Neurologic Complications
Shlomit Yust-Katz and Mark R. Gilbert
Incidence of Chemotherapy- and Radiation Therapy–Induced Neurotoxicity
• The actual incidence of treatment-related neurotoxicity is unknown, but the frequency is increasing.
• Improvements in supportive care, but not neuroprotective regimens, have allowed dose escalation for many drugs, and thus neurotoxicity often is the dose-limiting factor.
• Increased survival from cancer has resulted in an increasing prevalence of late-onset neurotoxicity.
• Newer treatments directed at tumors in the central nervous system often result in neurotoxicity, particularly with therapies administered directly into the cerebrospinal fluid.
• Direct effects on neurons, myelin, and supporting glial cells have been implicated.
• Effects on neuronal cytoskeleton and axonal transport, neuronal metabolism, and neurotransmitter function are the most commonly hypothesized mechanisms of toxicity. Alterations in specific ion channels have been reported in some cases of chemotherapy-induced peripheral neuropathy.
• In general, chemotherapy or radiation toxicity should be considered a diagnosis of exclusion.
• Specific diagnostic tests do not exist for treatment-induced toxicity from most agents and regimens in use.
• The diagnosis often is made by recognition of a neurotoxic syndrome temporally related to treatment and by exclusion of other causes of neurologic dysfunction.
• Grading scales are of limited value for monitoring individual patients and are used only for study populations.
• More refined grading for management is a component of neurologic and neuropsychological testing.
• With most neurotoxic syndromes, specific treatment is not available.
• Prevention or reduction of risk often is possible with proper monitoring or treatment planning.
• New agents are under development for management or prevention of neurotoxicity, but careful testing is required to ensure that the antineoplastic effect is not compromised.
Introduction
First, recent advances in supportive care allow the use of much higher doses of chemotherapeutic agents. For example, administration of granulocyte or granulocyte-macrophage colony-stimulating factors allows use of drug doses that previously would have caused severe bone marrow suppression. Unfortunately, similar factors for prevention of the development of neurotoxicity do not exist. In initial studies of paclitaxel, a novel antitubulin agent, myelosuppression was dose limiting. Use of colony-stimulating factors has allowed dose escalation, but the development of severe peripheral neuropathy has been described in many patients receiving these higher doses.1,2 Similarly, nephrotoxicity and myelosuppression limited dose escalation of cisplatin in the past. Innovative hydration schemes and the availability of colony-stimulating factors have reduced the risk of these toxicities. Dose escalation has resulted in severe neurotoxicity and ototoxicity, which are now considered the dose-limiting toxic effects.3
Second, improvements in cancer treatment have increased the duration of survival from many malignant diseases. As a consequence, treatment-related neurotoxicity with a long latency between treatment and onset of symptoms is being recognized with increasing frequency. Childhood acute lymphoid leukemia often was fatal until recognition of the central nervous system (CNS) as a sanctuary for leukemia cells. Treatment of cerebrospinal fluid (CSF) with direct administration of methotrexate and use of cranial irradiation resulted in a marked increase in long-term remission and cure. This treatment of the CNS, however, also caused delayed neurotoxicity. Severe dementia developed in some patients, and many others experienced a decline in cognitive function years after completion of treatment.4–8 Similarly, combined-modality treatment using both whole-brain irradiation and chemotherapy with high-dose methotrexate has markedly improved survival in patients with primary CNS lymphoma over that achieved with radiation therapy alone. A marked increase in leukoencephalopathy, however, has been observed with this combination regimen, particularly in elderly persons.9
Third, newer agents, including biological response modifiers, and novel routes of administration designed specifically to target the nervous system for management of brain metastases or primary brain tumors are very likely to result in an increase in neurotoxicity. Likewise, chemical disruption of the blood-brain barrier to improve drug delivery to brain tumors has led to a marked increase in neurotoxicity compared with standard systemic administration. Results of animal studies have demonstrated that chemical opening of the blood-brain barrier results in a marked increase in exposure of normal brain parenchyma to chemotherapy with a much smaller increase in delivery to the tumor.10 Implantation of carmustine (BCNU)-impregnated polymer wafers into the resection cavity has shown a survival benefit for patients with malignant glioma but with an increase in the incidence of brain necrosis and infection.11
Specific Agents
Cytosine Arabinoside
Cerebellar Toxicity
Systemic administration of high-dose (greater than 1 g/m2) intravenous ara-C can cause acute cerebellar toxicity.12–15 Onset of neurologic symptoms generally is acute and often is noticed during administration of a multiday regimen.12,13 Patients exhibit evidence of global cerebellar dysfunction, which manifests as truncal, limb, and gait ataxia, dysarthria, and nystagmus. In some cases, permanent cerebellar dysfunction results; in others, a mild cerebellar syndrome develops that resolves promptly after completion of the chemotherapy.12,13
Patients with irreversible cerebellar damage have a characteristic and selective loss of Purkinje cells in the cerebellum.15 The pathogenesis of the specific cellular damage is unknown, and no pathological findings have been described in the cerebella of patients who have recovered from transient cerebellar dysfunction.
The incidence of irreversible cerebellar toxicity with high-dose ara-C regimens has been reported to be 8% to 20%. Factors reported to affect the likelihood of development of cerebellar toxicity include size of current dose, cumulative dose, age (persons older than 50 years are at higher risk), abnormal alkaline phosphatase (alkaline phosphatase greater than or equal to three times normal) and renal status (renal dysfunction with impaired drug clearance is associated with increased risk).13,14,16–18 Whether patients in whom reversible cerebellar dysfunction develops with previous treatment are at higher risk for permanent dysfunction is unknown.
Recent data show that, in animal models, oral application of the antioxidant N-acetylcysteine is able to prevent ara-C–induced behavioral deficits and cellular alterations of the adult cerebellum in a rat model.19,20 However, this treatment has not yet been tested in humans.
Encephalopathy
Acute encephalopathy, often accompanied by seizures, occurs less frequently than cerebellar dysfunction in patients receiving high-dose intravenous ara-C.13 In most cases, somnolence and lethargy completely resolve soon after completion of chemotherapy. Patients with persistent encephalopathy usually have had additional medical problems, such as severe infection or metabolic abnormalities.18 In addition, leukoencephalopathy that is clinically and pathologically indistinguishable from the leukoencephalopathy associated with methotrexate has been reported as a late complication of high-dose intravenous ara-C administration and with administration of ara-C directly into the CSF.13
Spinal Cord Toxicity
Direct administration of ara-C into the lumbar thecal space has been reported to cause myeloradiculopathy.21–25 Patients exhibit evidence of both spinal cord and nerve root dysfunction. This complication is uncommon and usually is found only after an extensive course of intrathecal chemotherapy. In many instances, patients have received both intrathecal ara-C and methotrexate.26,27 When the exclusive use of intrathecal ara-C resulted in spinal cord injury, it was either administered at relatively high doses (100 to 150 mg, three cases) or in liposomal form (50 mg, two cases) where prolonged release of ara-C in the CSF overlapped with systemic high-dose methotrexate and high-dose ara-C.28
Several hypotheses have been proposed for the mechanism of chemotherapy-induced myelopathy. Focal damage from injection of hyperosmolar solution,29 barbotage from injection,30 and direct toxic effects of chemotherapy on the spinal cord parenchyma are the most common proposed mechanisms of subacute myelopathy. Histologic examination of the spinal cord shows focal areas of necrosis that are most marked along the periphery of the spinal cord.22,23,26 Microscopic examination shows axonal swelling with accompanying demyelination.22,23,26 With chemotherapy-induced spinal cord injury, myelin basic protein levels in the CSF may be elevated before marked neurologic damage occurs.26,27 For patients with early symptoms, such as paresthesia, back pain, or Lhermitte sign, the level of myelin basic protein in the CSF should be measured. If the level is elevated, further lumbar administration of chemotherapy should be avoided.27
Liposomal Ara-C
Liposomal cytarabine is frequently used because of its convenience, that is, every-2-week dosing because of a long CSF half-life (approximately 140 hours), and its potential improved efficacy compared with free cytarabine and methotrexate.29,30 Although the pharmacokinetics of drug delivery in the CSF is improving with the liposomal ara-C, an increase in the incidence of arachnoiditis has been observed, and isolated cranial nerve palsies may be more frequent. In a recently published retrospective series of 120 adult patients with lymphomatous meningitis who were treated with liposomal ara-C, serious treatment-related neurologic complications developed in 12.5% of patients. Toxicity included bacterial meningitis, chemical meningitis, communicating hydrocephalus, conus medullaris/cauda equine syndrome, decreased visual acuity, encephalopathy, leukoencephalopathy, myelopathy, radiculopathy, and seizures. Distribution of toxicity was similar regardless of the route of administration (ventricular vs. lumbar).31,32
Other Neurotoxicity Associated with Cytosine Arabinoside
Peripheral neuropathy has been reported after administration of high-dose ara-C.33 In this report, symmetrical sensorimotor polyneuropathy developed 2 weeks after completion of treatment. Nerve biopsy demonstrated axonal damage with patchy regions of demyelination. In addition, a case of reversible Parkinsonism has been reported after administration of high-dose ara-C.13 Onset of tremor, bradykinesia, and masklike facies were observed 3 weeks after completion of treatment. Treatment with carbidopa-levodopa provided only transient improvement. However, all Parkinsonian features resolved over 12 weeks.
L-Asparaginase
Cerebrovascular Events
Cerebrovascular events caused by l-asparaginase–induced coagulopathy constitute the most common form of neurotoxicity associated with l-asparaginase treatment. Both thrombotic and hemorrhagic strokes have been reported in patients receiving l-asparaginase.36–36 Thrombosis of cerebral venous sinuses also has occurred in patients receiving this agent.37
The clinical manifestations of sinus thrombosis, which usually are acute, include severe headache, nausea, and vomiting caused by the rapid increase in intracranial pressure. Changes in level of consciousness occur most frequently with sagittal sinus thrombosis with bilateral cerebral hemisphere involvement. Although most patients with sinus thrombosis demonstrate acute and rapidly progressive changes in neurologic function, some patients experience only headache and mild neurologic dysfunction.34
Neuropsychiatric Effects
Less frequently, l-asparaginase treatment has been associated with development of neuropsychiatric symptoms,38 most notably depression, delusions, hallucinations, disorientation, and altered level of consciousness. In the series described by Holland and colleagues,38 5 of 19 patients with acute leukemia experienced psychiatric symptoms. The onset of symptoms occurred 2 to 19 days after treatment, and the symptoms resolved completely in three patients who lived longer than 6 weeks. Neuropathological analysis of the brains in two cases revealed leukemic infiltration. The combination of leukemic involvement of the CNS and treatment-induced depletion of l-asparagine and l-glutamine in the brain has been proposed as a possible factor contributing to development of psychiatric symptoms.
Busulfan
Busulfan administration has been reported to cause generalized tonic-clonic seizures.39 This reaction has been reported with high-dose treatment as a preparative regimen for bone marrow transplantation. Prophylactic treatment with anticonvulsant agents, particularly phenytoin, has been shown to reduce the risk of seizures.40 However, phenytoin is contraindicated because of its ability to induce busulfan metabolism and because of possible toxicities. The existing clinical data support the use of benzodiazepines, most notably clonazepam and lorazepam, to prevent busulfan-induced seizures. The second-generation antiepileptic drug levetiracetam possesses the characteristics of optimal prophylaxis for busulfan-induced seizures, and early data of its efficacy are promising, although further study is needed.41
Methotrexate
Methotrexate can cause acute, subacute, or chronic neurotoxicity.
Acute Neurotoxicity
Acute MTX neurotoxicity ranges from 3% to 10% and varies with the dose and route of administration, occurring more often after intrathecal administration and higher doses.42 The acute encephalopathy is characterized primarily by somnolence, confusion, and seizures.5,43 Headache, chorea, Klüver-Bucy syndrome, blurred vision, aphasia, and transient or persistent hemiparesis have also been described.44
Although the pathogenesis of this syndrome is unknown, laboratory studies with rats have shown profound metabolic alteration in the brain after intravenous administration of high-dose methotrexate.45 In these experiments, a widespread decrease in glucose utilization and protein synthesis was found. In similar studies, folinic acid (leucovorin) markedly diminished these metabolic effects,46 a finding that suggested a possible role for leucovorin in decreasing the severity of methotrexate-induced somnolence syndrome. Methotrexate is known to cause inhibition of the enzyme dihydrofolate reductase, which prevents the conversion of folic acid to tetrahydrofolic acid and thereby increases the levels of homocysteine and excitotoxic neurotransmitters and inhibits cell replication. Furthermore, methotrexate neurotoxicity is associated with polymorphic mutations of folate/methionine metabolism. Other mechanisms that have been suggested include the direct toxic effect on myelin, disruption of mitochondrial energy metabolism resulting in oxidative stress, increased vulnerability of neurons to physiological glutamate concentrations, and breakdown of the blood-brain barrier.47,48 Although the somnolence and confusion that occur with acute methotrexate toxicity resolve completely, evidence shows that patients in whom this syndrome develops are at greater risk for chronic methotrexate-induced neurotoxicity.49 Cases have been reported in which, despite resolution of the clinical symptoms, white matter changes persist on MR images.50
Subacute Toxicity
Subacute methotrexate neurotoxicity is a rare syndrome, usually occurring in children, that is manifested by abrupt onset of focal neurologic deficits, such as aphasia and hemiparesis. It typically develops weeks after methotrexate administration and is associated with increased cumulative exposure, route of administration (intrathecal and intravenous), age (older than 10 years) and a high methotrexate : leucovorin ratio.5 Transient inhibition of myelin formation is thought to be the mechanism of toxicity. Methotrexate-associated subacute toxicity syndrome can be confidently diagnosed when diffusion-weighted imaging on MRI shows areas of restricted diffusion across multiple vascular beds and involving deep cerebral white matter, in the clinical context of waxing and waning neurologic signs and symptoms. Follow-up diffusion-weighted imaging typically shows resolution of restricted diffusion.51 The syndrome is completely reversible over weeks, and corticosteroid treatment may accelerate recovery.
Chronic Neurotoxicity
Chronic methotrexate neurotoxicity is known as leukoencephalopathy. This syndrome develops months to years after methotrexate administration and has been seen after both intravenous and intrathecal administration of methotrexate.4–8,52–62 Cranial radiation therapy, particularly when it precedes methotrexate administration, greatly increases the risk of leukoencephalopathy.5,63,64 In addition, elevated CSF methotrexate concentration has been associated with an increased risk of neurotoxicity.54 Younger patients are at higher risk for leukoencephalopathy.67–67 Clinically, patients show progressive loss of cognitive function and focal neurologic signs, which may progress to profound dementia, coma, or death.4–6,8,55 Some patients experience seizures as a consequence of widespread neuronal injury. No treatment is known, and the neurologic deficits generally are irreversible. Brain imaging with MRI or CT often shows large areas of abnormalities in cerebral white matter (Fig. 57-1). Elevated levels of myelin basic protein in CSF have been reported in patients with progressive neurologic dysfunction from methotrexate-induced leukoencephalopathy.56
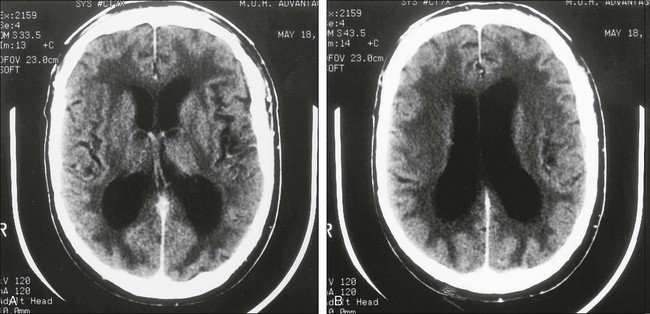
Neuropathological examination shows wide areas of coagulative necrosis with swollen axonal cylinders and demyelination.4,52 Regions with vascular changes, particularly microangiopathic calcifications, are characteristic.58,61 The pathogenesis of leukoencephalopathy is unknown, but results of laboratory studies with brain explant cultures suggest that the primary injury may be neuronal (axonal), and the characteristic demyelination may be a secondary phenomenon.68
Spinal Cord Toxicity
Chemotherapy-induced myelopathy is an uncommon toxicity of intrathecal treatment with methotrexate.69–74 Myelopathy generally develops only after extensive intrathecal treatment. Both methotrexate and ara-C have been associated with myelopathy. The clinical syndrome of methotrexate-induced myelopathy is identical to that with ara-C (see the earlier section “Cytosine Arabinoside”). Loss of neurologic function may be progressive; only approximately half of patients experience either complete or partial recovery. The onset of symptoms generally is subacute. Symptoms develop over days to weeks, usually beginning days to weeks after administration of chemotherapy. The histologic findings are identical to those with ara-C–induced myelopathy.
Vinca Alkaloids
Treatment with vinca alkaloids, particularly vincristine, commonly is associated with neurotoxicity.
Peripheral Neuropathy
Peripheral neuropathy is the toxicity associated most frequently with vincristine and correlates with the cumulative dose of the drug.75–80 Loss of deep tendon reflexes occur in nearly all patients who receive several vincristine treatments. Distal sensorimotor polyneuropathy develops with continued treatment.76,77 The predominant neurologic finding is loss of pain and temperature sensation in a stocking-and-glove distribution. Motor and vibration or proprioceptive loss usually is milder and occurs later with continued treatment.
The mechanism of toxicity is unknown but probably is related to the effects of the vinca alkaloids on microtubules. Vinca-induced disruption of axonal microtubules causes marked disarray of the axonal cytoskeleton and formation of neurofilamentous masses79,81–85 and reversible neurofilament-containing crystalloid inclusions.86 These effects are very likely to influence axonal transport, which depends on microtubules as the transport mechanism.87
A recently published study suggests that vincristine might cause an increase in nerve excitability and induce a state of glutamate excitotoxicity by enhancing N-methyl-D-aspartate receptor expression and diminishing calcitonin gene-related peptide expression. In this study erythropoietin had a neuroprotective effect, probably through decreasing N-methyl-D-aspartate receptor expression and increasing calcitonin gene-related peptide expression.88
In patients with underlying neuropathy, such as diabetic neuropathy or Charcot-Marie-Tooth disease (hereditary motor sensory neuropathy type 1), vinca-induced neuropathy may be severe even with low cumulative doses.89 Severe, even life-threatening neuropathy has been reported after administration of as little as 2 mg to patients with Charcot-Marie-Tooth disease, and evidence suggests that patients should be screened for this disorder before administration of vincristine.91–91 Previous radiation treatment of peripheral nerves also increases the neurotoxic effects of vincristine.92
In addition to peripheral neuropathy, autonomic neuropathy and cranial nerve palsy have been reported. Autonomic neuropathy most commonly manifests as gastrointestinal dysmotility (obstipation or constipation). In severe cases, paralytic ileus and intestinal perforation have resulted.93 Less frequently, orthostatic hypotension develops as a consequence of autonomic involvement. Vincristine-induced mononeuropathy involving the femoral nerve has been reported. Vincristine can cause cranial nerve palsies affecting the optic (II), oculomotor (III), trigeminal, abducens, facial, acoustic, and vagus nerves.96–96 In addition, patients occasionally report facial pain with vincristine treatment, which possibly is due to a transient effect on the trigeminal nerve or ganglion.80
Central Nervous System Effects
Vincristine has been reported to cause encephalopathy, coma, and seizures.97–101 These effects are rare and reversible. The underlying mechanism is unknown in most cases, although in some reports these neurologic effects have been attributed to vincristine-induced syndrome of inappropriate antidiuretic hormone secretion (SIADH) and hyponatremia.100,102–105 The mechanism of SIADH is unknown, but serum antidiuretic hormone levels are elevated.
Other Toxicity Associated with Vinca Alkaloids
Quadriplegia with vincristine treatment has been reported, in one case in association with Guillain-Barré syndrome.106–109 The time of onset has been variable. In some patients, quadriparesis develops soon after vincristine treatment, whereas in others, it occurs several weeks after treatment. In most instances, the weakness is partially reversible.106
Myopathy has occurred with vincristine therapy.110 No clinical correlate has been found in cases in which histopathological examination of muscle tissue has revealed spheromembranous degeneration.
Cisplatin
Peripheral Neuropathy
The most common neurotoxicity associated with cisplatin is peripheral neuropathy, which is so significant that it is a dose-limiting adverse effect. The neuropathy predominantly involves the large sensory fibers, which mediate vibration and proprioceptive function. Deep tendon reflexes are lost because of toxic effects on the large myelinated sensory fibers, which provide the afferent arm of the reflex arc. Involvement of motor function generally is mild and is seen only in patients with severe sensory neuropathy. Development of neuropathy is dose related. The earliest signs are detected when the cumulative dose exceeds 300 mg/m2.111 The schedule of administration may be a significant factor, because the reported incidence of neuropathy has been higher in patients receiving treatment on five consecutive days than in patients on a regimen with a shorter dosing schedule but the same cumulative dose.114–114
Continued treatment with cisplatin in patients with neuropathy can result in severe sensory ataxia that often impairs ambulation. The neuropathy is partially reversible, and patients with mild impairment generally are more likely to experience full recovery.113 A longitudinal study confirmed that there is often a delay in the onset of neuropathy. Eleven percent of the patients had neuropathy at the end of treatment, but the incidence had increased to 65% 3 months later. One year later, most of the patients had recovered, with only 17% having persistent symptoms.115 A recently published trial demonstrated that long-term serum platinum levels are significantly associated with the severity of neurotoxicity 5 to 20 years after cisplatin-based chemotherapy. Importantly, the relationship remained significant after adjustment for the initial cisplatin dose.116
The pathogenesis of cisplatin-induced neuropathy is unknown. Neuropathological studies have shown involvement of the large sensory fibers with regions of axonal swelling and myelin breakdown and, in more severe cases, axonal loss.117 The spinal cord shows almost exclusive involvement of myelinated axons in the dorsal columns—a finding consistent with the clinical features of vibratory and proprioceptive loss.118 Cisplatin has been shown to induce apoptosis in dorsal root ganglion sensory neurons by covalently binding to nuclear DNA, resulting in DNA damage, subsequent p53 activation, and BAX-mediated apoptosis via the mitochondria and also by causing a reduction in mitochondrial DNA transcription.119
Platinum concentration in peripheral nerve and spinal ganglia was 20 times greater than in the brain in patients at autopsy.115 This finding may explain the predilection of cisplatin for sensory fibers and sparing of the CNS.
Spinal Cord Toxicity
Cisplatin treatment has been associated with the development of Lhermitte sign, which is an electric shock–like sensation down the spine or into the extremities with neck flexion.120,121 The phenomenon most commonly is associated with spinal cord demyelinating lesions in persons with multiple sclerosis. A similar mechanism, cisplatin-induced demyelination, may be the cause in patients with this syndrome. Most patients achieve full recovery, although, as with recovery from cisplatin-induced neuropathy, improvement may take several months.
Other Neurotoxicity Associated with Cisplatin
Other neurotoxic effects reported with cisplatin include optic neuropathy, seizures, encephalopathy, and cortical blindness.122 These complications are rare. Patients with optic neuropathy may have prolonged vision loss and demonstrate pallor of the optic disk.123,124 The reported cases of seizures and cortical blindness have been self-limited, with all patients recovering fully.127–127 The cause of the seizures and cortical blindness from cisplatin treatment is unknown but may be similar to toxicity observed from other heavy metals (e.g., lead and thallium), although endovascular injury has been proposed as a possible mechanism.122,126 Many of these patients have white matter abnormalities on brain MRI, consistent with posterior reversible encephalopathy syndrome (PRES; see later under “Dementia and Encephalopathy”). A syndrome also has been described in which patients experience focal neurologic deficits and seizures after intravenous administration of cisplatin. In these patients, findings on brain MRI were normal. One patient experienced recurrence of encephalopathy with cisplatin rechallenge, and a second patient died of status epilepticus. At autopsy, the brain of the latter patient showed only focal gliosis.127
Toxicity Associated with Intraarterial Administration
Intraarterial administration of cisplatin causes focal toxicity. Administration into the internal carotid artery can cause severe retinal toxicity.128,129 Supraophthalmic administration can cause focal areas of brain parenchymal necrosis with resulting seizures and neurologic impairment.130
Ototoxicity
Ototoxicity is a dose-related effect of cisplatin. Long-term ototoxicity was found to be correlated to serum platinum levels.116 Patients receiving more than 200 mg/m2 are at high risk for the development of hearing loss.131 Hearing loss, particularly when it is moderate to severe, often is permanent.131,132 Results of most studies suggest that patients with underlying hearing loss are at greater risk for functional hearing loss, although a small series found no relation to previous hearing loss.131,133,134 Additional risk factors include age (older than 46 years) and previous cranial radiation therapy involving the ears or temporal lobes.132,133 Patients with normal hearing lose high-frequency hearing first, but with continued treatment, hearing may be lost in all frequency ranges.131 A rapid screening audiogram technique has been developed to monitor patients undergoing cisplatin treatment.134 Postmortem pathological examination of cochleae from patients with cisplatin-induced ototoxicity reveals extensive loss of the outer hair cells and less effect on the inner hair cells.131
Oxaliplatin
Oxaliplatin has been shown to cause two distinct types of neuropathy: acute and chronic.135 The acute neuropathy syndrome may begin during oxaliplatin infusion or up to 1 to 2 days after completion of treatment. Patients experience paresthesias and dysesthesias of the hands and feet, jaw tightness, and a sensation of loss of breathing without respiratory distress. This latter syndrome has been named pharyngolaryngodysesthesia.136 Dysesthesias constitute the most prominent symptom, although many patients describe pain that is more like muscle spasms of the jaw, tongue, and extremities. Hemibody paresthesias with muscle cramping have been described as an acute syndrome.137 Some patients are unable to relax a tensed muscle, such as a grasp, during these episodes. The incidence of acute neuropathy increases with continued dosing, and an increased incidence also has been noted with higher dosing regimens. Overall, acute, severe (grade 3 or 4) neurotoxicity has been estimated to occur in 10% of patients with the initial dose, but increases to 50% by the ninth cycle of treatment.138 Most patients experience some resolution of symptoms, but most patients have residual neuropathic symptoms up to 6 months after treatment cessation. The symptoms often return upon subsequent administration of oxaliplatin.139 The pathogenesis of acute oxaliplatin neuropathy is thought to be related to drug-induced alterations in voltage-gated sodium channels140,141 in response to oxalate, a metabolic byproduct of oxaliplatin. In addition, oxalate may interact indirectly with the voltage-gated sodium channels through chelation of calcium and magnesium.
The development of chronic neuropathy from oxaliplatin is related to cumulative dose, with most studies reporting that early neuropathy is noted after a total dose of greater than 540 mg/m2. As with cisplatin, chronic oxaliplatin peripheral neuropathy affects large-caliber sensory nerves, with the resultant loss of proprioceptive function as the predominant clinical manifestation. Additionally, the Lhermitte-like phenomenon described with cisplatin also has been reported with oxaliplatin.142,143 The chronic neuropathy may abate over several months after the cessation of treatment, although some reports suggest that symptoms may persist.144,145
Several approaches have been used to prevent oxaliplatin-induced neurotoxicity, including use of an intermittent oxaliplatin dosing schedule that reduced the incidence of grade 3 neurotoxicity146 and the concurrent use of neuromodulatory agents, such as antidepressant drugs, antiepileptic agents, or calcium and magnesium infusions.147
Cyclophosphamide
Cyclophosphamide can indirectly cause metabolic encephalopathy and seizures. High-dose cyclophosphamide can result in SIADH.150–150 When unrecognized, this syndrome can lead to severe hyponatremia, coma, and seizures. Although it is not reported in the literature in association with cyclophosphamide-induced SIADH, rapid correction of hyponatremia can result in central pontine myelinolysis, which is irreversible loss of the central pontine pathways.151 A locked-in syndrome may develop, or a chronic vegetative state can result from central pontine myelinolysis.
Ifosfamide
The most common manifestation of neurotoxicity associated with ifosfamide is encephalopathy. Severe ifosfamide-induced encephalopathy has been reported in children and adults.152–156 This neurotoxicity is dose dependent and may be fatal. Neurologic deterioration usually begins within hours of administration of ifosfamide.152,154,156 Confusion, hallucinations, and aphasia are the most common initial signs. Progression to coma generally is rapid. Some patients also exhibit clinical evidence of seizure activity or myoclonus with intermittent twitching of the extremities.152,155 Electroencephalography (EEG) shows severe slowing with delta wave activity and can display evidence of seizure activity.152,155 In most cases, encephalopathy completely resolves over several days after cessation of therapy, although in one study, investigators found persistent mental status changes in some patients 10 weeks after treatment.157 A recent study of 60 patients reported the incidence of ifosfamide neurotoxicity to be 26%.158 Several risk factors have been reported to predispose patients to development of neurotoxicity from ifosfamide. These factors include low serum albumin concentration,156 high serum creatinine concentration, pelvic cancer,159 and previous treatment with cisplatin.160 Altered mental status before treatment and bolus or rapid infusion have also been described as a predisposing factor to development of neurotoxicity from ifosfamide.161,162 A recent report, however, could not confirm any risk factor except age, with an increased incidence of encephalopathy in younger patients.158 Ifosfamide treatment has been associated with an extrapyramidal syndrome characterized by choreoathetosis, blepharospasm, and opisthotonic posturing.163,164 Data from case reports indicate that methylene blue is an option in the treatment of ifosfamide-induced encephalopathy. However, the lack of controlled clinical trials and the possibility of spontaneous resolution of encephalopathy calls the effectiveness of methylene blue into question.165 Thiamine and albumin have also been described as options for treatment and prophylaxis of ifosfamide-induced encephalopathy.166
5-Fluorouracil
Cerebellar Toxicity
5-Fluorouracil (5-FU) causes acute cerebellar dysfunction. Patients experience moderate to severe gait ataxia, scanning speech, appendicular ataxia marked by severe dysmetria, and often nystagmus.167–170 These neurologic abnormalities resolve completely within several days after completion of therapy. The incidence of cerebellar toxicity has been reported to be 3% to 7% and correlates with dose and the interval between treatments.170
Neuropsychiatric Symptoms
Organic brain syndrome has been reported with 5-FU treatment.172 Confusion and disorientation can develop without evidence of cerebellar dysfunction. In one patient, retreatment with 5-FU resulted in a similar episode of mental deterioration. Oculomotor disturbances, specifically vergence disturbances characterized by diplopia on viewing distant objects, were reported in two patients.173 Seizures have also been reported.174 A possible association of 5-FU treatment with recurrent acute toxic neuropathy has been reported.175
Fludarabine
Fludarabine can cause a variety of neurologic toxicities ranging from a mild peripheral neuropathy to severe altered mental status with hallucinations, motor weakness, paralysis, or seizures. At high doses, white matter changes, particularly in the occipital lobes and brainstem, have been reported.181 The severe neurotoxicity may produce progressive worsening to death over the course of weeks to months. Visual disturbances are the most commonly reported symptom and result from cortical blindness, visual pathway demyelination, and/or retinal bipolar cell loss. Resolution of neurotoxicity rarely occurs, with most patients experiencing irreversible and severe dysfunction. The risk factors for toxicity at recommended doses have not been identified.
Fludarabine is distinctive among agents causing central nervous system toxicity in that its clinical effects do not manifest until weeks to months after exposure.182 The risk of development of progressive multifocal leukoencephalopathy due to an infection with the JC virus may be increased with fludarabine treatment. Diagnosis requires biopsy of involved brain or polymerase chain reaction testing of CSF.183
Nitrosoureas
Central Nervous System Toxicity
Nitrosoureas, most commonly carmustine (BCNU), can cause encephalopathy characterized by a progressive decline in cognitive function, development of seizures, coma, and death.186–186 This complication most commonly is observed with intracarotid or supraophthalmic arterial delivery of nitrosourea, although a similar syndrome has been reported in patients who receive very high dose intravenous carmustine treatment.187 Histopathologically, the toxicity associated with intraarterial therapy is characterized by necrosis, regions of demyelination, edema, and axonal loss limited to the region perfused by the intraarterial therapy. Because the predominant changes have been seen in the white matter, the condition is called leukoencephalopathy; the changes are indistinguishable from those seen with methotrexate and ara-C treatment. Similar pathological changes are seen with high-dose intravenous carmustine, but both cerebral hemispheres are involved.187 In addition, focal brain necrosis has been reported with intraarterial nitrosourea treatment. This toxicity is thought to be a consequence of “streaming” of the drug along the vessel wall, without mixing with arterial blood.188 This stream of concentrated drug may flow into a small branch of the artery. A small region of brain (or tumor) thus receives an enormous dose of drug, and focal necrosis results.
A biodegradable polymer containing BCNU has been shown to be effective in the management of recurrent malignant glioma. The BCNU-impregnated polymer also has been approved for treatment of newly diagnosed glioblastoma. This treatment usually is followed by conventional external beam radiation therapy.189 The treatment combination may increase the incidence of treatment-associated necrosis. The wafers are placed into the tumor cavity after surgical resection. Results of clinical trials indicate that the local therapy is well tolerated, although an increase in peritumoral edema necessitates a temporary increase in corticosteroid dose, and reports of treatment-associated infections and necrosis have been made.192–192
Retinal Toxicity
Retinal toxicity has been reported with intracarotid administration of nitrosoureas, particularly carmustine.193 This retinopathy is painful and often results in permanent vision loss. Infusion above the ophthalmic artery eliminates this toxicity but may increase the likelihood of streaming (see the preceding section, “Central Nervous System Toxicity”).
Procarbazine
Central Nervous System Toxicity
Signs of CNS depression, ranging from mild drowsiness to profound stupor, have been reported.194,196 This toxicity worsens with use of phenothiazine to control emesis and may be related to the monoamine oxidase inhibitor–like qualities of procarbazine. CNS depression was reported to occur in 14% to 33% of patients in these series.196–196
Paclitaxel and Docetaxel
Paclitaxel is associated with a syndrome of subacute aches and pains, “paclitaxel-associated acute pain syndrome” (P-aps), and with peripheral neuropathy. This pain syndrome causes significant morbidity, with an incidence of up to 70% in patients receiving treatment with paclitaxel. It manifests with diffuse aching discomfort, most often in the legs, hips, and lower back, although it can be widespread.197
The nature and temporal profile of P-aps distinguishes it as a separate entity from chemotherapy-induced peripheral neuropathy, which can be a devastating long-term consequence of this drug. Chemotherapy-induced peripheral neuropathy has been described in up to 70% of patients receiving treatment with paclitaxel. An association exists between the presence and severity of P-aps and the eventual development of sensory neuropathy.198,199
In phases 1 and 2 testing of paclitaxel, myelosuppression was the dose-limiting toxicity. Since colony-stimulating factors (e.g., granulocyte macrophage and granulocyte-colony stimulating factors) have become widely available, however, peripheral neuropathy has become the dose-limiting toxicity.1,2
The neuropathy is predominantly sensory, particularly affecting small-caliber (pain and temperature) sensory fibers.199,200 The effect on motor fibers and large-caliber sensory fibers (vibration and proprioception) is less severe. Nerve conduction studies of large myelinated nerve fibers show evidence of both axonal injury and demyelination.200 Neuropathy generally occurs at doses greater than 200 mg/m2 and is more frequent when the drug is given every three weeks at a higher dose compared with once a week at a lower dose.201 Symptoms usually develop 1 to 3 days after treatment. Overall the prognosis is good, because much of the neurologic dysfunction reverses over several weeks. However, continued treatment in the setting of existing neuropathy causes progressive neurologic toxicity that may not resolve. The peripheral neuropathy associated with docetaxel appears to be similar to that with paclitaxel in preliminary reports, although in a randomized trial, the incidence and severity were less with docetaxel.204–204 Peripheral neuropathy also is being reported with other microtubule-stabilizing agents such as the epothilones.205,206
A case report of transient vocal cord paralysis resulting from use of paclitaxel has been described.207
Encephalopathy has also been described as an adverse effect of paclitaxel. In one series, transient encephalopathy was reported to occur within hours of administration of standard doses of paclitaxel. All patients had undergone previous brain radiation therapy, and all recovered within hours.208 Acute encephalopathy was reported to occur in six patients receiving a very high intravenous dose of paclitaxel (greater than 600 mg/m2). Encephalopathy developed between 7 and 23 days after treatment. Three patients recovered, and the other three patients died of progressive coma. Autopsy revealed generalized white matter atrophy.209 Chronic neurocognitive changes also have been reported with extensive administration of taxanes.210
Tamoxifen
Tamoxifen, as with other hormonal agents, is associated with headache.211 Tamoxifen can cause reversible retinal dysfunction at the conventional antiestrogen doses used for breast cancer therapy.214–214 At higher doses, reversible encephalopathy with delusions, somnolence, and cerebellar dysfunction has been reported.217–217 Several cases of sinus vein thrombosis in patients treated with tamoxifen have been described.218,219
Biological Response Modifiers
Interleukin-2
Central Nervous System Toxicity
The vascular leak associated with intravenous interleukin-2 (IL-2) administration can result in encephalopathy and coma.220 A high percentage of patients receiving both systemic IL-2 and lymphocyte-activated killer cells experience encephalopathy or a neuropsychiatric syndrome.220 In most cases, severe but reversible cognitive impairment occurs. In the presence of an intracranial mass lesion, the increase in brain edema leads to an asymmetric shift in the brain, resulting in herniation. In a single case report, development of multifocal white matter lesions was associated with intravenous administration of IL-2. The lesions resolved completely over several months.221 Another case report described fatal leukoencephalopathy associated with IL-2 treatment.222
Interferons
Systemic administration of interferon affects both the CNS and the peripheral nervous system.
Central Nervous System Toxicity
Development of neuropsychiatric symptoms, predominantly depression, is the most commonly reported adverse effect associated with use of interferons. The availability of preventive and treatment interventions suggests that neuropsychiatric toxicity can often be managed without needing to discontinue the treatment.225 Confusion, lethargy, loss of cognitive function, and encephalopathy with perseveration and aphasia have also been described.228–228 In addition, a Parkinsonian syndrome with bradykinesia, masklike facies, micrographia,229 retinopathy,230 and optic neuropathy231 have been reported.
The neurologic effects of interferon are dose related, but they are more severe in patients with underlying neurologic abnormalities.232 Although the neurotoxic effects of interferon therapy usually resolve completely within a few weeks, some patients may exhibit persistent behavioral changes.226 Intraventricular administration of interferon-alpha caused severe neurologic toxicity in one study. Effects ranged in severity from headache and confusion to coma. Additional adverse effects of intraventricular administration included Parkinsonism, hearing loss, and seizures.233
Thalidomide and Lenalidomide
Thalidomide has both immunomodulatory and antiangiogenic properties, has shown significant activity in multiple myeloma, and is being evaluated in the management of several other cancers.235 Somnolence, the predominant adverse effect, is dose-related, and the drug can be titrated in most patients to a level of tolerance. Peripheral neuropathy is treatment limiting. Rates of neuropathy after thalidomide treatment vary from 15% to 70%. The neuropathy has been characterized as sensory-motor axonal polyneuropathy manifesting as painful paresthesia or numbness.236 Severity of symptoms correlates with cumulative dose and duration of therapy. Factors influencing the risk of neurotoxicity include prior neuropathy, age, previous chemotherapy, and vitamin B12 and/or folate deficiency. The mainstay of neuropathy prevention is dose reduction or withdrawal of thalidomide, which can lead to symptom resolution in up to 16 weeks; however, in some cases, the neuropathy is irreversible.237,238
A person’s risk of developing a peripheral neuropathy after thalidomide treatment has been linked to single nucleotide polymorphisms in genes governing repair mechanisms and inflammation in the peripheral nervous system.239 Nerve biopsy and examination reveal distal axonal degeneration and demyelination. Studies of lenalidomide indicate that peripheral neuropathy may occur in a smaller percentage of patients compared with thalidomide.240,241
Bevacizumab
Bevacizumab is a humanized monoclonal antibody against vascular endothelial growth factor (VEGF) that has shown activity in a wide variety of cancer types including colon, lung, and renal cell cancers and glioblastoma. Exacerbation of hypertension is a commonly reported toxicity and may be related to the development of PRES, described in detail later in this chapter.242,243 Bevacizumab increases the risks of hemorrhage and thromboembolism, although an analysis identifying four trials of anti-VEGF therapy in patients with brain metastases found a negligible rate of intracranial bleeding.244 Patients with high-grade glioma who are undergoing anticoagulation in addition to treatment with bevacizumab have an elevated hemorrhage risk, although the serious hemorrhage rate remains low (3%), suggesting that bevacizumab can be safely administered with anticoagulation in most patients.245,246
In multiple studies, bevacizumab has been reported to increase the risk of stroke. A pooled analysis of five randomized trials showed an increase in stroke from 0.5% to 1.7%.247 In addition, two cases of dural sinus venous thrombosis have been reported in patients during treatment with bevacizumab.248,249
Bortezomib
Bortezomib is a proteosome inhibitor that has demonstrated activity in multiple myeloma. Peripheral neuropathy, predominantly a sensory neuropathy, initially was reported during early phase 1 trials. A prospective study evaluated the occurrence of neuropathy in a group of 256 patients with refractory myeloma who were treated with standard dosing schedules of bortezomib.252 The incidence of neuropathy was estimated to be approximately 35%, and neuropathy was more common in patients receiving bortezomib at 1.3 mg/m2 than in those who received a dosage of 1.0 mg/m2. The cumulative dose also correlated with severity of the neuropathy, as did the presence of neuropathy before the initiation of treatment. Most patients experienced either partial or complete resolution with cessation of treatment.
Two cases of acute axonal motor neuropathies associated with paralytic ileus, urinary retention, and impotence occurring early in the course of bortezomib treatment have been described. Both patients had previously been exposed to other neurotoxic agents including vincristine and thalidomide and had co-morbid conditions associated with neurologic damage, namely chronic renal failure and autoimmune hepatitis.253
Sunitinib
Sunitinib is a polytyrosine kinase inhibitor that inhibits the intracellular tyrosine kinase domain of the VEGF, platelet-derived growth factor, and c-KIT receptor; it is used for gastrointestinal stromal tumors and advanced kidney cancer. This drug may be related to the development of PRES.254
Imatinib
Imatinib is an oral tyrosine kinase inhibitor that is used in treating chronic myelogenous leukemia and gastrointestinal stromal tumors. The main neurologic side effects of imatinib are muscle cramping and myalgias.255 Intracranial bleeding is a rare complication of imatinib therapy. In a retrospective study of 121 patients with chronic myelogenous leukemia who were treated with imatinib, subdural hematomas developed in seven patients. In three of the patients the subdural hematomas were not associated with thrombocytopenia or other risk factors.256 In addition, in clinical trials when patients with high-grade glioma were treated with imatinib, significant intratumoral hemorrhages were observed.257,258 One case report described muscle edema, creatine kinase elevation, and rhabdomyolysis with myoglobinuria in a young woman treated with imitanib.259
Ipilimumab
Ipilimumab is a human monoclonal antibody against cytotoxic T lymphocyte antigen-4 that activates the immune system. It is approved for the treatment of metastatic melanoma. Ipilimumab is associated with multiple autoimmune adverse effects, including hypophysitis with hypopituitarism, which has been reported in 0% to 17% of patients involved in ipilimumab trials.262–262
Other immune-related adverse effects that have been described are inflammatory myopathy, as well as peripheral neuropathy and an autoimmune syndrome that emulates Guillain-Barré syndrome.263 A case report of PRES during ipilimumab therapy has also been described.264
Radiation Neurotoxicity
Radiation neurotoxicity is becoming an increasingly important and recognized complication of cancer therapy (see Chapter 39). New therapeutic strategies for systemic cancer and for CNS metastasis have improved survival, uncovering a greater incidence of late, chronic toxicity from radiation and combined chemoradiotherapy treatments. Advances in technology, such as radiosurgery and brachytherapy, allow local dose intensification of radiation treatment for brain tumors. Furthermore, advances in development of radiosensitizers will increase the effects of radiation therapy on tumor and surrounding normal tissue.
Central Nervous System Effects
Cranial radiation therapy can cause acute, subacute, and chronic neurotoxicity. The volume of brain treated, total dose, and dose fraction are the most important determinants of toxicity, although some variation in patient susceptibility is apparent. Additionally, radiotherapy can indirectly affect the CNS by damaging the blood vessels and increasing the risk of brain cancer.265
Acute Toxicity
Acute toxicity is characterized by headache, nausea, vomiting, and sometimes also somnolence. This disorder generally occurs within the first few days of initiation of cranial radiation therapy.266 The symptoms are thought to be a result of disruption of the blood-brain barrier and secondary edema.267 Acute toxicity is most common in patients receiving a large dose of radiation per fraction, mainly as whole-brain radiation therapy. Most patients experience complete recovery of neurologic function. Corticosteroids can treat and sometimes prevent the acute toxicity.267
Early-Delayed Toxicity
Early-delayed toxicity is noticed weeks to 3 months after completion of radiation treatment.268 Most commonly, patients have drowsiness, nausea, headaches, ataxia, and worsening of underlying neurologic dysfunction. Complete resolution is expected, although rare patients have an idiosyncratic reaction with widespread brain necrosis.269 Corticosteroids can speed recovery. The pathogenesis of early-delayed toxicity is unknown, but the reaction may be related to reversible demyelination. CT reveals decreased attenuation in the cerebral white matter; MRI reveals increased signal intensity in white matter on T2-weighted images.
An increase (up to 30%) in early treatment–related brain injury has been reported with the use of concurrent daily temozolomide with external beam radiation in patients with glioblastoma.270 Changes are seen on brain imaging studies that emulate tumor growth with increasing edema and an increase in uptake of contrast material. These changes, now referred to as “pseudoprogression,” may reflect an increase in blood-brain barrier breakdown that is the consequence of an augmentation of radiation effect from the co-administration of chemotherapy.270
Chronic, Late Radiation Injury
Chronic, late radiation injury usually appears 9 months to 2 years after completion of radiation treatment, although some patients have experienced onset of symptoms 10 years after treatment.271 Late radiation injuries are often progressive and irreversible. Patients exhibit focal areas of radiation necrosis or evidence of diffuse radiation injury. The incidence of each type of injury depends on the dose, fractionation schedule, and area of treatment.272,273
Radionecrosis
Focal necrosis primarily affects the white matter of the brain or spinal cord. It usually occurs within 18 months of radiation but can develop years later.274 The risk of developing radiation necrosis depends on the dose and the volume of radiation (mainly above 65 Gy and 2 Gy daily fractions). Focal delivery of one large radiation fraction during radiosurgery can lead to radiation necrosis adjacent to the irradiated lesion in 5% to 20% of cases. The latency of these changes can be as short as 3 months.275 Radiation necrosis often manifests as focal neurologic deficits, such as hemiparesis or aphasia. Global signs such as obtundation can occur when the localized necrosis causes increased intracranial pressure and herniation. Similarly, focal necrotic regions can cause seizures. Conventional MRI findings cannot discriminate radiation necrosis from recurrent tumor (in the case of treated brain metastases or primary brain tumors). Positron emission tomography, single-photon emission computed tomography, and new MRI techniques, including perfusion and spectroscopy, may help distinguish tumor from radiation necrosis, but their sensitivity and specificity are between 50% and 90%.276 Pathological examination of the necrotic region reveals vascular injury to the small arteries and arterioles and evidence of coagulative necrosis, with destruction of all elements of the nervous tissue.57 Treatment is directed at decreasing edema and mass effect. Patients generally respond to corticosteroids; however, the effect is short lived, and long-term steroid requirements are common.277 Antiplatelet agents, anticoagulants, and hyperbaric oxygen have been studied, but currently, high-quality evidence to support their use in routine clinical practice is minimal.278 Bevacizumab has also been suggested as a therapeutic option. A randomized controlled study that included 14 patients showed clinical and radiographic efficacy of bevacizumab with radiation necrosis.279 Surgical resection, if feasible, can be curative, although even with extensive resection, additional areas of necrosis may develop.
Diffuse Injury
Diffuse injury manifests as global neurologic dysfunction, with cognitive decline, personality change, confusion, and lethargy, and it can progress to dementia, obtundation, or coma.267,273 Clinically, mild to moderate forms of cognitive impairment are more common than severe dementia. Short-term memory, executive functions, attention, and judgment are mainly affected.280 Symptoms usually appear several years after the radiation exposure. Diffuse white matter changes are found on CT or MRI, manifesting as low attenuation on CT scans and high signal intensity in the periventricular and subcortical white matter on T2-weighted and proton-density MR images.283–283 Mass effect and focal neurologic signs are not common, although late in the course, seizures and motor dysfunction can occur.
The main risk factors for diffuse injury include the volume of brain treated, concurrent or adjuvant chemotherapy directed at the CNS, and use of short-course and high-dose fraction regimens. Other risk factors are increasing age and vascular risk factors such as diabetes and hypertension.284 The incidence is difficult to determine, but reports have shown neurologic and imaging changes in 32% to 50% of patients subjected to radiation therapy.282,283,285
The pathogenesis of diffuse radiation injury is not known, although distinctive neuropathological changes have been described. The most prominent pathological finding is vascular changes in the small arteries and arterioles. Hyalinization of the vessel walls with occlusion is common.283 In addition, areas of necrosis, gliosis, and demyelination are found. Impaired neuronal stem cell function and decreased neurogenesis have also been described.286 Modern radiation techniques have been developed to protect the hippocampus to reduce memory defect; however, the efficacy of these techniques has not yet been proven.287 No treatment has been established for global brain injury. However, donepezil may be useful for the cognitive decline.288
Necrotizing Leukoencephalopathy
Necrotizing leukoencephalopathy is the most severe form of neurologic toxicity associated with radiation therapy. It is most common when CNS-directed chemotherapy is combined with radiation therapy. This syndrome was described earlier in this chapter in the discussion of methotrexate-associated toxicity. The incidence of leukoencephalopathy is much greater in patients who receive chemotherapy after cranial radiation therapy than in patients receiving either treatment alone or in those receiving chemotherapy before the initiation of radiation therapy.289
Mineralizing angiopathy has occurred in pediatric patients receiving both intrathecal methotrexate and radiation therapy.290 The changes frequently are diagnosed as dystrophic calcifications during neuroradiologic examinations. Histopathological analysis reveals deposits of calcium in small blood vessels, often with surrounding regions of necrotic brain. The clinical significance of these changes is uncertain.
Endocrinologic Effects
Cranial radiation therapy has been associated with a wide spectrum of endocrinologic effects.291 Most of these effects are attributed to damage to the hypothalamic-pituitary axis. These effects are generally dose dependent and may manifest several years after completion of the radiation treatment. Results of several studies have suggested differing vulnerability among the various endocrine loops.292 Growth hormone deficiency and stimulation of precocious puberty have occurred at doses as low as 18 Gy.293 Deficiency of gonadotropins, thyroid-stimulating hormone, and corticotropin usually results only when the hypothalamic-pituitary axis receives more than 40 Gy.294 Similarly, hyperprolactinemia occurs most often in young women who receive more than 40 Gy of radiation therapy. Careful long-term monitoring of these patients is critical.295 Appropriate hormone replacement therapy or prolactin suppression treatment should prevent sequelae.
Indirect Effects of Radiation on the CNS
Radiation can damage the endothelia of cerebral vasculature, causing accelerated atherosclerosis, stenosis, and occlusion, leading to stroke. The typical latency of these phenomena is several years.296,297 In children, radiation can cause occlusion of blood vessels and development of a moyamoya-like pattern of collateral vessels. This phenomenon has been described mainly in association with neurofibromatosis 1, which is a known risk factor for vasculopathy.298 Brain and spinal vascular malformations, including cavernomas and, rarely, aneurysms, have also been described.299 The risk of developing other brain tumors in patients treated with CNS radiation is seven times that of the general population. Three types of brain tumors have been associated with prior cranial radiation: meningioma, glioma, and sarcoma.300
Radiation Myelopathy
The acute syndrome is rare with the dosing schedules currently being used. Case reports state that weakness evolves over a few hours or days.301 The syndrome probably is caused by acute necrotizing radiation injury due to rapidity of onset and failure of neurologic recovery.
The early transient form appears 6 to 12 weeks after treatment.302 Most patients exhibit Lhermitte sign—an electrical shock–like sensation down the spine with neck flexion. No neurologic deficits are found, imaging is usually intact, and the symptoms resolve spontaneously over several weeks. Transient demyelination is hypothesized to be the cause of the symptoms.
Delayed progressive radiation myelopathy is the most common radiation therapy–induced spinal cord disorder, although it is relatively rare. Signs and symptoms generally develop 6 months to 1 year after completion of radiation therapy, although some data suggest that the latent period may be as long as 18 months to 2 years.303 The clinical picture may vary from patient to patient. Some patients have monoplegia, and others may have paraplegia or quadriplegia. Sensory symptoms are generally more severe than motor symptoms. Some patients have Brown-Séquard syndrome. Progression may take several years.301 The pathogenesis of spinal radiation injury is not completely understood. Radiation might damage myelin production and might induce secretion of cytokines that might cause inflammatory response.301,302 Pathological examination reveals rarefaction of spinal cord white matter with small areas of necrosis. Degeneration of myelin sheaths and loss of oligodendrocytes have been noted. Other causes of spinal cord dysfunction need to be excluded, including intramedullary tumor or infection, multiple sclerosis, vitamin B12 deficiency, sarcoidosis, and Lyme disease. No treatment exists, and most patients experience progressive neurologic dysfunction over months to years.304
Lower motor neuron syndrome occurs after spinal cord radiation therapy. Patients exhibit pure motor signs of weakness, atrophy, and fasciculations.301 The syndrome manifests 3 months to 2 years after radiation treatment and is similar to polio. The pathogenesis is unknown. Hypotheses include loss of anterior horn cell neurons and radiation-induced injury to motor nerve roots. No treatment exists, with supportive care being the only option.
Peripheral Nerve Toxicity
Peripheral nerves are relatively resistant to the effects of radiation therapy. Early effects, which develop within 2 days of a single large-fraction treatment in an experimental animal system, include changes in vascular permeability of the nerve, changes in bioelectrical activity, and abnormal microtubule assembly in the axon.305 Late changes include fibrosis in the nerve sheath and angiopathic changes in the small arterioles that provide the vascular supply to the nerve.306 Use of large (>25 Gy) single doses or extended fractionated treatment to a very high total dose (>80 Gy) is thought to be necessary for injury to the peripheral nerves. Similar changes have been found for some cranial nerves.
Brachial and lumbar plexopathies warrant a separate discussion. These syndromes result from radiation injury to the nerve fibers in the plexus. Brachial plexopathy, caused by axillary radiation therapy for breast cancer, is most common.307 Lumbar plexopathy is more commonly associated with pelvic external beam radiation therapy, although local radioactive seed implantation (brachytherapy) may result in local nerve damage.305
A mid and reversible plexopathy can occur as a subacute complication of radiation therapy. Delayed brachial plexopathy manifests as numbness and weakness of the arm. Delayed lumbar plexopathy usually manifests as asymmetric bilateral leg weakness with predominance of the lower plexus (L5-S1) innervated muscles. The diagnosis of delayed radiation plexopathy can be difficult and requires excluding tumor infiltration as the cause of the symptoms. This process is most difficult with brachial plexus dysfunction and in patients with apical lung cancer or metastatic breast cancer. The distinguishing features in comparing radiation with tumor brachial plexopathy have been described extensively.307 None of the criteria is absolute, although tumor infiltration is more likely to be painful and involve the lower nerve roots (C7-T1) than is radiation injury, which is less likely to cause severe pain and generally involves higher roots (C5 and C6). Acute reversible radiation injury to the brachial plexus has been described. This condition is often painful, although the pain generally is mild. Patients experience weakness and atrophy in a C6-T1 distribution. Spontaneous but gradual recovery is typical.308
The treatment of radiation-induced brachial plexus neuropathy depends on the grade of severity of injury. In cases of mild injuries, conservative treatment is indicated, including nonnarcotic and narcotic analgesics and anesthetic interventions. Severe cases may require surgical exploration to allow the neural elements to be released from fibrotic tissue and prevent fibrosis of the vascular supply to the nerve.309 Radiation has also been reported to accelerate vascular injury, causing segmental obstruction of the subclavian artery.310 This condition usually is painless. The motor and sensory loss evolves quickly, without subsequent progression or improvement.
Lumbosacral plexopathy is less common than radiation-related brachial plexopathy.311 Onset of signs and symptoms usually is delayed until at least 1 year after radiation treatment. Pain is usual and mild. The pathogenesis is uncertain, although fibrosis causing nerve compression and ischemia is a likely cause. The diagnosis of radiation-related lumbosacral plexopathy is made by excluding tumor infiltration. Imaging (MRI or CT) often is useful, although in select cases, surgical exploration may be required.
Muscle Injury from Radiation Treatment
Muscle is thought to be relatively resistant to the effects of radiation therapy, although several studies have suggested that at higher doses (greater than 50 Gy), significant late toxicity may occur.222 Early effects are uncommon, symptoms generally are found after 1 year, and the onset of changes has been reported as late as 10 years after radiation therapy. Signs and symptoms include muscle contractures, loss of function, pain, extremity edema, and pathological fractures. The synergy of the toxic effects with those of chemotherapy is uncertain, although most patients have received both modalities.306 No treatment other than conservative measures, including physical therapy, have been established.
Differential Diagnosis
Dementia and Encephalopathy
Acute Encephalopathy
A common neurologic problem in patients with cancer is acute encephalopathy.312 The differential diagnosis is extensive. Most commonly, however, acute encephalopathy is caused by a toxic or metabolic derangement. Frequent causes include narcotic effects, electrolyte abnormalities, hypoxia, and renal or hepatic dysfunction. Neoplastic meningitis frequently manifests as mental status changes as a result of increased intracranial pressure, seizures, or infiltration of the cortex through the Virchow-Robin spaces surrounding surface blood vessels. Likewise, brain metastasis can cause an acute change in mental status when a sudden increase in tumor size (often hemorrhage) results in a rapid change in intracranial pressure or when tumor-induced seizures occur. A paraneoplastic syndrome, limbic encephalitis, can manifest as a progressive dementia, which often is subacute. This paraneoplastic syndrome most frequently is associated with small cell lung cancer, but it has been described in association with other tumors. Patients often are found to have serum anti-Hu antibodies, which also is a feature of paraneoplastic sensory neuropathy.315–315
Cancer treatment can directly or indirectly cause acute encephalopathy. High doses of intravenous methotrexate or ara-C can cause reversible encephalopathy, often accompanied by lethargy.5,13,43 Results of studies with animals and with patients in which positron emission tomography was used after administration of high-dose methotrexate showed temporary reduction in brain metabolic activity in both glucose utilization and protein synthesis.45 Folinic acid (leucovorin) administration improved metabolic activity in laboratory studies.46 Other agents known to cause encephalopathy include vincristine, ifosfamide, procarbazine, fludarabine, paclitaxel, cisplatin, and l-asparaginase. The immunostimulant levamisole combined with 5-FU causes reversible encephalopathy and has been associated with a demyelinating condition known as multifocal inflammatory leukoencephalopathy.176 The hormonal agent tamoxifen has been reported to cause encephalopathy at high doses. Biological response modifiers such as the interferons and interleukins commonly cause reversible encephalopathy, although permanent neurologic sequelae may result from prolonged use of interferon.226,227 Radiation therapy can cause an acute syndrome manifesting as lethargy that generally is reversible.
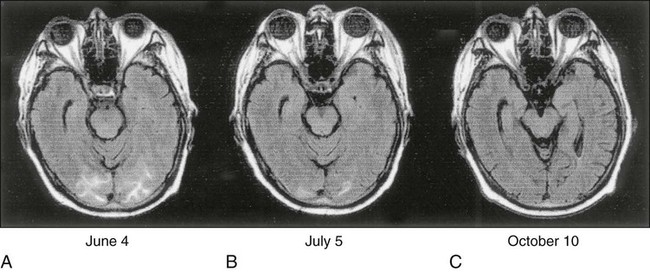
PRES has been seen most frequently with use of the immunomodulators cyclosporine and tacrolimus, but cisplatin, gemcitabine, paclitaxel, bortezomib, ipilimumab, and combination chemotherapy regimens also have been associated with the syndrome. Drugs that modulate the VEGF pathway, such as bevacizumab and sorafenib, have been associated with PRES as well. PRES typically manifests as changes in mental status, cortical blindness, headache, and variable loss of other higher cortical functions, such as aphasia and apraxia. The syndrome is often associated with hypertension. Brain imaging with CT and MRI reveals characteristic patchy white matter changes involving bilateral and occipital lobes bilaterally in a subcortical distribution316 (Fig. 57-2). The pathogenesis of the syndrome is unknown, although hypertension and hypomagnesemia have been associated with it. Full recovery has occurred with cessation of treatment or adjustment of the dose of the immunosuppressant.317–321
Many chemotherapeutic agents provoke a change in mental status by causing secondary metabolic derangement. Cyclophosphamide and vincristine can stimulate SIADH.105,106,150 The resulting hyponatremia can lead to seizures and encephalopathy. Hyponatremia secondary to cisplatin-induced salt-wasting nephropathy can cause the same neurologic problems. Treatment with l-asparaginase, corticosteroids, and streptozocin can lead to glucose intolerance. Left untreated, this condition can result in nonketotic hyperosmolar coma. Many chemotherapeutic agents can cause hepatic and renal dysfunction with consequent development of secondary neurologic symptoms.
Chronic Encephalopathy and Dementia
Chemobrain
In recent years, chemotherapy-induced cognitive decline in cancer survivors, especially those with breast cancer, has been described; however, the frequency and the severity of this complication are unclear.322 The term “chemobrain” or “chemofog” was defined as cognitive adverse effects of chemotherapeutic agents administered to treat solid tumors that patients experience during and after chemotherapy.323 Global impairment in attention, memory, and executive functions has been reported in 16% to 75% of adult survivors of solid tumors after chemotherapy in several studies.324–327
In contrast, other reports have not reported any cognitive dysfunction in survivors of breast cancer after adjuvant chemotherapy.328 Differences in the nature of the study and the methodology probably account for the discrepancies. Despite the controversy, there is growing evidence of an increased incidence of cognitive decline in cancer survivors, likely resulting from chemotherapy. The cognitive skills that are impaired with chemobrain include memory, executive function, processing speed, and reaction time.329 Risk factors for the development of chemobrain are increasing age, history of head trauma, vascular risk factors, and treatment with additional neurotoxic treatment. Cumulative dose, intensity, and duration of treatment have also been associated with chemobrain.330 The epsilon 4 allele of apolipoprotein E is a potential genetic marker to suggest an increased vulnerability to chemotherapy-induced cognitive decline.331 The pathogenesis of cognitive impairment in patients with cancer who are treated with chemotherapy is largely unknown. The etiology is probably multifactorial; one suggested mechanism is that chemotherapy can damage the neural progenitor cell function, which can lead to cognitive dysfunction.332
Standard brain imaging does not generally demonstrate substantial anatomic changes in patients treated with standard chemotherapy alone. However, a reduction of right prefrontal and parahippocampal gyrus volumes that were associated with attention (or concentration) and visual memory function have been reported.333 Functional neuroimaging studies showed greater recruitment of frontal cortical tissue or expanded spatial extent of brain activation during cognitive tasks in patients treated with conventional chemotherapy compared with nontreated control subjects.334
No medication has been clearly established to prevent or treat chemobrain. Methylphenidate was suggested as a possible therapy, but in a recently published randomized controlled trial, methylphenidate failed to improve the cognitive function of cancer survivors and was found only to be active in ameliorating chemotherapy-induced fatigue.335
Diagnostic Evaluation
A thorough search for a treatable underlying cause of encephalopathy should be undertaken in all affected patients. The diagnosis of chemotherapy-induced encephalopathy is made by excluding other potential causes. Ordering of diagnostic tests is dictated by findings on the physical and neurologic evaluations. Patients with focal neurologic abnormalities require early imaging studies (CT or MRI), although certain metabolic disorders (e.g., hypoglycemia) can manifest as focal neurologic deficits. A metabolic evaluation, including measurement of electrolytes, renal and liver function tests, measurement of oxygenation, serum calcium and magnesium, serum ammonia, and possibly thyroid and adrenal function testing, should be performed for most patients. Careful review of prescribed and over-the-counter medications may provide critical information. In some cases, blood and urine toxicology screening may be necessary. EEG often is helpful in directing the evaluation by indicating the presence of structural abnormalities (e.g., periodic localizing epileptiform discharges or focal slowing) or may indicate a global metabolic process (e.g., diffuse slowing with Delta waves or triphasic waves in hepatic encephalopathy). Patients who have subclinical status epilepticus may present with encephalopathy; in these cases, the EEG findings are diagnostic. Lumbar puncture often is needed to exclude infectious or neoplastic meningitis. In most cases, head MRI or CT should be performed before lumbar puncture to look for a mass lesion in the brain that would make lumbar puncture unsafe. The search for the underlying cause often is revealing. One group of investigators reported uncovering the cause of encephalopathy in 31 of 37 patients.313
Seizures
Clinical Manifestations and Differential Diagnosis
The differential diagnosis for seizures in patients with cancer includes an extensive list of possible causes. Causal factors can be broadly classified into toxic-metabolic and structural causes. Metabolic factors include hepatic and renal failure resulting from tumor growth or treatment toxicity, electrolyte abnormalities such as hypercalcemia from bone destruction or parathyroid hormone effect, hypomagnesemia from chemotherapy (cisplatin) or excessive vomiting, hyponatremia from SIADH or salt-wasting nephropathy (cisplatin), hypoxia, hyperglycemia from pancreatic failure (streptozocin), and glucose intolerance (corticosteroids). Several chemotherapeutic agents are known to be the direct cause of seizures (Table 57-1).
Table 57-1
Chemotherapeutic Agents that Cause Seizures
| Agent | Route of Administration | Comment(s) |
| l-Asparaginase | Intravenous | Associated with cerebrovascular events |
| Bevacizumab | Intravenous | Associated with PRES |
| Busulfan | Oral | Bone marrow transplantation–preparative regimen |
| Cisplatin, oxaliplatin | Intravenous | Rare; may be associated with cortical blindness, PRES |
| Cytosine arabinoside | High-dose intravenous (acute) | Late with leukoencephalopathy |
| Ifosfamide | Intravenous | Associated with encephalopathy |
| Methotrexate | High-dose intravenous (acute) | Late with leukoencephalopathy |
| Metronidazole | Intravenous, oral | |
| Nitrosoureas | Intracarotid or supraophthalmic arterial | Often associated with focal brain necrosis |
| Sorafenib | Oral | Associated with PRES |
| Vincristine | Intravenous |
PRES, as described in a previous section, can manifest with seizures, with typical reversible imaging appearance and symptomatology that is shared by a diverse array of causes. Structural causes of seizures include brain metastasis, dural metastasis, and meningeal carcinoma. Ischemic and hemorrhagic vascular events also may provoke seizures. A tumor-induced hypercoagulable state may greatly increase the risk of stroke or venous sinus thrombosis.338 A similar hypercoagulable state can be seen with l-asparaginase treatment. The paraneoplastic syndrome marantic endocarditis often results in cerebrovascular events, as can infectious endocarditis, a potential complication of treatment-induced neutropenia.232 Cisplatin can cause vasospasm of the intracranial vessels, producing a stroke.339 Furthermore, immunocompromised patients are at greater risk for CNS infection (including bacterial and fungal meningitis), brain abscess, and viral encephalitis (including progressive multifocal leukoencephalopathy).
Headache
Many chemotherapy drugs can cause headache as a component of PRES syndrome. Aseptic (chemical) meningitis might be caused by intrathecal therapy, as described earlier in this chapter. Chemotherapy agents that are frequently associated with headache are hormonal agents, interferon therapy, and high doses of fludarabine and carmustine. All trans-retinoic acid is highly associated with headache and can cause pseudotumor cerebri.340 The new tyrosine kinase inhibitors, imatinib, nilotinib, and dasatinib, cause headache in a relatively high percentage of patients.341 The diagnostic evaluation generally involves evaluation similar to that described for encephalopathy.
Cerebellar Dysfunction
Clinical Manifestations and Differential Diagnosis
Cerebellar dysfunction in patients who have cancer occurs most commonly with metastatic spread to the cerebellum or brainstem. Meningeal carcinomatosis occasionally causes cerebellar signs by infiltrating cerebellar pathways. Patients with structural cerebellar lesions often show asymmetric dysfunction, which can be useful in differentiating this condition from drug-induced cerebellar toxicity. Paraneoplastic cerebellar degeneration is characterized by subacute progressive loss of cerebellar function.342,343 Patients usually have symmetric loss of all cerebellar function and exhibit appendicular and truncal ataxia, nystagmus, and scanning speech. This paraneoplastic syndrome occurs most commonly with lung cancer but also has been associated with gynecologic cancers, breast cancer, and Hodgkin disease. Many patients have antibodies in the serum, including anti-Yo, anti-Ri, anti-Hu, anti-CV2/CRMP5, anti-Ma2, anti-Tr, and anti-Zic4. Only two antibodies, anti-Yo and anti-Tr, are predominantly associated with cerebellar dysfunction; the other paraneoplastic antibodies are often associated with symptoms involving other areas of the nervous system.342
5-FU and ara-C can cause reversible cerebellar toxicity. Ara-c can sometimes cause permanent irreversible damage to the cerebellar Purkinje cells. The result is truncal ataxia, unsteady gait, dysarthria, and nystagmus.12–15,167–170 The dysfunction usually is symmetric. MRI findings immediately after chemotherapy are normal, but in the irreversible cases, cerebellar atrophy often is detected by MRI months later.
Cranial Neuropathy
Clinical Manifestations and Differential Diagnosis
Cranial neuropathy in patients with cancer most commonly indicates the presence of meningeal carcinomatosis, tumor involvement of the bones of the cranial base with encroachment of neural foramina, or rarely, brainstem metastasis.343 Chemotherapy-induced cranial neuropathy is rare. Vincristine can cause cranial nerve palsy; extraocular eye movement abnormalities are most common.76,80,94 Intraventricular administration of drugs and of biologic response–modifying agents can cause transient cranial nerve palsy, which often is due to abnormalities in CSF circulation.344
Optic Neuropathy and Ocular Toxicity
Clinical Manifestations and Differential Diagnosis
Optic neuropathy can be seen in patients with neoplastic meningitis, lymphoma, or leukemia with infiltration of the optic nerve.345,346 In addition, compression from cranial base tumors and radiation therapy can cause similar loss of vision. Optic neuritis, characterized by unilateral or bilateral loss of vision, has been reported as a very rare paraneoplastic syndrome associated with small cell lung cancer.347 Retinopathy most commonly is associated with intracarotid administration of nitrosoureas and cisplatin.131,132,190,191 High concentrations of drug flow into the ophthalmic artery, which supplies the retina, causing severe pain and often complete loss of vision. Newer techniques, allowing administration above the ophthalmic artery, have reduced the incidence of this local toxicity.
Intravenous cisplatin administration has been associated with optic disk swelling and optic neuropathy.123,124 This reaction is idiosyncratic, and the mechanism is unknown.
Spinal Cord Toxicity
Clinical Manifestations and Differential Diagnosis
The most common cause of spinal cord dysfunction in patients is epidural cord compression, either from direct extension of bone metastasis or from paravertebral spread through the intervertebral foramina (see Chapter 49). Intramedullary tumor and extramedullary intradural tumors can have myelopathic manifestations.348 Similarly, epidural abscess and hematoma can be clinically indistinguishable from epidural tumor. Encephalomyelitis is a frequently described paraneoplastic syndrome associated with several types of tumors, particularly small cell lung cancer.349 A rare paraneoplastic myelopathy has been described with lung cancer, renal cell carcinoma, and Hodgkin disease. This syndrome is characterized by subacute progression of spinal cord dysfunction, usually in the thoracic region, although damage occasionally occurs at several levels. Pathological examination reveals segmental necrosis.
Lumbar intrathecal administration of methotrexate and ara-C can result in focal myelopathy.21–27 Symptoms and signs appear hours to days after treatment and can include sensory loss, upper and lower motor neuron dysfunction, radiating pain (similar to Lhermitte sign), and bowel and bladder incontinence. The myelopathy may be transient or progressive; complete irreversible paraplegia usually is seen with progressive dysfunction. Myelopathy also has been reported with spinal radiation therapy. A rare, acute syndrome develops over days. The more common chronic myelopathy with radiation therapy develops months after completion of treatment.
Peripheral Neuropathy
Clinical Manifestations and Differential Diagnosis
Peripheral neuropathy is a common paraneoplastic syndrome. Mild sensorimotor neuropathy is the most common neuropathy in patients with cancer.350 The cause is unknown, but the condition is a distinct entity, separate from the diffuse weakness associated with cachexia. Rare pure motor and pure sensory paraneoplastic neuropathies also have been described. Sensory neuronopathy, which is most common with small cell lung cancer, causes severe sensory loss.351,352 The intense inflammatory reaction and neuronal destruction in the dorsal root ganglia have led to use of the term dorsal root ganglionitis. Some patients are found to have anti-Hu and anti anti-CV2 antibodies in serum.352,353
Motor neuronopathy manifests as isolated lower motor neuron loss.354 This syndrome has been found in both patients with Hodgkin lymphoma and those with non-Hodgkin lymphoma. The pathogenesis of this pure motor syndrome is unknown. Other causes of neuropathy that should be considered are diabetes, thyroid dysfunction, vitamin B12 deficiency, and alcoholic neuropathy.
The incidence of chemotherapy-induced peripheral neuropathy varies from 30% to 40% of patients receiving chemotherapy and depends on the type of cytotoxic drug, the duration of administration, cumulative dose, and preexisting peripheral neuropathy. Several cancer chemotherapeutic agents (listed in Table 57-2) cause peripheral neuropathy. The peripheral neuropathy resulting from chemotherapy is temporally related to administration for most agents. Cisplatin is an exception, and delayed neuropathy occurs in some patients.
Table 57-2
Agents that Cause Peripheral Neuropathy
| Agent | Comment(s) |
| Bortezomib | Primarily sensory, dose related and cumulative dose related |
| Cisplatin | Large sensory fiber (vibration, proprioception); dose related and cumulative dose related; dose limiting |
| Cytosine arabinoside | Rare, symmetric sensorimotor polyneuropathy |
| Interferons | Transient paresthesia |
| Oxaliplatin | Large sensory fiber (vibration, proprioception); dose related and cumulative dose related; dose limiting; acute hyperexcitability syndrome |
| Paclitaxel, docetaxel | Dose-related sensorimotor; dose limiting |
| Procarbazine | Transient paresthesia |
| Thalidomide, lenalidomide | Sensory-motor axonal polyneuropathy; incidence related to duration and dose of treatment |
| Vincristine | Frequent; dose related; autonomic and sensorimotor; exacerbated in patients with underlying neuropathy; dose limiting |
Myopathy
Clinical Manifestations and Differential Diagnosis
Myopathy is a rare symptom in cancer, and is usually the consequence of treatment. Dermatomyositis and polymyositis are inflammatory myopathies differentiated by the presence and the absence of a rash, respectively.355,356 Dermatomyositis and polymyositis have been associated with many tumors, most commonly adenocarcinomas of the cervix, lung, ovaries, pancreas, bladder, and stomach. The relative risk of malignancy appears to be higher among patients with dermatomyositis than among those with polymyositis. Patients have an elevated serum creatine kinase (CK) level.
Although myopathy is a rare complication of chemotherapy, it has been described with the use of paclitaxel and vincristine.77,357
Grading of Neurotoxicity
None of the uniform grading systems of neurotoxicity is suitable for use in serial clinical evaluations. The most commonly used neurotoxicity measures are based on the National Cancer Institute’s Common Terminology Criteria for Adverse Events Version 4—Neurology, as shown in Table 57-3. Although these toxicity tables are useful in the evaluation of groups of patients, they are not suitable for monitoring the clinical status of individual patients. Standard neurologic testing with careful recording is best suited for individual patient care. The techniques required for performing an in-depth neurologic evaluation are reviewed in classic neurology textbooks.358,359 Quantitative measures of mental status function, motor examination results, and sensory function have been published.362–362
Table 57-3
National Cancer Institute Common Terminology Criteria for Adverse Events: Neurologic Toxicities
| Grade | ||||||
| Adverse Event | Definition | 1 | 2 | 3 | 4 | 5 |
| Abducens nerve disorder | A disorder characterized by involvement of the abducens nerve (sixth cranial nerve) | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | — | — |
| Accessory nerve disorder | A disorder characterized by involvement of the accessory nerve (eleventh cranial nerve) | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | — | — |
| Acoustic nerve disorder (NOS) | A disorder characterized by involvement of the acoustic nerve (eighth cranial nerve) | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | — | — |
| Akathisia | A disorder characterized by an uncomfortable feeling of inner restlessness and inability to stay still; this is an adverse effect of some psychotropic drugs | Mild restlessness or increased motor activity | Moderate restlessness or increased motor activity; limiting instrumental ADL | Severe restlessness or increased motor activity; limiting self-care ADL | — | — |
| Amnesia | A disorder characterized by systematic and extensive loss of memory. | Mild, transient memory loss | Moderate; short-term memory loss; limiting instrumental ADL | Severe; long-term memory loss; limiting self-care ADL | — | — |
| Aphonia | A disorder characterized by the inability to speak; it may result from injuries to the vocal cords or may be functional (psychogenic) | — | — | Voicelessness; unable to speak | — | — |
| Arachnoiditis | A disorder characterized by inflammation of the arachnoid membrane and adjacent subarachnoid space | Mild symptoms | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | Life-threatening consequences; urgent intervention indicated | Death |
| Ataxia | A disorder characterized by lack of coordination of muscle movements resulting in the impairment or inability to perform voluntary activities | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL; mechanical assistance indicated | — | — |
| Brachial plexopathy | A disorder characterized by regional paresthesia of the brachial plexus, marked discomfort and muscle weakness, and limited movement in the arm or hand | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | — | — |
| Central nervous system necrosis | A disorder characterized by a necrotic process occurring in the brain and/or spinal cord | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms; corticosteroids indicated | Severe symptoms; medical intervention indicated | Life-threatening consequences; urgent intervention indicated | Death |
| Cerebrospinal fluid leakage | A disorder characterized by loss of cerebrospinal fluid into the surrounding tissues | Postcraniotomy: asymptomatic; postlumbar puncture: transient headache; postural care indicated | Postcraniotomy: moderate symptoms; medical intervention indicated; postlumbar puncture: persistent moderate symptoms; blood patch indicated | Severe symptoms: medical intervention indicated | Life-threatening consequences; urgent intervention indicated | Death |
| Cognitive disturbance | A disorder characterized by a conspicuous change in cognitive function | Mild cognitive disability; not interfering with work/school/life performance; specialized education services/devices not indicated | Moderate cognitive disability; interfering with work/school/life performance but capable of independent living; specialized resources on part-time basis indicated | Severe cognitive disability; significant impairment of work/school/life performance | — | — |
| Concentration impairment | A disorder characterized by a deterioration in the ability to concentrate | Mild inattention or decreased level of concentration | Moderate impairment in attention or decreased level of concentration; limiting instrumental ADL | Severe impairment in attention or decreased level of concentration; limiting self-care ADL | — | — |
| Hypoglossal nerve disorder | A disorder characterized by involvement of the hypoglossal nerve (twelfth cranial nerve) | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | — | — |
| Intracranial hemorrhage | A disorder characterized by bleeding from the cranium | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms; medical intervention indicated | Ventriculostomy, ICP monitoring, intraventricular thrombolysis, or operative intervention indicated | Life-threatening consequences; urgent intervention indicated | Death |
| Ischemia cerebrovascular | A disorder characterized by a decrease or absence of blood supply to the brain caused by obstruction (thrombosis or embolism) of an artery resulting in neurologic damage. | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms | — | — | — |
| Fourth nerve disorder | A disorder characterized by involvement of the trochlear nerve (fourth cranial nerve) | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | — | — |
| Lethargy | A disorder characterized by a decrease in consciousness characterized by mental and physical inertness | Mild symptoms; reduced alertness and awareness | Moderate symptoms; limiting instrumental ADL | — | — | — |
| Leukoencephalopathy | A disorder characterized by diffuse reactive astrocytosis with multiple areas of necrotic foci without inflammation | Asymptomatic; small focal T2/FLAIR hyperintensities; involving periventricular white matter or < of susceptible areas of cerebrum ± mild increase in SAS and/or mild ventriculomegaly of susceptible areas of cerebrum ± mild increase in SAS and/or mild ventriculomegaly |
Moderate symptoms; focal T2/FLAIR hyperintensities involving periventricular white matter extending into centrum semiovale or involving  to to  of susceptible areas of cerebrum ± moderate increase in SAS and/or moderate ventriculomegaly of susceptible areas of cerebrum ± moderate increase in SAS and/or moderate ventriculomegaly |
Severe symptoms: extensive T2/FLAIR hyperintensities, involving periventricular white matter involving  or more of susceptible areas of cerebrum ± moderate to severe increase in SAS and/or moderate to severe ventriculomegaly or more of susceptible areas of cerebrum ± moderate to severe increase in SAS and/or moderate to severe ventriculomegaly |
Life-threatening consequences; extensive T2/FLAIR hyperintensities, involving periventricular white matter involving most of susceptible areas of cerebrum ± moderate to severe increase in SAS and/or moderate to severe ventriculomegaly | Death |
| Memory impairment | A disorder characterized by a deterioration in memory function | Mild memory impairment | Moderate memory impairment; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | — | — |
| Meningismus | A disorder characterized by neck stiffness, headache, and photophobia resulting from irritation of the cerebral meninges | Mild symptoms | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | Life-threatening consequences; urgent intervention indicated | Death |
| Movements involuntary | A disorder characterized by uncontrolled and purposeless movements | Mild symptoms | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | — | — |
| Myelitis | A disorder characterized by inflammation involving the spinal cord; symptoms include weakness, paresthesia, sensory loss, marked discomfort and incontinence | Asymptomatic; mild signs (e.g., Babinski reflex or Lhermitte sign) | Moderate weakness or sensory loss; limiting instrumental ADL | Severe weakness or sensory loss; limiting self-care ADL | Life-threatening consequences; urgent intervention indicated | Death |
| Neuralgia | A disorder characterized by intense painful sensation along a nerve or group of nerves | Mild pain | Moderate pain; limiting instrumental ADL | Severe pain; limiting self-care ADL | — | — |
| Nystagmus | A disorder characterized by involuntary movements of the eyeballs | — | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | — | — |
| Oculomotor nerve disorder | A disorder characterized by involvement of the oculomotor nerve (third cranial nerve) | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | — | — |
| Olfactory nerve disorder | A disorder characterized by involvement of the olfactory nerve (first cranial nerve) | — | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | — | — |
| Paresthesia | A disorder characterized by functional disturbances of sensory neurons resulting in abnormal cutaneous sensations of tingling, numbness, pressure, cold, and warmth and are experienced in the absence of a stimulus | Mild symptoms | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | — | — |
| Peripheral motor neuropathy | A disorder characterized by inflammation or degeneration of the peripheral motor nerves | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL; assistive device indicated | Life-threatening consequences; urgent intervention indicated | Death |
| Peripheral sensory neuropathy | A disorder characterized by inflammation or degeneration of the peripheral sensory nerves | Asymptomatic; loss of deep tendon reflexes or paresthesia | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | Life-threatening consequences; urgent intervention indicated | Death |
| Phantom pain | A disorder characterized by marked discomfort related to a limb or an organ that is removed from or is not physically part of the body | Mild pain | Moderate pain; limiting instrumental ADL | Severe pain; limiting self-care ADL | — | — |
| Presyncope | A disorder characterized by an episode of lightheadedness and dizziness which may precede an episode of syncope | — | Present (e.g., near fainting) | — | — | — |
| Pyramid tract syndrome | A disorder characterized by dysfunction of the corticospinal (pyramidal) tracts of the spinal cord; symptoms include an increase in the muscle tone in the lower extremities, hyperreflexia, positive Babinski sign and a decrease in fine motor coordination | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms; limiting instrumental ADL | Severe symptoms; limiting self-care ADL | Life-threatening consequences; urgent intervention indicated | Death |
| Radiculitis | A disorder characterized by inflammation involving a nerve root; patients experience marked discomfort radiating along a nerve path because of spinal pressure on the connecting nerve root | Mild symptoms | Moderate symptoms; limiting instrumental ADL; medical intervention indicated | Severe symptoms; limiting self-care ADL | Life-threatening consequences; urgent intervention indicated | Death |
| Recurrent laryngeal nerve palsy | A disorder characterized by paralysis of the recurrent laryngeal nerve | Asymptomatic, clinical or diagnostic observations only; intervention not indicated | Moderate symptoms | Severe symptoms; medical intervention indicated (e.g., thyroplasty, vocal cord injection) | Life-threatening consequences; urgent intervention indicated | Death |
| Reversible posterior leukoencephalopathy syndrome | A disorder characterized by headaches, mental status changes, visual disturbances, and seizures associated with imaging findings of posterior leukoencephalopathy; it has been observed in association with hypertensive encephalopathy, eclampsia, and immunosuppressive and cytotoxic drug treatment; it is an acute or subacute reversible condition | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Moderate symptoms; abnormal imaging studies; limiting instrumental ADL | Severe symptoms; very abnormal imaging studies; limiting self-care ADL | Life-threatening consequences; urgent intervention indicated | Death |
| Seizure | A disorder characterized by a sudden, involuntary skeletal muscular contractions of cerebral or brain stem origin | Brief partial seizure; no loss of consciousness | Brief generalized seizure | Multiple seizures despite medical intervention | Life threatening; prolonged repetitive seizures | Death |
| Sinus pain | A disorder characterized by marked discomfort in the face, between the eyes, or upper teeth originating from the sinuses | Mild pain | Moderate pain; limiting instrumental ADL | Severe pain; limiting self-care ADL | — | — |
| Somnolence | A disorder characterized by excessive sleepiness and drowsiness | Mild but more than usual drowsiness or sleepiness | Moderate sedation; limiting instrumental ADL | Obtundation or stupor | Life-threatening consequences; urgent intervention indicated | Death |
| Spasticity | A disorder characterized by increased involuntary muscle tone that affects the regions interfering with voluntary movement; it results in gait, movement, and speech disturbances | Mild or slight increase in muscle tone. | Moderate increase in muscle tone and increase in resistance through range of motion | Severe increase in muscle tone and increase in resistance through range of motion | Life-threatening; unable to move; active or passive range of motion | Death |
| Stroke | A disorder characterized by a sudden loss of sensory function due to an intracranial vascular event | Asymptomatic or mild neurologic deficit; radiographic findings only | Moderate neurologic deficit | Severe neurologic deficit | Life-threatening consequences; urgent intervention indicated | Death |
| Syncope | A disorder characterized by spontaneous loss of consciousness caused by insufficient blood supply to the brain | — | — | Fainting; orthostatic collapse | — | — |
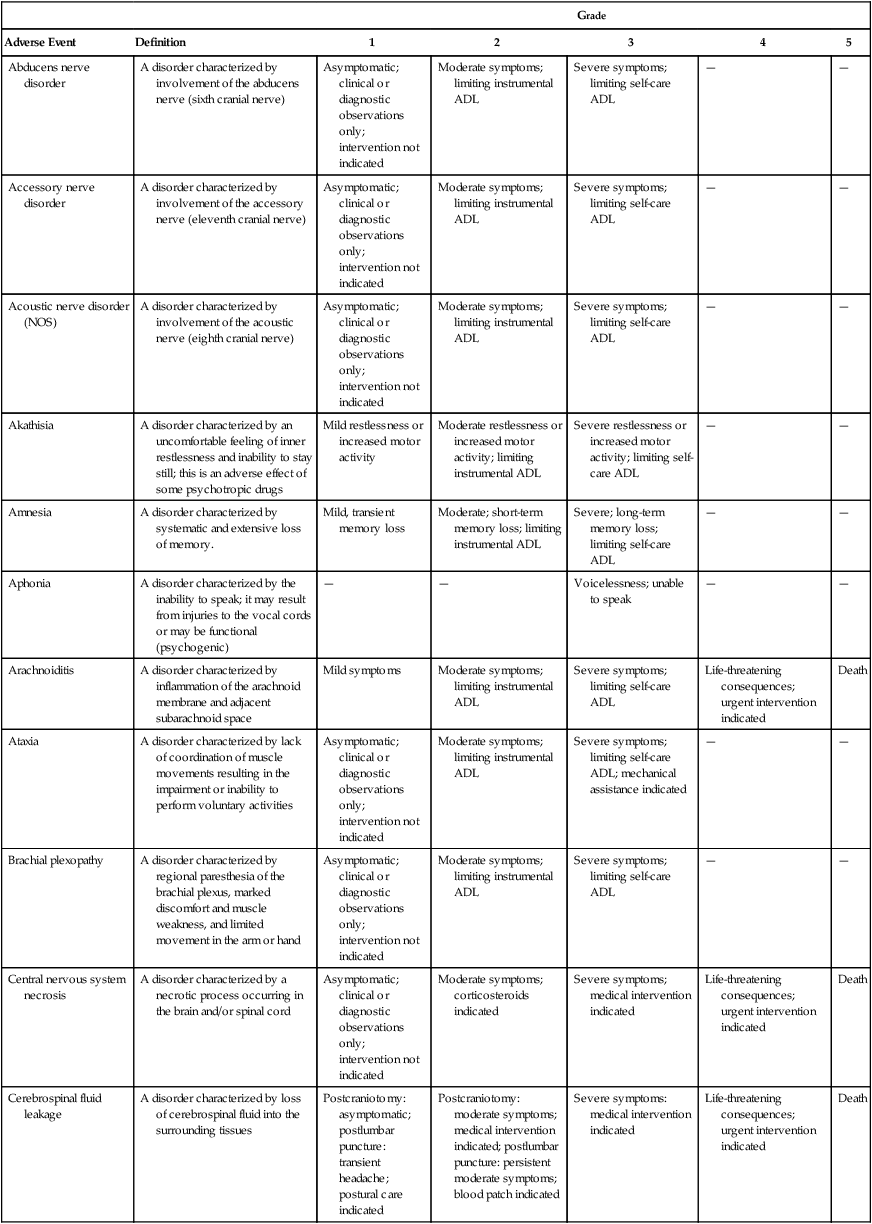
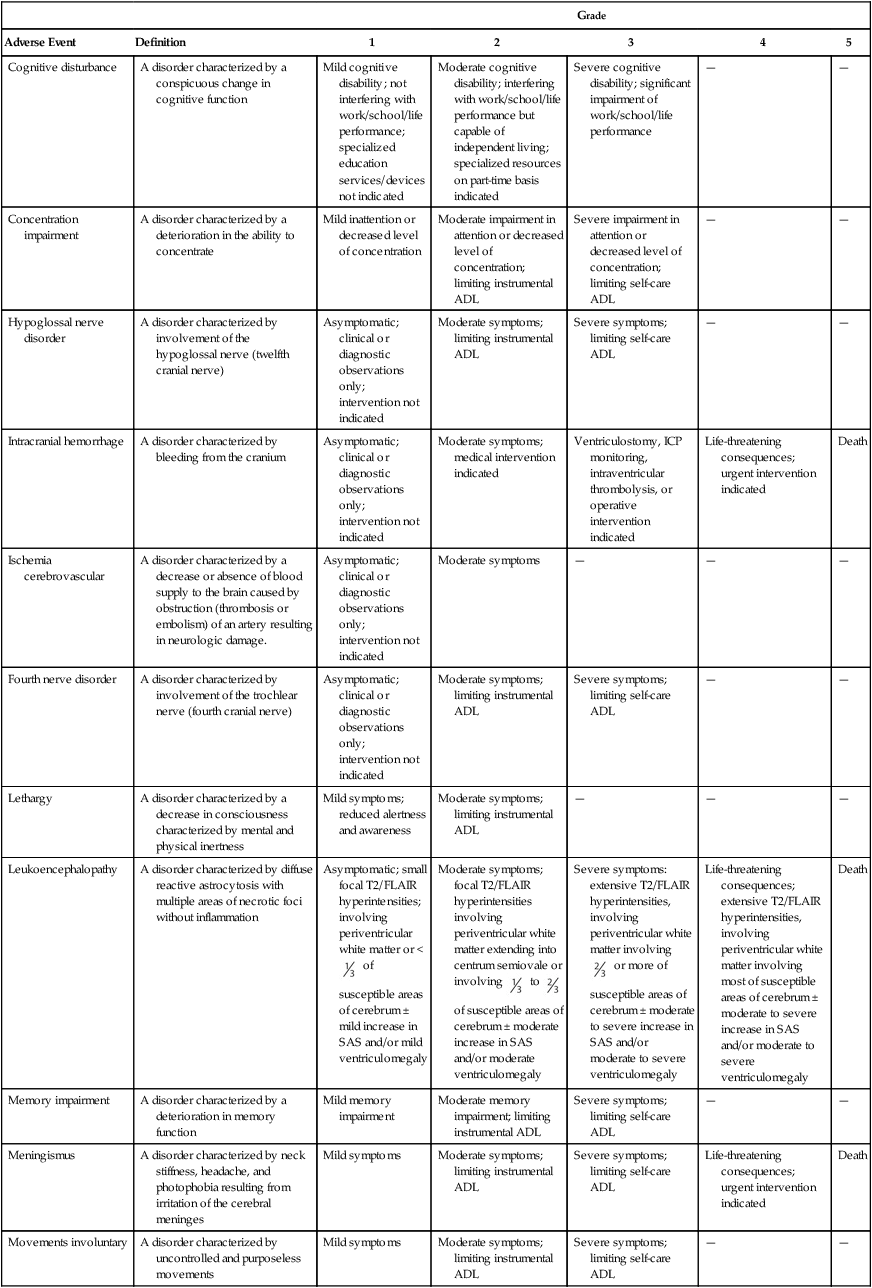
Treatment
Prevention
The key to prevention of permanent neurologic damage is to initiate a system for monitoring for toxicity. For example, patients receiving intrathecal chemotherapy are at risk of chemotherapy-induced myelopathy, an often irreversible toxicity. These patients may report vague neurologic symptoms or radicular back pain during the course of treatment that may be an early sign of neurologic damage. Findings at neurologic examination may not indicate loss of function, but an elevated CSF myelin basic protein level indicates that with subsequent treatment, the risk of myelopathy is high.26,27
Modification of Drug Dosing or Order
In certain treatment regimens, altering the order of the drugs administered can result in a marked decrease in the risk of neurotoxic complications. The risk of methotrexate-induced leukoencephalopathy after therapy for meningeal leukemia or carcinoma is reduced if the chemotherapy treatments, particularly intrathecal chemotherapy, precede cranial radiation therapy.5 The reason for this schedule-dependent toxicity is unknown. It has been suggested that radiation opens the blood-brain barrier, thereby increasing exposure of the brain to subsequent chemotherapy effects. Other regimens in which dose order seems important include combination chemotherapy with cisplatin and ifosfamide. Patients receiving cisplatin before ifosfamide are at greater risk for ifosfamide-induced encephalopathy.160 Discontinuation and reintroduction of oxaliplatin administration in a stop-and-go strategy showed the same response rate with a lower incidence of neuropathy in the OPTIMOX study. When the toxicity is severe, a dose reduction may be considered; however, it might reduce overall and disease-free survival, especially in the adjuvant setting, and thus it is necessary to carefully weigh the benefits with the risk of treatment toxicity.
Protective Agents
Interest continues in developing agents that either block development of certain chemotherapy-induced neuropathies or help reverse toxicity. However, clinical evidence for the efficacy of these strategies is sparse. Several neuroprotective strategies, including calcium/magnesium infusion, amifostine, glutathione (GSH), glutamine, acetyl-l-carnitine, and erythropoietin have been investigated.23–25,27–31,56–58 Published reports of studies of neuroprotection during chemotherapy have recently been reviewed.363,364 Erythropoietin has been shown to prevent cisplatin and docetaxel-induced neurotoxicity without compromising chemotherapeutic activity and increasing tumor growth in animal studies.365,366 These results are very promising, especially because of its concomitant use against hematologic toxicity; however, this neuroprotective effect requires additional confirmation.
The agent ORG 2766 has been reported to protect against cisplatin neuropathy. This drug, a synthetic analog of corticotropin, was tested in a double-blind, placebo-controlled trial in which patients with ovarian cancer underwent intensive cisplatin therapy. A dose-related protective effect was found.35,367,368 However, in a following study that included patients treated with vincristine, ORG 2766 did not provide protection from vincristine-induced neuropathy.369
Amifostine, an organic thiophosphate, serves as an antioxidant and protects normal cells from the effects of radiation. Amifostine has been tested as a neuroprotectant with paclitaxel therapy in two phase 3 studies. A study of non–small cell lung cancer found a reduction in the incidence of radiation esophagitis but not in paclitaxel-induced neuropathy.370 In a second study of ovarian cancer, the incidence of grade 3 and grade 4 peripheral neuropathy was significantly reduced.371 Amifostine is accompanied by serious adverse effects, such as hypotension; therefore more clinical evidence is needed before standard use with this agent can be recommended.
Glutamine also has been evaluated as a neuroprotectant for the peripheral nervous system.372 Two pilot studies suggested glutamine (10 g three times a day for 4 days) as a neuroprotective agent without interfering with chemotherapy response.372,373 Accordingly, a randomized trial with patients who had colorectal cancer reported significantly less neuropathy in the glutamine arm (15 g twice a day for 7 consecutive days) compared with control subjects.374 However, in this study no differences in electrophysiological examination were found between the groups. Another randomized pilot study revealed no difference in the use of glutamate at a dose of 500 mg.375
A wide variety of agents have been reported as putative toxicity-modulating agents. Acetyl-l-carnitine has been reported to lessen neuropathic symptoms in patients treated with paclitaxel or cisplatin.376 Because vitamin E is an antioxidant, it also was proposed as a neuroprotective agent; however, no convincing evidence indicates that vitamin E is effective in the prevention of chemotherapy-induced neuropathy. Alpha-lipoic acid, a broad-spectrum antioxidant, reportedly may help ameliorate symptoms from docetaxel and cisplatin or oxaliplatin neuropathy. Leucovorin may be helpful in reversing acute methotrexate-induced encephalopathy and somnolence.37 The administration of methylene blue has been reported to accelerate recovery from ifosfamide-induced encephalopathy.377 No known therapy exists for chemotherapy-induced leukoencephalopathy once brain injury has developed.
The acute neuropathic syndrome associated with oxaliplatin has generated special interest because of the painful nature of this toxicity. Various treatments including infusions of calcium gluconate, oral gabapentin, oral carbamazepine, intravenous amifostine, and intravenous glutathione have been used to prevent the development of this complication.132,378 Infusions of calcium gluconate and alpha-lipoic acid also have been used to treat the acute neuropathy once it has developed.
Recognition of Groups at High Risk for Development of Neurotoxicity
Certain patients are at high risk for the development of treatment-related neurotoxicity. Patients with underlying peripheral neuropathy (e.g., Charcot-Marie-Tooth disease and diabetic neuropathy) and patients who abuse alcohol and have a poor nutritional state have been reported to be much more susceptible to the development of severe neuropathy, including potentially fatal autonomic neuropathy.379,380 Such patients need careful monitoring with immediate cessation of treatment if autonomic dysfunction develops. Similarly, in patients in whom peripheral neuropathy may be a component of the cancer, such as multiple myeloma, the use of a neurotoxic agent such as bortezomib may increase the likelihood and severity of neuropathy.240
Patients with meningeal tumors (e.g., carcinoma, leukemia, and lymphoma) often require extensive intrathecal chemotherapy. An intraventricular reservoir often is placed for this purpose. Before the reservoir system is used to administer chemotherapy, it is essential that correct placement and normal CSF flow be confirmed. This procedure is most readily performed by injecting indium 111–labeled albumin into the lateral ventricle through the reservoir system.381,382 Not only does this test help confirm catheter placement, but also monitoring the flow of tracer at 6, 24, and 48 hours helps determine the presence of abnormalities of clearance from the ventricular system or drug resorption at the arachnoid granulations. Figure 57-3 shows normal CSF flow and resorption of tracer through the arachnoid granulations. By contrast, Figures 57-4 and 57-5 show poor outflow from the lateral ventricles persisting 24 hours after contrast injection. Poor clearance of drug markedly increases the neurotoxicity of intraventricular treatment. Patients with poor ventricular outflow may need local radiation therapy for restoration of normal flow before instillation of chemotherapeutic agents.
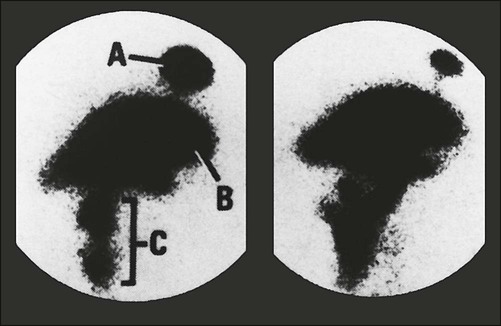
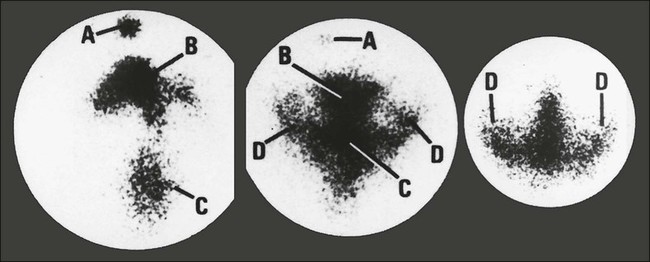
Irreversible ototoxicity from cisplatin administration is more severe in patients with underlying hearing loss. Hearing loss with cisplatin also may be accelerated when the inner ear region and temporal lobe are included in the irradiated field.118 Radiation treatment before or after cisplatin administration can cause this synergistic ototoxicity.