Chapter 14 Neurodegeneration in the Neonatal Brain
Introduction
Underlying mechanisms of neuronal cell death are common to both immature and mature brains; however, important developmental differences in propinquity to cell cycle, metabolic requirements, connectivity, basal and stimulated expression of cell death proteins, excitatory receptor expression and subunit composition, antioxidant mechanisms, synaptic density, and neurotrophin requirement account for significant differences in injury response between the immature and mature central nervous system (CNS) [Corbett et al., 1993; Martin, 2001, 2002; Ferriero, 2004; Johnston, 2005, 2009; Northington et al., 2005; Zhu et al., 2005; Kostovic and Jovanov-Milosevic, 2006]. In this chapter, we will review information obtained on basic cell death processes from both mature and immature model systems, but highlight findings from studies on the response of the immature brain to injury. While clearly important to this topic, a discussion of the contribution of inflammation to injury, recovery, and developmental neuroplasticity following neonatal brain injury is outside the scope of this chapter. A recent review of this topic is available elsewhere [Vexler and Yenari, 2009] and in Chapter 15.
Types of Cell Death
Cells can die by different processes. These processes have been classified generally into two distinct categories, called necrosis and apoptosis. These forms of cellular degeneration were classified originally as different because they appeared different morphologically under a microscope (Figure 14-1). Necrosis is a lytic destruction of individual cells or groups of cells, while apoptosis (derived from a Greek word for “dropping of leaves from trees”) is an orderly and compartmental dismantling of single cells or groups of cells into consumable components for nearby cells. Apoptosis is an example of programmed cell death (PCD) that is an adenosine triphosphate (ATP)-driven (sometimes gene transcription-requiring) form of cell suicide often committed by demolition enzymes called caspases, but other apoptotic and nonapoptotic, caspase-independent forms of PCD exist [Lockshin and Zakeri, 2002]. Apoptotic PCD is instrumental in developmental organogenesis and histogenesis and adult tissue homeostasis, functioning to eliminate excess cells. Each day in normal humans, estimates reveal that between 50 to 70 billion cells in adults and 20 to 30 billion cells in a child between the ages of 8 and 14 die due to apoptosis [Gilbert, 2006]. Another form of cell degeneration seen first with yeast and then in metazoans has been called autophagy [Klionsky and Emr, 2000]. Autophagy is an intracellular catabolic process that occurs by lysosomal degradation of damaged or expendable organelles. Necrosis and apoptosis both differ morphologically (see Figure 14-1) and mechanistically from autophagy [Klionsky and Emr, 2000; Lockshin and Zakeri, 2002].
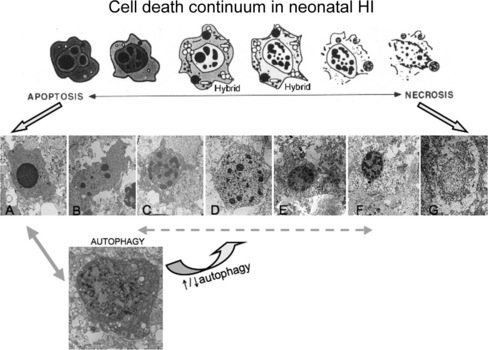
Fig. 14-1 Cell death continuum.
After its initial description by Portera-Cailliau et al., the continuum concept, in its original form, organized cell death as a linear spectrum with apoptosis and necrosis at the extremes and different syncretic hybrid forms in between (top) [Martin et al., 1998]. Subsequently we have found that this concept is fully realized in the neonatal rat brain following hypoxia-ischemia (HI), with clearly apoptotic (A) and necrotic (G) cells found intermixed with the hybrid forms (B–F). Cells at the extremes have the well-described structures of necrosis (G – swelling and vacuolation of organelles, loss of cell membrane integrity, maintenance of nuclear membrane integrity, random digestion of chromatin) and apoptosis (A – condensation and darkening of cytoplasm within intact cell membrane, intact organelles until late phases of apoptosis, compaction of chromatin into few uniformly dense and rounded aggregates and loss of nuclear membrane). Cells with irregular chromatin condensation, organized in a “clockface” pattern around an intact nuclear membrane (E, F), may or may not have preservation of the cytoplasmic membrane, and their cytoplasmic organelles are disrupted. This structure has commonly been reported in models of excitotoxicity. Increasing organization of chromatin into regular crescenteric or rounded aggregates, dissolution of the nuclear membrane, and preservation of the cytoplasmic membrane, with or without swelling of cytoplasmic organelles (B–D), is the rule as cell death forms more closely mimic apoptosis. Autophagocytic neurons with large numbers of cytoplasmic vacuoles, partially condensed nuclear chromatin, and preservation of cellular integrity are found after neonatal HI (H). Autophagocytic cell death and apoptosis exist on a continuum. Drugs known to inhibit or promote autophagy can also modulate cell death along the apoptosis–necrosis continuum.
More recently, the morphological and molecular regulatory distinctions between the different forms of cell death have become blurred and uncertain due to observations made on degenerating neurons in vivo and to a new concept that attempts to accommodate these observations. This concept, in its original form, posits that cell death exists as a continuum with necrosis and apoptosis at opposite ends of a spectrum, with hybrid forms of degeneration manifesting in between (see Figure 14-1) [Portera-Cailliau, et al., 1997a, b; Martin et al., 1998; Martin, 1999]. For example, the degeneration of neurons in diseased or damaged human and animal nervous systems is not always strictly necrosis or apoptosis, according to the traditional binary classification of cell death, but also occurs as intermediate or hybrid forms with coexisting morphological and biochemical characteristics that lie in a structural continuum, with necrosis and apoptosis at the two extremes [Portera-Cailliau et al., 1997a, b]. Thus, neuronal cell death can be syncretic. The different processes leading to the putative different forms of cell death can be activated concurrently with graded contributions of the different cell death modes to the degenerative process.
The in vivo reality of a neuronal cell death continuum was revealed first in neonatal and adult rat models of glutamate receptor excitotoxicity [Portera-Cailliau et al., 1997a, b] and then very nicely in rodent models of neonatal hypoxic-ischemic encephalopathy (HIE) [Nakajima et al., 2000; Northington et al., 2001b, 2007]. The hybrid cells can be distinguished cytopathologically by the progressive compaction of the nuclear chromatin into few, discrete, large, irregularly shaped clumps (see Figure 14-1). This morphology contrasts with the formation of few, uniformly shaped, dense, round masses in classic apoptosis and the formation of numerous, smaller, irregularly shaped chromatin clumps in classic necrosis. The cytoplasmic organelle pathology in hybrid cells has a basic pattern that appears more similar to necrosis than apoptosis, but is lower in amplitude than in necrosis (e.g., mitochondrial swelling). Toxicological studies of cultured cells have shown that stimulus intensity influences the mode of cell death [Lennon et al., 1991; Fernandes and Cotter, 1994; Bonfoco et al., 1995], such that apoptosis can be induced by injurious stimuli of lesser amplitude than insults causing necrosis [Raffray and Cohen, 1997], but the cell death modes were still considered distinct [Bonfoco et al., 1995].
Basic research is uncloaking the molecular mechanisms of cell death [Orrenius et al., 2003; Yuan et al., 2003], and, with this, the distinctiveness of different cell death processes, as well as the potential overlap among different cell death mechanisms. Experimental studies on cell death mechanisms, and particularly the cell death continuum, are important because they could lead to the rational development of molecular mechanism-based therapies for treating neonatal hypoxic-ischemic encephalopathy (HIE). The different categories of cell death are discussed below.
Necrosis
Cell death caused by cytoplasmic swelling, nuclear dissolution (karyolysis), and lysis has been classified traditionally as necrosis [Trump and Berezesky, 1996]. Cell necrosis (sometimes termed oncosis) [Majno and Joris, 1995], results from rapid and severe failure to sustain cellular homeostasis, notably cell volume control [Trump et al., 1965]. The process of necrosis involves damage to the structural and functional integrity of the cell plasma membrane and associated enzymes, e.g., Na+, K+ ATPase, abrupt influx and overload of ions (e.g., Na+ and Ca2+) and H2O, and rapid mitochondrial damage and energetic collapse [Bonfoco et al., 1995; Leist et al., 1997; Martin et al., 2000; Golden et al., 2001]. Metabolic inhibition and oxidative stress from reactive oxygen species (ROS) are major culprits in triggering necrosis. Inhibitory crosstalk between ion pumps causes pronecrotic effects when Na+, K+ ATPase “steals” ATP from the plasma membrane Ca2+ ATPase, resulting in Ca2+ overload [Castro et al., 2006].
The morphology and some biochemical features of classic necrosis in neurons are distinct (see Figure 14-1). The main features are swelling and vacuolation/vesiculation of organelles, destruction of membrane integrity, random digestion of chromatin due to activation of proteases and deoxyribonucleases (DNases), and dissolution of the cell. The overall profile of the moribund cell is maintained generally as it dissolves into the surrounding tissue parenchyma and induces an inflammatory reaction in vivo. In necrosis, dying cells do not bud to form discrete, membrane-bound fragments. The nuclear pyknosis and karyolysis appear as condensation of chromatin into many irregularly shaped, small clumps, sharply contrasting with the formation of few, uniformly dense and regularly shaped chromatin aggregates that occurs in apoptosis. In cells undergoing necrosis, genomic DNA is digested globally because protease that digest histone proteins that protect DNA, and DNases are coactivated to generate many randomly sized fragments seen as a DNA “smear.” These differences in the cytoplasmic changes and condensation and digestion of nuclear chromatin in pure apoptosis and pure necrosis are very diagnostic.
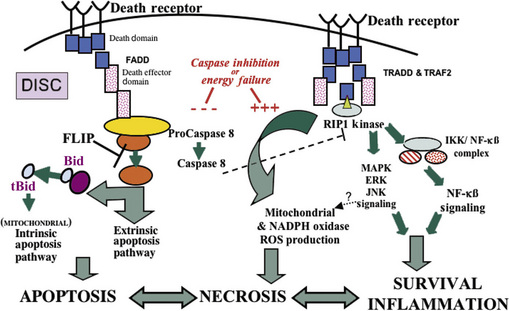
Recent work has shown that cell necrosis might not be as chaotic or random as envisioned originally, but can involve the activation of specific signaling pathways to eventuate in cell death [Proskuryakov et al., 2003]. For example, DNA damage can lead to poly(ADP-ribose) polymerase activation and ATP depletion, energetic failure, and necrosis [Ha and Snyder, 2000]. Other pathways for “programmed” necrosis involve death receptor signaling through receptor interacting protein 1 (RIP1) kinase and mitochondrial permeability transition (Figure 14-2) [Crompton, 1999; Festjens et al., 2006; Hitomi et al., 2008].
Mitochondrial Ca2+ overload, excessive oxidative stress, and decreases in the electrochemical gradient, ADP and ATP, can favor mitochondrial permeability transition, which is defined by disruption of the proton-motive force [Crompton et al., 1998; Crompton, 1999; van Gurp et al., 2003]. This disruption involves the so-called mitochondrial permeability transition pore (mPTP), which functions as a voltage, thiol, and Ca2+ sensor. The mPTP is a large polyprotein transmembrane channel formed at contact sites between the inner mitochondrial membrane and the outer mitochondrial membrane (Figure 14-3). The complete components of the mPTP are still controversial. The primary components of the mPTP are the voltage-dependent anion channel (VDAC; also called porin) in the outer mitochondrial membrane and the adenine nucleotide translocator (ANT) in the inner mitochondrial membrane [Crompton et al., 1998]. The VDAC makes the inner mitochondrial membrane permeable to most small hydrophilic molecules <5 kDa for free exchange of respiratory chain substrates. The ANT mediates the exchange of ADP for ATP. During normal mitochondrial function the intermembrane space separates the outer and inner mitochondrial membranes and the VDAC and the ANT do not interact, or interact only transiently in a state described as “flicker” [Crompton et al., 1998]. When the mPTP is in the open state, it permits influx of solutes of ≤1500 Da and H2O into the matrix, resulting in depolarization of mitochondria and dissipated proton electrochemical gradient. Consequently, the inner mitochondrial membrane loses its integrity and oxidative phosphorylation is uncoupled. When this occurs, oxidation of metabolites by O2 proceeds with electron flux not coupled to proton pumping, resulting in further dissipation of transmembrane proton gradient and ATP production, production of ROS, and large-amplitude mitochondrial swelling triggering necrosis or apoptosis [van Gurp et al., 2003]. Several proteins regulate the mPTP. Cyclophilin D is one of these proteins found in the mitochondrial matrix and it interacts reversibly with the ANT. Inactivation of cyclophilin D can block mitochondrial swelling and cellular necrosis induced by Ca2+ overload and ROS in the adult [Baines et al., 2005; Nakagawa et al., 2005] but cyclophilin D appears to have prosurvival functions in the hypoxia-ischemia (HI)-injured immature brain [Wang et al., 2009]. Another protein that causes mPTP opening is BNIP3, which can integrate into the outer mitochondrial membrane and can trigger necrosis [Vande Velde et al., 2000].
Apoptosis
Apoptosis is a form of PCD because it is carried out by active, intrinsic transcription-dependent [Tata, 1966] or transcription-independent mechanisms involving specific molecules. Apoptosis should not be used as a synonym for PCD because nonapoptotic forms of PCD exist [Schwartz et al., 1993; Amin et al., 2000]. Apoptosis is only one example of PCD. It is critical for the normal growth and differentiation of organ systems in vertebrates and invertebrates [see Jacobson, 1991, regarding Ernst’s discovery of developmental PCD; Glucksmann, 1951; Lockshin and Williams, 1964; Saunders, 1966]. The structure of apoptosis is similar to the type I form of PCD described by Clarke [Clarke, 1990]. In physiological settings in adult tissues, apoptosis is a normal process, occurring continuously in populations of cells that undergo slow proliferation (e.g., liver and adrenal gland) or rapid proliferation (e.g., epithelium of intestinal crypts) [Wyllie et al., 1980; Bursch et al., 1990]. Apoptosis is a normal event in the immune system when lymphocyte clones are deleted after an immune response [Nagata, 1999]. Kerr and colleagues were the first to describe apoptosis in pathological settings [Kerr et al., 1972], but many descriptions were made prior to this time in studies of developing animal systems [Lockshin and Zakeri, 2001].
Classical apoptosis has a distinctive structural appearance (see Figure 14-1). The cell condenses and is dismantled in an organized way into small packages that can be consumed by nearby cells. Nuclear breakdown is orderly. The DNA is digested in a specific pattern of internucleosomal fragments (see Figure 14-1), and the chromatin is packaged into sharply delineated, uniformly dense masses that appear as crescents abutting the nuclear envelope or as smooth, round masses within the nucleus (see Figure 14-1). The execution of apoptosis is linked to Ca2+-activated DNases [Wyllie, 1980], one being DNA fragmentation factor 45 (DFF45) [Liu et al., 1997], which digests genomic DNA at internucleosomal sites only (because proteases that digest histone proteins remain inactivated and the DNA at these sites is protected from DNases) to generate a DNA “ladder” (see Figure 14-1). However, the emergence of the apoptotic nuclear morphology can be independent of the degradation of chromosomal DNA [Sakahira et al., 1999]. Cytoplasmic breakdown is also orderly. The cytoplasm condenses, (as reflected by a darkening of the cell in electron micrographs, see Figure 14-1), and subsequently the cell shrinks in size, while the plasma membrane remains intact. During the course of these events, it is believed that the mitochondria are required for ATP-dependent processes. Subsequently, the nuclear and plasma membranes become convoluted, and then the cell undergoes a process called budding. In this process, the nucleus, containing smooth, uniform masses of condensed chromatin, undergoes fragmentation in association with the condensed cytoplasm, forming cellular debris (called apoptotic bodies) composed of pieces of nucleus surrounded by cytoplasm with closely packed and apparently intact organelles. Apoptotic cells display surface markers (e.g., phosphatidylserine or sugars) for recognition by phagocytic cells. Phagocytosis of cellular debris by adjacent cells is the final phase of apoptosis in vivo.
Variants of classical or nonclassical apoptosis can occur during nervous system development [Pilar and Landmesser, 1976; Clarke, 1990] and also frequently in pathophysiological settings of nervous system injury and disease [Portera-Cailliau et al., 1997a, b; Martin et al., 1998]. Axonal damage (axotomy) and target deprivation in the mature nervous system can induce apoptosis in neurons that is similar structurally, but not identical, to developmental PCD [Martin et al., 1998]. Excitotoxins can induce readily and robustly nonclassical forms of apoptosis in neurons [Portera-Cailliau et al., 1997a, b]. Types of cell death similar to those seen with excitotoxicity occur frequently in pathological cell death resulting from neonatal HI [Martin et al., 2000; Nakajima et al., 2000; Northington et al., 2001b, 2007].
Cells can die by PCD through mechanisms that are distinct from apoptosis [Jacobson, 1991; Schwartz et al., 1993]. The structure of nonapoptotic PCD is similar to the type II or type III forms of cell death described by Clarke [Clarke, 1990]. Interestingly, there is no internucleosomal fragmentation of genomic DNA in some forms of nonapoptotic PCD [Schwartz et al., 1993; Amin et al., 2000].
Autophagy
Autophagy is a mechanism whereby eukaryotic cells degrade their own cytoplasm and organelles [Klionsky and Emr, 2000]. The degradation of organelles and long-lived proteins is carried out by the lysosomal system. Although autophagy functions as a cell death mechanism, it is primarily a homeostatic nonlethal stress response mechanism for recycling proteins to protect cells from low supplies of nutrients. Autophagy is classified as type II PCD [Clarke, 1990]. A hallmark of autophagic cell death is accumulation of autophagic vacuoles of lysosomal origin. Autophagy has been seen in developmental and pathological conditions. For example, insect metamorphosis involves autophagy [Lockshin and Zakeri, 1994], and developing neurons can use autophagy as a PCD mechanism [Schweichel and Merker, 1973; Xue et al., 1999]. Degeneration of Purkinje neurons in the mouse mutant Lucher appears to be a form of autophagy, thus possibly linking excitotoxic and autophagic cell deaths to constitutive activation of the GluRδ2 glutamate receptor [Yue et al., 2002].
The molecular controls of autophagy appear common in eukaryotic cells from yeast to human, and it is believed that autophagy evolved before apoptosis [Yuan et al., 2003]. However, most of the work has been done on yeast, with detailed work on mammalian cells only beginning [Mizushima et al., 2002]. Double-membrane autophagosomes for sequestration of cytoplasmic components are derived from the endoplasmic reticulum (ER) or the plasma membrane. Tor kinase, phosphatidylinositol 3 (PI3) kinase, a family of cysteine proteases called autophagins, and death-associated proteins function in autophagy [Bursch, 2001; Inbal et al., 2002]. Autophagic and apoptotic cell death pathways crosstalk. The product of the tumor suppressor gene Beclin1 (the human homolog of the yeast autophagy gene APG6) interacts with the anti-apoptosis regulator Bcl-2 [Liang et al., 1998]. Autophagy can block apoptosis by sequestration of mitochondria. If the capacity for autophagy is reduced, stressed cells die by apoptosis, whereas inhibition or blockade of molecules that function in apoptosis can convert the cell death process into autophagy [Ogier-Denis and Codogno, 2003]. Thus, a mechanistic continuum between autophagy and apoptosis exists (see Figure 14-1).
Autophagy appears to have a significant role in neurodegeneration after neonatal HI that may be insult severity-, time-, and region-specific [Lockshin and Zakeri, 1994; Koike et al., 2008; Ginet, et al., 2009]. Genetic deletion of the atg7 gene results in near-complete protection from HI in adult mice [Koike et al., 2008], and pharmacologic inhibition of autophagy with 3-methyladenine up to 4 hours after focal ischemia is neuroprotective in p12 rats [Puyal et al., 2009]. Conversely, induction of autophagy immediately following neonatal global HI in mice appeared to be an endogenous neuroprotective mechanism in other studies [Carloni et al., 2008]. Pre-insult blockade of autophagy with methyladenine inhibited expression of autophagocytic proteins and switched cell death from apoptotic to necrotic; conversely, enhancing early autophagy with pre-insult administration of rapamycin provided neuroprotection in this model [Carloni et al., 2008]. Interestingly, known neuroprotective preconditioning strategies also increased markers of autophagy [Carloni et al., 2008] while providing the expected neuroprotection. Regional differences in induction of autophagocytic proteins, severity of insult, and timing of drug administration might account for these discrepant results.
Molecular and Cellular Regulation of Apoptosis
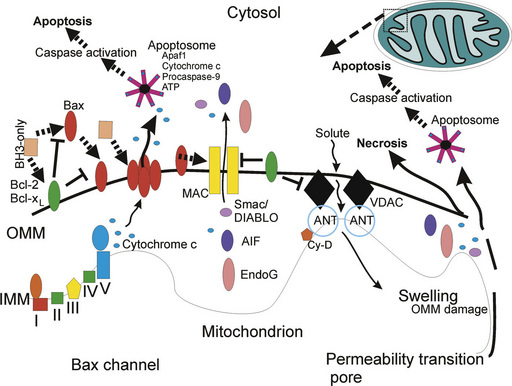
Apoptosis is a structurally and biochemically organized form of cell death. The basic machinery of apoptosis is conserved in yeast, hydra, nematode, fruitfly, zebrafish, mouse, and human [Ameisen, 2002]. Our current understanding of the molecular mechanisms of apoptosis in mammalian cells is built on studies by Robert Horvitz and colleagues on PCD in the nematode Caenorhabditis elegans [Metzstein et al., 1998]. They pioneered the understanding of the genetic control of developmental cell death by showing that this death is regulated predominantly by three genes (ced-3, ced-4, and ced-9) [Metzstein et al., 1998]. Several families of apoptosis-regulation genes have been identified in mammals, including the Bcl-2 family [Metzstein et al., 1998; Cory and Adams, 2002], the caspase family of cysteine-containing, aspartate-specific proteases [Wolf and Green, 1999], the p53 gene family [Levrero et al., 2000], cell surface death receptors [Nagata, 1999], and other apoptogenic factors, including cytochrome c, apoptosis inducing factor (AIF), and second mitochondrial activator of caspases (Smac) [Liu et al., 1996; Li et al., 1997; Hegde et al., 2002; Klein et al., 2002; Lockshin and Zakeri, 2002]. Moreover, a family of inhibitors of apoptosis proteins (IAPs) actively blocks cell death, and IAPs are inhibited mitochondrial proteases [Hegde et al., 2002]. Specific organelles have been identified as critical for the apoptotic process, including mitochondria and the ER (see Figure 14-3). In seminal work by Li, Wang, and colleagues, it was discovered that the mitochondrion integrates death signals mediated by proteins in the Bcl-2 family and releases molecules residing in the mitochondrial intermembrane space, such as cytochrome c, which complexes with cytoplasmic proteins (e.g., apoptotic protease activating factor-1 [Apaf-1]) to activate caspase proteases, leading to internucleosomal cleavage of DNA [Liu et al., 1996; Li et al., 1997]. The finding that cytochrome c has a function in apoptosis, in addition to its better-known role in oxidative phosphorylation, was astounding, although foreshadowing clues were available. The release of cytochrome c from mitochondria to the cytosol with concomitant reduced oxidative phosphorylation was described as the “cytochrome c effect” in irradiated cancer cells [van Bekkum, 1957]. The ER, which regulates intracellular Ca2+ levels, participates in a loop with mitochondria to modulate mitochondrial permeability transition and cytochrome c release through the actions of Bcl-2 protein family members [Scorrano et al., 2003].
Bcl-2 Family of Survival and Death Proteins
The bcl-2 proto-oncogene family is a large group of apoptosis regulatory genes encoding about 25 different proteins, defined by at least one conserved B-cell lymphoma (Bcl) homology domain (BH1–BH4 can be present) in their amino acid sequence that functions in protein–protein interactions [Metzstein et al., 1998; Cory and Adams, 2002]. Some of the protein products of these genes (e.g., Bcl-2, Bcl-xL, and Mcl-1) have all four BH1–BH4 domains and are anti-apoptotic. Other gene products, which are pro-apoptotic, are multidomain proteins possessing BH1–BH3 sequences (e.g., Bax and Bak), or proteins with only the BH3 domain (e.g., Bad, Bid, Bim, Bik, Noxa, and Puma) that contains the critical death domain. Bcl-xL and Bax have α-helices resembling the pore-forming subunit of diphtheria toxin [Muchmore et al., 1996]; thus, Bcl-2 family members appear to function by conformation-induced insertion into the outer mitochondrial membrane to form channels or pores that can regulate the release of apoptogenic factors (see Figure 14-3).
The expression of many of these proteins is regulated developmentally, and the proteins have differential tissue distributions and subcellular localizations. Most of these proteins are found in the CNS, but the relative quantities of pro- and anti-apoptotic family members change over time [Li et al., 1997; Shimohama et al., 1998]. It appears that in mouse brain, the relative abundance of the pro-apoptosis Bcl-2 family members declines markedly after the perinatal period, while the anti-apoptotic Bcl-2 family members exhibit stable expression over time in the brain [Shimohama et al., 1998]. The subcellular distributions of Bax, Bak, and Bad in healthy adult rodent CNS tissue [Martin et al., 2003] are consistent with in vitro studies of non-neuronal cells [Wolter et al., 1997; Nechushtan et al., 2001]. Bax, Bad, and Bcl-2 reside primarily in the cytosol, whereas Bak resides primarily in mitochondria (see Figure 14-3). Bcl-2 family members can form homodimers or heterodimers and higher-order multimers with other family members. Bax forms homodimers or heterodimers with Bak, Bcl-2, or Bcl-xL. When Bax and Bak are present in excess, the anti-apoptotic activity of Bcl-2 and Bcl-xL is antagonized. The formation of Bax homo-oligomers promotes apoptosis, whereas Bax heterodimerization with either Bcl-2 or Bcl-xL neutralizes its pro-apoptotic activity. Neonatal HI enhances the relative pro-apoptosis balance of Bcl-2 family proteins. Marked increases in mitochondrial Bax occur within 24 hours of HI in neonatal rats, during which time there is no change in the relative amount of Bcl-xL [Northington et al., 2001a] and changes in the subcellular distribution of Bax occur rapidly, prior to the activation of downstream apoptosis-effector mechanisms [Lok and Martin, 2002].
Release of cytochrome c from mitochondria may occur through mechanisms that involve the formation of membrane channels comprised of Bax or Bak [Antonsson et al., 1997] and Bax and the VDAC [Shimizu et al., 2000] (see Figure 14-3). Cytochrome c triggers the assembly of the cytoplasmic apoptosome (a protein complex of Apaf1, cytochrome c, and procaspase-9), which is the engine driving caspase-3 activation in mammalian cells [Li et al., 1997]. Bcl-2 and Bcl-xL block the release of cytochrome c [Kluck et al., 1997; Yang et al., 1997] from mitochondria, and thus the activation of caspase-3 [Liu et al., 1996; Li et al., 1997]. The blockade of cytochrome c release from mitochondria by Bcl-2 and Bcl-xL [Liu et al., 1996; Vander Heiden et al., 1997] is caused by inhibition of Bax channel-forming activity in the outer mitochondrial membrane [Antonsson et al., 1997] or by modulation of mitochondrial membrane potential and volume homeostasis [Vander Heiden et al., 1997]. Bcl-xL also has anti-apoptotic activity by interacting with Apaf-1 and caspase-9, and inhibiting the Apaf-1-mediated autocatalytic maturation of caspase-9 [Hu et al., 1998]. Boo can inhibit Bak- and Bik-induced apoptosis (but not Bax-induced cell death), possibly through heterodimerization and by interactions with Apaf-1 and caspase-9 [Song et al., 1999]. Bax and Bak double-knockout cells are completely resistant to mitochondrial cytochrome c release during apoptosis [Wei et al., 2001]. BH3-only proteins, such as Bim, Bid, Puma, and Noxa, appear to induce a conformational change in Bax or they serve as decoys for Bcl-xL, allowing Bax to form pores in the outer mitochondrial membrane [Letai et al., 2002]. The role of the BH3-only proteins in neonatal HI brain injury is unclear. There are no reports of Bid activation in neonatal HI, and in a single study using Bid-deficient mice no protection from neonatal HI brain injury was found [Ness et al., 2006].
Although many studies have focused on how Bcl-2 family members regulate mitochondrial apoptosis, it is now evident that Bcl-2 proteins localize to the ER and also translocate to the nucleus [Lithgow et al., 1994; Zhu et al., 2009]. This finding is relevant to neonatal HIE and excitotoxicity, where ER abnormalities may be particularly important to pathogenesis [Portera-Cailliau et al., 1997a, b; Martin et al., 2000; Puka-Sundvall et al., 2000]. The ER functions to fold proteins, and when this capacity is compromised, an unfolded protein response (UPR) is engaged. The UPR can lead to a return to homeostasis or to cell death. Bak and Bax also operate in the ER and function in the activation of ER-specific caspase-12 [Zong et al., 2003]. Cells lacking Bax and Bak are resistant to ER stress-induced apoptosis [Wei et al., 2001]. Translocation and accumulation of Bcl-2 in the nucleus occurs during a period when the total amounts of Bcl-2 decrease via calpain-dependent mechanisms following neonatal HI in mice [Hu et al., 1998; Zhu et al., 2009]. Overexpression of Bcl-2 and Bcl-xL can block ER stress-induced apoptosis [Murakami et al., 2007], but the function of nuclear Bcl-2 is not known.
Protein phosphorylation regulates the functions of some Bcl-2 family members. Bcl-2 loses its anti-apoptotic activity following serine phosphorylation, possibly because its antioxidant function is inactivated [Haldar et al., 1995]. Bcl-2 phosphorylation at serine 24 in the BH4 domain precedes caspase-3 cleavage following cerebral HI in neonatal rat [Hallin et al., 2006]. In addition to interacting with homologous proteins, Bcl-2 can associate with nonhomologous proteins, including the protein kinase Raf-1 [Wang et al., 1996]. Bcl-2 is thought to target Raf-1 to mitochondrial membranes, allowing this kinase to phosphorylate Bad at serine residues. The phosphatidylinositol 3-kinase (PI3-K)–Akt pathway also regulates the function of Bad [Datta et al., 1997; del Peso et al., 1997] and caspase-9 [Cardone et al., 1998] through phosphorylation. In the presence of trophic factors, Bad is phosphorylated. Phosphorylated Bad is sequestered in the cytosol by interacting with soluble protein 14-3-3 and, when bound to protein 14-3-3, Bad is unable to interact with Bcl-2 and Bcl-xL, thereby promoting survival [Zha et al., 1996]. Conversely, when Bad is dephosphorylated by calcineurin [Wang et al., 1999], it dissociates from protein 14-3-3 in the cytosol and translocates to the mitochondria, where it exerts pro-apoptotic activity. Nonphosphorylated Bad heterodimerizes with membrane-associated Bcl-2 and Bcl-xL, thereby displacing Bax from Bax-Bcl-2 and Bax-Bcl-xL dimers and promoting cell death [Yang et al., 1995]. The phosphorylation status of Bad helps regulate glucokinase activity, thereby linking glucose metabolism to apoptosis [Danial et al., 2003].
Caspases: Cell Demolition Proteases
Caspases (cysteinyl aspartate-specific proteinases) are cysteine proteases that have a near-absolute substrate requirement for aspartate in the P1 position of the peptide bond. Fourteen members have been identified [Wolf and Green, 1999]. Caspases exist as constitutively expressed inactive proenzymes (30–50 kDa) in healthy cells. The protein contains three domains: an amino-terminal prodomain, a large subunit (approximately 20 kDa), and a small subunit (approximately 10 kDa). The inactive proenzymes are present at detectable levels under basal conditions, but at least one study suggests that transcription and increased expression of the proenzyme occur following neonatal hypoxia [Delivoria-Papadopoulos et al., 2008]. By far the greatest control of caspase activity occurs through regulated proteolysis of the proenzyme with “initiator” caspases activating “executioner” caspases, although some caspase proenzymes (e.g., caspase-9) have low activity without processing [Stennicke et al., 1999]. Other caspase family members function in inflammation by processing cytokines [Wolf and Green, 1999]. The prodomain of initiator caspases contains amino acid sequences that are caspase recruitment domains (CARD) or death effector domains (DED), which enable the caspases to interact with other molecules that regulate their activation. Activation of caspases involves proteolytic processing between domains, and then association of large and small subunits to form a heterodimer with both subunits contributing to the catalytic site. Two heterodimers associate to form a tetramer that has two catalytic sites that function independently. Active caspases have many target proteins [Schwartz and Milligan, 1996] that are cleaved during regulated and organized cell death. Caspases cleave nuclear proteins (e.g., DNases, poly(ADP) ribose polymerase, DNA-dependent protein kinase, heteronuclear ribonucleoproteins, transcription factors or lamins), cytoskeletal proteins (e.g., actin and fodrin), and cytosolic proteins (e.g., other caspases, protein kinases, Bid).
In cell models of apoptosis using human non-neuronal cell lines, activation of caspase-3 occurs when caspase-9 proenzyme (also known as Apaf-3) is bound by Apaf-1, which then oligomerizes in a process initiated by cytochrome c (identified as Apaf-2) and either ATP or dATP [Li et al., 1997] (see Figure 14-3). Cytosolic ATP or dATP is a required co-factor for cytochrome c-induced caspase activation. Apaf-1, a 130 kDa cytoplasmic protein, serves as a docking protein for procaspase-9 (Apaf-3) and cytochrome c [Li et al., 1997]. Apaf-1 becomes activated when ATP is bound and hydrolyzed, with the hydrolysis of ATP and the binding of cytochrome c promoting Apaf-1 oligomerization [Zou et al., 1999]. This oligomeric complex recruits and mediates the autocatalytic activation of procaspase-9 (forming the apoptosome), which disassociates from the complex and becomes available to activate caspase-3. In the newborn brain, HI in rat pups and hypoxia in piglets induced caspase 9-mediated caspase 3 activation, which is modulated by neuronal and inducible nitric oxide synthase (nNOS and iNOS) derived-NO and by neuronal nuclear Ca2+ influx [Zhu et al., 2004a; Delivoria-Papadopoulos et al., 2008]. These effects appear to occur at both transcriptional and post-translational levels. Once activated, caspase-3 cleaves multiple proteins, including a protein with DNase activity (i.e., DFF-45). DFF-45 cleavage activates a process leading to the internucleosomal fragmentation of genomic DNA [Liu et al., 1997].
So far, three caspase-related signaling pathways have been identified that can lead to apoptosis [Liu et al., 1996, 1997; Liu et al., 1997, 1998], but crosstalk among these pathways is possible. The intrinsic mitochondria-mediated pathway is controlled by Bcl-2 family proteins. It is regulated by cytochrome c release from mitochondria, promoting the activation of caspase-9 through Apaf-1 and then caspase-3 activation. The extrinsic death receptor pathway involves the activation of cell-surface death receptors, including Fas and tumor necrosis factor (TNF) receptor, leading to the formation of the death-inducible signaling complex (DISC) and caspase-8 activation, which in turn cleaves and activates downstream caspases such as caspase-3, 6, and 7. Caspase-8 can also cleave Bid, leading to the translocation, oligomerization, and insertion of Bax or Bak into the mitochondrial membrane. Another pathway involves the activation of caspase-2 by DNA damage or ER stress as a premitochondrial signal [Robertson et al., 2002].
Caspases are also critical regulators of nondeath functions in cells, notably some maturational processes. These nonapoptotic functions include modulation of synaptic plasticity via involvement in long-term potentiation [Gulyaeva, 2003] and cleavage of AMPA receptor subunits [Lu et al., 2002], and normal differentiation and migration of neurons to the olfactory bulb [Yan et al., 2001]. Transient caspase inactivation for the purpose of neuroprotection following neonatal HI may interfere with ongoing, normal nondeath-related functions of caspases. It is unknown whether inhibition of caspase activity in the acute setting of injury impairs similar functions or interferes with axonal sprouting, and whether there are long-term changes in brain structure or function as a result of acute caspase inhibition. Because of the potential importance of the nonapoptotic functions of caspase-3, it might be more appropriate to block the formation of “stress-induced”, cleaved caspase-3 following injury with selective caspase-8 and 9 inhibitors that would not interfere with basal levels of caspase-3 activity. However, the nonapoptotic functions of caspase-8 and 9 are unknown, and the effects of acute caspase-8 and 9 inhibition on the developing brain, other than neuroprotection, are similarly unknown.
Caspases seem to be involved in the evolution of neonatal brain injury caused by HI, although this may vary between models. Caspase-3 cleavage and activation occur in brain after HI and trauma in neonatal rodents [Hu et al., 2000; Blomgren et al., 2001; Northington et al., 2001a; Felderhoff-Mueser et al., 2002], and after hypoxia in neonatal piglets [Delivoria-Papadopoulos et al., 2008]. The extent of caspase-3 cleavage and activation following brain injury is clearly greater in developing rodents compared to adults [Hu et al., 2000; Zhu et al., 2005]. This principle is replicated in immature and mature neuronal culture systems [Lesuisse and Martin, 2002]. Cerebroventricular injection of a pan-caspase inhibitor or intraperitoneal injection of a serine protease inhibitor 3 hours after neonatal HI in rat has neuroprotective effects [Cheng et al., 1998; Feng and LeBlanc, 2003]. Subsequent studies have shown 30–50 percent decreases in HI-induced tissue loss in neonatal rat brain at 15 days after the insult with non-selective inhibitors of caspase-8 and caspase-9 [Feng et al., 2003a, b]. However, the lack of enzyme specificity of caspase inhibitor drugs prevents unambiguous identification of caspases in mediating brain injury in most studies. The class of irreversible tetrapeptide caspase inhibitors covalently coupled to chloromethylketone, fluoromethylketone, or aldehydes efficiently inhibits other classes of cysteine proteases like calpains [Rozman-Pungercar et al., 2003]. Calpains, Ca2+-activated, neutral, cytosolic cysteine proteases, are highly activated following neonatal HI in rats [Ostwald et al., 1993; Blomgren et al., 2001]. MDL28170, a drug that inhibits calpains and caspase-3, exerts neuroprotective actions in the neonatal rat brain by decreasing necrosis and apoptosis [Kawamura et al., 2005] and thus may be a particularly valuable tool for the treatment of neonatal HI. Cathepsins, cysteine proteases concentrated in the lysosomal compartment, are also likely to be activated based on electron microscopy evidence of lysosomal and vacuolar changes found following neonatal HI [Martin et al., 2000]. More potent, selective, and reversible nonpeptide caspase-3 inhibitors have been developed [Han et al., 2005] and used to protect against brain injury following neonatal HI in rats [Han et al., 2002], but the protective effects were more modest compared to initial reports with nonselective pan-caspase inhibition [Cheng et al., 1998].
Not all forms of apoptotic cell death are caspase-dependent [Beresford et al., 2001; Fan et al., 2003]. The serine protease granzyme A (GrA) mediates a caspase-independent apoptotic pathway [Beresford et al., 2001]. GrA is delivered to target cells through Ca2+-dependent, perforin-generated pores, and activates a DNase (GrA-DNase, non-metastasis factor 23, NM23) that is sequestered in the cytoplasm. NM23 activity is inhibited by the SET complex, which is located in the ER and comprised of the nucleosome assembly protein SET, an inhibitor of protein phosphatase 2A, apurinic endonuclease-1, and a high-mobility group protein (a nonhistone DNA-binding protein that induces alterations in DNA architecture). GrA cleaves components of the SET complex to release activated NM23, which translocates to the nucleus to induce single-strand DNA nicks and cell death that can be apoptotic or nonapoptotic [Fan et al., 2003]. To date, there are no studies of this pathway in neonatal HI.
Inhibitor of Apoptosis Protein Family
The activity of pro-apoptotic proteins must be placed in check to prevent unwanted apoptosis in normal cells. Apoptosis is blocked by the IAP family in mammalian cells [Deveraux et al., 1998; LaCasse et al., 1998; Holcik, 2002]. This family includes X chromosome-linked IAP (XIAP), IAP1, IAP2, neuronal apoptosis inhibitory protein (NAIP), Survivin, Livin, and Apollon. These proteins are characterized by 1–3 baculoviral IAP repeat domains consisting of a zinc finger domain of approximately 70–80 amino acids [Holcik, 2002]. Apollon is a huge (530 kDa) protein that also has a ubiquitin-conjugating enzyme domain. The main identified anti-apoptotic function of IAPs is the suppression of caspase activity [Deveraux et al., 1998]. Procaspase-9 and procaspase-3 are major targets of several IAPs. IAPs reversibly interact directly with caspases to block substrate cleavage. Apollon also ubiquitylates and facilitates proteosomal degradation of active caspase-9 and Smac [Hao et al., 2004]. However, IAPs do not prevent caspase-8-induced proteolytic activation of procaspase-3. IAPs can also block apoptosis by reciprocal interactions with the nuclear transcription factor NF-κβ [LaCasse et al., 1998].
Scant information is available on IAPs in the nervous system and in neonatal brain injury. Survivin is essential for nervous system development in mouse because conditional deletion of survivin gene in neuronal precursor cells causes death shortly after birth, and reduced brain size and severe multifocal degeneration [Jiang et al., 2005]. NAIP is expressed throughout the CNS in neurons [Xu et al., 1997]. XIAP is highly enriched in mouse spinal motor neurons [Martin et al., 2007]. The importance of the IAP gene family in human pediatric neurodegeneration is underscored by the finding that NAIP is partially deleted in a significant proportion of children with spinal muscular atrophy [Roy et al., 1995]. Studies in transgenic XIAP mice indicate that XIAP plays an important role in regulating caspase activity after neonatal HI. Overexpression of XIAP virtually abolished activation of both caspase-9 and 3, and provided a 40 percent reduction in tissue loss in forebrain following neonatal HI in mouse [Wang et al., 2004].
Proteins exist that inhibit mammalian IAPs. A murine mitochondrial protein called Smac and its human ortholog, DIABLO (for direct IAP-binding protein with low pI), inactivate the anti-apoptotic actions of IAPs and thus exert pro-apoptotic actions [Du et al., 2000; Verhagen et al., 2000]. These IAP inhibitors are 23 kDa mitochondrial proteins (derived from 29 kDa precursor proteins processed in the mitochondria) that are released from the intermembrane space and sequester IAPs. High temperature requirement protein A2 (HtrA2), also called Omi, is another mitochondrial serine protease that exerts pro-apoptotic activity by inhibiting IAPs [Suzuki et al., 2004]. HtrA2/Omi functions as a homotrimeric protein that cleaves IAPs irreversibly, thus facilitating caspase activity. The intrinsic mitochondrial-mediated cell death pathway is regulated by Smac and HtrA2/Omi, and both of these proteins co-localize with XIAP in injured brain neurons after neonatal HI in mouse [Wang et al., 2004].
Apoptosis Inducing Factor
AIF is a mammalian cell mitochondrial protein identified as a flavoprotein oxidoreductase [Susin et al., 1999]. AIF has an N-terminal mitochondrial localization signal, and after import into the intermitochondrial membrane space the mitochondrial localization signal is cleaved off to generate a mature protein of 57 kDa. Under normal physiological conditions AIF might function as a ROS scavenger, targeting H2O2 [Klein et al., 2002], or in redox cycling with NAD(P)H [Mate et al., 2002]. With apoptotic stimuli (see Figure 14-3), AIF translocates to the nucleus [Susin et al., 1999]. Overexpression of AIF induces cardinal features of apoptosis, including chromatin condensation, high molecular weight DNA fragmentation, and loss of mitochondrial transmembrane potential [Susin et al., 1999]. Nuclear translocation of AIF plays a central role in neuronal loss induced by neonatal HI in mouse, particularly in males [Zhu et al., 2003, 2006, 2007]. These studies further showed that a substantial portion of the neurodegeneration following neonatal HI in mouse is caspase-independent [Zhu et al., 2003].
Cell Surface Death Receptors
Cell death by apoptosis can also be initiated at the cell membrane by surface death receptors of the tumor necrosis factor (TNF) receptor family. Fas (CD95/Apo-1) and the 75-kDa neurotrophin receptor (p75NTR) are members of the large TNF receptor family [Nagata, 1999]. The signal for apoptosis is initiated at the cell surface by aggregation (trimerization) of the death domain containing members of this receptor family by their specific ligand. Fas death receptor-mediated apoptosis is the best described of these death receptor signaling pathways. Activation of Fas is induced by binding of the multivalent Fas ligand (FasL), a member of the TNF-cytokine family. FasL is expressed on activated T cells and natural killer cells. Clustering of Fas on the target cell by FasL recruits Fas-associated death domain (FADD), a cytoplasmic adapter molecule that functions in the activation of the caspase-8–Bid pathway, thus forming the “death-induced signaling complex” (DISC) [Li et al., 1998] (see Figure 14-2). Signaling for apoptosis then proceeds via the extrinsic or intrinsic pathway. In the extrinsic pathway, active caspase-8 then directly cleaves caspase-3 [Nagata, 1999]. Activation of the mitochondrial or intrinsic pathway proceeds via caspase-8-mediated cleavage of cytosolic Bid (a pro-apoptotic Bcl2 family member) [Li et al., 1998] (see Figure 14-2). The truncated form of Bid then translocates to mitochondria, thereby functioning as a BH3-only transducer of Fas activation signal at the cell plasma membrane to mitochondria [Li et al., 1998]. Bid translocation from the cytosol to mitochondrial membranes is associated with a conformational change in Bax (that is prevented by Bcl-2 and Bcl-xL), and is accompanied by release of cytochrome c from mitochondria [Desagher et al., 1999]. Apoptosis through Fas is independent of new RNA or protein synthesis. Evidence for the importance of these signaling pathways in experimental neonatal brain injury is growing. Activation of multiple components of the Fas death receptor signaling pathway have been found in neonatal rat and mouse models of HI and hyperoxic brain injury [Northington et al., 2001a; Graham et al., 2004; Dzietko et al., 2008], and blocking Fas death receptor signaling by either pharmacologic or genetic means affords protection in these models [Feng et al., 2003a; Graham et al., 2004; Dzietko et al., 2008]. Concurrent with activation of Fas death receptors, levels of Bax in mitochondrial-enriched cell fractions increase, and cytochrome c accumulates in the soluble protein compartment. Increased levels of Fas death receptor and Bax, cytochrome c accumulation, and activation of caspase-8 precede the marked activation of caspase-3 and the occurrence of neuronal apoptosis in the thalamus in neonatal rat HI [Northington et al., 2001a]. This thalamic neuronal apoptosis in the neonatal rat brain after HI is identical structurally to the apoptosis of thalamic neurons after cortical trauma [Natale et al., 2002]. HI in the neonatal rat causes severe infarction of cerebral cortex [Northington et al., 2001b], and it is assumed that this thalamic neuron apoptosis is caused by target deprivation via a Fas-dependent pathway.
Apoptosis can also be mediated by p75NTR [Troy et al., 2002]. Activation of p75NTR occurs through binding of nerve growth factor. When p75NTR is activated without Trk receptors, neurotrophin binding induces homodimer formation and activates an apoptotic cascade. Activation of p75NTR leads to the generation of ceramide through sphingomyelin hydrolysis. Ceramide production is associated with the activation of Jun N-terminal kinase (JNK), which phosphorylates and activates c-Jun and other transcription factors. p75 mediates hippocampal neuron death in response to neurotrophin withdrawal, involving cytochrome c, Apaf1, and caspases-9, 6, and 3 (but not caspase-8), and thus is different from Fas-mediated cell death [Troy et al., 2002].
Information on the role of death receptor activation in human and experimental neonatal brain injury, although relatively sparse, has been recently reviewed [Northington et al., 2007]. Inflammatory cytokines, especially TNFα, clearly play a role in the pathogenesis of ischemic brain injury in the human newborn [Nelson et al., 1998; Savman et al., 1998; Shalak et al., 2002; Bartha et al., 2004], suggesting that understanding death receptor signaling pathways is critical. Furthermore, activation of Fas death receptor pathways has been found in hydrocephalus, hemorrhagic and focal ischemic brain injury, and following neonatal asphyxia in human newborns [Felderhoff-Mueser et al., 2001, 2003; Mehmet, 2001; Van Landeghem et al., 2002]. The importance of these pathways in human neonatal brain injury may become even more prominent as the contributions of programmed necrosis and on-going/delayed inflammation to initiation and propagation of injury in the developing brain are understood.
p53 Family of Tumor Suppressors
Cell death by apoptosis can be triggered by DNA damage. p53 and related DNA binding proteins identified as p73 and p63 are involved in this process [Levreroi et al., 2000]. p53, p73, and p63 function in apoptosis or growth arrest and repair. They can commit to death cells that have sustained DNA damage from ROS, irradiation, and other genotoxic stresses [Levrero et al., 2000]. p53 and p73 have similar oligomerization and DNA sequence transactivation properties. p73 exists as a group of full-length isoforms (including p73α and p73β), and as truncated isoforms that lack the transactivation domain ( N-p73). p53 is the best-studied of this family of proteins.
N-p73). p53 is the best-studied of this family of proteins.
p53 is a short-lived protein with a half-life of approximately 5–20 min in most types of cells studied. p53 rapidly accumulates several-fold in response to DNA damage. This rapid regulation is mediated by post-translational modification such as phosphorylation and acetylation, as well as intracellular redox state [Giaccia and Kastan, 1998]. The elevation in p53 protein levels occurs through stabilization and prevention of degradation that is under partial control of hypoxia-inducible factor-1α [Calvert et al., 2006; Fan et al., 2009]. p53 is degraded rapidly in a ubiquitination-dependent proteosomal pathway [Maki et al., 1996; Chang et al., 1998]. Murine double minute 2 (Mdm2; the human homolog is Hdm2) has a crucial role in this degradation pathway [Shieh et al., 1997]. Mdm2 functions in a feedback loop to limit the duration or magnitude of the p53 response to DNA damage. Expression of the Mdm2 gene is controlled by p53 [Shieh et al., 1997]. Mdm2 binds to the N-terminal transcriptional activation domain of p53 and regulates its DNA binding activity and stability by direct association. Mdm2 has ubiquitin ligase activity for p53 through the ubiquitin-conjugating enzyme E2. Stabilization of p53 is achieved through phosphorylation of serine15, resulting in inhibition of formation of Mdm2–p53 complexes. Activated p53 binds the promoters of several genes encoding proteins associated with growth control and cell cycle checkpoints (e.g., p21, Gadd45, Mdm2) and apoptosis (e.g., Bax, Bcl-2, Bcl-xL, and Fas). The BH3-only proteins Puma and Noxa are critical mediators of p53-mediated apoptosis [Villunger et al., 2003].
p53 and p73 regulate neuronal cell survival. p53 has a critical apoptotic role in cultured sympathetic ganglion neurons in response to neurotrophin withdrawal [Aloyz et al., 1998]. p53 deficiency protects against neuronal apoptosis induced by axotomy and target deprivation in vivo [Martin et al., 2001; Martin and Liu, 2002]. p53-mediated neuronal apoptosis can be blocked by the 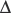 N-p73 isoform by direct binding and inactivation of p53 [Pozniak et al., 2000]. Little is known about the roles of p53 in neurodegeneration following neonatal HI, except that p53 levels increase [Calvert et al., 2006], and acute inhibition of NF-κβ following neonatal HI prevents both nuclear and mitochondrial accumulation of p53 and provides significant sustained neuroprotection [Nijboer et al., 2008].
N-p73 isoform by direct binding and inactivation of p53 [Pozniak et al., 2000]. Little is known about the roles of p53 in neurodegeneration following neonatal HI, except that p53 levels increase [Calvert et al., 2006], and acute inhibition of NF-κβ following neonatal HI prevents both nuclear and mitochondrial accumulation of p53 and provides significant sustained neuroprotection [Nijboer et al., 2008].
Excitotoxic Cell Death
Neuronal death can be induced by excitotoxicity. This observation was made originally in 1957 [Lucas and Newhouse, 1957], formulated into a concept by John Olney after showing that glutamate can kill neurons in brain [Olney, 1971], and then examined mechanistically by Dennis Choi [Choi, 1992]. This concept has fundamental importance to a variety of acute neurological insults, such as cerebral HI, epilepsy, trauma, and hypoxia [Delivoria-Papadopoulos and Mishra, 1998; Martin et al., 1998, 2000; Martin, 2003; Johnston, 2005]. This pathologic neurodegeneration is mediated by excessive activation of glutamate-gated ion channel receptors and voltage-dependent ion channels. Increased cytosolic free Ca2+ causes activation of Ca2+-sensitive proteases, protein kinases/phosphatases, phospholipases, and NOS when glutamate receptors are stimulated. The excessive interaction of ligand with subtypes of glutamate receptors causes pathophysiological changes in intracellular ion concentrations, pH, protein phosphorylation, and energy metabolism [Choi, 1992; Lipton and Rosenberg, 1994]. The precise mechanisms of excitotoxic cell death are still being examined intensively, driven by the hope of identifying therapeutic targets for neurological disorders with putative excitotoxic components. Yet, in vitro and in vivo experimental data are discordant with regard to whether excitotoxic neuronal death is apoptotic or necrotic, or perhaps even a peculiar form of cell death that is unique to excitotoxicity.
The contribution of apoptotic mechanisms to excitotoxic death of neurons has been examined in cultured neurons. However, these studies provide conflicting results. Excitotoxicity can cause activation of endonucleases and specific internucleosomal DNA fragmentation in cultures of cortical neurons [Kure et al., 1991; Gwag et al., 1997] and cerebellar granule cells [Ankarcrona et al., 1995; Simonian et al., 1996]. Internucleosomal fragmentation of DNA was not seen in other studies of cerebellar granule cell cultures [Dessi et al., 1993]. Excitotoxic cell death in neuronal cultures is prevented [Kure et al., 1991] or unaffected [Dessi et al., 1993; Simonian et al., 1996; Gwag et al., 1997] by inhibitors of RNA or protein synthesis, and sensitive [Kure et al., 1991; Simonian et al., 1996] or insensitive [Dessi et al., 1993] to the endonuclease inhibitor aurintricarboxylic acid. In primary cultures of mouse cortical cells, the non-N-methyl-d-aspartic acid (NMDA) glutamate receptor agonist kainic acid (KA) induces increases in Bax protein, and bax gene deficiency significantly protects cells against KA receptor toxicity [Xiang et al., 1998]. However, NMDA receptor toxicity in mouse cerebellar granule cells [Miller et al., 1997] and mouse cortical cells [Dargusch et al., 2001] was not Bax-related. These results support our expectation that non-NMDA glutamate receptor excitotoxicity is more likely than NMDA receptor-mediated excitotoxicity to induce apoptosis or continuum cell death [Portera-Cailliau et al., 1997a, b]. Glutamate (100 μM) stimulation of mouse cortical cells did not cause an increase in caspase activity [Johnson et al., 1999], but NMDA-treated rat cortical cells showed increased caspase activity [Tenneti and Lipton, 2000]. In cerebellar granule neurons, glutamate (100 μM to 1 mM) did not activate caspase activity, and adenoviral-mediated expression of IAPs did not influence excitotoxic cell death [Simons et al., 1999]. These conflicting results can also be related to the finding that activation of different subtypes of glutamate receptors appears to activate different modes of cell death [Portera-Cailliau et al., 1997a, b].
The morphological characteristics of excitotoxicity in many neurons in vivo include somatodendritic swelling, mitochondrial damage, and chromatin condensation into irregular clumps [Olney, 1971; van Lookeren Campagne et al., 1995; Portera-Cailliau et al., 1997a, b], features that are thought to be typical of cellular necrosis; however, in other neurons, excitotoxicity causes cytological features more like apoptosis [van Lookeren Campagne et al., 1995; Portera-Cailliau et al., 1997a, b]. Excitotoxic degeneration of CA3 neurons in response to KA is increased in NAIP-deleted mice, further supporting a contribution of apoptosis [Holcik et al., 2000]. Excitotoxic neurodegeneration in vivo has been shown to be either sensitive [Schreiber et al., 1993] or insensitive [Leppin et al., 1992] to protein synthesis inhibition; therefore, a role for de novo protein synthesis in the expression of a PCD cascade in excitotoxicity is uncertain.
The precise mechanisms of excitotoxic neuronal apoptosis in vivo have not been identified specifically. Neurons in the immature rodent CNS undergo massive apoptosis in response to glutamate receptor excitotoxicity [Portera-Cailliau et al., 1997a, b]. Apoptosis is much more prominent after excitotoxic injury in the immature brain compared to the mature brain [Portera-Cailliau et al., 1997a, b]. Intrastriatal administration of KA in newborn rodents causes copious apoptosis of striatal neurons [Ginsberg and Busto, 1989; Portera-Cailliau et al., 1997a, b], serving as an unequivocal model of apoptosis in neurons that are selectively vulnerable in HIE. This apoptosis has been verified structurally with light microscopy and electron microscopy, and by immunolocalization of cleaved caspase-3 [Lok and Martin, 2002]. Ubiquitous apoptosis is observed at 24 hours after the insult. DNA degradation by internucleosomal fragmentation further confirms the presence of apoptosis. Excitotoxic neuronal apoptosis is associated with rapid (within 2 hours after neurotoxin exposure) translocation of Bax and cleaved caspase-3 to mitochondria [Lok and Martin, 2002]. Moreover, this study revealed that the ratio of mitochondrial membrane-associated Bax to soluble Bax in normal developing striatum changes prominently with brain maturation. Newborn rat striatum has a much greater proportion of Bax in the mitochondrial fraction, with lower levels of soluble Bax. Mature rat striatum has a much larger proportion of Bax in the soluble fraction and low amounts of Bax in the mitochondrial fraction. With brain maturation there is a linear decrease in the ratio of mitochondrial Bax to soluble Bax. This developmental subcellular redistribution of Bax might be a reason why immature rodent neurons exhibit a more robust classical apoptosis response compared to adult neurons after brain damage [Martin, 2001].
The therapeutic potential of NMDA receptor blockade for neonatal HI and excitotoxic insults, in rat, was initially reported by McDonald et al. [McDonald et al., 1987]. MK-801 (dizocilpine, (5R,10S)-(+)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate), a noncompetitive antagonist of NMDA receptors, provided near-complete neuroprotection but only when given as an immediate pretreatment and during the insult [McDonald et al., 1987]. Pretreatment with MK-801 also protected against hippocampal neurodegeneration and improved performance in memory tasks following neonatal HI [Ford et al., 1989]. These studies were followed by evidence that MK-801, given immediately prior to excitotoxin injection, completely blocks NMDA and partially blocks quisqualic acid excitotoxicity in neonatal rats [McDonald et al., 1990b], and blocks both NMDA-mediated lesions and associated hyperemia in the newborn lamb [Taylor et al., 1995]. Although, at low doses, MK-801 is highly effective as a neuroprotectant in neonatal HI [Hagberg et al., 1994], it is not a therapeutically useful agent because it exacerbates injury when given 24 hours prior to HI [McDonald et al., 1990a], and even brief exposure causes massive apoptosis in the developing brain when given at p7 to the rat in the absence of an acute injury [Ikonomidou et al., 1999]. This neuroprotection/neurodevelopmental apoptosis paradox also extends to other NMDA receptor antagonists, including magnesium, which provides neuroprotection to the p7 rat receiving intrastriatal NMDA but results in widespread neuronal apoptosis when given at p3 and p7 to naïve uninjured mice [McDonald et al., 1990; Dribben et al., 2009].
Other attempts to interrupt NMDA excitotoxicity circumvent direct NMDA receptor blockade or use novel pharmacologic agents unrelated to MK-801. Xenon, an NMDA receptor antagonist, provides both in vivo and in vitro neuroprotection against neonatal HI in the rat and oxygen-glucose deprivation in neuronal-glial co-culture experiments, and these effects are accentuated by dexmedetomidine [Rajakumaraswamy et al., 2006]. Memantine, a low-affinity voltage-dependent channel NMDA receptor antagonist, shows promise in the neonatal rat HI model, in vitro in organotypic slices and neuronal culture models [Volbracht et al., 2006], and most recently in the neonatal rat HI model modified to produce a more selective white matter injury [Manning et al., 2008]. Unlike MK-801, memantine appears to permit neuronal signaling, which may be crucial to prevent the neurodevelopmental apoptosis caused by MK-801 [Volbracht et al., 2006]. Other potentially useful targets for neuroprotection after neonatal HI include non-NMDA glutamate receptors and sodium channels.
The Cell Death Continuum
Using animal models of neurodegeneration, we discovered that cell death exists as a continuum of necrosis and apoptosis at opposite ends of a degenerative spectrum; numerous hybrid forms of degeneration manifest between necrosis and apoptosis (see Figure 14-1) [Portera-Cailliau et al., 1997a, b; Martin et al., 1998]. The age or maturity of brain, and the subtype of excitatory glutamate receptor that is activated influence the mode and speed of neuronal cell death [Portera-Cailliau et al., 1997a, b; Martin, 2001; Natale et al., 2002] (Figure 14-4). This structural and temporal diversity of neuronal cell death is seen with a variety of brain injuries, including excitotoxicity, HI, target deprivation, and axonal trauma. Biochemical evidence for the existence of an intermediate “continuum” form of cell death is verified by the coexpression of markers for both apoptosis and necrosis in neurons in the injured forebrain at 3 hours following HI in neonatal rat [Ostwald et al., 1993; Blomgren et al., 2001], and evidence for crosstalk between calpain and caspase pathways as a fundamental mechanism contributing to “continuum” cell death is found [Blomgren et al., 2001]. The significance of this finding is evident from the demonstration that caspase-3 inhibition provides complete blockade of caspase activation but only partial neuroprotection. Caspase-3 inhibitors fail to prevent the necrotic mode of cell death induced by HI, as revealed by the presence of necrosis markers, and, thus, the forebrain still sustains significant injury [Han et al., 2002].
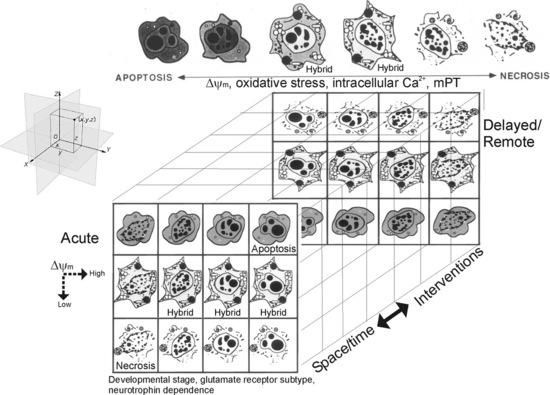
Fig. 14-4 Cell death matrix.
(Adapted from Martin, LJ. Excitotoxicity. Encyclopedia of Neuroscience, CD-ROM. G Adelman and BH Smith (eds). Elsevier, Philadelphia, PA 2004.)
This diversity of cell death is one of the fundamental differences in injury-associated neuronal death in immature and mature CNS, manifesting much more often in the injured immature brain [Northington et al., 2007]. Additionally, cell death is clearly pleiomorphic in neurons within the same brain [Sheldon et al., 2001; Blomgren et al., 2007; Northington et al., 2007]. To help explain these data we formulated the concept of the cell death continuum (see Figure 14-1). A fundamental cornerstone of the continuum is thought to be gradations in the responses of cells to stress. Some specific mechanisms thought to be driving the continuum are the developmental expression of different subtypes of glutamate receptors, mitochondrial energetics (see Figure 14-3), the propinquity of developing neurons to the cell cycle, neurotrophin requirements, DNA damage vulnerability, and the degree of axonal collateralization [Martin, 2001, 2002]. Although the molecular mechanisms that drive this cell death continuum in the brain are currently uncertain, cell culture data hint that ATP levels [Leist et al., 1997], intracellular Ca2+ levels [Trump and Berezesky, 1996], level of caspase activity [Lin et al., 1999], and mitochondrial permeability transition [Crompton, 1999] could be involved (see Figure 14-4). In vivo experiments so far suggest that the relative level of Bax in the outer mitochondrial membrane could regulate the cell death continuum in neurons [Lok and Martin, 2002] (see Figure 14-3). In the newborn brain with its proclivity to apoptotic cell death, the concept that energy failure interrupts and prevents successful completion of apoptosis with a resultant increase in necrotic cell death forms is an appealing explanation for the abundant expression of biochemical markers of apoptosis and the paucity of evidence for complete execution of apoptosis [Northington et al., 2007]. This suggests that the concept of the cell death continuum is particularly pertinent to neuron degeneration in the immature brain (see Figure 14-1), although it might be applicable to cytopathology in general.
Recent data now suggest that, under certain conditions and perhaps in specific brain regions, autophagy is also part of the complex continuum of neurodegeneration following neonatal HI in mice [Ginet et al., 2009]. Pharmacologic inhibition of autophagy prior to neonatal HI worsens injury and shifts the cell death continuum from apoptosis to necrosis in p7 rats [Carloni et al., 2008]. One possible explanation for these results is that autophagy may play an important role in maintaining cellular energy stores at adequate levels soon after a severe HI insult, and thus allows for successful completion of apoptosis. Clearly, autophagocytic and apoptotic markers are often coexpressed following neonatal HI [Ginet et al., 2009]. Early on, without the energy savings provided for by induction of autophagocytic mechanisms, ATP levels may fall below the critical threshold required for completion of apoptosis, and forms of necrosis ensue. Conversely, inhibition of autophagy appears to be neuroprotective at later time points and in less severe insults [Puyal et al., 2009]. In these settings, maintenance of cellular energy may not be as critical and autophagocytic mechanisms may be activated in stressed but possibly still viable neurons. Crosstalk between autophagic and apoptotic pathways may then result in the cells ultimately being eliminated by apoptotic mechanisms [Maiuri et al., 2007]. Under these conditions, inhibition of autophagy would provide much earlier and perhaps more effective interruption of cell death pathways. It remains to be seen whether inhibiting delayed autophagy provides functional benefits. Moreover, it is clearly evident that timing and energy state are critical variables in any attempt to modulate autophagy following neonatal HI.
Despite the mounting evidence for the cell death continuum, the concept has been challenged and deemed confusing by some investigators [Ishimaru et al., 1999; Fujikawa, 2000; Sloviter, 2002]. Opponents of the cell death continuum assume that morphology and underlying biochemical processes remain binary and discrete [Fujikawa, 2000]. While this is the case at the extremes of the cell death continuum, absolute discreteness ignores the observable features of cell degeneration seen in the injured and diseased CNS, particularly in the immature brain (see Figure 14-1). Experiments done by us [Martin, 2001; Northington et al., 2005] and others [Sheldon et al., 2001; Wei et al., 2004; Baille et al., 2005; Zhu et al., 2005] have shown that neuronal degeneration triggered by excitotoxicity and HI can be apoptotic, apoptosis-necrosis hybrids, necrotic, and possibly autophagocytic-apoptosis hybrids. Rigid conceptualization regarding cellular pathology is not realistic and is misleading, and can hinder our goal of the identification of relevant molecular mechanisms in complex biological systems, especially in the injured perinatal brain, and ultimately limit the realization of therapeutic opportunities. This view is now supported by recommendations of the Nomenclature Committee on Cell Death 2009 [Kroemer et al., 2009]. In comments reminiscent of the above reasoning for discarding the rigid/nonoverlapping conceptual framework of neuronal cell death, the primary conclusion, of the panel addressing cell death in general, is that, if there is no clear equivalence between morphology and biochemistry, then rigid morphologic classifications are not useful. Conversely, cell death nomenclature is most useful when it predicts the possibilities of pharmacologic or genetic modulation of cell death or when it predicts the consequences of cell death in vivo [Kroemer et al., 2009].
Programmed Cell Necrosis
Forms of neurodegeneration in the developing brain similar to “continuum” cell death have been previously described and termed “pathological apoptosis” [Blomgren et al., 2007] and excitotoxic neurodegeneration [Ishimaru et al., 1999]. In vitro, similar hybrid forms of neurodegeneration are termed necroptosis, programmed necrosis, paraptosis, oncosis, and aponecrosis [Degterev et al., 2008]. The important contribution of these regulated but morphologically hybrid forms of cell death to neurodegeneration and stroke has recently been extensively reviewed [Yuan et al., 2003; Festjens et al., 2006; Bredesen, 2008; Henriquez et al., 2008], but their importance in neonatal brain injury is just now emerging [Blomgren et al., 2007]. In 2007, we identified the morphologic hallmark of delayed-ongoing neurodegeneration in the neonatal brain as the “apoptotic–necrotic continuum cell” [Northington et al., 2007]. After its initial description by Portera-Cailliau et al. [Portera-Cailliau et al., 1997a, b], continuum cell and similar hybrid forms of neurodegeneration have been identified in neonatal animal models of excitotoxicity and HI [Northington et al., 2001b; Sheldon et al., 2001; Blomgren et al., 2007] and in cell culture models [Formigli et al., 2000]. “Continuum” cell neurodegeneration may fundamentally result from failure to complete caspase-dependent apoptotic cell death following neonatal HI [Northington et al., 2007] (see Figure 14-2). The abundance of biochemical evidence for activation of caspase-dependent pathways following neonatal HI [Nakajima et al., 2000; Blomgren et al., 2001; Northington et al., 2001a; Han et al., 2002; Feng et al., 2003a; Feng et al., 2003b; Zhu et al., 2006], and the paucity of evidence that apoptosis is fully executed following neonatal HI [Ishimaru et al., 1999; Martin et al., 2000; Blomgren et al., 2007; Northington et al., 2007], lend credence to this argument.
A recently described small-molecule inhibitor of programmed cell necrosis will likely shed light on mechanisms relevant to these regulated hybrid forms of cell death. Necrostatin was first described as a specific antagonist of programmed cell necrosis or necroptosis in cell culture models. This regulated form of “necrotic” cell death is a good example of molecular switching between modes of cell death and a “backup” mechanism generally avoided but activated when other less injurious cell death mechanisms fail [Henriquez et al., 2008]. Necroptosis is defined as “nonapoptotic” death with necrotic features, and occurs optimally when death receptors are activated in the presence of caspase inhibition [Yuan et al., 2003; Degterev et al., 2005]. The death domain containing kinase, RIP1, appears to be a key signaling intermediate in this programmed necrosis and is inhibited by necrostatin [Festjens et al., 2007; Degterev et al., 2008] (see Figure 14-2). Necrostatin has shown promise as a neuroprotectant in adult animal models of myocardial ischemia and traumatic and ischemic brain injury [Degterev et al., 2005; Lim et al., 2007; You et al., 2008], and we have shown that it provides robust sustained neuroprotection following neonatal HI in mice [Graham et al., 2009].
Necrostatin’s primary mechanism of action is allosteric inhibition of RIP1 kinase [Degterev et al., 2008] with possible downstream anti-inflammatory effects [You et al., 2008] and perhaps other mechanisms. Depending on the experimental conditions, RIP1 functions as the crucial adaptor kinase at the crossroads of a death receptor-stimulated cell’s decision to live or die, and also at a critical juncture determining whether programmed cell apoptosis or necrosis is predominant after death receptor activation [Lin et al., 1999; Festjens et al., 2007]. RIP1 is one of seven known members of a family of serine/threonine kinases, and is constitutively expressed in many tissues, including the brain [Festjens et al., 2007]. Importantly, RIP1 contains a caspase recruitment domain and a death domain within its C-terminal region. It is the death domain that allows RIP1 to be recruited to death receptor-initiated protein complexes, which then propagate either prosurvival, proinflammatory, pro-apoptotic, or pronecrotic signals [Festjens et al., 2007] (see Figure 14-2). RIP1-mediated activation of the regulated necrosis-signaling pathway preferentially occurs in the presence of caspase inhibition [Shen and Pervaiz, 2006; Festjens et al., 2007]. Inhibition of endogenous caspase-8-mediated cleavage of RIP1 is essential for maximal execution of death receptor-mediated programmed necrosis. Caspase inhibition can also occur in the setting of significant energy failure [Eguchi et al., 1997; Leist et al., 1997, 1999; Leist and Jaattela, 2001], such as that which occurs following neonatal HI (see Figure 14-2). It has been hypothesized that this energy failure interrupts the neonatal brain’s proclivity to apoptosis [Leist et al., 1999; Leist and Jaattela, 2001; Blomgren et al., 2007; Northington et al., 2007] and results in the hybrid, continuum cell death morphology.
Although necrostatin has no known direct antioxidant effect, and does not prevent cell death in vitro caused by hydrogen peroxide, it clearly modulates redox mechanisms in experimental systems [Xu et al., 2007]. Necrostatin inhibition of glutamate excitotoxicity occurs with an increase in glutathione levels and a decrease in ROS production [Xu et al., 2007]. It remains controversial, but necrostatin may also delay opening of mitochondrial permeability transition pores (see Figure 14-3) [Lim et al., 2007; Smith et al., 2007], or block the reduction in mitochondrial membrane potentials caused by excitotoxic stimuli [Hsu et al., 2009]. Furthermore, RIP1 is an essential component of the complex formed by TRADD, RIP1, and Rac1 upon death receptor activation [Shen et al., 2004] (see Figure 14-2). This complex then activates membrane-bound NOX1 NADPH oxidase to produce O2– [Kim et al., 2007]. Superoxide radical formation is critical to TNF and Fas-induced necrotic cell death in L929 (fibrosarcoma cells), and is potentiated in the presence of pan-caspase inhibition, the exact setting in which necrostatin has maximal effect to block cell death [Vercammen et al., 1998; Shen and Pervaiz, 2006].
Necrostatin appears to be a useful tool to direct therapy at “continuum” and other hybrid forms of neurodegeneration, and may allow improved understanding of some of the complex interactions and ordering of key components of death receptor-mediated injury mechanisms that result from HI injury to the immature brain. Importantly, it may additionally clarify the contribution of hybrid or continuum forms of neurodegeneration to HI neonatal brain injury. This task is likely to be simplified by the recent identification of the genes most important to the cellular signaling network that regulates necroptosis and the molecular bifurcation that controls the choice of apoptotic vs hybrid forms of cell death [Hitomi et al., 2008].
The Cell Death Matrix
Studies show that the morphologic appearance of the dying cell is a valuable tool for providing hints about the biochemical and molecular events responsible for cell death [Portera-Cailliau et al., 1997]. When studying mechanisms of cell death in human disease and in animal/cell models of disease, we believe that it is helpful to embrace the idea that apoptosis, necrosis, autophagy, and nonapoptotic PCD are not strictly “black and white.” For the nervous system, this complexity is overlaid with cell death mechanisms that are influenced by brain maturity, capacities for protein/RNA synthesis and DNA repair, antioxidant status, neurotrophin requirements, location in brain, and location relative to the primary sites of injury, as well as intensity of the insult. These factors that influence nervous system damage, at least in animal models, can make the pathobiology of perinatal HIE seem to abandon strict certainty and causality, thus yielding a neuropathology that is probabilistic and uncertain.
To help organize neurodegeneration and discover laws that determine causes and effects in neurodegenerative settings, the concept of the cell death continuum was extended to a hypothetical cell death matrix to embrace the “fuzziness” and spatiotemporal dynamics of cell death in the injured CNS (see Figure 14-4). A matrix might be a useful tool for pathology in general, and specifically for delineating and mathematically modeling the contributions and temporal/spatial emergence of the different forms of cell death, and the possible identification and prediction of previously unrecognized forms of cell death in human neurological disorders and in their animal/cell models. Our cell death matrix draws on the framework of biological space/time. It integrates space (location in brain, location of primary insult) and time into a continuum; thus, cell death manifests in a brain regional three-dimensional context, with time playing the role of a fourth dimension that is of a different context than the spatial dimension. By combining space and time into a single matrix we can potentially organize a large number of cell death phenotypes and mechanisms into a manageable frame of reference to reveal the potential early and delayed responses of the brain to injury.
A cell death matrix could also be useful for modeling how drugs and other treatments can influence outcomes. We need to identify better the relationships between mechanisms of cell death and the structure of dying cells in human pathology, in developing and adult CNS, as well as in animal and cell models of neurotoxicity in undifferentiated immature and terminally differentiated cells. It must be emphasized strongly that much more work needs to be done on the pathobiology of human HIE to define its cell death types better. To help with this necessity, perinatal intensivists and pediatric neurologists must encourage autopsy. The concept of a cell death matrix could be important for understanding neuronal degeneration in a variety of pathophysiological settings, and thus may be important for mechanism-based neuroprotective treatments in neurological disorders in infants, children, and adults. If brain maturity and brain location dictate how and when neurons die relative to the insult [Martin et al., 1998; Martin, 2001], then the molecular mechanisms responsible for neuronal degeneration in different brain regions (and at different times after the injury) in infants and children might be different from the mechanisms of neuronal degeneration in adults; hence, therapeutic targets will differ, and, thus, therapies will need to be customized for different brain regions, postinsult time, and age groups.
Effects of Hypothermia on Neuronal Cell Death Following HIE
Hypothermia is the only effective method of neuroprotection currently available for the clinical treatment of neonatal HIE [Gunn et al., 1998; Gluckman et al., 2005; Shankaran et al., 2005; Thoresen and Whitelaw, 2005; Jacobs et al., 2007; Azzopardi et al., 2009]. The neuroprotective clinical effects appear to derive from the ability of hypothermia to interrupt cell death cascades at multiple levels in non-neuronal and neuronal cells [Thoresen and Whitelaw, 2005; Zhao et al., 2007]. Specifically, hypothermia results in a reduction in cerebral metabolism that slows cell depolarization, prevents ATP depletion, reduces accumulation of excitotoxic neurotransmitters, preserves blood–brain barrier integrity, suppresses oxygen and nitrogen free radical release and lipid peroxidation of cell membranes, blocks postischemic inflammation by suppressing microglial activation and cytokine release, and may effect gene and protein expression in a positive manner [Mitani and Kataoka, 1991; Thoresen et al., 1997; Gunn and Bennet, 2002; Colbourne et al., 2003; Erecinska et al., 2003; Zhao et al., 2007; Gonzalez and Ferriero, 2008; Polderman, 2009]. The effect of cooling on non-neuronal cells in the brain is also likely important to its overall efficacy. Endothelial cells are a front-line target for reperfusion injury, and in vitro hypothermia blocks activation of both extrinsic and intrinsic apoptosis cascades, suppresses Fas-mediated caspase-8 activation, returns levels of Bax and Bcl-2 to control levels, blocks expression of cleaved caspase-3, and inhibits activation of JNK1/2 in endothelial cells [Yang et al., 2009]. A full review of the effects of hypothermia on non-neuronal cells is beyond the scope of this chapter; however, these new data suggest that many additional studies of the effect of hypothermia on non-neuronal cell death in the neonatal brain are needed.
Despite the effects of hypothermia on excitotoxic and oxygen free radical release, initial animal studies suggested that hypothermia specifically blocked apoptotic cell death but not necrotic cell death following transient HI in the neonatal piglet [Edwards et al., 1995]. Many subsequent mechanistic studies have followed this observation by focusing primarily on effects of hypothermia to block caspase-3 activation. Both intra-ischemic and postischemic hypothermia block cytochrome c release from mitochondria, caspase-3 activation and apoptotic cell death as determined with TUNEL (terminal deoxynucleotidyl transferance duTP nick end labelling) labeling [Zhu et al., 2004b, 2006], which is not specific for apoptosis [Martin et al., 1998]. Intra-ischemia hypothermia additionally blocks translocation of AIF to nuclei and supports cell survival by inhibiting dephosphorylation of Akt [Zhu et al., 2006]. Postischemic hypothermia may not have the same effect on Akt phosphorylation [Tomimatsu et al., 2001]. A combination of intra-ischemic hypothermia and pan-caspase inhibition pretreatment provides robust and additive neuroprotection in the Vannucci model [Adachi et al., 2001] suggesting that hypothermia is also blocking non-caspase-mediated cell death pathways. Recent studies now confirm that hypothermia blocks necrosis as well as apoptosis following neonatal HI in rat [Ohmura et al., 2005]. This is consistent with our findings that 24 hours of mild, whole-body hypothermia with sedation and paralysis has profound, perhaps sustained, neuroprotective effects on the HI piglet striatum where we find necrosis to be the major form of neurodegeneration and minimal evidence for apoptosis [Agnew et al., 2003]. Hypothermia-mediated striatal neuroprotection may result from blocking NMDA receptor activation and oxidative damage to proteins [Mueller-Burke et al., 2008], as well as preventing ischemia-induced downregulation of the GluR2 AMPA receptor subunit that limits Ca2+ influx [Colbourne et al., 2003].
In an important study, it was shown that experimental asphyxia causes long-lasting morphologic modification of postsynaptic densities. These damaged postsynaptic densities stained intensely for ubiquitin, and hypothermia effectively blocked both the morphological and molecular changes [Capani et al., 2009]. Whether neurons with damaged synapses are ultimately targeted for premature cell death is unknown. It is also important to note that it is not known if neurons salvaged by treatment with hypothermia are normal. We have found that neurons can exist in damaged atrophic states with few synaptic contacts for months after injury [Ginsberg and Martin, 1998]. Determining the functionality of rescued neurons will have important implications for neurologic function following neonatal HI. The panoply of neuroprotective effects of hypothermia suggests that it may work to prevent neuronal cell death along the cell death continuum and throughout our hypothetical cell death matrix.
Conclusion
It will be extremely important to combine data from biochemical studies with information on cell death structure following different degrees and types of perinatal brain injury to understand better which insults are most likely to respond to antinecrosis, anti-apoptosis, anti-autophagy, or combination therapies, and whether these therapies actually ameliorate injury or simply delay or change the mode of cell damage. Animal studies predict that apoptosis inhibitors alone will be inadequate to ameliorate most of the early brain damage following neonatal HI [Martin et al., 2000; Northington et al., 2001b], and the cell death continuum predicts that apoptosis inhibitor drugs administered at acute and perhaps delayed time points may push cell degeneration from apoptosis to apoptosis-variant or necrotic cell death, as seen in vitro with caspase inhibitors applied following chemical hypoxia [Formigli et al., 2000]. When given as a pretreatment, inhibitors of autophagy may also push neurodegeneration along the cell death continuum in a deleterious manner [Carloni et al., 2008]. No studies to date have shown this; however, inhibitors of programmed necrosis may actually afford the opportunity to push neurodegeneration from necrosis to apoptosis. In settings where postinjury inflammation contributes to delayed neurodegeneration, this may actually be beneficial. Using concepts from the cell death matrix, we predict that it will be difficult to pinpoint appropriate times for effective mechanism-based, spatially directed drug therapy, particularly for neonatal HIE. It is especially important to prepare for the possibility that pharmacological interventions directed against a single mechanism of injury might only delay, convert, or worsen the evolving brain damage associated with HIE in newborns. For this reason, hypothermia might be an ideal strategy because it appears to protect against necrosis and apoptosis [Ohmura et al., 2005], and may be working along both the cell death continuum and possibly the space/time gradient that defines the matrix of cell death. Clearly, much more experimental and clinical work needs to be done. In light of these difficulties, it is clear that combination therapies with broad neuroprotective properties, acting at early and late time points, and alternative therapeutic approaches using stem and progenitor cells should be investigated for the treatment of perinatal HIE.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Adachi M., Sohma O., et al. Combination effect of systemic hypothermia and caspase inhibitor administration against hypoxic-ischemic brain damage in neonatal rats. Pediatr Res. 2001;50(5):590-595.
Agnew D.M., Koehler R.C., et al. Hypothermia for 24 hours after asphyxic cardiac arrest in piglets provides striatal neuroprotection that is sustained 10 days after rewarming. Pediatr Res. 2003;54(2):253-262.
Aloyz R.S., Bamji S.X., et al. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol. 1998;143:1691-1703.
Ameisen J.C. On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ. 2002;9(4):367-393.
Amin F., Bowen I.D., et al. Apoptotic and non-apoptotic modes of programmed cell death in MCF-7 human breast carcinoma cells. Cell Biol Int. 2000;24(4):253-260.
Ankarcrona M., Dypbukt J.M., et al. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15(4):961-973.
Antonsson B., Conti F., et al. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277(5324):370-372.
Azzopardi D.V., Strohm B., et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med. 2009;361(14):1349-1358.
Baille V., Clarke P.G., et al. Soman-induced convulsions: the neuropathology revisited. Toxicology. 2005;215(1–2):1-24.
Baines C.P., Kaiser R.A., et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434(7033):658-662.
Bartha A.I., Foster-Barber A., et al. Neonatal encephalopathy: association of cytokines with MR spectroscopy and outcome. Pediatr Res. 2004;56(6):960-966.
Beresford P.J., Zhang D., et al. Granzyme A activates an endoplasmic reticulum-associated caspase-independent nuclease to induce single-stranded DNA nicks. J Biol Chem. 2001;276(46):43285-43293.
Blomgren K., Leist M., et al. Pathological apoptosis in the developing brain. Apoptosis. 2007;12(5):993-1010.
Blomgren K., Zhu C., et al. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J Biol Chem. 2001;276(13):10191-10198.
Bonfoco E., Krainc D., et al. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92(16):7162-7166.
Bredesen D.E. Programmed cell death mechanisms in neurological disease. Curr Mol Med. 2008;8(3):173-186.
Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8(6):569-581.
Bursch W., Paffe S., et al. Determination of the length of the histological stages of apoptosis in normal liver and in altered hepatic foci of rats. Carcinogenesis. 1990;11(5):847-853.
Calvert J.W., Cahill J., et al. Oxygen treatment after experimental hypoxia-ischemia in neonatal rats alters the expression of HIF-1alpha and its downstream target genes. J Appl Physiol. 2006;101(3):853-865.
Capani F., Saraceno G.E., et al. Protein ubiquitination in postsynaptic densities after hypoxia in rat neostriatum is blocked by hypothermia. Exp Neurol. 2009;219(2):404-413.
Cardone M.H., Roy N., et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282(5392):1318-1321.
Carloni S., Buonocore G., et al. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008;32(3):329-339.
Castro J., Ruminot I., et al. ATP steal between cation pumps: a mechanism linking Na+ influx to the onset of necrotic Ca2+ overload. Cell Death Differ. 2006;13(10):1675-1685.
Chang Y.C., Lee Y.S., et al. mdm2 and bax, downstream mediators of the p53 response, are degraded by the ubiquitin-proteasome pathway. Cell Growth Differ. 1998;9(1):79-84.
Cheng Y., Deshmukh M., et al. Caspase Inhibitor Affords Neuroprotection with Delayed Administration in a Rat Model of Neonatal Hypoxic-Ischemic Brain Injury. J Clin Invest. 1998;101(9):1992-1999.
Choi D.W. Excitotoxic cell death. J Neurobiol. 1992;23(9):1261-1276.
Clarke P.G. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl). 1990;181(3):195-213.
Colbourne F., Grooms S.Y., et al. Hypothermia rescues hippocampal CA1 neurons and attenuates down-regulation of the AMPA receptor GluR2 subunit after forebrain ischemia. Proc Natl Acad Sci USA. 2003;100(5):2906-2910.
Corbett R.J., Laptook A.R., et al. Energy reserves and utilization rates in developing brain measured in vivo by 31P and 1H nuclear magnetic resonance spectroscopy. J Cereb Blood Flow Metab. 1993;13(2):235-246.
Cory S., Adams J.M. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2(9):647-656.
Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341(Pt 2):233-249.
Crompton M., Virji S., et al. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem. 1998;258(2):729-735.
Danial N.N., Gramm C.F., et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424(6951):952-956.
Dargusch R., Piasecki D., et al. The role of Bax in glutamate-induced nerve cell death. J Neurochem. 2001;76(1):295-301.
Datta S.R., Dudek H., et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91(2):231-241.
Degterev A., Hitomi J., et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313-321.
Degterev A., Huang Z., et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112-119.
Delivoria-Papadopoulos M., Ashraf Q.M., et al. Nuclear mechanisms of hypoxic cerebral injury in the newborn: the role of caspases. Semin Perinatol. 2008;32(5):334-343.
Delivoria-Papadopoulos M., Mishra O.P. Mechanisms of cerebral injury in perinatal asphyxia and strategies for prevention. J Pediatr. 1998;132(3 Pt 2):S30-S34.
del Peso L., Gonzalez-Garcia M., et al. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278(5338):687-689.
Desagher S., Osen-Sand A., et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144(5):891-901.
Dessi F., Charriaut-Marlangue C., et al. Glutamate-induced neuronal death is not a programmed cell death in cerebellar culture. J Neurochem. 1993;60(5):1953-1955.
Deveraux Q.L., Roy N., et al. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. Embo J. 1998;17(8):2215-2223.
Dribben W.H., Creeley C.E., et al. High dose magnesium sulfate exposure induces apoptotic cell death in the developing neonatal mouse brain. Neonatology. 2009;96(1):23-32.
Du C., Fang M., et al. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102(1):33-42.
Dzietko M., Boos V., et al. A critical role for Fas/CD-95 dependent signaling pathways in the pathogenesis of hyperoxia-induced brain injury. Ann Neurol. 2008;64(6):664-673.
Edwards A.D., Yue X., et al. Specific inhibition of apoptosis after cerebral hypoxia-ischaemia by moderate post-insult hypothermia. Biochem Biophys Res Commun. 1995;217(3):1193-1199.
Eguchi Y., Shimizu S., et al. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57(10):1835-1840.
Erecinska M., Thoresen M., et al. Effects of hypothermia on energy metabolism in Mammalian central nervous system. J Cereb Blood Flow Metab. 2003;23(5):513-530.
Fan X., Heijnen C.J., et al. The role and regulation of hypoxia-inducible factor-1alpha expression in brain development and neonatal hypoxic-ischemic brain injury. Brain Res Rev. 2009.
Fan Z., Beresford P.J., et al. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell. 2003;112(5):659-672.
Felderhoff-Mueser U., Buhrer C., et al. Soluble Fas (CD95/Apo-1), soluble Fas ligand, and activated caspase 3 in the cerebrospinal fluid of infants with posthemorrhagic and nonhemorrhagic hydrocephalus. Pediatr Res. 2003;54(5):659-664.
Felderhoff-Mueser U., Herold R., et al. Increased cerebrospinal fluid concentrations of soluble Fas (CD95/Apo- 1) in hydrocephalus. Arch Dis Child. 2001;84(4):369-372.
Felderhoff-Mueser U., Sifringer M., et al. Pathways leading to apoptotic neurodegeneration following trauma to the developing rat brain. Neurobiol Dis. 2002;11(2):231-245.
Feng Y., Fratkin J.D., et al. Inhibiting caspase-8 after injury reduces hypoxic-ischemic brain injury in the newborn rat. Eur J Pharmacol. 2003;481(2–3):169-173.
Feng Y., Fratkin J.D., et al. Inhibiting caspase-9 after injury reduces hypoxic ischemic neuronal injury in the cortex in the newborn rat. Neurosci Lett. 2003;344(3):201-204.
Feng Y., LeBlanc M.H. Treatment of hypoxic-ischemic brain injury in newborn rats with TPCK 3 h after hypoxia decreases caspase-9 activation and improves neuropathologic outcome. Dev Neurosci. 2003;25(1):34-40.
Fernandes R.S., Cotter T.G. Apoptosis or necrosis: intracellular levels of glutathione influence mode of cell death. Biochem Pharmacol. 1994;48(4):675-681.
Ferriero D.M. Neonatal brain injury. N Engl J Med. 2004;351(19):1985-1995.
Festjens N., Vanden Berghe T., et al. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta. 2006;1757(9–10):1371-1387.
Festjens N., Vanden Berghe T., et al. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 2007;14(3):400-410.
Ford L.M., Sanberg P.R., et al. MK-801 prevents hippocampal neurodegeneration in neonatal hypoxic-ischemic rats. Arch Neurol. 1989;46(10):1090-1096.
Formigli L., Papucci L., et al. Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J Cell Physiol. 2000;182(1):41-49.
Fujikawa D.G. Confusion between neuronal apoptosis and activation of programmed cell death mechanisms in acute necrotic insults. Trends Neurosci. 2000;23(9):410-411.
Giaccia A.J., Kastan M.B. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev. 1998;12(19):2973-2983.
Gilbert S. Developmental Biology. Sunderland: Sinauer Associates; 2006.
Ginet V., Puyal J., et al. Enhancement of Autophagic Flux after Neonatal Cerebral Hypoxia-Ischemia and Its Region-Specific Relationship to Apoptotic Mechanisms. Am J Pathol. 2009.
Ginsberg M.D., Busto R. Rodent models of cerebral ischemia. Stroke. 1989;20(12):1627-1642.
Ginsberg S.D., Martin L.J. Ultrastructural analysis of the progression of neurodegeneration in the septum following fimbria-fornix transection. Neuroscience. 1998;86(4):1259-1272.
Gluckman P.D., Wyatt J.S., et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005;365(9460):663-670.
Glucksmann A. Cell deaths in normal vertebrate ontogeny. Biol Rev. 1951;26:59-86.
Golden W.C., Brambrink A.M., et al. Failure to sustain recovery of Na,K-ATPase function is a possible mechanism for striatal neurodegeneration in hypoxic-ischemic newborn piglets. Brain Res Mol Brain Res. 2001;88(1–2):94-102.
Gonzalez F.F., Ferriero D.M. Therapeutics for neonatal brain injury. Pharmacol Ther. 2008;120(1):43-53.
Graham E., Mack D.W., et al. Single dose necrostatin after neonatal hypoxia-ischemia provides prolonged neuroprotection to neocortex and hippocampus. Pediatr Res E-PAS. 2009:3215.1.
Graham E.M., Sheldon R.A., et al. Neonatal mice lacking functional Fas death receptors are resistant to hypoxic-ischemic brain injury. Neurobiol Dis. 2004;17(1):89-98.
Gulyaeva N.V. Non-apoptotic functions of caspase-3 in nervous tissue. Biochemistry (Mosc). 2003;68(11):1171-1180.
Gunn A.J., Bennet L. Cerebral Hypothermia in the Management of Hypoxic-Ischemic Encephalopathy. NeoReviews. 2002;3(6):e116.
Gunn A.J., Gluckman P.D., et al. Selective head cooling in newborn infants after perinatal asphyxia: a safety study. Pediatrics. 1998;102(4 Pt 1):885-892.
Gwag B.J., Koh J.Y., et al. Slowly triggered excitotoxicity occurs by necrosis in cortical cultures. Neuroscience. 1997;77(2):393-401.
Hagberg H., Gilland E., et al. Hypoxia-ischemia in the neonatal rat brain: histopathology after post-treatment with NMDA and non-NMDA receptor antagonists. Biol Neonate. 1994;66(4):205-213.
Ha H.C., Snyder S.H. Poly(ADP-ribose) polymerase-1 in the nervous system. Neurobiol Dis. 2000;7(4):225-239.
Haldar S., Jena N., et al. Inactivation of Bcl-2 by phosphorylation. Proc Natl Acad Sci USA. 1995;92(10):4507-4511.
Hallin U., Kondo E., et al. Bcl-2 phosphorylation in the BH4 domain precedes caspase-3 activation and cell death after neonatal cerebral hypoxic-ischemic injury. Neurobiol Dis. 2006;21(3):478-486.
Han B.H., Xu D., et al. Selective, reversible caspase-3 inhibitor is neuroprotective and reveals distinct pathways of cell death after neonatal hypoxic-ischemic brain injury. J Biol Chem. 2002;277(33):30128-30136.
Han Y., Giroux A., et al. Novel pyrazinone mono-amides as potent and reversible caspase-3 inhibitors. Bioorg Med Chem Lett. 2005;15(4):1173-1180.
Hao Y., Sekine K., et al. Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat Cell Biol. 2004;6(9):849-860.
Hegde R., Srinivasula S.M., et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J Biol Chem. 2002;277(1):432-438.
Henriquez M., Armisen R., et al. Cell death by necrosis, a regulated way to go. Curr Mol Med. 2008;8(3):187-206.
Hitomi J., Christofferson D.E., et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135(7):1311-1323.
Holcik M. The IAP proteins. Trends Genet. 2002;18(10):537.
Holcik M., Thompson C.S., et al. The hippocampal neurons of neuronal apoptosis inhibitory protein 1 (NAIP1)-deleted mice display increased vulnerability to kainic acid-induced injury. Proc Natl Acad Sci USA. 2000;97(5):2286-2290.
Hsu T.S., Yang P.M., et al. Attenuation of cadmium-induced necrotic cell death by necrostatin-1: potential necrostatin-1 acting sites. Toxicol Appl Pharmacol. 2009;235(2):153-162.
Hu B.R., Liu C.L., et al. Involvement of caspase-3 in cell death after hypoxia-ischemia declines during brain maturation. J Cereb Blood Flow Metab. 2000;20(9):1294-1300.
Hu Y., Benedict M.A., et al. Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc Natl Acad Sci USA. 1998;95(8):4386-4391.
Ikonomidou C., Bosch F., et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283(5398):70-74.
Inbal B., Bialik S., et al. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol. 2002;157(3):455-468.
Ishimaru M.J., Ikonomidou C., et al. Distinguishing excitotoxic from apoptotic neurodegeneration in the developing rat brain. J Comp Neurol. 1999;408(4):461-476.
Jacobson M. Developmental Neurobiology. New York: Plenum Press; 1991.
Jacobs S., Hunt R., et al. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev. (4):2007. CD003311
Jiang Y., de Bruin A., et al. Essential role for survivin in early brain development. J Neurosci. 2005;25(30):6962-6970.
Johnson M.D., Kinoshita Y., et al. Contribution of p53-dependent caspase activation to neuronal cell death declines with neuronal maturation. J Neurosci. 1999;19:2996-3006.
Johnston M.V. Excitotoxicity in perinatal brain injury. Brain Pathol. 2005;15(3):234-240.
Johnston M.V. Plasticity in the developing brain: implications for rehabilitation. Dev Disabil Res Rev. 2009;15(2):94-101.
Kawamura M., Nakajima W., et al. Calpain inhibitor MDL 28170 protects hypoxic-ischemic brain injury in neonatal rats by inhibition of both apoptosis and necrosis. Brain Res. 2005;1037(1–2):59-69.
Kerr J.F., Wyllie A.H., et al. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239-257.
Kim Y.S., Morgan M.J., et al. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell. 2007;26(5):675-687.
Klein J.A., Longo-Guess C.M., et al. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419(6905):367-374.
Klionsky D.J., Emr S.D. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717-1721.
Kluck R.M., Bossy-Wetzel E., et al. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275(5303):1132-1136.
Koike M., Shibata M., et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172(2):454-469.
Kostovic I., Jovanov-Milosevic N. The development of cerebral connections during the first 20–45 weeks’ gestation. Semin Fetal Neonatal Med. 2006;11(6):415-422.
Kroemer G., Galluzzi L., et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3-11.
Kure S., Tominaga T., et al. Glutamate triggers internucleosomal DNA cleavage in neuronal cells. Biochem Biophys Res Commun. 1991;179(1):39-45.
LaCasse E.C., Baird S., et al. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene. 1998;17(25):3247-3259.
Leist M., Jaattela M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2(8):589-598.
Leist M., Single B., et al. Inhibition of mitochondrial ATP generation by nitric oxide switches apoptosis to necrosis. Exp Cell Res. 1999;249(2):396-403.
Leist M., Single B., et al. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185(8):1481-1486.
Lennon S.V., Martin S.J., et al. Dose-dependent induction of apoptosis in human tumour cell lines by widely diverging stimuli. Cell Prolif. 1991;24(2):203-214.
Leppin C., Finiels-Marlier F., et al. Failure of a protein synthesis inhibitor to modify glutamate receptor-mediated neurotoxicity in vivo. Brain Res. 1992;581(1):168-170.
Lesuisse C., Martin L.J. Immature and mature cortical neurons engage different apoptotic mechanisms involving caspase-3 and the mitogen-activated protein kinase pathway. J Cereb Blood Flow Metab. 2002;22(8):935-950.
Letai A., Bassik M.C., et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2(3):183-192.
Levrero M., De Laurenzi V., et al. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci. 2000;113(Pt 10):1661-1670.
Li H., Zhu H., et al. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491-501.
Li P., Nijhawan D., et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479-489.
Liang X.H., Kleeman L.K., et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72(11):8586-8596.
Lim S.Y., Davidson S.M., et al. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc Drugs Ther. 2007;21(6):467-469.
Lin Y., Devin A., et al. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13(19):2514-2526.
Lipton S.A., Rosenberg P.A. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330(9):613-622.
Lithgow T., van Driel R., et al. The protein product of the oncogene bcl-2 is a component of the nuclear envelope, the endoplasmic reticulum, and the outer mitochondrial membrane. Cell Growth Differ. 1994;5(4):411-417.
Liu X., Kim C.N., et al. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86(1):147-157.
Liu X., Zou H., et al. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89(2):175-184.
Lockshin R.A., Williams C.M. Programmed cell death: II. J Insect Physiol. 1964;10:643-649.
Lockshin R.A., Zakeri Z. Caspase-independent cell deaths. Curr Opin Cell Biol. 2002;14(6):727-733.
Lockshin R.A., Zakeri Z. Programmed cell death and apoptosis: origins of the theory. Nat Rev Mol Cell Biol. 2001;2(7):545-550.
Lockshin R.A., Zakeri Z. Programmed cell death: early changes in metamorphosing cells. Biochem Cell Biol. 1994;72(11–12):589-596.
Lok J., Martin L.J. Rapid subcellular redistribution of Bax precedes caspase-3 and endonuclease activation during excitotoxic neuronal apoptosis in rat brain. J Neurotrauma. 2002;19(7):815-828.
Lu C., Fu W., et al. Direct cleavage of AMPA receptor subunit GluR1 and suppression of AMPA currents by caspase-3: implications for synaptic plasticity and excitotoxic neuronal death. Neuromolecular Med. 2002;1(1):69-79.
Lucas D.R., Newhouse J.P. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol. 1957;58(2):193-201.
Maiuri M.C., Zalckvar E., et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741-752.
Majno G., Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146(1):3-15.
Maki C.G., Huibregtse J.M., et al. In vivo ubiquitination and proteasome-mediated degradation of p53(1). Cancer Res. 1996;56(11):2649-2654.
Manning S.M., Talos D.M., et al. NMDA receptor blockade with memantine attenuates white matter injury in a rat model of periventricular leukomalacia. J Neurosci. 2008;28(26):6670-6678.
Martin L.J. Excitotoxicity. Encyclopedia of Neuroscience. In: Adelman G., H B., editors. CD-ROM. Smith: Elsevier Press, 2004.
Martin L.J. Mechanisms of brain damage in animals models of hypoxia-ischemia in newborns. In: Stevenson D.K., Benitz W.E., Sunshine P., editors. Fetal and Neonatal Brain Injury: Mechanisms, Management, and the Risks of Practice. New York: Oxford University Press; 2003:30-57.
Martin L.J. Neuronal cell death in nervous system development, disease, and injury (Review). Int J Mol Med. 2001;7(5):455-478.
Martin L.J. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol. 1999;58(5):459-471.
Martin L.J. The apoptosis-necrosis cell death continuum in CNS development, injury and disease: contributions and mechanisms. In: Lo E.H., Marwah J., editors. Neuroprotection. Prominent Press; 2002:379-412.
Martin L.J. The mitochondrial permeability transition pore: A molecular target for amyotrophic lateral sclerosis therapy. Biochim Biophys Acta. 2009.
Martin L.J., Al-Abdulla N.A., et al. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis. Brain Res Bull. 1998;46(4):281-309.
Martin L.J., Brambrink A.M., et al. Neuronal death in newborn striatum after hypoxia-ischemia is necrosis and evolves with oxidative stress. Neurobiol Dis. 2000;7(3):169-191.
Martin L.J., Kaiser A., et al. Injury-induced apoptosis of neurons in adult brain is mediated by p53-dependent and p53-independent pathways and requires Bax. J Comp Neurol. 2001;433(3):299-311.
Martin L.J., Liu Z. Injury-induced spinal motor neuron apoptosis is preceded by DNA single- strand breaks and is p53- and Bax-dependent. J Neurobiol. 2002;50(3):181-197.
Martin L.J., Liu Z., et al. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J Comp Neurol. 2007;500(1):20-46.
Martin L.J., Portera-Cailliau C., et al. Animal models and degenerative disorders of the human brain. Lab Anim (NY). 1998;27(10):18-25.
Martin L.J., Price A.C., et al. Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J Neurochem. 2003;85(1):234-247.
Martin L.J., Sieber F.E., et al. Apoptosis and necrosis occur in separate neuronal populations in hippocampus and cerebellum after ischemia and are associated with differential alterations in metabotropic glutamate receptor signaling pathways. J Cereb Blood Flow Metab. 2000;20(1):153-167.
Mate M.J., Ortiz-Lombardia M., et al. The crystal structure of the mouse apoptosis-inducing factor AIF. Nat Struct Biol. 2002;9(6):442-446.
McDonald J.W., Silverstein F.S., et al. Magnesium reduces N-methyl-D-aspartate (NMDA)-mediated brain injury in perinatal rats. Neurosci Lett. 1990;109(1–2):234-238.
McDonald J.W., Silverstein F.S., et al. MK-801 pretreatment enhances N-methyl-D-aspartate-mediated brain injury and increases brain N-methyl-D-aspartate recognition site binding in rats. Neuroscience. 1990;38(1):103-113.
McDonald J.W., Silverstein F.S., et al. MK-801 protects the neonatal brain from hypoxic-ischemic damage. Eur J Pharmacol. 1987;140(3):359-361.
McDonald J.W., Silverstein F.S., et al. Systemic administration of MK-801 protects against N-methyl-D-aspartate- and quisqualate-mediated neurotoxicity in perinatal rats. Neuroscience. 1990;36(3):589-599.
Mehmet H. Stroke treatment enters the Fas lane. Cell Death Differ. 2001;8(7):659-661.
Metzstein M.M., Stanfield G.M., et al. Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet. 1998;14(10):410-416.
Miller T.M., Moulder K.L., et al. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139(1):205-217.
Mitani A., Kataoka K. Critical levels of extracellular glutamate mediating gerbil hippocampal delayed neuronal death during hypothermia: brain microdialysis study. Neuroscience. 1991;42(3):661-670.
Mizushima N., Ohsumi Y., et al. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27(6):421-429.
Muchmore S.W., Sattler M., et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381(6580):335-341.
Mueller-Burke D., Koehler R.C., et al. Rapid NMDA receptor phosphorylation and oxidative stress precede striatal neurodegeneration after hypoxic ischemia in newborn piglets and are attenuated with hypothermia. Int J Dev Neurosci. 2008;26(1):67-76.
Murakami Y., Aizu-Yokota E., et al. Suppression of endoplasmic reticulum stress-induced caspase activation and cell death by the overexpression of Bcl-xL or Bcl-2. J Biochem. 2007;141(3):401-410.
Nagata S. Fas ligand-induced apoptosis. Annu Rev Genet. 1999;33:29-55.
Nakagawa T., Shimizu S., et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434(7033):652-658.
Nakajima W., Ishida A., et al. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci. 2000;20(21):7994-8004.
Natale J.E., Cheng Y., et al. Thalamic neuron apoptosis emerges rapidly after cortical damage in immature mice. Neuroscience. 2002;112(3):665-676.
Nechushtan A., Smith C.L., et al. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J Cell Biol. 2001;153(6):1265-1276.
Nelson K.B., Dambrosia J.M., et al. Neonatal cytokines and coagulation factors in children with cerebral palsy. Ann Neurol. 1998;44(4):665-675.
Ness J.M., Harvey C.A., et al. Selective involvement of BH3-only Bcl-2 family members Bim and Bad in neonatal hypoxia-ischemia. Brain Res. 2006;1099(1):150-159.
Nijboer C.H., Heijnen C.J., et al. Strong neuroprotection by inhibition of NF-kappaB after neonatal hypoxia-ischemia involves apoptotic mechanisms but is independent of cytokines. Stroke. 2008;39(7):2129-2137.
Northington F.J., Ferriero D.M., et al. Delayed neurodegeneration in neonatal rat thalamus after hypoxia-ischemia is apoptosis. J Neurosci. 2001;21(6):1931-1938.
Northington F.J., Ferriero D.M., et al. Early neurodegeneration after hypoxia-ischemia in neonatal rat Is necrosis while delayed neuronal death Is apoptosis. Neurobiol Dis. 2001;8(2):207-219.
Northington F.J., Ferriero D.M., et al. TNFRSF death receptor activation and signaling following neonatal brain injury. In: Mishra O.P., editor. Mechanisms of Hypoxic Brain Injury in the Newborn and Potential Strategies for Neuroprotection. Kerala: Transworld Research Network, 2007.
Northington F.J., Graham E.M., et al. Apoptosis in perinatal hypoxic-ischemic brain injury: How important is it and should it be inhibited? Brain Res Brain Res Rev. 2005;50(2):244-257.
Northington F.J., Zelaya M.E., et al. Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as continuum phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience. 2007;149(4):822-833.
Ogier-Denis E., Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta. 2003;1603(2):113-128.
Ohmura A., Nakajima W., et al. Prolonged hypothermia protects neonatal rat brain against hypoxic-ischemia by reducing both apoptosis and necrosis. Brain Dev. 2005;27(7):517-526.
Olney J.W. Glutamate-induced neuronal necrosis in the infant mouse hypothalamus. An electron microscopic study. J Neuropathol Exp Neurol. 1971;30(1):75-90.
Orrenius S., Zhivotovsky B., et al. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4(7):552-565.
Ostwald K., Hagberg H., et al. Upregulation of calpain activity in neonatal rat brain after hypoxic-ischemia. Brain Res. 1993;630(1–2):289-294.
Pilar G., Landmesser L. Ultrastructural differences during embryonic cell death in normal and peripherally deprived ciliary ganglia. J Cell Biol. 1976;68(2):339-356.
Polderman K.H. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. 2009;37(Suppl 7):S186-S202.
Portera-Cailliau C., Price D.L., et al. Excitoxic Neuronal Death in the Immature Brain Is an Apoptosis-Necrosis Morphological Continuum. J Comp Neurol. 1997;378:70-87.
Portera-Cailliau C., Price D.L., et al. Non-NMDA and NMDA Receptor-Mediated Excitoxic Neuronal Deaths in Adult Brain Are Morphologically Distinct: Further Evidence for an Apoptosis-Necrosis Continuum. J Comp Neurol. 1997;378:88-104.
Pozniak C.D., Radinovic S., et al. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289(5477):304-306.
Proskuryakov S.Y., Konoplyannikov A.G., et al. Necrosis: a specific form of programmed cell death? Exp Cell Res. 2003;283(1):1-16.
Puka-Sundvall M., Gajkowska B., et al. Subcellular distribution of calcium and ultrastructural changes after cerebral hypoxia-ischemia in immature rats. Brain Res Dev Brain Res. 2000;125(1–2):31-41.
Puyal J., Vaslin A., et al. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66(3):378-389.
Raffray M., Cohen G.M. Apoptosis and necrosis in toxicology: a continuum or distinct modes of cell death? Pharmacol Ther. 1997;75(3):153-177.
Rajakumaraswamy N., Ma D., et al. Neuroprotective interaction produced by xenon and dexmedetomidine on in vitro and in vivo neuronal injury models. Neurosci Lett. 2006;409(2):128-133.
Robertson J.D., Enoksson M., et al. Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J Biol Chem. 2002;277(33):29803-29809.
Roy N., Mahadevan M.S., et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell. 1995;80(1):167-178.
Rozman-Pungercar J., Kopitar-Jerala N., et al. Inhibition of papain-like cysteine proteases and legumain by caspase-specific inhibitors: when reaction mechanism is more important than specificity. Cell Death Differ. 2003;10(8):881-888.
Sakahira H., Enari M., et al. Apoptotic nuclear morphological change without DNA fragmentation. Curr Biol. 1999;9(10):543-546.
Saunders J.W.Jr. Death in embryonic systems. Science. 1966;154(749):604-612.
Savman K., Blennow M., et al. Cytokine response in cerebrospinal fluid after birth asphyxia. Pediatr Res. 1998;43(6):746-751.
Schreiber S.S., Tocco G., et al. Cycloheximide prevents kainate-induced neuronal death and c-fos expression in adult rat brain. J Mol Neurosci. 1993;4(3):149-159.
Schwartz L.M., Milligan C.E. Cold thoughts of death: the role of ICE proteases in neuronal cell death. Trends Neurosci. 1996;19(12):555-562.
Schwartz L.M., Smith S.W., et al. Do all programmed cell deaths occur via apoptosis? Proc Natl Acad Sci USA. 1993;90(3):980-984.
Schweichel J.U., Merker H.J. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7(3):253-266.
Scorrano L., Oakes S.A., et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300(5616):135-139.
Shalak L.F., Laptook A.R., et al. Clinical chorioamnionitis, elevated cytokines, and brain injury in term infants. Pediatrics. 2002;110(4):673-680.
Shankaran S., Laptook A.R., et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353(15):1574-1584.
Sheldon R.A., Hall J.J., et al. Delayed cell death in neonatal mouse hippocampus from hypoxia-ischemia is neither apoptotic nor necrotic. Neurosci Lett. 2001;304(3):165-168.
Shen H.M., Lin Y., et al. Essential roles of receptor-interacting protein and TRAF2 in oxidative stress-induced cell death. Mol Cell Biol. 2004;24(13):5914-5922.
Shen H.M., Pervaiz S. TNF receptor superfamily-induced cell death: redox-dependent execution. Faseb J. 2006;20(10):1589-1598.
Shieh S.Y., Ikeda M., et al. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91(3):325-334.
Shimizu S., Ide T., et al. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J Biol Chem. 2000;275(16):12321-12325.
Shimohama S., Fujimoto S., et al. Differential expression of rat brain bcl-2 family proteins in development and aging. Biochem Biophys Res Commun. 1998;252(1):92-96.
Simonian N.A., Getz R.L., et al. Kainate induces apoptosis in neurons. Neuroscience. 1996;74(3):675-683.
Simons M., Beinroth S., et al. Adenovirus-mediated gene transfer of inhibitors of apoptosis protein delays apoptosis in cerebellar granule neurons. J Neurochem. 1999;72(1):292-301.
Sloviter R.S. Apoptosis: a guide for the perplexed. Trends Pharmacol Sci. 2002;23(1):19-24.
Smith C.C., Davidson S.M., et al. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc Drugs Ther. 2007;21(4):227-233.
Song Q., Kuang Y., et al. Boo, a novel negative regulator of cell death, interacts with Apaf-1. Embo J. 1999;18(1):167-178.
Stennicke H.R., Deveraux Q.L., et al. Caspase-9 can be activated without proteolytic processing. J Biol Chem. 1999;274(13):8359-8362.
Susin S.A., Lorenzo H.K., et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397(6718):441-446.
Suzuki Y., Takahashi-Niki K., et al. Mitochondrial protease Omi/HtrA2 enhances caspase activation through multiple pathways. Cell Death Differ. 2004;11(2):208-216.
Tata J.R. Requirement for RNA and protein synthesis for induced regression of the tadpole tail in organ culture. Dev Biol. 1966;13(1):77-94.
Taylor G.A., Trescher W.H., et al. Experimental neuronal injury in the newborn lamb: a comparison of N-methyl-D-aspartic acid receptor blockade and nitric oxide synthesis inhibition on lesion size and cerebral hyperemia. Pediatr Res. 1995;38(5):644-651.
Tenneti L., Lipton S.A. Involvement of activated caspase-3-like proteases in N-methyl-D-aspartate-induced apoptosis in cerebrocortical neurons. J Neurochem. 2000;74(1):134-142.
Thoresen M., Satas S., et al. Post-hypoxic hypothermia reduces cerebrocortical release of NO and excitotoxins. Neuroreport. 1997;8(15):3359-3362.
Thoresen M., Whitelaw A. Therapeutic hypothermia for hypoxic-ischaemic encephalopathy in the newborn infant. Curr Opin Neurol. 2005;18(2):111-116.
Tomimatsu T., Fukuda H., et al. Effects of hypothermia on neonatal hypoxic-ischemic brain injury in the rat: phosphorylation of Akt, activation of caspase-3-like protease. Neurosci Lett. 2001;312(1):21-24.
Troy C.M., Friedman J.E., et al. Mechanisms of p75-mediated death of hippocampal neurons. Role of caspases. J Biol Chem. 2002;277(37):34295-34302.
Trump B.F., Berezesky I.K. The role of altered [Ca2+]i regulation in apoptosis, oncosis, and necrosis. Biochim Biophys Acta. 1996;1313(3):173-178.
Trump B.F., Goldblatt P.J., et al. Studies on Necrosis of Mouse Liver in Vitro. Ultrastructural Alterations in the Mitochondria of Hepatic Parenchymal Cells. Lab Invest. 1965;14:343-371.
van Bekkum D. The effect of x-rays on phosphorylations in vivo. Biochim Biophys Acta. 1957;25:487-492.
Vander Heiden M.G., Chandel N.S., et al. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria [see comments]. Cell. 1997;91(5):627-637.
Vande Velde C., Cizeau J., et al. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20(15):5454-5468.
van Gurp M., Festjens N., et al. Mitochondrial intermembrane proteins in cell death. Biochem Biophys Res Commun. 2003;304(3):487-497.
Van Landeghem F.K., Felderhoff-Mueser U., et al. Fas (CD95/Apo-1)/Fas Ligand Expression in Neonates with Pontosubicular Neuron Necrosis. Pediatr Res. 2002;51(2):129-135.
van Lookeren Campagne M., Lucassen P.J., et al. NMDA and kainate induce internucleosomal DNA cleavage associated with both apoptotic and necrotic cell death in the neonatal rat brain. Eur J Neurosci. 1995;7(7):1627-1640.
Vercammen D., Brouckaert G., et al. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188(5):919-930.
Verhagen A.M., Ekert P.G., et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102(1):43-53.
Vexler Z.S., Yenari M.A. Does inflammation after stroke affect the developing brain differently than adult brain? Dev Neurosci. 2009;31(5):378-393.
Villunger A., Michalak E.M., et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302(5647):1036-1038.
Volbracht C., van Beek J., et al. Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur J Neurosci. 2006;23(10):2611-2622.
Wang H.G., Pathan N., et al. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284(5412):339-343.
Wang H.G., Rapp U.R., et al. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996;87(4):629-638.
Wang X., Carlsson Y., et al. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J Neurosci. 2009;29(8):2588-2596.
Wang X., Zhu C., et al. X-linked inhibitor of apoptosis (XIAP) protein protects against caspase activation and tissue loss after neonatal hypoxia-ischemia. Neurobiol Dis. 2004;16(1):179-189.
Wei L., Ying D.J., et al. Necrosis, apoptosis and hybrid death in the cortex and thalamus after barrel cortex ischemia in rats. Brain Res. 2004;1022(1–2):54-61.
Wei M.C., Zong W.X., et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727-730.
Wolf B.B., Green D.R. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J Biol Chem. 1999;274(29):20049-20052.
Wolter K.G., Hsu Y.T., et al. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139(5):1281-1292.
Wyllie A.H. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284(5756):555-556.
Wyllie A.H., Kerr J.F., et al. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251-306.
Xiang H., Kinoshita Y., et al. Bax involvement in p53-mediated neuronal cell death. J Neurosci. 1998;18(4):1363-1373.
Xu D.G., Korneluk R.G., et al. Distribution of neuronal apoptosis inhibitory protein-like immunoreactivity in the rat central nervous system. J Comp Neurol. 1997;382(2):247-259.
Xue L., Fletcher G.C., et al. Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol Cell Neurosci. 1999;14(3):180-198.
Xu X., Chua C.C., et al. Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J Neurochem. 2007;103(5):2004-2014.
Yang D., Guo S., et al. Hypothermia attenuates ischemia/reperfusion-induced endothelial cell apoptosis via alterations in apoptotic pathways and JNK signaling. FEBS Lett. 2009;583(15):2500-2506.
Yang E., Zha J., et al. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80(2):285-291.
Yang J., Liu X., et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275(5303):1129-1132.
Yan X.X., Najbauer J., et al. Expression of active caspase-3 in mitotic and postmitotic cells of the rat forebrain. J Comp Neurol. 2001;433(1):4-22.
You Z., Savitz S.I., et al. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28(9):1564-1573.
Yuan J., Lipinski M., et al. Diversity in the mechanisms of neuronal cell death. Neuron. 2003;40(2):401-413.
Yue Z., Horton A., et al. A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35(5):921-933.
Zha J., Harada H., et al. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell. 1996;87(4):619-628.
Zhao H., Steinberg G.K., et al. General versus specific actions of mild-moderate hypothermia in attenuating cerebral ischemic damage. J Cereb Blood Flow Metab. 2007;27(12):1879-1894.
Zhu C., Hallin U., et al. Nuclear translocation and calpain-dependent reduction of Bcl-2 after neonatal cerebral hypoxia-ischemia. Brain Behav Immun. 2009.
Zhu C., Qiu L., et al. Involvement of apoptosis-inducing factor in neuronal death after hypoxia-ischemia in the neonatal rat brain. J Neurochem. 2003;86(2):306-317.
Zhu C., Wang X., et al. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 2007;14(4):775-784.
Zhu C., Wang X., et al. Intraischemic mild hypothermia prevents neuronal cell death and tissue loss after neonatal cerebral hypoxia-ischemia. Eur J Neurosci. 2006;23(2):387-393.
Zhu C., Wang X., et al. Nitrosylation precedes caspase-3 activation and translocation of apoptosis-inducing factor in neonatal rat cerebral hypoxia-ischaemia. J Neurochem. 2004;90(2):462-471.
Zhu C., Wang X., et al. Post-ischemic hypothermia-induced tissue protection and diminished apoptosis after neonatal cerebral hypoxia-ischemia. Brain Res. 2004;996(1):67-75.
Zhu C., Wang X., et al. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005;12(2):162-176.
Zhu C., Xu F., et al. Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxia-ischaemia. J Neurochem. 2006;96(4):1016-1027.
Zong W.X., Li C., et al. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol. 2003;162(1):59-69.
Zou H., Li Y., et al. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274(17):11549-11556.
