evident between 1 and 6 months of age. Tongue thrusting, ocular convergence spasm (Figure 39-2), myoclonic jerks, and episodes of sudden loss of head control or episodes resembling flexor spasms are common and frequently lead to a clinical diagnosis of epilepsy. Oculogyric crises, orofacial dystonia, torticollis, limb tremor with attempted voluntary movement, and blepharospasm are often the most compelling evidence supporting a defect in dopaminergic transmission. Breath-holding or apneic spells, paroxysmal sweating, nasal congestion, sudden respiratory or cardiorespiratory arrest, unresponsiveness associated with hypoglycemia, intermittent hypothermia, and feeding and gastrointestinal issues are manifestations of the often profound autonomic dysfunction these patients demonstrate [Swoboda et al., 2003]. First- and second-degree relatives of affected individuals have been shown to have a high incidence of psychiatric disorders, including depression, psychosis, suicide, or suicide attempts [Manegold et al., 2009].
Cerebrospinal fluid neurotransmitter metabolites demonstrate a characteristic pattern: low homovanillic acid and 5-hydroxyindoleacetic acid levels; markedly elevated 3-O-methyldopa, 5-hydroxytryptophan, and l-DOPA; and normal biopterin and neopterin levels. Plasma l-DOPA is markedly elevated. Urine catecholamines may be reduced or elevated, specifically with vanillactic acid, despite normal preliminary organic acid results [Manegold et al., 2009; Abeling et al., 1998, 2000].
Although some children benefit in terms of the underlying movement disorder, treatment is complex, and these patients are vulnerable to an array of medication-related side effects. Instead of replacement of neurotransmitter precursors, as in the BH4 deficiency-related disorders, the use of neurotransmitter receptor agonists or strategies to hinder reuptake or metabolism of endogenously produced neurotransmitters is necessary. Reported benefit has been noted in a subset of patients with monoamine oxidase inhibitors, dopamine receptor agonists, anticholinergic agents, pyridoxine, and, in rare cases associated with a defect in the gene encoding aromatic l-amino acid decarboxylase at the dopa-binding site, l-DOPA [Swoboda et al., 1999]. Dopamine receptor antagonists, such as clozapine, have been proposed as a new therapeutic option, given their increase in aromatic l-amino acid decarboxylase activity in a mouse model [Allen et al., 2009].
However, in spite of a variety of treatment interventions directed at ameliorating the effects of the associated neurotransmitter deficiency state, overall clinical outcomes in aromatic l-amino acid decarboxylase deficiency remain poor. All patients have had some degree of cognitive impairment, and there is increasing evidence of a gender-dependent discrepancy in severity, with females demonstrating the most severe phenotypes [Pons et al., 2004].
Sepiapterin Reductase Deficiency
Sepiapterin reductase catalyzes the (NADP) reduction of carbonyl derivatives, including pteridines, and plays an important role in BH4 biosynthesis. Somewhat surprisingly, the first identified cases had normal plasma phenylalanine and urine pterin levels [Bonafe et al., 2001b]. Blau and colleagues have hypothesized that peripheral tissues can use alternative carbonyl, aldose, and dihydrofolate reductases to perform the last two steps in BH4 biosynthesis. Therefore, BH4 levels in the liver are likely to be normal, probably explaining the absence of hyperphenylalaninemia in these patients. In addition, it is likely that low dihydrofolate reductase activity in the brain allows accumulation of dihydrobiopterin that inhibits tyrosine and tryptophan hydroxylases and uncouples neuronal nitric oxide synthase, leading to neurotransmitter deficiency and neuronal cell death. Thus, identification of low cerebrospinal fluid neurotransmitter levels and the presence of elevated cerebrospinal fluid dihydrobiopterin is crucial for making the diagnosis in these patients.
Few patients have been described to date [Neville et al., 2005; Blau et al., 2001; Bonafe et al., 2001a]. Dystonic posturing, oculogyric crises, spasticity, tremor, ataxia, and psychiatric disturbances have been reported. The largest and most recent study reviewed clinical phenotypes in seven children, showing uniform early motor delay and diurnal variation in motor symptoms, with a high frequency of oculogyric crises, dystonia, and hypotonia [Neville et al., 2005]. All had cognitive delay to some extent, and some had parkinsonian symptoms and bulbar involvement. Also, in all cases, significant motor improvement resulted from low-dose l-DOPA/carbidopa therapy (range 1–6 mg/kg/day), although cognitive outcomes were not changed by therapy. In addition to l-DOPA/carbidopa therapy, there has been anecdotal improvement with the use of selegiline, a monoamine oxidase B inhibitor that prolongs the half-life of dopamine at the synapse. Cerebrospinal fluid neurotransmitter metabolite and pterin analysis reveals low levels of homovanillic acid and 5-hydroxyindoleacetic acid and high levels of biopterin, dihydrobiopterin, and sepiapterin. Diagnosis can be confirmed by documenting low sepiapterin reductase activity in skin fibroblast cultures.
Tyrosine Hydroxylase Deficiency or Autosomal-Recessive Dopa-Responsive Dystonia
Tyrosine hydroxylase deficiency, sometimes referred to as autosomal-recessive Segawa’s disease, displays a diverse phenotype ranging from exercise-induced dystonia to progressive gait disturbance and tremor in childhood to severe infantile parkinsonism [Bodeau-Pean et al., 1999; De Lonlay et al., 2000; de Rijk-Van Andel et al., 2000]. A wide range of symptoms can be associated with tyrosine hydroxylase deficiency, with mild, moderate, and severe phenotypes. In a large review of dopa-responsive dystonia, tyrosine hydroxylase defiency accounted for less than 5 percent of all cases [Clot et al., 2009].
Patients variably respond to l-DOPA/carbidopa, and some have complete reversal of symptoms. The exception to this is the patient with the severe infantile parkinsonism form. These patients sometimes tolerate l-DOPA poorly, with excessive dyskinesia, irritability, and reflux, or have incomplete or inadequate response with regard to their motor manifestations of the disorder [Brautigam et al., 1999; Dionisi-Vici et al., 2000]. When diagnosis occurs late in such patients, motor development must be recapitulated, and continued slow improvement during months is to be expected. Typical features in infants affected by the severe infantile parkinsonism variant include rigidity, tremor, bradykinesia, oculogyric crises, and severe psychomotor delay.
Treatment with l-DOPA/carbidopa alone may lead to severe dyskinesias with marked on-off effects in such patients. Addition of dopamine agonists such as selegiline or anticholinergic agents like trihexyphenidyl can provide significant benefit and help promote the gradual on-going attainment of motor skills and ability to ambulate independently, but such achievements may occur over years, rather than months or weeks, in the most severely affected patients. Whereas more mildly affected patients may tolerate 1–3 mg/kg of l-DOPA per dose, 2–4 times per day, severely affected infants and younger children require l-DOPA quantities as low as 0.2 mg/kg/dose. Breaking standard formulations of l-DOPA/carbidopa into more than two doses per tablet is not advisable because the amount of carbidopa will then be insufficient to help enhance transport of l-DOPA across the blood–brain barrier and to minimize peripheral side effects, such as nausea, reflux, decreased appetite, and vomiting. Carbidopa should be maintained at a minimum of approximately 1 mg/kg/dose up to 25 mg per dose, no matter the l-DOPA requirement. Slow institution of small, compounded doses of l-DOPA/carbidopa, along with selegiline (monoamine oxidase B inhibitor) and an anticholinergic agent such as trihexyphenidyl, may be more beneficial than l-DOPA/carbidopa alone [Haussler et al., 2001]. Selegiline doses may also require compounding because low-dose formulations are not available for use in infants. Frequent side effects of excessive dosing include eating disorders, nightmares, and insomnia. Dosing should be started in the range of 0.1 mg/kg and increased as tolerated. Patients with a mild form of the disorder, such as an isolated gait disorder or exercise-induced dystonia, respond well to monotherapy with l-DOPA/carbidopa and rarely develop dyskinesia.
Although inheritance to date in most families seems to be recessive, at least one family has been described in which the father of the affected proband had mild exercise-induced dystonia responsive to therapy, raising the possibility that tyrosine hydroxylase deficiency could present in an autosomal-dominant fashion in some families with a milder phenotype [Furukawa et al., 2001].
Tryptophan Hydroxylase Deficiency
Tryptophan hydroxylase catalyzes the BH4-dependent hydroxylation of tryptophan to 5-hydroxytryptophan, which is then decarboxylated to form serotonin. Tryptophan hydroxylase expression is limited to certain cells in the CNS and periphery, including raphe neurons, pinealocytes, mast cells, mononuclear leukocytes, beta cells of the islets of Langerhans, and enterochromaffin cells of the gut. Patients with presumed tryptophan hydroxylase deficiency have been described, although it is not yet certain that their symptoms result from tryptophan hydroxylase deficiency [Raemaekers et al., 2001]. Clinical features, consisting of ataxia, speech delay, mild psychomotor retardation, and hypotonia, are nonspecific. Cerebrospinal fluid neurotransmitter metabolite and pterin studies demonstrate the expected low 5-hydroxyindoleacetic acid with normal homovanillic acid, neopterin, and biopterin levels. Mutations in the gene encoding tryptophan hydroxylase have not yet been identified, although identification of isoforms specific to nervous tissue will likely facilitate future research [Invernizzi, 2007].
Dopamine β-Hydroxylase Deficiency
Dopamine β-hydroxylase is the enzyme that converts dopamine to norepinephrine. Presenting symptoms of this disorder have been largely attributed to the importance of this enzyme in postganglionic sympathetic neurons [Robertson et al., 1986]. Patients with severe deficiency of this enzyme, however, cannot synthesize norepinephrine, epinephrine, and octopamine in CNS or peripheral autonomic neurons. Dopamine acts as a false neurotransmitter for noradrenergic neurons. Neonates with dopamine β-hydroxylase deficiency can have episodic hypothermia, hypoglycemia, and hypotension, leading to early death. Survivors do fairly well until late childhood, when overwhelming orthostatic hypotension profoundly limits their activities. The hypotension can be so severe as to lead to convulsive syncope with recurrent clonic seizures [Robertson et al., 1991].
Monoamine Oxidase Deficiency
Monoamine oxidase is a mitochondrial enzyme involved in the catabolism of biogenic amines. Monoamine oxidase A, the primary type in fibroblasts, preferentially degrades serotonin and norepinephrine. Monoamine oxidase B, the primary type in platelets and in the brain, preferentially degrades phenylethylamine and benzylamine. These enzymes are critical in the neuronal metabolism of catecholamine and indoleamine neurotransmitters. The genes are closely linked on the X chromosome, near the Norrie’s disease locus, and only affected boys have been identified to date [Schuback et al., 1999]. Comparisons of the neurochemical characteristics of previously described patients with combined monoamine oxidase A and B deficiency and selective monoamine oxidase A deficiency have led to an improved understanding of the roles of monoamine oxidase A and monoamine oxidase B in the metabolic degradation of catecholamines and other biogenic amines, including serotonin and the trace amines.
Monoamine Oxidase A Deficiency
Brunner described a family with an X-linked nondysmorphic mild mental retardation and a tendency to aggressive or violent behavior, including arson, attempted rape, exhibitionism, and attempted suicide [Brunner et al., 1993b]. Urine studies revealed marked disturbance of monoamine metabolism. Normal platelet monoamine oxidase B activity suggested that the unusual behavior pattern in this family might be caused by isolated monoamine oxidase A deficiency, which was later confirmed by identification in all affected males of a point mutation leading to premature termination of the protein [Brunner et al., 1993a].
Measurement of 3-methoxy-4-hydroxyphenylglycol, a metabolite of norepinephrine, in plasma is the most sensitive index of monoamine oxidase A activity in humans and can be used to screen potential cases. Monoamine oxidase A enzyme activity can be measured directly from fibroblasts. The inability to identify additional patients, despite screening of at-risk males with mental retardation or a behavioral phenotype, makes it likely that this disorder is rare [Segawa et al., 1976], although report of a knockout mice strain involving monoamine oxidase A with enhanced aggression will likely provide new directions for research [Scott et al., 2008]. Interestingly, a high-activity monoamine oxidase A promoter allele has been found with increased frequency in women with panic disorder [Deckert et al., 1999]. A possible association of decreased enzyme activity in women with bipolar disorder has also been reported [Preisig et al., 2000].
Monoamine Oxidase B Deficiency
Isolated monoamine oxidase B deficiency has not yet been reported in a patient. Two brothers with a microdeletion, including the Norrie locus and monoamine oxidase B, however, had features consistent with Norrie’s disease alone, with congenital blindness and progressive hearing loss caused by cochlear degeneration in adolescence. These patients had neither abnormal behavior nor mental retardation, leading the authors to conclude that monoamine oxidase A plays a more significant role than does monoamine oxidase B in the metabolism of biogenic amines, and monoamine oxidase B deficiency alone may have a primarily neurochemical phenotype: that of increased phenylethylamine in urine [Lenders et al., 1996].
Monoamine Oxidase A and B Deficiency
Conclusions about the phenotype of individuals with combined deficiency of monoamine oxidase A and monoamine oxidase B come primarily from a study of a single boy with a microdeletion at Xp14 involving the Norrie locus and documented severe deficiency of monoamine oxidase A and B activity [Collins et al., 1992]. He was severely mentally retarded and blind, and he had other neurologic features, including myoclonus and tendency for motor stereotypies. Because Norrie’s disease is an X-linked recessive disorder, obligate carriers would not be expected to have symptoms. In this family, two obligate carriers had normal intelligence. The proband’s mother had psychiatric symptoms characterized by “chronic hypomania and schizotypal features,” however, and both carriers had low monoamine oxidase activity.
Disorders of Amino Acid Neurotransmitters
Amino acid neurotransmitters are the main inhibitory and excitatory messengers in the nervous system. Although the list of proposed neurotransmitters is long, relatively few have been found to play a role in human neurologic disease. Among these, disorders of GABA and glycine have undergone the most study, yet current understanding is far from complete. Glycine, a simple amino acid with ubiquitous function, has both excitatory and inhibitory properties. Glycine encephalopathy, formerly referred to as nonketotic hyperglycinemia, is a disorder of glycine degradation that typically manifests with neonatal encephalopathy, lethargy, hypotonia, myoclonus, and apnea. It will be touched on briefly, as more extensive review occurs elsewhere in this text. Disorders of GABA degradation will also be reviewed (Figure 39-3), with specific emphasis on succinic semialdehyde dehydrogenase deficiency, the most common and best characterized. Previously, it was thought that disordered GABA synthesis might be related to the clinical entity of pyridoxine-responsive epilepsy; however, recent elucidation of the epileptogenic mechanism has proven it to be due to lysine degradation [Mills et al., 2006] and so it will not be discussed in this chapter.
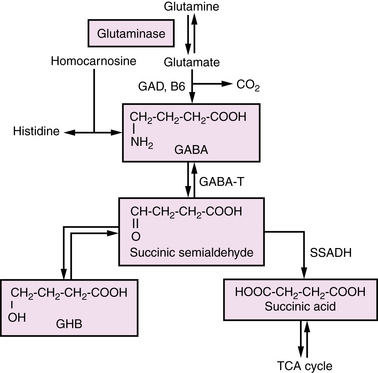
Fig. 39-3 The gamma-aminobutyric acid metabolism pathway.
(From Pearl PL, Gibson KM. Clinical aspects of the disorders of GABA metabolism in children. Curr Opin Neurol 2004;17:107–113.)
Gamma-Aminobutyric Acid Transaminase Deficiency
GABA is the major inhibitory neurotransmitter of the brain, derived primarily from glutamate, the major excitatory neurotransmitter. The first step of the GABA degradation pathway involves GABA transaminase, which removes an amino group from GABA and adds it to alpha-ketoglutarate, thus replenishing glutamate and re-establishing the closed loop system known as the GABA shunt. GABA transaminase deficiency is a rare, autosomal-recessive disorder characterized by abnormal development, seizures, and high levels of GABA in serum and cerebrospinal fluid [Pearl and Gibson, 2004]. Publications to date have presented limited family and case studies, all of which have had poor outcomes. Definitive diagnosis can be made by measurement of GABA transaminase activity in liver, lymphocytes isolated from whole blood, or Epstein–Barr virus-transformed cultured lymphocytes [Gibson et al., 1985].
Succinic Semialdehyde Dehydrogenase Deficiency
Succinic semialdehyde dehydrogenase deficiency is an autosomal-recessive inborn error of metabolism associated with a defect in the metabolism of GABA [Gibson et al., 1998]. Phenotypic features range from nonspecific global developmental delay and hypotonia to ataxia, severe mental retardation, visual impairment, and seizures. The course of the disease is somewhat unique in regard to other neurotransmitter abnormalities in that it is not intermittent or episodic, making distinction from static encephalopathies challenging [Pearl et al., 2009]. Neuropsychiatric symptoms are prominent and include sleep disorders, inattention, hyperactivity, and anxiety [Pearl et al., 2009]. Generalized epilepsy affects nearly half of patients, with over half having abnormal electroencephalograms (EEGs) [Pearl et al., 2009]. MRI is commonly abnormal in patients with succinic semialdehyde dehydrogenase deficiency, with increased T2-weighted signal in the globus pallidi, subcortical white matter, cerebellar dentate nucleus, and brainstem [Pearl et al., 2009].
Urine organic acid screening to detect elevated 4-hydroxybutyric acid is the most easily available screening strategy, but GABA levels in cerebrospinal fluid and urine are also elevated. Improvement of seizures in a mouse model of the disorder was demonstrated with treatment with vigabatrin or a GABA B receptor antagonist [Hogema et al., 2001]. Unfortunately, this medicine has not been consistently helpful in humans and there is considerable risk involved with its administration. Investigations into other potential therapies, including taurine, the ketogenic diet, and GABA B receptor antagonists is currently under way in mouse models [Pearl et al., 2009].
Secondary Neurotransmitter Deficiency States
Menkes’ disease is an X-linked recessive disorder in which affected males have progressive encephalopathy, spasticity, seizures, and sparse, brittle hair. The primary defect is reduced or absent function of the copper-transporting ATPase ATP7A. Multiple copper-dependent enzymes can be secondarily affected, including dopamine β-hydroxylase, leading to secondary autonomic involvement and norepinephrine deficiency. Recently, plasma monoamine monitoring has been posed as a method of early detection in Menkes’ disease [Kaler et al., 2008].
Hyperekplexia, or “startle disease,” is a heterogeneous disorder caused by defects in the a1 subunit of the glycine receptor [Shiang et al., 1993]. The disorder occurs in autosomal-dominant and autosomal-recessive forms, and is characterized predominantly by stimulus-sensitive myoclonus. Transient hypertonia and hypokinesia in infancy in some families with the disorder has led to the designation “stiff baby syndrome.” Dubowitz et al. [1992] described an infant with classic startle disease, in whom the cerebrospinal fluid concentrations of GABA were substantially lower than normal during the first weeks of life. Infants with hyperekplexia have higher than expected rates of sudden infant death syndrome.
Neurologic Disorders Characterized by Excess Neurotransmitter Levels
Glycine Encephalopathy
Glycine encephalopathy, formerly referred to as nonketotic hyperglycinemia, is a heterogeneous disorder associated with insufficient activity of various components of the mitochondrial glycine cleavage system. The enzyme system for cleavage of glycine is composed of four protein components: P protein, a pyridoxal phosphate-dependent glycine decarboxylase; H protein, a lipoic acid-containing protein; T protein, a tetrahydrofolate-requiring enzyme; and L protein, a lipoamide dehydrogenase. Nonketotic hyperglycinemia may be caused by a defect in any one of these enzymes. It is an autosomal-recessive disorder with several reported phenotypes, including the classic severe neonatal form, an infantile variant, a mild-episodic childhood variant, a late-onset form, and a benign reversible form [Hamosh and Johnston, 2001].
Diagnosis is best made by documenting an increased cerebrospinal fluid to plasma glycine ratio [Applegarth and Toone, 2001]. In the neonatal form of nonketotic hyperglycinemia, cerebrospinal fluid glycine level can be 30 times normal. Plasma glycine level is also typically high, but it can be in the normal range. A cerebrospinal fluid to plasma ratio above 0.08 is usually considered diagnostic, but mildly affected cases can have ratios of 0.04–0.1 [Applegarth and Toone, 2001]. (In rare cases, cerebrospinal fluid to plasma ratios are normal, and only elevations in plasma glycine occur [Jackson et al., 1999].) Confirmation of diagnosis requires enzyme analysis in liver or transformed lymphoblasts [Christodoulou et al., 1993]. Treatment with dextromethorphan and sodium benzoate has led to variable improvement in seizure control and behavioral problems in some patients [Boneh et al., 1996; Hamosh et al., 1998].
Leukoencephalopathy with Vanishing White Matter
Leukoencephalopathy with vanishing white matter (also known as childhood ataxia with central white matter hypomyelinization) is a heterogeneous autosomal-recessive leukodystrophy characterized by progressive ataxia, motor impairment, and encephalopathy, in which episodic deterioration is associated with infection or minor head trauma [van der Knaap et al., 1997]. A wide range of onset has been reported in the dozen patients described to date. Cerebrospinal fluid glycine is elevated and can be helpful in confirming the diagnosis [van der Knaap et al., 1999]. At least two genes have been noted to have mutations in patients with this disorder: EIF2B5 and EIF2B2, the first translation initiation factors implicated in human disease [Leegwater et al., 2001]. This disorder is described in more detail in Chapter 71.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Abeling N.G., Brautigam C., Hoffmann G.F., et al. Pathobiochemical implications of hyperdopaminuria in patients with aromatic l-amino acid decarboxylase deficiency. J Inherit Metab Dis. 2000;23:325.
Abeling N.G., van Gennip A.H., Barth P.G., et al. Aromatic l-amino acid decarboxylase deficiency: A new case with a mild clinical presentation and unexpected laboratory findings. J Inherit Metab Dis. 1998;21:240.
Adler C., Ghisla S., Rebrin I., et al. Suspected pterin-4a-carbinolamine dehydratase deficiency: Hyperphenylalaninaemia due to inhibition of phenylalanine hydroxylase by tetrahydro-7-biopterin. J Inherit Metab Dis. 1992;15:405.
Allen G.F.G., Land J.M., Heales S.J.R. A new perspective on the treatment of aromatic L-amino acid decarboxylase deficiency. Mol Gen Metab. 2009;97:6-14.
Applegarth D.A., Toone J.R. Nonketotic hyperglycinemia (glycine encephalopathy): Laboratory diagnosis. Mol Genet Metab. 2001;74:139.
Blau N., Bonafe L., Thony B. Tetrahydrobiopterin deficiencies without hyperphenylalaninemia: Diagnosis and genetics of dopa-responsive dystonia and sepiapterin reductase deficiency. Mol Genet Metab. 2001;74:172.
Blau N., Dhondt J.L., Guibaud P., et al. New variant of hyperphenylalaninaemia with excretion of 7-substituted pterins [Letter]. Eur J Pediatr. 1988;148:176.
Bodeau-Pean S., Ravassard P., Neuner-Jehle M., et al. A human tyrosine hydroxylase isoform associated with progressive supranuclear palsy shows altered enzymatic activity. J Biol Chem. 1999;274:3469.
Bonafe L., Blau N., Burlina A.P., et al. Treatable neurotransmitter deficiency in mild phenylketonuria. Neurology. 2001;57:908.
Bonafe L., Thony B., Penzien J.M., et al. Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. Am J Hum Genet. 2001;69:269.
Boneh A., Degani Y., Harari M. Prognostic clues and outcome of early treatment of nonketotic hyperglycinemia. Pediatr Neurol. 1996;15:137.
Brautigam C., Steenbergen-Spanjers G.C., Hoffmann G.F., et al. Biochemical and molecular genetic characteristics of the severe form of tyrosine hydroxylase deficiency. Clin Chem. 1999;45:2073.
Brunner H.G., Nelen M., Breakefield X.O., et al. Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science. 1993;262:578.
Brunner H.G., Nelen M.R., van Zandvoort P., et al. X-linked borderline mental retardation with prominent behavioral disturbance: Phenotype, genetic localization, and evidence for disturbed monoamine metabolism. Am J Hum Genet. 1993;52:1032.
Christodoulou J., Kure S., Hayasaka K., et al. Atypical nonketotic hyperglycinemia confirmed by assay of the glycine in lymphoblasts. J Pediatr. 1993;123:100.
Citron B.A., Kaufman S., Milstien S., et al. Mutation in the 4a-carbinolamine dehydratase gene leads to mild hyperphenylalaninemia with defective cofactor metabolism. Am J Hum Genet. 1993;53:768.
Clot F., Grabli D., Cazeneuve C., et al. Exhaustive analysis of BH4 and dopamine biosynthesis genes in patients with Dopa-responsive dystonia. Brain. 2009;132:1753-1763.
Collins F.A., Murphy D.L., Reiss A.L., et al. Clinical, biochemical, and neuropsychiatric evaluation of a patient with a contiguous gene syndrome due to a microdeletion Xp11.3 including the Norrie disease locus and monoamine oxidase (MAOA and MAOB) genes. Am J Med Genet. 1992;42:127.
Concolino D, Muzzi G, Rapsomaniki M et al. Serum prolaction as a tool for the follow up of treated DHPR-deficient patients. J lnherit Metab Dis, Apr 15 Epub only 2008
de Rijk-Van Andel J.F., Gabreels F.J., Geurtz B., et al. L-Dopa-responsive infantile hypokinetic rigid parkinsonism due to tyrosine hydroxylase deficiency. Neurology. 2000;55:1926.
Deckert J., Catalano M., Syagailo Y.V., et al. Excess of high activity monoamine oxidase A gene promoter alleles in female patients with panic disorder. Hum Mol Genet. 1999;8:621.
De Lonlay P., Nassogne M.C., van Gennip A.H., et al. Tyrosine hydroxylase deficiency unresponsive to l-dopa treatment with unusual clinical and biochemical presentation. J Inherit Metab Dis. 2000;23:819.
Dionisi-Vici C., Hoffmann G.F., Leuzzi V., et al. Tyrosine hydroxylase deficiency with severe clinical course: Clinical and biochemical investigations and optimization of therapy. J Pediatr. 2000;136:560.
Dubowitz L.M., Bouza H., Hird M.F., et al. Low cerebrospinal fluid concentration of free gamma-aminobutyric acid in startle disease. Lancet. 1992;340:80.
Dudesek A., Roschinger W., Muntau A.C., et al. Molecular analysis and long-term follow-up of patients with different forms of 6-pyruvoyl-tetrahydropterin synthase deficiency. Eur J Pediatr. 2001;160:267.
Furukawa Y., Graf W.D., Wong H., et al. Dopa-responsive dystonia simulating spastic paraplegia due to tyrosine hydroxylase (TH) gene mutations. Neurology. 2001;56:260.
Furukawa Y., Lang A.E., Trugman J.M., et al. Gender-related penetrance and de novo GTP-cyclohydrolase I gene mutations in dopa-responsive dystonia. Neurology. 1998;50:1015.
Gibson K.M., Hoffmann G.F., Hodson A.K., et al. 4-Hydroxybutyric acid and the clinical phenotype of succinic semialdehyde dehydrogenase deficiency, an inborn error of GABA metabolism. Neuropediatrics. 1998;29:14.
Gibson K.M., Sweetman L., Nyhan W.L., et al. Demonstration of 4-aminobutryic acid aminotransferase deficiency in lymphocytes and lymphoblasts. J Inherit Metab Dis. 1985;8:204-208.
Hamosh A., Johnston M.V.. Nonketotic hyperglycinemia. New York: McGraw-Hill; 2001:2065.
Hamosh A., Maher J.F., Bellus G.A., et al. Long-term use of high-dose benzoate and dextromethorphan for the treatment of nonketotic hyperglycinemia. J Pediatr. 1998;132:709.
Harwood G., Hierons R., Fletcher N.A., et al. Lessons from a remarkable family with dopa-responsive dystonia. J Neurol Neurosurg Psychiatry. 1994;57:460.
Haussler M., Hoffmann G.F., Wevers R.A. L-Dopa and selegiline for tyrosine hydroxylase deficiency. J Pediatr. 2001;138:451.
Hogema B.M., Gupta M., Senephansiri H., et al. Pharmacologic rescue of lethal seizures in mice deficient in succinate semialdehyde dehydrogenase. Nat Genet. 2001;29:212.
Hyland K. Inherited disorders affecting dopamine and serotonin: critical neurotransmitters derived from aromatic amino acids. J Nutr. 2007;137:1568S-1572S.
Hyland K., Surtees R.A., Rodeck C., et al. Aromatic l-amino acid decarboxylase deficiency: Clinical features, diagnosis, and treatment of a new inborn error of neurotransmitter amine synthesis. Neurology. 1992;42:1980.
Ichinose H., Ohye T., Takahashi E., et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet. 1994;8:236.
Invernizzi R.W. Role of TPH-2 in brain function: news from behavioral and pharmacologic studies. J Neurosci Res. 2007;85:3030-3035.
Jackson A.H., Applegarth D.A., Toone J.R., et al. Atypical nonketotic hyperglycinemia with normal cerebrospinal fluid to plasma glycine ratio. J Child Neurol. 1999;14:464.
Kaler S.G., Holmes C.S., Goldstein D.S., et al. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008;358:605-614.
Larnaout A., Belal S., Miladi N., et al. Juvenile form of dihydropteridine reductase deficiency in 2 Tunisian patients. Neuropediatrics. 1998;29:322.
Leegwater P.A., Vermeulen G., Konst A.A., et al. Subunits of the translation initiation factor eif2b are mutant in leukoencephalopathy with vanishing white matter. Nat Genet. 2001;29:383.
Lenders J.W., Eisenhofer G., Abeling N.G., et al. Specific genetic deficiencies of the A and B isoenzymes of monoamine oxidase are characterized by distinct neurochemical and clinical phenotypes. J Clin Invest. 1996;97:1010.
Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis. 2009;32:333-342.
Manegold C., Hoffmann G.F., Degen I., et al. Aromatic L-amino acid decarboxylase deficiency: clinical features, drug therapy and follow-up. J Inherit Metab Dis. 2009;32:371-380.
Mills P.B., Struys E., Jakobs C., et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. 2006;12:307-309.
Milstien S., Kaufman S., Summer G.K. Hyperphenylalaninemia due to dihydropteridine reductase deficiency: Diagnosed by measurement of oxidized and reduced pterins in urine. Pediatrics. 1980;65:806.
Neville B.G.R., Parascandalo R., Farrugia R., et al. Sepiapterin reductase deficiency: a congenital dopa-responsive motor and cognitive disorder. Brain. 2005;128:2291-2296.
Niederwieser A., Shintaku H., Leimbacher W., et al. Peripheral tetrahydrobiopterin deficiency with hyperphenylalaninaemia due to incomplete 6-pyruvoyl tetrahydropterin synthase deficiency or heterozygosity. Eur J Pediatr. 1987;146:228.
Pearl P.L., Gibson K.M. Clinical aspects of the disorders of GABA metabolism in chidren. Curr Opin Neurol. 2004;17:107-113.
Pearl P.L., Gibson K.M., Cortez M.A., et al. Succinic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. 2009;32:343-352.
Pons R., Ford B., Chiriboga C.A., et al. Aromatic l-amino acid decarboxylase deficiency: Clinical features, treatment, and prognosis. Neurology. 2004;62:1058. 13
Preisig M., Bellivier F., Fenton B.T., et al. An association between bipolar disorder and monoamine A gene polymorphisms: Results of a multicenter study. Am J Psychiatry. 2000;157:948.
Raemaekers V.T., Senderek J., Hausler M., et al. A novel neurodevelopmental syndrome responsive to 5-hydroxytryptophan and carbidopa. Mol Genet Metab. 2001;73:179.
Robertson D., Goldberg M.R., Onrot J., et al. Isolated failure of autonomic noradrenergic neurotransmission: Evidence for impaired beta-hydroxylation of dopamine. N Engl J Med. 1986;314:1494.
Robertson D., Haile V., Perry S.E., et al. Dopamine beta-hydroxylase deficiency. A genetic disorder of cardiovascular regulation. Hypertension. 1991;18:1.
Schuback D.E., Mulligan E.L., Sims K.B., et al. Screen for MAOA mutations in target human groups. Am J Med Genet. 1999;88:25.
Scott A.L., Bortolato M., Chen K., et al. Novel monoamine oxidase A knock out mice with human-like spontaneous mutation. Neuroreport. 2008;19:739-743.
Segawa M., Hosaka A., Miyagawa F., et al. Hereditary progressive dystonia with marked diurnal fluctuation. Adv Neurol. 1976;14:215.
Shiang R., Ryan S.G., Zhu Y.-Z., et al. Point mutations in the gene encoding the alpha-1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet. 1993;5:351.
Steinberger D., Weber Y., Korinthenberg R., et al. High penetrance and pronounced variation in expressivity of GCH1 mutations in five families with dopa-responsive dystonia. Ann Neurol. 1998;43:634.
Swoboda K.J., Hyland K., Goldstein D.S., et al. Clinical and therapeutic observations in aromatic l-amino acid decarboxylase deficiency. Neurology. 1999;53:1205.
Swoboda K.J., Saul J.P., McKenna C.E., et al. Aromatic l-amino acid decarboxylase deficiency: Overview of clinical features and outcomes. Ann Neurol. 2003;54(Suppl 6):S49.
Thony B., Blau N. Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydrobiopterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase. Hum Mutat. 2006;27:870-878.
Thony B., Neuheiser F., Kierat L., et al. Hyperphenylalaninemia with high levels of 7-biopterin is associated with mutations in the PCBD gene encoding the bifunctional protein pterin-4a-carbinolamine dehydratase and transcriptional coactivator (dcoh). Am J Hum Genet. 1998;62:1302.
van der Knaap M.S., Barth P.G., Gabreels F.J.M., et al. A new leukoencephalopathy with vanishing white matter. Neurology. 1997;48:845.
van der Knaap M.S., Wevers R.A., Kure S., et al. Increased cerebrospinal fluid glycine: A biochemical marker for a leukoencephalopathy with vanishing white matter. J Child Neurol. 1999;14:728.
Van Hove J.L.K., Steyaert J., Matthijs G., et al. Expanded motor and psychiatric phenotype in autosomal dominant Segawa syndrome due to GTP cyclohydrolase deficiency. J Neurol Neurosurg Psychiatry. 2006;77:18-23.
Woody R.C., Brewster M.A., Glasier C. Progressive intracranial calcification in dihydropteridine reductase deficiency prior to folinic acid therapy. Neurology. 1989;39:673.
